
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of iridium complexes of a macrocyclic PNP pincer ligand†
Thomas M.
Hood
 and
Adrian B.
Chaplin
*
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 20th January 2021
Abstract
Having recently reported on the synthesis and rhodium complexes of the novel macrocyclic pincer ligand PNP-14, which is derived from lutidine and features terminal phosphine donors trans-substituted with a tetradecamethylene linker (Dalton Trans., 2020, 49, 2077–2086 and Dalton Trans., 2020, 49, 16649–16652), we herein describe our findings critically examining the chemistry of iridium homologues. The five-coordinate iridium(I) and iridium(III) complexes [Ir(PNP-14)(η2:η2-cyclooctadiene)][BArF4] and [Ir(PNP-14)(2,2′-biphenyl)][BArF4] are readily prepared and shown to be effective precursors for the generation of iridium(III) dihydride dihydrogen, iridium(I) bis(ethylene), and iridium(I) carbonyl derivatives that highlight important periodic trends by comparison to rhodium counterparts. Reaction of [Ir(PNP-14)H2(H2)][BArF4] with 3,3-dimethylbutene induced triple C–H bond activation of the methylene chain, yielding an iridium(III) allyl hydride derivative [Ir(PNP-14*)H][BArF4], whilst catalytic homocoupling of 3,3-dimethylbutyne into Z-tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCHCHtBu could be promoted at RT by [Ir(PNP-14)(η2:η2-cyclooctadiene)][BArF4] (TOFinitial = 28 h−1). The mechanism of the latter is proposed to involve formation and direct reaction of a vinylidene derivative with HCCtBu outside of the macrocyclic ring and this suggestion is supported experimentally by isolation and crystallographic characterisation of a catalyst deactivation product.
CCHCHtBu could be promoted at RT by [Ir(PNP-14)(η2:η2-cyclooctadiene)][BArF4] (TOFinitial = 28 h−1). The mechanism of the latter is proposed to involve formation and direct reaction of a vinylidene derivative with HCCtBu outside of the macrocyclic ring and this suggestion is supported experimentally by isolation and crystallographic characterisation of a catalyst deactivation product.
1. Introduction
Phosphine-based pincers are robust ancillary ligands that continue to find notable applications in organometallic chemistry and homogenous catalysis.1,2 In particular, rhodium and iridium complexes of these ligands are associated with fundamental and applied breakthroughs in the chemistry of C–H bond activation reactions; exemplified by the characterisation of σ-alkane complexes and development of high-performance alkane dehydrogenation catalysts, respectively.3,4 These mer-tridentate ligands are evidently well-suited to supporting the formation of highly reactive M(I) derivatives necessary to bring about cleavage of strong C–H bonds,5 able to accommodate the changes in geometry associated with M(I)/M(III) redox shuttling, and suitably modular in composition to enable augmentation of metal-based reactivity through considered change of the central donor atom, wingtip substituents, or backbone constituents.2 As marked out by lower carbonyl stretching frequencies,6 the heavier iridium congeners are particularly notable for a more pronounced tendency for oxidative addition of H2,7,8 C(sp3)–H bonds,3,9 and C(sp2)–H bonds10 compared to their rhodium counterparts.Motivated by the potential to exploit additional reaction control though their unique steric profile, use in the construction of interlocked assemblies, and as an extension of our related work with NHC-based variants,11–13 we have recently become interested in the chemistry of macrocyclic phosphine-based pincers.14–16 Last year we reported on the synthesis and rhodium complexes of the lutidine-derived macrocyclic pincer PNP-14, where the chiral P-donors are trans-substituted with a tetradecamethylene linker (Chart 1).14,15 As a novel platform for exploring the organometallic chemistry of Group 9 pincer complexes, we now present our findings critically examining the chemistry of iridium PNP-14 homologues aided by reference to acyclic complexes of 2,6-(R2PCH2)2C5H3N (PNP-R; e.g. R = tBu, iPr).
 | ||
| Chart 1 Complexes of the macrocyclic pincer ligand PNP-14. | ||
2. Results and discussion
Mirroring synthetic strategies that we have successfully employed for rhodium homologues,14,15 the preparation of iridium(I) and iridium(III) complexes of PNP-14 was attempted through substitution reactions of [Ir(COD)2][BArF4]17 in 1,2-difluorobenzene (DFB) and [Ir(COD)(biph)Cl]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 in fluorobenzene, respectively, with the latter exploiting Na[BArF4] as a halide abstracting agent (COD = 1,5-cyclooctadiene, ArF = 3,5-(CF3)2C6H3, biph = 2,2′-biphenyl; Scheme 1). These reactions produced five-coordinate cationic derivatives [Ir(PNP-14)(η2:η2-COD)][BArF4] 1 and [Ir(PNP-14)(biph)][BArF4] 2 under mild conditions, which were subsequently isolated as analytically pure crystalline materials in good yield (ca. 80%) and fully characterised (Scheme 1).
18 in fluorobenzene, respectively, with the latter exploiting Na[BArF4] as a halide abstracting agent (COD = 1,5-cyclooctadiene, ArF = 3,5-(CF3)2C6H3, biph = 2,2′-biphenyl; Scheme 1). These reactions produced five-coordinate cationic derivatives [Ir(PNP-14)(η2:η2-COD)][BArF4] 1 and [Ir(PNP-14)(biph)][BArF4] 2 under mild conditions, which were subsequently isolated as analytically pure crystalline materials in good yield (ca. 80%) and fully characterised (Scheme 1).
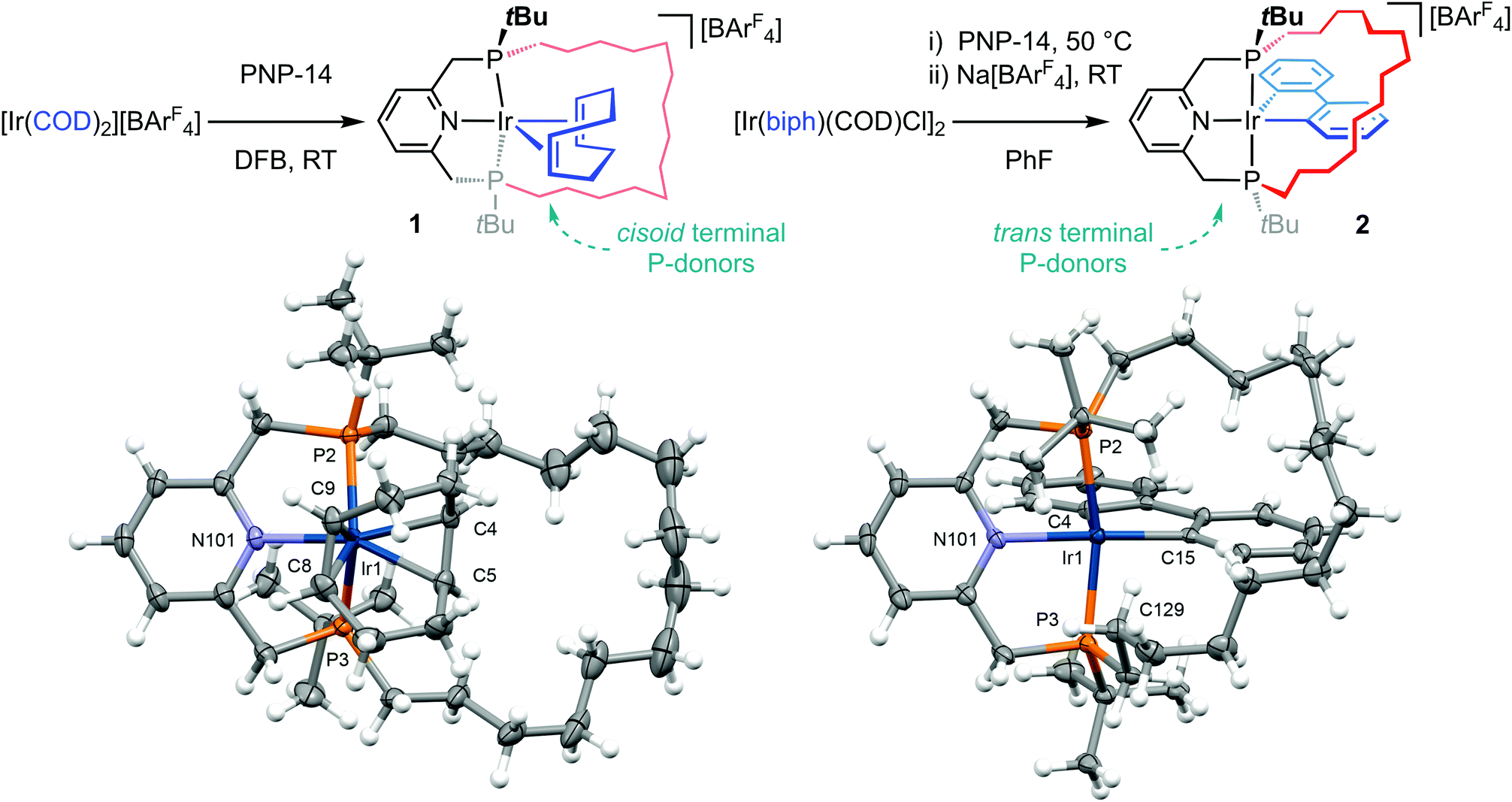 | ||
Scheme 1 Synthesis and solid-state structures of iridium pincer complexes 1 and 2: thermal ellipsoids at 50% and 30% probability, respectively; anions omitted. Selected bond lengths (Å) and angles (°): 1, Ir1–P2, 2.3743(7); Ir1–P3, 2.3846(7); P2–Ir1–P3, 115.24(2); Ir1–N101, 2.107(2); Ir1–Cnt(C4,C5), 2.083(2); Ir1–Cnt(C8, C9), 2.036(2); N101–Ir1–Cnt(C4, C5), 172.52(8); Cnt(C4, C5)–Ir–Cnt(C8, C9), 84.29(9); 2, Ir1–P2, 2.3253(15); Ir1–P3, 2.2849(15); P2–Ir1–P3, 163.24(5); Ir1–N101, 2.158(5); Ir1–C4, 2.048(6); Ir1–C15, 2.044(6); N101–Ir1–C15, 172.5(2); C4–Ir1–C15, 81.9(2); Ir1⋯C129, 3.152(7); ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ⋯ ⋯![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C129, 2.54; Cnt = centroid. C129, 2.54; Cnt = centroid. | ||
Iridium(I) complex 1 adopts a distorted trigonal bipyramidal metal geometry (18 VE), with the terminal phosphine donors positioned in the equatorial coordination sites conferring a distinctly puckered pincer ligand geometry. Distortion of the PNP ligand towards a fac coordination mode in this manner is associated with a compressed P–Ir–P bite angle of 115.24(2)° in the solid state and a pair of 31P resonances at δ 17.0 and 13.4 with no appreciable 2JPP coupling in DFB solution. While unusual, the formulation of 1 simply appears to be a consequence of COD chelation, although this is contingent upon the flexible lutidine-based backbone and asymmetric steric profile of the phosphine donors.19 Moreover, given the rhodium(I) homologue is instead characterised as a C1-symmetric square planar complex, viz. [Rh(PNP-14)(η2-COD)]+(1′, 16 VE; δ31P 57.4, 45.9, 2JPP = 312 Hz),15,20 the capacity of the heavier metal congener to from stronger metal–ligand bonds is clearly a decisive factor. Bulk purity was established by combustion analysis and the structure of 1 was fully corroborated in solution by NMR spectroscopy and HR ESI-MS.
