
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular self-assembly of 1D infinite polyiodide helices in a phenanthrolinium salt†
Tomasz
Poręba
 *a,
Marcin
Świątkowski
b and
Rafał
Kruszyński
b
*a,
Marcin
Świątkowski
b and
Rafał
Kruszyński
b
aEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: tomasz.poreba@esrf.fr
bInstitute of General and Ecological Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 26th January 2021
Abstract
A new linear polymeric polyiodide, catena-poly[tris(1,10-phenanthrolin-1-ium)tris(1,10-phenanthroline)heptaiodide], was prepared by one-step synthesis. Its formation is driven by hydrogen-bond assisted supramolecular assembly in the presence of chromium(III) acetate. Its structure has been characterized by the means of single-crystal X-ray diffraction. To date, this is only one of the few examples of organized linear infinite polyiodides with a known structure. The interplay between the interactions within the hypervalent iodine chain and its supramolecular environment is elucidated. The electrical, thermal, and spectroscopic properties of the studied compound were investigated and associated with the structural features. The infinite character of the polyiodide chain and its similarity to the blue starch–iodine complex has been additionally confirmed by Raman spectroscopy. Despite the apparent structural and spectroscopic similarities with the previously reported 1D polymeric polyiodide, its physical properties, i.e. electrical conductivity and thermal stability, differ significantly. This can be rationalized by the differences in the orbital overlap within the iodine chain, as well as the distinct interactions with the cation.
Introduction
Polyiodides are inorganic anions which can be often described with a general formula In−2m+n, where n indicates the number of iodide anions and m the number of iodine molecules. The n and m can be any positive cardinal number i.e. I3−, I5−, I82−etc. In the solid state, discrete polyiodide anions with higher m and n numbers exist, up to I293−.1 Structures of larger bulky polyiodides are stabilized by various intermolecular interactions within a crystal, such as hydrogen bond, electrostatic halogen–halogen, anion⋯π, and dispersion interactions.2–6 Generally, the bigger cations allow for the formation of longer stable polyiodide chains in the solid state.7 Additionally, the cation shape and its polarizability play an important role in the final length and geometry of the polyiodide anion.8–11 Employment of ammonium cations with long aliphatic constituents leads to polyiodide dimensional caging through dispersive interactions12,13 or even mechanical interlocking of the polyiodides in the cation cavity, as in the case of cyclobis(paraquat-p-phenylene) salt.14 On a larger supramolecular scale, a self-organization of polyiodide chains can be also executed within the channels of metal–organic frameworks.15,16 Appropriate cation tailoring and reaction conditions can lead to the formation of extended polyiodide systems. Formation of corrugated layers, branched zigzag chains and even interpenetrating chains has been reported.6,17–22Some examples of structures extending in only one direction include zigzag chains made of [I3−–I5−] units in a 1,1′-(propane-1,3-diyl)-ferrocenium complex23 or corrugated ribbons made of rectangular iodine loops in a bipyridinium salt.24 Moreover, polyiodide zigzag chains can form extensive structures through metal iodide–iodine complexes, such as Cd2I62− or Hg2I62−.25
Infinite linear chains are less common. They have been registered as weakly interacting I3−⋯I3− (intermolecular distance >4.0 Å),26 linear chains interconnected with I3− perpendicular to the propagation axis,27,28 or a pure linear chain with alternating I–I distances in (Me4Sb)3I8.29 In the latter example, however, doubts about its structure have been raised on the basis of theoretical calculations.30–32 Recently, an infinite linear polyiodide chain has been registered in a pyrroloperylene–iodine compound (prpi).33 The formation of this structure was achieved by the confinement of polyiodide units within channels along the stacks of the planar cations. This structure resembles the iodine arrangement in a blue starch–iodine complex.34–38 Another method of catenation of polyiodide subunits is the application of high pressure.39–42 It can force the polyiodide building blocks (e.g. I2 and I3−) to link through donor–acceptor interactions, which consequently allows the formation of a polymeric net.39
Compounds containing polyiodide ions are not only of structural curiosity, but also exhibit a wide range of electrical conductivity. Hence, they find applications in electrolytes, batteries, solar cells, and optical devices.16,43–46 The I3−/I− redox system is used not only in dye-sensitized solar cells but also in microbicidal and viricidal agents with a long release time.47,48 The major disadvantage of polyiodides as functional materials is their poor thermal stability. Most of the polyiodides lose iodine by sublimation, even at room temperature. They decompose to lower polyiodides and, eventually, to iodides.49 This process also complicates the analysis of polyiodides by spectroscopic methods. It was found that the laser beam used in Raman spectroscopy measurements can cause dissociation of polyiodides, leading to erroneous results.32,50,51
It is still challenging to predict the properties of a given polyiodide salt based on its structure. The polyiodide arrangement and even product stoichiometry often cannot be easily anticipated on the basis of the used cation type, reaction conditions, and stoichiometry. Herein, the design and synthesis of a polymeric 1D polyiodide organic salt are described and its structure, and spectroscopic, electrical, and thermal properties are elucidated. The presented data are compared to a very similar reported structure of the infinite polyiodide chain, and are related to the elusive structure of a starch–iodine system.52,53
Experimental
Synthesis procedures
All of the chemicals, except chromium(III) acetate hydrate, were analytical grade, purchased from Sigma Aldrich. 50 mg of chromium(III) acetate hydrate (0.2 mmol; POCh, Poland) and 108 mg of 1,10-phenanthroline monohydrate (phen, 0.6 mmol) were dissolved in 2.5 ml of dmf. Subsequently, 1 ml of 40% hydroiodic acid and 50 mg of iodine (0.2 mmol) were added. The solution was stirred until all of the reactants dissolved, and then 1 ml of the solution was transferred to a test tube. The dmf solution was topped slowly with 2 ml of methanol, avoiding mixing of the phases. The tube was sealed and left in the dark for solvent-diffusion crystallization at room temperature. After one week, long golden needles were formed in the methanolic layer.Crystal structure determination
Single crystal X-ray diffraction data were collected on a Synergy Dualflex Pilatus 300 K diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with a Pilatus 300 K detector and a CuKα1 radiation microsource (λ = 1.54184 Å). Measurements were performed at 100.0(1) K in ω-scan mode. The structure was solved with the dual-space algorithm, as implemented in SHELXT.54 All non-hydrogen atoms were refined anisotropically using the full-matrix, least-squares method on F2 by the SHELXL software.55 All hydrogen atoms were refined using the riding model. Isotropic displacement factors of hydrogen atoms were equal to 1.2 times the value of an equivalent displacement factor of the parent atoms. Crystal structure visualizations were performed using OLEX2.56 Powder diffraction (PXRD) experiments were carried out at the Materials Science Beamline at SLS.57 The sample was ground and tightly packed in a 0.3 mm borosilicate capillary. A monochromatic, collimated beam with an energy of 25.2 keV was utilized. The exact wavelength was calibrated using a NIST Si640d standard. The measured XRPD patterns were refined using TOPAS (version 6, Bruker AXS, Germany) software.Other physical measurements
The UV-vis diffuse reflectance spectra were recorded on a Jasco V-660 spectrophotometer (JASCO, Ishikawa-machi Hachioji-shi Tokyo Japan), in a 200–800 nm spectral range, using BaSO4 as a 100% reflectance standard. Thermal analyses were carried out with a Netzsch STA 449 F1 Jupiter thermoanalyzer coupled with a Netzsch Aeolos Quadro QMS 403 mass spectrometer. The samples were heated in corundum crucibles up to 800 °C, with a heating rate of 5 °C min−1 in synthetic air (20% O2, 80% N2) flow. The Raman spectra were recorded using an HR800 (Jobin Yvon-Horiba, France) spectrometer with a spectral resolution of 1.5 cm−1. The spectra were collected in reflection mode, by excitation with 532 nm laser light with a nominal power of 12 mW. The laser intensity was decreased to 1% and the exposure time was 10 s. Electrical conductivity measurements were carried out in two-probe mode using 25 μm thick golden wires as electrodes, and the data were recorded using a Keysight 34461A multimeter. The crystal used for the electrical measurements was needle-shaped with a 500 μm length. For the calculation of electrical conductivity, its shape was estimated (by examination under a microscope) to be a long cylinder with 100 μm radius.Results and discussion
The reaction among iodine, hydroiodic acid, and 1,10-phenanthroline in the presence of chromium(III) acetate resulted in the formation of long, golden needle-like crystals (Fig. 1 ESI†) of catena-poly[tris(1,10-phenanthrolin-1-ium)tris(1,10-phenanthroline) heptaiodide] (phenpi). The bulk purity of the product has been confirmed by means of synchrotron PXRD (Fig. 2 ESI†). The compound crystallizes in the non-centrosymmetric Pna21 space group (Table 1 ESI†) as a one-dimensional polymeric polyiodide. It is impossible to distinguish a discrete polyiodide moiety in the structure, as iodine–iodine distances within a chain vary between 3.129 and 3.351 Å (Fig. 1a). The asymmetric unit [(I7)3−] atoms were chosen based on the shortest distances between the constituting I atoms, i.e. a 3.351 Å distance exists between the symmetry generated [(I7)3−] building blocks. The iodine–iodine distances within a chain do not indicate any presence of short covalent I–I bonds (I–I bond lengths of the solid iodine58 and the mean value for I3− from the Cambridge Structural Database59 (CSD) are equal to 2.7179(2) and 2.92(5) Å [n = 934], respectively) or the secondary (>3.40 Å) intermolecular halogen–halogen interactions. It is impossible to ascribe the formal charges to the given iodine atoms. The formal charge of a [(I7)3−] fragment is 3-, which indicates [3I−·2I2] assembly.60 A polymeric iodine right-handed ellipse helix propagates along the crystallographic [0 0 1] axis with arc radii the along [1 0 0] and [0 1 0] directions equal to 0.619 Å and 0.304 Å, respectively. The pitch of this tightly twisted helix is 22.533 Å (length along the [0 0 1] axis, Fig. 1a). Distribution of the intramolecular I–I distances is much more narrower than that in the reported discrete I73− unit, where the bond alteration was more indicative of [I−–I2–I− I2–I−] assembly.60 It appears that the catenation into a linear chain balances the iodine bond lengths due to the more homogeneous charge distribution, as observed in prpi. | ||
| Fig. 1 Molecular structure and polyiodide geometry of phenpi. (a) Asymmetric part of the polymeric iodine chain with outlined dimensions and intramolecular I–I distances (Å) and (b) and (c) projections of the phenpi structure along [010] and [001], respectively. | ||
Two of the symmetry-related iodine helices are packed parallelly inside the channels created by the four hydrogen-bonded 1,10-phenanthrolinium dimers stacked into 1D piles along the crystallographic [0 0 1] axis (Fig. 1c). Each iodine atom is “pinned” by C–H⋯I hydrogen bonds with the hydrogen atom of the aromatic carbon atoms from the three directions (Fig. 2).61 The length of the hydrogen bonds is in a range of 3.02–3.26 Å. Due to different arrangements of the interaction for different I atoms of an asymmetric unit, totally, the In chains are “pinned” from all four directions (Fig. 2). Without the other strong hydrogen bonds in this structure, the interplay among π⋯π stacking, H⋯I, and I⋯I interactions dominates the supramolecular architecture in phenpi.
 | ||
| Fig. 2 Hirshfeld surface overlaid onto an asymmetric part of the polymeric polyiodide chain in phenpi with indicated hydrogen bonds “pinning” the chain.65 Colour code corresponds to the surface property de (the distance from the surface to the nearest nucleus external to the surface), as indicated in the colour scale. | ||
It is noteworthy that the templating reaction performed without the addition of chromium(III) acetate always resulted in 1,10-phenanthrolinium triiodide, and phenpi is not formed in such a system. The role of the chromium salt is not well defined, but it might be connected to a redox activity of in situ formed [Cr(phen)3]3+ moieties. These cationic moieties easily undergo one-electron reduction to chromium(II) coordination compounds.62 In the synthesis of phenpi the redox reaction can lead to the oxidation of I3− anions (formed instantly after mixing I− and I2), and subsequently larger polyiodides are formed and the final hydrogen-bond assisted supramolecular assembly of the (I73−)n chain occurs.
The phenanthrolinium cation forms a short hydrogen bond with one unprotonated nitrogen of another phenanthroline molecule (Fig. 1c). The length of the hydrogen bonds within the dimers is about 2.10 Å. The angle between the phen units engaged in the dimers is approximately equal to 45.5°, and deviates slightly (±0.5°) between the symmetry-unrelated piles, which is in agreement with the values observed in similar compounds.63 Distances between the parallel phen units are 3.84 and 3.66 Å in the respective piles. To date, there is only one reported structure of prpi which contains a similar 1D polymeric polyiodide unit.33 Similarly, the previously reported structure contains helical iodine chains stabilized in the channels between the stacked planar organic cations.
