
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the hyperpolarisation of EGTA-based ligands using SABRE†
Ben. J.
Tickner‡§
 a,
Yulia
Borozdina‡
b,
Simon B.
Duckett
*a and
Goran
Angelovski
*bc
a,
Yulia
Borozdina‡
b,
Simon B.
Duckett
*a and
Goran
Angelovski
*bc
aCentre for Hyperpolarisation in Magnetic Resonance (CHyM), Department of Chemistry, University of York, Heslington, York YO10 5NY, UK. E-mail: simon.duckett@york.ac.uk
bMR Neuroimaging agents, Max Planck Institute for Biological Cybernetics, Tuebingen, 72076, Germany. E-mail: goran.angelovski@tuebingen.mpg.de
cLaboratory of Molecular and Cellular Neuroimaging, International Center for Primate Brain Research (ICPBR), Center for Excellence in Brain Science and Intelligence Technology (CEBSIT), Chinese Academy of Sciences (CAS), Shanghai, PR China
First published on 22nd January 2021
Abstract
The design of molecules whose magnetic resonance (MR) signals report on their biological environment is receiving attention as a route to non-invasive functional MR. Hyperpolarisation techniques improve the sensitivity of MR and enable real time low concentration MR imaging, allowing for the development of novel functional imaging methodologies. In this work, we report on the synthesis of a series of EGTA-derived molecules (EGTA – ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid), whose core structures are known to bind biologically relevant metal ions in vivo, in addition to pyridyl rings that allow reversible ligation to an iridium dihydride complex. Consequently, they are amenable to hyperpolarisation through the parahydrogen-based signal amplification by reversible exchange (SABRE) process. We investigate how the proximity of EGTA and pyridine units, and the identity of the linker group, affect the SABRE hyperpolarisation attained for each agent. We also describe the effect of catalyst identity and co-ligand presence on these measurements and can achieve 1H NMR signal enhancements of up to 160-fold. We rationalise these results to suggest the design elements needed for probes amenable to SABRE hyperpolarisation whose MR signals might in the future report on the presence of metal ions.
Introduction
Many spectroscopic techniques have been developed to study the chemical structures of molecules and the morphology of materials. These include the development of methods that rely on the principles of magnetic resonance (MR) to probe nuclear spin without the need for approaches based on ionising radiation such as X-ray, computed or positron emission tomography (CT or PET respectively), or sample destruction as in mass spectrometry. These advantages have led to magnetic resonance imaging (MRI) becoming routine for the diagnosis of structural abnormalities in living tissue.Despite the diversity of its successes, MR is insensitive at the molecular level as the detected signal relies on small Boltzmann derived population differences across closely spaced nuclear spin energy levels. In fact, only around 1 in every 195![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 1H nuclei contribute positively to an MR signal recorded in a 1.5 T field common for clinical MRI scanners. It is not surprising therefore that MRI is usually concerned with detection of highly concentrated species, such as bulk water. Nevertheless, images with anatomical contrast can be produced by T1 or T2 weighted sequences to differentiate tissue morphologies. The information content of these approaches is often improved by the injection of a paramagnetic contrast agent.1 However, as such images normally convey little direct information about biological function, there are opportunities to improve the diagnostic ability of MRI.
000 1H nuclei contribute positively to an MR signal recorded in a 1.5 T field common for clinical MRI scanners. It is not surprising therefore that MRI is usually concerned with detection of highly concentrated species, such as bulk water. Nevertheless, images with anatomical contrast can be produced by T1 or T2 weighted sequences to differentiate tissue morphologies. The information content of these approaches is often improved by the injection of a paramagnetic contrast agent.1 However, as such images normally convey little direct information about biological function, there are opportunities to improve the diagnostic ability of MRI.
Many researchers are attempting to achieve this using biologically induced changes in the bulk water signal intensity.2,3 Among these are approaches such as functional MRI (fMRI), which attempts to probe the bulk water response as a function of the oxygenation state of haemoglobin,4 or chemical exchange saturation transfer (CEST), which modulates bulk water signals via proton exchange with an injected CEST agent.5 More recently, specific types of MRI contrast agents have been developed, being responsive to metal ions6 (Ca2+, K+, Mg2+, Zn2+ Fe2+, Fe3+, Cu2+ and others), neurotransmitters7 and proteins.8 The use of functional reporters that are able to respond to such changes may therefore have benefit in studying different biological processes in a non-invasive way.
Approaches that target Ca2+, both intracellular and extracellular, could have remarkable impact in neuroimaging due to the essential role of this ion in neuronal signalling.9 Other physiological processes that depend on Ca2+ could also be followed in real-time by means of MRI.10 For this approach to work, the preparation of appropriate functional markers is necessary and consequently, different types of Ca-responsive agents have been developed to date.11,12 Such reporters often consist of paramagnetic chelates coupled to a calcium sensing unit via a suitable linker.11 Conformational changes in the agent upon metal ion binding are used to increase the inner sphere hydration of the paramagnetic ions, most commonly gadolinium, thereby enhancing the agent's relaxivity and subsequently increasing the intensity of the MRI signal in T1 weighted images.12
While these functional imaging approaches have achieved great success, they do not fully address the low sensitivity of MRI. Over recent years, hyperpolarisation has been used to produce contrast agents with MR signals whose intensity is enhanced by up to five orders of magnitude relative to those derived from Boltzmann laws.13 With these hyperpolarised agents, successful imaging of low concentration biomolecules, drugs and metabolites in vivo was accomplished.13–15 While there are several possible experimental techniques for the production of hyperpolarised contrast agents, dissolution dynamic nuclear polarisation (d-DNP) has achieved the most success and a growing range of clinical applications are developing.13 However, d-DNP uses complex experimental apparatus to transfer polarisation from electrons to nuclei over a period of tens of minutes to several hours. The d-DNP procedure involves microwave irradiation of a target agent and organic radical in a 1–10 T field at temperatures between 1 and 5 K before rapid sample melting and transfer into the detection system.13 d-DNP hyperpolarisation has yielded 13C polarisation levels of up to 70% for pyruvate-1-[13C]14 and its subsequent metabolic imaging can identify cancer in humans in vivo.15
An alternative hyperpolarisation method uses parahydrogen (pH2), the antisymmetric nuclear spin isomer of dihydrogen, as its source of polarisation. pH2 reflects a potentially cheaper and faster route to a hyperpolarised contrast agent for in vivo injection and detection.16 Hydrogen gas exists as 25% pH2 at room temperature with the remaining 75% consisting of orthohydrogen. H2 can easily be enriched in the para state (>98%) by cooling to low temperature (28 K) in the presence of a spin exchange catalyst.17 While pH2 is NMR silent, its latent hyperpolarisation is typically unlocked in a hydrogenation reaction.13 Consequently, pH2 induced hyperpolarisation (PHIP) usually requires unsaturated functionality within the target agent.16 This can be alleviated by using a non-hydrogenative variant of PHIP, called signal amplification by reversible exchange (SABRE), which relies on a catalytic process to transfer pH2 derived spin order to a target molecule.18 While SABRE is clearly at a much earlier stage in its development than DNP, the simplicity of this approach suggest that its potential clinical use is highly desirable. This can be highlighted by the fact that SABRE works in seconds by creating an iridium-based active polarisation transfer complex, which exchanges both pH2 and a target substrate (Scheme 1).