Coordination of PNP-14 is more conventional in the formally 16 VE square pyramidal iridium(III) complex 2, as evidenced by a P–Ir–P bite angle of 163.24(5)° in the solid state and C1 symmetry in CD2Cl2 solution; with a pair of 31P resonances at δ 38.7 and 20.9 exhibiting a characteristically large trans-phosphine 2JPP coupling constant of 307 Hz.21 The crystal structure of 2 is isomorphous to the direct rhodium homologue 2′,14 with the tetradecamethylene linker skewed to one side of the basal plane away from the biph ligand and contorted in such a way as to enable adoption of a weak γ-agostic interaction (2, ⋯H–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif)
![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) = 3.152(7); cf. 3.184(2) Å for 2′). Previously reported five-coordinate complexes of the form [M(pincer)(biph)][BArF4] provide further structural precedent for 2 and the metal-based metrics of the acyclic analogue [Ir(PNP-tBu)(biph)][BArF4] (II) are similar.6,11,14 Moreover, as II is fluxional in solution as a result of facile biph pseudorotation on the NMR timescale,6 retention of C1 symmetry in solution suggests that buttressing with the methylene strap prevents such dynamics in 2.
= 3.152(7); cf. 3.184(2) Å for 2′). Previously reported five-coordinate complexes of the form [M(pincer)(biph)][BArF4] provide further structural precedent for 2 and the metal-based metrics of the acyclic analogue [Ir(PNP-tBu)(biph)][BArF4] (II) are similar.6,11,14 Moreover, as II is fluxional in solution as a result of facile biph pseudorotation on the NMR timescale,6 retention of C1 symmetry in solution suggests that buttressing with the methylene strap prevents such dynamics in 2.
Reaction of 2 with dihydrogen (1 atm) in DFB at RT resulted in immediate and full conversion into [Ir(PNP-14)(2-biphenyl)H][BArF4] 3 (δ31P 40.6, 36.5, 2JPP = 302 Hz; δ1H −21.6; Scheme 2). No further reaction was observed after 18 h, but heating at 85 °C for 6 h resulted in complete hydrogenolysis of the biph ligand and formation of dihydride dihydrogen complex [Ir(PNP-14)H2(H2)][BArF4] 4 in quantitative spectroscopic yield. Hydrogenolysis also occurs for 2′ and II, but longer reaction times are required under otherwise equivalent conditions (both ca. 2 days).14 Coordinatively saturated 1 rapidly affords 4 upon reaction with dihydrogen (1 atm) in DFB at RT (<5 min), invoking facile and reversible chelation of COD.
 | ||
| Scheme 2 Synthesis and reactivity of iridium dihydride dihydrogen complex 4. [BArF4]− counter anions omitted for clarity. | ||
Complex 4 was characterised in situ using NMR spectroscopy, with adoption of time-averaged C2 symmetry, a single 31P resonance at δ 42.1, and a broad 4H resonance at δ −9.26 (T1 = 88.8 ± 0.7 ms, 600 MHz, argon) the most diagnostic features at 298 K. The hydride signal remained broad upon cooling to 253 K but exhibits faster spin–lattice relaxation (δ −9.27, T1 = 50 ± 1 ms, 600 MHz, argon). The acyclic analogue [Ir(PNP-tBu)H2(H2)]BF4 (IV) is known and formulation as a dihydride dihydrogen complex was corroborated in a similar manner in situ by NMR spectroscopy.7 Whilst the data was recorded under difference conditions, the similarly of the hydride signatures is striking (IV, δ1H −9.31, T1 = 24 ms, 400 MHz, 233 K in CD3OD). In line with the reduced propensity for oxidative addition, the rhodium homologue of 4 is instead observed as a dihydrogen complex, viz. [Rh(PNP-14)(H2)][BArF4] 4′.14
Further supporting the assignment of 4, reaction with ethylene (1 atm) generated the corresponding C1-symmetric dihydride π-complex 5 (δ31P 33.4, 12.4, 2JPP = 314; δ1H −7.89, −17.80) within 5 min at RT (Scheme 2). Subsequent heating at 85 °C for 16 h yielded the bis(ethylene) complex 6 (δ31P 9.0) in quantitative spectroscopic yield, with concomitant formation of ethane. C2 symmetry and coordination of two molecules of ethylene was established in situ by NMR spectroscopy. The latter is associated with four chemically inequivalent 2H signals at δ 3.23, 2.80, 2.49 and 1.97 and two 13C resonances at δ 26.0 and 18.0, and reinforces the disposition of iridium(I) centres to adopt five-coordinate geometries: as seen in 2, but contrasting that observed under the same conditions in the rhodium(I) system, viz. [Rh(PNP-14)(C2H4)][BArF4] 6′.14 Structurally-related bis(ethylene) iridium(I) complexes of CNC- and pybox-based pincer ligands have been crystallographically characterised, exhibiting distorted trigonal bipyramidal metal geometries with the ethylene ligands located in the equatorial sites,22 but are unknown for PNP- and PONOP-based ligands.23
When 2 was instead treated with an excess of 3,3-dimethylbutene (5 equivalents) the allyl hydride derivative [Ir(PNP-14*)H][BArF4] 7 was produced in quantitative spectroscopic yield after 5 days heating at 100 °C, presumably through intramolecular transfer dehydrogenation of the methylene chain followed by allylic C–H activation (Scheme 2).24 As precedent for this reactivity, examples of cyclometallated rhodium(III) and iridium(III) pincer complexes can be found in the literature.25 Complex 7 was subsequentially isolated in 49% yield and fully characterised, including in the solid state by single crystal X-ray diffraction (Fig. 1). In solution 7 is distinctly C1-symmetric, with a pair of 31P resonances at δ 81.4 and 25.8 with 2JPP = 313 Hz, allyl 13C resonances at δ 81.9, 66.0, and 38.9, and a 1H hydride resonance at δ −8.49 (2JPH = 19.6, 9.7 Hz). The crystal structure demonstrates that 7 adopts a pseudo-octahedral metal geometry in the solid state, with κ6-coordination of PNP-14* creating iridacyclopentyl and iridacyclododecyl rings, and the hydride ligand was located from the Fourier difference map. Little distortion of the PNP-core is evident in 7 and the associated metal-based metrics are broadly comparable to those in 2 (e.g. P–Ir–P ca. 163°). There is considerable variance in the allyl Ir–C bond lengths, with the longest contact trans to the hydride ligand (Ir1–C119 = 2.311(4) Å, Ir1–C120, 2.155(3) Å, Ir1–C121, 2.199(3) Å), but all the internal carbon bond angles are >120°. The geometry of the iridacyclododecyl ring is reminiscent of the quadrilateral conformations adopted by 12-membered cycloalkanes.26
 | ||
| Fig. 1 Solid-state structure of 7: the hydride ligand was located from the Fourier difference map; thermal ellipsoids at 30% probability; solvent molecule and anion omitted. Selected bond lengths (Å) and angles (°): Ir1–P2, 2.2694(8); Ir1–P3, 2.3339(9); P2–Ir1–P3, 163.04(3); Ir1–N101, 2.126(2); Ir1–H1, 1.39(3); Ir1–C119, 2.311(4); Ir1–C120, 2.155(3); Ir1–C121, 2.199(3); C119–C120, 1.430(5); C120–C121, 1.409(5); C119–C120–C121, 124.4(3); N101–Ir1–C121, 169.59(11). | ||
The onward reactivity of 6 was harnessed to access the C2-symmetric Ir(I) carbonyl derivative 8 (δ31P 62.5) by reaction with carbon monoxide, which was isolated in 89% yield (overall from 2; Scheme 2). Complexes of this nature are of interest as the carbonyl ligand is a convenient spectroscopic reporter group for the electronic characteristics of the metal-pincer fragment.27,28 In this case, the ν(CO) band of 8 (1984 cm−1) is shifted to considerably lower frequency compared to the rhodium(I) homologue 8′ (1997 cm−1) under the same conditions (CH2Cl2 solution, Table 1). This is in line with expected periodic trends, which are also apparent from the IR data collected for tBu- and iPr-substituted analogues. These data suggest that PNP-14 is a marginally weaker net donor than PNP-tBu, but equivalent to PNP-iPr.14
Of the organometallic chemistry we have discovered so far using PNP-14, the capacity for rhodium complexes to promote the stoichiometric homocoupling of 3,3-dimethylbutyne through the annulus of the macrocyclic ligand stands out (1′ → 9′ in Scheme 3).15 Given that a structurally related Ir(PCP) system has also been shown to promote stoichiometric terminal alkyne coupling reactions,30,31 we were very interested to ascertain if similar reactivity could be brought about in iridium complexes of PNP-14. Iridium(I) complex 1 was selected as the most suitable precursor and initial screening studies using a twofold excess of HCCtBu in DFB at RT indicated rapid production of the corresponding Z-enyne (δ1H 5.53, 5.25; 3JHH = 11.9 Hz)32 without any observable consumption of 1 (Scheme 3). This stereochemistry contrasts that observed for the rhodium system and, as homocoupling through the macrocycle would be expected to result in an interpenetrated enyne complex, it appears that production of the enyne occurs catalytically outside the ring. Subsequent detailed investigation of this reaction using 100 equivalents HCCtBu confirmed that 1 is an effective precatalyst.33 Under these conditions, the dimerisation of HCCtBu into Z-tBuCCCHCHtBu proceeds with an initial TOF of 28 h−1. After 6 h, analysis by 1H NMR spectroscopy indicated complete consumption of HCCtBu and exclusive production of Z-tBuCCCHCHtBu. From the 31P{1H} NMR spectrum generation of a new organometallic species was apparent (10, δ 25.0, 16.3; 2JPP = 364 Hz), accounting for 88% of the metal-containing species with 1 making up 10%. Addition of a further 50 equivalents of HCCtBu induced complete conversion of 1 into 10 within 24 h but coincided with a halt in homocoupling, which plateaued at 65 TONs.
