
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chromatography-free total synthesis of a ferrocene-containing dendrimer exhibiting the property of recognizing 9,10-diphenylanthracene†
Artur
Kasprzak
 *a,
Monika K.
Nisiewicz
ab and
Anna M.
Nowicka
b
*a,
Monika K.
Nisiewicz
ab and
Anna M.
Nowicka
b
aDepartment Faculty of Chemistry, Warsaw University of Technology, Noakowskiego Str. 3, 00-664 Warsaw, Poland. E-mail: akasprzak@ch.pw.edu.pl
bFaculty of Chemistry, University of Warsaw, Pasteura Str. 1, 02-093 Warsaw, Poland
First published on 8th January 2021
Abstract
Molecules comprising several ferrocene residues constitute an intriguing group of compounds for various applications. Here, the total synthesis of a new example of a ferrocene-containing dendrimer is presented. The target compound was obtained in excellent combined yield (65%) employing facile, chromatography-free methods at each step. Interesting findings, meeting the dynamic covalent chemistry concept, are reported. Cyclic voltammetry analyses revealed one pair of current signals for the ferrocene moieties. Ultimately, the synthesized ferrocene-containing dendrimer has been used as an innovative recognition material for 9,10-diphenylanthracene, a polycyclic aromatic hydrocarbon, with the limit of detection value equal to 0.06 μM.
Introduction
Over the years, ferrocene (Fc) has been a coordination compound of wide interest in organic and materials chemistry.1,2 This metallocene has various prospective applications in a number of important fields, such as medicinal chemistry,3 self-healing materials,4 catalysis5, or ion receptors.6 Even though the discovery of Fc was accomplished 80 years ago, in recent years its chemistry has developed remarkably, and Fc-containing molecules have become important synthetic targets.The synthesis of compounds bearing several Fc residues has been one of the major goals of modern chemistry, since such molecules exhibit intriguing chemical structures, as well as unprecedented properties and applications. Some of the milestones achieved in recent years include, e.g. the reports by Albrecht & Long on the preparation of a Fc cyclic oligomer7 or Fc-bearing macrocycles.8 Special interest has been given to the synthesis of Fc-containing dendrimers. Dendrimers have advantages in comparison to their linear or branched macromolecular analogs, such as well-defined monodispersed structures, encouraging bio-physical features and the possibility of the control of the position and number of introduced substituents.1,9,10 These beneficial parameters of dendrimers are extremely important also from the viewpoint of their applied sciences, e.g., medicinal chemistry or organic electronics. The literature reports on Fc-containing dendrimers include the preparation of silicon-,11–13 tetraphenylmethane-,14a pyridylphenylene-,14b or poly(amidoamine)-based15,16a structures as well as metallodendrimers,16b hydrogen-bonded,16c or coordination-type16d–f assemblies. These dendrimers showed interesting features, such as water solubility,17 catalytic activity,13 ion recognition14b,16c or the property to form inclusion complexes with cyclodextrins.15 The chemistry of metallocene-tethered dendrimers has intensively developed over the years, and a number of these interesting molecules have been summarized in recent reviews.18a–e
Importantly, the dendrimer chemistry has been commonly merged with the π-conjugation concept, since π-conjugated aromatic compounds bearing a number of delocalized π-electrons were reported as interesting frameworks and ideal materials for various applications.18f–h,19a In general, the analysis of the literature examples of Fc-containing dendrimers brings a conclusion that the previous reports focused mostly on the installation of ferrocene units in the peripheral parts of the π-conjugated dendrimer cores. The reports dealing with aromatic dendrimers with Fc units located both in the peripheries and the core are sparse, as well as the synthesis of these molecules is challenging; in consequence there is limited number of reports on the successful synthesis of such organized compounds (see the graphical representation in Fig. 1). Even though the examples of such dendrimers can be found in the literature, one may conclude that the synthetic protocols are time-, cost- and solvent-consuming, such as in the case of the interesting phosphorous-containing ferrocene-tethered dendrimers.19b,c
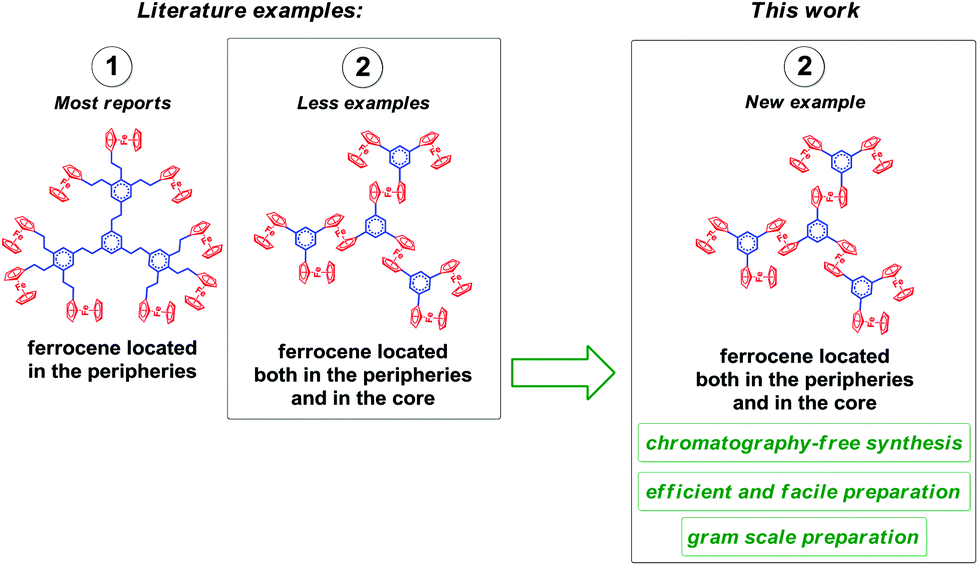 | ||
| Fig. 1 Graphical summary of the reported Fc-decorated dendrimers and the aim of this work. | ||
In pursuit of new dendrimers consisting of several Fc units and π-conjugated building blocks, herein a chromatography-free total synthesis of the novel Fc-containing dendrimer Fc-den is presented (Fig. 2). Our goal was to design a new, chromatography-free and efficient way for the synthesis of a Fc-tethered dendrimer of potential interest to various communities. The structure of Fc-den includes 1,3,5-triphenylbenzene cores and imine linkages. These structural motifs have been widely employed for the construction of organized π-conjugated molecules, including several examples of aromatic dendrimers with Fc units located in the peripheries of the dendrimer.5,20,21a,b We expect π-conjugation within a Fc-den framework because of enhancing the number of delocalized p-electrons of the 1,3,5-triphenylbenzene framework (bearing multiple bonds) with imine linkages (bearing C(sp2) and N(sp2) atoms) and cyclopentadienyl rings bound on the opposite sides of the iron atom (Fc units of Fc-den).
 | ||
| Fig. 2 Structure of the target Fc-containing dendrimer (Fc-den) and its retrosynthetic analysis. | ||
The herein presented synthesis of Fc-den bearing nine Fc residues lies in a convergent synthesis strategy, in which the key to success was the preparation of two π-conjugated building blocks 1 and 2 (Fig. 2). In general, we hypothesized that sequential imine-bond formation reactions shall provide the target dendrimer and intermediate compounds. Importantly, the success of this strategy would be also beneficial in terms of the green synthesis of Fc-den, since the previous studies on imine-containing triphenylbenzenes5,21,22 suggested that no chromatographic purification shall be required.
