
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening polymerisation of L- and rac-lactide using group 4 permethylpentalene aryloxides and alkoxides†
Zoë R.
Turner
 ,
Jessica V.
Lamb
,
Thomas P.
Robinson
,
Dipa
Mandal
,
Jean-Charles
Buffet
and
Dermot
O′Hare
*
,
Jessica V.
Lamb
,
Thomas P.
Robinson
,
Dipa
Mandal
,
Jean-Charles
Buffet
and
Dermot
O′Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12, Mansfield Road, OX1 3TA, Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk; Tel: +44 (0)1865 272686
First published on 16th March 2021
Abstract
A new family of group 4 permethylpentalene (C8Me62−; Pn*) aryloxide and alkoxide complexes have been synthesised and fully characterised by multinuclear NMR spectroscopy and single-crystal X-ray diffraction; (η8-C8Me6)Zr(OR)2 (R = tBu (1), 2,6-Me-C6H3 (2), 2,6-iPr-C6H3 (3) and 4-OMe-C6H4 (4)), (η8-C8Me6)Zr (OR) (R = 2,6-tBu-C6H3 (5) and 2,6-tBu-4-Me-C6H2 (6)), (η8-C8Me6)ZrCp(OR) (R = tBu (7), 2,6-Me-C6H3 (8) and 2,6-iPr-C6H3 (9)), (η8-C8Me6)TiCp(O-2,6-Me-C6H3) (10) and (η8-C8Me6)ZrCpMe(OR) (R = 2,6-Me-C6H3 (11), 2,6-iPr-C6H3 (12) and 2,4-tBu-C6H3 (13)). 2, 3, 6, 7, 9, 10 and 12 were studied as initiators for the ring-opening polymerisation (ROP) of L-lactide, and 2, 3, 6, 7 and 10 were studied as initiators for the ROP of rac-lactide. 3 was found to be the most active initiator for the ROP of L-lactide (kobs = 0.35 h−1) and 2 for the ROP of rac-lactide (kobs = 0.21 h−1). These initiators produced isotactic PLA for the ROP of L-lactide and moderately heterotactic enriched (maximum Pr of 0.69) or atactic PLA for the ROP of rac-lactide with polymer chains consisting of polylactic acid repeat units with –OR and –OH end groups.
Introduction
Polyolefins have diverse uses, including packaging, agriculture and medical applications.1 However, the resistance of polyolefins to chemical, physical and biological degradation has become a serious environmental concern.2 Therefore, there is a global necessity for biodegradable and biocompatible polymers derived from renewable feedstocks that can be broken down into smaller molecules (such as CO2, CH4 and H2O) naturally by microorganisms.1,3–7 Polylactides (PLAs) produced from the ring-opening polymerisation (ROP) of lactide are one the most versatile materials among biodegradable polymers due to their inherent biodegradability, biocompatibility, high mechanical strength, low toxicity and easy availability from renewable sources.3–5,8,9 The versatile properties of PLAs enables their use in a wide range of applications, from biomedical and pharmaceutical materials for tissue engineering and wound dressings to biodegradable materials for bags and cutlery.3,10–12 The stereochemistry of the lactide monomer units leads to PLA chains with diverse stereochemical arrangements. This results in variations in the mechanical, physical and thermal degradation properties of the polymers, which play a crucial role in determining the potential applications. Stereospecific, single-site catalysts with the ability to control polymer architectures are therefore highly desired.A range of metal complexes and ligand frameworks have been studied as single-site initiators6,13,14–17 for the stereoselective ROP of lactide (indium,18–21 scandium and yttrium,22–27 lanthanum28,29 samarium,29 and iron30). For the polymerisation of rac-lactide, high levels of isotactic stereocontrol were observed when using indium initiators (Pm up to 0.87), while highly heterotactic PLA was obtained for yttrium initiators (Pr > 0.87).20,23 Spassky and co-workers first reported chiral salen complexes affording isotactic PLA from rac-lactide.31,32 Aluminium complexes bearing tetradentate N,N′-disubstituted bis(amino-phenoxide)33–37 also show high degrees of stereocontrol for the polymerisation of rac-lactide, with polymer tacticity largely dependent on the ligand substituents; isotactic PLAs are produced with unsubstituted phenoxide groups (Pm up to 0.98), while highly heterotactic PLAs are obtained with 3,5-substituted phenoxide groups (Pr up to 0.96).38 β-Diiminate zinc complexes have also been shown to afford highly heterotactic PLA from the ROP of rac-lactide (Pr up to 0.94).39
Group 4 complexes are far less studied as initiators for the ROP of lactide. However, zirconium alkoxide compounds bearing a tridentate N-heterocyclic carbene-linked bis(phenolate) ligand have been reported to show controlled and highly stereoselective ROP of rac-lactide to yield heterotactic PLA under mild conditions (Pr > 0.95).40 Zirconium and hafnium amine tris(phenolate) alkoxides also show high activity and stereocontrol for the ROP of rac-lactide with heterotactic PLA produced under solvent-free conditions (Pr > 0.90).41 O'Hare and co-workers have also reported families of well-defined zirconium and hafnium biscyclopentadienyl, bisindenyl and unsymmetrical permethylindenyl complexes that were active for the ROP of L-, D- and rac-lactide with varying degrees of stereocontrol achieved.42,43 In addition, we recently reported a new family of chiral group 4 alkoxide and aryloxide half-sandwich η5-complexes of a chiral cyclopentadienyl-derived (hydro)-permethylpentalenyl ligand {(C8Me6H)ML3; Pn*(H)ML3} as very active initiators for the ROP of L- and rac-lactide.56
Herein, following on from our work on Pn*MX2,44,45 we report the development of a new family of group 4 η8-permethylpentalenyl complexes of the type {(η8-C8Me6)ML2; Pn*ML2} (Chart 2) as initiators for the ROP of L- and rac-lactide. By targeting Pn*ML2 complexes the stereoelectronic properties of the ancillary ligands and initiation groups can be varied to influence polymerisation activity and stereocontrol.
Results and discussion
Synthesis and characterisation of Pn*Zr(OR)2 and Pn*ZrCl(OR)
The reaction of one equivalent of previously synthesised [Pn*ZrCl2]2·LiCl(thf)x46 with four equivalents KOR (R = tBu, 2,6-Me-C6H3 and 2,6-iPr-C6H3) afforded two equivalents of the bis(alkoxide) complexes Pn*Zr(OtBu)2 (1), Pn*Zr(O-2,6-Me-C6H3)2 (2) and Pn*Zr(O-2,6-iPr-C6H3)2 (3) (Charts 1 and 2). 1 was synthesised in a Young's tap NMR spectroscopy tube as preparative-scale synthesis was hindered by its instability (formation of intractable mixture of species). Complexes 2 and 3 were synthesised on a preparative scale and isolated as yellow solids in 78 and 79% yields respectively. The mono(aryloxide) complex Pn*ZrCl(O-2,6-Me-C6H3) can also be formed by the addition of sub-stoichiometric amounts of KO-2,6-Me-C6H3 to [Pn*ZrCl2]2·LiCl(thf)x. The 1H NMR spectra of 1, 2 and 3 show two singlets with integration 6 and 12 between 1.86 and 2.13 ppm, which are diagnostic of the wing-tip and non-wing-tip methyl groups of the Pn* ligand respectively and are consistent with molecules of C2v symmetry (Fig. S1, S3 and S4†). For 1, a singlet at 1.24 ppm represents the protons of the t-butyl groups, while for 2 and 3, a doublet at approximately 7.00 ppm and a triplet at approximately 6.85 ppm define the aromatic protons of the aryloxide group. The methyl resonances of the aryloxide group of 2 are observed as a singlet at 2.16 ppm, while for 3, a doublet at 1.24 ppm and a septet at 3.20 ppm represent the isopropyl methyl groups and methine proton respectively.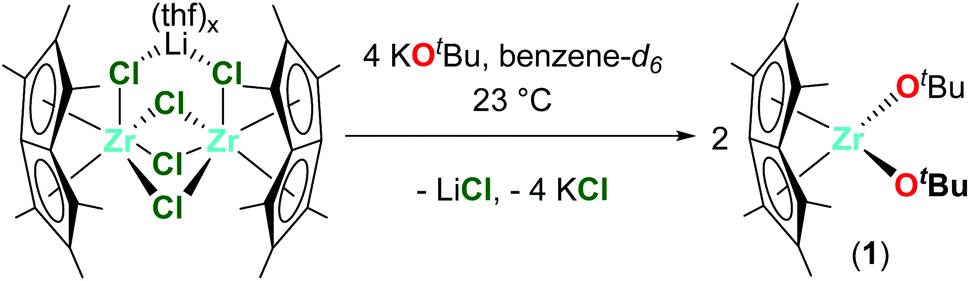 | ||
| Chart 1 Synthesis of Pn*Zr(OtBu)2 (1). | ||
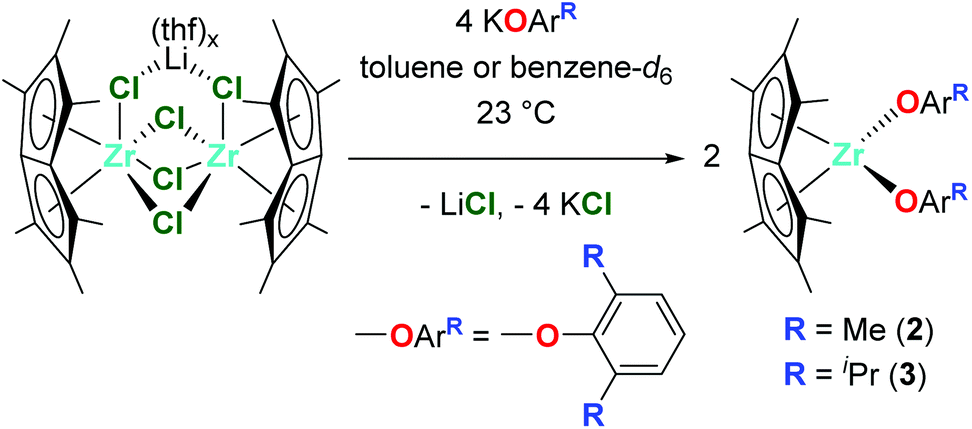 | ||
| Chart 2 Synthesis of Pn*Zr(O-2,6-Me-C6H3)2 (2) and Pn*Zr(O-2,6-iPr-C6H3)2 (3). | ||
Crystals of 2 and 3 suitable for single crystal X-ray diffraction studies were grown from hexane solutions at −30 °C. The solid–state molecular structures are depicted in Fig. 1, with selected bond lengths and angles presented in Table 1. Compound 2 suffers from disorder which have been fixed using SADI. Hence, caution should be applied when discussing the metrical parameters. Nevertheless, compounds 2 and 3 are isostructural in the solid state and adopt distorted tetrahedral geometries, exemplified by the O(1)–Zr(1)–O(2) angles of 98.6(4) and 101.18(5)° respectively.
