
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The degradation of phenol via in situ H2O2 production over supported Pd-based catalysts†
Alba
Santos‡
a,
Richard J.
Lewis‡
 *a,
David J.
Morgan
ab,
Thomas E.
Davies
a,
Euan
Hampton
c,
Paul
Gaskin
c and
Graham J.
Hutchings
*a
*a,
David J.
Morgan
ab,
Thomas E.
Davies
a,
Euan
Hampton
c,
Paul
Gaskin
c and
Graham J.
Hutchings
*a
aMax Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT), Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: LewisR27@Cardiff.ac.uk; Hutch@Cardiff.ac.uk
bHarwellXPS, Research Complex at Harwell (RCaH), Didcot, OX11 OFA, UK
cDŵr Cymru Welsh Water, Pentwyn Road, Nelson, Treharris, CF46 6LY, UK
First published on 8th November 2021
Abstract
The oxidative degradation of phenol via the in situ production of H2O2 from molecular H2 and O2 offers an attractive route to the destruction of organic contaminants in water streams, potentially overcoming the significant economic and environmental concerns associated with traditional water remediation technologies. Herein we demonstrate the efficacy of a series of bifunctional Pd-based catalysts, which offer appreciable rates of phenol degradation. In particular, the introduction of Fe into a supported Pd catalyst leads to a near four-fold increase in pollutant remediation. We ascribe this improvement in catalytic performance to the ability of Fe to catalyse the formation of oxygen-based radical species from in situ synthesised H2O2via Fenton's pathways and the promotion of Pd domains of mixed oxidation state, with a resulting inhibition of H2O2 degradation pathways.
Introduction
A major challenge of the coming decades will revolve around access to potable water, with increased competition driven largely by a combination of population growth, urbanisation and industrialisation. With the most severe risks effecting communities that are already facing water-scarcity and those who are not currently served by traditional water disinfection infrastructure, which typically relies on chlorination. As such there is a pressing need to develop alternative means of water treatment, with a particular focus on decentralized approaches.1 In particular there is a growing concern around the environmental and health impacts of organic pollutants released from industrial, agricultural and urban activities, including dyes, pharmaceuticals and pesticides.2The high resistance of many organic compounds found in industrial waste streams to conventional chemical or biological treatments has led to growing interest in the application of advanced oxidation processes (AOPs), which utilise oxygen-based radicals, primarily hydroxyl radicals (˙OH), for contaminant degradation.3 In particular the combination of pre-formed H2O2 with ozone (O3/H2O2) or ultraviolet light (UV/H2O2) offers an attractive route to the remediation of such recalcitrants. However, the high costs associated with reagents such O3 or H2O2 or indeed with energy light sources have precluded their application on an industrial scale.4 Similarly, the generation of ˙OH radials from H2O2via the use of Fenton's reagents, typically homogenous Fe2+ species,5 has suffered from the need to remove Fe or other Fenton-like metals from waste streams. In addition, there is a need to maintain a low pH to achieve optimal catalytic performance with such approaches.6 Which, when coupled with the additional costs associated with the neutralisation and purification of treated waste streams prior to discharge are likely to preclude the application of such approaches on a meaningful scale.7
Furthermore, there are added complications associated with the use of pre-formed H2O2. These largely result from the means by which H2O2 is generated on an industrial scale, the anthraquinone oxidation (AO) process. While highly efficient, economies of scale in addition to the complexity of the AO process preclude the production of H2O2 on-site via this route. As such H2O2 is typically transported and stored at concentrations (30–70 wt%) greatly exceeding that required by the end user (typically 1–10 wt%), prior to dilution, with this effectively wasting the energy utilised in the concentration process.8 Additionally, the low stability of H2O2, readily decomposing to H2O under mild temperatures or weakly basic conditions necessitates the use of acidic or halide stabilizing agents to prolong shelf-life.9 While effective in promoting H2O2 stability, the presence of such agents not only lead to reactor corrosion but also present a hazard to human and aquatic life.10 As such, if such processes were adopted for potable water treatment additional costly purification steps would be required to ensure their removal prior to the discharge of the treated water.
The in situ synthesis of H2O2 from molecular H2 and O2 offers an attractive alternative to the use of pre-formed H2O2, overcoming the considerable aforementioned concerns. In particular the activity of Pd catalysts towards H2O2 production has been well established.8,11,12 However, due to limited selectivity there is often a need to alloy Pd with a range of precious metals,13–17 although in recent years, a growing attention has been placed on the use of more abundant transition metals as promoters for Pd.18–23 Indeed, we have recently demonstrated the ability of bi-functional Pd-based bimetallic nanoparticles to catalyse the direct synthesis of H2O2 in addition to the selective oxidation of a range of substrates24–26 as well as the remediation of microorganisms through radical pathways.27 With these studies in mind, we now investigate the efficacy of supported Pd-based bimetallic catalysts that combine the H2O2 synthesising activity of Pd and the ability of a range of transition metals to generate reactive oxygen species through Fenton's pathways for the degradation of phenol.
Experimental
Catalyst synthesis
Mono- and bi-metallic 1% PdX/TiO2 (X = Au, Fe, Co, Cu) catalysts, prepared on a weight basis, with a Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :X ratio of 1:1 (wt/wt) have been synthesised via an excess chloride co-impregnation procedure, based on methodology previously reported in the literature.28,29 With catalysts produced via an impregnation procedure widely studied for the direct synthesis of H2O2 due to the simplicity and ease with which this approach can be scaled to meet industrial application. In particular catalysts prepared via the excess chloride co-impregnation route, where precious metal precursors (in this case the PdCl2) are acidified with HCl (0.58 M) have been shown to offer enhanced performance in a range of chemical transformations, including the direct synthesis of H2O2, compared to analogous materials prepared via a conventional wet co-impregnation procedure, where the metal precursors are not highly acidified.28 This has been attributed to a combination of improved alloy formation and enhanced nanoparticle dispersion.
