
Metal–ligand cooperation behaviour of Fe and Co complexes bearing a tetradentate phenanthroline-based PNNP ligand
Yumiko
Nakajima,  *ab
Nai-Yuan
Jheng
ab
*ab
Nai-Yuan
Jheng
ab
*ab
Nai-Yuan
Jheng
ab
Abstract
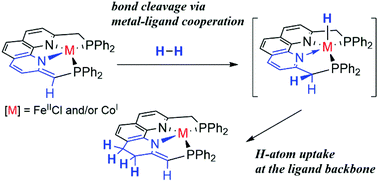
This perspective article describes the synthesis of a series of Fe and Co complexes coordinated with a phenanthroline-based meridional PNNP ligand (2,9-bis((diphenylphosphino)methyl)-1,10-phenanthroline). PNNP–iron(II) dichloride and –cobalt(I) chloride, [FeCl2(PNNP)] and [CoCl(PNNP)], underwent abstraction of the benzylic H-atom upon treatment with NaOtBu, forming the corresponding deprotonated products [FeCl(PNNP′)] (1) and [Co(PNNP′)] (2), respectively, each of which bears an asymmetrical PNNP′ ligand with a dearomatized phenanthroline backbone as a good metal–ligand cooperation (MLC) scaffold. Complex 2 achieved facile H–H bond cleavage mediated by unique long-range MLC, where the PNNP backbone acts as a H-atom reservoir.
 Please wait while we load your content...
Please wait while we load your content...