The Cambridge Structural Database (CSD)64 contains 37 structures of compounds in which (PheH⋯Phe)+ dimers exist (Fig. 3). Most of them are (nearly) parallel to each other due to π⋯π stacking, which provides higher stabilization energy than the intermolecular hydrogen bonds (Fig. 3, inset). The length of the hydrogen bonds and the interplanar spacing in these cases are above 3 Å. The second group includes the phenanthrolinium dimers where the planar units form an angle >35°, and rather variable hydrogen bonds with H⋯A distances in a range of 1.9–3.4 Å. In these cases, usually bulky cations separate the interacting phen units, weakening the stabilizing effect of the π⋯π stacking. This group contains phenanthrolinium–phenanthroline cations with soft anions such as NO3−, BF4−, and ClO4−. In each of these compounds, phenanthroline stacks similarly to the arrangement observed in phenpi, with the anions located in the channels between them. In literature-known compounds, these anions are well separated and do not interact mutually, which is in opposition to phenpi, in which iodine forms a 1D polymeric chain. phenpi can be ascribed to the latter group (Fig. 3, star) on the basis of the arrangement of phen units (forming dimers with a respective dihedral angle and close arrangement of molecules connected through the N–H⋯N hydrogen bond). It appears that in this group, the stabilization of the crystal structure by the hydrogen bonds and π⋯π stacking is energetically favoured than that from pure π⋯π stacking, even in the most privileged parallel arrangement.66
 | ||
| Fig. 3 Distribution of intermolecular distances and corresponding N–H⋯H hydrogen bonds between the phen and phenH+ units in bis-phenanthrolinium cations (according to the CSD data). Points represent contact lengths between two phenanthroline centroids as a function of phen ring inclination (within a dimer). Colour of the points represents the H⋯N distance of the hydrogen bond, according to the legend in the bottom right corner. Structural insets: different conformations of phen molecules found at different centroid separations, and the red dotted line represents the mentioned hydrogen bond. | ||
Electrical conductivity measurements were performed in four-probe mode along the direction of the iodine chains (importantly, with the exclusion of the use of silver paste serving as a sample–electrode contact). A silver paste in contact with the high-iodine content materials forms an insulating layer of silver iodide, which makes the experiment not reproducible and results in the lowering of values of measured conductivity. The recorded resistance (proportional to the inverse of conductivity) was >10 GΩ (<1.6 nS cm−1 for the tested crystal), which indicates an insulating character of phenpi. This is in contrast to the observations made for prpi, in which the conductivity at room temperature is about 0.01 S cm−1. This dissimilarity in the electrical properties of apparently structurally similar materials can be explained by the differences in iodine–iodine distances in these two compounds. The I–I distances in prpi are in the 3.046–3.174 Å range (measured at 100 K), which are shorter than the respective values in phenpi (Fig. 1a), and result in different electronic properties of the iodine chain. The shortening of I–I distances (in an organic polyiodide salt) below 3.32 Å can trigger a dramatic increase of electrical conductivity due to the formation of a more covalent iodine polymeric network.39 In the case of the conductive prpi compound the largest spacing between the adjacent iodine atoms within a chain is below this border value. In contrast, in the case of phenpi one of the in-chain distances is longer than 3.32 Å already at 100 K. Thus, it does not allow for electron transport due to the disruption of the continuity of the electronic band. Even though the In chain is polymeric in phenpi (forming an infinite discrete species), the electronic structure shows a discontinuity, as is the case of tetraetylammonium diiodine triiodide at high pressure.39 Apart from the potential conduction along the iodine chain, the effect of the cation has been demonstrated to play a major role in some of the organic conductors containing polyiodides. For example, in the case of tetrathiatetracene and perylene polyiodide complexes, the unusually high electrical conductivity has been mainly ascribed to the cation stacks.67–70 These salts show the non-stoichiometric iodine content and incommensurability of the cation and polyiodide sublattices, often coupled with the structural disorder. In the case of phenpi, none of the abovementioned effects were observed. It was additionally confirmed that the electrical conductivity of phenpi crystals after thermal annealing at 80 °C was virtually the same as in the fresh crystals. One cannot, however, rule out any electronic transitions at low temperature due to Peierls distortion.71,72
Thermochemistry
phenpi melts at around 90 °C (versus 113.7 and 117.0 °C for pure iodine and phenanthroline, respectively) and after 105 °C starts to decompose (Fig. 4). The decomposition is total and occurs in two stages. Mass spectra collected during the decomposition confirm that phen combustion and iodine release occur in both stages (Fig. 3 ESI†). The larger part of iodine is removed in the first stage, which is indicated by the high iodine content in volatile products (Fig. 3 ESI†). The proposed pathway of phenpi decomposition, which is best fitted to mass losses, assumes that in the first stage (105 °C–285 °C) two phen, one phenH+, and I5− are removed (experimental/theoretical mass loss: 59.62%/59.50%). The intermediate product is 2phenH+·phen·2I−, which decomposes totally directly after the first stage (285 °C–420 °C). The formation of iodides as intermediates is a known phenomenon, which was observed in the case of some higher organic polyiodides.49,73,74 | ||
| Fig. 4 TG (green) and DTA (red) curves for phenpi. | ||
The iodine content in the intermediate product may also indicate the presence of I2 adducts but at a temperature around 285 °C their presence is strongly unlikely. The absence of the mass signals higher than m/z = 127 in the mass spectra (Fig. 3 ESI†) is noteworthy. This confirms that during decomposition evaporating iodine does not form any organic derivatives with phen decomposition products. The immediate start of decomposition after the melting point proves the role of the crystal lattice in the thermal stabilization of the iodine moieties. Once the structure collapses, the weak hydrogen bonds between phen and iodine break, subsequently allowing the dissociation of iodine chains (which were kept in the channels existing between phenanthrolinium piles). Accordingly, crystals of phenpi do not lose a substantial amount of iodine in air at ambient temperature, as indicated by the iodometric titration of the samples kept in an open container for 14 days. It is noteworthy that the starch–iodine compound starts decomposing at the higher temperature, i.e. at 168 °C.74 The structure and multiple hydrogen bonds between the amylose helices allow higher sustainability of the iodine molecules within the helix channel than in molecularly more simple systems such as phenpi.
