
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chitosan as a chiral ligand and organocatalyst: preparation conditions–property–catalytic performance relationships†
Vanessza Judit
Kolcsár
a and
György
Szőllősi
 *b
*b
aDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, 6720 Szeged, Hungary
bStereochemistry Research Group, Eötvös Loránd Research Network, University of Szeged, Eötvös utca 6, 6720 Szeged, Hungary. E-mail: szollosi@chem.u-szeged.hu
First published on 15th October 2021
Abstract
Chitosan is an abundant and renewable chirality source of natural origin. The effect of the preparation conditions by alkaline hydrolysis of chitin on the properties of chitosan was studied. The materials obtained were used as ligands in the ruthenium-catalysed asymmetric transfer hydrogenation of aromatic prochiral ketones and oxidative kinetic resolution of benzylic alcohols as well as organocatalysts in the Michael addition of isobutyraldehyde to N-substituted maleimides. The degrees of deacetylation of the prepared materials were determined by 1H NMR, FT-IR and UV-vis spectroscopy, the molecular weights by viscosity measurements, their crystallinity by WAXRD, and their morphology by SEM and TEM investigations. The materials were also characterized by Raman spectroscopy. The biopolymers which have molecular weights in a narrow (200–230 kDa) range and appropriate (80–95%) degrees of deacetylation were the most efficient ligands in the enantioselective transfer hydrogenation, whereas in the oxidative kinetic resolution the activity of the complexes and the stereoselectivity increased with the degree of deacetylation. The chirality of the chitosan was sufficient to obtain enantioselection in the Michael addition of isobutyraldehyde to maleimides in the aqueous phase. Interestingly, the biopolymer afforded the opposite enantiomer in excess compared to the monomer, D-glucosamine. In this reaction, good correlation between the degree of deacetylation and the catalytic activity was found. These results are novel steps in the application of this natural, biocompatible and biodegradable polymer in developing environmentally benign methods for the production of optically pure fine chemicals.
Introduction
Strict environmental regulations require novel solutions in the chemical industry to reduce production wastes and pollution. Therefore, the application of renewable raw materials and auxiliaries is the focus of both bulk and fine-chemical industries. Asymmetric catalytic methods are advantageous for preparing enantiomerically pure chiral intermediates required mostly in the pharmaceutical industry.1–3 However, in these processes the application of natural materials as chirality sources, i.e. ligands or catalysts, often led to unsatisfactory performances compared to the expensive, synthetic, structurally tailored chiral compounds. Shell food industries generate a significant amount of wastes, as only 20–30% of the processed materials are edible; thus, marine arthropod shell deposits are a huge source of chitin.4,5 This carbohydrate polymer has diverse applications;6,7 among others, it serves as raw material for the production of glucosamine, a nonsteroidal anti-inflammatory drug.8,9 Deacetylation of chitin results in the formation of chitosan, a polysaccharide containing amine groups, having multiple biomedical, agricultural, and industrial applications.10–12 An effective way to prepare chitosan is the hydrolysis of chitin in aqueous alkaline medium (Scheme 1), widely applied to gain materials for various applications.10,12–18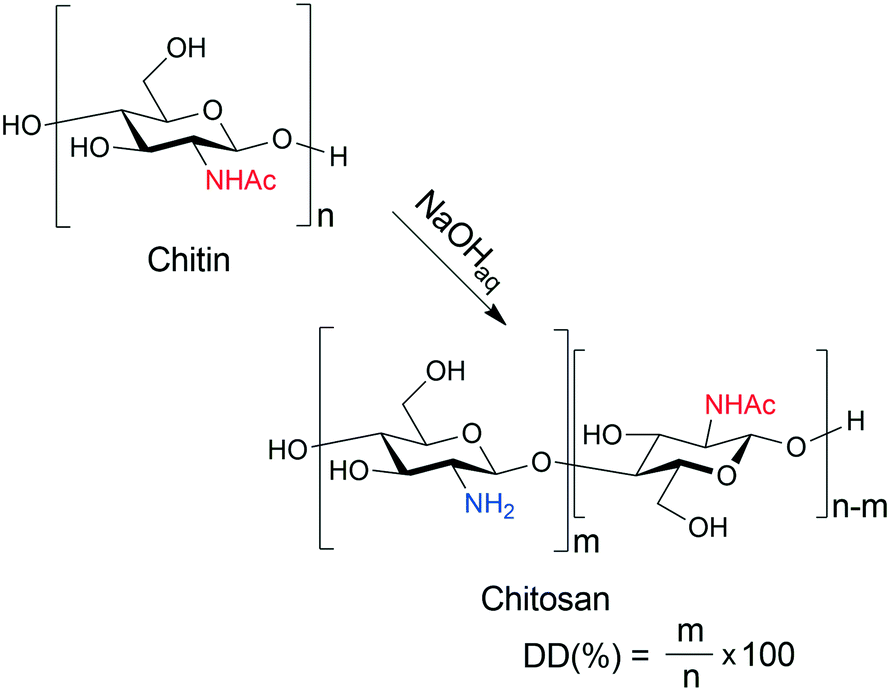 | ||
| Scheme 1 Preparation of chitosan from chitin by aqueous alkaline hydrolysis; DD: degree of deacetylation. | ||
The terms “chitin” and “chitosan” are used to denote polysaccharides built up by N-acetyl-D-glucosamine (D-GlcNAc) and D-glucosamine (D-GlcNH2) monomers of various ratios. Due to the availability of large amounts of chitosan, in the past few decades this material appeared in the focus of green chemistry and catalysis as well. Chitosan, as a chirality source, offers the possibility of developing environmentally benign, sustainable stereoselective chemical processes. However, in most catalytic applications, either as a support, ligand or organocatalyst, to achieve appropriate performances it was necessary to modify its structure by various functionalization.19–27 Thus, the catalytic efficiency in asymmetric catalytic processes of many chitosan derivatives has been studied, whereas that of unfunctionalized materials so far has received less attention.28–40
In our recent studies, we have examined the asymmetric transfer hydrogenation (ATH) of prochiral ketones catalysed by chiral Ru complexes formed in situ using commercially available chitosan as ligand,41,42 knowing from Noyori's pioneering work that chiral 1,2-aminoalcohols may be efficient in these reductions.43 Numerous chiral alcohols were prepared in good to high optical purity in this catalytic system using an aqueous solvent mixture, which even surpassed the values obtained by applying chitosan derivatives.31,34 The probable structure of the chiral complex and possible interactions leading to the transition states responsible for the enantioselection were suggested.41,42
Hence, highly efficient catalytic systems using chitosan as a natural chirality source was developed, which may lead to environmentally benign synthesis of valuable chiral compounds. However, a more detailed examination of the effect of the chitosan structure, i.e. degree of deacetylation (DD), molecular weight (MW), morphology or crystallinity would be of great value to tune the catalyst. Moreover, these studies, combined with the knowledge gathered on the mechanism of the ATH using the Noyori–Ikariya Ru catalyst in aqueous systems,44–49 may lead to novel information related to the structure of the active species and the interactions occurring during ATH. These can further aid the development of other catalytic applications of this natural chiral material. Thus, our aim in the present study was to prepare a chitosan series with progressively altering characteristics either from chitin or from commercially available chitosan to investigate the effect of their properties in test ATHs. We also intended to investigate the applicability of these materials in other reactions, such as in the Ru-catalysed oxidative kinetic resolution of racemic alcohols, or as organocatalysts in asymmetric Michael additions.
Results and discussion
Preparation and characterization of chitosan series
A structure–efficiency study on reactions utilizing unfunctionalized chitosan as a chiral ligand or organocatalyst is presently not available. Accordingly, we have prepared chitosan homologues by deacetylation of commercial chitin or chitosan in alkaline aqueous media.First, we examined the effect of the hydrolysis time of chitin on the properties of the materials obtained using reaction conditions, i.e. amount of chitin, NaOHaq concentration (40 wt%), temperature (120 °C), inert gas (N2), and autogenous pressure, based on a previous report.16 Yields (%), DD (%) determined by three spectroscopic methods and the molecular weight derived from viscosity measurements, MW (kDa) (for determination methods, see the ESI†), of samples obtained after different hydrolysis times, denoted as C1 (1 h), C2 (2 h), C3 (4 h), C4 (8 h), C5 (16 h), and C6 (32 h), are presented in Table 1 and in the ESI,† Fig. S3. The spectroscopic methods used to determine the DD of the products, i.e.1H NMR, FT-IR and UV-vis (denoted as DD(NMR), DD(IR) and DD(UV), respectively), gave similar results. Small differences using these analytical methods were found in the case of samples having lower DD (70–85%).
| Entry | Sample | Starting material | Time (h) | Temp. (°C) | Yieldb (%) | DD(NMR)c | DD(IR)c | DD(UV)c | MW (kDa) | Xcr; Dpd (%; nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 600 mg starting material, 40 wt% NaOHaq 12 cm3, closed vial flushed with N2, 120 °C, 4 h. b Yield of the product calculated using the DD(NMR) values. c Degree of deacetylation (%) obtained using 1H NMR, IR and UV-vis spectroscopy, respectively. d Degree of crystallinity (Xcr) and crystal sizes (Dp) in the direction perpendicular to the (110) plane calculated based on WAXRD measurements. e Hydrolysis in a round-bottom flask using a reflux condenser. f Using 45 wt% NaOHaq. g Commercial chitosan, see the Experimental section (Exp.). h An additional unknown, crystalline material also resulted. | ||||||||||
| 1 | C1 | Chitin | 1 | 120 | 88 | 81 | 80 | 82 | 229 | 32; 3.8 |
| 2 | C12 | C1 | 1 | 120 | 95 | 96 | 94 | 96 | 219 | 60; 4.5 |
| 3 | C13 | C12 | 1 | 120 | 98 | 99 | 99 | >99 | 190 | 45; 4.2 |
| 4 | C2 | Chitin | 2 | 120 | 88 | 85 | 81 | 85 | 230 | 50; 4.4 |
| 5 | C3 | Chitin | 4 | 120 | 88 | 87 | 86 | 86 | 209 | 58; 4.5 |
| 6 | C4 | Chitin | 8 | 120 | 88 | 88 | 88 | 89 | 208 | 36; 4.5 |
| 7 | C5 | Chitin | 16 | 120 | 87 | 92 | 90 | 91 | 182 | 28; 4.4 |
| 8 | C6 | Chitin | 32 | 120 | 88 | 94 | 97 | 95 | 97 | 25; 7.3h |
| 9 | C7 | Chitin | 64 | 120 | 80 | 95 | 98 | 98 | 39 | 23; 9.9h |
| 10 | C8 | Chitin | 4 | 80 | 63 | 71 | 67 | 73 | 229 | 44; 2.9 |
| 11 | C9 | Chitin | 4 | 100 | 74 | 82 | 80 | 81 | 221 | 48; 3.7 |
| 12 | C10 | Chitin | 4 | 140 | 91 | 92 | 90 | 89 | 173 | 42; 4.8 |
| 13 | C11 | Chitin | 4 | 160 | 94 | 96 | 94 | 97 | 45 | 40; 4.9 |
| 14e | C14 | Chitin | 4 | 100 | 94 | 82 | 78 | 79 | 274 | 32; 3.9 |
| 15e | C15 | C14 | 4 | 100 | 97 | 98 | 97 | 98 | 260 | 51; 4.7 |
| 16e | C16 | C15 | 4 | 100 | 98 | >99 | 98 | >99 | 253 | 48; 4.6 |
| 17 | C17 | Chitin | 16 | 80 | 91 | 84 | 82 | 79 | 214 | 41; 4.3 |
| 18f | C18 | Chitin | 4 | 120 | 94 | 91 | 87 | 90 | 219 | 30; 4.6 |
| 19 | CHMWg | — | — | — | — | 92 | 96 | 90 | 317 | 53; 4.5 |
| 20 | C19 | CHMWg | 4 | 120 | 97 | 99 | >99 | >99 | 217 | 46; 4.5 |
| 21 | CLvisg | — | — | — | — | 86 | 78 | 86 | 191 | 33; 3.5 |
| 22 | C20 | CLvisg | 4 | 120 | 97 | 99 | 99 | >99 | 138 | 36; 4.3 |
| 23 | CLMWg | — | — | — | — | 90 | 82 | 91 | 93 | 58; 4.1 |
| 24 | C21 | CLMWg | 4 | 120 | 97 | 98 | 99 | 99 | 88 | 38; 4.1 |
Under the above conditions, 1 h was enough to reach 80% DD (entry 1), which slowly increased upon applying longer reaction times. The MW decreased by prolonging the reactions up to 8 h (entry 6), followed by a more drastic drop in longer reactions. However, the yield was independent of the hydrolysis time, showing that the material is not lost by fragmentation to water-soluble, unrecoverable small molecules, even during long hydrolysis, i.e. fragmentation of the polymer chain occurs randomly. The above tendencies were confirmed by extending the hydrolysis to 64 h (C7, Table 1, entry 9). Materials having high DD (DD(NMR) 95%, DD(IR) 98%, DD(UV) 98%) and the lowest MW (39 kDa) were obtained in similar yield (80%). Next, we have studied the effect of the hydrolysis temperature (Table 1 and ESI† Fig. S4). The yield and the DD increased significantly by raising the temperature; however, high temperatures contributed to the degradation of the polymer, and products with lower MW were obtained at temperatures over 120 °C (C10 and C11; entries 12 and 13).
Based on these results the effect of the hydrolysis conditions was further examined. C1 was hydrolysed in a subsequent experiment to C12 (entry 2), which by repeating the deacetylation gave C13 (entry 3). This sequential method results in close to fully deacetylated material having slightly decreased MW; accordingly, a much higher DD was reached compared to the one-step protocol carried out for 4 h (C3, entry 5). Moreover, yields of the additional steps were progressively higher than that of the first one, showing that formation of soluble fragments causing material loss occurs predominantly in the first hydrolysis. This may be explained by the easier accessibility of the N-acetyl groups following the partial deacetylation due to initial predominant formation of D-GlcNAc and D-GlcNH2 block copolymers, as earlier suggested by Kurita and co-workers.15
By carrying out the deacetylation in an open flask under reflux, using otherwise identical conditions (100 °C, N2 atmosphere), product C14 was obtained with a similar DD to C9 (entries 11 and 14). However, the higher MW and better yield of C14 showed that under autogenous pressure the hydrolysis of the polymer chain is more extensive. The three-step reaction under reflux conditions was also efficient in preparing highly deacetylated chitosan, similarly to the reactions carried out under pressure, whereas the final product had higher MW (C16, 253 kDa, compared to C13, 190 kDa). Moreover, under reflux, yields of the individual steps were also higher (except that of the third). Accordingly, three deacetylation steps are needed to achieve fully deacetylated products and reactions carried out without pressurizing the reactor ensure better yields and less fragmentation of the biopolymer.
In a low-temperature 16 h reaction (C17, entry 17) the polymer chain has not fragmented much more compared to the 4 h reaction (C8). Nevertheless, only a small (∼10%) increase in the DD was detected during the final 12 h. Increasing the concentration of NaOH to 45 wt% (C18, entry 18) resulted in slightly increased yield and DD, whereas decreasing the alkali concentration to below 40 wt% (25 or 35 wt%) resulted in insufficiently deacetylated insoluble products. When commercially available chitosans of various MW (high molecular weight: CHMW, low viscosity: CLvis, low molecular weight: CLMW) and DD in the range 83–93% (average values of DD) were hydrolysed at 120 °C for 4 h, close to fully deacetylated materials were recovered in high yields (C19, C20 and C21; entries 20, 22 and 24) but had lower MW compared to the original materials.
The crystallinity of the samples was examined by wide-angle powder X-ray diffractometry (WAXRD). The sharp peaks detected at 9.5, 13.1, 19.2 and 26.5 and the shoulders at 21.0 and 23.5 2Θ values in the diffractogram of the pristine chitin were indexed as 020, 021, 110, 130, 120 and 101 lattice planes, respectively (Fig. 1).50,51 These reflections are characteristic of the α-chitin polymorph, which has a two-chain orthorhombic unit cell with P212121 symmetry and antiparallel arrangement of chains.52 In the diffraction patterns of chitosans the intensity of the 020 and 110 reflections decreased and became broader, and their maxima shifted to higher 2Θ values (see Fig. 1 and ESI† Fig. S72–S75).53–55 Degrees of crystallinity (Xcr) and the apparent crystallite size (Dp) in the direction perpendicular to the (110) plane were calculated according to known procedures (see the ESI†).52,56
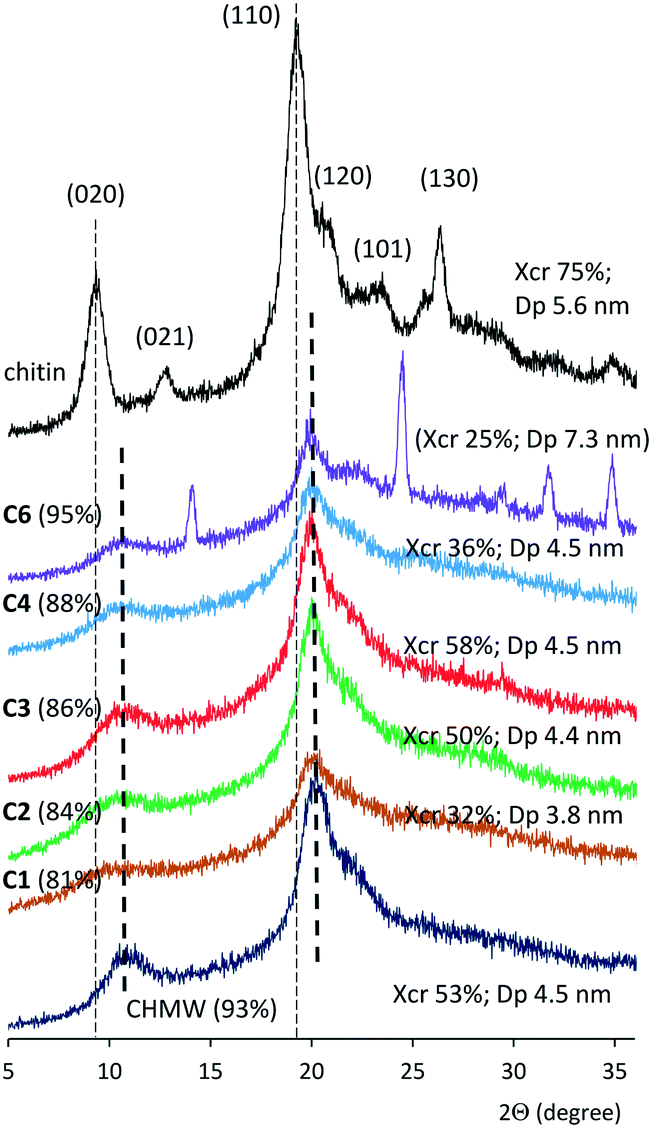 | ||
| Fig. 1 Wide-angle X-ray diffractograms of chitin, CHMW and products obtained by hydrolysis. Average DD values in parentheses; Xcr: crystallinity degree, Dp: crystallite size in the direction perpendicular to the (110) crystal plane. | ||
Low-crystallinity chitosans were obtained after a short hydrolysis time (C1) or lower than 120 °C temperatures (C8 and C9). The Xcr increased by extending the time up to 4 h (C3), reaching a value higher than that of the commercial CHMW (Table 1); however, further extending the reaction had a detrimental effect on the Xcr (C4–C6). Extending excessively the hydrolysis led to the appearance of a new crystalline material (see Fig. 1, C6, and Fig. S72,†C7). Small apparent crystallite sizes (3.8 nm) were obtained following 1 h of hydrolysis, which increased to about 4.5 nm. Further increase was observed only following long reaction times. By increasing the temperature to over 120 °C, Xcr decreased (C10 and C11), whereas the crystal sizes increased together with the DD value. The consecutive hydrolysis of C1 also increased the crystallinity and the crystal size (C12); however, during the third hydrolysis step the Xcr decreased. A similar behaviour was observed in reactions carried out under reflux (C14–C16, Table 1). According to the above it is not surprising that the crystallinity of the commercial chitosans having high DD (CHMW and CLMW, ≈90%) decreased by deacetylation (C19 and C21), whereas CLvis (DD 83%) afforded materials with slightly higher crystallinity.