Experimental section
Materials and methods
Chemical reagents and solvents were commercially purchased and purified according to the standard methods, if necessary. NMR experiments were carried out using a Varian VNMRS 500 MHz spectrometer (1H NMR at 500 MHz or 13C NMR at 125 MHz) equipped with a multinuclear z-gradient inverse probe head. Unless otherwise stated, the spectra were recorded at 25 °C. Standard 5 mm NMR tubes were used. 1H and 13C chemical shifts (δ) were reported in parts per million (ppm) relative to the solvent signal: CDCl3, δH (residual CHCl3) 7.26 ppm, δC 77.2 ppm. NMR spectra were analyzed with the MestReNova v12.0 software (Mestrelab Research S.L). 1H DOSY (Diffusion Ordered SpectroscopY) NMR experiments were performed using a stimulated echo sequence incorporating bipolar gradient pulses and with convection compensation. The gradient strength was logarithmically incremented in 15 steps from 25% up to 95% of the maximum gradient strength. The DOSY Toolbox software was used for DOSY NMR spectral processing (The DOSY Toolbox – version 2.5, 2014, Mathias Nilsson, School of Chemistry, University of Manchester, UK).Fourier-transform infrared (FT-IR) spectra were recorded in the attenuated total reflectance (ATR) mode with a Thermo Nicolet Avatar 370 spectrometer with a spectral resolution of 2 cm−1 (100 scans). The wavenumbers for the absorption bands ν were reported in cm−1.
UV-vis measurements were performed with the PerkinElmer spectrometer Lambda 25, at room temperature in a quartz cuvette with a 1 cm long optical window. For the UV-Vis measurements, the wavelengths for the absorption maxima λmax were reported in nm.
TOF-HRMS (ESI) measurements were performed with a Q-Exactive ThermoScientific spectrometer. Melting points were determined on Standford Research Systems MPA 100 and were uncorrected.
Elemental analyses were performed using CHNS Elementar Vario EL III apparatus. Each elemental composition was reported as an average of two analyses.
Electrochemical measurements were performed using an Autolab Eco Chemie potentiostat, model PGSTAT 12 controlled via software on a personal computer. All experiments were carried out in a three-electrode system. A disc glassy carbon electrode (GC, ∅ = 3 mm, BAS Instruments) was used as the working electrode, an Ag/AgCl/3 M KCl as the reference electrode, and a platinum wire as the auxiliary electrode. Before each experiment, the GC electrode was polished with 1 μm aluminium oxide powder on a wet pad, rinsed with water and then dried with argon. During all measurements, the electrochemical cell was kept in a Faraday cage to minimize the electrical noise. The measurements were carried out with deoxygenated solutions.
Synthesis of 1
To a stirred solution of 1,3,5-tris(4-aminophenyl)benzene (3) (140.6 mg, 0.40 mmol, 1.0 equiv.) and 9,10-diphenylantracene (528.6 mg, 1.60 mmol, 4.0 equiv.) in MeOH (15 mL), a solution of 1,1′-diformylferrocene (4) (871.4 mg, 3.60 mmol, 9.0 equiv.) in MeOH (13 mL) was added in one portion. Glacial acetic acid (250 μL) was added, and the mixture turned turbid within several minutes. The reaction mixture was stirred at room temperature for 24 hours. The solid was filtered off and washed with methanol (15 mL). The solid residue was suspended in methanol (80 mL), sonicated for 20 minutes at room temperature, filtered off, washed with methanol (10 mL) and dried at room temperature for 24 hours to give compound 1 (393.1 mg; 96% yield) as an orange-red solid.Mp: 262–263 °C; 1H NMR (CDCl3, 500 MHz, ppm), δH 9.95 (s, 3H), 8.13 (s, 3H), 7.41 (s, 3H), 7.24–7.22 (m, 6H), 6.82–6.81 (m, 6H), 4.96–4.88 (m, 12H), 4.58–4.52 (m, 12H); 13C{1H} NMR (CDCl3, 125 MHz, ppm), δC 193.0 (3C), 160.6 (3C), 141.3 (3C), 138.2 (3C), 127.5 (3C), 123.8 (3C), 121.4 (3C), 120.6 (6C), 84.7 (3C), 82.4 (3C), 72.6 (6C), 71.4 (6C), 70.5 (6C), 70.1 (6C); FT-IR (ATR), v 3082, 3014, 2868, 2830, 1682, 1614, 1588, 1508, 1364, 1242, 1178, 1038, 820, 744 cm−1; UV-Vis, λmax (CHCl3) 264, 333 nm; TOF-HRMS (ESI): calcd for C60H46Fe3N3O3 [M + H]+ = 1024.1582, found: m/z 1024.1561; Anal. calcd for C60H45Fe3N3O3: C, 70.41; H, 4.43; N, 5.65. Found: C, 70.50; H, 4.44; N, 4.10.
Synthesis of 2
A solution of 3 (1054.2 mg, 3.00 mmol, 1.0 equiv.) and ferrocenecarboxaldehyde (5) (1605.3 mg, 7.50 mmol, 2.5 equiv.) in MeOH (300 mL) was stirred for 5 minutes. Glacial acetic acid (1800 μL) was added, and the mixture turned turbid within several minutes. The reaction mixture was stirred at room temperature for 24 hours. The solid was filtered off and washed with methanol (15 mL). The solid residue was suspended in methanol (200 mL), sonicated for 20 minutes at room temperature, filtered off, washed with methanol (10 mL) and dried at room temperature for 24 hours to give compound 2 (2052.0 mg; 92% yield) as an orange solid.Mp: 225–226 °C; 1H NMR (CDCl3, 500 MHz, ppm), δH 8.44 (s, 2H), 7.82 (s, 3H), 7.75–7.71 (m, 6H), 7.31–7.28 (m, 4H), 6.82–6.79 (m, 2H), 4.86 (bs, 2H), 4.80–4.79 (m, 4H), 4.61–4.60 (m, 4H), 4.28 (s, 10H); 13C{1H} NMR (CDCl3, 125 MHz, ppm), δC 161.5 (2C), 146.0 (2C), 142.3 (1C), 142.1 (1C), 132.1 (2C), 128.4 (4C), 128.2 (2C), 123.4 (2C), 123.1 (2C), 121.2 (2C), 115.6 (4C), 115.5 (2C), 73.4 (2C), 71.6 (4C), 69.8 (10C), 69.5 (4C); FT-IR (ATR), v 3080, 3026, 2890, 1634, 1579, 1496, 1160, 1096, 954, 800, 740 cm−1; UV-Vis, λmax (CHCl3) 269, 340 nm; TOF-HRMS (ESI): calcd for C46H38Fe2N3 [M + H]+ = 744.1759, found: m/z 744.1760; Anal. calcd for C46H37Fe2N3: C, 74.31; H, 5.02; N, 5.65. Found: C, 74.62; H, 5.00; N, 5.67.
Synthesis of Fc-den
A solution of 1 (460.6 mg, 0.45 mmol, 1.0 equiv.) and 2 (1107.9 mg, 1.49 mmol, 3.3 equiv.) in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 1:1 v/v (80 mL) was stirred for 5 minutes. Glacial acetic acid (300 μL) was added, and the mixture turned turbid within several minutes. The reaction mixture was stirred at room temperature for 48 hours. The solid was filtered off and washed with THF:MeOH 1:1 v/v (10 mL). The solid residue was suspended in THF:MeOH 1:1 v/v (100 mL), sonicated for 20 minutes at room temperature, filtered off, washed with THF:MeOH 1:1 v/v (10 mL) and dried at room temperature for 24 hours to give Fc-den (1281.6 mg; 89% yield) as a red solid.
:MeOH 1:1 v/v (80 mL) was stirred for 5 minutes. Glacial acetic acid (300 μL) was added, and the mixture turned turbid within several minutes. The reaction mixture was stirred at room temperature for 48 hours. The solid was filtered off and washed with THF:MeOH 1:1 v/v (10 mL). The solid residue was suspended in THF:MeOH 1:1 v/v (100 mL), sonicated for 20 minutes at room temperature, filtered off, washed with THF:MeOH 1:1 v/v (10 mL) and dried at room temperature for 24 hours to give Fc-den (1281.6 mg; 89% yield) as a red solid.
Mp: >300 °C; 1H NMR (CDCl3, 500 MHz, ppm), δH 8.45 (s, 6H), 8.14 (s, 6H), 7.80 (s, 9H), 7.75–7.71 (m, 18H), 7.41 (s, 3H), 7.31–7.29 (m, 18H), 7.24–7.21 (m, 6H), 6.82–6.79 (m, 6H), 5.10–5.09 (m, 12H), 4.85–4.84 (m, 12H), 4.53–4.52 (m, 24H), 4.28 (s, 30H); 13C{1H} NMR (CDCl3, 125 MHz, ppm), δC 161.6 (6C), 161.5 (6C), 152.4 (3C), 151.2 (3C), 146.2 (3C), 146.1 (3C), 142.1 (3C), 141.3 (3C), 138.5 (6C), 138.3 (6C), 138.2 (6C), 131.9 (3C), 128.3 (6C), 128.4 (6C), 128.2 (6C), 128.1 (6C), 124.6 (3C), 124.2 (3C), 123.9 (3C), 123.7 (3C), 121.4 (6C), 121.3 (3C), 115.6 (6C), 115.5 (6C), 82.4 (6C), 80.6 (6C), 73.3 (6C), 71.6 (6C), 71.5 (12C), 70.0 (6C), 69.8 (6C), 69.5 (30C), 69.3 (12C); 1H DOSY NMR (500 MHz, CDCl3, 0.7 mM), D 0.901 × 10−10; FT-IR (ATR), v 3086, 3024, 2884, 1620, 1584, 1490, 1456, 1168, 1102, 954, 818, 746 cm−1; UV-Vis, λmax (CHCl3) 284, 295, 363, 492 nm; TOF-HRMS (ESI): calcd for C198H151Fe9N12 [M + H]+ = 3200.6318, found: m/z 3200.6330; Anal. calcd for C198H150Fe9N12: C, 74.32; H, 4.72; N, 5.25. Found: C, 74.40; H, 4.74; N, 5.26.
Results and discussion
The synthetic studies were initiated by synthesizing 1,3,5-(4-aminophenyl)triphenylbenzene (3; Fig. 3a) in two steps following a literature procedure.5 With 3 in hand, the preparations of 1 and 2, the key building blocks of Fc-den, were investigated.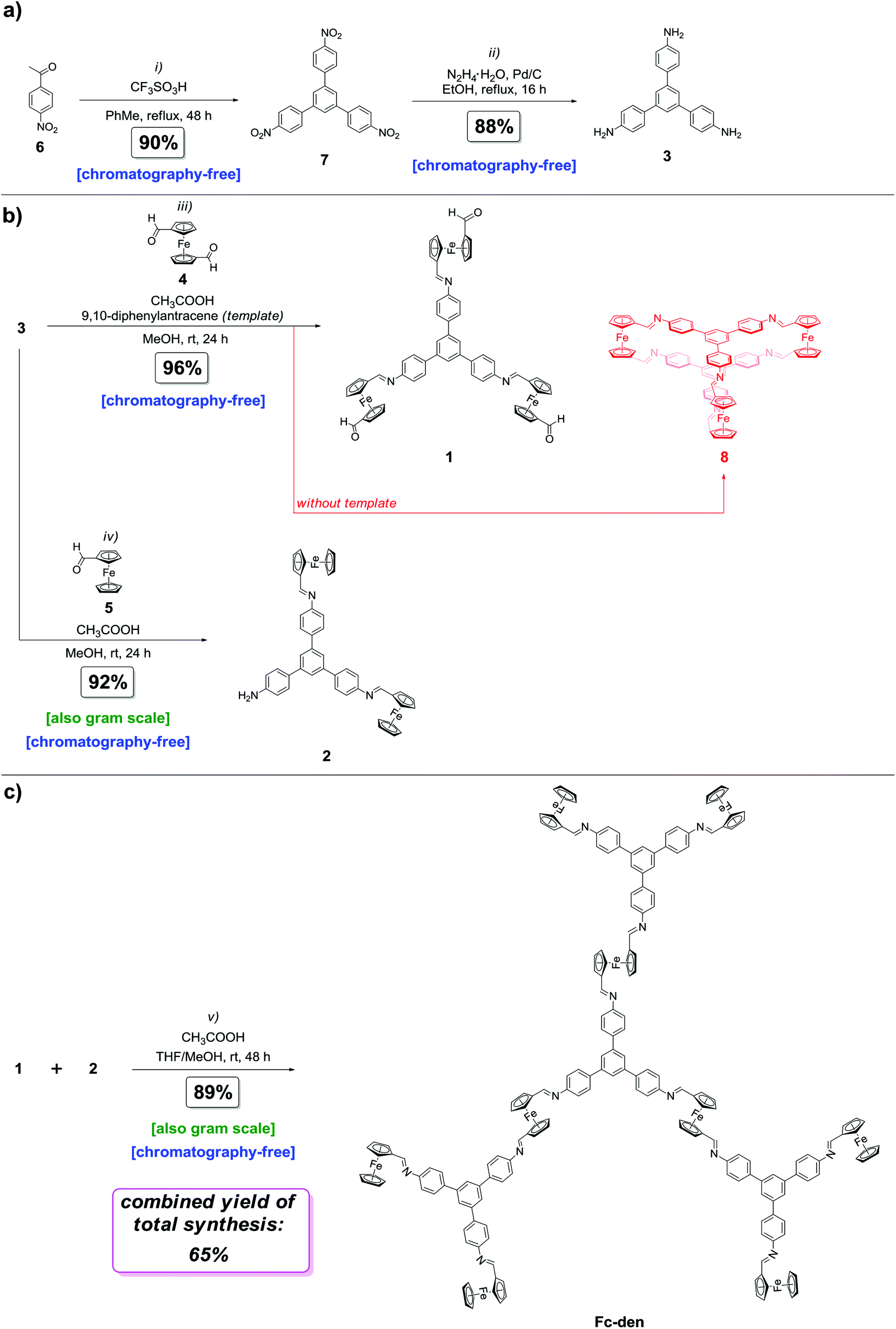 | ||
| Fig. 3 Total synthesis of Fc-den: (a) synthesis of the starting material 3; (b) synthesis of the key building blocks 1 and 2; (c) assembly of 1 and 2 to obtain Fc-den. Reagents and conditions: (i) 6 (1.0 equiv.), CF3SO3H (cat.), PhMe, reflux, 48 h; (ii) 7 (1.0 equiv.), N2H4 monohydrate (12.0 equiv.), Pd/C, EtOH, reflux, 16 h; (iii) 3 (1.0 equiv.), 4 (9.0 equiv.), 9,10-diphenylanthracene (4.0 equiv.), CH3COOH (cat.), MeOH, rt, 24 h; (iv) 3 (1.0 equiv.), 5 (2.5 equiv.), CH3COOH (cat.), MeOH, rt, 24 h; (v) 1 (1.0 equiv.), 2 (3.3 equiv.), CH3COOH (cat.), THF:MeOH 1:1 v/v, rt, 48 h. | ||
It was found that the major obstacle for the successful synthesis of the aldehyde-terminated triferrocenyl derivative 1 was not only the possible formation of some oligomers but also a cage compound 8 (Fig. 3b). In other words, one shall control the supramolecular arrangement formed, especially the rotation between Fc's cyclopentadienyl rings of 4 during the reaction course, in order to selectively obtain 1 instead of 8. Although the developed method was facile and chromatography-free (the solid product was filtered and washed off with methanol), numerous attempts had to be made to selectively obtain target compound 1 in good yield (Table 1a). In general, better results were found for higher molar excesses of 1,1′-diformylferrocene (4) and its higher concentrations; however, the yields did not exceed 32% (Table 1a, entries 1–4). A breakthrough within this extensive screening of reaction parameters occurred when the reaction was conducted with an excess of 4, in its high concentration and in the presence of 9,10-diphenylanthracene as a template (Table 1a, entries 5–7). The basics of template's action were as follows.23a,b Firstly, from a previous report5 it is known that the confined space of cage 8 is too small to incorporate an aromatic molecule (guest), such as 9,10-diphenylanthracene, between stacked 1,3,5-triphenylbenzene moieties. Secondly, the π–π stacking interaction, a dynamic process, between 9,10-diphenylanthracene and 3 and 1 were expected during the reaction course. For the effective formation of 8, the CHO moieties of 4 must be in the syn conformation; for the effective synthesis of 8, there should be no steric hindrance toward the formation of the stacked cage-type product 8. In other words, the anti-conformation of the substituents in the cyclopendatienyl rings of 4 limits the efficient formation of 8. Thus, we supposed that a molecule (9,10-diphenylanthracene) that interacts via dynamic π–π stacking non-covalent interactions with 1 or 3 (or their intermediate products with 4) might therefore increase this steric hindrance, which results in the inefficient formation of 8 because of facilitation of the discussed anti-conformation of CHO functionalities. In the light of these facts, this stacking phenomenon with 9,10-diphenylanthracene prevented the formation of cage 8 and enabled the selective synthesis of 1. Importantly, 9,10-diphenylanthracene, and unreacted 3 and 4 can be easily removed from the reaction mixture by washing with methanol to yield pure 1 in an excellent yield of 96% (Table 1a, entry 7). Elemental analysis confirmed the synthesis of pure 1. Finally, to confirm that 1 can interact with 9,10-diphenylanthracene (template) by means of non-covalent forces, at first 1H DOSY NMR analysis was performed (see the ESI† for the data). 