
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and electronic studies of substituted m-terphenyl lithium complexes†
Andrew J.
Valentine
 a,
Ana M.
Geer
b,
Laurence J.
Taylor
a,
Andrew M.
Teale
a,
Katherine E.
Wood
a,
Huw E. L.
Williams
c,
William
Lewis
d,
Stephen P.
Argent
a,
Jonathan
McMaster
*a and
Deborah L.
Kays
*a
a,
Ana M.
Geer
b,
Laurence J.
Taylor
a,
Andrew M.
Teale
a,
Katherine E.
Wood
a,
Huw E. L.
Williams
c,
William
Lewis
d,
Stephen P.
Argent
a,
Jonathan
McMaster
*a and
Deborah L.
Kays
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Deborah.Kays@nottingham.ac.uk; Jonathan.McMaster@nottingham.ac.uk
bDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC, Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain
cCentre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
dSchool of Chemistry, The University of Sydney, F11, Eastern Ave, Sydney, NSW 2006, Australia
First published on 8th December 2020
Abstract
The effect of para-substitution upon the structural and electronic properties of a series of m-terphenyl lithium complexes [R–Ar#–Li]2 (R = t-Bu 1, SiMe32, H 3, Cl 4, CF35; where R–Ar# = 2,6-{2,6-Xyl}2-4-R-C6H2 and 2,6-Xyl = 2,6-Me2C6H3) has been investigated. X-ray crystallography reveals the complexes to be structurally similar, with little variation in C–M–C bond lengths and angles across the series. However, in-depth NMR spectroscopic studies reveal notable electronic differences, showing a linear correlation between the 7Li{1H} NMR chemical shifts of the para-substituted complexes and their Hammett constants. The flanking methyl protons exhibit a similar electronic shift in the 1H NMR spectra, which has been rationalised by the presence of through-space Li⋯H interactions, as evidenced by two-dimensional 7Li–1H heteronuclear Overhauser spectroscopy (HOESY). In both cases, electron-withdrawing substituents are found to cause an upfield peak shift. A computational analysis is employed to account for these trends.
1. Introduction
The development of sterically demanding ligand systems has become a central research theme for the stabilisation of low-coordinate metal complexes. One important example is the m-terphenyl ligand, which has been repeatedly utilised in the isolation of low-coordinate main group and transition metal complexes.1–6 Although a plethora of m-terphenyl frameworks have been designed, most studies have been structurally focused, aimed at investigating the effects of steric bulk on the geometries, bonding modes, and reactivities of the resulting compounds.7–18For instance, the solid state structures observed for a series of m-terphenyl lithium complexes vary depending on the steric demands of the flanking ortho-aryl substituents.19,20 Increasing the steric bulk of 2,6-Ar2C6H3Li alters its aggregation state from a dimer,19 to a more crowded dimer featuring η6-arene coordination of the flanking aryl groups,20 to a monomer stabilised by a coordinated molecule of benzene,20 for Ar = Mes (2,4,6-Me3C6H2), Dipp (2,6-i-Pr2C6H3) and Tripp (2,4,6-i-Pr3C6H2) respectively.5,6
However, less work has been reported on varying the electronic effects of these m-terphenyl systems, with studies limited mainly to a handful of main group complexes21–25 and the quintuply-bonded Cr–Cr dimer [(2,6-Dipp2-4-R-C6H2)Cr]2 (R = H, SiMe3, OMe, F).26 Even their lithium precursors, while structurally characterised, have not been studied from an electronic viewpoint. Therefore, it is the objective of this research to develop a toolbox of m-terphenyl ligands featuring a range of para-substituents (R) to investigate the electronic effects of substitution upon their metal complexes.
To this end, a series of para-substituted m-terphenyl lithium complexes [R–Ar#–Li]2 (R–Ar# = 2,6-{2,6-Xyl}2-4-R-C6H2; R = t-Bu, SiMe3, Cl, CF3; 2,6-Xyl = 2,6-Me2C6H3) are reported, and discussed alongside the previously published unsubstituted analogue [H–Ar#–Li]2.27 The geometric and electronic structures of these compounds are elucidated through X-ray crystallographic and NMR spectroscopic studies, respectively. Specifically, 7Li NMR spectroscopy is employed to assess the electronic effects directly at the metal centre, with two-dimensional 7Li–1H heteronuclear Overhauser spectroscopy (HOESY) measurements demonstrating the presence of through-space 7Li⋯1H interactions. Computational modelling with density functional theory (DFT) is also employed to help rationalise the observed trends in NMR parameters across the series.
