
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ni(II) and Co(II) bis(acetylacetonato) complexes for alkene/vinylsilane silylation and silicone crosslinking†
Magali
Puillet‡
a,
James
Delorme‡
a,
Delphine
Crozet
a,
Matthieu
Humbert
a,
David
Gajan
 b,
Magali
Bousquié
c,
Delphine
Blanc
c,
Jean
Raynaud
*a and
Vincent
Monteil
*a
b,
Magali
Bousquié
c,
Delphine
Blanc
c,
Jean
Raynaud
*a and
Vincent
Monteil
*a
aLaboratory of Catalysis, Polymerization, Processes and Materials, CP2M (UMR 5128, CNRS/Université de Lyon 1- Claude Bernard/CPE Lyon), Université de Lyon, 43 Bd du 11 Nov. 1918, 69616 Villeurbanne cedex, France. E-mail: jean.raynaud@univ-lyon1.fr; vincent.monteil@univ-lyon1.fr
bCentre de Résonance Magnétique Nucléaire à Très Hauts champs (UMR 5082, CNRS/Ecole Normale Supérieure de Lyon/Université Claude Bernard Lyon 1), Université de Lyon, 5 rue de la Doua, 69100 Villeurbanne, France
cElkem Silicones, R&D Chemistry, 85 avenue des Fréres Perret, 69192 Saint-Fons cedex, France
First published on 22nd June 2021
Abstract
Commercially available Ni(II) and Co(II) complexes – M(acac)2 (acac = acetylacetonate) and M(tmhd)2 (tmhd = 2,2,6,6-tetramethyl-3,5-heptanedionato) – exhibit catalytic activity for alkene/vinylsilane dehydrogenative silylation (DS) and hydrosilylation (HS) with tertiary silanes without the use of any external reducing agents. Using the model compounds divinyltetramethylsiloxane a.k.a dvtms and vinylpentamethyldisiloxane a.k.a vpmds, different selectivities (HS, DS, undesired non-C–Si bond-forming reactions…) are observed whether nickel or cobalt catalysts are employed, with Ni being DS-selective and Co yielding bothHS and DS products. All four complexes are efficient at thermally inducing silicone-oil crosslinking under a non-inert atmosphere, and promote metal-dependent selectivity that is slightly different from model reactions, which HR-MAS NMR spectroscopy unveils. Additional observations as well as NMR studies of [Ni(tmhd)2 + reagent] mixtures provide some insights into the possible activation pathways.
Introduction
Alkene hydrosilylation is a key reaction to manufacture silicone lubricants, adhesives, rubbers, and other high-value products.1–3 Platinum compounds such as Speier's4 and Karstedt's5 complexes traditionally serve as efficient catalysts because of their high activity, stability and selectivity for alkene hydrosilylation. However, high and volatile prices of noble metals induce an important interest in tackling hydrosilylation using earth-abundant metal complexes. Nickel, cobalt and iron catalysts have already been reported and reviewed as relatively active and selective for alkene hydrosilylation (HS) and dehydrogenative silylation (DS).6–13 Two types of systems have been reported in the literature: low-oxidation state and high-oxidation state complexes. Among low oxidation state catalysts, Chirik's bis(imino)pyridine iron(0)14 and cobalt(I)15 are seminal examples, which are very active under strict inert conditions. More stable, high oxidation state nickel, iron and cobalt precursors have been developed to catalyze alkene HS (or DS) after an activation via reduction with Grignard reagents, lithium species or borohydride.16–22 New development in the field are systems which do not need external reducing agents and might harness the SiH inherent to the reaction medium for reduction of iron(II)15–17,19,24 and cobalt(II)23–27 precatalysts, though the exact activation mechanism remains elusive.10,27In the 90s, Marciniec reported the use of the commercially available Ni(II) acetylacetonate Ni(acac)2 without external reducing agent as a hydrosilylation catalyst.28–30 It was claimed that the SiH from the used silane allowed for reduction of the precursor. This catalyst was not selective for alkene hydrosilylation (HS) and gave several other products such as dehydrogenative silylation (DS), reduction (Red) and dimerization products (Dim) (see Fig. 1). Note that only HS and DS establish a Si–C linkage and are thus interesting in coupling or crosslinking reactions.
 | ||
| Fig. 1 Products of alkene silylation with a tertiary silane of the H–SiMe(O–R)2 type. HS represents the hydrosilylation product, DS the product of dehydrogenative silylation, Red the associated reduction product and Dim, the dimerization product. | ||
Herein we developed readily available and easy-to-handle precursors for vinyl silane/alkene silylation without the use of external strong reducing agent, yet promoting the curing of functional silicone oils. Our study is based on Marciniec's seminal work on Ni(acac)2.1–3,10,28–30 We extended the scope of catalysts with a solubility-enhancing ligand for Ni – 2,2,6,6-tetramethyl-3,5-heptanedionate (tmhd) – and with cobalt analogues.31 We were particularly interested in comparing bis(acetylacetonato)Ni(II) and Co(II) complexes (Fig. 2) and their respective activities and selectivities for alkene/vinylsilane hydrosilylation and dehydrogenative silylation using industrially relevant tertiary silanes. We also implemented these precatalysts in the crosslinking of vinyl- and Si–H-functionalized PDMS chains under non-inert atmosphere (presence of O2 and ambient moisture, see Experimental section).
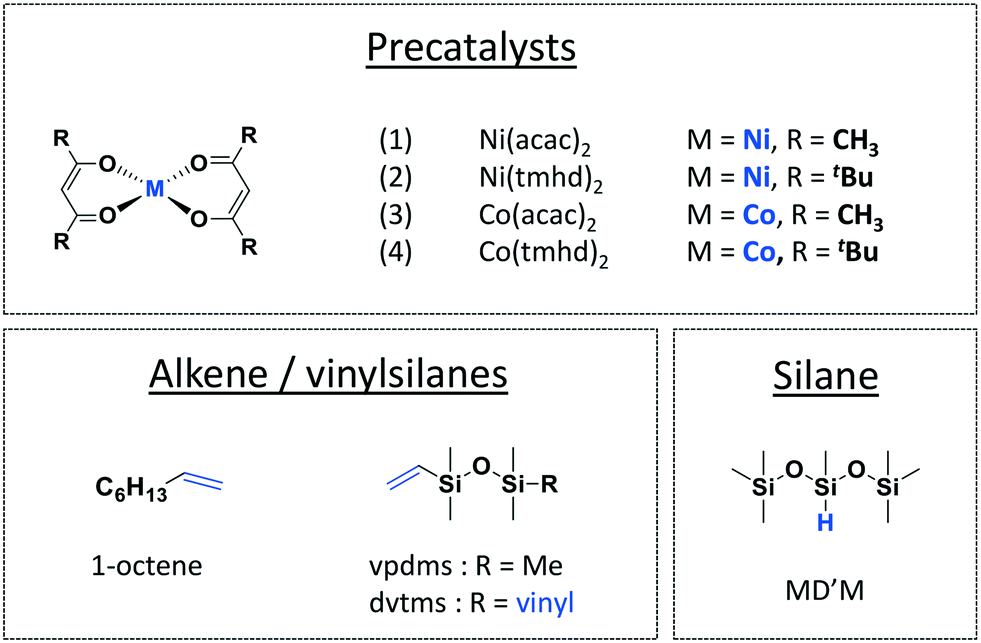 | ||
| Fig. 2 Precatalysts and substrates investigated for alkene/vinylsilane silylation using a tertiary silane. | ||
Results and discussion
We first studied the silylation of divinyltetramethylsiloxane (dvtms), vinylpentamethyldisiloxane (vpmds) and 1-octene (see Fig. 2, ESI† for all experiments, sections 1–4 for dvtms and vpdms & 5 for 1-octene) with MD′M (1,1,1,3,5,5,5-heptamethyltrisiloxane). Before quantifying the conversion and the selectivity, the major products of the reaction catalyzed by Ni(tmhd)2 were identified by NMR spectroscopy. A combination of 1H, 13C and 29Si NMR, as well as GC/GC-MS analyses (for confirmation, in 1-octene case in particular) revealed the major and minor products of the reaction depicted in Fig. 1 for vinylsilane model compounds and 1-octene (see Experimental section and ESI,† sections 1 to 5).Harnessing NMR spectroscopy, the starting substrates were characterized using 1H and 29Si nuclei. Then, the reaction medium was analyzed with 1H and 29Si NMR thanks to a 2D 29Si–1H INEPT. Using the 2D spectra (Fig. 3 and ESI,† sections 2 & 3), we could identify the various Si moieties, and in particular the characteristic signals of the dehydrogenative silylation (DS) and reduction products (Red). Thus, we can conclude that Ni(tmhd)2 is selective for dehydrogenative silylation and concomitant/subsequent reduction, as reported in the literature.1–10 On the contrary, Co proved to yield both HS and DS products, even slightly favouring the former. 1H-NMR was particularly helpful in the case of Co, to evidence all products of catalytic silylation (see ESI,† sections 1–4).
 | ||
| Fig. 3 2D 29Si–1H NMR spectra (700 MHz, methylcyclohexane (MCH) d14) using INEPT sequence to attribute all signals to the various reaction products. This example uses Ni(tmhd)2 as precatalyst (see Table 1 & corresponding scheme for conditions), selective for dehydrogenative silylation (DS). | ||
To further exemplify as well as compare Ni and Co, we summarized the catalytic performances of the precatalysts (1) to (4) in Table 1, using model reactions depicted in corresponding scheme. Reactions took place at 90 °C in neat substrates using 0.5 mol% of catalysts and with a ratio of SiH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiVi = 1:1 (mol ratio of MD′M:dvtms = 2:1 and for ratio of MD′M:vpmds = 1:1). All catalysts mainly yield alkene/vinyl silane dehydrogenative silylation (DS) giving also reduction products (Red) (entry 1, 3, 5 and 7 in Table 1 for dvtms), for both dvtms and vpdms substrate (comparison with entries 2, 4, 6 and 8 in Table 1 for vpdms). Cobalt catalysts yield some hydrosilylation product, which sets them apart from DS-selective nickel catalysts (entries 5–8 for Co compared to 1–4 for Ni), in particular for dvtms as substrate. However, only the Co(tmhd)2 precatalyst 4 is active for the silylation of vpdms. The acac-counterpart is likely quickly deactivated through formation of Co black (black residues are quickly observed in that case during catalysis).
:SiVi = 1:1 (mol ratio of MD′M:dvtms = 2:1 and for ratio of MD′M:vpmds = 1:1). All catalysts mainly yield alkene/vinyl silane dehydrogenative silylation (DS) giving also reduction products (Red) (entry 1, 3, 5 and 7 in Table 1 for dvtms), for both dvtms and vpdms substrate (comparison with entries 2, 4, 6 and 8 in Table 1 for vpdms). Cobalt catalysts yield some hydrosilylation product, which sets them apart from DS-selective nickel catalysts (entries 5–8 for Co compared to 1–4 for Ni), in particular for dvtms as substrate. However, only the Co(tmhd)2 precatalyst 4 is active for the silylation of vpdms. The acac-counterpart is likely quickly deactivated through formation of Co black (black residues are quickly observed in that case during catalysis).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Vinylsilane | Conversion (%) | Selectivity (% vinylsilane) | ||||
| MD′M | Vinylsilane | HS | DS | Red | Dim | |||
|
Reaction conditions: 90 °C, neat, 0.5 mol%, ratio SiH/SiVi = 1:1, 24 h reaction time. Conversion and selectivity determined by NMR analysis (see ESI,† sections 1–5).
|
||||||||
| 1 | (1) | Dvtms | 53 | 100 | 0 | 53 | 47 | 0 |
| 2 | Vpmds | 52 | 100 | 0 | 45 | 40 | 15 | |
| 3 | (2) | Dvtms | 52 | 100 | 0 | 52 | 48 | 0 |
| 4 | Vpmds | 47 | 100 | 0 | 43 | 43 | 14 | |
| 5 | (3) | Dvtms | 35 | 73 | 14 | 48 | 38 | 0 |
| 6 | Vpmds | 0 | 0 | 0 | 0 | 0 | 0 | |
| 7 | (4) | Dvtms | 35 | 73 | 14 | 48 | 38 | 0 |
| 8 | Vpmds | 30 | 45 | 70 | 15 | 15 | 0 | |

The catalytic activity for the hydrosilylation of 1-octene with MD′M was also investigated (ESI,† section 5) and all Ni and Co catalysts are mainly selective for isomerization. Notably, observed selectivities are different from Shimada and co-workers' publication22 where Ni(acac)2 is active for hydrosilylation with the use of NaHBEt3 as external reductant. Active-catalyst generation mechanism is thus probably different when the system is activated with a stronger external reducing agent than the inherent tertiary silane featuring H–SiMe(O–R)2 moieties.1–10
In order to study the catalyst activation mechanism, dvtms silylation was monitored in situ using 1H NMR at 90 °C with 12.5 mol% of Ni(tmhd)2 as catalyst and with a ratio of SiH:SiVi = 1:1 (molar ratio of MD′M:dvtms = 2:1). No catalytic intermediate has been observed when the catalyst is mixed with either MD′M or dvtms alone. Monitoring the silylation through an NMR-tube experiment has allowed us to follow the kinetics of the reaction, without significant perturbation from paramagnetism (see ESI,† sections 4 & 8), inferring specific environments for Ni(II) (since few environments are devoid of paramagnetism)32–36 and no reduction product (either paramagnetic for Ni(I) or even ferromagnetic for some nanoparticules of Ni(0)).37–39 We could evidence that the reaction is occurring under these conditions and some free ligand is released (small characteristic signals for keto/enol forms of free tmhd-derived 2,2,6,6-tetramethylheptane-3,5-dione can be observed on 1H-NMR spectra, see ESI,† sections 2, 4, 7 & 8). This was confirmed by pre-contacting the Ni complex with either Si–H or vinyl moieties under similar conditions in order to identify ligand-set alteration (see ESI,† section 2 & 4).
In his study, Marciniec assumed that Ni(acac)2 was reduced by the SiH from the silane before the Chalk–Harrod mechanism.1–3,28–30 From our study, it seems that the catalytic mechanism might be different, either needing both the silane and the alkene to reduce minute amounts of the precatalyst before either conventional or modified Chalk–Harrod mechanisms occurs (see Scheme 1). Reaction could also proceed through a concerted mechanism with neither fully-reduced species nor subsequent oxidative addition as suggested in recent reports,7–10 or alternatively bi-nuclear Ni(I) species might be at play.39
 | ||
| Scheme 1 Global putative mechanism explaining the formations of various products. The oxidation state of the metal and the generation of active species is omitted voluntarily since subjected to speculation (adapted from Marciniec1–3). | ||
In light of the absence of paramagnetism suggested by NMR spectroscopy, monitoring the color change of the reaction medium could suggest the formation of non-octahedral Ni(II) species via σ-bond metathesis, with release of acetylacetonate-type ligand (as clearly evidenced by 1H NMR for the tmhd ligand, see ESI,† sections 4, 7 & 8). The green Ni(acac)2 (due to octahedral geometry [paramagnetic] in the more favorable trimeric structure) and pink Ni(tmhd)2 (due to a monomeric square-planar geometry [diamagnetic] in that case)32–36 both rapidly turn bright orange, and NMR spectra do not display paramagnetic features (broad peaks or wide range of isotropic chemical shifts) even at high catalyst loading. This could suggest the formation of either square-planar, square-pyramidal trigonal or bipyramidal geometries with coordination of Si–H and vinyl moieties and subsequent release of one tmhd ligand as already suggested in the literature for such acetylactetonate-type ligands.1–10,28–30 The release of the acac ligand seems less clear in our study, which could be explained by a higher propensity towards hydrosilylation of the released acetylacetonate in the mixture making the detection complicated. This could also suggest a much easier nanoparticle formation (via complete reduction)37,38 in the case of Ni(acac)2, that could be detrimental to the reaction. That could explain the highest catalytic efficiency of the tmhd counterpart, which role would then not be limited to an enhanced solubility. It is likely that the increased sterics from the tert-butyl group play a role in the bidentate ligand release, and potentially hindered subsequent hydrosilylation, making its detection possible (see ESI,† sections 4, 7, 8 & 9 for pictures).
Co catalysis seems more difficult to rationalize, all systems are paramagnetic no matter the geometry and the darkening of the solution (see ESI,† section 9 – miscellaneous) could suggest reduction to Co(0) or Co(I) species, as previously proposed.10–12,40,41
In the second part of our study, we monitored the activity of these catalysts for the crosslinking of Si–H and Si-vinyl functionalized silicone oils. Harnessing alkene (hydro)silylation to cure silicone formulations is the cornerstone of silicone materials' manufacturing: multiple applications heavily rely on this reaction.1–10 The crosslinking is conventionally catalyzed by platinum complex that remain trapped in the final product, representing a large fraction of the cost of the material. As evidenced in Marciniec work,1–3,10,28–30 a catalyst which is selective for dehydrogenative silylation can become active for hydrosilylation as well when it reacts with functional polymeric chain, suggesting a selectivity switch that is substrate-dependent.
The crosslinking activity of the four precatalysts Ni(acac)2, Ni(tmhd)2, Co(acac)2 and Co(tmhd)2 was studied, and some curing parameters were changed to monitor the influence on the crosslinking time and on the final material properties. Crosslinking time is determined with a stop-stirring test (SST) consisting of mixing di-vinyl and poly-SiH silicone-oils with the catalyst heating in an oil bath and measuring time when the magnetic stirrer stops (see Experimental section). Here, we performed this test under non-inert atmosphere (presence of O2 and ambient moisture).31
Crosslinking of dvtms with poly-SiH oil MD′50M was first evaluated. The catalytic activity of complexes (1) to (4) is summarized in Table 2 (entries 1 to 4). Reference reactions were performed at 90 °C using 0.5 mol% of catalysts and with a ratio of SiH:SiVi = 1:1. Nickel catalysts are sluggish for the crosslinking of dvtms. On the contrary, cobalt catalysts are very efficient, and more particularly Co(acac)2 which allows a crosslinking in less than 15 minutes at 90 °C.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | y | Catalyst | Mol% | SiH:SiVi | T | SST |
| 1 | 0 | (1) | 0.5 | 1 | 90 | 3 h |
| 2 | 0 | (2) | 0.5 | 1 | 90 | 3 h |
| 3 | 0 | (3) | 0.5 | 1 | 90 | 15 min |
| 4 | 0 | (4) | 0.5 | 1 | 90 | 40 min |
| 5 | 20 | (1) | 1 | 3 | 110 | 30 min |
| 6 | 20 | (2) | 1 | 3 | 110 | 15 min |
| 7 | 20 | (3) | 1 | 3 | 110 | 25 min |
| 8 | 20 | (4) | 1 | 3 | 110 | 40 min |

The crosslinking of di-vinyl PDMS oil with poly Si–H oil was also surveyed and results are compiled in Table 2. Reference reactions were done at 110 °C using 1 mol% of catalyst and with a ratio of SiH:SiVi = 3:1, to ensure efficient crosslinking even through undesired reactions. All four catalysts are active for the crosslinking of di-vinyl and poly-SiH oil under non-inert atmosphere (entries 5 to 8, Table 2). Contrary to the results with dvtms (y = 0), nickel catalysts are more active than cobalt catalysts for more viscous silicone oils. Dvtms possesses a specific chelating structure and is the bidentate ligand of choice for Karstedt's type catalysts.5,42 Difference between Co- and Ni-based catalyst activities may arise from a variation of dvtms coordination on the active center, precluding activity in the case of Ni.3,42 It is possible that the chelating nature of dvtms is detrimental to the reactivity of Ni catalysts, maybe suggesting the formation of a stable octahedral Ni(II) complex, no longer able to interact efficiently with the Si–H moiety.
To evidence that crosslinking is really occurring, and to evaluate further crosslinking kinetics and compare gel properties, rheology was performed for Si–H and Si-vinyl PDMS oil curing. The catalytic crosslinking of di-vinyl and poly-SiH PDMS oils with complexes (1) to (4) is presented in Fig. 4. Reactions took place at 110 °C using 1 mol% of catalyst and with a selected ratio of SiH:SiVi = 3:1. First, we systematically observed an intersect of G′ and G′′ suggesting that crosslinking is effective, with gel times apparently shorter than observed during SST measurements. Ni catalysts are more active under this set of conditions and yield materials with slightly higher G′ moduli (close to 105 Pa for a reference formulation of Table 2, see also ESI,† sections 7 & 8). Co(acac)2 is less active as expected from SST, probably due to higher air instability leading to catalyst degradation, and gives lower modulus gel (∼103 Pa). Co(tmhd)2 has a gel time in accordance with SST and modulus higher than 104 Pa and consistently close to 105 Pa. Premature oxidation might be detrimental to Co catalysis. Adventitious contaminants in silicone oils might also be responsible for the lower efficiency of a slightly more sensitive Co catalysis, which could also explain the delay in crosslinking observed by rheometry.
 | ||
| Fig. 4 Rheology curves following crosslinking of di-vinyl and poly-SiH PDMS oil. Reaction conditions: 110 °C, 1 mol%, ratio SiH/SiVi = 3:1. Blue disks mark the intersects between G′ & G′′, characteristic of crosslinking. | ||
Interestingly DSC can be used to monitor these crosslinking reactions. Thermal kinetics of the catalyzed crosslinking of di-vinyl and poly-SiH PDMS oils was monitored using DSC analysis. Crosslinking of dvtms with poly-SiH oil MD′50M was first studied. Reference reactions were heated at 8 °C min−1 rate, using 0.5 mol% of complexes (1) to (4) with a ratio of SiH:SiVi = 1:1 (see ESI,† section 6 Fig. S21).
Crosslinking catalyzed by cobalt complexes starts at a lower temperature comparing to nickel complexes, and this is in accordance with the crosslinking time measured with SST (see Experimental section and ESI† section 8 for values of SST for crosslinking at 90 °C, Table S4† caption). Catalysts bearing the acac ligand set have a sharper reaction profile than with tmhd ligands, which reaction profile is broad. This activation could be linked to the removal of the acetylacetonate molecule during activation with the Si–H substrate in the presence of Si-vinyl moieties; with the ligand substitution being easier with the less bulky analogue.
Crosslinking of di-vinyl PDMS oil with poly-SiH oil MD′50M was also surveyed. Reference reactions were performed at a heating rate of 8 °C min−1, using 1 mol% of complexes (1) to (4) with a ratio of SiH:SiVi = 2:1 and thermograms are presented in Fig. S22 (see ESI†). Under these conditions, crosslinking catalyzed by Co(acac)2 starts at a lower temperature but the enthalpy is less important than with the other catalysts, suggesting incomplete reaction and possible catalyst decomposition due to air exposure. Overall, and contrary to dvtms crosslinking, Ni precatalysts seem to outperform Co counterparts for longer oil-based formulations. This could also be a consequence of the increased sensitivities of Co complexes.
DSC thus unveils that Co is more reactive at lower temperatures than Ni for dvtms formulations (see ESI,† section 6), probably due to the aforementioned chelating nature of dvtms, deactivating Ni (pre)catalysts. However, this statement does not hold true for longer oil-based industrial formulations with spaced-apart vinyl moieties.
These DSC results are consistent with rheology experiments with the observed G′ modulus is lower in the case of Co(acac)2 than with the other catalysts. Initiation temperatures are then lower for Ni catalysts than for Co(tmhd)2 for crosslinking (see ESI,† Fig. S22) and reaction profiles are very similar for these three catalysts confirming the rheology findings. These observations are in agreement with what was suggested by Marciniec:1,3–30 reaction kinetics and selectivities are strongly substrate-dependent and (hydro)silylation model reactions behave differently than crosslinking of silicone-based oils.3,43 Model reactions are thus interesting to rationally design regio- and stereoselective catalyst for silylation reactions, but crosslinking of polyfunctional oils might yield different product selectivities due to unique sterics and diffusion constraints. Therefore, another method is necessary to assess the different products when crosslinking is considered.
High-resolution magic angle spinning NMR (HR-MAS NMR) spectroscopy can advantageously characterize gels displaying satisfactory mobility, bridging the gap between liquid NMR and solid-state NMR. It consists in running liquid-like sequences in a rotor spun at high frequency at the magic angle to retain proper resolution (see ESI,† sections 7 & 8).
Very interestingly, HR-MAS NMR of crosslinking formulations allowed us to get a better understanding of the selectivities taking place in these 3D-polymer networks. If Ni still promotes DS linkage as a major crosslinking knot, Co displays a higher propensity towards HS linkage in the crosslinking of functionalized-polymer oils (see Fig. 5 and ESI† – [HR-MAS NMR analyses of crosslinking of di-vinyl PDMS and poly-SiH oils], section 7).
 | ||
| Fig. 5 HR-MAS NMR spectroscopy: comparison for all catalysts of a) 1H-NMR-HR-MAS spectra and b) 13C-NMR-HR-MAS spectra. Gels swollen with CDCl3 (see HR-MAS section 7, Fig. S43 and S44 for overlays, ESI†). | ||
More specifically, Fig. 5 clearly depicts a very selective Co(tmhd)2 precatalyst yielding species promoting hydrosilylation of vinylsilane moieties from the functional silicone oil. Co(acac)2 seems a bit less selective, since some DS product can be identified. Moreover, its lesser activity, probably due to premature deactivation precludes an efficient use in such systems. This ligand dependency could suggest that the active species might retain one acetylacetonate-type ligand, akin postulated species for Pt(acac)2-based precatalytic systems.43,44 It could also be the result of Co-based nanoparticles/colloids (Co(0)/Co(I) reduced species) formed in the case of Co(acac)2 that would induce a different catalytic pathway.11,12,38,40,41
On the contrary, Ni precatalysts still promote the DS pathway, with identifiable characteristic olefinic protons (Fig. 5a) and carbons (Fig. 5b) as well as Red associated products (aliphatic regions on both overlays). Noteworthy is that Ni(tmhd)2 and Ni(acac)2 seem to be leading to very similar selectivities, in agreement with the model reactions (see Table 1), with the obvious solubility edge of the tmhd-based analogue in silicone formulations.
The HR-MAS methodology could further be harnessed to identify new systems amenable to selectively yield new silicone networks from anti-Markovnikov & Markovnikov products,45 or develop new catalysts based on these plateforms.46,47
Conclusions
We have evaluated the activity and selectivity of nickel and cobalt acetylacetonate-based complexes as precatalysts for silylation and crosslinking using Si–H and Si-vinyl functionalized substrates without external reducing agents and under non-inert atmosphere. Those complexes promote the in situ formation of competent catalysts featuring different activities/selectivities depending on the chosen metal and ligand set. This choice consequently affects the crosslinking kinetics/efficiency and the mechanical properties of the final materials. Ni complexes promote dehydrogenative silylation selectively. Co complexes yield both hydrosilylation and DS products for model compounds, whereas the HS product is favored for crosslinking of functionalized oils, which was evidenced harnessing HR-MAS NMR spectroscopy. Co acetylacetonate-based complexes also tend to display activity at lower temperatures than Ni, while being more sensitive to non-inert conditions. When silicone curing formulations are employed, Ni(tmhd)2 is the most active precatalyst for crosslinking of silicone oils under non-inert atmosphere, probably due to an enhanced solubility in silicone media. The activation of these high-oxidation state complexes does not seem to proceed via Si–H-promoted reduction. Further investigations are currently underway to pinpoint the active-catalyst generation mechanism. Finally, these thermally-induced crosslinking reactions could prove key to develop efficient “one-pack” formulations, stable at room temperature for extended shelf lives and that could be triggered to form the silicone mastics/putties when heated.Experimental
General considerations
Reaction were performed under non-inert atmosphere. All commercially available reagents were used as received. Silicon oils were kindly provided by Elkem Silicones©. Silicon oils were degassed under vacuum, to remove traces of volatiles from synthetic process. All reaction vials were loaded with precisely weighted (0.1 mg precision) substrate and catalyst amounts, without precautions regarding the atmosphere (presence of O2 and ambient moisture). The chemicals were obtained from the following suppliers: Ni(acac)2 and Ni(tmhd)2 from Strem, Co(tmhd)2 from Alfa Aesar, Co(acac)2 from Sigma, 1-octene from Sigma, and CDCl3 and methylcyclohexane (MCH) d14 from Eurisotop. Elkem Silicones© provided divinyltetramethylsiloxane (dvtms), (MD′M), and all silicone oils. Vpmds was sourced from abcr.General procedure for catalytic silylation reactions
A 20 mL reaction vial was loaded with the catalyst (0.045 mmol) together with the chosen alkene/vinyl (9 mmol) and MD′M (9 mmol). Dodecane (standard) was added for GC quantification. The solution was stirred and heated to 90 °C for 24 hours. During the course of reaction, aliquots were withdrawn for analysis. After the reaction, NMR and and/or GC/GC-MS analyses were performed. 1H, 13C and 29Si NMR spectra were recorded on a Bruker 400 MHz and 700 MHz spectrometers in CDCl3 and MCH d14. The mass spectra of the products were evaluated by gas chromatography coupled with mass spectrometry (GC-MS) and quantification was determined with gas chromatography (GC) analysis using calibration curves with appropriate substrates. GC was performed on an HP 6890 instrument using a HP5 column made of 5% of diphenyl and 95% dimethyl polysiloxane (32 m length, 0.32 mm diameter, 0.25 μm film) and a flame ionization detector. The injection volume was 1 μL and nitrogen was used as mobile phase with a flow of 1 mL min−1. The temperature program was as follow: 40 °C plateau for 5 min; heating to 250 °C at 10 °C min−1; 250 °C plateau for 5 min. GC-MS analysis was performed on an Agilent 6850 instrument equipped with a HP5 column made of 5% of diphenyl and 95% dimethyl polysiloxane (32 m length, 0.32 mm diameter, 0.25 μm film) and an Agilent 5975C EI detector. The same temperature program as GC was used for GC-MS analysis. Alkene/vinyl silane conversions and products' selectivities are calculated from 1H NMR and double-checked using GC/GC-MS analysis (see ESI,† sections 1–5) in particular for 1-octene.General procedure for silicone oil crosslinking
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Elkem Silicones© for providing raw materials and funding for MP's and JD's PhD projects. DC thanks the FUI ECOMAT for funding her post-doctorate. JR and VM also thank Roger Spitz for enlightening discussions and Franck Collas for his DSC expertise, Sébastien Norsic for his technical support as well as Carlos Fernández de Alba Encinas for his HR-MAS NMR expertise.Notes and references
- B. Marciniec, Coord. Chem. Rev., 2005, 249(21), 2374–2390 CrossRef CAS.
- B. Marciniec, Comprehensive Handbook on Hydrosilylation - 1st Edition, Pergamon Press, 1992 Search PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, in “Hydrosilylation: A Comprehensive Review on Recent Advances Springer”, 2009, vol. 1 Search PubMed.
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79(4), 974–979 CrossRef CAS.
- B. Karstedt, US Pat., 3715334A, 1970 Search PubMed.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS , (and references within).
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5(26), 20603–20616 RSC , (and references within).
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255(13–14), 1440–1459 CrossRef CAS , (and references within).
- A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef , (and references within).
- L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed , (and references within).
- M. Jakoobi, V. Dardun, L. Veyre, V. Meille, C. Camp and C. Thieuleux, J. Org. Chem., 2020, 85(18), 11732–11740 CrossRef PubMed.
- M. Jakoobi, V. Dardun, C. Camp and C. Thieuleux, Catal. Sci. Technol., 2021, 11, 3176–3181 RSC.
- A. Sanagawa and H. Nagashima, Organometallics, 2018, 37(17), 2859–2871 CrossRef CAS , Advance Article.
- A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335(6068), 567–570 CrossRef CAS PubMed.
- C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136(34), 12108–12118 CrossRef CAS PubMed.
- R. Arevalo and P. J. Chirik, J. Am. Chem. Soc., 2019, 141(23), 9106–9123 CrossRef PubMed.
- A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hoyt, K. J. Weller, S. A. Nye, K. M. Lewis, J. L. Boyer and J. G. P. Delis, Organometallics, 2012, 31(13), 4886–4893 CrossRef CAS.
- M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356(2–3), 584–590 CrossRef CAS.
- K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31(10), 3825–3828 CrossRef CAS.
- B. Raya, S. Jing and T. V. RajanBabu, ACS Catal., 2017, 7(4), 2275–2283 CrossRef CAS PubMed.
- X. Chen and Z. Lu, Org. Lett., 2016, 18(18), 4658–4661 CrossRef CAS PubMed.
- V. Srinivas, Y. Nakajima, W. Ando, K. Sato and S. Shimada, J. Organomet. Chem., 2016, 809, 57–62 CrossRef CAS.
- T. Diao and P. J. Chiriket al., Patents WO2015077306, WO2015171881, WO2015077298, 2015.
- J. Boyer and A. K. Roy, WO2014186513 (A1), 2014.
- D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138(8), 2480–2483 CrossRef CAS PubMed.
- Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139(5), 1798–1801 CrossRef CAS PubMed.
- C. H. Schuster, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2016, 6, 2632–2636 CrossRef CAS.
- B. Marciniec, H. Maciejewski and J. Mirecki, J. Organomet. Chem., 1991, 418(1), 61–67 CrossRef CAS.
- B. Marciniec and H. Maciejewski, J. Organomet. Chem., 1993, 454(1), 45–50 CrossRef CAS.
- B. Marciniec, H. Maciejewski and I. Kownacki, J. Mol. Catal. A: Chem., 1998, 135(3), 223–231 CrossRef CAS.
- V. Monteil, J. Raynaud, M. Bousquie, D. Crozet and S. Marrot, Patents WO2016071651 and WO2016071652, 2016.
- G. J. Bullen, R. Mason and P. Pauling, Nature, 1961, 189, 291–292 CrossRef CAS.
- G. J. Bullen, R. Mason and P. Pauling, Inorg. Chem., 1965, 4(4), 456–462 CrossRef CAS.
- Ö. Metin, L. Tatar Yıldırım and S. Özkar, Inorg. Chem. Commun., 2007, 10(9), 1121–1123 CrossRef.
- Md. Kudrat-E-Zahan, Y. Nishida and H. Sakiyama, Inorg. Chim. Acta, 2010, 363(1), 168–172 CrossRef CAS.
- F. Emmenegger, C. W. Schlaepfer, H. Stoeckli-Evans, M. Piccand and H. Piekarski, Inorg. Chem., 2001, 40(16), 3884–3888 CrossRef CAS PubMed.
- T. Galeandro-Diamant, I. Suleimanov, L. Veyre, M. Bousquié, V. Meille and C. Thieuleux, Catal. Sci. Technol., 2019, 9, 1555–1558 RSC.
- J. M. Asensio, D. Bouzouita, P. W. N. M. van Leeuwen and B. Chaudret, Chem. Rev., 2020, 120(2), 1042–1084 CrossRef CAS PubMed.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 137(18), 6104–6110 CrossRef CAS PubMed.
- W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119(4), 2876–2953 CrossRef CAS PubMed.
- Y. Gao, L. Wang and L. Deng, ACS Catal., 2018, 8(10), 9637–9646 CrossRef CAS.
- H. Maciejewski, B. Marciniec and I. Kownacki, The Ni equivalent of Karstedt's Pt catalyst is an efficient catalyst of DS for very limited substrates, J. Organomet. Chem., 2000, 597(1–2), 175–181 CrossRef CAS.
- R. J. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9(10), 534–571 CrossRef PubMed.
- F. D. Lewis and G. D. Salvi, Inorg. Chem., 1995, 34, 3182–3189 CrossRef CAS.
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8(10), 9865–9876 CrossRef CAS , (and references within).
- V. Monteil, J. Raynaud, M. Bousquie, D. Crozet and S. Marrot, Patent WO2016071654, 2016.
- A. Kownacka, I. Kownacki, B. Marciniec, B. Nguyen, A. Surgenor, R. Taylor and M. S. Tzou, US2014296468 (A1), 2014.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00834j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |