
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular light-upconversion: we have had a problem! When excited state absorption (ESA) overcomes energy transfer upconversion (ETU) in Cr(III)/Er(III) complexes†
Bahman
Golesorkhi
*a,
Inès
Taarit
a,
Hélène
Bolvin
 *b,
Homayoun
Nozary
a,
Juan-Ramón
Jiménez
a,
Céline
Besnard
c,
Laure
Guénée
c,
Alexandre
Fürstenberg
ad and
Claude
Piguet
*a
*b,
Homayoun
Nozary
a,
Juan-Ramón
Jiménez
a,
Céline
Besnard
c,
Laure
Guénée
c,
Alexandre
Fürstenberg
ad and
Claude
Piguet
*a
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, 30 quai E. Ansermet, CH-1211 Geneva 4, Switzerland. E-mail: Bahman.Golesorkhi@berkeley.edu; Claude.Piguet@unige.ch
bLaboratoire de Chimie et Physique Quantiques, CNRS, Université Toulouse III, 118 route de Narbonne, F-31062 Toulouse, France. E-mail: bolvin@irsamc.ups-tlse.fr
cLaboratory of Crystallography, University of Geneva, 24 quai E. Ansermet, CH-1211 Geneva 4, Switzerland
dDepartment of Physical Chemistry, University of Geneva, 30 quai E. Ansermet, CH-1211 Geneva, Switzerland
First published on 20th April 2021
Abstract
Nine-coordinate [ErN9] or [ErN3O6] chromophores found in triple helical [Er(L)3]3+ complexes (L corresponds to 2,2′,6′,2′′-terpyridine (tpy), 2,6-(bisbenzimidazol-2-yl)pyridine (bzimpy), 2,6-diethylcarboxypyridine (dpa-ester) or 2,6-diethylcarboxamidopyridine (dpa-diamide) derivatives), [Er(dpa)3]3− (dpa is the 2,6-dipicolinate dianion) and [GaErGa(bpb-bzimpy)3]9+ (bpb-bzimpy is 2,6-bis((pyridin-2-benzimidazol-5-yl)methyl-(benzimidazol-2-yl))pyridine) exhibit NIR (excitation at 801 nm) into visible (emission at 542 nm) linear light upconversion processes in acetonitrile at room temperature. The associated quantum yields 5.5(6) × 10−11 ≤ ϕuptot(ESA) ≤ 1.7(2) × 10−9 appear to be 1–3 orders of magnitude larger than those predicted by the accepted single-center excited-state absorption mechanism (ESA). Switching to the alternative energy transfer upconversion mechanism (ETU), which operates in multi-centers [CrErCr(bpb-bzimpy)3]9+, leads to an improved quantum yield of ϕuptot(ETU) = 5.8(6) × 10−8, but also to an even larger discrepancy by 4–6 orders of magnitude when compared with theoretical models. All photophysical studies point to Er(4I13/2) as being the only available ‘long-lived’ (1.8 ≤ τ ≤ 6.3 μs) and emissive excited state, which works as an intermediate relay for absorbing the second photon, but with an unexpected large cross-section for an intrashell 4f → 4f electronic transition. With this in mind, the ETU mechanism, thought to optimize upconversion via intermetallic Cr → Er communication in [CrErCr(bpb-bzimpy)3]9+, is indeed not crucial and the boosted associated upconversion quantum yield is indebted to the dominant contribution of the single-center erbium ESA process. This curious phenomenon is responsible for the successful implementation of light upconversion in molecular coordination complexes under reasonable light power intensities, which paves the way for applications in medicine and biology. Its origin could be linked with the presence of metal–ligand bonding.
Introduction
Light upconversion represents a rather counter-intuitive energetic process, which was theoretically predicted in 1931 by Goeppert-Meyer1 when considering the non-linear dependence of the refractive index on light intensity (Kerr effect).2 Its experimental demonstration was delayed until the early sixties when sufficiently intense laser excitation beams became available for inducing second harmonic generation (SHG, a second-order non-linear optical (NLO) process)3 and two-photon absorption (TPA, a third-order NLO process).4 However, even for optimized polarized materials,5 these non-linear responses are so weak that NLO upconversion was found to be mainly useful for multiplying the frequency of intense laser beams. Consequently, NLO seems poorly adapted for the preparation of solar cell concentrators6 or for the design of upconverters able to transform deep penetrating low power near-infrared (NIR) beams into visible radiations of higher energy for biological or medical applications.7 The parallel discovery that light upconversion, relying strictly on successive linear optical response, is 5–8 orders of magnitude more efficient than NLO processes8 opened wide perspectives for technological applications based on (i) metal-based upconversion implemented in low-phonon ionic solids9 and (ii) triplet–triplet annihilation processes induced by the collision of two excited polyaromatic units.10 The common concept for linear light upconversion exploits a first efficient photonic excitation in order to reach long-lived intermediate excited states for energy storage prior to undergoing a second excitation (via photonic absorption or via collision), which gives finally access to an emissive excited state of higher energy. Focusing on metal-based upconversion, the second excitation process corresponds to the absorption of an additional photon with a non-negligible probability compared to the relaxation rate of the intermediate relay, a phenomenon referred to as excited state absorption (ESA).9b The scale of regularly spaced multiplets found for trivalent open-shell lanthanides (Ln3+ with electronic configurations [Xe]4fn, n = 1–13), and rationalized by the Russel–Saunders coupling scheme,11 offers a privileged access for (linear) upconversion operating within a single molecular unit as long as the lifetime of the intermediate excited state (level |1〉 in Fig. 1b) is long enough for being compatible with a reasonable competition between the absorption of a second photon kexc(1→2)A to reach the doubly excited state (level |2〉 in Fig. 1b) and the relaxation k1→0A to the ground state (level |0〉 in Fig. 1b).12,13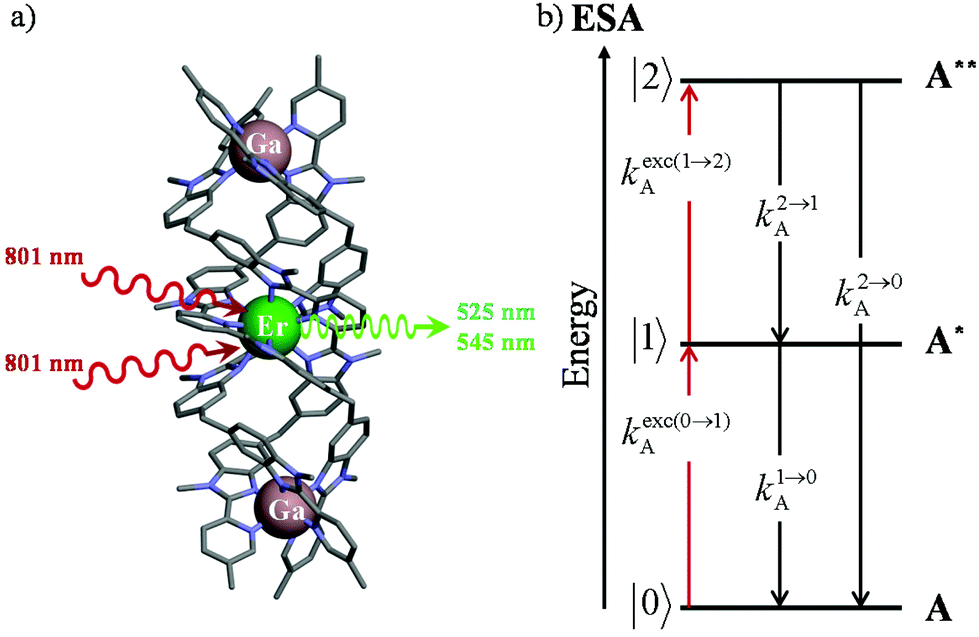 | ||
| Fig. 1 (a) Molecular structure of [GaErGa(bpb-bzimpy)3]9+ (ref. 12) and (b) associated kinetic scheme depicting the modelling of the one-ion excited state absorption (ESA) process occurring upon off-resonance irradiation into the activator-centered absorption band (A = Er) where kexc(i→j)A correspond to the excitation rate constants (eqn (1)) and ki→jA stand for the global decay rate constant of level i into level j.13 The pertinent kinetic matrix is given in Scheme S1a.† | ||
The low-energy phonons (a few tens of cm−1) available in ionic oxides and fluorides are poorly efficient for inducing non-radiative relaxation between the spectroscopic levels of lanthanide cations (separated by several hundreds/thousands of cm−1),9a–c which makes these ionic solids ideal hosts for welcoming Ln3+ as dopants with the ultimate goal of inducing efficient (linear) upconversion processes in the solid state (maximum reported quantum yields about ϕuptot = 9–12%).14 The recurrent need for miniaturizing within the frame of biological applications resulted in an intense scientific activity, which aimed at transforming Ln-doped upconverting ionic solids into nanoparticles.9d–h The unfavorable quenching due to the increase of the surface/volume ratio in the latter entities15 can be partially compensated (i) by coupling with plasmonic surfaces9f,k and/or (ii) by statistically introducing some efficient light-sensitizers16 compatible with the operation of the more efficient energy transfer upconversion (ETU) mechanism (Fig. 3b).9b,17 In this context, the design of molecular lanthanide coordination complexes for upconversion was attempted at the turn of the century by Reinhard and Güdel with a detailed photophysical investigation of Na3[Ln(dpa)3]·13H2O (Ln = Er, Tm, Yb; dpa = pyridine-2,6-dicaboxylate, see Fig. 5a).18 They concluded that the high-energy vibrations, characteristic for molecular objects, lead to intermediate metal-centered excited states with nano/microsecond lifetimes (instead of millisecond in ionic solids), which are not compatible with the detection of upconverted signals in these molecules.18 Synthetic chemists, probably unaware of this major physical deadlock, were nonetheless able to overcome this limitation, firstly with the preparation of multi-components supramolecular assemblies exhibiting light-upconversion assigned to the ETU mechanism (Fig. 3),12,19,20 secondly via the closely related cooperative upconversion (CU) mechanism21 and finally according to the basic excited state absorption pathway (ESA, Fig. 1).22 Reminiscent to the original analysis reported by Reinhard and Güdel,18 the modeling of the quantum yield for the ESA mechanism (ϕuptot in Fig. 2a) using standard experimental values for the different relaxation rate constants in a molecular Er3+ complex, as those found in [GaErGa(bpb-bzimpy)3]9+ (Fig. 1),12 indeed predicts faint quantum yields 10−13 ≤ ϕuptot ≤ 10−11 (Fig. 2b) under reasonable excitation power intensities 1 ≤ P ≤ 30 W cm−2 (Fig. 2b, the excitation rate constants kexc(i→j)A is obtained with eqn (1), where λP is the pump wavelength, P is the incident pump intensity (in W cm−2), σi→jA is the absorption cross section of the activator-centered i → j transition (in cm2) related to the decadic molar absorption coefficient εi→j (in M−1 cm−1) according to σi→j = 3.8 × 10−21εi→j,23h is the Planck constant and c is the speed of light in vacuum).24
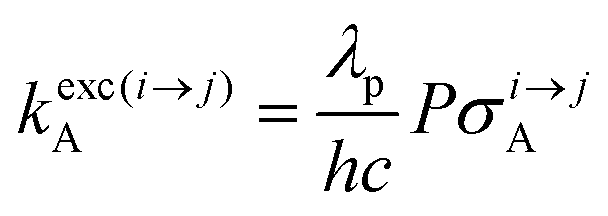 | (1) |
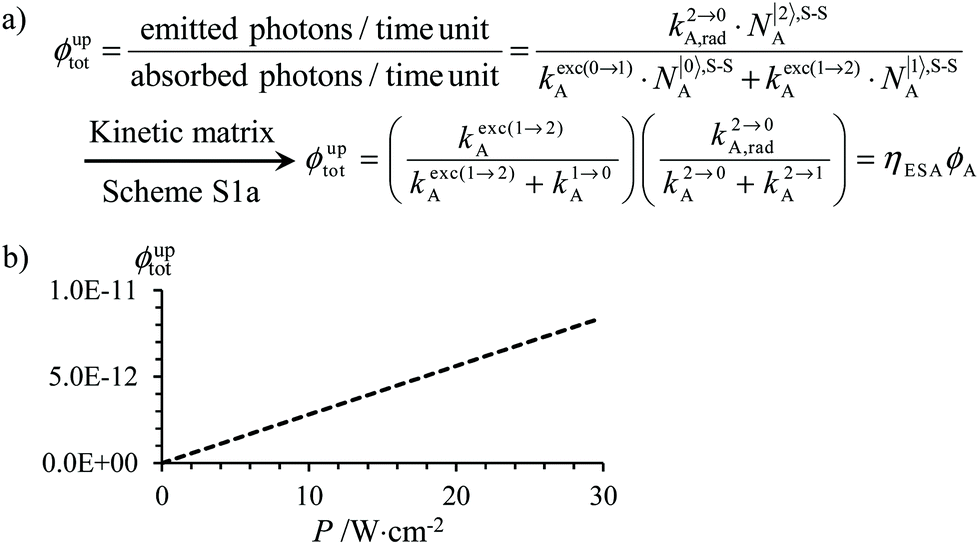 | ||
Fig. 2 (a) Definition and modeling of the global upconversion quantum yield (ϕuptot) obtained under steady-state (S-S) excitation for the ESA mechanism depicted in Fig. 1b. k2→0A, rad corresponds to the radiative decay constant, ηESA represents the efficiency of the ESA mechanism and ϕA stands for the activator-based intrinsic quantum yield. (b) Simulation of the upconversion quantum yield (ϕuptot) upon increasing incident pump intensity for the standard erbium activator found in [GaErGa(bpb-bzimpy)3]9+ at room temperature (Fig. 1a). Excitation fixed at λP = 801 nm Er(4I9/2 ← 4I15/2), absorption cross-section σ0→1A = 6.2 × 10−22 cm2 (ε801 = 0.163 M−1 cm−1), 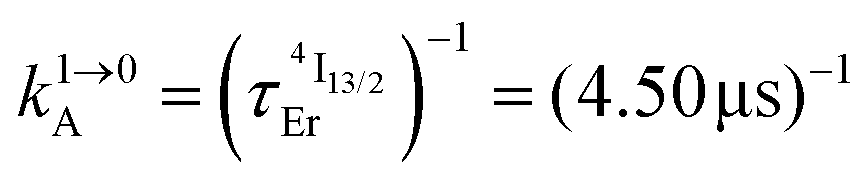 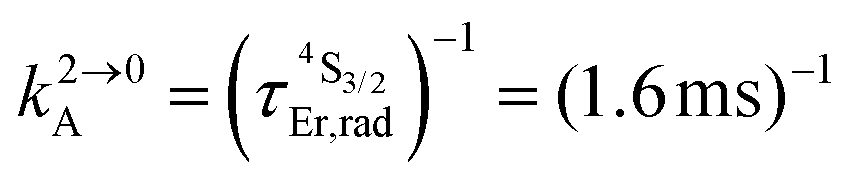 , , 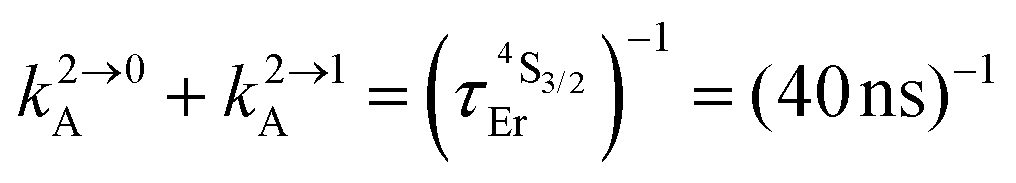 .12σ1→2A = σ0→1A = 6.2 × 10−22 cm2 (ε801 = 0.163 M−1 cm−1). σ1→2A = σ0→1A is arbitrarily (but reasonably) fixed for the simulation. .12σ1→2A = σ0→1A = 6.2 × 10−22 cm2 (ε801 = 0.163 M−1 cm−1). σ1→2A = σ0→1A is arbitrarily (but reasonably) fixed for the simulation. | ||
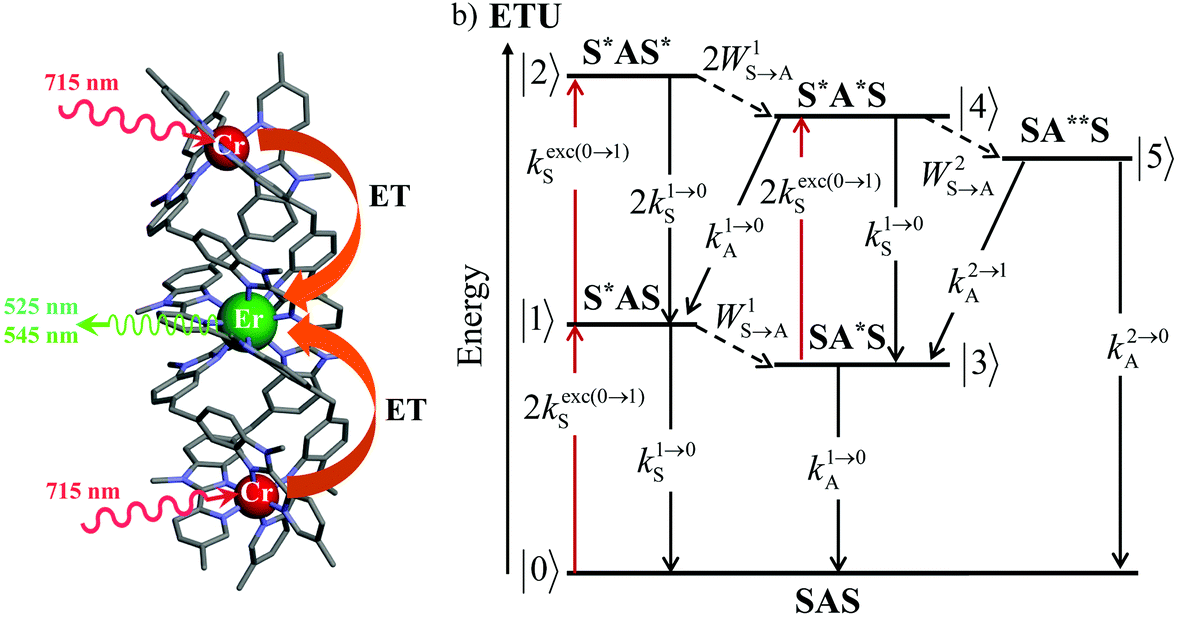 | ||
| Fig. 3 (a) Molecular structure of [CrErCr(bpb-bzimpy)3]9+ (ref. 19) and (b) associated kinetic scheme depicting the modelling of the sensitizer/activator energy transfer upconversion (ETU) process occurring upon off-resonance irradiation into the sensitizer-centered absorption band in a SAS system (S = Cr, A = Er) where kexc(0→1)S corresponds to the sensitized-based excitation rate constant (eqn (1)), ki→jS and ki→jA stand for the sensitizer-based, respectively activator-based global decay rate constants of level i into level j. WiS→A correspond to the first-order sensitizer-to-activator energy transfer (ET) rate constants.13 The pertinent kinetic matrix is given in Scheme S1b.† | ||
With these predictions in mind, only massive excitation intensities could give the lie to Reinhard and Güdel and the detection of a faint, but measurable green Er(4S3/2 → 4I15/2) upconverted signals (545 nm) from a 0.02 M solution of [Er(dpa)3]3− in D2O indeed required 109 W cm−2 (= 1 GW cm−2) near-infrared (800–980 nm) laser excitation.25 Similarly, Sorensen and Faulkner had to focus a high-power OPO tunable NIR femtosecond laser onto simple Tm3+ solvates in DMSO for inducing some weak visible luminescence, which could be unambigously assigned to second and third-order NLO responses whereas linear upconversion based on linear optics only negligibly contributed to the visible luminescence.26 Suprisingly, the few preliminar quantum yields determined experimentally for ESA occuring in mononuclear molecular erbium complexes with [ErN9] chromophores in solution lie in the 10−9 ≤ ϕuptot ≤ 10−8 range (measured for a fixed incident excitation power around 21 W cm−2)22 and appear to be 2–3 orders of magnitude larger than those predicted in Fig. 2b with the help of the accepted ESA mechanism.