 | ||
| Scheme 1 SABRE involves the in situ formation of active polarisation transfer catalysts that reversibly exchange both pH2 and substrate (Sub). Such species are typically of the type [Ir(H)2(NHC)(Sub)3]Cl where NHC is an N-heterocyclic carbene and are formed from the reaction of an iridium precatalyst with substrate and H2. Magnetisation is catalytically transferred from pH2 derived hydride ligands to ligated substrate at an optimum magnetic field. Typical substrate molecules contain N-, O- or S-donor groups. In this work, Sub contains an iridium binding and EGTA-derived motif separated by a linker (Fig. 1). We also show the structures of the NHC ligands for the iridium precatalysts used in this work. | ||
Hyperpolarisation transfer is catalytic at low magnetic field (∼65 G18 or 1–10 mG19,20 for transfer to 1H or 13C/15N nuclei respectively) from the pH2 derived hydride ligands in the catalyst to its bound target ligands through the associated J-coupling network. Subsequent ligand dissociation allows the build-up of chemically unchanged and yet hyperpolarised molecules in solution before their magnetisation decays slowly under relaxation back to the Boltzmann derived state. One of the most common substrates reported for hyperpolarisation using SABRE is the N-heterocycle pyridine and its derivatives.18,20–23 Indeed, pyridine was one of the first molecules to be hyperpolarised using SABRE18 and exhibits appropriate exchange kinetics to allow efficient polarisation transfer within the active [Ir(H)2(NHC)(pyridine)3]Cl catalyst, where NHC is an N-heterocyclic carbene.24 SABRE has achieved up to 65% 1H,22 4% 13C25 and 79% 15N26 polarisation for N-heterocycles, although other functionalities including nitriles,27 amines28 and even O-donors pyruvate19 and acetate29 can be hyperpolarised by SABRE. Relayed proton exchange effects are now expanding the scope to include non-ligating molecules such as alcohols,30 sugars31 and silanols.32 SABRE hyperpolarised molecules with MR signals responsive to factors such as pH33,34 NO concentration35 or H2O236 have been reported.
Considering the importance that hyperpolarization techniques can have for the potential development of molecular fMRI approaches, in this work we aimed to combine the areas of bio-responsive MR probes and SABRE hyperpolarisation. Namely, we sought to explore SABRE on prototype responsive agents, which contain Ca-binding chelators appended with pyridyl units. Therefore, we report on the synthesis of several probes that contain functionalities based on ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), which are known to ligate biologically relevant metal ions37,38 alongside pyridyl rings that can bind iridium as required for SABRE (Fig. 1).18 Moreover, we demonstrate the SABRE hyperpolarisation properties of the agents synthesised here and discuss a strategy for the rational design of responsive probes with enhanced MR signals.
 | ||
| Fig. 1 Structures of the substrates used in this work. These contain Ca-binding chelators based on EGTA appended with pyridyl arms, which can bind iridium polarisation transfer catalysts used in SABRE hyperpolarisation. | ||
Results and discussion
Design and synthesis of EGTA-based molecular probes suitable for SABRE hyperpolarisation
Development of Ca-sensitive hyperpolarized 13C and 15N contrast agents for MRI using the d-DNP technique has been reported recently.37,39,40 One of these hyperpolarisation approaches has been applied to 13C-EGTA, which is similar to the ligands used in this work. In this example, changes in hyperpolarised 13C-labelled carboxylate chemical shifts in response to coordination to various metals including calcium, magnesium and zinc were reported.37 Here, we did not use 13C or 15N isotopically labelled molecules to improve the NMR signal, but functionalised EGTA, a typical calcium binding motif,38 with pyridyl arms to create a series of molecules amenable to SABRE hyperpolarisation. This strategy of molecular functionalisation with a pyridyl group allowed us to create molecules compatible for SABRE hyperpolarisation and has been applied previously to fentanyl derivatives,41 synthetic oligopeptides42 and others.35 The relative geometry of the pyridyl binding site and the EGTA-derived unit is expected to play an important role in determining both metal sensitivity and SABRE efficiency. The steric properties of the target agents are particularly important in controlling binding affinity to iridium, as the hyperpolarisation of bulky ligands is known to be limited by steric repulsion between the catalyst and target agent.43 Therefore, we prepared a set of agents that had ortho (o), meta (m) and para (p) relationships between the pyridyl nitrogen and the EGTA unit (Scheme 2).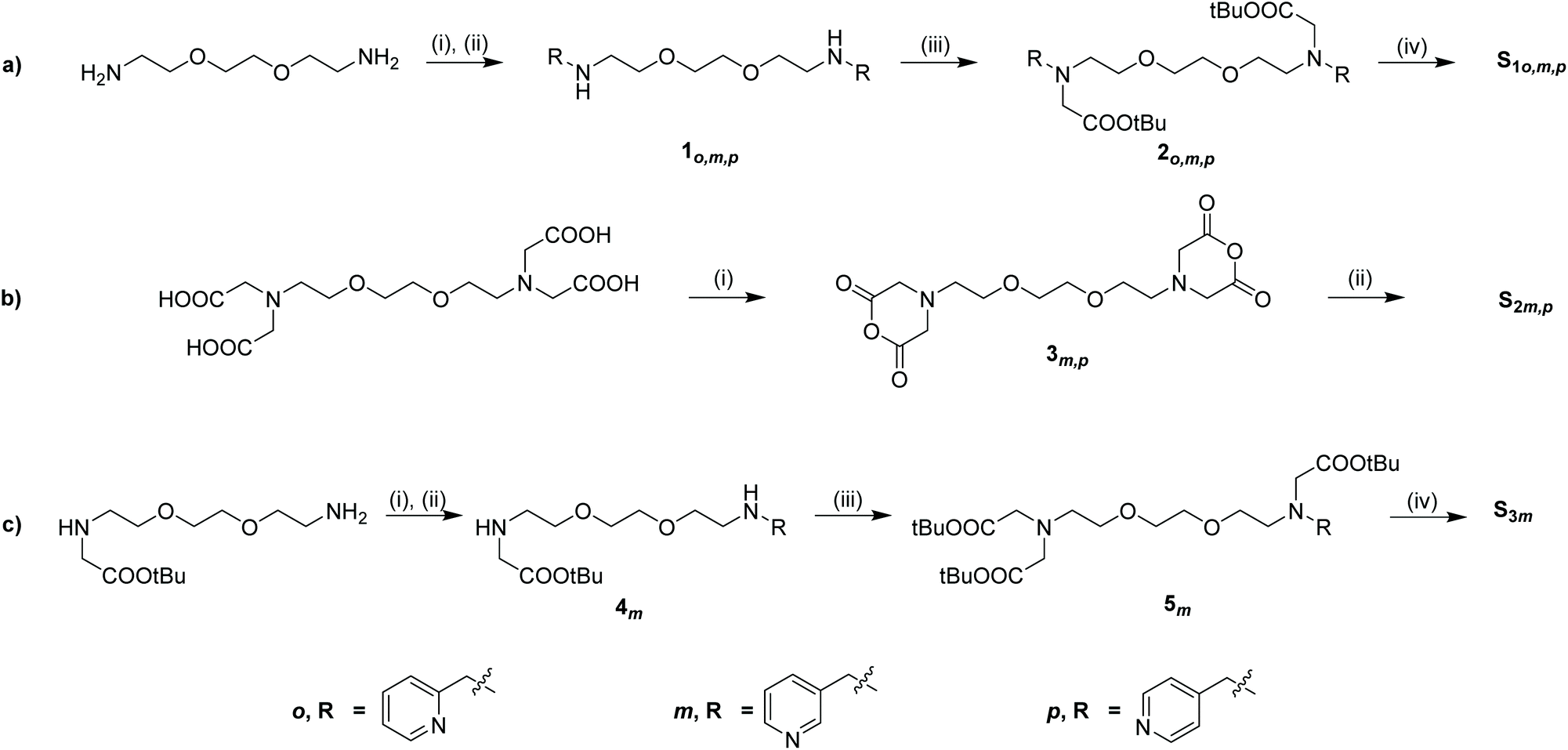 | ||
| Scheme 2 Structures of (a) S1o,m,p (b) S2m,p and (c) S3m, and a summary of their synthetic preparation: (a) (i) PyCHO, CH2Cl2, N2; (ii) NaBH4, EtOH, 0 °C, N2; (iii) BrCH2COOtBu, K2CO3, KI, DMF, N2; (iv) TFA, CH2Cl2, 0 °C, N2 (b) (i) Ac2O, CH2Cl2, N2; (ii) PyNH2, Py (or Py/DMF), N2 and (c) (i)–(iii) same as (a), (iv) HCl, dioxane, 0 °C, N2. | ||
The synthesis of the desired products was performed in different 2- or 3-step procedures. Briefly, a reductive amination procedure was used to modify a 2,2′-(ethylenedioxy)bis(ethylamine) core with ortho-, meta-, or para-pyridinecarboxaldehydes in a step that proceeded quantitatively. The subsequent successive alkylation of the resulting pyridyl ethanamines, 1o,m,p with tert-butyl bromoacetate was tested using several reaction conditions. Reasonable yields of 2o,m,p were obtained in dry acetonitrile with the base potassium carbonate and potassium iodide. After their isolation and the subsequent acidic hydrolysis of the esters, the target substrates S1m,p were crystallized by slow diffusion of methanol solutions into diethyl ether, while the acid derivative S1o was isolated as a dark-red oil.
