
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsertion of single-ion magnets based on mononuclear Co(II) complexes into ferromagnetic oxalate-based networks†
M.
Palacios-Corella
,
V.
García-López
,
C.
Sánchez-Sánchez
,
J. M.
Clemente-Juan
 ,
M.
Clemente-León
* and
E.
Coronado
*
,
M.
Clemente-León
* and
E.
Coronado
*
Instituto de Ciencia Molecular (ICMol), Universidad de Valencia, C/Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: miguel.clemente@uv.es; eugenio.coronado@uv.es; Fax: +34 963543273; Tel: +34 963544419
First published on 23rd March 2021
Abstract
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:1 Co(II) complexes of the L ligand (L = 6-(3,5-diamino-2,4,6-triazinyl)2,2′-bipyridine) with formulas [CoII(L)2](ClO4)2·0.5MeCN·Et2O (1) and [CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN (2) have been prepared. The structural and magnetic characterization of the two compounds shows that they contain octahedral high-spin Co(II) and present a field-induced slow relaxation of the magnetization. 1 has been inserted into a bimetallic oxalate-based network leading to a novel achiral 3D compound of formula [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (3) exhibiting ferromagnetic ordering below 4.6 K. EPR measurements suggest a weak magnetic coupling between the two sublattices.
:2 and 1:1 Co(II) complexes of the L ligand (L = 6-(3,5-diamino-2,4,6-triazinyl)2,2′-bipyridine) with formulas [CoII(L)2](ClO4)2·0.5MeCN·Et2O (1) and [CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN (2) have been prepared. The structural and magnetic characterization of the two compounds shows that they contain octahedral high-spin Co(II) and present a field-induced slow relaxation of the magnetization. 1 has been inserted into a bimetallic oxalate-based network leading to a novel achiral 3D compound of formula [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (3) exhibiting ferromagnetic ordering below 4.6 K. EPR measurements suggest a weak magnetic coupling between the two sublattices.
Introduction
In the area of coordination chemistry, a strategy that has gained increasing interest in the last few years is the control of the magnetic properties of molecular magnetic units by their insertion into coordination polymers (CPs). For instance, lanthanide- and transition metal-based single-ion magnets (SIMs) and single-molecule magnets (SMMs) have been incorporated into the nodes or encapsulated into porous CPs.1 Another example is that of assembling single-chain magnets (SCMs) inside the pores of CPs.2 This hybrid approach has resulted in the creation of multifunctional materials with multiple physical properties. In this context, oxalate-based networks have been widely used to prepare multifunctional compounds. They are formed by 2D3 or 3D4 anionic magnetic networks, which can host different functional cations.5,6 These compounds combine the cooperative magnetism of the oxalate network with paramagnetism,7 photochromism,8 electrical conductivity,9 proton conductivity,10 ferroelectricity,11 chirality,12 spin-crossover (SCO)13 or fluorescence14 from the counter-cation. In these structures the dimensionality of the coordination network is controlled by the relative configuration of the adjacent metal ions linked by an oxalate bridge. Thus, a 2D honeycomb structure is obtained when the relative configurations are opposite, e.g. (Δ, Λ), leading to non-chiral networks. In contrast, if they are identical, (Λ, Λ) or (Δ, Δ), a chiral 3D network is formed.15 The counter-ions associated with the formation of these 3D chiral networks have been essentially chiral dicationic complexes such as [ZII(bpy)3]2+ (Z = Ru, Fe, Co and Ni; bpy = bipyridine)4 or [FeII(pyimH)3]2+ (pyimH = 2-(1H-imidazol-2-yl)pyridine),13j while non-chiral 2D networks can be formed with a larger variety of templating monocations with different shapes and sizes. Apart from these two types of structures, other atypical 3D networks have also been obtained. It is worth mentioning that the non-chiral networks formed by metals in the two configurations have been obtained with a variety of MnIII,7d,16 FeII (ref. 13a) and FeIII complexes13c,d,i and triethylammonium cations17 or organic radicals,18 together with chiral 3D networks of lower symmetry (see Table 1).7d,13e In addition, other topologies have been obtained in oxalate networks with hepta-13i or octacoordinated metal ions.10b,e,h,i One of these hybrids exhibits coexistence and even coupling of ferromagnetism and SMM behaviour. Thus, the insertion of the SMM [MnIII(salen)(H2O)]22+ (salen2− = N,N′-ethylenebis-(salicylideneiminate)) in an oxalate-based 3D network gives rise to the compound [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·(CH3OH)·(CH3CN)2 in which the coupling between the spin anisotropy of the SMM with the soft ferromagnetic network results in the enhancement of the magnetic hysteresis of the compound.16| T C (K) | |
|---|---|
| a H2PPD2+ = p-phenylenediammonium. b ppy = 2-phenylpyridine. | |
| Achiral | |
| (C2H5)3NH+ (ref. 17) | |
| [MIII(sal2-trien)]+ (M = Fe or In)13b | 5.2 |
| [Mn((R/S)-salmen)(CH3OH)2]+ (ref. 7d) | 5.2 |
| (H2PPD)2+(benzo[18]crown-6)2a4i |
5.5 |
| [MnIII(salen)(H2O)]22+ (ref. 16) | 5.5 |
| [FeIII(5-X-sal2-trien)]+ (X = MeO)13d | 5.2 |
| [Fe(bpp)2]2+ (ref. 13a) | 3 |
| Chiral | |
| [Z(bpy)3]2/3+ (Z = CrIII, CoIII, RuII, FeII, CoII and NiII)4a–e | 2.2–4.2 |
| [RuII(ppy)(bpy)2]+b12b |
5.8 |
| [IrIII(ppy)2(bpy)]+ (ref. 4g) | 5.1 |
| [MIII(5-X-sal2-trien)]+ (M = Fe or In, X = Cl, Br)13e | 4.8–5 |
| [FeII(L)2]2+ (ref. 20) | 4.2 |
| [FeII(pyimH)3]2+ (ref. 13j) | 4.5 |
| [MnIII(salpn)(solvent)2]+ (ref. 7d) | 5.4 |
In a previous work, we showed that the [FeII(L)2]2+ complex with L = (3,5-diamino-2,4,6-triazinyl)2,2′-bipyridine (see Scheme 1)19 stabilized both a 2D oxalate network and a chiral 3D structure.20 Interestingly, the [FeII(L)2](ClO4)2·CH3CN precursor exhibited a photo-induced spin transition.20
 | ||
| Scheme 1 Structural view of the L ligand. | ||
In this paper, we have explored the preparation of analogous precursors with Co(II) instead of Fe(II) and their insertion into an oxalate-based network. These Co(II) analogues present interesting magnetic properties such as SMM behavior21,22 or SCO,23 which could interact or coexist with that of the oxalate-based network. In this work, the syntheses, structures and magnetic properties of the 1:1 and 1:2 Co(II):L complexes are reported together with those of the bimetallic oxalate-based compounds obtained with the 1:2 complex.
Results
Synthesis
L was synthesized as described in the literature.19a It was reacted with Co(II) in acetonitrile with 1:2 and 1:1 metal:ligand ratios to give the mononuclear Co(II) complexes [CoII(L)2](ClO4)2·0.5MeCN·Et2O (1) and [CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN (2), respectively, which were crystallized by layering with diethyl ether. This method is similar to that used to obtain the analogous Fe(II) complexes.19,20 In a further step, the hybrid compound [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (3) formed by an achiral 3D network was prepared using a protocol similar to that reported for other bimetallic oxalate-based compounds.13c–j,20 The preparation method is based on slow diffusion of a solution containing the Co(II) complex 1 dissolved in nitromethane into another solution with the precursors of the oxalate network dissolved in methanol. Long diffusion times led to the formation of crystals of a different compound with the same formula [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (4) but with a 2D oxalate network. This last compound appears as a minor product always mixed with 3. A similar diffusion method using 2 in place of 1 led to the formation of crystals of compound 3. Therefore, the synthesis conditions used for the growth of the bimetallic oxalate network provoked the decomposition of the 1:1 Co(II):L complex and the formation of the 1:2 Co(II):L one. The crystal structures of 1, 2, 3 and 4 were solved by single-crystal X-ray diffraction at 120 K. The composition of crystals of these compounds, examined by energy dispersive X-ray spectroscopy (EDAX), shows a Co:Cl ratio of 1:2 for 1 and 2 and a Co:Mn:Cr: ratio of 1:2:2 for 3 and 4. Elemental analysis indicates the possible replacement of the MeCN and Et2O molecules of 1 by water molecules after filtering the crystals (see the Experimental section). In the case of 3, elemental analysis is more consistent with the presence of H2O after filtering the crystals (see the Experimental section). The structure of 4 is reported in the ESI† (Fig. S10 and S11, Table S1† and the associated text).