The situation becomes even more critical when one considers that Charbonnière reported ϕuptot = 1.4 × 10−8 (at P = 10.3 W cm−2) for a [Tb(YbL)2] assembly dissolved in deuterated water,21a,b and recently ϕuptot = 10−7 (at P = 2.9 W cm−2) for a nonanuclear Yb8Tb cluster,21c in which only a poorly efficient cooperative energy (CU) transfer mechanism9b may explain the feeding of the high-energy emissive Tb(5D4) level. A simulation of the steady-state quantum yields expected for the ETU mechanism pertinent to upconversion implemented in [CrErCr(bpb-bzimpy)3]9+ (Cr = sensitizer, Er = activator, Fig. 4a) indeed results in negligible upconversion quantum yields at room temperature (10−15 ≤ ϕuptot ≤ 10−14, red trace in Fig. 4b), which are improved at 150 K (10−14 ≤ ϕuptot ≤ 10−12, blue trace in Fig. 4b) because the lifetime of the intermediate excited state of the chromium sensitizer increases by one order of magnitude. Again, the predicted quantum yields are much smaller (4–6 orders of magnitude) than the few pertinent experimental data reported for the less efficient CU mechanism.
 | ||
Fig. 4 (a) Definition and modeling of the global upconversion quantum yield (ϕuptot) obtained under steady-state (S-S) excitation for the ETU mechanism depicted in Fig. 3b. ηETU represents the efficiency of the ETU mechanism and ϕA stands for the activator-based intrinsic quantum yield. (b) Simulation of the upconversion quantum yield (ϕuptot) upon increasing incident pump intensity simulated for the erbium activator found in [CrErCr(bpb-bzimpy)3]9+ (Fig. 3a). Excitation fixed at λP = 718 nm Cr(2T1 ← 4A2), absorption cross-section σ0→1S = 3.84 × 10−22 cm2 (ε718 = 0.101 M−1 cm−1), 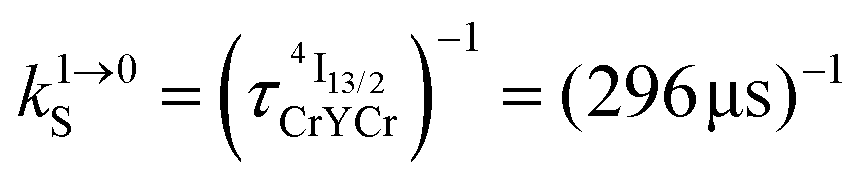 at 293 K and (2.81 ms)−1 at 150 K, at 293 K and (2.81 ms)−1 at 150 K, 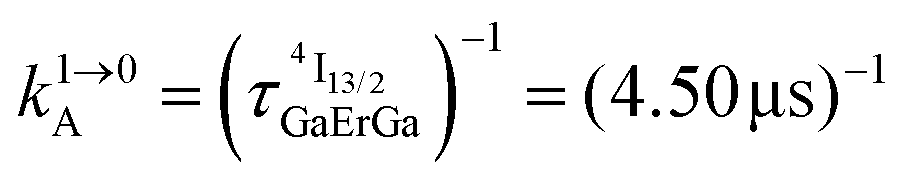 at 293 K and (4.30 μs)−1 at 150 K, at 293 K and (4.30 μs)−1 at 150 K, 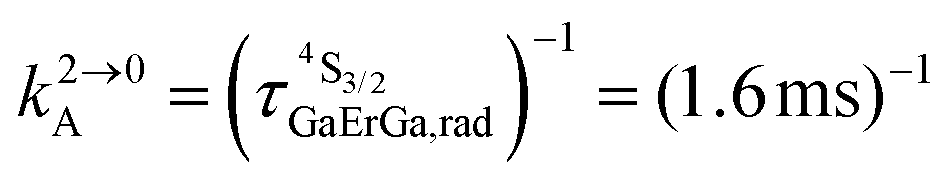 , , 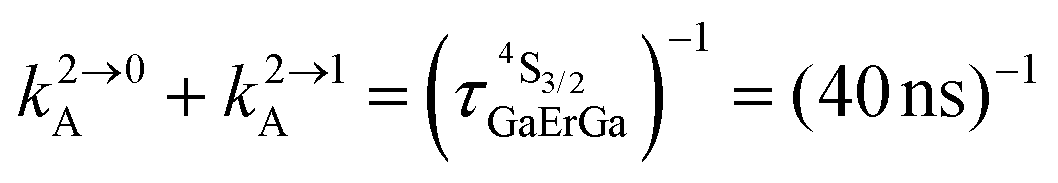 at 3 K, W1S→A = 232 s−1 at 293 K and 169 s−1 at 150 K.12W2S→A = 1000 s−1 is arbitrarily (but reasonably) fixed for the simulation. at 3 K, W1S→A = 232 s−1 at 293 K and 169 s−1 at 150 K.12W2S→A = 1000 s−1 is arbitrarily (but reasonably) fixed for the simulation. | ||
Paraphrasing astronaut Jim Lovell, who confirmed the discovery of the explosion that severely damaged the Appolo 13 spacecraft by saying “Ah, Houston, we have had a problem”, we report here our efforts for recording reliable and accurate experimental quantum yields for the ESA mechanism operating in a series of triple-helical [Er(L)3] complexes possessing nine-coordinate [ErN6O3] (Fig. 5a) and [ErN9] chromophores (Fig. 5b) with tunable crystal fields and variable protections of the erbium activator. A thorough exploration of the origin of the discrepancy between modelling and experiments is described together with some cures compatible with a pertinent rationalization of single-site ESA, but also multi-centered ETU and CU upconversion mechanisms operating in multimetallic molecules and metallosupramolecular assemblies.
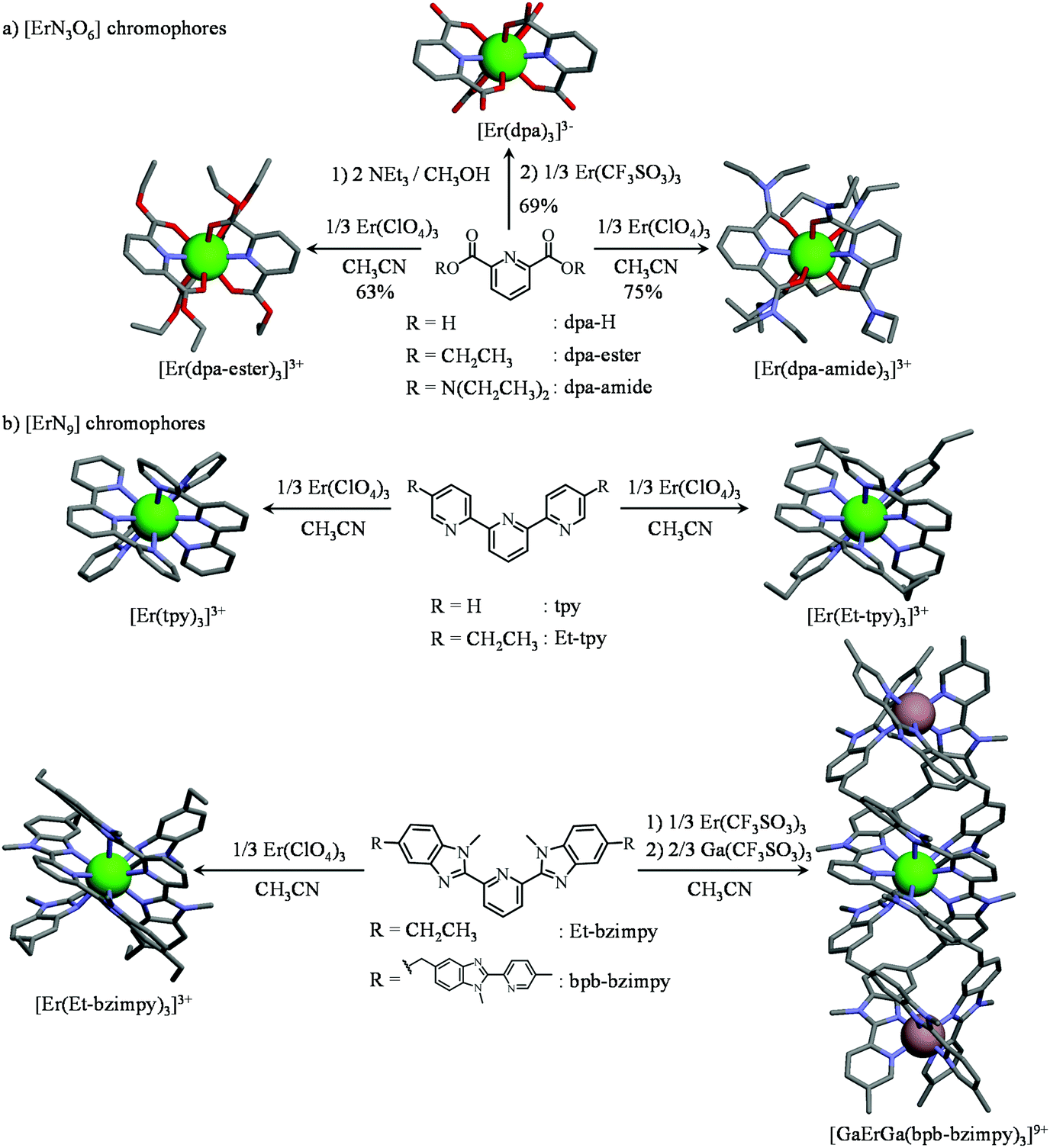 | ||
| Fig. 5 Synthesis and molecular structures of the triple-helical complexes possessing (a) [ErN6O3] and (b) [ErN9] chromophores considered in this work. The molecular structures are taken from the crystal structures of (NHEt3)5[Er(dpa)3](CF3SO3)2 (1), [Er(dpa-ester)3](ClO4)3 (2), [Er(dpa-diamide)3](ClO4)3 (3), [Er(Et-bzimpy)3](ClO4)3·2CH3CN (4),28 [Er(tpy)3](ClO4)3 (5),28 [Er(Et-tpy)3](ClO4)3·1.5CH3CN28 and [GaErGa(bpb-bzimpy)3]2(CF3SO3)18·30C3H5N.12 Green sphere = Er(III), greyisch-red sphere = Ga(III). | ||
Results and discussion
Synthesis, molecular structures, crystal field parameters and ‘phonon bath’ in triple-helical erbium complexes
According to (i) the considerable cumulative thermodynamic stability constants measured for the formation of triple-helical [Er(dpa)3]3− in water (log(βEr, L1,3) = 22.13)27 and for [Er(L)3]3+ in acetonitrile (L = dpa-ester with log(βEr,L1,3) = 17.3,29L = dpa-amide with log(βEr,L1,3) ≈ log(βY,L1,3) = 22.7,30L = tpy with log(βEr,L1,3) = 22.5,28L = Et-tpy with log(βEr,L1,3) = 21.8,28L = Et-bzimpy with log(βEr,L1,3) = 26;28Fig. 5) and (ii) the extreme kinetic inertness of [GaErGa(bpb-bzimpy)3]9+,31 we conclude that all these complexes (Fig. S1a†), except [Er(dpa-diester)3]3+ (Fig. S1b†), are quantitatively formed in solution (>99%) for |Er|tot/|L|tot = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and total ligand concentrations of 3–10 mM, the specific conditions that are used for recording the photophysical studies. For the less stable triple-helical [Er(dpa-diester)3]3+ complex, its speciation corresponds to more than 80% of the total ligand content in the same conditions (Fig. S1b†). These complexes can be also isolated in the solid state and the crystal structures of those containing [ErN9] chromophores have been previously solved by X-ray diffraction (Fig. 5b).12,28 For [ErN3O6] units, X-ray quality crystals of (NHEt3)5[Er(dpa)3](CF3SO3)2 (1), [Er(dpa-ester)3](ClO4)3 (2), [Er(dpa-amide)3](ClO4)3 (3) could be obtained by slow diffusion of diethylether into concentrated butyronitrile solutions (Fig. 5a and Fig. S2–S4, Tables S1–S7†).
:3 and total ligand concentrations of 3–10 mM, the specific conditions that are used for recording the photophysical studies. For the less stable triple-helical [Er(dpa-diester)3]3+ complex, its speciation corresponds to more than 80% of the total ligand content in the same conditions (Fig. S1b†). These complexes can be also isolated in the solid state and the crystal structures of those containing [ErN9] chromophores have been previously solved by X-ray diffraction (Fig. 5b).12,28 For [ErN3O6] units, X-ray quality crystals of (NHEt3)5[Er(dpa)3](CF3SO3)2 (1), [Er(dpa-ester)3](ClO4)3 (2), [Er(dpa-amide)3](ClO4)3 (3) could be obtained by slow diffusion of diethylether into concentrated butyronitrile solutions (Fig. 5a and Fig. S2–S4, Tables S1–S7†).
As expected, the molecular structure of the triple-helical [Er(dpa)3]3− anion exactly mirrors those reported for Na3[Ln(dpa)3]·13H2O18,32 and for (imidazol-H)3[Ln(dpa)3]·3H2O,33 but the crystals of (NHEt3)5[Er(dpa)3](CF3SO3)2 are soluble in acetonitrile with no sign of significant water content. The [Er(dpa-amide)3]3+ building block found in 3 is almost superimposable with that previously reported for [La(dpa-amide)3](ClO4)3·2.5C2H5CN30 while [Er(dpa-ester)3]3+, to the best of our knowledge, is the first reported crystal structure along the 2,6-diesterpyridine series. All nine-coordinate Er(III) centers adopt slightly distorted tricapped trigonal prismatic geometries (SHAPE's factors 1.59 ≤ S ≤ 3.30, Table S8†)34 with the pyridine nitrogen atom of each wrapped ligand occupying a capping position in the final polyhedra (Fig. S5†). The Er–N and Er–O bond lengths are poorly dispersed (Table S8†) and correspond to those expected for triple-helical [ErN9] and [ErN3O6] complexes with tridentate ligands.12,19,20b,28 Given that the existence of ‘long’ Er-centered excited state lifetimes in molecular complexes, which are critical for implementing linear upconversion, requires (i) a minimum splitting of the J manifolds produced by the crystal field effect and (ii) a global lack of energy matching between the high-energy oscillators of the ligands and the average energy gap between the successive 2S+1LJ spectroscopic levels,35,36 the crystal field parameters (Table S9†) and associated energy splitting of the J manifolds in complexes 1–5 have been described by SO-CASSCF and SO-CASPT2 calculations (Fig. S6†).37 Interestingly, the computed global crystal field strengths S38 are larger for [ErN3O6] units (S(Er(dpa)3) = 217(16) cm−1 > S(Er(dpa-amide)3) = 186 cm−1 > S(Er(dpa-ester)3) = 171 cm−1) than for [ErN9] chromophores (S(Er(Et-bzimpy)3) = 157 cm−1 ≈ S(Er(tpy)3) = 155 cm−1, Table S9†). Consequently the total splitting of the Er(2S+1LJ) manifolds is broader when tridentate NO2 ligands are bound to Er(III) (Tables S10–S14†), thus offering more probabilities for non-radiative relaxation induced by high-energy vibrations (and shorter intermediate excited lifetimes) in [ErN3O6] units than in [ErN9] analogues. Although less pertinent for optical39 than for magnetic properties,40 the main difference between [ErN3O6] and [ErN9] chromophores lies in the sign of B20 which is negative for [ErN9] (oblate arrangement of the donor atoms with the principal magnetic axis parallel to the pseudo-threefold Z axis) and positive for [ErN6O3] (prolate arrangement of the donor atoms with the principal magnetic axis perpendicular to the Z axis).33 Finally, according to Reinhard and Güdel,18 the weighted average of the vibrations participating in the nonradiative relaxation process (= ‘phonon bath’) in molecular [Ln(dpa)3]3−can be set to hνeff ≈ 2000 cm−1, an approximation which can be extended for complexes 1–5 according to the vibrational IR spectra (Fig. S7†).
Molecular light-upconversion operating in single-center triple-helical erbium complexes
The NIR-Visible absorption spectra of triple-helical [GaErGa(bpb-bzimpy)3]9+ (Fig. 6a and b), [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, tpy, dpa-amide, dpa-ester) and [Er(dpa)3]3− in acetonitrile (Fig. S8 and S9†) are all similar and display weak metal-centered Er(2S+1LJ ← 4I15/2) transitions (0.1 ≤ εmax ≤ 5 M−1 cm−1) characteristic of the well-known energy diagram depicted in Fig. 6c.11,12 The radiative rate constant kJ’→Jrad, and related radiative lifetime τJ’→Jr = 1/kJ’→Jrad, associated with the emission between each excited Er(2S+1LJ) level and the ground Er(4I15/2) level can be calculated from the absorption spectrum ε(ṽ) (in M−1 cm−1) using eqn (2), where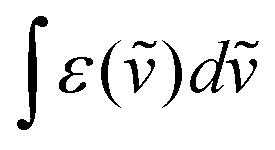 is the integrated spectrum of the incriminated absorption transition recorded in solution, J and J′ refer to the ground (J = 15/2) and excited states, respectively, n is the refractive index of the medium, NA is Avogadro's number (in mol−1), c is the speed of light in vacuum (in cm s−1) and ṽm is the barycenter of the transition (in cm−1) given in eqn (3).41
is the integrated spectrum of the incriminated absorption transition recorded in solution, J and J′ refer to the ground (J = 15/2) and excited states, respectively, n is the refractive index of the medium, NA is Avogadro's number (in mol−1), c is the speed of light in vacuum (in cm s−1) and ṽm is the barycenter of the transition (in cm−1) given in eqn (3).41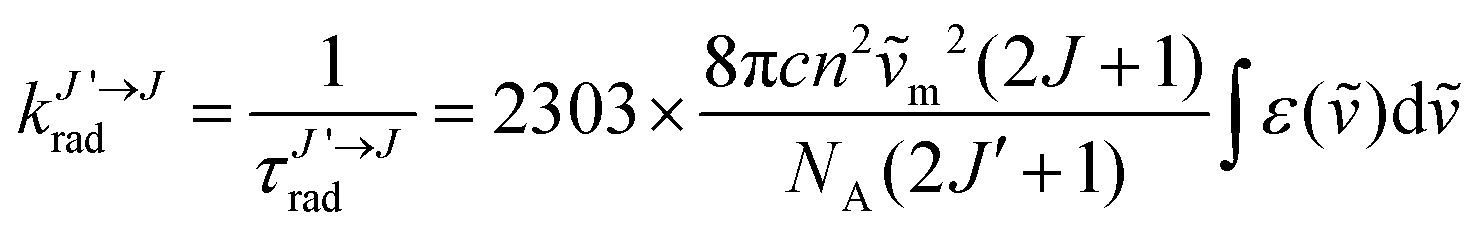 | (2) |
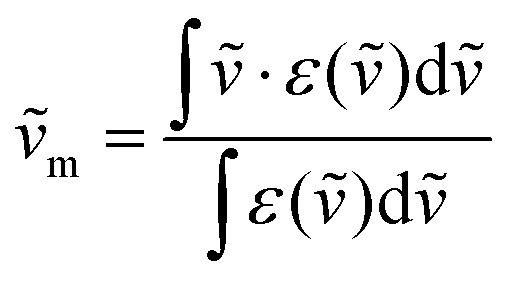 | (3) |
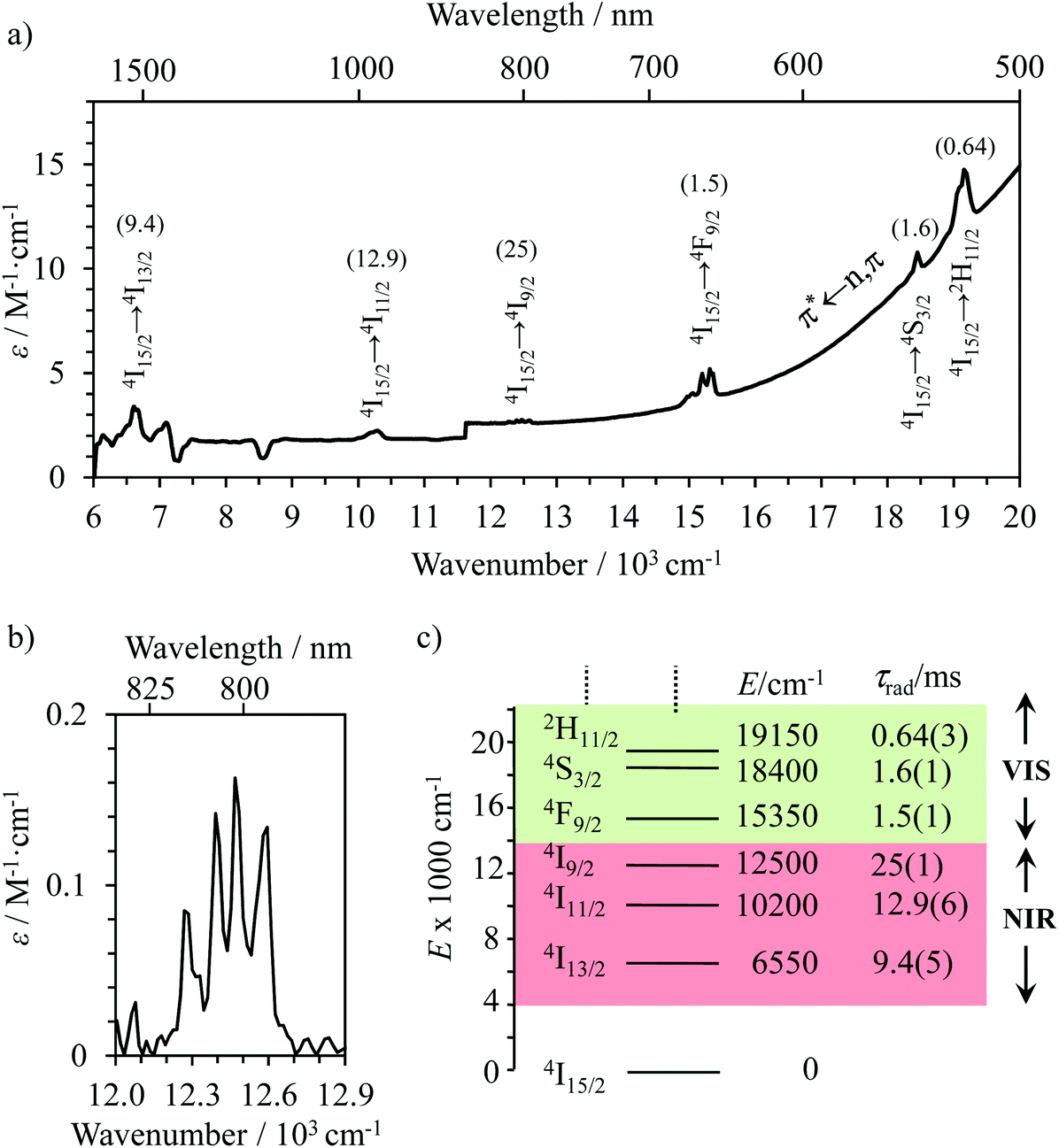 | ||
| Fig. 6 (a) NIR-VIS absorption spectrum of [GaErGa(bpb-bzimpy)3]9+ (0.01 M in acetonitrile at 293 K) showing the Er(2S+1LJ ← 4I15/2) transitions and the associated radiative lifetimes (in ms) between parenthesis, (b) highlight of the Er(4I9/2 ← 4I15/2) centered at 801 nm and (c) energy diagram of low-energy erbium-centered levels. | ||
The experimental τJ’→J=15/2rad extracted for the Er(2S+1LJ′) excited levels located within the 6000–20000 cm−1 domain cover the 1–20 ms range in agreement with the symmetry-forbidden character of the intrashell (f–f) electric dipole transitions (Fig. 6a and c and S8† and Tables 1 and S15†). As expected from the dependence of the Einstein coefficient for spontaneous emission with ṽm,23a,41 the global radiative lifetimes decrease with increasing energy gaps. The allowed ligand-centered π* ← n,π absorption bands (2 × 104 ≤ ε ≤ 20 × 104 M−1 cm−1) cover the UV part of the absorption spectra (24000–40000 cm−1)12,28 and mask the Er-centered transitions expected to occur in this domain. In this context, the low-energy tail of the latter ligand-based absorption can be easily detected in the visible part of the absorption spectra recorded for [GaErGa(bpb-bzimpy)3]9+ (Fig. 6a) or for [Er(Et-bzimpy)3]9+ (Fig. S8†).
| Complexes | λ exc/nm | 405 | 405 | 355 | 355 | 805 | 975 | ||
|---|---|---|---|---|---|---|---|---|---|
| Level | Er(4S3/2) | Er(4S3/2) | Er(4S3/2) | Er(4I13/2) | Er(4I13/2) | Er(4I13/2) | Er(4I13/2) | Er(4I13/2) | |
| τ rad/ms | τ tot/ns | ϕ Er | τ rad/ms | τ tot/μs | ϕ Er | τ tot/μs | τ tot/μs | ||
a In acetonitrile.
b Too weak to be measured.20b
c Recorded at 3–10 K.20b
d Computed by using 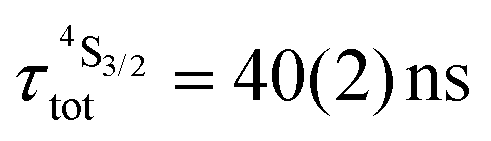 . .