Our WAXRD study indicated that mild conditions usually provide materials with lower Xcr and Dp, which may be increased by applying harsher conditions or a successive reaction. However, excessively increasing the time, temperature, and NaOH concentration or employing further additional steps, although increasing the DD, leads to loss of crystallinity. These observations show the disappearance of the chitin crystalline phases in materials with low DD (75–80%) and the gradual crystallization of chitosan as the remaining N-acetyl groups were cleaved. However, over 90% DD, along with the hydrolysis of the residual N-acetyl groups, bond breaking between chitosan chains may occur, resulting in a decrease in crystallinity, although the size of the crystals increases.
The morphologies of the materials were examined by scanning electron microscopy (SEM, Fig. 2(a) and ESI† Fig. S76–S83). Particles of a wide size distributions (20–200 μm) were detected with irregular shape and relatively rough, striated surfaces, also having small smooth arrays.57,58 At higher magnifications, puckers and foldings are visible on the surface of particles with rounded edges. SEM detected no systematic change in the morphology of the samples. As the post-hydrolysis processing, which may also influence the morphology,59 was identical, the examined properties could not be correlated with morphological changes. The transmission electron microscopy (TEM) investigation of C16 (Fig. 2(b)) showed irregular, superimposed plates. At high magnification (inset), cracks or crinkling of the surface are visible.
 | ||
Fig. 2 SEM (a) and TEM (b) image of C16; SEM magnification: 200× (inset: 5000×); TEM magnification: 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× (inset: 360000×). 000× (inset: 360000×). | ||
Raman spectra of selected materials in comparison with that of chitin are presented in Fig. 3 and the ESI,† Fig. S84–S86. Bands in the Raman spectra of chitin and deacetylated materials were identified based on previous reports.60,61 Bands centred at 1657 and 1621 cm−1 in the spectrum of chitin, assigned to ν(CO) amide 1 and to δ(NH) vibrations, disappeared from the spectra of chitosans, while broad bands appeared at 1676 and 1592 cm−1, which may be assigned to ν(CO) and δ(NH2) vibrations. The intensities of the bands positioned at 1414 cm−1 assigned to δs(CH3) + δ(CH) and that of the sharp one at 1327 cm−1 attributed to ν(CN) + δ(CH) also decreased by deacetylation. The band positioned at 1205 cm−1 in the spectrum of chitin disappears from that of chitosans. The one assigned to ν(C–O–C)ether vibration at 1108 cm−1 is shifted to higher wavenumbers upon deacetylation (∼1112 cm−1), similarly to that situated at 1059 cm−1 corresponding to ν(C–O–C)ring vibration, which appears at 1091 cm−1. The sharp intense band at 954 cm−1 in the chitin spectrum corresponds to the ν(CN) vibration, which in the obtained materials shifted to 931 cm−1 and has very low intensity. Bands at 2963, 2936, 2880 and 2724 cm−1 in the spectrum of chitin (see the ESI†) are attributable to ν(CH3) (the first two), ν(CH2) and ν(CH) vibrations, respectively. The intensity of the former two decreased as the acetyl group was cleaved.
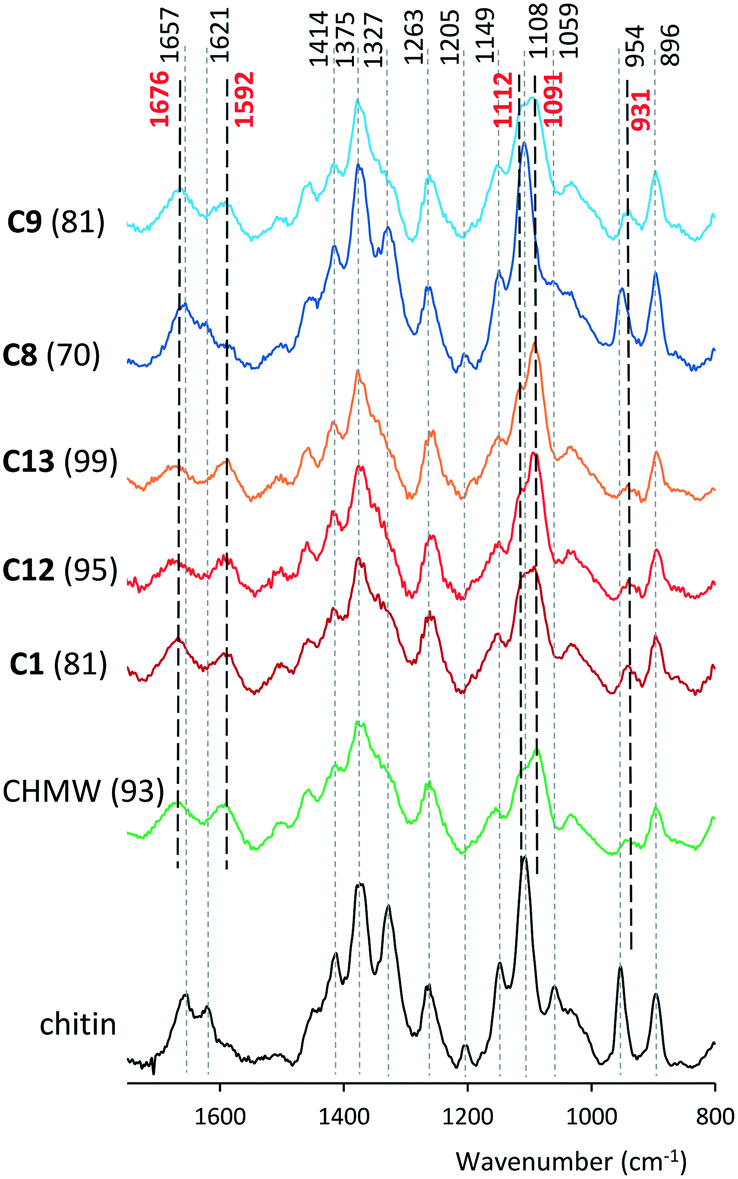 | ||
| Fig. 3 Raman spectra of selected materials. DD (%) values are indicated in parentheses. | ||
The strong residual bands at 1621, 1327, 1205 and 954 cm−1 in the spectrum of the sample prepared at 80 °C (C8) showed low DD values. The progressive deacetylation of chitin (C1, C12 and C13) is traceable in the Raman spectra by the gradual decrease of the shoulder at 1327 cm−1 and the alteration of the relative intensities of bands at 1112 and 1091 cm−1. A decrease in the ratio of the latter's intensities (ν(C–O–C)ether/ν(C–O–C)ring vibrations) also indicates gradual fragmentation of the macromolecules, i.e. decrease in the MW, as detected by viscosity measurements. Decrease of the 2936 cm−1 band in the C1, C12, and C13 series (Fig. S84†) pointed to a similar conclusion. The Raman spectra also confirmed the significant MW decrease of CLvis during hydrolysis and the progressive increase of DD and simultaneous decrease of MW in the series C14, C15 and C16 (ESI,† Fig. S85 and S86).
Application of chitosan as a ligand in Ru-catalysed asymmetric transfer hydrogenation
We tested the prepared materials as chiral ligands in the ATH of 4-chromanone (1a) catalysed by the in situ formed Ru complexes using [Ru(p-cym)Cl2]2 as a metal precursor (p-cym: para-cymene). According to our previous report, (S)-4-chromanol (S-1b) may be obtained in high yield and up to 97% ee in 46 h using CHMW as ligand.41 The reaction conditions for the present study were selected based on these previous observations. Results obtained in 8 and 24 h are summarized in Table 2.|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Conv8b (%) | Conv24b (%) | eec (%) |
| a Reaction conditions: 0.00625 mmol [Ru(p-cym)Cl2]2, 3 mg ligand, 0.25 mmol 1a, 1.25 mmol HCOONa, 1 cm3 H2O/iPrOH 4/1, rt, 8 or 24 h. b Conversions after 8 h (Conv8) and 24 h (Conv24) determined by gas chromatography (GC). c Enantiomeric excess (ee) at 24 h by GC; the configuration of the excess enantiomer was S.41 | ||||
| 1 | Chitin | — | 14 | 82 |
| 2 | C1 | 58 | 98 | 97 |
| 3 | C12 | 58 | 99 | 97 |
| 4 | C13 | 45 | 82 | 97 |
| 5 | C3 | 62 | 90 | 96 |
| 6 | C4 | 54 | 93 | 96 |
| 7 | C5 | 49 | 92 | 96 |
| 8 | C6 | 47 | 88 | 96 |
| 9 | C7 | 25 | 75 | 95 |
| 10 | C8 | 28 | 84 | 96 |
| 11 | C9 | 53 | 97 | 96 |
| 12 | C10 | 55 | 97 | 96 |
| 13 | C11 | 53 | 99 | 96 |
| 14 | C14 | 41 | 90 | 96 |
| 15 | C15 | 33 | 94 | 96 |
| 16 | C16 | 31 | 78 | 95 |
| 17 | C17 | 64 | 99 | 96 |
| 18 | C18 | 72 | 99 | 96 |
| 19 | CHMW | 28 | 71 | 96 |
| 20 | C19 | 45 | 97 | 97 |
| 21 | CLvis | 39 | 94 | 96 |
| 22 | C20 | 35 | 76 | 96 |
| 23 | CLMW | 9 | 40 | 93 |
| 24 | C21 | 35 | 71 | 96 |
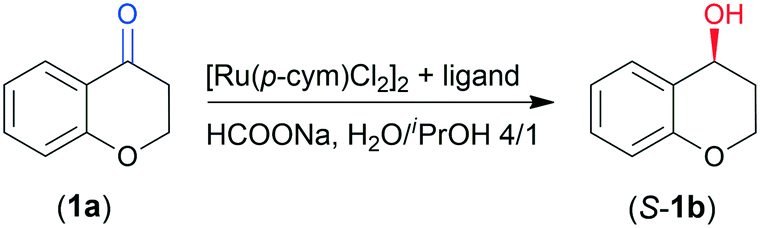
In 24 h, good to high conversions (Conv24: 71–99%) and high ee values (95–97%) were reached with all these materials with the exception of CLMW and chitin. Several biopolymers were exceptionally efficient, such as C1, C9, C10, C11, C12, C17, C18 and C19, giving 97–99% Conv24. No clear correlations between these performances and the materials' properties were found. Furthermore, good Conv24 (91 ± 3%) gave several other materials, too, such as C3, C4, C5, C6, C14, C15 and CLvis, covering the whole interval of each determined property. However, examination of the conversions after 8 h (Conv8) revealed significant differences. Thus, high activities have the catalysts formed with C1, C3, C12, C17 and C18 ligands, all having relatively close MW (210–230 kDa) and DD values in the 81–95% range. The crystallinity of these materials varied largely (30–60%), indicating no major effect on the catalytic performance. Materials having higher DD values, such as C7, C13, C16, C19, C20 or C21, gave lower Conv8, similarly to C8 having the lowest DD obtained in the present work (70%). Low MW materials (C6, C7, C20, C21 and CLMW) provided decreased Conv8, likewise chitosans having high MW (>250 kDa; C14, C15, C16 and CHMW) irrespective of their DD.
Thus, although chitosans with properties in a wide range may form a Ru complex active in the ATH of 1a, in order to reach outstanding performances the DD and MW must be in certain intervals. Materials having very high DD are not among the most efficient. This calls attention to a possible role of the residual N-acetyl groups influencing either the formation of the complex or the interaction of the catalytically active species with the ketone. However, these groups may not be directly involved in the complex formation or the reaction, as they could also be responsible for a proper arrangement of the polymer chain or the active site isolation. Moreover, they may also coordinate to the metal and hinder reduction of the Ru(II) species to less active and unselective Ru nanoparticles.
The above correlations were established in the ATH of 1a, having an O-heterocyclic ring fused with the aromatic moiety and thus is especially prone to enantiodiscriminative interaction with the complex.41 Next, we have selected the ATH of the more flexible 3′-(trifluoromethyl)acetophenone (2a) as another test reaction. Results obtained with this ketone, providing S-2b in 85% ee in two days with CHMW,41 are presented in Table 3. All chitosans gave the product in good ee (82–85%). However, significant variations in the conversions following 24 and 48 h (Conv24 and Conv48) were observed. In this ATH, C17 and C18 gave the best Conv48; however, high values (86–95%) were also obtained using C1, C3, C6, C12, C13 and C19. These materials were among those which also provided good Conv24 (42–49%), along with C9, C10 and C11. C3 and C6 also afforded the best ee (85%) together with C11, C20 and C21. The latter biopolymers gave satisfactory conversions (Conv24: 33–48%, Conv48: 66–83%). In the C1, C12 and C13 series the conversion decreased with the third member, similarly to the ATH of 1a, as the MW of the latter was low. In contrast, using the C14, C15 and C16 series resulted in a gradual increase of the conversion, attributable to the high MW of these materials.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Conv24b (%) | Conv48b (%) | eec (%) |
| a Reaction conditions: 0.00625 mmol [Ru(p-cym)Cl2]2, 3 mg ligand, 0.25 mmol 2a, 1.25 mmol HCOONa, 1 cm3 H2O/iPrOH 4/1, rt, 24 or 48 h. b Conversions after 24 h (Conv24) and 48 h (Conv48) determined by GC. c Enantiomeric excess (ee) at 48 h by GC; the configuration of the excess enantiomer was S.41 | ||||
| 1 | C1 | 49 | 90 | 84 |
| 2 | C12 | 48 | 87 | 84 |
| 3 | C13 | 42 | 88 | 84 |
| 4 | C3 | 49 | 88 | 85 |
| 5 | C6 | 49 | 86 | 85 |
| 6 | C7 | 45 | 64 | 84 |
| 7 | C8 | 34 | 71 | 84 |
| 8 | C9 | 52 | 75 | 82 |
| 9 | C10 | 50 | 81 | 82 |
| 10 | C11 | 48 | 83 | 85 |
| 11 | C14 | 28 | 78 | 83 |
| 12 | C15 | 30 | 80 | 82 |
| 13 | C16 | 33 | 81 | 82 |
| 14 | C17 | 48 | 99 | 84 |
| 15 | C18 | 46 | 99 | 83 |
| 16 | CHMW | 39 | 78 | 82 |
| 17 | C19 | 45 | 95 | 83 |
| 18 | CLvis | 37 | 76 | 82 |
| 19 | C20 | 34 | 74 | 85 |
| 20 | CLMW | — | 28 | 82 |
| 21 | C21 | 33 | 66 | 85 |
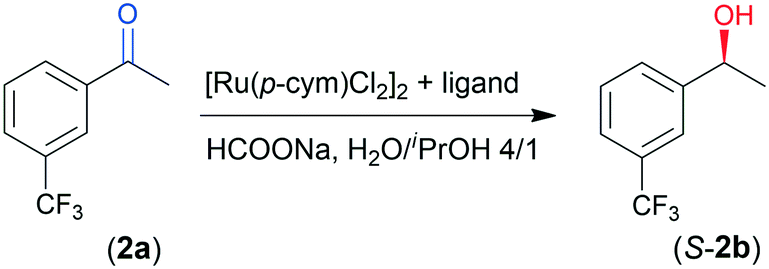
Although differences were detected in the effect of the chitosan properties on the ATH of 1a and 2a, generally, we reached similar conclusions as to the characteristics of the most efficient biopolymer. Thus, in both reactions, close to completely deacetylated materials (DD > 98%) are less efficient than those having an intermediate DD value (80–95%). No correlations between the crystallinity or primary crystallite sizes and results of the reactions were observed. We found that the MW of the efficient materials fall in a rather narrow range. The most efficient catalysts resulted using chitosan with MW in the 200–230 kDa interval; however, not all the chitosans having the MW in this range were among the most efficient. Biopolymers having the proper MW accompanied by certain values of DD, as given above, ensured the formation of both highly active and enantioselective catalyst.
In order to examine the interactions in which the prochiral ketone is involved we carried out a deuterium tracer study investigating the asymmetric transfer deuteration (ATD) of 1a using CHMW ligand in D2O or D2O/iPrOH. The results are summarized in the ESI,† Table S1.
Reactions were carried out for 24 h for examination of the kinetic isotope effect (KIE) and the simultaneous H/D exchange with the ATD and for 72 h to investigate processes occurring during the late stages of the reactions. No significant KIE was observed when the solvent was changed from H2O to D2O, whereas a decrease in the rate to almost half was detected in D2O using DCOONa (KIE 1.94). These KIE values indicate the involvement in the rate-determining step (RDS) of the Ru–H(D) hydride(deuteride) generated by decomposition of formate without the influence of the solvent. In H2O/iPrOH 4/1 the use of DCOONa afforded a smaller KIE compared to D2O, which may be attributed to scrambling of the deuterium in the presence of iPrOH, as suggested in a previous study,46 which was partially eliminated in D2O/iPrOH 4/1 (KIE 2.37). As the rates obtained in D2O and D2O/iPrOH 4/1 with DCOONa were close, we reached the conclusion that the iPrOH has no role in the catalytic cycle, influencing the outcome mainly by dissolving the reactants and products. These observations indicated that the Ru–chitosan complex works similarly to the Noyori–Ikariya metal–ligand bifunctional catalyst, i.e. the hydride is involved in the RDS44 of the reaction in which its transfer precedes the addition of the proton.46,47,62
The position of deuterium in the recovered 1a and isolated 1b was examined by NMR spectroscopy (see the ESI,†1H, 2H and 13C NMR spectra, Fig. S93–S124†). Incorporation of small amounts of D in position 3 of 1a in D2O (see Table S1† and Fig. 4) with the HCOONa donor indicated that ketone–enol tautomerization and exchange at the enolic –OH occurs during ATH; the former may be catalysed by chitosan. Interestingly, a much higher amount of D was detected in this position using a D2O/iPrOH 4/1 solvent rather than D2O, whereas in the latter, H–D exchange in positions 8 and 5 also occurred. The use of DCOONa afforded the alcohol 1b deuterated in position 4 even in a non-labelled solvent, which was confirmed by the 2H NMR spectra (see the ESI,† Fig. S5). A small amount of D in position 3 was detected in the 2H NMR spectra of samples obtained in 24 h, which increased in the 72 h reaction to a slightly higher value as registered in 1a after 24 h (Fig. S5†). According to the above, besides transfer deuteration, previous partial H–D exchange (ATD) also occurs, responsible for the products deuterated in position 3. The D detected in the aromatic positions 5 and 8, in both 1a and 1b, showed that the molecule is bonded to the catalyst in a manner that allows the H–D exchange of these H, i.e. it is likely that both the keto group and the ring heteroatom may anchor the molecule to the catalyst.
