
Highly dispersed Pt nanoparticles in the Cs-modified X zeolite with enhancement for toluene side-chain alkylation with methanol†
Zhe
Hong
 ,
Xiaoxia
Wang
,
Fangtao
Huang
,
Lei
Miao
,
Yanqing
Huang
and
Zhirong
Zhu
*
,
Xiaoxia
Wang
,
Fangtao
Huang
,
Lei
Miao
,
Yanqing
Huang
and
Zhirong
Zhu
*
School of Chemical Science and Engineering, TongJi University, Shanghai, PR China. E-mail: zhuzhirong@tongji.edu.cn
First published on 16th December 2020
Abstract
Modification of Cs/X with highly dispersed Pt nanoparticles (average size = 1.8 nm) improved the reaction activity and durability in the side-chain alkylation of toluene with methanol. The high catalytic performance could be attributed to the Pt species promoting C–H bond activation in the methyl group of toluene and inhibiting the formation of coke.
Styrene is an essential chemical for production of commercial polymers with significant application value.1,2 The most conventional route for the production of styrene is based on a well-known Friedel–Crafts alkylation process, typically starting from benzene alkylation with ethylene and followed by the dehydrogenation of ethylbenzene.3,4 However, the dehydrogenation step is a highly endothermic process and usually operated at high temperature above 540 °C.5,6 Side-chain alkylation of toluene with methanol has been proposed as a potential one-step alternative for the production of styrene under mild conditions. Sidorenko et al. firstly reported this reaction by using K+ and Rb+ ion exchanged FAU zeolites as catalysts in 1967.7 From then on, intensive study on the toluene side-chain alkylation has been carried out, including the catalyst design and reaction mechanism.
It is well accepted that the side-chain alkylation of toluene is an acid–base synergistic catalysis process.8–10 Generally, the base sites are proposed to activate the C–H bond on the side chain of toluene and catalyse dehydrogenation of methanol to formaldehyde (HCHO), while the role of acid sites is to adsorb and stabilize toluene. Towards the development of side-chain alkylation catalysts, the studies mainly focus on alkali metal modified zeolites. Among them, the Cs ion-exchanged X (CsX) has been most frequently studied and various modifications were made based on CsX to improve the activity. Han et al.11 investigated the different performances of Cs-modified zeolite X by ion-exchange and impregnation method and proposed the synergistic effect of cesium ions and cesium oxide for the formation of styrene. Zhang et al.12 reported that the BPO4/CsX catalyst provided high methanol utilization efficiency and exhibited lower amount of basic sites in comparison to CsX. Jiang et al.13 found that the KRbCsX catalyst exhibited higher selectivity of styrene and ethylbenzene than KX or CsX catalyst. In addition, diverse Cs-based catalyst such as metal borate modified CsX (ZrB2O5/CsX),14 transition metal (Cu, Ag, Zn, Fe, Mo) deposited CsX,15–18 Na2B4O7–CsX composite catalysts,19 and others have also been investigated for this reaction. However, the catalytic conversion of toluene is still unsatisfactory due to the low efficiency of the side-chain methyl group activation. Therefore, it is still a challenging task to develop a catalyst with high activity and long-term stability.
Platinum species has been intensively reported to play a key role in the C–H bond activation. For example, Conejero et al. reported the T-shaped PtII complexes for intermolecular C–H bond activation of arenes.20 Iglesia et al. studied the selectivity of chemisorbed oxygen in C–H bond activation and kinetic consequences for CH4–O2 catalysis on Pt clusters.21 In addition, the good hydrogenation properties of Pt may minimize the amount of alkenes, thereby likely bring excellent stability.22 It is reasonable to speculate that the Pt nanoparticles in the Cs-modified X may enhance the catalytic activity for toluene side-chain alkylation. In this work, CsX catalysts are promoted with platinum to enhance their performance during toluene side-chain alkylation process. A systematic approach was followed, where a series of catalysts were screened, by varying Pt loadings, calcination and reduction procedures. Catalytic tests evaluate the effect of Pt on the catalysts' activity, selectivity, and stability. Detailed characterization help identify the nature, location, and state of the Pt promoter. The detailed preparation processes of the samples are given in the ESI.†
Textural properties of the catalysts are shown in Table S1.† The BET surface area of CsX decreased about 25% compared with NaX, which should be attributed to the introduction of cesium ion which has a large ion radius. After loading CsX with 0.5 wt% Pt, the BET surface area decreased only slightly, which indicated that the presence of Pt did not affect significantly the textural properties of X zeolite. The N2 adsorption/desorption isotherms of Pt modified samples were almost coincident with those of CsX and NaX (Fig. S1†). Moreover, all of the XRD patterns exhibited the characteristic diffraction peaks of X, indicating that the framework of X zeolite was well preserved upon Cs ion-exchange and Pt impregnation (Fig. S2a†). It is worth noting that a new low intensity diffraction peak at around 25.5° was observed on CsX and Pt modified CsX catalysts, which was attributed to Cs2O phase (Fig. S2b†). In addition, no diffraction peaks corresponding to Pt species can be observed, which may be due to the low loading and good dispersion of Pt species. SEM images further demonstrated that almost all the samples exhibited similar size distribution and morphologies, which mostly consist of crystallites in the range of 2 to 3 μm (Fig. S3†). Fig. S4† showed the CO2-TPD curves of different samples. It can be seen that all the samples showed a pronounced CO2 desorption peak ranged from 200 to 250 °C, which can be attributed to the framework oxygen atoms with negative charge (Oδ−) in the zeolite.23 NaX showed a weak CO2 desorption peak centered at 214 °C. By Cs exchange, the peak area and desorption temperature increased, indicating that both the amount and strength of the base sites increased. In the case of alkali modified X, the origin of basicity attributed to the framework oxygen atoms of zeolite having partial negative charge. Thus, the basicity increases in the presence of more electropositive cations (Na+ < Cs+).8 With addition of Pt, the peak intensity and desorption temperature slightly decreased, indicating that some of available base sites are blocked by platinum. However, the differences in the basicity between CsX and Pt modified CsX catalysts were not significant (Table S1†).
To acquire further insight into the nanostructures of three Pt modified catalysts, the samples were subjected to TEM analysis. Fig. 1a–c clearly showed that the Pt nanoparticles were located on the surface of the crystals. The Pt nanoparticles average sizes of 0.5Pt/CsX-540 and 0.5Pt/CsX-DR250 were around 5.4 and 6.6 nm, respectively. Remarkably, for 0.5Pt/CsX-540-R250, the ultra-small Pt nanoparticles with an average particle size of 1.8 nm were uniformly dispersed on the surface of X zeolite (red circle in Fig. 1b). Meanwhile, the Pt dispersion was ranked as follows: 0.5Pt/CsX-540-R250 (62.3%) > 0.5Pt/CsX-540 (20.9%) > 0.5Pt/CsX-DR250 (17.1%). As there was almost no difference in the actual Pt loading among these catalysts (determined by ICP-OES), it can be considered that the calcination and reduction procedures influenced the Pt nanoparticle sizes and Pt species dispersion. It should be noted that even in 0.5Pt/CsX-540-R250, the Pt particle is bigger than the micropore of X zeolite (around 0.74 nm), and therefore these particles cannot be trapped or confined in the micropore of X zeolite. Fig. 1d showed the HRTEM images of 0.5Pt/CsX-540-R250. Two clear lattice fringes with inter-planar distances at ∼0.23 and ∼0.20 nm were observed and matched the values of the d-spacing of Pt (111) and Pt (200).24,25 In addition, the EDS mapping of 0.5Pt/CsX-540-R250 confirmed that other elements, especially Cs was uniformly distributed over the entire X zeolite crystallites without appreciable zoning (Fig. S5†).
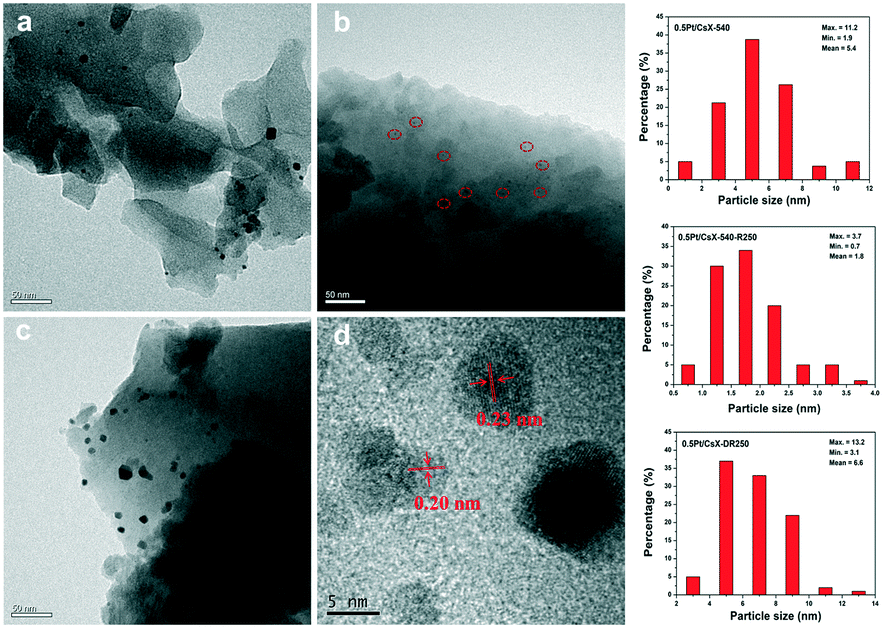 | ||
| Fig. 1 TEM of images of the samples: (a) 0.5Pt/CsX-540; (b) 0.5Pt/CsX-540-R250; (c) 0.5Pt/CsX-DR250. (d) HRTEM of 0.5Pt/CsX-540-R250. | ||
XPS analysis was performed based on the different Pt modified samples (Fig. 2). Firstly, all the samples showed two Cs 3d5/2 peaks, indicating the existence of two kinds of Cs species: the peak centred at around 724.2 eV was attributed to Cs+, while the other signal ranged from 724.8 to 725.0 eV was assigned to Cs2O species. For 0.5Pt/CsX-540, Pt2+ (binding energy at 72.2 eV and 74.9 eV) and Pt4+ (binding energy at 76.8 eV) were observed on X support (Fig. 2a).26 No Pt0 species were observed, which evidenced that all Pt in this catalyst was in the oxidized state, mainly as Pt2+ (77.3%) and to a lesser extent as Pt4+ (22.7%). For 0.5Pt/CsX-540-R250 and 0.5Pt/CsX-DR250, metallic Pt0 (binding energy at 71.2 eV and 73.9 eV)27 and Pt2+ were observed on the material, while no Pt4+ species were observed. As shown in Table S2,† the majority of the oxidized Pt species were reduced to Pt0 after reduction at 250 °C. Obviously, the H2 reduction procedure plays a decisive role on the existence status of Pt species. In addition, the introduction of Pt species to CsX resulted in no significant changes in the existence status of Cs species (Fig. 2d). The detailed surface valence distribution of Pt (4f) and Cs (3d5/2) species over different catalysts are summarized in Table S2.†
 | ||
| Fig. 2 Pt 4f XPS signals of (a) 0.5Pt/CsX-540; (b) 0.5Pt/CsX-540-R250; (c) 0.5Pt/CsX-DR250; (d) Cs 3d5/2 XPS signals of various Pt modified CsX catalysts. | ||
The catalytic performances for toluene side-chain alkylation are shown in Fig. 3 and Table 1. Initially, CsX showed a relatively low catalytic activity. After the introduction of platinum, 0.5Pt/CsX-540 (without reduction) showed 5.9% conversion of toluene with 85.2% selectivity of styrene. It can be seen that the toluene conversion on 0.5Pt/CsX-540 catalyst was very similar to the original CsX, while the styrene selectivity was enhanced by addition of oxidized Pt species. When the catalyst was reduced at 250 °C in H2 flow, 0.5Pt/CsX-540-R250 showed significantly higher conversion of toluene (9.7%) than CsX (5.1%). However, the styrene selectivity slightly decreased comparing with CsX, which might be attributed to the good hydrogenation property of Pt. Moreover, no side-chain alkylation was observed over NaX and 0.5Pt/NaX-DR250 samples (Fig. 3b), xylenes and polyalkylates were the main product, indicating that Cs species is necessary to the reaction of toluene side-chain alkylation. It can be inferred that the oxidized Pt species, Pt2+ and Pt4+ (as determined by XPS), which are most abundant on the unreduced catalyst 0.5Pt/CsX-540, are probably not favor the activation of side chain methyl group of toluene. Conversely, the metallic Pt nanoparticles (Pt0) on 0.5Pt/CsX-540-R250 could effectively help the toluene conversion. On the other hand, when the Pt modified catalyst was directly reduced at 250 °C after impregnation without prior calcination (0.5Pt/CsX-DR250), it showed much lower catalytic performance compared to CsX. Combining the catalytic result with the TEM results, the activity trend can be correlated to the much larger and fewer Pt nanoparticles present on the surface of 0.5Pt/CsX-DR250 sample. In addition, all of the Pt modified samples exhibited higher methanol conversion than CsX (Fig. S6†), indicating the promotion of Pt species for methanol conversion.
 | ||
| Fig. 3 (a) Toluene conversion; (b) styrene selectivity over NaX, CsX and Pt-modified CsX catalysts in the side-chain alkylation reaction; (c) Arrhenius plots of the intrinsic reaction rate constants with CsX and Pt modified CsX catalysts; (d) natural log of the toluene reaction rate at 430 °C as a function of the toluene mol% on 0.5Pt/CsX-540-R250 catalyst. Reaction conditions: 430 °C (390 to 450 °C in (c)), toluene/methanol ratio = 3.0, WHSV = 2.0 h−1. | ||
| Catalysts | Methanol conversiona (%) | Toluene conversiona (%) | Product selectivity (%) | ST/EB | E app (kJ mol−1) | ||
|---|---|---|---|---|---|---|---|
| Styrene | Ethylbenzene | Othersb | |||||
| Reaction conditions: 430 °C, toluene/methanol ratio = 3.0, WHSV = 2.0 h−1. a Determined at 1 h time-on-stream. b Major byproducts are xylenes, TMB, and benzene. c The apparent activation energy (Eapp) was tested by tuning the temperature between 390 °C and 450 °C. | |||||||
| CsX | 41.2 | 5.1 | 77.1 | 18.4 | 4.5 | 4.2 | 65.3 |
| 0.5Pt/CsX-540 | 46.6 | 5.7 | 85.2 | 12.5 | 2.3 | 6.8 | 60.2 |
| 0.5Pt/CsX-540-250 | 52.1 | 9.7 | 72.4 | 24.7 | 2.9 | 2.9 | 51.9 |
| 0.5Pt/CsX-DR250 | 45.9 | 3.2 | 74.6 | 22.3 | 3.1 | 3.3 | 70.9 |
The effect of Pt loading amount in Pt/CsX-540-R250 catalysts was studied. The conversion of toluene and the selectivity of styrene improved upon an increase in the loading of Pt from 0.1 to 0.5%, whereas further increase of the loading to 1 and 2% caused a drop of activity (Fig. S7†). The results can be rationalized considering that at lower loading of Pt (<0.5%), the larger total mass of support employed might hinder the accessibility of the Pt nanoparticles, whereas at higher loading of Pt (1 and 2%) the larger size and worse dispersion of Pt nanoparticles on the X zeolite surface account for the lower activity of the catalysts.28 The intermediate loading of Pt (0.5%) provided the best balance between these two factors, leading to the observed highest activity with 0.5Pt/CsX-540-R250.
In order to further verify the intrinsic activity of these catalysts, the apparent activation energy (Eapp) was determined in the temperature range of 390–450 °C. As depicted in Fig. 3c, the 0.5Pt/CsX-540-R250 catalyst gave the lowest activation energy (51.9 ± 1.1 kJ mol−1), followed by the 0.5Pt/CsX-540, CsX and 0.5Pt/CsX-DR250 (60.2 ± 0.9, 65.3 ± 2.3 and 70.9 ± 1.5 kJ mol−1, respectively). The results suggested that highly dispersed Pt nanoparticles on the surface of CsX could lower the Eapp value, which was easily initiated due to its low activation energy. The effect of the concentration of toluene on the normalized rate was investigated by independently varying the toluene flowrates while maintaining constant total flowrate by varying the N2 flowrate. The correlative apparent reaction order for toluene was obtained on both CsX and 0.5Pt/CsX-540-R250 and catalyst. As shown in Fig. 3d, the reaction order of CsX for toluene was calculated to be 0.8 ± 0.03. Notably, the 0.5Pt/CsX-540-R250 catalyst exhibited greater reaction order for toluene than did CsX, which was calculated to be 1.4 ± 0.06. The difference in toluene reaction order observed on CsX and 0.5Pt/CsX-540-R250 suggested that the toluene adsorption and activation should play the key role in the difference in activity between CsX and 0.5Pt/CsX-540-R250. In addition, the transport effect and product inhibition effect was discussed to ensure the accuracy of kinetic data (Fig. S8†).
According to the mechanism of the side chain alkylation of toluene, the C–H bond in the methyl group of toluene was activated by Lewis base sites (Oδ− in zeolite framework), thus the methyl carbon carrying partial negative charge. In consideration of the enhancement of Pt species for toluene conversion, we speculated that Pt-mediated C–H bond cleavage may exist together with Oδ−. Meanwhile, the carbon in formaldehyde has a sp2-hybridized structure with partial positive charge. Damte et al.29 reported the B, N-co-doped graphene-supported Pt clusters for C–C coupling by using the density functional theory (DFT) method. Hence, we suspected that the Pt species may link to the Si–OH groups on the surface of zeolites, and cooperated with the base sites on the zeolites to facilitate the C–C bond formation. The possible reaction mechanism is shown in Scheme S1.†
Additionally, the durability of 0.5Pt/CsX-540-R250 and CsX catalysts was investigated (Fig. S9†). The 8.3% toluene conversion on 0.5Pt/CsX-540-R250 was preserved for 100 h in the toluene side-chain alkylation reaction. In the case of CsX, the toluene conversion decreased from 5.1 to 0.8% after 100 h on reaction stream. Obviously, the 0.5Pt/CsX-540-R250 catalyst was more stable than CsX, indicating a better potential for practical applications. Meanwhile, the deactivation constant kd (h−1) is used to determine the catalyst deactivation. The kd values for 0.5Pt/CsX-540-R250 and CsX were 1.9 × 10−3 and 18.7 × 10−3 h−1, respectively. The results highlight the promoting effect of Pt on improving reaction durability during toluene side-chain alkylation reaction. To acquire further insight into the catalyst deactivation, the spent 0.5Pt/CsX-540-R250 and CsX catalysts were subjected to TG analysis (Fig. S10†). The initial weight loss in the range of 35 to 200 °C was attributed to desorption of physically adsorbed water and organics.11 For CsX sample, obvious weight losses (8.58%) above 200 °C were observed, which was ascribed to decomposition of deposited coke species on the CsX. However, 0.5Pt/CsX-540-R250 showed a gradual weight loss (1.87%) with increasing temperature from 200 to 700 °C. It implied that only a small quantity of coke species was formed on the 0.5Pt/CsX-540-R250, which explained why the Pt modified catalyst demonstrated the higher stability than parent CsX. The inhibition of coke formation on the catalyst modified by Pt could be ascribed to the hydrogenation of alkenes to alkanes, and then suppress the formation of multi-substituted alkylbenzenes.
In summary, a series of heterogeneous catalysts consisting of highly dispersed Pt nanoparticles supported on CsX was synthesized and investigated for the reaction of toluene side-chain alkylation with methanol. Among which, the 0.5Pt/CsX-540-250R catalyst exhibited the best catalytic activity, leading to a toluene conversion of 9.7% and styrene selectivity of 72.4%. Meanwhile, this Pt modified catalyst exhibited better reaction durability than CsX. The high performance mainly lies in two aspects: (1) the highly dispersed Pt nanoparticles could assist C–H bond activation in the methyl group of toluene more efficiently to promote the toluene conversion; (2) the inhibition of coke formation by Pt modification to retard the catalyst deactivation.
This work was supported by the National Natural Science Foundation of China (Grants 91534115).
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Arrighi, I. J. McEwen, H. Qian and M. B. S. Prieto, Polymer, 2003, 44, 6259–6266 CrossRef CAS.
- E. Le Grognec, R. Claverie and R. Poli, J. Am. Chem. Soc., 2001, 123, 9513–9524 CrossRef CAS.
- S. Smeds, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2000, 201, 55–59 CrossRef CAS.
- J. B. Zheng, Z. F. Wu, Z. Y. Yan, J. J. Li, W. K. Lai, X. D. Yi, B. H. Chen, W. Fang and H. L. Wan, Fuel, 2013, 104, 547–552 CrossRef CAS.
- I. Rossetti, E. Bencini, L. Trentini and L. Forni, Appl. Catal., A, 2005, 292, 118–123 CrossRef CAS.
- B. A. Vaughan, M. S. Webster-Gardiner, T. R. Cundari and T. B. Gunnoe, Science, 2015, 348, 421–424 CrossRef CAS.
- Y. N. Sidorenko, P. N. Galich, V. S. Gutyrya, V. G. Ilin and I. E. Neimark, Dokl. Akad. Nauk SSSR, 1967, 173, 132 CAS.
- Z. Hong, C. F. Xiong, G. Q. Zhao and Z. R. Zhu, Catal. Sci. Technol., 2019, 9, 6828–6840 RSC.
- A. E. Palomares, G. EderMirth and J. A. Lercher, J. Catal., 1997, 168, 442–449 CrossRef CAS.
- H. Itoh, A. Miyamoto and Y. Murakami, J. Catal., 1980, 64, 284–294 CrossRef CAS.
- H. Han, M. Liu, F. S. Ding, Y. R. Wang, X. W. Guo and C. S. Song, Ind. Eng. Chem. Res., 2016, 55, 1849–1858 CrossRef CAS.
- Z. H. Zhang, W. L. Shan, H. Li, W. C. Zhu, N. Zhang, Y. Tang, J. H. Yu, M. J. Jia, W. X. Zhang and C. L. Zhang, J. Porous Mater., 2015, 22, 1179–1186 CrossRef CAS.
- J. Jiang, G. Z. Lu, C. X. Miao, X. Wu, W. H. Wu and Q. Sun, Microporous Mesoporous Mater., 2013, 167, 213–220 CrossRef CAS.
- W. O. Alabi, B. B. Tope, R. B. Jermy, A. M. Aitani, H. Hattori and S. S. Al-Khattaf, Catal. Today, 2014, 226, 117–123 CrossRef CAS.
- N. K. Das and K. Pramanik, J. Sci. Food Agric., 1997, 74, 701–704 CAS.
- H. Hattori, A. A. Amusa, R. B. Jermy, A. M. Aitani and S. S. Al-Khattaf, J. Mol. Catal. A: Chem., 2016, 424, 98–105 CrossRef CAS.
- L. L. Song, Y. Yu, Z. R. Li, S. Q. Guo, L. F. Zhao and W. Li, J. Braz. Chem. Soc., 2014, 25, 1346–1354 CAS.
- Z. H. Zhang, X. L. Yuan, S. S. Miao, H. Li, W. L. Shan, M. J. Jia and C. L. Zhang, Catalysts, 2019, 9, 829–842 CrossRef CAS.
- Q. Han, P. D. Li, Y. F. Zhang, P. Lu, L. Xu, H. C. Guo and L. Xu, ChemCatChem, 2019, 11, 1610–1614 CrossRef CAS.
- O. Rivada-Wheelaghan, M. A. Ortuno, J. Diez, A. Lledos and S. Conejero, Angew. Chem., Int. Ed., 2012, 51, 3936–3939 CrossRef CAS.
- Y. H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Catal., 2011, 283, 10–24 CrossRef CAS.
- Y. Zhao, H. Y. Wu, W. Tan, M. M. Zhang, M. Liu, C. S. Song, X. S. Wang and X. W. Guo, Catal. Today, 2010, 156, 69–73 CrossRef CAS.
- Y. Okamoto, M. Ogawa, A. Maezawa and T. Imanaka, J. Catal., 1988, 112, 427–436 CrossRef CAS.
- Z. Hong, X. Y. Sun, Z. Wang, G. Q. Zhao, X. B. Li and Z. R. Zhu, Catal. Sci. Technol., 2019, 9, 3994–4001 RSC.
- Z. Hong, Z. Wang, D. Chen, Q. Sun and X. B. Li, Appl. Surf. Sci., 2018, 440, 1037–1046 CrossRef CAS.
- W. Y. Teoh, L. Madler and R. Amal, J. Catal., 2007, 251, 271–280 CrossRef CAS.
- L. D. Li, P. Wu, Q. Yu, G. J. Wu and N. J. Guan, Appl. Catal., B, 2010, 94, 254–262 CrossRef CAS.
- Z. C. Tang, S. L. Fiorilli, H. J. Heeres and P. P. Pescarmona, ACS Sustainable Chem. Eng., 2018, 6, 10923–10933 CrossRef CAS.
- J. Y. Damte, Z. J. Zhu, P. J. Lin, C. H. Yeh and J. C. Jiang, J. Comput. Chem., 2020, 41, 194–202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy02002h |
| This journal is © The Royal Society of Chemistry 2021 |