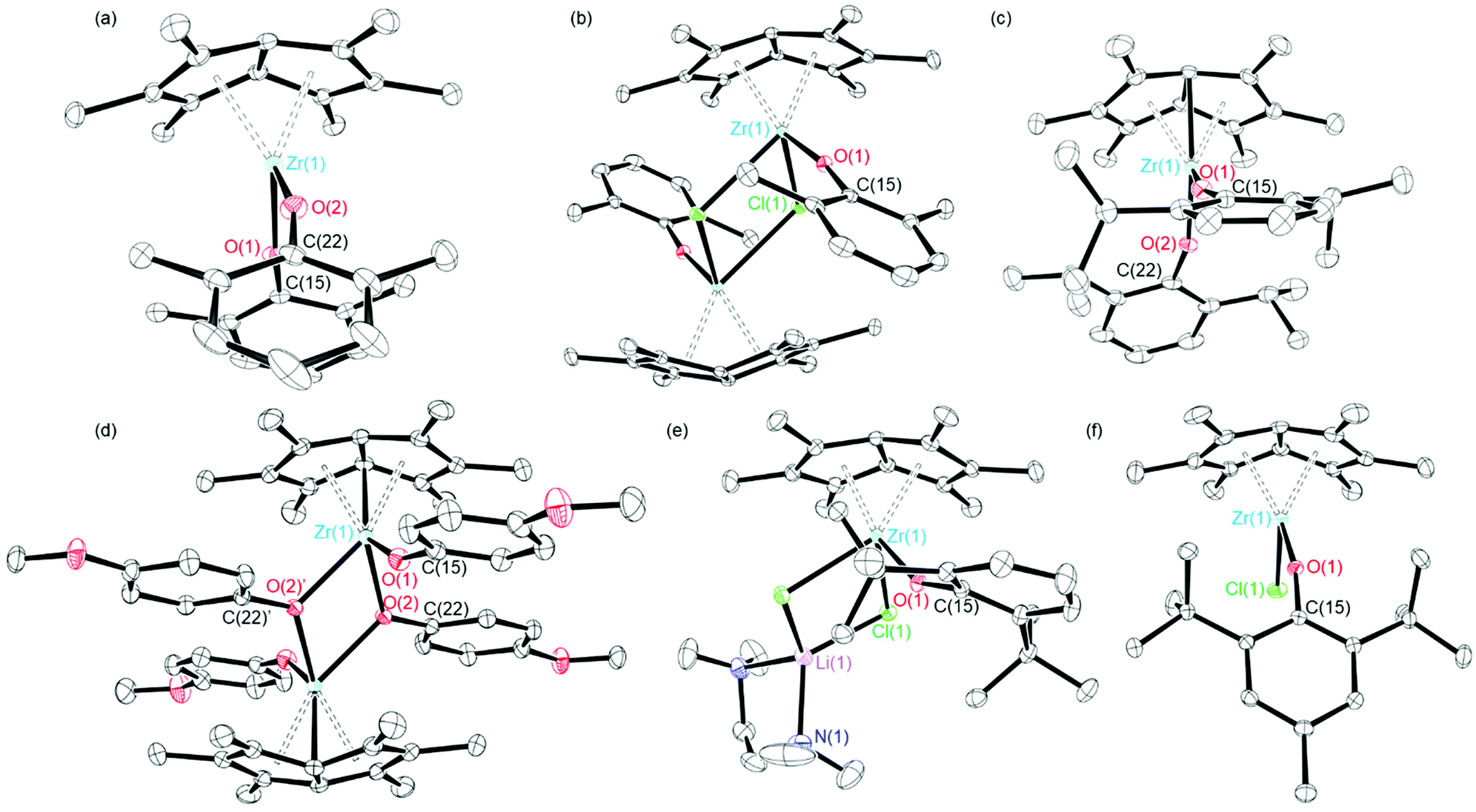 | ||
| Fig. 1 Solid-state molecular structures of (a) Pn*Zr(O-2,6-Me-C6H3)2 (2), (b) Pn*ZrCl(O-2,6-Me-C6H3)2, (c) Pn*Zr(O-2,6-iPr-C6H3)2 (3), (d) [Pn*Zr(O-4-OMe-C6H4)2]2 (4′), (e) Pn*Zr(O-2,6-tBu-C6H3)2Cl·tmeda and (f) Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6). H atoms omitted for clarity. Ellipsoids given at 30% probability. | ||
| Complex | Zr(1)–O(1) (Å) | Zr(1)–O(2) (Å) | Zr(1)–Cl(1) (Å) | Zr(1)–Pn*cent (Å) | Zr(1)–O(1)–C(15) (°) | Zr(1)–O(2)–C(22) (°) |
|---|---|---|---|---|---|---|
| a Solid-state parameters are given as average of the two independent molecules in the asymmetric unit. b Average of Zr(1)–O(2) and Zr(1)–O(2)′. c Average of Zr(1)–O(2)–C(22) and Zr(1)–O(2)′–C(22)′. | ||||||
| Pn*Zr(O-2,6-Me-C6H3)2 (2) | 2.015(6) | 1.963(3) | — | 2.090, 2.087 | 164.2(9) | 164.2(7) |
| Pn*ZrCl(O-2,6-Me-C6H3)a | 1.9844(14) | — | 2.5767(5) | 2.1116(1), 2.1161(6) | 174.38(6) | — |
| Pn*Zr(O-2,6-iPr-C6H3)2 (3) | 1.9896(11) | 1.9901(11) | — | 2.0932(9), 2.0940(9) | 165.20(11) | 158.69(10) |
| [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) | 2.0190(13) | 2.2176(6)b | 2.1423(1), 2.1557(1) | 154.07(12) | 124.43(4)c | |
| Pn*ZrCl(O-2,6-tBu-C6H2)·LiCl(tmeda) | 2.0399(9) | — | 2.5635(3) | 2.1412(8), 2.1479(7) | 152.49(9) | — |
| Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6) | 1.9822(11) | — | 2.4586(4) | 2.1020(7), 2.1130(8) | 159.47(10) | — |
| Pn*ZrCpMe(O-2,6-iPr-C6H3) (12) | 2.072(2) | — | — | 2.1302(13), 2.1457(13) | 150.01(19) | — |
Both complexes show similar Zr–O distances; 2.015(6) and 1.963(3) Å for 2 and 1.9896(11) and 1.9901(11) Å for 3, which are slightly longer than the Ti–O distances of analogous Pn*Ti(O-2,6-Me-C6H3)2 (1.8712 and 1.8890 Å),47 likely due to the larger size of zirconium compared to titanium. The Zr–O bond lengths of 2 and 3 are comparable to average values reported for other zirconium aryloxide complexes: 1.9628(1), 1.97(1), 1.9873(1) and 1.997 Å for Me2SB(Cp,I*)ZrCl(O-2,6-Me-C6H3), Cp2ZrCl(O-2,6-iPr-C6H3), Me2SB(Cp,I*)ZrCl(O-2,6-iPr-C6H3) and Cp2Zr(O-2,6-Me-C6H3)2 respectively.43,48–50 The Zr–O bond lengths for 2 and 3 are significantly shorter than the sum of the covalent radii of zirconium and oxygen (1.75 and 0.66 Å respectively), indicating that there is a partial ionic character to the Zr–O bond.51 The two Zr–O–C angles of 2 are very similar, however the aryloxide ligands point in different directions; Zr(1)–O(1)–C(15) points towards the Pn* ligand with an angle of 164.2(9)°, while Zr(1)–O(1)–C(22) points away from the Pn* ligand with an angle of 164.2(7)°. This is in agreement with Pn*Ti(O-2,6-Me-C6H3)2, where one aryloxide group points towards the Pn* ligand with Ti–O–C of 159.15° and the other aryloxide group points away from the Pn* ligand with Ti–C–O of 143.87°.47 For 3, the Zr–O–C angles of the two aryloxide ligands are slightly different (165.20(11) and 158.69(10)°); however, both aryloxide groups point towards the Pn* ligand. Both 2 and 3 have similar Zr–Pn*cent distances (2.089 and 2.096 Å respectively) and similar Pn* fold angles, defined as the angle by which the Pn* ligand deviates from planarity,52 (32.28 and 32.53° respectively), which are similar to those reported for Pn*Ti(O-2,6-Me-C6H3)2; average Ti–Pn*cent of 1.9492 Å and fold angle of 34.46°.47
Crystals of Pn*ZrCl(O-2,6-Me-C6H3) suitable for a single crystal X-ray diffraction study were obtained from benzene-d6 at 25 °C (Fig. 1 and Table 1). The relatively low steric demands of the aryloxide ligand allow dimerisation to occur in the solid-state. In addition, the solid–state structure shows two molecules in the asymmetric unit, akin to Pn*TiCl(O-2,6-Me-C6H3).47 The Zr–O bond length of Pn*ZrCl(O-2,6-Me-C6H3) is similar to the average Zr–O distance of bis(aryloxide) 2 (1.9844(14) and 1.989(5) Å respectively), however is longer than the Ti–O distance of Pn*TiCl(O-2,6-Me-C6H3) (1.838(2) Å).47 The Zr–O–C angle is larger than for 2 and 3 (170.87(13), 164.27 and 158.69(10)° respectively), likely due to the increased steric bulk caused by dimerisation, with the aryloxide group pointing away from the Pn* ligand. The near-linear nature of the Zr–O–C angle of Pn*ZrCl(O-2,6-Me-C6H3) suggests some π-orbital interaction between Zr and O, and is in good agreement with the values reported for mono(aryloxides) Cp2ZrCl(O-2,6-Me-C6H3) and Cp2ZrCl(O-2,6-iPr-C6H3) (171.6 and 172(1)° respectively).42,48
Dimeric [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) was synthesised by the reaction of one equivalent [Pn*ZrCl2]2·LiCl·(thf)2 with four equivalents KO-4-OMe-C6H4 (Scheme 1). Dissolution of [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) followed by heating to 65 °C resulted in dissociation of the dimer to afford Pn*Zr(O-4-OMe-C6H4)2 (4). The 1H NMR spectrum of 4 shows the diagnostic wing-tip and non-wing tip Pn* methyl group resonances at 2.01 and 1.99 ppm respectively (Fig. S6†). A singlet at 3.63 ppm corresponds to the methyl group of the aryloxide ligand, while doublets at 6.64 and 6.50 ppm represent the aromatic protons. Crystals of [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) suitable for a single crystal X-ray diffraction study were obtained from benzene-d6 at 25 °C (Fig. 1 and Table 1). The structure contains two bridging aryloxide ligands and two terminal aryloxide ligands, where the Zr–O bridging bond lengths (2.2665(1) and 2.1688(11) Å) are longer than the Zr–O terminal bond lengths (2.0190(13) Å). The Zr–O–C angles of the two bridging aryloxide ligands are significantly smaller than the Zr–O–C angle of the terminal aryloxide ligands (126.80(9), 122.05(9) and 154.07(12)° respectively), likely due to the steric constraints of dimerisation. By increasing the steric bulk in the 2,6-positions of the phenyl ring, mono-substituted Pn*ZrCl(O-2,6-tBu-C6H3) (5) and Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6) were obtained from the reaction of one equivalent [Pn*ZrCl2]2·LiCl(thf)x (TMEDA is present in 5 due to its presence in the starting material) with four equivalents KOAr (R = 2,6-tBu-C6H3 and 2,6-tBu-4-Me-C6H2) at 25 and 70 °C, respectively, in quantitative yield (Chart 3). 6 was also isolated on a preparative scale in 83% yield.
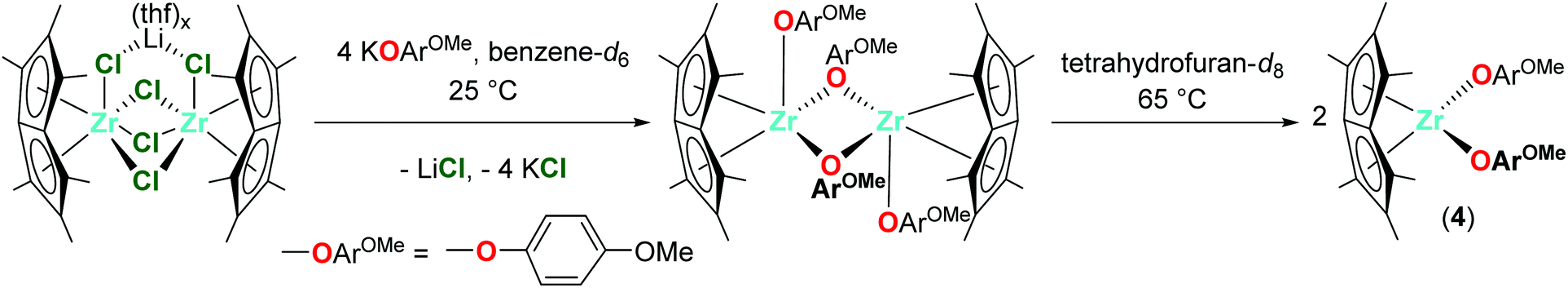 | ||
| Scheme 1 Synthesis of Pn*Zr(O-4-Me-C6H4)2 (4) via [Pn*Zr(O-4-Me-C6H4)2]2 (4′). | ||
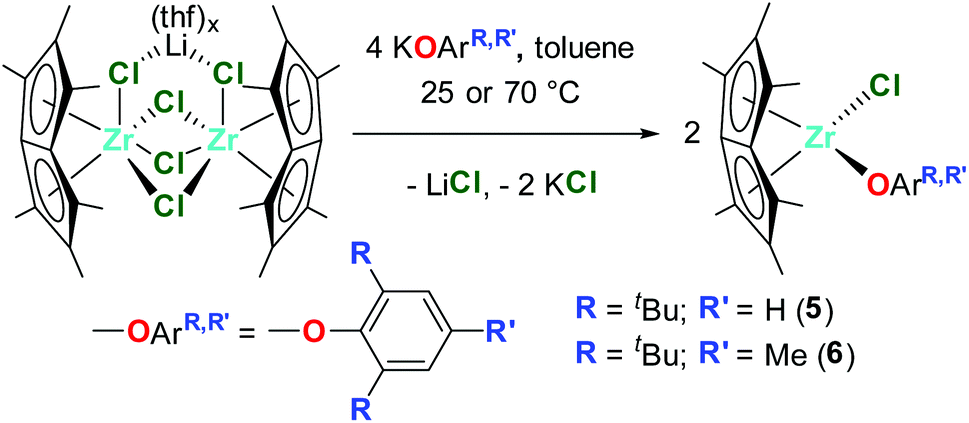 | ||
| Chart 3 Synthesis of Pn*ZrCl(O-2,6-tBu-C6H3) (5) and Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6). | ||
The 1H NMR spectrum of 6 contains two diagnostic singlets in an 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio at 1.44 and 2.34 ppm corresponding to the tert-butyl and methyl groups of the aryloxide ligand (Fig. S8†). The Pn* methyl resonances appear as two singlets at 2.06 and 1.84 ppm in a 12:6 ratio (two resonances juxtaposing). At temperatures below 213 K these resonances split into three singlets in a 6:6:6 ratio (Fig. S10†), as would typically be expected for a complex with molecular Cs symmetry, Pn*MXY.
