
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatial activity profiling along a fixed bed of powder catalyst during selective oxidation of propylene to acrolein†
Matthias
Stehle
 a,
Thomas L.
Sheppard
ab,
Michael
Thomann
c,
Achim
Fischer
c,
Heino
Besser
d,
Wilhelm
Pfleging
d and
Jan-Dierk
Grunwaldt
*ab
a,
Thomas L.
Sheppard
ab,
Michael
Thomann
c,
Achim
Fischer
c,
Heino
Besser
d,
Wilhelm
Pfleging
d and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Engesserstr. 20, 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cEvonik Operations GmbH, Rodenbacher Chaussee 4, 63457 Hanau-Wolfgang, Germany
dInstitute for Applied Materials - Applied Materials Physics, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
First published on 3rd June 2021
Abstract
Spatial profiling of the reactant and product concentration including the gas phase temperature during the selective oxidation of propylene to acrolein along a catalyst bed allowed locating and distinguishing between specific processes occurring in each individual point of a chemical reactor. For this purpose, a lab-scale testing setup capable of resolving concentration and temperature gradients in a fixed-bed reactor was developed. The local gas phase composition and temperature were determined using a sampling capillary and mass spectrometry along a multicomponent Bi–Mo–Co–Fe oxide catalyst bed during selective oxidation of propylene to acrolein under high conversion conditions. In this way, the reaction progress in terms of conversion, selectivities and yields along the reactor was revealed. While ca. 66% of the integral propylene conversion occurred in the first third of the catalyst bed with high selectivity towards acrolein, the latter third of the bed was dominated by the formation of acrylic acid and CO2 as further and total oxidation products, respectively. Acrylic acid, which originates from the sequential oxidation of propylene to acrolein, was the by-product with the highest yield and especially formed above 440 °C. CO and CO2 were observed directly from propylene, along with consecutive pathways of propylene oxidation, which favor CO2 formation. The numerous insights obtained by even a single profile highlight the strong capabilities of spatially resolved activity and temperature measurements for diagnostics of packed-bed reactors and identifying the reaction pathways occurring within.
Introduction
Selective oxidation reactions are highly relevant in the chemical industry as they form the basis of numerous chemical production processes.1,2 A prominent example is the oxidation of propylene to acrolein, which serves as an intermediate for e.g., acrylic acid and methionine.3–5Alongside continuous improvements on catalytic systems, several mechanistic and kinetic investigations have been performed to obtain a detailed understanding of selective propylene oxidation.5,6 It is generally accepted that the initial α-H abstraction leading to a symmetric π-allyl intermediate is the rate limiting step.7–13 Subsequently, a second H abstraction and insertion of oxygen take place. For bismuth molybdate based catalysts, which represent the majority of propylene oxidation catalysts in use today,1418O labeling studies have shown that the inserted oxygen originates from the catalyst itself (bulk oxygen).6,15–18 Finally, the reduced catalyst is reoxidized by gas phase oxygen. Hence, the oxidation reaction from propylene to acrolein proceeds via a Mars-van Krevelen mechanism19 with nucleophilic lattice oxygen as the oxidizing agent.14,20 Besides the target product acrolein, various by-products originating from parallel and/or consecutive reactions are known. Thus, complex reaction networks20–27 have been proposed to elucidate the origin of by-products including carbon monoxide, carbon dioxide, acrylic acid, acetic acid, acetaldehyde, formaldehyde, and others.
From an industrial point of view, knowledge of reaction networks is essential to understand the processes taking place inside the reactor. This further allows optimization of process parameters and reactor design for efficient and selective synthesis of desired products. In many cases, such reaction networks are built by a combination of simulation and kinetic measurements. In the laboratory, such kinetic datasets are typically obtained in ideal plug flow reactors operated under differential conditions (i.e., low conversion) to prevent gradients along the catalyst bed.28 However, such conditions may not accurately represent industrial reactors, which may be operated at high conversion for economic reasons. This situation is especially problematic if existing gradients (e.g., heat, concentration, pressure) also affect the structural properties or stability of the catalyst.29
Hence, efforts were made to follow the reaction progress along the catalytic bed,30–33 so that deeper knowledge about reaction networks could be obtained. An overview of bench- and lab-scale testing units capable of resolving concentration and/or temperature gradients along the catalyst bed is provided by Morgan et al.29 Many of the developed devices were designed to obtain profiles along structured catalyst bodies such as monoliths,31,34–36 foams,33,37,38 spheres,39,40 or pellets.41 Horn et al. have presented spatially resolved investigations fulfilling different experimental needs (e.g., high pressure and temperature reactors,38 profile reactors combined with Raman spectroscopy,39,40 pilot scale reactors41) and employing the results for reactor modelling. Very recently, they could even resolve concentration profiles in a single porous pellet.42
However, there are very few studies dealing with a fixed bed of powder sieve fractions, even though this shape is often used in early catalyst research studies.29 Touitou et al.43–45 developed the first setup for axially resolved profiles along a packed bed of sieved powder and demonstrated its application in the model reaction of CO oxidation over a Pd/Al2O3 catalyst.43 Finally, the measured temperature profile of a Pt/Al2O3 catalyst was employed for a semi-empirical model, which was able to depict the measured concentration profiles far better than the fully empirical one.46
These examples underline how spatially resolved data provides a powerful alternative to the ‘black box’ interpretation assumed by conventional measurements in catalytic reactors, which are advantageous to validate or develop corresponding reactor models.29 In general, such data are rare in the case of selective oxidation of hydrocarbons.41 Nevertheless, Ganzer and Freund27 recently showed that spatially resolved, non-isothermal data enable the development of detailed reaction networks for selective propylene oxidation. The data were obtained during complex experiments in a pilot plant with irregularly positioned side-inserted sampling devices. Alternatively, such data could be collected in lab reactors that are easier to handle and operate with flexible reaction conditions, then later compared to real data on pilot/industrial plant reactors.
This study demonstrates activity and temperature profiling along a fixed-bed lab reactor during selective oxidation of propylene to acrolein under industrially relevant conditions (i.e., high conversion). For this purpose, a dedicated, multifunctional setup capable of resolving gradients along a fixed bed of a sieved powder fraction was developed. It enables the simultaneous determination of the local gas phase composition and temperature in the axial direction, together with integral activity measurements for comparison. Based on the obtained concentration profiles, the reaction progress in terms of propylene conversion as well as product selectivities and yields was resolved. Moreover, (by-)product formation was investigated with respect to propylene conversion, concentration of substances, and reaction temperature, thus enabling fundamental validation of proposed reaction networks. Thereby, an improved understanding of the various processes taking place inside the reactor is provided and possibilities for process optimization are envisioned.
Experimental
Setup for axially resolved profile measurements of the gas phase composition and temperature
For the acquisition of spatially resolved concentration and temperature profiles along packed beds of sieved powder catalysts, a capillary sampling setup inspired by those described in the literature was developed.31,33,35,37,38,43,44 Subsequently, it was implemented in an existing lab-scale testing unit, which is described in detail elsewhere.47 The reactor schematic and the investigated packed bed are shown in Fig. 1.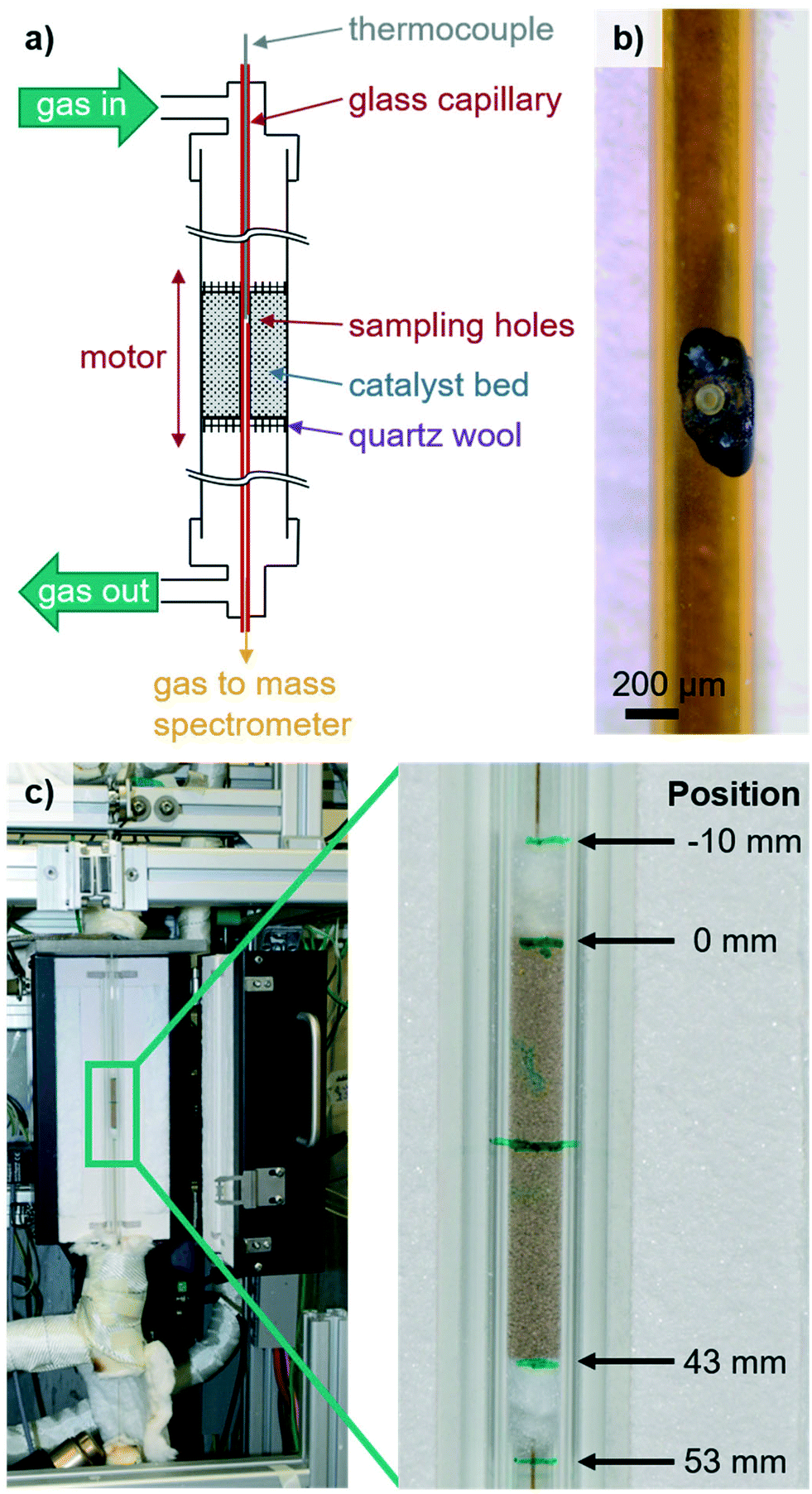 | ||
| Fig. 1 Scheme of the setup for simultaneous acquisition of gas phase concentration and temperature profiles along fixed catalyst beds (a), light microscope image of the glass capillary used for local sampling showing the laser drilled sampling holes (ø = 50 μm) with black marking for alignment during mounting (b), and photo of the setup with zoom in onto the catalyst bed with inserted sampling capillary and selected position indications (c). | ||
The developed apparatus (scheme given in Fig. 1a) enables the simultaneous determination of integral (via gas chromatography (GC)) and local catalytic activity (via mass spectrometry (MS)) as well as the acquisition of the local temperature of the gas phase at the sampling spot (via a thermocouple) within a single experiment. As shown in Fig. 1c, the setup consists of a quartz glass reactor (øouter = 10 mm, øinner = 6 mm, l = 33 cm) heated by and fixed to a small oven (l = 27 cm, HTM Reetz GmbH). The oven is mounted on a vertically operated linear stage (X-LRT0100AL-E08C, Zaber Technologies Inc.) to move the oven/reactor unit along the fixed sampling capillary. Reaction gases are supplied via mass flow controllers (Bronkhorst). The oxygen concentration at the outlet is monitored by an on-line oxygen sensor (PAROX 1200 H, MBE AG) and product gases are analyzed using an on-line GC (7890B, Agilent). All gas lines are heated to 220 °C to prevent product condensation (e.g., water, acrolein, acrylic acid).
For local sampling, a glass capillary (øouter = 450 μm, øinner = 320 μm, TSP320450, Polymicro Technologies) with two opposing orifices (ø = 50 μm, see Fig. 1b) is inserted. As the capillary diameter is in the same size range as individual particles, the impact of the sampling system is minimal.45 This is considered the best compromise between brittleness and an acceptable influence in the reactor. For profile acquisition, the sampling capillary is fixed at both ends by external mountings (stainless steel, VICI). During catalytic tests, a type-K thermocouple (ø = 150 μm, l = 400 mm, Reckmann GmbH) is inserted from the top end of the capillary and the tip aligned with the sampling orifices, while the bottom end is connected to a mass spectrometer (ThermoStar GSD320, Pfeiffer Vacuum GmbH) to determine the local gas phase composition. Thus, spatially resolved concentration and temperature profiles are acquired simultaneously at the same spot.
Junctions between the capillary and reactor or external holders are sealed by graphite ferrules (VICI). The bottom end of the capillary outside the reactor is heated by a hot air blower. Connections of the movable oven/reactor system to gas dosing and integral on-line analytics are realized by stainless steel convoluted hoses (Swagelok) heated via heating cords. Hence, gas-tight operation at elevated temperatures is achieved while the whole reactor/oven system is moved via the linear stage along the fixed sampling capillary for profile acquisition.
Processing of the glass capillary
Ultrafast laser structuring is applied to generate micro-structures without crack formation or significant heat impact on the surrounding material. Furthermore, it offers the possibility of contactless rapid manufacturing of functional designs with high flexibility and process reliability.48,49 The lateral holes of the glass capillary were therefore produced using an ultrafast fiber laser (Tangerine, Amplitude Systèmes) with a pulse length of 400 fs, a laser wavelength of 1030 nm, and an average laser power of 4.5 W. For drilling of each hole, a rotating laser hatch of 10 μm line pitch within a 50 μm circle contour was applied. The used glass capillary with the corresponding laser-drilled holes is shown in Fig. 1b.Catalytic testing
The concentration and temperature profiles were recorded on a Bi–Mo–Co–Fe oxide catalyst prepared by co-precipitation and spray drying, thus representing a representative catalyst model system. The catalyst powder was pressed, granulated, and sieved to a fraction of 300–450 μm.First, quartz glass wool was loaded into the reactor to fix and center the sampling capillary. Then, 1.60 g of the sieved catalyst was added and subsequently the other end of the catalyst bed was fixed with quartz glass wool, resulting in a catalyst bed length of 43 mm (c.f.Fig. 1c). After mounting the reactor, the sampling capillary was fixed to the external holders, the thermocouple was inserted and the tip aligned with respect to the sampling holes, and finally the MS was connected. Subsequently, the catalyst was activated and integral performance tests at Toven = 345 °C, 380 °C and 400 °C (N2/O2/C3H6/H2O = 70/14/8/8 vol%, WHSVC3H6 = 1.14 h−1) were performed for internal comparison. Due to the small catalyst sieve fraction, the small capillary size and as the integral performance during these tests was in line with the results obtained where no capillary was inserted (±3%), the influence of the capillary on catalytic performance (i.e., propylene conversion and (by-)product selectivity) is considered minimal. Although there is a (local) disturbance of the flow properties (e.g., wall effect, sucking of gas) by insertion of a sampling capillary compared to conventional integral testing without a capillary, we could not observe a significant effect of this disturbance on catalytic performance.
Finally, the total gas flow was reduced to reach high propylene conversion (>90%). After a total of 5 days on stream and after reaching a stable integral catalytic performance at high propylene conversion (c.f. ESI†), profile acquisition was performed (N2/O2/C3H6 = 78/14/8 vol%, WHSVC3H6 = 0.29 h−1, Toven = 400 °C). The reactor was moved stepwise (Δx = 3 mm) along the holes of the sampling capillary from 9 mm in front (position −9 mm) to 11 mm behind the catalyst bed (position 54 mm), resulting in 22 individual measurement points (c.f. ESI†). Data acquisition at each measurement point lasted for 20 min, resulting in a total time of ∼7.5 h for a single profile. At the same time, the integral activity was continuously monitored by on-line GC (c.f. ESI†).
Calculation of the concentration, temperature, and reaction progress profiles
To visualize the concentration and temperature profiles, the time-logged temperature and MS data were correlated according to the position log of the translation stage (see ESI†). At each measurement point, temperature and MS signals for the final five minutes of the 20 min acquisition period were averaged (60 temperature points, 5–6 points for each m/z ratio, see ESI†). For MS data evaluation, preferentially unique fragments for each species (e.g., m/z = 22 for CO2) or fragments with a low contribution from other species (e.g., m/z = 42 for propylene) were selected. The fragmentation pattern for the reaction mixture and an overview of the selected m/z ratios are given in the ESI.†Subsequently, the averaged signals of the three measurement points in front of the catalyst bed (−9, −6, −3 mm) were correlated to the bypass composition (i.e., 0% propylene conversion), and the averaged signals for the last three measurement points after the catalyst bed (48, 51, 54 mm) were correlated to the averaged integral activity (determined by GC, compositions given in the ESI†). In this way, MS device specific sensitivity factors for each m/z ratio were obtained, leading finally to the quantified concentration profiles reported in the results part (additional profiles are given in the ESI†).
As the amount of water was not quantified by GC, it was calculated based on the stoichiometry of the individual reactions (c.f. ESI†). The reaction progress along the catalyst bed was calculated on the basis of the individual amounts of the species. Local reaction rates, which report the conversion/formation of species between two consecutive points, were derived from the concentration profiles with respect to the total gas flow (c.f. ESI†). Propylene conversion was calculated with respect to the propylene amount at the first measurement point (−9 mm). The selectivity towards individual products was calculated based on the product amounts at each measurement point. More information about the calculations is provided in the ESI.†
Results and discussion
Setup for concentration and temperature profiling of fixed-bed reactors
The setup was designed to perform both conventional integral testing and to acquire axially resolved profiles. Thus, obtained conventional (i.e., integral) testing results can be directly compared to those where local sampling was performed. The reactor is positioned vertically with the gas flow from top to bottom to reduce the risk of bypassing of the gases and the risk of swirling of the catalyst grains in the case of shrinking of the bed. Heating via an oven ensures uniform heat input along the catalyst bed, which is considered problematic if hot air blowers are used,50,51 especially in the case of larger reactors as in the present study.A sampling capillary fixed at both ends was used as it allows the acquisition of multiple profiles within one testing run (e.g., different reaction conditions) and exhibits higher stability during movement of the bed. In general, glass capillaries enable easy visual alignment of the thermocouple within the capillary but exhibit a higher risk of rupture compared to steel capillaries. Sampling via side orifices reduces the risk of gas turbulence and backmixing39 as well as blocking of the orifices compared to open-end capillaries.43 As integral and local activity measurements are performed simultaneously, possible changes during the course of profile acquisition (e.g., catalyst deactivation) can be resolved. This is especially relevant for time consuming profile acquisitions, i.e., high spatial resolution profiles.
In order to obtain profiles under industrially relevant process conditions, selective propylene oxidation over a multicomponent Bi–Mo–Co–Fe oxide catalyst was investigated. This multicomponent system is known for its superior catalytic activity compared to the basic system of bismuth molybdates.5,6 Prior to acquiring temperature and concentration gradients along the bed, the catalyst reached a quasi-steady state as tracked by the on-line oxygen sensor (ΔO2 ≤ 0.4% relative to O2 concentration) and subsequently confirmed by integral GC measurements.
During the course of the following profile acquisition (∼7.5 h), the variation in integral amounts of the species determined by on-line GC was less than ±5% relative to the individual amounts (c.f. ESI†). The deviation of integral propylene conversion, product selectivities and product yields was less than 3% relative to the individual values. Thus, there was no statistically meaningful deactivation of the catalyst during profile acquisition.
The local monitoring by MS enabled an on-line visualization of gas phase equilibration within the sampling system (c.f. ESI†) with high time resolution (∼1 min for all m/z ratios acquired) compared to GC data acquisition (∼20 min for a single run). Hence, the stepwise movement of the catalyst bed could be clearly observed in the MS data. This allowed a direct assessment of profile acquisition parameters (e.g., spatial resolution, acquisition time per step). In general, the use of a MS as a local sampling device enables a high time resolution, and thus a more detailed look into the catalytic reactor. However, in the present study, the time resolution was limited by the equilibration time of the acrylic acid signal (m/z = 72) even though all gas lines were carefully heated. On the other hand, GC offers easy deconvolution and quantification of the gas phase composition by choosing appropriate column and detector combinations, but with lower time resolution.
As expected, the respective ratios of individual species measured at the positions in front (−9, −6, and −3 mm) and behind the catalyst bed (45, 48, 51, and 54 mm), respectively, were constant (c.f.Fig. 2). The reactant concentration only decreased as soon as the gases were in contact with the catalyst bed. In fact, reactant conversion and product formation were exclusively observed within the catalyst bed. Furthermore, the hotspot of the reaction was at the same position as the highest gradients in propylene and oxygen concentration (6–9 mm). Thus, the heat load was highest where the highest reaction rate of the exothermic oxidation of propylene occurred (c.f.Fig. 2). Moreover, the calculated carbon balance along the catalyst bed closed within 99 ± 3% (for carbon balance of each profile point see Fig. S4c, ESI†), which shows that most probably all main product species relevant for the overall catalytic performance were detected.
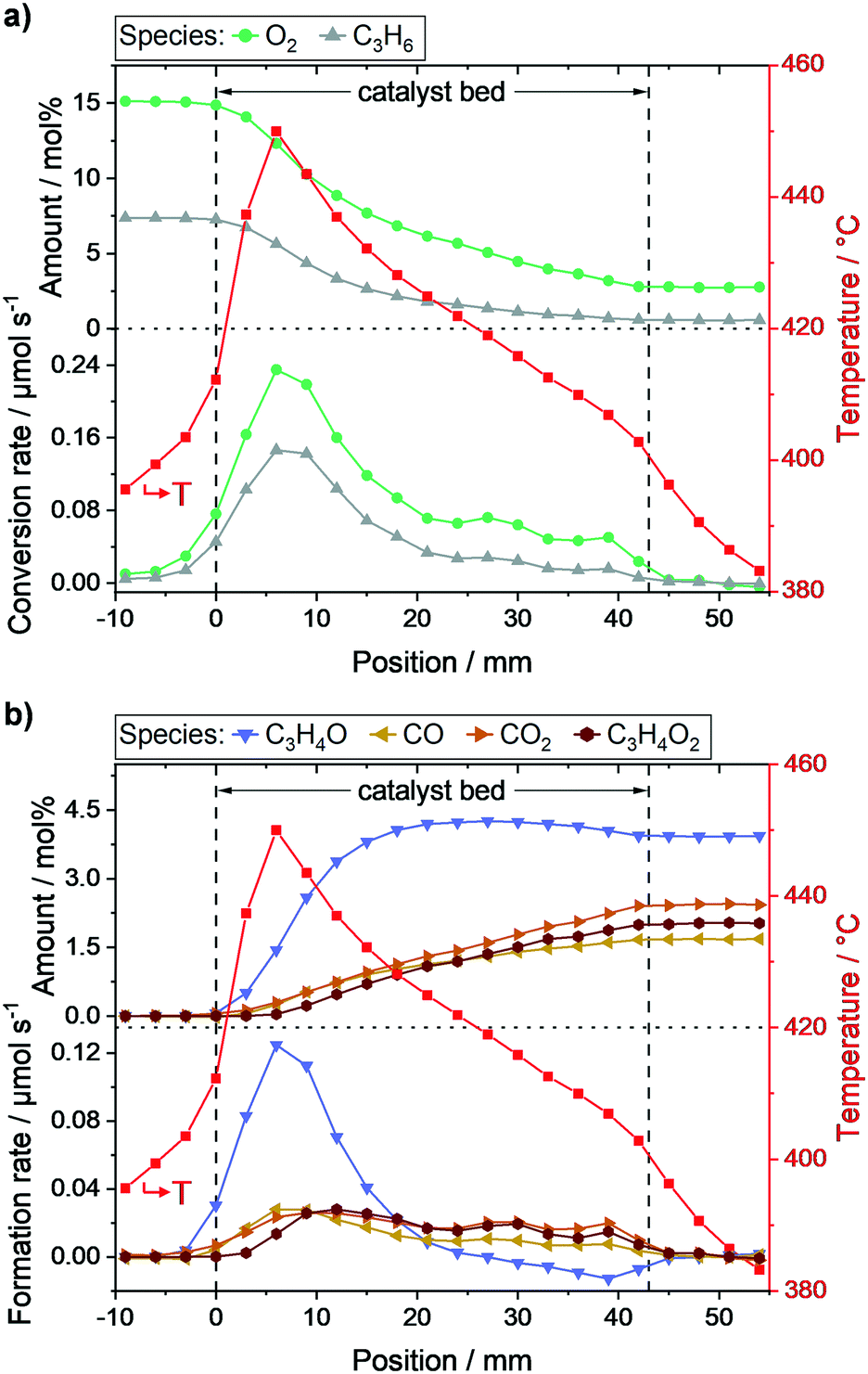 | ||
| Fig. 2 Concentration profiles, local reaction rates, and temperature profile along the packed catalyst bed during selective propylene oxidation (N2/O2/C3H6 = 78/14/8 vol%, WHSVC3H6 = 0.29 h−1, Toven = 400 °C) derived from local MS and integral GC data. Concentration profiles of reactants, corresponding conversion rates and overlaid temperature profile (a). Concentration profiles of quantified carbon-containing products, corresponding formation rates and overlaid temperature profile (b). Species: O2: oxygen, C3H6: propylene, C3H4O: acrolein, CO: carbon monoxide, CO2: carbon dioxide, C3H4O2: acrylic acid. | ||
Overall, these points verify that the developed setup is fully functional and does not exhibit a pronounced backmixing around the local sampling orifices. Hence, the system allows the simultaneous acquisition of quantified concentration and temperature profiles along fixed beds of a powder sieved fraction. Moreover, the positioning system enables even higher spatial resolution but with the drawback of longer profile acquisition times, at least in the present case. So far, spatially resolved concentration and temperature profiles of packed powder catalysts have only been reported for the model reaction of CO oxidation.43,44 With the developed multifunctional setup, various heterogeneously catalyzed reactions are now accessible on the powdered catalyst level as demonstrated here for the selective oxidation of propylene.
Axially resolved concentration and temperature profiles
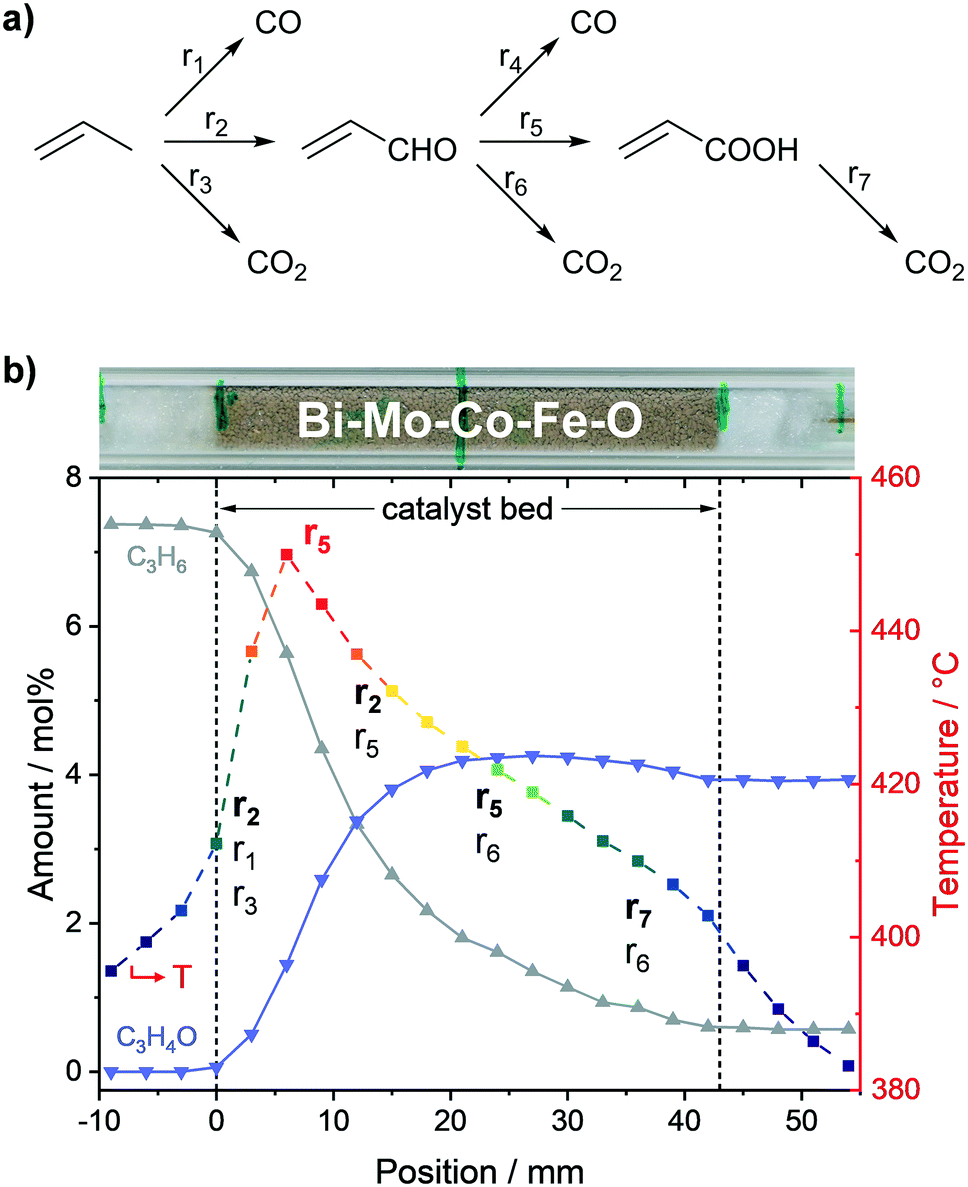
The obtained concentration profiles of reactants and carbon-containing products along the catalyst bed and the corresponding temperature profile are shown in Fig. 2 (see additional profiles in the ESI†). For comparison, the temperature profile under an inert gas flow (50 mL min−1 N2) is shown in Fig. S5, ESI.†As described above, the individual species amounts only changed within the catalyst bed. The strongest gradients in reactants (propylene, oxygen) as well as the target product acrolein, and thus highest conversion and formation rates, respectively, were observed within the range 6–9 mm. According to the temperature measurements, the hotspot was located at the same position and amounted to 450 °C. The hotspot formation can be traced back to the high exothermicity of the oxidation reactions taking place. Hence, the catalyst exhibited its highest activity within the first 20% of the bed, where the relative concentration of reactants was highest.
Based on both concentration and rate profiles shown in Fig. 2, the catalyst bed can be divided into two parts. Within the range 0–15 mm, high propylene and oxygen conversion rates were observed leading to an acrolein (C3H4O) amount of ∼3.8 mol% of the total gas feed at position 15 mm. At the same spot, about 1 mol% carbon dioxide (CO2), 0.9 mol% carbon monoxide (CO), and 0.7 mol% acrylic acid (C3H4O2) were detected as carbon containing by-products (see Fig. 2b). Acrolein, CO, and CO2 formation started at the beginning of the catalyst bed, whereas the formation of acrylic acid was significantly shifted towards the end. This indicates that acrylic acid is formed from further oxidation of acrolein, whereas CO and CO2 are primary products initially formed from propylene. Furthermore, as the amounts of CO and CO2 increased nearly equally in the first part of the catalyst bed, their formation rates from propylene are similar.
In the second part of the catalyst bed (range 18–42 mm), the propylene concentration decreased from 6.8 mol% to 2.8 mol%, and the oxygen concentration from 2.2 mol% to 0.6 mol%, both with only minor changes in the conversion rate (c.f.Fig. 2a). Additionally, a constant decrease in temperature was observed from 428 °C at 18 mm to 403 °C at 42 mm. In contrast, the acrolein concentration within this range was nearly constant (c.f.Fig. 2b), as it increased from 4.1 mol% at 18 mm to its maximum amount of 4.3 mol% at 27 mm, and subsequently decreased to 3.9 mol% at 42 mm. Thus, after position 27 mm acrolein conversion was higher than acrolein production from propylene, which resulted in a negative acrolein formation rate (c.f.Fig. 2b).
Furthermore, the CO concentration showed a pronounced dependency on the propylene concentration. The CO formation in the second part of the catalyst bed was significantly lowered compared to the first part, represented by the lower CO formation rate. Hence, it can be concluded that CO is mostly formed from propylene. For CO2 and acrylic acid such a clear tendency could not be observed. However, the rate profiles for CO2 and acrylic acid showed similar courses in the second part with almost same rates. This together with the slight increase in the CO2 formation rate compared to the acrylic acid formation rate indicates that acrylic acid is oxidized to CO2.
Reaction progress, product selectivity and yield along the catalyst bed
To follow the reaction progress along the catalyst bed, propylene conversion (X), individual product selectivities (S), and product yields (Y) were calculated (c.f. ESI†) and are shown in Fig. 3 together with the overlaid temperature profile.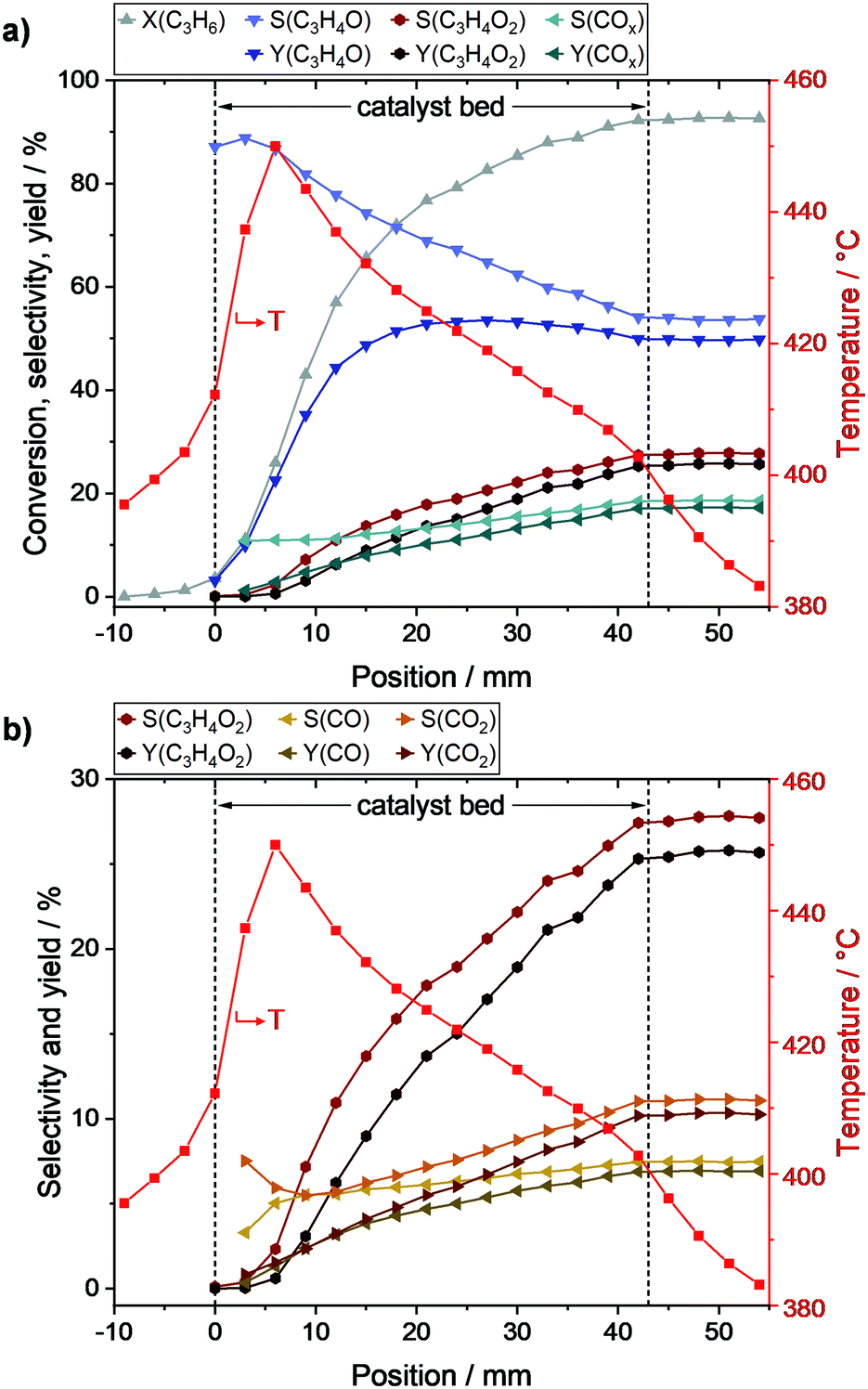 | ||
| Fig. 3 Determined conversion, product selectivity, and yield profiles along the packed catalyst bed during selective propylene oxidation (N2/O2/C3H6 = 78/14/8 vol%, WHSVC3H6 = 0.29 h−1, Toven = 400 °C) derived from local MS and integral GC data. Reaction progress for all quantified carbon containing products with overlaid temperature profile (a), and reaction progress for by-products with the differentiation of COx in CO and CO2 with overlaid temperature profile (b). Species: C3H6: propylene, C3H4O: acrolein, C3H4O2: acrylic acid, CO: carbon monoxide, CO2: carbon dioxide. | ||
As shown in Fig. 3a, the propylene conversion strongly increased within the first part of the catalyst bed and reached 66% at position 15 mm. In the second part (18–43 mm), the rate of propylene conversion was much lower and tapered off towards the end of the catalyst bed, where a propylene conversion of 92% was reached. Hence, two thirds of the reaction progress was achieved within the first third of the catalyst bed.
The acrolein selectivity continuously decreased from initially ∼88% to 54% at the end of the catalyst bed, indicating the presence of at least one reaction that consumes acrolein. The acrolein yield strongly increased within the first part of the bed to 49% at position 15 mm. Subsequently, only minor changes were observed as the maximum yield of 54% was reached at position 27 mm before it declined to its final value of 50%. A different trend was observed for COx (i.e., CO and CO2) as its selectivity increased from initially ∼11% to finally ∼19%. Within the first part, the COx selectivity was nearly constant (∼11%), while it increased linearly in the second part, where the acrolein amount only slightly changed.
The by-product selectivities and yields with the discrimination of COx into CO and CO2 are shown in Fig. 3b. Starting from position 9 mm, it could be observed that the selectivity towards CO only slightly increased from ∼5.5% to finally ∼7.5%, while a stronger increase could be observed in the case of CO2 (from ∼5.5% to ∼11.0%). Due to the low concentration of the by-products in front of position 9 mm, the selectivity and yield profiles might be more prone to errors in this range. However, it could still be observed that the acrylic acid selectivity, and thus the yield, were almost zero in this range. Nevertheless, the acrylic acid selectivity subsequently increased along the bed reaching a final value of 27% (yield of 25%). Hence, acrylic acid is only formed if acrolein is present, and thus is a secondary oxidation product.
No direct influence of the hotspot on the by-product formation could be observed as the profiles did not show an increased by-product selectivity in the corresponding range (around 6 mm). However, with decreasing temperature along the catalyst bed also a decreasing gradient in acrylic acid selectivity was observed. This indicates that after ignition of the reaction and hotspot formation within the first 6 mm of the bed, high temperatures promote the further oxidation of acrolein to acrylic acid, which is in line with the results of Ganzer and Freund.27
Moreover, as the profiles in the present study match quite well with the pilot plant data,27 it seems that in such a small lab reactor data close to pilot/industrial plants can be derived. Notably, similar profiles for all quantified gas phase species were obtained and the temperature profile including the location of the hotspot could be reproduced. This shows that the setup is capable of providing industrially relevant data on the lab-scale. While in the case of pilot plant data the profiles were assembled by seven stationary sampling points along the 1 m reactor filling, 22 individual data points were acquired in the present study over a range of 64 mm. Hence, a far better spatial resolution was obtained even though the reactor was much smaller.
Reaction network and influence of gradients
To better resolve the main- and by-product formation along the packed bed, corresponding yield ratios were calculated and are depicted in Fig. 4. As shown in Fig. 4a, acrolein formation was especially favored at the beginning of the catalyst bed. With increasing bed length, acrylic acid formation became more and more relevant, reflected by a drastic decrease in the ratio of acrolein to acrylic acid yield from 806 (0 mm) to 11 (9 mm). Subsequently, the ratio further declined to 2 at the end of the bed.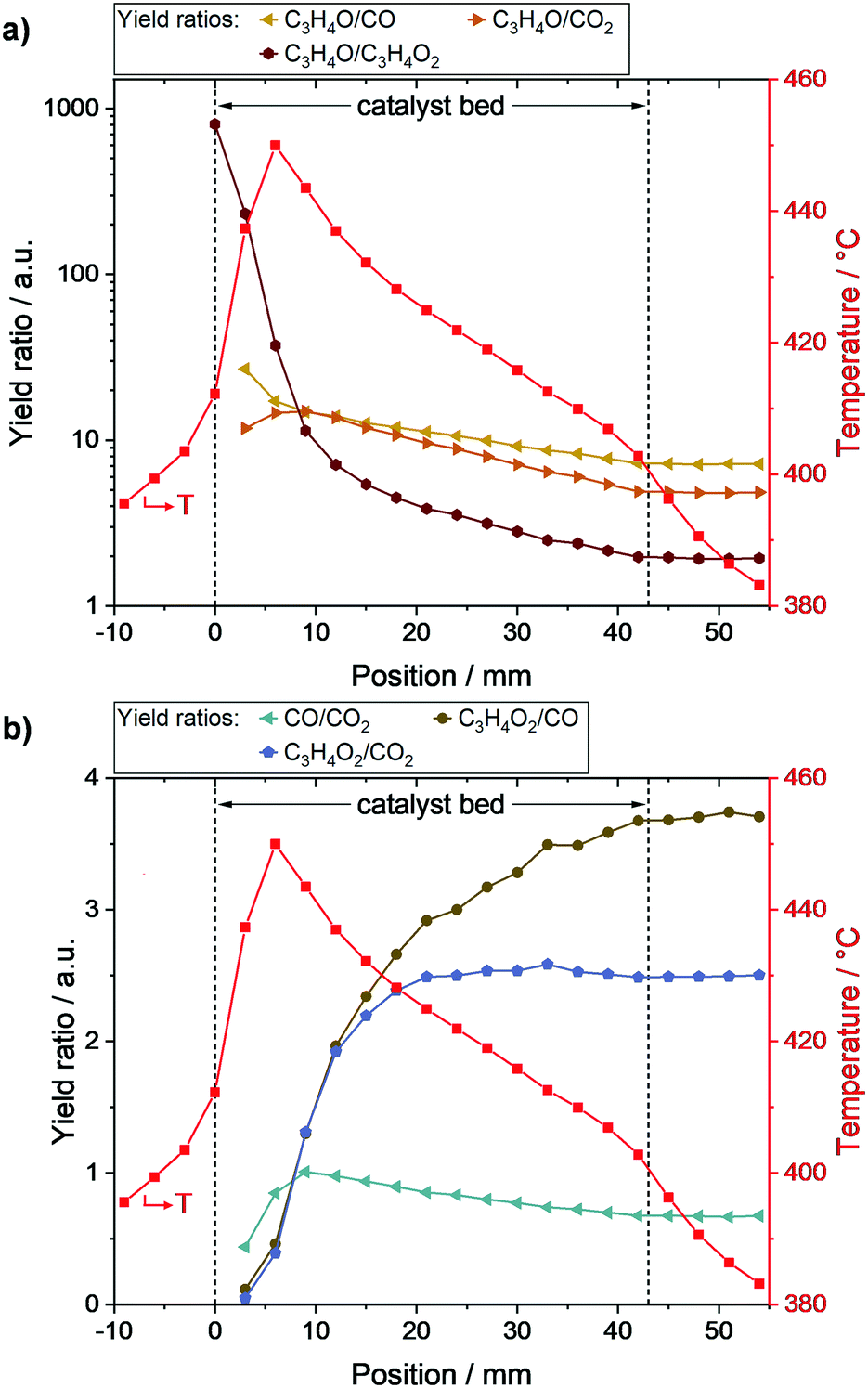 | ||
| Fig. 4 Product yield ratios along the packed bed during selective propylene oxidation (N2/O2/C3H6 = 78/14/8 vol%, WHSVC3H6 = 0.29 h−1, Toven = 400 °C) derived from local MS and integral GC data. Acrolein to by-product yield ratios with overlaid temperature profile (a), and by-product yield ratios with overlaid temperature profile (b). Species: C3H4O: acrolein, C3H4O2: acrylic acid, CO: carbon monoxide, CO2: carbon dioxide. | ||
The ratios of acrolein to CO and acrolein to CO2, respectively, were nearly the same at the beginning of the bed (∼15), which is in line with the local CO and CO2 formation rates shown in Fig. 2b. With increasing catalyst bed length, and thus increasing acrolein amount, the acrolein to CO and acrolein to CO2 ratios declined to 7 and 5, respectively. As the decline of the acrolein to CO2 ratio was more pronounced, the oxidation of acrolein led preferably to the formation of CO2 compared to CO.
The by-product yield ratios are given in Fig. 4b. The decreasing ratio of CO to CO2 along the bed underlines the preferred CO2 formation of acrolein oxidation steps. Furthermore, the acrylic acid to CO ratio continuously increases along the bed, while the acrylic acid to CO2 ratio reaches its final value of ∼2.5 already at position 21 mm. Hence, acrylic acid is mainly oxidized to CO2 as indicated by the rate profiles (c.f.Fig. 2b).
Overall, the obtained profiles enable several insights into the complex reaction network of propylene oxidation. On the one hand, formation of CO and CO2 started at the beginning of the catalyst bed resulting in similar amounts. Thus, both are initially formed from propylene with nearly similar rates, confirming the results of Ganzer and Freund.27 On the other hand, the yield profiles of CO and CO2 differed along the bed, which reveals the presence of consecutive reactions attributing to their formation. The higher tendency of these reactions towards CO2 was reflected by a stronger increase in CO2 selectivity compared to CO. As the yield ratio of acrylic acid to CO2 is constant in the second half of the catalyst bed, while that of acrylic acid to CO increased, acrylic acid is mainly oxidized to CO2 after the hotspot region (6–9 mm). Furthermore, the initial absence of acrylic acid confirms its formation from acrolein. Hence, the reaction network shown in Fig. 5a can be derived.
 | ||
| Fig. 5 Reaction network for selective oxidation of propylene to acrolein derived from the obtained concentration, reaction rate, selectivity, and yield profiles along the Bi–Mo–Co–Fe oxide catalyst (a). Influence of gradients in temperature and gas phase composition (for clarity only propylene and the target product acrolein are given) on the reaction network and thus on the reactions taking place along the packed bed (b). The main reaction for each zone is in bold print, red color indicates reactions promoted by high temperature. | ||
The derived reaction scheme (Fig. 5a) is in line with several reaction networks proposed in the literature22–27 with respect to the formation of acrylic acid from acrolein and the formation of COx directly from propylene as well as by consecutive reactions. However, other by-products or intermediates included in these networks (e.g., allyl alcohol, acetaldehyde, acetic acid) were not observed in the present study. This might be due to a very small amount of such products and/or a fast and selective consecutive reaction like in the case of allyl alcohol.27,52 Nevertheless, a closer look into the formation of such by-products can be achieved by fine tuning of the analytics and a different design of the experiment. As a propylene conversion of 26% was already reached within the first three sampling points (position 6 mm), a higher spatial resolution at the beginning of the catalyst bed would enable a closer look into the manifold pathways of the reaction network. In general, it is possible to establish a detailed kinetic model based on spatially resolved activity data as recently shown by Ganzer and Freund,27 which allows modelling of industrial reactors. However, more experimental data are required to determine the kinetic parameters of the individual reaction pathways.
Moreover, as the gradients in the gas phase composition and temperature along the catalyst bed occurring under industrial conditions have been revealed in the present study, their influence on the reactions taking place along the packed bed was resolved. The corresponding scheme is depicted in Fig. 5b. At the beginning of the reactor, propylene oxidation to acrolein (r2) is dominant while CO (r1) and CO2 (r3) are formed as by-products. Due to the high rate of these highly exothermic oxidation reactions, a hotspot is formed (6–9 mm) promoting the oxidation of acrolein to acrylic acid (r5). Even though the acrolein concentration subsequently further increases, the rate of acrylic acid formation remains constant before it declines towards the end of the bed (c.f.Fig. 2b) due to the decreasing temperature. When reaching high acrolein concentration (∼18 mm), its oxidation to acrylic acid (r5) and CO2 (r6) becomes dominant as acrolein formation from propylene (r2) is rather low due to the low amount of propylene. At the end of the bed, CO2 formation (r7 and r6) is the main reaction taking place.
Based on the obtained profiles along the fixed-bed, suggestions for process optimization can be made. As described above, two thirds of the reaction progress was accomplished in the first third of the catalyst bed (0–15 mm). In this range, the catalyst exhibited its highest acrolein selectivity while especially the selectivity towards acrylic acid was fairly low. However, in the second part (18–43 mm), the acrolein yield stagnated more or less. This was mainly caused by acrolein oxidation to acrylic acid. Hence, from the perspective of acrolein yield, high propylene conversion (>85%) is unfavorable. In order to still operate under high conversion conditions, a two- or even multi-zone catalyst bed with catalysts of different activities could be applied.53–55 A catalyst for the second part should exhibit a high acrolein and low acrylic acid selectivity while a relatively high acrolein amount is present. Furthermore, the reaction temperature should be lowered to decrease the formation of acrylic acid. As recently shown by Dong et al.41 for n-butane oxidation over vanadyl pyrophosphate, measured reactor profiles combined with reactor modeling enable the improvement of catalytic processes, even of well-established ones.
Conclusion
A lab-scale testing setup capable of resolving axial concentration and temperature gradients along a fixed-bed of sieved powder catalysts was developed and applied in the selective oxidation of propylene to acrolein. The combination of mass spectrometry for local gas phase sampling with integral on-line gas analysis by gas chromatography allowed easy calibration of MS signals. Resolving gradients in lab reactors is particularly attractive as it enables detailed insights into the reaction and the reactor at an early stage of catalyst research.In general, the multifunctional design of the setup allows conventional integral testing as well as advanced axially resolved profile acquisition for a broad range of reactions. By the combination of several on-line analytical tools, the local gas phase composition and temperature as well as integral activity could be monitored simultaneously. Using a sampling capillary fixed at both ends with side orifices for local product sampling, the acquisition of multiple profiles within a single test run is achieved.
In the case of selective propylene oxidation to acrolein, the reaction progress along a multicomponent Bi–Mo–Co–Fe oxide catalyst bed, serving as a representative catalyst model system, was monitored. Highest gradients in propylene and oxygen concentration coincided with the location of the hotspot. Around two thirds of the integral propylene conversion was reached after the first third of the catalyst bed with strikingly high selectivity towards acrolein. Even though propylene conversion increased within the last two thirds up to 92%, the acrolein yield remained almost constant (∼50%) due to the increased formation of overoxidation products (i.e., acrylic acid and CO2). Furthermore, the comparison with profiles obtained in a pilot plant showed that the lab reactor is capable of reproducing axial gradients present in industrial reactors while providing easier operability and superior spatial resolution.
Moreover, several insights into the complex reaction network of propylene oxidation were obtained. The results show that CO and CO2 are formed from propylene and additionally by consecutive reactions, which favor CO2 formation. Additionally, acrylic acid was identified as the secondary oxidation product and is especially formed at high temperatures (T > 440 °C).
Overall, the results show the importance of spatially resolved reactor diagnostics. The measured profiles revealed the various reactions occurring during selective propylene oxidation and their relevance along the reactor, thus uncovering the influence of concentration as well as temperature gradients on the integral catalytic performance. Hence, possibilities for process optimization could be envisioned (e.g., multizone catalyst bed, lower process temperature) but need to be combined with reactor modelling to fully exploit the potential of spatially resolved measurements. The herein obtained spatially resolved data provide important input for reactor modelling as existing axial gradients in the reactor are resolved. In the future, structural investigations along catalyst beds should be performed, e.g., by Raman or X-ray spectroscopy, to resolve the influence of the observed gradients in the gas phase on the structural properties of the catalyst. Finally, it would be rewarding to extend the reactor diagnostic tools to resolve radial gradients and thus to verify existing models in the literature.
Author contributions
M. S.: methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization, project administration; T. L. S.: conceptualization, writing – review & editing, supervision; M. T.: conceptualization, resources, writing – review & editing, supervision; A. F.: conceptualization, resources, writing – review & editing, supervision; H. B.: methodology, resources, writing – review & editing; W. P.: resources, writing – review & editing, funding acquisition; J.-D. G.: conceptualization, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the host institutions for financial support of this research project and acknowledge the Helmholtz Research Program STN for financial support related to laser drilling of the glass capillaries. We thank Patrick Lott (ITCP, KIT) for discussion of the technical implementation, Jan Pesek (ITCP, KIT) and the ITCP workshop team for help during build-up and technical realization as well as Dmitry E. Doronkin (ITCP, KIT) for software programming.Notes and references
- G. Centi, F. Cavani and F. Trifirò, in Selective Oxidation by Heterogeneous Catalysis, Springer, Boston, MA, 2001, pp. 1–24 Search PubMed.
- J. Haber, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3359–3384 Search PubMed.
- R. K. Grasselli and J. D. Burrington, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3479–3489 Search PubMed.
- L. Liu, X. P. Ye and J. J. Bozell, ChemSusChem, 2012, 5, 1162–1180 CrossRef CAS PubMed.
- P. Sprenger, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 5628–5642 CrossRef CAS.
- Y. Moro-Oka and W. Ueda, Adv. Catal., 1994, 40, 233–273 CAS.
- C. R. Adams and T. J. Jennings, J. Catal., 1963, 2, 63–68 CrossRef CAS.
- W. M. H. Sachtler, Recl. Trav. Chim. Pays-Bas, 1963, 82, 243–245 CrossRef CAS.
- C. McCain, G. Gough and G. Godin, Nature, 1963, 198, 989–990 CrossRef CAS.
- C. Adams and T. Jennings, J. Catal., 1964, 3, 549–558 CrossRef CAS.
- J. L. Callahan, R. K. Grasselli, E. C. Milberger and H. A. Strecker, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 134–142 CrossRef CAS.
- G. W. Keulks, L. D. Krenzke and T. M. Notermann, Adv. Catal., 1979, 27, 183–225 Search PubMed.
- A. B. Getsoian, V. Shapovalov and A. T. Bell, J. Phys. Chem. C, 2013, 117, 7123–7137 CrossRef CAS.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, in Ullmann's encyclopedia of industrial chemistry, Wiley-VCH, Weinheim, 2007, pp. 329–346 Search PubMed.
- G. W. Keulks, J. Catal., 1970, 19, 232–235 CrossRef CAS.
- R. Wragg, P. Ashmore and J. Hockey, J. Catal., 1971, 22, 49–53 CrossRef CAS.
- L. C. Glaeser, J. F. Brazdil, M. A. Hazle, M. Mehicic and R. K. Grasselli, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2903–2912 RSC.
- Y. Moro-Oka, W. Ueda and K.-H. Lee, J. Mol. Catal. A: Chem., 2003, 199, 139–148 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- J. Haber and W. Turek, J. Catal., 2000, 190, 320–326 CrossRef CAS.
- H. Tan, J. Downie and D. Bacon, Can. J. Chem. Eng., 1989, 67, 412–417 CrossRef CAS.
- H. Redlingshöfer, O. Kröcher, W. Böck, K. Huthmacher and G. Emig, Ind. Eng. Chem. Res., 2002, 41, 1445–1453 CrossRef.
- H. Redlingshöfer, A. Fischer, C. Weckbecker, K. Huthmacher and G. Emig, Ind. Eng. Chem. Res., 2003, 42, 5482–5488 CrossRef.
- T. Franzke, Kinetic investigation of the heterogeneously catalysed selective oxidation of propene to acrolein on molybdenum based mixed oxides, Doctoral thesis, Ruhruniversität Bochum, 2011 Search PubMed.
- L. Bui, R. Chakrabarti and A. Bhan, ACS Catal., 2016, 6, 6567–6580 CrossRef CAS.
- L. Bui and A. Bhan, Appl. Catal., A, 2017, 546, 87–95 CrossRef CAS.
- G. Ganzer and H. Freund, Ind. Eng. Chem. Res., 2019, 58, 1857–1874 CrossRef CAS.
- O. Deutschmann, H. Knözinger, K. Kochloefl and T. Turek, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011, pp. 483–549 Search PubMed.
- K. Morgan, J. Touitou, J.-S. Choi, C. Coney, C. Hardacre, J. A. Pihl, C. E. Stere, M.-Y. Kim, C. Stewart and A. Goguet, ACS Catal., 2016, 6, 1356–1381 CrossRef CAS.
- W. P. Partridge, J. M. Storey, S. A. Lewis, R. W. Smithwick, G. L. DeVault, M. J. Cunningham, N. W. Currier and T. M. Yonushonis, SAE Trans., 2000, 109, 2983–2991 Search PubMed.
- J. Sá, D. L. A. Fernandes, F. Aiouache, A. Goguet, C. Hardacre, D. Lundie, W. Naeem, W. P. Partridge and C. Stere, Analyst, 2010, 135, 2260–2272 RSC.
- M. Bosco and F. Vogel, Catal. Today, 2006, 116, 348–353 CrossRef CAS.
- R. Horn, K. Williams, N. Degenstein and L. Schmidt, J. Catal., 2006, 242, 92–102 CrossRef CAS.
- J.-S. Choi, W. P. Partridge and C. S. Daw, Appl. Catal., A, 2005, 293, 24–40 CrossRef CAS.
- A. Donazzi, D. Livio, M. Maestri, A. Beretta, G. Groppi, E. Tronconi and P. Forzatti, Angew. Chem., Int. Ed., 2011, 50, 3943–3946 CrossRef CAS PubMed.
- D. Livio, C. Diehm, A. Donazzi, A. Beretta and O. Deutschmann, Appl. Catal., A, 2013, 467, 530–541 CrossRef CAS.
- R. Horn, N. J. Degenstein, K. A. Williams and L. D. Schmidt, Catal. Lett., 2006, 110, 169–178 CrossRef CAS.
- R. Horn, O. Korup, M. Geske, U. Zavyalova, I. Oprea and R. Schlögl, Rev. Sci. Instrum., 2010, 81, 064102 CrossRef CAS PubMed.
- O. Korup, S. Mavlyankariev, M. Geske, C. F. Goldsmith and R. Horn, Chem. Eng. Process., 2011, 50, 998–1009 CrossRef CAS.
- M. Geske, O. Korup and R. Horn, Catal. Sci. Technol., 2013, 3, 169–175 RSC.
- Y. Dong, M. Geske, O. Korup, N. Ellenfeld, F. Rosowski, C. Dobner and R. Horn, Chem. Eng. J., 2018, 350, 799–811 CrossRef CAS.
- B. Sosna, O. Korup and R. Horn, J. Catal., 2020, 381, 285–294 CrossRef CAS.
- J. Touitou, K. Morgan, R. Burch, C. Hardacre and A. Goguet, Catal. Sci. Technol., 2012, 2, 1811–1813 RSC.
- J. Touitou, R. Burch, C. Hardacre, C. McManus, K. Morgan, J. Sá and A. Goguet, Analyst, 2013, 138, 2858–2862 RSC.
- A. Goguet, C. Stewart, J. Touitou and K. Morgan, in Advances in Chemical Engineering, ed. A. G. Dixon and O. Deutschmann, Academic Press, 2017, pp. 131–160 Search PubMed.
- J. Touitou, F. Aiouache, R. Burch, R. Douglas, C. Hardacre, K. Morgan, J. Sá, C. Stewart, J. Stewart and A. Goguet, J. Catal., 2014, 319, 239–246 CrossRef CAS.
- P. Sprenger, M. Stehle, A. Gaur, A. M. Gänzler, D. Gashnikova, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2018, 8, 6462–6475 CrossRef CAS.
- E. Mottay, X. Liu, H. Zhang, E. Mazur, R. Sanatinia and W. Pfleging, MRS Bull., 2016, 41, 984–992 CrossRef.
- W. Pfleging, NANO, 2018, 7, 549–573 CAS.
- D. E. Doronkin, H. Lichtenberg and J.-D. Grunwaldt, in XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer International Publishing, Cham, 2017, pp. 75–89 Search PubMed.
- M. A. Newton, S. Checchia, A. J. Knorpp, D. Stoian, W. van Beek, H. Emerich, A. Longo and J. A. van Bokhoven, Catal. Sci. Technol., 2019, 9, 3081–3089 RSC.
- J. D. Burrington, C. T. Kartisek and R. K. Grasselli, J. Catal., 1980, 63, 235–254 CrossRef CAS.
- K. Kadowaki, K. Sarumaru and T. Shibano, U.S. Pat., US4837360A, 1989 Search PubMed.
- M. Tanimoto, H. Yunoki, H. Hironaka and N. E. P. Kimura, E.P. Pat., EP1074538A2, 2000 Search PubMed.
- M. Tanimoto, U.S. Pat., US6781013B2, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00553g |
| This journal is © The Royal Society of Chemistry 2021 |