Structure of [CoII(L)2](ClO4)2·0.5MeCN·Et2O (1)
1 crystallizes in the monoclinic P21/c space group. The unit cell of this compound is different from that of the Fe(II):L 1:2 complex [FeII(L)2](ClO4)2·CH3CN.19a The asymmetric unit at 120 K is composed of two [CoII(L)2]2+ cations, four perchlorate anions, some of them disordered, one acetonitrile molecule, and two disordered diethyl ether molecules (see Fig. S1 in the ESI†). The central cobalt(II) ions (Co1 and Co2) of the two crystallographically independent Co(II) complexes are coordinated by six nitrogen atoms from two tridentate L ligands with a distorted octahedral coordination geometry with two shorter Co–N axial bond distances to the inner pyridine rings (2.065(2) Å (Co1) and 2.046(2) Å (Co2)) and four longer equatorial Co–N bond distances (2.164(2) and 2.171(2) Å (Co1) and 2.184(2) and 2.185(2) Å (Co2) for the external pyridine and triazine rings, respectively) (Fig. 1). A similar distortion is observed in another octahedral Co(II) SIM such as [CoII(dmphen)(NCS)2].21a These distances are typical of Co(II) in the high-spin state although Co–N distances in this type of compound are not very sensitive to the metal ion spin state. Calculation of the degree of distortion with respect to an ideal six-vertex polyhedron using SHAPE software24 lead to shape measures relative to the octahedron (OC-6) and trigonal prism (TPR-6) with the values of 4.8 and 9.0 for Co1 and 4.5 and 9.0 for Co2, respectively. SHAPE measures relative to other reference polyhedra are significantly larger. SHAPE calculations show that the coordination geometry of the two crystallographically independent Co presents a deviation close to 25% from the minimal distortion path between a perfect octahedron and a perfect trigonal prism. Due to this, the generalized coordinates between the two structures have not been calculated, as they are approximate for structures that significantly deviate from that path. The trans N(pyridyl)–Co–N(pyridyl) angles between the two ligands (167.47(10)° for Co1 and 167.64(10)° for Co2) deviate from the ideal octahedron. This deviation is more important for the trans N(triazine)–Co–N(pyridyl) intraligand angles (150.88(9) and 151.77(9)° for Co1 and 153.07(9) and 151.47(10)° for Co2).
 | ||
| Fig. 1 Structural view of the [CoII(L)2]2+ complex in 1. | ||
Two neighbouring [CoII(L)2]2+ complexes form hydrogen bonds between the NH2 groups of complexes with Co2 and a N atom from the triazine ligand of a complex with Co1 (dN2⋯N20 = 3.02 Å). Besides these interactions, the two neighbouring complexes are linked through several short contacts involving their two triazine rings. These dimers of complexes are linked through short contacts between a CH group from the central pyridine ring and a triazine ring (see Fig. S2 in the ESI†). This gives rise to chains of complexes running along the c axis, which are surrounded by perchlorate anions and solvent molecules. Perchlorates form hydrogen bonds with the triazine rings of [CoII(L)2]2+ complexes. Three of the four crystallographically independent perchlorate anions with Cl2, Cl3 and Cl4 are disordered. The powder X-ray diffraction (PXRD) pattern measured in contact with the mother liquor agrees with the simulated pattern for the structure at 120 K (Fig. S3 in the ESI†).
[CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN (2)
The structure of 2 is similar to that of the Fe(II):L 1:1 compound of formula [FeII(L)(CH3CN)2(H2O)](ClO4)2·CH3CN.20 Both compounds crystallize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with similar unit cells. The asymmetric unit is composed of one [CoII(L)(CH3CN)2(H2O)]2+ cation, two perchlorate anions and one acetonitrile solvent molecule (Fig. S4 in the ESI†). The cobalt(II) ion of the complex present a distorted N5O octahedral coordination. It is coordinated by three nitrogen atoms from a tridentate L ligand, one water molecule in trans to the N atom from the inner pyridine ring of L and two nitrogen atoms from two acetonitrile molecules in trans positions (Fig. 2). The Co–N bond distances are 2.0644(17), 2.1340(18) and 2.1564(17) Å for the inner pyridine ring, the external pyridine ring and the triazine ring of L, respectively. The Co–N distances to the two acetonitrile coordinating molecules are similar (2.1108(18) and 2.1368(19) Å), while the Co–O distance to the water molecule is the shortest one (2.0423(17) Å). As in 1, there are two axial ligands (N from the inner pyridine ring and the water molecule) at shorter distances than the four equatorial ones. These distances are typical high-spin Co(II) bond lengths. The trans N(acetonitrile)–Co–N(acetonitrile) and O(water)–Co–N(pyridyl) angles (167.72(7) and 174.32(7)°, respectively) are closer to the ideal one for a perfect octahedron (180°) than the trans N(triazine)–Co–N(pyridyl) intraligand angle (151.27(7)°), which presents a stronger distortion similar to that of 1. Calculations using SHAPE software24 lead to shape measures relative to the octahedron (OC-6) and trigonal prism (TPR-6) with the values of 2.4 and 10.6, respectively, while those relative to other reference polyhedra are significantly larger. This indicates that the Co(II) coordination polyhedron in 2 is closer to a perfect octahedron than that in 1. A possible explanation is that the two L ligands per complex in 1 impose more important geometrical restrictions than the one L ligand found in 2. SHAPE calculations show that the generalized coordinate between these two ideal polyhedra is 36.8%. However, as it presents a deviation close to 15% from the minimal distortion path between the two ideal geometries, this value has to be taken with caution.
space group with similar unit cells. The asymmetric unit is composed of one [CoII(L)(CH3CN)2(H2O)]2+ cation, two perchlorate anions and one acetonitrile solvent molecule (Fig. S4 in the ESI†). The cobalt(II) ion of the complex present a distorted N5O octahedral coordination. It is coordinated by three nitrogen atoms from a tridentate L ligand, one water molecule in trans to the N atom from the inner pyridine ring of L and two nitrogen atoms from two acetonitrile molecules in trans positions (Fig. 2). The Co–N bond distances are 2.0644(17), 2.1340(18) and 2.1564(17) Å for the inner pyridine ring, the external pyridine ring and the triazine ring of L, respectively. The Co–N distances to the two acetonitrile coordinating molecules are similar (2.1108(18) and 2.1368(19) Å), while the Co–O distance to the water molecule is the shortest one (2.0423(17) Å). As in 1, there are two axial ligands (N from the inner pyridine ring and the water molecule) at shorter distances than the four equatorial ones. These distances are typical high-spin Co(II) bond lengths. The trans N(acetonitrile)–Co–N(acetonitrile) and O(water)–Co–N(pyridyl) angles (167.72(7) and 174.32(7)°, respectively) are closer to the ideal one for a perfect octahedron (180°) than the trans N(triazine)–Co–N(pyridyl) intraligand angle (151.27(7)°), which presents a stronger distortion similar to that of 1. Calculations using SHAPE software24 lead to shape measures relative to the octahedron (OC-6) and trigonal prism (TPR-6) with the values of 2.4 and 10.6, respectively, while those relative to other reference polyhedra are significantly larger. This indicates that the Co(II) coordination polyhedron in 2 is closer to a perfect octahedron than that in 1. A possible explanation is that the two L ligands per complex in 1 impose more important geometrical restrictions than the one L ligand found in 2. SHAPE calculations show that the generalized coordinate between these two ideal polyhedra is 36.8%. However, as it presents a deviation close to 15% from the minimal distortion path between the two ideal geometries, this value has to be taken with caution.
 | ||
| Fig. 2 Structural view of the [CoII(L)(CH3CN)2(H2O)]2+ complex in 2. | ||
The [CoII(L)(CH3CN)2(H2O)]2+ complexes present short contacts with the four neighbouring complexes (Fig. S5 in the ESI†). These contacts involve the triazine rings of two of them, the coordinated acetonitrile molecules and the triazine or pyridine rings. Finally, the L rings of all complexes are almost parallel but they do not present intermolecular contacts shorter than the sum of the van der Waals radii (shortest contacts dC4–C8 = 3.63 Å and dC5–C7 = 3.57 Å). Due to this, significant π–π stacking interactions could be excluded. The coordinated water molecule form hydrogen bonds with the two perchlorate anions. On the other hand, one of the two amino groups from triazine forms hydrogen bonds with an acetonitrile solvent molecule and perchlorate anions. The PXRD pattern agrees with the simulated pattern for the structure at 120 K (see Fig. S6 in the ESI†).