1H DOSY NMR is a powerful method to track non-covalent interactions, such as host–guest complexation or π–π stacking.24a–c The non-covalent phenomena occurring between two molecules in the sample lead to a change in their diffusion coefficients in 1H DOSY NMR. The diffusion coefficient for 9,10-diphenylanthracene in the presence of 1 (D = 7.112 × 10−10 m2 s−1) was found to be lower in comparison to the sample of native 9,10-diphenylanthracene in the absence of 1 (D = 9.621 × 10−10 m2 s−1; see the ESI† for the data). This change was ascribed to the non-covalent π–π stacking interactions between 1 and 9,10-diphenylanthracene. Although the apparent binding constant (Kapp) was found to be ca. 1.3 × 102 M−1 (because this interaction is a dynamic process) the 9,10-diphenylanthracene:1 system has been clearly detected by cold-spray ESI-MS analysis (see the ESI† for data). This further confirms that 9,10-diphenylanthracene can interact with 1via non-covalent π–π stacking interactions and supports the role of 9,10-diphenylanthracene as a template in the synthesis of 1.
| Entry | 1 (equiv.) | 2 (equiv.) | Solvent | Time (hours) | Yield of Fc-denb |
|---|---|---|---|---|---|
| a CH3COOH (cat.), MeOH, rt, 24 h. b Isolated yields. c Based on 1H NMR. d Reaction scale: 0.02 mmol of 3. e Reaction scale: 0.40 mmol of 3. f CH3COOH (cat.), MeOH. g Gram scale reaction (3.00 mmol of 3). h CH3COOH (cat.), rt. i Reaction scale: 0.015 mmol of 1. j Impurities of 1 and/or 2 were found in the product (based on 1H NMR). k Gram scale reaction (1.49 mmol of 2). | |||||
| (c) | |||||
| 1 | 1.0 | 3.3 | THF | 48 | 18i,j |
| 2 | 1.0 | 3.3 | THF:MeOH 4:1 v/v |
48 | 30i,j |
| 3 | 1.0 | 3.3 | THF:MeOH 2:1 v/v |
48 | 71i,j |
| 4 | 1.0 | 3.3 | THF:MeOH 1:1 v/v |
48 | 89i |
| 5 | 1.0 | 3.0 | THF:MeOH 1:1 v/v |
48 | 85i |
| 6 | 1.0 | 3.3 | THF:MeOH 1:1 v/v |
72 | 84i |
| 7 | 1.0 | 3.3 | THF:MeOH 1:1 v/v |
48 | 89k |
With this milestone achieved, the synthesis of 2, a diferrocenyl derivative of 3, was investigated (Fig. 3b). The method was based on an acid-catalyzed imine-bond formation reaction between 3 and ferrocenecarboxaldehyde (5). As expected, synthesizing 2 without traces of monoferrocenyl or triferrocenyl triphenylbenzenes has been the biggest challenge. Various reaction parameters were tested (Table 1b). Gratifyingly, the selective and efficient formation of 2 has been achieved by tuning the molar equivalent of 5 and its concentration. Similarly to the synthesis of 1, pure 2 has been obtained in very high yield (90–92%; Table 1b, entries 6 and 7) employing an easy-to-perform, chromatography-free method (the solid product was filtered and washed off with methanol). Ultimately, 2 can also be prepared on a gram scale (Table 1b, entry 7). Elemental analysis confirmed the synthesis of pure 2.
All that remained to construct Fc-den was merging 1 and 2. In the light of the successful, facile syntheses of 1 and 2, the ultimate goal was to design a facile, chromatography-free methodology to obtain Fc-den. This would make the developed total synthesis chromatography-free in all stages. However, the major issue in this synthetic step was the similar solubility profile for Fc-den to those for 1 and 2, i.e. similarly to 1 and 2, Fc-den is only soluble in chloroalkanes (CH2Cl2, CHCl3) and THF. Seeking a possible reaction parameter, the reaction in the THF/MeOH system was exposed (Fig. 3c; Table 1c). This trial originated from the prediction that 1 and 2 shall remain dissolved when their THF solution is diluted with MeOH, whilst Fc-den shall precipitate from such a reaction mixture. Through control experiments (Table 1c, entries 1–6), it was indeed found that on the contrary to 1 and 2, Fc-den is not soluble in the THF:MeOH solvent system. Through the application of these conditions, Fc-den has been successfully prepared in a chromatography-free way in an excellent yield of 89% under mild conditions (Table 1c, entry 4). Importantly, the large-scale access to 2 provided the possibility of the chromatography-free gram-scale preparation of Fc-den (89%; Table 1c, entry 7). The presence of one set of signals in 1H NMR (12 groups of signals in total; see Fig. S5†) reveals that, in solution, all building subunits of Fc-den, i.e. monosubstituted ferrocenes, 1,1′-disubstituted ferrocenes, terminal 1,3,5-triphenylbenzene cores, and imine linkages (two sets), are equivalent on the NMR timescale. The selective formation of one system has been further confirmed by 1H DOSY NMR analysis (see Fig. S7†). Finally, elemental analysis confirmed that pure Fc-den has been synthesized. It is worth noting that 1H NMR analyses revealed that Fc-den is stable in air (the solid sample was kept for three months in air and the 1H NMR spectrum was acquired, see Fig. S19†), as well as in water (see Fig. S20†).
The UV-Vis spectra of 1, 2 and Fc-den in CHCl3 are presented in Fig. 4. Fc-den exhibited incomparably higher absorbance values in comparison to the starting reactants 1 and 2 (see Fig. S15†). The spectra featured two major absorption bands at 262–294 nm and 330–362 nm. The first band was ascribed to the π–π* transition of 1,3,5-triphenylbenzene cores, whilst the latter originated from the presence of Fc and imine moieties. The significant red shifts for these bands for Fc-den (294 nm, 362 nm) in comparison to the starting materials 1 (262 nm, 330 nm) and 2 (266 nm, 338 nm) were ascribed to the π-conjugation within the dendrimer framework. A significantly smaller peak at 488 nm for Fc-den (observed only for this compound) could be attributed to the dynamic E–Z isomerization behavior within imine-type linkages25 or some metal-to-triphenylbenzene charge transfers.26
 | ||
| Fig. 4 UV-Vis spectra (CHCl3) of 1, 2 and Fc-den. | ||
Fc and its derivatives easily undergo one electron oxidation to form a ferrocenium cation in a reversible manner.27–29a Thus, we investigated the electrochemical behavior of Fc-den in dichloromethane (DCM) by cyclic voltammetry (CV) using a conventional glassy carbon electrode. The cyclic voltammogram of 1 mM Fc-den recorded in DCM with 100 mM tetrabutylammonium hexafluorophosphate (TBAHFP) as a supporting electrolyte for 100 mV s−1 scan rate is presented in the bottom inset of Fig. 5. The recorded voltammogram shows only one pair of current signals. This behavior suggests that the redox (Fc) units of Fc-den are electrochemically equivalent and are oxidized and reduced at the same potential. The number of current signals observed in cyclic voltammograms depends not only on the number of Fc units, but also on the distance between them. The closer the Fc units are to the central benzene core, and therefore closer to each other, the more significant is the electrostatic factor (interaction of the ferrocene cation with the ferrocene cation of the other molecule); therefore multiple CV waves were observed.29b–e Taking into account the structure of the Fc-den molecule presented in Fig. 3 the distance between Fc–Fc units as well as the Fc–central benzene core is large enough for the electrostatic interactions to not affect the shape and number of current signals. Moreover, the separation of CV signals in the case of compounds with few identical redox centers also depends on the type of electrolyte anions.29b–e The anions strongly bonded to ferricinium cations such as PF6− or BF4− reduce the ability of CV current signal separation, as well as slow down the electron exchange process. Fc-den contains two types of Fc units in the structure (inner and peripheral one). The presence of two types of redox centres should be visible on the CV curve in the form of two pairs of currents signals, but it only occurs when the Fc–Fc distance is very short. Probably in the case of Fc-den, only one type of Fc unit undergoes an electron exchange reaction with the electrode surface. This hypothesis was confirmed by the comparison of the charge values under cathodic or anodic peaks obtained for Fc-den and pure Fc. This ratio (QFc-den:QFc) is 2:1. Moreover, the difference in the position of anodic and cathodic peaks (ΔEp) is 350 mV; the value was determined for scan rates ≤100 mV s−1, which suggests relatively slow electron transfer and solution resistance. It should be stressed that the value of ΔEp for Fc under the same conditions is equal to 160 mV; in the case of aqueous solution, the ΔEp for Fc and its derivatives in aqueous solutions is roughly equal to 88 mV. This significant difference in the separation of the anode and cathode current signals in relation to aqueous solutions is most likely related to the solution resistance.
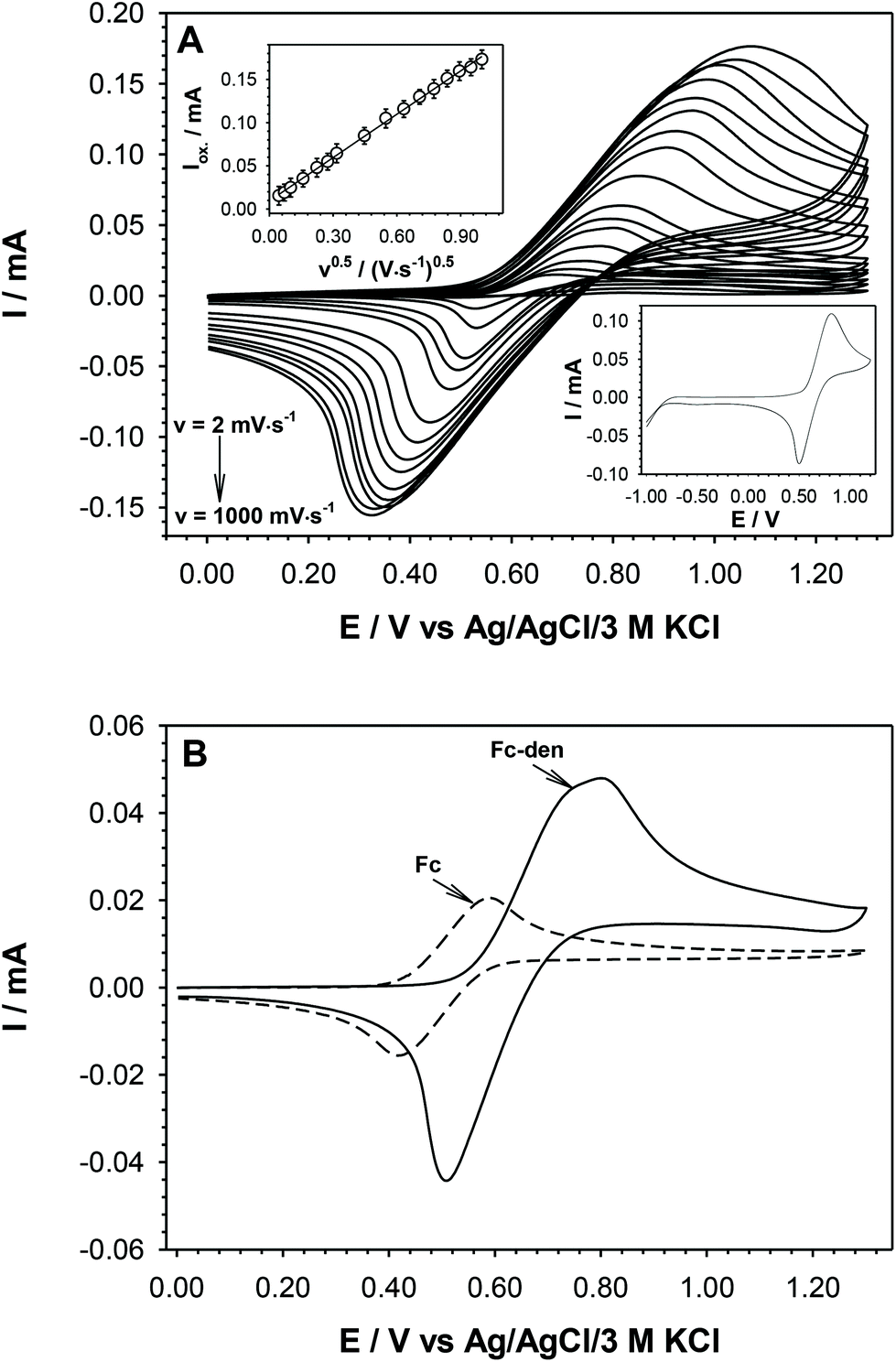 | ||
| Fig. 5 (A) Cyclic voltammograms of Fc-den recorded in DCM at various scan rates. Top inset: Plot of Fc-den oxidation currents versus the square root of the scan rate. Bottom inset: the CV voltammogram in a wider potential range (v = 100 mV s−1). (B) Cyclic voltammograms of Fc-den and Fc recorded in DCM at 50 mV s−1. Experimental conditions: CFc-den = 1.0 mM, CTBAHFP = 100 mM, T = 21 °C. | ||
In order to determine the value of the diffusion coefficient of Fc-den, CVs were recorded in an 1 mM solution of Fc-den at different scan rates in the range of 2–1000 mV s−1. The linear dependence of the current signal intensity versus square root of the scan rate (see the top inset in Fig. 5) confirmed the diffusional character of the electrochemical reaction. Therefore, from the slope of the plot Ipa = f(v0.5), the diffusion coefficient for the studied ferrocene derivative according to the Randles–Sevcik equation was determined and was equal to 1.31 × 10−9 cm2·s−1. In turn, from the intercept of the dependence ln(Ipa) = f(Epa − Ef), the electron-transfer rate constant (k0 = 3.01 × 10−4 cm s−1) was determined according to the formula:

 is the concentration of the electroactive species, Epa is the potential of the anodic peak, Ef is the formal potential, R is the gas constant, T is temperature, and α is the transition coefficient.
is the concentration of the electroactive species, Epa is the potential of the anodic peak, Ef is the formal potential, R is the gas constant, T is temperature, and α is the transition coefficient.
The Fc units were attached to the 1,3,5-triphenylbenzene skeletons via imine bonds. It is known that by applying too high a potential >1.1 V such bonds can be broken relatively easily, as representatively shown in our previous report on Fc-functionalized 1,3,5-triphenylbenzene.30 However, in the case of this compound, the decomposition of the imine bond was not observed even when the applied potential was higher than 1.3 V.
Due to the presence of phenyl rings in the Fc-den structure, this compound can be an excellent receptor for other aromatic compounds. The ability of Fc-den in the detection of aromatic compounds was checked electrochemically. To form a recognition layer, the glassy carbon electrode surface was modified by placing a 10 μL droplet of 1 mM Fc-den in DCM with addition of 100 mM TBAHFP and 5% Nafion® and left to dry in a desiccator. Before the voltammetric experiments, the recognition layer was first cycled between 0 and 1 V in an 100 mM aqueous solution of tetrabutylammonium perchlorate (TBAP) until a stable voltammogram was obtained. In order to prove the analytical potential of the proposed compound, the measurements were carried out using the following aromatic compounds: 1,4-terphenyl, pyrene, phenanthrene, fluorene and 9,10-diphenylanthracene (9,10-DPA). In the presence of 200 μM analyte in the solution, a drastic decrease in the intensity of the Fc-den oxidation signal was observed only in the case of 9,10-DPA (Fig. 6). In the case of the other compounds, the registered changes in the intensity of the Fc-den current signal were insignificant. This high sensitivity and selectivity of Fc-den to 9,10-DPA are probably due to the steric fit; it is possible to form a complex by π–π interactions.
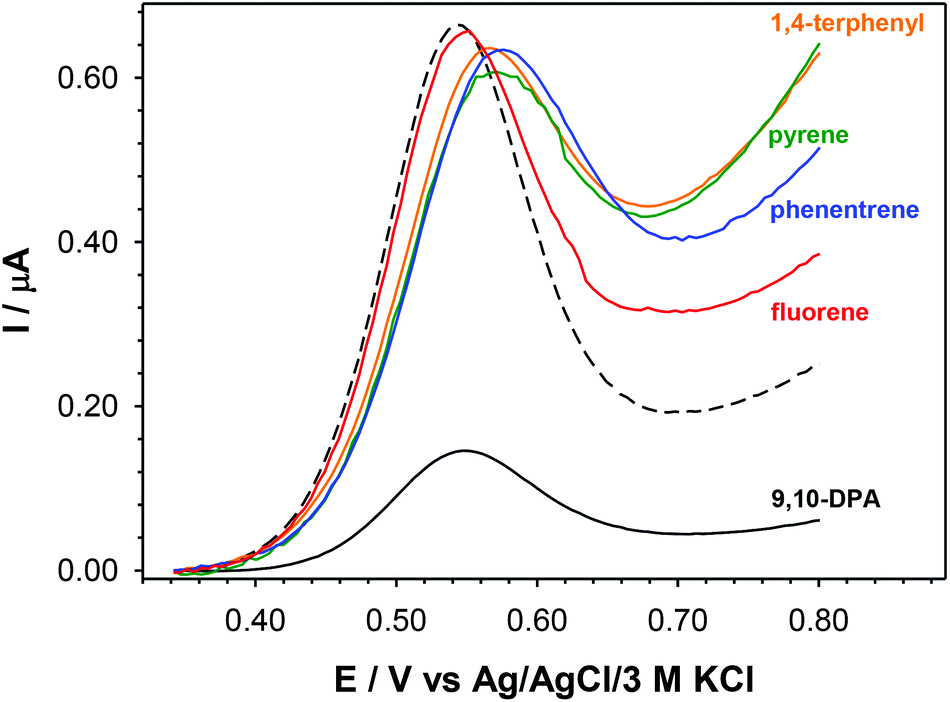 | ||
| Fig. 6 DPV voltammograms of GC/Fc-den-TBAHFP-Nafion® recorded in the presence of various aromatic analytes in an aqueous solution of 100 mM TBAP. Canalyte = 200 μM. The dotted line stands for the spectrum of native Fc-den. | ||
Having known the above discussed specificity and inspired by the fact that 9,10-diphenylantracene can be used as a template during the synthesis steps of Fc-den (Fig. 3b), we began to engineer the application of Fc-den as the building unit of an analytical device dedicated to the specific recognition of 9,10-DPA, a polycyclic aromatic hydrocarbon (PAH) commonly used in, e.g. the technologies of organic light emitting diodes.31,32 The analytical characteristics of the sensor (GC/Fc-den-TBAHFP-Nafion®), which has been fabricated as noted above, were determined electrochemically on the basis of the changes in the Fc-den oxidation current signal. The representative differential pulse voltammograms (DPV) plotted as a function of the 9,10-DPA concentration are presented in Fig. 7. After the addition of 9,10-DPA to the solution, a decrease in the intensity of the Fc-den current signal was observed. This decrease was greater when a greater concentration of the analysed compound was introduced into the solution. The signal stabilization after the addition of 9,10-DPA took place after three consecutive voltammograms were recorded in succession (total time: 4 minutes). The detection limit (LOD) was estimated from the calibration plot as the three standard deviations of the five independent controls (measurements of the current in the pure buffer solution). The regression equations describing the linear responses of IFc-den = f(C9,10-DPA) as well as the analytical ranges and LODs are presented in Table 2A, while the calculated inter- and intraday precisions are given in Table 2B.
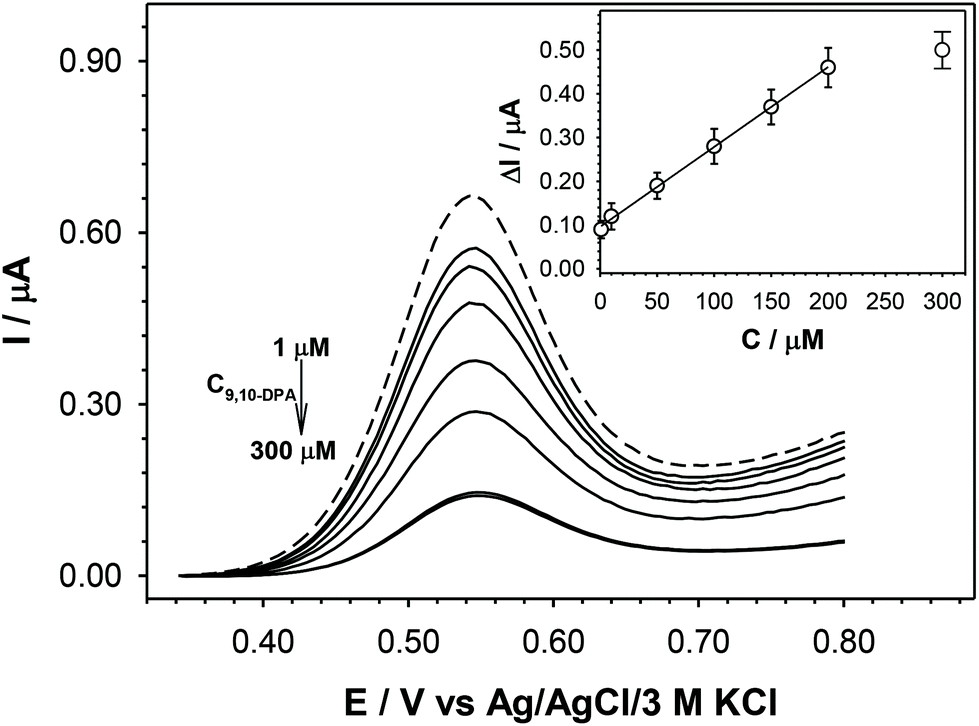 | ||
| Fig. 7 DPV voltammograms of a receptor (GC/Fc-den-TBAHFP-Nafion®) recorded at various concentrations of 9,10-DPA in an aqueous solution of 100 mM TBAP. Inset: Calibration plots. | ||
| Regression equation | R 2 | Analytical range [μM] | LOD [μM] | RSD (%) |
|---|---|---|---|---|
| A: Calibration equation obtained in an aqueous solution of 100 mM TBAP (n = 3) | ||||
| I = (1.83 ± 0.03)·10-3C9,10DPA + (0.096 ± 0.003) | 0.9995 | 0.1–200 | 0.06 | 4.5 |
| 9,10-DPA added [μM] | 9,10-DPA found [μM] | RSD (%) intraday | RSD (%) interday | Recovery (%) |
|---|---|---|---|---|
| B: Recovery results of TBAP solution spiked by 9,1-DPA (n = 3) | ||||
| 5.00 | 4.90 | 3.3 | 5.2 | 98 |
| 50.0 | 50.5 | 4.2 | 5.0 | 99 |
| 100 | 101 | 4.8 | 4.9 | 99 |
A great advantage of the proposed sensor is that 9,10-DPA did not form too stable complexes with Fc-den, so the receptor layers did not require regeneration. It was enough to immerse the sensor (GC/Fc-den-TBAHFP-Nafion®) after the measurement in a pure 100 mM TBAP aqueous solution, and the sensor was ready for re-use, which is a very important factor from the viewpoint of the application of the designed methodology. It should be also stressed that analytical studies were performed in an aqueous environment with small addition of DCM (not higher than 3%). Finally, it is worth noting that no significant interaction between the parent subunit structure of Fc-den, i.e., 1 without formyl functionalities, which was recently reported by our group,30 was observed during the electrochemical assays. This means that the organized, π-conjugated structure of Fc-den is indeed essential for providing the studied electrochemical recognition ability.
Conclusions
In conclusion, herein it is shown that the π-conjugated ferrocene-containing dendrimer (Fc-den) can be successfully prepared employing facile, efficient, chromatography-free processes that proceed under mild conditions. Noteworthily, the presented total synthesis can be regarded as time-, solvent- and cost-saving since there is no need for chromatographic purification at each synthetic step. While the imine-bond formation reaction has been demonstrated to be a convenient way for the synthesis of many valuable π-conjugated molecules, the presented total synthesis provides important tools for chromatography-free methods in total synthesis as well as for controlling the dynamic covalent chemistry behaviours with ferrocene-tethered systems. Electrochemical studies with Fc-den revealed one pair of ferrocenes’ current signals. Ultimately, the title dendrimer has been employed for the construction of an electrochemical sensor dedicated to the recognition of 9,10-diphenylanthracene. The limit of detection value equalled 0.06 μM, and importantly receptor layers consisting of Fc-den did not require regeneration. Taking into account the easiness of the preparation of the target dendrimer under mild conditions, the possibility of its gram-scale synthesis, as well as its property to recognize selected aromatic species and to track this process under electrochemical conditions, it is believed that this work would open up new avenues in the chemistry of ferrocene-bearing molecules. Our future work in this topic will include, e.g. the preparation of further generations of Fc-den and new Fc-den-derived highly ordered molecules, as well as the evaluation of their properties and applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Warsaw University of Technology (WUT) is acknowledged. This work was implemented as a part of Operational Project Knowledge Education Development 2014–2020 co-financed by the European Social Fund, project no. POWR.03.02.00-00-I007/16-00 (POWER 2014–2020). We would like to thank Dr Magdalena Poplawska (WUT) for fruitful discussions and for sharing her knowledge in organic synthesis over the years of our scientific collaboration.Notes and references
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef CAS.
- K. Heinze and H. Lang, Organometallics, 2013, 32, 5623–5625 CrossRef CAS.
- S. Daum, V. F. Chekhun, I. N. Todor, N. Y. Lukianova, Y. V. Shvets, L. Sellner, K. Putzker, J. Lewis, T. Zenz, I. A. M. de Graaf, G. M. M. Groothuis, A. Casini, O. Zozulia, F. Hampel and A. Mokhir, J. Med. Chem., 2015, 58, 2015–2024 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- A. Kasprzak and P. A. Guńka, Dalton Trans., 2020, 49, 6974–6979 RSC.
- T. Romero, A. Caballero, A. Tarraga and P. Molina, Org. Lett., 2009, 11, 3466–3469 CrossRef CAS PubMed.
- M. S. Inkpen, S. Scheerer, M. Linseis, A. J. P. White, R. F. Winter, T. Albrecht and N. J. Long, Nat. Chem., 2016, 8, 825–830 CrossRef CAS PubMed.
- L. E. Wilson, C. Hassenrgck, R. F. Winter, A. J. P. White, T. Albrecht and N. J. Long, Angew. Chem., Int. Ed., 2017, 56, 6838–6842 CrossRef CAS PubMed.
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabani, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef.
- (a) K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800–5835 RSC; (b) W.-J. Li, W. Wang, X.-Q. Wang, M. Li, Y. Ke, R. Yao, J. Wen, G.-Q. Yin, B. Jiang, X. Li, P. Yin and H.-B. Yang, J. Am. Chem. Soc., 2020, 142, 8473–8482 CrossRef CAS PubMed; (c) B. Jiang, L.-J. Chen, Y. Zhang, H.-W. Tan, L. Xu and H.-B. Yang, Chin. Chem. Lett., 2016, 27, 607–612 CrossRef CAS.
- S. Nlate, J. Ruiz, J.-C. Blais and D. Astruc, Chem. Commun., 2000, 417–418 RSC.
- B. Alonso, I. Cuadrado, M. Moran and J. Losada, J. Chem. Soc., Chem. Commun., 1994, 2575–2576 RSC.
- (a) C.-O. Turrin, E. Manoury and A.-M. Caminade, Molecules, 2020, 25, 447 CrossRef CAS PubMed; (b) C. Ornelas, J. R. Aranzaes, E. Cloutet, S. Alves and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 872–877 CrossRef CAS PubMed; (c) J. Camponovo, J. Ruiz, E. Cloutet and D. Astruc, Chem. – Eur. J., 2009, 15, 2990–3002 CrossRef CAS PubMed.
- (a) S. Sengupta and S. K. Sadhukhan, Organometallics, 2001, 20, 1889–1891 CrossRef CAS; (b) E. S. Serkova, A. A. Chamkin, K. L. Boldyrev and Z. B. Shifrina, Macromolecules, 2020, 53, 2735–2743 CrossRef CAS.
- B. J. Ravoo, Dalton Trans., 2008, 1533–1537 RSC.
- (a) C. M. Cardona and A. E. Kaifer, J. Am. Chem. Soc., 1998, 120, 4023–4024 CrossRef CAS; (b) L. Wang, L.-J. Chen, J.-Q. Ma, C.-H. Wang, H. Tan, J. Huang, F. Xiao and L. Xu, J. Organomet. Chem., 2016, 823, 1–7 CrossRef CAS; (c) M.-C. Daniel, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2003, 125, 1150–1151 CrossRef CAS PubMed; (d) Q.-J. Li, G.-Z. Zhao, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2012, 31, 7241–7247 CrossRef CAS; (e) Q. Han, Q.-J. Li, J. He, B. Hu, H. Tan, Z. Abliz, C.-H. Wang, Y. Yu and H.-B. Yang, J. Org. Chem., 2011, 76, 9660–9669 CrossRef CAS PubMed; (f) G.-Z. Zhao, L.-J. Chen, C.-H. Wang, H.-B. Yang, K. Ghosh, Y.-R. Zheng, M. M. Lyndon, D. C. Muddiman and P. J. Stang, Organometallics, 2010, 29, 6137–6140 CrossRef CAS.
- A. Salmon and P. Jutzi, J. Organomet. Chem., 2001, 637–639, 595–608 CrossRef CAS.
- (a) L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC; (b) S. Ø. Scottwell and J. D. Crowley, Chem. Commun., 2016, 52, 2451–2464 RSC; (c) E. Perris, Coord. Chem. Rev., 2004, 248, 279–297 CrossRef; (d) L. Xu, L.-J. Chen and H.-B. Yang, Chem. Commun., 2014, 50, 5156–5170 RSC; (e) G.-Y. Wu, L.-J. Chen, L. Xu, X.-L. Zhao and H.-B. Yang, Coord. Chem. Rev., 2018, 369, 39–75 CrossRef CAS; (f) M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC; (g) W. Wang, L.-J. Chen, X.-Q. Wang, B. Sun, X. Li, Y. Zhang, J. Shi, Y. Yu, L. Zhang, M. Liu and H.-B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597–5601 CrossRef CAS PubMed; (h) D. Astruc, C. Ornelas and J. Ruiz, Acc. Chem. Res., 2008, 41, 841–856 CrossRef CAS PubMed.
- (a) C. Schubert, J. T. Margraf, T. Clark and D. M. Guldi, Chem. Soc. Rev., 2015, 44, 988–998 RSC; (b) C.-O. Turrin, J. Chiffre, D. de Montauzon, J.-C. Daran, A.-M. Caminade, E. Manoury, G. Balavoine and J.-P. Majoral, Macromolecules, 2000, 33, 7328–7336 CrossRef CAS; (c) C.-O. Turrin, J. Chiffre, J.-C. Daran, D. de Montauzon, G. Balavoine, É. Manoury, A.-M. Caminade and J.-P. Majoral, C. R. Chim., 2002, 5, 309–318 CrossRef CAS.
- E. David, K. Thirumoorthy and N. Palanisami, Appl. Organomet. Chem., 2018, 32, e4522 CrossRef.
- (a) D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159–7171 RSC; (b) J. Palomero, J. A. Mata, F. Gonzalez and E. Peris, New J. Chem., 2002, 26, 291–297 RSC.
- E. G. Percastegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006–2018 RSC.
- (a) Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, WILEY-VCH Verlag GmbH, Weinhein, Germany, 2007, ISBN: 978-3-527-61352-6 Search PubMed; (b) Y. Xu, R. Kaur, B. Wang, M. B. Minameyer, S. Gsanger, B. Meyer, T. Drewello, D. M. Guldi and M. von Delius, J. Am. Chem. Soc., 2018, 140, 13413–13420 CrossRef CAS PubMed.
- (a) Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed; (b) F. Parenti, F. Tassinari, E. Libertini, M. Lanzi and A. Mucci, ACS Omega, 2017, 2, 5775–5784 CrossRef CAS PubMed; (c) L. Avram and Y. Cohen, J. Org. Chem., 2002, 67, 2639–2644 CrossRef CAS PubMed.
- T. Berbasova, E. M. Santos, M. Nosrati, C. Vasileiou, J. H. Geiger and B. Borhan, ChemBioChem, 2016, 17, 407–414 CrossRef CAS PubMed.
- A. J. King, Y. V. Zatsikha, T. Blessener, F. Dalbec, P. C. Goff, M. Kayser, D. A. Blank, Y. P. Kovtun and V. N. Nemykin, J. Organomet. Chem., 2019, 887, 86–97 CrossRef CAS.
- P. Molina, A. Tárraga, D. Curiel and M. D. Velasco, J. Organomet. Chem., 2001, 637–639, 258–265 CrossRef CAS.
- T. Yamaguchi, K. Takahashi and T. Komura, Electrochim. Acta, 2001, 46, 2527–2535 CrossRef CAS.
- (a) M. Vilas-Boas, E. M. Pereira, C. Freire and A. R. Hillman, J. Electroanal. Chem., 2002, 538, 47 CrossRef; (b) R. J. LeSuer and W. E. Geiger, Angew. Chem., Int. Ed., 2000, 39, 248–250 CrossRef CAS; (c) F. Barrière, N. Camine and W. E. Geiger, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef PubMed; (d) F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef PubMed; (e) F. Barrière and W. E. Geiger, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef PubMed.
- A. Kasprzak, P. A. Guńka, A. Kowalczyk and A. M. Nowicka, Dalton Trans., 2020, 49, 14807–14814 RSC.
- S. B. Lee, S. N. Park, C. Kim, H. W. Lee, H. W. Lee, Y. K. Kim and S. S. Yoon, Synth. Met., 2015, 203, 174–179 CrossRef CAS.
- H. Chen, W. Liang, Y. Chen, G. Tian, Q. Dong, J. Huang and J. Su, RSC Adv., 2015, 5, 70211–70219 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Compound characterization data and spectra. See DOI: 10.1039/d0dt04261g |
| This journal is © The Royal Society of Chemistry 2021 |