:3 ratio at 1.44 and 2.34 ppm corresponding to the tert-butyl and methyl groups of the aryloxide ligand (Fig. S8†). The Pn* methyl resonances appear as two singlets at 2.06 and 1.84 ppm in a 12:6 ratio (two resonances juxtaposing). At temperatures below 213 K these resonances split into three singlets in a 6:6:6 ratio (Fig. S10†), as would typically be expected for a complex with molecular Cs symmetry, Pn*MXY.
Crystals grown from a benzene solution of 5 at 25 °C were identified as Pn*ZrCl(O-2,6-tBu-C6H3)·LiCl(tmeda) by a single crystal X-ray diffraction study, where tmeda binds to the chloride ligands to create a stable diamond core (Fig. 1 and Table 1). The Zr–O distance is comparable to Pn*ZrCl(O-2,6-Me-C6H3) and Cp2ZrCl(O-2,6-tBu-C6H3) (2.0399(9), 1.9844(14) and 2.008(2) Å respectively), with comparable Zr–Cl distances (2.5635(3), 2.5767(5) and 2.4642(11) Å respectively).53 The Pn*cent distances are longer than for 2, 3 and Pn*ZrCl(O-2,6-Me-C6H3) (average values of 2.1446(3), 2.089, 2.0936(9) and 2.1138(9) Å respectively), likely due to the increased steric bulk of the aryloxide ligand. The Zr–O–C angle is smaller than for 2, 3 and Pn*ZrCl(O-2,6-Me-C6H3), with the aryloxide group directed towards the Pn* ligand (152.49(9), 164.2(9), 165.20(11) and 174.38(6)° respectively). Single crystals of 6 suitable for a single crystal X-ray diffraction study were grown from a −30 °C toluene solution (Fig. 1 and Table 1). The Zr–O bond length is comparable to Pn*ZrCl(O-2,6-Me-C6H3) (1.9822(11) and 1.9796(14) Å respectively), as are the Zr–Pn*cent distances (average values of 2.1075(8) and 2.1138(9) Å), which is likely due to the similar size of the aryloxide ligands. The aryloxide group points away from the Pn* ligand with a Zr–C–O angle of 159.47(10)°.
Synthesis and characterisation of Pn*MCpR(OR)
Pn*ZrCp(OtBu) (7), Pn*ZrCp(O-2,6-Me-C6H3) (8) and Pn*ZrCp(O-2,6-iPr-C6H3) (9) were prepared by the addition of 1 equivalent KOR (R = tBu, 2,6-Me-C6H3 and 2,6-iPr-C6H3) to 1 equivalent Pn*ZrCp(Cl), (Chart 4). The 1H NMR spectra display three sharp singlets in a ratio of 6:6:6 between 1.84 and 2.05 ppm corresponding to the Pn* methyl protons, where two singlets define the non–wingtip methyl groups, and one singlet defines the wingtip methyl groups (Fig. S11, S13 and S15†). This splitting pattern is consistent with molecules of Cs symmetry and has been reported previously for Pn*TiCl(OAr) and Pn*ZrCpR(Cl) complexes.44,47 The cyclopentadienyl protons are observed as singlets at approximately 5.6 ppm, with other features of the –OR groups present as expected; a singlet at 1.19 ppm for OtBu, a singlet at 2.05 ppm, a triplet at 6.81 ppm and a doublet at 7.18 ppm for O-2,6-Me-C6H3, and doublets at 1.21 and 1.33 ppm, a septet at 2.93 ppm, a triplet at 6.97 ppm and a doublet at 7.17 ppm for O-2,6-iPr-C6H3.
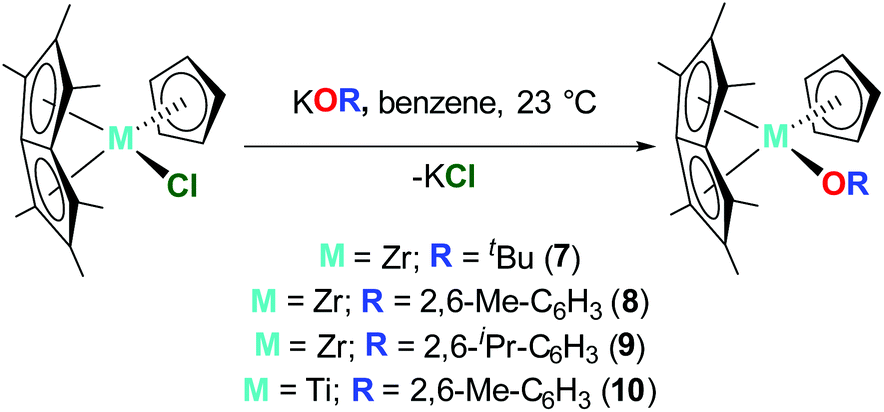 | ||
| Chart 4 Synthesis of Pn*ZrCp(OtBu) (7), Pn*ZrCp(O-2,6-Me-C6H3) (8), Pn*ZrCp(O-2,6-iPr-C6H3) (9) and Pn*TiCp(O-2,6-Me-C6H3) (10). | ||
Pn*TiCp(O-2,6-Me-C6H3) (10) was prepared by the reaction of 1 equivalent Pn*TiCp(Cl) with 1.2 equivalents KO-2,6-Me-C6H3. Following work up, 10 was isolated as a brown crystalline solid in 34% yield. The 1H NMR spectrum shows two singlets at 1.70 and 1.97 ppm, corresponding to two overlapping Pn* methyl resonances and an overlapping Pn* methyl and aryloxide methyl resonance respectively (Fig. S17†), as confirmed by 2D NMR spectroscopy. The 1H NMR spectrum also shows a diagnostic singlet resonance at 5.25 ppm corresponding to the cyclopentadienyl protons, and a triplet and doublet at 6.85 and 7.21 ppm respectively corresponding to the aryloxide aromatic protons.
Pn*ZrCpMe(O-2,6-Me-C6H3) (11), Pn*ZrCpMe(O-2,6-iPr-C6H3) (12) and Pn*ZrCpMe(O-2,4-tBu-C6H3) (13) were prepared by the addition of 1 equivalent KOR (R = 2,6-Me-C6H3, 2,6-iPr-C6H3 and 2,4-tBu-C6H3) to 1 equivalent Pn*ZrCpMe(Cl) (Chart 5). The 1H NMR spectra of 11 and 12 show the diagnostic Pn* resonances as three singlets in a 6:6:6 ratio between 1.84 and 2.01 ppm (Fig. S19 and S21†). The cyclopentadienyl proton resonances are observed as two multiplets between 5.20 and 5.64 ppm while a singlet at approximately 1.85 ppm corresponds to the cyclopentadienyl methyl group. The aryloxide protons are observed as a triplet and doublet at approximately 6.85 and 7.15 ppm respectively, with the aryloxide methyl groups of 11 observed as a singlet at 2.08 ppm and the isopropyl groups of 12 observed as two doublets at 1.21 and 1.35 ppm and a septet at 2.93 ppm. Due to changes in symmetry, the 1H NMR spectrum of 13 shows the Pn* resonances as six singlets integration 3 between 1.85 and 2.19 ppm and the cyclopentadienyl proton resonances as three multiplets integration 2:1:1 between 5.30 and 5.65 ppm (Fig. S23†). The aryloxide protons are observed as a doublet, doublet and triplet at 5.99, 7.27 and 7.57 ppm respectively, with the tert-butyl groups appearing as two singlets at 1.42 and 1.60 ppm.
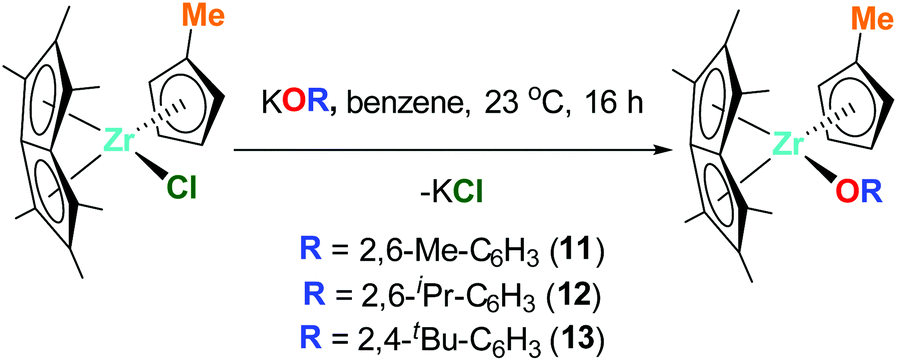 | ||
| Chart 5 Synthesis of Pn*ZrCpMe(O-2,6-Me-C6H3) (11), Pn*ZrCpMe(O-2,6-iPr-C6H3) (12) and Pn*ZrCpMe(O-2,4-tBu-C6H3) (13). | ||
Single crystals of 11 and 12 suitable for single crystal X-ray diffraction studies were grown from a pentane solution at −30 °C. The solid-state molecular structures are shown in Fig. 2. For 11, the crystallographic data clearly confirms the connectivity of the structure and agrees with other experimental data. However, due to the low quality of the X-ray crystallography data, discussion of the metrical parameters is not discussed. 12 shows a longer Zr–O bond length and smaller Zr–O–C angle than the complexes discussed previously (2.072(2) Å and 150.01(19)°), with the aryloxide group pointing away from the Pn* ligand. The Zr–Pn*cent distances (2.1302(13) and 2.1457(13) Å) are slightly longer than the parent Pn*ZrCpMe(Cl) complex (2.1062(6) and 2.1065(7) Å), likely due to the increased size of the aryloxide ligand compared to chloride.44 The average Zr(1)–Pn*cent distance (2.1379(63) Å) is shorter than the Zr(1)–Cpcent distance (2.2173(16) Å), which may be due to the increased electron donating ability of the η8-permethylpentalene ligand compared to the η5-cyclopentadienyl ligand. The same trend is also observed for Pn*ZrCpMe(Cl) and Pn*TiCpMe(Cl).44 The Zr–Cpcent distance of 12 (2.2173(16) Å) is similar to those reported for Pn*ZrCpMe(Cl) (2.2219(7) Å) and Pn*ZrCpMe(Me) (2.2262(11) Å).44 The fold angle of the Pn* ligand of 12 is smaller than for 2 and 3 (29.72, 32.28 and 32.53° respectively), which is likely due to the additional electron density provided by the Cp ligand, and is similar to the Pn*ZrCpMe(Cl) parent compound (30.57°).44 The fold angle is also smaller than PnZrCp(Cl) (32.74°), caused by the increased inductive donation of Pn* compared to Pn (η8-C8H6).54
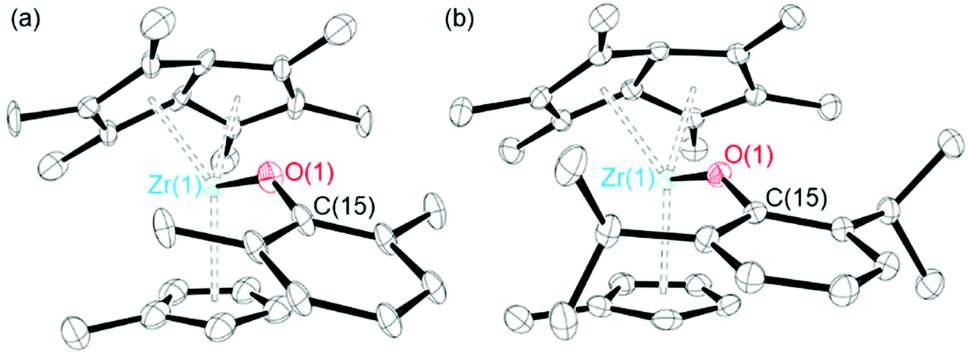 | ||
