
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-dependent surface modification of uranium oxides exposed to water plasma†
Ghada
El Jamal
 *a,
Thomas
Gouder
b,
Rachel
Eloirdi
b and
Mats
Jonsson
a
*a,
Thomas
Gouder
b,
Rachel
Eloirdi
b and
Mats
Jonsson
a
aSchool of Engineering Sciences in Chemistry, Biotechnology and Health (CBH), Department of Chemistry, Applied Physical Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: ghadaej@kth.se
bEuropean Commission, Joint Research Centre, Directorate for Nuclear Safety and Security, Postfach 2340, DE-76215 Karlsruhe, Germany
First published on 16th March 2021
Abstract
Thin UO2 films exposed to water plasma under UHV conditions have been shown to be interesting models for radiation induced oxidative dissolution of spent nuclear fuel. This is partly attributed to the fact that several of the reactive oxidizing and reducing species in a water plasma are also identified as products of radiolysis of water. Exposure of UO2 films to water plasma has previously been shown to lead to oxidation from U(IV) to U(V) and (VI). In this work we have studied the dynamics of water plasma induced redox changes in UO2 films by monitoring UO2 films using X-Ray photoelectron Photoemission (XPS) and Ultra-Violet Photoemission (UPS) spectroscopy as a function of exposure time. The surface composition in terms of oxidation states obtained from U4f7/2 peak deconvolution could be retraced along the exposure time, and compared to the valence band. The spectral analysis showed that U(IV) is initially oxidized to U(V) which is subsequently oxidized to U(VI). For extended exposure times it was shown that U(VI) is slowly reduced back to U(V). UPS data show that, unlike the U(V) formed on the surface upon oxidation of U(IV), the U(V) formed upon reduction of U(VI) is localized in the bulk of the film. It also displays a different reactivity than the initially formed U(V). The experiments can be reproduced using a simple kinetic model describing the redox processes involved.
Introduction
In many countries, management of used nuclear fuel is based on isolating the fuel from the biosphere by placing it in a deep geological repository.1 In the safety assessment of such a repository, the worst-case scenario is failure of all barriers leading to groundwater intrusion.1,2 The major component of the used fuel is UO2, which has very low solubility in groundwater.3 Oxidation of U(IV) to U(VI), will lead to significantly increased solubility of the fuel matrix and thereby enable release and migration of radionuclides (fission products and actinides) incorporated in the fuel matrix into the environment.4 While groundwater at potential repository sites is usually reducing, oxidative dissolution of the fuel matrix can only be initiated by the ionizing radiation emitted from the fuel itself. When ionizing radiation (e.g., alpha, beta and gamma radiation) is absorbed by water, the water undergoes radiolysis and both oxidizing (H2O2, HO˙, HO2˙ and O2) and reducing (H2, H˙, and e−aq) species are formed.5 Initially, the oxidants will dominate the surface reactions for kinetic reasons.6 Assessment of the relative impact of radiolytic oxidants under repository conditions revealed that H2O2 is the oxidant of main importance.7 In addition to oxidizing UO2, H2O2 can also undergo catalytic decomposition on the UO2 surface.8,9 The final product of the latter reaction is O2 and H2O. Interestingly, both reactions appear to have the surface bound hydroxyl radical as a common intermediate.10Research on oxidative dissolution of UO2 has been conducted quite extensively using various material types as powder, pellets, crystals, films of UO2 or simulated spent fuels.7,11–17 Such studies include interactions between aqueous radiolysis products and UO2 surfaces, conducted in chemical and electrochemical experiments.1,18–22 Some studies were made by continuously exposing uranium oxide to low steady-state concentrations of oxidants produced by irradiating suspensions of uranium oxide or pellets immersed in aqueous solution.8 Other studies focused on the leaching of pellets (pure or doped with non-radioactive isotopes of fission products)23–27 and even of thin films.18,28,29 The impact of dose rate, pH, temperature, oxidant or reductant concentration/pressure, bicarbonate and other groundwater constituents, have been assessed.14,19,25,26,30–32
During the last decade, considerable progress has been made in the use of surface spectroscopy to study thin films, including uranium oxide films under UHV conditions.18,28,33 Very recently, X-ray Photoelectron Spectroscopy (XPS) and UV Photoelectron Spectroscopy (UPS) were used to study changes in uranium oxide thin films exposed to O2, H2 and H2O plasmas.34 The plasmas were used to mimic the impact of various water radiolysis products, thereby providing a novel model system for radiation induced oxidation of uranium oxide. This model system is particularly useful for studies of the mechanism of oxidation of UO2 (and other oxidation states) as it does not involve dissolution of oxidized uranium oxide or deposition of secondary phases due to solubility limitations. The oxides previously under study were UO2, U2O5 and UO3. Interestingly, upon exposure to H2O-plasma at 20 °C, UPS showed that the final state of the surface layer of the oxide was U(VI) regardless of starting material. However, at 400 °C, XPS reveal differences depending on the starting material.
UO2 has a fluorite lattice structure, which can accommodate oxygen at high temperature to form non-stoichiometric oxides UO2+x.32 For x < 0.5 the fluorite structure is preserved, while for x > 0.5 a layered structure forms. For example, U3O8 is a thermodynamically stable phase consisting of a mixture of U5+ and U6+.35 U2O5 was previously assumed to be characterized by a mixed valence state.36 However, recent XPS characterization of thin U2O5 films produced by reducing UO3 with H2 plasma shows that U(V) is the only oxidation state.37 UO3 exists in seven different crystallographic phases and an amorphous phase.32 Recent studies on radiation induced oxidation of UO2-pellets reveal that considerable amounts of U(V) can be found on the surface of the pellet after exposure to alpha-irradiated aqueous solutions.38 This implies that the UO2-surface has been one-electron oxidized and that complete oxidation of UO2 is most probably attributed to two consecutive one-electron oxidation steps, very much in line with previous mechanistic studies.10,11 To shed more light on the detailed mechanism of radiation induced oxidation of UO2 we have studied ECR water plasma-induced transformation of thin UO2 films produced by sputter deposition as a function of exposure time. The water plasma is largely composed of the same constituents as are formed in radiolysis of water and the experiment therefore serves as a model for radiation induced oxidation of UO2.39 The majority of the experiments were carried out at 400 °C to allow fast diffusion and formation of homogeneous compounds under equilibrium conditions. For the same reason thin films of 20 nm thickness were used.