2. Results and discussion
2.1. Synthesis
A series of m-terphenyl iodides R–Ar#–I (R = t-Bu, SiMe3, Cl, CF3)28 was synthesised via similar procedures to other terphenyl compounds,22,25–27,29–33 and H–Ar#–I was obtained following a previously reported method15,32 (see ESI, section S2†). Lithium-halogen exchange reactions performed on these iodide compounds with excess n-butyllithium in isohexane at 0 °C afforded the m-terphenyl lithium complexes [R–Ar#–Li]2 (R = t-Bu 1, SiMe32, H 3, Cl 4, CF35) as white powders, according to Scheme 1. A range of yields was obtained (38%, 60%, 99%, quantitative and 95% for 1–5 respectively); the low yield of 1 can be ascribed to the greater solubility of the t-Bu substituted complex in hexane. Recrystallisation of 1, 2, 4 and 5 from a −30 °C isohexane solution produced colourless crystals suitable for X-ray diffraction analysis. The crystal structure of 3 has previously been reported.27 Complexes 1–5 have been characterised by multinuclear (1H, 13C{1H}, 19F, 29Si, 7Li{1H}) NMR spectroscopies, diffusion-ordered spectroscopy (DOSY), and 7Li–1H HOESY experiments. | ||
| Scheme 1 Synthesis of lithium complexes [R–Ar#–Li]2 (R = t-Bu 1, SiMe32, H 3, Cl 4, CF35). Reaction conditions: (i) isohexane, 0 °C to room temperature, 16 h, – n-BuI. | ||
2.2. Crystallographic characterisation
Single crystal X-ray diffraction data were collected for all new iodide ligand precursors, as well as the new lithium complexes 1, 2, 4, and 5. The crystal structure of 1 is presented in Fig. 1 and full crystallographic data are provided in the ESI (section S4, Fig. S30–S32†). The crystallographic data confirms that all complexes adopt a dimeric structure featuring two anionic m-terphenyl units linked by two lithium cations to form a planar Li2C2 core (see Fig. S31, S32 and Table S1 in the ESI†). Similar Li2C2 cores are observed across the series, with Li(1)⋯Li(2) distances of 2.304(3)–2.332(4) Å, Cipso⋯Cipso distances between 3.6893(18)–3.702(3) Å, and Cipso–Li–Cipso angles in the range of 114.59(13)–116.60(10)° [ΣLi–C–Li–C = 360°]. Furthermore, all Cipso–Li bond lengths occur within a narrow range [2.158(3)–2.1985(18) Å] and are comparable to the unsubstituted analogue 3 [2.143(5)–2.187(6) Å] and other m-terphenyl lithium dimers.15,19,27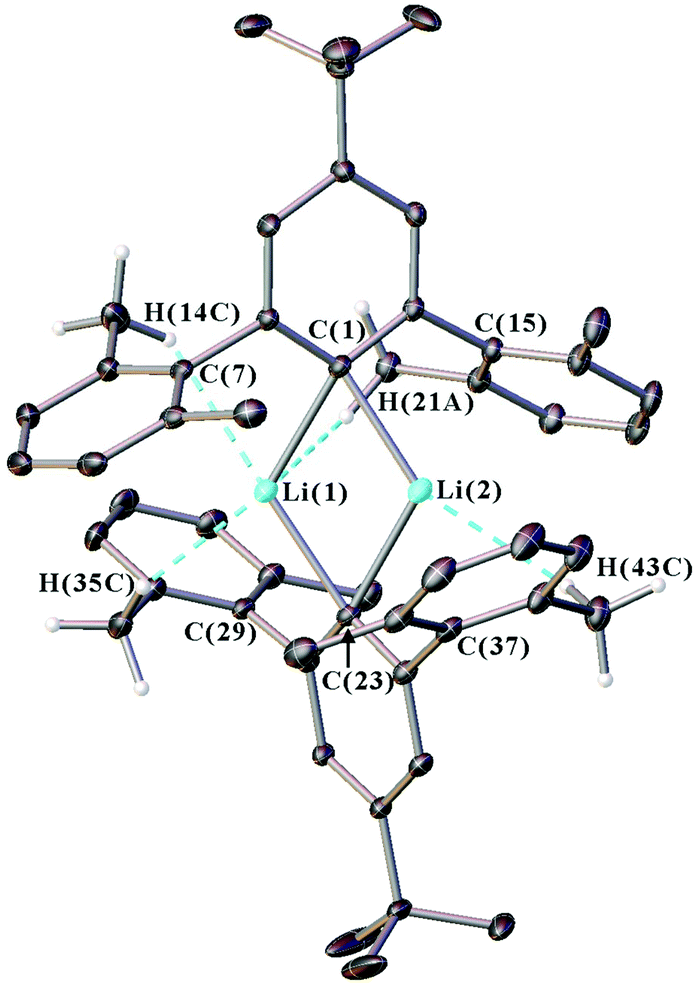 | ||
| Fig. 1 Crystal structure of the m-terphenyl lithium complex 1. Dashed lines indicate the short Li⋯H–C anagostic contacts. Ellipsoids set at 40% probability, and the hydrogen atoms are placed in idealised positions for structure refinement. All non-anagostic hydrogen atoms and residual solvent molecules are omitted for clarity. | ||
Weaker intramolecular interactions are also observed between the lithium ions and the xylyl flanking groups, with secondary contacts that are shorter than the sum of the van der Waals radii for lithium and hydrogen (3.02 Å) or lithium and carbon (3.52 Å).34,35 A summary of these contacts is given in Table S2 of the ESI.† For complexes 1, 2, 4 and 5, the lithium ions interact with the ipso-carbons of the flanking aryls [2.4244(15)–2.729(4) Å], and also form multiple Li⋯H–C anagostic interactions36 with the hydrogens of the flanking methyl groups [2.338(2)–2.920(3) Å].