| Fig. 2 Solid-state molecular structures of (a) Pn*ZrCpMe(O-2,6-Me-C6H3) (11) and (b) Pn*ZrCpMe(O-2,6-iPr-C6H3) (12). H atoms omitted for clarity. Ellipsoids given at 30% probability. | ||
Polymerisation of L– and rac-lactide
Pn*Zr(O-2,6-Me-C6H3)2 (2), Pn*Zr(O-2,6-iPr-C6H3)2 (3), Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6), Pn*ZrCp(OtBu) (7), Pn*ZrCp(O-2,6-iPr-C6H3) (9), Pn*TiCp(O-2,6-Me-C6H3) (10) and Pn*ZrCpMe(O-2,6-iPr-C6H3) (12) were studied as initiators for the ring-opening polymerisation (ROP) of L-lactide, and 2, 3, 6, 7 and 10 were studied as initiators for ROP of rac-lactide. The results are summarised in Fig. 3–7, S25–S34† and Tables 2, 3 and S3–S32.† Polymerisations were conducted in Young's tap NMR spectroscopy tubes at 80 °C in benzene-d6 with an initial lactide to catalyst ratio ([LA]0/[M]0) of 50 or 200 and an initial lactide monomer concentration ([LA]0) of 0.5 or 2.0 M. Plots of ln([LA]0/[LA]t) vs. time revealed linear relationships indicating first-order dependence with respect to monomer concentration (Fig. 3–6). The gradients of the ln([LA]0/[LA]t) vs. time plots afforded the observed first order rate constants, kobs.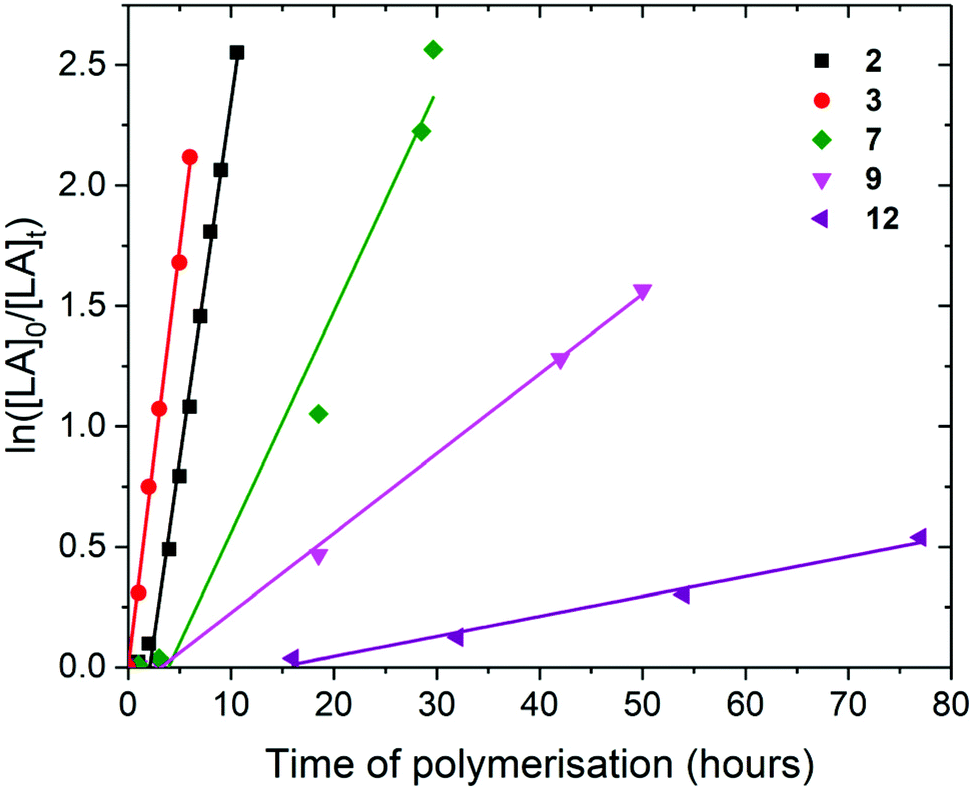 | ||
| Fig. 3 ln(([LA]0/[LA]t) as a function of time of polymerisation for the ROP of L-lactide using Pn*Zr(O-2,6-Me-C6H3)2 (2, black square, kobs = 0.30 ± 0.01 h−1), Pn*Zr(O-2,6-iPr-C6H3)2 (3, red circle), kobs = 0.35 ± 0.01 h−1), Pn*ZrCp(OtBu) (7, green diamond, kobs = 0.09 ± 0.01 h−1), Pn*ZrCp(O-2,6-iPr-C6H3) (9, pink down triangle, kobs = 0.03 ± 0.001 h−1) and Pn*ZrCpMe(O-2,6-iPr-C6H3) (12, purple left triangle, kobs = 0.01 ± 0.001 h−1). Polymerisation conditions: 80 °C, [LA]0/[M]0 = 50, [LA]0 = 0.5 M and benzene-d6. | ||
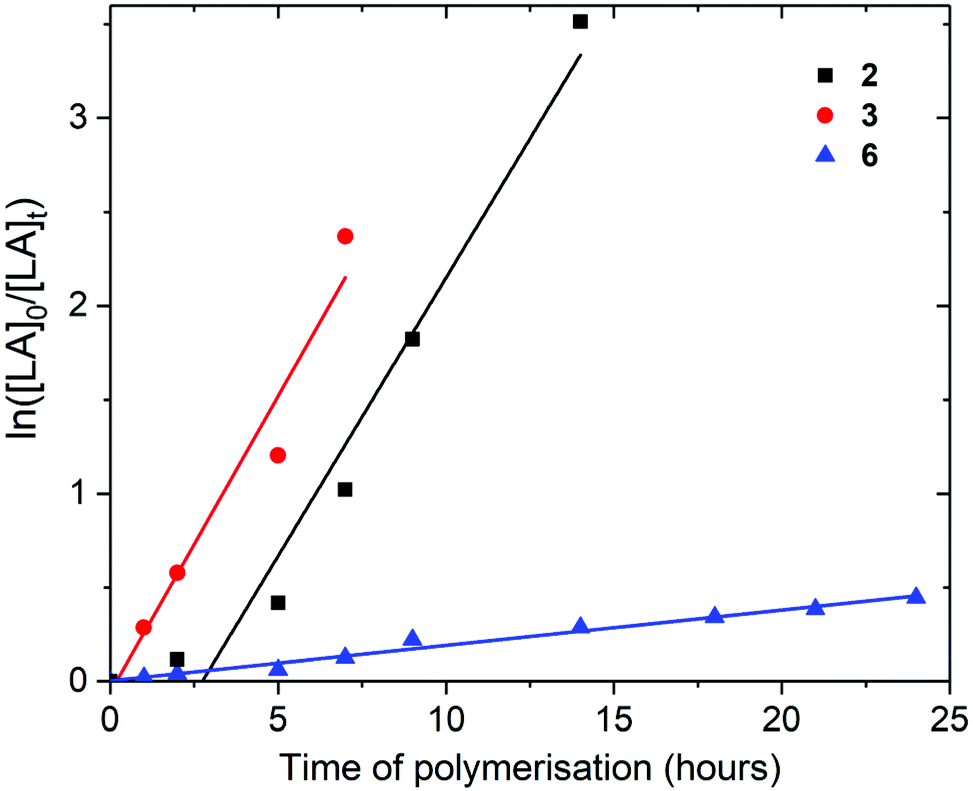 | ||
| Fig. 4 ln([LA]0/[LA]t) as a function of time of polymerisation for the ROP of L-lactide using Pn*Zr(O-2,6-Me-C6H3)2 (2, black square, kobs = 0.30 ± 0.03 h−1), Pn*Zr(O-2,6-iPr-C6H3)2 (3, red circle, kobs = 0.32 ± 0.04 h−1), and Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6, blue triangle, kobs = 0.02 ± 0.001 h−1). Polymerisation conditions: 80 °C, [LA]0/[M]0 = 200, [LA]0 = 2.0 M and benzene-d6. | ||
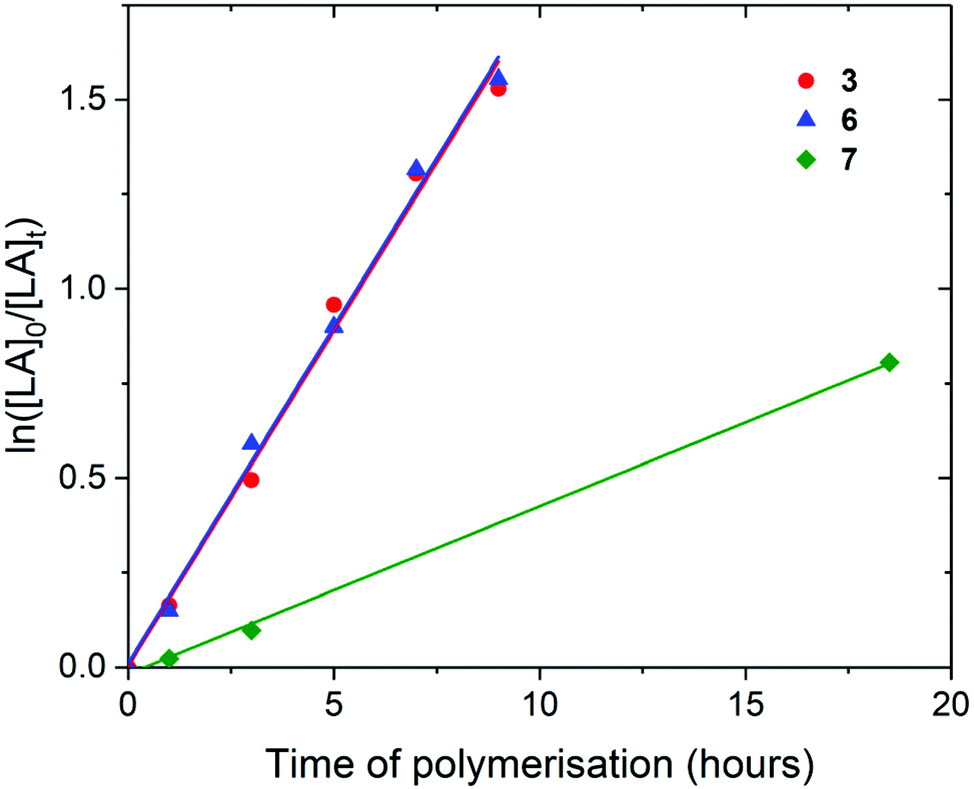 | ||
| Fig. 5 ln([LA]0/[LA]t) as a function of time of polymerisation for the ROP of rac-lactide using Pn*Zr(O-2,6-iPr-C6H3)2 (3, red circle, kobs = 0.18 ± 0.01 h−1), Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6, blue triangle, kobs = 0.18 ± 0.01 h−1) and Pn*ZrCp(OtBu) (7, green diamond, kobs = 0.04 ± 0.001 h−1). Polymerisation conditions: 80 °C, [LA]0/[M]0 = 50, [LA]0 = 0.5 M and benzene-d6. | ||
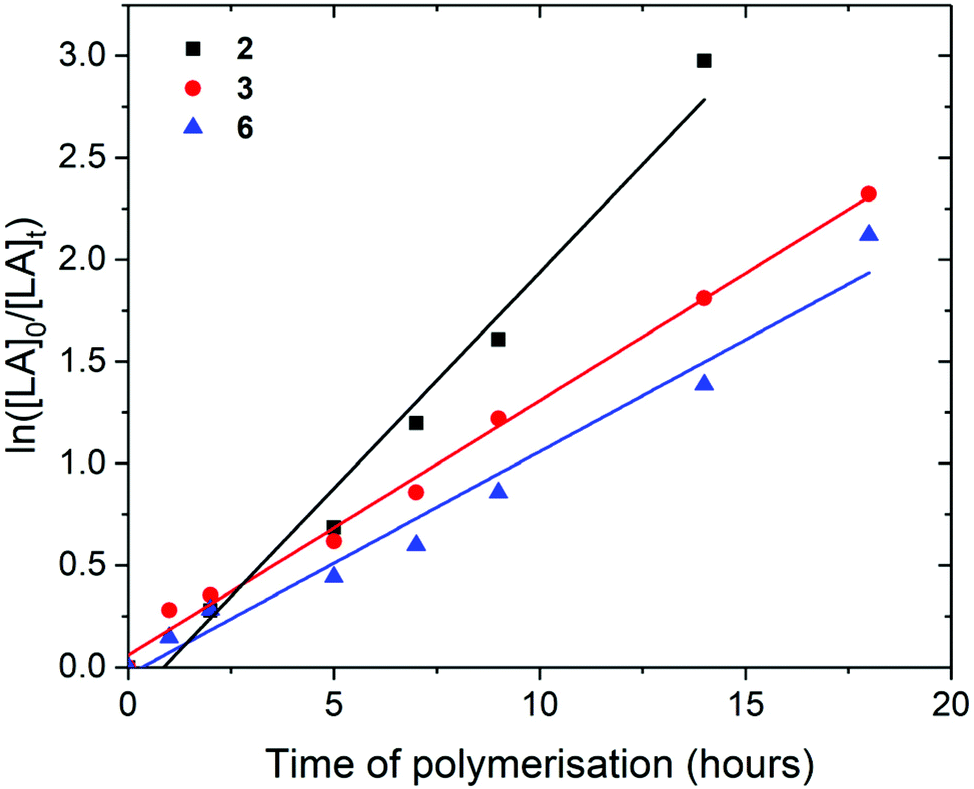 | ||
| Fig. 6 ln([LA]0/[LA]t) as a function of time of polymerisation for the ROP of rac-lactide using Pn*Zr(O-2,6-Me-C6H3)2 (2, black square, kobs = 0.21 ± 0.02 h−1), Pn*Zr(O-2,6-iPr-C6H3)2 (3, red circle, kobs = 0.12 ± 0.01 h−1), and Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6, blue triangle, kobs = 0.11 ± 0.01 h−1). Polymerisation conditions: 80 °C, [LA]0/[M]0 = 200, [LA]0 = 2.0 M and benzene-d6. | ||
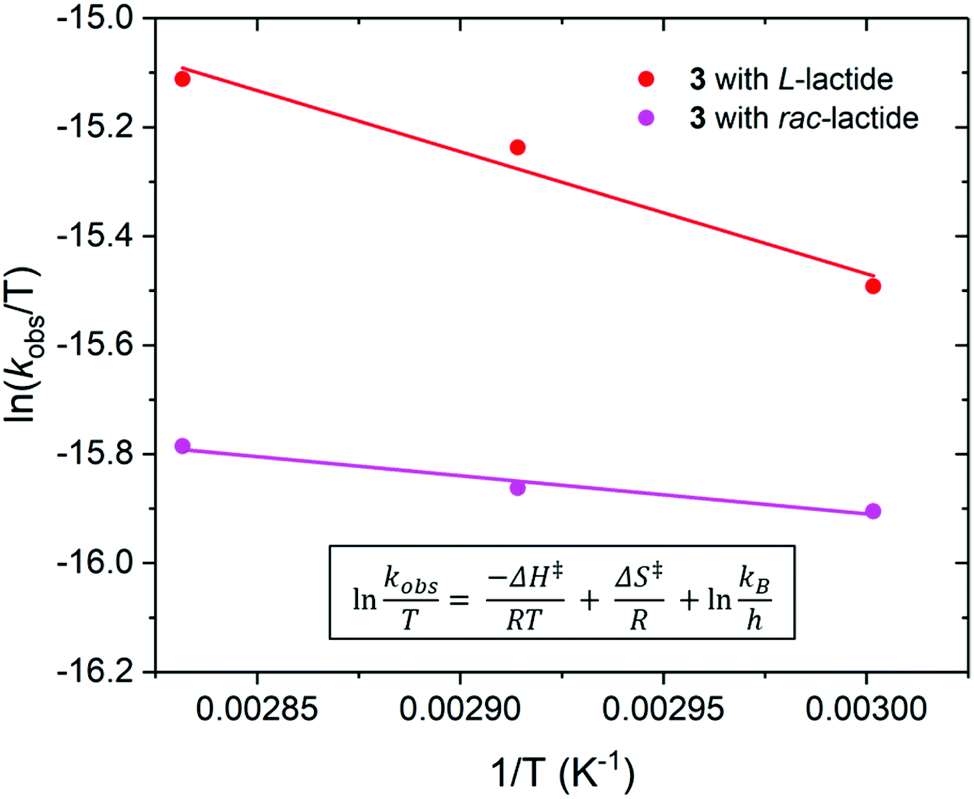 | ||
| Fig. 7 Eyring plot of ln(kobs/T) as a function of 1/T for the polymerisation of L- (red) and rac-lactide (pink) using Pn*Zr(O-2,6-iPr-C6H3)2 (3). L-Lactide: ΔH‡ = 19 ± 3 kJ mol−1 and ΔS‡ = −270 ± 10 J mol−1 K−1. rac-Lactide: ΔH‡ = 6 ± 1 kJ mol−1 and ΔS‡ = −312 ± 3 J mol−1 K−1. Polymerisation conditions: [LA]0/[M]0 = 200, [LA]0 = 2.0 M and benzene-d6. | ||
| Initiator | [LA]0/[M]0 | [LA]0 (M) | Time (h) | Conversionb (%) | k obs (h−1) | M n (calcd)c (g mol−1) | M n (GPC)d (g mol−1) | M w/Mnd |
|---|---|---|---|---|---|---|---|---|
| a Polymerisation conditions: 80 °C and benzene-d6. b Measured by 1H NMR spectroscopic analysis. c M n (calcd) = (MLA × [LA]0/[M]0 × (conv.(%)/100) + Mend group. d Determined by GPC in chloroform at 30 °C against polystyrene standards (Mn values are corrected by a factor of 0.58).55 | ||||||||
| 2 | 50 | 0.5 | 10.6 | 92 | 0.30 ± 0.01 | — | — | — |
| 3 | 50 | 0.5 | 6 | 88 | 0.35 ± 0.01 | 6520 | 17398 |
1.62 |
| 7 | 50 | 0.5 | 30 | 92 | 0.09 ± 0.01 | — | — | — |
| 9 | 50 | 0.5 | 50 | 79 | 0.03 ± 0.001 | — | — | — |
| 12 | 50 | 0.5 | 164 | 85 | 0.01 ± 0.001 | — | — | — |
| 2 | 200 | 2.0 | 21 | 98 | 0.30 ± 0.03 | 28374 |
30709 |
1.37 |
| 3 | 200 | 2.0 | 28 | 98 | 0.32 ± 0.04 | 28430 |
31327 |
1.28 |
| 6 | 200 | 2.0 | 24 | 36 | 0.02 ± 0.001 | 10598 |
12950 |
1.45 |
| Initiator | [LA]0/[M]0 | [LA]0 (M) | Time (h) | Conversion(%) | k obs (h−1) | M n (calcd)b (g mol−1) | M n (GPC)c (g mol−1) | M w/Mnd | P r |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerisation conditions: 80 °C and benzene-d6. b M n (calcd) = (MLA × [LA]0/[M]0 × (conv.(%)/100) + Mend group. c Determined by GPC in chloroform at 30 °C against polystyrene standards (Mn values are corrected by a factor of 0.58).55 | |||||||||
| 3 | 50 | 0.5 | 9 | 78 | 0.18 ± 0.01 | 5800 | 9930 | 1.57 | — |
| 6 | 50 | 0.5 | 9 | 79 | 0.18 ± 0.01 | 5914 | 6617 | 1.56 | — |
| 7 | 50 | 0.5 | 30 | 87 | 0.04 ± 0.001 | — | — | — | — |
| 2 | 200 | 2.0 | 21 | 98 | 0.21 ± 0.02 | 28374 |
25275 |
1.23 | 0.66 |
| 3 | 200 | 2.0 | 30 | 97 | 0.12 ± 0.01 | 28141 |
25509 |
1.14 | 0.54 |
| 6 | 200 | 2.0 | 24 | 95 | 0.11 ± 0.01 | 27607 |
26747 |
1.38 | 0.68 |
For the polymerisation of L-lactide with [LA]0/[M]0 of 50 and [LA]0 of 0.5 M, rate of polymerisation followed the order 3, 2, 7, 9 and 12. Complex 2 required a 2 hours initiation period, after which 2 and 3 showed very similar rates for the ROP of L-lactide; kobs of 0.30 and 0.35 h−1 respectively, taking 10.6 and 6 hours to reach full conversion (92 and 88%) (Fig. 3). This suggests that the increase in the steric bulk of the aryloxide substituent from methyl to isopropyl does not hinder L-lactide monomer insertion. When compared to the corresponding Pn*(H)Zr(OAr) complexes, 2 shows polymerisation activity 37% lower than Pn*(H)Zr(O-2,6-Me-C6H3)3 under similar conditions (kobs of 0.48 h−1) and 3 shows polymerisation activity 10% lower than Pn*(H)Zr(O-2,6-iPr-C6H3)3 (kobs of 0.39 h−1).56
These initiators show polymerisation activities lower than zirconium complexes bearing OSSO-type ligands (up to 74% conversion with kobs up to 0.50 h−1 at 80 °C after 3 hours with [LA]/[M]0 of 100)57 and lower than the neutral bis(ester enolate) complex Ph2CB(Cp,Flu)Zr(OC(OiPr)CMe2)2 (92% conversion after 105 minutes at 80 °C with [LA]0/[M]0 = 50).58 However, they showed faster polymerisation activities than aryloxide Cp2ZrMe(O-2,6-Me-C6H3), which displayed kobs of 0.029 h−1 at 80 °C with [LA]0/[M]0 = 50.59