Surface speciation was done by XPS and UPS, following U 4f, O 1s and valence band regions. The oxidation state of uranium was determined by deconvolution of the U 4f7/2 core level peak into its U(IV), U(V) and U(VI) components. Surface characterization is also based on the binding energy shift and peak broadening of U 4f5/2 and O 1s, on the U 4f5/2 satellites, and on the valence band region with the O 2p valence band and the U 5f level.
Experimental
Sample preparation
Uranium oxide films were prepared in situ by direct current (DC) sputtering from a uranium metal target in a gas mixture of Ar (6 N) and O2 (5 N), at 5 × 10−3 mbar and 2 × 10−6 mbar, respectively. The conditions were optimized to form pure 20 nm thick UO2.0 films. The uranium target voltage was fixed at −700 V. The plasma in the diode source was maintained by injection of electrons of 25–50 eV energy (triode setup), allowing working at low Ar pressure in the absence of stabilizing magnetic fields. The deposition was done on a polycrystalline Au foil substrate heated at 250 °C. Gold was chosen because of its electrical conductivity, preventing surface charging during photoemission, and its high melting point, preventing any chemical reaction or diffusion of atoms (O, H, U) from the film into the substrate at the elevated temperatures.ECR plasma source
The electrons were excited by an ECR (Electron Cyclotron Resonance) discharge based on stochastic heating of electrons by microwave radiation with permanent magnets. When the resonance condition between the electrons and the microwave electric field is fulfilled, the electrons gain sufficient energy to ionize the gas and sustain the plasma. In addition, they produce excited species, free radicals, and ions providing a reactive plasma environment. More details about the ECR source and different gas plasma characterization can be found in a previous study.34 The impact of oxygen or hydrogen plasma on uranium oxide surface was followed by XPS. Based on the comparison with molecular oxygen or molecular hydrogen exposure it was concluded that the reactive gas plasmas are generating atomic oxygen or hydrogen, respectively. The plasma of water is more complex than that of the H2 and O2. The RGA-MS results revealed that the H2O-plasma contains a mixture of oxidizing and reducing species. The detected products (H2 and O2) are the result of recombination reactions between the atomic species formed. The total pressure of the feed gas introduced was 2.00 × 10−5 mbar.34A Pyrolytic Boron Nitride (PBN) heater installed below the sample holder maintained the sample temperature at 400 °C during the exposure to water plasma.
Surface characterization
High-resolution X-ray photoelectron spectroscopy analyses were done with a Specs Phoibos 150 hemispherical analyzer, using Al-Kα (1486.6 eV) radiation, produced by a XRC-1000 μ-focus source, equipped with a monochromator and operating at 120 W. The background pressure in the analysis chamber was 2 × 10−10 mbar. UPS spectra were taken with He II (40.81 eV) UV light, produced by a high intensity windowless discharge lamp. The spectrometer was calibrated using the Au 4f7/2 line of Au metal (83.9 eV BE) and the Cu 2p3/2 line of Cu metal (932.7 eV BE). Photoemission spectra were taken at room temperature. The fitting routine of the U 4f7/2 peak was based on a Lorentzian–Gaussian contribution with a Shirley background using XPSpeak41 software version 4.1.Results and discussion
Spectral analysis
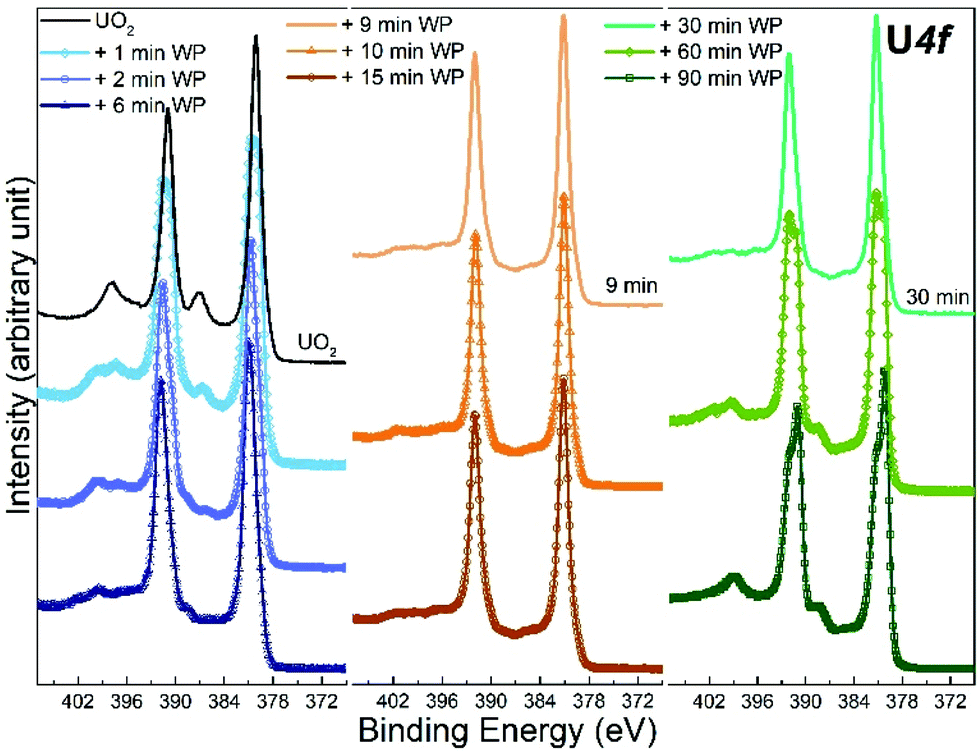 | ||
| Fig. 1 Core level X-ray photoemission spectra of U 4f recorded for unexposed UO2 film, and 9 other UO2 films exposed to water plasma (WP) for different length of time. | ||
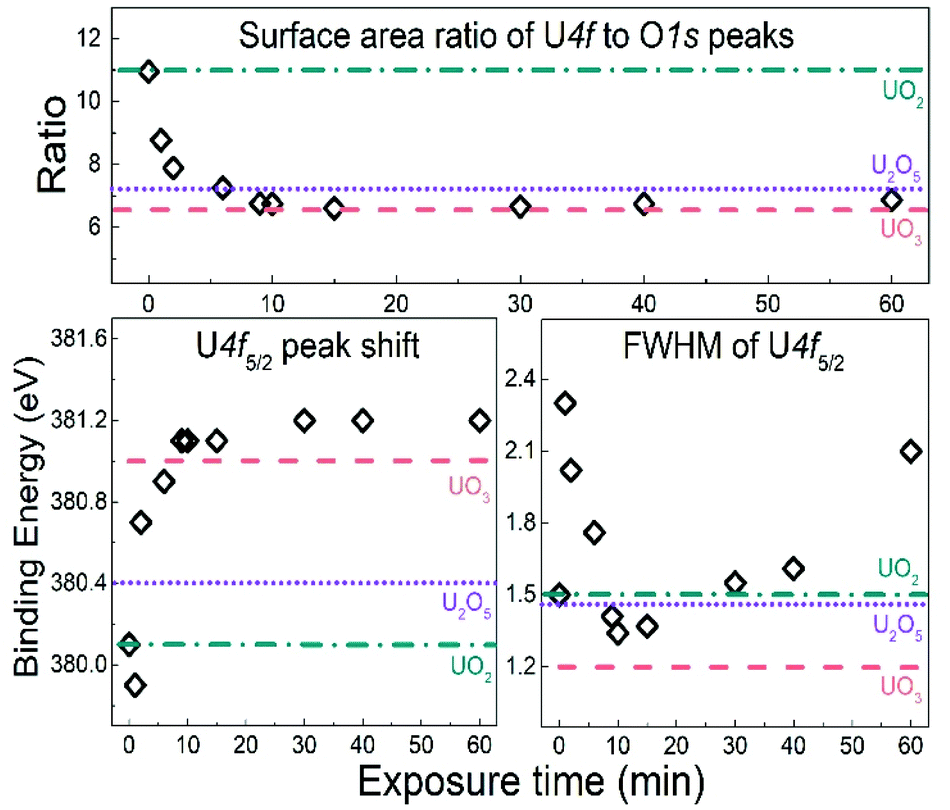 | ||
| Fig. 2 Peak position, FWHM of U 4f5/2 lines and surface area ratio of U 4f lines to O 1s peak for UO2 samples exposed to water plasma for different time length. The same characteristics for UO2, U2O5 and UO3 reference films are inserted as horizontal lines. | ||
