
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron transfer pathways in photoexcited lanthanide(III) complexes of picolinate ligands†
Daniel
Kovacs
,
Daniel
Kocsi
,
Jordann A. L.
Wells
 ,
Salauat R.
Kiraev
and
K. Eszter
Borbas
*
,
Salauat R.
Kiraev
and
K. Eszter
Borbas
*
Department of Chemistry, Ångström Laboratory, Box 523, Uppsala University, 75120, Uppsala, Sweden. E-mail: eszter.borbas@kemi.uu.se
First published on 1st March 2021
Abstract
A series of luminescent lanthanide(III) complexes consisting of 1,4,7-triazacyclononane frameworks and three secondary amide-linked carbostyril antennae were synthesised. The metal binding sites were augmented with two pyridylcarboxylate donors yielding octadentate ligands. The antennae carried methyl, methoxymethyl or trifluoromethyl substituents in their 4-positions, allowing for a range of excited state energies and antenna electronic properties. The 1H NMR spectra of the Eu(III) complexes were found to be analogous to each other. Similar results were obtained in the solid-state by single-crystal X-ray crystallography, which showed the structures to have nine-coordinate metal ions with heavily distorted tricapped trigonal prismatic geometries. Steady-state and time-resolved luminescence spectroscopy showed that the antennae could sensitize both Tb(III) and Eu(III), however, quantum yields were lower than in other octadentate complexes lacking pyridylcarboxylate. Complexes with more electron-poor pyridines were less emissive even when equipped with the same antenna. The oxidation and reduction potentials of the antennae and the pyridinecarboxylates, respectively, were determined by cyclic voltammetry. The obtained values were consistent with electron transfer from the excited antenna to the pyridine providing a previously unexplored quenching pathway that could efficiently compete with energy transfer to the lanthanide. These results show the crucial impact that photophysically innocent ligand binding sites can have on lanthanide luminescence.
Introduction
Pyridines are among the most versatile ligands for metal coordination. Transition metal pyridine and bipyridine complexes promote self-assembly1,2 and a variety of transformations,3–6 while their photophysical properties are used for both analyte detection7,8 and photocatalysis.9 The trivalent lanthanide (Ln) ions have coordination requirements that are distinct from those of the d-block metals. The 4f orbitals are shielded by the 5s and 5p orbitals, therefore, ligand binding is governed by coulombic forces. Since the confirmation of bidentate binding of α-picolinate and its N-oxide to Ln(III) ions a large variety of pyridine-based ligands have been developed for Ln(III) coordination (Fig. 1).10–15 Ln(III) ions commonly have large coordination numbers, typically 8–9,16 with some notable exceptions.17,18 Complex stability is improved by the introduction of additional coordinating groups onto the α-picolinate core, e.g. carboxylates,19 phosphonates,20,21 tetrazolates,22 and oxazolines23 (LI–LIV, Fig. 1a). Integration of the pyridine into a polyazamacrocycle (LV–LVII, Fig. 1a) can further increase stability,24–28 as demonstrated by the Eu(III) complex of locked-in cyclam LV incorporating two bidentate picolinates which is stable for over 167 days in 2 M aqueous HCl; under the same conditions the analogous DOTA chelate has a half-life shorter than 7 h.29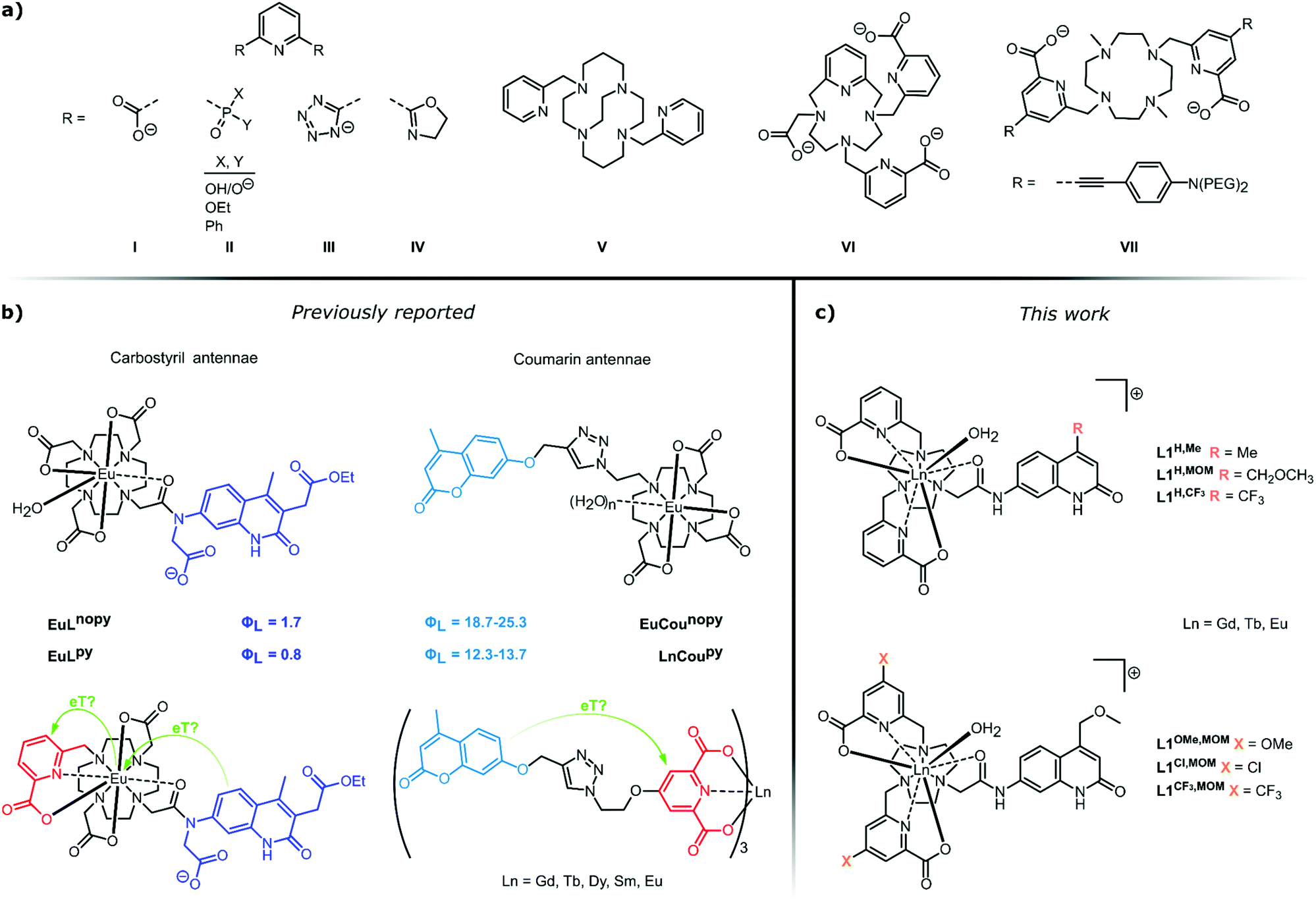 | ||
| Fig. 1 (a) A selection of pyridine-containing ligands for Ln(III) coordination and sensitisation. (b) Putative electron transfer steps in photoexcited Ln(III) complexes carrying carbostyril (left)55 or coumarin (right)52 light-harvesting antennae. (c) Complexes prepared in this work for the investigation of antenna-pyridine ligand interactions. | ||
Ln(III) emitters have now reached a level of maturity, indicated by the commercial availability, and industrial and medical application of several luminescent cryptates.30,31 However, this does not imply a full understanding of their photophysical behaviour. Ln(III) emissions originate from f–f transitions, and their direct excitation is inefficient. Indirect excitation is possible by placing a light-harvesting moiety (antenna) close to the Ln(III). Pyridine-based ligands allow for the introduction of sensitizing antennae in a variety of constellations (Fig. 1a). Simple pyridines, picolinates, and bipyridines are suitable sensitizers for multiple Lns.13,23,32–34 Cs3Eu(LI)3 and Cs3Tb(LI)3 have found use as standards for luminescent quantum yield (Φ) determinations35 and as invisible inks.36 Tb(LVI)3 has a luminescence quantum yield of 90%.37 The heterocycle absorption spectrum can be red-shifted through judiciously chosen para-substituents, or by fusion of additional aryl rings onto the pyridine.21 The push–pull systems obtained by para-functionalization with electron-donating alkynes yield chromophores that are excellent sensitisers for Eu(III),38–42 and have large two-photon absorption cross-sections.43–47 Thus, the near infrared-emitting YbLVII and related structures can be excited in the red at λex = 760 nm.48,49 Sensitising antennae can also be grafted onto the pyridine using a linker,50–54 or both pyridine ligand and antenna can be assembled around a macrocyclic core.55 The coordination and sensitising properties of the ligand are often assumed to work independently of each other.
Energy transfer (EnT) can occur from the ligand triplet or singlet via several alternative mechanisms, and both the antenna and the lanthanide excited states are subject to quenching by a variety of processes. We have recently reported that photoinduced electron transfer (PeT) from the antenna to the Ln(III) is a prominent quencher for several lanthanides. The largest effect was seen for Eu(III), with the most positive reduction potential.56 PeT quenching was in some cases comparable in effect to that of the well-known X–H quenching.57 Vibronic quenching of the lanthanide excited states due to coupling to X–H (X = O, N, C) oscillators has long been known, and provides the basis for the determination of the hydration state of lanthanide complexes.58–60 In an effort to eliminate a coordinated water quencher from octadentate DO3A-type Eu complexes we prepared a nonadentate ligand (Fig. 1b, left).55 Much to our surprise no improvement was seen in the Eu(III) luminescence quantum yield (ΦEu) compared to the octadentate parent. Analysis of the sensitization efficiency (ηsens) and the residual antenna fluorescence quantum yield (ΦL) suggested the presence of an additional quenching process depleting the antenna. We hypothesised that the pyridine engaged in a competitive PeT from the excited antenna. This suggestion was somewhat supported by re-analysis of the photophysical properties of lanthanide complexes in picolinate-linked oxycoumarin antennae.51,52 Specifically, we noted a marked decrease in the coumarin emission when the ligand contained a picolinate binding site (Fig. 1b, right). This difference was observable irrespective of the nature of the lanthanide, including in non-photoactive, redox-inactive Gd(III), photophysically active, redox-inactive Tb(III), and luminescent and reducible Eu(III). Interestingly, in the nonadentate ligands binding Tb(III) or Gd(III) there was only a minor difference (10 and 5%, respectively) in ΦL compared to the octadentate ones (Fig. 1b, left).
Here, we investigate the fate of photoexcited carbostyril antennae in ligands containing pyridines. The structures and the compound numbering are shown in Fig. 1c. All ligands are octadentate, the Ln(III) coordination sphere is completed by a water molecule. As metal-bound water molecules quench the Ln excited state, keeping this number constant facilitates comparison of the lanthanide-based emissions. Sensitisation is performed by carbostyril antennae carrying either electron-donating Me, or slightly or strongly electron-withdrawing methoxymethyl (MOM) or CF3 groups in the 4-positions. The abovementioned class of ligands contains two unsubstituted carboxypyridine groups and the electronically different carbostyrils (LH,Me, LH,MOM, LH,CF3). In the second set of ligands, four donor atoms are provided by two carboxypyridines with electron-donating (OMe, LOMe,MOM), electron-neutral (H, LH,MOM) and electron-withdrawing (Cl, LCl,MOM and CF3, LCF3,MOM) para-substituents; the antenna in these ligands is only the MOM-substituted carbostyril. These ligands test the electron-accepting ability of the pyridines. In both sets of ligands the antenna and the coordination sphere are linked through a secondary amide.
Results and discussion
Synthesis
Complexes were synthesised as shown in Scheme 1. Additional details, experimental procedures, and characterization for all new compounds are given in the ESI.† Triazacyclononane (TACN) 1 was dialkylated with picolinate benzylic bromide 2. Alkylation of the secondary nitrogen in the macrocycle was possible using the appropriately substituted carbostyril chloroacetate 3. Basic hydrolysis of the methyl esters yielded the ligands L, which could be complexed by exposure to LnCl3 (Ln = Eu, Tb, Gd) in a warm EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture. The complexes were isolated in quantitative yield as white solids.
:H2O mixture. The complexes were isolated in quantitative yield as white solids.
 | ||
| Scheme 1 Preparation of LnLH,R (left) and LnLX,MOM (right). | ||
To investigate the hypothesized communication between the excited antenna and the pyridines, a series of complexes was prepared where the picolinate arms were para-substituted with electron-donating OMe, or electron-withdrawing Cl or CF3 groups (Scheme 1, right). TACN 1 was monoalkylated with the MOM substituted carbostyril chloroacetate 3b yielding intermediate 5. Dialkylation of the two secondary nitrogens was possible using p-substituted picolinate benzylic bromides, giving 6X,MOM. Methyl ester hydrolysis and complexation were carried out as described above, and the complexes were isolated in 73% to quantitative yield as white solids.
1H NMR spectroscopy
The ligands with the variously p-substituted picolinates had similar 1H NMR spectra. The chemical shifts of H-3 and H-5 of the protected ligands 6X,MOM were indicative of the electronic properties of the pyridine.61 H-3 were observed at 7.38, 7.78–7.89, 7.88, and 8.05 ppm, and H-5 at 7.12, 7.57–7.66, 7.70, and 7.95 ppm for the increasingly electron-withdrawing OMe, H, Cl, and CF3 substituents, respectively (Fig. S1†).Full assignment of the EuL1H NMR spectra was not possible as we could not obtain good quality 2D NMR spectra. At r.t. in CD3OD the spectra were consistent with the presence of two enantiomers of a single diastereomer. Upon heating to 50 °C the 1H NMR signals slightly broadened, as did the 19F NMR signals of EuLCF3,MOM. Cooling of the solutions to 0 °C also caused broadening relative to what was seen at room temperature, suggesting the presence of dynamic equilibria that are being slowed down (Fig. S2–S14†).
Crystallography
Single crystals suitable for X-ray diffraction analysis were obtained by vapour diffusion of glyme into concentrated aqueous solutions of GdLH,MOM–F, TbLCF3,MOM–Cl, GdLCl,MOM–F, and EuLCl,MOM–Cl. The fluoride structures were obtained from solutions containing one equiv. KF, which was added to facilitate crystallization. All four compounds have a nine-coordinate Ln centre with (heavily) distorted tricapped trigonal prismatic geometry (Fig. 2 and S16–S18†). The planes of the trigonal prism are formed by the triazacyclononane N-donors (N3PL), and the pyridine N- and antenna amide O-donors (NNOPL). The remaining two carboxylate ligands and a fluoride or chloride ion cap the trigonal prism (Fig. 2). The two planes are not co-planar with N3PL–Ln–NNOPL angles ranging 114–120°, owing to the significantly distorted geometry. The lanthanide centres are just below NNOPL (∼0.3 Å), and the Ln–N3PL distances range 2.017(4) to 2.065(4) Å. The complexes are racemic in the solid state, with both Δ and Λ isomers present in the unit cell.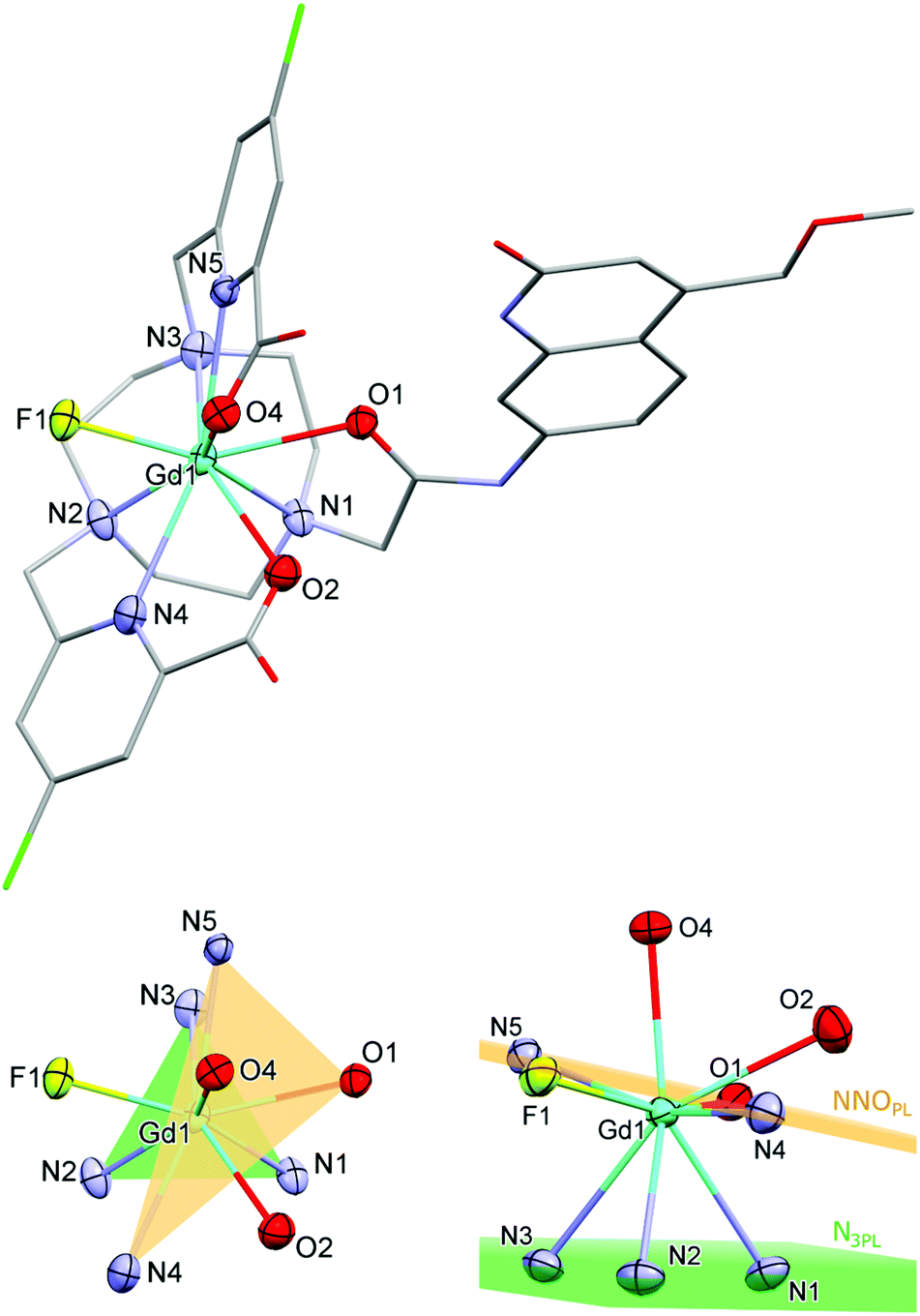 | ||
| Fig. 2 X-ray crystal structure of GdLCl,MOM-F (top) and the Gd coordination environment (bottom). H atoms and solvent molecules are omitted for clarity. The first coordination sphere of the metal centre is depicted with ellipsoids at 50% probability, the rest of the atoms are displayed as capped sticks. | ||
Overall, the bond distances are comparable across this series of complexes, with no significant changes between fluoride- or chloride-bound complexes. The Ln–NTACN range 2.630(6)–2.671(7) Å, comparing well with related lanthanide complexes reported previously. The Ln–Npy distances for GdLH,MOM and TbLCF3,MOM (2.557(2) Å and 2.532(6) Å, respectively) are comparable to those of related complexes (mean 2.533 Å) while those for GdLCl,MOM–F and EuLCl,MOM–Cl are slightly elongated (2.581(5) Å and 2.594(8) Å, respectively).62 The carboxylate Ln–O distances range 2.403(2)–2.436(3) Å, which is slightly longer than those reported for related TACN62 and cyclen57 complexes (mean 2.338 Å), and could be due to the steric demand of the carbostyril antenna and the smaller TACN ligand platform. The antenna Ln–O1 distances range 2.450(5)–2.474(2) Å, comparing well to related carbostyril-substituted cyclen complexes (mean 2.444 Å).57,62–65 The Ln–F distances are shorter than in the square antiprismatic Eu cyclen complex previously reported (range 2.117(3)–2.185(2) Å versus 2.225(2) Å), and are shorter than the Ln–Cl (TACN) and Ln–O (cyclen) distances of related complexes.‡
Electrochemistry
The ability of the pyridines to accept electrons from the photoexcited antenna was evaluated by determining the reduction potentials of the p-substituted pyridines and the oxidation potential of the MOM-substituted carbostyril antenna. The reduction potentials of the unsubstituted picolinate fragments were measured by cyclic voltammetry in the model compounds GdLX (X = CF3, Cl, H, OMe, Fig. 3) to account for the effect of Ln(III) binding but avoid potential interference from the antenna. The CF3-substituted pyridines were expected to be the easiest to reduce providing a well characterised irreversible reduction wave at −1.13 V (vs. NHE), while the Cl-substituted ones being less reducible showed diminished response at −1.21 V (vs. NHE). Along the series the H-substituted analogues revealed faint reduction wave at −1.29 V (vs. NHE), while the reduction of the OMe-substituted one was outside of the solvent window (Fig. 4a).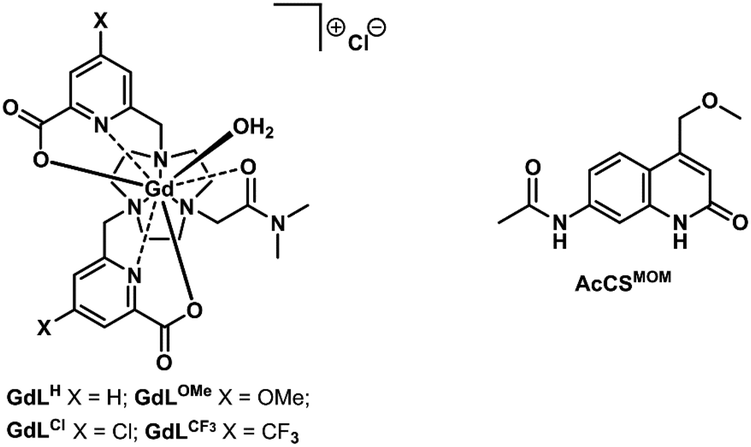 | ||
| Fig. 3 Model compounds for electrochemistry. | ||
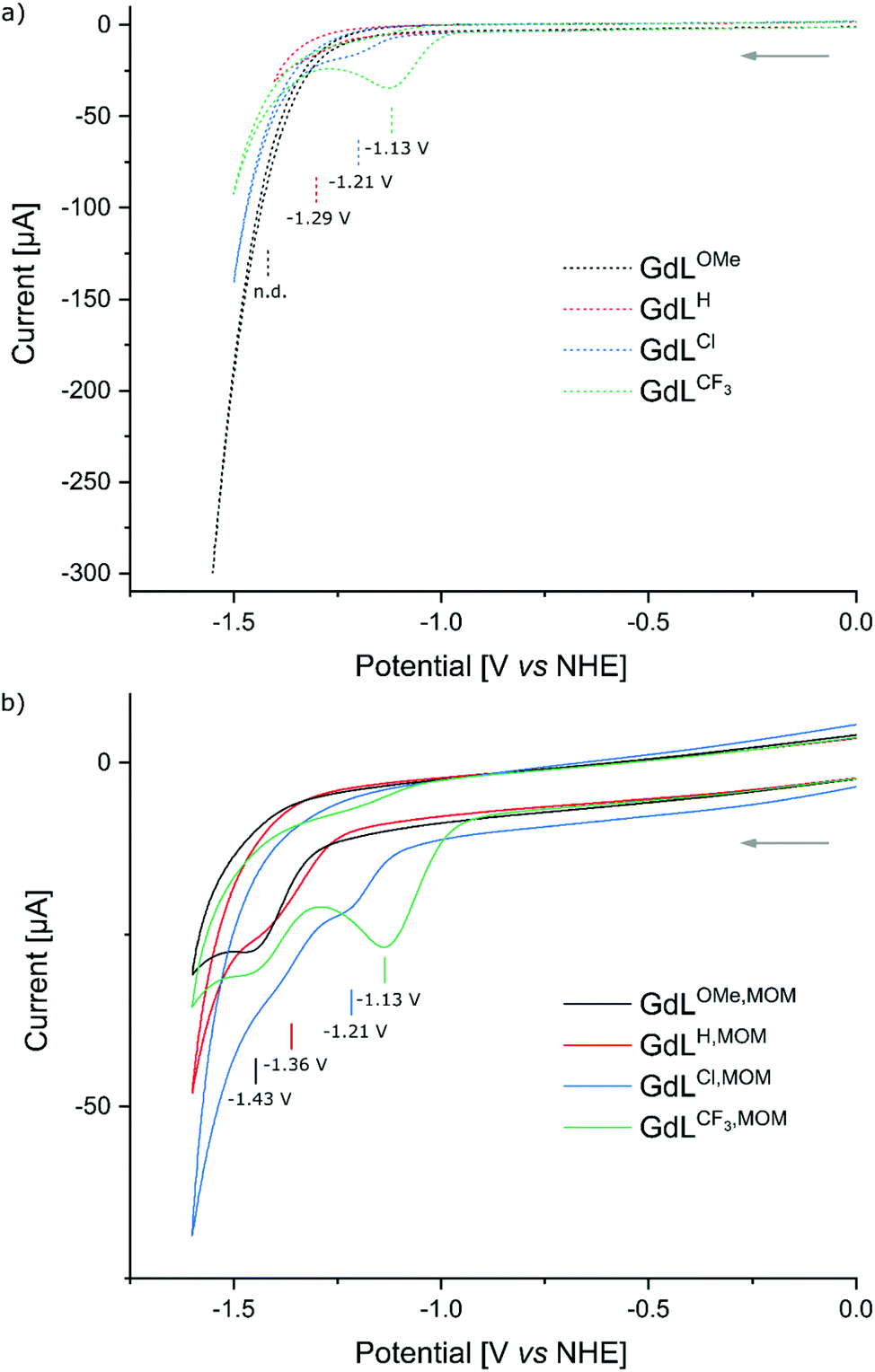 | ||
| Fig. 4 Cyclic voltammograms of 0.2 mM solutions of complexes in 100 mM aqueous NH4Cl solution under Ar using a glassy carbon working electrode, Ag/AgCl/KCl(sat.) reference electrode, and a Pt wire counter electrode at 100 mV s−1 scan rate. (a) Overlaid cyclic voltammograms of GdLX complexes. (b) Overlaid cyclic voltammograms of GdLX,MOM complexes. | ||
The cyclic voltammograms of GdLX,MOM showed poorly defined irreversible reduction waves at −1.13 V, −1.21 V, −1.36 V, and −1.43 V (vs. NHE) for X = CF3, Cl, H, and OMe, respectively (Fig. 4b). Irreversible reductions at comparable potentials (−1.54 V vs. Ag/AgCl/KCl(sat.), −1.34 V vs. NHE) have been observed during CV analysis of pyridine under acidic conditions.66 Therefore, these waves were assigned to picolinate-based reduction events. The influence of the para-substituent on the pyridine reduction potential mirrors what has been observed in Pd(II)–C^N^C pincer complexes.61 The first reduction event in the latter series occurred at ∼0.2 V more positive values than in the Gd complexes, which is consistent with their lacking the fully negatively-charged carboxylate. The reduction potential of the model compound GdLH was more positive by ∼70 mV than that of the analogous complex with the antenna, GdLH,MOM. Variations in the amide substituents could influence the reduction potentials of the encapsulated metals, and likely for the rest of the ligand. However, the effect of such substitution is expected to be much smaller than seen here and of the opposite direction.67
The excited state oxidation potential of the MOM-substituted carbostyril was estimated from the singlet excited states of the ligand (3.53 eV, vide infra) and the oxidation potential of the ground state antenna (1.76 V vs. NHE, AcCSMOM, Fig. 3 and S19–S21†). Using eqn (1), the free energy of the electron transfer (ΔGET) can be calculated from the oxidation potential of the carbostyril (Eox), the reduction potential of the pyridine (Ered), the excited state of the antenna (ES), and the attraction between the radical ion pair (e02/ε). The last term is ∼0.15 eV for an exciplex,68 and this is the value we will use.
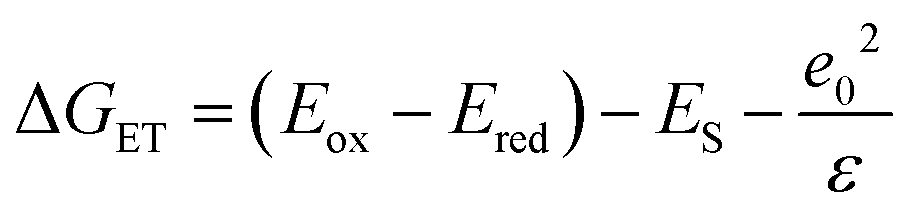 | (1) |
Negative ΔGET was calculated for PeT from S1 for GdLH (−0.63 eV) and the GdLX,MOM (−0.79, −0.71, −0.56, −0.49 eV for X = CF3, Cl, H, and OMe, respectively), PeT from the first triplet excited state (T1) was not thermodynamically favoured. Linear correlations were found between the substituent constants described by Hammett and the measured reduction potentials of the pyridine units. The picolinate electron accepting ability increases with increasing electron withdrawing ability of the para-substituent (Fig. 5). This observation supports our hypothesis that as the electron accepting ability of the pyridines increase, greater PeT quenching of the antenna is observed. Therefore, lower sensitization efficiency, and thus weaker Ln luminescence was expected.
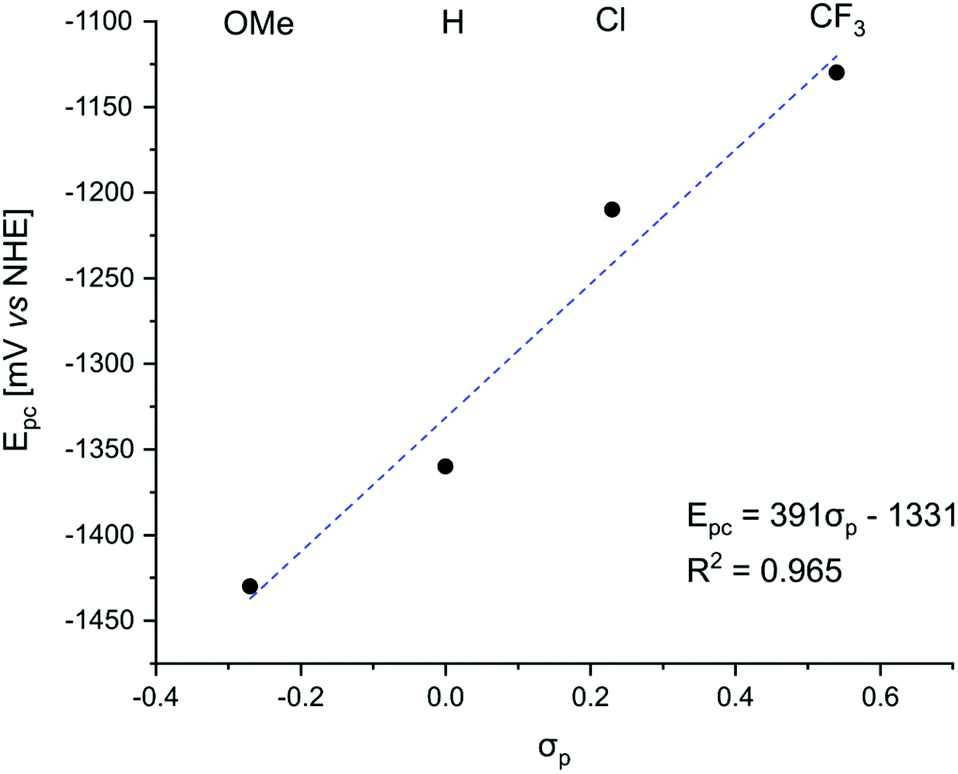 | ||
| Fig. 5 Values for linear fit of Hammett σp constants vs. pyridine reduction potentials. | ||
Photophysical studies
The expected effect of the pyridine electronic properties on Ln(III) luminescence were investigated using UV-Vis absorption, and steady-state and time-resolved emission spectroscopies. The photophysical properties of LnL were determined in PIPES-buffered aqueous solutions (pH 6.5) at complex concentrations in the 10–15 μM range. Changes in the absorption spectra were observed both upon changing the picolinate p-substituent, and the 4-substituent of the antenna. Pyridine substitution caused absorption changes only in the 250–310 nm range. Above 310 nm only the carbostyril antennae absorb, thus, emission spectra collected with λex > 330 nm are due to carbostyril-based sensitisation. Absorption (λabs) and emission (λem) maxima were red-shifted with increasing electron-withdrawing ability (CF3 > MOM > Me) of the carbostyril 4-substituent (Table 1). For the same type of antenna λem were at the same wavelength irrespective of the rest of the ligand structure. Thus, λem was shortest for the Me-substituted carbostyril (366 nm), followed by the MOM (375 nm) and finally the CF3-substituted antenna had the lowest-energy emission (391 nm).a
| Complex | λ abs [nm] |
λ
em [ms]b |
E
00(S1) [cm−1]c |
E
00(T1) [cm−1]c |
|---|---|---|---|---|
| a In aqueous PIPES buffer (10 mM), pH 6.5. b λ ex = 329 nm (GdLH,Me), 331 nm (GdLH,MOM, GdLH,CF3). c Calculated from the 0–0 phonon transitions observed in the Gd-complex at 77 K. | ||||
| GdLH,Me | 329 | 366 | 28900 |
22900 |
| GdLOMe,MOM | 331 | 375 | 28500 |
22500 |
| GdLH,MOM | 331 | 375 | 28500 |
22500 |
| GdLCl,MOM | 331 | 375 | 28500 |
22500 |
| GdLCF3,MOM | 331 | 375 | 28500 |
22500 |
| GdLH,CF3 | 342 | 391 | 27400 |
21700 |
The T1 energies of the antennae were determined from the 77 K steady-state emission spectra of the Gd-complexes (Table 1 and Fig. S42†). The CF3-substituted antenna had the lowest energy, while the highest value was found for the Me-substituted one. The antenna fluorescence excitation spectra matched the absorption spectrum attributed to the carbostyril unit (Fig. S30–S41†). The excitation spectra of the phosphorescence bands, however, matched the absorption spectrum of the entire complex. The pyridine pendant arms have observable triplet energy levels in this region (Fig. S44†).50 The antenna triplets are reasonably well-placed for energy transfer to Eu(III), as they are more than 2000 cm−1 but not more than 5000 cm−1 above its 5D0 excited state. The T1 of the CF3-substituted antennae at 21700 cm−1 (Table 1) were within 2000 cm−1 above the Tb(III) 5D4 excited state at 20490 cm−1.69
Antenna excitation at λex = 330 nm resulted in Tb(III) and Eu(III) emission. For Tb the 5D4 → 7FJ (J = 6–0) transitions were seen at 488, 543, 582, 620, 652, 668 and 678 nm, respectively. The J = 5 transition was found to be more than twice as high as the second most intense peak. In the Eu spectra the 5D0 → 7FJ (J = 0–5) transitions were located at 579, 593, 614, 649, 693 and 751 nm, along with the hypersensitive and higher energy 5D1 → 7FJ (J = 0, 1) transitions (537, 554 nm, respectively) (Fig. 6). The shape of the spectra and intensity of the peaks were different from those of the cyclen-based complexes carrying the same antenna/linker due to the different ligand environment (Fig. S49 and S50†). The most intense peak corresponds to the ΔJ = 2 transition in contrast to the cyclen-based Eu complexes, where the ΔJ = 4 transition is the strongest.
 | ||
| Fig. 6 Steady-state emission spectra of TbLH,MOM (top) and EuLH,MOM (bottom) with the transitions indicated. | ||
The overall luminescence quantum yields of the complexes were determined in 10 mM aqueous PIPES buffer (pH = 6.5) using quinine sulfate (Φ = 0.59) in H2SO4 (0.05 M)70 as the reference (Table 2). Tb(III) quantum yields were 25.4–30.6% for the complexes carrying the 4-Me or the 4-MOM-substituted carbostyril and OMe, Cl and H-substituted pyridines. A CF3-substituent in either the antenna 4-position or in the pyridine para-position resulted in diminished ΦTb. The Tb(III) emission intensity of TbLH,CF3 increased from ΦTb = 3.3% upon deoxygenation; ligand emission was not affected. The oxygen sensitivity of ΦTb is consistent with back energy transfer from the Tb(III) excited state to the antenna T1 and the quenching of the antenna T1 by dissolved 3O2 (Fig. S54 and S55†). ΦEu values were in the 0.8–8.0% range, which is comparable to complexes sensitised by carbostyril antennae in DO3A frameworks.
| Complex |
Φ
L [%]a |
Φ
Ln [%]a |
|---|---|---|
| a Relative to quinine sulfate (Φ = 0.59) in H2SO4 (0.05 M).70 b Mean ± standard deviation for two independent measurements. c Mean ± standard deviation for three independent measurements. | ||
|
GdLH,Meb |
4.40 ± 0.10 | — |
|
GdLOMe,MOMb |
6.85 ± 0.19 | — |
|
GdLH,MOMb |
6.42 ± 0.28 | — |
|
GdLCl,MOMb |
4.64 ± 0.03 | — |
|
GdLCF3,MOMb |
2.11 ± 0.07 | — |
|
GdLH,CF3b |
4.53 ± 0.17 | — |
|
TbLH,Meb |
3.50 ± 0.10 | 27.05 ± 1.05 |
|
TbLOMe,MOMb |
5.23 ± 0.12 | 30.55 ± 1.75 |
|
TbLH,MOMb |
4.92 ± 0.01 | 28.05 ± 0.95 |
|
TbLCl,MOMb |
3.60 ± 0.08 | 25.35 ± 1.25 |
|
TbLCF3,MOMb |
1.79 ± 0.02 | 13.10 ± 0.20 |
|
TbLH,CF3b |
4.16 ± 0.24 | 3.24 ± 0.14 |
|
EuLH,Mec |
0.42 ± 0.03 | 0.83 ± 0.02 |
|
EuLOMe,MOMc |
0.12 ± 0.03 | 3.61 ± 0.15 |
|
EuLH,MOMc |
0.16 ± 0.01 | 2.44 ± 0.07 |
|
EuLCl,MOMc |
0.08 ± 0.03 | 1.34 ± 0.04 |
|
EuLCF3,MOMc |
0.06 ± 0.04 | 0.75 ± 0.03 |
|
EuLH,CF3c |
0.61 ± 0.02 | 7.95 ± 0.42 |
The intensity of Ln(III) luminescence was dependent on the pyridine donors in the ligand. For complexes carrying the same antenna ΦL decreased dramatically with decreasing pyridine electron density (Fig. S51–S53†). The ligand emission was only a third (2.1%) in GdLCF3,MOM compared to the ΦL of GdLOMe,MOM (6.9%). The trend was reproduced in the Tb(III) complexes. Within the series, a smaller ΦL was accompanied by smaller ΦLn, further confirming that the pyridines promote quenching of the antenna. The data for Eu(III) follow the same trend, albeit ΦL of EuLH,MOM is somewhat larger than that of EuLOMe,MOM. Given the small ΦL of the Eu(III) complexes, these variations carry a large uncertainty.
The luminescent lifetimes of EuLX,R and TbLX,R were determined using time-resolved luminescence spectroscopy (Table 3). In H2O EuLX,R had lifetimes (τH2O) ∼0.50 ms, in D2O these (τD2O) were lengthened to ∼1.30 ms. The number of Ln(III)-coordinated water molecules (q) were calculated using the following equations: q = (5 ms)·(1/τH2O − 1/τD2O − 0.06 ms−1) for Tb, and q = (1.2 ms)·(1/τH2O − 1/τD2O − 0.25 ms−1 − m·0.075 ms−1) for Eu, where m is the number of nearby N–H oscillators.58,60 The complexes had one Ln(III)-bound water molecule, which is in line with an expected nine-coordinate structure. The q-values obtained for CF3-substituted TbL1H,CF3 and TbL1CF3,MOM were unrealistic, presumably due to the presence of additional Ln(III)-quenching pathways. Deoxygenation of the samples increased the integrated luminescence intensity (315–800 nm) by 50 and 28%, respectively. The oxygen sensitivity of these complexes suggests that energy back transfer to the antenna triplet is taking place in both. Given the high structural similarities of the complexes, even for these two complexes q is most likely 1. An alternative energy transfer from the Tb(III) to the picolinates was also conceivable. Substitutions in more extensively conjugated dipicolinate-type ligands have been found to dramatically shift T1, from 27050 cm−1 in unsubstituted dipicolinate71 to 22080 cm−1 in p-aminopicolinate72 and 21080 cm−1 in the p-thienylated derivative.23 The energies of T1 of the picolinate fragments in the current complexes were determined from the 77 K luminescence spectra of GdLX (Fig. S44†). Due to the lack of vibrational structure in the spectra T1 were located by deconvolution of the emission bands into Gaussian functions. This analysis yielded T1 at 25600, 25600, 25400, and 25100 cm−1, for GdLX (for X = OMe, H, Cl, and CF3, respectively; Fig. S45–S48†). These values are higher than that of the antenna T1, therefore any energy back transfer would likely involve the latter (located at 22500 cm−1). Thus, Tb(III) to picolinate EnT is unlikely.
| Complex |
τ
H2O [ms]b |
τ
D2O [ms]b |
q |
|---|---|---|---|
| a In PIPES buffered (10 mM, pH 6.5), non-deaerated aqueous solutions at nominally 10 μM complex concentrations. b Lifetime values were averaged from three independent measurements and are subject to an error of ±10%.60 | |||
| TbLH,Me | 1.12 | 1.67 | 1.16 |
| TbLOMe,MOM | 0.76 | 1.01 | 1.29 |
| TbLH,MOM | 0.82 | 1.08 | 1.19 |
| TbLCl,MOM | 0.73 | 0.99 | 1.48 |
| TbLCF3,MOM | 0.60 | 0.94 | — |
| TbLH,CF3 | 0.09 | 0.16 | — |
| EuLH,Me | 0.50 | 1.27 | 1.06 |
| EuLOMe,MOM | 0.51 | 1.34 | 1.09 |
| EuLH,MOM | 0.50 | 1.33 | 1.13 |
| EuLCl,MOM | 0.49 | 1.20 | 1.06 |
| EuLCF3,MOM | 0.48 | 1.14 | 1.05 |
| EuLH,CF3 | 0.51 | 1.28 | 1.04 |
The reasons behind the different ΦL and ΦLn in LnLX,R were next investigated. ΦLn is a product of the efficiency of the sensitization (ηsens) and the intrinsic quantum yield (ΦLnLn) of the Ln (eqn (2)).73 The former is influenced by processes that are disruptive to the antenna excited state, e.g. X–H quenching of the antenna or PeT to or from the antenna. The intrinsic quantum yield is mostly determined by the coordination environment of the Ln(III). For Eu(III), ηsens and ΦLnLn can be calculated from the corrected Eu(III) luminescence spectrum using eqn (2) and (3).74 Here, τrad is the Eu(III) radiative lifetime, Itot and IMD are the integrated full spectrum (521–800 nm) and the area for the 5D0 → 7F1 transition (582–603 nm), respectively. AMD,0 (14.65 s−1) is the spontaneous emission probability for the 5D0 → 7F1 transition of Eu(III) in vacuo; τobs is the same as τH2O. The refractive index of the solution is taken as n = 1.333 in H2O.75 The results are summarised in Table 4.
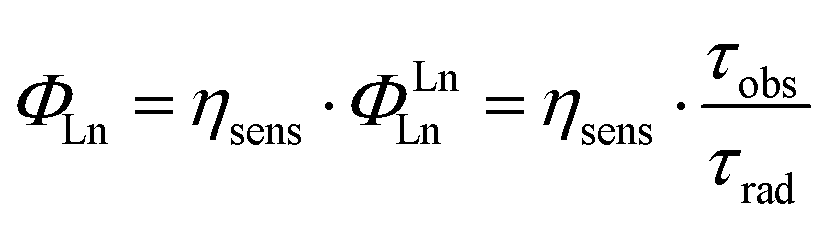 | (2) |
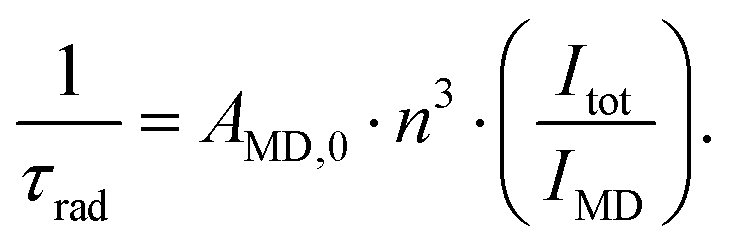 | (3) |
| Complex | τ rad [ms] | τ obs [ms] | Φ EuEu [%] | η sens [%] |
|---|---|---|---|---|
| a Measurements were performed in 10 mM aqueous PIPES buffer solutions at pH 6.5 at 10 μM concentrations. | ||||
| EuLH,Me | 2.87 | 0.50 | 17.4 | 5 |
| EuLOMe,MOM | 2.67 | 0.51 | 19.0 | 20 |
| EuLH,MOM | 2.86 | 0.50 | 17.5 | 14 |
| EuLCl,MOM | 2.69 | 0.49 | 18.2 | 8 |
| EuLCF3,MOM | 2.85 | 0.48 | 16.9 | 5 |
| EuLH,CF3 | 2.87 | 0.51 | 17.8 | 46 |
The ligand may influence ηsens indirectly, by modulating the Eu(III) reduction potential, or via a separate quenching pathway that does not involve the Ln(III). More electron-rich ligands would be expected to stabilize Eu(III), and thus result in less PeT. Thus, for the same antenna ηsens should decrease in the following order: EuLOMe,MOM > EuLH,MOM > EuLCl,MOM > EuLCF3,MOM; which is what is seen, with ηsens = 20, 14, 8, and 5%, respectively. However, electron transfer to Eu(III) does not explain the ΦL and ΦTb variations in TbLX,MOM, which decrease in the same order from 5.3% to 1.8% and 30.6% to 13.1%, respectively. As Tb(III) is one of the most difficult to reduce Ln(III),77 and is stable under these conditions, these data are consistent with the picolinates directly quenching the excited antenna, which was shown above to be thermodynamically favoured. Additional support is provided by the ΦL values of GdLX,MOM, which follow the same trend, going from 7.0% to 2.2% with increasingly electron poor picolinates (Table 2).
The extent to which the pyridine electronic properties were affecting the excited state were then analysed by plotting ΦL, ΦLn, and ηsens for LnLX,MOM (Ln = Eu, Tb, Gd) vs. the picolinate reduction potential (Fig. 7 and S56–S60†). Good linear relationships with R2 close to 1 were found for ΦL(GdLX,MOM), ΦEu, and ηsens (EuLX,MOM), and a somewhat poorer one for ΦTb. No correlation was found between ΦL (EuLX,MOM) and the pyridine reduction potential, which is likely due at least in part to the large uncertainty associated with measuring such low values. However, there are at least 2 competing electron transfer pathways in Eu complexes, from the antenna to pyridine, and from the antenna to Eu(III). The electron poorer pyridines presumably destabilize Eu(III) thus also increasing PeT to Eu(III). The combination of these may cause a deviation from the trend seen in GdLX,MOM and TbLX,MOM.
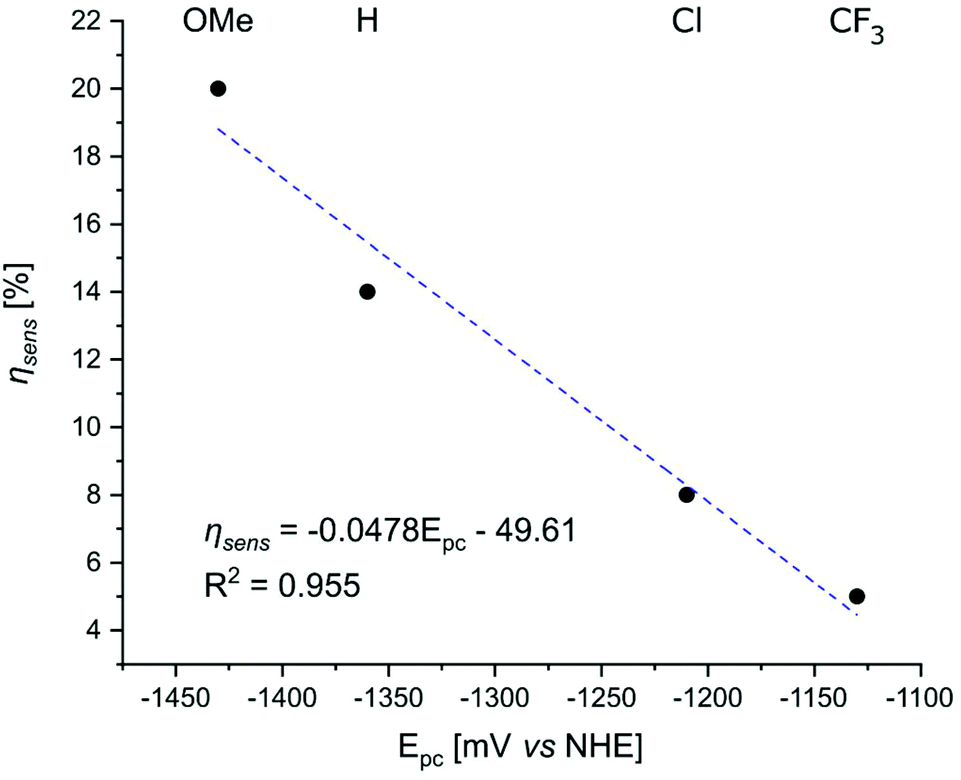 | ||
| Fig. 7 Correlation between the reduction potential of the p-substituted picolinates in the Gd complexes and ηsens in the Eu complexes of the same ligands. | ||
A conceivable photochemical reaction would be the oxidation of the photoexcited Tb(III) to Tb(IV) by the electron-poor pyridine. Tb(IV),78–80 along with Ce(IV)81–84 and Pr(IV)85 is more stable than the tetravalent ions of the other Ln. However, the Tb(III)/Tb(IV) oxidation potential is still quite positive, 1.3 V vs. NHE in concentrated aqueous carbonate solution,86 and most examples of Tb(IV) are reported in the solid state rather than as coordination complexes.87 Applying the values of 1.3 V for Eox (TbIII/TbIV oxidation potential), −1.25 V for Ered (picolinate reduction potential), 2.54 eV for ES (Tb(III) excited state) to eqn (1), a ΔGET of 0.01 eV is obtained without any coulombic stabilization. For more electron-poor picolinates ΔGET < 0 eV may be possible, which could account for the deviation from linearity for ΦTb (TbLX,MOM) vs. Epc(py/py˙−) and the lack of such deviation for the ΦL (TbLX,MOM) vs. Epc(py/py˙−) relationship. However, given the variation in measured Eox (TbIII/TbIV) and the expected large influence of the ligand, Tb(III) is unlikely to be easily oxidized in +1 charged TbLX,MOM.
Conclusions
A set of structurally related TACN-based Ln(III) complexes equipped with carbostyril sensitising antennae were prepared with the aim of studying the effect picolinate binding sites have on the luminescent properties. The similarities of the solution and solid-state geometries of the species carrying H, OMe, Cl, or CF3 substituents in the pyridine p-positions was supported by paramagnetic 1H NMR spectroscopy and single crystal X-ray crystallography. Further support was provided by the similar radiative and observed lifetimes (average values: 2.80 ms and 0.50 ms, respectively), and intrinsic quantum yields (mean value: 17.8%) for all the Eu(III)-based emitters irrespective of pyridine or carbostyril substituents.Despite the similarities in structure and certain photophysical properties, the complexes showed remarkable differences in their photon outputs. It was revealed that lowering the electron density on the pyridine binding units promote their electron accepting ability from the excited antenna decreasing the overall quantum yields. The phenomenon is thermodynamically favoured from the antenna S1, and in Eu(III) complexes it competes with processes such as energy transfer and ligand-to-metal PeT. This intra-ligand electron transfer also impacts the emission of non-redox active Ln complexes, as shown by the large degree of quenching in residual ligand fluorescence of Gd(III) and Tb(III) complexes.
Pyridine-bearing TACN-based ligands enjoy increasing popularity due to their ease of synthesis, versatility, and stability. These systems can trace their origins to tris-picolinate-based emitters, which are among the best understood and most widely employed Ln chelates. This study demonstrates that the pyridine structural units may participate in unintended background reactions. Notably, many of these quenching processes can be difficult to notice without in-depth structure–property relationship analyses, and their relevance is likely dependent on the arrangement of the pyridines vis-a-vis the sensitizing antenna, as suggested by the excellent luminescence properties of several picolinate-carrying TACN-based Eu(III) chelates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (project grant 2017-04077 for K. E. B.), and the Knut och Alice Wallenbergs Foundation (Dnr: 2018.0066 and Dnr: KAW 2019.0071).Notes and references
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- G. de Ruiter, M. Lahav and M. E. van der Boom, Acc. Chem. Res., 2014, 47, 3407–3416 CrossRef CAS PubMed.
- G. Tseberlidis, D. Intrieri and A. Caselli, Eur. J. Inorg. Chem., 2017, 3589–3603 CrossRef CAS.
- H.-L. Kwong, H.-L. Yeung, C.-T. Yeung, W.-S. Lee, C.-S. Lee and W.-L. Wong, Coord. Chem. Rev., 2007, 251, 2188–2222 CrossRef CAS.
- A. J. Pardey and C. Longo, Coord. Chem. Rev., 2010, 254, 254–272 CrossRef CAS.
- D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef PubMed.
- Z. Dai, Molecules, 2016, 21, 1647–1666 CrossRef CAS PubMed.
- V. Guerchais and J.-L. Fillaut, Coord. Chem. Rev., 2011, 255, 2448–2457 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- H. Yoneda, G. R. Choppin, J. L. Bear and A. J. Graffeo, Inorg. Chem., 1965, 4, 244–246 CrossRef CAS.
- J. E. Powell and J. W. Ingemanson, Inorg. Chem., 1968, 7, 2459–2461 CrossRef CAS.
- P. G. Manning, Can. J. Chem., 1966, 44, 1471–1473 CrossRef CAS.
- C. Doffek, N. Alzakhem, M. Molon and M. Seitz, Inorg. Chem., 2012, 51, 4539–4545 CrossRef CAS PubMed.
- J. Scholten, G. A. Rosser, J. Wahsner, N. Alzakhem, C. Bischof, F. Stog, A. Beeby and M. Seitz, J. Am. Chem. Soc., 2012, 134, 13915–13917 CrossRef CAS PubMed.
- C. Bischof, J. Wahsner, J. Scholten, S. Trosien and M. Seitz, J. Am. Chem. Soc., 2010, 132, 14334–14335 CrossRef CAS PubMed.
- S. Cotton, in Lanthanide and Actinide Chemistry, 2006, ch. 4, pp. 35–60 Search PubMed.
- P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- I. Grenthe, J. Am. Chem. Soc., 1961, 83, 360–364 CrossRef CAS.
- J. Andres and A.-S. Chauvin, Inorg. Chem., 2011, 50, 10082–10090 CrossRef CAS PubMed.
- C. Wei, B. Sun, Z. Cai, Z. Zhao, Y. Tan, W. Yan, H. Wei, Z. Liu, Z. Bian and C. Huang, Inorg. Chem., 2018, 57, 7512–7515 CrossRef CAS PubMed.
- E. S. Andreiadis, D. Imbert, J. Pecaut, R. Demadrille and M. Mazzanti, Dalton Trans., 2012, 41, 1268–1277 RSC.
- A. de Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef CAS PubMed.
- A. Nonat, C. Gateau, P. H. Fries and M. Mazzanti, Chem. – Eur. J., 2006, 12, 7133–7150 CrossRef CAS PubMed.
- A. Nonat, M. Giraud, C. Gateau, P. H. Fries, L. Helm and M. Mazzanti, Dalton Trans., 2009, 8033–8046 RSC.
- A. Bourdolle, M. Allali, J.-C. Mulatier, B. Le Guennic, J. M. Zwier, P. L. Baldeck, J.-C. G. Bunzli, C. Andraud, L. Lamarque and O. Maury, Inorg. Chem., 2011, 50, 4987–4999 CrossRef CAS PubMed.
- A. D'Aléo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- M. Regueiro-Figueroa, B. Bensenane, E. Ruscsák, D. Esteban-Gómez, L. J. Charbonnière, G. Tircsó, I. Tóth, A. D. Blas, T. Rodríguez-Blas and C. Platas-Iglesias, Inorg. Chem., 2011, 50, 4125–4141 CrossRef CAS PubMed.
- A. Rodriguez-Rodriguez, D. Esteban-Gomez, R. Tripier, G. Tircso, Z. Garda, I. Toth, A. de Blas, T. Rodriguez-Blas and C. Platas-Iglesias, J. Am. Chem. Soc., 2014, 136, 17954–17957 CrossRef CAS PubMed.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef CAS PubMed.
- J. M. Zwier and N. Hildebrandt, Rev. Fluoresc., 2016, 9, 17–43 CAS.
- A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987–6994 CrossRef CAS PubMed.
- J. Wahsner and M. Seitz, Inorg. Chem., 2013, 52, 13301–13303 CrossRef CAS PubMed.
- J. W. Walton, R. Carr, N. H. Evans, A. M. Funk, A. M. Kenwright, D. Parker, D. S. Yufit, M. Botta, S. De Pinto and K.-L. Wong, Inorg. Chem., 2012, 51, 8042–8056 CrossRef CAS PubMed.
- A. S. Chauvin, F. Gumy, D. Imbert and J. C. G. Bünzli, Spectrosc. Lett., 2004, 37, 517–532 CrossRef CAS.
- J. Andres, R. D. Hersch, J.-E. Moser and A.-S. Chauvin, Adv. Funct. Mater., 2014, 24, 5029–5036 CrossRef CAS.
- M. Le Fur, E. Molnar, M. Beyler, O. Fougere, D. Esteban-Gomez, O. Rousseaux, R. Tripier, G. Tircso and C. Platas-Iglesias, Inorg. Chem., 2018, 57, 6932–6945 CrossRef CAS PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef CAS PubMed.
- S. J. Butler, B. K. McMahon, R. Pal, D. Parker and J. W. Walton, Chem. – Eur. J., 2013, 19, 9511–9517 CrossRef CAS PubMed.
- J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Delbianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600–1602 RSC.
- B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2013, 49, 5363–5365 RSC.
- A. T. Bui, M. Beyler, Y.-Y. Liao, A. Grichine, A. Duperray, J.-C. Mulatier, B. L. Guennic, C. Andraud, O. Maury and R. Tripier, Inorg. Chem., 2016, 55, 7020–7025 CrossRef CAS PubMed.
- N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed.
- J. Mendy, T. B. Anh, A. Roux, J.-C. Mulatier, D. Curton, A. Duperray, A. Grichine, Y. Guyot, S. Brasselet, F. Riobe, C. Andraud, B. Le Guennic, V. Patinec, P. R. Tripier, M. Beyler and O. Maury, ChemPhysChem, 2020, 21, 1036–1043 CrossRef CAS PubMed.
- N. Hamon, M. Galland, M. Le Fur, A. Roux, A. Duperray, A. Grichine, C. Andraud, B. Le Guennic, M. Beyler, O. Maury and R. Tripier, Chem. Commun., 2018, 54, 6173–6176 RSC.
- A. D'Aleo, A. Picot, P. L. Baldeck, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10269–10279 CrossRef PubMed.
- A. T. Bui, M. Beyler, A. Grichine, A. Duperray, J.-C. Mulatier, Y. Guyot, C. Andraud, R. Tripier, S. Brasselet and O. Maury, Chem. Commun., 2017, 53, 6005–6008 RSC.
- A. D'Aleo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- J. Andres and A.-S. Chauvin, Eur. J. Inorg. Chem., 2010, 2700–2713 CrossRef CAS.
- J. Andres and A.-S. Chauvin, Phys. Chem. Chem. Phys., 2013, 15, 15981–15994 RSC.
- J. Andres and K. E. Borbas, Inorg. Chem., 2015, 54, 8174–8176 CrossRef CAS PubMed.
- M. Andrews, J. E. Jones, L. P. Harding and S. J. A. Pope, Chem. Commun., 2011, 47, 206–208 RSC.
- Y.-W. Yip, Z. Yan, G.-L. Law and W.-T. Wong, Eur. J. Inorg. Chem., 2019, 813–820 CrossRef CAS.
- D. Kovacs, S. R. Kiraev, D. Phipps, A. Orthaber and K. E. Borbas, Inorg. Chem., 2020, 59, 106–117 CrossRef CAS PubMed.
- D. Kovacs, X. Lu, L. S. Mészáros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 CrossRef CAS PubMed.
- D. Kovacs, E. Mathieu, S. R. Kiraev, J. A. L. Wells, E. Demeyere, A. Sipos and K. E. Borbas, J. Am. Chem. Soc., 2020, 142, 13190–13200 CrossRef CAS PubMed.
- W. D. Horrocks and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef CAS.
- R. M. Supkowski and W. D. Horrocks Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 RSC.
- J. A. Therrien and M. O. Wolf, Inorg. Chem., 2017, 56, 1161–1172 CrossRef CAS PubMed.
- C. Gateau, M. Mazzanti, J. Pécaut, F. A. Dunand and L. Helm, Dalton Trans., 2003, 2428–2433 RSC.
- K. Mason, A. C. Harnden, C. W. Patrick, A. W. J. Poh, A. S. Batsanov, E. A. Suturina, M. Vonci, E. J. L. McInnes, N. F. Chilton and D. Parker, Chem. Commun., 2018, 54, 8486–8489 RSC.
- M. Vonci, K. Mason, E. R. Neil, D. S. Yufit, E. J. L. McInnes, D. Parker and N. F. Chilton, Inorg. Chem., 2019, 58, 5733–5745 CrossRef CAS PubMed.
- G. Nocton, A. Nonat, C. Gateau and M. Mazzanti, Helv. Chim. Acta, 2009, 92, 2257–2273 CrossRef CAS.
- M. D. C. Teixeira, F. S. Felix, S. S. Thomasi, Z. M. Magriotis, J. M. da Silva, L. L. Okumura and A. A. Saczk, Microchem. J., 2019, 148, 66–72 CrossRef CAS.
- S. R. Kiraev, E. Mathieu, F. Siemens, D. Kovacs, E. Demeyere and K. E. Borbas, Molecules, 2020, 25, 5282 CrossRef CAS.
- A. Beeby, S. Faulkner and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 1918–1922 RSC.
- T. J. Soerensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- J. H. S. K. Monteiro, D. Machado, L. M. de Hollanda, M. Lancellotti, F. A. Sigoli and A. de Bettencourt-Dias, Chem. Commun., 2017, 53, 11818–11821 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- A. H. Harvey, J. S. Gallagher and J. M. H. Levelt Sengers, J. Phys. Chem. Ref. Data, 1998, 27, 761–774 CrossRef CAS.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef CAS.
- S. Cotton, in Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd, 2006, pp. 9–22 Search PubMed.
- A. R. Willauer, C. T. Palumbo, R. Scopelliti, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, Angew. Chem., 2020, 59, 3549–3553 CrossRef CAS PubMed.
- C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS PubMed.
- N. T. Rice, I. A. Popov, D. R. Russo, J. Bacsa, E. R. Batista, P. Yang, J. Telser and H. S. La Pierre, J. Am. Chem. Soc., 2019, 141, 13222–13233 CrossRef CAS.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS.
- J. R. Levin, W. L. Dorfner, P. J. Carroll and E. J. Schelter, Chem. Sci., 2015, 6, 6925–6934 RSC.
- N. A. Piro and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS.
- J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS.
- D. E. Hobart, K. Samhoun, J. P. Young, V. E. Norvell, G. Mamantov and J. R. Peterson, Inorg. Nucl. Chem. Lett., 1980, 16, 321–328 CrossRef CAS.
- Z. Lin, M. L. Shelby, D. Hayes, K. A. Fransted, L. X. Chen and M. J. Allen, Dalton Trans., 2014, 43, 16156–16159 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional syntheses, photophysical, crystallographic, and electrochemical characterization, 1H, 13C and 19F NMR spectra. CCDC 2057882 and 2057884–2057886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00616a |
| ‡ Due to the Cl/H2O disorder modelling in the TACN ligand complexes TbLCF3,MOM−Cl and EuLCl,MOM−Cl, the Ln-OH2 and Ln-Cl distances are not reliable. A more reliable comparison is obtained when compared with the related carbostyril cyclen complexes.57 |
| This journal is © The Royal Society of Chemistry 2021 |