Spectrometric studies
The electronic spectrum of solid phenpi shows three distinct broad absorption maxima (Fig. 5). The first (in the UV region) originates from a set of π → π* transitions within neutral phenanthroline organic molecules. This set is composed of three different π5 → π1*, π6 → π1*, and π7 → π1* transitions,75 broadened and overlapping due to the presence of three structurally different phenanthroline molecules in a slightly different environment. The second maximum is even broader and more complex. It comprises three different n → π* transitions within neutral phenanthroline and charge transitions75 within a polyiodide chain (as was proven above on the basis of electrical conductivity measurements, the molecular orbitals cover only a finite number of atomic centers within a chain, which allows respective charge transitions). The third absorption maximum originates from the sets of n → π* transitions within the phenanthrolinium ion and phenanthrolinium/phenanthroline interacting systems. This third maximum contains an arm at longer wavelengths (at about 500 nm) caused by π* → σ* transitions within the polyiodide chain.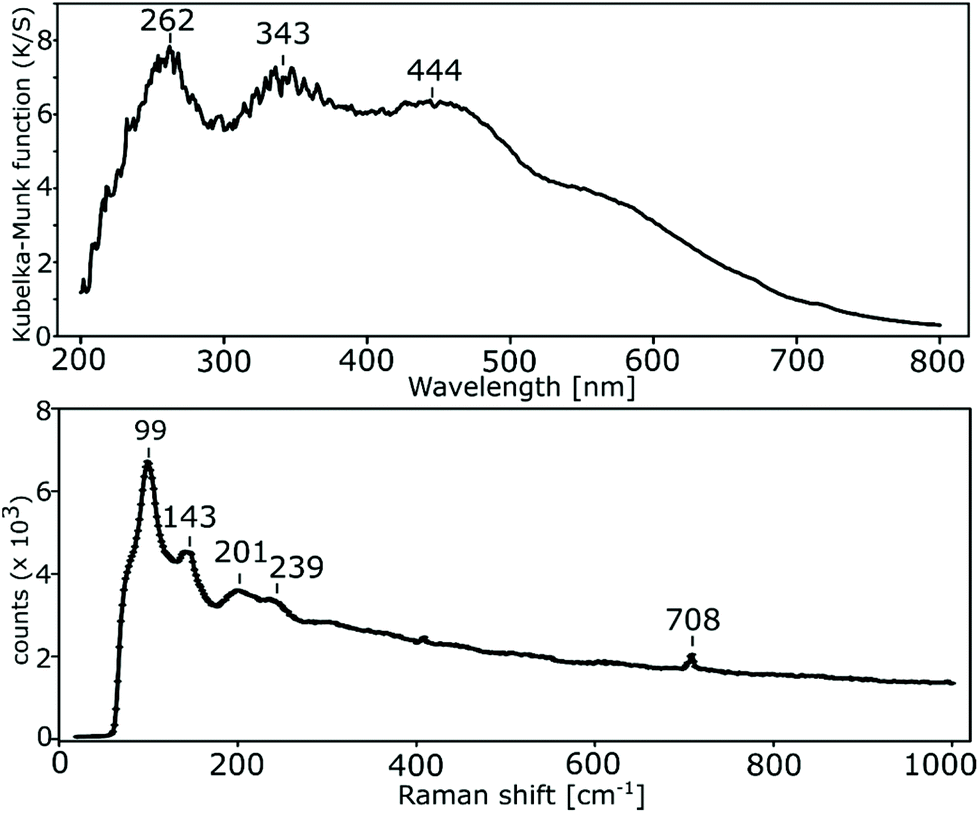 | ||
| Fig. 5 UV-vis (DRS) electronic spectrum (top) and Raman spectrum (bottom) for phenpi. | ||
Raman spectroscopy of polyiodides allows for easier exploration of the iodine–iodine vibrational mode region, in comparison with infrared spectroscopy. The wavelength of the incident radiation during Raman spectrum acquisition was chosen on the basis of visible radiation absorption (polyiodide moieties exhibit a resonant Raman signal enhancement).50,76 The available laser with 532 nm wavelength was selected, as its radiation was mostly absorbed by the iodine in the studied crystal. Unfortunately, protonated phen also partially absorbs the utilized laser light and emits fluorescence with a maximum at about 410 nm.77 It is visible in the Raman spectrum as a background characterized by the high exponential component (Fig. 5). Apart from this obstacle, the vibrational modes arising from the iodine fragments are observed (Fig. 5). The single band at 99 cm−1, with an overtone (2ν1) at 201 cm−1 points to the presence of the symmetric I3− ν1 mode.50 Usually, this mode is found at around 110 cm−1. Strong wavenumber lowering has been observed in some N-alkylurotropinium L-shaped pentaiodides (100 cm−1).78 Such a red shift exemplifies the influence of the strong donor–acceptor interaction between I2 and I3− on the force constants. In fact, some of the segments of the polyiodide building blocks in phenpi can be regarded as asymmetric I3− forming such interactions with the adjacent iodine fragments, as described above. The presence of asymmetric triiodides can be indicated by the ν3 mode at around 143 cm−1. The other clearly distinguishable bands at 239 and 708 cm−1 are caused by the presence of two non-bonded phen.79 It is noteworthy that as in the case of 1D polymeric prpi and the starch–iodine compound, the signal from ν(I2) (at around 180 cm−1 for the solid iodine51) is absent, which implies that the polymeric polyiodide chains are not only extended donor–acceptor (e.g. (I3−⋯I2)n) adducts, but also form a real In entity. This observation contradicts some contemporary views that polyiodides higher than I3− are not real moieties, but should be considered as adducts.51
Conclusions
The synthesized compound consists of ordered linear polymeric Inδ− chains and stacks of phenanthrolinium cations and phenanthroline molecules. Even though its structure resembles the one found in prpi, their physical properties are different. The title compound is an electrical insulator, while another one shows electrical conductivity. It can be explained by the variation of molecular orbitals formed in the respective polymers (different covalent characters of I–I bonds). Spectrometric studies show that the homoleptic iodine chain acts as a distinct multinuclear system, and its Raman spectrum resembles the one produced by the starch–iodine compound. This observation supports the model of infinite polyiodide chains embedded into an amylose helix. It was proven that the hydrogen bonding between the iodine chain and cations can contribute to the retainment of the supramolecular architecture of the iodine helices, influencing their thermal stability. Even though the vibrational spectra are similar, the stability and electrical conductivity of different infinite polyiodides are in contrast and depend on the fine supramolecular alteration of their structures.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Dr N. Casati and Dr M. Wilke for their help with PXRD data acquisition and refinement.Notes and references
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS.
- M. Savastano, Á. Martínez-Camarena, C. Bazzicalupi, E. Delgado-Pinar, J. Llinares, P. Mariani, B. Verdejo, E. García-España and A. Bianchi, Inorganics, 2019, 7, 48 CrossRef CAS.
- M. Savastano, C. Bazzicalupi, C. García, C. Gellini, M. D. López de la Torre, P. Mariani, F. Pichierri, A. Bianchi and M. Melguizo, Dalton Trans., 2017, 46, 4518–4529 RSC.
- M. Giese, M. Albrecht, C. Bohnen, T. Repenko, A. Valkonen and K. Rissanen, Dalton Trans., 2014, 43, 1873–1880 RSC.
- G. J. Reiss, Z. Kristallogr. - New Cryst. Struct., 2019, 234, 899–902 CAS.
- Á. Martínez-Camarena, M. Savastano, J. M. Llinares, B. Verdejo, A. Bianchi, E. García-España and C. Bazzicalupi, Inorg. Chem. Front., 2020, 7, 4239–4255 RSC.
- C. Walbaum, I. Pantenburg, P. Junk, G. B. Deacon and G. Meyer, Z. Anorg. Allg. Chem., 2010, 636, 1444–1446 CrossRef CAS.
- M. D. García, J. Martí-Rujas, P. Metrangolo, C. Peinador, T. Pilati, G. Resnati, G. Terraneo and M. Ursini, CrystEngComm, 2011, 13, 4411 RSC.
- A. J. Blake, F. A. Devillanova, R. O. Gould, W.-S. Li, V. Lippolis, S. Parsons, C. Radek and M. Schroder, Chem. Soc. Rev., 1998, 27, 195–205 RSC.
- G. J. Reiß and J. S. Engel, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1114–1117 Search PubMed.
- K. Shimizu and J. Ferreira da Silva, Molecules, 2018, 23, 2959 CrossRef.
- P. H. Svensson, M. Gorlov and L. Kloo, Inorg. Chem., 2008, 47, 11464–11466 CrossRef CAS.
- G. Manca, A. Ienco and C. Mealli, Cryst. Growth Des., 2012, 12, 1762–1771 CrossRef CAS.
- M. Savastano, C. Bazzicalupi, C. Gellini and A. Bianchi, Chem. Commun., 2020, 56, 551–554 RSC.
- S. Jiang, N. Holtgrewe, S. S. Lobanov, F. Su, M. F. Mahmood, R. S. McWilliams and A. F. Goncharov, Nat. Commun., 2018, 9, 2624 CrossRef.
- Z. Yin, Q. X. Wang and M. H. Zeng, J. Am. Chem. Soc., 2012, 134, 4857–4863 CrossRef CAS.
- Z. Wang, Y. Cheng, C. Liao and C. Yan, CrystEngComm, 2001, 3, 237–242 RSC.
- K. Lamberts, P. Handels, U. Englert, E. Aubert and E. Espinosa, CrystEngComm, 2016, 18, 3832–3841 RSC.
- X. Dong, A. R. Oganov, A. F. Goncharov, E. Stavrou, S. Lobanov, G. Saleh, G. R. Qian, Q. Zhu, C. Gatti, V. L. Deringer, R. Dronskowski, X. F. Zhou, V. B. Prakapenka, Z. Konôpková, I. A. Popov, A. I. Boldyrev and H. T. Wang, Nat. Chem., 2017, 9, 440–445 CrossRef CAS.
- P. H. Svensson, J. Rosdahl and L. Kloo, Chem. – Eur. J., 1999, 5, 305–311 CrossRef CAS.
- G. J. Reiss, Z. Naturforsch., B: J. Chem. Sci., 2015, 70, 735–739 CAS.
- J. Hu, J. Zhou and S. Cao, Dalton Trans., 2018, 47, 17216–17220 RSC.
- T. Y. Dong, H. M. Lin, M. Y. Hwang, T. Y. Lee, L. H. Tseng, S. M. Peng and G. H. Lee, J. Organomet. Chem., 1991, 414, 227–244 CrossRef CAS.
- K.-F. Tebbe and M. Bittner, Z. Anorg. Allg. Chem., 1995, 621, 218–224 CrossRef CAS.
- P. H. Svensson and L. Kloo, Inorg. Chem., 1999, 38, 3390–3393 CrossRef CAS.
- L. Kloo, P. H. Svensson and M. J. Taylor, J. Chem. Soc., Dalton Trans., 2000, 1061–1065 RSC.
- T. Akutagawa, Y. Abe, Y. Nezu, T. Nakamura, M. Kataoka, A. Yamanaka, K. Inoue, T. Inabe, C. A. Christensen and J. Becher, Inorg. Chem., 1998, 37, 2330–2331 CrossRef CAS.
- I. Pantenburg and K.-F. Tebbe, Z. Anorg. Allg. Chem., 2002, 628, 1780 CrossRef CAS.
- U. Behrens, H. J. Breunig, M. Denker and K. H. Ebert, Angew. Chem., Int. Ed. Engl., 1994, 33, 987–989 CrossRef.
- L. J. Sæthre, O. Gropen, J. Sletten, T. Pedersen, L. B. Zinner, F. Lehrich, C. J. Nielsen, D. L. Powell and M. Trætteberg, Acta Chem. Scand., Ser. A, 1988, 42, 16–26 CrossRef.
- K. N. Robertson, P. K. Bakshi, T. S. Cameron and O. Knop, Z. Anorg. Allg. Chem., 1997, 623, 104–114 CrossRef CAS.
- P. Deplano, F. A. Devillanova, J. R. Ferraro, M. L. Mercuri, V. Lippolis and E. F. Trogu, Appl. Spectrosc., 1994, 48, 1236–1241 CrossRef CAS.
- S. Madhu, H. A. Evans, V. V. T. Doan-Nguyen, J. G. Labram, G. Wu, M. L. Chabinyc, R. Seshadri and F. Wudl, Angew. Chem., Int. Ed., 2016, 55, 8032–8035 CrossRef CAS.
- R. C. Teitelbaum, S. L. Ruby and T. J. Marks, J. Am. Chem. Soc., 1978, 100, 3215–3217 CrossRef CAS.
- W. Saenger, Naturwissenschaften, 1984, 71, 31–36 CrossRef CAS.
- R. E. Rundle, J. F. Foster and R. R. Baldwin, J. Am. Chem. Soc., 1944, 66, 2116–2120 CrossRef CAS.
- J. Hollo and J. Szejtli, Period. Polytech., Chem. Eng., 1957, 1, 141–145 CAS.
- R. E. Rundle and R. R. Baldwin, J. Am. Chem. Soc., 1943, 65, 554–558 CrossRef CAS.
- T. Poręba, M. Ernst, D. Zimmer, P. Macchi and N. Casati, Angew. Chem., Int. Ed., 2019, 58, 6625–6629 CrossRef.
- S. S. Lobanov, J. A. Daly, A. F. Goncharov, X. Chan, S. K. Ghose, H. Zhong, L. Ehm, T. Kim and J. B. Parise, J. Phys. Chem. A, 2018, 122, 6109–6117 CrossRef CAS.
- L. Alvarez, J.-L. Bantignies, R. Le Parc, R. Aznar, J.-L. Sauvajol, A. Merlen, D. Machon and A. San Miguel, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205403 CrossRef.
- A. Sengupta, E. L. Quitevis and M. W. Holtz, J. Phys. Chem. B, 1997, 101, 11092–11098 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS.
- G.-M. Weng, Z. Li, G. Cong, Y. Zhou and Y.-C. Lu, Energy Environ. Sci., 2017, 10, 735–741 RSC.
- I. Turkevych, S. Kazaoui, N. A. Belich, A. Y. Grishko, S. A. Fateev, A. A. Petrov, T. Urano, S. Aramaki, S. Kosar, M. Kondo, E. A. Goodilin, M. Graetzel and A. B. Tarasov, Nat. Nanotechnol., 2019, 14, 57–63 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS.
- L. R. Fina, N. Hassouna, G. L. Horacek, J. P. Lambert and J. L. Lambert, Appl. Environ. Microbiol., 1982, 44, 1370–1373 CrossRef CAS.
- C. He, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2014, 20, 6699–6706 CrossRef CAS.
- I. D. Yushina, D. G. Pikhulya and E. V. Bartashevich, J. Therm. Anal. Calorim., 2020, 139, 1017–1023 CrossRef CAS.
- E. M. Nour, L. H. Chen and J. Laane, J. Phys. Chem., 1986, 90, 2841–2846 CrossRef CAS.
- P. Deplano, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, Coord. Chem. Rev., 1999, 188, 71–95 CrossRef CAS.
- J. M. Reddy, K. Knox and M. B. Robin, J. Chem. Phys., 1964, 40, 1082–1089 CrossRef CAS.
- S. Moulay, J. Polym. Eng., 2013, 33, 389–443 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Böge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. Flechsig, F. Gozzo, B. Henrich, S. Jäggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Lüdeke, R. Lüscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitins, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS.
- F. Bertolotti, A. V. Shishkina, A. Forni, G. Gervasio, A. I. Stash and V. G. Tsirelson, Cryst. Growth Des., 2014, 14, 3587–3595 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- J. Lin, J. Martí-Rujas, P. Metrangolo, T. Pilati, S. Radice, G. Resnati and G. Terraneo, Cryst. Growth Des., 2012, 12, 5757–5762 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- G. A. Lawrance and D. F. Sangster, J. Chem. Soc., Dalton Trans., 1987, 1425–1429 RSC.
- L. Maresca, G. Natile, F. P. Fanizzi and F. Stasi, J. Am. Chem. Soc., 1989, 111, 1492–1493 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef.
- R. Kruszynski and T. Sierański, Cryst. Growth Des., 2016, 16, 587–595 CrossRef CAS.
- J. Kommandeur and F. R. Hall, J. Chem. Phys., 1961, 34, 129–133 CrossRef CAS.
- S. K. Khanna, S. P. S. Yen, R. B. Somoano, P. M. Chaikin, C. L. Ma, R. Williams and S. Samson, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 655–663 CrossRef CAS.
- M. R. Bryce and L. C. Murphy, Nature, 1984, 309, 119–126 CrossRef CAS.
- L. C. Isett and E. A. Perez-Albuerne, Solid State Commun., 1977, 21, 433–435 CrossRef CAS.
- P. A. Albouy, P. Le Guennec, J. P. Pouget and C. Noguera, Synth. Met., 1987, 19, 687–692 CrossRef CAS.
- V. F. Kaminskii, M. L. Khidekel, R. B. Lyubovskii, I. F. Shchegolev, R. P. Shibaeva, E. B. Yagubskii, A. V. Zvarykina and G. L. Zvereva, Phys. Status Solidi, 1977, 44, 77–82 CrossRef CAS.
- I. Yushina, B. Rudakov, I. Krivtsov and E. Bartashevich, J. Therm. Anal. Calorim., 2014, 118, 425–429 CrossRef CAS.
- P. P. Danilovas, R. Rutkaite and A. Zemaitaitis, Carbohydr. Polym., 2014, 112, 721–728 CrossRef CAS.
- R. Kruszynski, Inorg. Chim. Acta, 2011, 371, 111–123 CrossRef CAS.
- E. Mulazzi, I. Pollini, L. Piseri and R. Tubino, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 3555–3563 CrossRef CAS.
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, Chem. Soc. Rev., 2009, 38, 1690 RSC.
- H. Mittag, H. Stegemann, H. Füllbier and G. Irmer, J. Raman Spectrosc., 1989, 20, 251–255 CrossRef CAS.
- K. Zawada and J. Bukowska, J. Mol. Struct., 2000, 555, 425–432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectra and crystallographic parameters. CCDC 2041514. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt04042h |
| This journal is © The Royal Society of Chemistry 2021 |