
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The catalytic decomposition of nitrous oxide and the NO + CO reaction over Ni/Cu dilute and single atom alloy surfaces: first-principles microkinetic modelling†
Konstantinos G.
Papanikolaou
 and
Michail
Stamatakis
*
and
Michail
Stamatakis
*
Thomas Young Centre and Department of Chemical Engineering, University College London, Roberts Building, Torrington Place, London WC1E 7JE, UK. E-mail: m.stamatakis@ucl.ac.uk
First published on 22nd February 2021
Abstract
The development of platinum group metal-free (PGM-free) catalysts, which can efficiently reduce pollution-causing emissions, is an important task for overcoming major environmental challenges. In particular, nitrogen oxides (NOx) are major contributors to air pollution, being one of the culprits for smog and ozone depletion. In this work, we employ density functional theory (DFT) and microkinetic modelling to investigate the decomposition of N2O and the NO + CO reaction over two PGM-free Ni/Cu dilute alloys. On the first surface, Ni atoms are isolated on the host Cu(111), thereby forming a single atom alloy surface (i.e. Ni/Cu(111) SAA), while on the second, the same atoms are organised as Ni–Ni dimers (i.e. Ni2Cu(111)). The same reactions are also simulated on pure Cu(111) (i.e. the host surface), and on Rh(111), which is used for benchmarking as Rh is a well-established PGM in emissions control catalysis. Our results suggest that the addition of trace amounts of Ni on Cu(111) may bring about significant improvement to the catalytic performance with regard to the catalytic decomposition of N2O. Additionally, we determine that Ni2Cu(111) shows equivalent, or under some circumstances even better, performance as compared to Rh(111) for the NO + CO reaction. This work contributes to the long-standing efforts toward the design of efficient PGM-free catalytic materials for the reduction of noxious gases.
1. Introduction
The catalytic reduction of nitric oxide (NO) and the decomposition of nitrous oxide (N2O) are reactions of central significance for the prevention and mitigation of critical environmental problems. The emissions of these molecules are, to a large extent, associated with automobiles,1 which are equipped with the so-called three way catalyst (TWC). TWCs are composed of a complex mixture of oxides (e.g. γ-Al2O3, BaO), whereon noble metals Rh, Pt and Pd are deposited and “undertake” the task of converting noxious gases (e.g. CO, NO, N2O, CxHy) into environmentally acceptable products (i.e. N2, H2O, CO2).The catalytic reduction of NO by CO is a crucial reaction for controlling automobile emissions, and Rh is regarded as the most promising platinum group metal (PGM) to this end.1 By and large, this is because Rh can activate the N–O bond at relatively low temperatures2–4 (the cleavage of this bond is in many cases the rate determining step of the NO + CO reaction5,6), and also because of its high resistance to common poisons (e.g. sulphur).7 As a result, the mechanism and kinetics of the NO + CO reaction over Rh catalysts have been the subject of extensive research for several experimental,8–14 and theoretical studies.15–19
Although Rh exhibits the best performance among other PGMs toward the reduction of NO, its high cost and limited resources are major shortcomings.1 Unsurprisingly, these downsides have turned the attention of the catalysis community into the search of TWCs that are either PGM-free20–29 or utilise minimal amounts of PGMs.30–33 For example, Asakura et al. showed that a NiCu/Al2O3 alloy catalyst exhibits distinct catalytic behaviour compared to its Cu/Al2O3 and Ni/Al2O3 monometallic counterparts.21 The three materials were subject to alternating lean–rich cycles similar to those a TWC may experience during operation. The performance of the Ni-based catalyst deteriorated considerably within the first lean–rich cycle; the Cu-based catalyst was found to be susceptible to oxidation, thereby losing its activity within short time under lean conditions. By contrast, the NiCu/Al2O3 catalyst retained very high N2 productivity for large time intervals even under lean conditions, and could rapidly self-regenerate (i.e. transition from an oxide state to the corresponding metallic state) in the beginning of each rich period. The authors ascribed the self-regenerative property of NiCu/Al2O3 to the coexistence of Ni and Cu oxide species that remained in close contact at the end of lean periods.21 Tanaka and co-workers reported that the same bimetallic alloy supported on a Mg–Al mixed oxide serves as an efficient catalyst for hydrocarbon oxidation (i.e. another important reaction that happens over TWCs), under both reducing and oxidative atmospheres.20 Xing et al. synthesised a highly dilute PdCu/Al2O3 catalyst, whereby Pd atoms were atomically dispersed on the Cu host.33 This catalyst not only showed excellent stability during the NO + CO reaction, but also was able to convert fully NO to N2 at relatively low temperatures (473 K).
In our recent theoretical work, we screened a number of dilute alloys for their performance on catalysing important “elementary” steps for the NO + CO reaction (e.g. direct NO dissociation, N2 association and CO oxidation).4 According to our results, a Ni2Cu alloy, where Ni atoms are organised as Ni–Ni dimers over the Cu host surface, is promising in activating the N–O bond and is capable of performing facile N2 association.4 In particular, Ni2Cu exhibited the best performance among the investigated bimetallic surfaces, and similar, or in many cases even better, performance than the PGMs in TWCs (i.e. Rh, Pd and Pt). Finally, we argued that this alloy might, in practice, exhibit bifunctional behaviour,34 where Ni sites cleave N–O bonds, while Cu sites serve as the loci for the oxidation of CO.35
In this paper, we employ density functional theory (DFT) and investigate in detail two very relevant reactions to the NO + CO chemistry over Ni/Cu(111) single atom and Ni2Cu(111) dilute alloy surfaces; these are the formation and decomposition of N2O*, in particular, NO* + N* ↔ N2O* and N2O* ↔ 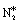 + O*, respectively, where * denotes an adsorbed species. Besides their relevance to the catalytic reduction of NO by CO (N2O is an exhaust gas and an adduct of catalytic surface chemistry),36 these reactions are also of general interest.37 This is because N2O is a potent greenhouse gas and an undesired by-product of large-scale processes like the production of adipic and nitric acid.38 The same reactions are studied over Rh(111), which is used for benchmarking, and also over Cu(111), which is the corresponding host metal surface. We identify different pathways for the activation of N2O* over all surfaces, and we demonstrate that the selectivity of this reaction can be tuned based on the size of the Ni cluster. Importantly, our calculations imply that the presence of small amounts of Ni on Cu(111) strengthens the binding of N2O* to the surface, thereby preventing its desorption and promoting its dissociation. Finally, using the obtained DFT energetics we parameterise a microkinetic model for the NO + CO reaction over the four (111) surfaces. Our theoretical studies aim at providing a first assessment for the performance of the Ni/Cu dilute alloys toward the aforementioned reaction. These simulations reveal that the performance of Ni2Cu(111) is certainly superior compared to that of Cu(111) and closely comparable to that of Rh(111). On this basis, the present study highlights the potential of well-engineered Ni2Cu alloys, which are composed of inexpensive and abundant metals, for emissions control technologies.
+ O*, respectively, where * denotes an adsorbed species. Besides their relevance to the catalytic reduction of NO by CO (N2O is an exhaust gas and an adduct of catalytic surface chemistry),36 these reactions are also of general interest.37 This is because N2O is a potent greenhouse gas and an undesired by-product of large-scale processes like the production of adipic and nitric acid.38 The same reactions are studied over Rh(111), which is used for benchmarking, and also over Cu(111), which is the corresponding host metal surface. We identify different pathways for the activation of N2O* over all surfaces, and we demonstrate that the selectivity of this reaction can be tuned based on the size of the Ni cluster. Importantly, our calculations imply that the presence of small amounts of Ni on Cu(111) strengthens the binding of N2O* to the surface, thereby preventing its desorption and promoting its dissociation. Finally, using the obtained DFT energetics we parameterise a microkinetic model for the NO + CO reaction over the four (111) surfaces. Our theoretical studies aim at providing a first assessment for the performance of the Ni/Cu dilute alloys toward the aforementioned reaction. These simulations reveal that the performance of Ni2Cu(111) is certainly superior compared to that of Cu(111) and closely comparable to that of Rh(111). On this basis, the present study highlights the potential of well-engineered Ni2Cu alloys, which are composed of inexpensive and abundant metals, for emissions control technologies.
2. Methods
Density functional theory
Periodic DFT calculations were performed using the Vienna ab initio simulation package (VASP) version 5.4.1.39,40 Exchange and correlation effects were treated with the optB86b-vdW functional,41,42 which captures van der Waals (vdW) interactions.43,44 The latter are important to our work and recent studies have shown that the inclusion of dispersion forces in DFT calculations may increase the binding strength of loosely bound adsorbates of the NO + CO reaction (i.e. N2O*,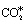 ) by as much as 0.7 eV.16 A kinetic energy cut-off of 400 eV was used for the plane wave basis set that was adopted to describe the wave functions of valence electrons. The interactions between core and valence electrons were modelled by the projector augmented wave (PAW) method.45 The electronic wave function was converged to 10−7 eV, and the structures were relaxed until the forces on each atom were less than 0.01 eV Å−1. The optB86b-vdW-computed lattice constants are 3.608 Å and 3.829 Å for Cu and Rh, respectively; these values agree well with the corresponding experimental values (3.596 Å and 3.793 Å for Cu and Rh, respectively).46 The metal surfaces were modelled by a 3 × 3 cell with 5 layers, of which the two bottom ones were fixed at the corresponding lattice constant, thereby simulating the bulk of the material, while the three top layers and any adsorbate atoms were relaxed during geometry optimisation. The Brillouin zone was sampled using a 9 × 9 × 1 Monkhorst–Pack k-point mesh.47 The adsorption energy of N2O was computed based on the following equation:
) by as much as 0.7 eV.16 A kinetic energy cut-off of 400 eV was used for the plane wave basis set that was adopted to describe the wave functions of valence electrons. The interactions between core and valence electrons were modelled by the projector augmented wave (PAW) method.45 The electronic wave function was converged to 10−7 eV, and the structures were relaxed until the forces on each atom were less than 0.01 eV Å−1. The optB86b-vdW-computed lattice constants are 3.608 Å and 3.829 Å for Cu and Rh, respectively; these values agree well with the corresponding experimental values (3.596 Å and 3.793 Å for Cu and Rh, respectively).46 The metal surfaces were modelled by a 3 × 3 cell with 5 layers, of which the two bottom ones were fixed at the corresponding lattice constant, thereby simulating the bulk of the material, while the three top layers and any adsorbate atoms were relaxed during geometry optimisation. The Brillouin zone was sampled using a 9 × 9 × 1 Monkhorst–Pack k-point mesh.47 The adsorption energy of N2O was computed based on the following equation: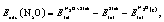 | (1) |
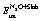 , ESlabtot and
, ESlabtot and 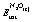 are the total DFT energies for a slab with an N2O* thereon, a clean slab, and an N2O molecule in the gas phase, respectively (the pertinent results are reported in Table 1). The reported transition states were first approached using the dimer method,48 fully converged with Newton's method, and verified by vibrational analyses, making sure that all the reported transition states had only one imaginary vibrational frequency. The reported activation barriers were computed as Ea = ETS − EIS, where ETS and EIS are the DFT energies of the transition and initial states, respectively. Vibrational frequencies were computed within the harmonic approximation where the energy of the system is expressed as a Taylor expansion that includes up to second order terms, and the second derivative was estimated within the finite–difference approximation with a displacement of 0.02 Å.
are the total DFT energies for a slab with an N2O* thereon, a clean slab, and an N2O molecule in the gas phase, respectively (the pertinent results are reported in Table 1). The reported transition states were first approached using the dimer method,48 fully converged with Newton's method, and verified by vibrational analyses, making sure that all the reported transition states had only one imaginary vibrational frequency. The reported activation barriers were computed as Ea = ETS − EIS, where ETS and EIS are the DFT energies of the transition and initial states, respectively. Vibrational frequencies were computed within the harmonic approximation where the energy of the system is expressed as a Taylor expansion that includes up to second order terms, and the second derivative was estimated within the finite–difference approximation with a displacement of 0.02 Å.
| Adsorption structure | Property | Rh(111) | Cu(111) | Ni/Cu(111) SAA | Ni2Cu(111) |
|---|---|---|---|---|---|
| η1-(Nt{top}) (denoted as η1) | E ads(N2O) | −0.71 | −0.21 | −0.70 | −0.68 |
| d N–O | 1.20 | 1.20 | 1.20 | 1.21 | |
| d N–N | 1.15 | 1.15 | 1.15 | 1.15 | |
| η2-f(Nt{bridge},Nc{top}) (denoted as η2NbNt) | E ads(N2O) | −0.83 | +0.15 | −0.43 | −0.74 |
| d N–O | 1.22 | 1.23 | 1.23 | 1.23 | |
| d N–N | 1.35 | 1.29 | 1.29 | 1.31 | |
| η2-(Nt{top},O{top})(denoted as η2NtOt) | E ads(N2O) | −0.72 | −0.20 | −0.53 | −0.68 |
| d N–O | 1.33 | 1.28 | 1.30 | 1.32 | |
| d N–N | 1.20 | 1.19 | 1.20 | 1.20 | |
| η2-(Nt{hcp},O{top}) | E ads(N2O) | — | −0.25 | −0.44 | −0.73 |
| d N–O | — | 1.30 | 1.31 | 1.32 | |
| d N–N | — | 1.27 | 1.25 | 1.27 |
Microkinetic modelling
The microkinetic model for the NO + CO reaction included 16 reaction steps for Cu(111), Ni/Cu(111) single atom alloy (SAA), and Ni2Cu(111) surfaces and 14 reaction steps for Rh(111) – (see Table 2). On the monometallic surfaces there was only one site type, while in the bimetallic surfaces there were Cu and Ni sites, denoted as Cu* and Ni*, respectively. Therefore, for the latter surfaces, we define the “local” coverages as follows: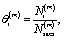 | (2) |
 | (3) |
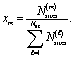 | (4) |
| Reaction & reaction number | Rh(111) | Cu(111) | Ni/Cu(111) SAA | Ni2Cu(111) | |||||
|---|---|---|---|---|---|---|---|---|---|
| E fwd | E rev | E fwd | E rev | E fwd | E rev | E fwd | E fwd | ||
| NO(g) + * ↔ NO* | (R1) | 0.00 | 2.87 | 0.00 | 1.55 | 0.00 | 2.10 | 0.00 | 2.65 |
| CO(g) + * ↔ CO* | (R2) | 0.00 | 2.20 | 0.00 | 0.78 | 0.00 | 1.51 | 0.00 | 1.71 |

|
(R3) | 0.00 | 0.79 | 0.00 | 0.15 | 0.00 | 0.79 | 0.00 | 0.77 |
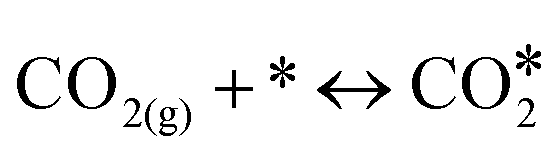
|
(R4) | 0.00 | 0.27 | 0.00 | 0.30 | 0.00 | 0.28 | 0.00 | 0.37 |
| N2O(g) + * ↔ N2O* η2NbNt | (R5) | 0.00 | 0.83 | 0.00 | 0.00 | 0.00 | 0.43 | 0.00 | 0.74 |
| N2O(g) + * ↔ N2O* η2NtOt | (R6) | 0.00 | 0.72 | 0.00 | 0.20 | 0.00 | 0.53 | 0.00 | 0.68 |
| N2O(g) + * ↔ N2O* η1 | (R7) | 0.00 | 0.71 | 0.00 | 0.21 | 0.00 | 0.70 | 0.00 | 0.68 |
| NO* + * ↔ N* + O* | (R8) | 1.42 | 2.03 | 1.57 | 1.43 | 1.47 | 1.43 | 1.30 | 1.24 |
| NO* + N* ↔ N2O* η2NbNt + * | (R9) | 1.50 | 0.40 | 0.44 | 0.94 | 0.51 | 0.45 | 0.60 | 0.68 |
| N2O* η2NbNt ↔ N2O* η1 | (R10) | 0.46 | 0.30 | 0.14 | 0.56 | 0.24 | 0.50 | 0.37 | 0.31 |
| N2O* η1 ↔ N2O* η2NtOt | (R11) | 0.24 | 0.40 | 0.06 | 0.05 | 0.29 | 0.12 | 0.32 | 0.32 |
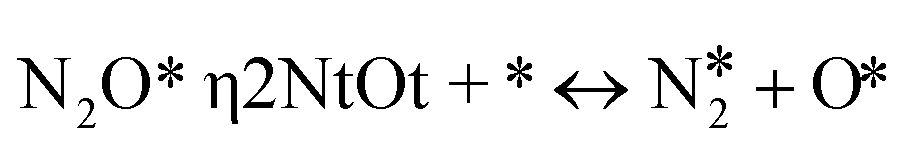
|
(R12) | 0.07 | 2.54 | 0.05 | 2.12 | 0.03 | 2.26 | 0.09 | 2.48 |
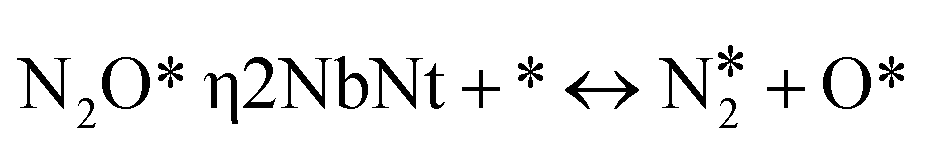
|
(R13) | – | – | 0.19 | 2.20 | 0.23 | 2.56 | 0.23 | 2.51 |

|
(R14) | 1.17 | 0.41 | 0.48 | 1.22 | 0.71 | 0.60 | 0.88 | 0.48 |
| NO* + NO* ↔ N2O* η1 + O* | (R15) | – | – | 0.84 | 1.82 | 1.27 | 1.60 | 1.30 | 1.69 |
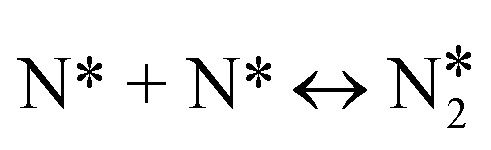
|
(R16) | 1.85 | 2.14 | 0.64 | 3.6 | 0.88 | 3.40 | 0.62 | 2.81 |
All reactions were considered reversible, and the forward/reverse rates were given by the typical mass-action law expressions used in microkinetic models, which contain the partial pressures of gas-phase species (considered as constants) and the surface coverages. The gas-phase species taken into account were NO, CO, N2, CO2, N2O, while the surface species were O*, CO*, N*, NO*,  ,
, 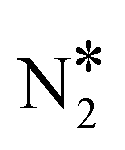 and N2O*, as well as the vacant site pseudo-species denoted as *. Regarding the N2O* species, three different adsorption geometries were taken into account (see the next section). The transitions from one adsorption geometry to another could happen through transformation reactions that were included in the reaction mechanism (see R10–R11 in Table 2). The forward rate for reaction j on site-type m is formulated as follows:
and N2O*, as well as the vacant site pseudo-species denoted as *. Regarding the N2O* species, three different adsorption geometries were taken into account (see the next section). The transitions from one adsorption geometry to another could happen through transformation reactions that were included in the reaction mechanism (see R10–R11 in Table 2). The forward rate for reaction j on site-type m is formulated as follows:
 | (5) |
 is the set of gas-phase reactant species of reaction j; Pg is the partial pressure of gas species g; and vgj is the stoichiometric coefficient of that gas phase species in reaction j. By convention, stoichiometric coefficients are negative for reactants and positive for products; if a species does not appear in a certain reaction, the corresponding stoichiometric coefficient is zero. Moreover,
is the set of gas-phase reactant species of reaction j; Pg is the partial pressure of gas species g; and vgj is the stoichiometric coefficient of that gas phase species in reaction j. By convention, stoichiometric coefficients are negative for reactants and positive for products; if a species does not appear in a certain reaction, the corresponding stoichiometric coefficient is zero. Moreover,  is the set of surface reactant species of reaction j; vij is the stoichiometric coefficient of surface species i in reaction j; and
is the set of surface reactant species of reaction j; vij is the stoichiometric coefficient of surface species i in reaction j; and 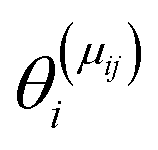 is the local coverage of surface species i on sites of type μij. The latter term may or may not be equal to m, since, a reaction that is said to happen on site m (e.g. Ni*), may well involve another species adsorbed on a neighbouring site type (e.g. Cu*). For instance, when reaction R9 of Table 2 (NO* + N* ↔ N2O* + *) happens on a Ni site, NO* is found on the Ni site, while N* is on Cu; therefore, the rate would be:
is the local coverage of surface species i on sites of type μij. The latter term may or may not be equal to m, since, a reaction that is said to happen on site m (e.g. Ni*), may well involve another species adsorbed on a neighbouring site type (e.g. Cu*). For instance, when reaction R9 of Table 2 (NO* + N* ↔ N2O* + *) happens on a Ni site, NO* is found on the Ni site, while N* is on Cu; therefore, the rate would be:| R(Ni*)fwd,9 = k(Ni*)fwd,jθ(Ni*)NO*θ(Cu*)N*. |
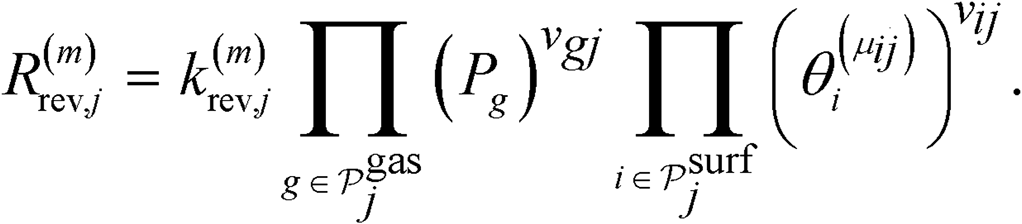 | (6) |
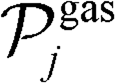 and
and 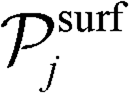 denote sets of products of reaction j, and the stoichiometric coefficients appear with their “original” positive signs, because of the convention mentioned earlier.
denote sets of products of reaction j, and the stoichiometric coefficients appear with their “original” positive signs, because of the convention mentioned earlier.
The rate constant calculations for the surface reactions (k(m)fwd,j and k(m)rev,j) are calculated after invoking widely used transition state theory approximations. If a reaction cannot happen on a certain site, then k(m)fwd,j = k(m)rev,j = 0. We further define the net rate of reaction j on site m as:
| R(m)j = R(m)fwd,j − R(m)rev,j. | (7) |
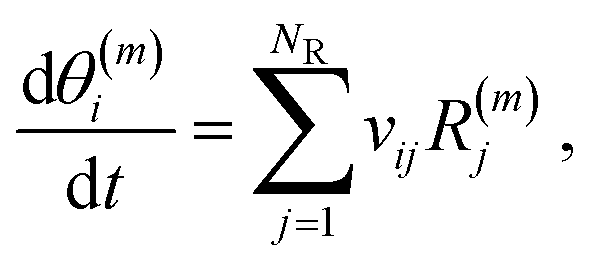 | (8) |
 | (9) |
 | (10) |
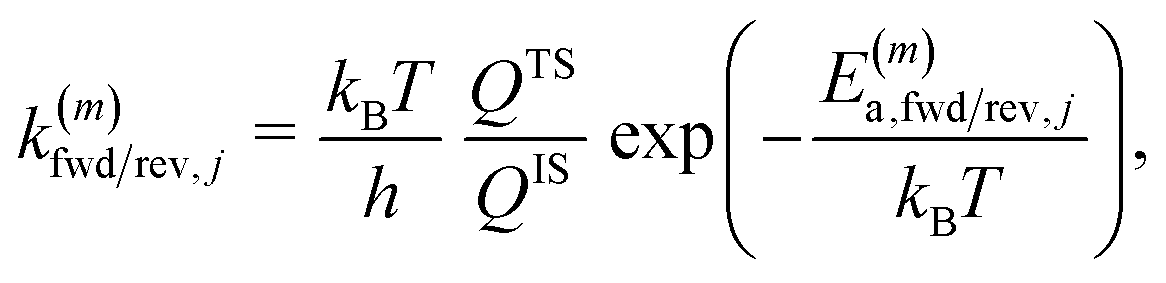 | (11) |
 | (12) |
The net rates for N2 and N2O are calculated as follows:
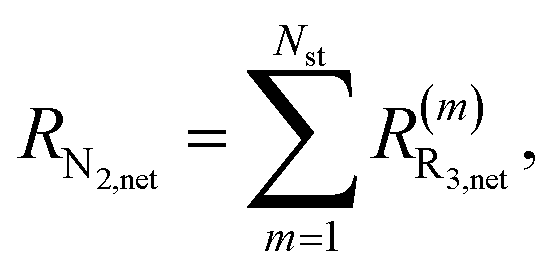 | (13) |
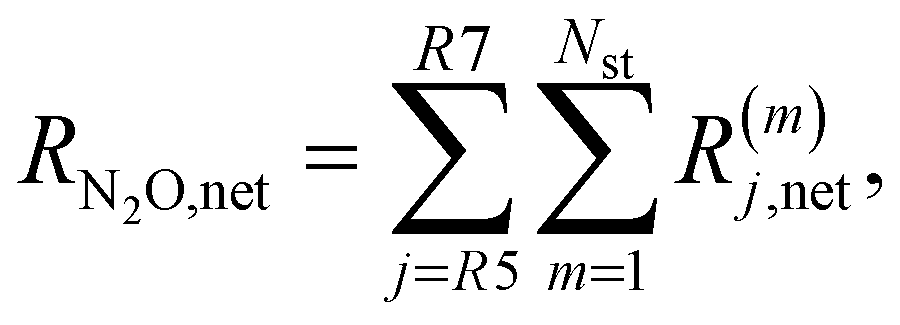 | (14) |
 | (15) |
![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) is the equilibrium constant of reaction = 1,…, NR on site m = 1,…, Nst; k(n≠m)≠j are the rate constants for all other steps than j that take place on either Cu* or Ni* (the site other than m). The larger the absolute value of X(m)RC,j the larger the influence of that reaction step to the overall reaction rate; also when X(m)RC,j > 0, the reaction is rate-limiting, whereas for X(m)RC,j < 0 the reaction is rate-inhibiting.
is the equilibrium constant of reaction = 1,…, NR on site m = 1,…, Nst; k(n≠m)≠j are the rate constants for all other steps than j that take place on either Cu* or Ni* (the site other than m). The larger the absolute value of X(m)RC,j the larger the influence of that reaction step to the overall reaction rate; also when X(m)RC,j > 0, the reaction is rate-limiting, whereas for X(m)RC,j < 0 the reaction is rate-inhibiting.
3. Results and discussion
3.1. Adsorption of N2O on Ni/Cu dilute alloy surfaces
Gas-phase nitrous oxide is a linear molecule (C∞V symmetry) and a harmful by–product of industrial processes (e.g. nitric acid production). Its catalytic decomposition has been investigated over many transition metals, including Rh,53 Cu,54 Ru,55 Pd,56 Fe,57 Ni,58 Pt,59 PdAu,60 and PdCu.61 Here, we first examine the adsorption of nitrous oxide on Cu(111), Ni/Cu(111) SAA and Ni2Cu(111) surfaces, but also on our “benchmarking surface” Rh(111).It is known that N2O* may adopt a number of different adsorption geometries upon its interaction with metal surfaces.56,62 Accordingly, we identify six stable adsorption geometries out of which four are displayed in Fig. 1, while the full list is given in the first section of the ESI.† These four adsorption structures are important because they are adopted by N2O* upon its decomposition to either to  + O* or NO* + N* (see sections 3.2 and 3.3), and are denoted as: η1-(Nt{top}), η2-f(Nt{bridge},Nc{top}), η2-(Nt{top},O{top}), and η2-(Nt{hcp},O{top}) – (Fig. 1). Since we will be referring often to the first three throughout this paper, we adopt the following abbreviations for them: η1, η2NbNt and η2NtOt, respectively.
+ O* or NO* + N* (see sections 3.2 and 3.3), and are denoted as: η1-(Nt{top}), η2-f(Nt{bridge},Nc{top}), η2-(Nt{top},O{top}), and η2-(Nt{hcp},O{top}) – (Fig. 1). Since we will be referring often to the first three throughout this paper, we adopt the following abbreviations for them: η1, η2NbNt and η2NtOt, respectively.
 | ||
| Fig. 1 Top and side views of (A) η1-(Nt{top}); (B) η2-f(Nt{bridge},Nc{top}); (C) η2-(Nt{top},O{top}) and (D) η2-(Nt{hcp},O{top}) adsorption structure. On the side view of (A) we highlight the terminal (Nt) and central (Nc) nitrogen atoms. Ni, Cu, N and O atoms are shown in purple, orange, blue and red, respectively. The adsorption geometries are shown over Ni2Cu(111), but they are representative for all surfaces. | ||
The computed adsorption energies for the four geometries, along with the N–O (dN–O) and N–N (dN–N) bond distances are summarised in Table 1. We note that the most preferred N2O* adsorption structure on Rh(111) is the η2NbNt mode (Eads(N2O) = −0.83 eV) – (Table 1). This type of adsorption can be considered as a weak chemisorption because: (1) the geometry of N2O* deviates noticeably from the gas-phase geometry, which is linear; and (2) because the N–N bond is considerably elongated (dN–N = 1.14 Å and 1.35 Å for gas-phase N2O and η2NbNt N2O*, respectively). The following most stable adsorption structures are the η1 and η2NtOt with Eads(N2O) = −0.71 eV and Eads(N2O) = −0.72 eV, respectively. The former structure can be characterised as a strong physisorption owing to the unaffected geometry and bond lengths of η1 N2O* as compared to gas–phase N2O (dN–N = 1.14 Å and dN–O = 1.20 Å for gas N2O) – (Table 1).
The activation of the N–N bond in the η2NbNt structure can be elucidated by careful examination of the electronic structure of this geometry (Fig. S2†).62 Our density of states (DOS) analyses indicate that in η2NbNt the 2π and 3π orbitals of N2O* become broader as a result of their interaction with the metal states, whilst the same is not true for the η1 structure where the same orbitals appear rather localised (Fig. S2†). The broadening of the 3π orbitals is indicative of electron back-donation, which in turn leads to the activation of the N–N bond. This result is in qualitative agreement with the work of Paul et al.62 where the authors, by means of DFT calculations using the PW91 functional, found that the η2NbNt and η1 are ca. equally stable on Rh(111) (Eads(N2O) = −0.35 eV and Eads(N2O) −0.39 eV, respectively). Moreover, our calculations suggest that N2O* is bound stronger by ca. 0.5 eV on Rh(111) compared to the work of Paul et al.62 and this discrepancy may be attributed to the inclusion of nonlocal electron correlation effects in our calculations.16
We proceed by investigating the adsorption of N2O* over Cu(111) and the Cu-based alloy surfaces where Ni atoms are either distributed as isolated atoms or as Ni–Ni dimers. In general, we find that N2O* interacts weakly with Cu(111) (Table 1) and that the most stable adsorption geometries thereon are η2-(Nt{hcp},O{top}) and η1 for which Eads(N2O) = −0.25 eV and −0.21 eV, respectively. Yet, the presence of a small amount of Ni on the surface layer of Cu(111) brings about drastic changes with regard to the binding strength of N2O* (Table 1). Thus, the most stable adsorption geometry on the Ni/Cu(111) SAA surface is η1 (Eads(N2O) = −0.70 eV), where the Nt atom of N2O* interacts closely with the isolated Ni atom. By contrast, the η2NbNt and η2-(Nt{hcp},O{top}) are the most favourable adsorption modes for Ni2Cu(111), with Eads(N2O) = −0.74 eV Eads(N2O) = −0.73 eV, respectively. Crucially, the corresponding adsorption processes are about 0.5 eV more exothermic than the η1 and η2-(Nt{hcp},O{top}) modes on Cu(111), thereby highlighting the potential of the Ni/Cu dilute alloys for the decomposition of N2O*. With this in mind, we examine the latter reaction over Cu(111) and the Cu-based surfaces.
3.2. N2O* formation and activation on Cu-based surfaces – the “conventional” reaction path
In order to verify the reliability of our data, we first perform calculations in relation to the activation of N2O* on the Rh(111) surface, and compare our results to those reported in previous theoretical works. The computed reaction pathway for the decomposition of N2O* to either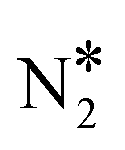 + O* or NO* + N* is displayed in Fig. S3.† In this “conventional” reaction pathway the transformation of NO* + N* to
+ O* or NO* + N* is displayed in Fig. S3.† In this “conventional” reaction pathway the transformation of NO* + N* to  + O* proceeds via the η1 adsorption structure (Fig. S3†), and our computed activation barriers are congruent with previously calculated values. For example, Paul et al.62 reported an activation barrier of 0.34 eV for the transformation of the η2NtOt structure to the η1 structure; this number is in good agreement with our computed barrier (Ea = 0.38 eV from state (4) to state (3) in Fig. S3†). Another example is the required barrier for the decomposition of the η2NbNt (state (1) in Fig. S3†) structure to NO* + N*. The values for this work and ref. 62 are 0.36 eV and 0.41 eV, respectively. Consequently, we use our computational setup and study the decomposition of N2O* on Cu(111), Ni/Cu(111) SAA and Ni2Cu(111) surfaces.
+ O* proceeds via the η1 adsorption structure (Fig. S3†), and our computed activation barriers are congruent with previously calculated values. For example, Paul et al.62 reported an activation barrier of 0.34 eV for the transformation of the η2NtOt structure to the η1 structure; this number is in good agreement with our computed barrier (Ea = 0.38 eV from state (4) to state (3) in Fig. S3†). Another example is the required barrier for the decomposition of the η2NbNt (state (1) in Fig. S3†) structure to NO* + N*. The values for this work and ref. 62 are 0.36 eV and 0.41 eV, respectively. Consequently, we use our computational setup and study the decomposition of N2O* on Cu(111), Ni/Cu(111) SAA and Ni2Cu(111) surfaces.
Fig. 2(A) shows the “conventional” decomposition pathway for Cu(111), where the η1 structure “connects” the NO* + N* and  + O* states. During the NO + CO reaction, the combination of NO* and N* species may result in the formation of N2O*, which ideally should be decomposed to
+ O* states. During the NO + CO reaction, the combination of NO* and N* species may result in the formation of N2O*, which ideally should be decomposed to 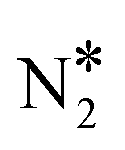 + O*. Once formed, N2O* adopts the η2NbNt structure, and starting from this geometry on Cu(111) (state (1) in Fig. 2(A)), we realise that the formation of
+ O*. Once formed, N2O* adopts the η2NbNt structure, and starting from this geometry on Cu(111) (state (1) in Fig. 2(A)), we realise that the formation of  and O* is thermodynamically and kinetically favoured over the formation of NO* and O*. In particular, the decomposition of η2NbNt N2O* to NO* + O* requires the traversing of a barrier of 0.94 eV, while the three barriers to be traversed for the formation of
and O* is thermodynamically and kinetically favoured over the formation of NO* and O*. In particular, the decomposition of η2NbNt N2O* to NO* + O* requires the traversing of a barrier of 0.94 eV, while the three barriers to be traversed for the formation of 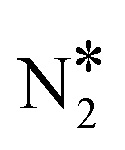 + O* are only 0.14 eV, 0.06 eV and 0.06 eV. Yet, we conjecture that Cu(111) will be susceptible to the production of N2O during the NO + CO reaction. This is because of the following reasons: (1) N2O* can be formed from NO* and N* species with a relatively small kinetic barrier of 0.44 eV (Fig. 2(A)); once N2O* is formed from NO* and N* in the η2NbNt structure (state (1) in Fig. 2(A)), its desorption is the most probable scenario (Table 1); and (3) even in the η1 and η2NtOt geometries, N2O* binds weakly on Cu(111) and its desorption will be proceeding at considerable rates even at moderate reaction temperatures.
+ O* are only 0.14 eV, 0.06 eV and 0.06 eV. Yet, we conjecture that Cu(111) will be susceptible to the production of N2O during the NO + CO reaction. This is because of the following reasons: (1) N2O* can be formed from NO* and N* species with a relatively small kinetic barrier of 0.44 eV (Fig. 2(A)); once N2O* is formed from NO* and N* in the η2NbNt structure (state (1) in Fig. 2(A)), its desorption is the most probable scenario (Table 1); and (3) even in the η1 and η2NtOt geometries, N2O* binds weakly on Cu(111) and its desorption will be proceeding at considerable rates even at moderate reaction temperatures.
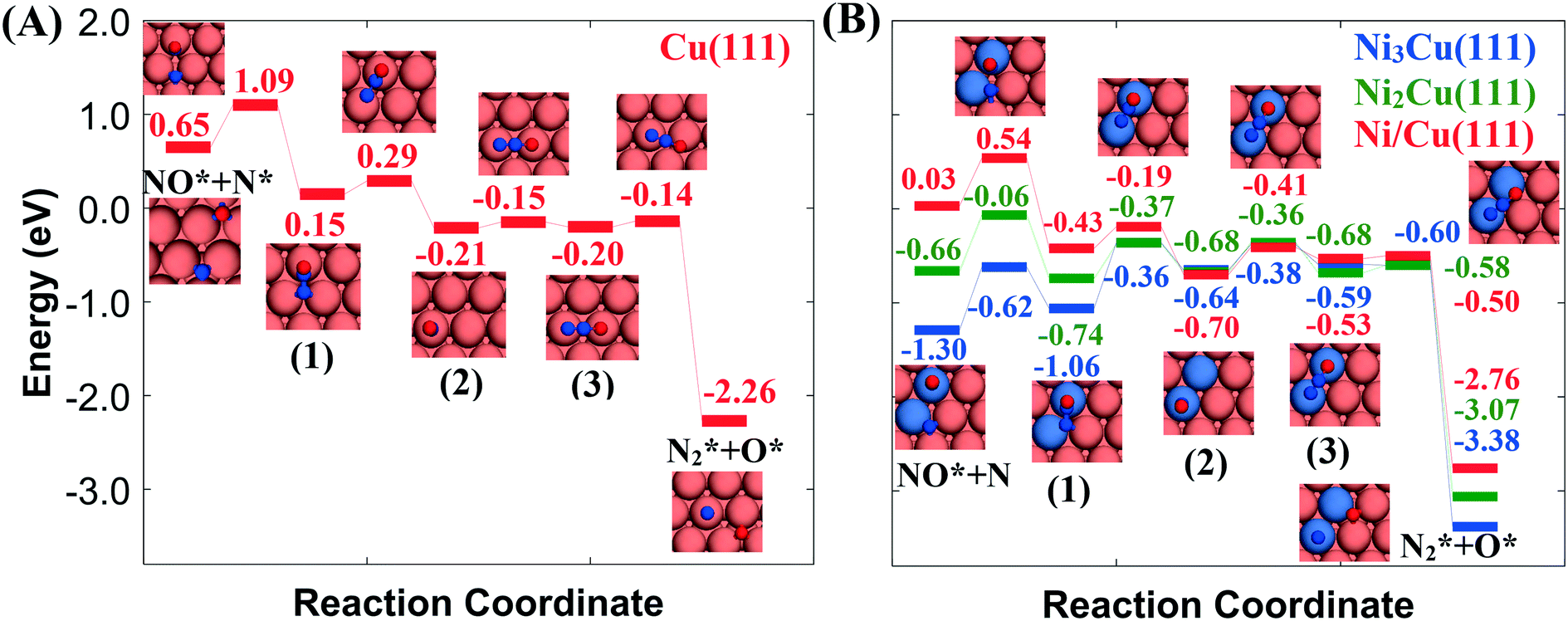 | ||
Fig. 2 Reaction path for the decomposition of N2O* to NO* + N* or  + O* over (A) Cu(111) surface; (B) Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111) surfaces. The numbering of the adsorbed configurations of N2O is as follows: (1) η2NbNt, (2) η1 and (3) η2NtOt. The zero level corresponds to infinitely separated (and thus non-interacting) N2O molecule and clean slab. States without any labelling are transition states. Side views of the different states are shown in the ESI.† Ni, Cu, O and N atoms are shown in purple, orange, red and blue, respectively. + O* over (A) Cu(111) surface; (B) Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111) surfaces. The numbering of the adsorbed configurations of N2O is as follows: (1) η2NbNt, (2) η1 and (3) η2NtOt. The zero level corresponds to infinitely separated (and thus non-interacting) N2O molecule and clean slab. States without any labelling are transition states. Side views of the different states are shown in the ESI.† Ni, Cu, O and N atoms are shown in purple, orange, red and blue, respectively. | ||
On the contrary, we find that the decomposition of N2O* may be significantly promoted by embedding one or two Ni atoms on Cu(111), thereby forming a single atom alloy or a dilute alloy surface,64–71 where in the latter case, Ni atoms are organised as dimers or trimers. We note that ab initio Monte Carlo simulations predict that small Ni clusters (e.g. Ni–Ni dimers) are abundant in Ni/Cu dilute alloy surfaces under vacuum conditions, while their thermodynamic stability can be further enhanced by exposing the alloy surface to CO at a range of partial pressures that lead to dopant fractional coverages less than 1.72 The computed desorption energies for η2NbNt N2O* (state (1) in Fig. 2(B)) on Ni/Cu(111) SAA and Ni2Cu(111) are 0.43 eV and 0.74 eV, respectively. By considering this adsorption structure as the starting point, we note that the transformation of N2O* to structure η1 (i.e. state (2)) and η2NtOt (i.e. state (3)), and the decomposition of the latter to 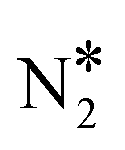 + O would generally traverse small barriers, which are always less than 0.30 eV and 0.40 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively. Thus, η2NbNt N2O* (state (1)) would prefer to decompose to
+ O would generally traverse small barriers, which are always less than 0.30 eV and 0.40 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively. Thus, η2NbNt N2O* (state (1)) would prefer to decompose to  + O*, than desorb to the gas phase (Fig. 2(B)).
+ O*, than desorb to the gas phase (Fig. 2(B)).
The exothermic adsorption of η2NbNt N2O* (i.e. the first adopted structure after N2O* formation from NO* and N*) will, to certain extent, prevent the N2O* desorption to the gas phase. This will increase the probability of “trapping” N2O* to the catalyst surface and therefore the probability for its decomposition. Moreover, even stronger η2NbNt N2O* binding should be expected on Ni–Ni dimers and Ni single atoms that are embedded on more open surfaces than the densely packed (111) and on undercoordinated sites that can be found in catalytic nanoparticles.
Another point that merits consideration is that the selectivity toward the decomposition products (NO* + N* or  + O*) can be altered by tuning the size of the Ni cluster. To better illustrate this point, we present the corresponding N2O* decomposition pathway over a Cu(111) with an embedded Ni trimer (Ni3Cu(111) in Fig. 2(B)). Interestingly, the kinetic barrier for the formation of the η2NbNt geometry (state (1) in Fig. 2(B)) from NO* and N* increases monotonically at increasing size of the Ni cluster (Ea = 0.51 eV, 0.60 eV and 0.68 eV for Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111), respectively). The opposite is true for the reverse reaction (i.e. η2NbNt N2O* to NO* + N*) for which Ea = 0.97 eV, 0.68 eV and 0.44 eV for Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111), respectively. This result underlines the importance of developing ways to control the architecture of dilute alloy surfaces and former studies discuss that this may be achieved under reactive conditions.72–75
+ O*) can be altered by tuning the size of the Ni cluster. To better illustrate this point, we present the corresponding N2O* decomposition pathway over a Cu(111) with an embedded Ni trimer (Ni3Cu(111) in Fig. 2(B)). Interestingly, the kinetic barrier for the formation of the η2NbNt geometry (state (1) in Fig. 2(B)) from NO* and N* increases monotonically at increasing size of the Ni cluster (Ea = 0.51 eV, 0.60 eV and 0.68 eV for Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111), respectively). The opposite is true for the reverse reaction (i.e. η2NbNt N2O* to NO* + N*) for which Ea = 0.97 eV, 0.68 eV and 0.44 eV for Ni/Cu(111) SAA, Ni2Cu(111) and Ni3Cu(111), respectively. This result underlines the importance of developing ways to control the architecture of dilute alloy surfaces and former studies discuss that this may be achieved under reactive conditions.72–75
3.3. N2O formation and activation on Cu-based surfaces – an alternative reaction path
Besides the “conventional” route for the decomposition of N2O* (Fig. 2), we have identified an alternative reaction pathway which, to the best of our knowledge, has not been reported before. This path exists only on Cu(111) and on the Ni/Cu dilute alloy surfaces. The decomposition of N2O* to + O* happens without transformation to the η1 structure as in the conventional pathway. In contrast, in this pathway the two decomposed states (i.e. NO* + N* and
+ O* happens without transformation to the η1 structure as in the conventional pathway. In contrast, in this pathway the two decomposed states (i.e. NO* + N* and  + O*) are “connected” via the η2-(Nt{fcc},O{top}) adsorption structure (this is as Fig. 1(D) but over an fcc site; the two adsorption structures exhibit the same binding strength – Eads(N2O) = −0.74 eV for Ni2Cu(111)). After performing a number of test simulations, we could not identify the same path on Rh(111), and this might explain why it was not reported in previous studies.62
+ O*) are “connected” via the η2-(Nt{fcc},O{top}) adsorption structure (this is as Fig. 1(D) but over an fcc site; the two adsorption structures exhibit the same binding strength – Eads(N2O) = −0.74 eV for Ni2Cu(111)). After performing a number of test simulations, we could not identify the same path on Rh(111), and this might explain why it was not reported in previous studies.62
For all the Cu-based surfaces, Fig. 3 shows that η2NbNt N2O* is formed in the same way as in the reaction path of Fig. 2. Then the η2NbNt N2O* rotates around the axis of the N–N bond, thereby bringing the more electronegative O closer to the surface. Interestingly, once O is closer to the Ni/Cu(111) SAA, the N–O bond is immediately cleaved and the kinetic barrier for this process is only 0.23 eV (Fig. 3(B)). The ease by which the N–O is broken over the Ni/Cu SAA surface may be associated with the sharp and narrow distribution of the electron density of the single Ni atom close to the Fermi level,4,76 and it is expected that back-donation to the 3π antibonding orbital of N2O* enables the facile activation of the N–O bond.
 | ||
| Fig. 3 A second reaction path for the decomposition of N2O* on (A) Cu(111); (B) Ni/Cu(111) SAA and (C) Ni2Cu(111). State (1) corresponds to a η2NbNt structure in all panels. The η2-(Nt{fcc},O{top}) structure is state (2) and state (3) in panels (A) and (C), respectively. The zero level corresponds to infinitely separated (and thus non-interacting) N2O molecule and clean slab. States without any labelling are transition states. Side views of the different states are shown in the ESI.† Ni, Cu, O and N atoms are shown in purple, orange, red and blue, respectively. | ||
By contrast, the decomposition of N2O* to  and O* is taking place through the η2- (Nt{fcc},O{top}) geometry (state (2) and (3) in Fig. 3(A) and (C), respectively) over Cu(111) and Ni2Cu(111). The intervening barriers between the η2NbNt and
and O* is taking place through the η2- (Nt{fcc},O{top}) geometry (state (2) and (3) in Fig. 3(A) and (C), respectively) over Cu(111) and Ni2Cu(111). The intervening barriers between the η2NbNt and  + O* states are small (≤0.23 eV). Irrespective of these low kinetic barriers, Cu(111) is still expected to be prone to releasing N2O* to the gas phase given the generally weak N2O*–Cu(111) interaction (Fig. 3(A)). The same is not true for Ni2Cu(111) where the N2O* desorption energy is in the range of 0.65–0.74 eV, while the kinetic barriers that lead to
+ O* states are small (≤0.23 eV). Irrespective of these low kinetic barriers, Cu(111) is still expected to be prone to releasing N2O* to the gas phase given the generally weak N2O*–Cu(111) interaction (Fig. 3(A)). The same is not true for Ni2Cu(111) where the N2O* desorption energy is in the range of 0.65–0.74 eV, while the kinetic barriers that lead to 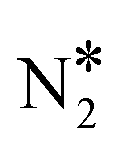 + O* are between 0.06 eV and 0.23 eV (Fig. 3(C)). Given the similar energetics between the pathway of Fig. 3 and the “conventional” one, we conclude that both of them need to be considered in the reaction mechanism of the NO + CO reaction. Importantly, the existence of alternative N2O* decomposition paths may provide an explanation of the high selectivity to N2 exhibited by dilute Cu-based alloys.33
+ O* are between 0.06 eV and 0.23 eV (Fig. 3(C)). Given the similar energetics between the pathway of Fig. 3 and the “conventional” one, we conclude that both of them need to be considered in the reaction mechanism of the NO + CO reaction. Importantly, the existence of alternative N2O* decomposition paths may provide an explanation of the high selectivity to N2 exhibited by dilute Cu-based alloys.33
3.4. N2O formation and activation on Cu-based surfaces through the formation of 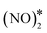
Thus far, the formation of nitrous oxide was assumed to proceed through the coupling of NO* and N* species (Fig. 2 and 3). NO* is of course the product of the molecular adsorption of gas-phase nitric oxide. On the other hand, the existence of N* species implies prior scission of the N–O bond. In general, low-index coinage metal surfaces exhibit large kinetic barriers for the direct dissociation of NO* (Ea = 1.57 eV for Cu (111) and (100), and Ea > 2.5 eV for Ag and Au (111) and (100) surfaces),4 thereby being ineffective at activating the N–O bond of NO*. Yet, they are known to be active for the reduction of NO, which is mainly converted to N2O. The activity of coinage metal surfaces is ascribed to the formation of NO* dimer species (i.e.
 ) whose N–O bonds are more easily activated than those of monomeric NO*.33,54,77–79 This species is formed owing to vdW interactions between neighbouring NO* species,80 and may be observed even at relatively low NO* coverages over Cu(111).43 On the contrary, our calculations indicate that NO* is adsorbed as a monomer on Rh(111) and this is corroborated by near edge X-ray absorption fine structure spectroscopy.81
) whose N–O bonds are more easily activated than those of monomeric NO*.33,54,77–79 This species is formed owing to vdW interactions between neighbouring NO* species,80 and may be observed even at relatively low NO* coverages over Cu(111).43 On the contrary, our calculations indicate that NO* is adsorbed as a monomer on Rh(111) and this is corroborated by near edge X-ray absorption fine structure spectroscopy.81
The most energetically favoured adsorption structure of NO* on Cu(111) is an N-down geometry where the N–O bond axis is perpendicular to the surface, and N is above an fcc hollow site (Eads(NO) = −1.55 eV).4 A stable NO dimer is formed when two NO* species are adsorbed on adjacent fcc sites (NO* + NO* state in Fig. 4(A)). We note that in the relaxed geometry of this state, the O atoms of the neighbouring nitric oxide adspecies are slightly tilted towards each other (Fig. 4(A)). The thermodynamic stability of this configuration has been confirmed by other DFT studies, as well as in scanning tunnelling microscopy experiments.43,80 The two neighbouring NO* species can be converted to N2O* (with an η1 structure) and O* (see state (1) in Fig. 4(A)). This is happening via a transition state where one of the two NO* adsorbates bends down to the Cu(111) surface, while the other is slightly lifted (Fig. 4(A)). Once η1 N2O* is formed, its decomposition occurs in the same way as in Fig. 2(A), namely through the formation of the η2NtOt structure. We note that the structure of the 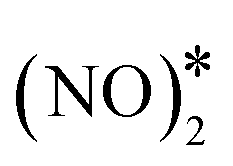 transition state, and the computed barrier for the scission of the N–O bond via the
transition state, and the computed barrier for the scission of the N–O bond via the 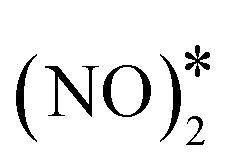 precursor (Ea = 0.84 eV) are in excellent agreement with the DFT calculations by Bogicevic and Hass (Ea = 0.82 eV),54 thereby furnishing further evidence for the reliability of our calculations.
precursor (Ea = 0.84 eV) are in excellent agreement with the DFT calculations by Bogicevic and Hass (Ea = 0.82 eV),54 thereby furnishing further evidence for the reliability of our calculations.
 | ||
Fig. 4 Reaction pathway for the formation and decomposition of N2O* via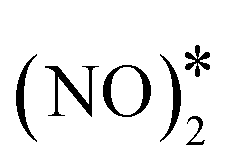 for (A) Cu(111); (B) Ni/Cu(111) SAA; and (C) Ni2Cu(111). The zero level corresponds to two non-interacting gas-phase NO molecules and a clean slab. States without any labelling are transition states. Side views of the different states are shown in the ESI.† Ni, Cu, O and N atoms are shown in purple, orange, red and blue, respectively. for (A) Cu(111); (B) Ni/Cu(111) SAA; and (C) Ni2Cu(111). The zero level corresponds to two non-interacting gas-phase NO molecules and a clean slab. States without any labelling are transition states. Side views of the different states are shown in the ESI.† Ni, Cu, O and N atoms are shown in purple, orange, red and blue, respectively. | ||
We continue by investigating the same reaction pathway over the Ni/Cu(111) SAA and Ni2Cu(111) surfaces. Our calculations show that the formation of N2O* via the dimerisation route is indeed possible over small Ni clusters. In contrast to Cu(111), on these dilute alloys 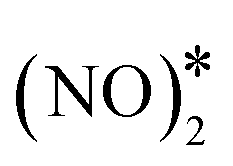 adopts a flat geometry parallel to the surfaces in the transition state, and the computed kinetic barriers are 1.27 eV and 1.30 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively (Fig. 4(A) and (B)). These values are higher than the computed barrier for Cu(111) – (Ea = 0.84 eV), and this may be attributed to the extra energy cost required for bending down both NO* species. Nevertheless, they are lower than or equal to the corresponding kinetic barriers for the direct dissociation of NO* (Ea = 1.47 eV and 1.30 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively).4 Therefore, the formation and decomposition of N2O* through dimerisation is another pathway that should be included in the reaction mechanism of the NO + CO reaction over the Cu-based alloy surfaces.
adopts a flat geometry parallel to the surfaces in the transition state, and the computed kinetic barriers are 1.27 eV and 1.30 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively (Fig. 4(A) and (B)). These values are higher than the computed barrier for Cu(111) – (Ea = 0.84 eV), and this may be attributed to the extra energy cost required for bending down both NO* species. Nevertheless, they are lower than or equal to the corresponding kinetic barriers for the direct dissociation of NO* (Ea = 1.47 eV and 1.30 eV for Ni/Cu(111) SAA and Ni2Cu(111), respectively).4 Therefore, the formation and decomposition of N2O* through dimerisation is another pathway that should be included in the reaction mechanism of the NO + CO reaction over the Cu-based alloy surfaces.
To elucidate the effect of the Ni cluster size to the formation rate of η1 N2O* and O* via the 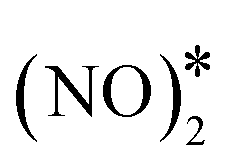 intermediate, we perform additional calculations for the Ni3Cu(111) surface (Fig. S5†). On this surface, we compute Ea = 1.77 eV and Ea = 1.37 eV for the splitting of the N–O bond via dimerisation (Fig. S5†) and via the direct NO* dissociation,4 respectively. Additionally, we note that on Ni dimers and trimers the formed η1 N2O* can be transformed to the η2NtOt (state (3) in Fig. 4(C) for Ni2Cu), and decompose to
intermediate, we perform additional calculations for the Ni3Cu(111) surface (Fig. S5†). On this surface, we compute Ea = 1.77 eV and Ea = 1.37 eV for the splitting of the N–O bond via dimerisation (Fig. S5†) and via the direct NO* dissociation,4 respectively. Additionally, we note that on Ni dimers and trimers the formed η1 N2O* can be transformed to the η2NtOt (state (3) in Fig. 4(C) for Ni2Cu), and decompose to 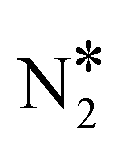 + 2O* only after O* spillover to Cu(111). The barrier for O* diffusion from a mixed hollow site to a Cu hollow site over Ni2Cu(111) (from state (1) to state (2) in Fig. 4(C)) is 0.61 eV. Therefore, this extra energy cost in conjunction with the large kinetic barrier for the scission of the N–O bond of
+ 2O* only after O* spillover to Cu(111). The barrier for O* diffusion from a mixed hollow site to a Cu hollow site over Ni2Cu(111) (from state (1) to state (2) in Fig. 4(C)) is 0.61 eV. Therefore, this extra energy cost in conjunction with the large kinetic barrier for the scission of the N–O bond of 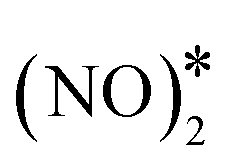 render the decomposition of N2O* through the dimerisation pathway less likely on Ni clusters with more than two Ni atoms.
render the decomposition of N2O* through the dimerisation pathway less likely on Ni clusters with more than two Ni atoms.
3.5. The microkinetics of the NO + CO reaction over Ni/Cu dilute alloys
Using the computed energetics for the decomposition of N2O* in conjunction with previous results for the formation of , CO* oxidation and NO* decomposition over Ni/Cu surfaces,4 we parameterise a microkinetic model for the NO + CO reaction. Our studies include one site type for Cu(111) and Rh(111) surfaces, and two site types for the bimetallic surfaces (see section 2). The goal is a preliminary assessment of the catalytic performance of the Ni/Cu dilute alloys and a comparison to Cu(111) and Rh(111). Accordingly, the microkinetic simulations are performed in the absence of adsorbate–adsorbate interactions,82–85 whose effects on the coverage profiles, and consequently on the catalytic performance of the Cu-based surfaces may be important (this is part of ongoing research). Regarding the bimetallic surfaces, we assume that Ni* species (single atoms or dimers) can be occupied by one adspecies (e.g. CO*), which can react with another adspecies on a Cu site (e.g. O*) and form a product on the Ni site (e.g.
, CO* oxidation and NO* decomposition over Ni/Cu surfaces,4 we parameterise a microkinetic model for the NO + CO reaction. Our studies include one site type for Cu(111) and Rh(111) surfaces, and two site types for the bimetallic surfaces (see section 2). The goal is a preliminary assessment of the catalytic performance of the Ni/Cu dilute alloys and a comparison to Cu(111) and Rh(111). Accordingly, the microkinetic simulations are performed in the absence of adsorbate–adsorbate interactions,82–85 whose effects on the coverage profiles, and consequently on the catalytic performance of the Cu-based surfaces may be important (this is part of ongoing research). Regarding the bimetallic surfaces, we assume that Ni* species (single atoms or dimers) can be occupied by one adspecies (e.g. CO*), which can react with another adspecies on a Cu site (e.g. O*) and form a product on the Ni site (e.g.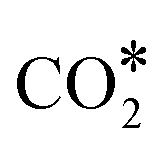 ). Such events are treated as reactions that take place on the dopant site, and follow the energetics computed over the Ni site of the Ni/Cu surfaces. On the contrary, the Cu(111) energetics are used if the reaction involves two adspecies that are both on Cu sites (Table S3†). Despite their simplicity, such microkinetic models are capable of capturing the salient features of experimental trends,86 providing mechanistic insights,87 and aiding in the identification of the active site during catalysis.88,89
). Such events are treated as reactions that take place on the dopant site, and follow the energetics computed over the Ni site of the Ni/Cu surfaces. On the contrary, the Cu(111) energetics are used if the reaction involves two adspecies that are both on Cu sites (Table S3†). Despite their simplicity, such microkinetic models are capable of capturing the salient features of experimental trends,86 providing mechanistic insights,87 and aiding in the identification of the active site during catalysis.88,89
The NO + CO reaction mechanism is composed of 16 reversible reaction steps, shown in Table 2 along with their forward and reverse barriers. For all simulations the total pressure is set to 16.0 Torr with PNO = PCO = 8.0 Torr, thereby replicating the experimental conditions of Belton and co-workers.13 At this point, we note that the associative desorption of O2 and the formation of 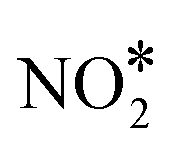 are reactions through which O* may be removed from the surface and they could be included in the microkinetic model. However, both of them exhibit very high kinetic barriers, and on this basis are excluded from the reaction mechanism. For example, the computed barrier for the
are reactions through which O* may be removed from the surface and they could be included in the microkinetic model. However, both of them exhibit very high kinetic barriers, and on this basis are excluded from the reaction mechanism. For example, the computed barrier for the  association reaction on Cu(111) is 2.10 eV, while the barrier for the reverse process is just 0.16 eV (see section 7 in the ESI†); these values are in reasonable agreement with former DFT calculations.90 Along the same lines, we find that the dissociation of
association reaction on Cu(111) is 2.10 eV, while the barrier for the reverse process is just 0.16 eV (see section 7 in the ESI†); these values are in reasonable agreement with former DFT calculations.90 Along the same lines, we find that the dissociation of 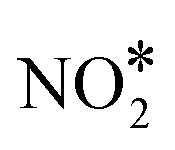 to NO* + O* is significantly more facile than its formation and its desorption (see section 7 in the ESI†).
to NO* + O* is significantly more facile than its formation and its desorption (see section 7 in the ESI†).
We first simulate the NO + CO reaction on Rh(111). The total coverages of the surface species and the DRC analysis for this surface are shown in Fig. 5(A). The coverage profiles reveal that the catalyst surface is saturated with NO* species up to temperatures of 1000 K. Under these conditions, the high surface coverage gives rise to steric hindrance effects, which prevent the dissociation of NO*. This behaviour has been reported in the experimental work of Herman et al., and is in qualitative agreement with the fact that Rh(111) is catalytically active only at temperatures higher than 625 K.13 Moreover, our model predicts that surface sites are freed up by NO adspecies only at T > 1000 K; this high “T threshold” can be attributed to (1) the absence of the repulsive NO*–NO* interactions in our microkinetics (see Fig. S4 in the ESI†); and (2) the very strong NO*–Rh(111) interaction predicted by the optB86b-vdW functional. In particular, NO*–NO* interactions may contribute to the reduction of the surface coverage, and in turn, this will generate free sites whereon the dissociation of NO* can happen at lower temperatures than those predicted by our model.91,92 Regarding the second point, we find that the most stable adsorption site for NO* on Rh(111) is hcp, in line with previous computational and experimental works.5,93,94 However, we compute Eads(NO) = −2.85 eV, which is larger than the PW91 values of Mavrikakis et al.94 (−2.39 eV – 2 × 2 cell) and of González et al.93 (−2.62 eV – 3 × 3 cell). Unfortunately, at coverages of 0.11 ML, like in our DFT calculations, accurate experimental measurement of Eads(NO) is challenging because of the tendency of NO* to dissociate on Rh(111).94 The reduction of the NO* surface coverage gives rise to the formation of N* and O* at T > 1000 K. The accumulation of N* species in the temperature range of 1000–1200 K, is associated with the inefficiency of Rh(111) in forming η2NbNt N2O* (R9, Ea = 1.50 eV) and 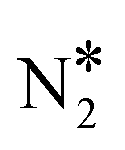 (R16, Ea = 1.85 eV) – (Table 2). Both reaction steps are rate-limiting with a positive DRC coefficient (0.19 ≤ XDRC,R9 ≤ 0.30) between 1100 K and 1300 K (Fig. 5(A)). Along the same lines, the build-up of O* is ascribed to the moderate activation barrier for the CO* oxidation reaction (Ea = 1.17 eV – R14 in Table 2), which is the only reaction that exhibits a reasonable activation barrier for the removal of O* from the surface.
(R16, Ea = 1.85 eV) – (Table 2). Both reaction steps are rate-limiting with a positive DRC coefficient (0.19 ≤ XDRC,R9 ≤ 0.30) between 1100 K and 1300 K (Fig. 5(A)). Along the same lines, the build-up of O* is ascribed to the moderate activation barrier for the CO* oxidation reaction (Ea = 1.17 eV – R14 in Table 2), which is the only reaction that exhibits a reasonable activation barrier for the removal of O* from the surface.
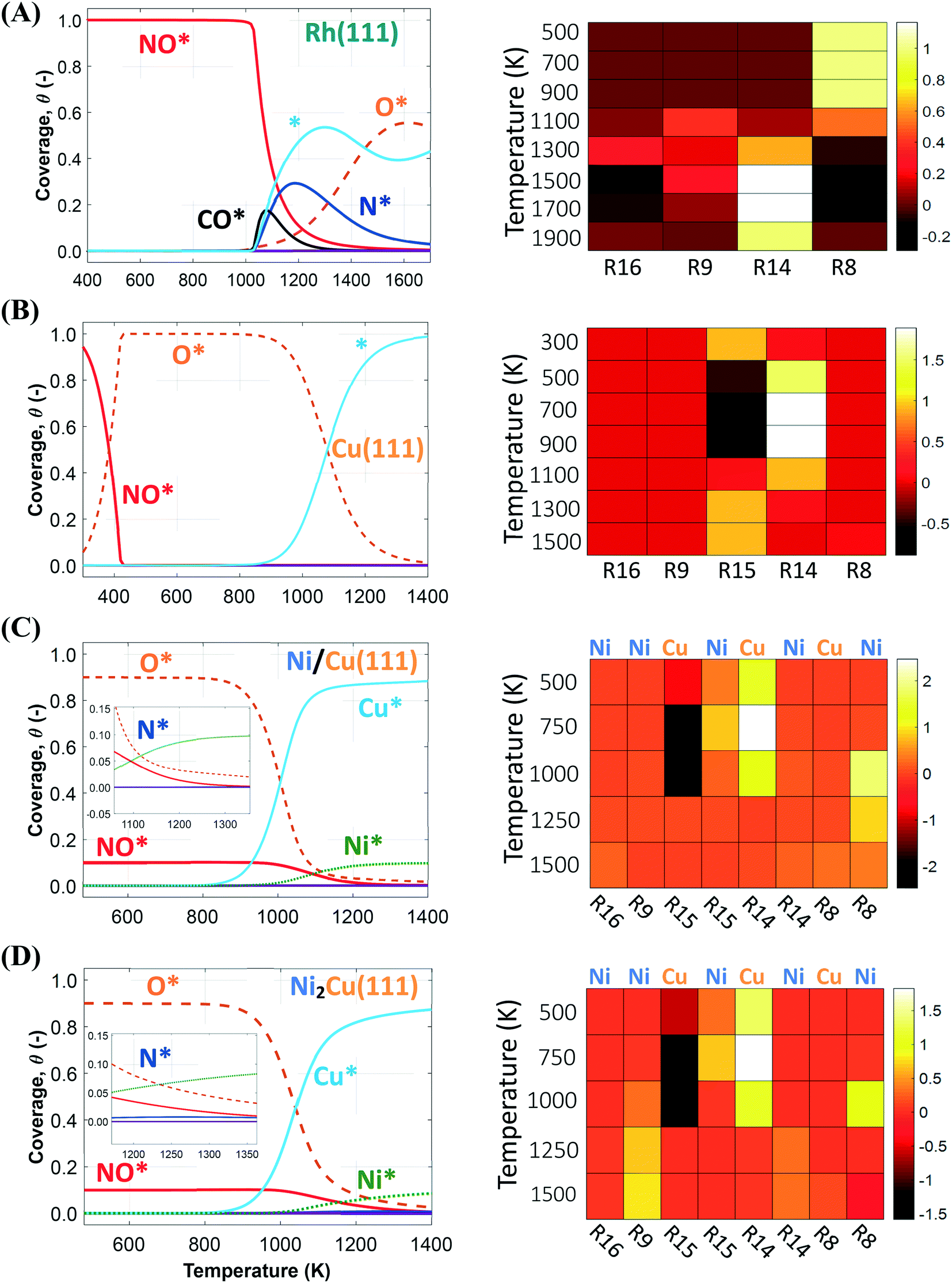 | ||
| Fig. 5 Total coverage (eqn (3)) profiles and DRC coefficients (XRC) for the NO + CO reaction steps for (A) Rh(111); (B) Cu(111); (C) Ni/Cu(111) SAA; and (D) Ni2Cu(111). XRC values are presented at various temperatures by means of heatmaps; the site whereon the reaction occurs is shown on the top of the heatmaps for the bimetallic surfaces. | ||
The corresponding coverage profiles and DRC analysis for Cu(111) are displayed in Fig. 5(B). Cu(111) exhibits rather different behaviour than Rh(111). In particular, at T < 350 K all surface sites are occupied by NO*, but at T > 350 K there is a sharp increase in the coverage of the O* species. This sharp transition is attributed to the activation of the N–O bond via the NO* dimerisation reaction (Ea = 0.84 eV) – (R15 in Table 2), which converts 2NO* to O* and η1 N2O*. The catalyst surface remains fully covered by O* within the temperature range of 420–900 K. Accordingly, our DRC analysis shows that under these conditions, the oxidation of CO* controls the reaction rate, and that the NO* dimerisation is an inhibiting step as it adds more O* onto the surface (Fig. 5(B)).
The last two coverage profiles shown in Fig. 5 (panels (C) and (D)) are those for the Ni/Cu alloy surfaces. These surfaces contain a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sites, out of which 9000 are Cu sites (Cu*) and 1000 are Ni sites (Ni*). The coverage profiles are very similar in both cases, and indicate that Cu* sites are covered with O* up to ca. 900 K (similar to Cu(111)), while Ni* sites are poisoned by NO*. A disparity between the two surfaces is seen between 1000 K and 1500 K, where we observe a small build-up of N* over Ni2Cu(111) only (Fig. 5(C) and (D)). The presence of N* on the latter surface is indicative of the direct NO* dissociation (R8, Ea = 1.30 eV – Table 2), which happens to a smaller extent on the SAA surface (R8, Ea = 1.47 eV – Table 2). Markedly, the N* accumulation remains at low levels thanks to the efficiency of Ni2Cu(111) in forming
000 sites, out of which 9000 are Cu sites (Cu*) and 1000 are Ni sites (Ni*). The coverage profiles are very similar in both cases, and indicate that Cu* sites are covered with O* up to ca. 900 K (similar to Cu(111)), while Ni* sites are poisoned by NO*. A disparity between the two surfaces is seen between 1000 K and 1500 K, where we observe a small build-up of N* over Ni2Cu(111) only (Fig. 5(C) and (D)). The presence of N* on the latter surface is indicative of the direct NO* dissociation (R8, Ea = 1.30 eV – Table 2), which happens to a smaller extent on the SAA surface (R8, Ea = 1.47 eV – Table 2). Markedly, the N* accumulation remains at low levels thanks to the efficiency of Ni2Cu(111) in forming  and η2NbNt N2O* (Table 2). The latter can either decompose to
and η2NbNt N2O* (Table 2). The latter can either decompose to 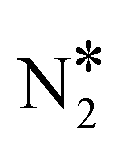 + O* (R10, R11, R12 and R13) or desorb (R5).
+ O* (R10, R11, R12 and R13) or desorb (R5).
Next, we examine the activity and selectivity to N2 of the four surfaces. The latter metric is computed as
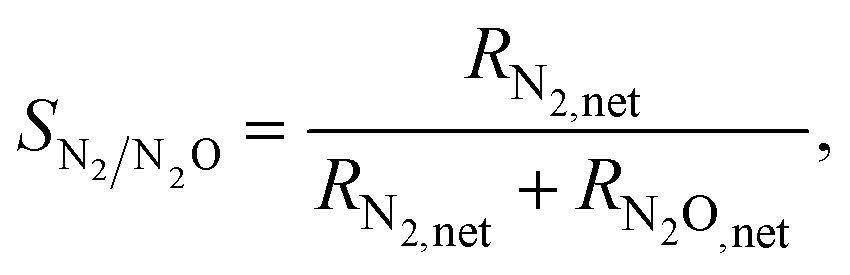 | (16) |
Fig. 6 displays the activity plots for the studied surfaces, where the catalytic rate is provided by the computed turnover frequency (TOF) at various temperatures. The observed trend for Rh(111) (Fig. 6(A)) can be rationalised based on the corresponding coverage plot (Fig. 5(A)). As seen in Fig. 6(A), the activity of Rh(111) is low below 950 K owing to the high NO* coverage, which hinders the direct NO* dissociation (Fig. 5(A)). On the contrary, for T > 950 K there is an increase in the catalytic activity. Initially the rate of N2O production is greater than that of N2, and only at T > 1200 K the two production rates become equal (Fig. 6(A)).
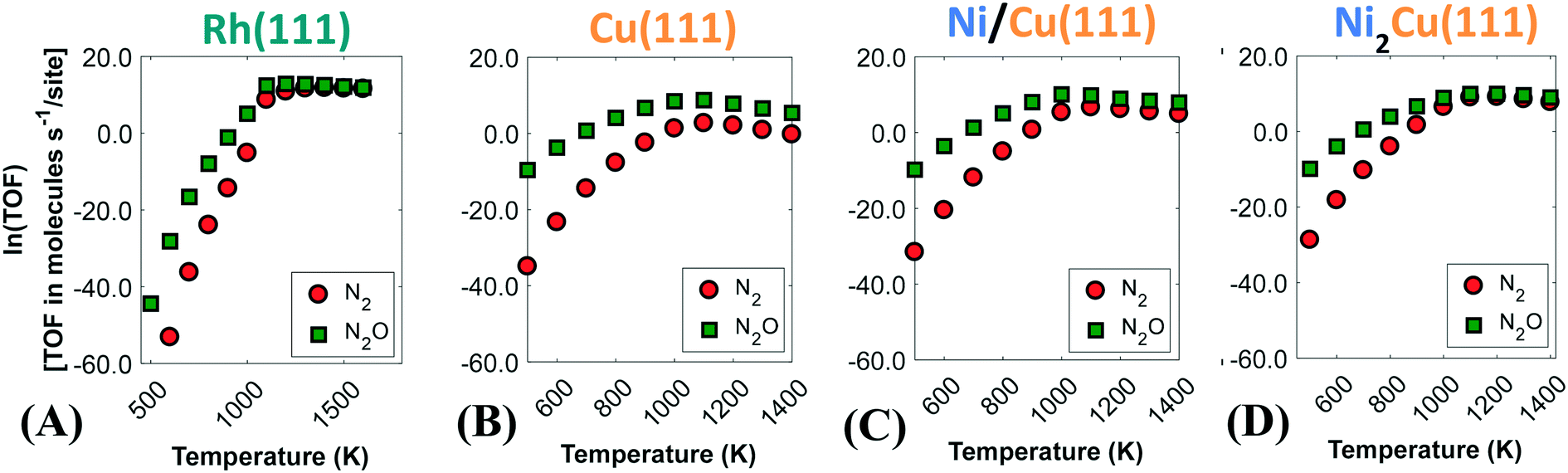 | ||
| Fig. 6 Rates of production of nitrogen-containing products for (A) Rh(111); (B) Cu(111); (C) Ni/Cu(111) SAA; and (D) Ni2Cu(111). | ||
Similarly to Rh(111), the catalytic activity of Cu(111) can be explained from the coverage profile plot in Fig. 5(B). For this surface, low (i.e. 300–500 K) and high (i.e. 500–1400 K) temperatures can be discussed separately. Between 300 K and 420 K, we observe that the catalytic activity increases steadily (see Fig. S11 in the ESI†), and the surface transitions from a NO*-rich phase to an O*-rich phase. As discussed earlier, this transition is associated with the dimerisation reaction (R15 in Table 2), which consumes NO*, releases N2O and yields O*. At ca. 420 K, there is a sharp reduction in the catalytic activity (Fig. S11††), and this is the result of the poisoning of Cu(111) by O* species. The surface remains in the poisoned state for temperatures up to ca. 700 K, where the removal of O* species happens efficiently and the dimerisation reaction begins to take place again at considerable rates (see Fig. 6(B) and the heatmap in Fig. 5(B)). Finally, for T > 1000 K there is a decrease in the catalytic activity (Fig. 6(B)) because under these conditions, the gaseous state of the reactants is preferred over adsorption on the catalytic surface. Throughout the investigated temperature range, the production rate of N2O is far greater than the production rate of N2, and this is attributed to the inability of Cu(111) to directly dissociate NO* as well as to the weak binding of the η1 N2O* produced by the dimerisation reaction.
On the other hand, enhanced catalytic activity can be achieved when Ni* species are present in Cu(111) (Fig. 6(C) and (D)). Remarkably, the production rate of N2 is considerably larger on Ni/Cu(111) SAA than on Cu(111) and even more so on Ni2Cu(111), where the N2 and N2O production rates become equal beyond 1000 K.
Given the importance of SN2/N2O, this section concludes with an investigation on this metric, followed by suggestions for further improvements in this regard. Regarding Rh(111), our microkinetic model predicts that the main nitrogen-containing product from Rh(111) at T < 1000 K is N2O, whilst the production of N2 exhibits a substantial increase beyond 1100 K (Fig. 7(A)). The latter temperature corresponds to the point where the surface sites are freed up (Fig. 5(A)), and the dissociation of NO* is enabled. Notably, this trend is qualitatively in line with the reactor experiments of Peden et al.12 on Rh(111). The experiments showed that Rh(111) exhibits poor selectivity to N2 for reaction temperatures up to 700 K; yet, the authors observed a sharp increase in SN2/N2O at temperatures higher than that. One should expect that closer quantitative agreement can be achieved by accounting for coverage effects, which will tend to decrease the surface coverage at T < 1000 K (see Fig. S4†), thereby freeing up sites and shifting the profiles of Fig. 5 to lower temperatures.
 | ||
| Fig. 7 (A) Selectivity to N2 for Cu(111), Rh(111) and the Ni/Cu alloys. (B) Maximum of the N2 selectivity peak at various N2O* adsorption energies. Values on the right correspond to smaller N2O* binding strength on Ni* than the DFT-computed (red shade); values on the left correspond to larger N2O* binding strength on Ni* than the DFT-computed (green shade). In the simulations of panel (B) only the desorption energy of N2O* was varied, whilst all the other kinetic barriers were kept fixed at their DFT-computed values. | ||
The same analysis for Cu(111) reveals that this surface is indeed susceptible to the formation of N2O (Fig. 7(A)). We find that the main way of forming N2O* (in the η1 structure) on Cu(111) is via the formation of the 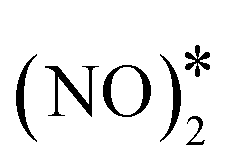 intermediate followed by N–O activation (R15). This is in line with molecular beam/infrared spectroscopy studies on other Cu low-index surfaces.77 The η1 N2O* can go through one of the following paths: (1) desorb directly (Ea = 0.21 eV); (2) transform to η2NtOt N2O* (Ea = 0.06 eV) and either desorb (Ea = 0.20 eV) or dissociate to
intermediate followed by N–O activation (R15). This is in line with molecular beam/infrared spectroscopy studies on other Cu low-index surfaces.77 The η1 N2O* can go through one of the following paths: (1) desorb directly (Ea = 0.21 eV); (2) transform to η2NtOt N2O* (Ea = 0.06 eV) and either desorb (Ea = 0.20 eV) or dissociate to 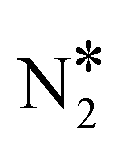 + O* (Ea = 0.05 eV); (3) transform to η2NbNt N2O* (Ea = 0.56 eV) and desorb spontaneously. Therefore, N2O* can easily undergo transformations over Cu(111), but in every new state there is a high probability for desorption, thereby explaining the poor N2 selectivity of this surface.
+ O* (Ea = 0.05 eV); (3) transform to η2NbNt N2O* (Ea = 0.56 eV) and desorb spontaneously. Therefore, N2O* can easily undergo transformations over Cu(111), but in every new state there is a high probability for desorption, thereby explaining the poor N2 selectivity of this surface.
Interestingly, the catalytic behaviour Ni/Cu(111) SAA and Ni2Cu(111) appears to be more similar to Rh(111), which is well established for the NO + CO reaction, than to Cu(111), which is the host metal (Fig. 7(A)). In more precise terms, it is observed that on each of the dilute alloy surfaces the selectivity to N2 remains low at T < 900 K but increases sharply at higher temperatures similarly to Rh(111). SN2/N2O for both Ni/Cu(111) SAA and Ni2Cu(111) exhibits an interesting behaviour by which it first increases for T > 900 K and then decreases at 1200 K.
To shed light on this behaviour, we have carried out additional microkinetic simulations for Ni2Cu(111) where the activation barrier of one of the following events on Ni* is assigned a very large value (e.g. 2.5 eV): (1) NO* direct dissociation (R8); (2) 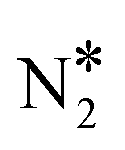 formation (R16); (3) NO* dimerisation (R15); and (4) η2NbNt N2O* formation (R9). In doing so, we record how the selectivity peak responds to the obstruction of the aforementioned events (see section 9 in the ESI†). We determine that the selectivity spike in the two bimetallic surfaces is associated with the direct dissociation of NO* and the formation of η2NbNt N2O*, which could subsequently decompose to
formation (R16); (3) NO* dimerisation (R15); and (4) η2NbNt N2O* formation (R9). In doing so, we record how the selectivity peak responds to the obstruction of the aforementioned events (see section 9 in the ESI†). We determine that the selectivity spike in the two bimetallic surfaces is associated with the direct dissociation of NO* and the formation of η2NbNt N2O*, which could subsequently decompose to 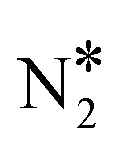 + O* (see section 9 in the ESI†). Therefore, it is the ability of Ni/Cu alloys to form and process η2NbNt N2O* that gives rise to the selectivity peak in Fig. 7(A). The selectivity to N2 enters a downturn because at T > 1200 K, there is a rise in the N2O* desorption rate. On the other hand, the formation of N2 on Rh(111) is solely relying on the direct dissociation of NO* and such a selectivity spike is not observed (Fig. 7(A)).
+ O* (see section 9 in the ESI†). Therefore, it is the ability of Ni/Cu alloys to form and process η2NbNt N2O* that gives rise to the selectivity peak in Fig. 7(A). The selectivity to N2 enters a downturn because at T > 1200 K, there is a rise in the N2O* desorption rate. On the other hand, the formation of N2 on Rh(111) is solely relying on the direct dissociation of NO* and such a selectivity spike is not observed (Fig. 7(A)).
Accordingly, the higher intensity of the N2 selectivity peak on Ni2Cu(111) than on Ni/Cu(111) SAA can be explained by: (1) the higher concentration of N* species on Ni2Cu(111) owing to its better ability to dissociate NO* as compared to Ni/Cu(111) SAA (see Table 2 and Fig. 5(D)); (2) the generally stronger interaction between N2O* and Ni2Cu(111) than that between N2O* and Ni/Cu(111) SAA (Table 2), noting that strong interaction will favour the decomposition of N2O* over its desorption.
Moreover, we explore the effect of the N2O* binding strength on the height of the selectivity peak on Ni2Cu(111) by performing a sensitivity analysis with respect to Eads(N2O) – (Fig. 7(B)). Remarkably, the adsorption energy of N2O* appears to have a great impact upon the N2 selectivity at 1100–200 K (Fig. 7(B)). For example, shifting the adsorption energy of all N2O* adsorption structures to more negative values by 0.15 eV and 0.30 eV (i.e. stronger binding) results to an increase in the maximum of the peak by 0.31 (from 0.33 to 0.67) and 0.52 (from 0.33 to 0.85), respectively. We conjecture that binding strengths of this magnitude may be provided by sites on more open low-index surfaces (e.g. (100) and (110)) but also on stepped surfaces, and if this is true, the presence of such sites will contribute dramatically to the N2 selectivity at low temperatures. Therefore, this result underscores the potential of well-engineered dilute Ni/Cu alloys for the NO + CO reaction and creates motivation for further investigations.
Finally, given the importance of Eads(N2O) we have computed the binding energy of N2O* using other vdW functionals, including optPBE-vdW, BEEF-vdW,95 and the Tkatchenko–Scheffler method (DFT-TS) – (see Table S4 in the ESI†). The latter method is similar to the DFT-D2 method of Grimme,96 with the difference that the dispersion coefficients and the damping function in the dispersion correction are dependent on the charge density.97 These additional calculations highlight that significant variations in the predicted Eads(N2O) should be expected when treating vdW interactions based on different approaches,98 thereby influencing the predictions of ab initio microkinetic and kinetic Monte Carlo models (see section 11 in the ESI†).
4. Concluding remarks
By means of DFT calculations, we performed a thorough investigation of the formation and decomposition of N2O* over Rh(111), Cu(111), a Ni/Cu(111) SAA surface and a Ni2Cu(111) surface, where Ni atoms form dimer clusters. The DFT–derived energetics, in conjunction with results from our previous work,4 were then used to parameterise a microkinetic model for the NO + CO reaction.Our DFT calculations showed that the presence of a small amount of Ni over Cu(111) strengthens significantly the interaction between N2O* and the catalyst surface. This enhanced interaction is desirable because it prevents the desorption of N2O*, thereby benefiting the selectivity to N2 during the NO + CO reaction. Regarding the decomposition of N2O*, we explored three competing reaction paths. In the first pathway the decomposition products (i.e. NO* + O* and 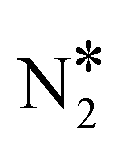 + O*) are connected through the η1 adsorption structure of N2O*. In the second, the same products are connected through another N2O* adsorption structure (i.e. η2-(Nt{fcc},O{top})), and the third involves the formation of an
+ O*) are connected through the η1 adsorption structure of N2O*. In the second, the same products are connected through another N2O* adsorption structure (i.e. η2-(Nt{fcc},O{top})), and the third involves the formation of an  intermediate. These paths exhibit comparable energetics and therefore merit consideration when modelling the kinetics of the NO + CO reaction. We also demonstrated that the selectivity of the Ni/Cu dilute alloy surfaces can be manipulated by tuning the size of the Ni cluster; generally, the formation of NO* and atomic nitrogen is kinetically favoured over “large clusters” (e.g. trimers), whereas small clusters (i.e. dimers) and single atoms promote the dissociation of N2O* to
intermediate. These paths exhibit comparable energetics and therefore merit consideration when modelling the kinetics of the NO + CO reaction. We also demonstrated that the selectivity of the Ni/Cu dilute alloy surfaces can be manipulated by tuning the size of the Ni cluster; generally, the formation of NO* and atomic nitrogen is kinetically favoured over “large clusters” (e.g. trimers), whereas small clusters (i.e. dimers) and single atoms promote the dissociation of N2O* to  and atomic oxygen.
and atomic oxygen.
Finally, the performance of the Ni/Cu dilute alloy surfaces was assessed by means of microkinetic simulations for the NO + CO reaction. Our studies highlighted the potential of Ni2Cu(111), which showed considerably improved catalytic performance as compared to Cu(111) and comparable performance to the best transition metal for the reduction of NO (i.e. Rh(111)). Future work could focus on the effect of adsorbate–adsorbate interactions on the reaction kinetics of the NO + CO reaction,99–101 and explore the behaviour of other facets of the Ni2Cu catalyst in an effort to quantify potential structure-sensitivity effects.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. G. P. is funded by the Department of Chemical Engineering at University College London. The authors acknowledge the use of the UCL High Performance Computing Facilities Kathleen@UCL in the completion of the simulations of this work. We are also grateful to the U.K. Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/P020194/1), for computational resources (HPC facility Thomas). K. G. P. thanks Sai Yadavalli for fruitful discussions on the performance of DFT exchange and correlation functionals.References
- V. P. Zhdanov and B. Kasemo, Surf. Sci. Rep., 1997, 29, 31–33 CrossRef.
- W. T. Wallace, Y. Cai, M. S. Chen and D. W. Goodman, J. Phys. Chem. B, 2006, 110, 6245–6249 CrossRef CAS.
- D. Loffreda, D. Simon and P. Sautet, J. Catal., 2003, 213, 211–225 CrossRef CAS.
- K. G. Papanikolaou and M. Stamatakis, Catal. Sci. Technol., 2020, 10, 5815–5828 RSC.
- H. J. Borg, J. F. C.-J. M. Reijerse, R. A. Van Santen and J. W. Niemantsverdriet, J. Chem. Phys., 1994, 101, 10052–10063 CrossRef CAS.
- D. N. Belton, C. L. Dimaggio, S. J. Schmieg and K. Y. S. Ng, J. Catal., 1995, 157, 559–568 CrossRef CAS.
- K. C. Taylor, Catal. Rev.: Sci. Eng., 1993, 35, 457–481 CrossRef CAS.
- F. Zaera and C. S. Gopinath, Phys. Chem. Chem. Phys., 2003, 5, 646–654 RSC.
- C. S. Gopinath and F. Zaera, J. Catal., 2001, 200, 270–287 CrossRef CAS.
- C. H. F. Peden, D. W. Goodman, D. S. Blair, P. J. Berlowitz, G. B. Fisher and S. H. Oh, J. Phys. Chem., 1988, 92, 1563–1567 CrossRef CAS.
- L. H. Dubois, P. K. Hansma and G. A. Somorjai, J. Catal., 1980, 65, 318–327 CrossRef CAS.
- C. H. F. Peden, D. N. Belton and S. J. Schmieg, J. Catal., 1995, 155, 204–218 CrossRef CAS.
- G. S. Herman, C. H. F. Peden, S. J. Schmieg and D. N. Belton, Catal. Lett., 1999, 62, 131–138 CrossRef CAS.
- H. Permana, K. Y. Simon Ng, C. H. F. Peden, S. J. Schmieg, D. K. Lambert and D. N. Belton, Catal. Lett., 1997, 47, 5–15 CrossRef CAS.
- L. Tan, L. Huang, Y. Liu and Q. Wang, Appl. Surf. Sci., 2018, 444, 276–286 CrossRef CAS.
- A. Ishikawa and Y. Tateyama, J. Phys. Chem. C, 2018, 122, 17378–17383 CrossRef CAS.
- J. Liu, L. Tan, L. Huang, Q. Wang and Y. Liu, Langmuir, 2020, 36, 3127–3140 CrossRef CAS PubMed.
- V. P. Zhdanov and B. Kasemo, Catal. Lett., 1996, 40, 197–202 CrossRef.
- J. Cortés and E. Valencia, J. Phys. Chem. B, 2006, 110, 7887–7897 CrossRef PubMed.
- C. Y. Kang, M. Taniguchi, M. Uenishi and H. Tanaka, Chem. Lett., 2012, 41, 822–824 CrossRef CAS.
- H. Asakura, T. Onuki, S. Hosokawa, N. Takagi, S. Sakaki, K. Teramura and T. Tanaka, Phys. Chem. Chem. Phys., 2019, 21, 18816–18822 RSC.
- K. Ueda, C. A. Ang, Y. Ito, J. Ohyama and A. Satsuma, Catal. Sci. Technol., 2016, 6, 5797–5800 RSC.
- H. Yoshida, Y. Okabe, N. Yamashita, S. Hinokuma and M. Machida, Catal. Today, 2017, 281, 590–595 CrossRef CAS.
- K. Koizumi, H. Yoshida, M. Boero, K. Tamai, S. Hosokawa, T. Tanaka, K. Nobusada and M. Machida, Phys. Chem. Chem. Phys., 2018, 20, 25592–25601 RSC.
- H. Yoshida, N. Yamashita, S. Ijichi, Y. Okabe, S. Misumi, S. Hinokuma and M. Machida, ACS Catal., 2015, 5, 6738–6747 CrossRef CAS.
- T. Hirakawa, Y. Shimokawa, W. Tokuzumi, T. Sato, M. Tsushida, H. Yoshida, J. Ohyama and M. Machida, ACS Appl. Nano Mater., 2020, 3, 9097–9107 CrossRef CAS.
- N. Takagi, K. Ishimura, H. Miura, T. Shishido, R. Fukuda, M. Ehara and S. Sakaki, ACS Omega, 2019, 4, 2596–2609 CrossRef CAS PubMed.
- H. Yoshida, Y. Kawakami, W. Tokuzumi, Y. Shimokawa, T. Hirakawa, J. Ohyama and M. Machida, Bull. Chem. Soc. Jpn., 2020, 93, 1050–1055 CrossRef CAS.
- C. B. Jones, I. Khurana, S. H. Krishna, A. J. Shih, W. N. Delgass, J. T. Miller, F. H. Ribeiro, W. F. Schneider and R. Gounder, J. Catal., 2020, 389, 140–149 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- S. Hosokawa, T. Shibano, H. Koga, M. Matsui, H. Asakura, K. Teramura, M. Okumura and T. Tanaka, ChemCatChem, 2020, 12, 4276–4280 CrossRef CAS.
- L. Zhang, I. A. W. Filot, Y.-Q. Su, J.-X. Liu and E. J. M. Hensen, J. Catal., 2018, 363, 154–163 CrossRef CAS.
- F. Xing, J. Jeon, T. Toyao, K. Shimizu and S. Furukawa, Chem. Sci., 2019, 10, 8292–8298 RSC.
- R. Réocreux, P. L. Kress, R. T. Hannagan, V. Çınar, M. Stamatakis and E. C. H. Sykes, J. Phys. Chem. Lett., 2020, 11, 8751–8757 CrossRef PubMed.
- S. Kandoi, A. A. Gokhale, L. C. Grabow, J. A. Dumesic and M. Mavrikakis, Catal. Lett., 2004, 93, 93–100 CrossRef CAS.
- F. Zaera and C. S. Gopinath, Chem. Phys. Lett., 2000, 332, 209–214 CrossRef CAS.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS PubMed.
- H. Tian, R. Xu, J. G. Canadell, R. L. Thompson, W. Winiwarter, P. Suntharalingam, E. A. Davidson, P. Ciais, R. B. Jackson, G. Janssens-Maenhout, M. J. Prather, P. Regnier, N. Pan, S. Pan, G. P. Peters, H. Shi, F. N. Tubiello, S. Zaehle, F. Zhou, A. Arneth, G. Battaglia, S. Berthet, L. Bopp, A. F. Bouwman, E. T. Buitenhuis, J. Chang, M. P. Chipperfield, S. R. S. Dangal, E. Dlugokencky, J. W. Elkins, B. D. Eyre, B. Fu, B. Hall, A. Ito, F. Joos, P. B. Krummel, A. Landolfi, G. G. Laruelle, R. Lauerwald, W. Li, S. Lienert, T. Maavara, M. MacLeod, D. B. Millet, S. Olin, P. K. Patra, R. G. Prinn, P. A. Raymond, D. J. Ruiz, G. R. van der Werf, N. Vuichard, J. Wang, R. F. Weiss, K. C. Wells, C. Wilson, J. Yang and Y. Yao, Nature, 2020, 586, 248–256 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- A. Shiotari, S. Hatta, H. Okuyama and T. Aruga, J. Chem. Phys., 2014, 141, 134705 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- P. Haas, F. Tran and P. Blaha, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 085104 CrossRef.
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B: Solid State, 1977, 16, 1748–1749 CrossRef.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- K. W. Kolasinski, in Surface Science, John Wiley & Sons, Ltd, Chichester, UK, 2012, pp. 51–114 Search PubMed.
- W. F. K. Wynne-Jones and H. Eyring, J. Chem. Phys., 1935, 3, 492–502 CrossRef CAS.
- C. T. Campbell, ACS Catal., 2017, 7, 2770–2779 CrossRef CAS.
- C. Stegelmann, A. Andreasen and C. T. Campbell, J. Am. Chem. Soc., 2009, 131, 8077–8082 CrossRef CAS PubMed.
- Y. Li and M. Bowker, Surf. Sci., 1996, 348, 67–76 CrossRef CAS.
- A. Bogicevic and K. C. Hass, Surf. Sci., 2002, 506, L237–L242 CrossRef CAS.
- V. G. Komvokis, M. Marti, A. Delimitis, I. A. Vasalos and K. S. Triantafyllidis, Appl. Catal., B, 2011, 103, 62–71 CrossRef CAS.
- A. Kokalj, I. Kobal and T. Matsushima, J. Phys. Chem. B, 2003, 107, 2741–2747 CrossRef CAS.
- S. Y. Wu, C. H. Su, J. G. Chang, H. T. Chen, C. H. Hou and H. L. Chen, Comput. Mater. Sci., 2011, 50, 3311–3314 CrossRef CAS.
- H. Orita and N. Itoh, Surf. Sci., 2004, 550, 166–176 CrossRef CAS.
- R. Burch, S. T. Daniells and P. Hu, J. Chem. Phys., 2004, 121, 2737 CrossRef CAS PubMed.
- X. Wei, X. F. Yang, A. Q. Wang, L. Li, X. Y. Liu, T. Zhang, C. Y. Mou and J. Li, J. Phys. Chem. C, 2012, 116, 6222–6232 CrossRef CAS.
- K. Kim, S. Baek, J. J. Kim and J. W. Han, Appl. Surf. Sci., 2020, 510, 145349 CrossRef CAS.
- J.-F. Paul, J. Pérez-Ramírez, F. Ample and J. M. Ricart, J. Phys. Chem. B, 2004, 108, 17921–17927 CrossRef CAS.
- J. L. Teffo and A. Chédin, J. Mol. Spectrosc., 1989, 135, 389–409 CrossRef CAS.
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed.
- M. B. Boucher, B. Zugic, G. Cladaras, J. Kammert, M. D. Marcinkowski, T. J. Lawton, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2013, 15, 12187–12196 RSC.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed.
- D. A. Patel, R. T. Hannagan, P. L. Kress, A. C. Schilling, V. Çınar and E. C. H. Sykes, J. Phys. Chem. C, 2019, 123, 28142–28147 CrossRef CAS.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. a Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- K. G. Papanikolaou, M. T. Darby and M. Stamatakis, ACS Catal., 2020, 10, 1224–1236 CrossRef CAS.
- K. G. Papanikolaou, M. T. Darby and M. Stamatakis, J. Phys. Chem. C, 2019, 123, 9128–9138 CrossRef CAS.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS PubMed.
- M. Ouyang, K. G. Papanikolaou, A. Boubnov, A. S. Hoffman, G. Giannakakis, S. R. Bare, M. Stamatakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2021, 12, 1549 CrossRef CAS PubMed.
- H. Thirumalai and J. R. Kitchin, Top. Catal., 2018, 61, 462–474 CrossRef CAS.
- W. A. Brown, R. K. Sharma, D. A. King and S. Haq, J. Phys. Chem., 1996, 100, 12559–12568 CrossRef.
- Y. Wang, D. Zhang, Z. Yu and C. Liu, J. Phys. Chem. C, 2010, 114, 2711–2716 CrossRef CAS.
- Z. P. Liu, S. J. Jenkins and D. A. King, J. Am. Chem. Soc., 2004, 126, 7336–7340 CrossRef CAS PubMed.
- T. N. Pham, Y. Hamamoto, K. Inagaki, D. N. Son, I. Hamada and Y. Morikawa, J. Phys. Chem. C, 2020, 124, 2968–2977 CrossRef CAS.
- I. Nakai, H. Kondoh, T. Shimada, R. Yokota, T. Katayama, T. Ohta and N. Kosugi, J. Chem. Phys., 2007, 127, 024701 CrossRef PubMed.
- K. G. Papanikolaou, M. T. Darby and M. Stamatakis, J. Chem. Phys., 2018, 149, 184701 CrossRef PubMed.
- M. Stamatakis, J. Phys.: Condens. Matter, 2015, 27, 013001 CrossRef CAS PubMed.
- K. G. Papanikolaou and M. Stamatakis, in Computational Modelling of Nanomaterials, 2020, pp. 95–125 Search PubMed.
- M. Stamatakis and S. Piccinin, ACS Catal., 2016, 6, 2105–2111 CrossRef CAS.
- J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, in Fundamental Concepts in Heterogeneous Catalysis, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2014, pp. 68–84 Search PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- S. Li, S. Singh, J. A. Dumesic and M. Mavrikakis, Catal. Sci. Technol., 2019, 9, 2836–2848 RSC.
- S. Bhandari, S. Rangarajan and M. Mavrikakis, Acc. Chem. Res., 2020, 53, 1893–1904 CrossRef CAS PubMed.
- Y. Xu and M. Mavrikakis, Surf. Sci., 2001, 494, 131–144 CrossRef CAS.
- A. C. Lausche, A. J. Medford, T. S. Khan, Y. Xu, T. Bligaard, F. Abild-Pedersen, J. K. Nørskov and F. Studt, J. Catal., 2013, 307, 275–282 CrossRef CAS.
- J. Schumann, A. J. Medford, J. S. Yoo, Z. J. Zhao, P. Bothra, A. Cao, F. Studt, F. Abild-Pedersen and J. K. Nørskov, ACS Catal., 2018, 8, 3447–3453 CrossRef CAS.
- S. González, C. Sousa and F. Illas, J. Catal., 2006, 239, 431–440 CrossRef.
- M. Mavrikakis, J. Rempel, J. Greeley, L. B. Hansen and J. K. Nørskov, J. Chem. Phys., 2002, 117, 6737–6744 CrossRef CAS.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- R. B. Getman, Y. Xu and W. F. Schneider, J. Phys. Chem. C, 2008, 9559–9572 CrossRef CAS.
- S. Matera, W. F. Schneider, A. Heyden and A. Savara, ACS Catal., 2019, 9, 6624–6647 CrossRef CAS.
- D. J. Schmidt, W. Chen, C. Wolverton and W. F. Schneider, J. Chem. Theory Comput., 2012, 8, 264–273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00011j |
| This journal is © The Royal Society of Chemistry 2021 |