Structure of [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (3)
3 crystallises in the centrosymmetric orthorhombic space group Pnna. In contrast to [FeII(L)2]2+, which gives rise to a chiral 3D bimetallic oxalate-based network under the same synthesis conditions,20 the structure of 3 contains a 3D network involving metals of both chiralities. This network contains two crystallographically independent Mn(II) and Cr(III) ions with occupancies of 0.5. The Mn–O distances lie between 2.137(5) and 2.199(4) Å, while the Cr–O ones lie between 1.970(4) and 1.988(4) Å. These are typical MnII–O and CrIII–O distances similar to those found in other oxalate networks. [CoII(L)2]2+ and disordered solvent molecules occupy the cavities of this network (Fig. 3). Similar achiral 3D oxalate networks have been found in the compounds of formula [FeII(bpp)2][MnCr(ox)3]2·bpp·CH3OH that crystallizes in the centrosymmetric P21/n space group,13a [FeIII(sal2-trien)][MnIICrIII(ox)3]·(CH3OH) and [InIII(sal2-trien)][MnIICrIII(ox)3]·0.25(H2O)·0.25(CH3OH)·0.25(CH3CN) that crystallize in the acentric Cc space group,13c [InIII(sal2-trien)][MnIICrIII(ox)3]·(CH3NO2)·0.5(H2O)13c and [FeIII(5-CH3Osal2-trien)][MnIICrIII(ox)3]13d that crystallize in the acentric orthorhombic space group Pna21 or [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·(CH3OH)·(CH3CN)2 that crystallizes in the centrosymmetric C2/c space group (see Table 1).16 This network is formed by bis-chelating oxalate ligands connecting MnII and CrIII ions in such a way that each MnII is surrounded by three CrIII and vice versa, building ten-membered rings in a (10, 3) topology. The (10, 3) decagon rings perpendicular to the a axis are formed by metal centers with the same chirality (see Fig. 4) forming layers along the bc plane. In these layers, the Cr and Mn ions are not in the same plane. Two neighbouring (10, 3) decagon rings belonging to different layers are linked through two oxalate ligands and present opposite chirality (see Fig. 4). Projection of the oxalate network on the ac plane is almost identical to that of the chiral 3D oxalate network in the ab, bc or ac plane (see Fig. S7 in the ESI†). However, projection on the bc (Fig. 3) or ab (Fig. 4) plane is different as a result of the different chiralities of the neighbouring (10, 3) decagon rings. Thus, in the bc projection an eclipsed disposition of the neighbouring oxalate rings is observed as a result of the heterochiral oxalate junctions among them (see Fig. 3). | ||
| Fig. 3 Projection on the bc plane of the structure of 3; Co (dark blue), C (black), N (blue), O (red), Mn (pink) and Cr (green); hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 4 Two (10, 3) decagon rings perpendicular to the a axis in the structure of 3; C (black), O (red), Mn (pink) and Cr (green). | ||
The [CoII(L)2]2+ complexes and solvent molecules are enclosed in the channels described by this 3D oxalate network. There is half crystallographically independent [CoII(L)2]2+ complex. Co(II) presents a distorted N6 octahedral coordination to the two tridentate L ligands similar to that of 1. The Co–N bond lengths of the inner pyridine and triazine rings are 2.089(6) and 2.152(5) Å, respectively, while that of the external pyridine ring is 2.168(6) Å. These distances are very similar to those of 1 and typical of Co(II) in the high-spin state. The trans N(pyridyl)–Co–N(pyridyl) angle between the two ligands (165.2(3)°) deviates from the ideal octahedron. This deviation is more important for the trans N(triazine)–Co–N(pyridyl) intraligand angle (150.5(2)°) as in 1. Calculations performed using SHAPE software24 lead to similar results to those of 1 for shape measures with respect to a perfect octahedron and a trigonal prism with the values of 5.0 and 7.1, respectively. The insertion into the 3D oxalate-based network does not give rise to a higher distortion of the complex than that of 1 (see above).
[CoII(L)2]2+ complexes present two short contacts with the two neighbouring ones. These interactions involve a CH group from the external pyridine ring and a N atom from the triazine ring (see Fig. S8 in the ESI†). This gives rise to chains of [CoII(L)2]2+ complexes running along the a axis in the channels formed by the bimetallic oxalate-based network. Therefore, the insertion of the complex into the 3D oxalate-based network restricts the intermolecular interactions among the complexes to one dimension. Indeed, these chains of [CoII(L)2]2+ complexes present many short contacts with the surrounding oxalate-based network. Thus, there are hydrogen bonds between the NH2 groups from the triazine rings and the oxygen atoms from the oxalate network. As in the oxalate-based compounds obtained with [FeII(L)2]2+,203 contains disordered solvent molecules in the structure. This is a consequence of the 2+ charge of these Co(II) and Fe(II) complexes used as templating cations of the oxalate-based network. Thus, when In(III) or Fe(III) complexes with the 1+ charge are inserted into similar MnIICrIII 3D achiral oxalate networks, there is a double number of inserted complexes.13b Due to this, in 3, as in similar compounds,13a,i,j there is more empty space available for solvent molecules. Due to disorder in the solvent molecules they could not be modelled. This disorder has been treated with PLATON/SQUEEZE (see the Experimental section).25 Indeed, the crystals of 3 lose solvent very quickly after filtering, leading to a featureless PXRD pattern (data not shown). In contrast to this, the PXRD pattern measured in contact with the mother liquor shows a good agreement with the simulated pattern for the structure at 120 K (see Fig. S9 in the ESI†).
Magnetic properties
The thermal dependence of the product of the molar magnetic susceptibility and the temperature (χT) of 1 and 2 is shown in Fig. 5. In both cases χT shows the value of 2.7 emu K mol−1 at 300 K, which is higher than the spin-only value for a high-spin Co(II) (d7) due to considerable orbital angular momentum contribution to the magnetic moment. Upon cooling, χT of 1 and 2 decreases continuously with a more abrupt decrease below 50 K due to the strong zero-field splitting (ZFS) to reach the values of 1.6 for 1 and 1.7 emu K mol−1 for 2 at 2 K. The isothermal field (H) dependence of the magnetization (M) was measured up to 5 T at temperatures of 2 and 5 K (the inset of Fig. 5). The magnetization at 5 T (2.1 and 2.3 B. M. for 1 and 2, respectively) is significantly lower than the expected saturation for a system with S = 3/2 and g > 2. In general, for six-coordinated Co(II) complexes, the orbital moment is either not quenched or partially quenched. In that case the Griffith Hamiltonian should be considered, but in the case of large positive axial orbital splitting (easy-plane) a spin Hamiltonian with D and E parameters can be used.26 Simultaneous fit of both the susceptibility and magnetization data was performed using the MAGPACK program.27 The best fit values are g = 2.37, D = 48 cm−1 and R = 2.7 × 10−4 for 1 and g = 2.47, D = 55 cm−1 and R = 7.8 × 10−5 for 2. For E/D different from zero, other solutions with less quality can be found (an increase in the residual value of less than an order of magnitude). These solutions give rise to E/D less than 10−3 and a small decrease in the D values and an increase in the g value. This correlation between the parameters is due to the mixing in the composition of the Kramers doublet functions (pure for E = 0 but mixture [−3/2, 1/2] and [−1/2, 3/2] for E ≠ 0). More precise spectroscopic measurements should be necessary to avoid this correlation and to determine more precisely the E parameters.28 These parameters are similar to those found for the related compounds.21,22,28,29 | ||
| Fig. 5 Temperature dependence of the product of the molar magnetic susceptibility and temperature (χT) at 0.1 T for compounds 1 (a) and 2 (b). Inset: Magnetization vs. magnetic field for both compounds measured at T = 2 and 5 K. The blue solid lines are the best-fitting curves. | ||
The relaxation properties of 1 and 2 were studied by susceptibility measurements performed with an alternating magnetic field (AC susceptibility). In the absence of a magnetic field, no signal in the out of phase molar susceptibility (χ′′) is observed. This behavior is not uncommon in mononuclear SMMs and has been attributed to tunnelling, dipolar interactions and/or hyperfine interactions.29 When magnetic dc fields of 0.1 T are applied for 1 and 2, strong frequency-dependent peaks in both the in phase molar susceptibility (χ′) and χ′′ appear with clear maxima of χ′′ below 4 K for 1 and 7.5 K for 2 (Fig. 6). This is a clear indication that the two compounds present a field-induced slow relaxation of magnetization. The variable-frequency AC data at different temperatures of 1 and applied fields of 0.1 T show a single relaxation event with a maximum at 6000 Hz at 3.5 K (see Fig. S12 in the ESI†). There is likely no quantum tunneling occurring, as the relaxation frequency moves to lower values as temperature is decreased with a maximum near 3000 Hz at 2.5 K. In the case of 2 with an applied field of 0.1 T, maxima at 8200 Hz at 8 K and 115 Hz at 2.5 K are observed. The Cole–Cole plots (χ′′ vs. χ′) of 1 and 2 at higher temperatures confirm the presence of a single relaxation process (Fig. 7). Thus, at fixed temperatures between 2.5 and 4.5 K for 1 and 2.5 and 8.0 K for 2, the semi-circular plots were obtained and fitted using a generalized Debye model, yielding the α parameter in the range of 0.02–0.03 at 2.5–4.5 K for 1 and values of 0.14 at the lowest temperature and 0.02 at 8 K for 2. This indicates that the range of distribution of relaxation times increases when the temperature decreases. Additional relaxation mechanisms become inoperative and therefore there is a deviation from the Debye model.
 | ||
| Fig. 6 Temperature dependence of χ′ and χ′′ of 1 (a) and 2 (b) in an applied dc field of 0.1 T at frequencies in the range 100 to 10000 Hz. | ||
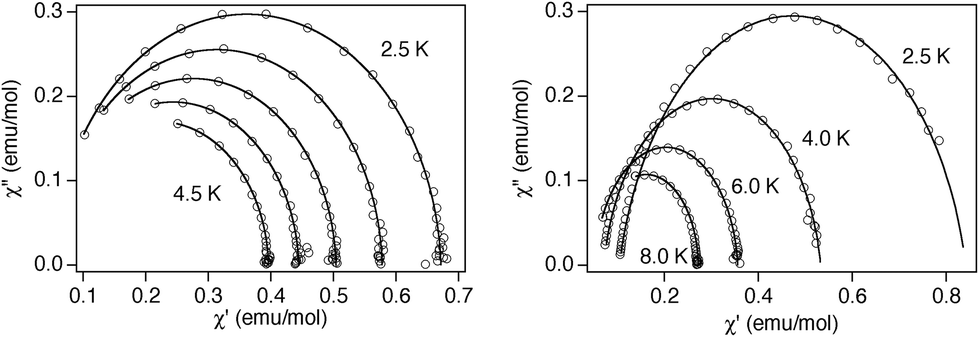 | ||
| Fig. 7 Cole–Cole plots of 1 (left) and 2 (right) measured at the same temperatures as those of Fig. 7 under an applied dc field of 0.1 T. The solid lines represent the best fit. | ||
The relaxation times (τ) of 1 were determined from the maximum of χ′′ at a given frequency (τ = 1/2πν) fitting to the Arrhenius expression for a thermally activated process (Orbach, τ = τ0exp(Ueff/kBT)) leading to τ0 = 1.8 × 10−6 s and Ueff = 5.8 cm−1 at 0.1 T. The Ueff values are smaller than the experimental energy gap between two Kramers doublets. This may be indicative of the existence of other relaxation mechanisms. In fact, the slow relaxation of magnetization for this type of Kramers ions shows a combination of a direct mechanism assisted by very effective hyperfine coupling (I = 7/2), spin-lattice relaxation and Raman processes.21d This temperature dependence of relaxation can be fitted with an expression containing two terms: a field-dependent mechanism and Raman terms (τ−1 = τFDM−1 + CTn). The best fit for 1 is obtained with C = 1207 s−1 K−2.9, n = 2.92 and τFDM−1 = 3589 s−1 at 0.1 T; n parameters are far from the expected value for the Raman term (n = 9), but similar values have been obtained recently for other Co(II) complexes with large positive D values (see Fig. S13 in the ESI†).21c,22e,f,30–32 This reduction in the value of n can be due to different mixtures in the eigenvector of the Kramers doublets and the consideration of other phonons.33
In the case of 2, the higher value of Ueff obtained from the Arrhenius expression (τ0 = 2.7 × 10−7 s and Ueff = 20.5 cm−1 at 0.1 T) allows the addition of the other two relaxation processes (field-dependent mechanism and Raman terms) to the Orbach process. The best fit obtained indicates that only two processes have an important contribution, the Orbach and the field-dependent processes. The best fit is obtained with τ0 = 1.1 × 10−7 s, Ueff = 25.8 cm−1 and τFDM−1 = 6060 s−1 with a Raman contribution that is very weak due to a significantly small C parameter, C = 0.21 s−1 K−4.7 (see Fig. S13 in the ESI†). The values for the pre-exponential factor and activation energy are consistent with those of the other Co(II) complexes showing this behavior.34–36
The magnetic properties of 3 were measured in contact with the mother liquor in view of the loss of crystallinity observed in PXRD measurements after extracting the crystals from the mother liquor. The thermal dependence of χT shows a value of 15.3 cm3 K mol−1 at 300 K (see Fig. 8). This value is approximately equal to the sum of the contributions from the two magnetic components: the bimetallic oxalate-based network and the isolated paramagnetic Co(II) ions (expected spin-only value of χT = 12.5 cm3 K mol−1 for two Mn(II) and two Cr(III)) plus the χT of 1 at 300 K (2.7 cm3 K mol−1)). At lower temperatures, χT shows a gradual increase, which is very sharp below 50 K. This indicates the presence of ferromagnetic interactions between neighboring MnII–CrIII magnetic centers leading to long-range ferromagnetic ordering at low temperatures, as observed for other 3D bimetallic oxalate networks. The M vs. H plot of 3 at 2 K shows a very abrupt increase at low fields, as expected for a ferromagnet, reaching a saturation value of 19.5 B. M. at 5 T (see Fig. 8). This increase is more gradual at higher fields, which is in agreement with the presence of a paramagnetic Co(II) component. This saturation value is close to the expected contributions for the ferromagnetic oxalate network (16 B. M. for two MnII and two CrIII ferromagnetically coupled) plus one paramagnetic Co(II) (2.1 B. M. for 1 at 2 K). This indicates that M of 3 is a superposition of the magnetization of each of the two components, with no significant interactions between them. The hysteresis loop at 2 K shows a coercive field of ca. 3 mT (see Fig. S14 in the ESI†), which is typical of a soft ferromagnet and very close to that of other oxalate-based compounds with similar achiral 3D networks and inserted diamagnetic or paramagnetic cations.13c
 | ||
| Fig. 8 Temperature dependence of the product of the molar magnetic susceptibility and temperature (χT) at 0.1 T for compound 3. Inset: Magnetization vs. magnetic field for 3 measured at T = 2 K. The continuous line is a guide to the eye. | ||
To confirm the presence of long-range magnetic ordering and to obtain a more accurate determination of the Curie temperature, AC susceptibility measurements were carried out. A maximum of χ′ near 4.6 K and a χ′′ signal that starts to appear at temperatures just below this temperature are observed (Fig. 9). From this data the Curie temperature (TC) of 3 is found to be 4.6 K, a value that is similar to those reported for other MnIICrIII 3D networks (see Table 1).
