
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape selectivity effects in the hydroconversion of perhydrophenanthrene over bifunctional catalysts†
Larissa
Brito
a,
Gerhard D.
Pirngruber
 *a,
Javier
Perez-Pellitero
a,
Emmanuelle
Guillon
a,
Florian
Albrieux
a and
Johan A.
Martens
b
*a,
Javier
Perez-Pellitero
a,
Emmanuelle
Guillon
a,
Florian
Albrieux
a and
Johan A.
Martens
b
aRond Point de l'échangeur de Solaize, IFP Energies Nouvelles, BP-3, 69360 Solaize, France. E-mail: gerhard.pirngruber@ifpen.fr
bCenter for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
First published on 21st October 2021
Abstract
Hydroconversion of perhydrophenanthrene was performed over Pt/Beta and Pt/ASA bifunctional catalysts and compared to results obtained on the Pt/USY zeolite catalyst, under the same operating conditions. Perhydrophenanthrene resulted from the hydrogenation of the parent aromatic, phenanthrene, on a Pt/alumina pre-catalyst. All three bifunctional catalysts followed the general reaction pathway observed previously, i.e. isomerization of perhydrophenanthrene by ring-shift and ring-contraction, followed either by the formation of alkyladamantanes or by ring-opening. The ring opening products cracked to smaller naphthenes. Yet, the intermediates of this general reaction network differed very clearly from one catalyst to another. These shape selectivity effects could be explained by GCMC simulations of the adsorption selectivities of different intermediates. Bulky intermediates were preferentially adsorbed on USY zeolites, whereas Beta zeolites adsorbed preferentially linear structures. The product distribution on Beta zeolites was explained through the formation of a central ring-opening intermediate which cracked rapidly to C7 naphthenes. USY-based catalysts were the most active among the solids tested, while Beta zeolites were slightly more selective to generate cracked products.
1. Introduction
In petroleum refining, hydrocracking plays an important role in upgrading heavy oil fractions, such as vacuum gas oil (VGO), to more valuable fractions, such as middle distillates.1–4 Hydrocracking employs bifunctional catalysts, which comprise acid and metal functions. In order to process heavy fractions such as VGO, large-pore catalysts must be employed to avoid diffusional limitations.5–8 Zeolites and amorphous silica–alumina (ASA) are commonly used in hydrocracking units as the acid function of bifunctional catalysts.9–12 The acid function works in synergy with a hydrogenating component, such as Pt or promoted MoS2. The latter becomes more active than Pt if the environment contains high H2S concentrations.13–15VGO fractions are rich in cyclic molecules, such as naphthenes and aromatics.16 Hydroconversion of small monocyclic naphthenes has been well described in the literature,17–21 as well as the reaction pathways of alkanes.22–31 The knowledge of reaction routes involved in the conversion of heavy and/or polycyclic naphthenes containing more than two cycles is scarcer.11,32–38 In a preceding study, we established hydroisomerization and hydrocracking pathways of perhydrophenanthrene (PHP), a 3-cycle naphthene, over a bifunctional Pt/USY catalyst.39 The model molecule was first isomerized to ring-shift and ring-contraction compounds. These isomers generated ring-opening products or went through further isomerization to produce alkyladamantanes. The latter are very stable molecules and resisted hydrocracking. The ring opening products underwent cracking, resulting in a broad distribution of carbon atoms.
Since PHP is a rather bulky reactant, we can expect shape selectivity effects to play a role in its conversion. It is, therefore, important to analyze how the reactivity depends on the pore size and structure. Indeed, previous work by Leite et al. demonstrated that the reaction pathway over zeolite Beta differed from the one over USY. Moreover, large pore sizes (mesopores in USY and silica–alumina) favored isomerization over cracking products.40,41
The very high resolutive power of GCxGC analysis that we have developed in our previous work allows us to push Leite's preliminary analysis further. In this paper we, therefore, present a detailed analysis of the reaction pathways of the hydroconversion of PHP over Pt/USY, Pt/Beta and Pt/ASA (amorphous silica–alumina) bifunctional catalysts. We identify the nature of reaction intermediates and of cracked products generated on each solid and relate the differences to shape selective effects. Monte Carlo simulations show that these shape selectivity effects can be attributed to different adsorption selectivities of the intermediates in the FAU and BEA topologies. We also address the question whether the formation of adamantanes is limited to the mesopores/external surface of zeolites or whether it can occur in the micropores of large-pore zeolites.
2. Materials and methods
2.1. Preparation and characterization of catalysts
Ultra-stable-Y (CBV720) and Beta (CP814e) zeolites were provided by Zeolyst. The powder was mixed with an alumina binder (Pural SB3), furnished by Sasol, and extruded, according to a protocol described elsewhere.39,42 Zeolite loading corresponded to 1 or 3 wt% of the dried support. The low zeolite loadings were chosen in order to obtain activity values of the same order of magnitude as a silica–alumina catalyst. Amorphous silica–alumina (Siralox 30) was supplied by Sasol in its extrudate form. All supports were calcined at 600 °C, under an air flow of 1.5 NL h−1 gsupport−1. For a good control of the hydrodynamics in the reactor, the extrudates were sorted to a length from 3 to 6 mm. The Pt precursor (H2PtCl6 from Sigma-Aldrich) was deposited on the extrudates by incipient wetness impregnation, in the presence of hydrochloric acid as a competitor. Further details on the preparation of catalysts may be found in previous work.39 Pt-Based catalysts were calcined at 520 °C for 2 h, under an air flow of 1 NL h−1 gcatalyst−1. A 0.8 wt% Pt/Al2O3 catalyst was prepared using the same procedure and used as a pure hydrogenation catalyst.The content of Pt in the catalysts was determined with X-ray fluorescence and the dispersion of Pt was measured by H2–O2 chemisorption. Pt particles were considered to be spherical for the calculation of their size. The concentration of Brønsted acid sites (BAS) of the parent zeolites was determined from pyridine adsorption followed by FTIR. The concentration of BAS in USY and BETA parent zeolites corresponded to ca. 200 and 220 μmol g−1, respectively.43 The BAS concentration of the final catalysts was estimated by a rule of proportionality, considering that extrusion did not affect the acidity of the shaped support.44 The BAS acidity of ASA was below the limit of reliable quantification. We also tested the H/D exchange method proposed by Hensen and co-workers,45 but ASA did not show any O–D bands which could be attributable to strongly acidic sites when exposed to C6D6 at room temperature. The properties are summarized in Table 1.
| Type of support | Zeolite content in binder (wt%) | Pt loading (wt%) | Pt dispersion (%) | Pt particle size (nm) | n Pt (μmol g−1) | BAS (μmol g−1) |
|---|---|---|---|---|---|---|
| USY (CBV720) | 1 | 0.60 | 48 | 2.3 | 9.0 | 2.0 |
| Beta (CP814e) | 1 | 0.65 | 45 | 2.5 | 9.0 | 2.2 |
| USY (CBV720) | 3 | 0.66 | 89 | 1.3 | 18.8 | 6.0 |
| Beta (CP814e) | 3 | 0.69 | 85 | 1.3 | 15.8 | 6.6 |
| ASA (Siralox 30) | — | 0.60 | 84 | 1.3 | 15.8 | n.d. |
2.2. Catalytic tests
A downflow fixed-bed reactor was used to perform catalytic tests at 280 or 300 °C and 60 bar. The reactor was loaded with a stacking of 2 g of Pt/Al2O3 and 2 g of bifunctional catalyst. Prior to the tests, the solids were reduced in situ, at 450 °C for 2 h under a H2 flow of 1 NL h−1 gcatalyst−1. The feedstock was composed of 3 wt% phenanthrene (Alfa Aesar) diluted in 97 wt% n-heptane (AnalaR Normapur). N-Heptane was chosen as a solvent because it does not crack readily. A high hydrogen to hydrocarbon molar ratio of 7 mol mol−1 was applied for all the tests and ensured the total hydrogenation of the parent aromatic to the naphthenic molecule over the Pt/Al2O3 pre-catalyst. This ratio also avoided catalyst deactivation throughout the tests. Different conversion degrees of PHP were obtained by changing the feed flow entering the reactor. Gas phase reaction products were analyzed online with a GC-FID apparatus (Varian). Liquid phase reaction products were collected for offline identification and quantification, with GCxGC-FID/MS. Further details on the analysis of reaction products are available elsewhere.39The molar flow of a carbon number fraction (Ni) along with catalytic descriptors comprising the conversion of perhydrophenanthrene (X), yield of reaction products (Yi) and distribution of a given product i within a family J (Di,J) was calculated according to eqn (1)–(4).
 | (1) |
 | (2) |
 | (3) |
 | (4) |
The contact time was expressed as 1/WHSV, and WHSV was defined as the liquid mass flow divided by the mass of the catalyst.
2.3. Grand Canonical Monte Carlo (GCMC) simulations
Grand Canonical Monte Carlo simulations combined with a bias scheme for the insertion of the center of mass of the guest molecules were performed at 280 °C to calculate the adsorbed amounts and selectivities of the different mixtures of hydrocarbons in both zeolites Beta and faujasite. The structure was represented by a 4 × 4 × 2 replication of the purely siliceous unit cell of the A polymorph of *BEA (taken from the IZA database46). The same strategy was employed for the faujasite by implementing a 2 × 2 × 2 unit cell replication system. The simulations were performed using the code GIBBS 9.3.47 The adsorbate molecules were described according to the anisotropic united atoms potential (AUA) for hydrocarbons,48,49 while the description of the zeolite was based on a “Kiselev type” potential.50 The adsorbed amounts were determined from production runs of at least 50 million Monte Carlo steps. Further details about the model parameters and methodologies are provided in the ESI.†3. Results
3.1. Identification and quantification of reaction products with GCxGC-FID/MS
The reaction products resulting from hydroisomerization and hydrocracking of PHP over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts were identified and quantified with a two-dimensional GC coupled to FID and MS detectors. The chromatograms at similar PHP conversion are illustrated in Fig. 1. The conversion of phenanthrene, the parent aromatic, to the tricyclic naphthene was complete under the operating conditions applied during the catalytic tests. The method to distinguish PHP stereoisomers, i.e. the reactant, from reaction products was described in ref. 39. | ||
| Fig. 1 GCxGC-FID analysis of reaction products from hydroconversion of perhydrophenanthrene obtained over A) Pt/USY zeolite catalyst at 48% conversion, B) Pt/Beta zeolite catalyst at 45% conversion, and C) Pt/ASA catalyst at 45% conversion. | ||
The reaction products were lumped into families, according to their number of carbon atoms and chemical similarity. The six stereoisomers of PHP, obtained over the pre-catalyst Pt/Al2O3, were considered as the reactants, which underwent hydroisomerization and hydrocracking over the zeolite or silica–alumina catalysts.
Isomerization products (C14H24) were then divided into substituted adamantanes and other skeletal PHP isomers (Fig. 2). Among the latter, two subgroups were distinguished: ring-shift (perhydroanthracene and methylperhydrophenalenes) and ring-contraction isomers (having one 5-ring). Ring-opening products (ROPs, C14H26) were compounds presenting the same number of carbon atoms as PHP, but only two naphthenic cycles. Ring-opening of two out of three cycles of PHP was not observed among the reaction products.
 | ||
| Fig. 2 Families and typical examples of molecules in the reaction products of hydroconversion of perhydrophenanthrene over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. | ||
Finally, cracked products comprised all the molecules with less than 14 carbon atoms and were mainly constituted of naphthenes.
3.2. Hydroconversion of perhydrophenanthrene
The conversion of PHP over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts, as a function of contact time at 280 °C, is depicted in Fig. 3. The catalysts containing 3 wt% of zeolite were the most active among the solids tested. Results obtained with 1 wt% zeolite loading are available in the ESI.† There was a fairly good proportionality between zeolite content and activity (i.e. similar conversion at the same “zeolite” residence time). USY-based catalysts gave higher PHP conversion than the Beta zeolite. The activity of Pt/ASA at 280 °C was very low compared to the zeolite catalysts. We, therefore, raised the temperature to 300 °C, in order to achieve a reasonable range of PHP conversions (30 to 45%, see the ESI†). In the rest of the manuscript, the 300 °C data will be presented for ASA and compared with the zeolite data at 280 °C. | ||
| Fig. 3 Evolution of PHP conversion with contact time at 280 °C, on Pt/USY and Pt/Beta catalysts with 3 wt% zeolite in the alumina binder, and the Pt/ASA bifunctional catalyst. | ||
The yield of isomerization, ROP and cracking products (on a carbon molar basis) over Pt/USY, Pt/Beta and Pt/ASA catalysts is illustrated in Fig. 4. Isomerization products comprised skeletal PHP isomers and alkyladamantanes. Isomerization was the predominant reaction at low conversion. Over Pt/USY, a maximum yield of isomerization of ca. 43% was obtained at 68% conversion. Pt/Beta and Pt/ASA followed a similar profile, but the yield of isomerization products over zeolite Beta was slightly lower.
 | ||
| Fig. 4 Yield of isomerization, ring-opening and cracked products as a function of PHP conversion over Pt/USY and Pt/Beta at 280 °C and the Pt/ASA bifunctional catalyst at 300 °C. | ||
Ring-opening and cracking products were clearly secondary products. The ROP yield was quite low and reached a maximum at ∼80% conversion. The yield of cracked products increased strongly above 40% PHP conversion. For the Pt/ASA catalyst, the contribution of cracking was very low. At medium conversion, Beta zeolites were slightly more selective to cracking products than USY and ASA, which goes in hand with the lower yield of isomerization products of Beta.
The distribution of PHP isomers, represented by skeletal isomers and alkyladamantanes, over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts is revealed in Fig. 5. The proportion of alkyladamantanes increased with PHP conversion, whereas the proportion of skeletal PHP isomers decreased. Among the solids tested, USY zeolite seemed to be the most suitable for alkyladamantane production, in particular at high conversions. ASA-based catalysts were the least selective towards the formation of substituted adamantanes.
 | ||
| Fig. 5 Distribution of C14H24 reaction products from hydrocracking of PHP over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts as a function of PHP conversion. Skeletal PHP isomers (▲), alkyladamantanes (■). | ||
The distribution of skeletal PHP isomers over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts at about 45% conversion is depicted in Fig. 6. We point out that structure I4, corresponding to a ring-contraction product of PHP, was completely absent on zeolite Beta. In contrast, this molecule was the major intermediate on USY zeolite and ASA at this conversion. This difference between USY and Beta had already been reported by Leite.41 Structure I1, a ring-shift isomer of PHP (perhydroanthracene, PHA), was strongly favored over Pt/Beta at this conversion level, in comparison to the other catalysts. Precursors of alkyladamantanes, characterized by methylperhydrophenalenes (I3), were more abundant over Beta and ASA catalysts than over USY. Finally, spiro-type compounds, like isomer I2, were hardly detected over USY zeolites, but formed to a small extent over Beta and ASA based catalysts. At this conversion, the unknown fraction accounted for 20% of the distribution.
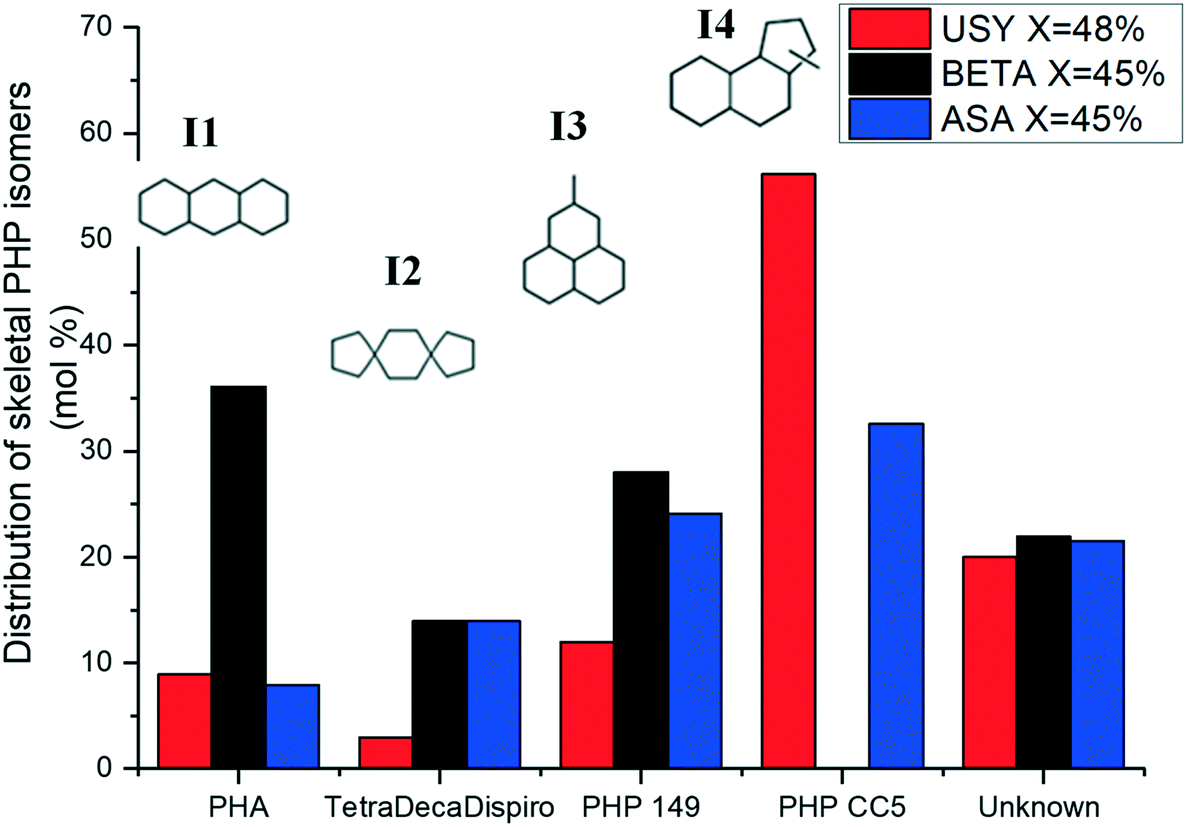 | ||
| Fig. 6 Distribution of skeletal isomers of PHP (excluding substituted adamantanes) at isoconversion over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. PHP 149 stands for methylperhydrophenalene. PHP CC5 represents a skeletal PHP isomer containing a C5-ring. | ||
The distribution of substituted adamantanes over Pt/USY, Pt/Beta and Pt/ASA catalysts at similar conversion is presented in Fig. 7. USY zeolite formed preferentially multibranched isomers, such as tetramethyladamantanes (A1), the thermodynamically most stable substituted adamantane,51 and dimethyl-ethyladamantanes (A3). Zeolite Beta produced mainly isomers A1 and A2 (diethyladamantanes). Methylpropyladamantanes (A4), in addition to isomers A3 and A1, were the dominating isomers over ASA. The unknown fraction accounted for about 20% of the distribution of alkyladamantanes on each solid tested.
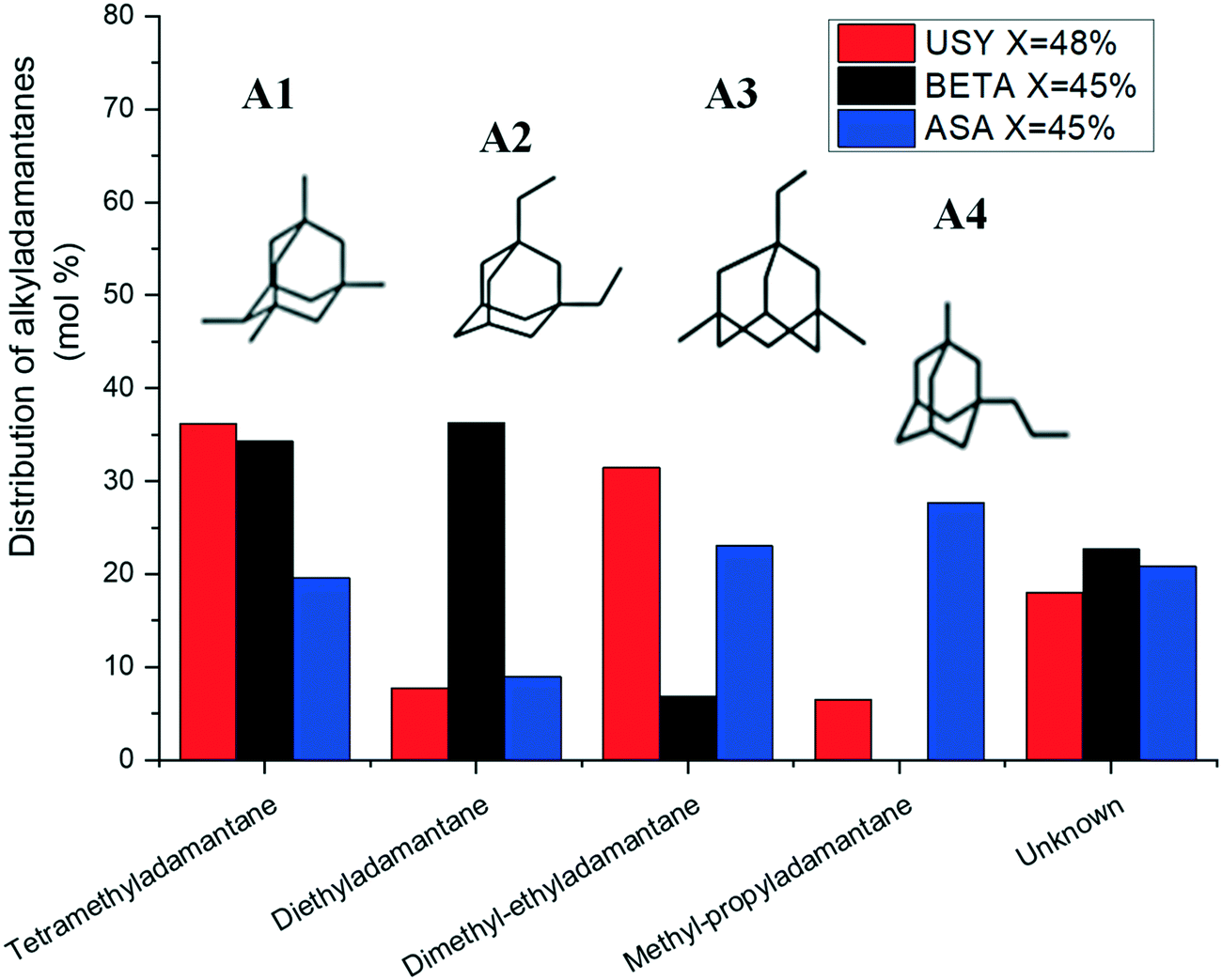 | ||
| Fig. 7 Distribution of alkyladamantanes at isoconversion over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. | ||
Ring-opening products were represented by molecules presenting different degrees of branching, resulting from the opening of the external or central cycle of the model molecule and ring-contraction intermediates (Fig. 8). At ∼45% PHP conversion, USY zeolites produced preferentially structures R5, R1 (opening of the central cycle of PHP) and R6, R7 (opening of the external cycle of PHP). Beta zeolites mainly generated structures R1, R4 (cyclopentyl-cyclohexane structures) and R6. ASA catalysts produced mostly R3 and R6 intermediates, corresponding to the opening of the central and external cycle of PHP, respectively. The unknown fraction varied from 10% to 25% of the distribution.
 | ||
| Fig. 8 Distribution of ring-opening products at isoconversion over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. | ||
The selectivity to cracked products resulting from hydroconversion of PHP over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts is illustrated in Fig. 9. At 37% conversion, a predominant cracking to C7 compounds was observed over the three catalysts, followed by the production of C12. Note that the production of C12 was not accompanied by the formation of light components, such as methane and ethane. Cracking to C12 molecules has been discussed in a previous work,39 where reaction pathways leading to such products were understood as a contribution of disproportionation and addition–cracking reactions. When the conversion increased, USY catalysts gave a broader distribution of carbon atoms as cracked products, while over zeolite Beta and ASA a central cracking of PHP to C7 molecules remained predominant. The main other cracking products were C6 and C8 compounds (formed in equimolar amounts), as well as C4 (mainly isobutane) and C10 molecules.
 | ||
| Fig. 9 Selectivity to cracked products according to the number of carbon atoms at similar conversion of PHP over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. | ||
Most of the cracked products were naphthenes (>80%). Iso-alkanes were minor products (less than 20% of the cracking products) and only very small amounts of n-alkanes were formed. The main alkane product was isobutane. The proportion of alkanes in the cracking products did not change much as a function of conversion. It is worth mentioning that C7 isoalkanes were not counted in the distribution of cracked products, since it was not possible to determine whether they resulted from hydrocracking of the tricyclic model molecule or from isomerization of the solvent.
We examined the composition of the main cracking product, i.e. C7 naphthenes, in more detail. The distribution of C7 naphthenes over Pt/USY, Pt/Beta and Pt/ASA is presented in Fig. 10. Over Beta zeolites and ASA, primary C7 cyclic compounds were composed of methylcyclohexanes, whereas dimethylcyclopentanes seemed to be formed in a primary route over Pt/USY. The proportion of dimethylcyclopentanes increased with conversion over Pt/Beta and Pt/ASA catalysts. Over the three catalysts, ethylcylopentane was observed to a small extent within this subfamily. These results indicate that Beta zeolites and silica–alumina supports differ from USY catalysts not only in terms of carbon number distribution, but also with respect to the nature of primary products formed.
 | ||
| Fig. 10 Distribution of C7 naphthenes as a function of PHP conversion over Pt/USY, Pt/Beta and Pt/ASA bifunctional catalysts. C7 naphthenes were identified as dimethylcyclopentanes (triangles), methylcyclohexanes (squares) and ethylcyclopentanes (circles). | ||
The detailed composition of each family of cracked products will not be presented herein, since the results are similar to the ones reported in our previous work regarding PHP conversion on Pt/USY.39 C6 and C8 reaction products were mainly dimethylcyclopentanes and ethylcyclohexanes, while C10 structures were identified as decalin (decahydronaphthalene) and its isomers.
3.3. Molecular simulation
Fig. 6 and 8 show that the distribution of PHP isomers and of ring opening products is quite different on the three catalysts, i.e. we observe shape selectivity. Shape selectivity is often related to the fact that one isomer is preferentially adsorbed over another in a given zeolite.52,53 In order to explore this hypothesis, a series of molecular simulations of the adsorption of selected intermediates were performed in the crystal structure of USY (FAU) and Beta (*BEA) zeolites. Taking into account the low polarity of the studied adsorbates and the main goal of the study (identifying the potential presence of shape selectivity), the purely siliceous forms of the mentioned structures were considered. The case of the amorphous silica–alumina was not evaluated with this method due to the uncertainty that could be introduced due to the lack of an accurate structural model as compared to the perfectly known zeolitic crystallographic frameworks.54 For the molecular simulation study, we selected the most representative molecules found at ca. 70% PHP conversion.As a first step, we investigated the competitive adsorption of the reactant PHP, skeletal PHP isomers and adamantanes on each zeolite. The choice of representative intermediates was made based on the distribution of skeletal PHP isomers and alkyladamantanes (Fig. 6, 7 and 11). Isomer I4 (PHP CC5) was selectively formed over USY zeolites and ASA. In contrast, PHA isomers (I1) seemed to be favored over Beta zeolites, especially at high PHP conversion (Fig. 11). 1,3,5,7-Tetramethyladamantane was chosen as representative of the alkyladamantane family.
 | ||
| Fig. 11 Distribution of skeletal isomers of PHP (excluding substituted adamantanes) at isoconversion over Pt/USY and Pt/Beta bifunctional catalysts. | ||
We, thus, carried out a simulation of the adsorption of an equimolar mixture of the reactant PHP, PHA (I1), the ring contraction isomer I4 and 1,3,5,7-tetramethyladamantane, in the presence of n-heptane as a solvent. The chosen partial pressures were representative of the reaction conditions. The quantity of molecules adsorbed per volume is provided in Table 2. Clearly, both zeolites preferred the adsorption of PHP isomers I1 and I4 over the adsorption of alkyladamantanes. The adsorption of alkyladamantane was especially unfavorable (practically zero) on zeolite Beta.
| q i (molecules per nm3) | |||||
|---|---|---|---|---|---|
| Zeolite topology | PHP reactant | A1 1,3,5,7-tetramethyl-adamantane | I4 (PHP CC5) | I1 (PHA) | n-C7 |
| FAU | 0.144 | 0.026 | 0.067 | 0.063 | 0.681 |
| BEA | 0.114 | 0.000 | 0.030 | 0.190 | 0.753 |
Furthermore, adsorption of the ring-shift isomer PHA (I1) was more favored in zeolite BEA, in comparison to bulky molecules such as isomer I4 (the ring contraction isomer of PHP). These trends were confirmed by carrying out simulations with a mixture containing only isomers I1 and I4 (Table 3). The I1/I4 selectivity was ca. 3.2 for BEA, but only 1.06 for FAU. We can, therefore, assume that the preferential adsorption of PHA in BEA is partially responsible for the abundant formation of this intermediate in zeolite Beta.
| q i (molecules per nm3) | Selectivity | ||
|---|---|---|---|
| Zeolite topology | I4 (PHP CC5) | I1 (PHA) | I1/I4 |
| FAU | 0.310 | 0.328 | 1.06 |
| BEA | 0.215 | 0.684 | 3.18 |
Table 2 further demonstrates that the PHP reactant was less adsorbed in zeolite BEA than in the FAU structure.
4. Discussion
4.1. Global hydrocracking pathways
USY zeolite-based catalysts were the most active among the solids tested, followed by Beta and ASA catalysts. We note that a comparison of the same USY and Beta zeolites in the hydrocracking of n-hexadecane showed that zeolite Beta was more active than USY.55 We presume that shape selectivity issues explain the inversion of activity ranking between USY and Beta. The molecular simulations showed that PHP isomers adsorbed much less in the pores of Beta zeolite than linear structures (Table 2). Hence, the access of the reactant to the acid sites in the micropores may be more restricted than in the faujasite structure.All three catalysts globally followed the same reaction pathways (Fig. 4): ring-shift and ring-contraction PHP isomers were formed first as primary products. They either converted into alkyladamantanes or into ring-opening products. The latter underwent cracking. Cracking could only take place after the opening of at least one cycle of skeletal PHP isomers. The yield of ROPs was very low over USY, Beta and ASA, indicating that cracking occurred rapidly after the opening of a central or external cycle of PHP isomers. Alkyladamantanes, on the other hand, were not easily converted through hydrocracking to C10 adamantanes and isobutane, which is coherent with their high thermodynamic stability. Although this global scheme was common to all catalysts, the main members of each product family differed from one catalyst to another, as illustrated in Fig. 12. The reasons for these selectivity differences will be discussed in the following paragraphs.
 | ||
| Fig. 12 Simplified reaction pathway for hydrocracking of perhydrophenanthrene over bifunctional Pt/Beta, Pt/USY zeolite catalysts and Pt/ASA catalyst. Main reaction intermediates obtained on each catalyst are illustrated. | ||
4.2. Selectivity to adamantanes
We first take a closer look at the formation of adamantanes. As discussed before,39 they are presumably formed from methylperhydrophenalene intermediates (I3).51,56 Isomer I3, which is formed by ring-shift of PHP, was indeed observed over the three catalysts at all conversion levels. The selectivity to adamantane formation decreased in the order USY ≧ Beta > ASA. We will first discuss the difference between zeolite catalysts and ASA and then turn to the differences between USY and Beta zeolites.Molecular simulations showed that alkyladamantanes were not easily adsorbed in the micropores of USY and Beta zeolites. Still, both zeolites produced significant amounts of alkyladamantanes, whereas ASA had a much lower selectivity to these products. The low adamantane selectivity of ASA is coherent with previous work:37 the hydroconversion of fluorene over Pt/ASA produced only very little adamantane products. This result is somewhat surprising, since ASA may accommodate large molecules such as alkyladamantanes thanks to its large pore size (pore diameter of 70–80 Å). On the other hand, the tendency to generate alkyladamantanes in USY zeolites was previously attributed to the presence of supercages of 7.4 Å diameter, which corresponds to the van der Waals diameter of non-substituted adamantane,37,57 but our simulations do not confirm that hypothesis of a good fit. We may infer from our results that a large pore size is not a sufficient criterion to form alkyladamantanes. The weak acid sites in amorphous silica–alumina are apparently not good catalysts for adamantane formation. Their production might be favored on the more strongly acidic zeolites, although the very bulky molecules cannot be easily accommodated in the zeolite micropores. We assume that alkyladamantane formation probably takes place in the pore mouths of the zeolites. The cavity effect of the pore mouth must make the difference compared to the silica–alumina, which does not exert any confinement effect.58
We note that Iglesia and co-workers59 recently offered an alternative explanation for the different catalytic properties of zeolites and silica–alumina. They argued that the low selectivity of mesoporous silica–alumina catalysts to secondary/tertiary products (adamantanes and cracking products in our case) was due to the absence of diffusional constraints: primary intermediates were quickly hydrogenated before undergoing secondary reactions. Zeolites would favor the formation of secondary/tertiary products not because of higher acid strength, but because of a longer intracrystalline residence time of the primary intermediates in the micropores with high acid site density. In our case, we do not believe that this reasoning is applicable, since the formation of adamantanes presumably takes place in the pore mouths.
Let us now have a closer look at the adamantane formation on zeolite Beta. In spite of zero adsorption of adamantanes in BEA (according to the simulations), the selectivity to alkyladamantanes over Pt/Beta was non-zero, but still significantly lower than that of Pt/USY, at least at high conversion (Fig. 4 and 5). The higher alkyladamantane selectivity of USY vs. Beta is coherent with the literature. In a previous study, Rollmann and coworkers compared the effectiveness to convert perhydrofluorene,57 a tricyclic naphthene, to trimethyladamantanes over Pt or Pd/USY and Pt or Pd/Beta zeolite catalysts. Under similar operating conditions, the yield of alkyladamantanes obtained over USY-based catalysts corresponded to 27%, whereas a yield of only 3% was acquired with Beta zeolites. Wang et al. studied the hydroconversion of fluorene on Pt-supported catalysts.37 Higher yields of perhydrophenalene, precursors of alkyladamantanes, and propyladamantanes were obtained over the USY zeolite, in comparison to Beta. In our case, as shown in Fig. 6 and 11, methylperhydrophenalene (I3) was more abundant over Beta zeolites. It is likely that Beta zeolites did not easily convert these intermediates, hindering the further isomerization to alkyladamantanes.
4.3. Isomerization and ring opening products
We now turn to the PHP isomers, which subsequently lead to the formation of ring opening products. In the Results section, we had already pointed out the remarkable result that ring-contraction PHP isomer I4 (Fig. 6) was exclusively formed in the presence of USY and ASA. In accordance with Leite,41 it was completely absent on zeolite Beta. Our molecular simulations showed that the adsorption of I4 in zeolite Beta was quite unfavorable compared to more linear isomers. BEA adsorbed preferentially more linear structures, represented by PHA stereoisomers. It is clear that shape-selectivity impeded the production of bulky molecules on zeolite Beta, due to its more strained pore system. This different selectivity to PHP isomers in turn leads to different ring opening products.As shown in Fig. 13, a very favorable ring opening pathway of I4 leads to R3, which can subsequently rearrange to R5 by very easy ring contraction reactions. An alternative favorable ring opening pathway of I4 leads via β-scission in the external ring to butyl-decalin (R6), which can subsequently rearrange to R7. According to Fig. 8, the sum of R3 + R5 decreases in the order USY > ASA ≫ Beta. The sum of R6 + R7 follows the same order. We can deduce that the ring opening products, which can be easily formed from I4, were less preferred in zeolite Beta.
 | ||
| Fig. 13 Favorable ring opening pathways of the isomers a) I4 and b) PHA (I1). | ||
Beta prefers the formation of PHA over I4; a favorable ring opening pathway of PHA (I1) leads to R2 (Fig. 13b), which can subsequently rearranged to R1 and R4. The sum of R2 + R1 + R4 follows the order Beta > USY > ASA (Fig. 8). This trend can be linked to the preferential formation of PHA on zeolite Beta. We will discuss the implication on the distribution of cracking products in the next section.
4.4. Distribution of cracking products
In order to explain the distribution of cracked products, it is necessary to analyze the hydrocracking pathways of the major ROPs. The basis for this analysis was laid in our previous work on Pt/USY.39Beta zeolites produced mainly structures R1, R4 and R6, according to the distribution presented in Fig. 8. The cracking of butyldecalin (R6) to C10 naphthenes, represented by decalin and its isomers, and isobutane was observed at high PHP conversion. Yet, the major cracking products of zeolite Beta were not C10 and C4, but C7, followed by C6 and C8 (Fig. 9).
We tried to construct reaction pathways for the cracking of intermediates R1 and R4 to C7 naphthenes (Fig. 14A and B). Several rearrangements, including a slow step of ring-contraction (type B isomerization), were necessary to achieve a configuration allowing fast beta-scission. In both cases, cracking leading to C6 and C8 was more easily obtained through exocyclic alkyl shifts, generating dimethylcyclopentanes and ethylcyclohexanes. We, therefore, presume that the cracking of R1 and R4 explains the formation of C6 and C8 naphthene products.
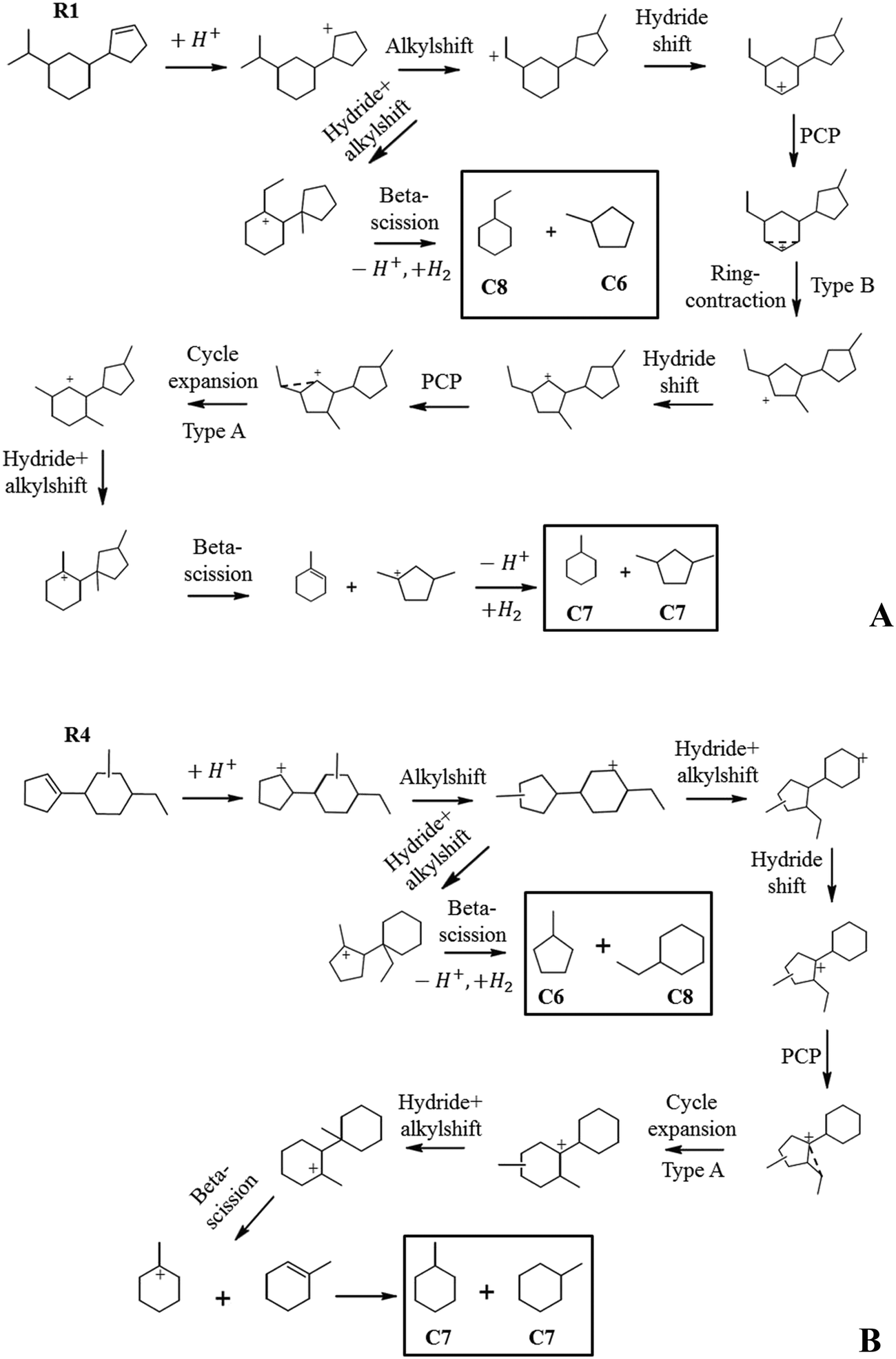 | ||
| Fig. 14 A) Suggested hydrocracking pathway to C7, C6 and C8 naphthenes resulting from hydroconversion of ring-opening intermediate R1. B) Suggested hydrocracking pathway to C7, C6 and C8 naphthenes resulting from hydroconversion of ring-opening intermediate R4. | ||
Preferential cracking to C7 naphthenes over Beta zeolites can only be rationalized from the consumption of the R2 intermediate, which is the favored ring opening product of PHA (Fig. 15). The distribution of ROPs (Fig. 8) shows that R2 accounts for ca. 10% of the distribution at low PHP conversion, and its content decreases to virtually zero at high PHP conversion. R2 can very easily crack to C7 naphthenes by a fast tertiary–tertiary beta-scission, once the alkyl group has been shifted to a favorable position, as illustrated in Fig. 15 (we note that this pathway is more favorable than the routes leading to the C7 isomers preferred on USY or to C6 + C8, which had been proposed in ref. 39). We presume that the R2 intermediate is very quickly consumed by this very favorable cracking pathway, which explains its low abundance in the products. The pathway shown in Fig. 15 must be the main hydrocracking pathway over the Pt/Beta zeolite catalyst, since it can explain the exclusive formation of methylcyclohexane as the primary C7 product on Beta zeolites (Fig. 10). Since ASA exhibits the same high selectivity to methylcyclohexane, this pathway must also play an important role in silica–alumina. The similarity in the product distribution of ASA and Beta reinforces the idea that a lot of catalysis on zeolite Beta may be going on in the pore mouth or on its outer surface.
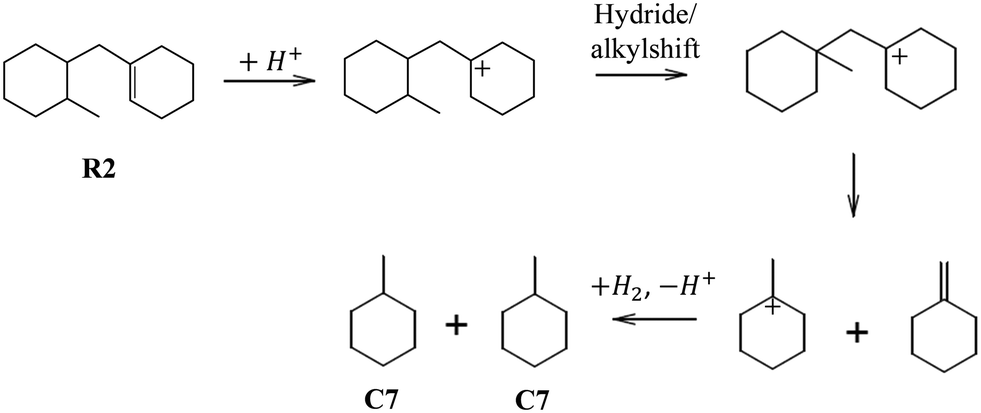 | ||
| Fig. 15 Very favorable hydrocracking pathway of ring-opening product R2 to C7 naphthenes. | ||
We note that the ROPs, which dominate in the product distribution, are not necessarily the ones which explain best the cracking pattern, because they may actually correspond to the most stable intermediates, i.e. those which crack least readily.
Finally, let us examine the question why USY does not as selectively crack to C7 naphthenes as zeolite Beta. In USY, the preferred isomer was the ring-contraction isomer I4, which could favorably ring-open to R3. A cracking pathway from R3 to C7 products (involving dimethyl-cyclopentane, which is the main C7 isomer on USY) had been proposed in ref. 39, but this pathway involves a type B isomerization step, shifting an alkyl group from the ring to the central alkyl chain. Moreover, the beta scission leads to a secondary carbenium ion. It will, thus, have a higher activation energy than the cracking pathway shown in Fig. 15 and will be less dominating compared to other possible reaction pathways leading to C6 + C8 naphthenes.
5. Conclusions
Hydroisomerization and hydrocracking of perhydrophenanthrene were performed over Pt/Beta and Pt/ASA bifunctional catalysts and compared to results obtained with the Pt/USY zeolite catalyst, under the same operating conditions. The common reaction pathway consists of converting the reactant into skeletal perhydrophenanthrene isomers, which are further transformed into substituted adamantanes or ring-opening products. The ring-opening products undergo rapid hydrocracking, whereas alkyladamantanes are stable.USY-based catalysts were more active than Beta. Molecular simulations point out a weak adsorption of the PHP reactant on the pores of the BEA structure, indicating that the lower activity of Beta is linked to a limited access of the reactant to the micropores of zeolite Beta. ASA was by far the least active catalyst, due to its low acidity.
On top of the activity difference, shape selectivity effects were evidenced by comparing zeolites USY and Beta. The bulky ring-contraction isomers of PHP, which were dominating in USY, do not easily fit into the pores of zeolite Beta. The Beta structure prefers the adsorption of the ring-shift isomer perhydroanthracene (PHA). The preferential adsorption of PHA can explain the very selective cracking to methyl-cyclohexane, which was observed over zeolite Beta: PHA offers a very favorable central ring opening pathway to methyl-(cyclohexylmethyl)-cyclohexane (R2), which can then readily crack to two methyl-cyclohexane molecules, leading to a peak at C7 in the distribution of cracked products. For zeolite USY, on the other hand, the dominating ring-contraction isomer of PHP does not offer an equally favorable cracking pathway and, thus, leads to a broader distribution of cracking products.
Quite remarkably, zeolite Beta also produced significant amounts of alkyl-adamantanes (albeit less than USY), although these products absolutely do not fit into the pores of the BEA structure. Their formation is, therefore, attributed to pore mouth catalysis. ASA catalysts, on the other hand, were not very selective to alkyl-adamantanes, although the formation of these bulky molecules should be favored in the large pores of ASA. We tentatively attribute the difficulty to from adamantanes on ASA to the absence of confinement effects and/or of strong acid sites on the ASA surface.
With respect to PHP isomers, ring opening intermediates and cracking products, ASA showed a quite particular behavior. The main intermediates of the ASA catalyst resemble those of USY, but the distribution of cracking products is closer to Beta. The similarity in the distribution of cracking products on ASA suggests that a lot of catalysis on zeolite Beta may be going on in the pore mouth or on its outer surface.
From an application point of view, the results presented in this work will be a useful guide for catalyst selection in hydroconversion processes, especially when dealing with feeds with a high content of polyaromatic molecules. It will also help in improving the kinetic models of the hydrocracking process, where naphthene conversion is often treated in a very rudimentary manner, due to the lack of analytic information.60,61
Conflicts of interest
There are no conflicts to declare.References
- M. S. Rana, V. Samano, J. Ancheyta and J. A. I. Diaz, A review of recent advances on process technologies for upgrading of heavy oils and residua, Fuel, 2007, 86, 1216–1231 CrossRef CAS.
- G. Valavarasu, M. Bhaskar and K. S. Balaraman, Mild hydrocracking - A review of the process, catalysts, reactions, kinetics, and advantages, Pet. Sci. Technol., 2003, 21, 1185–1205 CrossRef CAS.
- F. Bertoncini, A. Bonduelle, J. Francis and E. Guillon, in Catalysis by Transition Metal Sulfides, ed. H. Toulhoat and P. Raybaud, Technip, Paris, 2013, p. 609 Search PubMed.
- J. Scherzer and A. J. Gruia, Hydrocracking science and technology, M. Dekker, New York, 1996 Search PubMed.
- A. Corma, M. J. Diaz-Cabanas, C. Lopez and A. Martinez, Hydrocracking catalysts based on the new large-pore ITQ-21 zeolite for maximizing diesel products, Stud. Surf. Sci. Catal., 2004, 154, 2380–2386 CrossRef.
- A. Chica and A. Corma, Comparison of large pore zeolites for n-octane hydroisomerization: Activity, selectivity and kinetic features, Chem. Ing. Tech., 2007, 79, 857–870 CrossRef CAS.
- T. F. Degnan, Jr, Applications of zeolites in petroleum refining, Top. Catal., 2000, 13, 349–356 CrossRef.
- W. M. Zhang and P. G. Smirniotis, Effect of zeolite structure and acidity on the product selectivity and reaction mechanism for n-octane hydroisomerization and hydrocracking, J. Catal., 1999, 182, 400–416 CrossRef CAS.
- M. A. Ali, T. Tatsumi and T. Masuda, Development of heavy oil hydrocracking catalysts using amorphous silica-alumina and zeolites as catalyst supports, Appl. Catal., A, 2002, 233, 77–90 CrossRef CAS.
- K. Sato, Y. Nishimura, K. Honna, N. Matsubayashi and H. Shimada, Role of HY Zeolite Mesopores in Hydrocracking of Heavy Oils, J. Catal., 2001, 200, 288–297 CrossRef CAS.
- H. W. Haynes, J. F. Parcher and N. E. Helmer, Hydrocracking Polycyclic Hydrocarbons over a Dual-Functional Zeolite (Faujasite)-Based Catalyst, Ind. Eng. Chem. Process Des. Dev., 1983, 22, 401 CrossRef CAS.
- V. Y. Pereyma, P. P. Dik, O. V. Klimov, S. V. Budukva, K. A. Leonova and A. S. Noskov, Hydrocracking of vacuum gas oil in the presence of catalysts NiMo/Al2O3–amorphous aluminosilicates and NiW/Al2O3–amorphous aluminosilicates, Russ. J. Appl. Chem., 2015, 88, 1969–1975 CrossRef CAS.
- C.-E. Hédoire, C. Louis, A. Davidson, M. Breysse, F. Maugé and M. Vrinat, Support effect in hydrotreating catalysts: hydrogenation properties of molybdenum sulfide supported on β-zeolites of various acidities, J. Catal., 2003, 220, 433–441 CrossRef.
- N. Guernalec, T. Cseri, P. Raybaud, C. Geantet and M. Vrinat, Influence of H2S on the hydrogenation activity of relevant transition metal sulfides, Catal. Today, 2004, 98, 61–66 CrossRef.
- N. Guernalec, C. Geantet, T. Cseri, M. Vrinat, H. Toulhoat and P. Raybaud, Compensation effect and volcano curve in toluene hydrogenation catalyzed by transition metal sulfides, Dalton Trans., 2010, 39, 8420–8422 RSC.
- T. Dutriez, M. Courtiade, D. Thiébaut, H. Dulot, F. Bertoncini, J. Vial and M.-C. Hennion, High-temperature two-dimensional gas chromatography of hydrocarbons up to nC60 for analysis of vacuum gas oils, J. Chromatogr. A, 2009, 1216, 2905–2912 CrossRef CAS PubMed.
- R. Nageswara Rao, N. You, S. Yoon, D. P. Upare, Y.-K. Park and C. W. Lee, Selective Ring Opening of Methylcyclopentane and Methylcyclohexane Over Iridium Bifunctional Catalysts Supported on Surface Modified γ-Al2O3, SiO2 and Ultra Stable Y Zeolites, Catal. Lett., 2011, 141, 1047–1055 CrossRef CAS.
- H. Ziaei-Azad and A. Sayari, Bifunctional MCM-41 aluminosilicate supported Ir with adjusted metal and acid functionality for catalytic ring opening of 1,2-dimethylcyclohexane, J. Catal., 2016, 344, 729–740 CrossRef CAS.
- E. Gutierrez-Acebo, C. Leroux, C. Chizallet, Y. Schuurman and C. Bouchy, Metal/Acid Bifunctional Catalysis and Intimacy Criterion for Ethylcyclohexane Hydroconversion, ACS Catal., 2018, 8, 6035–6046 CrossRef CAS.
- S. A. D'Ippolito, C. Especel, L. Vivier, F. Epron and C. L. Pieck, Influence of the Brønsted acidity, SiO2/Al2O3 ratio and Rh–Pd content on the ring opening. Part II. Selective ring opening of methylcyclohexane, Appl. Catal., A, 2014, 469, 541–549 CrossRef.
- J. Weitkamp and S. Ernst, in Catalysis by Acids and Bases, ed. B. Imelik, C. Naccache, G. Coudurier, Y. Ben Taarit and J. C. Vedrine, Elsevier, 1985, pp. 419–426 Search PubMed.
- F. Alvarez, G. Giannetto, M. Guisnet and G. Perot, Hydroisomerization and hydrocracking of n-Alkanes. 2. n-Heptane transformation on a Pt-dealuminated Y zeolite - comparison with a Pt-Y zeolite, Appl. Catal., 1987, 34, 353–365 CrossRef CAS.
- G. E. Giannetto, G. R. Perot and M. R. Guisnet, Hydroisomerization and hydrocracking of n-alkanes. 1. Ideal hydroisomerization PtHY catalysts, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25, 481–490 CrossRef CAS.
- F. Alvarez, F. R. Ribeiro, G. Giannetto, F. Chevalier, G. Perot and M. Guisnet, in Studies in Surface Science and Catalysis, ed. A. Galarneau, F. Fajula, F. Di Renzo and J. Vedrine, Elsevier, 2001, pp. 1339–1348 Search PubMed.
- N. Batalha, L. Pinard, Y. Pouilloux and M. Guisnet, Bifunctional Hydrogenating/Acid Catalysis, Catal. Lett., 2013, 143, 587–591 CrossRef CAS.
- G. Burnens, C. Bouchy, E. Guillon and J. A. Martens, Hydrocracking reaction pathways of 2,6,10,14-tetramethylpentadecane model molecule on bifunctional silica-alumina and ultrastable Y zeolite catalysts, J. Catal., 2011, 282, 145–154 CrossRef CAS.
- M. C. Claude and J. A. Martens, Monomethyl-branching of long n-alkanes in the range from decane to tetracosane on Pt/H-ZSM-22 bifunctional catalyst, J. Catal., 2000, 190, 39–48 CrossRef CAS.
- J. Weitkamp, P. A. Jacobs and J. A. Martens, Isomerization and hydrocracking of C-9 through C-16 n-alkanes on Pt/HZSM-5 zeolite, Appl. Catal., 1983, 8, 123–141 CrossRef CAS.
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, n-Hexadecane hydroisomerization over Pt-HBEA catalysts. Quantification and effect of the intimacy between metal and protonic sites, J. Catal., 2013, 307, 122–131 CrossRef CAS.
- R. Kenmogne, A. Finiels, C. Cammarano, V. Hulea and F. Fajula, Hydroconversion of n-hexadecane over bifunctional microporous and mesoporous model catalysts. Influence of pore architecture on selectivity, J. Catal., 2015, 329, 348–354 CrossRef CAS.
- P. S. F. Mendes, F. M. Mota, J. M. Silva, M. F. Ribeiro, A. Daudin and C. Bouchy, A systematic study on mixtures of Pt/zeolite as hydroisomerization catalysts, Catal. Sci. Technol., 2017, 7, 1095–1107 RSC.
- S.-U. Lee, Y.-J. Lee, J.-R. Kim, E.-S. Kim, T.-W. Kim, H. J. Kim, C.-U. Kim and S.-Y. Jeong, Selective ring opening of phenanthrene over NiW-supported mesoporous HY zeolite catalyst depending on their mesoporosity, Mater. Res. Bull., 2017, 96, 149–154 CrossRef CAS.
- L. Leite, E. Benazzi and N. Marchal-George, Hydrocracking of phenanthrene over bifunctional Pt catalysts, Catal. Today, 2001, 65, 241–247 CrossRef CAS.
- A. T. Lapinas, M. T. Klein and B. C. Gates, Catalytic Hydrogenation and Hydrocracking of Fluorene: Reaction Pathways, Kinetics, and Mechanisms, Ind. Eng. Chem. Res., 1991, 30, 42–50 CrossRef CAS.
- T. Isoda, S. Maemoto, K. Kusakabe and S. Morooka, Hydrocracking of Pyrenes over a Nickel-Supported Y-Zeolite Catalyst and an Assessment of the Reaction Mechanism Based on MD Calculations, Energy Fuels, 1999, 13, 617–623 CrossRef CAS.
- S. C. Korre, M. T. Klein and R. J. Quann, Polynuclear Aromatic Hydrocarbons Hydrogenation. 1. Experimental Reaction Pathways and Kinetics, Ind. Eng. Chem. Res., 1995, 34, 101–117 CrossRef CAS.
- L. Wang, Y. Chen, S. Jin, X. Chen and C. Liang, Selective Ring-Shift Isomerization in Hydroconversion of Fluorene over Supported Platinum Catalysts, Energy Fuels, 2016, 30, 3403–3412 CrossRef CAS.
- W. Souverijns, R. Parton, J. A. Martens, G. F. Froment and P. A. Jacobs, Mechanism of the paring reaction of naphtenes, Catal. Lett., 1996, 37, 207–212 CrossRef CAS.
- L. Brito, G. D. Pirngruber, E. Guillon, F. Albrieux and J. A. Martens, Hydroconversion of perhydrophenanthrene over bifunctional Pt/H-USY zeolite catalyst, ChemCatChem, 2020, 12, 3477–3488 CrossRef CAS.
- E. Benazzi, L. Leite, N. Marchal-George, H. Toulhoat and P. Raybaud, New insights into parameters controlling the selectivity in hydrocracking reactions, J. Catal., 2003, 217, 376–387 CrossRef CAS.
- L. Leite, PhD thesis, Université Paris VI, 2000 Search PubMed.
- P. S. F. Mendes, J. M. Silva, M. Filipa Ribeiro, A. Daudin and C. Bouchy, From powder to extrudate zeolite-based bifunctional hydroisomerization catalysts, J. Ind. Eng. Chem., 2018, 62, 72–83 CrossRef CAS.
- P. S. Mendes, G. Lapisardi, C. Bouchy, M. Rivallan, J. M. Silva and M. F. Ribeiro, Hydrogenating activity of Pt/zeolite catalysts focusing acid support and metal dispersion influence, Appl. Catal., A, 2015, 504, 17–28 CrossRef CAS.
- Y. Bi, G. Xia, W. Huang and H. Nie, Hydroisomerization of long chain n-paraffins, RSC Adv., 2015, 5, 99201–99206 RSC.
- D. G. Poduval, J. A. R. van Veen, M. S. Rigutto and E. J. M. Hensen, Brønsted acid sites of zeolitic strength in amorphous silica-alumina, Chem. Commun., 2010, 46, 3466–3468 RSC.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/.
- P. Ungerer, B. Tavitian and A. Boutin, Applications of Molecular Simulation in the Gas Industry: Monte Carlo Methods, Editions Technip - IFP Publications, Paris, 2005 Search PubMed.
- P. Ungerer, C. Beauvais, J. Delhommelle, A. Boutin, B. Rousseau and A. H. Fuchs, Optimization of the anisotropic united atoms intermolecular potential for n -alkanes, J. Chem. Phys., 2000, 112, 5499–5510 CrossRef CAS.
- E. Bourasseau, P. Ungerer and A. Boutin, Prediction of Equilibrium Properties of Cyclic Alkanes by Monte Carlo Simulation New Anisotropic United Atoms Intermolecular Potential New Transfer Bias Method, J. Phys. Chem. B, 2002, 106, 5483–5491 CrossRef CAS.
- P. Pascual, P. Ungerer, B. Tavitian, P. Pernot and A. Boutin, Development of a transferable guest–host force field for adsorption of hydrocarbons in zeolites, Phys. Chem. Chem. Phys., 2003, 5, 3684–3693 RSC.
- A. Schneider, R. W. Warren and E. J. Janoski, Formation of Perhydrophenalenes and Polyalkyladamantanes by Isomerization of Tricyclic Perhydroaromatics, J. Org. Chem., 1966, 31, 1617–1625 CrossRef CAS.
- B. Smit and T. L. M. Maesen, Towards a molecular understanding of shape selectivity, Nature, 2008, 451, 671–678 CrossRef CAS PubMed.
- P. Bai, M. Y. Jeon, L. Ren, C. Knight, M. W. Deem, M. Tsapatsis and J. I. Siepmann, Discovery of optimal zeolites for challenging separations and chemical transformations using predictive materials modeling, Nat. Commun., 2015, 6, 5912 CrossRef CAS PubMed.
- C. Chizallet, Toward the Atomic Scale Simulation of Intricate Acidic Aluminosilicate Catalysts, ACS Catal., 2020, 10, 5579–5601 CrossRef CAS.
- P. S. F. Mendes, J. M. Silva, M. F. Ribeiro, P. Duchêne, A. Daudin and C. Bouchy, Quantification of metal-acid balance in hydroisomerization catalysts, AIChE J., 2017, 63, 2864–2875 CrossRef CAS.
- M. A. McKervey, Adamantane rearrangements, Chem. Soc. Rev., 1974, 3, 479 RSC.
- L. Rollmann, L. A. Green, R. A. Bradway and H. K. C. Timken, Adamantanes from petroleum with zeolites, Catal. Today, 1996, 31, 163–169 CrossRef CAS.
- L. Treps, A. Gomez, T. D. Bruin and C. Chizallet, Environment, Stability and Acidity of External Surface Sites of Silicalite-1 and ZSM-5 Micro and Nano Slabs, Sheets, and Crystals, ACS Catal., 2020, 10, 3297–3312 CrossRef CAS.
- G. Noh, Z. Shi, S. I. Zones and E. Iglesia, Isomerization and β-scission reactions of alkanes on bifunctional metal-acid catalysts, J. Catal., 2018, 368, 389–410 CrossRef CAS.
- P. Agarwal, M. Sahasrabudhe, S. Khandalkar, C. Saravanan and M. T. Klein, Molecular-Level Kinetic Modeling of a Real Vacuum Gas Oil Hydroprocessing Refinery System, Energy Fuels, 2019, 33, 10143–10158 CrossRef CAS.
- B. E. Browning, I. Pitault, F. Couenne and M. Tayakout-Fayolle, Effects of Bifunctional Catalyst Geometry on Vacuum Gas Oil Hydrocracking Conversion and Selectivity for Middle Distillate, Ind. Eng. Chem. Res., 2018, 57, 16579–16592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01556g |
| This journal is © The Royal Society of Chemistry 2021 |