The U 4f/O 1s surface area ratio swiftly decreases, reflecting incorporation of oxygen atoms into the lattice of the oxide and pronounced oxidation of the UO2 film. All these observations confirm progressive quantitative oxidation of UO2 by the water plasma. The oxidation of UO2 involves incorporation of O2− ions into vacant interstitial locations in the UO2 cubic fluorite structure accompanied by the charge-balancing conversion of adjacent U(IV) cations to U(V).1
In the time interval of 9–15 minutes of exposure to the water plasma, the U 4f main peaks shift to slightly higher BE. The U 4f/O 1s intensity ratio passes through a minimum corresponding to that of UO3. In addition, the FWHM reaches a minimum. This stage presents the maximal oxidation of the film (to almost pure UO3). The small FWHM proves that only one compound is present. Two oxidation states with their shifted U 4f peaks would produce a broader composite peak. The maximum surface oxidation of UO2 is reached after ca. 10 minutes of exposure to the water plasma. After an exposure longer than 10–15 minutes, the U 4f/O 1s intensity ratio increases indicating that the oxide is being slowly reduced.
The U 4f/O 1s intensity ratio increase is accompanied by a FWHM increase, indicating superposition of U 4f peaks with different binding energies. After 60 minutes of water plasma exposure, the two oxidation states become clearly distinct as shown in Fig. 1. The BE difference is about 0.8 eV which is close to the BE difference between U(VI) and U(V) reference lines. After 90 minutes exposure to water plasma the peak assigned to U(V) becomes predominant, indicating that further reduction of U(VI) has taken place.
Fig. 3 shows the O 1s region of the starting UO2 film and the films exposed to water plasma for 2, 10 and 60 minutes. The reference line of U2O5 was added for comparison. As can be seen, it is almost overlapping with the spectrum of the film exposed to water plasma for 2 minutes. The exact values of binding energy peak position and FWHM are displayed as functions of exposure time with reference peaks of UO2, U2O5 and UO3 from the literature.37 Upon exposure to water plasma, the FWHM of the O 1s peak (after 2 minutes) first increases to the value corresponding to U2O5 and then (after 10 minutes) increases to the value corresponding to UO3. After 60 minutes, the FWHM has decreased again to a value between UO3 and U2O5. This is consistent with UO3 formation and the intensity increase of the U(V) satellite peak in the U 4f scans of the same film as will be shown below. Evolution of the peak binding energy is less informative. The shift is mainly due to changing work function and Fermi-energy in the different oxides. This is a rigid shift affecting all photoemission peaks (see e.g. the valence region in Fig. 5) of both U and O. In all cases, oxygen is at the state O2− in contrast to uranium, whose oxidation state varies from +4 to +6. So, there is no reason to expect significant chemical BE shifts (used for speciation in photoemission) for the O 1s.
 | ||
| Fig. 3 Core level X-ray photoemission spectra of O 1s recorded for unexposed UO2 film, and 3 other UO2 films exposed to water plasma (WP) for different time. The peak position and FWHM are plotted as function of exposure time and compared to UO2, U2O5 and UO3 reference values inserted as horizontal lines. | ||
A close plot of the satellite region after different water plasma exposure times is displayed in Fig. 4. The satellite for the unexposed UO2 film is characteristic for U(IV). After only 1 minute of water plasma exposure, the U(V) satellite peak appears and partly replaces the U(IV) satellite. The early stage of oxidation of UO2 by water plasma (from 1 to 6 minutes) produces a mixture of U(IV) and U(V). For slightly longer exposures (from 9 to 15 minutes), the satellite lines of U(IV) and U(V) gets weaker with increasing exposure time. This is accompanied by the appearance of the U(VI) satellite peak. For even longer exposures to water plasma (30 to 90 minutes), the U(VI) satellite peaks gradually become weaker and after 60 minutes of plasma exposure, the U(V) satellite peak becomes the dominating satellite peak.