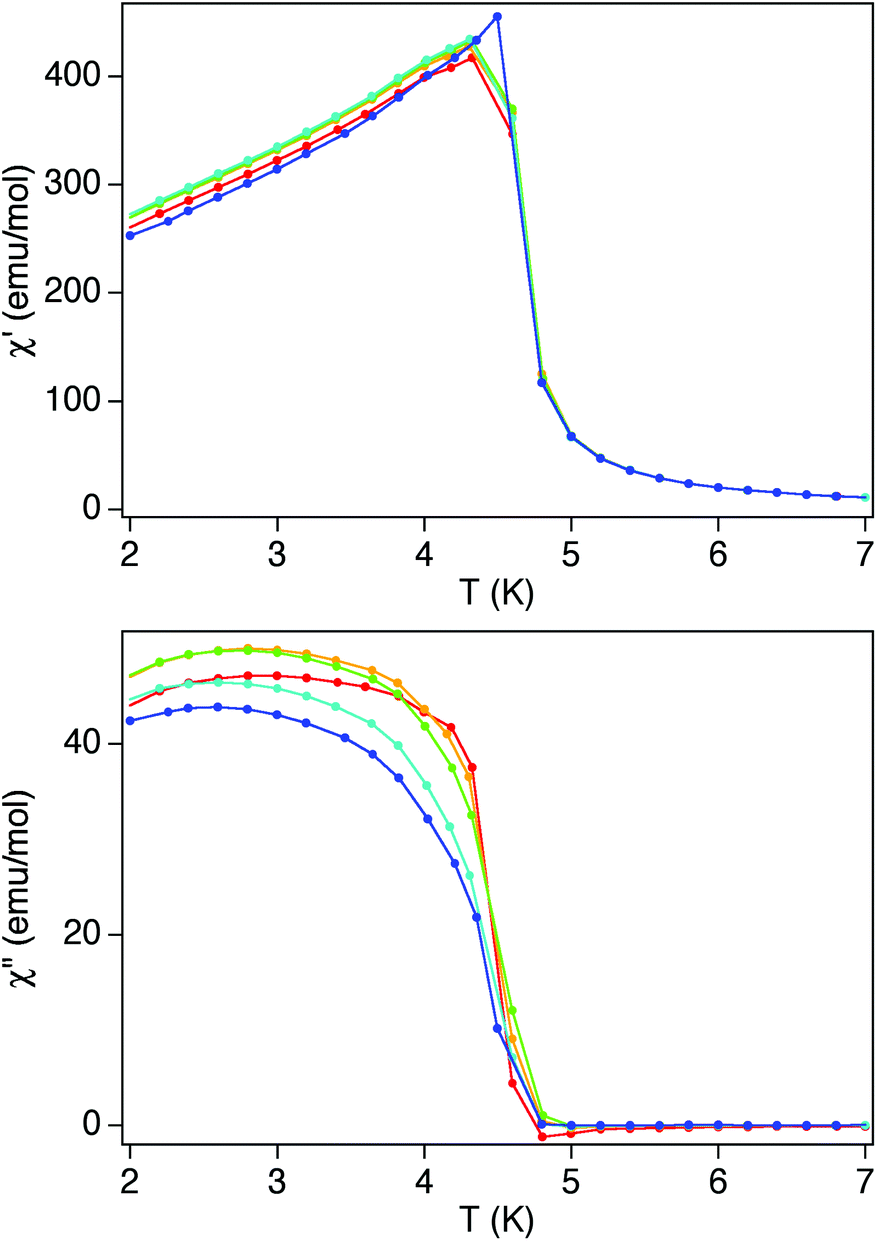 | ||
| Fig. 9 Temperature dependence of χ′ (top) and χ′′ (bottom) of 3 at frequencies in the range 1 (blue) to 997 Hz (red). | ||
These results indicate that the magnetic behavior at low temperatures is dominated by the ferromagnetic bimetallic oxalate network. This is an expected result if one takes into account that the main contribution to χ′ and χ′′ arises from the strong response of the ferromagnetic fraction. To confirm this, we studied the magnetic field dependence of χ′ and χ′′ (see Fig. S15 in the ESI†). Even relatively weak dc magnetic fields H = 100 G suppress the χ′ divergence, as expected for a ferromagnetic phase transition. Notice that χ′′ tends to vanish in the presence of a magnetic field, showing that magnetic relaxation processes become also considerably faster. In addition to this, the magnetic measurements of the published compounds [In(sal2-trien)][MnIICrIII(ox)3]·solv and [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv (solv = solvents) with similar 3D ferromagnetic networks and diamagnetic or SMM cations, respectively, have been used as the reference. As shown in Fig. S16 in the ESI,† clear frequency dependence of χ′ and χ′′ and the presence of additional maxima in χ′′ are observed in the compound containing [MnIII(salen)(H2O)]22+ SMM and not in that containing diamagnetic [In(sal2-trien)]+. As the ac magnetic properties of 3 are more similar to those of [In(sal2-trien)][MnIICrIII(ox)3]·solv, we conclude that the effect of the inserted [CoII(L)2]2+ complexes on the magnetic properties of 3 is negligible.
Finally, the magnetic properties of the filtered crystals of 3 change drastically with respect to those measured in contact with the mother liquor (see Fig. S17 to S19 and related discussion in the ESI†), which is probably due to the structural changes in the bimetallic oxalate network arising from the loss of solvent and the collapse of the structure.
EPR spectroscopy
X-band EPR spectra of the polycrystalline samples of 1 and 3 in contact with the mother liquor were measured to study the magnetic interactions between the two magnetic subnetworks in 3.In the case of 1, the spectra below 20 K are similar to those of the other octahedral Co(II) complexes reported in the literature presenting a rhombic symmetry and large positive D values (geff = [5.62, 4.11, 2.15] at 4 K. See Fig. S20a in the ESI†).21a,30,37,38 A similar behavior is shown by the EPR spectrum of 2 (geff = [6.17, 3.34, 2.04] at 4 K. See Fig. S20b in the ESI†).
The EPR spectrum of 3 at room temperature shows a single line centered at g = 2.00 with a peak-to-peak width ΔHpp = 500 G. Upon lowering the temperature, the intensity of the signal increases. Below TC (see the spectrum at 4 K in Fig. S21 in the ESI†), the signal splits into two components centered at g = 1.69 and 2.54. These features coincide with that previously reported for the 2D MnIICrIII oxalate-based compounds [FeIIICp*2][MnIICrIII(ox)3] (Cp* = pentamethylcyclopentadienyl)7c and p-rad[MnIICrIII(ox)3]·H2O (p-rad = N-methylpyridinium cation bearing a nitronyl nitroxide moiety at position 4 of the pyridine ring),18 which also show splitting of the signal into two components below TC. The characteristic intrinsic signal of the inserted Co(II) complex is not observed. Comparing the EPR spectrum of the cobalt monomer 1 with that of compound 3, both normalized to one mole of each compound, we can see that the monomer signal is not observed when it is intercalated in the oxalate network (see Fig. S22 in the ESI† and the Experimental section for details). This suggests the presence of a weak coupling between both sublattices, as observed in the 2D compound p-rad[MnIICrIII(ox)3]·H2O,18 and in contrast to [FeIIICp*2][MnIICrIII(ox)3],7c which shows the intrinsic signals of the two sublattices.
Discussion
The magnetic properties of 3 are very similar to those of other compounds with the same type of 3D network and diamagnetic countercation, with the contribution of the inserted Co(II) complex being almost negligible. This could be explained by the presence of extremely weak magnetic interactions, which are not strong enough to induce the coupling of the magnetic moments of the countercation and bimetallic oxalate network observed in the compound [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv with a similar achiral 3D network and a Mn(III) SMM dimer countercation. However, these magnetic interactions are strong enough to modify the EPR spectra, which is not the sum of the contributions from the oxalate network and the inserted cation as observed in the 2D compound [FeIIICp*2][MnIICrIII(ox)3]. Therefore, 3 shows an intermediate behavior between those two compounds, in which the magnetic exchange interactions between the moments of the two subnetworks do not modify drastically the magnetic properties of the oxalate network as in [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv, but they lead to a unique EPR signal in contrast to [FeIIICp*2][MnIICrIII(ox)3].The stronger magnetic interactions found in [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv with respect to those of 3 could be explained by the intermolecular interactions between the two subnetworks in the structures of both compounds. Indeed, the intermolecular contacts between the atoms from the oxalate network and those of the countercation are closer to the magnetic center in [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv than in 3. Thus, [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv presents a hydrogen bond between O from the oxalate ligands and a water molecule directly coordinated to the magnetic Mn(III) from the SMM dimer. In 3, the closest contact between the two magnetic networks is a hydrogen bond between O from the oxalate ligands and a NH2 group from the L ligand, which is not directly coordinated to the magnetic center of the countercation as in the case of [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv. This suggests a better magnetic isolation of the magnetic cation in the case of 3, which is in agreement with the magnetic properties of both compounds.
Conclusions
In this work, we have prepared two Co(II) complexes of the tridentate 6-diaminotriazyl-2,2-bipyridine (L) ligand. The magnetic and structural characterization shows that [CoII(L)2]2+ in 1 and [CoII(L)(CH3CN)2(H2O)]2+ in 2 exhibit a field-induced slow relaxation of the magnetization due to the magnetic anisotropy of high-spin Co(II), as observed for other octahedral Co(II) complexes with similar tridentate ligands. This result demonstrates that the Co(II) complexes of this ligand may behave as SIMs. The main advantage of these complexes is that they open the way to study the interplay between the ferromagnetic ordering and SIM behavior in oxalate-based compounds containing [CoII(L)2]2+ templating cations in an analogous way to that reported for the Fe(II) complexes with the same ligand. Interestingly, the structures of the oxalate-based compounds obtained with this cation change drastically with respect to those obtained with [FeII(L)2]2+. Thus, a 3D achiral oxalate-based network is obtained with [CoII(L)2]2+ in compound 3, in contrast to the compounds obtained with [FeII(L)2]2+, which present regular 2D or chiral 3D oxalate-based networks. This demonstrates that subtle changes in the geometry of the templating cation can lead to a different structure of the oxalate-based network.The magnetic properties of 3 are dominated by the ferromagnetic ordering of the oxalate-based network. This indicates that the magnetic isolation provided by the L ligand prevents strong magnetic interactions between the two subnetworks. However, EPR spectra suggest the presence of weak magnetic exchange interactions. Magnetic measurements at lower temperatures are needed to verify if these weak magnetic interactions could cause an increase in the coercive field of 3 with respect to similar networks containing cations with less magnetic anisotropy as observed in [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·solv.
Experimental
Synthesis
L was synthesized as reported in the literature.19a Ag3[Cr(ox)3] was prepared by metathesis from the corresponding potassium salt.39[CoII(L)2](ClO4)2·0.5MeCN·Et2O (1)
L (70.0 mg, 0.264 mmol) was suspended in MeCN (40 mL). This was followed by the addition of Co(ClO4)2·xH2O (35.0 mg, 0.096 mmol). After stirring for 15 minutes the reaction mixture was saturated with a few drops of Et2O. This was followed by vapour–liquid diffusion of diethyl ether. After three days orange crystals were obtained and they were filtered (56 mg, 74%). Anal. Calcd for [CoII(L)2](ClO4)2·2H2O: C, 37.88; H, 3.18; N, 23.79%. Found: C, 38.25; H, 3.15; N, 23.64%.[CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN (2)
L (70.0 mg, 0.264 mmol) was suspended in MeCN (40 mL). This was followed by the addition of Co(ClO4)2·xH2O (96.0 mg, 0.264 mmol). After stirring for 15 minutes the reaction mixture was saturated with a few drops of Et2O. This was followed by vapour–liquid diffusion of diethyl ether. After three days orange crystals were obtained and they were filtered (86 mg, 42%). Anal. Calcd for [CoII(L)(CH3CN)2(H2O)](ClO4)2·MeCN: C, 34.35; H, 3.34; N, 21.09%. Found: C, 34.55; H, 3.27; N, 21.18%.[CoII(L)2][MnIICrIII(ox)3]2·(solvate) (3)
Crystals of this compound were obtained by slow diffusion of two solutions. The first solution was prepared by adding MnCl2·4H2O (22 mg, 0.11 mmol) to a suspension of Ag3Cr(ox)3 (44.6 mg, 0.07 mmol) in 7 mL of methanol. The AgCl precipitate was filtered off. The second solution was obtained by dissolving 1 (16 mg, 0.02 mmol) in 4 mL of nitromethane. After 3 days purple crystals of 3 were obtained. If the diffusion process was continued for longer times (two weeks), the crystals of the 2D compound [CoII(L)2][MnIICrIII(ox)3]2·(solvate) (4) appeared together with those of 3. Anal. Calcd for [CoII(L)2][MnIICrIII(ox)3]2·10H2O: C, 30.19; H, 2.80; N, 12.97%. Found: C, 30.72; H, 3.01; N, 12.57%.Structural characterization
Single crystals of all compounds were mounted on glass fibers using a viscous hydrocarbon oil to coat the crystal and then transferred directly to a cold nitrogen stream for data collection. The reflection data of 1, 2, 3 and 4 were collected at 120 K using a Supernova diffractometer equipped with a graphite-monochromated enhance (Mo) X-ray source (λ = 0.7107 Å). The CrysAlisPro, Oxford Diffraction Ltd, program was used for unit cell determination and data reduction. The empirical absorption correction was performed using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The structures were solved with the ShelXT40 structure solution and refined with the SHELXL-2013 program,41 using Olex2.42 Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed in the calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters. One molecule of diethyl ether in 1 was found to be disordered over several positions and could not be modelled satisfactorily. It was removed from the electron density map using the OLEX solvent mask command. The subroutine SQUEEZE from PLATON has been used to calculate and remove the diffracting component of disordered solvents in 3 and 4.25 This results for 3 in a total potential solvent accessible void volume per unit cell of 2790 Å3 (40.2% of the total unit cell volume). This volume is distributed in one void in 0.091 0.015 0.409, which is occupied by 1190 electrons. This roughly corresponds to 25 MeNO2 and 35 MeOH molecules per unit cell. In 4, the total potential solvent accessible void volume per unit cell is 3495 Å3 (32.5% of the total unit cell volume) in a void of 3349 Å3 in −0.121 0.624 0.079 and other smaller voids. The largest void is occupied by 1142 electrons, which roughly correspond to 30 MeNO2 and 10 MeOH molecules per unit cell. Due to this disorder and the small size of the crystals, the overall quality of the data of 3 and 4 is poor and this results in high R1 factors (0.0846, and 0.1444, respectively) with scan widths of 1 degree and exposure times of 17.5 s for 3 and 5 s for 4. CCDC-2063853–56† contain the supplementary crystallographic data for this paper.The collected data and refinement statistics are reported in Tables 2 and S1 in the ESI.† A 1 mm glass capillary was filled with the polycrystalline samples of 1, 2 and 3 in the mother liquor and mounted and aligned on an Empyrean PANalytical powder diffractometer, using CuKα radiation (λ = 1.54177 Å). A total of 3 scans were performed at room temperature in the 2θ range of 5–40°.
| a R1(F) = ∑||Fo| − |Fc||/∑|Fo|. b wR2(F2) = [∑w(Fo2 − Fc2)2/∑wFo4]½. c S(F2) = [∑w(Fo2 − Fc2)2/∑(n + r − p)]½. | |||
|---|---|---|---|
| Compound | 1 | 2 | 3 |
| Empirical formula | C27H23.5Cl2Co N14.5O8 | C38H44Cl4Co2N20O18 | C39H25CoCr2Mn2N15O33 |
| Formula weight | 808.93 | 1328.59 | 1504.55 |
| Crystal colour | Orange | Orange | Violet |
| Crystal size | 0.16 × 0.13 × 0.12 | 0.11 × 0.09 × 0.07 | 0.14 × 0.03 × 0.03 |
| Temperature (K) | 120(2) | 120(2) | 120(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system, Z | Monoclinic, 8 | Triclinic, 2 | Orthorhombic, 4 |
| Space group | P21/c |
P |
Pnna |
| a (Å) | 14.8849(2) | 10.0029(4) | 19.4566(8) |
| b (Å) | 33.6403(5) | 11.8949(4) | 15.5539(6) |
| c (Å) | 15.7252(2) | 12.5420(4) | 22.9545(9) |
| α (°) | 90 | 65.515(3) | 90 |
| β (°) | 98.895(2) | 78.397(3) | 90 |
| γ (°) | 90 | 82.037(3) | 90 |
| V (Å3) | 7779.42(19) | 1327.79(9) | 6946.5(5) |
| ρ calc (Mg m−3) | 1.381 | 1.662 | 1.439 |
| μ(MoKα) (mm−1) | 0.641 | 0.917 | 0.981 |
| θ range (°) | 3.299–27.515 | 3.346–27.486 | 3.264–27.913 |
| Reflns collected | 64541 |
12287 |
149747 |
| Independent reflns (Rint) | 17831(0.055) |
6074 (0.0232) | 8086 (0.3327) |
| L. S. parameters | 1046 | 417 | 368 |
| R1(F),aI > 2σ(I) | 0.0542 | 0.0375 | 0.0846 |
| wR2(F2),b all data | 0.1455 | 0.0884 | 0.2988 |
| S(F2),c all data | 1.043 | 1.028 | 1.017 |
Physical measurements
Magnetic measurements were performed with Quantum Design MPMS-XL-5 SQUID and PPMS-9 magnetometers on powdered polycrystalline samples in contact with the mother liquor in the case of 2 and 3. EPR measurements of 1 and 3 were recorded using a Bruker ELEXYS E580 spectrometer under X-band irradiation (∼9.4 GHz). To compare the EPR signal of both compounds, they were measured with similar setup parameters and each signal was normalized per mole of compound. The Co/Mn/Cr and Co/Cl ratios were measured with a Philips ESEM X230 scanning electron microscope equipped with an EDAX DX-4 microsonde. Elemental analyses (C, H, and N) were performed with a CE Instruments EA 1110 CHNS elemental analyzer.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the EU (ERC advanced grant MOL-2D 788222 and FET-OPEN COSMICS 766726), the Spanish MCIU (MAT-2017-89993-R, and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the Generalitat Valenciana (PROMETEO program and iDiFEDER/2018/061) and the Ministry of Education and Science of Russian Federation (agreement no. 14.W03.31.0001) is gratefully acknowledged. We all thank A. Soriano-Portillo, J. M. Martínez-Agudo and G. Agustí for PXRD and magnetic measurements.Notes and references
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533 RSC.
- M. Wang, X. Gou, W. Shi and P. Cheng, Chem. Commun., 2019, 55, 11000 RSC.
- (a) H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, J. Am. Chem. Soc., 1992, 114, 6974 CrossRef CAS; (b) H. Tamaki, M. Mitsumi, N. Nakamura, N. Matsumoto, S. Kida, H. Okawa and S. Ijima, Chem. Lett., 1992, 1975 CrossRef CAS; (c) C. Mathonière, J. Nutall, S. G. Carling and P. Day, Inorg. Chem., 1996, 35, 1201 CrossRef PubMed; (d) R. Pellaux, H. W. Schmalle, R. Huber, P. Fisher, T. Hauss, B. Ouladdiaf and S. Decurtins, Inorg. Chem., 1997, 36, 2301 CrossRef CAS PubMed; (e) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. M. Martínez-Agudo, E. Martínez-Ferrero, J. C. Waerenborgh and M. Almeida, J. Solid State Chem., 2001, 159, 391 CrossRef CAS; (f) K. S. Min, A. L. Rhinegold and J. S. Miller, Inorg. Chem., 2005, 44, 8433 CrossRef CAS PubMed; (g) E. Coronado, J. R. Galán-Mascarós and C. Martí-Gastaldo, J. Mater. Chem., 2006, 16, 2685 RSC; (h) M. Sadakiyo, T. Yamada, K. Kato, M. Takata and H. Kitagawa, Chem. Sci., 2016, 7, 1349 RSC.
- (a) S. Decurtins, H. W. Schmalle, P. Schneuwly and H. R. Oswald, Inorg. Chem., 1993, 32, 1888 CrossRef CAS; (b) S. Decurtins, H. W. Schmalle, P. Schneuwly, J. Ensling and P. Gütlich, J. Am. Chem. Soc., 1994, 116, 9521 CrossRef CAS; (c) S. Decurtins, H. W. Schmalle, R. Pellaux, P. Schneuwly and A. Hauser, Inorg. Chem., 1996, 35, 1451 CrossRef CAS PubMed; (d) M. Hernández-Molina, F. Lloret, C. Ruiz-Pérez and M. Julve, Inorg. Chem., 1998, 37, 4131 CrossRef PubMed; (e) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and J. M. Martínez-Agudo, Inorg. Chem., 2001, 40, 113 CrossRef CAS PubMed; (f) F. Pointillart, C. Train, M. Gruselle, F. Villain, H. W. Schmalle, D. Talbot, P. Gredin, S. Decurtins and M. Verdaguer, Chem. Mater., 2004, 16, 832 CrossRef CAS; (g) M. Clemente-León, E. Coronado, C. J. Gómez-García and A. Soriano-Portillo, Inorg. Chem., 2006, 45, 5653 CrossRef PubMed; (h) M. Juric, D. Pajic, D. Zilic, B. Ravkin, K. Molcanov and J. popovic, Dalton Trans., 2015, 44, 20626 RSC; (i) T. Endo, K. Kubo, M. Yoshitake, S.-i. Noro, N. Hoshino, T. Akutagawa and T. Nakamura, Cryst. Growth Des., 2015, 15, 1186 CrossRef CAS; (j) M. Mon, T. Grancha, M. Verdaguer, C. Train, D. Armentano and E. Pardo, Inorg. Chem., 2016, 55, 6845 CrossRef CAS PubMed.
- M. Clemente-León, E. Coronado, C. Martí-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473 RSC.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716 CrossRef CAS PubMed.
- (a) M. Clemente-León, E. Coronado, J. R. Galán-Mascarós and C. J. Gómez-García, Chem. Commun., 1997, 1727 RSC; (b) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and J. M. Martínez-Agudo, Adv. Mater., 1999, 11, 558 CrossRef CAS; (c) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. Ensling and P. Gutlich, Chem. – Eur. J., 2000, 6, 552 CrossRef CAS PubMed; (d) M. Clemente-León, E. Coronado and M. López-Jordà, Dalton Trans., 2013, 42, 5100 RSC.
- (a) S. Bénard, P. Yu, J. P. Audière, E. Rivière, R. Clèment, J. Ghilhem, L. Tchertanov and K. Nakatami, J. Am. Chem. Soc., 2000, 122, 9444 CrossRef; (b) S. M. Aldoshin, N. A. Sanina, V. I. Minkin, N. A. Voloshin, V. N. Ikorskii, V. I. Ovcharenko, V. A. Smirnov and N. K. Nagaeva, J. Mol. Struct., 2007, 826, 6 CrossRef.
- (a) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447 CrossRef CAS PubMed; (b) A. Alberola, E. Coronado, J. R. Galán-Mascarós, C. Giménez-Saiz and C. J. Gómez-García, J. Am. Chem. Soc., 2003, 125, 10774 CrossRef CAS PubMed; (c) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, E. Martínez-Ferrero and S. Van Smaalen, Inorg. Chem., 2004, 43, 4808 CrossRef CAS PubMed; (d) B. Zhang, Y. Zhang and D. Zhu, Chem. Commun., 2012, 48, 197 RSC.
- (a) H. Okawa, A. Shigematsu, M. Sadakiyo, T. Miyagawa, K. Yoneda, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 13516 CrossRef CAS PubMed; (b) E. Pardo, C. Train, G. Contard, K. Boubekeur, O. Fabelo, H. Liu, B. Dkhil, F. Lloret, K. Nakagawa, H. Tokoro, S.-I. Ohkoshi and M. Verdaguer, J. Am. Chem. Soc., 2011, 133, 15328 CrossRef CAS PubMed; (c) M. Sadayiko, H. Okawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472 CrossRef PubMed; (d) H. Okawa, M. Sadakiyo, T. Yamada, M. Maesato, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 2256 CrossRef CAS PubMed; (e) C. Maxim, S. Ferlay, H. Tokoro, S. I. Ohkoshi and C. Train, Chem. Commun., 2014, 50, 5629 RSC; (f) M. Sadayiko, T. Yamada, K. Honda, H. Matsui and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7701 CrossRef PubMed; (g) H. Okawa, M. Sadayiko, K. Otsubo, K. Yoneda, T. Yamada, M. Ohba and H. Kitagawa, Inorg. Chem., 2015, 54, 8529 CrossRef CAS PubMed; (h) M. Mon, J. Vallejo, J. Pasán, O. Fabelo, C. Train, M. Verdaguer, S. I. Ohkoshi, H. Tokoro, K. Nakagawae and E. Pardo, Dalton Trans., 2017, 46, 15130 RSC; (i) H. Okawa, Y. Yoshida, K. Otsubo and H. Kitagawa, Inorg. Chem., 2020, 59, 623 CrossRef CAS PubMed.
- (a) T. Endo, T. Akutagawa, S. I. Noro and T. Nakamura, Dalton Trans., 2011, 40, 1491 RSC; (b) E. Pardo, C. Train, H. Liu, L.-M. Chamoreau, B. Dkhil, K. Boubekeur, F. Lloret, K. Nakatani, H. Tokoro, S.-I. Ohkoshi and M. Verdaguer, Angew. Chem., Int. Ed., 2012, 51, 8356 CrossRef CAS PubMed.
- (a) R. Andrés, M. Gruselle, B. Malézieux, M. Verdaguer and J. Vaissermann, Inorg. Chem., 1999, 38, 4637 CrossRef PubMed; (b) R. Andrés, M. Brissard, M. Gruselle, C. Train, J. Vaissermann, B. Malézieux, J. P. Jamet and M. Verdaguer, Inorg. Chem., 2001, 40, 4633 CrossRef PubMed; (c) M. Clemente-León, E. Coronado, J. C. Dias, A. Soriano-Portillo and R. D. Willett, Inorg. Chem., 2008, 47, 6458 CrossRef PubMed; (d) C. Train, R. Gheorghe, V. Krstic, L. M. Chamoreau, N. S. Ovanesyan, G. L. J. A. Rikken, M. Gruselle and M. Verdaguer, Nat. Mater., 2008, 7, 729 CrossRef CAS PubMed; (e) C. Train, T. Nuida, R. Gheorghe, M. Gruselle and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 16838 CrossRef CAS PubMed; (f) M. Gruselle, Y. Li, N. Ovanesyan, V. Markhaev, G. Shilov, F. Mushenok, C. Train and S. Aldoshin, Chirality, 2013, 25, 444 CrossRef CAS PubMed; (g) C. Maxim, S. Ferlay and C. Train, C. R. Chim., 2019, 22, 534 CrossRef CAS.
- (a) E. Coronado, J. R. Galán-Mascarós, M. C. Giménez-López, M. Almeida and J. C. Waerenborgh, Polyhedron, 2007, 26, 1838 CrossRef CAS; (b) M. Clemente-León, E. Coronado, M. C. Giménez-López, A. Soriano-Portillo, J. C. Waerenborgh, F. S. Delgado and C. Ruiz-Pérez, Inorg. Chem., 2008, 47, 9111 CrossRef PubMed; (c) M. Clemente-León, E. Coronado, M. López-Jordà, G. Mínguez Espallargas, A. Soriano-Portillo and J. C. Waerenborgh, Chem. – Eur. J., 2010, 16, 2207 CrossRef PubMed; (d) M. Clemente-León, E. Coronado and M. López-Jordà, Dalton Trans., 2010, 39, 4903 RSC; (e) M. Clemente-León, E. Coronado, M. López-Jordà and J. C. Waerenborgh, Inorg. Chem., 2011, 50, 9122 CrossRef PubMed; (f) M. Clemente-León, E. Coronado, M. López-Jordà, C. Desplanches, S. Asthana, H. Wang and J.-F. Létard, Chem. Sci., 2011, 2, 1121 RSC; (g) M. Clemente-León, E. Coronado and M. López-Jordà, Eur. J. Inorg. Chem., 2013, 753 CrossRef; (h) M. Clemente-León, E. Coronado, M. López-Jordà, J. C. Waerenborgh, C. Desplanches, H. Wang, J.-F. Létard, A. Hauser and A. Tissot, J. Am. Chem. Soc., 2013, 135, 8655 CrossRef PubMed; (i) A. Ben Djamâa, M. Clemente-León, E. Coronado and M. López-Jordà, Polyhedron, 2013, 64, 142 CrossRef; (j) M. López-Jordà, M. Giménez-Marqués, C. Desplanches, G. Mínguez Espallargas, M. Clemente-León and E. Coronado, Eur. J. Inorg. Chem., 2016, 2016, 2187 CrossRef.
- S. Benmansour, C. Cerezo-Navarrete, J. Canet-Ferrer, G. Muñoz-Matutano, J. Martínez-Pastor and C. J. Gómez-García, Dalton Trans., 2018, 47, 11909 RSC.
- C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3279 RSC.
- M. Clemente-León, E. Coronado, C. J. Gómez-García, M. López-Jordà, A. Camón, A. Repollés and F. Luis, Chem. – Eur. J., 2014, 20, 1669 CrossRef PubMed.
- B. Zhang, Y. Zhang and D. Zhu, Dalton Trans., 2012, 14, 8509 RSC.
- A. Alberola, E. Coronado, C. Giménez-Saiz, C. J. Gómez-García, F. M. Romero and A. Tarazón, Eur. J. Inorg. Chem., 2005, 2005, 389 CrossRef.
- (a) M. C. Young, E. Liew, J. Ashby, K. E. McCoy and R. J. Hooley, Chem. Commun., 2013, 49, 6331 RSC; (b) M. C. Young, E. Liew and R. J. Hooley, Chem. Commun., 2014, 50, 5043 RSC.
- C. Sánchez-Sánchez, C. Desplanches, J. M. Clemente-Juan, M. Clemente-León and E. Coronado, Dalton Trans., 2017, 46, 2680 RSC.
- (a) J. Vallejo, I. Castro, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, G. De Munno, W. Wernsdorfer and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15704 CrossRef CAS PubMed; (b) E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130 CrossRef CAS PubMed; (c) R. Herchel, L. Váhovská, I. Potocnák and Z. Trávnícek, Inorg. Chem., 2014, 53, 5896 CrossRef CAS PubMed; (d) S. Gómez-Coca, A. Urtizberea, E. Cremades, P. J. Alonso, A. Camón, E. Ruiz and F. Luis, Nat. Commun., 2014, 5, 4300 CrossRef PubMed; (e) S. Roy, I. Oyarzabal, J. Vallejo, J. Cano, E. Colacio, A. Bauza, A. Frontera, A. M. Kirillov, M. G. B. Drew and S. Das, Inorg. Chem., 2016, 55, 8502 CrossRef CAS PubMed; (f) A. Switlicka-Olszewska, J. Palion-Gazda, T. Klemens, B. Machura, J. Vallejo, J. Cano, F. Lloret and M. Julve, Dalton Trans., 2016, 45, 10181 RSC; (g) G. Novitchi, S. Jiang, S. Shova, F. Rida, I. Hlavicka, M. Orlita, W. Wernsdorfer, R. Hamze, C. Martins, N. Suaud, N. Guihéry, A. L. Barra and C. Train, Inorg. Chem., 2017, 56, 14809 CrossRef CAS PubMed.
- (a) F. Habib, O. R. Luca, V. Vieru, M. Shiddiq, I. Korobkov, S. I. Gorelsky, M. K. Takase, L. F. Chibotaru, S. Hill, R. H. Crabtree and M. Murugesu, Angew. Chem., Int. Ed., 2013, 52, 11290 CrossRef CAS PubMed; (b) L. Rigamonti, N. Bridonneau, G. Poneti, L. Tesi, L. Sorace, D. Pinkowicz, J. Jover, E. Ruiz, R. Sessoli and A. Cornia, Chem. – Eur. J., 2018, 24, 8857 CrossRef CAS PubMed; (c) R. F. Higgins, B. N. Livesay, T. J. Ozumerzifon, J. P. Joyce, A. K. Rappé and M. P. Shores, Polyhedron, 2018, 143, 193 CrossRef CAS; (d) D. Sertphon, K. S. Murray, W. Phonsri, J. Jover, E. Ruiz, S. G. Telfer, A. Alkas, P. Harding and D. J. Harding, Dalton Trans., 2018, 47, 859 RSC; (e) V. García-López, F. Orts-Mula, M. Palacios-Corella, J. M. Clemente-Juan, M. Clemente-León and E. Coronado, Polyhedron, 2018, 150, 54–60 CrossRef; (f) B. Brachnakova, S. Matejová, J. Moncol, R. Herchel, J. Pavlik, E. Moreno-Pineda, M. Ruben and I. Salitros, Dalton Trans., 2020, 49, 1249 RSC.
- (a) H. A. Goodwin, Top. Curr. Chem., 2004, 234, 23 CrossRef CAS; (b) S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981 CrossRef CAS; (c) M. G. Cowan, J. Olguin, S. Narayanaswamy, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2012, 134, 2892 CrossRef CAS PubMed; (d) Y. Guo, X. L. Yang, R. J. Wei, L. S. Zheng and J. Tao, Inorg. Chem., 2015, 54, 7670 CrossRef CAS PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE (2.1), Universitat de Barcelona, Barcelona, Spain, 2013 Search PubMed.
- J. Spek, Appl. Crystallogr., 2003, 36, 7 CrossRef.
- (a) F. Lloret, M. Julve, J. Cano, R. Ruiz-García and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432 CrossRef CAS; (b) J. Titiš and R. Boča, Inorg. Chem., 2011, 50, 11838 CrossRef PubMed.
- J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblar, J. Comput. Chem., 2001, 22(9), 985 CrossRef.
- E. Y. Misochko, A. V. Akimov, D. V. Korchagin, J. Nehrkorn, M. Ozerov, A. V. Palii, J. M. Clemente-Juan and S. M. Aldoshin, Inorg. Chem., 2019, 58, 16434 CrossRef CAS PubMed.
- Y. Z. Zhang, S. Gómez-Coca, A. J. Brown, M. R. Saber, X. Zhang and K. R. Dunbar, Chem. Sci., 2016, 7, 6519 RSC.
- E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130 CrossRef CAS PubMed.
- M. A. Palacios, J. Nehrkorn, E. A. Suturina, E. Ruiz, S. Gómez-Coca, K. Holldack, A. Schnegg, J. Krzystek, J. M. Moreno and E. Colacio, Chem. – Eur. J., 2017, 23, 11649 CrossRef CAS PubMed.
- F. Kobayashi, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Inorg. Chem., 2019, 58, 7409 CrossRef CAS PubMed.
- (a) A. Singh and K. Shrivastava, Phys. Status Solidi B, 1979, 95, 273 CrossRef CAS; (b) K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437 CrossRef CAS.
- Y. Wu, D. Tian, J. Ferrando-Soria, J. Cano, L. Yin, Z. Ouyang, Z. Wang, S. Luo, X. Liu and E. Pardo, Inorg. Chem. Front., 2019, 6, 848 RSC.
- J. Vallejo, M. Viciano-Chumillas, F. Lloret, M. Julve, I. Castro, J. Krzystek, M. Ozerov, D. Armentano, G. De Munno and J. Cano, Inorg. Chem., 2019, 58, 15726 CrossRef CAS PubMed.
- L. S. Mariano, I. M. L. Rosa, N. R. De Campos, A. C. Doriguetto, D. F. Dias, W. D. do Pim, A. K. S. M. Valdo, F. T. Martins, M. A. Ribeiro, E. E. B. De Paula, E. F. Pedroso, H. O. Stumpf, J. Cano, F. Lloret, M. Julve and M. V. Marinho, Cryst. Growth Des., 2020, 20, 2462 CrossRef CAS.
- S. Roy, I. Oyarzabal, J. Vallejo, J. Cano, E. Colacio, A. Bauza, A. Frontera, A. M. Kirillov, M. G. B. Drew and S. Das, Inorg. Chem., 2016, 55, 8502–8513 CrossRef CAS PubMed.
- G. K. Gransbury, R. W. Gable, B. Moubaraki, K. S. Murray, L. Sorace, A. Soncini and C. Boskovic, Chem. Sci., 2019, 10, 8855 RSC.
- J. C. Baylar and E. M. Jones, in Inorganic Synthesis, ed. H. S. Booth, McGraw-Hill, New York, 1939, vol. 5, p. 35 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural views, PXRD patterns, magnetic properties and crystallographic data of the compounds. CCDC 2063853–2063856. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00595b |
| This journal is © The Royal Society of Chemistry 2021 |