Pn*ZrCpR(OR′) complexes 7, 9 and 12 showed much lower kobs than Pn*Zr(OAr)2 complexes 2 and 3 (0.09, 0.03 and 0.01 h−1 respectively), and required longer initiation periods (3, 3 and 16 hours respectively), indicating that the addition of a non–initiating cyclopentadienyl ligand significantly reduces polymerisation activity. This is opposite to the trend found for ethylene polymerisation using these types of complexes, where polymerisation activity increased with increasing electron donating ability of the ancillary ligands due to increased stabilisation of the positively charged olefin polymerisation intermediates.44 For lactide polymerisation, it may be that the increased electron donating ability of the cyclopentadienyl ligands reduces the Lewis acidity of the metal centre, leading to lower rates of lactide insertion and decreases in polymerisation activity. This is in contrast to some yttrium initiators, where initiators bearing more electron-donating phosphasalen ligands showed higher activities than initiators bearing salen ligands.23,60 Alkoxide 7 shows a faster rate of polymerisation than aryloxides 9 and 12 (92, 79 and 85% conversion after 30, 50 and 164 h respectively), as has been seen before for (Ind)2ZrMe(OR) complexes (kobs of 0.06 and 0.22 h−1 for R = 2,6-Me-C6H3 and tBu respectively), and is likely due to the increased steric bulk of the aryloxide substituent inhibiting monomer coodination.59
The faster polymerisation rate of 9 compared to 12 is likely due to the increased electron donating ability of methylcyclopentadienyl compared to unsubstituted cyclopentadienyl, which further reduces the Lewis acidity of the metal centre and inhibits initiation. The titanium analogue Pn*TiCp(O-2,6-Me-C6H3) (10) shows a very slow rate of polymerisation compared to 9 (24 days to react 89% conversion) and requires a 3 day initiation period (Fig. S25†). This trend has also been observed for Pn*M(O-2,6-Me-C6H3)3 initiators where M = Ti showed a much lower rate of polymerisation than M = Zr (kobs = 0.11 and 0.48 h−1 respectively),56 and for well–defined alkoxotitanium and alkoxozirconium complexes of tetradentate amine-phenolate ligands where polymerisation activity was up to 30 times faster for zirconium initiators compared to titanium.61 This effect is attributed the larger, less crowded zirconium centre facilitating approach and coordination of lactide monomers.
For the polymerisation of L-lactide with [LA]0/[M]0 of 200 and [LA]0 of 2.0 M, rate of polymerisation followed the order 3, 2 and 6 (Fig. 4). As previously discussed for [LA]0/[M]0 of 50 and [LA]0 of 0.5 M, 2 and 3 show very similar rates for the polymerisation of L-lactide (0.30 and 0.32 h−1 respectively). Mono(aryloxide) 6 showed a much slower rate of polymerisation than bis(aryloxides) 2 and 3 (0.02 h−1). These initiators show faster rates of L-lactide polymerisation than the neutral bis(ester enolate)s Cp2Zr[OC(OiPr)CMe2]2 and rac-EBIZr[OC(CiPr)CMe2]2, which showed 7% conversion after 26 and 18 hours respectively at 80 °C with [LA]0/[M]0 = 200.58
The experimental number averaged molecular weights (Mn) of the poly-L-lactides produced using 2, 3 and 6 with [LA]0/[M]0 of 200 show good agreement with the calculated values, suggesting that all the metal centres were active during polymerisation and that only one aryloxide group is involved in polymerisation using 2 and 3; Mn (calcd) of 28374 g mol−1 and Mn (GPC) of 30709 g mol−1 for 2 (Table 2). The experimental Mn for the poly-L-lactides produced using 3 with [LA]0/[M]0 of 50 are approximately triple the calculated Mn, suggesting that only a third of complex 3 initiate polymerisation at this concentration. This is confirmed by the molecular weight distributions (MWD = Mw/Mn) being relatively large in all cases, indicating transesterification processes may be occurring (Mw/Mn = 1.28–1.62). The homonuclear decoupled 1H NMR spectra of the poly-L-lactides produced using 2, 3 and 6 with [LA]0/[M]0 of 200 demonstrated that no epimerisation occurred and confirmed the production of isotactic PLA (Fig. S35–S37†).
For the polymerisation of rac-lactide with [LA]0/[M]0 of 50 and [LA]0 of 0.5 M, rate of polymerisation followed the order 3, 6 and 7, with 3 and 6 showing identical rates of polymerisation (kobs of 0.18 h−1) (Fig. 5). This suggests that the increase in the steric bulk of the aryloxide substituent from isopropyl to tertbutyl does not hinder rac-lactide monomer insertion, neither does the decrease in the number of aryloxide initiation groups. As for the polymerisation of L-lactide, the rate of polymerisation of 7 is slower than 3 (0.04 h−1), likely due to the decrease in Lewis acidity at the metal centre and the presence of a non-initiating cyclopentadienyl ligand. When compared to the ROP of L-lactide, 3 and 7 displayer slower rates of polymerisation for rac-lactide. As for the polymerisation of L-lactide, the titanium initiator Pn*TiCp(O-2,6-Me-C6H3) (10) shows a much slower rate of polymerisation than 3, 6 and 7 (24 days to react 79% conversion) and requires a 3 day initiation period (Fig. S25†). The slower polymerisation activity of Ti compared to Zr for the ROP of rac-lactide has also been observed for the ROP of rac–lactide using the tetracarbamato complexes M(O2CNEt2)4, where 96% conversion was achieved after 13 hours at 100 °C for M = Zr and only 31% for M = Ti.62
These initiators display significantly lower polymerisation activities for the ROP of rac-lactide than Pn*(H)Zr(rac-OCH{CH3}2C6H5)3 and Pn*(H)Zr(S-OCH{CH3}2C6H5)3 under similar conditions (kobs = 1.67 and 1.34 h−1 respectively).56 They are also slower rac-lactide polymerisation initiators than some unsymmetrical zirconium metal complexes based on ONNO salalen-type ligands, where up to 99% conversion of rac-lactide was achieved after 2 hours at 80 °C with [LA]0/[M]0 of 100.63,64 Complexes 3, 6 and 7 complexes show polymerisation activities more similar to coordinatively unsaturated cationic zirconium benzyl/alkoxide complexes with phosphasalen ligands (up to 93% conversion after 15 hours at 70 °C and up to 99% conversion after 14 hours at 90 °C with [LA]0/[M]0 of 100),65 and are faster than zirconium complexes of bipyrrolidine derived salan ligands (60% conversion after 8 hours at 70 °C with [LA]0/[M]0 of 100).66
The polymerisation of rac-lactide using 3 and 6 was also carried out with [LA]0/[M]0 of 25 and 10, keeping [LA]0 constant at 0.5 M (Fig. S26 and S27 and Table S4†). As expected, kobs increased with increasing initiator concentration; kobs of 0.18, 0.34 and 0.49 h−1 for polymerisation using 3 with [LA]0/[M]0 of 50, 25 and 10 respectively. The rates of polymerisation of 3 and 6 remained similar with varying initiator concentrations; kobs of 0.49 and 0.46 h−1 respectively with [LA]0/[M]0 of 10. Plots of −ln(kobs) vs. −ln([M]0 is shown in Fig. S28,† gradients of 0.62 ± 0.15 and 0.58 ± 0.16 for 3 and 6 respectively are indicative of first-order dependence on the concentration of 3 and 6. The propagation rate constant (kp) of 7.17 ± 2.29 M−1 h−1 and 6.37 ± 2.37 M−1 h−1 for 3 and 6 respectively was calculated from plot of kobsvs. [M]0 (Fig. S29†). The overall rate laws were determined as −d[rac-LA]/dt = kp[rac-LA][M].
For the polymerisation of rac-lactide with [LA]0/[M]0 of 200 and [LA]0 of 2.0 M, rate of polymerisation followed the order 2, 3 and 6; kobs of 0.21, 0.12 and 0.11 h−1 with 95, 84 and 75% conversion respectively after 14 hours at 80 °C (Fig. 6). The catalysts show faster rates of polymerisation than Zr(O2CNR2)4 tetracarbamato complexes; 87 and 88% for Zr(O2CNEt2)4 and Zr(O2CNiPr2)4 respectively after 13 hours at 100 °C with [LA]0/[M]0 of 200.62 They show similar rates of polymerisation to some zirconium complexes bearing ONSO ligands where up to 98% conversion of rac-lactide was achieved after 20 hours at 70 °C with [LA]0/[M]0 of 300,67 and to some zirconium complexes bearing phenylene–salalen ligands where up to 99% conversion was achieved after 24 hours at 70 °C with [LA]0/[M]0 of 300.68 In contrast to the ROP of L-lactide, 2 shows a faster rate of polymerisation than 3 for the ROP of rac-lactide, which suggests that 2 may have a more preferential ligand environment for D-lactide monomer insertion than 3. Similar to the polymerisation of L-lactide, 6 shows a slower rate of polymerisation than 2 and 3, which may be due to its increased steric bulk or the reduction in the number of aryloxide groups. Unlike 2 and 3, 6 shows a faster rate of polymerisation for rac-lactide compared to L-lactide (0.11 and 0.02 h−1 respectively).
Similar to the polymerisation of L-lactide, the experimental Mn of the poly-rac-lactides produced using 2, 3 and 6 with [LA]0/[M]0 of 200 show good agreement with the calculated values, suggesting that all the metal centres of were active during polymerisation; Mn (calcd) of 27607 g mol−1 and Mn (GPC) of 26747 g mol−1 for 6 (Table 3). The experimental Mn of the poly–rac–lactides produced using 6 with [LA]0/[M]0 of 50 and 25 are also in good agreement with the calculated values. However, similar to the polymerisation of L-lactide, poly-rac-lactides produced using 3 with [LA]0/[M]0 of 50 and 25 show experimental Mn approximately 2 and 0.25 times larger than the calculated values respectively, suggesting that not all of the metal sites were active during polymerisation at these concentrations. Mw/Mn for the polymers produced using 2 and 3 with [[LA]0/[M]0 of 200 are relatively narrow, indicating controlled polymerisation (Mw/Mn = 1.23 and 1.14 respectively). However, Mw/Mn for the polymers produced under all other conditions are relatively large, indicating that transesterification processes may be occurring (Mw/Mn = 1.38–1.57).
The homonuclear decoupled 1H NMR spectra of the poly-rac-lactides produced using 2 and 6 show the production of moderately heterotactic enriched PLA (Pr = 0.66 and 0.69 respectively, where Pr is the probability of forming racemic linkages (Fig. S38 and S40†). Hence, we can expect that the polymerisation occurs by a chain-end mechanism. This indicates some degree of sterocontrol provided by the ligand environment, which is in contrast to Pn*(H)OR systems where atactic PLA was formed.56 However, the homonuclear decoupled 1H NMR spectrum of the poly-rac-lactide produced using 3 shows the production of atactic PLA (Pr = 0.54) and indicates a lack of stereocontrol for this catalyst system (Fig. S39†). The MALDI-TOF mass spectrum for the poly-rac-lactides produced using 3 shows peak envelopes Δm/z = 144 apart, demonstrating controlled polymerisation with no transesterification; as indicated by the narrow dispersity mentioned previously (Fig. S41†). The spectrum also reveals polymer chains consisting of polylactic acid repeat units with -O-2,6-iPr-C6H3 and –OH end groups, demonstrating lactide monomer insertion into the aryloxide bond and suggesting a coordination–insertion polymerisation mechanism where the aryloxide group initiates polymerisation. The MALDI-TOF mass spectrum for the poly-rac-lactides produced using 6 shows peak envelopes Δm/z = 72 apart, indicative of intermolecular transesterification and highlighting that polymerisation using 6 is less controlled than 3 (Fig. S42†). This lesser degree of control is consistent with the larger Mw/Mn recorded for 6 when compared to 3 (Mw/Mn of 1.38 and 1.14 respectively). Similar to polymerisation using 3, the MALDI-TOF mass spectrum of 6 reveals polymer chains consisting of polylactic acid repeat units with -O-2,6-tBu-4-Me-C6H2 and –OH end groups.
A study to investigate the effects of solvent on polymerisation rate was conducted for the ROP of rac-lactide using 3 and 6 at 80 °C with [LA]0/[M]0 of 200 and [LA]0 of 2.0 M (Fig. S30 and S31 and Table S4†). For 3, the rates of polymerisation in benzene-d6 and chloroform-d1 were similar (kobs of 0.12 and 0.13 h−1 respectively). However, for 6, polymerisation in benzene-d6 was much faster than in chloroform-d1 (0.11 and 0.01 h−1 respectively). For both catalysts, tetrahydrofuran-d8 was found to inhibit polymerisation, as was also observed for the ROP of L-lactide using an unsymmetrical permethylindenyl zirconocene,43 and is attributed to coordination of the tetrahydrofuran-d8 molecules to the metal centres inhibiting the coordination of lactide monomers. The experimental Mn of the poly-rac-lactides produced using 3 and 6 in chloroform-d1 are lower than the calculated values (26988 and 9930 g mol−1 respectively for 3 and 6851 and 4337 g mol−1 respectively) with broad Mw/Mn (1.86 for 6), which suggests a greater degree of transesterification reactions occur in chloroform-d1 than in benzene-d6.
The temperature of polymerisation was varied for the ROP of L-lactide using 2 and 3, and for the ROP of rac-lactide using 3 with [LA]0/[M]0 of 50 and [LA]0 of 0.5 M (Fig. S32–S34 and Tables S3 and S4†). As expected, the rate of polymerisation increased with an increase in polymerisation temperature as the system had more energy to overcome the activation barriers for polymerisation; kobs of 0.30 and 0.79 h−1 at 80 and 100 °C respectively for the ROP of L-lactide using 2, kobs of 0.22, 0.30 and 0.35 h−1 at 60, 70 and 80 °C respectively for the ROP of L-lactide using 3, and kobs of 0.15, 0.16 and 0.18 h−1 at 60, 70 and 80 °C respectively for the ROP of rac-lactide using 3. Akin to polymerisation at 80 °C, the experimental Mn are approximately triple and double the calculated Mn for the polymerisation of L- and rac-lactide respectively at 60 and 70 °C. Mn and Mw/Mn were also observed to increase with an increase in polymerisation temperature, indicating less controlled polymerisation and an increase in transesterification processes with an increase in temperature; Mn of 7999, 9800 and 9930 g mol−1 and Mw/Mn of 1.27, 1.51 and 1.57 for the polymerisation of rac–lactide using 3 at 60, 70 and 80 °C respectively.
By varying the temperature of polymerisation of L- and rac-lactide using 3, the enthalpy (ΔH‡) and entropy (ΔS‡) of activation were calculated from an Eyring plot of ln(kobs/T) vs. (1/T) (Fig. 7). For the polymerisation of L-lactide, ΔH‡ = 19 kJ mol−1 and ΔS‡ = −270 J mol−1 K−1, and for the polymerisation of rac-lactide ΔH‡ = 6 kJ mol−1 and ΔS‡ = −312 J mol−1 K−1. ΔH‡ are slightly lower than those reported for the ROP of L- and rac-lactide using Pn*(H)Zr(OR)3 under similar conditions (30 < ΔH‡ < 76 kJ mol−1), with much less favourable ΔS‡ (411 < ΔS‡ < 1847 J K−1 mol−1).56 The ΔS‡ values are more similar to those observed when using catalysts based on the permethylindenyl ligand (C9Me6, I*); −155 < ΔS‡ < −40 J mol−1 K−1 for the ROP of L- and rac-lactide using Me2SB(CpR,I*)ZrCl2 and ΔS‡ = −86 J mol−1 K−1 for the ROP of L-lactide using Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF).43,69 Both ΔH‡ and ΔS‡ are similar to, although albeit lower than, the values reported for the ROP of rac–lactide using a chiral alkoxy–bridged dinuclear indium catalyst (ΔH‡ = 49 kJ mol−1 and ΔS‡ = −140 J K−1 mol−1).70 The low enthalpy and negative entropy for the polymerisation of L- and rac-lactide using 3 are indicative of a more ordered transition state in the coordination–insertion polymerisation mechanism.71
Conclusions
A new family of group 4 permethylpentalene (Pn*) aryloxide and alkoxide complexes (Pn*ML2; Zr, Ti) have been synthesised and structurally characterised. Seven complexes have been investigated as initiators for the ring-opening polymerisation (ROP) of L- and rac-lactide. The initiators displayed first-order dependence on monomer concentration. In general, polymerisation followed the trend Pn*Zr(OAr)2 > Pn*ZrCp(OR) > Pn*ZrCp(OAr), with Pn*Zr(O-2,6-iPr-C6H3) (3) showing the fastest rate of polymerisation of L-lactide (kobs = 0.35 h−1) and Pn*Zr(O-2,6-Me-C6H3) (2) showing the fastest rate of polymerisation of rac-lactide (kobs = 0.21 h−1).Isotactic PLA was produced for the ROP of L-lactide (demonstrating no epimerisation had occurred) and moderately heterotactic enriched PLA (Pr = 0.66 and 0.69 for Pn*Zr(O-2,6-Me-C6H3)2 (2) and Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6) respectively) or atactic PLA (Pr = 0.54 for Pn*Zr(O-2,6-iPr-C6H3)2 (3)) was produced for the ROP of rac–lactide, demonstrating that some degree of stereocontrol can be achieved with these initiators. In general, the molecular weights of the polylactides showed good agreement with the calculated values, suggesting that all metal centres were active during polymerisation. However, the molecular weight distributions were relatively large (maximum Mw/Mn of 1.62), indicating that intermolecular transesterification processes may be occurring. Polymer chains consisting of polylactic acid repeat units with aryloxide and hydroxy end groups were identified, suggesting that polymerisation follows a coordination-insertion where the aryloxide group initiates polymerisation.
Experimental
Synthesis of Pn*Zr(OtBu)2 (1)
1.0 equivalent [Pn*ZrCl2]·LiCl·(thf)1.85 (15.0 mg, 0.017 mmol) and 4.0 equivalents KOtBu (7.7 mg, 0.069 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube to give Pn*Zr(OtBu)2 (1) as a yellow solution. The instability of the complex prevented isolation on a preparative scale. 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 2.14 (1,3,5,7-Me-Pn*, 12H, s), 1.91 (2,6-Me-Pn*, 6H, s), 1.24 (OCMe3, 18H, s). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 131.8 (2,6-Pn*), 130.5 (4,8-Pn*), 106.9 (1,3,5,7-Pn*), 74.4 (OCMe3), 33.7 (OCMe3), 12.6 (1,3,5,7-Pn*), 11.2 (2,6-Pn*).Synthesis of Pn*Zr(O-2,6-Me-C6H3)2 (2)
1.0 equivalent [Pn*ZrCl2]2·LiCl(thf)x (250 mg, 0.285 mmol) and 4.0 equivalents KO-2,6-Me-C6H3 (247 mg, 1.14 mmol) were combined in toluene (10 mL) in a Schlenk tube. The reaction was stirred for 24 hours at room temperature, filtered through Celite and dried in vacuo to yield Pn*Zr(O-2,6-Me-C6H3)2 (2) as a yellow solid in 78% yield (282 mg, 0.446 mmol). Crystals suitable for a single crystal X-ray diffraction study were grown from a hexane solution at −30 °C. 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.00 (OArH, 4H, d, 3JH–H = 8 Hz), 6.75 (OArH, 2H, t, 3JH–H = 8 Hz), 2.16 (OArMe, 12H, s), 1.90 (2,6-Me-Pn*, 6H, s), 1.86 (1,3,5,7-Me-Pn*, 12H, s).Synthesis of Pn*Zr(O-2,6-iPr-C6H3)2 (3)
1.0 equivalent [Pn*ZrCl2]2·LiCl(thf)1.85 (200 mg, 0.229 mmol) and 4.0 equivalents KO-2,6-iPr-C6H3 (198 mg, 0.915 mmol) were combined in toluene (5 mL) in a Schlenk tube. The solution was sonicated for 10 minutes followed by stirring for 3 h at room temperature. The resulting pale yellow solution was dried in vacuo, the product extracted with hexane (5 mL) and concentrated to ∼2 mL. Storage −30 °C yielded Pn*Zr(O-2,6-iPr-C6H3)2 (3) as yellow crystals, suitable for a single crystal X-ray diffraction study, in 79% yield (229 mg, 0.362 mmol). 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.05 (ArH, 4H, d, 3JH–H = 8 Hz), 6.93 (ArH, 2H, t, 3JH–H = 8 Hz), 3.20 (OArCHMe, 4H, sept, 3JH–H = 8 Hz), 1.95 (2,6-Me-Pn*, 6H, s), 1.88 (1,3,5,7-Me-Pn*, 12H, s), 1.24 (OArCHMe, 24H, d, 3JH–H = 8 Hz). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 156.2 (OAr), 136.4 (OAriPr), 133.4 (2,6-Pn*), 132.4 (4,8-Pn*), 123.2 (ArH), 120.0 (ArH), 110.4 (1,3,5,7-Pn*), 27.3 (OArCH), 24.2 (OArCHMe), 11.5 (1,3,5,7-Me-Pn*), 11.1 (2,6-Me-Pn*).Synthesis of Pn*Zr(O-4-OMe-C6H4)2 (4)
1.0 equivalent [Pn*ZrCl2]2·LiCl(thf)1.85 (15.0 mg, 0.017 mmol) and 4.0 equivalents KO-4-OMe-C6H4 (11.1 mg, 0.069 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube to give a cloudy orange solution. Dimeric [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) was observed after 20 h at room temperature and dark orange crystals suitable for a single crystal X–ray diffraction study were obtained from the benzene-d6 solution. [Pn*Zr(O-4-OMe-C6H4)2]2 (4′) was dissolved in THF by sonication and heating at 65 °C. Filtration and removal of solvent in vacuo afforded Pn*Zr(O-4-OMe-C6H4)2 (4) as an orange solid. 1H NMR (tetrahydrofuran-d8, 400 MHz, 23 °C) δ (ppm): 6.64 (OArH, 4H, d, 3JH–H = 8 Hz), 6.50 (OArH, 4H, d, 3JH–H = 8 Hz), 3.63 (OArOMe, 3H, s), 2.01 (2,6-Me-Pn*, 6H, s), 1.99 (1,3,5,7-Me-Pn*, 12H, s). 13C{1H} NMR (tetrahydrofuran-d8, 101 MHz, 23 °C) δ (ppm): 158.6 (OArOMe), 152.9 (OArOMe), 132.7 (Pn*), 128.9 (Pn*), 128.7 (Pn*), 128.4 (Pn*), 119.7 (OArH), 115.2 (OArH), 107.6 (Pn*), 55.9 (OArOMe), 12.0 (1,3,4,5-Me-Pn*), 11.4 (2,6-Me-Pn*).Synthesis of Pn*ZrCl(O-2,6-tBu-C6H3) (5)
1.0 equivalent [Pn*ZrCl2]2·LiCl(thf) and 4.0 equivalents KO-2,6-tBu-C6H3 were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube. Pn*ZrCl(O-2,6-tBu-C6H3) (5) was observed in seconds at room temperature in quantitative yield. Crystals of Pn*ZrCl(O-2,6-tBu-C6H3)·LiCl(tmeda) suitable for a single crystal X-ray diffraction study were grown from a benzene solution of 5 at room temperature.Synthesis of Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6)
1.0 equivalent [Pn*ZrCl2]2·LiCl(thf)x (0.150 g, 0.171 mmol) and 4.0 equivalents KO-2,6-tBu-4-Me-C6H2 (0.089 g, 0.344 mmol) were combined in toluene (3 mL) in a gas-tight ampoule. The solution was stirred for 12 h at 70 °C, and the resulting bright yellow solution dried in vacuo, extracted into toluene (3 × 30 mL), filtered through a Celite and concentrated to approximately 1 mL. Storage at −30 °C yielded Pn*ZrCl(O-2,6-tBu-4-Me-C6H2) (6) as large yellow crystals, suitable for a single crystal X-ray diffraction study, in 83% yield (0.152 g, 0.286 mmol). 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.19 (ArH, 2H, s), 2.34 (OArMe, 3H, s), 2.06 (1,3,5,7-Me-Pn*, 12H, s), 1.84 (2,6-Me-Pn*, 6H, s), 1.44 (OArCMe3, 18H, s). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 159.6 (OAr), 137.8 (OArCMe3), 133.0 (Pn*), 128.4 (Pn*), 125.9 (OArH), 34.7 (OArCMe3), 31.9 (OArCMe3), 21.5 (OArMe), 12.2 (1,3,5,7-Me-Pn*), 11.9 (2,6-Me-Pn*); 2 × quaternary carbons missing (Pn* and OArMe). CHN Analysis (%): expected C 65.43, H 7.76, observed C 65.35, H 7.86.Synthesis of Pn*ZrCp(OtBu) (7)
1.0 equivalent Pn*ZrCp(Cl) (34.0 mg, 0.0899 mmol) and 1.0 equivalent KOtBu (10.1 mg, 0.0899 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated at room temperature for 20 minutes to afford Pn*ZrCp(OtBu) (7). 1H NMR (benzene-d6, 500 MHz, 23 °C): 5.82 (CpH, 5H, s), 2.05 (Pn*Me, 6H, s), 1.91 (Pn*Me, 6H, s), 1.89 (Pn*Me, 6H, s), 1.19 (OCMe3, 9H, s). 13C{1H} NMR (benzene-d6, 125 MHz, 23 °C) δ (ppm): 129.7 (Pn*), 127.4 (Pn*), 122.3 (Pn*), 109.7 (Cp), 107.4 (Pn*), 102.5 (Pn*), 74.1 (OCMe3), 34.2 (OCMe3), 13.4 (Pn*Me), 12.9 (Pn*Me), 11.6 (Pn*Me).Synthesis of Pn*ZrCp(O-2,6-Me-C6H3) (8)
1.0 equivalent Pn*ZrCp(Cl) (48.7 mg, 0.129 mmol) and 1.0 equivalent KO-2,6-Me-C6H3 (25.2 mg, 0.129 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated at room temperature until KO-2,6-Me-C6H3 fully dissolved, yielding Pn*ZrCp(O-2,6-Me-C6H3) (8). 1H NMR (benzene-d6, 500 MHz, 23 °C) δ (ppm): 7.18 (OArH, 2H, d, 3JH–H = 10 Hz), 6.81 (OArH, 1H, t, 3JH–H = 10 Hz), 5.50 (CpH, 5H, s), 2.05 (OArMe, 3H, s), 1.90 (Pn*Me, 3H, s), 1.85 (Pn*Me, 3H, s), 1.84 (Pn*Me, 3H, s). 13C{1H} NMR (benzene-d6, 125 MHz, 23 °C) δ (ppm): 165.1 (OAr), 130.7 (OArH), 128.7 (Pn*), 128.0 (Pn*) 127.0 (Pn*), 124.8 (OArMe), 122.09, 116.5 (OArH), 110.7 (CpH), 110.3 (Pn*), 102.9 (Pn*), 17.2 (OArMe), 13.1 (Pn*Me), 11.5 (Pn*Me), 11.0 (Pn*Me).Synthesis of Pn*ZrCp(O-2,6-iPr-C6H3) (9)
1.0 equivalent Pn*ZrCp(Cl) (50.9 mg, 0.135 mmol) and 1.0 equivalent KO-2,6-iPr-C6H3 (29.1 mg, 0.135 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated at room temperature for 20 minutes to afford Pn*ZrCp(O-2,6-iPr-C6H3) (9). 1H NMR (benzene-d6, 500 MHz, 23 °C) δ (ppm): 7.17 (OArH, 2H, d, 3JH–H = 10 Hz), 6.97 (OArH, 1H, t, 3JH–H = 10 Hz), 5.61 (CpH, 5H, s), 2.93 (OArCHMe2, 2H, sept, 3JH–H = 10 Hz), 1.98 (Pn*Me, 3H, s), 1.89 (Pn*Me, 3H, s), 1.87 (Pn*Me, 3H, s), 1.33 (OArCHMe2, 6H, d, 3JH–H = 10 Hz), 1.21 (OArCHMe2, 6H, d, 3JH–H = 10 Hz). 13C{1H} NMR (benzene-d6, 125 MHz, 23 °C) δ (ppm): 161.6 (OAr), 136.4 (OArCHMe2), 130.6 (Pn*), 127.3 (Pn*), 123.8 (ArH), 122.0 (Pn*), 117.8 (ArH), 111.1 (Pn*), 110.9 (CpH), 103.7 (Pn*), 27.0 (OArCHMe2), 25.7 (OArCHMe2), 24.3 (OArCHMe2), 13.7 (Pn*Me), 11.9 (Pn*Me), 11.2 (Pn*Me).Synthesis of Pn*TiCp(O-2,6-Me-C6H3) (10)
1.0 equivalent Pn*TiCp(Cl) (50 mg, 0.15 mmol) and 1.2 equivalents KO-2,6-Me-C6H3 (29 mg, 0.18 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube. The solution was left for 24 h at room temperature, and was then filtered, dried in vacuo and the resulting dark orange solid extracted with pentane (3 × 10 mL). Storage at −80 °C yielded Pn*TiCp(O-2,6-Me-C6H3) (10) as a dark brown crystalline solid in 23% yield (15 mg, 0.04 mmol). 1H NMR (benzene-d6, 500 MHz, 23 °C) δ (ppm): 7.21 (OArH, 2H, d, 3JH–H = 10 Hz), 6.85 (OArH, 1H, t, 3J = 10 Hz), 5.25 (CpH, 5H, s), 1.97 (OArMe, 6H, s), 1.97 (2,6-Me-Pn*, 6H, s), 1.70 (1,3,5,7-Me-Pn*, 12H, s). 13C{1H} NMR (benzene-d6, 125 MHz, 23 °C) δ (ppm): 129.2 (OArH), 128.4 (Pn*), 128.0 (Pn*), 126.1 (OArMe), 124.7 (Pn*), 120.8 (Pn*), 116.1 (OArH), 112.1 (Pn*), 110.8 (CpH), 17.8 (OArMe), 13.9 (1,3,5,7-Me-Pn*), 13.0 (2,6-Me-Pn*), 11.2 (1,3,5,7-Me-Pn*). 1 × quaternary carbon missing (OAr).Synthesis of Pn*ZrCpMe(O-2,6-Me-C6H3) (11)
1.0 equivalent Pn*ZrCpMe(Cl) (18.0 mg, 0.0459 mmol) and 1.0 equivalent KO-2,6-Me-C6H3 (8.96 mg, 0.0459 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated for 2 × 30 minutes at room temperature to afford a yellow solution and colourless precipitate. After 16 h at room temperature, filtration followed by drying of the filtrate in vacuo afforded Pn*ZrCpMe(O-2,6-Me-C6H3) (11) as a pale yellow solid. Crystals, suitable for a single crystal X-ray diffraction study, grown from a pentane solution at −30 °C. 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.17 (OArH, 2H, d, 3JH–H = 8 Hz), 6.82 (OArH, 1H, t, 3JH–H = 8 Hz), 5.47 (CpH, 2H, app. t, 3JH–H = 3 Hz), 5.20 (CpH, 2H, app. t, 3JH–H = 3 Hz), 2.08 (OArMe, 6H, s), 1.92 (Pn*Me, 6H, s), 1.88 (Pn*Me, 6H, s), 1.84 (Pn*Me, 6H, s) 1.83 (CpMe, 3H, s). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 164.3 (OAr), 130.8 (Pn*), 129.0 (OArH), 127.4 (Pn*), 125.9 (OArMe), 123.7 (CpMe), 122.4 (Pn*), 116.7 (OArH), 111.0 (Pn*), 110.7 (CpH), 110.2 (CpH), 102.7 (Pn*), 17.6 (OArMe), 13.5 (CpMe), 13.4 (Pn*Me), 11.9 (Pn*Me), 10.8 (Pn*Me).Synthesis of Pn*ZrCpMe(O-2,6-iPr-C6H3) (12)
1.0 equivalent Pn*ZrCpMe(Cl) (19.9 mg, 0.0508 mmol) and 1.0 equivalent KO-2,6-iPr-C6H3 (11.0 mg, 0.0508 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated for 2 × 30 minutes at room temperature to afford a yellow solution and colourless precipitate. After 16 h at room temperature, filtration followed by drying of the filtrate in vacuo afforded Pn*ZrCpMe(O-2,6-iPr-C6H3) (12) as a pale yellow solid. 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.17 (OArH, 2H, d, 3JH–H = 8 Hz), 6.96 (OArH, 1H, t, 3JH–H = 8 Hz), 5.64 (CpH, 2H, app. t, 3JH–H = 3 Hz), 5.23 (CpH, 2H, app. t, 3JH–H = 3 Hz), 2.93 (OArCHMe2, 1H, sept., 3JH–H = 8 Hz) 2.01 (Pn*Me, 6H, s), 1.90 (Pn*Me, 6H, s), 1.89 (CpMe, 3H, s), 1.87 (Pn*Me, 6H, s), 1.35 (OArCHMe2, 6H, d, 3JH–H = 8 Hz), 1.21 (OArCHMe2, 6H, d, 3JH–H = 8 Hz). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 160.7 (OAr), 136.5 (OArCHMe2), 130.3 (Pn*), 127.4 (Pn*), 123.9 (OArH), 123.8 (CpMe), 121.8 (Pn*), 117.7 (OArH), 111.2 (Pn*), 110.5 (CpH), 109.5 (CpH), 103.2 (Pn*), 27.4 (OArCHMe2), 25.7 (OArCHMe2), 24.5 (OArCHMe2), 13.4 (Pn*Me), 13.4 (CpMe), 12.1 (Pn*Me), 10.7 (Pn*Me).Synthesis of Pn*ZrCpMe(O-2,4-tBu-C6H3) (13)
1.0 equivalent Pn*ZrCpMe(Cl) (32.1 mg, 0.0819 mmol) and 1.0 equivalent KO-2,4-tBu-C6H3 (20.0 mg, 0.0819 mmol) were combined in benzene-d6 (0.5 mL) in a Young's tap NMR spectroscopy tube and sonicated for 2 × 30 minutes at room temperature to afford a yellow solution and colourless precipitate. After 16 h at room temperature, filtration followed by drying of the filtrate in vacuo afforded Pn*ZrCpMe(O-2,4-tBu-C6H3) (13) as a pale yellow solid. 1H NMR (benzene-d6, 400 MHz, 23 °C) δ (ppm): 7.57 (ArH, 1H, s), 7.27 (ArH, 1H, dd, 3JH–H = 4, 8 Hz), 5.99 (ArH, 1H, d, 3JH–H = 8 Hz), 5.65 (CpH, 1H, m), 5.57 (CpH, 1H, m), 5.30 (CpH, 2H, m), 2.19 (Pn*Me, 3H, s), 1.99 (Pn*Me, 3H, s), 1.93 (CpMe, 3H, s), 1.92 (Pn*Me, H, s), 1.90 (Pn*Me, 3H, s), 1.89 (Pn*Me, 3H, s), 1.85 (Pn*Me, 3H, s), 1.60 (ArCMe3, 9H, s), 1.42 (ArCMe3, 9H, s). 13C{1H} NMR (benzene-d6, 101 MHz, 23 °C) δ (ppm): 163.2 (OAr), 138.0 (OArCMe3), 136.5 (OArCMe3), 127.30 (Pn*), 126.83 (Pn*), 123.9 (ArH), 122.7 (ArH), 121.7 (ArH), 121.26 (Pn*), 121.21 (Pn*), 112.4 (CpMe), 112.2 (Pn*), 112.1 (Pn*), 111.6 (CpH), 109.7 (CpH), 108.8 (CpH), 104.4 (Pn*), 103.3 (Pn*), 35.7 (OArCMe3), 34.3 (OArCMe3), 32.3 (OArCMe3), 31.5 (OArCMe3), 13.7 (Pn*Me), 13.6 (Pn*Me), 13.5 (Pn*Me), 13.1 (Pn*Me), 11.9 (Pn*Me), 11.0 (CpMe), 10.8 (Pn*Me).Lactide polymerisation procedure ([LA]0/[M]0 = 50, [LA]0 = 0.5 M)
50 equivalents L– or rac-lactide (40 mg, 0.278 mmol) were weighed into Young's tap NMR spectroscopy tubes and 1.0 equivalent initiator in benzene-d6 (0.56 mL), corresponding to an initial lactide concentration ([LA]0) of 0.5 M, added. Polymerisations were run at 60–100 °C and halted at certain time intervals by submerging the NMR tubes in an ice bath. Polymerisation was monitored by 1H NMR spectroscopy. On completion, the reaction mixture was decanted into −5 °C pentane (10 mL), the solution allowed to settle, and the solvent removed. The polymer was then washed with diethyl ether (2 × 10 mL) and dried under vacuum at 30 °C for 18 hours.Lactide polymerisation procedure ([LA]0/[M]0 = 200, [LA]0 = 2.0 M)
1.0 equivalent initiator (3 mg) was dissolved in deuterated solvent (0.5 mL) and added to Young's tap NMR spectroscopy tubes. 200 equivalents L– or rac-lactide, corresponding to an initial lactide concentration ([LA]0) of 2 M, was then added. Polymerisations were run between 80 °C and halted at certain time intervals by submerging the NMR tubes in an ice bath. Polymerisation was monitored by 1H NMR spectroscopy. On completion, the reaction mixture was decanted into −5 °C pentane (10 mL), the solution allowed to settle, and the solvent removed. The polymer was then washed with diethyl ether (2 × 10 mL) and dried under vacuum at 30 °C for 18 hours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z.R.T., J.V.L., T.P.R., D.M. and J.-C.B. thank SCG Chemicals Co., Ltd (Thailand) for financial support and for a research Fellowship (Z.R.T). Chemical Crystallography (University of Oxford) is thanked for use of the diffractometers and Prof. Charlotte K. Williams (University of Oxford) is thanked for use of the GPC.Notes and references
- G. E. Luckachan and C. K. S. Pillai, J. Polym. Environ., 2011, 19, 637–676 CrossRef CAS.
- C. Romera-Castillo, M. Pinto, T. M. Langer, X. A. Alvarez-Salgado and G. J. Herndl, Nat. Commun., 2018, 9, 7 CrossRef PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1–16 CrossRef CAS PubMed.
- P. J. Dijkstra, H. Z. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS.
- H. R. Kricheldorf, Chemosphere, 2001, 43, 49–54 CrossRef CAS PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- R. Dattatray Karande, V. K. Abitha, A. Vasudeo Rane and R. K. Mishra, J. Mater. Sci. Eng. Adv. Tech., 2015, 12, 1–37 Search PubMed.
- R. W. F. Kerr, P. M. D. A. Ewing, S. K. Raman, A. D. Smith, C. K. Williams and P. L. Arnold, ACS Catal., 2021, 11, 1563–1569 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC.
- M. Normand, E. Kirillov, T. Roisnel and J. F. Carpentier, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS PubMed.
- S. Praban, P. Piromjitpong, V. Balasanthiran, S. Jayaraj, M. H. Chisholm, J. Tantirungrotechai and K. Phomphrai, Dalton Trans., 2019, 48, 3223–3230 RSC.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204–2212 CrossRef CAS PubMed.
- C. Bakewell, T. P. A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed.
- C. Bakewell, T. P. A. Cao, X. F. Le Goff, N. J. Long, A. Auffrant and C. K. Williams, Organometallics, 2013, 32, 1475–1483 CrossRef CAS.
- E. Grunova, E. Kirillov, T. Roisnel and J. F. Carpentier, Organometallics, 2008, 27, 5691–5698 CrossRef CAS.
- A. Alaaeddine, C. M. Thomas, T. Roisnel and J. F. Carpentier, Organometallics, 2009, 28, 1469–1475 CrossRef CAS.
- W. K. Gu, P. F. Xu, Y. R. Wang, Y. M. Yao, D. Yuan and Q. Shen, Organometallics, 2015, 34, 2907–2916 CrossRef CAS.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS PubMed.
- Y. F. Yu, D. Yuan, Y. R. Wang and Y. M. Yao, J. Organomet. Chem., 2016, 819, 37–45 CrossRef CAS.
- Y. Liang, R. L. Duan, C. Y. Hu, L. L. Li, X. Pang, W. X. Zhang and X. S. Chen, Chin. J. Polym. Sci., 2018, 36, 185–189 CrossRef CAS.
- M. Wisniewski, A. L. Borgne and N. Spassky, Macromol. Chem. Phys., 1997, 198, 1227–1238 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Rapid Commun., 1996, 197, 2627–2637 CrossRef CAS.
- C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS PubMed.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed.
- M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. Zhou, Inorg. Chem., 2008, 47, 2613–2624 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS PubMed.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- C. Romain, B. Heinrich, S. Bellemin-Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- J.-C. Buffet, G. R. Harris, J. J. Coward, T. A. Q. Arnold, Z. R. Turner and D. O'Hare, J. Organomet. Chem., 2016, 801, 87–95 CrossRef CAS.
- J. V. Lamb, J. C. Abell, J. E. McLaren, J.-C. Buffet, Z. R. Turner and D. O'Hare, Mol. Catal., 2020, 484, 110735 CrossRef CAS.
- D. A. X. Fraser, Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2016, 35, 2664–2674 CrossRef CAS.
- F. M. Chadwick, R. T. Cooper and D. O'Hare, Organometallics, 2016, 35, 2092–2100 CrossRef CAS.
- R. T. Cooper, F. M. Chadwick, A. E. Ashley and D. O'Hare, Organometallics, 2013, 32, 2228–2233 CrossRef CAS.
- D. D. Clement, S. C. Binding, T. A. Q. Arnold, F. M. Chadwick, I. J. Casely, Z. R. Turner, J.-C. Buffet and D. O'Hare, Polyhedron, 2019, 157, 146–151 CrossRef CAS.
- A. V. Firth, J. C. Stewart, A. J. Hoskin and D. W. Douglas, J. Organomet. Chem., 1999, 591, 185–193 CrossRef CAS.
- P. Angpanitcharoen, J. V. Lamb, Z. R. Turner, J.-C. Buffet and D. O'Hare, Mol. Catal., 2020, 498, 111275 CrossRef CAS.
- F. Benetollo, G. Cavinato, L. Crosara, F. Milani, G. Rossetto, C. Scelza and P. Zanella, J. Organomet. Chem., 1998, 555, 177–185 CrossRef CAS.
- W. A. Howard, T. M. Trnka and G. Parkin, Inorg. Chem., 1995, 34, 5900–5909 CrossRef CAS.
- O. T. Summerscales and F. G. N. Cloke, Coord. Chem. Rev., 2006, 250, 1122–1140 CrossRef CAS.
- T. Repo, G. Jany, M. Salo, M. Polamo and M. Leskela, J. Organomet. Chem., 1997, 541, 363–366 CrossRef CAS.
- K. Jonas, P. Kolb, G. Kollbach, B. Gabor, R. Mynott, K. Angermund, O. Heinemann and C. Krüger, Angew. Chem., Int. Ed., 1997, 36, 1714–1718 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2014, 33, 3891–3903 CrossRef CAS.
- A. Meduri, M. Mazzeo, M. Lamberti, C. Capacchione and S. Milione, Mol. Catal., 2019, 471, 54–59 CrossRef.
- Y. Ning, Y. Zhang, A. Rodriguez-Delgado and E. Y. X. Chen, Organometallics, 2008, 27, 5632–5640 CrossRef CAS.
- J.-C. Buffet, G. R. Harris, J. J. Coward, T. A. Q. Arnold, Z. R. Turner and D. O'Hare, J. Organomet. Chem., 2016, 801, 87–95 CrossRef CAS.
- T. P. A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 CrossRef CAS PubMed.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed.
- F. Marchetti, G. Pampaloni, C. Pinzino, F. Renili, T. Repo and S. Vuorinen, Dalton Trans., 2013, 42, 2792–2802 RSC.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS PubMed.
- A. T. Normand, R. Malacea-Kabbara, R. Lapenta, A. Dajnak, P. Richard, H. Cattey, A. Bolley, A. Grassi, S. Milione, A. Auffrant, S. Dagorne and P. Le Gendre, Dalton Trans., 2020, 49, 6989–7004 RSC.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
- A. Stopper, T. Rosen, V. Venditto, I. Goldberg and M. Kol, Chem. – Eur. J., 2017, 23, 11540–11548 CrossRef CAS PubMed.
- N. Diteepeng, J.-C. Buffet, Z. R. Turner and D. O'Hare, Dalton Trans., 2019, 48, 16099–16107 RSC.
- A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS PubMed.
- D. Mandal, D. Chakraborty, V. Ramkumar and D. K. Chand, RSC Adv., 2016, 6, 21706–21718 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General details, NMR spectroscopy, crystallographic data and definitions of structural parameters, and additional polymerisation data and characterisation information. CCDC 2057993–2057999. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00252j |
| This journal is © The Royal Society of Chemistry 2021 |