 | ||
| Fig. 4 U 4f5/2 satellite region recorded for unexposed UO2 film, and initial fresh UO2 films exposed to water plasma (WP) for different times. | ||
For intermediate exposure times (9 and 30 minutes), the satellite peaks are much less pronounced than in pure, crystalline oxides. Obviously, their intensity not only depends on the oxidation state but also on additional factors, in particular defect concentration affecting the lifetime induced broadening. Ion sputtering of a surface is known for inducing structural disorder and amorphisation.42–44 It has been previously observed that uranium oxide surfaces sputtered with Ar ions, amorphise the surface and introduces defects.43 Also a previous study on UO2 film showed that its oxidation (into UO2+x) leads to less defined U 4f satellite intensity peaks.45 Conversely, sample annealing, which restores the crystal order,46 intensifies the satellite peaks.46 In the present case by oxidation, the plasma alters the initial fluorite structured UO2 into layer structured UO3. This process destroys crystal order. The longer water plasma exposure times (60 to 90 minutes) at 400 °C work as annealing and allow the film to order again. As a direct consequence, the satellites become more pronounced. Hence, satellite intensities are useful, but not unambiguous criteria for oxidation states. It is therefore important to combine the analysis of satellite peaks with other sources of information, such as valence region spectra, which give an independent indication on the oxidation state via the intensity and width of the U 5f emission (see below).
Valence band region
Analysis of the valence region (Fig. 5) reveals the same dynamics due to water plasma exposure as mentioned above. Here, we will only analyze spectra recorded after 2, 10 and 60 minutes of exposure. The valence region spectrum of a pure U2O5 film is added to the plots as a reference. | ||
| Fig. 5 Core level X-ray photoemission spectra of valence band region recorded with XPS in (a) and (b) and with UPS in (c) for unexposed UO2, U2O5 and 3 other UO2 films exposed to water plasma (WP) for different time length. | ||
The black curve shows the spectrum of the unexposed UO2 film. The peak from 2 to 8 eV is due to the O 2p valence band, with two prominent features at 4.5 and 7 eV, related to the band structure.37 The peak at 1.2 eV is attributed to the U 5f emission. In UO2 this peak corresponds to the U 4f2 level, which is a localized (non-bonding) state. After water plasma exposure the U 5f peak shifts to lower BE, together with the O 2p valence band signal. This is again due to a rigid BE shift related to work function and Fermi-energy change, as mentioned for the O 1s line. The U 5f peak narrows (already after 2 minutes) and its intensity has drastically decreased after 10 and 60 minutes of exposure. For further discussion, we show two representations of the spectra, one normalized on the O 2p intensity (Fig. 5a) and one on the U 5f intensity (Fig. 5b).
The films exposed to water plasma had almost the same peak width for all three exposure times. This width corresponds to that of pure 5f1 of U2O5, which is narrower than that of the 5f2 line of UO2. This is due to the different electron configurations (5f2 and 5f1) leading to different final state multiplet structures for UO2 and U2O5. Photoemission from the 5f2 initial state of UO2 leads to the 5f1 final state with a doublet structure (5f5/2 and 5f7/2) due to spin–orbit interaction. Photoemission from the 5f1 initial state of U2O5 leads to the 5f0 final state, with a singlet structure (no spin–orbit interaction). Therefore, the 5f emission in U2O5 is narrower than in UO2. Also, the intensity of 5f1 is lower than that of 5f2. One would expect a reduction by a factor of about 5, but it is hard to obtain a quantitative analysis from the figure, because there is no intensity reference: the 5f line decreases with exposure while the O 2p grows because of O incorporation and change in the type of bonding of the U–O bond (affecting the photoemission cross-section of the valence band). The overall effect is a strong decrease of the U 5f/valence band intensity ratio. For longer surface exposures (>2 minutes) the U 5f decreases in intensity but keeps the same width, (5f1). We conclude that U4+ disappears very fast (mostly after 2 minutes exposure) and the oxidized films are a mixture of the two oxidation states: U(V) and U(VI). The change in intensity is due to a change in the proportion of each valence state: U(V) has a 5f1 signal while U(VI) has a 5f0 initial state configuration, i.e. no 5f line at all.
As mentioned above, the water plasma exposures were done at 400 °C to ensure fast diffusion of oxygen through the film and homogeneity of compounds. However, UPS measurements reveal that the top layers are not homogeneous. This is shown in Fig. 5, where the same energy region (0 to 12 eV) as the valence region monitored with XPS has been monitored with the significantly more surface sensitive techniques UPS (information depth of 1 monolayer instead of 6 monolayers). This is due to the mean free path of UV photons which is much smaller than that of X-ray photons due to the relatively small optical depth (the scattering cross section of UV photons is much larger than that of X-ray photons).47 UPS shows the same trends in intensity and position of the U 5f and the valence band as XPS. The different 5f/VB intensity ratios for the same water plasma exposure times are due to the different cross-sections in UPS and XPS.48 However, the complete disappearance of the 5f after 60 (90) minutes exposure cannot be explained by cross-section effects but is due to the complete oxidation of the outermost surface layer. This difference in oxidation is not due to a lack of diffusion (kinetics) but to the difference in oxide stability at the surface and in the bulk. Oxygen is well known to accumulate to the surface of incompletely oxidized compounds because it lowers their surface energy.
Peak fitting
The U 4f7/2 peaks in Fig. 1 were resolved and fitted using a combination of Gaussian and Lorentzian peak shapes (30–70%) together with a Shirley background in which the background intensity at any given binding energy is proportional to the intensity of the total peak area above the background in the lower binding energy peak.47 The deconvolution was made using reference peaks of pure UO2, U2O5 and UO3 films.37 The films represent the oxidation states of U(IV), U(V), and U(VI) respectively. The characteristic peak position and FWHM are stated in Table 1.| Oxidation states | Binding energy of U 4f7/2 (eV) | Binding energy of U 4f5/2 (eV) | Spin orbit splitting of U 4f main lines (eV) | FWHM of U 4f7/2 (eV) | Satellite peaks separation from U 4f5/2 (eV) |
|---|---|---|---|---|---|
| U(IV) | 380.1 | 390.9 | 10.7 | 1.5 | 6.7 |
| U(V) | 380.4 | 391.3 | 11.2 | 1.46 | 8.1 |
| U(VI) | 381.1 | 391.8 | 10.7 | 1.2 | 4.4 and 9.9 |
Fig. 6 shows results from peak deconvolution for the UO2 films after 2, 10 and 60 minutes water plasma exposure, respectively. The XPS scans were better fitted using all three reference peaks, even when the concentration of one of the valence states was very low. A larger number of parameters (peaks) always provides a better fit, still we tentatively apply it to have all three oxidation states in our consideration (even if valence spectra point to the preponderance of only two, (V) and (VI), in most cases).