In summary, the crystal structures of 1, 2, 4 and 5 show little structural variation as the para-substituent is changed, which suggests that the geometries of m-terphenyl organolithiums are dominated by steric rather than electronic factors.5,6,19,20 The structures do, however, show weak Li⋯H–C anagostic contacts between the lithium ions and the flanking methyl groups. These interactions have been explored further in our NMR spectroscopic investigations, as discussed below.
2.3. NMR spectroscopic characterisation
The electronic properties of 1–5 were studied by 1H, 13C{1H} and 7Li{1H} NMR spectroscopies in d6-benzene. Due to their poor solubilities the solutions were saturated, except for 1 whose t-Bu group aided dissolution. To determine the aggregation state of the complexes in solution, DOSY experiments were performed.37,38 Whilst the unsubstituted iodide H–Ar#–I gave a hydrodynamic radius (rH) of 3.0 Å, the equivalent lithium complex 3 gave a larger rH of 4.8 Å, which correlates with its approximate crystallographic dimensions (9.4 × 9.9 Å). The other para-substituted complexes yield similarly large radii that, except for 5, increase with bulkier substituents. In all cases, only one set of NMR spectroscopic resonances is observed, suggesting that only one aggregation state is present in solution for each of the complexes. It should be noted that the rH values for 1, 2, 4, and 5 deviate more from the crystallographic dimensions than 3, which is likely due to the more ellipsoidal shape of the molecules (see Fig. S33, ESI†).39 However, the overall increase of hydrodynamic radii for 1–5 compared to the monomeric iodide H–Ar#–I suggests that all these lithium complexes are dimeric in d6-benzene solution. The results are summarised in Table 1.| Compounda | Diffusion coefficient, D (10−10 m2 s−1) | Hydrodynamic radius, rH (Å) | Crystallographic diameters, dc (Å) | |
|---|---|---|---|---|
| Lengthb | Widthc | |||
| a All samples were prepared as 24 mM solutions in d6-benzene. b Length along the compound measured between R⋯I (H–Ar#–I) and R⋯R (1–5), where R is the outermost nucleus of the para-substituent. c Width across the compound measured between C⋯C for the outermost carbon atoms of the flanking aryl rings on each m-terphenyl ligand. All widths averaged at 9.9 Å. | ||||
| H–Ar#–I | 12.30 | 3.0 | 4.9 | 9.9 |
| [H–Ar#–Li]2 (3) | 7.54 | 4.8 | 9.4 | 9.9 |
| [Cl–Ar#–Li]2 (4) | 6.72 | 5.4 | 12.8 | 9.9 |
| [F3C–Ar#–Li]2 (5) | 7.59 | 4.8 | 13.4 | 9.9 |
| [t-Bu–Ar#–Li]2 (1) | 6.59 | 5.5 | 13.6 | 9.9 |
| [Me3Si–Ar#–Li]2 (2) | 6.27 | 5.8 | 14.4 | 9.9 |
The key 1H, 13C{1H}, and 7Li{1H} NMR spectroscopic signals of 1–5 are summarised in Table 2, with the numbering scheme presented in Fig. 2. The 1H NMR spectra show four characteristic peaks, although in some cases the signals for the aromatic xylyl protons (H-7 and H-8) overlap. Relative to the iodides (R–Ar#–I), all resonances are shifted upfield due to the greater shielding provided by the anionic ligands. A comparison of the 1H NMR spectra across the series reveals three key features. Firstly, the meta-hydrogens on the central rings exhibit small variations in chemical shift (H-3 = 6.85, 7.04, 6.77, 6.78, 6.99 ppm for 1–5, respectively) with no overall trend, see Table 2. Secondly, the xylyl aryl protons, H-7 (6.86–7.01 ppm) and H-8 (6.94–7.01 ppm), remain essentially unshifted, suggesting that the substituent in the para-position has minimal electronic communication with the flanking aryl rings. Thirdly, the xylyl methyl protons shift upfield (H-9 = 1.83, 1.81, 1.80, 1.61, 1.55 ppm for 1–5, respectively) with increased electron-withdrawing strength of the para-substituent. A graph of these chemical shifts (δ) against the corresponding Hammett constants (σpara)40 reveals a linear correlation, see Fig. 4 (blue line; R2 = 0.92). This trend is not observed in the respective iodide compounds (H-9 = 2.06, 2.05, 2.06, 2.07 and 2.02 ppm for R–Ar#–I, where R = t-Bu, SiMe3, H, Cl and CF3, respectively) and proceeds in the opposite direction to what one might expect, with electron-withdrawing groups causing an apparent shielding effect. This suggests that the para-substituent is exerting a direct electronic influence on the environment of the H-9 methyl protons.
 | ||
| Fig. 2 NMR numbering scheme for the m-terphenyl lithium complexes 1–5. | ||
| [R–Ar#–Li]2 | R group | 1H, 13C{1H} and 7Li{1H} NMR Chemical Shifts, δ (ppm) | Li | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H-3 | H-9 | C-1 | C-2 | C-3 | C-4 | C-9 | |||
| a Peak C-4 for 5 not observed (obscured by d6-benzene solvent peak). | |||||||||
| 1 | t-Bu | 6.85 | 1.83 | 168.1 | 152.0 | 120.4 | 148.7 | 21.8 | 1.60 |
| 2 | SiMe3 | 7.04 | 1.81 | 174.2 | 151.6 | 128.1 | 136.5 | 21.9 | 1.47 |
| 3 | H | 6.77 | 1.80 | 172.7 | 152.0 | 123.6 | 126.3 | 21.8 | 1.46 |
| 4 | Cl | 6.78 | 1.61 | 170.7 | 153.6 | 123.6 | 132.9 | 21.5 | 1.10 |
| 5 | CF3 | 6.99 | 1.55 | 180.2 | 152.4 | 119.5 | —a | 21.5 | 0.93 |
The 13C{1H} NMR spectra of 1–5 show nine peaks for the carbons of the ligand framework, with additional peaks for the t-Bu, SiMe3 and CF3 groups in 1, 2 and 5, respectively. Compared to the iodides (R–Ar#–I), the largest change is seen in the ipso-carbons, which are shifted downfield (C-1 = 106.9 vs. 172.7 ppm for H–Ar#–I and 3, respectively) owing to a large deshielding effect in the plane perpendicular to the C–Li bond.19,41–44 In all cases, the NMR resonances for the ipso-carbons of 1–5 were of low intensity and therefore assigned from 1H–13C HMBC spectra. For 3, a longer 13C{1H} NMR experiment with d1 of 10 s was performed to achieve better resolution of the ipso-carbon peak (Fig. 3). This revealed a seven-line splitting pattern (blue) centred at 172.7 ppm with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:4:3:2:1 intensity ratio (1J13C7Li = 23.3 Hz) characteristic of quadrupolar coupling to two 7Li nuclei (I = 3/2, natural abundance: 92.41%), providing further evidence that the lithium complexes are dimeric in d6-benzene solution.45–48 The experimental spectrum was well simulated (red, see ESI, section S1†). Multiplets arising from 6Li (I = 1, natural abundance = 7.59%) were too low intensity to resolve experimentally.
:2:3:4:3:2:1 intensity ratio (1J13C7Li = 23.3 Hz) characteristic of quadrupolar coupling to two 7Li nuclei (I = 3/2, natural abundance: 92.41%), providing further evidence that the lithium complexes are dimeric in d6-benzene solution.45–48 The experimental spectrum was well simulated (red, see ESI, section S1†). Multiplets arising from 6Li (I = 1, natural abundance = 7.59%) were too low intensity to resolve experimentally.
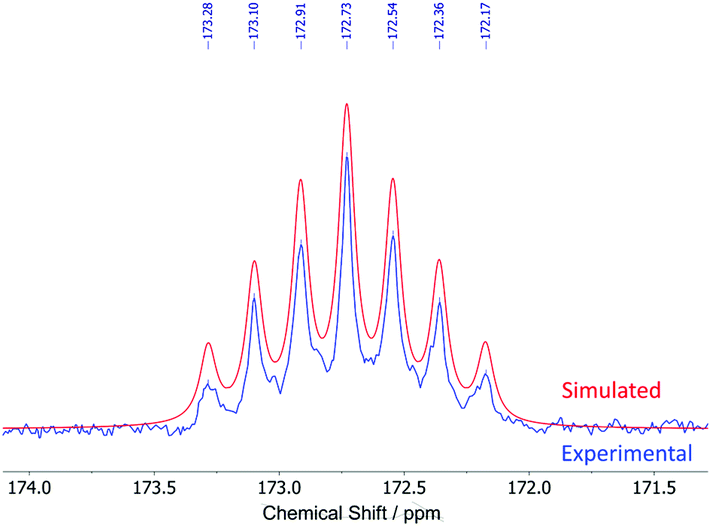 | ||
| Fig. 3 Seven-line splitting pattern of the ipso-carbons in the experimental (blue) and simulated (red) 13C{1H} NMR spectra of [H–Ar#–Li]2 (3). | ||
A comparison of the 13C{1H} NMR spectra of 1–5 reveals that all 13C signals of the xylyl flanking groups remain essentially unshifted across the series. The shift observed for the xylyl methyl groups in the 1H NMR spectrum (H-9) is not replicated in the 13C{1H} NMR spectrum (C-9). The peaks for the central ring carbons vary considerably across the series (except for C-2, see Table 2): larger variations are noted for C-4 (126.3–148.7 ppm) than for C-1 (168.1–180.2 ppm) and C-3 (119.5–128.1 ppm). Though no reliable trends can be identified, a general downfield shift is observed for C-1 with increasing σpara.
The 7Li{1H} NMR spectra of 1–5 also provided valuable information on the complexes. Although 7Li NMR spectroscopy is typically sensitive to the analyte concentration, aggregation, and solvent effects, these factors appear to be negligible here.49–51 This was demonstrated by the spectra for the more soluble t-Bu complex (1) whose 7Li{1H} NMR resonance remains unshifted over a wide concentration range (1.60 ppm at 24, 48 and 72 mM, respectively). This may be due to the steric bulk of the ligands shielding the metal centre from the surrounding solvent. For complexes 2–5, saturated samples were used for NMR measurements due to their poor solubilities in d6-benzene. The steric influence of the xylyl flanking groups on the lithium ions is assumed to remain consistent throughout the series and, therefore, not to contribute to changes in the 7Li{1H} NMR chemical shifts. This is in contrast to previous work on m-terphenyl lithium complexes, where changing the steric bulk of the flanking groups varies the 7Li{1H} NMR signal.15
In all cases, the 7Li{1H} NMR spectra show a single environment for the two lithium ions of the dimer. The position of these peaks shifts upfield (1.60, 1.47, 1.46, 1.10 and 0.93 ppm for 1–5, respectively) as the electron-withdrawing strength of the para-substituent is increased (Table 2). Once again, plotting a graph of these chemical shifts (δ) against the Hammett constants (σpara) reveals a linear correlation, see Fig. 4 (red line; R2 = 0.96),40 indicating a direct influence on the electronic properties of the lithium ions by the para-substituent. As with the H-9 protons, the trend is counterintuitive, since one might expect electron-withdrawing groups to deshield the nuclei and cause a downfield shift. However, a similar observation has been reported for the series of para-substituted aryllithium complexes [4-R-C6H4]2Li (R = OMe, Me, H, F, Cl, CF3).52–54
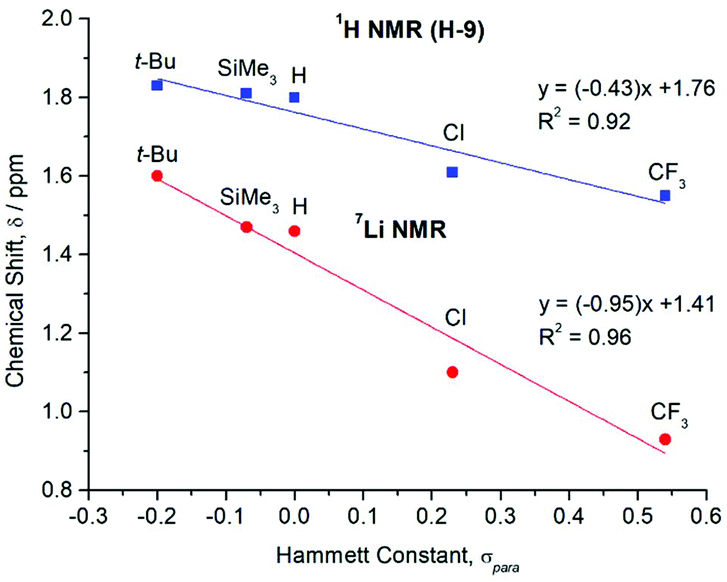 | ||
| Fig. 4 Plot of the 1H (for flanking methyl protons, H-9) and 7Li{1H} NMR chemical shifts (δ) for the para-substituted lithium complexes [R–Ar#–Li]2 (1–5) versus their corresponding literature Hammett constants (σpara).40 | ||
These 7Li{1H} NMR spectroscopic results help to rationalise the trend observed for the flanking methyl protons (H-9) (Fig. 4). Since all other atoms on the xylyl rings remain unshifted, this suggests that electronic communication occurs via a through-space interaction between the methyl protons and lithium atoms. Some evidence of this is seen in the crystal structures, with weak Li⋯H–C anagostic interactions observed in the solid state (Fig. S31 and S32†). In solution, this is further corroborated by two-dimensional 7Li–1H HOESY experiments,55,56 which reveal short 7Li⋯1H contacts in 1–5 as evidenced by the intense cross-peak between the lithium signals and the H-9 protons (Fig. 5 and ESI, section 3†).
 | ||
| Fig. 5 Superimposed 2D 7Li–1H HOESY spectra of the m-terphenyl lithium complexes [R–Ar#–Li]2 (1–5) showing strong cross-peaks between the flanking methyl protons, H-9, and the 7Li nuclei. | ||
Similar Li⋯H interactions have been reported for the monomeric [Mes*-Li] species, whose 6Li–1H HOESY spectrum gave a cross-peak for the ortho tert-butyl protons, but not for the para tert-butyl protons.42 Moreover, the ortho groups (1.61 ppm) occurred at lower field than the para groups (1.56 ppm), owing to the electric field produced by lithium that deshields the protons within close proximity.42 Comparable findings have been described for other aryllithium complexes [Ar–Li] (Ar = Naph, 2-{t-BuS}C6H4).57,58
From these results we suggest that the electronic influence of the para-substituents in complexes 1–5 is relayed through the central m-terphenyl ring, onto the lithium atoms, then through-space onto the nearby methyl protons. Evidence of similar through-space interactions within a m-terphenyl framework has already been reported for the m-terphenyl carboxylic acids (2,6-Ar2C6H3)COOH (Ar = 4-R-C6H4; R = OMe, Me, H, F, Cl, Br, C{O}Me) where para-substitution of the flanking aryl rings influences the pKa value, owing to through-space effects.59 In addition, the m-terphenyl silane (2,6-Ar2C6H3)SiMe2H (Ar = 2,6-F2C6H3) possesses flanking aryl fluorine atoms that couple through-space to the Si–H proton.60 To further investigate the origin of this effect we turned to computational analysis and DFT studies.