:X ratio of 1:1 (wt/wt) have been synthesised via an excess chloride co-impregnation procedure, based on methodology previously reported in the literature.28,29 With catalysts produced via an impregnation procedure widely studied for the direct synthesis of H2O2 due to the simplicity and ease with which this approach can be scaled to meet industrial application. In particular catalysts prepared via the excess chloride co-impregnation route, where precious metal precursors (in this case the PdCl2) are acidified with HCl (0.58 M) have been shown to offer enhanced performance in a range of chemical transformations, including the direct synthesis of H2O2, compared to analogous materials prepared via a conventional wet co-impregnation procedure, where the metal precursors are not highly acidified.28 This has been attributed to a combination of improved alloy formation and enhanced nanoparticle dispersion.
The procedure to produce 0.5% Pd–0.5% Fe/TiO2 (2 g) is detailed below, with a similar methodology utilized for all mono- and bi-metallic catalysts using chloride-based metal precursors in all cases (see Table S.1† for further details). In all cases catalysts have been prepared using PdCl2 (0.58 M HCl, 6 mg mL−1, Merck).
Aqueous acidified PdCl2 solution (1.667 mL, 0.58 M HCl, 6 mg mL−1, Merck) and aqueous FeCl3·6H2O solution (0.0484 mL, 6 mg mL−1, Merck) were mixed in a 50 mL round-bottom flask and heated to 65 °C with stirring (1000 rpm) in a thermostatically controlled oil bath, with total volume fixed to 16 mL using H2O (HPLC grade). Upon reaching 65 °C, TiO2 (1.98 g, Degussa, P25) was added over the course of 5 min with constant stirring. The resulting slurry was stirred at 65 °C for a further 15 min, following this the temperature was raised to 95 °C for 16 h to allow for complete evaporation of water. The resulting solid was ground prior to a reductive heat treatment (5% H2/Ar, 400 °C, 4 h, 10 °C min−1).
Total metal loading, as determined by MP-AES analysis of aqua regia digested catalysts can be seen in Table S.2,† with the corresponding surface area measurements, as determined by five-point N2 adsorption in Table S.3.†
Catalyst testing
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 50 mL, equipped with a glass liner so that nominal volume is reduced to 33 mL, and a maximum working pressure of 2000 psi. To test each catalyst for H2O2 synthesis, the autoclave liner was charged with catalyst (0.01 g) and solvent (8.5 g H2O, HPLC Grade, Fischer Scientific). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5% H2/CO2 to a pressure of 420 psi, followed by the addition of 25% O2/CO2 (160 psi). Pressure of 5% H2/CO2 and 25% O2/CO2 are given as gauge pressures. The reaction was conducted at a temperature of 2 °C, for 0.5 h with stirring (1200 rpm). H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.0085 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1. Reactant gases were not continually supplied.Reactor temperature was controlled using a HAAKE K50 bath/circulator using an appropriate coolant.
Similar reactions were carried out under reaction conditions identical to those used for the oxidative degradation of phenol via in situ H2O2 synthesis, namely in the absence of the methanol co-solvent and at 30 °C.
Degradation of H2O2
Catalytic activity towards H2O2 degradation (via hydrogenation and decomposition pathways) was determined in a similar manner to the direct synthesis activity of a catalyst. The autoclave liner was charged with water (7.81 g, HPLC grade, Fischer Scientific), H2O2 (50 wt% 0.69 g, Merck), and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution 2 aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The autoclave was pressurised with 420 psi 5% H2/CO2 (gauge pressure). The reaction was conducted at a temperature of 2 °C, for 0.5 h with stirring (1200 rpm). After the reaction was complete the catalyst was removed from the reaction mixture and two aliquots of 0.05 g were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.Similar reactions were carried out under reaction conditions identical to those used for the oxidative degradation of phenol via in situ H2O2 synthesis, namely in the absence of the methanol co-solvent and at 30 °C.
Oxidative degradation of phenol via the in situ production of H2O2
Catalytic activity towards the degradation of phenol was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 50 mL, equipped with a glass liner so that nominal volume is reduced to 33 mL, and a maximum working pressure of 2000 psi. In a typical test the autoclave was charged with catalyst (0.01 g) and phenol (8.5 g, 1000 ppm aqueous phenol). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5% H2/CO2 to a pressure of 420 psi, followed by the addition of 25% O2/CO2 (160 psi). Pressure of 5% H2/CO2 and 25% O2/CO2 are given as gauge pressures. The reactor was then heated to 30 °C followed by stirring (1200 rpm) typically for 2 h. After 2 h gas mixtures were sampled and analysed via GC (Varian 3800 GC fitted with a TCD and equipped with a Porapak Q column). The reaction solution was collected and catalyst removed via filtration, the post-reaction solution was analysed by high performance liquid chromatography (HPLC) fitted with an Agilent Poroshell 120 SB-C18 column. Reactant gases were not continually supplied.Throughout product distribution is of phenol oxidation have been grouped into two categories, namely phenol oxygenated derivatives (catechol, hydroquinone etc.) and organic acids (oxalic acid, formic acid etc.). While it is theoretically possible for the completed oxidation of phenol to occur, the presence of water as a reaction medium and carbon dioxide as a reagent gas diluent prevents the detection of these total oxidation products.