|
|||||||||
| [Er(Et-bzimpy)3]3+ | Solid | — | — | — | 5.57(6) | — | 4.786(7) | — | |
| Solutiona | 1.31(9) | — | 3.0(3) × 10−5d |
7.12(5) | — | 7.8(1) × 10−4 | 6.299(5) | 5.8(2) | |
| [GaErGa(bpb-bzimpy)3]9+ | Solid | — | 40(2)c | — | — | 4.04(4) | — | 3.955(7) | — |
| Solution | 1.6(1) | — | 2.5(2) × 10−5d |
9.4(5) | — | 4.3(2) × 10−4 | 5.109(5) | 4.8(1) | |
| [Er(tpy)3]3+ | Solid | — | — | — | 1.88(2) | — | 2.092(3) | — | |
| Solution | 0.75(5) | — | 5.3(4) × 10−5d |
8.1(6) | — | 2.3(2) × 10−4 | 2.005(1) | 1.9(1) | |
| [Er(Et-tpy)3]3+ | Solid | — | — | — | 1.94(2) | — | 2.309(3) | — | |
| Solution | 0.38(3) | — | 1.0(1) × 10−4d |
7.01(5) | — | 2.77(4) × 10−4 | 2.250(1) | 2.16(3) | |
| [Er(dpa)3]3− | Solid | — | — | — | 2.217(1) | — | 1.772(2) | — | |
| Solution | 0.98(7) | — | 4.1(4) × 10−5d |
6.9(5) | — | 3.2(2) × 10−4 | 2.450(1) | 2.39(6) | |
| [Er(dpa-ester)3]3+ | Solid | — | — | — | 3.270(3) | — | 2.78(4) | — | |
| Solution | 1.01(5) | — | 4.0(3) × 10−5d |
9.2(6) | — | 3.6(2) × 10−4 | 3.919(2) | 3.2(1) | |
| [Er(dpa-amide)3]3+ | Solid | — | — | — | 3.067(1) | — | 3.118(2) | — | |
| Solution | 0.81(6) | — | 4.9(4) × 10−5d |
7.4(5) | — | 4.1(3) × 10−4 | 3.441(3) | 3.03(9) | |
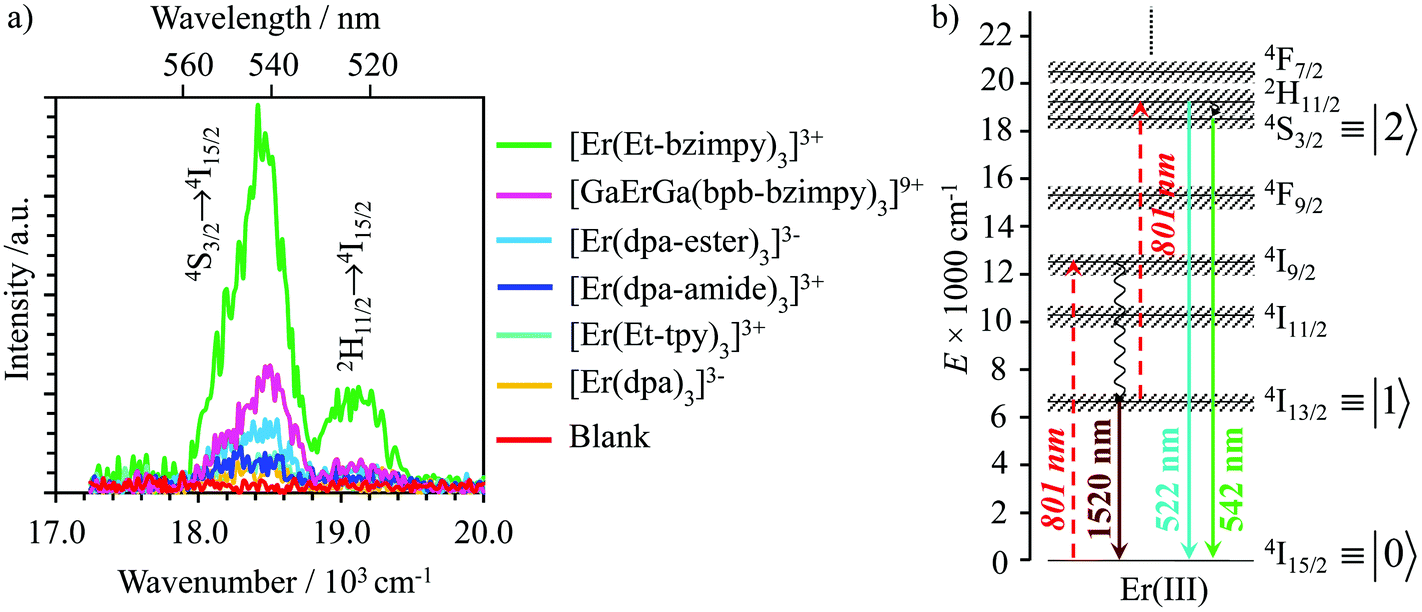
Ligand-centered UV-excitation (355 to 400 nm) of [GaErGa(bpb-bzimpy)3]9+,12 [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, tpy, dpa-amide, dpa-ester)28 or [Er(dpa)3]3− (ref. 18) sensitizes the Er(III) metal via the antenna effect, which provides some rare dual Er-based emissions in these molecular complexes (Fig. S10–S13†).28,35 The (very) weak visible band (λem = 540–560 nm, Fig. S10 and S12†) can be assigned to the Er(4S3/2 → 4I15/2) transition, while the more intense near infrared band (λem = 1500–1540 nm, Fig. S11 and S13†) corresponds to the common Er(4I13/2 → 4I15/2) luminescent transition.28
Both transitions display linear log(I)–log(P) plots between the emitted intensity (I) and the incident UV excitation power (P) with slopes close to one, which is diagnostic for the operation of linear light-downshifting in these complexes (Fig. S10–S13†).9c Because of the only faint visible (green) Er-centered emission, the determination of experimental lifetimes for the Er(4S3/2) excited level represents a real technical challenge, which could be addressed by a time-gated CCD-camera only for the ‘most intense’ emitter along the series at low temperature (3–10 K), namely [GaErGa(bpb-bzimpy)3](CF3SO3)9 with 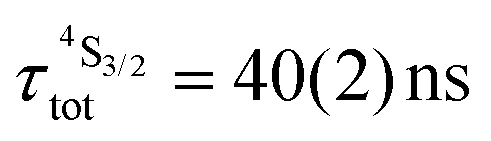 ; a value confirmed for its dinuclear analogue [GaEr(pb-bzimpy)3](CF3SO3)6 with
; a value confirmed for its dinuclear analogue [GaEr(pb-bzimpy)3](CF3SO3)6 with 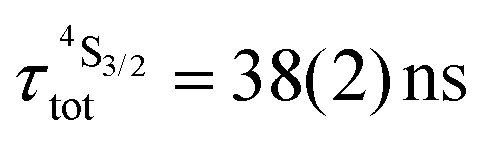 .20b The associated intrinsic quantum yields
.20b The associated intrinsic quantum yields  calculated for [GaErGa(bpb-bzimpy)3](CF3SO3)9 can be thus taken as a valuable estimate for the maximum efficiency of
calculated for [GaErGa(bpb-bzimpy)3](CF3SO3)9 can be thus taken as a valuable estimate for the maximum efficiency of 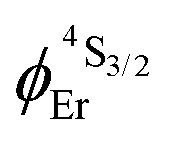 in these complexes (Table 1, column 5). Although weak, the intensity of the near infrared Er(4I13/2 → 4I15/2) transition is compatible with standard time-gated detection techniques and systematically gives mono-exponential decay traces with
in these complexes (Table 1, column 5). Although weak, the intensity of the near infrared Er(4I13/2 → 4I15/2) transition is compatible with standard time-gated detection techniques and systematically gives mono-exponential decay traces with 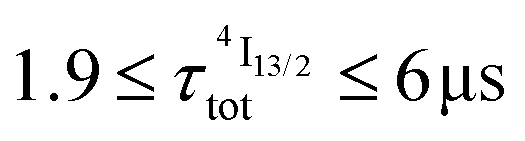 characteristic lifetimes (Table 1 column 7 and Fig. S14†) and
characteristic lifetimes (Table 1 column 7 and Fig. S14†) and 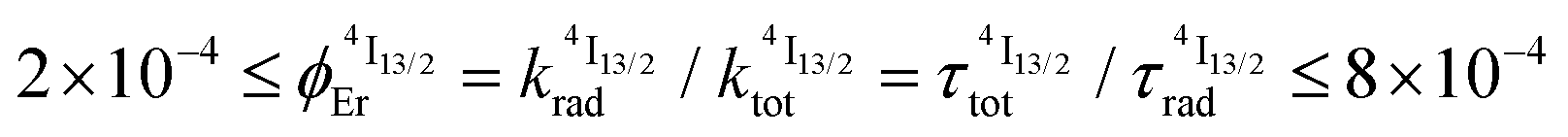 intrinsic quantum yields (Table 1, column 8). In line with the hypothesis that erbium complexes with smaller crystal field strength are less prone to undergo efficient non-radiative vibrational relaxation processes,35,36 the lifetimes measured for the Er(4I13/2) level are maximum for [Er(Et-bzimpy)3]3+ and [GaErGa(bpb-bzimpy)3]9+ (Table 1, column 7 and Fig. S15†).
intrinsic quantum yields (Table 1, column 8). In line with the hypothesis that erbium complexes with smaller crystal field strength are less prone to undergo efficient non-radiative vibrational relaxation processes,35,36 the lifetimes measured for the Er(4I13/2) level are maximum for [Er(Et-bzimpy)3]3+ and [GaErGa(bpb-bzimpy)3]9+ (Table 1, column 7 and Fig. S15†).
Upon continuous near-infrared diode laser excitation at 801 nm (12480 cm−1) into the Er(4I9/2 → 4I15/2) transition at reasonable power intensities, the [GaErGa(bpb-bzimpy)3]9+, [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, dpa-amide, dpa-ester) and [Er(dpa)3]3− complexes exhibit upconverted visible Er(2H11/2 → 4I15/2) and Er(4S3/2 → 4I15/2) emissions in the solid state (Fig. S16–S18†) and in solution (Fig. 7a). The associated log(I)–log(P) plots are linear with slopes close to 2.0, which is diagnostic for the operation of light-upconversion. Since all the absorption coefficients at 801 nm are comparable 0.07 ≤ ε801 ≤ 0.15 M−1 cm−1 (i.e.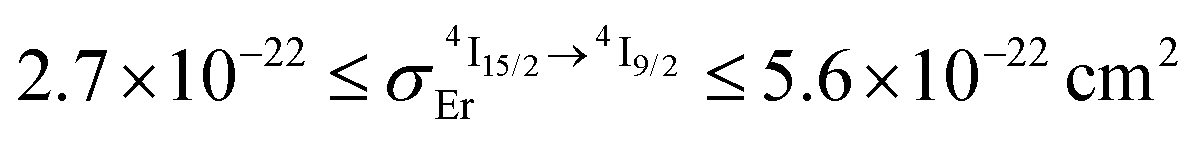 , Fig. 6b and S19† and Table 2 column 2),23 the upconverted intensities monitored in solution at the same concentration (Fig. 7a) suggest the following decreasing order for the upconversion efficiencies: [Er(Et-bzimpy)3]3+ > [GaErGa(bpb-bzimpy)3]9+ > [Er(dpa-ester)3]3+ > [Er(dpa-amide)3]3+ ≈ [Er(Et-tpy)3]3+ ≈ [Er(dpa)3]3−. This trend is confirmed by the total upconversion quantum yields ϕuptot determined in acetonitrile at room temperature with the help of the relative method using indocyanin green as a reference (Table 2, column 6; λexc = 801 nm and P = 25 W cm−2, see the Experimental section).42
, Fig. 6b and S19† and Table 2 column 2),23 the upconverted intensities monitored in solution at the same concentration (Fig. 7a) suggest the following decreasing order for the upconversion efficiencies: [Er(Et-bzimpy)3]3+ > [GaErGa(bpb-bzimpy)3]9+ > [Er(dpa-ester)3]3+ > [Er(dpa-amide)3]3+ ≈ [Er(Et-tpy)3]3+ ≈ [Er(dpa)3]3−. This trend is confirmed by the total upconversion quantum yields ϕuptot determined in acetonitrile at room temperature with the help of the relative method using indocyanin green as a reference (Table 2, column 6; λexc = 801 nm and P = 25 W cm−2, see the Experimental section).42
 | ||
| Fig. 7 (a) Upconverted visible Er(2H11/2 → 4I15/2) and Er(4S3/2 → 4I15/2) emissions observed for [GaErGa(bpb-bzimpy)3]9+, [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, dpa-amide, dpa-ester) and [Er(dpa)3]3− recorded upon laser excitation of the Er(4I9/2 ← 4I15/2) transition at λexc = 801 nm (ṽexc = 12284 cm−1) and using incident pump intensity P = 25 W cm−2 in acetonitrile solution at 298 K (c ∼ 10 mM with similar optical density at 801 nm, Fig. S19†). The blank (red curve) was recorded from pure acetonitrile solvent using the same incident pump intensity P = 25 W cm−2. (b) Associated energy diagram with the proposed kinetic mechanism. | ||
| Complexes |
σ
0→1Era |
k
1→0Er/105b |
k
2→0Er/102c |
k
2→1Er/107d |
ϕ
uptote |
η ESA |
σ
1→2Era,g |
|---|---|---|---|---|---|---|---|
a
ε
i→j = 2.6 × 1020σi→j are given in M−1 cm−1 between brackets.23
b
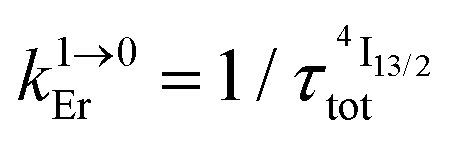 from Table 1 column 7.
c from Table 1 column 7.
c
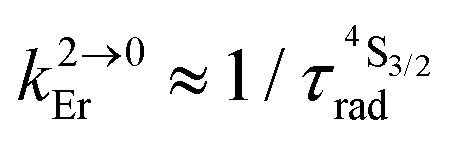 (see text).
d (see text).
d
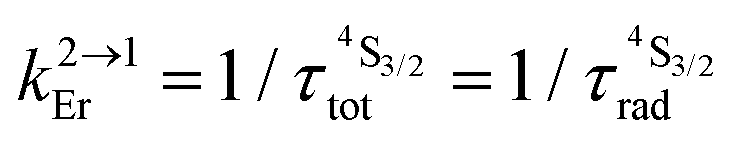 with with 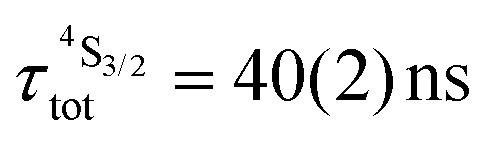 (Table 1, column 4).
e
λ
exc = 801 nm (ṽexc = 12284 cm−1) and P = 25 W cm−2.
f (Table 1, column 4).
e
λ
exc = 801 nm (ṽexc = 12284 cm−1) and P = 25 W cm−2.
f
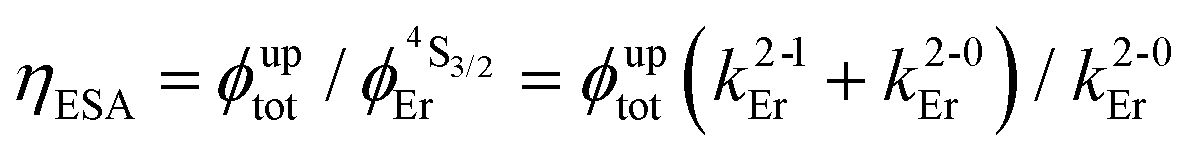 (see Fig. 3a).
g Computed by using (see Fig. 3a).
g Computed by using 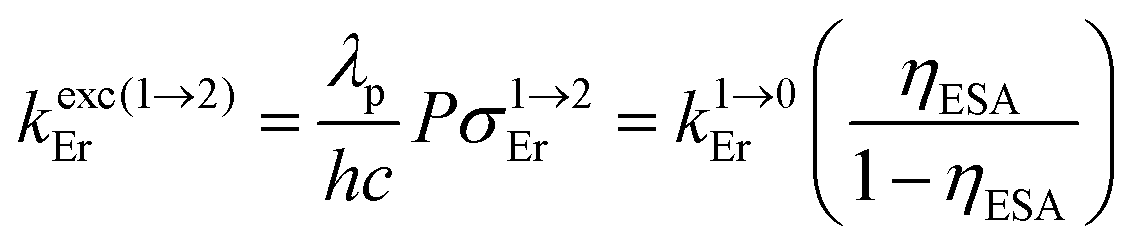 (eqn (4)). (eqn (4)).
|
|||||||
| [Er(Et-bzimpy)3]3+ | 4.6(2) × 10−22 [0.125(6)] | 1.80(2) | 7.6(5) | 2.5(1) | 1.7(2) × 10−9 | 5.6(7) × 10−5 | 10(1) × 10−20 [26(3)] |
| [GaErGa(bpb-bzimpy)3]9+ | 2.7(1) × 10−22 [0.074(4)] | 2.48(2) | 6.3(4) | 2.5(1) | 1.7(2) × 10−9 | 6.8(9) × 10−5 | 17(2) × 10−20 [43(6)] |
| [Er(Et-tpy)3]3+ | 5.6(3) × 10−22 [0.146(7)] | 5.32(6) | 26.3(2.1) | 2.5(1) | 5.5(6) × 10−11 | 5.2(7) × 10−7 | 2.8(4) × 10−21 [0.7(1)] |
| [Er(dpa)3]3− | 2.7(2) × 10−22 [0.070(4)] | 4.511(2) | 10.2(7) | 2.5(1) | 2.2(2) × 10−10 | 5.4(7) × 10−6 | 2.4(3) × 10−20 [6.3(8)] |
| [Er(dpa-ester)3]3+ | 2.8(1) × 10−22 [0.074(4)] | 3.058(3) | 9.9(5) | 2.5(1) | 5.1(5) × 10−10 | 1.3(2) × 10−5 | 3.9(5) × 10−20 [10(1)] |
| [Er(dpa-amide)3]3+ | 3.8(2) × 10−22 [0.099(5)] | 3.260(1) | 12.3(9) | 2.5(1) | 1.9(2) × 10−10 | 3.9(5) × 10−6 | 1.2(2) × 10−20 [3.2(4)] |
Having significantly improved both accuracy and reliability of the latter technique for measuring weak emitters thanks to the thorough procedures described by Charbonnière and co-workers21 and by Wurth et al.,42c we ultimately found ϕuptot([Er(Et-bzimpy)3]3+) = 1.7(2) × 10−9 and ϕuptot([Er(Et-tpy)3]3+) = 5.5(6) × 10−11 for the upper and lower limits in these erbium complexes (Table 2, column 6; preliminary estimations in the 1.6 × 10−8 and 4.1 × 10−9 range).22 As expected for the ESA mechanism (Fig. 1 and 2), the upconversion quantum yields ϕuptot (Table 2, column 6 for λexc = 801 nm and P = 25 W cm−2) and the associated ESA efficiencies 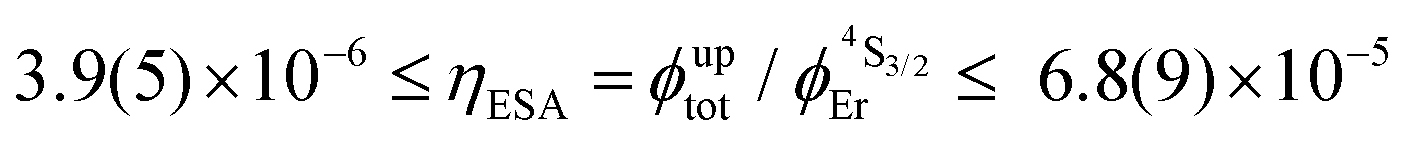 , Table 2, column 7) are found to be correlated with the increasing lifetimes of the intermediate Er(4I13/2) excited level (Fig. S20†). Moreover, the unusual temperature dependence of the upconverted signals observed in these complexes (i.e. IupEr increases with increasing temperature until reaching a maximum, Fig. S21 and S22†) is diagnostic for the operation of thermally-activated relaxation to reach the intermediate excited relays according to the upconversion mechanism proposed in Fig. 7b.22 The three-levels kinetic model depicted in Fig. 1 thus applies with |0〉 = Er(4I15/2) corresponding to the ground state, |1〉 = Er(4I13/2) being the intermediate excited relay (fed by fast internal conversion from 4I9/2) and |2〉 = Er(4S3/2) being the doubly excited emissive level. Since all the pertinent rate constants are at hand (Table 2, columns 3–5), the only unknown parameter
, Table 2, column 7) are found to be correlated with the increasing lifetimes of the intermediate Er(4I13/2) excited level (Fig. S20†). Moreover, the unusual temperature dependence of the upconverted signals observed in these complexes (i.e. IupEr increases with increasing temperature until reaching a maximum, Fig. S21 and S22†) is diagnostic for the operation of thermally-activated relaxation to reach the intermediate excited relays according to the upconversion mechanism proposed in Fig. 7b.22 The three-levels kinetic model depicted in Fig. 1 thus applies with |0〉 = Er(4I15/2) corresponding to the ground state, |1〉 = Er(4I13/2) being the intermediate excited relay (fed by fast internal conversion from 4I9/2) and |2〉 = Er(4S3/2) being the doubly excited emissive level. Since all the pertinent rate constants are at hand (Table 2, columns 3–5), the only unknown parameter  can be fitted (Table 2, column 8) to the experimental ESA efficiencies ηESA with eqn (4) (derived from eqn (1) and Fig. 2a)
can be fitted (Table 2, column 8) to the experimental ESA efficiencies ηESA with eqn (4) (derived from eqn (1) and Fig. 2a)
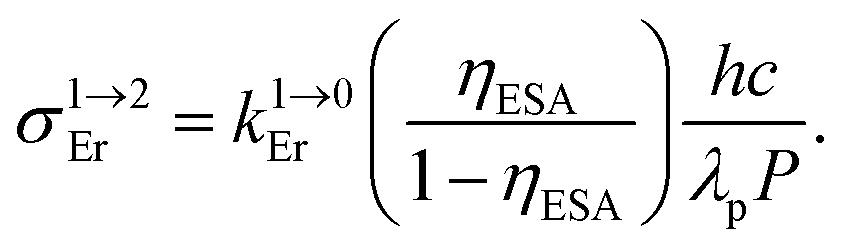 | (4) |
Translated into decadic molar absorption coefficients 0.7 ≤ ε1→2 ≤ 43 M−1 cm−1 (Table 2, column 8), the excited state Er(2H11/2,4S3/2 ← 4I13/2) absorptions appear to be two orders of magnitude more efficient than the ground state Er(4I9/2 ← 4I15/2) absorption process and around one order of magnitude larger than the other Er(2S+1LJ ← 4I15/2) transitions recorded for the ground state absorption spectra of these complexes (Fig. 6a and S8, S9†). In this context, the [ErN9] chromophores, produced by the binding of three bulky 2,6-bis(benzimidazol-2-yl)pyridine ligand strands possessing low-lying π* orbitals in [Er(Et-bzimpy)3]3+ and [GaErGa(bpb-bzimpy)3]9+, give the most efficient excited state absorptions with 26 ≤ ε1→2 ≤ 43 M−1 cm−1, which are at least one order of magnitude larger than those expected for standard intrashell f–f transitions. Recently, some non-negligible mixing of 4f-metal with ligand π orbitals have been demonstrated to significantly boost the efficiency of energy transfer processes in related europium tris-diketonate complexes,43 and a similar mechanism might be responsible for this unexpected improvement for molecular upconversion. For testing this hypothesis, the oscillator strengths fij, which are proportional to the molar absorption coefficient ε,44 of the electric-dipole (ED), magnetic-dipole (MD) and electric-quadrupole (EQ) contributions to the ligand field Er(2S+1LJ ← 4I15/2) (Table S16†) and Er(2S+1LJ ← 4I13/2) (Table S17†) transitions intensities have been evaluated from SO-CASSCF calculations (see computational details in the ESI†).45 As expected from the Judd–Ofelt theory,44 the contributions of EQ transitions are negligible except for the hypersensitive Er(2H11/2 ← 4I15/2) transition (fEQ = 6.2 × 10−8), which possess a large Judd–Ofelt U(2) matrix element.46 The oscillator strengths of the two MD-allowed transitions Er(4I13/2 ← 4I15/2) and Er(4I11/2 ← 4I13/2), which obey the selection rules ΔS = 0, ΔL = 0 and ΔJ = ±1, and of Er(2H11/2 ← 4I13/2) with only ΔJ = ±1, are found to compete with the forced ED transitions, the computed intensity of which roughly follow the trend reported for the aquo-ion.46 Focusing on λexc = 801 nm (ṽexc = 12284 cm−1) used in our studies, the absorption of the first photon is associated with the Er(4I9/2 ← 4I15/2) transition (Fig. 6 and S9†). Its small experimental absorption coefficient (ε < 0.2 M−1 cm−1 in all studied complexes) is mirrored by the weak computed oscillator strengths 3.5 ≤ f ≤ 5.7 × 10−8 (Table S16†) assigned to small Judd–Ofelt U(6) matrix elements, a curious trend which is characteristic for the transition to the highest-lying J-level in terms with Smax.46 After relaxation to the intermediate Er(4I13/2) level, the second excitation process reaches Er(2H11//2), the energy of which (18600–19400 cm−1. Fig. S9†) matches well Er(4I13/2) (6300–6800 cm−1) + ṽexc (12284 cm−1) ≈ 18600–19000 cm−1. Interestingly, the oscillator strength computed for the pertinent Er(2H11//2 ← 4I13/2) excited state absorption (f = 1.3 × 10−5, Table S17†) is 2–3 orders of magnitude larger than that computed for the first Er(4I9/2 ← 4I15/2) absorption process in agreement with the experimental ESA absorption coefficients (Table 2, column 8), which are 2–3 orders of magnitude larger than their GSA analogues (Table 2, column 2). However, the possibility that the non-emissive Er(4I9/2) and Er(4I11/2) excited state are indeed long-lived (i.e.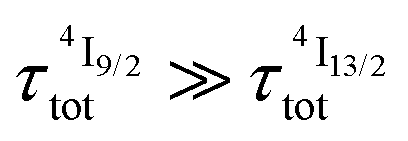 or
or 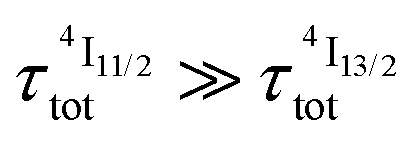 ) and may act as better relay than Er(4I13/2) for upconversion, cannot be ruled out without being explored prior to reach any conclusion (see Fig. 7b for the energy diagram).
) and may act as better relay than Er(4I13/2) for upconversion, cannot be ruled out without being explored prior to reach any conclusion (see Fig. 7b for the energy diagram).
Looking for a long-lived intermediate excited state working as relay for ESA in molecular erbium complexes
Continuous laser excitation into the Er(4I9/2 ← 4I15/2) transitions at λexc = 801 nm for [GaErGa(bpb-bzimpy)3]9+, [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, dpa-amide, dpa-ester) and [Er(dpa)3]3− does not only induce the weak upconverted signals Er(2H11/2,4S3/2 → 4I15/2) discussed above (Fig. 7a), but also downshifted Er(4I13/2 → 4I15/2) emissions at 1520 nm characterized by linear log(I)–log(P) plots with slopes close to one at low-to-medium intensity powers (Fig. 8 and Fig. S23, S24†).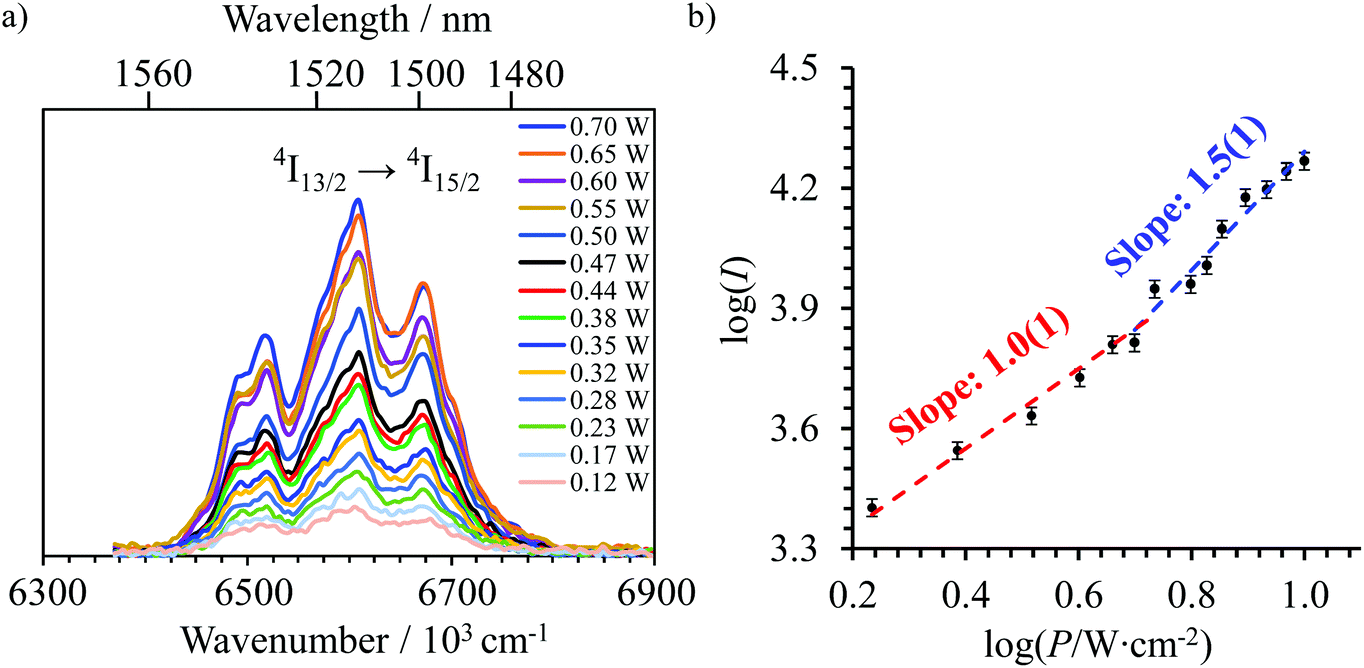 | ||
| Fig. 8 (a) Near-infrared downshifted Er(4I13/2 → 4I15/2) emission observed for [GaErGa(bpb-bzimpy)3] (solid state, 298 K) upon laser excitation of the Er(4I9/2 ← 4I15/2) transition at λexc = 801 nm (ṽexc = 12284 cm−1) and for different incident pump intensities focused on a spot size of ≈0.07 cm2. (b) Corresponding log–log plot of downshifted intensities I as a function of incident pump intensities P (in W cm−2). | ||
We thus conclude that the detected emissive Er(4I13/2) level is fed by internal conversion from the initially non-emissive excited Er(4I9/2) level via internal conversion prior to emitting its characteristic NIR photons upon relaxing to the ground Er(4I15/2) state (see Fig. 7b). The time-dependent normalized population densities 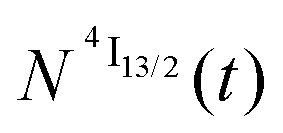 for the intermediate emissive Er(4I13/2) level thus follows a simplified sequence of two consecutive kinetic reactions
for the intermediate emissive Er(4I13/2) level thus follows a simplified sequence of two consecutive kinetic reactions 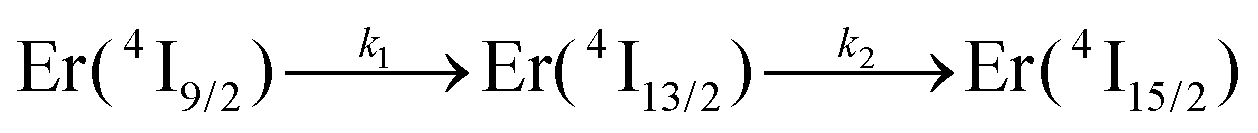 described in eqn (5) and (6), where
described in eqn (5) and (6), where 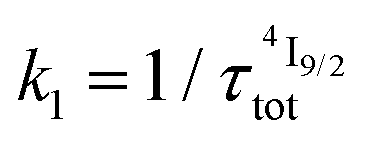 and
and 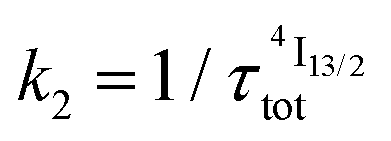 .47
.47
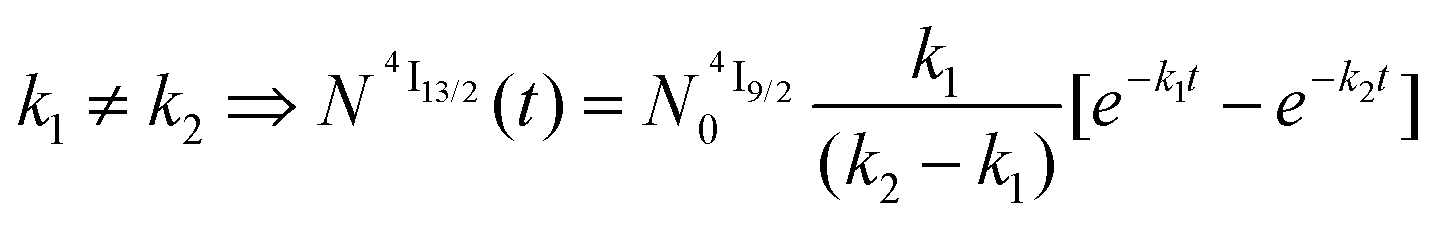 | (5) |
 | (6) |
When k1 ≫ k2 (i.e.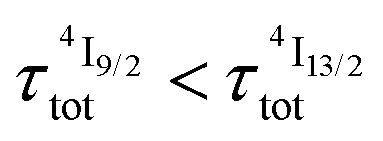 ), the time decay of the emissive Er(4I13/2) level approximately corresponds to a single exponential trace with its characteristic lifetime
), the time decay of the emissive Er(4I13/2) level approximately corresponds to a single exponential trace with its characteristic lifetime 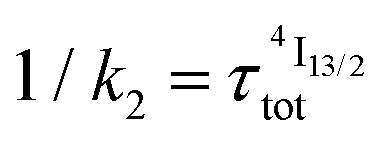 . When k1 ≈ k2 (i.e.
. When k1 ≈ k2 (i.e.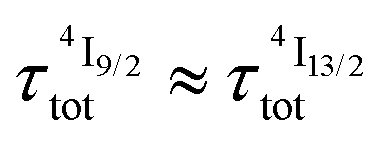 ), the time-dependence of the population density of the emissive Er(4I13/2) level corresponds to a two-phase process with a rising period controlled by
), the time-dependence of the population density of the emissive Er(4I13/2) level corresponds to a two-phase process with a rising period controlled by 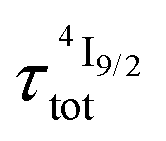 (exponential in eqn (5) or linear in eqn (6)), followed by an exponential decay period controlled by
(exponential in eqn (5) or linear in eqn (6)), followed by an exponential decay period controlled by 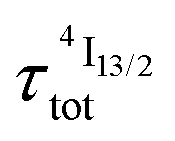 . Finally, k1 ≪ k2 (i.e.
. Finally, k1 ≪ k2 (i.e.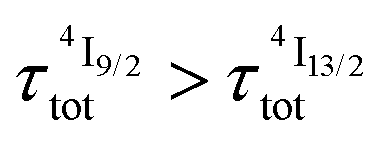 ) results in a decay of the emissive Er(4I13/2) level showing a rough single exponential trace with a characteristic
) results in a decay of the emissive Er(4I13/2) level showing a rough single exponential trace with a characteristic  lifetime reminiscent to that of the feeding Er(4I9/2) level. Pulsed-laser excitation into the Er(4I9/2 ← 4I15/2) transition at λexc = 805 nm systematically leads to single exponential emission decays arising from the Er(4I13/2) level (Fig. S25 and S26†) with characteristic microsecond lifetimes, which exactly mirror those obtained upon ligand-centered excitation at 355 nm (Table 1, column 9) and therefore assigned to
lifetime reminiscent to that of the feeding Er(4I9/2) level. Pulsed-laser excitation into the Er(4I9/2 ← 4I15/2) transition at λexc = 805 nm systematically leads to single exponential emission decays arising from the Er(4I13/2) level (Fig. S25 and S26†) with characteristic microsecond lifetimes, which exactly mirror those obtained upon ligand-centered excitation at 355 nm (Table 1, column 9) and therefore assigned to 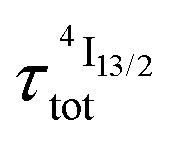 . Similar results were obtained upon alternative excitation into the Er(4I11/2 ← 4I15/2) transition at λexc = 975 nm (Table 1, column 10 and Fig. S27 and S28†), which confirms that the lifetimes of the non-emissive Er(4I9/2) and Er(4I11/2) levels are significantly shorter than
. Similar results were obtained upon alternative excitation into the Er(4I11/2 ← 4I15/2) transition at λexc = 975 nm (Table 1, column 10 and Fig. S27 and S28†), which confirms that the lifetimes of the non-emissive Er(4I9/2) and Er(4I11/2) levels are significantly shorter than 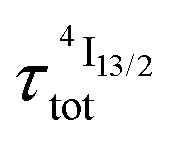 . This leaves Er(4I13/2) as the only available ‘long-lived’ intermediate relay for ESA in these complexes. It is worth noting here that the erbium-centered excitations at 801 nm (Er(4I9/2 ← 4I15/2), Fig. 8b) or at 966 nm (Er(4I11/2 ← 4I15/2), Fig. S29†) systematically exhibit convex log(Idown)–log(P) plots with slopes of 1.0 only at low excitation powers for the downshifted Er(4I13/2 → 4I15/2). At high incident NIR power intensities, the two-photon ESA process is efficient enough (via k2→1Er illustrated in Fig. 1b) to compete with the direct internal Er(4I9/2) → Er(4I13/2) (Fig. 7b) or Er(4I11/2) → Er(4I13/2) conversions for feeding the emissive Er(4I13/2) level.
. This leaves Er(4I13/2) as the only available ‘long-lived’ intermediate relay for ESA in these complexes. It is worth noting here that the erbium-centered excitations at 801 nm (Er(4I9/2 ← 4I15/2), Fig. 8b) or at 966 nm (Er(4I11/2 ← 4I15/2), Fig. S29†) systematically exhibit convex log(Idown)–log(P) plots with slopes of 1.0 only at low excitation powers for the downshifted Er(4I13/2 → 4I15/2). At high incident NIR power intensities, the two-photon ESA process is efficient enough (via k2→1Er illustrated in Fig. 1b) to compete with the direct internal Er(4I9/2) → Er(4I13/2) (Fig. 7b) or Er(4I11/2) → Er(4I13/2) conversions for feeding the emissive Er(4I13/2) level.
Molecular light-upconversion operating in multi-center triple-helical chromium–erbium complexes
Having now a reliable model for ESA operating in mononuclear complexes, we finally re-considered the original proposal made a decade ago for justifying the molecular upconversion process detected in [CrErCr(bpb-bzimpy)3]9+ and tentatively assigned to an ETU mechanism (Fig. 3 and 4).12,19 Direct excitation into the Cr(2T1 ← 4A2) transition at λexc = 718 nm (ṽexc = 13986 cm−1, Fig. 9a) in acetonitrile solution, where no Er-centered absorption occurs (Fig. 6a), indeed confirms the pioneerly reported12 upconverted visible Er(2H11/2 → 4I15/2) and Er(4S3/2 → 4I15/2) emissions (Fig. 9b).
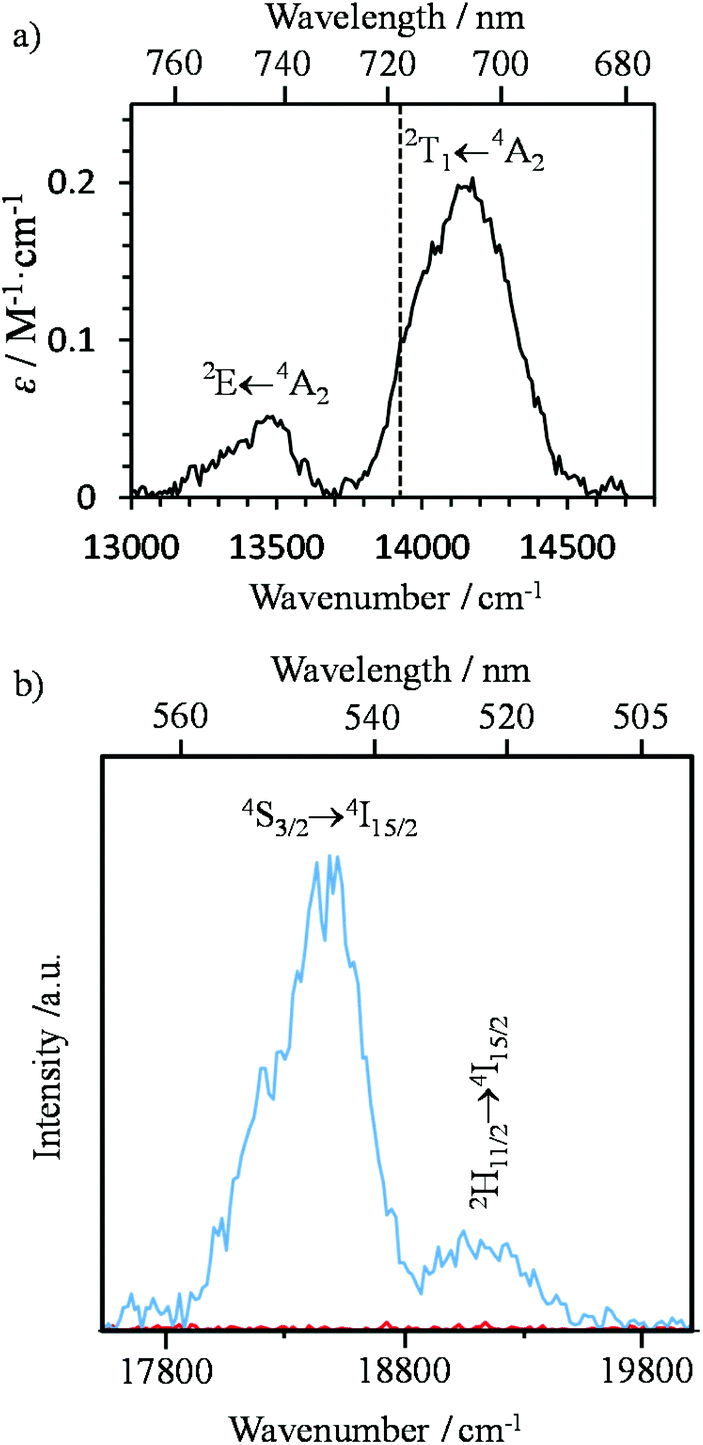 | ||
| Fig. 9 (a) NIR absorption spectrum of [CrErCr(bpb-bzimpy)3]9+ (0.01 M in acetonitrile at 298 K) showing Cr-centered transitions with decadic molar absorption coefficient ε per chromium center and (b) upconverted visible Er(2H11/2 → 4I15/2) and Er(4S3/2 → 4I15/2) emissions recorded upon laser excitation of the Cr(2T1 ← 4A2) transition at λexc = 718 nm (ṽexc = 13927 cm−1) and using incident pump intensity P = 38.2 W cm−2 in acetonitrile solution at 298 K (c ∼ 10 mM). The blank (red curve) was recorded from pure acetonitrile solvent using the same incident pump intensity. | ||
Combining the Er-centered rate constants measured for the [GaErGa(bpb-bzimpy)3]9+ complex (Tables 1 and 2) with the Cr-centered rate constants extracted from previous detailed studies of a series of isostructural [MLnM(bpb-bzimpy)3]9+ complexes (M = Cr, Ga; Ln = Er, Y; 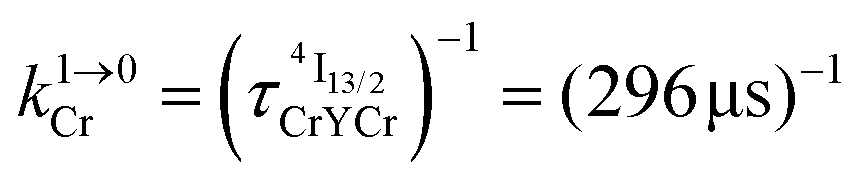 , W1S→A = 232 s−1 at 293 K)12 lead to the conclusion that W2S→A is the only missing parameter for computing the total upconversion quantum yield (ϕuptot, Fig. 4a) according to the ETU mechanism illustrated in Fig. 3b. The experimental upconversion quantum yields, determined in acetonitrile for [CrErCr(bpb-bzimpy)3]9+ at room temperature with the help of the accurate relative method using indocyanin green as a reference, eventually amounts to ϕuptot = 5.8(6) × 10−8 (λexc = 718 nm and P = 38.2 W cm−2), a value which is one order of magnitude larger than that found for the ESA process occurring in [GaErGa(bpb-bzimpy)3]9+ (ϕuptot = 1.7(2) × 10−9; λexc = 801 nm and P = 25 W cm−2). The latter result confirms the pioneering reports12 claiming that, by using a tunable Ti-sapphire excitation laser, an upconverted signal could be detected only for ETU operating in [CrErCr(bpb-bzimpy)3]9+,19 whereas no signal could be detected with the same setup for ESA in [GaErGa(bpb-bzimpy)3]9+.
, W1S→A = 232 s−1 at 293 K)12 lead to the conclusion that W2S→A is the only missing parameter for computing the total upconversion quantum yield (ϕuptot, Fig. 4a) according to the ETU mechanism illustrated in Fig. 3b. The experimental upconversion quantum yields, determined in acetonitrile for [CrErCr(bpb-bzimpy)3]9+ at room temperature with the help of the accurate relative method using indocyanin green as a reference, eventually amounts to ϕuptot = 5.8(6) × 10−8 (λexc = 718 nm and P = 38.2 W cm−2), a value which is one order of magnitude larger than that found for the ESA process occurring in [GaErGa(bpb-bzimpy)3]9+ (ϕuptot = 1.7(2) × 10−9; λexc = 801 nm and P = 25 W cm−2). The latter result confirms the pioneering reports12 claiming that, by using a tunable Ti-sapphire excitation laser, an upconverted signal could be detected only for ETU operating in [CrErCr(bpb-bzimpy)3]9+,19 whereas no signal could be detected with the same setup for ESA in [GaErGa(bpb-bzimpy)3]9+.
However, the upconversion quantum yield for the ETU mechanism (Fig. 4b with λexc = 718 nm and P = 38.2 W cm−2) is predicted to be ϕuptot(ETU) = 2.5 × 10−14 with the reasonable assumption that W2Cr→Er = W1Cr→Er 232 s−1. It can be expanded to ϕuptot(ETU) = 2.0 × 10−11 upon suspicious saturation W2Cr→Er ≥ 106 s−1 while W1Cr→Er = 232 s−1. In consequence, whatever the magnitude of W2Cr→Er, the computed ϕuptot(ETU) are at least three orders of magnitude smaller than the experimental value. The situation becomes much less critical if one considers that the absorption of the second photon at 718 nm (13927 cm−1) may be performed either (inefficiently) by a chromium sensitizer (kexc(0→1)Cr highlighted in red in Fig. 10a) followed by the second energy transfer of magnitude W2Cr→Er according to the ETU mechanism or (efficiently) by the erbium cation in its ‘long-lived’ intermediate Er(4I13/2) excited state via the ESA mechanism 6500(300) + 13927 ≈ 20400(300) cm−1 to reach either the highest crystal field sublevels of the Er(2H11/2) manifold or the lowest sublevels of the Er(4F7/2) manifold (kexc(1→2)Er highlighted in blue in Fig. 10a).
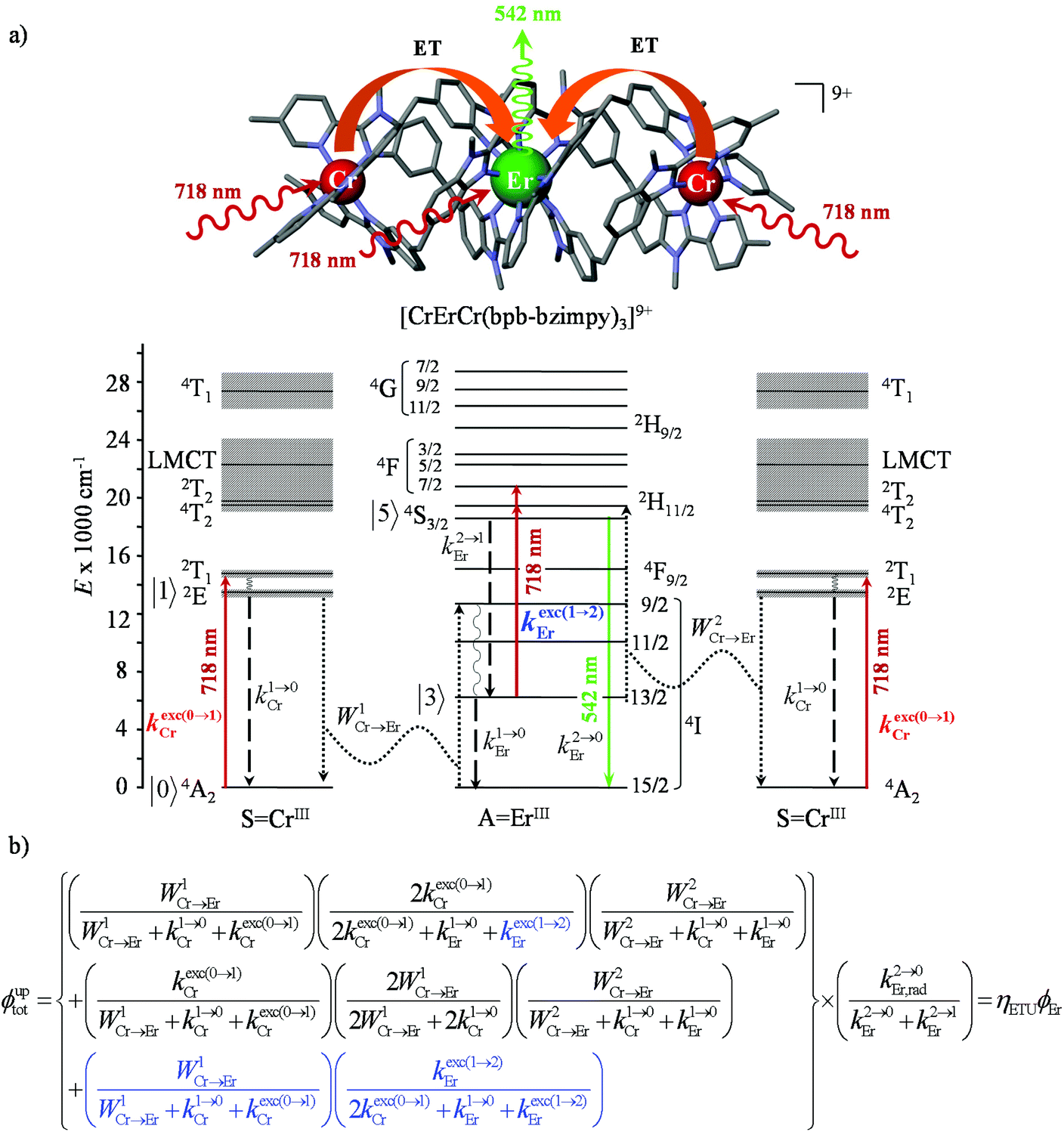 | ||
| Fig. 10 (a) Energy scheme for the upconversion mechanism operating in [CrErCr(bpb-bzimpy)3]9+ upon Cr(2T1 ← 4A2) excitation at λexc = 718 nm (ṽexc = 13927 cm−1) and (b) associated modeling of the upconversion quantum yield (ϕuptot) obtained under steady-state (S-S) excitation. The additional competitive Er-centered ESA mechanism is highlighted in blue. Full upward arrows = photonic excitation, full downward arrows = photonic emission, dashed downward arrow = global (radiative + non-radiative) relaxation processes, dotted lines = energy transfer processes, wavy lines: non-radiative internal conversions. | ||
Introducing ε1→2Er = 50 M−1 cm−1 (λexc = 718 nm), inspired by ε1→2Er = 43(6) M−1 cm−1 (λexc = 801 nm) deduced for ESA operating in [GaErGa(bpb-bzimpy)3]9+, into the adapted equation (Fig. 10b and S30†) gives ϕuptot(ETU) = 1.9 × 10−10, which results in a noticeable gain of four orders of magnitude whatever the value for the second Cr → Er energy transfer rate constant (100 ≤ W2Cr→Er ≤ 106 s−1). The remaining gap by a factor 100 with respect to the experimental quantum yield ϕuptot(ETU) = 5.8(6) × 10−8 is difficult to unambiguously assign, but it could be related to some improved intrinsic erbium-centered quantum yield 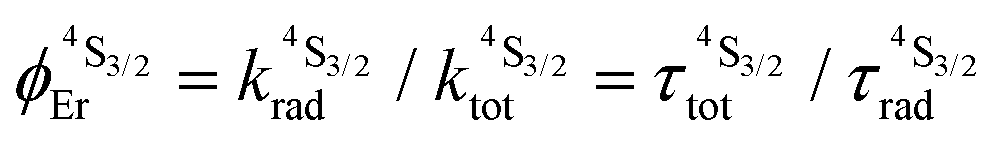 in going from [GaErGa(bpb-bzimpy)3]9+
in going from [GaErGa(bpb-bzimpy)3]9+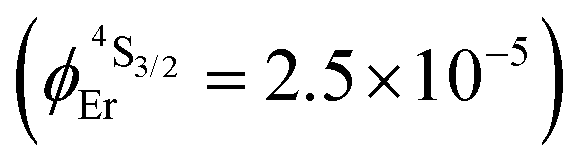 to [CrErCr(bpb-bzimpy)3]9+ where minor mixing with low-lying Cr-based LMCT states may severely reduce
to [CrErCr(bpb-bzimpy)3]9+ where minor mixing with low-lying Cr-based LMCT states may severely reduce 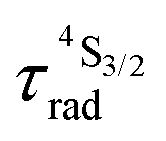 .43 We conclude that the main upconversion mechanism operating in [CrErCr(bpb-bzimpy)3]9+ starts with an initial Cr(2T1 ← 4A2) excitation (718 nm), followed by fast internal conversion to reach the Cr(2E) level, from which a Cr(2E)-to-Er(4I9/2) energy transfer occurs (W1Cr→Er). The major pathway for superexcitation is associated with an efficient Er(2H11/2, 4F7/2 ← 4I13/2) absorption of the second photon at 718 nm, followed by internal conversion to Er(4S3/2) and ultimate green Er(4S3/2 → 4I15/2) photoluminescence. The experimental upconversion quantum yield of ϕuptot(ETU) = 5.3(5) × 10−8 obtained experimentally for the dinuclear analogue [CrEr(pb-bzimpy)3](CF3SO3)6 in the same conditions (acetonitrile, 298 K, λexc = 718 nm and P = 38.2 W cm−2) is a very strong support for the proposed mixed ETU/ESA mechanism since a pure ETU mechanism should be accompanied by a decrease of the upconverted emission by a factor 102–103 in going from CrErCr to CrEr due to the removal of the contribution provided by the concerted Cr-centered ETU mechanism in going from SAS = CrErCr to SA = CrEr systems.13,20b
.43 We conclude that the main upconversion mechanism operating in [CrErCr(bpb-bzimpy)3]9+ starts with an initial Cr(2T1 ← 4A2) excitation (718 nm), followed by fast internal conversion to reach the Cr(2E) level, from which a Cr(2E)-to-Er(4I9/2) energy transfer occurs (W1Cr→Er). The major pathway for superexcitation is associated with an efficient Er(2H11/2, 4F7/2 ← 4I13/2) absorption of the second photon at 718 nm, followed by internal conversion to Er(4S3/2) and ultimate green Er(4S3/2 → 4I15/2) photoluminescence. The experimental upconversion quantum yield of ϕuptot(ETU) = 5.3(5) × 10−8 obtained experimentally for the dinuclear analogue [CrEr(pb-bzimpy)3](CF3SO3)6 in the same conditions (acetonitrile, 298 K, λexc = 718 nm and P = 38.2 W cm−2) is a very strong support for the proposed mixed ETU/ESA mechanism since a pure ETU mechanism should be accompanied by a decrease of the upconverted emission by a factor 102–103 in going from CrErCr to CrEr due to the removal of the contribution provided by the concerted Cr-centered ETU mechanism in going from SAS = CrErCr to SA = CrEr systems.13,20b
Comparison with ETU/ESA mechanisms operating in ionic solids and in nanoparticles doped with Cr/Er is rather tricky because the low-field [CrO6] chromophores found in these oxides are rarely used as sensitizers for upconversion.9g,48–50 In most studies dealing with Cr/Er mixtures, the elected solid garnet is co-doped with Cr3+, Er3+ and Yb3+, where Yb3+ is used as a near-infrared sensitizer (via its Yb(2F5/2 ← 2F7/2) transition at 980 nm).9g In absence of Yb3+, Er3+ is itself usually exploited as the sensitizer (via its Er(4F7/2 ← 4I15/2) transition at 488 nm (ref. 48) or its Er(4IJ ← 4I15/2) transitions in the near-infrared range (J = 15/2, 13/2, 11/2 and 9/2),49 whereas Cr3+ contributes to improve the upconversion properties by working as a relay via energy transfers from the erbium centers. The photoluminescent properties of Cr:Cr:GGG (GGG = gadolinium gallium garnet) represent an exception since direct Cr-centered excitation at 633 nm into the Cr(4T2 ← 4A2) transition is followed by Cr(4T2) → Er(4I9/2) energy transfer and subsequent multi-erbium cross-relaxation processes, which eventually promote a dual Er(4I11/2 → 4I13/2) at 2800 nm and Er(4I13/2 → 4I15/2) at 1600 nm emission.50
Conclusions
When Reinhard and Güdel concluded in 2002 that ‘there is no chance to induce and observe upconversion luminescence in [Ln(dpa)3]3−molecular compounds’,18 (understood that only reasonable incident intensity powers are considered)25,26 their completely pertinent reasoning was based on (i) the observation of intermediate Ln(2S+1LJ) levels with only sub-microsecond lifetimes in Na3[Ln(dpa)3]·13H2O and (ii) the reasonable hypothesis that all f–f absorptions possess cross sections within the 10−24 to 10−22 cm2 range. As synthetic chemists, it was rather obvious to find a way to remove the unfavorable high-energy water oscillators which limit the Er(4I13/2) lifetime in Na3[Ln(dpa)3]·13H2O with the preparation of (NHEt3)5[Er(dpa)3](CF3SO3)2 (1). The latter complex can be directly used in the solid state, but it also gives water-free [Er(dpa)3]3− anions in acetonitrile and displays Er(4I13/2) excited lifetimes reaching a few microseconds at room temperature as found in closely related nine-coordinate Er(III) complexes fitted with more sophisticated organic ligands in [GaErGa(bpb-bzimpy)3]9+ and [Er(L)3]3+ (L = Et-bzimpy, Et-tpy, tpy, dpa-amide, dpa-ester). With this in mind, the predicted upconversion quantum yields produced under reasonable excitation intensity powers (1–30 W cm−2) for the ESA mechanism (Fig. 1) should not exceed ϕuptot = 10−11 (Fig. 2b). However, our experimental data, that we believe to be as accurate as possible, point to 5 × 10−11 ≤ ϕuptot ≤ 2 × 10−9 (Table 2). After a thorough and unfruitful look for the existence of alternative long-lived non-emissive excited relays, we conclude that the only acceptable explanation relies on unusally large Er-based cross sections for the excited-state absorption in the 2 × 10−21 ≤ σ1→2Er ≤ 2 × 10−19 cm2 (i.e. 1 ≤ ε1→2Er ≤ 50 M−1 cm−1). This phenomenon also plays a crucial role in boosting the apparent ETU mechanism initially assigned to [CrErCr(bpb-bzimpy)3]9+ where Cr(III) act as sensitizers for Er(III). Interestingly, chromophores for which the excited-state absorption (ESA) cross section is larger than the ground state absorption (GSA) in a target spectral region may display reverse saturable absorption (RSA) which finds applications in optical limiting devices.51 This behavior is observed in many organic molecules, including metal-containing porphyrin and phthalocyanine chromophores,52 but might be extended to metallosupramolecular assemblies with 4f-block cations. Finally, the decomposition of the total upconversion quantum yields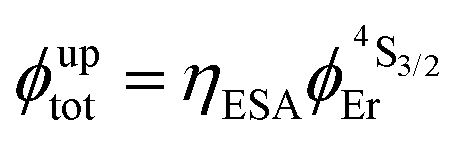 or
or 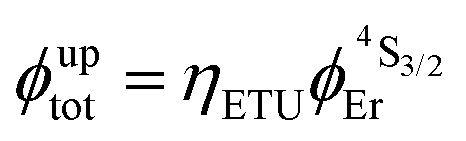 demonstrate that the intrinsic quantum yield
demonstrate that the intrinsic quantum yield 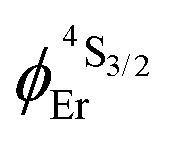 , which is very low for the molecular erbium complexes (
, which is very low for the molecular erbium complexes (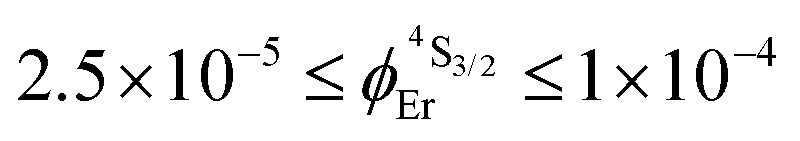 , Table 1), represents a crucial parameter to be optimized since it counts for approximately 50–70% of ϕuptot. In this context, Charbonnière and coworkers cleverly exploited Tb(III) as an alternative activator for molecular upconversion, since its associated rough intrinsic quantum yield of
, Table 1), represents a crucial parameter to be optimized since it counts for approximately 50–70% of ϕuptot. In this context, Charbonnière and coworkers cleverly exploited Tb(III) as an alternative activator for molecular upconversion, since its associated rough intrinsic quantum yield of 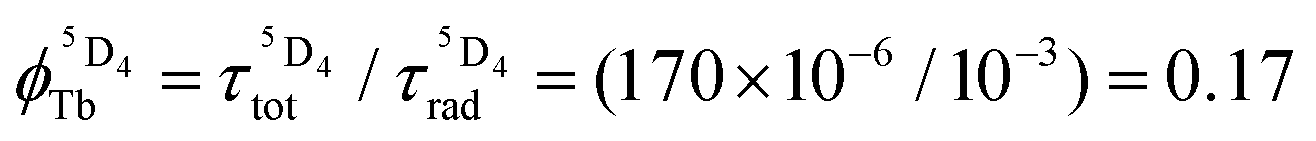 ,53 estimated for soluble heteronuclear Yb/Tb assemblies, may overcome the limited efficiency of the associated cooperative upconversion mechanism (ηCU). This approach lead to the currently largest reported molecular light upconversion quantum yield of
,53 estimated for soluble heteronuclear Yb/Tb assemblies, may overcome the limited efficiency of the associated cooperative upconversion mechanism (ηCU). This approach lead to the currently largest reported molecular light upconversion quantum yield of 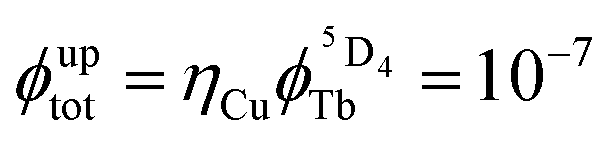 (CD3OD, 298 K, λexc = 980 nm and P = 2.86 W cm−2).21c Further efforts should be now focused on theoretical justifications for the unusually large absorption cross sections found for the excited state absorption occurring in molecular erbium complexes.