2.4. Computational studies
The calculation of NMR chemical shift parameters was carried out using the ReSpect program61–66 on structures obtained directly from the crystal structures, with optimisation of only the H atom locations at the PBE0 level;67,68 see ESI, section S5† for full computational details. NMR shielding constants were calculated using the KT2 density-functional approximation,69 in the pcS-n basis sets70 (n = 1, 2). This functional was specifically designed for the calculation of NMR shielding constants, as were the pcS-n basis sets. Comparisons of the results for a range of other commonly used functionals (BLYP, BP86, PBE, PBE0, PP86)67,68,71–74 may be found in the ESI.†Tables summarising the calculated shifts for H-9 and Li are given in the ESI, Tables S6 and S7.† Because of the relatively narrow chemical shift range, it is difficult to accurately model the trends shown in Fig. 3. Furthermore, both the H-9 and Li shifts are composed from absolute shielding constants containing large paramagnetic components (ESI, Table S5†). It is known that when the paramagnetic components are dominant, as in these cases, then density-functional methods often fail to achieve high accuracy. It is also known that density-functionals tend to be more accurate for 1H and 13C{1H} NMR resonances than those for other nuclei.75 In line with these observations the calculated values for the 7Li{1H} NMR spectrum do not accurately capture the trend in Fig. 3. However, the trend for the 1H NMR of the flanking H-9 nuclei is more adequately reproduced, particularly for the KT2 functional in the larger pcS-2 basis. As shown in Fig. S34 of the ESI† the general trend for an upfield shift with increasing σpara is reproduced, see also Table S7.† Linear regression of the calculated 1H NMR shifts for the H-9 nuclei as a function of σpara yields a fit with R2 = 0.92, a somewhat steeper slope of −0.62 and a y-intercept of +1.82 ppm. Given the challenges associated with the relatively narrow range of chemical shifts and large paramagnetic contributions for these nuclei, these results are in reasonable agreement with the experimental observations. However, based on these results, it seems likely that the counterintuitive trends observed in both the 1H and 7Li{1H} NMR resonances is due to a large paramagnetic contribution to the shielding constant. Finally, we note that the more challenging 7Li{1H} NMR spectroscopy may provide a useful test case for the development of more refined density-functionals for NMR studies.
3. Conclusions
A series of para-substituted m-terphenyl lithium complexes [R–Ar#–Li]2 (R = t-Bu 1, SiMe32, H 3, Cl 4, CF35) have been reported. Whilst crystallographically similar, and all displaying Li⋯H–C anagostic contacts in the solid state, NMR spectroscopic studies reveal significant electronic differences at the metal centre. A linear correlation was observed between the Hammett constants of the para-substituent and the 7Li and 1H NMR shifts of the lithium atoms and flanking methyl protons (H-9). Electronic interaction between these atoms was confirmed by 7Li–1H HOESY measurements. In both cases, increasing the electron-withdrawing power of the para-substituent results in a counterintuitive upfield peak shift. Computational analysis suggests this effect is due to an electric field generated about the lithium atoms, which is influenced by the para-substituent, and results in large paramagnetic and diamagnetic contributions to chemical shifts. While the observed trend in the 1H NMR spectra of the H-9 protons was reproduced relatively well, it was difficult to accurately model the 7Li NMR spectroscopic trend by DFT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the EPSRC [grant number EP/R004064/1], the Leverhulme Trust [grant number RPG-2014-317], and the University of Nottingham for financial support of this research. We also thank the University of Nottingham Analytical Services Team for elemental analyses, mass spectrometry, and NMR spectroscopy measurements. We are also grateful for access to the University of Nottingham's Augusta High Performance Computing service. AMT is grateful for support from the European Research Council under H2020/ERC Consolidator Grant “topDFT” (Grant No. 772259).References
- D. L. Kays, Organomet. Chem., 2010, 36, 56–76 CAS.
- E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS.
- L. J. Taylor and D. L. Kays, Dalton Trans., 2019, 48, 12365–12381 RSC.
- C. Ni and P. P. Power, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 59–111 Search PubMed.
- B. Twamley, S. T. Haubrich and P. P. Power, Adv. Organomet. Chem., 1999, 44, 1–65 CrossRef CAS.
- J. A. C. Clyburne and N. McMullen, Coord. Chem. Rev., 2000, 210, 73–99 CrossRef CAS.
- D. L. Kays and A. R. Cowley, Chem. Commun., 2007, 1053–1055 RSC.
- C. Stanciu, A. F. Richards, J. C. Fettinger, M. Brynda and P. P. Power, J. Organomet. Chem., 2006, 691, 2540–2545 CrossRef CAS.