 | ||
| Scheme 3 Terminal alkyne coupling reactions promoted by 1 and 1′: [M] = M(PNP-14)+. | ||
Repeating the homocoupling reaction on a larger scale under similar conditions enabled isolation of 10 from solution in 76% yield, which was subsequently identified as iridium(III) bis(alkenyl) complex [Ir(PNP-14)(η3-E-C(CCtBu)CHtBu)(η1-E-CHCHtBu)][BArF4] (Scheme 4). In the solid state, 10 adopts a very distorted octahedral geometry with the alkenyl ligands in a cis configuration (C4–Ir1–C6 = 102.50(9)°) and coordination of the σ-organyl derived from Z-tBuCCCHCHtBu reinforced by π-complexation of the alkyne (Ir1–alkyne = 2.505(2) Å): a binding mode, for which there are no crystallographically characterised Group 9 precedents to our knowledge (CSD 5.41).34 The respective alkenyl Ir–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths are not statistically different (Ir1–C4 = 2.043(2) Å, Ir1–C6 = 2.053(2) Å; C4–C5 = 1.330(3) Å, C6–C9 = 1.329(3) Å). The structure of 10 determined by X-ray crystallography was fully corroborated in solution using NMR spectroscopy. For instance, the alkenyl 1H resonances are located at δ 7.81 (IrCCHtBu; 3JHH = 15.0 Hz), 5.68 (IrCCtBu), and 4.78 (IrCHCtBu; 3JHH = 15.2 Hz) in a 1:1:1 ratio, with the associated 13C resonances at δ 143.5 (IrCHHtBu), 142.2 (IrCHtBu), 105.3 (IrCHtBu), and 95.7 (IrHCHtBu); the α-carbons exhibiting coupling to 31P (2JPC = 5–10 Hz). The HR-ESI MS of 10 is also notable for a strong [M]+ ion signal at 916.5623 (calcd 916.5628) m/z and bulk purity of was confirmed by combustion analysis.
C bond lengths are not statistically different (Ir1–C4 = 2.043(2) Å, Ir1–C6 = 2.053(2) Å; C4–C5 = 1.330(3) Å, C6–C9 = 1.329(3) Å). The structure of 10 determined by X-ray crystallography was fully corroborated in solution using NMR spectroscopy. For instance, the alkenyl 1H resonances are located at δ 7.81 (IrCCHtBu; 3JHH = 15.0 Hz), 5.68 (IrCCtBu), and 4.78 (IrCHCtBu; 3JHH = 15.2 Hz) in a 1:1:1 ratio, with the associated 13C resonances at δ 143.5 (IrCHHtBu), 142.2 (IrCHtBu), 105.3 (IrCHtBu), and 95.7 (IrHCHtBu); the α-carbons exhibiting coupling to 31P (2JPC = 5–10 Hz). The HR-ESI MS of 10 is also notable for a strong [M]+ ion signal at 916.5623 (calcd 916.5628) m/z and bulk purity of was confirmed by combustion analysis.
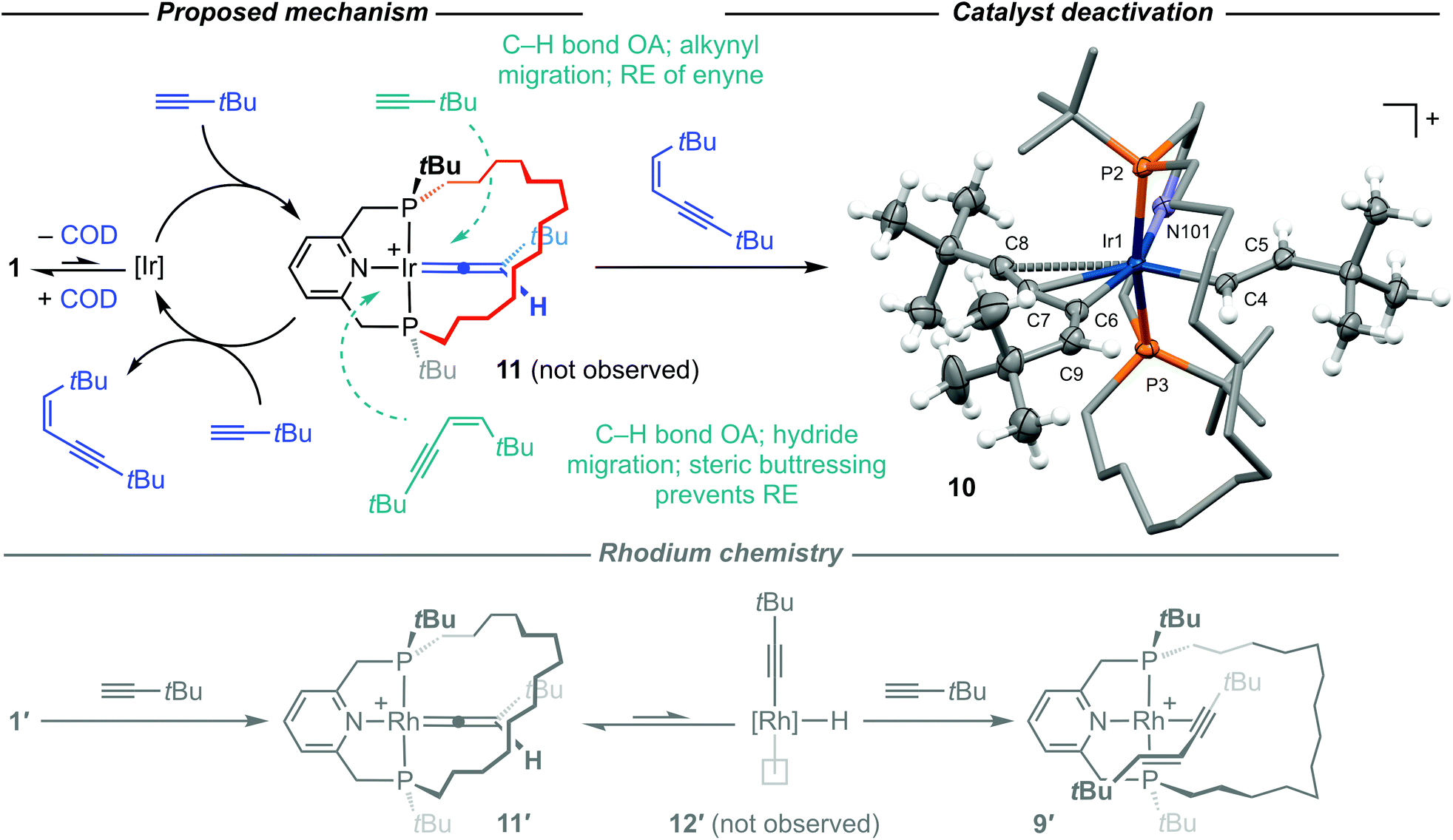 | ||
| Scheme 4 Mechanistic features of the terminal alkyne coupling reactions promoted by 1 and 1′: [M] = M(PNP-14)+ and [BArF4]− counter anions omitted. Solid-state structure of 10 depicted with thermal ellipsoids at 30% probability for selected atoms; minor disordered component (IrP2 core), most H atoms, and anion omitted; structural diagram provided in the experimental section. Selected bond lengths (Å) and angles (°): Ir1–P2, 2.3141(5); Ir1–P3, 2.3659(5); P2–Ir1–P3, 163.55(2); Ir1–N101, 2.122(2); Ir–C4, 2.043(2); Ir–C4–C5, 129.59(15); C4–C5, 1.330(3); Ir1–C6, 2.053(2); Ir1–C6–C9, 147.4(2); C6–C9, 1.329(3); Ir1–Cnt(C7, C8), 2.505(2); C6–C7–C8, 161.9(2); C7–C8, 1.216(3); N101–Ir1–C6, 159.35(8); C4–Ir1–Cnt(C7, C8), 153.16(7); C4–Ir1–C6, 102.50(9); Cnt = centroid. | ||
Terminal alkyne homocoupling reactions that produce Z-enyne products are generally understood to proceed via vinylidene intermediates, with 11 implicated in this case (Scheme 4).35,36 Indeed, the rhodium homologue 11′ is produced initially upon reaction of 1′ with HCCtBu.15 Reaction with the second alkyne equivalent, by net 1,2-addition of the constituent C(sp)–H bond across the vinylidene MC linkage (concerted or step-wise) followed by reductive elimination, would thereafter confer the enyne product. E-Enyne isomers such as that observed in the rhodium system can also be produced in this manner, although an indirect route involving equilibrium generation of the rhodium(III) alkynyl hydride 12′ is instead invoked in the formation of 9′ from 11′ (Scheme 4). Whilst 11 was not detected during the formation of Z-tBuCCCHCHtBu, the generation of 10 provides strong circumstantial evidence for its intermediate presence. No reaction between 1 and independently synthesised Z-tBuCCCHCHtBu in DFB was observed, even upon heating at 50 °C for 1 h. The formation of the bis(alkenyl) is, therefore, most reasonably reconciled by irreversible reaction of Z-tBuCCCHCHtBu with 11; involving net 1,2-addition of the {CC}C(sp2)–H bond across the IrC linkage. The addition evidently takes place through the ring in this instance, with the macrocycle preventing subsequent reductive elimination.12 We therefore attribute the generation of 10 to catalyst deactivation by irreversible product inhibition. The postulated reactivity of the Group 9 vinylidenes derived from 1 and 1′ is clearly nuanced by the nature of the metal and impact of the unique steric constraints imposed by the tetradecamethylene linker. We believe that the more facile Ir(I)/Ir(III) redox couple and propensity of the {Ir(PNP-14)}+ fragment to adopt geometries with the pincer ligand in a non-meridional conformation are the decisive factors. Specifically, we propose that the Z-selective homocoupling of HCCtBu proceeds catalytically outside the ring via C(sp)–H bond oxidative addition of HCCtBu to 11 affording fac-[Ir(PNP-14)(CCHtBu)(CCtBu)H]+, alkynyl migration yielding an enynyl hydride, and finally release of Z-tBuCCCHCHtBu by reductive elimination. In contrast, 1′ mediates the stoichiometric E-selective homocoupling of HCCtBu through the ring ultimately via a pathway bypassing the vinylidene intermediate 11′. Further computational analysis would be required to corroborate these suggestions, although accurately modelling the effect of the methylene chain is non-trivial.
3. Conclusions
The organometallic chemistry of iridium complexes of the macrocyclic PNP-14 pincer ligand has been explored. The five-coordinate iridium(I) and iridium(III) complexes [Ir(PNP-14)(η2:η2-COD)][BArF4] 1 and [Ir(PNP-14)(biph)][BArF4] 2 are readily prepared and fully characterised derivatives, with the former notable for a distorted trigonal bipyramidal metal geometry in which the pincer ligand adopts an unusual non-meridional confirmation, and the latter for adoption of a square pyramidal metal geometry stabilised by a weak γ-agostic interaction between the metal and the tetradecamethylene linker. These well-defined complexes have been shown to be effective precursors for the generation of iridium(III) dihydride dihydrogen (4), iridium(I) bis(ethylene) (6), and iridium(I) carbonyl (8) derivatives that highlight important periodic trends by comparison to rhodium counterparts: i.e. the more facile oxidative addition of dihydrogen, propensity to from five-coordinate d8 complexes, and greater π-basicity of the heavier metal conger, respectively.Onward reactivity of the {Ir(PNP-14)}+ fragment was also explored with the bulky unsaturated substrates 3,3-dimethylbutene and 3,3-dimethylbutyne. Reaction of 4 with 3,3-dimethylbutene induced triple C–H bond activation of the methylene chain yielding an iridium(III) allyl hydride complex [Ir(PNP-14*)H][BArF4] 7, whilst 1 is an effective pre-catalyst for the homocoupling of 3,3-dimethylbutyne into Z-tBuCCCHCHtBu under mild conditions. The latter is particularly remarkable given that reaction of the homologous rhodium precursor 1′ results in the formation of an interpenetrated E-enyne complex (9′). The mechanism of the homocoupling promoted by 1 is proposed to involve formation and direct reaction of the (unobserved) vinylidene derivative [Ir(PNP-14)(CCHtBu)][BArF4] (11) with HCCtBu outside of the macrocyclic ring. This suggestion is supported experimentally by isolation and crystallographic characterisation of [Ir(PNP-14)(η3-E-C(CCtBu)CHtBu)(η1-E-CHCHtBu)][BArF4] (10), which results from deactivation of the catalyst by product inhibition.
4. Experimental
4.1 General methods
All manipulations were performed under an atmosphere of argon using Schlenk and glove box techniques unless otherwise stated. Dihydrogen and ethylene were dried by passage through a column of activated 3 Å molecular sieves. Glassware was oven dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves were activated by heating at 300 °C in vacuo overnight. Fluorobenzene and DFB were pre-dried over Al2O3, distilled from calcium hydride and dried twice over 3 Å molecular sieves.37 CD2Cl2 was freeze–pump–thaw degassed and dried over 3 Å molecular sieves. C6D6 was distilled from sodium and stored over 3 Å molecular sieves. SiMe4 was distilled from liquid Na/K alloy and stored over a potassium mirror. Other anhydrous solvents and liquid reagents were purchased from Acros Organics or Sigma-Aldrich, freeze–pump–thaw degassed and stored over 3 Å molecular sieves. PNP-14,14 [Ir(COD)2][BArF4],17 [Ir(biph)(COD)Cl]2,18 Na[BArF4],38 and [Ir(PNP-tBu)(biph)][BArF4] (II)6 were synthesized according to published procedures. All other solid reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz. Virtual coupling constants are reported as the separation between the first and third lines. NMR spectra in DFB were recorded using an internal capillary of C6D6 or acetone-d6.37 High resolution ESI-MS were recorded on Bruker Maxis Plus instrument. Infrared spectra were recorded on a Jasco FT-IR-4700 using a KBr transmission cell in CH2Cl2. Microanalyses were performed at the London Metropolitan University by Stephen Boyer or Elemental Microanalysis Ltd.4.2 Preparation of [Ir(PNP-14)(η2:η2-COD)][BArF4] (1)
A solution of [Ir(COD)2][BArF4] (29.3 mg, 23.0 μmol) and PNP-14 (11.0 mg, 23.0 μmol) in DFB (0.5 mL) was mixed for 5 min at RT, the volatiles were removed in vacuo and the resulting orange oil washed with pentane (2 × 2 mL). The analytically pure product was obtained as a yellow crystalline solid by slow diffusion of SiMe4 (ca. 10 mL) into a DFB solution (0.5 mL) at −30 °C. Yield: 29.0 mg (17.7 μmol, 77%).
1
H NMR (500 MHz, DFB): δ 8.11–8.16 (m, 8H, ArF), 7.50 (br, 4H, ArF), 7.33 (t, 3JHH = 7.7, 1H, py), 7.08 (d, 3JHH = 7.7, 1H, py), 7.03 (obscured, py), 4.25–4.34 (m, 1H, Ir(CHCH){axial}), 4.34–4.44 (m, 1H, Ir(CHCH){axial}), 3.72 (d, 2JPH = 6.6, 2H, pyC2), 3.51 (dd, 2JHH = 18.2, 2JPH = 6.0, 1H, pyC2), 3.34 (dd, 2JHH = 18.2, 2JPH = 9.7, 1H, pyC2), 2.54–2.66 (m, 2H, CH2), 2.15–2.45 (m, 6H, CH2 + 1 × Ir(CHCH){equatorial} [δ 2.39]), 1.38–2.04 (m, 9H, CH2 + 1 × Ir(CHCH){equatorial} [δ 1.83]), 1.38 (d, 3JPH = 12.7, 9H, tBu), 0.94–1.32 (m, 19H, CH2), 0.65 (d, 3JPH = 12.5, 9H, tBu).
13
C{
1
H} NMR (126 MHz, DFB): δ 163.7 (dd, JPC = 5, 4, py), 162.5 (q, 1JCB = 50, ArF), 162.4 (obscured, py), 138.2 (s, py), 135.1 (s, ArF), 129.7 (qq, 2JFC = 32, 3JCB = 3, ArF), 124.9 (q, 1JFC = 272, ArF), 122.0 (d, 3JPC = 6, py), 120.7 (d, 3JPC = 8, py), 117.6 (sept, 3JFC = 4, ArF), 64.7 (br, Ir(CHCH){axial}), 60.9 (d, 2JPC = 3, Ir(CHCH){axial}), 60.2 (dd, 2JPC = 27, 4, Ir(CHCH){equatorial}), 50.1 (dd, 2JPC = 29, 2JPC = 5, Ir(CHCH){equatorial}), 45.6 (dd, 1JPC = 27, 3JPC = 4, pyH2) 42.3 (d, 1JPC = 22, pyH2), 35.8 (d, 3JPC = 10, CH2), 35.5 (br, CH2), 35.2 (dd, 1JPC = 19, 3JPC = 4, tBu{C}), 34.1 (d, 1JPC = 7, PCH2), 33.4 (br, tBu{C}), 31.1 (d, 3JPC = 10, CH2), 29.8 (d, 3JPC = 9, CH2), 29.7 (s, CH2), 29.5 (s, CH2), 29.3 (d, 1JPC = 17, PCH2), 29.0 (s, CH2), 28.8 (s, CH2), 28.7 (s, CH2), 28.6 (br, CH2), 28.3 (s, CH2), 28.12 (s, CH2), 28.05 (s, CH2), 27.8 (d, 2JPC = 4, tBu{CH3}), 27.1 (d, 3JPC = 7, CH2), 27.0 (s, CH2), 26.6 (s, CH2), 26.4 (d, 2JPC = 5, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, DFB): δ 17.0 (s, 1P), 13.4 (s, 1P).
HR ESI-MS (positive ion 4 kV): 778.4221 ([M]+, calcd 778.4218) m/z.
Anal. calcd for C69H77BF24IrNP2 (1641.32 g mol−1): C, 50.49; H, 4.73; N, 0.85. Found: C, 50.40; H, 4.64; N, 0.87.
4.3 NMR scale reaction of 1 with dihydrogen
A solution of 1 (12.2 mg, 7.43 μmol) in DFB (0.5 mL) within a J. Young valve NMR tube was freeze–pump–thaw degassed and placed under an atmosphere of dihydrogen (1 atm). Analysis by NMR spectroscopy indicated quantitative formation of 4 with concomitant formation of COD within 5 min at RT.4.4 Preparation of [Ir(PNP-14)(biph)][BArF4] (2)
A suspension of PNP-14 (13.3 mg, 27.8 μmol) and [Ir(biph)(COD)Cl]2 (13.7 mg, 14.0 μmol) in fluorobenzene (0.50 mL) was stirred for 2 days at 50 °C to give a pale-yellow solution. Na[BArF4] (24.7 mg, 27.9 μmol) was added and the suspension stirred for a further 4 h at RT. The volatiles were removed in vacuo and the resulting purple oil was washed with pentane (2 × 1 mL), dried in vacuo and then the product extracted into CH2Cl2 (2 mL). The analytically pure product was obtained as a purple crystalline solid by recrystallisation from CH2Cl2:hexane (1:20) at −30 °C. Yield: 36.4 mg (21.6 μmol, 78%).
1
H NMR (500 MHz, CD2Cl2): δ 8.00 (t, 3JHH = 7.9, py, 1H), 7.70–7.76 (m, 10H, py + ArF), 7.64 (d, 3JHH = 7.6, 1H, biph), 7.60 (d, 3JHH = 7.5, 1H, biph), 7.56 (br, 4H, ArF), 7.39 (d, 3JHH = 7.6, 1H, biph), 7.19 (t, 3JHH = 7.3, 1H, biph), 7.10 (t, 3JHH = 7.5, 1H, biph), 6.88 (t, 3JHH = 7.4, 1H, biph), 6.33 (t, 3JHH = 7.8, 1H, biph), 5.35 (d, 3JHH = 8.2, 1H, biph), 4.02 (dd, 2JHH = 19.4, 2JPH = 9.6, 1H, pyC2), 3.75–3.89 (m, 2H, 2 × pyC2), 3.50 (dd, 3JHH = 17.0, 3JHH = 9.2, 1H, pyC2), 3.02–3.15 (m, 1H, PCH2), 2.78–2.88 (m, 1H, PCH2), 1.88–1.98 (m, 1H, CH2), 0.66–1.73 (m, 23H, CH2), 1.16 (d, 3JPH = 14.0, 9H, tBu), 0.49 (d, 3JPH = 16.1, 9H, tBu), 0.22–0.37 (m, 2H, CH2).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 164.6 (app t, JPC = 4, py), 163.3 (br, py), 162.3 (q, 1JCB = 50, ArF), 150.6 (d, 3JPC = 2 biph), 149.6 (s, biph), 145.3 (dd, 2JPC = 8, 6, biph{IrC}), 139.9 (s, py), 135.4 (s, ArF), 135.2 (s, biph), 129.42 (qq, 2JFC = 32, 3JCB = 3, ArF), 129.39 (s, biph), 126.3 (s, biph), 125.6 (s, biph), 125.3 (s, biph), 125.1 (q, 1JFC = 272, ArF), 123.5 (s, biph), 123.2 (d, 3JPC = 10, py), 123.1 (d, 3JPC = 9, py), 122.1 (s, biph), 121.3 (s, biph), 121.2 (app t, 2JPC = 6, biph{IrC}), 118.0 (sept, 3JFC = 4, ArF), 40.9 (d, 1JPC = 29, pyH2), 39.5 (d, 1JPC = 26, pyH2), 35.0 (d, 1JPC = 23, tBu{C}), 32.9 (d, 2JPC = 14, CH2), 32.8 (obscured, tBu{C}), 30.4 (s, CH2), 29.6 (s, CH2), 29.5 (s, CH2), 29.42 (s, CH2), 29.35 (s, CH2), 29.2 (s, CH2), 29.1 (d, 2JPC = 3, tBu{CH3}), 28.1 (s, CH2), 27.9 (d, 1JPC = 28, PCH2), 27.3 (s, CH2), 25.8 (br, CH2), 25.5 (s, tBu{CH3}), 24.7 (s, CH2), 24.1 (s, CH2), 19.7 (dd, 1JPC = 22, 3JPC = 3, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 38.7 (d, 2JPP = 307, 1P), 20.9 (d, 2JPP = 307, 1P).
HR ESI-MS (positive ion, 4 kV): 822.3912 ([M]+, calcd 822.3906) m/z.
Anal. calcd for C73H73BF24IrNP2 (1685.33 g mol−1): C, 52.03; H, 4.37; N, 0.83; found: C, 51.88; H, 4.28; N, 0.81.
4.5 NMR scale reactions of 2
The following reactions were carried out starting with a solution of 2 (16.9 mg, 10.0 μmol) in DFB (0.5 mL) within a J. Young valve NMR tube and analysed in situ by NMR spectroscopy.
1
H NMR (400 MHz, DFB, H2, selected data): δ 7.61 (t, 3JHH = 7.8, 1H py), 3.66–3.82 (m, 2H, 2 × pyC2), 3.06 (dd, 2JHH = 17.3, 2JPH = 6.4, 1H, pyC2), 0.79 (d, 3JPH = 13.4, 9H, tBu), 0.67 (d, 3JPH = 14.1, 9H, tBu), −21.6 (app t, 2JPH = 15, 1H, IrH).
31 P{partial 1 H} NMR (162 MHz, DFB, H2): δ 40.6 (dd, 2JPP = 302, 2JPH = 14, 1P), 36.5 (dd, 2JPP = 302, 2JPH = 14, 1P).
1
H NMR (500 MHz, DFB, H2): δ 8.10–8.16 (m, 8H, ArF), 7.49 (br, 4H, ArF), 7.47 (t, 3JHH = 7.8, 1H, py), 7.19 (d, 3JHH = 7.8, 2H, py), 3.96 (dvt, 2JHH = 17.6, JPH = 8, 2H, pyC2), 3.12 (dvt, 2JHH = 17.6, JPH = 10, 2H, pyC2), 2.11–2.23 (m, 2H, PCH2), 1.12–1.69 (m, 26H, CH2), 0.91 (vt, JPH = 16, 18H, tBu), −9.27 (br, fwhm = 24 Hz, 4H, IrH4).
13
C{
1
H} NMR (126 MHz, DFB, H2): δ 162.3 (q, 1JCB = 50, ArF), 162.4 (vt, JPC = 6, py), 138.9 (s, py), 135.1 (s, ArF), 129.7 (qq, 2JFC = 32, 3JCB = 3, ArF), 124.9 (q, 1JFC = 272, ArF), 120.4 (vt, JPC = 10, py), 117.6 (sept, 3JFC = 4, ArF), 44.4 (vt, JPC = 28, pyH2), 30.0 (vt, JPC = 32, tBu{C}), 29.0 (s, CH2), 28.9 (vt, JPC = 8, CH2), 28.3 (s, CH2), 28.2 (s, CH2), 27.5 (s, CH2), 26.3 (s, CH2), 25.8 (vt, JPC = 32, PCH2), 24.5 (vt, JPC = 6, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, DFB, H2,): δ 42.1 (s, 2P).
1 H NMR (600 MHz, DFB, Ar, 298 K, selected data): δ −9.26 (br, fwhm = 29 Hz, T1 = 88.8 ± 0.7 ms, 4H, IrH).
1 H NMR (600 MHz, DFB, Ar, 253 K, selected data): δ −9.27 (br, fwhm = 21 Hz, T1 = 50 ± 1, 4H, IrH).
1
H NMR (500 MHz, DFB, C2H4, selected data): δ 7.43 (t, 3JHH = 7.8, 1H, py), 3.71–3.84 (m, 2H, 2 × pyC2), 3.18–3.37 (m, 5H, 1 × pyC2 + 4 × C2H4), 3.08 (dd, 2JHH = 17.6, 2JPH = 10.4, 1H, pyC2), 0.89 (d, 3JPH = 14.9, 9H, tBu), 0.87 (d, 3JPH = 13.9, 9H, tBu), −7.89 (dd, 2JPH = 17.6, 2JPH = 13.1, 1H, IrH), −17.80 (app t, JPH = 11, 1H, IrH).
13 C{ 1 H} NMR (126 MHz, DFB, C2H4, selected data): δ 48.7 (s, C2H4).
31 P{ 1 H} NMR (162 MHz, DFB, C2H4): δ 33.4 (d, 2JPP = 314, 1P), 12.4 (d, 2JPP = 314, 1P).
1
H NMR (600 MHz, DFB, C2H4): δ 8.11–8.15 (m, 8H, ArF4), 7.50 (br, 4H, ArF4), 7.46 (t, 3JHH = 7.8, 1H, py), 7.15 (d, 3JHH = 7.8, 2H, py), 3.63 (dvt, 2JHH = 16.8, JPH = 8, 2H, pyC2), 3.28 (dvt, 2JHH = 16.8, JPH = 8, 2H, pyC2), 3.23 (br, 2H, C2H4), 2.80 (br, 2H, C2H4), 2.49 (br, 2H, C2H4), 1.97 (br, 2H, C2H4), 1.03–1.48 (m, 28H, CH2), 0.79 (br, 18H, tBu).
13
C{
1
H} NMR (126 MHz, DFB, C2H4): δ 164.1 (d, 3JPC = 3, py), 162.6 (q, 1JCB = 50, ArF), 139.1 (s, py), 135.1 (s, ArF), 129.7 (qq, 2JFC = 32, 3JCB = 3, ArF), 124.9 (q, 1JFC = 272, ArF), 119.8 (vt, JPC = 8, py), 117.6 (sept, 3JFC = 4, ArF), 42.6 (vt, JPC = 28, pyH2), 33.5 (vt, JPC = 26, tBu{C}), 31.0 (s, CH2), 29.1 (vt, JPC = 12, CH2), 27.8 (s, CH2), 27.3 (s, CH2), 27.2 (s, CH2), 26.3 (s, tBu{CH3}), 26.0 (s, C2H4), 24.6 (s, CH2), 18.0 (s, C2H4), 14.3 (vt, JPC = 24, PCH2).
31 P{ 1 H} NMR (162 MHz, DFB, C2H4): δ 9.0 (s, 2P).
1
H NMR (500 MHz, CD2Cl2): δ 7.87 (t, 3JHH = 7.8, 1H, py), 7.70–7.76 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.51 (d, 3JHH = 7.9, 2H, py), 3.94 (dvt, 2JHH = 17.6, JPH = 8, 2H, pyC2), 3.47 (dvt, 2JHH = 17.6, JPH = 8, 2H, pyC2), 2.18–2.27 (m, 4H, PCH2), 1.94–2.07 (m, 2H, CH2), 1.76–1.89 (m, 2H, CH2), 1.64–1.74 (m, 2H, CH2), 1.54–1.64 (m, 2H, CH2), 1.23–1.48 (m, 16H, CH2), 1.14 (vt, JPH = 16, 18H, tBu)
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 182.5 (vt, 2JPC = 18, CO), 165.4 (vt, JPC = 10, py), 162.3 (q, 1JCB = 50, ArF), 141.9 (s, py), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 122.0 (vt, JPC = 10, py), 118.0 (sept, 3JFC = 4, ArF), 39.4 (vt, JPC = 24, pyH2), 34.7 (vt, JPC = 30, tBu{C}), 30.3 (vt, JPC = 10 CH2), 29.3 (s, CH2), 29.0 (s, CH2), 28.9 (s, CH2), 28.3 (s, CH2), 27.5 (vt, JPC = 6, tBu{CH3}), 26.1 (s, CH2), 23.4 (vt, JPC = 30, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 64.6 (s, 2P).
31 P{ 1 H} NMR (162 MHz, DFB): δ 62.5 (s, 2P).
IR (CH2Cl2): ν(CO) 1984 cm−1.
HR ESI-MS (positive ion, 4 kV): 698.3217 ([M]+, calcd 698.3228) m/z.
Anal. calcd for C62H65BF24IrNOP2 (1561.14 g mol−1): C, 47.70; H, 4.20; N, 0.90; found: C, 47.89; H, 4.13; N, 0.97.
4.6 NMR scale reaction of [Ir(PNP-tBu)(biph)][BArF4] (II) with dihydrogen
A solution of II (16.0 mg, 9.98 μmol) in DFB (0.5 mL) within a J. Young valve NMR tube was freeze–pump–thaw degassed and placed under dihydrogen (1 atm) and then heated at 80 °C for 2 days. Analysis by NMR spectroscopy indicated quantitative formation of [Ir(PNP-tBu)H2(H2)][BArF4]. The spectroscopic data are consistent with the literature for the related BF4− salt.7
1
H NMR (400 MHz, CD2Cl2, selected data): δ 8.11–8.17 (m, 8H, ArF), 7.50 (s, 4H, ArF), 3.52 (vt, JPH = 8, 4H, pyC2), 1.11 (vt, JPH = 14, 36H, tBu), −9.48 (br, 4H, IrH).
31 P{ 1 H} NMR (162 MHz, DFB): δ 64.6 (s, 2P).
4.7 Preparation of [Ir(PNP-14*)H][BArF4] (7)
A solution of 4 (13.6 μmol, generated in situ as described above) in DFB (0.5 mL) within a J. Young valve NMR tube was treated with 3,3-dimethylbutene (8.8 μL, 71.4 μmol) and the solution heated at 100 °C for 5 days. Analysis by NMR spectroscopy indicated quantitative formation of the product. The volatiles were removed in vacuo and the resulting pink/red oil washed with SiMe4 (2 × 0.5 mL). The analytically pure product was obtained as pale red blocks by the slow diffusion of excess SiMe4 into a DFB solution at −30 °C. Yield: 10.2 mg (6.66 μmol, 49%).
1
H NMR (500 MHz, CD2Cl2): δ 7.70–7.75 (m, 8H, ArF), 7.65 (t, 3JHH = 7.8, 1H, py), 7.56 (br, 4H, ArF), 7.34 (d, 3JHH = 7.8, 1H, py), 7.31 (d, 3JHH = 7.8, 1H, py), 4.86 (app q, JHH = 9, 1H, IrCH), 4.47 (app t, 3JHH = 7, 1H, IrCH), 3.71–3.81 (m, 2H, 2 × pyC2), 3.41 (dd, 2JHH = 17.1, 2JPH = 9.3, 1H, pyC2), 3.26 (dd, 2JHH = 17.4, 2JPH = 9.2, 1H, pyC2), 2.35–2.52 (m, 1H, CH2), 2.15–2.29 (m, 1H, CH2), 1.98–2.11 (m, 1H, CH2), 0.94–1.95 (m, 20H, IrCH [δ 1.89] + CH2), 1.17 (d, 3JPH = 15.4, 9H, tBu), 1.01 (d, 3JPH = 13.5, 9H, tBu), −8.49 (dd, 2JPH = 19.6, 9.7, 1H, IrH).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 163.4 (br, py), 162.3 (q, 1JCB = 50, ArF), 161.9 (br, py), 138.5 (s, py), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.1 (q, 1JFC = 272, ArF), 121.0 (d, 3JPC = 9, py), 120.8 (d, 3JPC = 9, py), 118.0 (sept, 3JFC = 4, ArF), 81.9 (s, IrCH), 66.0 (d, 2JPC = 5, IrCH), 41.9 (d, 1JPC = 30, pyH2), 39.2 (d, 1JPC = 29, pyH2), 38.9 (s, IrCH), 35.9 (s, CH2), 35.0 (dd, 1JPC = 22, 3JPC = 5, tBu{C}), 30.4 (dd, 1JPC = 22, 3JPC = 5, tBu{C}), 29.6 (s, CH2), 27.8 (d, 2JPC = 14, CH2), 27.4 (s, CH2), 27.0 (d, 2JPC = 3, tBu{CH3}), 25.7 (d, 2JPC = 4, tBu{CH3}), 24.8 (dd, 1JPC = 28, 3JPC = 1, PCH2), 24.4 (s, CH2), 24.3 (d, 2JPC = 5, CH2), 22.9 (s, CH2), 22.3 (s, CH2), 21.6 (s, CH2), 20.8 (dd, 1JPC = 24, 3JPC = 4, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 81.4 (d, 2JPP = 313, 1P), 25.8 (d, 2JPP = 313, 1P).
31 P{ 1 H} NMR (162 MHz, DFB): δ 80.9 (d, 2JPP = 313, 1P), 25.5 (d, 2JPP = 313, 1P).
HR ESI-MS (positive ion 4 kV): 668.3128 ([M]+, calcd 668.3122) m/z.
Anal. calcd for C62H67BF24IrNP2 (1531.12 g mol−1): C, 47.85; H, 4.15; N, 0.91. Found: C, 47.65; H, 4.03; N, 1.01.
4.8 NMR scale reaction of 7 with dihydrogen
A solution of 7 (7.7 mg, 5.03 μmol) in DFB (0.5 mL) within a J Young valve NMR tube was freeze–pump–thaw degassed, placed under dihydrogen (1 atm) and heated at 80 °C for 16 h. Analysis by NMR spectroscopy indicated quantitative formation of 4.4.9 Catalytic homocoupling of 3,3-dimethylbutyne promoted by 1
A solution of 1 (8.2 mg, 5.0 μmol) in DFB (400 μL) within a J. Young NMR tube was treated with a solution of 3,3-dimethylbutyne (62 μL, 503 μmol) and the resulting homocoupling reaction producing only Z-tBuCCCHCHtBu followed at RT in situ using NMR spectroscopy. The solution was mixed constantly when not in the spectrometer. Analysis after 1 hour indicated 28 TONs, with complete conversion observed within 6 h. At this point, 10 was observed as the major organometallic species (88%), with 1 the minor organometallic species (10%). Further 3,3-dimethylbutyne (31 μL, 252 μmol) was added, resulting in further homocoupling, totalling 65 TONs and complete conversion of 1 to 10 after 24 h. Spectroscopic data for Z-tBuCCCHCHtBu is consistent with literature values.32
Data for Z-tBuCCCHCHtBu:1H NMR (400 MHz, DFB, selected data): δ 5.53 (d, 3JHH = 11.9, 1H, CHCH), 5.25 (d, 3JHH = 11.9, 1H, CHCH), 1.12 (s, 9H, tBu), 1.11 (s, 9H, tBu).
4.10 Preparation of [Ir(PNP-14)(η3-E-C(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CtBu)CHtBu)(η1-E-CHCHtBu)][BArF4] (10)
CtBu)CHtBu)(η1-E-CHCHtBu)][BArF4] (10)
A solution of 1 (14.0 mg, 8.53 μmol) in DFB (0.5 mL) within a J Young valve NMR tube was treated with 3,3-dimethylbutyne (210 μL, 1.71 mmol) and stirred for 6 h at RT. Analysis by NMR spectroscopy indicated quantitative formation of the product. The volatiles were removed in vacuo, the resulting yellow oil washed with SiMe4 (2 × 0.5 mL) and dried in vacuo to afford the analytically product as a yellow solid. Yield: 11.6 mg (6.52 μmol, 76%). Crystals suitable X-ray crystallography were obtained by slow diffusion of excess SiMe4 into an Et2O solution. From the SiMe4 washings Z-tBuCCCHCHtBu was obtained as a colourless oil in low yield. Yield: 4.6 mg (28.0 μmol, 2%).
 CHtBu), 7.70–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.52 (d, 3JHH = 7.9, 1H, py), 7.49 (d, 3JHH = 7.9, 1H, py), 5.68 (t, 4JPC = 2, 1H, IrCCtBu), 4.78 (dt, 3JHH = 15.2, 4JPH = 2, 1H, IrCHCtBu), 4.28 (ddd, 2JHH = 16.5, 2JPH = 10.3, 4JPH = 3.0, 1H, pyC2), 3.69 (ddd, 2JHH = 17.4, 2JPH = 10.1, 4JPH = 1.3, 1H, pyC2), 3.64 (dd, 2JHH = 17.4, 2JPH = 9.9, 1H, pyC2), 3.22 (dd, 2JHH = 16.5, 2JPH = 7.9, 1H, pyC2), 2.28–2.43 (m, 2H, CH2), 2.08–2.19 (m, 1H, CH2), 0.88–1.96 (m, 25H, CH2), 1.24 (s, 9H, IrCCH
CHtBu), 7.70–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.52 (d, 3JHH = 7.9, 1H, py), 7.49 (d, 3JHH = 7.9, 1H, py), 5.68 (t, 4JPC = 2, 1H, IrCCtBu), 4.78 (dt, 3JHH = 15.2, 4JPH = 2, 1H, IrCHCtBu), 4.28 (ddd, 2JHH = 16.5, 2JPH = 10.3, 4JPH = 3.0, 1H, pyC2), 3.69 (ddd, 2JHH = 17.4, 2JPH = 10.1, 4JPH = 1.3, 1H, pyC2), 3.64 (dd, 2JHH = 17.4, 2JPH = 9.9, 1H, pyC2), 3.22 (dd, 2JHH = 16.5, 2JPH = 7.9, 1H, pyC2), 2.28–2.43 (m, 2H, CH2), 2.08–2.19 (m, 1H, CH2), 0.88–1.96 (m, 25H, CH2), 1.24 (s, 9H, IrCCH![[t with combining low line]](https://www.rsc.org/images/entities/i_char_0074_0332.gif)
![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) ), 1.17 (s, 9H, CC),1.02 (d, 3JPH = 14, 18H, 2 × PtBu), 1.00 (s, 9H, IrCHCH).
), 1.17 (s, 9H, CC),1.02 (d, 3JPH = 14, 18H, 2 × PtBu), 1.00 (s, 9H, IrCHCH).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 164.2 (app t, JPC = 3, py), 163.3 (app t, JPC = 3, py), 162.3 (q, 1JCB = 50, ArF), 143.5 (app t, 3JPC = 4, IrCHHtBu), 142.2 (br, IrCHtBu), 139.4 (s, py), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.1 (q, 1JFC = 272, ArF), 121.7 (d, 3JPC = 8, py), 121.5 (d, 3JPC = 8, py), 118.0 (sept, 3JFC = 4, ArF), 116.9 (s, CtBu), 105.3 (dd, 2JPC = 8, 2JPC = 5, IrCHtBu), 95.7 (app t, 2JPC = 10, IrHCHtBu), 60.1 (s, CtBu), 46.0 (d, 1JPC = 25, pyH2), 40.8 (d, 1JPC = 30, pyH2), 37.3 (s, CHCHtBu{C}), 37.1 (s, CCHtBu{C}), 36.6 (dd, 1JPC = 21, 3JPC = 4, PtBu{C}), 34.6 (dd, 1JPC = 21, 3JPC = 6, PtBu{C}), 33.3 (d, 2JPC = 15, CH2), 32.2 (s, CH2), 31.8 (s, CCtBu{C}), 31.4 (s, CH2), 31.28 (s, tBu{CH3}), 31.26 (s, tBu{CH3}), 31.1 (s, CH2), 30.5 (s, CH2), 30.3 (s, CH2), 30.2 (s, CH2), 29.7 (s, CHCHtBu{CH3}), 28.8 (br, CH2), 28.20 (d, 2JPC = 4, CH2), 28.17 (s, CH2), 27.1 (d, 2JPC = 3, 2 × PtBu{CH3}), 26.6 (s, CH2), 24.9 (s, CH2), 21.9 (dd, 1JPC = 30, 3JPC = 2, PCH2), 19.5 (dd, 1JPC = 19, 3JPC = 3, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 25.5 (d, 2JPP = 364, 1P), 16.8 (d, 2JPP = 364, 1P).
31 P{ 1 H} NMR (121 MHz, DFB): δ 25.0 (d, 2JPP = 364, 1P), 16.3 (d, 2JPP = 364, 1P).
HR ESI-MS (positive ion, 4 kV): 916.5623 ([M]+, calcd 916.5628) m/z.
Anal. calcd for C79H95BF24IrNP2 (1779.57 g mol−1): C, 53.35; H, 5.38; N, 0.79. Found: C, 53.26; H, 5.09; N, 0.77.
4.11 NMR scale reaction of 1 with Z-tBuCCCHCHtBu
A solution of 1 (16.1, 9.8 μmol) in DFB (0.5 mL) within a J. Young valve NMR tube was treated with Z-tBuCCCHCHtBu (4.6 mg, 28.0 μmol) and then heated at 50 °C for 1 h. No reaction was apparent upon analysis by NMR spectroscopy.
4.12 Crystallography
Data were collected on a Rigaku Oxford Diffraction SuperNova AtlasS2 CCD diffractometer using graphite monochromated Mo Kα (λ = 0.71073 Å) or CuKα (λ = 1.54184 Å) radiation and an Oxford Cryosystems N-HeliX low temperature device [150(2) K]. Data were collected and reduced using CrysAlisPro and refined using SHELXL,39 through the Olex2 interface.40 Full details about the collection, solution, and refinement are documented in CIF format, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 2051203 (1), 2051204 (2), 2051205 (7), 2051206 (10), 2051207 ([Rh(PNP-14)(η2-norbornene)][BArF4]).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A.B.C.) for financial support. High-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 637313).References
- (a) Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, 2018, vol. 1 Search PubMed; (b) R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC; (c) The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, ed. G. van Koten and R. A. Gossage, Topics in Organometallic Chemistry, Springer, 2016, vol. 45 Search PubMed; (d) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (e) Pincer and Pincer–Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. F. Wendt, Wiley-VCH, 2014 Search PubMed; (f) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Topics in Organometallic Chemistry, Springer, 2013, vol. 40 Search PubMed; (g) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed; (h) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- (a) M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed; (b) W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed.
- (a) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed; (b) W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS PubMed; (c) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (d) A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS PubMed; (e) M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, Chem. Commun., 1996, 2083–2084 RSC.
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (c) A. A. Bengali, R. H. Schultz, C. B. Moore and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 9585–9589 CrossRef CAS; (d) J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3723–3725 CrossRef CAS; (e) A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352–354 CrossRef CAS.
- T. M. Hood, B. Leforestier, M. R. Gyton and A. B. Chaplin, Inorg. Chem., 2019, 58, 7593–7601 CrossRef CAS PubMed.
- A. J. Holmes, P. J. Rayner, M. J. Cowley, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2015, 44, 1077–1083 RSC.
- (a) M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4467 CrossRef CAS PubMed; (b) A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466–4469 CrossRef CAS.
- W. H. Bernskoetter, S. K. Hanson, S. K. Buzak, Z. Davis, P. S. White, R. Swartz, K. I. Goldberg and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603–8613 CrossRef CAS PubMed.
- D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 CrossRef CAS.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Organometallics, 2018, 37, 3963–3971 CrossRef CAS PubMed.
- (a) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Chem. – Eur. J., 2020, 26, 14715–17723 CrossRef CAS PubMed; (b) C. M. Storey, A. Kalpokas, M. R. Gyton, T. Krämer and A. B. Chaplin, Chem. Sci., 2020, 11, 2051–2057 RSC; (c) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed.
- (a) R. E. Andrew, C. M. Storey and A. B. Chaplin, Dalton Trans., 2016, 45, 8937–8944 RSC; (b) R. E. Andrew, D. W. Ferdani, C. A. Ohlin and A. B. Chaplin, Organometallics, 2015, 34, 913–917 CrossRef CAS; (c) R. E. Andrew and A. B. Chaplin, Inorg. Chem., 2015, 54, 312–322 CrossRef CAS PubMed; (d) R. E. Andrew and A. B. Chaplin, Dalton Trans., 2014, 43, 1413–1423 RSC.
- T. M. Hood, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2077–2086 RSC.
- T. M. Hood and A. B. Chaplin, Dalton Trans., 2020, 49, 16649–16652 RSC.
- (a) B. Leforestier, M. R. Gyton and A. B. Chaplin, Angew. Chem., Int. Ed., 2020, 59, 23500–23504 CrossRef CAS PubMed; (b) B. Leforestier, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2087–2101 RSC.
- G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258–10261 CrossRef CAS PubMed.
- Z. Lu, C.-H. Jun, S. R. de Gala, M. P. Sigalas, O. Eisenstein and R. H. Crabtree, Organometallics, 1995, 14, 1168–1175 CrossRef CAS.
- For structurally related examples see: (a) P. Hermosilla, P. López, P. Garcia-Orduna, F. J. Lahoz, V. Polo and M. A. Casado, Organometallics, 2018, 37, 2618–2629 CrossRef CAS; (b) N. Grüger, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2013, 2013, 5358–5365 CrossRef; (c) P. Sánchez, M. Hernández-Juárez, E. Álvarez, M. Paneque, N. Rendón and A. Suárez, Dalton Trans., 2016, 45, 16997–17009 RSC; (d) J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 697–711 CrossRef CAS; (e) M. Yamashita, Y. Moroe, T. Yano and K. Nozaki, Inorg. Chim. Acta, 2011, 369, 15–18 CrossRef CAS; (f) R. Sablong and J. A. Osborn, Tetrahedron Lett., 1996, 37, 4937–4940 CrossRef CAS.
- Generation of [{Rh(PNP-14)}2(μ2–η2:η2-COD)]2+ under equilibrium is also implied in this case, as this dication was the species ultimately obtained upon crystallisation (see ref. 15). Mononuclear formulation in solution is, however, substantiated by synthesis of [Rh(PNP-14)(η2-norbornene)][BArF4] (δ31P 71.2, 55.9; 2JPP = 244 Hz). Details, including solid-state structure, are provided in the ESI.†.
- P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, 2012, pp. 258–264 Search PubMed.
- (a) A. A. Danopoulos, D. Pugh and J. A. Wright, Angew. Chem., Int. Ed., 2008, 47, 9765–9767 CrossRef PubMed; (b) P. Paredes, J. Díez and M. P. Gamasa, Organometallics, 2008, 27, 2597–2607 CrossRef CAS; (c) J. Díez, M. P. Gamasa, J. Gimeno and P. Paredes, Organometallics, 2005, 24, 1799–1802 CrossRef.
- Mono-ethylene complexes have instead been reported, see: ref. 8a and 25f.
- Reaction of 7 with dihydrogen (1 atm) in DFB regenerates 4 in quantitative spectroscopic yield, within 16 h at 80 °C.
- (a) S. Gu, R. J. Nielsen, K. H. Taylor, G. C. Fortman, J. Chen, D. A. Dickie, W. A. Goddard III and T. B. Gunnoe, Organometallics, 2020, 39, 1917–1933 CrossRef CAS; (b) M. Rimoldi, A. Nakamura, N. A. Vermeulen, J. J. Henkelis, A. K. Blackburn, J. T. Hupp, J. F. Stoddart and O. K. Farha, Chem. Sci., 2016, 7, 4980–4984 RSC; (c) S. Kundu, J. Choi, D. Y. Wang, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 5127–5143 CrossRef CAS PubMed; (d) K. S. Lokare, R. J. Nielsen, M. Yousufuddin, W. A. Goddard III and R. A. Periana, Dalton Trans., 2011, 40, 9094 RSC; (e) A. V. Polukeev, S. A. Kuklin, P. V. Petrovskii, A. S. Peregudov, F. M. Dolgushin, M. G. Ezernitskaya and A. A. Koridze, Russ. Chem. Bull., 2010, 59, 745–749 CrossRef CAS; (f) T. Yano, Y. Moroe, M. Yamashita and K. Nozaki, Chem. Lett., 2008, 37, 1300–1301 CrossRef CAS; (g) F. Novak, B. Speiser, H. A. Y. Mohammad and H. A. Mayer, Electrochim. Acta, 2004, 49, 3841–3853 CrossRef CAS; (h) H. A. Y. Mohammad, J. C. Grimm, K. Eichele, H.-G. Mack, B. Speiser, F. Novak, M. G. Quintanilla, W. C. Kaska and H. A. Mayer, Organometallics, 2002, 21, 5775–5784 CrossRef CAS; (i) S. Nemeh, C. Jensen, E. Binamira-Soriaga and W. C. Kaska, Organometallics, 1983, 2, 1442–1447 CrossRef CAS.
- S. Khorasani, M. A. Fernandes and C. B. Perry, Cryst. Growth Des., 2012, 12, 5908–5916 CrossRef CAS.
- G. L. Parker, S. Lau, B. Leforestier and A. B. Chaplin, Eur. J. Inorg. Chem., 2019, 2019, 3791–3798 CrossRef CAS.
- (a) L. Maser, C. Schneider, L. Vondung, L. Alig and R. Langer, J. Am. Chem. Soc., 2019, 141, 7596–7604 CrossRef CAS; (b) J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS PubMed.
- S. M. Chapp and N. D. Schley, Inorg. Chem., 2020, 59, 7143–7149 CrossRef CAS PubMed.
- R. Ghosh, X. Zhang, P. Achord, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2007, 129, 853–866 CrossRef CAS PubMed.
- For examples of iridium catalysed terminal alkyne coupling reactions see: (a) M. Ez-Zoubir, F. Le Boucher d'Herouville, J. A. Brown, V. Ratovelomanana-Vidal and V. Michelet, Chem. Commun., 2010, 46, 6332–6334 RSC; (b) K. Ogata and A. Toyota, J. Organomet. Chem., 2007, 692, 4139–4146 CrossRef CAS; (c) T. Ohmura, S.-I. Yorozuya, Y. Yamamoto and N. Miyaura, Organometallics, 2000, 19, 365–367 CrossRef CAS; (d) C.-H. Jun, Z. Lu and R. H. Crabtree, Tetrahedron Lett., 1992, 33, 7119–7120 CrossRef CAS.
- J. Alós, T. Bolaño, M. A. Esteruelas, M. Oliván, E. Oñate and M. Valencia, Inorg. Chem., 2013, 52, 6199–6213 CrossRef PubMed.
- The bis(ethylene) complex 6 is also an effective precatalyst for this reaction (complete conversion within 6 h using 1 mol% loading). Full details are provided in the ESI.†.
- For a crystallographically characterised iridium η1-enynyl complexes see: (a) J. S. Merola, T. L. Husebo and K. E. Matthews, Organometallics, 2012, 31, 3920–3929 CrossRef CAS; (b) K. Ilg and H. Werner, Chem. – Eur. J., 2002, 8, 2812–2820 CrossRef CAS; (c) T. X. Le and J. S. Merola, Organometallics, 1993, 12, 3798–3799 CrossRef CAS.
- For reviews see: (a) O. N. Temkin, Kinet. Catal., 2020, 60, 689–732 CrossRef; (b) Q. Liang, K. Hayashi and D. Song, ACS Catal., 2020, 10, 4895–4905 CrossRef CAS; (c) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC.
- For recent or notable examples see: (a) N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 7973–7982 CrossRef CAS; (b) O. Rivada-Wheelaghan, S. Chakraborty, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 6942–6945 CrossRef CAS PubMed; (c) J. Alós, T. Bolaño, M. A. Esteruelas, M. Oliván, E. Oñate and M. Valencia, Inorg. Chem., 2014, 53, 1195–1209 CrossRef PubMed; (d) H. Katayama, H. Yari, M. Tanaka and F. Ozawa, Chem. Commun., 2005, 4336–4338 RSC; (e) C. Bianchini, M. Peruzzini, F. Zanobini, P. Frediani and A. Albinati, J. Am. Chem. Soc., 1991, 113, 5453–5454 CrossRef CAS ; see also ref. 32.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- (a) A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC; (b) W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalytic homocoupling of 3,3-dimethylbutyne promoted by 6, synthesis and characterisation of [Rh(PNP-14)(η2-norbornene)][BArF4]; NMR, IR and ESI-MS spectra of new compounds, and selected reactions (PDF). Primary NMR data (MNOVA). CCDC 2051203–2051207. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt04303f |
| This journal is © The Royal Society of Chemistry 2021 |