
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient methylation of anilines with methanol catalysed by cyclometalated ruthenium complexes†
Patrick
Piehl
a,
Roberta
Amuso
ab,
Anke
Spannenberg
a,
Bartolo
Gabriele
 b,
Helfried
Neumann
a and
Matthias
Beller
*a
b,
Helfried
Neumann
a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e.V, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Matthias.Beller@catalysis.de
bLaboratory of Industrial and Synthetic Organic Chemistry (LISOC), Department of Chemistry and Chemical Technologies, University of Calabria, Via Pietro Bucci 12/C, 87036 Arcavacata di Rende, (CS), Italy
First published on 4th February 2021
Abstract
Cyclometalated ruthenium complexes 4–10 allow the effective methylation of anilines with methanol to selectively give N-methylanilines. This hydrogen autotransfer procedure proceeds under mild conditions (60 °C) in a practical manner (NaOH as base). Mechanistic investigations suggest an active homogenous ruthenium complex and β-hydride elimination of methanol as the rate determining step.
Introduction
The selective N-alkylation of amines continues to be an important and widely applied chemical transformation in organic synthesis,1–3 especially for the synthesis of bio-active compounds, e.g. in the pharmaceutical industry.4–6 In this respect, particularly N-methylation of amines is interesting, since this transformation is regularly used to influence the lipophilicity of such compounds, thus making them more biologically accessible.7,8 While classical methods for N-alkylation rely mainly on toxic and waste-generating alkylating agents like formaldehyde and alkyl halides, modern hydrogen autotransfer (also called hydrogen borrowing) reactions offer an attractive alternative. This latter methodology consists of a multistep reaction sequence starting from easily accessible and often inexpensive alcohols. Using a transition metal catalyst, the alcohol is dehydrogenated in a first reaction step, which leads to the formation of a more reactive aldehyde or ketone that can then undergo reactions like aldol condensation or imine formation. In the final step of the reaction sequence, the hydrogen abstracted in the first step is used to hydrogenate the intermediate product resulting in the overall formation of new C–C or C–N single bonds, while producing water as the sole stoichiometric by-product.9–14 Unfortunately, especially the aforementioned methylation of amines is difficult, since the dehydrogenation of methanol, that is needed as starting material, has a considerably higher energy barrier compared to the dehydrogenation of higher alcohols.In line with this, in recent years methylation of anilines with methanol was typically achieved at elevated temperatures in the presence of either molecularly-defined complexes or heterogenous materials. Most of the known catalysts are based on noble transition metal complexes, e.g. iridium15–17 and specifically ruthenium.18–23 However, more recently, first row transition metals like iron24,25 or manganese26–29 and others were developed, too.30–33
Notably, most of the currently known catalysts rely on sophisticated ligands and only work above 100 °C in the presence of large amounts of strong bases, which create additional environmental concern (see Scheme 1).
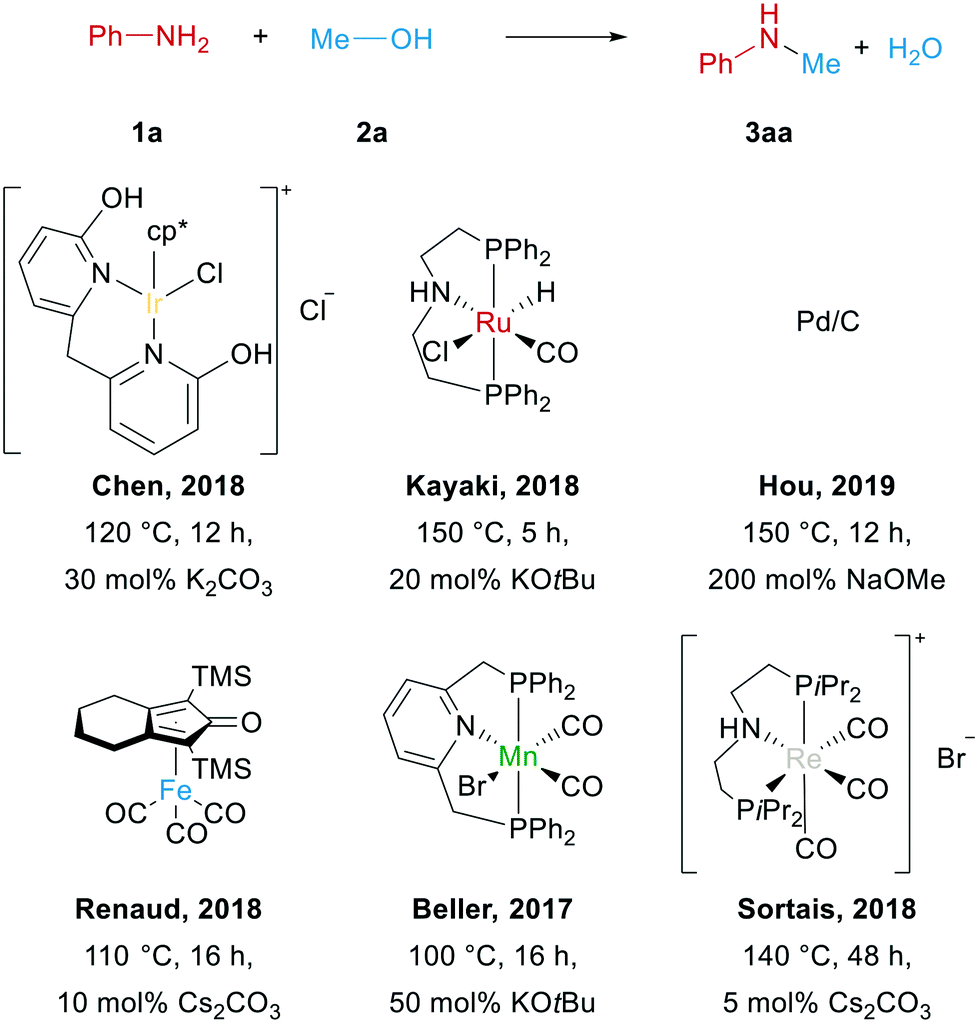 | ||
| Scheme 1 Selected catalysts used for the N-methylation of aniline. | ||
Very recently, our group discovered that cyclometalated ligands can have a positive influence on the reactivity of ruthenium pincer complexes,34 so we were interested in discovering more applications for such catalysts. Indeed, cyclometalated ruthenium complexes gained increasing attention for applications in hydrogen transfer catalysis, recently.35,36 Mainly in the transfer hydrogenation of ketones, ruthenium half-sandwich complexes bearing bidentate ligands with a carbon and a classical donor atom have been established.37–41 Moreover, cyclometalated ruthenium complexes were used in the dehydrogenation of alcohols,42 the direct hydrogenation of olefins43–45 and the α-alkylation of amides using hydrogen autotransfer.46 In addition to that, the implementation of a C-donor atom into pincer ligands was accomplished by the group of Baratta and others for efficient transfer hydrogenation, direct hydrogenation or acceptorless dehydrogenation reactions.47–60 With this potential in mind, we were interested in developing simple cyclometalated ruthenium complexes for the N-methylation of anilines with methanol.
Results and discussion
At the start of our investigations, different cyclometalated ruthenium complexes of the general structure Ru(C^N)(p-cymene)Cl (4–10 in Scheme 2) were synthesized following modified literature procedures.34,61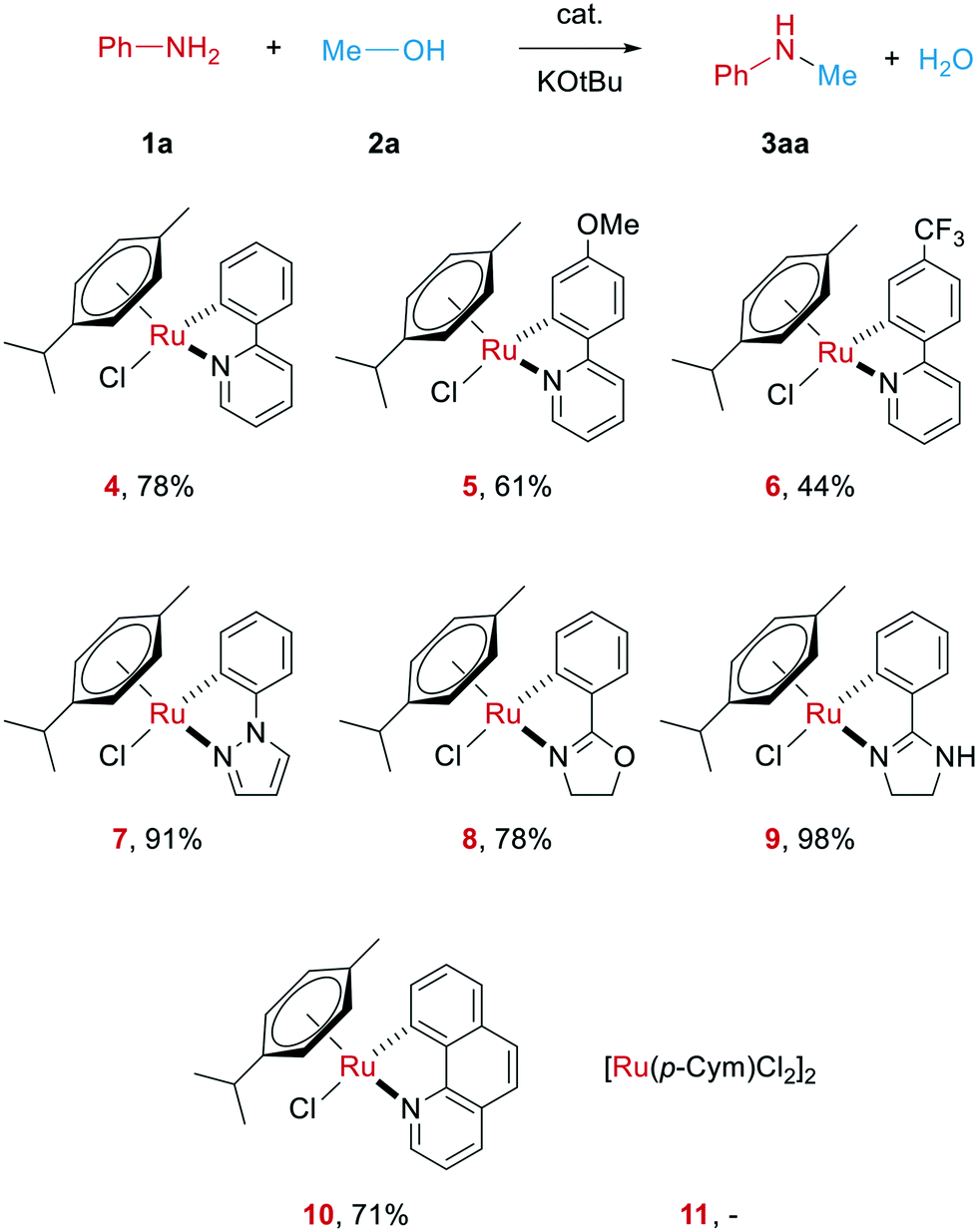 | ||
| Scheme 2 Cyclometalated Ru-complexes tested in the N-methylation of aniline. All reactions were carried out with 1a (1.0 mmol), Ru catalyst (0.02 mmol), KOtBu (1.0 mmol) in MeOH (1.5 ml) at 70 °C for 22 h. Yields determined by GC using hexadecane as internal standard. | ||
All these complexes were then tested in the N-methylation of aniline as a model system. In order to get an idea on the catalytic potential of these complexes, initial test reactions were carried out at 70 °C with 2 mol% of the respective complex, albeit in the presence of 1 equiv. of KOtBu as strong base (see Scheme 2). Surprisingly, all the investigated Ru complexes were able to deliver the desired product. Notably, the main precursor of these complexes, [Ru(p-cymene)Cl2]2, does not catalyse this reaction. The highest yield herein was gained when using the novel ruthenium complex 9 bearing a phenyl imidazoline ligand. The molecular structure of this complex is depicted in Fig. 1.
 | ||
| Fig. 1 Molecular structure of ruthenium complex 9. Displacement ellipsoids correspond to 50% probability. Carbon-bound hydrogen atoms are omitted for clarity. | ||
Next, we performed more systematic variations of critical reaction parameters. Gratifyingly, the base loading could be reduced to only 10 mol% without a significant decrease in reactivity (Table 1, entry 2). Furthermore, it was possible to replace the strong base KOtBu with cheaper sodium hydroxide (Table 1, entry 5). Interestingly, the reaction turned out to work best in only 0.5 ml of undried methanol (Table 1, entry 12). In addition, after defining 60 °C as the optimal reaction temperature, control experiments revealed that no product was formed when the reaction was carried out in the absence of catalyst or base. Interestingly, by scaling up the reaction to 5 mmol, the catalyst loading could be decreased to only 0.4 mmol%, while retaining a high yield of 88% N-methylaniline (Table 1, entry 17).
| # | Catalyst loading [mol%] | Base | Base loading [mol%] | Temp. [°C] | MeOH [ml] | Yielda [%] |
|---|---|---|---|---|---|---|
| Unless otherwise stated, reactions were carried out with 1a (1.0 mmol), catalyst 9 and base in methanol for 22 h.a Yields determined by GC using hexadecane as internal standard.b Technical grade methanol was used without drying prior to use.c Reaction was performed in 5 mmol scale. | ||||||
| 1 | 2 | KOtBu | 100 | 70 | 1.5 | 98 |
| 2 | 2 | KOtBu | 10 | 70 | 1.5 | 91 |
| 3 | 2 | KOtBu | 5 | 70 | 1.5 | 85 |
| 4 | 2 | Cs2CO3 | 10 | 70 | 1.5 | 88 |
| 5 | 2 | NaOH | 10 | 70 | 1.5 | 87 |
| 6 | 2 | K2CO3 | 10 | 70 | 1.5 | 84 |
| 7 | 2 | NEt3 | 10 | 70 | 1.5 | — |
| 8 | 3 | NaOH | 10 | 70 | 1.5 | 87 |
| 9 | 1 | NaOH | 10 | 70 | 1.5 | 75 |
| 10 | 2 | NaOH | 10 | 70 | 0.5 | 88 |
| 11 | 2 | NaOH | 10 | 70 | 2.0 | 68 |
| 12 | 2 | NaOH | 10 | 70 | 0.5b | 91 |
| 13 | 2 | NaOH | 10 | 60 | 0.5b | 96 |
| 14 | 2 | NaOH | 10 | 50 | 0.5b | 51 |
| 15 | — | NaOH | 10 | 60 | 0.5b | — |
| 16 | 2 | — | — | 60 | 0.5b | — |
| 17c | 0.4 | NaOH | 10 | 80 | 2.5b | 88 |
With optimal reaction conditions (1 mmol aniline 1a, 0.02 mmol of catalyst 9 and 0.1 mmol of sodium hydroxide in 0.5 ml of methanol at 60 °C for 22 h) in hand, we compared the new reaction system to some state-of-the-art catalysts for this transformation (see Scheme 3). Surprisingly, only a Mn PNP pincer complex previously developed in our group27 was able to produce N-methylaniline under such mild conditions, while other investigated complexes based on Ru, Pd, Fe or Re gave little or no product at all. This clearly shows that the here presented catalyst works under significantly milder conditions than most other catalysts known for this reaction.
 | ||
| Scheme 3 Performance of state-of-the-art catalysts under optimised reaction conditions. All reactions were carried out with 1a (1.0 mmol), catalyst (0.02 mmol), NaOH (0.1 mmol) in MeOH (0.5 ml) at 60 °C for 22 h. Yields determined by GC using hexadecane as internal standard. | ||
Next, to understand this superior performance compared to other known catalysts, we performed several mechanistic experiments. First, we wanted to rule out the possibility of nanoparticles or heterogeneous materials formed under reaction conditions to be the main active catalyst species. Hence, the reaction was performed in the presence of one drop of mercury, which had no negative impact on the reaction (see ESI† for details).62,63 Because amalgam formation is not necessarily taking place for Ru particles, further control experiments under addition of strongly coordinating ligands were performed. Here, the addition of sub-stoichiometric amounts of triphenylphosphine as well as 1,10-phenantroline led to slightly decreased yields of 78% or 68%, respectively. The addition of over-stoichiometric amounts of these ligands with respect to the catalyst however resulted in a complete shutdown of its reactivity. Overall, this behaviour is typical for homogenous catalysts, so that nanoparticles or even heterogenous materials are unlikely to be the active catalysts in this transformation.
Instead, we believe the pre-catalyst becomes activated by the base as shown by an 1H NMR experiment were only the catalyst and sodium hydroxide are suspended in MeOH-d4 (see ESI† for details). This immediately led to the formation of a clear orange solution (pre-catalyst 9 is insoluble in methanol), that was investigated via1H NMR spectroscopy. Here, the phenyl imidazole ligand as well as η6 coordinated p-cymene could be detected. The aromatic signals of p-cymene were relatively broad which indicated that the complex is dynamic in behaviour likely because the chloride of the precursor was abstracted and replaced by fluctuant solvent molecules. The possibility that phenyl imidazoline as well as p-cymene remained bound to the metal centre over the whole course of the reaction was additionally supported by other experiments in which 10 mol% of these ligands were added under otherwise optimised reaction conditions. Would any of these ligands be released to form the active catalyst, the additional ligand should have suppressed this reaction by shifting its equilibrium. Instead, for p-cymene this did not lead to any decrease in product formation, while the yield only dropped to 79% when phenyl imidazoline was added. We assume this slight decrease in product yield likely stems from the phenyl imidazoline acting as a weak additional ligand concurring for the catalysts active site.
To further investigate the mechanism, the model reaction was carried out with differently deuterated methanol derivatives and we investigated the deuteration pattern of the resulting N-methylaniline (see Fig. S1†). While the amount of deuterium at the NH position can easily scramble with solvent molecules during workup, the deuteration pattern at the methyl group is a clear indication for the reaction mechanism. Surprisingly, when methanol with a deuterated methyl group was the starting material, a fully deuterated methyl group was found in the product (>98%), while methanol without deuterium at the methyl group did not produce any deuteration at the product's methyl group (<1%). Overall, this means that there is no exchange between the hydrogen atoms stemming from methanol's CH3 group and any other H atoms in the reaction solution. This occurs either if no actual methanol dehydrogenation takes place during the reaction or if the Ru hydride (deuteride) species formed by methanol dehydrogenation only transfers this hydride (deuteride) to the imine's α-carbon and is not able to release it as H2.
To distinguish between these two possibilities, yield time plots of the reaction with differently deuterated methanol derivatives were recorded. As shown in Fig. 2, there is a clear reactivity difference between the reactions with CH3OH and CH3OD and the ones with CD3–OH and CD3–OD. More specifically, a kinetic isotope effect of 1.8 for MeOH-d3 was found. This indicates that the C–H bond of the methyl group is involved in the rate determining step of the reaction, which likely corresponds to the β-hydride elimination of methanol bound to the catalyst forming a Ru hydride species and formaldehyde.
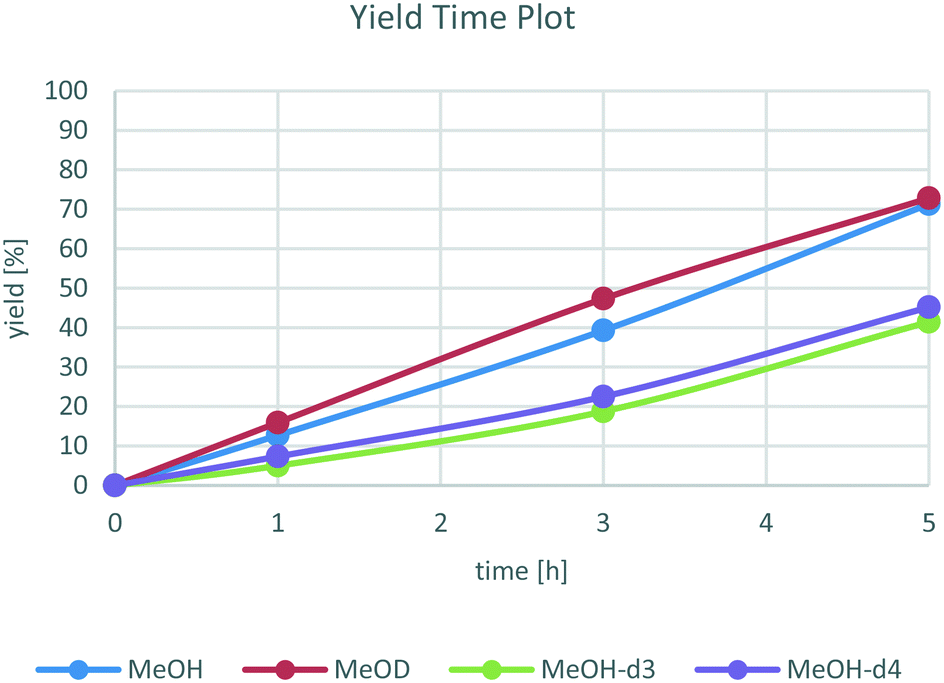 | ||
| Fig. 2 Yield time plot of the reaction for differently deuterated methanol derivatives. All reactions were carried out with 1a (1.0 mmol), catalyst 9 (0.02 mmol), NaOH (0.1 mmol) in MeOH (0.5 ml) at 60 °C for the stated time. Yields determined by GC using hexadecane as internal standard. | ||
On the other hand, for MeOD no significant KIE was found, so the O–H bond does not play a significant role in the rate determining step, ruling out a concerted H2 transfer from methanol to the catalyst. We believe that the reactivity differences between CH3OH/CH3OD as well as CD3–OH/CD3–OD are insignificant and can be explained by minor variations in the respective experiments.
Besides these findings, the yield time plots show a nearly linear progress without a significant induction period. This again hints towards a homogenously catalysed reaction that probably is in a state of catalyst saturation and therefore shows zero-order influence of the substrate aniline.
Overall, we explain these surprising results from the reactions with the different forms of deuterated methanol by a mechanistic proposal shown in Scheme S1 (see ESI† for details). The selective hydrogenation/deuteration of the in situ generated imine can be explained by the attractive interaction of the aryl groups of the cyclometalated ligand and the imine. Notably, H/D-exchange is easily possible on the nitrogen atom of the imidazole ring while this is not possible for the Ru–H/D bond.
Finally, we explored the substrate scope of the reaction (Scheme 4). First, different toluidines were used. While the methyl group in ortho position caused a lower yield due to steric hinderance even when the temperature is elevated and more NaOH is used (3ba), meta- or para-methylation had no negative impact and the corresponding products were formed quantitatively (3ca, 3da). An electron-withdrawing nitro group in aniline's 4-position led to lower 28% yield even with higher temperature and base loading (3ea). Interestingly, no significant hydrogenation of the nitro group was observed here. Halide-substituted anilines including 4-iodoaniline (3fa) were selectively methylated without significant dehalogenation side-reactions. Additionally, electron-rich 4-methoxyaniline gave the corresponding product with a high yield or 85% (3ha). Furthermore, 2-aminopyridine allowed for methylation, albeit with lower reactivity, while the corresponding 2-amino pyrimidine was not methylated at all (3ja). On the other hand, 1-amino naphthalene did not cause many problems, so that the corresponding product was obtained in 65% yield (3ka). Interestingly, 4-amidoaniline gave very good 92% of the corresponding product without methylation of the amido-group (3la). Unfortunately testing primary aliphatic amines as substrates under analogous conditions revealed only negligible reactivity for methylation.
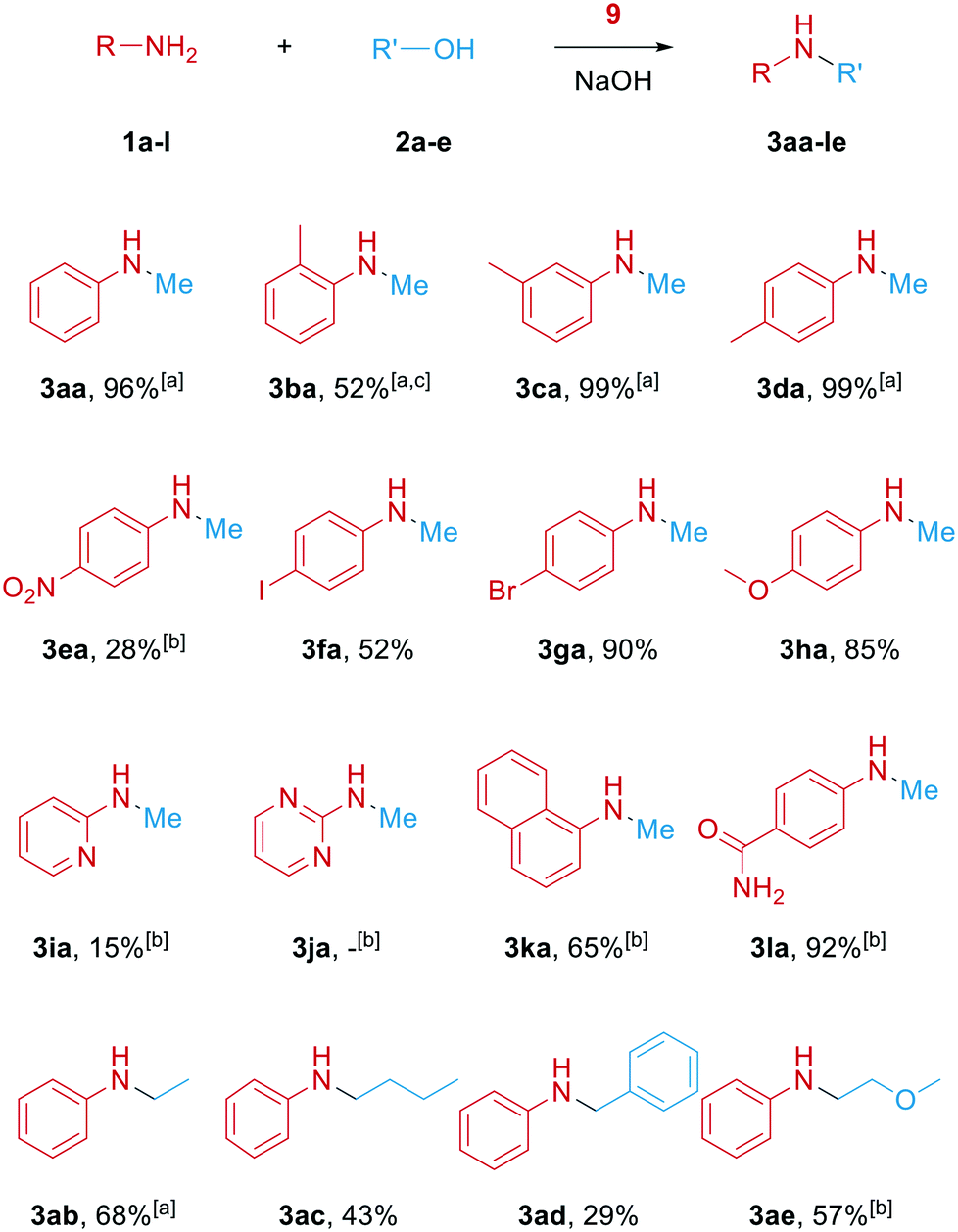 | ||
| Scheme 4 Scope of the N-alkylation of anilines with alcohols. Unless otherwise stated, reactions were carried out with 1a–l (1.0 mmol), catalyst 9 (0.02 mmol) and NaOH (0.1 mmol) in 2a–e (0.5 ml) at 60 °C for 22 h. Isolated yields. [a] Yields determined by GC using hexadecane as internal standard; [b] NaOH (0.3 mmol), 80 °C reaction temperature; [c] NaOH (0.3 mmol), 100 °C reaction temperature. | ||
Lastly, other aliphatic and benzylic alcohols were tested as alkylating agents. Without further optimization the corresponding products were formed in yields up to 68% (3ab–3ae).
Conclusions
To sum up, in this work cyclometalated ruthenium complexes have been established as novel catalysts for the methylation of anilines using methanol via a hydrogen autotransfer procedure. The optimal system 9 bearing a phenyl imidazoline as bidentate ligand likely works in a homogenous manner with β-hydride elimination of methanol to form a Ru–H species being the rate determining step. Compared to other known catalysts for this transformation, 9 allows to work under very mild reaction conditions without the necessity of strong/expensive bases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the state of Mecklenburg-Vorpommern, the Bundesministerium für Bildung und Forschung (BMBF), and the European Research Council (ERC Advanced grant NoNaCat) for financial support. We also thank the analytical department of LIKAT for technical support.Notes and references
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- A. Ricci, Modern Amination Methods, Wiley-VCH, Weinheim, 2000 Search PubMed.
- A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS.
- F. Li, J. Xie, H. Shan, C. Sun and L. Chen, RSC Adv., 2012, 2, 8645–8652 RSC.
- D. Deng, B. Hu, M. Yang and D. Chen, Organometallics, 2018, 37, 3353–3359 CrossRef CAS.
- M. González-Lainez, M. V. Jiménez, V. Passarelli and J. J. Pérez-Torrente, Catal. Sci. Technol., 2020, 10, 3458–3467 RSC.
- T. T. Dang, B. Ramalingam and A. M. Seayad, ACS Catal., 2015, 5, 4082–4088 CrossRef CAS.
- G. Choi and S. H. Hong, Angew. Chem., Int. Ed., 2018, 57, 6166–6170 CrossRef CAS.
- O. Ogata, H. Nara, M. Fujiwhara, K. Matsumura and Y. Kayaki, Org. Lett., 2018, 20, 3866–3870 CrossRef CAS.
- G. Choi and S. H. Hong, ACS Sustainable Chem. Eng., 2019, 7, 716–723 CrossRef CAS.
- S. N. R. Donthireddy, P. Mathoor Illam and A. Rit, Inorg. Chem., 2020, 59, 1835–1847 CrossRef CAS.
- M. Maji, K. Chakrabarti, B. Paul, B. C. Roy and S. Kundu, Adv. Synth. Catal., 2018, 360, 722–729 CrossRef CAS.
- K. Polidano, B. D. W. Allen, J. M. J. Williams and L. C. Morrill, ACS Catal., 2018, 8, 6440–6445 CrossRef CAS.
- A. Lator, S. Gaillard, A. Poater and J.-L. Renaud, Org. Lett., 2018, 20, 5985–5990 CrossRef CAS.
- S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef.
- J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Chem. Commun., 2019, 55, 6213–6216 RSC.
- Z. Liu, Z. Yang, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Adv. Synth. Catal., 2017, 359, 4278–4283 CrossRef CAS.
- D. Wei, O. Sadek, V. Dorcet, T. Roisnel, C. Darcel, E. Gras, E. Clot and J.-B. Sortais, J. Catal., 2018, 366, 300–309 CrossRef CAS.
- L. Jiang, F. Guo, Y. Wang, J. Jiang, Y. Duan and Z. Hou, Asian J. Org. Chem., 2019, 8, 2046–2049 CrossRef CAS.
- M. A. R. Jamil, A. S. Touchy, M. N. Rashed, K. W. Ting, S. M. A. H. Siddiki, T. Toyao, Z. Maeno and K.-i. Shimizu, J. Catal., 2019, 371, 47–56 CrossRef CAS.
- P. Piehl, R. Amuso, E. Alberico, H. Junge, B. Gabriele, H. Neumann and M. Beller, Chem. – Eur. J., 2020, 26, 6050–6055 CrossRef CAS.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS.
- J.-P. Djukic, J.-B. Sortais, L. Barloy and M. Pfeffer, Eur. J. Inorg. Chem., 2009, 817–853 CrossRef CAS.
- R. Sun, X. Chu, S. Zhang, T. Li, Z. Wang and B. Zhu, Eur. J. Inorg. Chem., 2017, 3174–3183 CrossRef CAS.
- N. U. Din Reshi, D. Senthurpandi and A. G. Samuelson, J. Organomet. Chem., 2018, 866, 189–199 CrossRef CAS.
- W.-G. Jia, T. Zhang, D. Xie, Q.-T. Xu, S. Ling and Q. Zhang, Dalton Trans., 2016, 45, 14230–14237 RSC.
- S. Bauri, S. N. R. Donthireddy, P. M. Illam and A. Rit, Inorg. Chem., 2018, 57, 14582–14593 CrossRef CAS.
- G. Balamurugan, R. Ramesh and J. G. Malecki, ChemistrySelect, 2017, 2, 10603–10608 CrossRef CAS.
- D. Gong, B. Hu and D. Chen, Dalton Trans., 2019, 48, 8826–8834 RSC.
- L. N. Lewis, J. Am. Chem. Soc., 1986, 108, 743–749 CrossRef CAS.
- S. M. Islam and C. R. Saha, J. Mol. Catal. A: Chem., 2004, 212, 131–140 CrossRef CAS.
- B. Bagh, A. M. McKinty, A. J. Lough and D. W. Stephan, Dalton Trans., 2015, 44, 2712–2723 RSC.
- D. Gong, B. Hu, W. Yang and D. Chen, ChemCatChem, 2019, 11, 4841–4847 CrossRef CAS.
- W. Baratta, G. Chelucci, S. Gladiali, K. Siega, M. Toniutti, M. Zanette, E. Zangrando and P. Rigo, Angew. Chem., Int. Ed., 2005, 44, 6214–6219 CrossRef CAS.
- W. Baratta, M. Bosco, G. Chelucci, A. Del Zotto, K. Siega, M. Toniutti, E. Zangrando and P. Rigo, Organometallics, 2006, 25, 4611–4620 CrossRef CAS.
- W. Baratta, M. Ballico, S. Baldino, G. Chelucci, E. Herdtweck, K. Siega, S. Magnolia and P. Rigo, Chem. – Eur. J., 2008, 14, 9148–9160 CrossRef CAS.
- W. Baratta, F. Benedetti, A. Del Zotto, L. Fanfoni, F. Felluga, S. Magnolia, E. Putignano and P. Rigo, Organometallics, 2010, 29, 3563–3570 CrossRef CAS.
- W. Baratta, K. Siega and P. Rigo, Adv. Synth. Catal., 2007, 349, 1633–1636 CrossRef CAS.
- W. Baratta, S. Baldino, M. J. Calhorda, P. J. Costa, G. Esposito, E. Herdtweck, S. Magnolia, C. Mealli, A. Messaoudi, S. A. Mason and L. F. Veiros, Chem. – Eur. J., 2014, 20, 13603–13617 CrossRef CAS.
- S. Facchetti, V. Jurcik, S. Baldino, S. Giboulot, H. G. Nedden, A. Zanotti-Gerosa, A. Blackaby, R. Bryan, A. Boogaard, D. B. McLaren, E. Moya, S. Reynolds, K. S. Sandham, P. Martinuzzi and W. Baratta, Organometallics, 2016, 35, 277–287 CrossRef CAS.
- S. Giboulot, S. Baldino, M. Ballico, R. Figliolia, A. Pöthig, S. Zhang, D. Zuccaccia and W. Baratta, Organometallics, 2019, 38, 1127–1142 CrossRef CAS.
- G. Bossi, E. Putignano, P. Rigo and W. Baratta, Dalton Trans., 2011, 40, 8986–8995 RSC.
- P. Dani, T. Karlen, R. A. Gossage, S. Gladiali and G. van Koten, Angew. Chem., Int. Ed., 2000, 39, 743–745 CrossRef CAS.
- M. Gagliardo, P. A. Chase, S. Brouwer, G. P. M. van Klink and G. van Koten, Organometallics, 2007, 26, 2219–2227 CrossRef CAS.
- E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826–3833 CrossRef CAS.
- S. Tang, N. von Wolff, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 7554–7561 CrossRef CAS.
- Q. Wang, H. Chai and Z. Yu, Organometallics, 2018, 37, 584–591 CrossRef CAS.
- B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC.
- L. Kathuria, N. Reshi and A. Samuelson, Chem. – Eur. J., 2020, 26, 7622–7630 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full optimisation table, experimental procedures, analytical data of synthesised compounds. See DOI: 10.1039/d0cy02210a |
| This journal is © The Royal Society of Chemistry 2021 |