- B. M. Gridley, A. J. Blake, A. L. Davis, W. Lewis, G. J. Moxey and D. L. Kays, Chem. Commun., 2012, 48, 8910–8912 RSC.
- B. M. Gridley, T. J. Blundell, G. J. Moxey, W. Lewis, A. J. Blake and D. L. Kays, Chem. Commun., 2013, 49, 9752–9754 RSC.
- B. Schiemenz and P. P. Power, Organometallics, 1996, 15, 958–964 CrossRef CAS.
- L. G. Perla, J. M. Kulenkampff, J. C. Fettinger and P. P. Power, Organometallics, 2018, 37, 4048–4054 CrossRef CAS.
- J. Su, X. W. Li, R. C. Crittendon and G. H. Robinson, J. Am. Chem. Soc., 1997, 119, 5471–5472 CrossRef CAS.
- R. J. Wehmschulte, W. J. Grigsby, B. Schiemenz, R. A. Bartlett and P. P. Power, Inorg. Chem., 1996, 35, 6694–6702 CrossRef CAS PubMed.
- T. J. Blundell, F. R. Hastings, B. M. Gridley, G. J. Moxey, W. Lewis, A. J. Blake and D. L. Kays, Dalton Trans., 2014, 43, 14257–14264 RSC.
- B. Twamley, C. D. Sofield, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1999, 121, 3357–3367 CrossRef CAS.
- S. Wang, M. L. McCrea-Hendrick, C. M. Weinstein, C. A. Caputo, E. Hoppe, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2017, 139, 6586–6595 CrossRef CAS.
- S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS.
- K. Ruhlandt-Senge, J. J. Ellison, R. J. Wehmschulte, F. Pauer and P. P. Power, J. Am. Chem. Soc., 1993, 115, 11353–11357 CrossRef CAS.
- B. Schiemenz and P. P. Power, Angew. Chem., Int. Ed. Engl., 1996, 35, 2150–2152 CrossRef CAS.
- R. C. Fischer, L. Pu, J. C. Fettinger, M. A. Brynda and P. P. Power, J. Am. Chem. Soc., 2006, 128, 11366–11367 CrossRef CAS PubMed.
- E. Rivard, R. C. Fischer, R. Wolf, Y. Peng, W. A. Merrill, N. D. Schley, Z. Zhu, L. Pu, J. C. Fettinger, S. J. Teat, I. Nowik, R. H. Herber, N. Takagi, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2007, 129, 16197–16208 CrossRef CAS PubMed.
- Z. Zhu, R. C. Fischer, B. D. Ellis, E. Rivard, W. A. Merrill, M. M. Olmstead, P. P. Power, J. D. Guo, S. Nagase and L. Pu, Chem. – Eur. J., 2009, 15, 5263–5272 CrossRef CAS PubMed.
- Y. Peng, R. C. Fischer, W. A. Merrill, J. Fischer, L. Pu, B. D. Ellis, J. C. Fettinger, R. H. Herber and P. P. Power, Chem. Sci., 2010, 1, 461–468 RSC.
- P. Wilfling, K. Schittelkopf, M. Flock, R. H. Herber, P. P. Power and R. C. Fischer, Organometallics, 2015, 34, 2222–2232 CrossRef CAS.
- R. Wolf, C. Ni, T. Nguyen, M. Brynda, G. J. Long, A. D. Sutton, R. C. Fischer, J. C. Fettinger, M. Hellman, L. Pu and P. P. Power, Inorg. Chem., 2007, 46, 11277–11290 CrossRef CAS PubMed.
- S. Hino, M. M. Olmstead, J. C. Fettinger and P. P. Power, J. Organomet. Chem., 2005, 690, 1638–1644 CrossRef CAS.
- It should be noted that t-Bu-Ar#-I has been reported once before. However, it was only briefly mentioned as an intermediate in a multi-step reaction and very little characterisation data were given. Please see: S. Watanabe, K. Goto, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1995, 36, 7677–7680 CrossRef CAS.
- M. Tian, L. Liu, Y. Li, R. Hu, T. Liu, H. Liu, S. Wang and Y. Li, Chem. Commun., 2014, 50, 2055–2057 RSC.
- T. C. Bedard and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 10662–10671 CrossRef CAS.
- D. Wrobel and U. Wannagat, J. Organomet. Chem., 1982, 225, 203–210 CrossRef CAS.
- A. Saednya and H. Hart, Synthesis, 1996, 1455–1458 CrossRef CAS.
- D. K. Miller, C. A. Bailey and R. E. Sammelson, Synthesis, 2015, 2791–2798 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- Mercury User Guide and Tutorials, Cambridge Crystallographic Data Centre, 2018, pp. 306–307 Search PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- T. D. W. Claridge, in High-Resolution NMR Techniques in Organic Chemistry, ed. T. D. W. Claridge, Elsevier, 2nd edn, 2009, vol. 27, pp. 303–334 Search PubMed.
- I. Keresztes and P. G. Williard, J. Am. Chem. Soc., 2000, 122, 10228–10229 CrossRef CAS.
- L. Allouche, A. Marquis and J. M. Lehn, Chem. – Eur. J., 2006, 12, 7520–7525 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. Seebach, R. Hässig and J. Gabriel, Helv. Chim. Acta, 1983, 66, 308–337 CrossRef CAS.
- W. Bauer, W. R. Winchester and P. V. R. Schleyer, Organometallics, 1987, 6, 2371–2379 CrossRef CAS.
- S. Berger, U. Fleischer, C. Geletneky and J. C. W. Lohrenz, Chem. Ber., 1995, 128, 1183–1186 CrossRef CAS.
- H. Dahn, J. Chem. Educ., 2000, 77, 905–909 CrossRef CAS.
- J. T. B. H. Jastrzebski, G. van Koten, M. Konijn and C. H. Stam, J. Am. Chem. Soc., 1982, 104, 5490–5492 CrossRef CAS.
- M. H. P. Rietveld, I. C. M. Wehman-Ooyevaar, G. M. Kapteijn, D. M. Grove, W. J. J. Smeets, H. Kooijman, A. L. Spek and G. van Koten, Organometallics, 1994, 13, 3782–3787 CrossRef CAS.
- E. Wehman, J. T. B. H. Jastrzebski, J. M. Ernsting, D. M. Grove and G. van Koten, J. Organomet. Chem., 1988, 353, 133–143 CrossRef CAS.
- E. Wehman, J. T. B. H. Jastrzebski, J. M. Ernsting, D. M. Grove and G. van Koten, J. Organomet. Chem., 1988, 353, 145–155 CrossRef CAS.
- P. A. Scherr, R. J. Hogan and J. P. Oliver, J. Am. Chem. Soc., 1974, 96, 6055–6059 CrossRef CAS.
- H. Günther, D. Moskau, P. Bast and D. Schmalz, Angew. Chem., Int. Ed. Engl., 1987, 26, 1212–1220 CrossRef.
- H. Günther, eMagRes, 2007, 1–21 Search PubMed.
- J. A. Ladd, Spectrochim. Acta, 1966, 22, 1157–1163 CrossRef CAS.
- J. Parker and J. A. Ladd, J. Organomet. Chem., 1969, 19, 1–7 CrossRef CAS.
- J. A. Ladd and J. Parker, J. Chem. Soc., Dalton Trans., 1972, 930–934 RSC.
- W. Bauer, Magn. Reson. Chem., 1996, 34, 532–537 CrossRef CAS.
- W. Bauer, G. Müller, R. Pi and P. von Ragué Schleyer, Angew. Chem., Int. Ed. Engl., 1986, 25, 1103–1104 CrossRef.
- W. Bauer, M. Feigel, G. Mueller and P. V. R. Schleyer, J. Am. Chem. Soc., 1988, 110, 6033–6046 CrossRef CAS PubMed.
- W. Bauer, P. A. A. Klusener, S. Harder, J. A. Kanters, A. J. M. Duisenberg, L. Brandsma and P. V. R. Schleyer, Organometallics, 1988, 7, 552–555 CrossRef CAS.
- C.-T. Chen and J. S. Siegel, J. Am. Chem. Soc., 1994, 116, 5959–5960 CrossRef CAS.
- P. Romanato, S. Duttwyler, A. Linden, K. K. Baldridge and J. S. Siegel, J. Phys. Org. Chem., 2014, 27, 277–283 CrossRef CAS.
- M. Repisky, S. Komorovsky, V. G. Malkin, O. L. Malkina, M. Kaupp and K. Ruud, with contributions from R. Bast, R. Di Remigio, U. Ekström, M. Kadek, S. Knecht, L. Konecny, E. Malkin and I. Malkin Ondik, ReSpect 5.1.0, relativistic spectroscopy DFT program, http://www.respectprogram.org, 2019.
- S. Komorovský, M. Repiský, O. L. Malkina, V. G. Malkin, I. Malkin Ondík and M. Kaupp, J. Chem. Phys., 2008, 128, 104101 CrossRef PubMed.
- S. Komorovský, M. Repiský, O. L. Malkina and V. G. Malkin, J. Chem. Phys., 2010, 132, 154101 CrossRef PubMed.
- S. Komorovsky, M. Repisky, E. Malkin, T. B. Demissie and K. Ruud, J. Chem. Theory Comput., 2015, 11, 3729–3739 CrossRef CAS PubMed.
- M. Repisky, S. Komorovsky, R. Bast and K. Ruud, in New Developments in NMR No. 6, ed. M. J. Karol Jackowski, The Royal Society of Chemistry, 2016, pp. 267–303 Search PubMed.
- M. Repiský, S. Komorovský, O. L. Malkina and V. G. Malkin, Chem. Phys., 2009, 356, 236–242 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. W. Keal and D. J. Tozer, J. Chem. Phys., 2003, 119, 3015–3024 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef PubMed.
- A. M. Teale, O. B. Lutnæs, T. Helgaker, D. J. Tozer and J. Gauss, J. Chem. Phys., 2013, 138, 024111 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2045214–2045221. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03972a |
| This journal is © The Royal Society of Chemistry 2021 |