Phenol conversion (eqn (1)), H2 conversion (eqn (2)) and selectivity towards phenolic derivatives (eqn (3)) or organic acids (eqn (4)) are defined as follows:
 | (1) |
 | (2) |
 | (3) |
 | (4) |
Catalytic hydrogenation of phenol
In order to determine the ability of the Pd-based catalysts to convert phenol to hydrogenation products under in situ oxidation reaction conditions an identical procedure to that outlined above for the for the degradation of phenol was followed for a reaction time of 2 h, with the 25% O2/CO2 component typically used exchanged for N2 (160 psi).Gas replacement experiments for the oxidative degradation of phenol via the in situ production of H2O2
An identical procedure to that outlined above for the degradation of phenol was followed for a reaction time of 2 h. After this, stirring was stopped and the reactant gas mixture was vented prior to replacement with the standard pressures of 5% H2/CO2 (420 psi) and 25% O2/CO2 (160 psi). The reaction mixture was then stirred (1200 rpm) for a further 2 h. To collect a series of data points, as in the case of Fig. 2, it should be noted that individual experiments were carried out and the reactant mixture was not sampled on-line.Hot filtration experiments for the oxidative degradation of the oxidation of phenol via the in situ production of H2O2
An identical procedure to that outlined above for the oxidative degradation of phenol was followed for a reaction time of 1 h. Following this, the stirring was stopped, and the reactant gas mixture vented prior to the removal of the solid catalyst via filtration. The post-reaction solution was returned to the reactor to identify the contribution of leached species to the observed activity, with both steps of the reaction conducted at a temperature of 30 °C. Further experiments were conducted, where a fresh 1% Pd/TiO2 catalyst (0.01 g) or preformed H2O2, comparable to that if all the H2 converted in the first 1 h reaction was converted selectively to H2O2, was added to the reaction mixture prior to running the reaction for a further 1 h.Catalyst characterisation
A Thermo Scientific K-Alpha+ spectrometer was used to collect X-ray photoelectron spectra utilizing a microfocused monochromatic Al Kα X-ray source operating at 72 W (6 mA × 12 kV). Data were collected over a X-ray spot defined elliptical area with a radius of approximately 400 μm2 at pass energies of 40 and 150 eV for high-resolution and survey spectra, respectively. Sample charging effects were minimized through a combination of low-energy electrons and Ar+ ions, and a C(1s) line at 284.8 eV was present for all samples. All data were processed using CasaXPS v2.3.24 (ref. 31) using a Shirley background, Scofield sensitivity factors, and an electron energy dependence of −0.6. Fitting was achieved using models taken from bulk compounds (Pd and Au metal foils and PdO). The Au(4f) peaks fit on top of the Ti loss structure and also the Pd(4s) signal; to account for this background, a broad, constrained Voight function was used to model these underlying peaks.The bulk structure of the catalysts was determined by powder X-ray diffraction using a (θ–θ) PANalytical X'pert Pro powder diffractometer using a Cu Kα radiation source, operating at 40 KeV and 40 mA. Standard analysis was carried out using a 40 min run with a back filled sample, between 2θ values of 10–80°. Phase identification was carried out using the International Centre for Diffraction Data (ICDD).
Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dispersion in ethanol by sonication and deposited on 300 mesh copper grids coated with holey carbon film. Energy dispersive X-ray analysis (EDX) was performed using an Oxford Instruments X-MaxN 80 detector and the data analysed using the Aztec software.
Total metal loading and metal leaching from catalyst supported was quantified using microwave plasma – atomic emission spectroscopy (MP-AES). Fresh catalysts were digested (25 mg catalyst, 2.5 ml aqua regia, 24 h) prior to analysis using an Agilent 4100 MP-AES, while post reaction solutions were also analysed after filtration of the solid material. The concentration of leached metals in product streams was quantified through microwave plasma atomic emission spectroscopy (MP-AES) using an Agilent MP-AES 4100. Post-reaction solutions were filtered using PTFE syringe filters (0.45 μm) prior to analysis for Fe and Pd. Emission lines were calibrated using commercial standards (Merck), in all cases r2 > 0.999.
Brunauer–Emmett–Teller (BET) surface area measurements were conducted using a Quadrasorb surface area analyzer. A five-point isotherm of each material was measured using N2 as the adsorbate gas. Samples were degassed at 250 °C for 2 h prior to the surface area being determined by five-point N2 adsorption at −196 °C, and data were analyzed using the BET method.
1H NMR spectra were recorded on a Bruker Ultrashield 500 MHz spectrometer, using a H2O solvent suppression program. Filtered post-hydrogenation reaction solvent (0.7 mL) was added to an NMR tube containing D2O (0.1 mL). In a similar manner cyclohexanone and cyclohexanol standards (0.7 mL, both Merck) were added to an NMR tube containing CDCl3 (0.1 mL, Merck).