 | ||
| Fig. 6 Deconvolution of U 4f7/2 main peak of UO2 films exposed to water plasma for (a) 2 min, (b) 10 min and (c) 90 min. The deduced fractions of uranium oxidation states are plotted below each case. | ||
The deconvolution of the U 4f7/2 peak after 2 minutes of water plasma exposure, Fig. 6a, implies that U(IV), U(V) and U(VI) are concomitant. When the exposure time is extended to 10 minutes, Fig. 6b, the dominant oxidation state according to the deconvolution is U(VI) (∼80%) with slight contributions of U(IV) and U(V). This is consistent with the shape of the satellite peaks of the U 4f scan of the same film plotted in Fig. 4 (even though the satellite point to a smaller (IV) and (VI) contribution after 2 minutes). The satellites after 10 minutes of water plasma exposure are matching with U(VI) satellite lines, rising at 4.4 eV and 9.9 eV higher binding energy than the U 4f5/2 main line.
The fitting result somewhat deviates from the XPS valence band spectrum, which shows principally U(V) – however curve fitting also indicates a majority of U(V) (70% versus 30% U(IV)). So there is qualitative agreement. In Fig. 6c, the deconvolution of the peak shows that the fraction of U(V) has increased significantly after 90 minutes of water plasma exposure when compared to the film exposed for 10 minutes.
The results of all the spectral analyses presented above are internally very consistent and show that when exposing a UO2 film to a water plasma under UHV conditions, U(IV) is rapidly converted to U(V) and subsequently to U(VI). For extended exposures to water plasma, the fraction of U(VI) slowly decreases and the fraction of U(V) increases with the same rate. To verify that the slow reduction of U(VI) is indeed caused by the exposure to the water plasma and not just by the prolonged heating of the film, a control experiment was performed. In this experiment a UO3 film was exposed to water plasma for 10 minutes at 400 °C. Thereafter, the water plasma was switched off and the film was kept at 400 °C under UHV conditions for 50 minutes. During this time, the U(VI) fraction decreased by ca. 10% which is considerably less than the reduction observed upon exposure to the water plasma for the same time period. This verifies that the slow reduction of U(VI) to U(V) observed in the plasma exposure experiments should mainly be attributed to the plasma exposure itself and not to thermal decomposition of UO3.
Mechanism and kinetics
Fig. 7 shows the evolution of the three oxidation states with exposure time based on the deconvolution results and the numerical simulation method described below.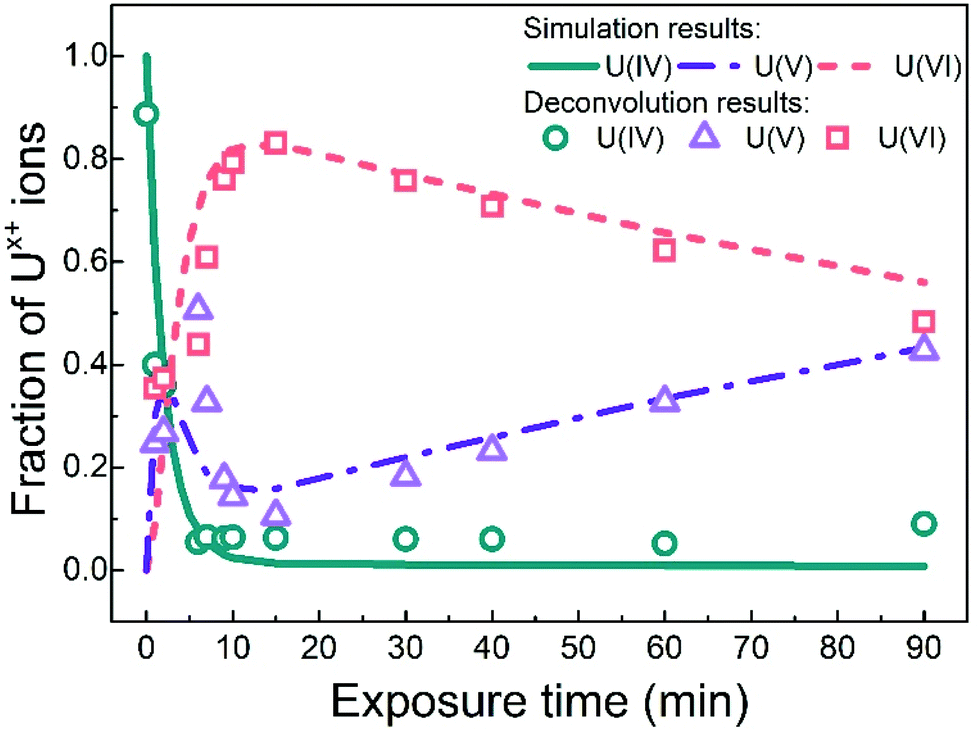 | ||
| Fig. 7 The fraction of uranium oxidation states deduced from the simulation and the U 4f7/2 peaks deconvolution results, plotted as function of exposure time to water plasma. | ||
To derive a mechanism and possibly to account for the dynamics of this process, we must first look at the properties of the water plasma. The water plasma contains H2, H˙, HO˙ and O and recombination products thereof, including O2. Hence, both reducing and oxidizing species are present. It has recently been shown that oxidation of UO2 by molecular O2 is much less pronounced than the oxidation by water plasma under comparable conditions. Consequently, O2 can be ruled out as the main oxidant here. However, a pure oxygen plasma where the main constituent is atomic oxygen, is considerably more efficient than the water plasma in oxidizing the UO2 film. The oxidation of the UO2 film exposed to water plasma can therefore, at least partly, be attributed to atomic oxygen but also to hydroxyl radicals. The actions of the oxidants are partly balanced by the actions of the reducing species and the magnitude of oxidation is also related to the concentration of oxidants in the plasma. For a tentative one-electron oxidation step we could write the following general reactions:
| U(IV) + PlasmaOx → U(V) | (1) |
| U(V) + PlasmaRed → U(IV) | (2) |
It has previously been shown that U(VI) as well as U(V) exposed to H2-plasma are quantitatively reduced to U(IV) within 10 minutes at 400 °C and that U(IV) is quantitatively oxidized to U(VI) by an O2-plasma under the same conditions. Considerably shorter exposures at lower temperature reveal that U(V) is formed as an intermediate between U(IV) and U(VI). U(V) exposed to water plasma is partly converted to U(VI) as shown by XPS. At the surface, the conversion is quantitative as shown by UPS. From these observations we can conclude that hydrogen atoms are capable of reducing U(VI) to U(V) and U(IV) and U(V) to U(IV) and that atomic oxygen can oxidize U(IV) to U(V) and U(VI) and U(V) to U(VI). As atomic oxygen as well as hydrogen atoms are also constituents of the water plasma, it is clear that a water plasma has the potential to drive oxidation of U(IV) all the way up to U(VI) and reduction of U(VI) all the way down to U(IV). The dynamics of these processes would then depend on the relative rate constants of the reactions involved and on the plasma composition (which is temperature dependent). The simplest mechanistic approach would be to assume that we can apply the general scheme presented above (i.e., that plasma oxidants can drive a one-electron oxidation step and that this is counteracted by plasma reductants reducing the one-electron oxidized product back to the original oxidation state). For the system studied here we would get the following reaction scheme:
| U(IV) + PlasmaOx → U(V) | (3) |
| U(V) + PlasmaRed → U(IV) | (4) |
| U(V) + PlasmaOx → U(VI) | (5) |
| U(VI) + PlasmaRed → U(V) | (6) |
Admittedly, the kinetics for adsorption and desorption of the plasma products on the surface should also be included in a complete mechanism. The surface is being continuously exposed to a flux of the plasma and there is no accumulation of plasma products. For this reason, the rate of each step can be expressed as being proportional to the flux of the active plasma constituent multiplied with the surface fraction of uranium in the oxidation state of relevance. In our system we start with pure U(IV) and if we apply the mechanism above, we would reach a state where the fractions of the three different oxidation states become constant (i.e. steady-state). Depending on the relative fluxes of the plasma products and the relative rate constants for each step, the final state could either contain all oxidation states or only U(VI).
As it is quite clear from the deconvolution results in Fig. 7 (symbols) and from the spectral analyses presented above that we do not reach this steady-state situation, we must conclude that the mechanism presented above is not sufficiently describing the system. What we can see in Fig. 7 is that U(VI) is slowly being reduced to U(V) and that this process does not appear to be balanced by oxidation of U(V) back to U(VI). It is interesting to note that UPS-data (in Fig. 5) show that the surface of the film is initially oxidized from U(IV) to U(V) and then to U(VI) but no slow reduction of U(VI) to U(V) is observed for longer exposure times. This implies that the slowly produced U(V) is formed deeper in the film and therefore displays a different reactivity. This can be accounted for by introducing an additional reaction to the overall mechanism. In Fig. 7 (lines), the results of a numerical simulation (using the software MAKSIMA49) taking this mechanism into account is also presented. As the exact plasma composition is not known at this point, the mechanism only includes the five redox reactions stated above (3 to 6). As a consequence, the contributions from adsorption and desorption and the possible diffusion from the film surface to deeper layers of the film are all assumed to be included in the rate coefficients used for the five redox reactions.
As can be seen, the numerical simulation reproduces the experimental (XPS) results fairly well. In these simulations the two oxidation steps have the same first order rate constant (i.e. the oxidant concentration multiplied with the second order rate constant for oxidation, kox[Ox]) and the two reduction steps also have the same first order rate constant (kred[Red]). The simulation results are obtained with a ratio of 8.6 between the first order rate constants for oxidation and reduction. The first order rate constant for the reaction leading to formation of the U(V) species inside the film (i.e. U(V)b) is a factor of 10 lower than for the other reduction steps. The overall dynamics is fairly insensitive to the ratio between the first order rate constants for oxidation and reduction (in the reversible part) as long as it is higher than 8.6, provided that the rate of oxidation is not changed. However, a higher ratio than 8.6 (i.e. slower reversible reduction) leads to higher maximum U(VI)-concentration than what is found experimentally. This verifies that the water plasma is overall oxidative in the uranium oxide system. The overall mechanism is depicted in Scheme 1.
 | ||
| Scheme 1 | ||
Conclusions
This study describes the surface modification of UO2 films exposed to a water plasma at 400 °C explored using XPS and UPS. Spectral analysis based on the U 4f and O 1s peaks, satellite peaks and valence bands unambiguously show the following evolution of the uranium oxidation state with increasing water plasma exposure time: U(IV) is rapidly (within a couple of minutes) consumed and this consumption is accompanied by rapid formation of U(V) and U(VI). The fraction of U(V) reaches a maximum after less than 10 minutes where after the fraction is rapidly decreased. The U(VI) fraction continues to increase and reaches a maximum after ca. 15 minutes. The maximum U(VI) fraction is just above 80%. After reaching the maximum fraction, the U(VI) fraction is slowly decreasing and at the same time the U(V) fraction is increasing. After 90 minutes exposure the fractions of U(VI) and U(V) are equal. The dynamics of the system can be accounted for by considering the oxidizing and reducing properties of the plasma constituent. However, reversible redox reactions connecting the three oxidation states do not account for the observed behaviour upon long-term exposure to the water plasma. To account for the slow transformation of U(VI) to U(V), an additional process must be included where the U(V)-species is different from that formed upon one-electron oxidation of U(IV). UPS data indicates that the slow transformation of U(VI) produces U(V) deeper in the film since the spectrum shows no traces of U(V). A simple kinetic model reproduces the experimental data fairly well which shows that the water plasma is, from a kinetic point of view, overall oxidizing to UO2-films under UHV conditions. The data presented here show that oxidation of U(IV) to U(VI) by a water plasma under UHV conditions can mechanistically be accounted for by two consecutive one-electron transfer steps. This further supports the validity of using thin films exposed to water plasma as a model for radiation induced oxidation of UO2-based nuclear fuel in aqueous systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Nuclear and Fuel Waste Management Company (SKB) is gratefully acknowledged for financial support.This work has been partially supported by the ENEN+ project that has received funding from the Euratom research and training Work Programme (2016-2017-1#755576).
We thank Frank Huber for excellent technical assistance.
Notes and references
- D. W. Shoesmith, Fuel corrosion processes under waste disposal conditions, J. Nucl. Mater., 2000, 282(1), 1–31 CrossRef CAS.
- T. E. Eriksen, D. W. Shoesmith and M. Jonsson, Radiation induced dissolution of UO2 based nuclear fuel – A critical review of predictive modelling approaches, J. Nucl. Mater., 2012, 420(1), 409–423 CrossRef CAS.
- A. S. Kertes and R. Guillaumont, Solubility of UO2. A comparative review, Nucl. Chem. Waste Manage., 1985, 5(3), 215–219 CrossRef CAS.
- D. W. Shoesmith and S. Sunder, The prediction of nuclear fuel (UO2) dissolution rates under waste disposal conditions, J. Nucl. Mater., 1992, 190, 20–35 CrossRef CAS.
- J. W. T. Spinks and R. J. Woods, An Introduction to Radiation Chemistry. John Wiley & Sons, New York, 1964 Search PubMed.
- Y. Kumagai, A. Barreiro Fidalgo and M. Jonsson, Impact of Stoichiometry on the Mechanism and Kinetics of Oxidative Dissolution of UO2 Induced by H2O2 and γ-Irradiation, J. Phys. Chem. C, 2019, 123(15), 9919–9925 CrossRef CAS.
- E. Ekeroth, O. Roth and M. Jonsson, The relative impact of radiolysis products in radiation induced oxidative dissolution of UO2, J. Nucl. Mater., 2006, 355(1), 38–46 CrossRef CAS.
- S. Nilsson and M. Jonsson, H2O2 and radiation induced dissolution of UO2 and SIMFUEL pellets, J. Nucl. Mater., 2011, 410(1), 89–93 CrossRef CAS.
- M. G. Bailey, L. H. Johnson and D. W. Shoesmith, The effects of the alpha-radiolysis of water on the corrosion of uo2, Corros. Sci., 1985, 25(4), 233–238 CrossRef CAS.
- A. Barreiro Fidalgo, Y. Kumagai and M. Jonsson, The role of surface-bound hydroxyl radicals in the reaction between H2O2 and UO2, J. Coord. Chem., 2018, 71(11–13), 1799–1807 CrossRef CAS.
- E. Ekeroth and M. Jonsson, Oxidation of UO2 by radiolytic oxidants, J. Nucl. Mater., 2003, 322(2), 242–248 CrossRef CAS.
- O. Roth, T. Bönnemark and M. Jonsson, The influence of particle size on the kinetics of UO2 oxidation in aqueous powder suspensions, J. Nucl. Mater., 2006, 353(1), 75–79 CrossRef CAS.
- M. M. Hossain, E. Ekeroth and M. Jonsson, Effects of HCO3- on the kinetics of UO2 oxidation by H2O2, J. Nucl. Mater., 2006, 358(2), 202–208 CrossRef CAS.
- O. Roth and M. Jonsson, Oxidation of UO2(s) in aqueous solution, Cent. Eur. J. Chem., 2008, 6(1), 1–14 CAS.
- A. J. Popel, S. Le Solliec, G. I. Lampronti, J. Day, P. K. Petrov and I. Farnan, The effect of fission-energy Xe ion irradiation on the structural integrity and dissolution of the CeO2 matrix, J. Nucl. Mater., 2017, 484, 332–338 CrossRef CAS.
- J. Giménez, E. Baraj, M. E. Torrero, I. Casas and J. de Pablo, Effect of H2O2, NaClO and Fe on the dissolution of unirradiated UO2 in NaCl 5 mol kg−1. Comparison with spent fuel dissolution experiments, J. Nucl. Mater., 1996, 238(1), 64–69 CrossRef.
- S. Sunder, N. H. Miller and D. W. Shoesmith, Corrosion of uranium dioxide in hydrogen peroxide solutions, Corros. Sci., 2004, 46(5), 1095–1111 CrossRef CAS.
- A. J. Popel, S. R. Spurgeon, B. Matthews, M. J. Olszta, B. T. Tan, T. Gouder, R. Eloirdi, E. C. Buck and I. Farnan, An Atomic-Scale Understanding of UO2 Surface Evolution during Anoxic Dissolution, ACS Appl. Mater. Interfaces, 2020, 12(35), 39781–39786 CrossRef CAS PubMed.
- S. Sunder, D. W. Shoesmith and N. H. Miller, Oxidation and dissolution of nuclear fuel (UO2) by the products of the alpha radiolysis of water, J. Nucl. Mater., 1997, 244(1), 66–74 CrossRef CAS.
- F. Clarens, J. de Pablo, I. Díez-Pérez, I. Casas, J. Giménez and M. Rovira, Formation of Studtite during the Oxidative Dissolution of UO2 by Hydrogen Peroxide: A SFM Study, Environ. Sci. Technol., 2004, 38(24), 6656–6661 CrossRef CAS PubMed.
- B. T. Tan, A. J. Popel, R. J. Wilbraham, J. Day, G. I. Lampronti, C. Boxall and I. Farnan, Surface and electrochemical controls on UO2 dissolution under anoxic conditions, J. Nucl. Mater., 2019, 520, 41–55 CrossRef CAS.
- L. Wu and D. W. Shoesmith, An Electrochemical Study of H2O2 Oxidation and Decomposition on Simulated Nuclear Fuel (SIMFUEL), Electrochim. Acta, 2014, 137, 83–90 CrossRef CAS.
- B. Muzeau, C. Jégou, F. Delaunay, V. Broudic, A. Brevet, H. Catalette, E. Simoni and C. Corbel, Radiolytic oxidation of UO2 pellets doped with alpha-emitters (238/239Pu), J. Alloys Compd., 2009, 467(1), 578–589 CrossRef CAS.
- L. Bauhn, N. Hansson, C. Ekberg, P. Fors, R. Delville and K. Spahiu, The interaction of molecular hydrogen with α-radiolytic oxidants on a (U,Pu)O2 surface, J. Nucl. Mater., 2018, 505, 54–61 CrossRef CAS.
- M. Odorowski, C. Jegou, L. De Windt, V. Broudic, G. Jouan, S. Peuget and C. Martin, Effect of metallic iron on the oxidative dissolution of UO2 doped with a radioactive alpha emitter in synthetic Callovian-Oxfordian groundwater, Geochim. Cosmochim. Acta, 2017, 219, 1–21 CrossRef CAS.
- A. C. Maier, A. Barreiro Fidalgo and M. Jonsson, Impact of H2 and Consecutive H2O2 Exposures on the Oxidative Dissolution of (U1−xGdx)O2 Pellets Under Deep Repository Conditions for Spent Nuclear Fuel, Eur. J. Inorg. Chem., 2020, 2020(20), 1946–1950 CrossRef CAS.
- M. Trummer, S. Nilsson and M. Jonsson, On the effects of fission product noble metal inclusions on the kinetics of radiation induced dissolution of spent nuclear fuel, J. Nucl. Mater., 2008, 378(1), 55–59 CrossRef CAS.
- R. Springell, S. Rennie, L. Costelle, J. Darnbrough, C. Stitt, E. Cocklin, C. Lucas, R. Burrows, H. Sims, D. Wermeille, J. Rawle, C. Nicklin, W. Nuttall, T. Scott and G. Lander, Water corrosion of spent nuclear fuel: radiolysis driven dissolution at the UO2/water interface, Faraday Discuss., 2015, 180(0), 301–311 RSC.
- C. M. Lousada, M. Trummer and M. Jonsson, Reactivity of H2O2 towards different UO2-based materials: The relative impact of radiolysis products revisited, J. Nucl. Mater., 2013, 434(1), 434–439 CrossRef CAS.
- V. Metz, A. Loida, E. Bohnert, D. Schild and K. Dardenne, Effects of hydrogen and bromide on the corrosion of spent nuclear fuel and γ-irradiated UO2(s) in NaCl brine, in Radiochimica Acta International journal for chemical aspects of nuclear science and technology, 2008, vol. 96, p. 637 Search PubMed.
- R. J. McEachern and P. Taylor, A review of the oxidation of uranium dioxide at temperatures below 400 °C, J. Nucl. Mater., 1998, 254(2), 87–121 CrossRef CAS.
- H. Idriss, Surface reactions of uranium oxide powder, thin films and single crystals, Surf. Sci. Rep., 2010, 65(3), 67–109 CrossRef CAS.
- T. Gouder, Thin layers in actinide research, J. Alloys Compd., 1998, 271–273, 841–845 CrossRef CAS.
- G. El Jamal, T. Gouder, R. Eloirdi and M. Jonsson, X-Ray and ultraviolet photoelectron spectroscopy studies of Uranium(iv),(v) and(vi) exposed to H2O-plasma under UHV conditions, Dalton Trans., 2021, 50(2), 729–738 RSC.
- K. Sanyal, A. Khooha, G. Das, M. K. Tiwari and N. L. Misra, Direct Determination of Oxidation States of Uranium in Mixed-Valent Uranium Oxides Using Total Reflection X-ray Fluorescence X-ray Absorption Near-Edge Spectroscopy, Anal. Chem., 2017, 89(1), 871–876 CrossRef CAS PubMed.
- Y. A. Teterin, V. M. Kulakov, A. S. Baev, N. B. Nevzorov, I. V. Melnikov, V. A. Streltsov, L. G. Mashirov, D. N. Suglobov and A. G. Zelenkov, A study of synthetic and natural uranium oxides by X-ray photoelectron spectroscopy, Phys. Chem. Miner., 1981, 7(4), 151–158 CrossRef CAS.
- T. Gouder, R. Eloirdi and R. Caciuffo, Direct observation of pure pentavalent uranium in U2O5 thin films by high resolution photoemission spectroscopy, Sci. Rep., 2018, 8(1), 8306 CrossRef CAS PubMed.
- N. L. Hansson, P. L. Tam, C. Ekberg and K. Spahiu, XPS study of external α-radiolytic oxidation of UO2 in the presence of argon or hydrogen, J. Nucl. Mater., 2021, 543, 152604 CrossRef CAS.
- R. S. Dixon, Dissociation of water vapour by photolytic, radiolytic, and electron impact methods, Radiat. Res. Rev., 1970, 2, 237–296 CAS.
- E. S. Ilton and P. S. Bagus, XPS determination of uranium oxidation states, Surf. Interface Anal., 2011, 43(13), 1549–1560 CrossRef CAS.
- P. S. Bagus, C. J. Nelin, E. S. Ilton, M. Baron, H. Abbott, E. Primorac, H. Kuhlenbeck, S. Shaikhutdinov and H. J. Freund, The complex core level spectra of CeO2: An analysis in terms of atomic and charge transfer effects, Chem. Phys. Lett., 2010, 487(4), 237–240 CrossRef CAS.
- D. Gupta, M. Goyal, R. Singhal, A. Sharma and S. Aggarwal, High flux Ar+ sputtering induced structural disorder and amorphization in Si (111), AIP Conf. Proc., 2019, 2093(1), 020013 Search PubMed.
- S. D. Senanayake, G. I. N. Waterhouse, A. S. Y. Chan, T. E. Madey, D. R. Mullins and H. Idriss, Probing Surface Oxidation of Reduced Uranium Dioxide Thin Film Using Synchrotron Radiation, J. Phys. Chem. C, 2007, 111(22), 7963–7970 CrossRef CAS.
- G. Greczynski and L. Hultman, Towards reliable X-ray photoelectron spectroscopy: Sputter-damage effects in transition metal borides, carbides, nitrides, and oxides, Appl. Surf. Sci., 2021, 542, 148599 CrossRef CAS.
- Z. Bao, Croissance Epitaxiale, Caractérisation Structurale et Etudes Magnétiques de Couches Minces d'UO2, L'UNIVERSITÉ DE GRENOBLE, 2013 Search PubMed.
- D. Gupta, G. R. Umapathy, R. Singhal, S. Ojha and S. Aggarwal, Nano-scale depth-varying recrystallization of oblique Ar+ sputtered Si(111) layers, Sci. Rep., 2020, 10(1), 11905 CrossRef CAS PubMed.
- D. Briggs and M. P. Seah, Practical surface analysis by auger and photo-electron spectroscopy, Wiley, Chichester, 1983 Search PubMed.
- C. S. Fadley and D. A. Shirley, Electronic Densities of States from X-Ray Photoelectron Spectroscopy, J. Res. Natl. Bur. Stand., Sect. A, 1970, 74A(4), 543–558 CrossRef CAS PubMed.
- M. B. Carver, D. V. Hanley and K. R. Chaplin, MAKSIMA-CHEMIST a program for mass action kinetics simulation by automatic chemical equation manipulation and integration using stiff techniques. AECL, Technical Reports, Ontario, 1979.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt00486g |
| This journal is © The Royal Society of Chemistry 2021 |