(CD3OD, 298 K, λexc = 980 nm and P = 2.86 W cm−2).21c Further efforts should be now focused on theoretical justifications for the unusually large absorption cross sections found for the excited state absorption occurring in molecular erbium complexes.
Author contributions
Conceptualization, B.G., H.B, H.N. and C.P.; methodology and practical chemical and spectroscopic studies, B.G., I.T., J.-R.J. and A.F.; crystallography, L.G. and C.B.; theoretical calculations H.B. (crystal-field and electronic transitions) and C.P. (kinetics); writing draft report, B.G., I.T. and H.B.; writing of ms and editing C.P. and A.F.; project administration and funding acquisition C.P.Conflicts of interest
They are no conflict to declare.Acknowledgements
This work is supported through grants from the Swiss National Science Foundation (grant 200020_178758). HB thanks Trond Saue (Toulouse) for fruitful discusions.References
- M. Goeppert-Mayer, Ann. Phys., 1931, 401, 273–294 CrossRef.
- (a) J. A. Kerr, Philos. Mag. Ser. 4, 1875, 50, 337–348 CrossRef; (b) J. Kerr, J. Phys. Theor. Appl., 1879, 8, 414–418 CrossRef.
- P. A. Franken, A. E. Hill, C. W. Peters and G. Weinreich, Phys. Rev. Lett., 1961, 7, 118–119 CrossRef.
- W. Kaiser and C. G. B. Garrett, Phys. Rev. Lett., 1961, 7, 229–231 CrossRef CAS.
- R. Medishetty, J. K. Zareba, D. Mayer, M. Samoc and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- (a) X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC; (b) J.-C. G. Bünzli and A.-S. Chauvin, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier North Holland, Amsterdam, 2014, vol. 44, pp. 169–281 Search PubMed.
- (a) V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed; (b) L. D. Sun, Y.-F. Wang and C.-H. Yan, Acc. Chem. Res., 2014, 47, 1001–1009 CrossRef CAS PubMed; (c) J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- (a) N. Bloembergen, Phys. Rev. Lett., 1959, 2, 84–85 CrossRef CAS; (b) J. F. Porter, Phys. Rev. Lett., 1961, 7, 414–415 CrossRef CAS; (c) C. A. Parker and C. G. Hatchard, Proc. Chem. Soc., 1962, 386–387 CAS; (d) C. A. Parker, Proc. R. Soc. London, Ser. A, 1963, 276, 125–135 CrossRef CAS.
- (a) D. R. Gamelin and H. U. Güdel, Acc. Chem. Res., 2000, 33, 235–242 CrossRef CAS PubMed; (b) F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed; (c) B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC; (d) M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed; (e) P. Ramasamy, P. Manivasakan and J. Y. Kim, RSC Adv., 2014, 4, 34873–34895 RSC; (f) D. M. Wu, A. Garcia-Etxarri, A. Salleo and J. A. Dionne, J. Phys. Chem. Lett., 2014, 5, 4020–4031 CrossRef CAS PubMed; (g) S. Ye, E.-H. Song and Q.-Y. Zhang, Adv. Sci., 2016, 3, 1600302 CrossRef PubMed; (h) M. Kaiser, C. Wurth, M. Kraft, I. Hyppanen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC; (i) J. Zhou, J. L. Leano Jr., Z. Liu, K.-L. Wong, R.-S. Liu and J.-C. G. Bünzli, Small, 2018, 1801882 CrossRef PubMed; (j) Z. Yi, Z. Luo, X. Qin, Q. Chen and X. Liu, Acc. Chem. Res., 2020, 53, 2692–2704 CrossRef CAS PubMed; (k) X. Qin, A. N. Carneiro Neto, R. L. Longo, Y. Wu, O. L. Malta and X. Liu, J. Phys. Chem. Lett., 2021, 12, 1520–1541 CrossRef CAS PubMed.
- (a) A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC; (b) V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC; (c) P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529–6554 RSC.
- C.-G. Ma, M. G. Brik, D.-X. Liu, B. Feng, Y. Tian and A. Suchocki, J. Lumin., 2016, 170, 369–374 CrossRef CAS.
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CrossRef CAS.
- Y. Suffren, B. Golesorkhi, D. Zare, L. Guénée, H. Nozary, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Inorg. Chem., 2016, 55, 9964–9972 CrossRef CAS PubMed.
- (a) R. Martin-Rodriguez, S. Fischer, A. Ivaturi, B. Froehlich, K. W. Krämer, J. C. Goldschmidt, B. S. Richards and A. Meijerink, Chem. Mater., 2013, 25, 1912–1921 CrossRef CAS; (b) E. I. Madirov, V. A. Konyushkin, A. N. Nakladov, P. P. Fedorov, T. Bergfeldt, D. Busko, I. A. Howard, B. S. Richards, S. V. Kuznetsov and A. Turshatov, J. Mater. Chem. C, 2021, 9, 3493–3503 RSC.
- J. C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- (a) X. Cheng, D. Tu, W. Zheng and X. Chen, Chem. Commun., 2020, 56, 15118–15132 RSC; (b) G. Bao, S. Wen, G. Lin, J. Yuan, J. Lin, K.-L. Wong, J.-C. G. Bünzli and D. Jin, Coord. Chem. Rev., 2021, 213642 CrossRef CAS.
- (a) F. Auzel, C. R. Acad. Sci. Paris, 1966, B262, 1016–1019 Search PubMed; (b) F. Auzel, C. R. Acad. Sci. Paris, 1966, B263, 819–821 Search PubMed.
- C. Reinhard and H. U. Güdel, Inorg. Chem., 2002, 41, 1048–1055 CrossRef CAS PubMed.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed.
- (a) I. Hyppänen, S. Lahtinen, T. Ääritalo, J. Mäkelä, J. Kankare and T. Soukka, ACS Photonics, 2014, 1, 394–397 CrossRef; (b) D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, Dalton Trans., 2015, 44, 2529–2540 RSC; (c) A. Nonat, C. F. Chan, C. Platas-Iglesias, Z. Liu, W.-T. Wong, W.-K. Wong, K.-L. Wong and L. J. Charbonnière, Nat. Commun., 2016, 11978 CrossRef CAS PubMed.
- (a) N. Souri, P. Tian, C. Platas-Iglesias, K.-L. Wong, A. Nonat and L. J. Charbonnière, J. Am. Chem. Soc., 2017, 139, 1456–1459 CrossRef CAS PubMed; (b) A. Nonat, S. Bahamyirou, A. Lecointre, F. Przybilla, Y. Mély, C. Platas-Iglesias, F. Camerel, O. Jeannin and L. J. Charbonnière, J. Am. Chem. Soc., 2019, 141, 1568–1576 CrossRef CAS PubMed; (c) R. C. Knighton, L. K. Soro, A. Lecointre, G. Pilet, A. Fateeva, L. Pontille, L. Frances-Soriano, N. Hildebrandt and L. J. Charbonniere, Chem. Commun., 2021, 57, 53–56 RSC.
- (a) B. Golesorkhi, H. Nozary, L. Guénée, A. Fürstenberg and C. Piguet, Angew. Chem., Int. Ed., 2018, 57, 15172–15176 CrossRef CAS PubMed; (b) B. Golesorkhi, A. Fürstenberg, H. Nozary and C. Piguet, Chem. Sci., 2019, 10, 6876–6885 RSC.
- (a) R. C. Hilborn, Am. J. Phys., 1982, 50, 982–986 CrossRef CAS; (b) T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi and H. U. Güdel, Phys. Rev., 2000, B61, 3337–3346 Search PubMed.
- X. Xiao, J. P. Haushalter and G. W. Faris, Opt. Lett., 2005, 30, 1674–1676 CrossRef CAS PubMed.
- O. A. Blackburn, M. Tropiano, T. J. Sorensen, J. Thom, A. Beeby, L. M. Bushby, D. Parker, L. S. Natrajan and S. Faulkner, Phys. Chem. Chem. Phys., 2012, 14, 13378–13384 RSC.
- I. Grenthe, J. Am. Chem. Soc., 1961, 83, 360–364 CrossRef CAS.
- B. Golesorkhi, L. Guénée, H. Nozary, A. Fürstenberg, Y. Suffren, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Chem. – Eur. J., 2018, 24, 13158–13169 CrossRef CAS PubMed.
- F. Renaud, C. Piguet, G. Bernardinelli, J.-C. G. Bünzli and G. Hopfgartner, Chem. – Eur. J., 1997, 3, 1660–1667 CrossRef CAS.
- F. Renaud, C. Piguet, G. Bernardinelli, J.-C. G. Bünzli and G. Hopfgartner, Chem. – Eur. J., 1997, 3, 1646–1659 CrossRef CAS.
- D. Zare, Y. Suffren, H. Nozary, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2017, 56, 14612–14617 CrossRef CAS PubMed.
- J. Albertsson, Acta Chem. Scand., 1972, 26, 985–1004 CrossRef CAS.
- M. Autillo, L. Guerin, T. Dumas, M. S. Grogoriev, A. M. Fedoseev, S. Cammelli, P. L. Solari, D. Guillaumont, P. Guilbaud, P. Moisy, H. Bolvin and C. Berthon, Chem. – Eur. J., 2019, 25, 4435–4451 CrossRef CAS PubMed.
- (a) SHAPE is a free software developed by M. Llunell, D. Casanova, J. Cirera, P.[:] Alemany and S. Alvarez and available online at http://www.ee.ub.edu/; (b) M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed; (c) D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed; (d) S. Alvarez, Dalton Trans., 2005, 2209–2233 RSC.
- B. Golesorkhi, H. Nozary, A. Fürstenberg and C. Piguet, Mater. Horiz., 2020, 7, 1279–1296 RSC.
- G. A. Kumar, R. E. Riman, L. A. D. Torres, O. B. Garcia, S. Banerjee, A. Kornienko and J. G. Brennan, Chem. Mater., 2005, 17, 5130–5135 CrossRef CAS.
- (a) F. Aquilante, L. De Vico, N. Ferre, G. Ghigo, P. A. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andres, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed; (b) J. Jung, M. A. Islam, V. L. Pecoraro, T. Mallah, C. Berthon and H. Bolvin, Chem. – Eur. J., 2019, 25, 15112–15122 CrossRef CAS PubMed.
- (a) N. C. Chang, J. B. Gruber, R. P. Leavitt and C. A. Morrison, J. Chem. Phys., 1982, 76, 3877–3889 CrossRef CAS; (b) T. A. Hopkins, J. P. Bolender, D. H. Metcalf and F. S. Richardson, Inorg. Chem., 1996, 35, 5356–5362 CrossRef CAS; (c) T. A. Hopkins, D. H. Metcalf and F. S. Richardson, Inorg. Chem., 1998, 37, 1401–1412 CrossRef CAS PubMed.
- K. Binnemans and C. A. Görller-Walrand, Chem. Phys. Lett., 1995, 245, 75–78 CrossRef CAS.
- (a) B. Bleaney, J. Magn. Reson., 1972, 8, 91–100 CAS; (b) V. S. Mironov, Y. G. Galyametdinov, A. Ceulemans, C. Görller-Walrand and K. Binnemans, Chem. Phys. Lett., 2001, 345, 132–140 CrossRef CAS; (c) D. Parker, E. A. Suturina, I. Kuprov and N. F. Chilton, Acc. Chem. Res., 2020, 53, 1520–1534 CrossRef CAS PubMed.
- (a) L. D. Carlos, R. A. S. Ferreira, V. de Zea Bernidez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509–534 CrossRef CAS; (b) J.-C. G. Bünzli, A.-S. Chauvin, H. K. Kim, E. Deiters and S. V. Eliseeva, Coord. Chem. Rev., 2010, 254, 2623–2633 CrossRef; (c) J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- (a) J. W. Verhoeven, Pure Appl. Chem., 1996, 68, 2223–2286 CAS; (b) K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed; (c) C. Wurth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- P. P. Ferreira da Rosa, S. Miyazaki, H. Sakamoto, Y. Kitagawa, K. Miyata, T. Akama, M. Kobayashi, K. Fushimi, K. Onda, T. Taketsugu and Y. Hasegawa, J. Phys. Chem. A, 2021, 125, 209–217 CrossRef CAS.
- (a) C. Görller-Walrand and K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner and L. Eyring, Elsevier Science, Amsterdam, 1998, vol. 25, pp. 101–264 Search PubMed; (b) C. Görller-Walrand and L. Fluyt, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science, Amsterdam, 2010, vol. 40, ch. 244, pp. 1–107 Search PubMed.
- (a) S. Bernadotte, A. J. Atkins and C. R. Jacob, J. Chem. Phys., 2012, 137, 204106 CrossRef; (b) G. Ganguly, H. D. Ludowieg and J. Autschbach, J. Chem. Theory Comput., 2020, 16, 5189–5202 CrossRef CAS PubMed.
- (a) C. K. Jorgensen and B. R. Judd, Mol. Phys., 1964, 8, 281–290 CrossRef; (b) R. Reisfeld and C. K. Jorgensen, Lasers and Excited States of Rare Earths, Springer-Veralg, Berloin Heidelberg New York, 1977, ch. 3, pp 123–156 CrossRef.
- M. E. Starzak, Mathematical Methods in Chemistry and Physics, Plenum Press, New York and London, 1989, pp. 300–309 Search PubMed.
- (a) M. A. Noginov, V. A. Smirnov and I. A. Shcherbakov, Opt. Quantum Electron., 1990, 22, S61–S74 CrossRef CAS; (b) M. A. Noginov, H. J. Caulfield, P. Venkateswarlu and M. Mahdi, Opt. Mater., 1996, 5, 97–103 CrossRef CAS.
- (a) R. Micheletti, P. Minguzzi, M. A. Noginov and M. Tonelli, J. Opt. Soc. Am. B, 1994, 11, 2095–2099 CrossRef CAS; (b) M. Pokhrel, G. A. Kumar, P. Samuel, K. I. Ueda, T. Yanagitani, H. Yagi and D. K. Sardar, Opt. Mater. Express, 2011, 1, 1272–1285 CrossRef CAS; T.-H. Kim, D.-J. Kim, W.-Y. Jang, A. Moon, K.-S. Lim and M. Lee, Jpn. J. Appl. Phys., 2011, 50, 06G11-01–06G11-04 Search PubMed; (c) Z. Gerelkhuu, B. T. Huy, J. W. Chung, T.-L. Phan, E. Conte and Y.-I. Lee, J. Lumin., 2017, 187, 40–45 CrossRef CAS.
- H. Lundt and H. Weidner, Opt. Commun., 1991, 82, 484–487 CrossRef CAS.
- (a) L. W. Tutt and T. F. Boggess, Prog. Quantum Electron., 1993, 17, 299–338 CrossRef CAS; (b) D. N. Bowman, J. C. Asher, S. A. Fischer, C. J. Cramer and N. Govind, Phys. Chem. Chem. Phys., 2017, 19, 27452–27462 RSC.
- (a) M. Calvete, G. Y. Yang and M. Hanack, Synth. Met., 2004, 141, 231–243 CrossRef CAS; (b) Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346–1353 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2059291–2059293. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D1DT01079D |
| This journal is © The Royal Society of Chemistry 2021 |