Results and discussion
Our initial studies investigated the efficacy of a range of supported bimetallic catalysts, prepared by a co-impregnation, excess chloride procedure towards the direct synthesis and subsequent degradation of H2O2 (Table 1). These experiments were carried out under conditions previously optimised to enhance H2O2 stability, namely in the presence of a methanol co-solvent, CO2 gaseous diluent and sub-ambient reaction temperatures, all of which have been shown to inhibit H2O2 degradation pathways.29| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. | ||
| 1% Pd/TiO2 | 30 | 198 |
| 0.5% Pd–0.5% Au/TiO2 | 97 | 258 |
| 0.5% Pd–0.5% Cu/TiO2 | 11 | 80 |
| 0.5% Pd–0.5% Co/TiO2 | 42 | 109 |
| 0.5% Pd–0.5% Fe/TiO2 | 38 | 51 |
In keeping with numerous studies,32–34 the introduction of Au into a supported Pd catalyst was found to significantly improve catalytic performance towards H2O2 synthesis, with H2O2 synthesis rates (97 molH2O2 kgcat−1 h−1) far greater than that observed over the Pd-only catalyst (30 molH2O2 kgcat−1 h−1). The introduction of a range of secondary base-metals, (Fe, Cu, Co) which are well-known to catalyse H2O2 decomposition to H2O via Fenton pathways35 offered lower H2O2 synthesis to that observed for the PdAu analogue, although with the exception of the 0.5% Pd–0.5% Cu/TiO2 catalyst, rates of H2O2 synthesis were comparable to the 1% Pd/TiO2 catalyst. Interestingly, the introduction of Fenton or Fenton-like metals did not result in an enhancement in activity towards H2O2 degradation (via hydrogenation or decomposition pathways), which may have been expected, with these materials offering H2O2 degradation rates significantly lower than either the PdAu (258 molH2O2 kgcat−1 h−1) or Pd-only catalysts (198 molH2O2 kgcat−1 h−1). This is in keeping with our previous studies24,36 and may be ascribed to a combination of (i) the reaction conditions chosen to determine H2O2 synthesis and degradation activity, with the dissolution of the CO2 reactant gas diluent forming carbonic acid in situ, resulting in H2O2 stabilisation30 or (ii) the possible modification of Pd oxidation state as a result of secondary metal introduction. With the formation of mixed domains, consisting of Pd0 and Pd2+ well reported to offer enhanced catalytic performance towards H2O2 synthesis, compared to purely Pd0 or Pd2+ analogues.14,37 However, our analysis by XPS (Fig. S.1†) reveals that while the introduction of base-metals (Fe, Cu, Co) does enhance Pd2+ content, a similar shift in Pd speciation is similarly observed with the introduction of Au, and as such we cannot definitively attribute the enhanced catalytic H2O2 synthesis activity of the Pd-X (Fe, Cu, Co) catalysts to the increased presence of Pd0–Pd2+ species. It should be noted that a number of previous experimental38,39 and computational40 studies have revealed the ability of high concentrations of Cu to inhibit catalytic activity towards H2O2 production. Indeed, Joshi et al. have revealed the thermodynamic instability of intermediate hydroperoxyl (OOH*) species and in turn H2O2 over Cu-containing surfaces.40
The generation of hydroxyl radicals via Fenton's or photo-Fenton's pathways has been well reported to offer high efficacy in the remediation of organic contaminants,41–43 with this in mind we next investigated the efficacy of these Pd-based bi-metallic catalysts towards the oxidative degradation of phenol via in situ H2O2 production (Table 2). It should be noted that it was not possible to measure residual H2O2via standard titration or colorimetric procedures given the strong reddish colour that results from the formation of the aromatic oxidation products (catechol, hydroquinone etc.).44 However, our previous studies have determined the ability of H2O2 to be synthesised under reaction conditions comparable to those used within this study.45
| Catalyst | Phenol conversion/% | Selectivity towards phenolic derivatives/% | Selectivity towards organic acids/% |
|---|---|---|---|
| Phenol degradation reaction conditions: catalyst (0.01 g), phenol (1000 ppm, 8.5 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 2 h, 30 °C, 2 h. a The mass of each catalyst is reduced to 0.005 g, to ensure total moles of metal are identical to those in the 0.5% Pd–0.5% Fe/TiO2 catalyst. | |||
| 1% Pd/TiO2 | 11.0 | 11.0 | 89.0 |
| 0.5% Pd–0.5% Au/TiO2 | 12.0 | 10.0 | 90.0 |
| 0.5% Pd–0.5% Cu/TiO2 | 6.0 | 6.0 | 94.0 |
| 0.5% Pd–0.5% Co/TiO2 | 6.0 | 43.0 | 57.0 |
| 0.5% Pd–0.5% Fe/TiO2 | 39.0 | 31.0 | 69.0 |
| 1% Pd/TiO2 + 1% Fe/TiO2a | 12.0 | 37.0 | 63.0 |
Interestingly, a direct correlation between catalytic performance towards H2O2 synthesis and phenol degradation was not observed. With the 0.5% Pd–0.5% Fe/TiO2 catalyst seen to be significantly more active towards phenol degradation (48%) compared to the alternative Pd-based catalysts, including the Pd-only (21%) and PdAu (5%) analogues, despite both these materials displaying H2O2 formation rates comparable to or greater than the 1% PdFe/TiO2 catalyst. This is perhaps unsurprising, given the ability of Fe to catalyse the formation of reactive oxygen species, such as ˙OH, from H2O2.46 Further studies revealed that there is a requirement for both Pd and Fe to be immobilised onto the same grain of support, with the bi-metallic catalyst greatly outperforming a physical mixture of the monometallic analogues (12%).
The significant improvement in phenol degradation in the presence of H2 and O2 in comparison to that observed when molecular H2 (6% phenol conversion) or O2 (4% phenol conversion) were used alone, or when commercial H2O2 (<1% phenol conversion) was used should also be noted (Table S.4†). With the relatively high rate of phenol conversion observed in the presence of a reductive atmosphere alone (5% H2/CO2 and N2) ascribed to the formation of low concentrations of H2O2, with dissolved O2 present in the reaction solution, rather than phenol hydrogenation. While numerous studies have reported the ability of Pd surfaces to catalyse the reduction of phenol to cyclohexanone, typically temperatures far exceeding that used within this work are required.47 Indeed, our analysis by 1H NMR (Fig. S.2, ESI†) does not indicate the formation of phenol hydrogenation products, such as cyclohexanol and cyclohexanone.
With the high catalytic efficacy of the 0.5% Pd–0.5% Fe/TiO2 catalyst established we next investigated the effect of Fe:Pd ratio on phenol degradation activity (Fig. 1). Given the separate and distinct roles of Pd and Fe, with the former primarily catalysing the synthesis of H2O2, and the latter subsequently activating the synthesised H2O2 through Fenton's pathways to oxygen-centred radicals, which are considered to be the key species responsible for phenol degradation, it is imperative to balance both reactions to achieve maximal oxidant efficiency.
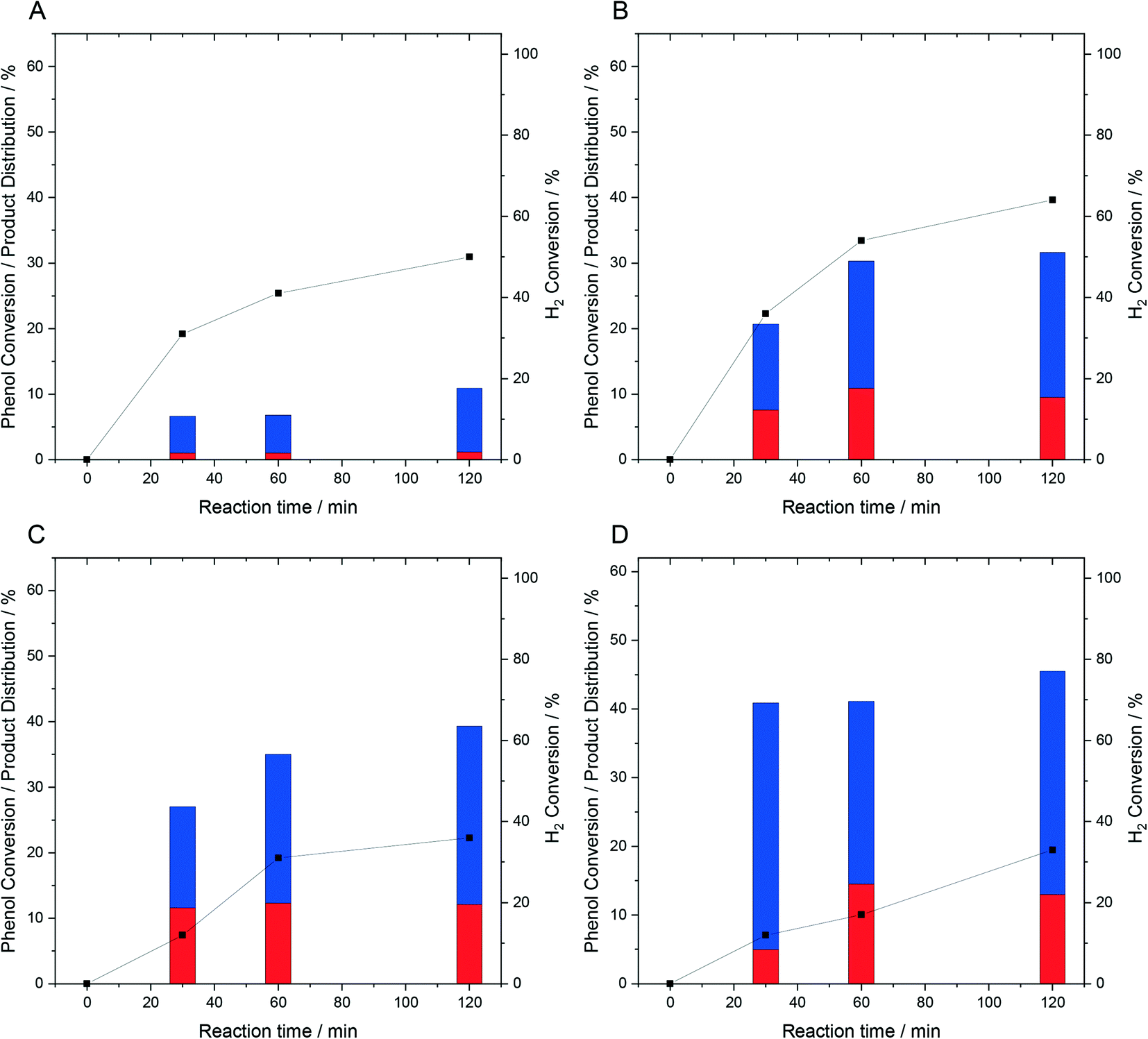 | ||
| Fig. 1 Catalytic activity of 1% PdFe/TiO2 catalysts towards the oxidative degradation of phenol, via in situ H2O2 production, as a function of Pd:Fe ratio. (A) 1% Pd/TiO2, (B) 0.75% Pd–0.25% Fe/TiO2, (C) 0.5% Pd–0.5% Fe/TiO2 and (D) 0.25% Pd-0.75% Fe/TiO2 phenol degradation reaction conditions: catalyst (0.01 g), phenol (1000 ppm, 8.5 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 2 h, 30 °C. Key; selectivity towards phenolic derivatives (red bar), selectivity towards organic acids (blue bar), H2 conversion (black squares). Note: the activity of the 1% Fe/TiO2 catalyst was found to be within experimental error of the blank reaction. | ||
As indicated by our previous studies into the direct synthesis of H2O2 under conditions optimised for H2O2 stability (Table 1) the addition of Fe, regardless of total content does not significantly alter catalytic activity towards H2O2 synthesis under reaction conditions identical to those utilised for the oxidative degradation of phenol, i.e., under near-ambient reaction temperatures and in the absence of the methanol co-solvent (Fig. S.3†). However, with increasing Pd content, activity towards H2O2 degradation is seen to decrease substantially (Table S.5†), in keeping with our previous studies into analogous AuPd catalysts.29 Subsequent investigation into the performance of the 1% PdFe/TiO2 catalysts towards the oxidative degradation of phenol revealed an optimal catalyst formulation of 0.25% Pd–0.75% Fe/TiO2, with this catalyst displaying the highest rate of phenol conversion (46%) over a 2 h reaction (Fig. 1).
Analysis of the as-prepared 1% PdFe/TiO2 catalysts by XPS is reported in Fig. S.4.† In the case of the 1% Pd/TiO2 catalyst Pd is seen to exist almost entirely as Pd0, which may be expected given the reductive heat treatment (4 h, 400 °C, 5%H2/Ar) applied to these materials prior to use. The introduction of Fe was found to result in a clear shift in Pd oxidation state, towards Pd2+, with a strong relationship between Fe content and the proportion of Pd2+ observed. Indeed, in the case of the 0.25% Pd–0.75% Fe/TiO2 catalyst Pd was found to exist predominantly as Pd2+. Meanwhile for all bi-metallic catalysts Fe was found to exist primarily as Fe2+, as evidenced by a signal at approximately 709–710 eV and satellite signal at 715 eV. Contrastingly, Fe was found to be present predominantly as Fe3+ in the monometallic Fe catalyst, as evidenced by a signal at 711 eV and a corresponding satellite at approximately 719 eV, although as in the bi-metallic PdFe catalysts some Fe2+ is also observed. Given the increased selectivity of domains of mixed Pd oxidation state towards H2O2,14 it is therefore reasonable to attribute the increased phenol degradation activity of the PdFe catalysts, in part, to an inhibition of competitive H2O2 degradation pathways and improved utilisation of H2O2 in the formation of reactive oxygen species.
With the strong correlation between catalytic selectivity and nanoparticle size well known,48 we subsequently established the high dispersion of the PdFe nanoparticles, by XRD (Fig. S.5†) and TEM (Fig. S.6†), with no observable nanoparticles detected regardless of Pd:Fe ratio. This is in keeping with our previous work into supported Pd catalysts prepared by an analogous excess chloride procedure, with this route to catalyst synthesis well known to offer improved metal dispersion compared to a conventional wet co-impregnation methodology.29
The relatively high H2 conversion rates observed over the bi-metallic PdFe catalysts at extended reaction times (Fig. 1), in addition to the concurrent plateau in phenol conversion may be indicative of the reaction becoming limited by H2 availability. This can be understood through the first order dependence of H2O2 production with respect to H2 (ref. 49) and in turn the limited generation of reactive oxygen species, responsible for phenol degradation, when H2 availability decreases. To determine if this was the case and with a focus on the 0.5% Pd–0.5% Fe/TiO2 catalyst we next conducted a series of sequential phenol oxidation tests, where the reagent gases were replenished at 2 h intervals (Fig. 2). It is observed that, while it is possible to enhance phenol conversion through replacement of gaseous reagents (67% over four sequential reactions) it is clear that catalytic performance decreases over multiple phenol degradation reactions, possibly indicative of catalyst deactivation rather than limited reagent availability.
 | ||
| Fig. 2 Comparison of the catalytic activity toward the oxidative degradation of phenol via in situ H2O2 synthesis over sequential reaction number. Phenol degradation reaction conditions: catalyst (0.01 g), phenol (1000 ppm, 8.5 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 2 h, 30 °C, 2 h. key; selectivity towards phenolic derivatives (red bar), selectivity towards organic acids (blue bar), H2 conversion (black squares). | ||
For any heterogeneous catalyst operating in a three-phase system the possibility of the leaching of the active phase and resulting homogeneous contribution to observed catalytic performance is of great concern, with the activity of homogeneous Fe and Pd species known to catalyse the formation of oxygen-based radicals (via Fenton's pathways) and the direct synthesis of H2O2 respectively.49–53 Analysis of post reaction solutions via MP-AES (Table S.6†) reveals minimal leaching of Pd in the post-reaction solution. On the other hand, a significant amount of leached Fe is observed, regardless of catalyst composition. This is in keeping with previous investigations that established the ability of phenolic oxidative products, such as oxalic acid and catechol, to chelate to heterogeneous Fe species and promote their dissolution.54 With MP-AES analysis of post-reaction solutions in the absence of phenol, or under conditions where H2O2 is not generated (Table S.7†), providing further evidence of the role of phenol oxidation products to promote the leaching of supported metals.