It has been reported that the substitution of two carboxylic groups by amide units in EGTA significantly influences its coordination properties and the consequent MR behaviour of the corresponding Gd-complexes.44 Consequently, we also synthesised substrate S2 to investigate whether an amide group linking the pyridyl and EGTA units would have an effect on SABRE. S2 was synthesised in both meta (S2m) and para (S2p) forms in two steps by following a literature procedure with only minor modifications (Scheme 2b).44 Finally, the substrate S3m, which contains one pyridyl ring, was prepared in three steps (Scheme 2c). The hydrolysis step used conditions adapted from Zalupski et al.45 The ortho isomer of S2 (S2o) and the ortho and para derivatives of S3 (S3o and S3p, respectively) were not synthesised due to the prediction that they would exhibit poor SABRE or challenging synthetic preparation.
Formation of SABRE active complexes with substrates S1 and S2
SABRE requires an active polarisation transfer catalyst that undergoes both pH2 and substrate exchange.18 Typically, this is achieved by activating solutions containing [IrCl(COD)(IMes)], A where COD = cis,cis-1,5-cyclooctadiene and IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazol-2-ylidene and 4–20 equivalents (relative to Ir) of substrate with 3 bar pH2 in solvents such as methanol-d4 or dichloromethane-d2. This process typically results in the formation of catalysts of the type [Ir(H)2(IMes)(substrate)3]Cl, which then undergo the required ligand exchange processes needed for SABRE.18The SABRE compatibility of substrates S1o,m,p, S2m,p and S3m was investigated by preparing samples of [IrCl(COD)(IMes)] (A, 2.5 mM) and 4 equiv. of the substrate in 0.6 mL of methanol-d4. 1H NMR spectra were then recorded at 298 K several hours after the addition of 3 bar H2. These measurements revealed in each case that hydride containing complexes form, but their signals were extremely weak indicating poor catalyst activation (Fig. S1, ESI†).
When the sample containing S1o was shaken with 3-bar pH2 for 10 seconds at 65 G, and then examined by high-field (9.4 T) NMR, PHIP enhanced hydride signals were visible at δ −20.56 and −31.21 (Fig. 2a). These signals are consistent with hydride environments trans to nitrogen- and oxygen-donor sites respectively. It is therefore likely that they arise from a complex of the type [Ir(H)2(IMes)(κ2-N,O-S1o)(L)], in which S1o is bound through N- and O-sites. This is possible due to the ortho arrangement of the pyridyl ring and the carboxylate motif. Related complexes containing bidentate H2NCH2COO− and RNC(R)COO− ligands have been reported with their hydride resonances yielding similar chemical shifts.46,47
 | ||
| Fig. 2 Partial single scan 1H NMR spectra recorded at 9.4 T and 298 K after a sample containing [IrCl(COD)(IMes)] (2.5 mM) and 4 equiv. (a) S1o (b) S1m (c) S1p (d) S2m (e) S2p and (f) S3m in 0.6 mL of methanol-d4 were shaken with 3 bar pH2 for 10 seconds at 65 G. | ||
In contrast, when the same experiments were repeated using S1m and S1p, hyperpolarised hydride NMR signals for analogous [Ir(H)2(IMes)(κ2-N,O-S1)(L)] complexes are no longer observed. This is likely the result of the arrangement between the pyridyl ring and the EGTA unit leading to steric strain in such products. Consequently, S1m and S1p can no longer act as N-,O-donors in the same way as S1o. In the case of S1m and S1p, hyperpolarised signals at δ −21.30 and −23.79 are observed instead. For S2m and S2p, the corresponding hyperpolarised signals appear at δ −22.53 and −23.54 (Fig. 2d and e respectively). The chemical shifts of these signals are characteristic of hydride ligands lying trans to chloride19,48,49 or pyridyl nitrogen.21,22,26–28,43,50 Therefore, these 1H NMR signals are expected to arise from [IrCl(H)2(IMes)(N-S1–2)(L)] where L, located cis to hydrides and trans to the NHC, is expected to correspond to S1–2. Full NMR characterisation and structural elucidation of these complexes was hampered by their low NMR signal intensity, which may reflect low stability. No PHIP enhanced hydride signals were observed using S3m suggesting that in this case an active polarisation transfer catalyst is not formed. We attribute this to the large steric size of S3m, which in this case would likely prevent binding of the two S3m molecules necessary to form an analogous [IrCl(H)2(IMes)(N-S3m)(L)] complex, where L is S3m.
No NMR signal enhancements corresponding to the agents S1o,m,p, S2p or S3m themselves are observed suggesting that while iridium-agent complexes exhibiting PHIP enhanced hydride resonances are formed using S1o,m,p and S2p, these complexes do not act as SABRE polarisation transfer catalysts in these cases. In contrast, hyperpolarised 1H NMR resonances for the pyridyl ring of S2m were observed (Fig. 3b–d); the ortho sites were enhanced by 16 and 13-fold relative to their Boltzmann derived signal strengths. The more distant meta and para1H NMR sites were enhanced by 9 and 12-fold respectively. The corresponding signal gains achieved at 273 K were comparable to these, although they increased to 32, 37, 12 and 21-fold for the two ortho, meta and para sites respectively at 318 K. This is likely to be the result of faster ligand exchange and suggests the resulting complex is reasonably stable.21,24
 | ||
| Fig. 3 Partial single scan 1H NMR spectra of the aromatic region of a sample containing (a) thermally polarised [IrCl(COD)(IMes)] (2.5 mM) and S2m (4 equiv.) in 0.6 mL of methanol-d4 (b)–(d) the same sample after shaking with 3 bar pH2 at 65 G for 10 seconds after being left in a thermostatically controlled water bath at (b) 273 K (c) 298 K and (d) 318 K for 60 seconds prior to pH2 shaking. | ||
Use of co-ligands to form stable SABRE active complexes
Agents S1–3 reflect some of the most structurally complex molecules investigated for hyperpolarisation using SABRE, which is usually applied to low molecular weight molecules with less than 20 atoms.23 The large size of S1–3 is likely to hamper the formation of typical SABRE catalysts of the type [Ir(H)2(IMes)(S1–3)3]Cl. In previous studies using sterically large targets, substrate coordination can be favoured by using SABRE catalysts with sterically smaller carbene ligands.43 The addition of a co-ligand to support the formation of active polarisation transfer catalysts with sterically large substrates35 or weakly donating O-donor ligands has also been used.19,29 We therefore added the co-ligands acetonitrile27,51 or benzylamine26,28 to see if suitable stable polarisation transfer catalysts form with agents S1–3.The effect of acetonitrile was tested by its addition (0.5 μL, and then a further 2 μL) to solutions of preactivated [IrCl(COD)(IMes)] (A, 2.5 mM) and S1 (4 equiv.) with 3 bar H2 in 0.6 mL of methanol-d4. No change in the appearance of the hydride region of the corresponding 1H NMR spectra were observed when compared to the spectra observed without addition of this co-ligand (Fig. 2). This is surprising given the known stability of mono-substituted acetonitrile complexes of this type27,51 and supports our earlier hypothesis that the complexes formed in these cases likely do not undergo the ligand exchange needed for SABRE.18,23,24
Therefore, as predicted, when a fresh sample was prepared containing A (4 mM), S1m (3 equiv.), acetonitrile (2 equiv.) and 3 bar H2 in 0.6 mL of methanol-d4, a different hydride-containing complex forms, exhibiting resonances at δ −20.77 and −22.29. These resonances are comparable to those previously reported for [Ir(H)2(IMes)(NCCH3)(pyridine)2]Cl, which appear at δ −20.56 and −22.12.27 The formation of an analogous complex is supported by the observation of three sets of aromatic 1H NMR resonances for three distinct types of pyridyl ring. The first of these has resonances at δ 8.79, 8.65, 7.62 and 8.17 and they match those of the free agent, S1m. Additional sets of resonances at δ 8.43, 8.45, 7.25, 7.95 and δ 8.38, 8.72, 7.15, 7.85 are visible corresponding to the pyridyl groups of S1m bound to iridium (Fig. 4b). 2D NMR characterisation data for this sample confirms the relative orientation of IMes, hydride, pyridyl and acetonitrile ligands in the immediate coordination sphere of the metal (Table S2 in ESI† for full characterisation details). These measurements cannot, however, confirm whether the two distinct bound pyridyl rings of S1m arise from the same or separate molecules of bound S1m. We note that high resolution mass spectrometry did not yield molecular ion peaks corresponding to intact complexes due to severe molecular fragmentation, even with liquid injection field desorption ionization (LIFDI) techniques. However, the appearance of these bound aromatic 1H NMR signals appears similar when an analogous sample is prepared using 0.9 equiv. of S1m relative to catalyst (Fig. S6, ESI†). We therefore deduce the complex formed from S1m is [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl, in which S1m acts as a bidentate ligand coordinating through both pyridyl rings. We note that when a sample of S3m (4 equiv.) and A (2.5 mM) with 3 bar pH2 in methanol-d4 (0.6 mL) was prepared, no PHIP enhanced hydride containing complexes were formed, which suggests that both pyridyl rings of S1m are required to form the hydride signals assigned as [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl.
 | ||
| Fig. 4 (a) Signal averaged 1H NMR spectrum of S1m in 0.6 mL of methanol-d4 recorded at 9.4 T and 298 K (b) partial single scan 1H NMR spectrum of a sample containing [IrCl(COD)(IMes)] (4 mM), S1m (3 equiv.) and acetonitrile (2 equiv.) in 0.6 mL of methanol-d4 recorded under Boltzmann conditions at 9.4 T and 298 K (c) SABRE hyperpolarised 1H NMR spectrum after shaking the sample described in (b) with 3 bar pH2 for 10 seconds at 65 G. | ||
When such a sample containing S1m was shaken for 10 seconds with pH2 at 65 G, prior to recording a high-field 1H NMR spectrum, the observed hydride resonances exhibit PHIP enhancement (Fig. S3, ESI†). To this end, some resonances in the aromatic region are now enhanced as a consequence of SABRE (Fig. 4). These hyperpolarised resonances correspond to one of the bound pyridyl rings in [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl, which must occupy a binding site trans to hydride in order to receive polarisation transfer via SABRE.18 The 1H NMR signal gains for the two ortho sites in this bound pyridyl ring (labelled as o and b in Fig. 4) are enhanced by a factor of 63-fold, compared to those recorded under Boltzmann derived conditions, although they cannot be distinguished from each other due to overlap. Signals for the meta and para sites in this pyridyl ring are enhanced by 3- and 53-fold respectively. Much weaker enhancements (<10-fold) are observed for a second set of resonances, which are attributed to a pyridyl ring that is located cis to the hydride ligands of [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl. Due to slow exchange, only a 3-fold enhancement is observed for the para resonance of the free agent. The NMR signal gains for each site have been compiled and summarized (Fig. 5 and Table S6 in ESI†).
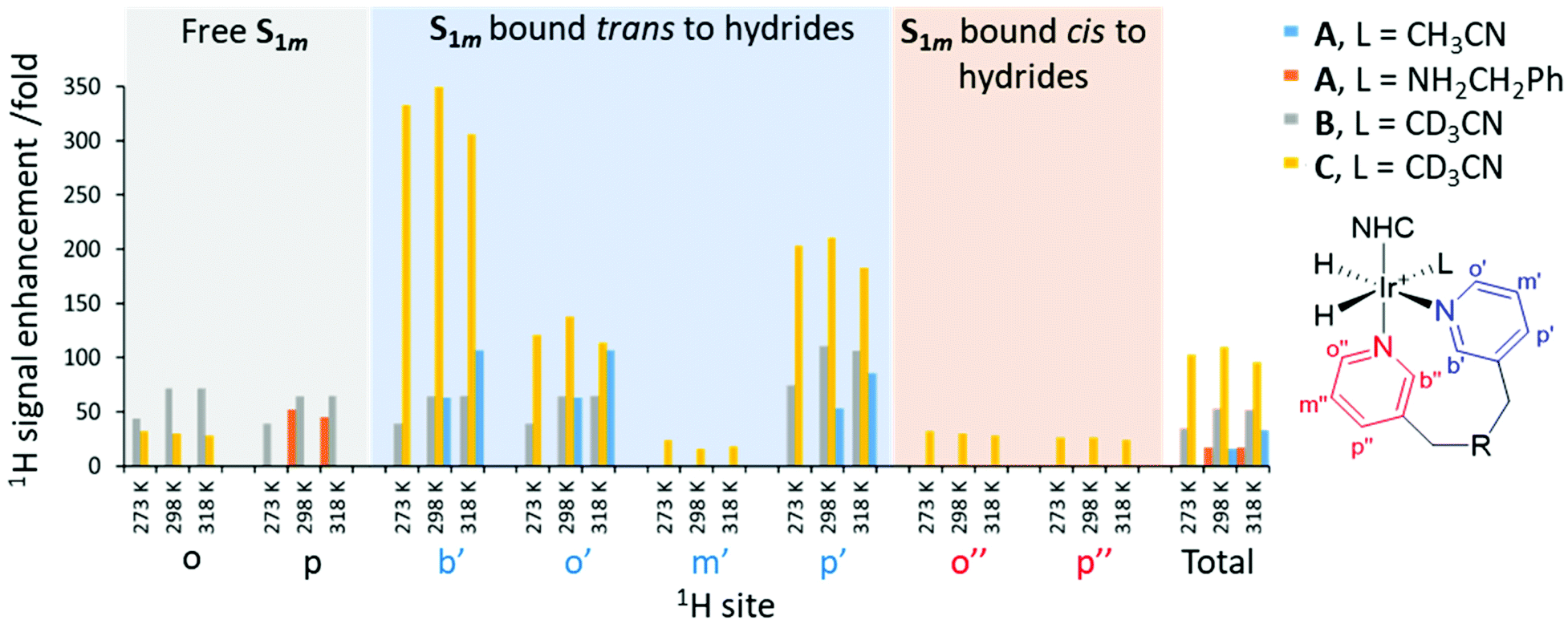 | ||
| Fig. 5 SABRE 1H NMR signal enhancements for S1m (3 equiv.) when shaken with precatalyst A (4 mM), the indicated co-ligand (2 equiv.) and 3 bar pH2 in 0.6 mL of methanol-d4 for 10 seconds at 65 G. For measurements recorded at 273 K and 318 K the samples were placed in a thermostatically controlled water bath for 60 seconds prior to pH2 shaking and detection at 298 K. For individual sites, signal enhancements lower than 25-fold were omitted for clarity. A full table showing all signal enhancements for all sites, including errors, is given in the ESI Table S6.† Note that resonances omitted from the figure gave no signal enhancements. The structure of the NHC ligand and substrate S1m are shown in Scheme 1 and Fig. 1 respectively. | ||
When these SABRE measurements were repeated after leaving the sample in a water bath at 318 K for 60 seconds, prior to pH2 shaking and detection at 298 K, the resulting 1H NMR signal gains were roughly double those recorded when the shaking process was performed at 298 K (Fig. 5). In these measurements, NMR signal gains for the 1H acetonitrile sites were also observed (enhanced by 27- and 48-fold per proton at 298 and 318 K respectively). A hyperpolarised 13C NMR response for acetonitrile at δ 116 could also be discerned in a single scan when shaken in a mu-metal shield, while no enhanced 13C signals corresponding to S1m were visible (Fig. S4, ESI†). These enhanced acetonitrile signals are consistent with its dissociation from [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl providing a route to pH2 exchange.27,52 These results suggest that in this system, an active SABRE catalyst of type [Ir(H)2(IMes)(NCCH3)(κ2-N,N-S1m)]Cl is able to catalyse polarisation transfer from pH2 to S1m. However, the high relative proportion of polarisation of S1m bound to iridium suggests that exchange of this ligand is very slow.
The analogous polarisation transfer complex [Ir(H)2(IMes)(NCCD3)(κ2-N,N-S2m)]Cl forms when A (4 mM), S2m (3 equiv.), acetonitrile-d3 (2 equiv.) and 3 bar H2 react in 0.6 mL of methanol-d4. However, when shaken with pH2 at 298 K, 1H NMR signal enhancements of less than 20-fold were seen for the corresponding bound ligand resonances, which did not improve at 273 or 318 K (see ESI, Table S3†). These signal gains for S2m are comparable to those achieved in the absence of a co-ligand. To improve the signal gains for these agents further an active polarisation transfer catalyst needs to be formed, which exchanges the target substrate more rapidly, whilst reducing polarisation wastage into the acetonitrile co-ligand.
Optimisation of SABRE enhancement of S1m by co-ligand and catalyst variation
The optimisation of SABRE performance is typically achieved by varying factors including the co-ligand50,53 and the carbene ligand of the catalyst,21,54 which serve to tune pH2 and substrate exchange within the active catalyst. When analogous SABRE measurements were performed using samples containing A (4 mM), S1m (3 equiv.), and the known SABRE co-ligand benzylamine,26,28 a major hydride containing product is formed with signals at δ −22.25 and −22.80. This species is expected to correspond to the related [Ir(H)2(IMes)(κ2-N,N-S1m)(ND2CH2Ph)]Cl; as whilst these measurements start with NH2CH2Ph, rapid H/D exchange in methanol-d4 forms ND2CH2Ph.28 When these samples were shaken with pH2, SABRE enhancements for the pyridyl groups of S1m bound within the catalyst were again observed; however, the effect is now much smaller than when acetonitrile was used as the co-ligand (Fig. 5). In these measurements, the presence of additional pyridyl resonances from the amine complicates spectral interpretation due to peak overlap. In fact, hyperpolarised signals for the pyridyl ring coordinated cis to hydrides are no longer discerned. In these cases, a more significant enhancement of the free para resonance of S1m is achieved (52-fold with benzylamine compared to 3-fold for acetonitrile), which does not increase at elevated temperatures.Deuteration of 1H sites in co-ligands, catalyst, or of target molecules, is often employed to optimise SABRE signal gains by preventing unwanted polarisation leakage and reducing relaxation.21,22 Catalyst design has also been used to fine tune substrate exchange rates with bulkier carbene ligands generally giving faster exchange.21,54 In order to encourage faster exchange of S1m within the active [Ir(H)2(NHC)L(κ2-N,N-S1m)]Cl species, the bulkier iridium precatalysts B and C (Scheme 1) were used. Analogous samples were prepared containing B or C (4 mM), acetonitrile-d3 (2 equiv.) and S1m (3 equiv.) with 3 bar H2 in 0.6 mL of methanol-d4. Upon shaking mixtures containing precatalyst B with pH2 at 298 K, total 1H signal enhancements per proton for the pyridyl resonances of S1m free in solution and bound to the SABRE catalyst were estimated. These are now much higher at 49- and 43-fold, respectively, when compared to 3- and 26-fold, respectively, using catalyst A (see above). Similar increases relative to A were observed when catalyst C was used (total 1H NMR signal enhancements per proton of 19- and 117-fold for free and bound S1m, respectively). Interestingly, while precatalyst B gives the highest response for the free agent (an enhancement of 72-fold is observed for one of the free ortho sites), C gives higher signal gains for the bound agent (signal enhancements of 350-, 138- and 211-fold were observed for the two ortho and para pyridyl sites of S1m bound trans to the hydrides in the SABRE polarisation transfer catalyst, respectively). This suggests that the use of precatalysts B and C can yield more efficient SABRE hyperpolarisation of S1m relative to A. This is likely the effect of incorporation of the deuterium labels, which are expected to reduce relaxation in the active catalyst.22 The greater proportion of polarisation on the bound ligand using C suggests that polarisation transfer or hydrogen exchange within C is more efficient, but that relaxation or substrate exchange effects are less optimal compared to B. These signal gains are summarised in Fig. 5 and Table 1 (the Table S6 in ESI† contains a full table of signal enhancements).
| Agent (3 equiv.) | Conditions | 1H NMR signal enhancements/fold | |||||
|---|---|---|---|---|---|---|---|
| Catalyst (4 mM) | Co-ligand (2 equiv.) | T/K | Total free enhancement | Total bound enhancement | Highest enhancement for individual free site | Highest enhancement for individual bound site | |
| S1o | C | CD3CN | 318 | 25 ± 3 | 0 | 30 ± 2 (o) | 0 |
| S1m | B | CD3CN | 298 | 49 ± 3 | 43 ± 3 | 72 ± 3 (o) | 111 ± 6 (p′) |
| S1m | C | CD3CN | 298 | 19 ± 1 | 117 ± 6 | 30 ± 1 (o, overlaps with o′′) | 350 ± 21 (b′ or o′) |
| S1p | C | CD3CN | 273 | 127 ± 19 | 0 | 159 ± 20 (o) | 0 |
| S2m | C | None | 318 | 33 ± 5 | 0 | 45 ± 8 (b) | 0 |
| S2p | C | CD3CN | 298 | 5 ± 1 | 77 ± 3 | 5 ± 1 (o) | 124 ± 6 (o′) |
| S3m | C | CD3CN | 298 | 0 | 0 | 0 | 0 |
Comparison of SABRE efficiency of S1–3
Signal enhancements of S1o, S1p, S2p and S3m were also recorded under the same conditions as those which gave the highest signal enhancements for S1m. This involved shaking samples containing precatalyst C (4 mM), acetonitrile-d3 (2 equiv.) and substrate (3 equiv.). SABRE performance of S2m (3 equiv.) was tested for a sample containing C (4 mM) with 3 bar pH2 in methanol-d4 (0.6 mL). No co-ligand was used in this case as our previous tests (see above) found comparable signal enhancements for conditions with and without acetonitrile for this agent. Under these conditions, precatalyst C delivers 45-fold 1H NMR signal gain for one of the ortho sites of free S2m at 318 K (Table 1) which is slightly higher than the largest signal gain (37-fold) for the equivalent site at the same temperature using precatalyst A (2.5 mM).For the remaining agents that were tested in the presence of acetonitrile-d3, total hyperpolarised 1H NMR responses for the free agent S1o are 20-fold. Interestingly, the hydride region of these NMR spectra reveal the presence of PHIP enhanced signals at δ −20.16, −20.75, −28.58 and −31.31 (Fig. S21, ESI†). This result suggests that even in the presence of this co-ligand, S1o can bind through both N and O sites to likely form [Ir(H)2(IMes)(κ2-N,O-S1o)(CH3CN)], which may hamper its SABRE efficiency.
Interestingly, S1p performs well under these conditions as the ortho and meta1H NMR resonances of the free agent are enhanced at 298 K by 138- and 68-fold respectively. These signal gains are higher than those achieved under analogous conditions for S1m free in solution (corresponding sites <30-fold). 1H NMR signal enhancements can be increased to 159- and 92-fold for the ortho and meta resonances, respectively, for free S1p when polarisation transfer takes place at 273 K. A summary of these 1H NMR signal gains is presented in Table 1 (a full table showing the NMR signal gains for each site is presented in the Table S8 in ESI†). There are no visible hyperpolarised signals for S1p bound to the catalyst, which suggests that polarisation transfer efficiency or hydrogen exchange using catalyst C must be less efficient for S1p compared to using agent S1m.
In contrast, SABRE 1H NMR signal gains for free S2p were low (<10-fold for the ortho site). Enhancements for the bound ligand were much higher at 77-fold suggesting that in this case polarisation transfer and ligand exchange processes are less efficient in comparison to S1m and S1p. The presence of a dominant PHIP enhanced hydride product at δ −20.9 and −22.2 appears when S1m, S1p, or S2p are used (Fig. S21, ESI†), which suggests that these differences in SABRE efficiency for each agent are not due to a change in the identity of the active magnetisation transfer catalyst.
We also note that tests involving the mono-pyridyl containing agent, S3m, did not yield discernible signals for any hydride containing complexes and no enhanced 1H NMR signals were observed. This suggests that while the presence of two pyridyl rings in agents S1–2 may lead to slow exchange kinetics, it appears that both pyridyl rings in the sterically large agents reported in this work are important for ligation to form SABRE active complexes.
Conclusions
In this work we prepared and studied a series of novel agents that contain a pyridyl ring to allow hyperpolarisation using SABRE and a metal ligating unit derived from EGTA. The introduction of the pyridyl motif allows the prepared agents to coordinate to the iridium SABRE polarisation transfer catalyst. The hydride ligands of the resultant complexes were enhanced by parahydrogen and in the case of agent S2m up to 33-fold enhancement is achieved for 1H sites of the free pyridyl ring.SABRE active complexes of the form [Ir(H)2(NHC)(NCCH3)(κ2-N,N-S1–2)]Cl can be formed using agents S1m, S1p, and S2p when acetonitrile was used as a co-ligand. We find that 1H NMR signal gains of S1m can be increased by using bulkier catalysts and in some cases increasing the temperature to encourage substrate exchange and deuteration of the catalyst and co-ligand to reduce polarisation wastage. 1H NMR signal gains of 72-fold for the ortho pyridyl site of free S1m can be achieved using precatalyst B, although higher 1H NMR signal gains of up to 350-fold can be achieved for pyridyl sites bound to precatalyst C. We find that in many cases 1H NMR signal gains for agents bound to the catalyst were much higher than those free in solution, which indicates slow ligand exchange and is perhaps expected given the large nature of these ligands and their ability to act as bidentate donors. Nevertheless, the molecules tested here reflect some of the sterically largest targets to be hyperpolarised using SABRE and 1H NMR signal gains of up to 159-fold are observed for the ortho pyridyl sites of S2m free in solution. We note that, to the best of our knowledge, the largest molecules successfully hyperpolarised using SABRE have molecular masses of 337 Da,41 and ∼320 Da,35,42 both lower than the molecules reported in this work (<471 Da).
Furthermore, this work presents a rational to design functional molecules that can be hyperpolarised using SABRE. The obtained results indicate that an ortho relationship between the pyridyl nitrogen atom and the EGTA unit should be avoided, as agents with this steric arrangement can act as mixed N-,O- donors to form [Ir(H)2(IMes)(κ2-N,O-S1o–2o)(L)] with reduced SABRE activity, even in the presence of a co-ligand. For the large substrates presented in this work, it appears the presence of two pyridyl rings is necessary for SABRE, as mono-pyridyl containing S3m was not found to form any PHIP or SABRE active iridium adducts. We therefore highlight the tension between inclusion of iridium ligating groups to form SABRE active complexes, and labile exchange kinetics. We find no inherent significant difference in SABRE efficiency of meta (S1m, S2m) or para (S1p, S2p) substituted agents, which is consistent with other studies.42 Generally, higher 1H NMR signal gains for agents S1m,p compared to S2m,p suggest that the increase in agent size upon inclusion of an additional amide linker group is not advantageous for SABRE hyperpolarisation.
However, it should be concluded that SABRE efficiency for all substrates strongly varies due to their various structural, as well as coordination, properties and exchange rates with the SABRE catalyst. Therefore, it is likely that different polarisation conditions (catalyst, co-ligand, concentration etc.) will have to be applied for each agent; this would require further optimisation, including increased pH2 pressure or the use of related catalysts that have recently allowed the coordination of sterically larger43,50 or weakly coordinating19,29 substrates to increase the NMR signal gain. Removal of the SABRE catalyst will be required before any in vivo applications; the routes for its separation,55,56 and the development of water soluble catalysts, have already been reported.57 Parallel progress in these areas will be required to produce a wider range of molecules whose MR signal(s) can respond to biological conditions. Consequently, the application of SABRE hyperpolarisation to address emerging molecular imaging questions could become feasible in the future.
Experimental section
General remarks
General procedure for reductive amination to form 1
2,2′-(Ethylenedioxy)bis(ethylamine) (13.5 mmol) was placed into a Schlenk flask with activated molecular sieves (3 Å) under a stream of nitrogen. Dry CH2Cl2 (15 mL) was added via a rubber septum. To the resulting solution, a pyridinecarboxaldehyde (28.3 mmol, 2.1 equiv.) was added dropwise. The flask was sealed with parafilm and the mixture was left stirring at room temperature under nitrogen for 18–20 h. The progress of the reaction was monitored by mass spectrometry and TLC analysis. After the reaction was completed, the mixture was filtered through a Celite layer and the solvent was distilled on a rotary evaporator untill dry.The flask with the residue was flushed with nitrogen and dry EtOH (15 mL) was added. The mixture was cooled in an ice bath. To the resulting solution, sodium borohydrate (56.7 mmol, 4.2 equiv.) was added in portions. After stirirng for ∼1 hour, the ice bath was removed and the mixture was left stirring at room temperature overnight. The mixture was filtered through a glass filter and the solvent was evaporated. The residue was washed with water, extracted with CH2Cl2 (5–6 × 40 mL), and dried over sodium sulfate. The inorganic salt was filtered, solvent evaporated, and the oily product dried on a high-vacuum pump. The purity of the so-obtained precursors 1 (according to NMR analysis) was found appropriate to continue without additional purification.
General procedure for alkylation of 1 to form 2
The flask containing diamine 1 (7.8 mmol) was flushed with nitrogen and dry DMF (40 mL) was added. Then potassium carbonate (62.6 mmol, 8 equiv.) and potassium iodide (4.7 mmol, 0.6 equiv.) were added under a stream of nitrogen. To the resulting mixture, t-butyl bromoacetate (31.3 mmol, 4 equiv.) was added dropwise in four portions. The flask was sealed with parafilm and left stirring at room temperature for 24 h. DMF was dried on a high-vacuum pump. The residue was dissolved in MeOH and dried with silica gel in vacuo. The derivatives 2o,m,p were isolated using column chromatography on silica gel with gradient solvent mixtures: EtOAc/10% hexane, EtOAc, EtOAc + 1.5% EtOH.General procedure for acidic hydrolysis of 2 to form S1
A solution of corresponding derivative 2 (0.48 mmol) in CH2Cl2 (6 mL) was flushed with nitrogen, cooled down in an ice-bath, and cold TFA (3.5 mL) was added dropwise. The progress of the reaction was monitored by mass spectrometry and TLC analysis. In the case of the diacid S1m, an additional amount of TFA (0.5 mL) was required to complete the process. The solvents were removed using a rotary evaporator. MeOH was then added to the residue, and the solution was dried in vacuo. The procedure was repeated multiple times in order to remove the remaining TFA.General procedure for the synthesis of S2 derivatives
Into a Schlenk flask containing the anhydride 3 (0.5 g, 1.5 mmol), dry pyridine (4 mL) was added and the mixture was purged with nitrogen for 15 min. In a separate Schlenk flask a solution of the corresponding aminopyridine (0.3 g, 3.2 mmol) in 3–4 mL of dry solvent (pyridine or DMF) was prepared. Due to limited solubility of 4-aminopyridine in pyridine, DMF was used in this case. The resulting solution was added to the anhydride 3via a rubber septum. The flask was sealed and left stirring under nitrogen for 14 h. The solvent was removed under reduced pressure. The residue was re-dissolved in water, the pH was adjusted to 10 and the mixture was washed with diethyl ether (4 × 30 mL). The aqueous fraction was dried under reduced pressure and the residue was purified by preparative HPLC.SABRE procedures
Parahydrogen (pH2) was produced by passing hydrogen gas over a spin-exchange catalyst (Fe2O3) and used for all hyperpolarisation experiments. This method produces constant pH2 with ca. 98% purity. 1H (400 MHz) and 13C (100.6 MHz) NMR spectra were recorded with an internal deuterium lock. Chemical shifts are quoted as parts per million and referenced to the solvent. 13C NMR spectra were recorded with broadband proton decoupling. Coupling constants (J) are quoted in Hertz.Samples were prepared in a 5 mm NMR tube that was fitted with a J. Young's tap. [IrCl(COD)(IMes)] was synthesized according to a literature procedure.59 The resulting solutions were degassed by two freeze–pump–thaw cycles before the addition of 3-bar H2.
The shake and drop method was employed for recording hyperpolarised NMR spectra. This involves filling NMR tubes with pH2 at 3 bar pressure and shaking them vigorously for 10 seconds in a 65 G magnetic field (stray field of the 9.4 T spectrometer). Typically, three shake and drop measurements are recorded and average 1H NMR signal enhancement values are quoted. The typical variation among these measurements is <±10%. Signal enhancements are calculated by dividing the integrated signal intensities from a single scan hyperpolarised spectrum by its thermal counterpart recorded under the same spectral conditions. These values are presented per fold, i.e. a 100-fold enhancement means that hyperpolarised signals are 100 times more intense than those recorded using Boltzmann controlled NMR.
Author contributions
Ben. J. Tickner: conceptualization, methodology, investigation (SABRE measurements), writing – original draft, visualization. Yulia Borozdina: investigation (Agent synthesis), writing – original draft. Simon B. Duckett: methodology, writing – review & editing, supervision, funding acquisition. Goran Angelovski: conceptualization, methodology, writing – review & editing, visualization, supervision, funding acquisition.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank Dr Peter Rayner and Dr Victoria Annis for synthesis of the iridium precatalysts and Dr Marianna Fekete for helpful discussions. Financial support from the Wellcome Trust (Grants 092506 and 098335), the MRC (MR/M008991/1), the EPSRC (B. J. T. studentship) and the Shanghai Municipal Science and Technology Major Project (Grant No. 2019SHZDZX02) is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- G. Angelovski, Angew. Chem., Int. Ed., 2016, 55, 7038–7046 CrossRef CAS.
- N. K. Logothetis, Nature, 2008, 453, 869–878 CrossRef CAS PubMed.
- B. Wu, G. Warnock, M. Zaiss, C. Lin, M. Chen, Z. Zhou, L. Mu, D. Nanz, R. Tuura and G. Delso, EJNMMI Phys., 2016, 3, 19 CrossRef CAS.
- E. L. Que and C. J. Chang, Chem. Soc. Rev., 2010, 39, 51–60 RSC.
- G. Angelovski and É. Tóth, Chem. Soc. Rev., 2017, 46, 324–336 RSC.
- P. Caravan, Acc. Chem. Res., 2009, 42, 851–862 CrossRef CAS PubMed.
- G. G. Somjen, Ions in the brain : normal function, seizures, and strokes, Oxford University Press, New York; Oxford, 2004 Search PubMed.
- H. Li and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 17025–17041 CrossRef CAS PubMed.
- G. Angelovski, Acc. Chem. Res., 2017, 50, 2215–2224 CrossRef CAS PubMed.
- W.-H. Li, G. Parigi, M. Fragai, C. Luchinat and T. J. Meade, Inorg. Chem., 2002, 41, 4018–4024 CrossRef CAS.
- P. Nikolaou, B. M. Goodson and E. Y. Chekmenev, Chem. – Eur. J., 2015, 21, 3156–3166 CrossRef CAS.
- J. H. Ardenkjær-Larsen, S. Bowen, J. R. Petersen, O. Rybalko, M. S. Vinding, M. Ullisch and N. C. Nielsen, Magn. Reson. Med., 2019, 81, 2184–2194 CrossRef PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. Van Criekinge, J. W. Chang, R. Bok and I. Park, Sci. Transl. Med., 2013, 5, 198ra108–198ra108 Search PubMed.
- J. B. Hövener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glöggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier and A. Jerschow, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef.
- D. Canet, C. Aroulanda, P. Mutzenhardt, S. Aime, R. Gobetto and F. Reineri, Concepts Magn. Reson., Part A, 2006, 28, 321–330 CrossRef.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., 2019, 131, 10377–10381 CrossRef.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 CrossRef CAS PubMed.
- P. J. Rayner, P. Norcott, K. M. Appleby, W. Iali, R. O. John, S. J. Hart, A. C. Whitwood and S. B. Duckett, Nat. Commun., 2018, 9, 4251 CrossRef PubMed.
- P. J. Rayner, M. J. Burns, A. M. Olaru, P. Norcott, M. Fekete, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3188–E3194 CrossRef CAS PubMed.
- D. A. Barskiy, S. Knecht, A. V. Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114, 33–70 CrossRef PubMed.
- D. A. Barskiy, A. N. Pravdivtsev, K. L. Ivanov, K. V. Kovtunov and I. V. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 89–93 RSC.
- D. A. Barskiy, R. V. Shchepin, C. P. N. Tanner, J. F. P. Colell, B. M. Goodson, T. Theis, W. S. Warren and E. Y. Chekmenev, ChemPhysChem, 2017, 18, 1493–1498 CrossRef CAS PubMed.
- M. Fekete, F. Ahwal and S. B. Duckett, J. Phys. Chem. B, 2020, 124, 4573–4580 CrossRef CAS PubMed.
- R. E. Mewis, R. A. Green, M. C. Cockett, M. J. Cowley, S. B. Duckett, G. G. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- W. Iali, P. J. Rayner, A. Alshehri, A. J. Holmes, A. J. Ruddlesden and S. B. Duckett, Chem. Sci., 2018, 9, 3677–3684 RSC.
- M. Gemeinhardt, M. Limbach, T. Gebhardt, C. Eriksson, S. Eriksson, J. Lindale, E. Goodson, W. Warren, E. Chekmenev and B. Goodson, Angew. Chem., 2020, 132, 426–431 CrossRef.
- P. J. Rayner, B. J. Tickner, W. Iali, M. Fekete, A. D. Robinson and S. B. Duckett, Chem. Sci., 2019, 10, 7709–7717 RSC.
- P. M. Richardson, W. Iali, S. S. Roy, P. J. Rayner, M. E. Halse and S. B. Duckett, Chem. Sci., 2019, 10, 10607–10619 RSC.
- P. J. Rayner, P. M. Richardson and S. B. Duckett, Angew. Chem., 2020, 132, 2732–2736 CrossRef.
- A. M. Olaru, M. J. Burns, G. G. R. Green and S. B. Duckett, Chem. Sci., 2017, 8, 2257–2266 RSC.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, T. Theis, F. Shi, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, ACS Sens., 2016, 1, 640–644 CrossRef CAS PubMed.
- F. F. Diaz-Rullo, F. Zamberlan, R. E. Mewis, M. Fekete, L. Broche, L. A. Cheyne, S. Dall'Angelo, S. B. Duckett, D. Dawson and M. Zanda, Bioorg. Med. Chem., 2017, 25, 2730–2742 CrossRef PubMed.
- B. J. Tickner, P. J. Rayner and S. B. Duckett, Anal. Chem., 2020, 92, 9095–9103 CrossRef CAS.
- A. Mishra, G. Pariani, T. Oerther, M. Schwaiger and G. G. Westmeyer, Anal. Chem., 2016, 88, 10790–10794 CrossRef CAS PubMed.
- M. Naraghi, Cell Calcium, 1997, 22, 255–268 CrossRef CAS PubMed.
- R. Hata, H. Nonaka, Y. Takakusagi, K. Ichikawa and S. Sando, Chem. Commun., 2015, 51, 12290–12292 RSC.
- H. Nonaka, R. Hata, T. Doura, T. Nishihara, K. Kumagai, M. Akakabe, M. Tsuda, K. Ichikawa and S. Sando, Nat. Commun., 2013, 4, 2441 CrossRef.
- T. B. R. Robertson, L. H. Antonides, N. Gilbert, S. L. Benjamin, S. K. Langley, L. J. Munro, O. B. Sutcliffe and R. E. Mewis, ChemistryOpen, 2019, 8, 1375–1382 CrossRef CAS PubMed.
- T. Ratajczyk, T. Gutmann, P. Bernatowicz, G. Buntkowsky, J. Frydel and B. Fedorczyk, Chem. – Eur. J., 2015, 21, 12616–12619 CrossRef CAS PubMed.
- C. M. Wong, M. Fekete, R. Nelson-Forde, M. R. D. Gatus, P. J. Rayner, A. C. Whitwood, S. B. Duckett and B. A. Messerle, Catal. Sci. Technol., 2018, 8, 4925–4933 RSC.
- S. Aime, A. Barge, M. Botta, L. Frullano, U. Merlo and K. I. Hardcastle, J. Chem. Soc., Dalton Trans., 2000, 3435–3440 RSC.
- T. S. Grimes, C. R. Heathman, S. Jansone-Popova, A. S. Ivanov, S. Roy, V. S. Bryantsev and P. R. Zalupski, Inorg. Chem., 2018, 57, 1373–1385 CrossRef CAS PubMed.
- S. Glöggler, R. Müller, J. Colell, M. Emondts, M. Dabrowski, B. Blümich and S. Appelt, Phys. Chem. Chem. Phys., 2011, 13, 13759–13764 RSC.
- B. J. Tickner, W. Iali, S. S. Roy, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2019, 20, 241–245 CrossRef CAS PubMed.
- B. J. Tickner, R. R. Parker, A. C. Whitwood and S. B. Duckett, Organometallics, 2019, 38, 4377–4382 CrossRef CAS PubMed.
- S. Knecht, S. Hadjiali, D. A. Barskiy, A. Pines, G. Sauer, A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya and G. Buntkowsky, J. Phys. Chem. C, 2019, 123, 16288–16293 CrossRef CAS.
- J. Colell, A. W. J. Logan, Z. Zhou, J. R. Lindale, R. Laasner, R. Shchepin, E. Chekmenev, V. Blum, W. S. Warren and S. J. Malcolmson, Chem. Commun., 2020, 56, 9336–9339 RSC.
- C. P. N. Tanner, J. R. Lindale, S. L. Eriksson, Z. Zhou, J. F. P. Colell, T. Theis and W. S. Warren, J. Chem. Phys., 2019, 151, 044201 CrossRef PubMed.
- B. J. Tickner, R. O. John, S. S. Roy, S. J. Hart, A. C. Whitwood and S. B. Duckett, Chem. Sci., 2019, 10, 5235–5245 RSC.
- B. J. Tickner, O. Semenova, W. Iali, P. J. Rayner, A. C. Whitwood and S. B. Duckett, Catal. Sci. Technol., 2020, 10, 1343–1355 RSC.
- B. J. A. van Weerdenburg, S. Glöggler, N. Eshuis, A. H. J. Engwerda, J. M. M. Smits, R. de Gelder, S. Appelt, S. S. Wymenga, M. Tessari and M. C. Feiters, Chem. Commun., 2013, 49, 7388–7390 RSC.
- D. A. Barskiy, L. A. Ke, X. Li, V. Stevenson, N. Widarman, H. Zhang, A. Truxal and A. Pines, J. Phys. Chem. Lett., 2018, 9, 2721–2724 CrossRef CAS PubMed.
- W. Iali, A. M. Olaru, G. R. R. Green and S. B. Duckett, Chem. – Eur. J., 2017, 23, 10491–10495 CrossRef CAS.
- P. Spannring, I. Reile, M. Emondts, P. P. M. Schleker, N. K. J. Hermkens, N. G. J. van der Zwaluw, B. J. A. van Weerdenburg, P. Tinnemans, M. Tessari, B. Bluemich, F. P. J. T. Rutjes and M. C. Feiters, Chem. – Eur. J., 2016, 22, 9277–9282 CrossRef CAS.
- M. Šebelaa, K. Jarkovskáa, R. Lenobelb, R. Meddac, A. Padigliac, G. Floris and P. Peča, ARKIVOC, 2007, 7, 222–232 Search PubMed.
- L. D. Vazquez-Serrano, B. T. Owens and J. M. Buriak, Inorg. Chim. Acta, 2006, 359, 2786–2797 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional SABRE and NMR experiments. See DOI: 10.1039/d0dt03839c |
| ‡ These authors contributed equally to this work. |
| § Current address: NMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, 90014 Oulu, Finland. |
| This journal is © The Royal Society of Chemistry 2021 |