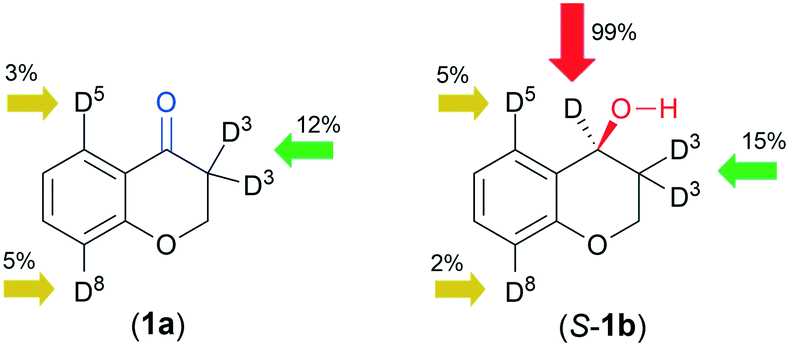 | ||
| Fig. 4 Highest amounts of D incorporated in the indicated positions of the recovered 1a and isolated 1b using D2O (or D2O/iPrOH 4/1) and DCOONa donor (see Table 4). | ||
Oxidative kinetic resolution (OKR) of alcohols with Ru(II)–chitosan complexes
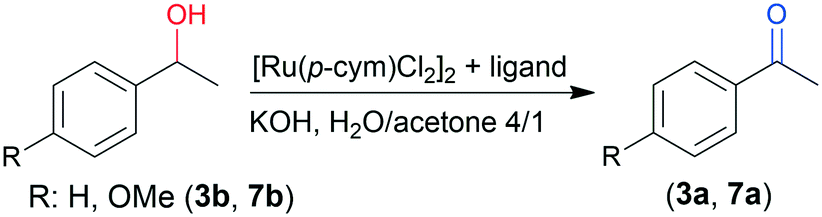
Noyori and co-workers reported that the ATH of ketones with Ru complexes is reversible when alcohols are the hydrogen donors, which makes possible the OKR of secondary benzylic alcohols.63,64 Thus, we examined the possibility of using Ru–chitosan complexes for the OKR of racemic alcohols. For comparison, initially we examined the ATH of 2a using iPrOH as donor and KOH as co-catalyst; the latter is necessary for the in situ formation of the active catalytic species.44 According to our results, 1 eq. base is necessary to reach high conversion in H2O/iPrOH 4/1 after two days at 50 °C (ESI,† Fig. S6). The reaction in iPrOH was complete in 48 h even with 0.1 mmol (0.4 eq.) KOH; however, it afforded a racemic product. Probably, in the absence of water, highly active but unselective chitosan-supported Ru–RuO particles are formed due to the low solubility of chitosan and the Ru–chitosan complex. The enantioselectivities in the H2O/iPrOH solvent were not sensitive to the amount of KOH. The highest value (75%) approached that obtained with HCOONa; the lower value may be ascribed to the effect of the higher temperature (50 °C).Next we have attempted the OKR of rac-2b and other benzylic alcohols (Fig. 5) using CHMW as ligand, KOH additive and acetone as acceptor in aqueous mixture (Table 4). The use of water was found necessary for obtaining enantioselection in OKR as well (entry 5). The ring substituents on 1-phenylethanol had a significant effect on the results due to their influence on the reduction potential of the alcohols.63 Thus, the low conversion of 2b (entry 1) is in contrast with the close to 50% conversion of 7b possessing high reduction potential (entries 12–14). The selectivity factor (s) was also much higher in reactions of the latter alcohol. The Br substituent in the ortho position hindered the interaction with the complex (entry 8). Following the OKRs the S enantiomers reacted faster, leading to recovery of R enantiomer-enriched alcohols in up to 50% ee (s up to 3.00). The conversion of 3b, 6b and 7b slowed down after reaching 40–47% (24 h), and only minor further transformation occurred in an additional day, without notably altering the ee. The substituted 1-phenylethanols 6b and 7b were recovered in better ee values compared to 3b. The difference in rate of oxidation of the two enantiomers was also evidenced by reactions of the pure 3b antipodes (initial ee values >99%, entries 6 and 7). The rate ratio determined in the reaction of the pure enantiomers after 24 h was 3.3.
 | ||
| Fig. 5 Racemic alcohols used in oxidative kinetic resolutions. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alcohol | Time (h) | Convb (%) | eec (%) | s |
| a Reaction conditions: 0.00625 mmol [Ru(p-cym)Cl2]2, 3 mg CHMW, 0.25 mmol 2b–8b, 0.25 mmol KOH, 1 cm3 H2O/acetone 4/1, 50 °C. b Conversion of alcohol determined by GC. c Enantiomeric excess (ee) of the unreacted alcohol by GC, excess of R enantiomer. d Selectivity factor = ratio of the reaction rates of the S and R enantiomers. e Reaction at rt. f Reaction in 1 cm3 acetone. g Reactions using optically pure 3b. h Excess of S enantiomer. | |||||
| 1 | 2b | 48 | 11 | 9 | 1.20 |
| 2 | 3b | 24 | 40 | 39 | 2.28 |
| 3 | 3b | 48 | 41 | 37 | 2.17 |
| 4e | 3b | 48 | 37 | 37 | 2.17 |
| 5f | 3b | 48 | 34 | 0 | 1.00 |
| 6g | S-3b | 24 | 49 | 97h | — |
| 7g | R-3b | 24 | 15 | 98 | — |
| 8 | 4b | 72 | <1 | nd | — |
| 9 | 5b | 48 | 37 | 38 | 2.23 |
| 10 | 6b | 24 | 38 | 49 | 2.92 |
| 11 | 6b | 48 | 43 | 50 | 3.00 |
| 12 | 7b | 24 | 47 | 42 | 2.45 |
| 13 | 7b | 48 | 49 | 43 | 2.51 |
| 14e | 7b | 48 | 49 | 47 | 2.77 |
| 15 | 8b | 6 | 33 | 47 | 2.77 |
| 16 | 8b | 24 | 35 | 53 | 3.26 |
| 17 | 8b | 48 | 39 | 64 | 4.56 |
Although the OKR of the cyclic 8b was also decelerated after 6 h, in contrast, with the 1-phenylethanol derivatives, the ee increased by extending the reaction to 24 or 48 h (entries 15–17), indicating a more stereospecific interaction of the heterocyclic alcohol compared to phenylethanols. This was confirmed by reactions carried out with 3b and 8b of various optical purities (ESI,† Fig. S7 and S8). The plot of the ee vs. the initial optical purity was closer to the theoretical plot of the completely selective reactions (calculated for 50% conversions) in case of 8b compared to 3b. Although the reactions were carried out for identical reaction times, i.e. the conversions varied with the initial ee, in reactions of the two compounds (3b and 8b) similar conversions were obtained at a given starting composition. The more specific interaction of the heterocyclic compound could be responsible for this kinetic effect, which may be due to additional interaction of 8b with the catalyst compared to 1-phenylethanol derivatives, as was suggested earlier.41,42
Next, we have examined the influence of the chitosan properties on the results of the OKR. Results obtained in reactions of 3b and 7b are presented in Table 5. Variations in the conversions and especially in the ee of both alcohols showed that the biopolymer structure has a more significant effect on the OKR, compared to the ATH. The complex formed with chitin afforded low conversion and ee (Table 5, entry 1). Although the reactions of the two compounds were explored under different conditions (50 °C, 24 h, or rt, 48 h), similarities in the effect of the MW or DD were found. Thus, complexes obtained with materials with low DD (such as C1, C2, and C8) were less efficient in inducing enantiodifferentiation (entries 3–5). Although C14 also has low DD (80%), it afforded higher ee compared to the above materials, especially in the reaction of 7b (entry 6). One may presume that the relatively high MW of this material compensates for the detrimental effect of the low DD partially. The close to completely deacetylated chitosans obtained by deacetylation of CHMW (C19) or that of C14 (C15 and C16) afforded higher conversion and ee values compared to the commercial material in the OKR of 7b (entries 7–9). The successively deacetylated chitosans afforded progressively better results, giving ee of up to 68%.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | OKR of 3b | OKR of 7b | ||
| Convb (%) | eec (%) | Convb (%) | eec (%) | ||
| a Reaction conditions: 0.00625 mmol [Ru(p-cym)Cl2]2, 0.03 mmol ligand, 0.25 mmol 3b or 7b, 0.25 mmol KOH, 1 cm3 H2O/acetone 4/1, 50 °C 24 h (3b) or rt 48 h (7b). b Conversion determined by GC. c Enantiomeric excess (ee) of the unreacted alcohol by GC, excess of R enantiomer. | |||||
| 1 | Chitin | 12 | 9 | 9 | 7 |
| 2 | CHMW | 40 | 39 | 49 | 47 |
| 3 | C1 | 30 | 23 | 42 | 40 |
| 4 | C2 | 33 | 28 | 45 | 43 |
| 5 | C8 | 27 | 22 | 36 | 32 |
| 6 | C14 | 34 | 32 | 53 | 58 |
| 7 | C15 | 35 | 33 | 56 | 62 |
| 8 | C16 | 41 | 40 | 61 | 68 |
| 9 | C19 | 42 | 41 | 50 | 52 |
Importantly, the Ru–chitosan complex was able to differentiate between the enantiomers of benzylic alcohols, though moderate ee values were obtained (3b up to 41%, 7b up to 68%), unlike with the Noyori–Ikariya complexes, which afforded high ee values.63 The much higher ee values obtained in the ATH of ketones compared to the OKR of the alcohols indicated that the enantioface differentiation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is more efficient than that of the two alcohol enantiomers. Thus, the planar scaffold may be more efficiently hindered from one side (re face), whereas the flexibility of the alcohol enantiomers allows a less stereospecific interaction with the catalyst. This led us to propose that the polymer network is able to create a chiral surrounding around the metal centre, which is not attributable solely to the chirality of the Ru-bonded monomer, but the neighbouring structure may also be responsible for stereospecific interaction with the complex.
O bond is more efficient than that of the two alcohol enantiomers. Thus, the planar scaffold may be more efficiently hindered from one side (re face), whereas the flexibility of the alcohol enantiomers allows a less stereospecific interaction with the catalyst. This led us to propose that the polymer network is able to create a chiral surrounding around the metal centre, which is not attributable solely to the chirality of the Ru-bonded monomer, but the neighbouring structure may also be responsible for stereospecific interaction with the complex.
Application of chitosan in asymmetric Michael addition
During the past twenty years, asymmetric organocatalysis has emerged as a viable alternative of metal-catalysed enantioselective reactions. Primary amines are an important class of chiral organocatalysts.65,66 Chitosan, owing to its free amino groups and the chirality of the carbohydrate backbone, is a promising material for these purposes. Although this biopolymer was already used to prepare chiral organocatalysts,21,26,27,67 only a few publications reported solely its catalytic activity and chirality.29,32 Conjugate additions are among the most versatile C–C coupling reactions, which may be catalysed by primary amines.68 So far, chitosan has been used as a support of cinchona alkaloid derivatives found efficient in asymmetric Michael additions.30,40 In our present work we attempted the application of the prepared materials in a selected Michael addition.As a test reaction, we chose the addition of isobutyraldehyde (9) to N-methylmaleimide (10) (Table 6). In organic solvents, such as CHCl3 or iPrOH, low conversion and ee values (both <10%) were obtained, probably due to the low solubility of chitosan.
|
|
||||
|---|---|---|---|---|
| Entry | Additiveb | Temp. (°C) | Convc (%) | eed (%) |
| a Reaction conditions: CHMW 0.03 mmol –NH2 (according to DD), 0.3 mmol 10, 1.2 mmol 9, 0.03 mmol additive, 1 cm3 H2O, 24 h. b Abbreviations: para-toluenesulfonic acid (p-TsOH), benzoic acid (BzOH). c Conversion determined by GC. d Enantiomeric excess (ee) by GC, excess of S enantiomer.69 e Yield of the product purified by flash chromatography. f Reaction time 48 h. g Using 0.06 mmol BzOH. h Using CHMW 0.06 mmol –NH2 and 0.06 mmol BzOH. | ||||
| 1 | — | 25 | 82 | 43 |
| 2 | HCl | 25 | 20 | 50 |
| 3 | p-TsOH | 25 | 20 | 53 |
| 4 | BzOH | 25 | 77 | 55 |
| 5 | BzOH | 5 | 58 | 58 |
| 6 | BzOH | 50 | 93 (80)e | 51 |
| 7f | BzOH | 25 | 88 | 54 |
| 8g | BzOH | 25 | 74 | 55 |
| 9h | BzOH | 25 | 94 (82)e | 54 |
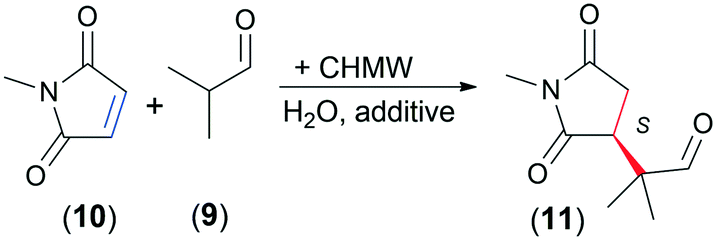
Better transformation was reached in water accompanied by low ee (Table 6, entry 1). Acidic additives, which besides solubilizing chitosan in water may affect the formation of the reaction intermediates,69 decreased the conversion and had a beneficial effect on the ee (entries 2–4). Thus, we carried out further experiments with addition of benzoic acid (BzOH), which afforded only slightly decreased conversion and moderate ee (55%). The effect of the temperature, amount of BzOH and catalyst, and reaction time was studied (Table 6 and ESI† Fig. S9–S12). Decrease of the temperature (5 °C) led to slightly higher ee, whereas higher temperature (50 °C) afforded over 90% transformation of 10 and decreased ee. Extending the reaction to 48 h also led to higher conversion, approaching 90%, while maintaining the ee value (entry 7). Using 2 eq. BzOH further decreased the conversion (entry 8), whereas with 20 mol% catalyst and additive we were able to reach over 90% conversion and similar ee in 24 h (entry 9).
Next, we carried out the reaction using a series of selected chitosans by applying 10 mol% catalyst and 1 eq. BzOH at 25 °C for 24 h (Table 7). Chitin provided low conversion and decreased ee (compared to CHMW, entries 2 and 5). The chitin monomer, D-GlcNAc, was inactive, whereas the hydrochloride of the chitosan monomer, D-GlcNH2·HCl and its stereoisomer D-galactosamine hydrochloride (D-GalNH2·HCl) afforded low conversions (entries 3 and 4). The ee obtained with GlcNH2·HCl was similar to that with CHMW, and, unexpectedly, the product enantiomer having opposite configuration was in excess with both monomer hydrochlorides (D-GlcNH2·HCl and D-GalNH2·HCl, excess of R-11). Accordingly, in this Michael addition the biopolymer chain is necessary to have increased activity and also determines the sense of enantioselection.
| Entry | Catalyst | Convb (%) | eeb (%) |
|---|---|---|---|
| a Reaction conditions: chitosan 0.03 mmol –NH2 (according to DD), 0.3 mmol 10, 1.2 mmol 9, 0.03 mmol BzOH, 1 cm3 H2O, 25 °C, 24 h. b Conversion and enantiomeric excess determined by GC, excess of S enantiomer. c Without addition of BzOH. d R configuration of the excess enantiomer. | |||
| 1 | D-GlcNAc | 1 | 5 |
| 2 | Chitin | 12 | 43 |
| 3c | D-GlcNH2·HCl | 10 | 54d |
| 4c | D-GalNH2·HCl | 5 | 37d |
| 5 | CHMW | 77 | 55 |
| 6 | C19 | 78 | 54 |
| 7 | CLvis | 72 | 56 |
| 8 | C20 | 82 | 46 |
| 9 | CLMW | 64 | 55 |
| 10 | C21 | 70 | 49 |
| 11 | C1 | 62 | 55 |
| 12 | C12 | 65 | 54 |
| 13 | C13 | 76 | 53 |
| 14 | C3 | 64 | 55 |
| 15 | C4 | 68 | 56 |
| 16 | C5 | 72 | 56 |
| 17 | C6 | 70 | 55 |
| 18 | C7 | 67 | 54 |
| 19 | C8 | 54 | 54 |
| 20 | C10 | 68 | 55 |
| 21 | C11 | 81 | 53 |
| 22 | C14 | 55 | 56 |
| 23 | C15 | 72 | 56 |
| 24 | C16 | 70 | 55 |
| 25 | C17 | 68 | 55 |
Moderate conversions (54–62%) were reached with materials having low DD and high MW (C1, C8 and C14). Most of the chitosans providing 60–70% conversions had moderate DD values, up to 90% (except C12, DD 95%). Materials having over 90% DD usually afforded ≥70% conversions (CHMW, C5, C6, C11, C13, C15, C16, C19, C20 and C21), despite the MW of these polymers varying in a wide range (45–317 kDa). Almost all the chitosans afforded similar ee values, i.e. 53–56%. The above observations indicated that there is a direct correlation between the DD of the chitosan and its catalytic activity in this Michael addition; however, the MW may also influence its performance. In contrast, these properties have less effect on the enantioselectivity. Thus, similar ee values can be obtained within a relatively wide DD and MW range. The N-substituent on the maleimide scaffold also had a small effect on the enantioselectivity, slightly decreasing the ee as the size of the substituent increases (Scheme 2). However, conversions reached using 10, N-ethylmaleimide (12) and N-benzylmaleimide (14) increased in this order.
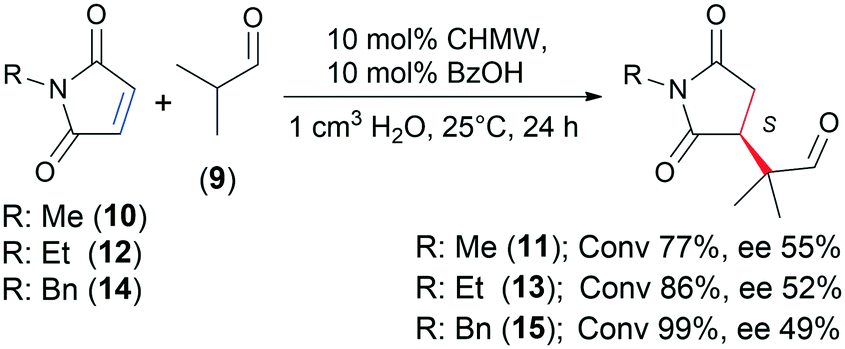 | ||
| Scheme 2 Asymmetric Michael addition of 9 to N-substituted maleimides catalysed by CHMW. | ||
Finally, we emphasize that the chirality of the chitosan is sufficient to obtain enantioselection in this Michael addition; moreover, the biopolymer chain has a decisive role in determining the direction of the enantiodifferentiation. Furthermore, the reaction proceeds in water and thus may pave the way to developing new environmentally friendly methods.
It is known that the Michael addition catalysed by primary amines proceeds through an enamine intermediate.69 In contrast with the reaction catalysed by the monomer, this nucleophilic moiety will approach preferentially the opposite side of the maleimide in the presence of the polymer chain. A plausible explanation of this inversion is that the polymeric framework allows multiple anchoring of the activated olefin, which may also account for the more efficient activation of the electrophilic reactant, thus for the faster reaction, compared to D-GlcNH2. The possible transition states formed with the use of the monomer and polymer are sketched in Fig. 6. However, other groups may also play a role in anchoring the maleimide, such as the C4–OH or the glycosidic –OH group in the monomer, which will be investigated in our forthcoming studies.
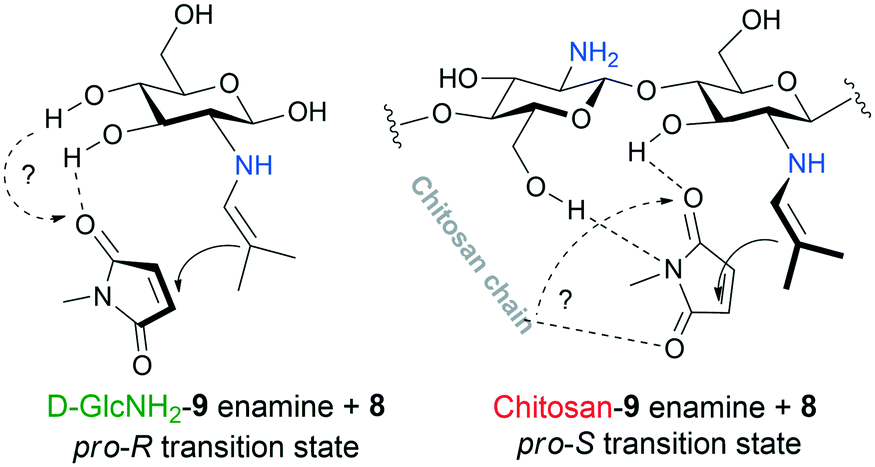 | ||
| Fig. 6 Possible structures of the transition states in Michael addition leading to opposite enantiomers in excess. | ||
Conclusions
Chitosan is a renewable source of chirality of natural origin, which may be prepared from the second most abundant biopolymer, chitin. Exploiting the chirality of this biopolymer in asymmetric synthesis is among the goals of catalytic studies. Recently we have reported the application of commercial chitosan as a chiral ligand in the Ru-catalysed ATH of ketones.41,42 In the present study we prepared a series of chitosans by alkaline hydrolysis of chitin in order to examine the effect of the properties of this biopolymer on its performance in ATH. Under various hydrolysis conditions we obtained materials having a DD in the range of 70–99% and MW of 37–317 kDa. At least three hydrolysis steps were needed to achieve fully deacetylated products. WAXRD study of the materials evidenced their partial crystalline character.Studying the ATH of two test compounds, we reached similar conclusions as regards the characteristics of the most efficient biopolymer, which had a MW in the 200–230 kDa and a DD in the 80–95% ranges. A deuterium tracer study showed that besides transfer deuteration, sequential H–D exchange-deuteration occurs. The H–D exchange on the aromatic ring of 4-chromanone indicated that the ring heteroatom also anchors this molecule to the complex. The reversibility of the ATH with alcohol donors provided the possibility of the OKR of racemic alcohols. In these reactions, the biopolymer structure had a more significant effect than in ATH. The much higher ee values obtained in the ATH compared to OKR indicated that the interaction of the catalyst with the CO bond is more stereospecific than with the alcohol enantiomers. This led us to the conclusion that not only the chirality of the Ru-bonded monomer is responsible for the high ee reached in the ATH but also the polymer network around the metal centre.
The prepared materials were applied as organocatalysts in the Michael addition of isobutyraldehyde to maleimides in aqueous systems. It was found that the chirality of the chitosan is sufficient to obtain enantioselection in this Michael addition. Moreover, the biopolymer chain has a major role in determining the activity and the direction of the enantiodifferentiation. Correlation between the DD of the chitosan and its catalytic activity was observed, whereas the enantioselectivity was less affected. We note that the latter application is the first in which the natural chirality of chitosan is sufficient to promote an enantioselective Michael addition, which opens the possibility of using these materials in other asymmetric conjugate additions to develop environmentally benign asymmetric catalytic methods in aqueous systems.
Finally, the preparation conditions–property–catalytic performance relationships revealed in this study are novel steps in the application of this natural, biodegradable polymer in asymmetric catalytic processes.
Experimental
Materials and methods
Commercial chitosans CHMW, CLvis, CLMW and chitin from shrimp shell were purchased from Aldrich. The materials used in catalytic reactions, [Ru(p-cym)Cl2]2, 4-chromanone (1a), 3′-trifluoromethylacetophenone (2a), acetophenone (3a), 2′-bromoacetophenone (4a), 3′-bromoacetophenone (5a), 4′-bromoacetophenone (6a), 4′-methoxycetophenone (7a), 4-thiochromanone (8a), sodium formate, sodium formate-d, isobutyraldehyde (9), N-methylmaleimide (10), N-ethylmaleimide (12), and N-benzylmaleimide (14), were obtained from commercial sources (Aldrich) and used as received. Racemic (2b–8b) and optically pure (S-3b, R-3b, S-8b, R-8b) alcohols were prepared by reduction of the corresponding ketones with NaBH4 or by ATH of 3a or 8a with a catalyst in situ formed from [Ru(p-cym)Cl2]2 and S,S- or R,R-Ts-Dpen ligand and HCOOH/Et3N 5/2 donor. Solvents and reagents of analytical grade were used without further purification.1H, 2H and 13C NMR spectra were recorded on a Bruker DRX-500 spectrometer at 500 (1H), 77 (2H) and 125 (13C) MHz. The chitosan polymers were dissolved in D2O by addition of CF3COOH. Spectra of compounds obtained in the catalytic reactions were recorded in CDCl3 solvent using TMS as internal standard. FT-IR measurements were recorded on a Bio-Rad Digilab Division FTS-65A/896 spectrometer operating in diffuse reflectance mode (DRIFT) between 4000 and 400 cm−1 using 2 cm−1 resolution by averaging 256 scans. UV-vis spectra were obtained on a Jenway 6850 UV/vis double beam spectrophotometer with 0.1 nm wavelength resolution using a deuterium lamp and 10 mm path length quartz cuvettes. Wide-angle powder X-ray diffractograms (WAXRDs) were recorded on a Rigaku Miniflex-II diffractometer using Cu Kα radiation (λ = 0.1548 nm) at a scanning rate of 4° min−1 between 5° and 40° 2Θ angles. Scanning electron microscopy (SEM) measurements using 10 kV accelerating voltage were performed on a Hitachi S-4700 Type II FE-SEM instrument. Transmission electron microscopy (TEM) measurements were obtained on a FEI Tecnai G2 X-Twin type microscope operating at 200 kV acceleration voltage. Raman spectra were recorded using a confocal Bruker Senterra II Raman Microscope equipped with a 50× magnification objective lens. A 785 nm laser for excitation and 100 mW laser power was applied. An average of 16 scans with an exposition time of 6 s in the 400–4000 cm−1 spectral range and 4 cm−1 spectral resolution were recorded.
Products resulting from catalytic reactions were analysed by gas chromatography using an Agilent Technologies 6890N GC-5973 MSD system (GC-MSD) equipped with a 30 m long DB-1MS UI (Agilent, J&W) capillary column for mass spectrometric identification of compounds and an Agilent 7890A GC-FID (GC-FID) instrument equipped with chiral capillary columns (Cyclosil-B, 30 m, Agilent, J&W; Cyclodex-B, 30 m, Agilent, J&W; HP-Chiral-20B, 30 m, Agilent, J&W or Hydrodex g-TBDAc, 25 m, Macherey-Nagel) for quantitative analysis.
Preparation of chitosan: general procedure
Unless otherwise noted reactions were carried out in 20 cm3 closed glass vials. 600 mg chitin was added to 12 cm3 40 wt% NaOH aqueous solution. The mixture was vigorously stirred and flushed with N2. The slurries were stirred magnetically (500 rpm). The vials were immersed in a heated oil bath to maintain the required temperature. After reaction the mixture was cooled to rt and the obtained solid material was filtered and washed with distilled water to neutral pH. The material was air-dried; the yield was determined by weight measurement using the determined DD and was stored in sealed vials until further use. Reactions under reflux conditions were carried out similarly in a 25 cm3 round-bottom flask connected to a reflux condenser.Characterization of the obtained chitosans
The degree of deacetylation (DD, %) of the products was determined by three spectroscopic methods, i.e.1H NMR, FT-IR and UV-vis spectroscopy, based on reported procedures.70–74 The calculation methods and the spectra of materials prepared in this study are included in the ESI.† The average molecular weight (MW) was determined based on the viscosity of chitosan solutions measured using an Ubbelohde capillary viscometer of 1C size.75,76 The calculation method and plots of ηred against chitosan concentration are included in the ESI.† The degree of crystallinity (Xcr) and crystallite sizes (Dp) in the direction perpendicular to the (110) crystal plane were determined by wide-angle powder X-ray diffractometry (WAXRD). Diffractograms of the materials are presented in the ESI.† Xcr values were calculated based on the method described by Ioelovich, as detailed in the ESI.†52 The apparent crystallite sizes in the direction perpendicular to the (110) plane were obtained using the Scherrer equation (see the ESI†) following deconvolution of the peak corresponding to the reflection on the 110 faces and its shoulder at higher 2Θ values.56Catalytic measurements
Author contributions
The authors contributed equally to the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Hungarian Ministry of Human Capacities through NTP-NFTÖ-20-B-0186 (V. J. Kolcsár) and through grant 20391-3/2018/FEKUSTRAT is highly appreciated. Financial support of the Hungarian National Science Foundation through OTKA Grant K 138871 is acknowledged. The authors thank Dr. Gábor Varga for his valuable help in recording the FT-IR and Raman spectra.References
- Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 3rd edn, 2010 Search PubMed.
- Catalytic Methods in Asymmetric Synthesis, Advanced Materials, Techniques, and Applications, ed. M. Gruttadauria and F. Giacalone, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2011 Search PubMed.
- G. Szőllősi, Catal. Sci. Technol., 2018, 8, 389–422 RSC.
- S. Santosh and P. T. Matthew, J. Appl. Polym. Sci., 2008, 107, 280–285 CrossRef.
- Chitin and Chitosan: History, Fundamentals and Innovations, ed. G. Crini and E. Lichtfouse, Sustainable Agriculture Reviews, Springer Nature, Cham, Switzerland, 2019, vol. 35 Search PubMed.
- E. Khor and A. C. A. Wan, Chitin Fulfilling a Biomaterial Promise, Elsevier, Ltd., Oxford, UK, 2nd edn, 2014 Search PubMed.
- Chitin and Chitosan Derivatives: Advances in Drug Discovery and Developments, ed. S.-K. Kim, CRC Press, Taylor & Francis Group, Boca Raton, USA, 2014 Search PubMed.
- A. Ajavakom, S. Supsvetson, A. Somboot and M. Sukwattanasinitt, Carbohydr. Polym., 2012, 90, 73–77 CrossRef CAS PubMed.
- D. L. Bertuzzi, T. B. Becher, N. M. R. Capreti, J. Amorim, I. D. Jurberg, J. D. Megiatto Jr. and C. Ornelas, Global Challenges, 2018, 2, 180046–180051 CrossRef PubMed.
- Chitosan: Manufacture, Properties, and Usage, ed. S. P. Davis, Nova Science Publ., Inc., New York, USA, 2011 Search PubMed.
- Chitin and Chitosan for Regenerative Medicine, ed. P. K. Dutta, Springer, New Delhi, India, 2016 Search PubMed.
- Chitosan Based Materials and its Applications, ed. G. L. Dotto, S. P. Campana-Filho and L. A. de Almeida Pinto, Frontiers in Biomaterials, Bentham Sci. Publ., Sharjah, UAE, 2017, vol. 3 Search PubMed.
- V. Y. Novikov, Russ. J. Appl. Chem., 2004, 77, 484–487 CrossRef CAS.
- M. Z. Abidin, M. P. Junqueira-Gonçalves, V. V. Khutoryansky and K. Niranjan, J. Chem. Technol. Biotechnol., 2017, 92, 2787–2798 CrossRef CAS.
- K. Kurita, T. Sannan and Y. Iwakura, Makromol. Chem., 1977, 178, 3197–3202 CrossRef CAS.
- K. Kurita, K. Tomita, T. Tada, S. Ishii, S.-I. Nishimura and K. Shimoda, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 485–491 CrossRef CAS.
- I. Peker, F. N. Oktar, M. Senol and M. Eroglu, Key Eng. Mater., 2006, 309–311, 473–476 CAS.
- I. Younes, O. Ghorbel-Bellaaj, M. Chaabouni, M. Rinaudo, F. Souard, C. Vanhaverbeke, K. Jellouli and M. Nasri, Int. J. Biol. Macromol., 2014, 70, 385–390 CrossRef CAS PubMed.
- D. J. Macquarrie and J. J. E. Hardy, Ind. Eng. Chem. Res., 2005, 44, 8499–8520 CrossRef CAS.
- E. E. Guibal, Prog. Polym. Sci., 2005, 30, 71–109 CrossRef CAS.
- J. Agarwal, Org. Biomol. Chem., 2016, 14, 10747–10752 RSC.
- Á. Molnár, Coord. Chem. Rev., 2019, 388, 126–171 CrossRef.
- F. Rafiee, Curr. Org. Chem., 2019, 23, 390–408 CrossRef CAS.
- D. Pan and J. Ganguly, Curr. Organocatal., 2019, 6, 106–138 CrossRef CAS.
- R. S. Varma, ACS Sustainable Chem. Eng., 2019, 7, 6458–6470 CrossRef CAS.
- D. A. Aguilera, N. Tanchoux, M. Fochi and L. Bernardi, Eur. J. Org. Chem., 2020, 3779–3795 CrossRef CAS.
- S. Meninno, ChemSusChem, 2020, 13, 439–468 CrossRef CAS PubMed.
- H. Zhang, W. Zhao, J. Zou, Y. Liu, R. Li and Y. Cui, Chirality, 2009, 21, 492–496 CrossRef CAS PubMed.
- A. Ricci, L. Bernardi, C. Gioia, S. Vierucci, M. Robitzer and F. Quignard, Chem. Commun., 2010, 46, 6288–6290 RSC.
- Y. Qin, W. Zhao, L. Yang, X. Zhang and Y. Cui, Chirality, 2012, 24, 640–645 CrossRef CAS PubMed.
- M. Babin, R. Clément, J. Gagnon and F.-G. Fontaine, New J. Chem., 2012, 36, 1548–1551 RSC.
- T. Heckel, D. D. Konieczna and R. Wilhelm, Catalysts, 2013, 3, 914–921 CrossRef CAS.
- C. Gioia, A. Ricci, L. Bernardi, K. Bourahla, N. Tanchoux, M. Robitzer and F. Quignard, Eur. J. Org. Chem., 2013, 588–594 CrossRef CAS.
- B. Liu, H. Zhou, Y. Li and J. Wang, Chin. J. Org. Chem., 2014, 34, 2554–2558 CrossRef CAS.
- W. Zhao, C. Qu, L. Yang and Y. Cui, Chin. J. Catal., 2015, 36, 367–371 CrossRef CAS.
- H. Dong, J. Liu, L. Ma and L. Ouyang, Catalysts, 2016, 6, 186 CrossRef.
- C. Shen, J. Qiao, L. Zhao, K. Zheng, J. Jin and P. Zhang, Catal. Commun., 2017, 92, 114–118 CrossRef CAS.
- J. M. Andrés, F. González, A. Maestro, R. Pedrosa and M. Valle, Eur. J. Org. Chem., 2017, 3658–3665 CrossRef.
- G. de Gonzalo, A. Franconetti, R. Fernández, J. M. Lassaletta and F. Cabrera-Escribano, Carbohydr. Polym., 2018, 199, 365–374 CrossRef CAS PubMed.
- M. A. Abdelkawy, E.-S. A. Aly, M. A. El-Badawi and S. Itsuno, Catal. Commun., 2020, 146, 106132 CrossRef CAS.
- G. Szőllősi and V. J. Kolcsár, ChemCatChem, 2019, 11, 820–830 CrossRef.
- V. J. Kolcsár, F. Fülöp and G. Szőllősi, ChemCatChem, 2019, 11, 2725–2731 CrossRef.
- J. Takehara, S. Hashiguchi, A. Fujii, S.-I. Inoue, T. Ikariya and R. Noyori, Chem. Commun., 1996, 233–234 RSC.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS PubMed.
- X. Wu, J. Liu, D. Di Tommaso, J. A. Iggo, C. R. A. Catlow, J. Bacsa and J. Xiao, Chem. – Eur. J., 2008, 14, 7699–7715 CrossRef CAS PubMed.
- P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756–6781 RSC.
- A. M. R. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, ACS Catal., 2019, 9, 2079–2090 CrossRef CAS.
- F. Feng, Y. Liu and K. Hu, Carbohydr. Res., 2004, 339, 2321–2324 CrossRef CAS PubMed.
- J. N. I. Balitaan, J.-M. Yeh and K. S. Santiago, Int. J. Biol. Macromol., 2020, 154, 1565–1575 CrossRef CAS PubMed.
- M. Ioelovich, Res. Rev.: J. Chem., 2014, 3, 7–14 Search PubMed.
- Y. Zhang, C. Xue, Y. Xue, R. Gao and X. Zhang, Carbohydr. Res., 2005, 340, 1914–1917 CrossRef CAS PubMed.
- A. Osorio-Madrazo, L. David, S. Trombotto, J.-M. Lucas, C. Peniche-Covas and A. Domard, Biomacromolecules, 2010, 11, 1376–1386 CrossRef CAS PubMed.
- R. S. C. M. de Queiroz Antonio, B. R. P. Lia Fook, V. A. de Oliveira Lima, R. Í. de Farias Rached, E. P. N. Lima, R. J. da Silva Lima, C. A. P. Covas and M. V. Lia Fook, Mar. Drugs, 2017, 15, 141 CrossRef PubMed.
- B. Focher, P. L. Beltrame, A. Naggi and G. Torri, Carbohydr. Polym., 1990, 12, 405–418 CrossRef CAS.
- K. D. Trimukhe and A. J. Varma, Carbohydr. Polym., 2008, 71, 698–702 CrossRef CAS.
- M. Mende, D. Schwarz, C. Steinbach, R. Boldt and S. Schwarz, Colloids Surf., A, 2016, 510, 275–282 CrossRef CAS.
- M. C. Pinto Cruz, S. P. Ravagnani, F. M. S. Brogna, S. P. Campana, G. Cardenas Triviño, A. C. Luz Lisboa and L. H. Innocenti Mei, Biotechnol. Appl. Biochem., 2004, 40, 243–253 CrossRef PubMed.
- A. Zając, J. Hanuza, M. Wandas and L. Dymińska, Spectrochim. Acta, Part A, 2015, 134, 114–120 CrossRef PubMed.
- D. Biniaś, W. Biniaś and J. Janicki, Fibres Text. East. Eur., 2016, 24, 27–38 CrossRef.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- S. Hashiguchi, A. Fujii, K.-J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 288–290 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- L. Chen and S. Luo, in Enantioselective Organocatalyzed Reactions I: Enantioselective Oxidation, Reduction, Functionalization and Desymmetrization, ed. R. Mahrwald, Springer Sci., Dordrecht, 2011, ch. 5, pp. 147–184 Search PubMed.
- M. Tsakos and C. G. Kokotos, Tetrahedron, 2013, 69, 10199–10222 CrossRef CAS.
- O. Mahé, J.-F. Brière and I. Dez, Eur. J. Org. Chem., 2015, 2559–2578 CrossRef.
- Organocatalytic Enantioselective Conjugate Addition Reactions, A Powerful Tool for the Stereocontrolled Synthesis of Complex Molecules, ed. J. L. Vicario, D. Badía, L. Carrillo and E. Reyes, RSC Catal. Series No. 5, RSC Publ., Cambridge, UK, 2010 Search PubMed.
- V. Kozma, F. Fülöp and G. Szőllősi, Adv. Synth. Catal., 2020, 362, 2444–2458 CrossRef CAS.
- A. Hirai, H. Odani and A. Nakajima, Polym. Bull., 1991, 26, 87–94 CrossRef CAS.
- M. Lavertu, Z. Xia, A. N. Serreqi, M. Berrada, A. Rodrigues, D. Wang, M. D. Buschmann and A. Gupta, J. Pharm. Biomed. Anal., 2003, 32, 1149–1158 CrossRef CAS PubMed.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Argüelles-Monal, J. Desbrières and M. Rinaudo, Polymer, 2001, 42, 3569–3580 CrossRef CAS.
- J. Qu, Q. Hu, K. Shen, K. Zhang, Y. Li, H. Li, Q. Zhang, J. Wang and W. Quan, Carbohydr. Res., 2011, 346, 822–827 CrossRef CAS PubMed.
- D. Liu, Y. Wei, P. Yao and L. Jiang, Carbohydr. Res., 2006, 341, 782–785 CrossRef CAS PubMed.
- G. A. Vikhoreva and L. S. Gal'braikh, Fibre Chem., 1997, 29, 287–291 CrossRef CAS.
- N. Yacob, N. Talip, M. Mahmud, N. A. I. M. Sani, N. A. Samsuddin and N. A. Fabillah, J. Nucl. Sci. Technol., 2013, 10, 40–44 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra and characterization data of chitosans, NMR spectra obtained in the deuterium tracer study and additional results obtained in the OKR and the Michael addition are given. See DOI: 10.1039/d1cy01674a |
| This journal is © The Royal Society of Chemistry 2021 |