With a particular focus on the 0.5% Pd–0.5% Fe/TiO2 catalyst, we next conducted a series of hot-filtration experiments to identify the contribution of leached metal species to catalytic activity (Fig. 3). In the absence of the heterogeneous catalyst, minimal additional phenol conversion was observed (41%) after a two-part, 2 h experiment, where the heterogeneous catalyst was removed by filtration after 1 h prior to the post-reaction solution being returned to the reactor for a further 1 h. This value was nearly identical to that observed for the 0.5% Pd–0.5% Fe/TiO2 catalyst over a 1 h reaction (35%), with the limited additional conversion of phenol possibly attributed to the contribution from residual H2O2 generated in the initial 1 h reaction.
 | ||
| Fig. 3 Efficacy of leached species in oxidative degradation of phenol as identified by a hot filtration experiment using the 0.5% Pd/3% Fe-ZSM-5 catalyst. Phenol oxidation reaction conditions: catalyst (0.01 g), phenol (1000 ppm), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 1200 rpm, 2 h, 30 °C. key: 0.5% Pd–0.5% Fe/TiO2 catalysed reaction (black squares); 1% Pd/TiO2 catalysed reaction (green inverted triangles); hot filtration reaction where the 0.5% Pd–0.5% Fe/TiO2 catalyst is removed by filtration after 1 h (red circles); hot filtration reaction where 0.5% Pd–0.5% Fe/TiO2 catalyst removed by filtration after 1 h and replaced by 1% Pd/TiO2 catalyst for final 1 h of reaction (blue triangles), hot filtration reaction where 0.5% Pd–0.5% Fe/TiO2 catalyst removed by filtration after 1 h and replaced by commercial H2O2 (concentration identical to that if all H2 utilised in a standard in situ reaction was selectively converted to H2O2) catalyst for final 1 h of reaction (purple diamonds). | ||
To determine if the inactivity observed in the 0.5% Pd–0.5% Fe/TiO2 catalyst hot-filtration experiment was due to the limited ability of the homogeneous component to synthesize H2O2, which may be reasonable given our previous studies which identified the stability of Pd during the phenol degradation reaction (Tables S.6 and S.7†) a further hot-filtration experiment was conducted whereby, after the initial 1 h reaction, the 0.5% Pd–0.5% Fe/TiO2 catalyst was replaced with a 1% Pd/TiO2 analogue, ensuring that the total moles of Pd was equal to that in the 0.5% Pd–0.5% Fe/TiO2 catalyst. Perhaps unexpectedly, given the ability of the 1% Pd/TiO2 catalyst to catalyse the oxidative degradation of phenol (Fig. 1) an increase in phenol conversion was observed (41%), similar to the sum of the 0.5% Pd–0.5% Fe/TiO2 (35%) and 1% Pd/TiO2 (7%) components when they were used independently over 1 h. Indeed, the extent of phenol conversion was found to be nearly identical to that observed over the analogous two-part, 2 h duration experiment conducted over the 0.5% Pd–0.5% Fe/TiO2 catalyst alone (39%). Given the ability of the 1% Pd/TiO2 catalyst to promote the degradation of phenol this experiment was unable to confirm a contribution from homogeneous Fe species. In a final experiment, after the initial 1 h reaction utilising the 0.5% Pd–0.5% Fe/TiO2 catalyst alone, commercial H2O2, at a concentration equivalent to if all H2 in the in situ reaction was selectively converted to H2O2, was added to the reaction mixture. After a further 1 h reaction (carried out in the presence of an atmosphere of O2/CO2 and the absence of a heterogenous catalyst) a small improvement in phenol conversion was observed (39%). When coupled with the known ability of homogenous Fe species to catalyse the formation of oxygen based radical species, via the Fenton process, this may be indicative of a homogenous component. However, if there is a homogeneous component to catalytic activity we consider these experiments, in addition to those which illustrate the need for close contact between Pd and Fe species (Table 2), to indicate that such a contribution is minimal.
Conclusion
We have demonstrated that the introduction of Fe into supported Pd catalysts can enhance catalytic performance, not only towards the direct synthesis of H2O2 but also the oxidative degradation of phenol via in situ H2O2 production. This is observed under reaction conditions where conversion is limited when using commercial H2O2. The enhanced catalytic performance of the 1% PdFe/TiO2 catalyst, observed to be almost four times more active than the Pd-only analogue, is attributed to decreased H2O2 degradation pathway and the ability of Fe to promote the formation of highly reactive oxygen-based radicals via Fenton's pathways. While catalyst stability is still of concern, with substantial leaching of Fe observed, which is promoted by phenol oxidation products observed we consider that these catalysts represent a promising basis for further exploration for the oxidative degradation of a range of recalcitrants.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors wish to acknowledge the financial support of and research discussion with Dŵr Cymru Welsh Water. The authors also acknowledge Cardiff University and the Max Planck centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support. We thank the EPSRC (Grant EP/L016443/1) for support. The Cardiff University electron microscope facility is also acknowledged for the transmission electron microscopy.References
- T. A. Larsen, S. Hoffmann, C. Lüthi, B. Truffer and M. Maurer, Science, 2016, 352, 928–933 CrossRef PubMed.
- S. B. Grant, J. Saphores, D. L. Feldman, A. J. Hamilton, T. D. Fletcher, P. L. M. Cook, M. Stewardson, B. F. Sanders, L. A. Levin, R. F. Ambrose, A. Deletic, R. Brown, S. C. Jiang, D. Rosso, W. J. Cooper and I. Marusic, Science, 2012, 337, 681–686 CrossRef PubMed.
- A. Fernandes, P. Makoś, J. A. Khan and G. Boczkaj, J. Cleaner Prod., 2019, 208, 54–64 CrossRef.
- S. Esplugas, J. Giménez, S. Contreras, E. Pascual and M. Rodríguez, Water Res., 2002, 36, 1034–1042 CrossRef PubMed.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- F. J. Potter and J. A. Roth, Hazard. Waste Hazard. Mater., 1993, 10, 151–170 CrossRef.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodríguez, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS PubMed.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- J. R. Scoville and I. A. Novicova, (Cottrell Ltd.), US 5900256, 1996.
- Y. T. G. Gao, X. Gong, Z. Pan, K. Yong and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef.
- N. M. Wilson and D. W. Flaherty, J. Am. Chem. Soc., 2016, 138, 574–586 CrossRef CAS PubMed.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- G. Han, X. Xiao, J. Hong, K. Lee, S. Park, J. Ahn, K. Lee and T. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 6328–6335 CrossRef CAS PubMed.
- S. Sterchele, P. Biasi, P. Centomo, P. Canton, S. Campestrini, T. Salmi and M. Zecca, Appl. Catal., A, 2013, 468, 160–174 CrossRef CAS.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Appl. Catal., A, 2008, 339, 130–136 CrossRef.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef PubMed.
- S. Maity and M. Eswaramoorthy, J. Mater. Chem. A, 2016, 4, 3233–3237 RSC.
- D. A. Crole, R. Underhill, J. K. Edwards, G. Shaw, S. J. Freakley, G. J. Hutchings and R. J. Lewis, Philos. Trans. R. Soc. London, 2020, 378, 20200062 Search PubMed.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y.-F. Han, Chin. J. Catal., 2018, 39, 673–681 CrossRef.
- K. Cao, H. Yang, S. Bai, Y. Xu, C. Yang, Y. Wu, M. Xie, T. Cheng, Q. Shao and X. Huang, ACS Catal., 2021, 11, 1106–1118 CrossRef.
- H. Xu, D. Cheng and Y. Gao, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2021, 151, 164–171 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. J. A. Slater, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2021, 151, 2762–2774 CrossRef CAS.
- R. Underhill, M. Douthwaite, R. J. Lewis, P. J. Miedziak, R. D. Armstrong, D. J. Morgan, S. J. Freakley, T. Davies, A. Folli, D. M. Murphy, Q. He, O. Akdim, J. K. Edwards and G. J. Hutchings, Res. Chem. Intermed., 2021, 47, 303–324 CrossRef CAS.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley, D. W. Knight, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6600–6613 CrossRef CAS PubMed.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388 RSC.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci., 2021, 5, 100112 CrossRef.
- J. Brehm, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catal. Lett., 2021 DOI:10.1007/s10562-021-03632-6.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- T. García, S. Agouram, A. Dejoz, J. F. Sánchez-Royo, L. Torrente-Murciano and B. Solsona, Catal. Today, 2015, 248, 48–57 CrossRef.
- R. C. C. Costa, M. F. F. Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. V. A. Rios, C. N. Silva and R. M. Lago, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- L. Ouyang, P. Tian, G. Da, X. Xu, C. Ao, T. Chen, R. Si, J. Xu and Y. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- F. Alotaibi, S. Al-Mayman, M. Alotaibi, J. K. Edwards, R. J. Lewis, R. Alotaibi and G. J. Hutchings, Catal. Lett., 2019, 149, 998–1006 CrossRef CAS.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins, J. A. Lopez-Sanchez, S. H. Taylor and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 3410–3418 RSC.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. C, 2007, 111, 7384–7395 CrossRef CAS.
- S. Varnagiris, M. Urbonavicius, S. Sakalauskaite, R. Daugelavicius, L. Pranevicius, M. Lelis and D. Milcius, Sci. Total Environ., 2020, 720, 137600 CrossRef CAS PubMed.
- M. Abidi, A. Hajjaji, A. Bouzaza, K. Trablesi, H. Makhlouf, S. Rtimi, A. A. Assadi and B. Bessais, J. Photochem. Photobiol., A, 2020, 400, 112722 CrossRef CAS.
- S. Y. Yang, D. Kim and H. Park, Environ. Sci. Technol., 2014, 48, 2877–2884 CrossRef CAS PubMed.
- F. Mijangos, F. Varona and N. Villota, Environ. Sci. Technol., 2006, 40, 5538–5543 CrossRef CAS PubMed.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Commun., 2007, 8, 247–250 CrossRef CAS.
- F. J. Benitez, J. Beltran-Heredia, J. L. Acero and F. J. Rubio, Ind. Eng. Chem. Res., 1999, 38, 1341–1349 CrossRef CAS.
- H. Zhou, B. Han, T. Liu, X. Zhong, G. Zhuang and J. Wang, Green Chem., 2017, 19, 3585–3594 RSC.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y.-F. Han, J. Catal., 2017, 349, 30–40 CrossRef.
- Q. Liu and J. H. Lunsford, Appl. Catal., A, 2006, 314, 94–100 CrossRef.
- V. Kavitha and K. Palanivelu, Chemosphere, 2004, 55, 1235–1243 CrossRef PubMed.
- V. Kavitha and K. Palanivelu, Int. J. Environ. Sci. Technol., 2016, 13, 927–936 CrossRef.
- Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619–621 CrossRef PubMed.
- S. J. Freakley, N. Agarwal, R. U. McVicker, S. Althahban, R. J. Lewis, D. J. Morgan, N. Dimitratos, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 5935–5944 RSC.
- S. O. Lee, T. Tran, Y. Y. Park, S. J. Kim and M. J. Kim, Int. J. Miner. Process., 2006, 80, 144–152 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01897c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |