
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Basic ionic liquids for catalysis: the road to greater stability
Peter
McNeice
 ,
Patricia C.
Marr
and
Andrew C.
Marr
*
,
Patricia C.
Marr
and
Andrew C.
Marr
*
Queen's University Ionic Liquids Laboratories and, The School of Chemistry and Chemical Engineering, Queen's University Belfast, The David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: a.marr@qub.ac.uk
First published on 12th January 2021
Abstract
Basic ionic liquids can replace organic or inorganic bases in a wide range of reactions. In this minireview the synthesis of homogeneous and heterogeneous basic ionic liquids and their applications in catalysis are discussed. The dual properties of tuneability and wide spectrum solubility make basic ionic liquids unique amongst bases. Acidic ionic liquids have been applied in large scale industrial processes, but in contrast the application of basic ionic liquids has been hampered by ion instability. This problem is now surmountable and strategies for stabilising ionic liquids within basic solutions have been developed that are molecular and material in basis. The review investigates the potential to apply these stabilised basic ionic liquids as catalysts, emphasising facile recovery from the reaction mixture.
 Peter McNeice | Dr Peter McNeice completed his masters degree at Queen's University Belfast, during which he conducted a research project under Dr Patricia Marr on low molecular weight ionic liquid gels. He stayed at QUB, joining the Queen's University Ionic Liquids Laboratories (QUILL) for a PhD on the development of base stable and basic ionic liquids for catalysis, supervised by Drs Andrew and Patricia Marr, and Prof. Kenneth Seddon. He is currently a postdoctoral researcher in industrial catalysis within the research group of Prof. Matthias Beller at the Leibniz Institute for Catalysis in Rostock. |
 Patricia C. Marr | Dr Patricia C. Marr specialises in functional materials (in particular ionic liquid gels) for green chemistry and sustainability. She obtained her PhD in functional gels and hybrids from the University of St. Andrews. This was followed by postdoctoral research on materials in supercritical fluids at the University of Nottingham, and on colloids, polymers and interfaces for medicinal applications at the University of Bristol. She is a senior faculty member in the School of Chemistry & Chemical Engineering. |
 Andrew C. Marr | Dr Andrew C. Marr is a green chemist specialising in catalysis with organometallic complexes, biocatalysts and ionic liquids. His PhD training was in industrial homogeneous catalysis in the University of St. Andrews. He then conducted postdoctoral research on biocatalysis at The University of Nottingham and ligand synthesis at The University of Bristol, before being appointed to the staff of Queen's University Belfast, where he is a senior faculty member in the School of Chemistry & Chemical Engineering. |
1. Introduction
1.1 Properties and uses of ionic liquids
Ionic liquids (ILs) are materials composed entirely of ions that can form stable liquids under operating conditions. Ionic liquids with melting points below room temperature are known as room temperature ionic liquids (RTILs).1,2 Ionic liquids are often prepared from a large organic cation combined with a multiatom inorganic anion (Fig. 1). The large irregular size of the ions disrupts ion packing, whilst charge delocalisation reduces the attraction between the ions.3,4 This results in lower lattice enthalpies and therefore lower melting points.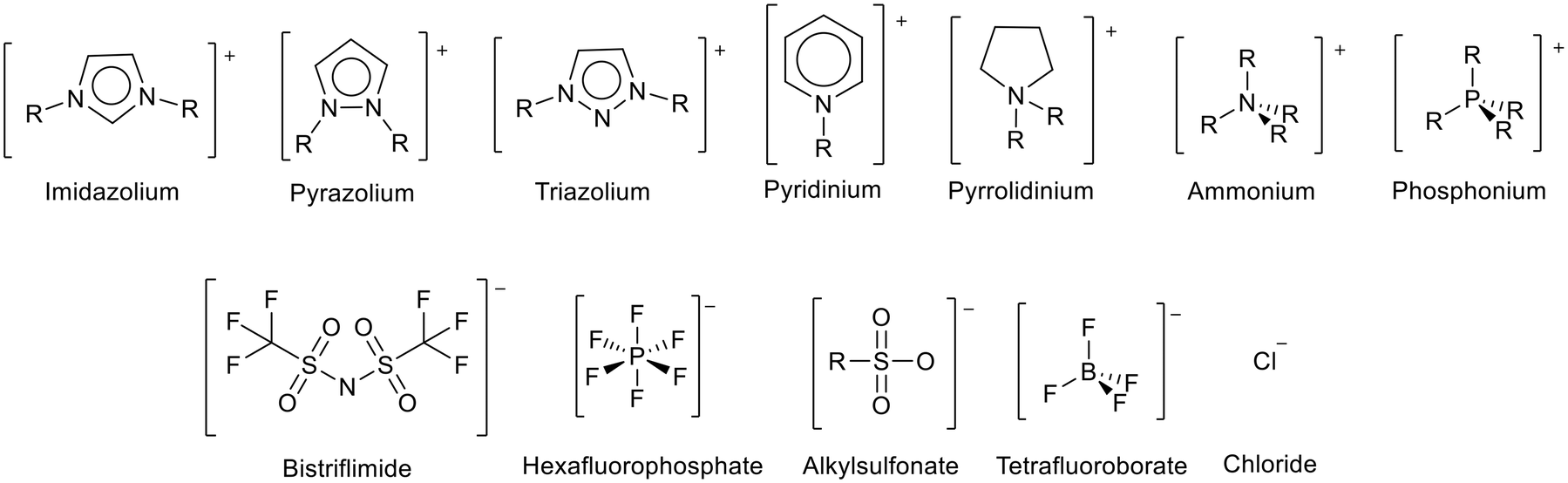 | ||
| Fig. 1 Some common cations and anions used to prepare ionic liquids. PF6− and BF4− have fallen out of general use due to concerns over their relative instability through hydrolysis to produce HF.5–7 | ||
Ionic liquids have diverse applications in catalysis2 including uses in electrochemistry,8,9 separations10 and biocatalysis.11 The adaptability of ionic liquids stems from their unique and tuneable properties. Ionic liquids often have high thermal stability and are essentially non-volatile and non-flammable, although they can ignite when they contain high energy moieties such as tetrazoles or nitro groups, or at temperatures near to their decomposition temperature.12,13 Therefore the more thermally stable an ionic liquid, the less likely it is to ignite. This improves the safety of chemical processes and facilitates the recovery and reuse of the ionic liquid after a reaction, limiting waste. The ability to tune the properties of an ionic liquid makes them remarkable amongst solvents. Exchanging one ion for another allows the properties, such as melting point, viscosity, hydrophobicity/hydrophilicity, acidity/basicity, to be tailored for a specific application. This has led to the creation of functionalised ionic liquids (previously called task specific ionic liquids).14 Acid functionalised ionic liquids are used on an industrial scale in the production of high-octane compounds for fuels via alkylation. Lewis acidic ionic liquids provide a safer and more recyclable alternative to HF and H2SO4 catalysts and have been applied in PetroChina's “Ionikylation” process,15,16 and Chevron and Honeywell UOP's ISOALKYL process.17,18
1.2 The base instability of common ionic liquids
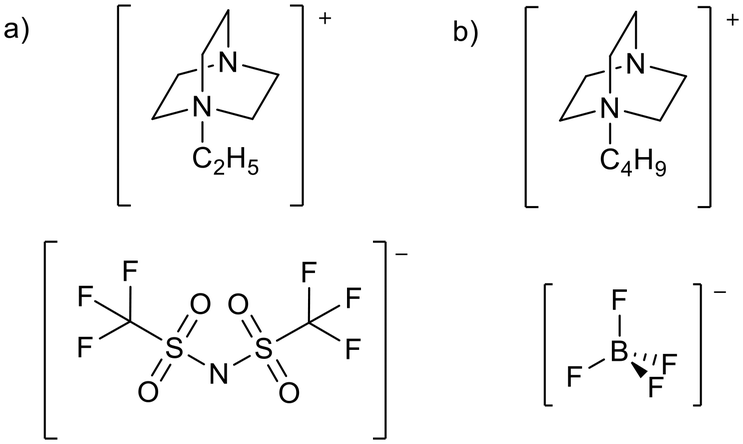
Despite the many attractive properties of ionic liquids, their use in catalysis can be retarded by base instability, as discussed in a recent review.19 This limits the use of ionic liquids as solvents for base catalysed reactions and the development of basic ionic liquids. The imidazolium cation, the cation of choice in much early ionic liquid research, is particularly sensitive to basic conditions and can be deprotonated at the C2 position20 to form an N-heterocyclic carbene (NHC, Fig. 2a). Studies in which the authors claim to have synthesised 1,3-butylmethylimidazolium hydroxide ([BMIM][OH]) and used it for catalysis have been discussed in a review article by Maschmeyer and co-workers.21 The authors state that there is often poor characterisation of [BMIM][OH] and the reactions may be catalysed by halide impurities or a decomposition product of [BMIM][OH] such as an NHC. For this reason the use of [BMIM][OH], which is inherently unstable in basic solution, will not be discussed in this review. Other cations are also vulnerable under basic conditions, acyclic ammonium cations are known to undergo Hofmann elimination (Fig. 2b),22 and pyridinium cations can decompose via ring-opening (Fig. 2c).23,24 Phosphonium ionic liquids have intermediate base stability. Small bases such as OH− can lead to decomposition via the formation of a phosphine oxide25,26 or phosphorus ylide27,28 (Fig. 2d and e). Phosphonium ionic liquids can be stable to hindered bases such as Grignard reagents27,29 and NHCs.30,31 This is aided by attaching bulky or electron donating groups to the phosphonium centre.27,29–32 Alternatives such as triazolium,33 pyrazolium,34 phosphonium, and cyclic ammonium (such as pyrrolidinium)22 cations (Fig. 1) are often more stable to basic conditions, and are therefore more appropriate for the preparation of basic ionic liquids. Base stability is demonstrated by resistance to strong bases,22,29,33 electrochemical stability,34 and resistance of the cation to being reduced.35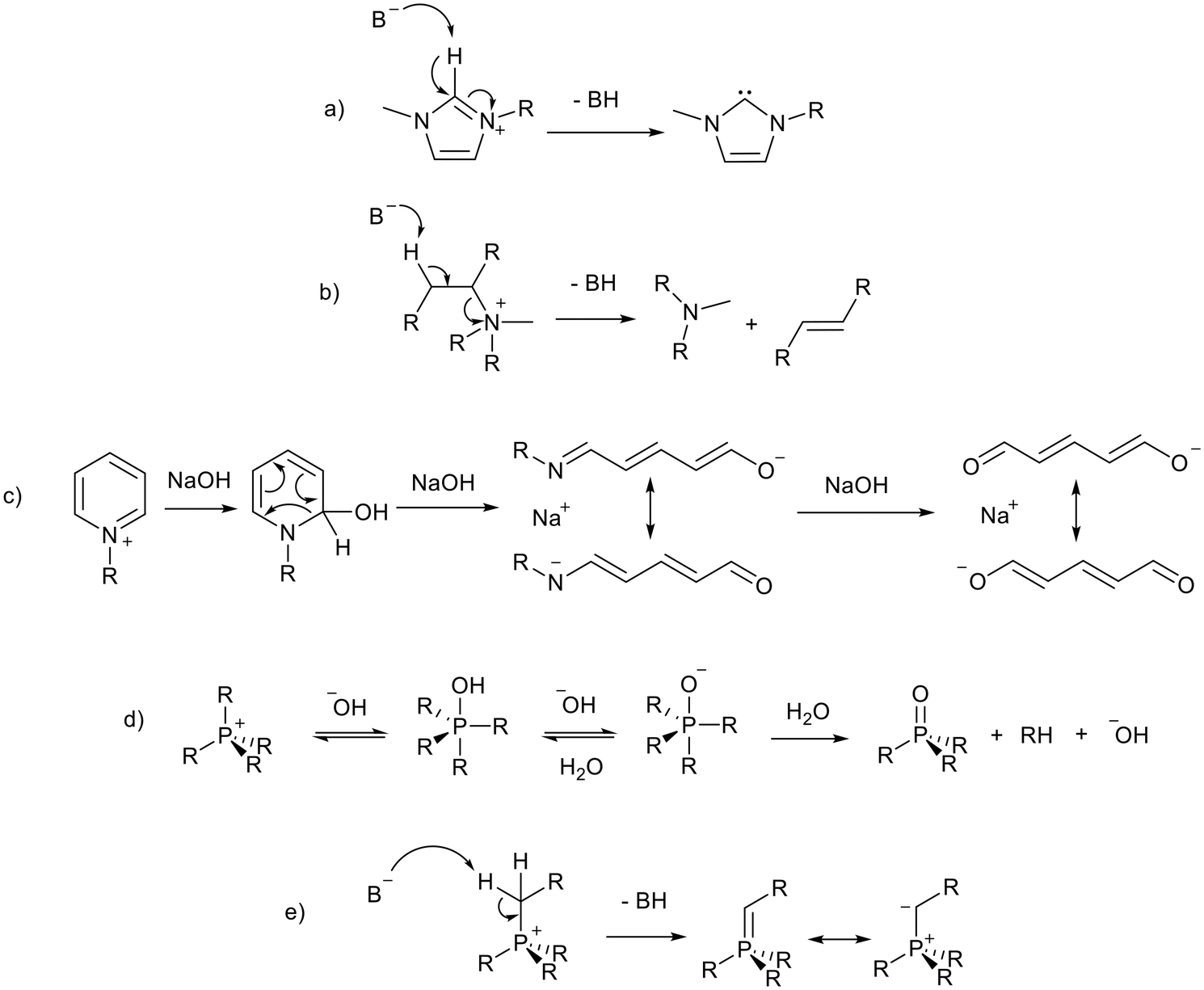 | ||
| Fig. 2 Base catalysed decomposition of an imidazolium cation, B− represents an external base or a basic anion (a),20 Hofmann elimination of an acyclic ammonium cation (b),22 ring opening decomposition of a pyridinium cation (c),23,24 and formation of a phosphine oxide (d)25,26 and phosphorus ylide (e)27 from a phosphonium ionic liquid. Kekulé structures are drawn for clarity. | ||
1.3 Basic ionic liquids
Basic ionic liquids can be prepared by associating basicity with the anion or the cation (Fig. 3). As discussed above, the use of strong anionic bases such as [OH]− can lead to ionic liquid instability due to attack of the cation by the anion (Fig. 2).21 Anions with higher basicity can also lead to ionic liquids with lower thermal stability than those containing anions of lower basicity.36 Basic cations often contain weaker bases such as amine groups (Fig. 3). The basicity can be further weakened as the positive charge of the cation reduces the availability of the amine to use an electron pair as a base.37,38 This, and the fact that cations tend to be more bulky/hindered, and less mobile than the anion, are reasons that ionic liquids containing basic cations are often more stable than those containing basic anions.39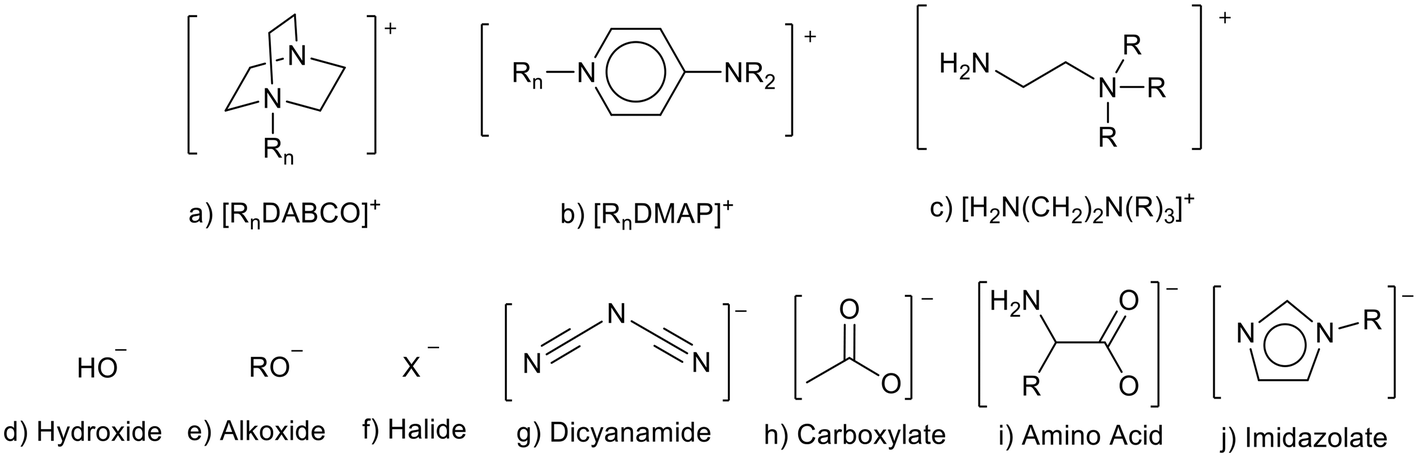 | ||
| Fig. 3 Some cations and anions that can be used to prepare basic ionic liquids (a,40 b,41 c,37 g,42 i43 and j44). | ||
A reliable method to assess the basicity of an ionic liquid is important in order to predict behaviour in reactions. The standard way to assess basicity is to measure the pH. This is possible with water soluble ionic liquids45,46 but is not a universal method as many ionic liquids are insoluble or unstable in water. The Hammett function (H0)47 has been used to measure the acidity of non-aqueous systems and has been further applied to acidic48 and basic49,50 ionic liquids. The Hammett function relies on the relative dissociation of acids in solution. In solvents, acids dissociate to a certain extent by releasing a proton. If the same acid is dissolved in an ionic liquid and its dissociation does not change, the ionic liquid can be assumed to be neutral. If the dissociation decreases, it is likely that the ionic liquid is acidic and is preventing the acid from dissociating to its previous extent. If the acid dissociation increases, then it is likely that the ionic liquid is basic and is accepting a proton, thereby increasing the relative dissociation of the acid.51 The extent of dissociation of acidic indicator solutions allows the Hammett basicity (H_, also called the reverse Hammett function) of an ionic liquid to be measured via UV spectrophotometry.48,50 This method is limited by the assumption that the indicator is fully dissociated, and by the need to dissolve the ionic liquid, making the values solvent dependant.52 A less quantitative method is to simply add ionic liquid into indicator solutions of different pKa values and visually note any colour change. If a colour change occurs the ionic liquid has a higher pKa than the indicator, and if no change occurs the ionic liquid has a lower pKa than the indicator.53 This allows the ionic liquid to be placed within a pKa range but, unlike the Hammett measurement, does not give a specific value. These indicator methods can also determine the relative basicity of heterogeneous basic ionic liquids by adding a fixed amount of material into the indicator solution and monitoring the change visually or by UV analysis.53 The basicity of heterogeneous basic ionic liquids can also be determined by alternative techniques. One is a back-titration method where the solid is reacted with a known amount of acid before the remaining acid is titrated with base in the presence of an indicator.54,55 A second method is CO2 temperature programmed desorption (TPD).56 The sample is saturated with CO2 before the desorption at different temperatures is monitored. CO2 desorption peaks at low temperatures indicate weakly basic sites, whilst peaks at higher temperatures indicate the presence of more strongly basic sites. The amount of CO2 desorbed can be calculated to compare the basicity of one heterogeneous ionic liquid to another.
All methods discussed are useful indicators of the basic strength of an ionic liquid, but a true assessment of a basic catalyst can only be obtained through application. Despite the potential instability of basic ionic liquids, the number of reactions catalysed by basic ionic liquids has expanded rapidly. In this review we provide selected recent literature examples of homogeneous and heterogeneous basic ionic liquids for base catalysed reactions, emphasising developments in ionic liquid and support technologies. Basic ionic liquids have been reviewed recently for the conversion of CO2 to cyclic carbonates.57 Reviews that feature basic ionic liquid catalysed reactions have been published previously.39,58,59
2. Homogeneous basic ionic liquids
2.1 Knoevenagel condensation
Knoevenagel condensation (Fig. 4) is an important C–C bond coupling reaction which is used to prepare drugs,60 fragrances61 and dyes.62 It was the first reported base catalysed reaction performed in an ionic liquid.63 Traditionally this reaction uses stoichiometric quantities of amine bases which can be harmful, difficult to remove, and/or flammable.64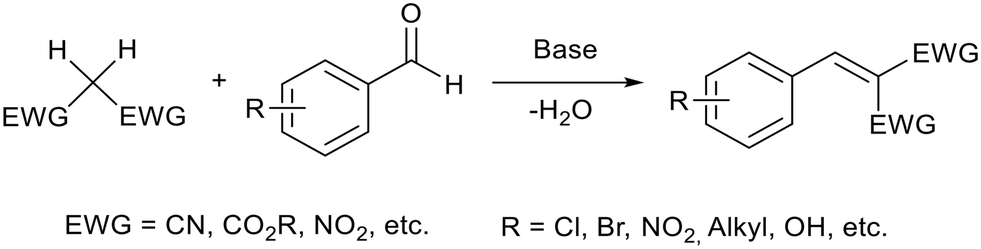 | ||
| Fig. 4 A general reaction scheme for a Knoevenagel condensation between an aryl aldehyde and a reagent with acidic hydrogens. | ||
As a replacement for traditional amine bases, Seddon and co-workers appended amines including Hunig's base to an ammonium cation using linkers of different chain lengths (Fig. 5).37 The quaternisation was performed in diethyl ether to precipitate the desired halide salt and prevent self-quaternisation of 2-diisopropylaminoethyl chloride hydrochloride (Fig. 5). Universal indicator was used to determine the relative basicity of the ionic liquids (section 1.3). A longer distance between the ammonium cation and the amine base produced more strongly basic ionic liquids. This was attributed to the greater stability of the dicationic species formed upon protonation. 10 mol% ionic liquid was applied to the solvent free Knoevenagel condensation between benzaldehyde and ethyl cyanoacetate at room temperature (Fig. 4). With reaction times ranging from 20 minutes to 24 hours the conversions achieved were between 12 and 91%. The more strongly basic ionic liquids produced higher yields within a shorter time. Universal indicator showed that the ionic liquids had reduced basicity relative to free Hunig's base in neutral 1,3-butylmethylimidazolium bistriflimide ([BMIM][NTf2]), which also gave quantitative conversion. However the basic ionic liquid could be recovered after the reaction by extracting the products into cyclohexane, whereas the Hunig's base was lost during solvent extraction or distillation.
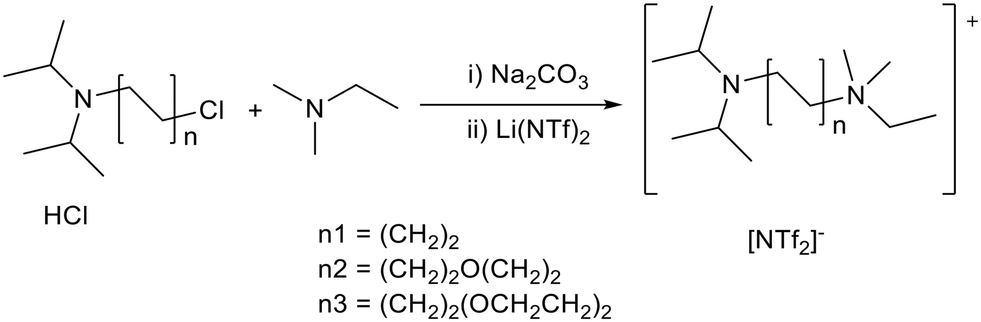 | ||
| Fig. 5 The synthesis of Hunig's base containing ionic liquids.37 | ||
Molecules with two amine groups have been utilised to prepare ionic liquids. Important examples include ionic liquids derived from DABCO (1,4-diazabicyclo[2.2.2]octane, (Fig. 3a)). MacFarlane and co-workers were the first to report [C2DABCO][NTf2] (Fig. 6a).40 In this IL one nitrogen of DABCO is alkylated, leaving the other to act as a base. This ionic liquid exhibited good thermal and electrochemical stability.40 The synthesis conditions had to be carefully controlled to minimize overalkylation. Low reaction temperatures (25–50 °C) and excess DABCO can be used to inhibit formation of the dialkylated species.38,65 The basicity of these ionic liquids is lower than DABCO.38 When 15 mole% of [C4DABCO][BF4] (Fig. 6b) was applied to the Knoevenagel condensation between malononitrile and benzaldehyde in water (Fig. 4), 100% yield was achieved within 1 minute at room temperature.66 Pure product precipitated out of the reaction whilst the ionic liquid remained in solution, enabling separation by filtration and subsequent reuse of the ionic liquid. The yield for the second reaction catalysed by the ionic liquid was reduced to 95%, but this was maintained for an additional 5 cycles without further decrease. The catalyst loading could be reduced to 5 mole% and still achieve 100% yield if the reaction time was increased to 20 minutes. Deactivated aromatic aldehydes, aliphatic aldehydes, aliphatic ketones, and also the less reactive ethyl cyanoacetate could be used as substrates and afforded yields higher than 90% once the reaction time had been modified appropriately.
Quinine has been used as a starting material to synthesise basic ionic liquids (Fig. 7). Quinine is a natural molecule which can be derived from Cinchona bark.67 The greater nucleophilicity of the non-aromatic quinuclidine position provides an easier route to the selective synthesis of ionic liquids. McNeice et al. prepared six quinine-derived ILs, including the first example of a room-temperature ionic liquid based on quinine (Fig. 7).68 Spartan computational calculations showed the basicity of the ionic liquids to be similar to neutral quinine. These ionic liquids were applied to the Knoevenagel condensation between malononitrile and benzaldehyde (Fig. 4). Yields of 78–94% were achieved within 1 h at room temperature. Ether functionalised ionic liquids achieved the highest yields of 88 and 94%. The yields achieved were higher than those measured for DABCO (62%), DBU (1,8-diazabicyclo[5.4.0]undec-7-en) (74%) and neutral quinine (63%), all of which have higher basicity than the quinine ionic liquids. Yields obtained were not simply a product of the basicity of the IL, and were postulated to depend also on inter-“molecular” interactions between the catalyst and the substrates. Catalyst recycling was tested when R = CH3 (Fig. 7) and consistent activity for a further three reactions was demonstrated.
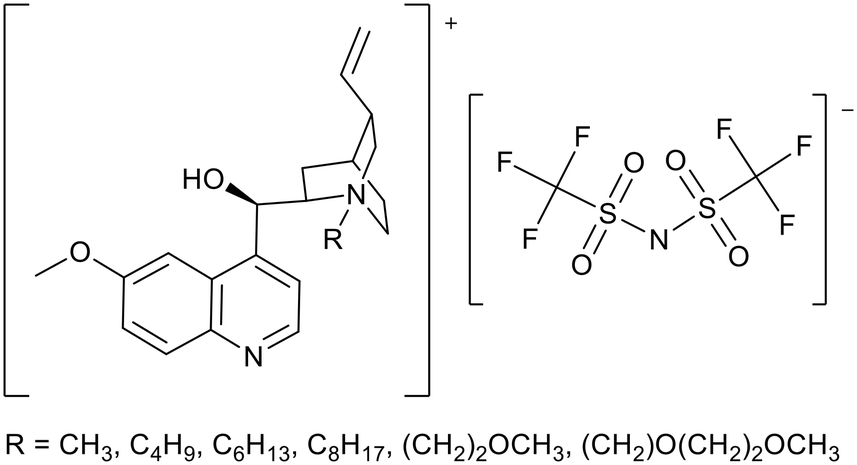 | ||
| Fig. 7 The general structure of quinine ionic liquids.68 | ||
Strongly basic ionic liquids can be prepared using Brønsted basic anions, but these generally have low stability due to attack on the cation by the anion, as discussed in section 1.2. McNeice et al. stabilized ionic liquids containing a strongly basic anion by preparing binary ionic liquids, combining a relatively base stable pyrrolidinium cation with iso-propoxide and the neutral anion [NTf2]− to make [Pyrr1,4][NTf2]x[OiPr]1−x (0.57 ≤ x ≥ 0.84, Fig. 8).69 The ionic liquid basicity was measured using the inverse Hammett method,50,57 where the ability of the ionic liquid to abstract a proton from an acidic indicator is assessed, as discussed in the section 1.3. The ionic liquids showed higher Hammett basicity (H_) than pyridine and triethylamine, and similar basicity to DBU, KOH and NaOMe. This class of ionic liquid was shown to be stable for at least 8 months under ambient conditions. The stability was attributed to lowering the proportion of iso-propoxide. Another factor thought to be important was the relative mobility of the basic anion, which is affected by the viscosity and ionic/coulombic interactions, with a lower mobility reducing the likelihood of cation decomposition. The ionic liquids catalysed Knoevenagel and aldol condensations (section 2.2). When 1 mole% ionic liquid was applied to the room temperature solvent free Knoevenagel condensation between malononitrile and benzaldehyde (Fig. 4) the yield increased with the proportion of alkoxide anion, from 24% up to 89%. A flexible preparation method was used to synthesise these ionic liquids affording variable basicity by changing the ratio of anions. The best performing catalyst [Pyrr1,4][NTf2]0.58[OiPr]0.42 had the same activity as pyridine, a traditional Knoevenagel catalyst.
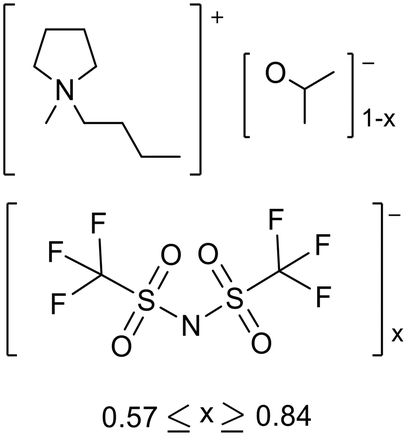 | ||
| Fig. 8 The structure of binary alkoxide ionic liquids ([Pyrr1,4][NTf2]x[OiPr]1−x).69 | ||
2.2 Aldol condensation
Aldol condensation is an important C–C bond forming reaction in organic chemistry. It is traditionally catalysed by aqueous NaOH,70–72 which can lead to reactor corrosion and the production of salt waste.Binary alkoxide ionic liquids ([Pyrr1,4][NTf2]x[OiPr]1−x, Fig. 8, discussed in section 2.1)69 catalysed the aldol condensation between acetone and benzaldehyde (Fig. 9). This reaction is a good mimic for the condensation between furfural and acetone which could be an important step in upgrading lignin and cellulose to valuable chemicals.73,74 The best performing ionic liquid ([Pyrr1,4][NTf2]0.57[OiPr]0.43) achieved a higher total yield (75%) than NaOH (69%). The total yield increased from 12% to 75%, while the yield of unsaturated ketone product 2 (Fig. 9) increased from 8% to 60% when a higher proportion of alkoxide anion was present in the ionic liquid. This highlights the tuneable nature of these binary ionic liquids which could be prepared according to the reaction requirements.
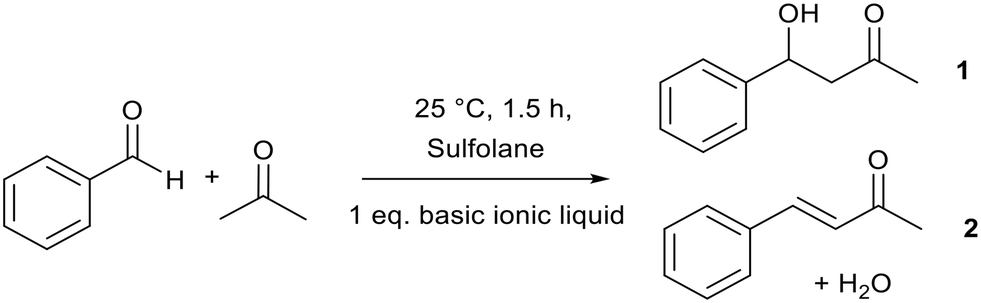 | ||
| Fig. 9 The aldol reaction catalysed by binary alkoxide ionic liquids.69 | ||
Davis and co-workers reported the synthesis of basic ionic liquids using sultones (Fig. 10).75 An amine group was tethered to an anion via nucleophilic addition of the amine to 1,3-propane sultone or 1,4-butane sultone, which undergoes ring opening to form a zwitterion. A solution of tetraalkylammonium hydroxide can be used to abstract a proton from the ammonium nitrogen to give the final ionic liquid.
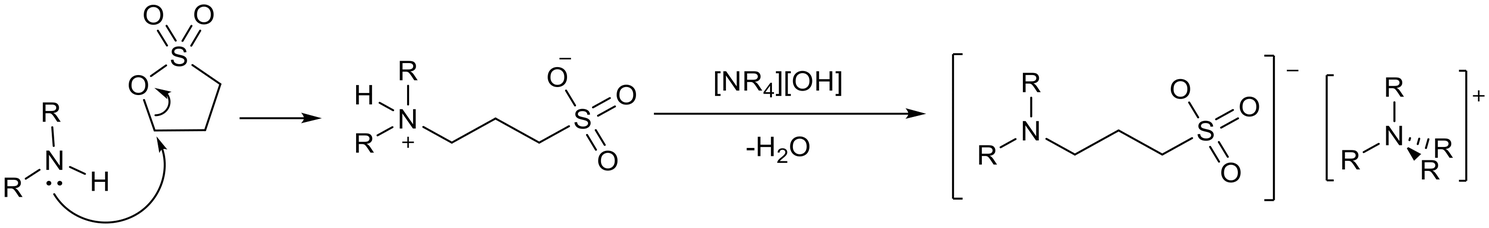 | ||
| Fig. 10 An example of the synthesis of ionic liquids containing neutral amine anions.75 | ||
Gao and co-workers synthesised analogous ionic liquids and applied them to aldol condensation reactions in water to produce α,α′-unsaturated ketones and α,β-unsaturated ketones (Fig. 11), which are intermediates for a range of natural products and drug molecules.76,77 Yields of over 90% were achieved for a range of substrates. The application of strong inorganic bases such as sodium hydroxide can lead to the generation of side products through the reverse aldol reaction, further aldol reactions at the carbonyl position, or Michael addition at the alkene position. Improved selectivity for the basic ILs was attributed to lower basicity. In some cases isolation was facile as solid products precipitated and were obtained by filtration.76 Soluble products were extracted into chloroform.77 The ionic liquid remained dissolved in the aqueous phase and was reused. Upon reuse high yields were maintained for a further four76 or five77 cycles but unreacted starting material remained in the aqueous phase after each reaction.
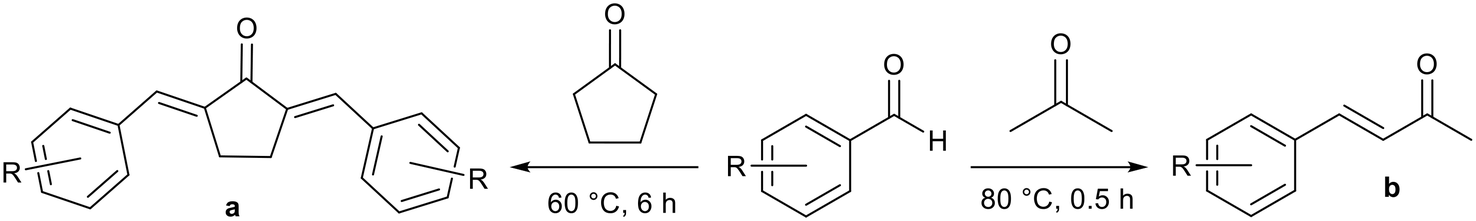 | ||
| Fig. 11 The Aldol synthesis of α,α′-unsaturated ketones (a)76 and α,β-unsaturated ketones (b)77 catalysed by basic ionic liquids. | ||
Basic ionic liquids have been applied to the production of fuels from renewable sources. Li and co-workers prepared a choline prolinate ([Ch][Pro]) ionic liquid via the deprotonation of proline with choline hydroxide.78 In this ionic liquid both the anion and cation are derived from nature. The initial step in fuel production was an aldol reaction between angelica lactone and furfural (Fig. 12). These are derived from cellulose and hemicellulose respectively and can therefore be considered as renewable. The ionic liquid was separated from the aldol product using a mixture of water and ethyl acetate, where the ionic liquid was soluble in water and the product in ethyl acetate. The aldol condensation product was then hydrogenated over palladium on carbon in a tubular reactor, before being hydrodeoxygenated in a fixed bed reactor using a palladium–copper catalyst supported on silica, to yield C8–C10 alkanes which could be used as fuels (Fig. 12). The authors report that [Ch][Pro] was more efficient than heterogeneous Mn2O3 for this aldol condensation.79 Compared to other ammonium ionic liquids containing the prolinate anion tested [Ch][Pro] was more active and selective, achieving 96% furfural conversion and 90% selectivity towards the unsaturated aldol product (Fig. 12). The authors proposed that short alkyl chains facilitate the reaction with furfural, whereas the reaction was inhibited by ammonium cations with longer alkyl chains. There was some loss of selectivity due to the self-Diels Alder reaction of furfural. This may be promoted by the strong hydrogen bonding of [Ch][Pro].80 Reuse of the recovered ionic liquid was not demonstrated but the authors cited the reuse of [Ch][Pro] with similar activity (ca. 90% yield) for the aldol reaction between 4-nitrobenzene and acetone.81
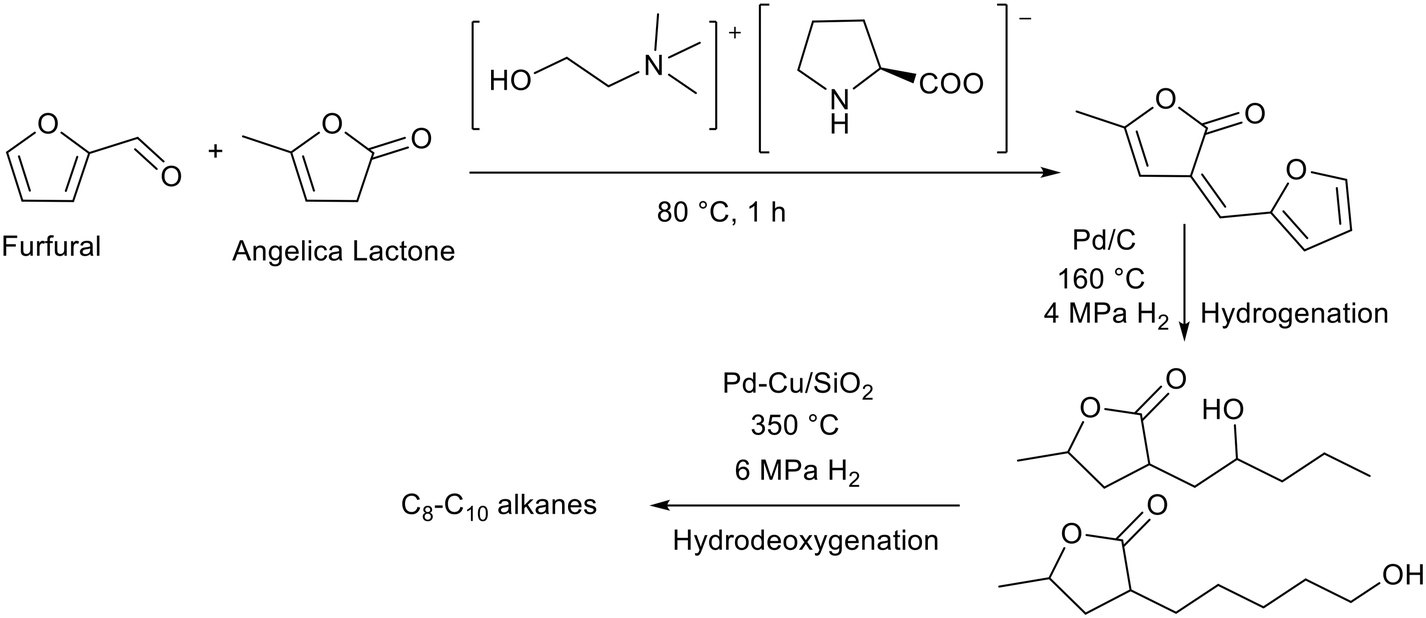 | ||
| Fig. 12 Generation of fuel alkanes by the [Ch][Pro] catalysed aldol condensation between furfural and angelica lactone followed by hydrogenation and hydrodeoxygenation.78 | ||
2.3 Esterification
The production of biofuels via esterification is becoming increasingly important as petroleum reserves are depleted. Ionic liquids could be utilised to overcome difficult catalyst separations that can occur in traditional homogeneous processes.59 Luo et al.36 used 1,3-butylmethylimidazolium imidazolate ([BMIM][Im]) to synthesise monoacylglycerol from triacylglycerol and glycerol (Fig. 13). Soybean oil was used as the triacylglycerol source. During initial comparative tests [BMIM][Im] achieved higher conversion and yield, 80% and 60% respectively, than both KOH, 70% and 30% respectively, and [BMIM][OH], which showed no activity. The authors attributed this to the better dispersibility of [BMIM][Im] in the reaction mixture compared to KOH, and the higher stability of [BMIM][Im] compared to [BMIM][OH]. The authors note that solubilising the hydrophilic glycerol in the hydrophobic triacylglycerol phase is key for this transformation. Therefore, increasing the reaction temperature from 170 °C to 200 °C improved the conversion of triacylglycerol and the yield of monoacylglycerols. Under optimised conditions, 95% conversion of triacylglycerol and 70% yield of monoacylglycerols was achieved after 3 hours. After the reaction the products formed a separate upper layer to the ionic liquid and glycerol lower layer. The products were obtained by washing with water whilst the ionic liquid and glycerol were recovered by washing with toluene and then drying. The recovered ionic liquid/glycerol layer was reused for a further six cycles with only a 1% decrease in monoacylglycerol yield.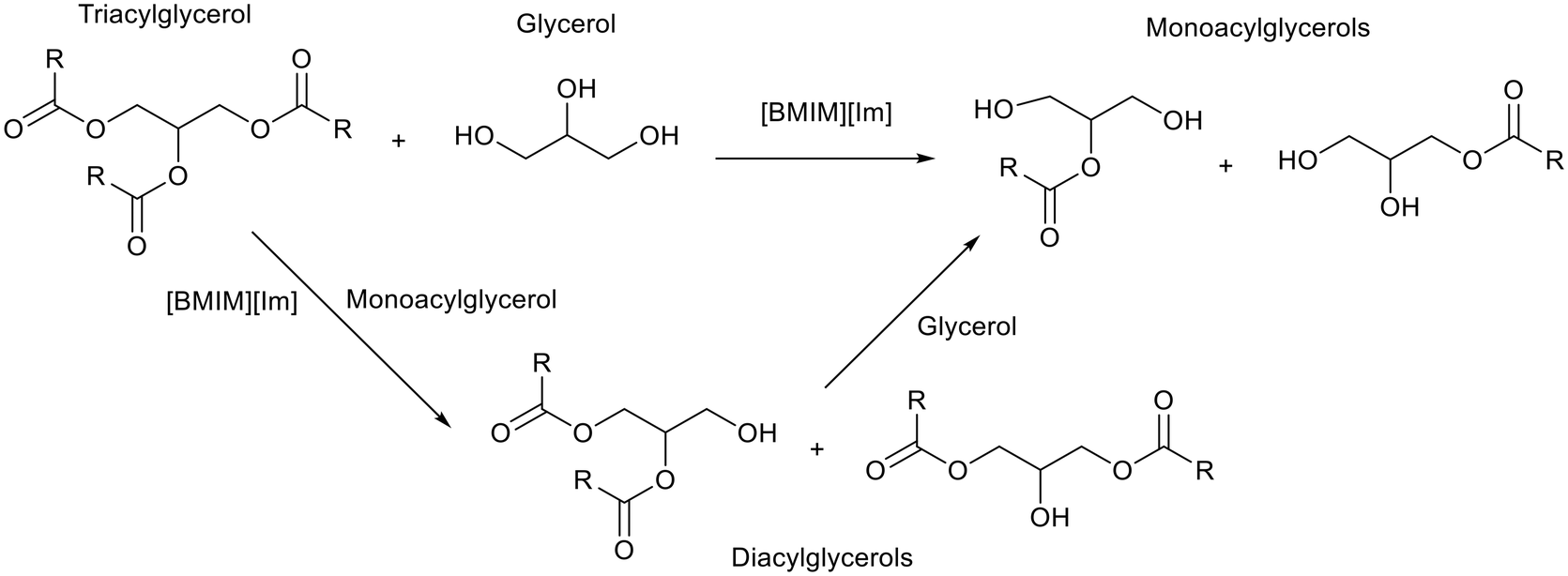 | ||
| Fig. 13 The synthesis of monoacylglycerols and diacylglycerols from triacylglycerol and glycerol catalysed by [BMIM][Im].36 | ||
An alternate esterification strategy was reported by Han and co-workers using 1,3-ethylmethylimidazolium acetate ([EMIM][OAc]) as the solvent and catalyst.82 A 94% yield was achieved for the self-esterification of benzyl alcohol and 70–94% yields were achieved for the cross-esterification of a range of benzylic and aliphatic alcohols after 12 hours at 80 °C. Molecular O2 was used as an oxidant to generate the carbonyl product (Fig. 14), with the basic ionic liquid catalyst replacing the metal catalyst that is normally required for this transformation. Both anion and cation were essential for the reaction, and hydrogen bonding to the alcohol substrate was implicated (Fig. 14a). An equilibrium between the ionic liquid and NHC initiated activation of the aldehyde formed in situ (Fig. 14b). After extracting the products into toluene the ionic liquid could be reused for a further 4 cycles with little change in activity. In this case the acidity of the imidazolium cation was used to promote the reaction without ionic liquid decomposition.
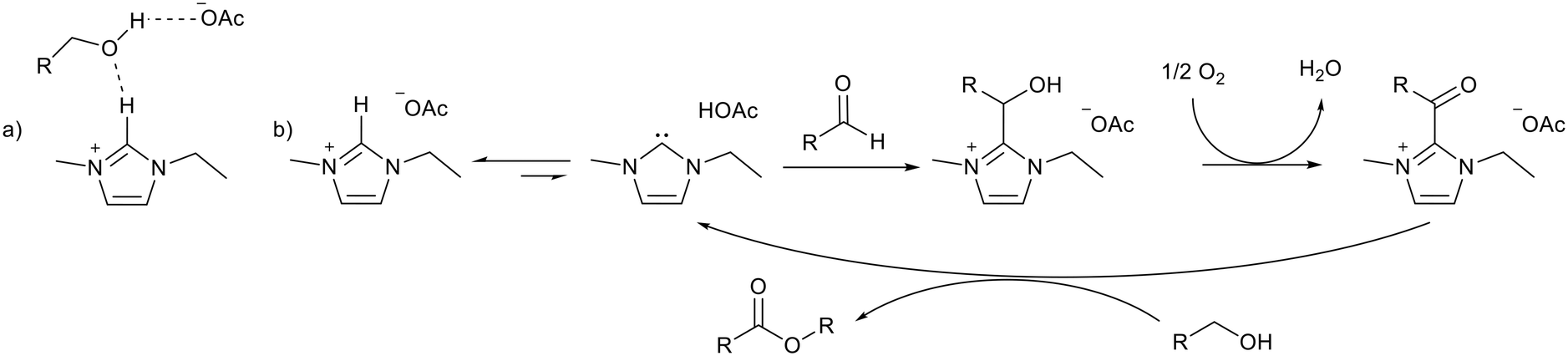 | ||
| Fig. 14 a) Hydrogen bonding between [EMIM][OAc] and an alcohol substrate. b) Activation of an aldehyde by an NHC formed from [EMIM][OAc] leading to esterification.82 | ||
2.4 Metal catalysed reactions
The higher stability of trihexyl tetradecylphosphonium hydroxide ([P6,6,6,14][OH]) compared to [BMIM][OH] was utilised by Rogers and co-workers who applied [P6,6,6,14][OH]·4MeOH as the basic promoter for cross-coupling reactions.83 MeOH was proposed to be important for the stability of ionic liquid as it caused a downfield shift in the 1H NMR of the alpha protons of the phosphonium cation compared to [P6,6,6,14][OH]. This could indicate that MeOH is interacting with the protons more strongly than the [OH]− anion, therefore [OH]− is less likely to attack the cation and cause decomposition. The authors claimed that [P6,6,6,14][OH]·4MeOH can prevent salt build-up during flow reactions as the chloride anion produced during the process exchanges with the [OH]− anion to form liquid [P6,6,6,14][Cl] (Fig. 15). When the [P6,6,6,14][OH]·4MeOH ionic liquid was used as base and solvent in a batch reaction, the yields for Buchwald–Hartwig reactions reached 44% and the yield for Suzuki–Miyaura reactions could reach 56%. This is comparable to KOtBu, 44% for Buchwald–Hartwig, and K2CO3, 50% for Suzuki–Miyaura. The authors stated that unlike the inorganic bases, under flow conditions the basic ionic liquid could be regenerated and reused using anion exchange resin, but data was not provided.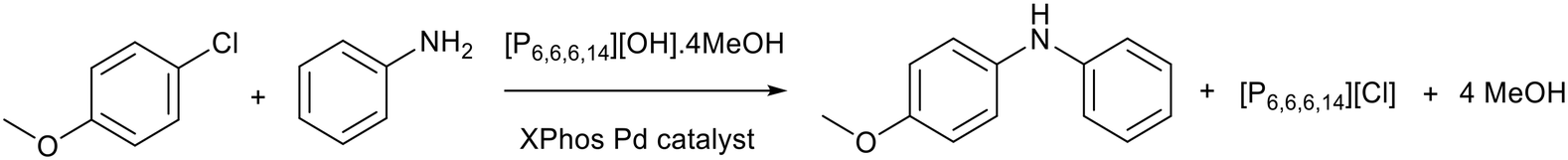 | ||
| Fig. 15 An example of a Buchwald–Hartwig cross-coupling promoted by [P6,6,6,14][OH]·4MeOH.83 XPhos is 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl. | ||
A reusable system for Suzuki reactions promoted by choline hydroxide ([Ch][OH]) was reported by Joo et al.84 Several palladium catalysts were tested for the reaction between 3-methoxyphenylboronic acid and 3-iodophenol (Fig. 16), with higher yields reported for [Pd(OAc)2] (65%) compared to [Pd(PPh3)] (51%), [Pd2(dba)3] (dba = dibenzylideneacetone) (38%) and [Pd(dppf)Cl2] (dppf = 1,1′-ferrocenediyl-bis(diphenylphosphine)) (63%). The use of [Pd(OAc)2] simplified the separation process after the reaction. Aqueous Pd/[Ch][OH] was reused 5 times with a small decrease in yield (62% to 59%). Lower yields were obtained for arylbromides (21–51%) and no reactivity were observed for arylchlorides.
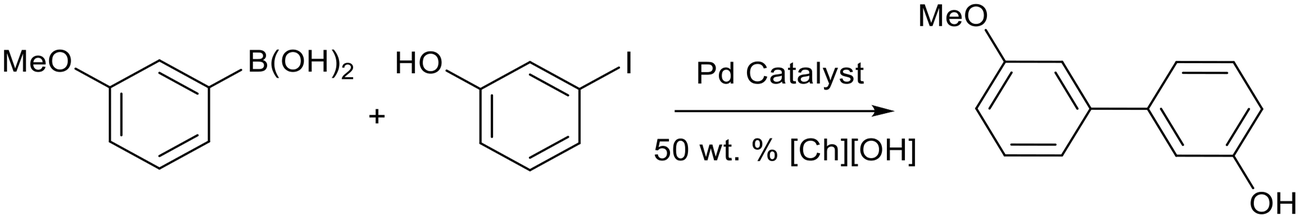 | ||
| Fig. 16 The palladium catalysed Suzuki cross-coupling reaction between 3-methoxyphenylboronic acid and 3-iodophenol promoted by choline hydroxide.84 | ||
2.5 CO2 capture and valorisation
Global warming due to a rise in atmospheric levels of CO2 is an increasing concern. Global accords, such as the Paris Agreement, are aimed at preventing catastrophic temperature rises by limiting CO2 release.85 A recent UN report highlights the discrepancy between these plans and projected CO2 reductions.86 In addition to reducing CO2 release, methods are being developed to capture CO2 from the atmosphere or at source, for example in energy plants. The traditional industrial method for CO2 capture is the use of ethanolamines, but these processes suffer from high energy costs for CO2 release and amine degradation.87 Ionic liquids have been explored as alternatives to ethanolamines. COSMO-RS calculations were used to compare CO2 capture processes between [BMIM][NTf2] and methyldiethanolamine. It was found that despite an increase in electrical energy usage, the ionic liquid could reduce the total energy consumption by 66%.88 The first use of functionalised ionic liquids to capture CO2 was reported by Davis and co-workers who used an amine functionalised imidazolium ionic liquid to chemically capture CO2 (Fig. 17).14 Reaction with almost 0.5 mole CO2 per mole ionic liquid was achieved, which is comparable to standard amines. Unlike many amines which decompose during CO2 release, the ionic liquid could be reused for five cycles by heating to 80–100 °C under vacuum for several hours.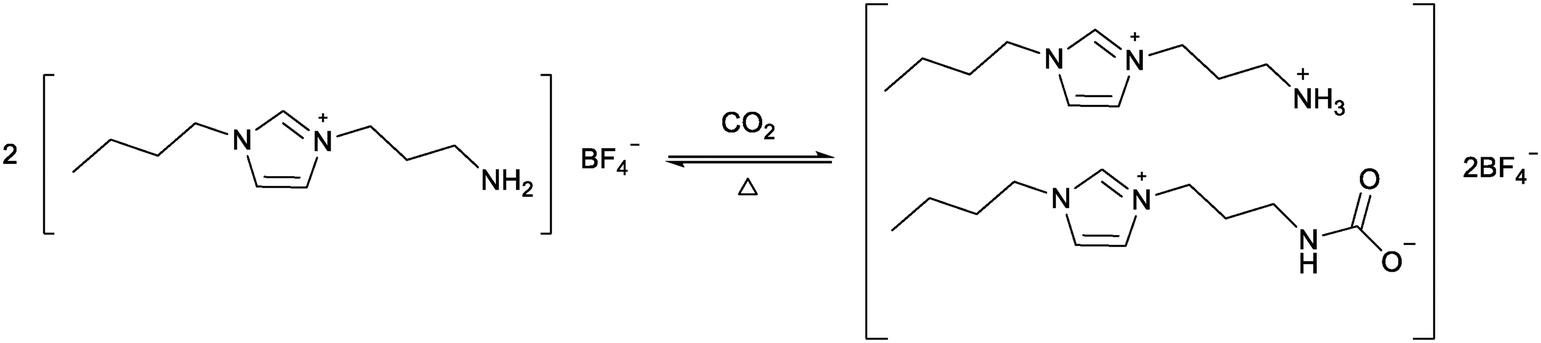 | ||
| Fig. 17 The reaction between CO2 and an amine functionalised ionic liquid proposed by Davis and co-workers.14 | ||
The use of ionic liquids for physical absorption, and the use of basic ionic liquids for chemical absorption, have been reviewed recently by Cui et al.89 and Zeng et al.90 These reviews include the design of ionic liquids based on their molecular structure and properties as well as the scale up and industrialisation of these ionic liquids and new ionic liquid based materials. In this review a brief update on CO2 capture and the use of CO2 as a C1 building block will be provided, focusing solely on the use of basic ionic liquids.
Ionic liquids with amine groups generally absorb CO2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:1 molar ratio by forming a carbamate. Luo et al.91 have prepared ionic liquids with amino functionalised anions which can capture CO2 in almost a 1:2 ratio. The best performing ionic liquid, bis(trihexyl tetradecylphosphonium) aspartate ([P6,6,6,14]2[Asp]), reacted with 1.96 moles of CO2 per mole of ionic liquid. This occurs through a proton transfer process which causes intramolecular hydrogen bond formation and allows the reaction of the ionic liquid with a second molecule of CO2 (Fig. 18). The formation of this intramolecular hydrogen bond also decreased the viscosity of the ionic liquid from 4903 cP to 3946 cP. This is unusual as the viscosity of amine functionalised ionic liquids tend to increase after reacting with CO214,89,90 which limits the ionic liquids' practicality and industrial relevance. [P6,6,6,14]2[Asp] released the captured CO2 when the temperature was increased from 30 °C to 80 °C and the CO2 pressure decreased from 1 atm to 0.01 atm. These are mild conditions compared to the traditional amine processes in which the recovery temperatures often exceed 200 °C.87 [P6,6,6,14]2[Asp] was recycled 20 times and the CO2 molar uptake remained consistent throughout.
:1 or 1:1 molar ratio by forming a carbamate. Luo et al.91 have prepared ionic liquids with amino functionalised anions which can capture CO2 in almost a 1:2 ratio. The best performing ionic liquid, bis(trihexyl tetradecylphosphonium) aspartate ([P6,6,6,14]2[Asp]), reacted with 1.96 moles of CO2 per mole of ionic liquid. This occurs through a proton transfer process which causes intramolecular hydrogen bond formation and allows the reaction of the ionic liquid with a second molecule of CO2 (Fig. 18). The formation of this intramolecular hydrogen bond also decreased the viscosity of the ionic liquid from 4903 cP to 3946 cP. This is unusual as the viscosity of amine functionalised ionic liquids tend to increase after reacting with CO214,89,90 which limits the ionic liquids' practicality and industrial relevance. [P6,6,6,14]2[Asp] released the captured CO2 when the temperature was increased from 30 °C to 80 °C and the CO2 pressure decreased from 1 atm to 0.01 atm. These are mild conditions compared to the traditional amine processes in which the recovery temperatures often exceed 200 °C.87 [P6,6,6,14]2[Asp] was recycled 20 times and the CO2 molar uptake remained consistent throughout.
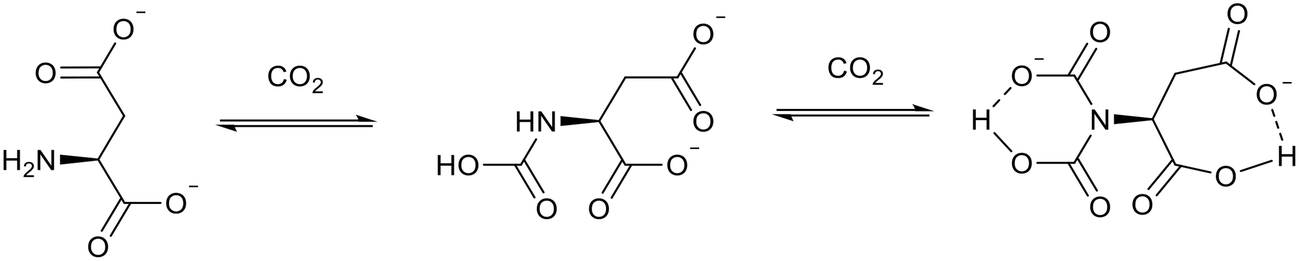 | ||
| Fig. 18 CO2 capture by the [Asp]− anion of [P6,6,6,14]2[Asp].91 | ||
Despite the high mol/mol CO2 uptake of [P6,6,6,14]2[Asp], the high molecular weight (1098.83 gmol−1) results in a poor gravimetric capacity (8 wt%).90 Higher gravimetric capacity was achieved by Huang et al.92 using tributyl ethylphosphonium succinimide ([P4,4,4,2][Suc]). They proposed “Preorganisation” of the diacetamide anion to form the succinimide anion (Fig. 19) which allowed the CO2 uptake to reach 22 wt% and 25 wt% when using 10 vol% CO2 in N2, and 100 vol% CO2 respectively. This mechanism of CO2 uptake was challenged by Dupont and co-workers, who proposed an alternative pathway via the formation of bicarbonate anions.93 Further work by Huang et al.94 showed that the mechanism could proceed through preorganisation when the ionic liquid had a low water content and through bicarbonate at higher water content. This conclusion was supported by Brennecke and co-workers.95 The ionic liquid was regenerated by bubbling N2 through it and the high absorption capacity rate and was maintained over 16 cycles.92
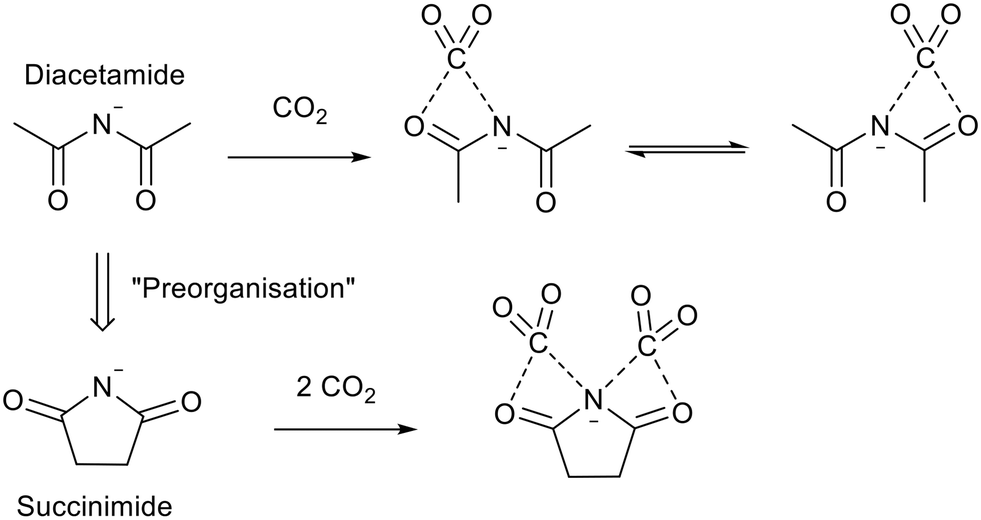 | ||
| Fig. 19 The “preorganisation” of diacetamide and CO2 capture by the succinimide anion.92 | ||
Basic ionic liquids can also be used to convert CO2 into value added products. A major focus of research to date has centred on the reaction of CO2 with epoxides to form cyclic carbonates. These transformations catalysed by ionic liquids, many of which were basic, have featured in several reviews in recent years.57,96,97 Other reactions have also been studied and some notable examples will be discussed here.
Li et al.98 used choline imidazolate ([Ch][Im]) based ionic liquids to promote the N-formylation of secondary amines at 120 °C in the presence of CuCl and a CO2/H2 mixture (1:2.6). The N-formylation of amines was found to proceed with 99% yield after 24 h using CuCl and [Ch][Im]. The anion was found to be key to the high yield. Ionic liquids with phenolate or pyridine-2-olate anions produced only 32 and 58% yield respectively. The authors attributed this to good coordination of [Im]− with CuCl. High yields were achieved when the system was applied to aliphatic cyclic amines (78–99%) and acyclic di-n-propylamine (91%). However only 31% yield was achieved for aromatic N-methylaniline showing the system may be limited to aliphatic amines.
Similarly, acetylcholine carboxylate ionic liquids were used by Zhao et al.99 to selectively promote either the N-methylation or N-formylation of amines with CO2 (Fig. 20). The ionic liquids used by Zhao et al. are constructed from cations and anions that have demonstrated lower toxicity and good biodegradability. The nature of the ionic liquid was found to influence the reaction yield in the N-methylation of methylaniline. The acetylcholine acetate ionic liquid (Fig. 20) was found to have the best balance between high basicity and good CO2 permittivity. A yield of 96% could be achieved after 6 h compared to the analogous propionate (77%), valerate (42%), and formate (12%) ionic liquids. The cation also played a role with the acetylcholine cation working to regulate the anion's basicity and effectively promote the reaction. Polar solvents increased the yield of N-methylation and performing the reaction under solvent free conditions at a reduced temperature promoted the N-formylation reaction. The catalyst system promoted the N-methylation of a range of aromatic amines and N-formylation of both aromatic and aliphatic amines. This improved substrate scope must be carefully weighed up against the requirement for 3 equivalents of phenylsilane in this protocol.
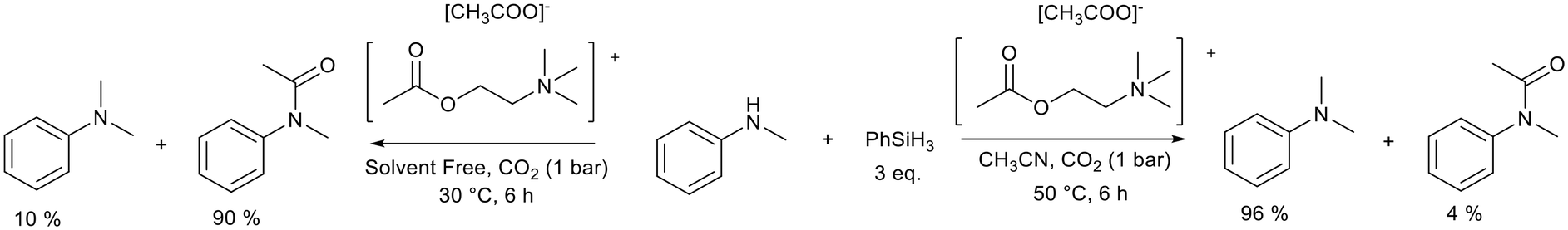 | ||
| Fig. 20 The N-methylation and N-formylation of N-methylaniline promoted by choline carboxylate ionic liquids.99 | ||
Water can be a very sustainable hydrogen source. H2O was used by Yuan, Verpoort, and co-workers for the semi-hydrogenation of propargylic alcohols to α-hydroxy ketones at 80 °C, using 1-ethyl-3-methylimidazolium acetate ([EMIM][OAc]) with [AgOAc] as a catalyst (Fig. 21).100 α-Hydroxy ketones are found in pharmaceutical products and are building blocks for fine chemicals.101,102 Good yields and chemoselectivity was obtained for a variety of substrates including those with olefin functionality (Fig. 21, reaction 4). The authors state that the metal loading of 0.25 mol% is the lowest for this class of reaction. The metal loading was further reduced to 0.005 mol% which gave an excellent turnover number of 9200, although the reaction time had to be increased from 6 h to 84 h and the yield was reduced from 95% to 46%. Screening various conditions, the authors found both neutral and acidic ionic liquids to be ineffective and proposed that the acetate anion was involved in activating both the alcohol hydroxy group and CO2. [KOAc] was also ineffective as it did not activate the hydroxyl group, as determined by 1H NMR. The imidazolium cation was also crucial as it formed a bis N-heterocyclic carbene (NHC) with silver which activated the triple bond for reduction. After extracting the products into ether, the ionic liquid and silver salt were dried and reused without a significant decrease in activity over five cycles. A comparison of the reaction metrics (atom economy {AE}, E-factor, carbon efficiency {CE}, reaction mass efficiency {RME}, mass intensity {MI}, and mass productivity {MP}) showed this system to be “greener” than other procedures used to achieve the semi-hydrogenation of propargylic alcohols.
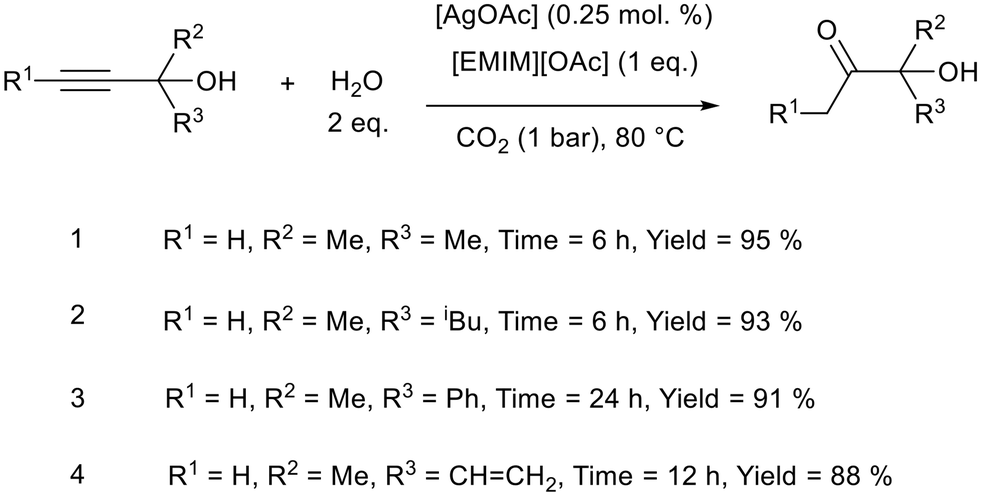 | ||
| Fig. 21 The general reaction scheme for semi-hydrogenation of propargylic alcohols and a selection of reactions performed by Yuan, Verpoort, and co-workers.100 | ||
3. Heterogenized ionic liquids
Associating a basic ionic liquid catalyst with a solid support can improve stability as well as separation. Marr and Bothwell showed that a solution of trihexyl tetradecylphosphonium hydroxide ([P6,6,6,14][OH]) decomposed within 2–3 weeks as a liquid, but when the same ionic liquid was used entrapped within a silica gel it remained stable for over a year.103 Ideally heterogenized ionic liquids will retain the favourable properties of the incorporated ionic liquid whilst being facile to separate and recycle. Homogeneous ionic liquids are often separated by solvent extraction or distillation, which can be wasteful and energy intensive.36,37,68,77,82 The following sections provide an introduction to methods of heterogenizing ionic liquids with illustrative examples of heterogenized basic ionic liquids and their applications.3.1 Supported ionic liquid phase (SILPs)
SILPs, also known as supported ionic liquid catalysts, or SILCs (Fig. 22) are the simplest form of a heterogenized ionic liquid. SILPs can be formed via impregnation or tethering of the ionic liquid to a porous support. Metal complexes can be incorporated before or after ionic liquid immobilisation.104,105 Simple impregnation can be problematic in liquid phase reactions as the ionic liquid can be stripped off the support (leaching) if the solvent is not chosen carefully.105 SILPs can dramatically reduce the amount of ionic liquid required for a reaction as only a thin layer is used. The ionic liquid layer can offer high activity as viscosity problems encountered in bulk ionic liquids are reduced due to the small diffusion distance offered to substrates and products (Fig. 22).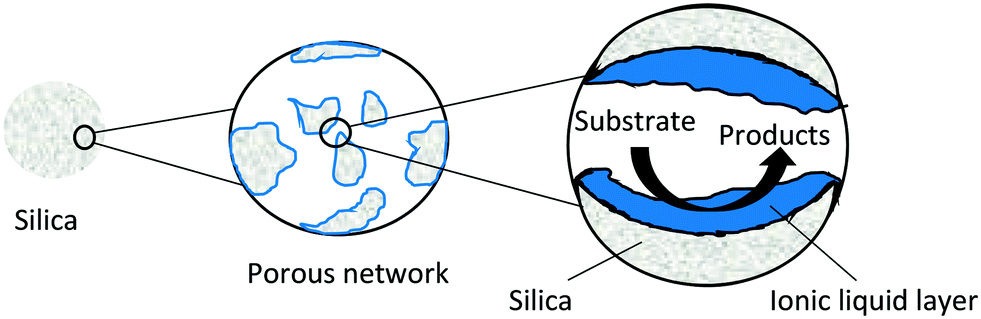 | ||
| Fig. 22 Schematic representation of a SILP. | ||
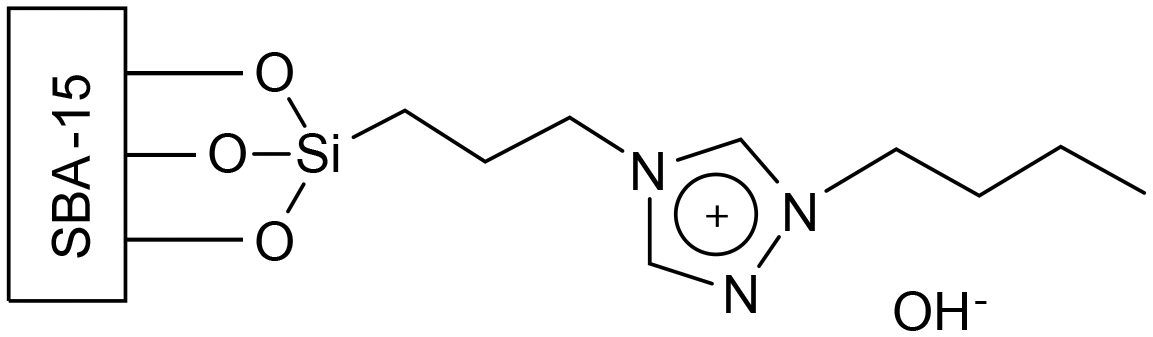 | ||
| Fig. 23 The structure of the tethered ionic liquid prepared by Xie et al.55 | ||
Catalyst recycling can be improved when the support material protects the ionic liquid. Two catalysts were prepared by Zhang et al.106 The support materials were synthesised by either dispersing CoFe2O4 onto mesoporous silica, or by coating a CoFe2O4 nanoparticle with silica. An imidazolium hydroxide ionic liquid was then tethered to the silica of both support materials (Fig. 24). These SILPs were applied to the esterification of glycerol trioleate using methanol. Higher activity was achieved using the SILP catalysts rather than NaOH and this was maintained upon catalyst reuse. The mesoporous silica catalyst exhibited the best activity when recycled. The authors propose that leaching of the ionic liquid caused by the substrate is unlikely due to the need for the substrate to diffuse through the catalyst pores in order to interact with the ionic liquid (Fig. 24a). In contrast, the substrate can flow freely around the nanoparticle catalyst and strip the ionic liquid more easily.
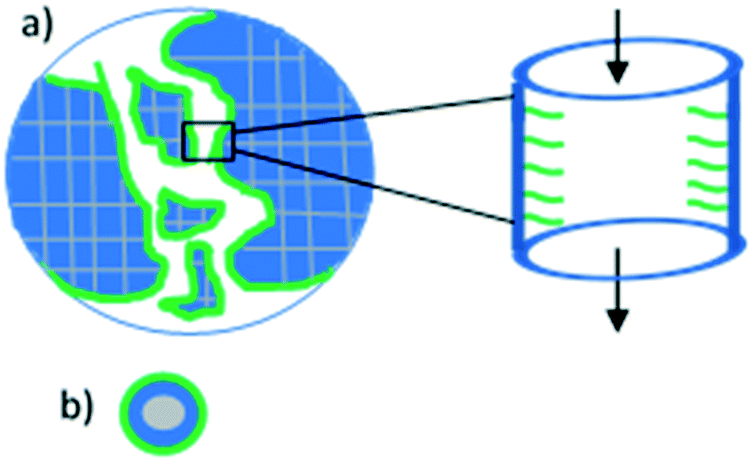 | ||
| Fig. 24 a) A representation of ionic liquid tethered to mesoporous silica, and the diffusion of the substrate through the SILP pores. b) A representation of ionic liquid coated on a magnetic silica nanoparticle. Blue is silica, grey is CoFe2O4 and green is ionic liquid.106 | ||
Despite the good activity and low levels of degradation in the SILPs discussed in this section, there are disadvantages to their use. Forces of attraction between the support surface and the ions tend to be too weak to prevent leaching to a liquid reactant phase and therefore tethering is necessary. The extra chemical steps required to tether the ions increases costs, consumption of materials and safety concerns. The best SILPs are often based on highly ordered supports such as SBA-15 and these also require more expensive templated syntheses.
3.2 Ionic liquid gel catalysis
Gels comprise a liquid phase entrapped within a solid matrix. Forces of attraction between the liquid and matrix in a gel are higher than in surface adsorption and therefore entrapment of an ionic liquid within a gel does not require covalent tethering.107 A SILP and a silica ionic liquid gel were compared for acidic ionic liquid catalysis under the same conditions. The acidic ionic liquid gel provided heterogeneous leach free dehydration of 1-phenylethanol to styrene in toluene and could be recycled without a loss in activity, whereas the SILP suffered from significant leaching.108 Despite the widespread use of ionic liquids to form gels, there are very few examples of basic ionic liquid gels. In this case the instability of basic ionic liquids is not the only limiting factor, in fact the relative speed of the base catalysed sol–gel process is most problematic. In alkaline solution the initiation of silica formation is very fast, and this tends to lead to oxide gels with poor structural properties, low porosity and poor mechanical stability.109,110 This problem was addressed by Bothwell and Marr who utilised the high viscosity of basic ionic liquid trihexyl tetradecylphosphonium hydroxide ([P6,6,6,14][OH]) to slow the sol–gel process and form stable silica and titania gels.103 An additional sol–gel catalyst was not added and the process was catalysed by the ionic liquid. These gels were found to have good transparency and were easy to prepare and handle. Unfortunately, they were not found to be stable enough to support reliable base catalysed reactions. Further work by Marr, Marr, and co-workers employed the binary approach (section 2.1) to stabilise the basic ionic liquid gels and enable catalytic applications of basic ionic liquid gels.111 Mixtures of [P6,6,6,14][OH] and neutral, hydrophobic 1-butyl-2,3-dimethylimidazolium bistriflimide ([BMMIM][NTf2]) were combined with the silica precursor tetraethyl orthosilicate (TEOS) to form gels which acted as the basic promoter in the hydrogen transfer initiated dehydration (HTID) of 1,3-propanediol (Fig. 25). Performing the reaction using an iridium cyclopentadienyl N-heterocyclic carbene complex [Cp*IrCl2(NHC)] as a hydrogen transfer catalyst in 1-ethyl-2,3-dimethylimidazolium bistriflimide ([EMMIM][NTf2]) allowed in situ distillation of the desired product propionaldehyde (Fig. 25). When a gel containing 1:3 [BMMIM][NTf2]:[P6,6,6,14][OH] was used, the gel, catalyst, and ionic liquid could be recycled with consistent activity over 5 reactions. The basic ionic liquid gel was found to be more stable than K2CO3 and KOH.112,113
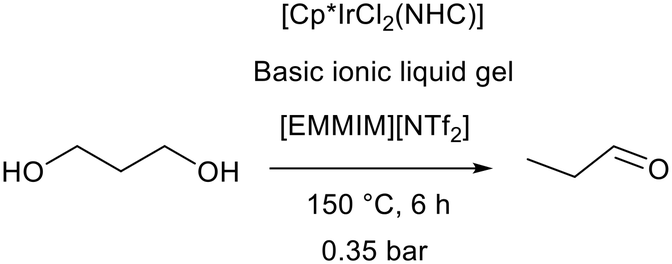 | ||
| Fig. 25 The hydrogen transfer initiated dehydration of 1,3-propanediol promoted by a basic ionic liquid gel.111 | ||
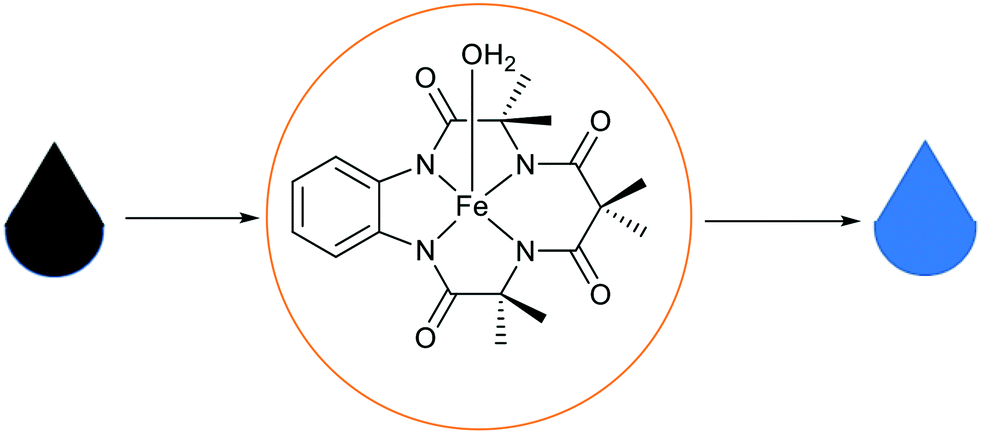 | ||
| Fig. 26 A representation of water purification using Fe-TAML entrapped in basic ionic liquid silica materials.115 | ||
3.3 Microcapsules, core shells, and spheres
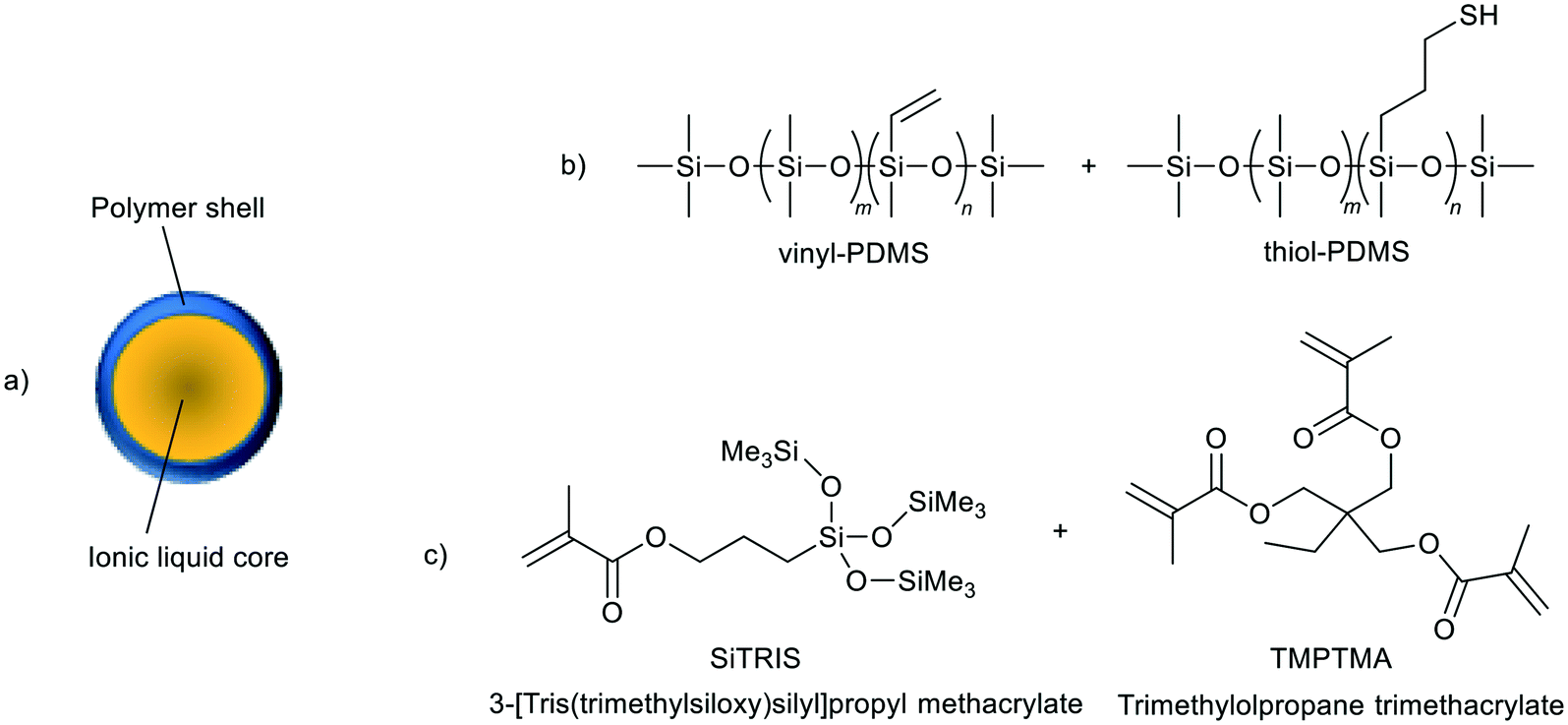 | ||
| Fig. 27 A representation of an ionic liquid microcapsule as prepared by Stolaroff et al. (a), and the components of the different polymer shell materials (b and c).118 | ||
Brennecke and co-workers adopted the process of Stolaroff et al. to encapsulate basic ionic liquids (Fig. 28) and showed the microcapsules had an almost stoichiometric uptake of CO2 above 0.4 bar, matching the free ionic liquid.119 After CO2 uptake and release, the capsules were broken open. 1H NMR analysis of the ionic liquids did not show any degradation products. The capsules could be used for 7 cycles with similar uptake and kinetics in each cycle.
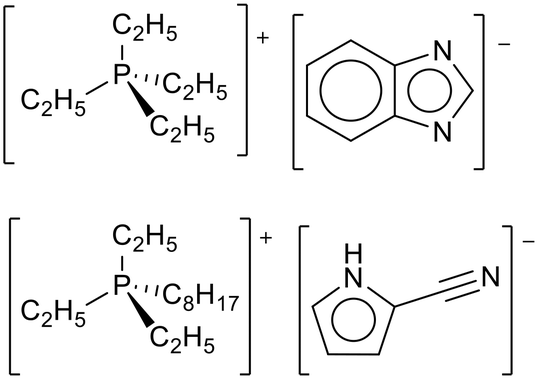 | ||
| Fig. 28 The basic ionic liquids used by Brennecke and co-workers for entrapment in polymer microcapsules.119 | ||
Encapsulating ionic liquids can also improve the kinetics of CO2 absorption and desorption, as demonstrated for trihexyl tetradecylphosphonium 2-cyanopyrrolide (Fig. 29).120 Brennecke and co-workers used a sol–gel process with TEOS and octadecyltrimethoxysilane to form a silica sphere with a solid core which was pyrolysed. A phenyl-formaldehyde carbon shell was formed around the silica sphere, followed by a second pyrolysis step and removal of the silica template by washing with HF to leave carbon spheres. The spheres were dispersed in a solution of ionic liquid dissolved in acetone. Removal of the acetone under vacuum dispersed the ionic liquid within the carbon microspheres. The spheres had a wall thickness of 70 nm which, combined with the increased liquid gas surface interface, allowed total CO2 absorption at 1 bar to be complete in 20 minutes rather than 12 hours for the bulk ionic liquid. The capsules were used for three cycles with similar uptake and kinetics. These ionic liquid microcapsules showed promise for post-combustion CO2 capture and could therefore be a solution to the poor permeability which can limit ionic liquid membranes.
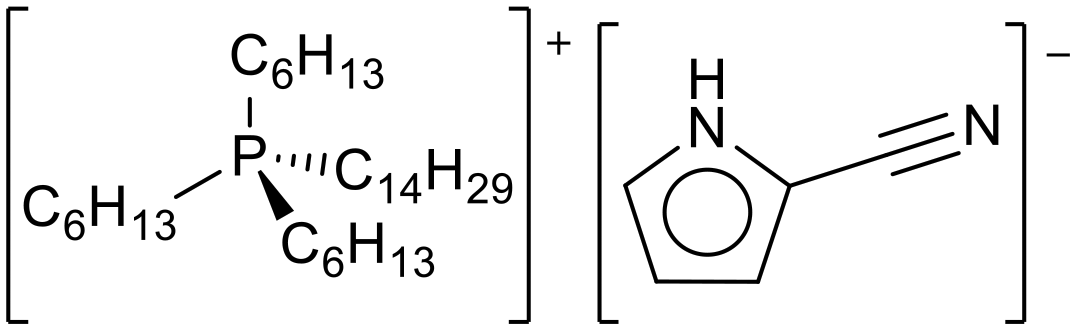 | ||
| Fig. 29 The structure of trihexyl tetradecylphosphonium 2-cyanopyrrolide, as used for entrapment in carbon shells for CO2 capture.120 | ||
As with homogeneous basic ionic liquids (section 2.5), the reaction between epoxides and CO2 to form cyclic carbonates is often reported catalysed by heterogeneous ionic liquids. Wang et al.121 have recently reviewed the use of heterogenized ionic liquids, many of which are basic, applied to this class of reactions.
4. Conclusions
Basic ionic liquids have been shown to catalyse a wide variety of organic reactions including Knoevenagel condensations, aldol reactions, esterification, and cross-coupling reactions. A recent trend is to use basic ionic liquids to promote reactions that could form the basis of more sustainable energy and chemical production such as biofuel production, biomass upgrading, and CO2 capture. Ionic liquids can be safer than traditional bases due to their low volatility and flammability. However traditional inorganic and organic bases are generally cheaper than ionic liquids. An ionic liquid must solve significant problems in the conventional process for it to be adopted as a feasible alternative. These improvements can be based on the tuneability of ionic liquids, allowing them to be designed for a particular reaction to improve the yield. Alternatively, or preferably additionally, employing an ionic liquid can lead to simplification of the separation of the product from the reaction mixture. The potential to recover and reuse a basic ionic liquid is a clear advantage over traditional bases and this reduces the cost of ionic liquid processes. A limiting factor in the recycle of ionic liquids is their stability, which can prevent industrial application. Understanding of the side-reactions and decomposition pathways of ionic liquids has led to the molecular design of more stable basic ILs. Weakly basic ionic liquids have been known for a while, and strongly basic stable ionic liquids are now being reported. The stability of a basic ionic liquid can be further enhanced by associating it with a solid material. Heterogenizing basic ionic liquids on solid supports is a powerful method to improve their stability and also facilitate simple recycling. Ionic liquids can be supported on porous supports such as silica (e.g. SILPs) through physical or covalent means, entrapped in porous soft materials such as gels, or entrapped in microcapsules, spheres, and core shells. Heterogenization of basic ionic liquids could also allow incorporation of nanoparticles, molecular catalysts, or enzymes for the development of more complex catalytic systems. Combining the molecular design of stable ionic liquid structures with an appropriate solid support should lead to industrial examples of basic ionic liquids in the near future, particularly when the best materials methods are combined with the most robust basic ionic liquids.Conflicts of interest
There are no conflicts to declare.Acknowledgment
Queen's University Ionic Liquid Laboratories for a studentship (PM). Dr Martyn J. Earle for illuminating discussions. This article is dedicated to our late collaborators and friends Prof. Ken Seddon and Prof. Paul Kamer.References
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- D. R. MacFarlane, J. Sun, J. Golding, P. Meakin and M. Forsyth, Electrochim. Acta, 2000, 45, 1271–1278 CrossRef.
- R. P. Swatloski, J. D. Holbry and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- G. A. Baker and S. N. Baker, Aust. J. Chem., 2005, 58, 174–177 CrossRef CAS.
- C. Villagrán, M. Deetlefs, W. R. Pitner and C. Hardacre, Anal. Chem., 2004, 76, 2118–2123 CrossRef.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS.
- Electrochemical Aspects of Ionic Liquids, ed. H. Ohno, John Wiley and Sons, Hoboken, New Jersey, 2005 Search PubMed.
- X.-H. Liu, M. Rebroš, I. Dolejš and A. C. Marr, ACS Sustainable Chem. Eng., 2017, 5, 8260–8268 CrossRef CAS.
- O. Verho and J. E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS.
- M. Smiglak, W. M. Reichert, J. D. Holbry, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rodgers, Chem. Commun., 2006, 2554–2556 RSC.
- D. M. Fox, J. W. Gilman, A. B. Morgan, J. R. Shields, P. H. Maupin, R. E. Lyon, H. C. De Long and P. C. Trulove, Ind. Eng. Chem. Res., 2008, 47, 6327–6332 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis Jr., J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- Z. Liu, C. Xu and C. Huang, Method for manufacturing alkylate oil with composite ionic liquid used as catalyst, US Pat., 0133056, 2004 Search PubMed.
- E. M. Broderick, A. Bhattacharyya, S. Yang and B. J. Mezza, Contaminant removal from hydrocarbon streams with lewis acidic ionic liquids, US Pat., 9574139, 2017 Search PubMed.
- R. Brelsford, Chevron's Utah refinery lets contract for alkylation technology retrofit, https://www.ogj.com/refining-processing/article/17290342/chevrons-utah-refinery-lets-contract-for-alkylation-technology-retrofit, (accessed 17 July 2020) Search PubMed.
- S. Raiguel, W. Dehaen and K. Binnemans, Green Chem., 2020, 22, 5225–5252 RSC.
- V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC.
- A. K. L. Yuen, A. F. Masters and T. Maschmeyer, Catal. Today, 2013, 200, 9–16 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS.
- A. N. Kost, S. P. Gromov and R. S. Sagitullin, Tetrahedron, 1981, 37, 3423–3454 CrossRef CAS.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, John Wiley and Sons, Chichester, 5th edn, 2011, ch. 8 Search PubMed.
- M. Zanger, C. A. Vander Werf and W. E. McEwen, J. Am. Chem. Soc., 1959, 81, 3806–3807 CrossRef CAS.
- M. Grayson and P. T. Keough, J. Am. Chem. Soc., 1960, 82, 3919–3924 CrossRef CAS.
- T. Ramnial, D. D. Ino and J. A. C. Clyburne, Chem. Commun., 2005, 325–327 RSC.
- T. R. Gohndrone, T. B. Lee, M. A. DeSilva, M. Quiroz-Guzman, W. F. Schneider and J. F. Brennecke, ChemSusChem, 2014, 7, 1970–1975 CrossRef CAS.
- T. Itoh, K. Kude, S. Hayase and M. Kawatsura, Tetrahedron Lett., 2007, 48, 7774–7777 CrossRef CAS.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Chem. Commun., 2004, 1972–1973 RSC.
- T. Ramnial, S. A. Taylor, M. L. Bender, B. Gorodetsky, P. T. K. Lee, D. A. Dickie, B. M. McCollum, C. C. Pye, C. J. Walsby and J. A. C. Clyburne, J. Org. Chem., 2008, 73, 801–812 CrossRef CAS.
- B. Zhang, R. B. Kaspar, S. Gu, J. Wang, Z. Zhuang and Y. Yan, ChemSusChem, 2016, 9, 2374–2379 CrossRef CAS.
- S. Raiguel, J. Thomas, K. Binnemans and W. Dehaen, Eur. J. Org. Chem., 2018, 35, 4850–4856 CrossRef.
- D. Vasilyev, E. Shirzadi, A. V. Rudnev, P. Broekmann and P. J. Dyson, ACS Appl. Energy Mater., 2018, 1, 5124–5128 CAS.
- M. Hayyan, F. S. Mjalli, M. A. Hashim, I. M. AlNashef and T. X. Mei, J. Ind. Eng. Chem., 2013, 19, 106–112 CrossRef CAS.
- H. Luo, Z. Zhai, W. Fan, W. Cui, G. Nan and Z. Li, Ind. Eng. Chem. Res., 2015, 54, 4923–4928 CrossRef CAS.
- S. A. Forsyth, U. Fröhlich, P. Goodrich, H. Q. N. Gunaratne, C. Hardacre, A. McKeown and K. R. Seddon, New J. Chem., 2010, 34, 723–731 RSC.
- A. Wykes and S. L. MacNeil, Synlett, 2007, 1, 0107–0110 Search PubMed.
- M. Yoshizawa-Fujita, K. Johansson, P. Newman, D. R. MacFarlane and M. Forsyth, Tetrahedron Lett., 2006, 47, 2755–2758 CrossRef CAS.
- M. Yoshizawa-Fujita, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Electrochem. Commun., 2006, 8, 445–449 CrossRef CAS.
- J. M. Crosthwaite, M. J. Muldoon, J. K. Dixon, J. L. Anderson and J. F. Brennecke, J. Chem. Thermodyn., 2005, 37, 559–568 CrossRef CAS.
- S. A. Forsyth, D. R. MacFarlane, R. J. Thomson and M. von Itzstein, Chem. Commun., 2002, 714–715 RSC.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS.
- C. Wang, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS.
- X. Cui, S. Zhang, F. Shi, Q. Zhang, X. Ma, L. Lu and Y. Deng, ChemSusChem, 2010, 3, 1043–1047 CrossRef CAS.
- C. A. Ober and R. B. Gupta, Ind. Eng. Chem. Res., 2012, 51, 2524–2530 CrossRef CAS.
- L. P. Hammett and A. J. Deyrup, J. Am. Chem. Soc., 1932, 54, 2721–2739 CrossRef CAS.
- C. Thomazeau, H. Olivier-Bourbigou, L. Magna, S. Luts and B. Gilbert, J. Am. Chem. Soc., 2003, 125, 5264–5265 CrossRef CAS.
- W. Li, Z. Zhang, B. Han, S. Hu, J. Song, Y. Xie and X. Zhou, Green Chem., 2008, 10, 1142–1145 RSC.
- C. Rizzo, F. D'Anna and R. Noto, RSC Adv., 2016, 6, 58477–58484 RSC.
- D. R. MacFarlane, J. M. Pringle, K. M. Johansson, S. A. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905–1917 RSC.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 3rd edn, 2003 Search PubMed.
- W. Xie and X. Huang, Catal. Lett., 2006, 107, 53–59 CrossRef CAS.
- J.-I. Take, N. Kikuchi and Y. Yoneda, J. Catal., 1971, 21, 164–170 CrossRef CAS.
- W. Xie, L. Hu and X. Yang, Ind. Eng. Chem. Res., 2015, 54, 1505–1512 CrossRef CAS.
- W. Y. Hernández, F. Aliç, A. Verberckmoes and P. Van Der Voort, J. Mater. Sci., 2017, 52, 628–642 CrossRef.
- J. Hu, H. Liu and B. Han, Sci. China: Chem., 2018, 61, 1486–1493 CrossRef CAS.
- Q. Miao, J. Liang, X. Yang, W. Xu. Y. Shu and J. Zhu, Shiyou Xuebao, 2015, 31, 188–193 CAS.
- B. Y. Han, W. D. Zhang, Y. B. Chen, F. Yin, S. Q. Liu and X. L. Zhao, Adv. Mater. Res., 2013, 772, 303–308 Search PubMed.
- N. N. E. El-Sayed, M. A. Abdelaziz, W. W. Wardakhan and R. M. Mohareb, Steroids, 2016, 107, 98–111 CrossRef CAS.
- F. Flachsmann, Fragrance compounds, US Pat., US20150247109A1, 2015 Search PubMed.
- A. M. Asiri, Dyes Pigm., 1999, 42, 209–213 CrossRef CAS.
- D. W. Morrison, D. C. Forbes and J. H. Davis Jr., Tetrahedron Lett., 2001, 42, 6053–6055 CrossRef CAS.
- G. Jones, Org. React., 1967, 15, 204–599 CAS.
- C. Chiappe, B. Melai, A. Sanzone and G. Valentini, Pure Appl. Chem., 2009, 81, 2035–2043 CAS.
- D.-Z. Xu, Y. Liu, S. Shi and Y. Wang, Green Chem., 2010, 12, 514–517 RSC.
- P. J. Pelletier and J. B. Caventou, Ann. Chim. Phys., 1820, 15, 289 Search PubMed.
- P. McNeice, F. M. F. Vallana, S. J. Coles, P. N. Horton, P. C. Marr, K. R. Seddon and A. C. Marr, J. Mol. Liq., 2020, 297, 111773 CrossRef CAS.
- P. McNeice, A. C. Marr, P. C. Marr, M. J. Earle and K. R. Seddon, ACS Sustainable Chem. Eng., 2018, 6, 13676–13680 CrossRef CAS.
- A. T. Nielsen and W. J. Houlihan, Org. React., 2011, 16, 1–438 Search PubMed.
- P. Gradeff, Process for preparation of methyl ionones, US Pat., US3840601, 1974 Search PubMed.
- P. W. D. Mitchell, Preparation of pseudoionones, US Pat., US4874900A, 1989 Search PubMed.
- T. Werpy and G. Peterson, Top Value Added Chemicals from Biomass: volume 1-results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab Report DOE/GO-102004-1992, 2004 Search PubMed.
- J. Julis and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8615–8619 CrossRef CAS.
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis Jr., Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS.
- C. Wang, X. Liu, M. Yang, H. Ma, P. Yan, J. M. Slattery and Y. Gao, RSC Adv., 2013, 3, 8796–8804 RSC.
- C. Wang, J. Liu, W. Leng and Y. Gao, Int. J. Mol. Sci., 2014, 15, 1284–1299 CrossRef.
- J. Xu, L. Li, G. Li, A. Wang, Y. Cong, X. Wang and N. Li, ACS Sustainable Chem. Eng., 2018, 6, 6126–6134 CrossRef CAS.
- J. Xu, N. Li, X. Yang, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ACS Catal., 2017, 7, 5880–5886 CrossRef.
- M. J. Earle, P. B. McCormac and K. R. Seddon, Green Chem., 1999, 1, 23–25 RSC.
- S. Hu, T. Jiang, Z. Zhang, A. Zhu, B. Han, J. Song, Y. Xie and W. Li, Tetrahedron Lett., 2007, 48, 5613–5617 CrossRef CAS.
- M. Liu, Z. Zhang, H. Liu, Z. Xie, Q. Mei and B. Han, Sci. Adv., 2018, 4, eaas9319 CrossRef CAS.
- H. Choudhary, P. Berton, G. Gurau, A. S. Myerson and R. D. Rogers, Chem. Commun., 2018, 54, 2056–2059 RSC.
- S.-R. Joo, G.-T. Kwon and S.-H. Kim, Asian J. Org. Chem., 2020, 9, 584–587 CrossRef CAS.
- United Nations Climate Change, The Paris Agreement, https://unfccc.int/process-and-meetings/the-paris-agreement/the-paris-agreement, (accessed July 22, 2020) Search PubMed.
- United Nations Environment Programme, Emissions Gap Report 2019, UNEP, Nairobi, 2019, (https://www.unenvironment.org/resources/emissions-gap-report-2019), (accessed July 22, 2020) Search PubMed.
- A. Kohl and R. Nielsen, Gas Purification, Gulf Publishing Company, Houston, Texas, 5th edn, 1997 Search PubMed.
- X. Liu, Y. Huang, Y. Zhao, R. Gani, X. Zhang and S. Zhang, Ind. Eng. Chem. Res., 2016, 55, 5931–5944 CrossRef CAS.
- G. Cui, J. Wang and S. Zhang, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS.
- X. Y. Luo, X. Y. Lv, G. L. Shi, Q. Meng, H. R. Li and C. M. Wang, AIChE J., 2019, 65, 230–238 CrossRef CAS.
- Y. Huang, G. Cui, Y. Zhao, H. Wang, Z. Li, S. Dai and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 13293–13297 CrossRef CAS.
- M. Zanatta, N. M. Simon, F. P. dos Santos, M. C. Corvo, E. J. Cabrita and J. Dupont, Angew. Chem., Int. Ed., 2019, 58, 382–385 CrossRef CAS.
- Y. Huang, G. Cui, Y. Zhao, H. Wang, Z. Li, S. Dai and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 386–389 CrossRef CAS.
- G. M. Avelar Bonilla, O. Morales-Collazo and J. F. Brennecke, ACS Sustainable Chem. Eng., 2019, 7, 16858–16869 CrossRef CAS.
- Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 RSC.
- A. A. Chaugule, A. H. Tamboli and H. Kim, Fuel, 2017, 200, 316–332 CrossRef CAS.
- R. Li, Y. Zhao, Z. Li, Y. Wu, J. Wang and Z. Liu, Sci. China: Chem., 2019, 62, 256–261 CrossRef CAS.
- W. Zhao, X. Chi, H. Li, J. He, J. Long, Y. Xu and S. Yang, Green Chem., 2019, 21, 567–577 RSC.
- D. Li, Y. Gong, M. Du, C. Bu, C. Chen, S. Chaemcheun, J. Hu, Y. Zhang, Y. Yuan and F. Verpoort, ACS Sustainable Chem. Eng., 2020, 8, 8148–8155 CrossRef CAS.
- Y. Fu, P. Wu, J. Xue and X. Wei, J. Nat. Prod., 2014, 77, 1791–1799 CrossRef CAS.
- C. Palomo, M. Oiarbide and J. M. García, Chem. Soc. Rev., 2012, 41, 4150–4164 RSC.
- K. M. Bothwell and P. C. Marr, ACS Sustainable Chem. Eng., 2017, 5, 1260–1263 CrossRef CAS.
- C. P. Mehnert, Chem. – Eur. J., 2005, 11, 50–56 CrossRef.
- C. Van Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. De Vos, Dalton Trans., 2010, 39, 8377–8390 RSC.
- Y. Zhang, Q. Jiao, B. Zhen, Q. Wu and H. Li, Appl. Catal., A, 2013, 453, 327–333 CrossRef CAS.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- Y.-M. Wang, V. Ulrich, G. F. Donnelly, F. Lorenzini, A. C. Marr and P. C. Marr, ACS Sustainable Chem. Eng., 2015, 3, 792–796 CrossRef CAS.
- R. K. Iler, The chemistry of silica: Solubility, polymerisation, colloid and surface properties and Biochemistry, John Wiley and Sons, New York, 1979 Search PubMed.
- M. A. Gelesky, S. X. Chiaro, F. A. Pavan, J. H. Z. dos Santos and J. Dupont, Dalton Trans., 2007, 5549–5553 RSC.
- K. M. Bothwell, F. Lorenzini, E. Mathers, P. C. Marr and A. C. Marr, ACS Sustainable Chem. Eng., 2019, 7, 2686–2690 CrossRef CAS.
- Y.-M. Wang, F. Lorenzini, M. Rebros, G. C. Saunders and A. C. Marr, Green Chem., 2016, 18, 1751–1761 RSC.
- Y. Ma, Y.-M. Wang, P. J. Morgan, R. E. Jackson, X.-H. Liu, G. C. Saunders, F. Lorenzini and A. C. Marr, Catal. Today, 2018, 307, 248–259 CrossRef CAS.
- G. R. Warner, Y. Somasundar, K. C. Jansen, E. Z. Kaaret, C. Weng, A. E. Burton, M. R. Mills, L. Q. Shen, A. D. Ryabov, G. Pros, T. Pintauer, S. Biswas, M. P. Hendrich, J. A. Taylor, F. S. Vom Saal and T. J. Collins, ACS Catal, 2019, 9, 7023–7037 CrossRef CAS.
- P. McNeice, A. Reid, H. T. Imam, C. McDonagh, J. D. Walby, T. J. Collins, A. C. Marr and P. C. Marr, Environ. Sci. Technol., 2020, 54, 14016–14035 CrossRef.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS.
- H. Gao, L. Bai, J. Han, B. Yang, S. Zhang and X. Zhang, Chem. Commun., 2018, 54, 12671–12685 RSC.
- J. K. Stolaroff, C. Ye, J. S. Oakdale, S. E. Baker, W. L. Smith, D. T. Nguyen, C. M. Spadaccini and R. D. Aines, Faraday Discuss., 2016, 192, 271–281 RSC.
- T. Song, G. M. Avelar Bonilla, O. Morales-Collazo, M. J. Lubben and J. F. Brennecke, Ind. Eng. Chem. Res., 2019, 58, 4997–5007 CrossRef CAS.
- C. Moya, N. Alonso-Morales, J. de Riva, O. Morales-Collazo, J. F. Brennecke and J. Palomar, J. Phys. Chem. B, 2018, 122, 2616–2626 CrossRef CAS.
- Y. Wang, L. Guo and L. Yin, Catal. Lett., 2019, 149, 985–997 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |