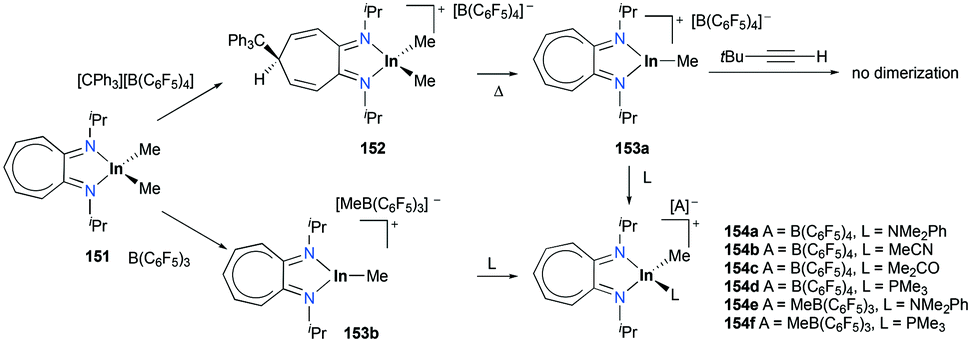
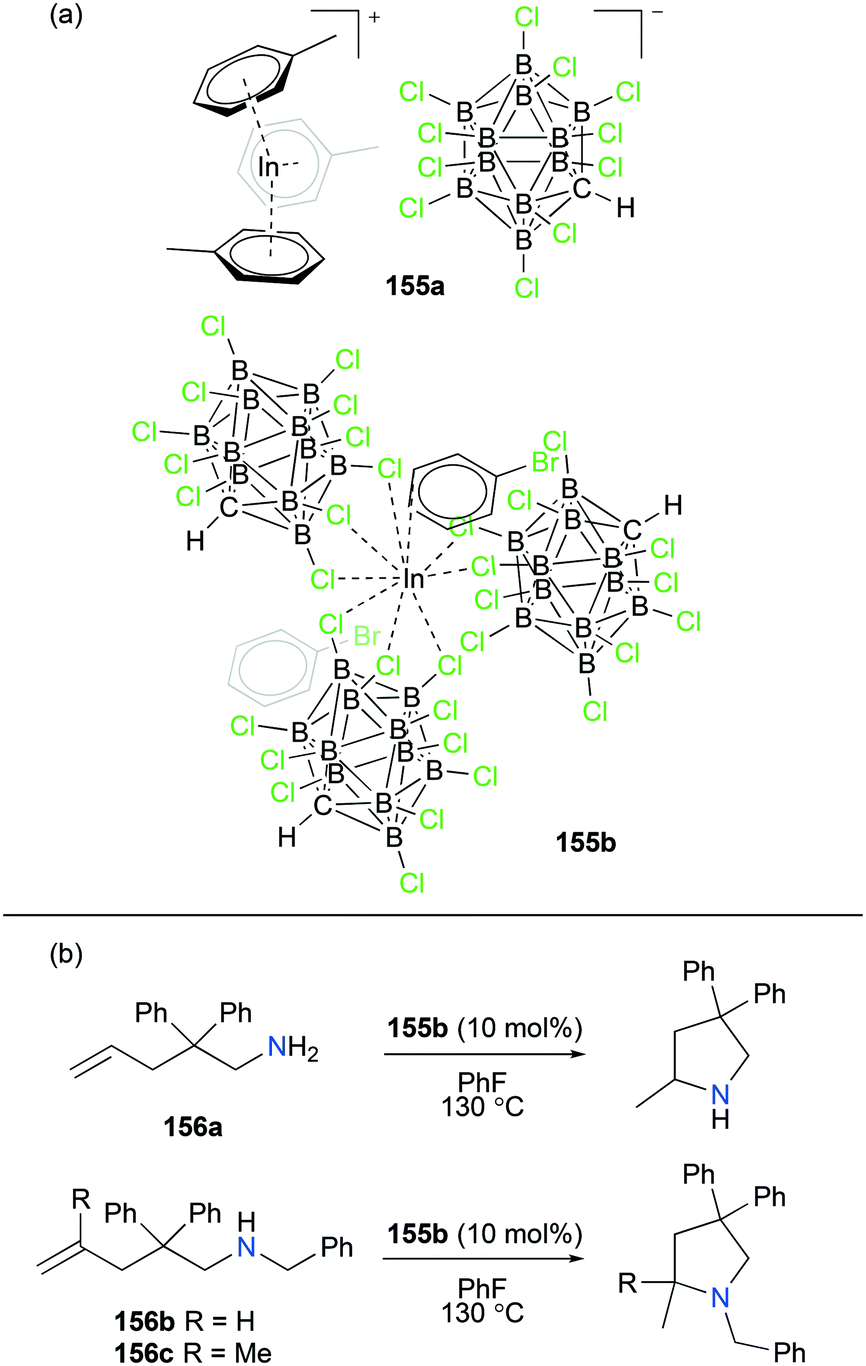
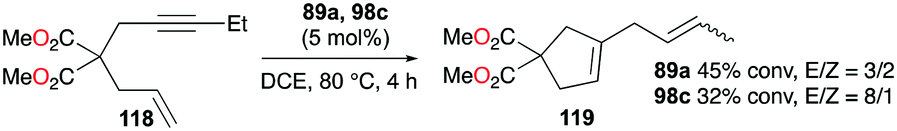Cationic aluminum, gallium, and indium complexes in catalysis
Received
5th September 2020
, Accepted 14th October 2020
First published on 10th November 2020
Abstract
Neutral heavier group 13 metals aluminum, gallium, and indium have been utilized as Lewis acid catalysts in various organic transformations ranging from classical organic reactions to polymerization reactions. The introduction of cationic charge can enhance the Lewis acidity of metal centers and allow cationic group 13 complexes to be excellent catalysts in Lewis acid catalysis, including most of the transformations achieved with neutral group 13 complexes. While cationic aluminum complexes have been investigated extensively in catalysis, there is a more recent push to explore the catalytic reactivities of cationic gallium and indium complexes. The field of cationic group 13 complexes has been expanding with discrete cationic complexes supported by purposely designed ligands. This review aims to provide an overview of what has been done to date and ideas of what can be possibly done from now in the growing field of cationic group 13 complexes as catalysts.
 Hyuk-Joon Jung | Hyuk-Joon Jung obtained his BSc (2013) and MSc (2015) in inorganic chemistry from Sogang University (Seoul, The Republic of Korea) under the supervision of Prof. N. H. Hur, working in nanomaterials. In 2016, he joined Prof. P. Mehrkhodavandi's research group as a PhD student at the Department of Chemistry, University of British Columbia (Vancouver, Canada). During his studies, his research has expanded from nanomaterials and solid-state reactions to metal complex-catalyzed reactions and biodegradable polymers. Currently, he is focusing on the development and reactivity investigation of neutral and cationic group 13 complexes. |
 Youngjung Cho | Youngjung Cho is a research undergraduate student at the University of British Columbia (UBC). He started studying chemistry at Okanagan College in Kelowna, Canada. Then, he transferred to UBC. He joined Prof. P. Mehrkhodavandi's research group as a summer research student in 2019. During his undergraduate career, he received outstanding international award in 2017 and summer research awards in 2019 and 2020. His current research interests include ligand design for group 13 metals and the synthesis of bimetallic organometal compounds. |
 Diana Kim | Diana Kim obtained her BSc in Chemistry from the University of British Columbia in 2019. In the same year, she joined Prof. Parisa Mehrkhodavandi's research group as an MSc student at the Inorganic Chemistry Laboratory, Department of Chemistry, University of British Columbia. She has research interests in functionalizing green polymers produced by metal catalysts and developing bio-derived electrospun nanofibers. |
 Parisa Mehrkhodavandi | Parisa Mehrkhodavandi is a Professor at the Department of Chemistry at the University of British Columbia, Vancouver, Canada. She completed her Ph.D. work at MIT (R. R. Schrock) and her postdoctoral work at Caltech (J. E. Bercaw) before starting her independent career at UBC in 2005. Prof. Mehrkhodavandi has garnered a number of awards including the Killam Research Fellowship and the Alexander von Humboldt Fellowship. Her research interests are focused on the cross section of inorganic chemistry, catalysis, polymer science, and green chemistry. |
1. Introduction
The chemistry of the heavier group 13 elements aluminum, gallium and indium has been dominated by their use as neutral Lewis acid salts in a variety of organic transformations such as Friedel–Crafts, aldol, cycloisomerization1 and Diels–Alder reactions.2 The synthesis of discrete group 13 complexes supported by appropriately-designed ligands has enabled the tuning of the Lewis acidity of metal centers and has improved the catalytic reactivity and selectivity in organic and polymer synthesis.3 The use of chiral ligands enables group 13 complexes to efficiently achieve asymmetric synthesis and control the tacticity of polymers.4 Although this research has been dominated by aluminum complexes, more recently, heavier element gallium and indium complexes have garnered increasing attention.5 The use of neutral aluminum, gallium, and indium in catalysis has been heavily reviewed elsewhere.5c,6
Cationic group 13 complexes, potentially excellent Lewis acid catalysts, have been less widely studied. Cationic group 13 complexes stabilized by weakly coordinating anions can be generated via a variety of synthetic routes such as halide, alkyl and hydride abstraction, asymmetric disproportionation, and protonolysis. The first review on cationic group 13 complexes by Atwood in 1998 classifies the complexes based on the types of formation reactions7 and weakly coordinating anions are comprehensively summarized in several reviews.8 Although the next review of cationic group 13 complexes published in 2008 classifies the complexes based on the coordination numbers including their applications, the review focuses more on the synthesis and structure of the complexes.9 The use of some representative neutral and cationic group 13 complexes as catalysts between 2012 and 2018 was reviewed in 2018.10
Given that there has been a growing interest in cationic group 13 complexes as catalysts and newly reported investigations of these complexes, there is a necessity for an updated and comprehensive review on cationic group 13 complexes in catalysis. The aim of the present review is to provide an overview of cationic aluminum, gallium, and indium catalysts. Cationic clusters, bimetallic complexes consisting of group 13 metals with/without other metals, and neutral group 13 complexes are excluded from the present review unless the catalytic reactivities of cationic complexes are compared with those of neutral analogues.11
2. Cationic aluminum complexes
Aluminum is an abundant, low cost, and low toxicity metal and thus a highly attractive potential substitute for cationic transition metals in some catalytic transformations. Since the first report of reactive cationic aluminum–salen complexes,12 a variety of discrete cationic complexes have emerged and been applied to a broad range of catalysis reactions. A range of discrete aluminum complexes supported by a wide range of ligand supports with coordination numbers ranging from two to six have been used as catalysts.
2.1. Ring opening polymerization (ROP) of cyclic ethers
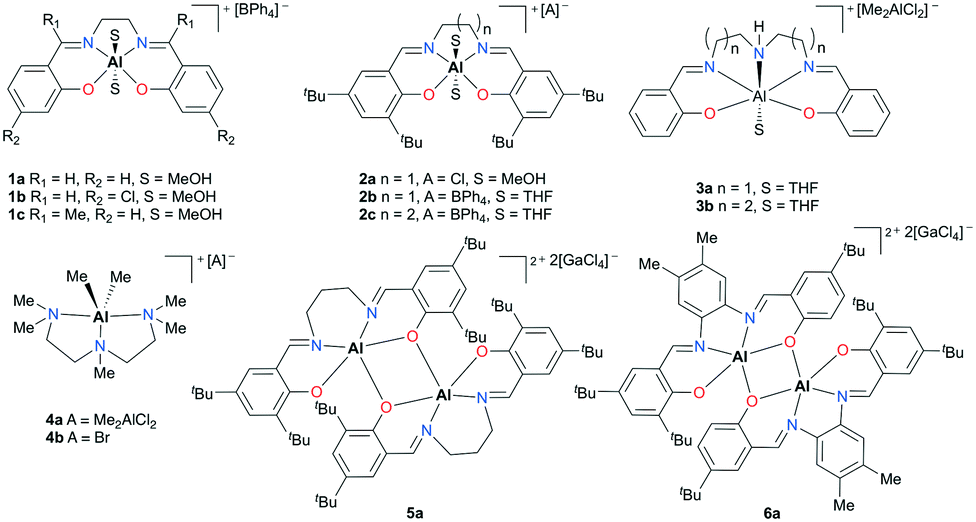
The groundwork for synthesizing cationic aluminum complexes has been established by Atwood and co-workers.12,13 Halide displacement or abstraction assisted by solvents, amines, or Lewis acids has been used as a general procedure to form salen- or amine-based cationic complexes. In addition, many of their discrete complexes have been examined for catalytic activity (Fig. 1). A handful of monomeric (1a–c, 2a–c, 3a–b, and 4a–b) and dimeric complexes (5a and 6a) exhibited mild activity for propylene oxide (PO) transformations to form polymers and oligomers.
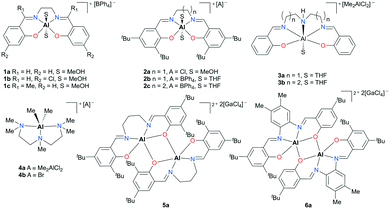 |
| | Fig. 1 Salen- and N,N′,N′′,N′′′-pentamethyldiethylenetriamine (PMDETA)-based cationic aluminum complexes that initiate PO oligomerization and polymerization. | |
In particular, salen-based complexes 2b and 2c bearing two axial tetrahydrofuran (THF) moieties afforded polypropylene oxides (PPOs) with high number average molecular weight (Mn) and narrow dispersity (Đ) values (Mn = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Đ = 1.32 for 2b, 180000 g mol−1 and Đ = 1.16 for 2c).13e Theoretical studies revealed that the activation energy of a PO ring-opening step decreases as the carbocation terminus in a growing chain moves away from an aluminum center. Furthermore, it was believed that the balance between reactivity and stabilization of the carbocation can lead to high molecular weight polymers.
000 g mol−1 and Đ = 1.32 for 2b, 180000 g mol−1 and Đ = 1.16 for 2c).13e Theoretical studies revealed that the activation energy of a PO ring-opening step decreases as the carbocation terminus in a growing chain moves away from an aluminum center. Furthermore, it was believed that the balance between reactivity and stabilization of the carbocation can lead to high molecular weight polymers.
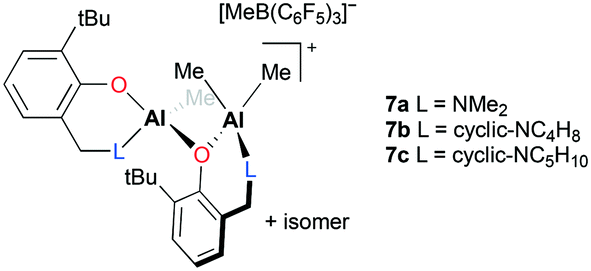
Dagorne and co-workers reported well-defined dinuclear cationic complexes 7a–c for PO polymerization (Fig. 2).14 The cationic species supported by bidentate aminophenolate ligands were generated from their monomeric neutral precursors and B(C6F5)3via methyl abstraction. Separately, they reported the role of B(C6F5)3 in hydroalumination reactions.15
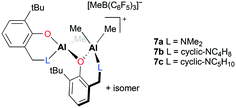 |
| | Fig. 2 Diastereomeric cationic aluminum catalysts 7a–c based on aminophenolate ligands for PO polymerization. | |
The two chiral environments, created by the point chirality of one Al center and the configurational stability of an AlMe2 chelate ring, gave rise to a mixture of diastereomers. Although pure diastereomers were not separated and examined toward PO polymerization, the diastereomeric mixtures successfully underwent catalysis, resulting in low-molecular-weight PPOs (Mn = 2560 to 3076 g mol−1) and good dispersity (Đ = 1.50–1.61).
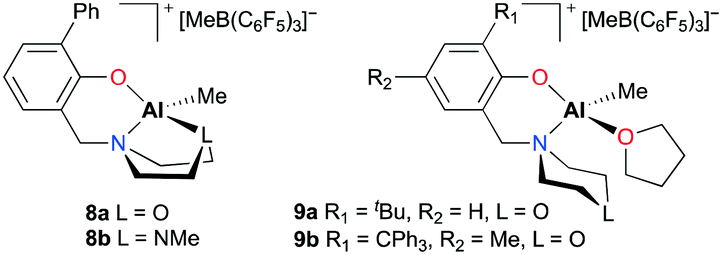
After a modification of aminophenolate ligands, mononuclear complexes 8a–b and 9a–b were developed by the same group for PO polymerization (Fig. 3).14b,16 It was envisioned that another heteroatom at a six-membered amine moiety can have a potential interaction with an aluminum metal center. Indeed, κ3-O,N,O and O,N,N were observed in the solution-state structures of 8a and 8b, respectively. However, for complexes 9a and 9b which incorporated one equivalent of THF, the ligand was bidentate. Regardless of the type of coordination mode, all complexes showed comparable activities, generating PPOs in the Mn range from 2530 to 3960 g mol−1.
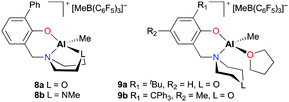 |
| | Fig. 3 Modified aminophenolate-based aluminum complexes 8a–b and 9a–b. | |
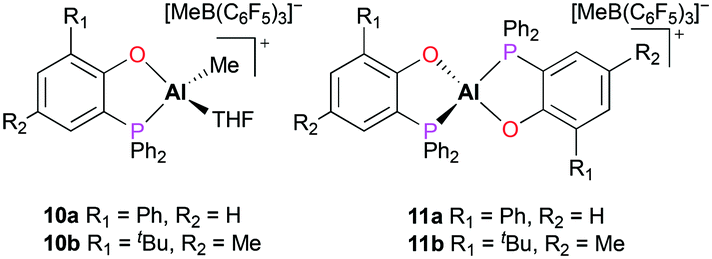
A series of cationic alkyl aluminum complexes supported by bidentate phosphinophenolate ligands were reported by Dagorne and co-workers.17 The mono(phosphinophenolate) and bis(phosphinophenolate) aluminum cationic complexes (10a–b and 11a–b) were generated by alkyl abstraction of the corresponding neutral aluminum complexes with B(C6F5)3 at room temperature in dichloromethane. In particular, the cationic complexes 10a and 10b were synthesized in the presence of THF to form Al–THF cationic adducts (Fig. 4). While all the neutral phosphinophenolate aluminum complexes were inactive for the polymerization of PO, all the cationic complexes polymerized PO under the same reaction conditions. The cationic monophosphinophenolate aluminium complexes 10a and 10b polymerized 100 equiv. of PO via a cationic ring-opening mechanism, producing atactic PPO in 15 minutes with low molecular weights and moderate dispersities (Mn = 3120 g mol−1, Đ = 1.45 and Mn = 3540 g mol−1, Đ = 1.63, respectively). For both cationic complexes, a longer reaction time did not affect the conversion of PO, suggesting deactivation of catalysts. When the reaction temperature was lowered to 0 °C, cationic complex 10b achieved a better conversion of PO and a higher molecular weight of PPO (Mn = 7420 g mol−1, Đ = 1.68). Although the cationic bisphosphinophenolate aluminum complexes 11a–b were less reactive, these complexes also obtained atactic PPO with higher molecular weights compared to 10a–b.
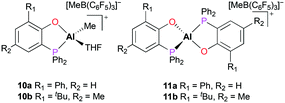 |
| | Fig. 4 The mono(phosphinophenolate) and bis(phosphinophenolate) aluminum cationic complexes for polymerization of propylene oxide. | |
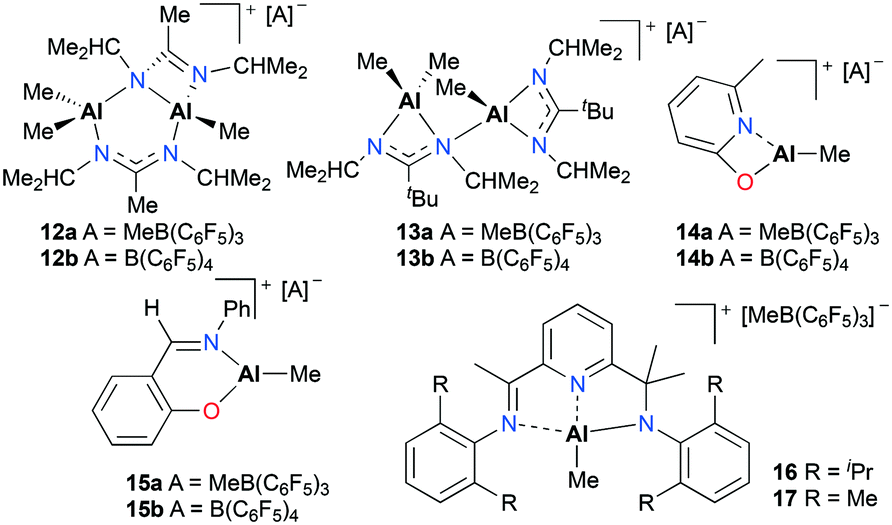
Baugh and co-workers reported in situ generated cationic alkyl aluminum complexes bearing N,N′- (12a–b, 13a–b), N,O-bidentate (14a–b, 15a–b) or N,N′,N′′-tridentate (16 and 17) ligands by alkyl abstraction of the corresponding neutral aluminum complexes with B(C6F5)3 or [Ph3C][B(C6F5)4] in non-coordinating solvents (Fig. 5).18 The catalytic reactivities of these aluminum complexes towards the polymerization of PO, ε-CL, and methyl methacrylate (MMA) were investigated (see below). For the polymerization of PO, the cationic aluminum complexes 12a–15a and 12b–15b were tested in toluene at 25 °C and showed moderate catalytic reactivities, producing low molecular weight PPO with a broad or bimodal molecular weight distribution (12a–b, 13a–bMn = 910–1750 g mol−1, Đ = 1.00–1.55 and 14a–b, 15a–b, Mn = 460–780 g mol−1, Đ = 1.37–2.19). However, boranes themselves proved to be active species for the polymerization and there was a possibility that decomposed species of 12a–b and 13a–b were active for the polymerization.
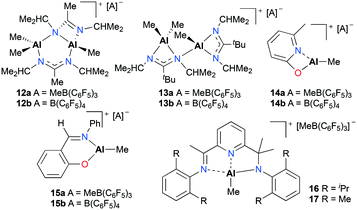 |
| | Fig. 5
N,N′-, N,O- and N,N′,N′′-ligated cationic alkyl aluminum complexes (12–17) generated in situ for the polymerization of PO and ε-CL. | |
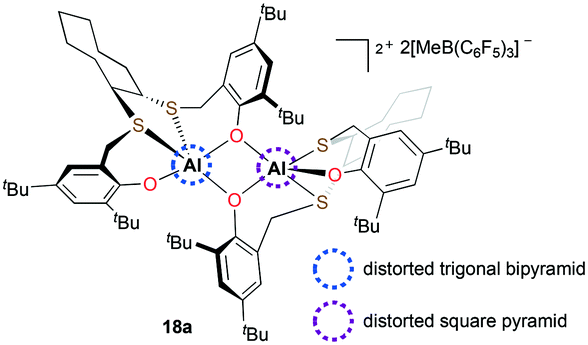
Ishii and co-workers reported dicationic complex 18a bearing both trans-1,2-cyclooctanediyl frameworks and phenolate moieties (Fig. 6).19 In the course of studying its solid-state structure, the τ (ref. 20) values supported that 18a is the first example of a cationic aluminum complex having a different geometry at each aluminum center. The investigation of reactivity showed that 18a can initiate PO polymerization at 25 °C. Although the catalytic system resulted in low-molecular-weight PPOs (Mn = 2500 g mol−1), a narrow dispersity was obtained (Đ = 1.04).
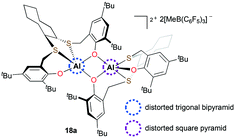 |
| | Fig. 6 Dicationic complex 18a with distinct geometries at each aluminum center. | |
2.2. Ring opening polymerization of cyclic esters
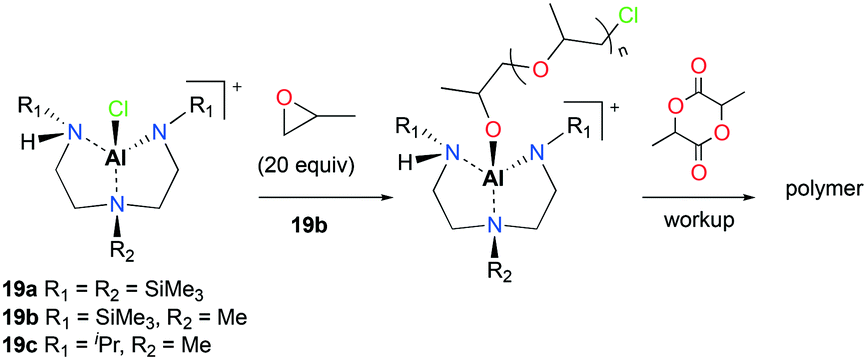
The first attempt of the ring opening polymerization (ROP) of cyclic esters using cationic aluminum complexes was reported by Bertrand and co-workers (Fig. 7).21 The chiral tetracoordinated aluminum cations 19a–c were synthesized via the reaction of the corresponding neutral aluminum complexes and HCl, followed by halide abstraction with AlCl3. The neutral and cationic aluminum complexes were inert towards polymerization of racemic lactide (rac-LA). However, sequential polymerization of PO and rac-LA with 19b produced low molecular weight PLA in 5 days (Mn = 1476 g mol−1, Đ = 1.23). Higher molecular weight PLA was obtained by increasing the equivalence of rac-LA (500 equiv., 80 °C, C6H6, 25 days; Mn = 18504 g mol−1, Đ = 1.39). The authors proposed the aluminum alkoxide species generated in situ as the initiators for rac-LA ROP.
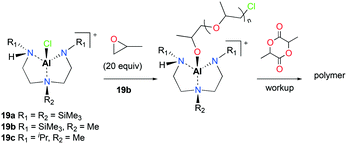 |
| | Fig. 7 The first chiral tetracoordinated aluminum cations (19a–c) and proposed pathway for the ROP of rac-LA with 19b. | |
Baugh and co-workers also investigated the catalytic reactivities of cationic alkyl aluminum complexes 12a–15a, 12b–15b and 17 for the ROP of ε-caprolactone (ε-CL) (Fig. 5).18 The cationic complexes 12a–15b and 17 were inactive towards ε-CL, whereas the cationic complexes 14a–b and 15a–b produced low molecular weight polycaprolactone (PCL) with broad dispersity values.
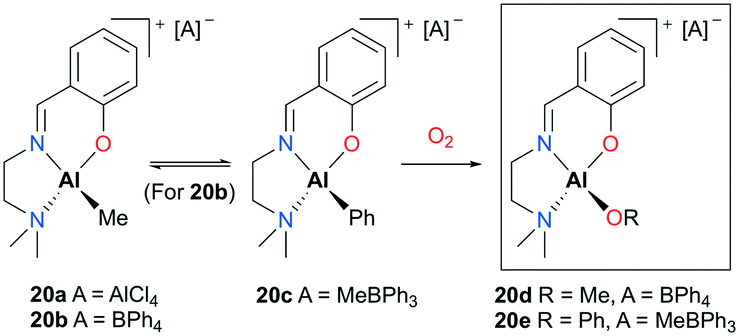
Lewiński and co-workers reported alkylaluminum and active alkoxyaluminum cationic species supported by ATI (N,N-diisopropylamino-troponiminate)22 for the ROP of ε-CL (Fig. 8).23 A series of cationic alkyl aluminum complexes 20a–c were generated by halide abstraction of the neutral aluminum analogue with AlCl3 or NaBPh4, and the cationic alkoxy aluminum complexes were prepared by oxygenation of 20b–c. Among these cationic complexes, 20d and 20e were investigated for the ROP of ε-CL and found to polymerize 50 equiv. of ε-CL at 40 °C in DCM via a coordination insertion mechanism in 95% conversion. End-group analysis by MALDI-TOF revealed that only the terminal alkoxide or aryloxide group is involved in the polymerization.
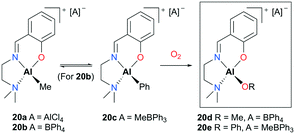 |
| | Fig. 8 Synthesis of the cationic alkoxy aluminum complexes 20d–20e. | |
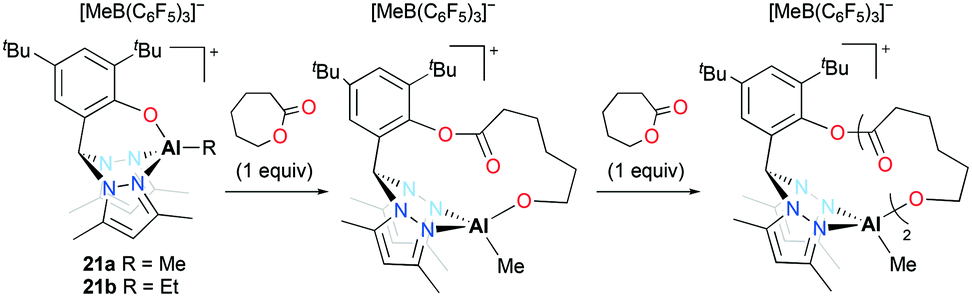
Cationic alkyl aluminum complexes 21a–b bearing a tridentate heteroscorpionate ligand were reported by Milione and co-workers (Fig. 9).24 They were synthesized by the reaction of the corresponding dialkyl complexes (bpzmp)AlR2 with B(C6F5)3. Both cationic complexes were tested for the ROP of ε-CL; however, only 21a polymerized ε-CL (50–150 equiv.) at 50 °C in toluene and achieved full conversion of the monomer in 2 hours. The discrepancy between experimental and theoretical molecular weights implied that approximately 30% of catalysts was involved in the polymerization. The reaction of an equimolar mixture of 21a and ε-CL was monitored by 1H NMR spectroscopy and the first insertion product was observed. The ROP of ε-CL with 21a followed a coordination and insertion mechanism, and the first monomer insertion occurred in the Al–O bond rather than the Al–Me bond. The faster second monomer insertion was observed in the newly formed Al–O bond and the Al–Me bond was inert during the reaction.
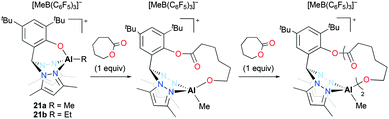 |
| | Fig. 9 A series of cationic heteroscorpionate aluminum complexes 21a–b and sequential insertion of ε-CL into the Al–O bond of 21a. | |
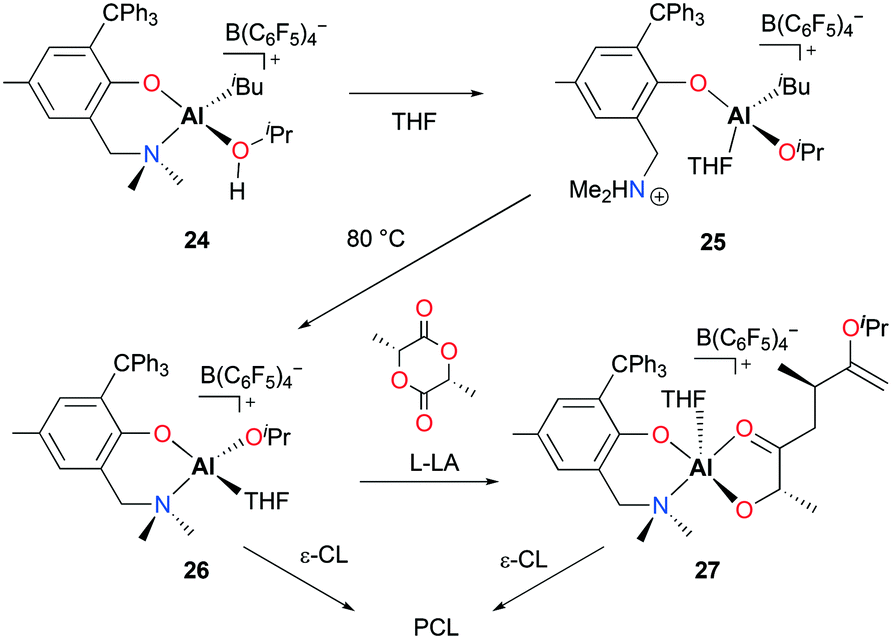
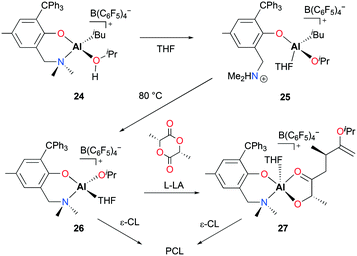
Dagorne and co-workers reported cationic alkyl and alkoxide aluminum complexes incorporating a sterically bulky bidentate aminophenolate ligand and explored the catalytic reactivities of those complexes towards ROP of ε-CL and lactide.25 The reaction of neutral complex (NO)Al(iBu)2 (22) with [Ph3C][B(C6F5)4] formed [(NO)Al(iBu)(PhBr)][B(C6F5)4] (23) in bromobenzene. The further reaction of 23 with iPrOH forms the adduct [(NO)Al(iBu)(iPrOH)][B(C6F5)4] (24) (Fig. 10). Intramolecular proton transfer in 24 forms ammonium–Al–alkyl complex 25 which can undergo alkane elimination at higher temperature to form Al–alkoxide complex [(NO)Al(OiPr)(THF)][B(C6F5)4] (26). While 26 is active for the ROP of ε-CL to form polycaprolactone (PCL), it is inactive for lactide polymerization. The authors isolated an Al–lactate cation 27 by reacting 26 with one equiv. of L-LA. Complex 27 was also active in the polymerization of ε-CL, albeit with lower activity due to chelation by the lactate initiator.
 |
| | Fig. 10 A series of cationic alkyl and alkoxy aluminum complexes [(NO)Al(OiPr)(THF)][B(C6F5)4] and their reactivity with ε-CL and L-LA. | |
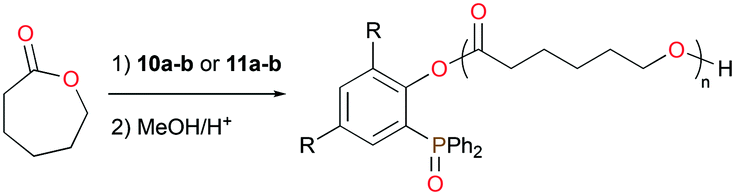
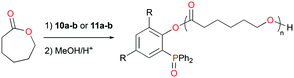
The mono- and bis(phosphinophenolate) aluminum cationic complexes 10a–b and 11a–b (Fig. 4) were also investigated for the ROP of rac-LA and ε-CL.17 None of these cationic complexes polymerized 100 equiv. of rac-LA at 75 °C in toluene, whereas they readily initiated the ROP of ε-CL under the same reaction conditions. Unusually, PCL with a (phosphine oxide)phenolate end-group was isolated, suggesting that ε-CL was polymerized via an initial insertion of ε-CL into Al–Ophenolate (Fig. 11). Thus, the chelating phosphinophenolate moiety functioned as both the ancillary ligand and the initiating group.
 |
| | Fig. 11 The ring-opening polymerization of ε-CL initiated by the phosphinophenolate moiety in 10a–b and 11a–b. | |
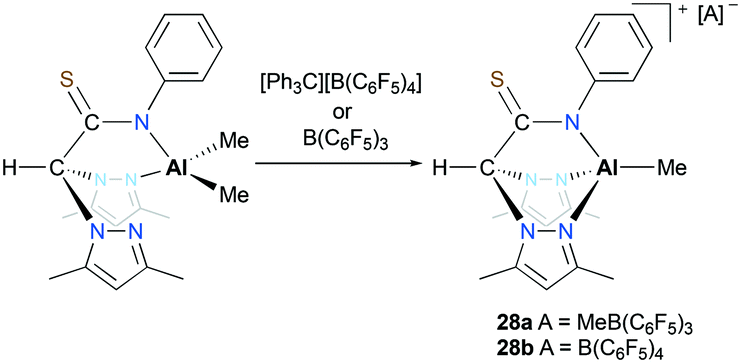
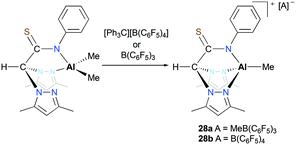
Otero, Lara-Sánchez and co-workers reported cationic alkyl aluminum complexes supported by tridentate thioacetamidate heteroscorpionate ligands.26 The cationic complexes [(κ3-pbptam)AlMe][A] (A = MeB(C6F5)3 (28a), A = B(C6F5)4 (28b)) were prepared by alkyl abstraction of the neutral dialkyl aluminum complex (κ2-pbptam)AlMe2 with B(C6F5)3 or [Ph3C][B(C6F5)4] in THF (Fig. 12). A significant counterion effect was observed for the ROP of ε-CL: complex 28a achieved 40% conversion, while 28b achieved 50% monomer conversion under the same reaction conditions. Although these cationic complexes showed enhanced reactivities compared to that of the neutral analogue, the discrepancy between experimental and theoretical molecular weights was larger with the cationic complexes.
 |
| | Fig. 12 Cationic alkyl aluminum complexes supported by tridentate thioacetamidate heteroscorpionate ligands for the ROP of ε-CL. | |
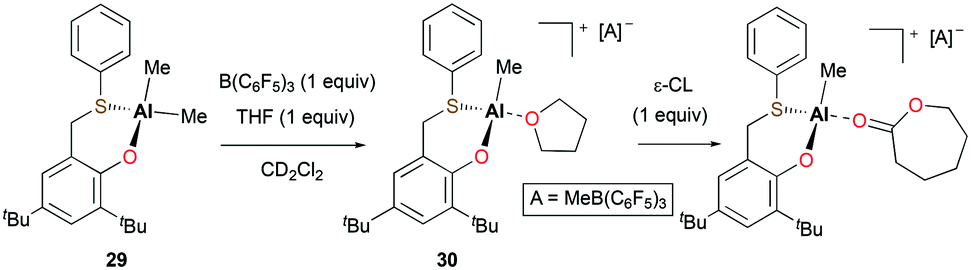
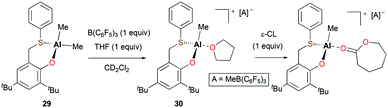
Lamberti, Mazzeo, and co-workers reported the cationic alkyl aluminum complex [(OS)Al(Me)(THF)][MeB(C6F5)3] (30) bearing a bidentate phenoxy-thioether ligand from the neutral species (OS)AlMe2 (29) (Fig. 13).27 In the ROP of 360 equiv. of ε-CL at 50 °C in toluene, cationic species 30 showed very low activity in comparison with 29. Full conversion of ε-CL to PCL in 3 hours was achieved by 29 (Mn = 79660 g mol−1, Đ = 1.77) via a coordination and insertion mechanism, whereas cationic complex 30 only converted 15% of ε-CL in 3 hours. The lack of activity of the cationic species was ascribed to the formation of a robust adduct with ε-CL.
 |
| | Fig. 13 Formation of cationic alkyl aluminum complex 30 and the adduct with ε-CL. | |
Dagorne and co-workers reported NHC-stabilized Al(III) alkyl cations for the ROP of rac-LA as analogues of cationic Ga complexes (Fig. 49).28 The cationic complexes [(NHC)Al(Me)2(THF)][MeB(C6F5)3] (NHC = Mes or Dipp, Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-di-isopropylphenyl) were prepared by alkyl abstraction of the neutral trialkyl aluminum complexes (NHC)AlMe3 with B(C6F5)3 in THF. The cationic complex [(Mes)Al(Me)2(THF)][MeB(C6F5)3] was inactive in the ROP of rac-LA at room temperature and achieved a moderate conversion (40%) at 80 °C in 30 hours. However, the resulting polymer had a bimodal molecular weight distribution (Mn = 1500 and 17500 g mol−1) and broad dispersity (Đ = 2.5), suggesting that the ROP process is poorly controlled. The addition of BnOH significantly improved the activity and control of the ROP process. The cationic complex with BnOH effectively achieved 82% conversion of rac-LA in 80 minutes at room temperature and a narrow dispersity (Mn = 7200 g mol−1, Đ = 1.1). The cationic complex [(Dipp)Al(Me)2(THF)][MeB(C6F5)3] showed a similar ROP activity and control to [(Mes)Al(Me)2(THF)][MeB(C6F5)3].
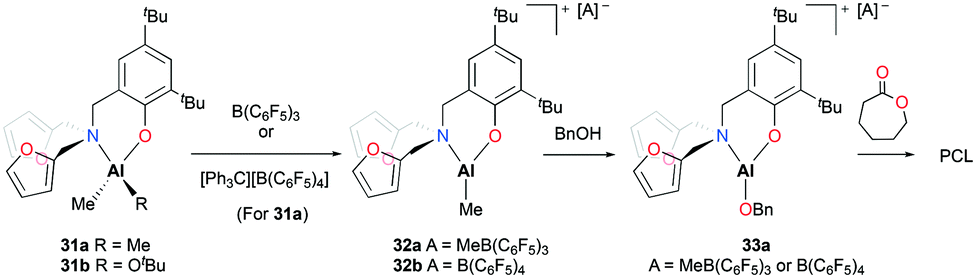
Phomphrai and co-workers reported cationic alkyl aluminum complexes supported by aminophenolate ligands containing a furfuryl group.29 The cationic complexes [(NO)Al(Me)][A] (A = MeB(C6F5)3 (32a), A = B(C6F5)4 (32b)) were prepared by alkyl abstraction of the neutral dialkyl aluminum complex (31a) with the corresponding methide abstracting reagents in non-coordinating solvents (Fig. 14). The neutral complexes (31a–b) and the cationic complex (32a) were evaluated as catalysts for the ROP of 10 equiv. of ε-CL at room temperature in DCM. Complexes 31a–b were inactive for the ROP of ε-CL. In contrast, the ROP of ε-CL in the presence of benzyl alcohol (BnOH) achieved 88% conversion of the monomer in 15 minutes. Given that PCL had benzyl alcohol as an end-group, the polymerization was proposed to progress through the benzyloxide aluminum intermediate (33a). Complex 32b showed slightly slower polymerization compared to 32a possibly due to a larger counterion blocking the active site.
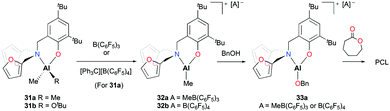 |
| | Fig. 14 Cationic alkyl aluminum complexes supported by aminophenolate ligands containing a furfuryl group (32a–b) and the ROP of ε-CL with 32a/BnOH. | |
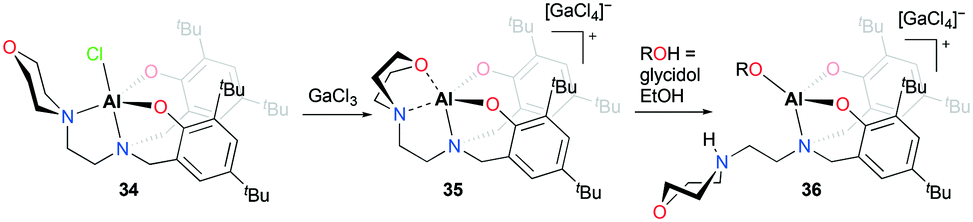
The first cationic aluminum complexes exhibiting hemilabile bidentate (N,O) chelation of a morpholine group were reported by Kerton and co-workers (Fig. 15).30 A series of cationic aluminum aminobis(phenolate) complexes containing morpholinyl donors were prepared by halide abstraction of the neutral complex 34 with various anhydrous Lewis acids. Among them, the cationic complex having a tetrachlorogallate anion 35 was investigated for the ROP of ε-CL. In the presence of one equiv. of EtOH, the cationic complex 35 achieved 50% conversion of ε-CL in 20 hours and produced PCL with a narrow dispersity (Đ = 1.02), whereas the neutral complex 34 showed higher reactivity under the same reaction conditions (99% conversion, Đ = 1.05). When glycidol was used as the alcohol, the reaction rate improved significantly; however, the obtained molecular weights were significantly higher than the theoretical values. Complex 35 was not active for the ROP of rac-LA.
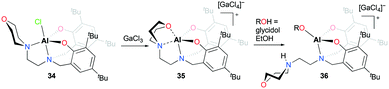 |
| | Fig. 15 Synthesis of cationic aluminum aminobis(phenolate) complexes containing morpholinyl donors with various weakly coordinating anions, and the reactivity of 35 with alcohol. | |
2.3. Polymerization of olefins
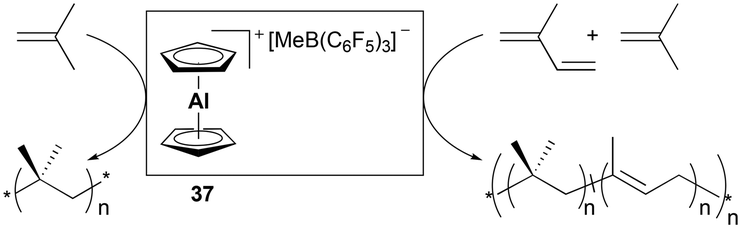
Bochmann and Dawson reported the first application of cationic aluminum sandwich complex 37 to the polymerization of isobutene and isoprene (Fig. 16).31 Complex 37 was readily generated in situ from Cp2AlMe and B(C6F5)3. High-molecular-weight poly(isobutene)s were produced at a lower temperature, whereas a reduced molecular weight was observed at increased temperature (Mw = 618000 g mol−1 at −50 °C vs. Mw = 289000 g mol−1 at −25 °C). The copolymerization of isobutene with a varying quantity of isoprene (2–5 vol%) produced copolymers bearing 1,4-trans structures of isoprene units (Mw = 200000–780000 g mol−1).
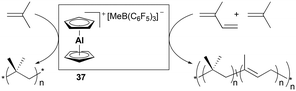 |
| | Fig. 16 Co- and homo-polymerization of isobutyl/isoprene and isobutyl with 37. | |
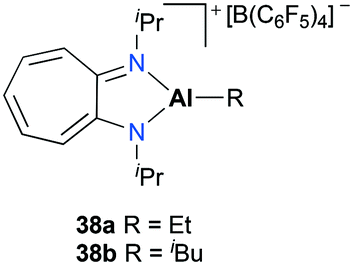
Jordan and co-workers introduced mononuclear cationic complexes 38a–b supported by an N,N′-diisopropylaminotroponiminate ligand (Fig. 17).32 Discrete complexes 38a–b were obtained by the reaction of the corresponding neutral species and [C3Ph][B(C6F5)4]. Complexes 38a–b were investigated for ethylene and propylene oxide polymerization. In particular, they found that the more reactive ethylene polymerization was observed with 38a; however, it was reduced by increasing the ethylene pressure and reaction temperature. The different reactivities between 38a and 38b indicated the strong influence of Al-R in ethylene polymerization.
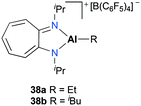 |
| | Fig. 17
N,N′-Diisopropylaminotroponiminate ligand-based complexes 38a–b. | |
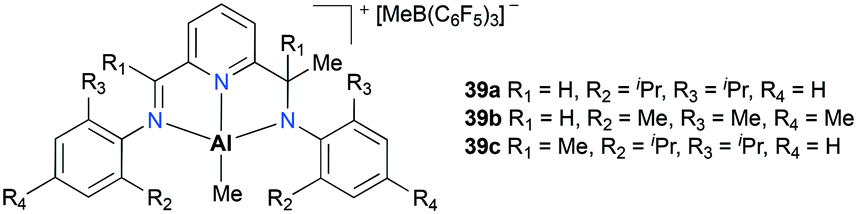
Gibson and co-workers have reported non-palindromic33 pincer-based complexes 39a–c (Fig. 18).34 These complexes showed a notable difference in their ethylene polymerization activity depending on the substituents at aryl moieties as well as at flaking branches (R1 and Me). For example, 39b showed the lowest activity, while doubled reactivity was observed with 39c. The overall polymerization resulted in low-molecular-weight polymers (Mw = 13000 to 33000 g mol−1).
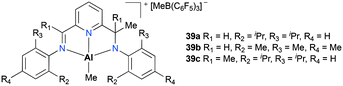 |
| | Fig. 18 Non-palindromic pincer-type aluminum complexes 39a–c. | |
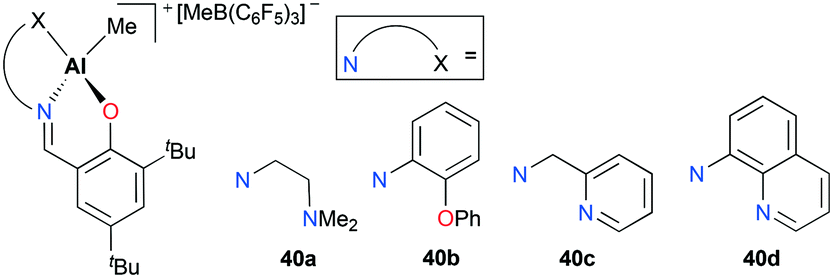
Cationic aluminum complexes supported by Schiff base ligands bearing a pendant donor arm 40a–d were also developed by Gibson and co-workers (Fig. 19).35 Contrary to the results shown with 39a–c, polymerization of ethylene with complexes 40a–b yielded polyethylene with significantly increased molecular weights (Mw = 172000 g mol−1 for 40a; 210000 g mol−1 for 40b). This improvement was rationalized by the labile nature of pendent donor arms as well as their potential stabilizing effects on aluminum metal centers. Unlike 40a–b, complexes 40c–d were inactive towards ethylene polymerization. This was attributed to the decreased lability associated with pyridyl nitrogen atoms.
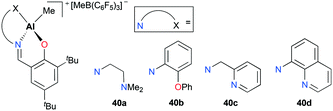 |
| | Fig. 19 Complexes 40a–d bearing a pendent donor arm. | |
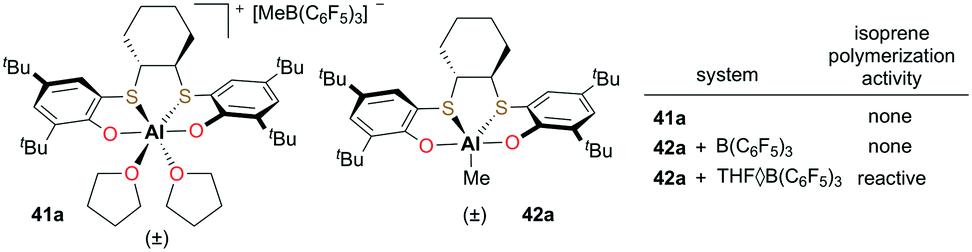
Okuda and co-workers introduced cationic and neutral aluminum complexes supported by (OSSO)-type bis(phenolato) ligands (Fig. 20).36 While attempts at the polymerization of isoprene with discrete cationic complex 41a or the cationic species generated from the reaction of neutral 42a and B(C6F5)3 did not form polymer, in situ generation of the cation with THF·B(C6F5)3 resulted in low-molecular-weight polyisoprene (Mn = 6500 g mol−1). The extent of electronic saturation around the metal center may be an important factor for the reactivity.
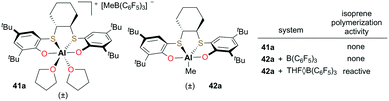 |
| | Fig. 20 Cationic complex 41a and corresponding neutral complex 42a. | |
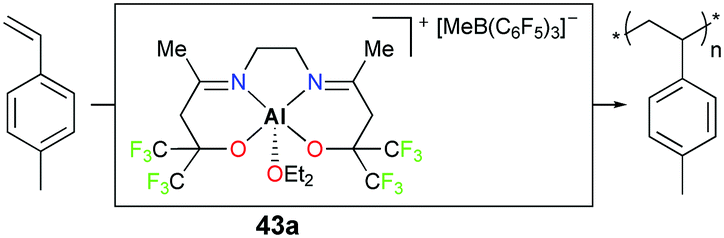
Carpentier, Dagorne, and co-workers reported cationic complex 43a bearing fluorinated moieties adjacent to alkoxides (Fig. 21).37 Complex 43a is an efficient initiator for the polymerization of p-methylstyrene (PMSt). The full conversion of 110 equivalents of PMSt at −20 °C resulted in a relatively narrow dispersity (Đ = 1.6) and the observed molecular weight was in close agreement with the calculated value (Mn,obs = 14900 g mol−1vs. Mn,cal = 13100 g mol−1).
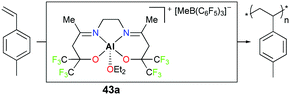 |
| | Fig. 21 Polymerization of PMSt with 43a. | |
2.4. Polymerization of methyl methacrylate (MMA)
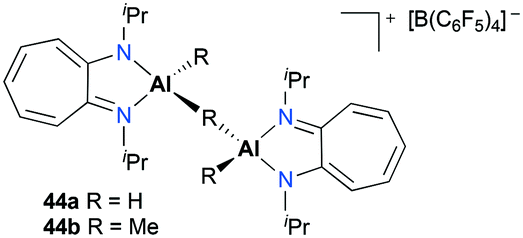
Jordan and co-workers reported a series of cationic aluminum complexes supported by diisopropylaminotroponiminate (ATI) ligands (Fig. 22).32 In particular, dinuclear monocationic complexes 44a–b were generated by the reaction of (iPr2-ATI)Al(R)2 (R = H, Me) with 0.5 equiv. of [Ph3C][B(C6F5)4] and stable base-free complexes 38a–b (Fig. 17) were generated in a similar fashion in C6D6. Only dinuclear hydride cation 44a polymerized methyl methacrylate (MMA) to form predominantly syndiotactic poly(methyl methacrylate) (PMMA) at 23 °C in toluene.
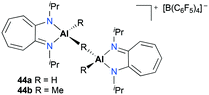 |
| | Fig. 22 Complexes 44a–b incorporating aminotroponiminate ligands for MMA polymerization. | |
2.5. Carbonylation of epoxides
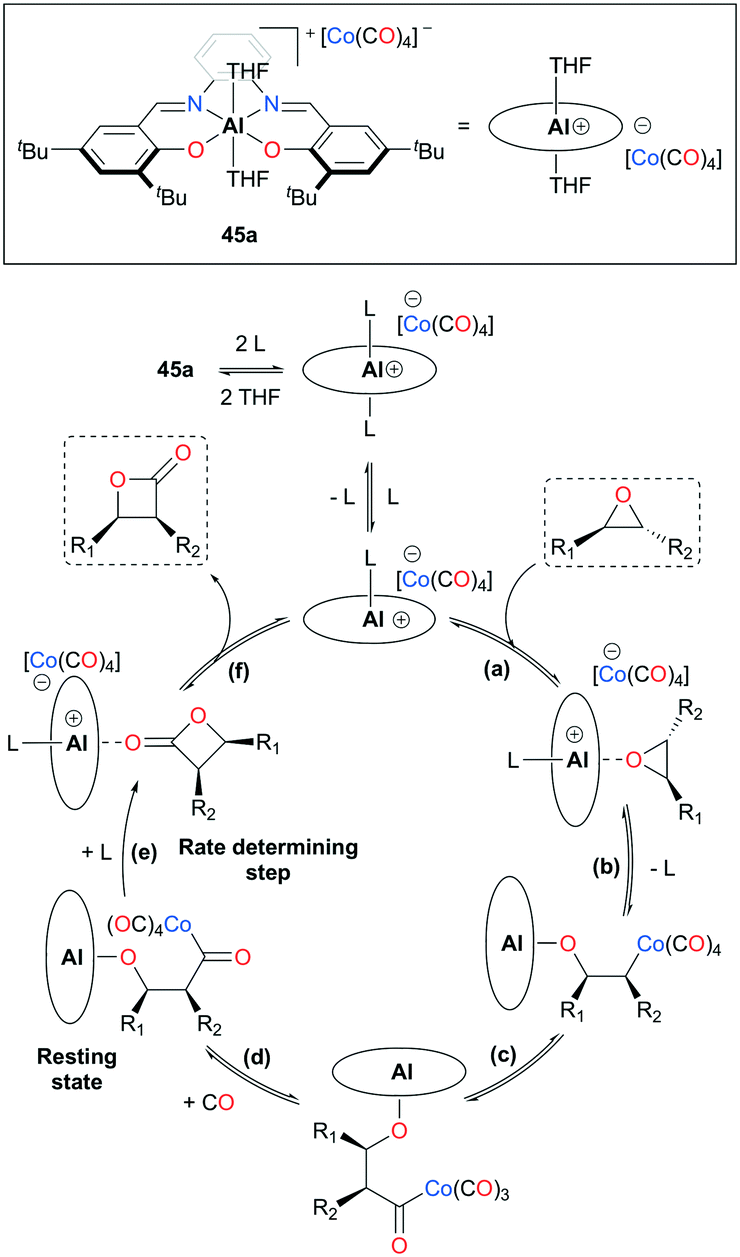
In the past two decades, studies by Coates and co-workers have established a methodology for facile carbonylation of heterocycles by exploiting a variety of salen- and porphyrin-based cationic aluminum complexes (Fig. 23).38 Each complex with a general formula of [(Lewis acid)][Co(CO)4] has shown optimized reactivity for a particular class of epoxides, which can be classified into trans-,38gcis-,38j,k terminal-,38f and 2,2-disubstituted38l epoxides. These reactions afford synthetically useful β-lactones or succinic anhydrides. In addition, catalytic systems have shown reliable applications in natural product syntheses38j,39 and multicomponent reactions.38e
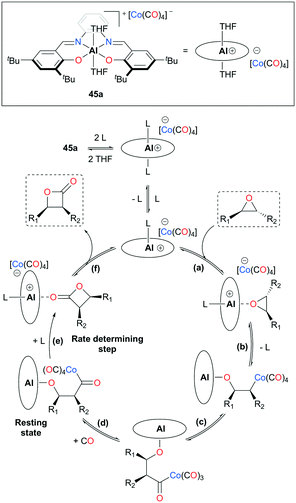 |
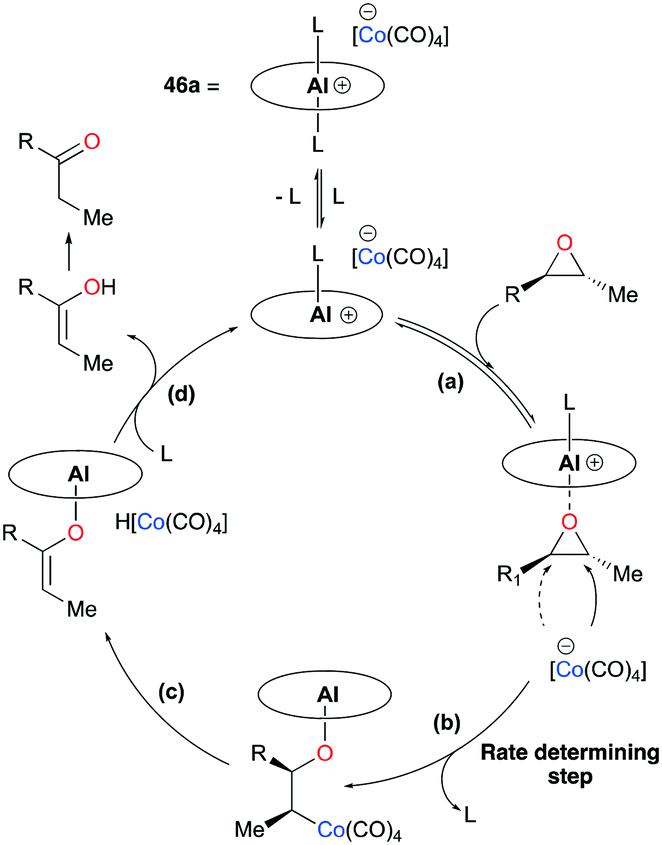
| | Fig. 23 Catalytic cycle of epoxide carbonylation (L stands for a coordinating solvent, epoxide, or β-lactone; L = THF if [THF] ≫ [epoxide] or [lactone]). | |
Initial studies using 45a have shown notable changes in stereochemistry, such that trans- and cis-epoxides were converted to cis- and trans-β-lactones, respectively.38b,c,40 These stereochemical transformations resulted from the inversion of a stereocenter caused by the nucleophilic attack of Co(CO)4− (Fig. 23). The mechanism was further elucidated by observing the solvent-assisted rate-determining step and trapping the resting state of 45a,38d along with corroboration from other sources.11a,13e,41 The catalytic cycle has six major steps: (a) substrate activation, (b) ring-opening via nucleophilic attack, (c) CO insertion, (d) CO uptake, (e) Lewis base-assisted ring-closure, and (f) dissociation of lactone.
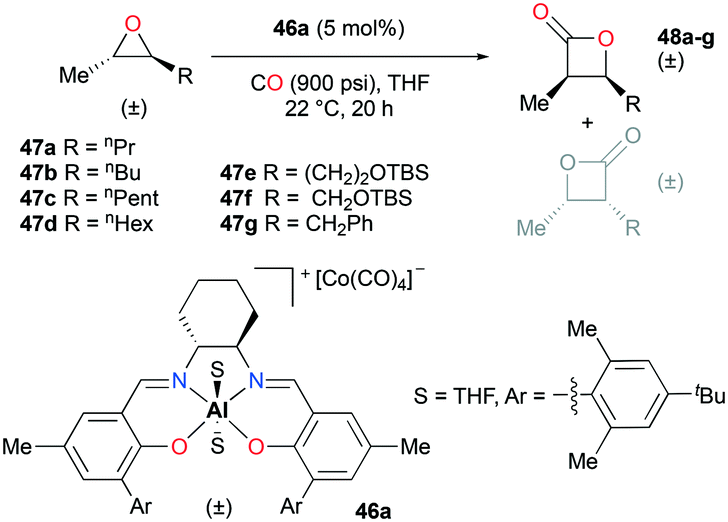
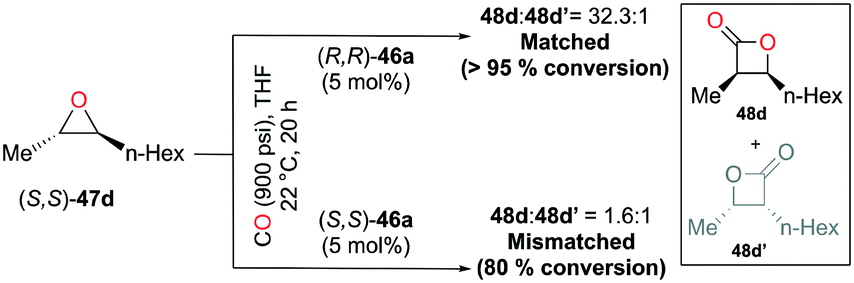
Regioselective carbonylations of racemic trans-epoxides were optimized by exploiting salen-based complex 46a (Fig. 24).38g This reaction resulted in the formation of racemic cis-β-lactones 48a–g (regioselectivity ratio of 10:1 for epoxides 47a–g). Interestingly, different regioselectivities were observed when enantioenriched epoxides were carbonylated by each enantiomer of 46a. For example, complex (S,S)-46a showed reduced selectivity and reactivity toward enantioenriched epoxide 47d, whereas both aspects were highly improved when (R,R)-46a reacted with the same epoxide, affording enantioenriched product 48d predominantly (Fig. 25). These results implied that (R,R)-46a has a propensity to form matched pairs with (S,S)-trans-epoxides.
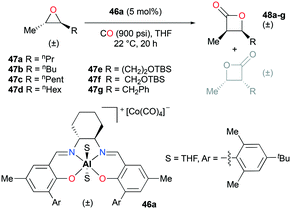 |
| | Fig. 24 Carbonylation of trans-epoxides 47a–g using 46a. | |
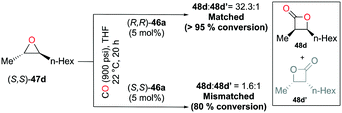 |
| | Fig. 25 Regioselective carbonylation of enantioenriched epoxide 47d with each enantiomer of 46a. | |
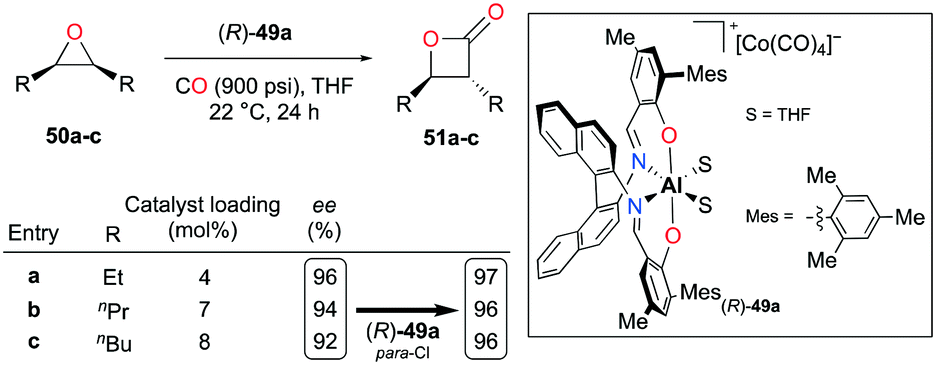
Enantiopure complex (R)-49a containing an (R)-binaphthyl diimine moiety allowed the enantioselective carbonylation of meso-epoxides (Fig. 26).38j Although more catalytic loadings were required as alkyl substituents on the epoxides became longer, epoxides 50a–c were successfully converted to trans-β-lactones 51a–c with a high enantiomeric excess. The electronic variation at para-positions of phenoxide moieties further enhanced the enantioselectivity.
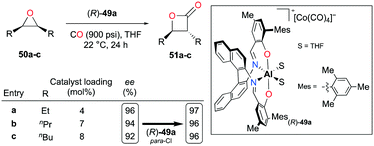 |
| | Fig. 26 Enantioselective carbonylation of meso-epoxides 50a–c with (R)-49a. | |
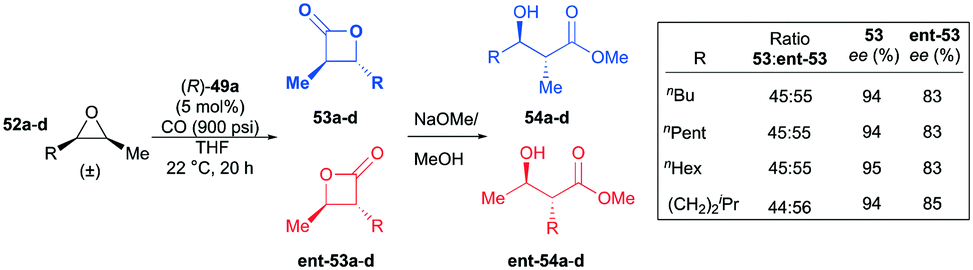
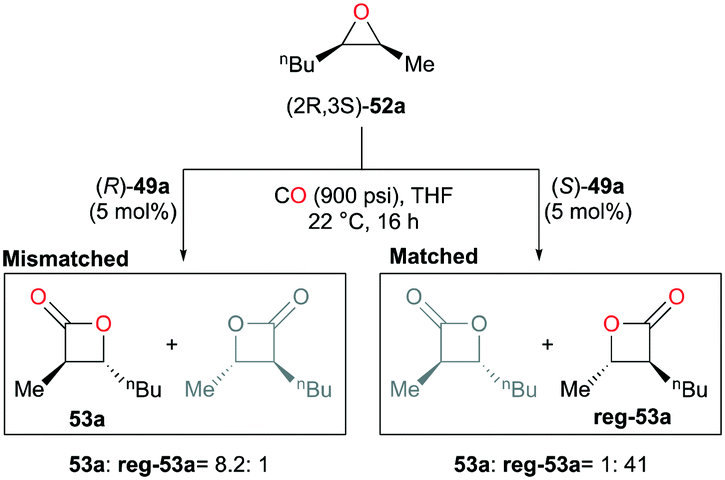
Interestingly, in contrast to the results shown in the meso-epoxide carbonylation, (R)-49a carbonylated racemic cis-epoxides 52a–d in a regiodivergent manner.38k As a consequence of this regiodivergent catalysis,42 the racemic epoxides were converted to the opposite regioisomers of trans-β-lactones 53a–d and ent-53a–d with high enantioenrichment (Fig. 27). A mixture of the enantioenriched regiomers further reacted with methoxide via a one-pot procedure, affording the corresponding β-hydroxy methyl esters. The highly enantioenriched methyl esters 54a–d were readily isolated by a flush column technique.
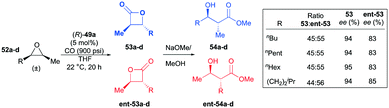 |
| | Fig. 27 Regio-divergent catalysis of racemic cis-epoxides with (R)-49a. | |
As another way to demonstrate the regiodivergency, enantiopure epoxide (2R,3S)-52a was carbonylated with each enantiomer of 49a (Fig. 28).38j Enantioenriched lactone 53a was preferentially produced by (R)-49a with a regioselectivity ratio of 8.2:1, while (S)-49a showed a five-fold increase in the selectivity towards the opposite enantioenriched regioisomer reg-53a. In addition, the ratios indicated the presence of matched and mismatched pairs between the epoxide and 49a. Additional kinetic analysis showed that (R)-49a consumes (2S,3R)-52a faster than (2R,3S)-52a by a factor of approximately four, thereby forming the opposite enantiomer reg-53a faster at the beginning of the carbonylation. This implies that the same or similar kinetic preference would be observed between the matched pair of (2R,3S)-52a and (S)-49a.
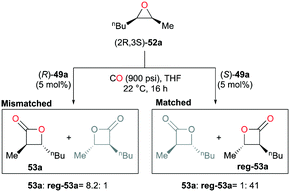 |
| | Fig. 28 Regiodivergent catalysis with racemic 49a. | |
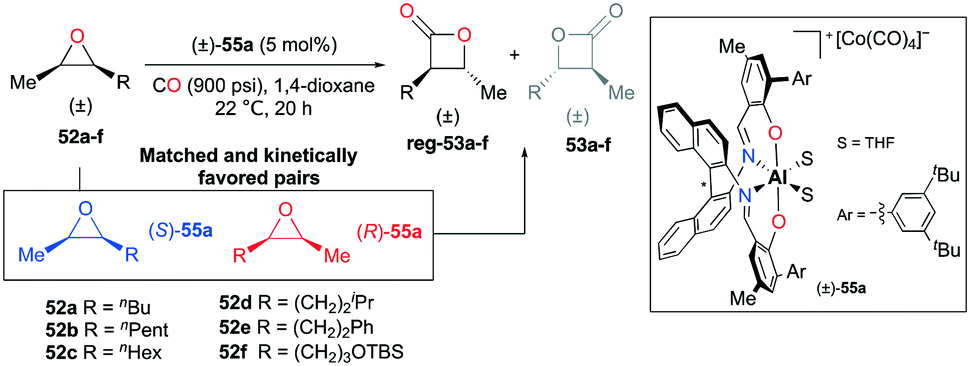
A slight modification of the para-aryl moieties at (R)-49a gave rise to complex rac-55a while retaining both the regiodivergent ability and kinetic preference towards matched substrates (Fig. 29).38j The harmony of the two features enabled the regioselective transformation of cis-epoxides to trans-β-lactones. For epoxides 52a–f, regioselectivity ratios were observed in the range from 13.3:1 to 24.0:1.
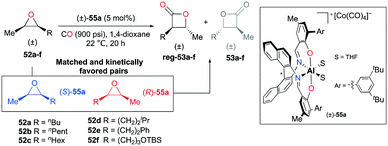 |
| | Fig. 29 Regioselective carbonylation of cis-epoxides with racemic 55a. | |
2.6. Catalytic isomerization of epoxides to ketones
Coates and co-workers observed the generation of ketones as a byproduct and confirmed their presence in the course of the previous mechanistic studies.38d This finding has further allowed them to develop the catalytic isomerization of epoxides that can overcome the limitations relative to transition metals.43 Similar to the aforementioned carbonylation systems, complexes of the type [(Lewis acid)][Co(CO)4] show optimized reactivity and selectivity for a particular class of epoxides. These catalytic systems can also be useful in other applications such as kinetic resolution and isotope labeling reactions.44
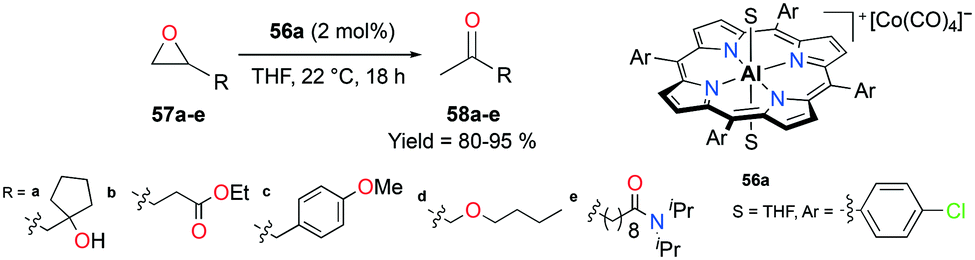
The porphyrin-based cationic complex 56a isomerized terminal epoxides into the corresponding ketones, avoiding a mixture of other possible aldehyde and vinyl alcohol byproducts (Fig. 30).45 Among a variety of terminal epoxides, the functionalized substrates 57a–e were selectively transformed to methyl ketones 58a–e, containing ester, ether, amide, alcohol, and aryl functional groups, in good to excellent yields. These outcomes indicated the great functional group comparability of 56a, which was observed in previous applications.38j,39
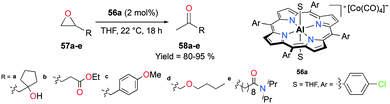 |
| | Fig. 30 Isomerization of functionalized terminal epoxides 57a–e with 56a. | |
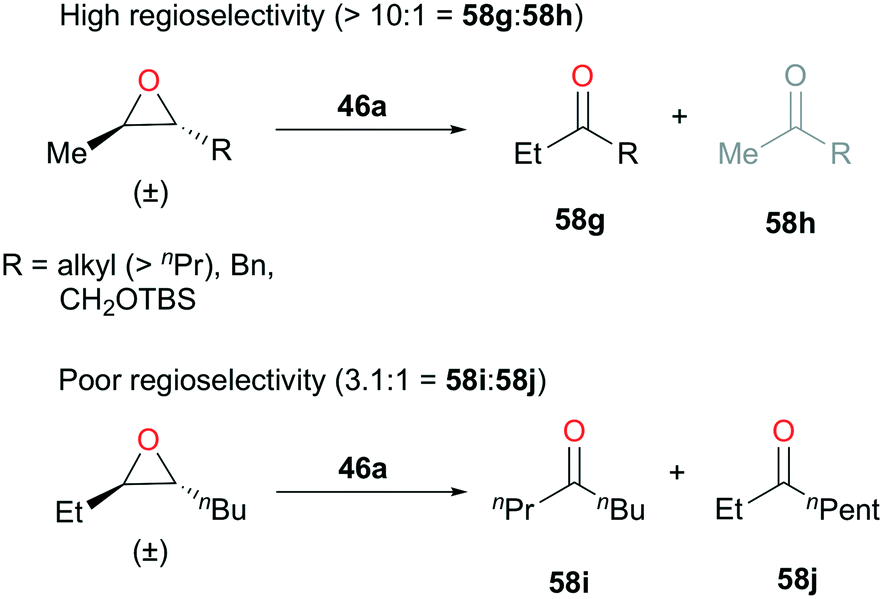
Coates and coworkers also introduced a similar catalytic system optimized for a set of 2,3-trans-epoxides. In the absence of carbon monoxide, 46a (Fig. 24) proved to be a great initiator for isomerization (Fig. 31).44 By optimizing the concentration of diethyl ether and catalyst loading, 46a showed highly functional group dependent regioselective isomerization of trans-epoxides to ethyl ketones. For the trans-epoxides with a methyl and a long aliphatic chain (R > nPr), silyl, or benzyl group, greater than ten-to-one ratios of regioselectivities were observed (Fig. 31). However, for the more symmetrical trans-2-ethyl-3-butyloxirane, ketone 58i was obtained as a major product, but with a regioselectivity ratio of 3.1:1.
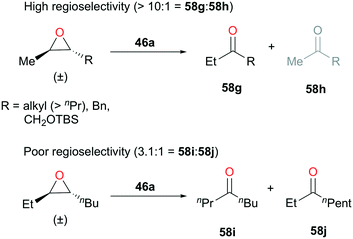 |
| | Fig. 31 Regioselective isomerization of 2,3-trans-epoxides with 46a. | |
The proposed mechanism for isomerization, based on previous mechanistic observations45 and other reports,43b,46 had four major steps: (a) substrate binding, (b) ring-opening via nucleophilic attack, (c) β-hydride elimination, and (d) dissociation of vinyl alcohol, followed by tautomerization of the enol to the ketone product (Fig. 32). Kinetic and isotope labeling studies supported that the nucleophilic ring-opening step is likely to be the rate-determining and regioselectivity-determining step for the isomerization of trans-epoxides.
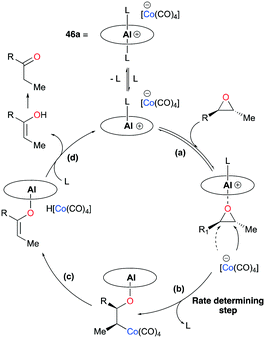 |
| | Fig. 32 Mechanism of ketone isomerization with 46a (L = epoxide, THF, or Et2O). | |
2.7. Silylation
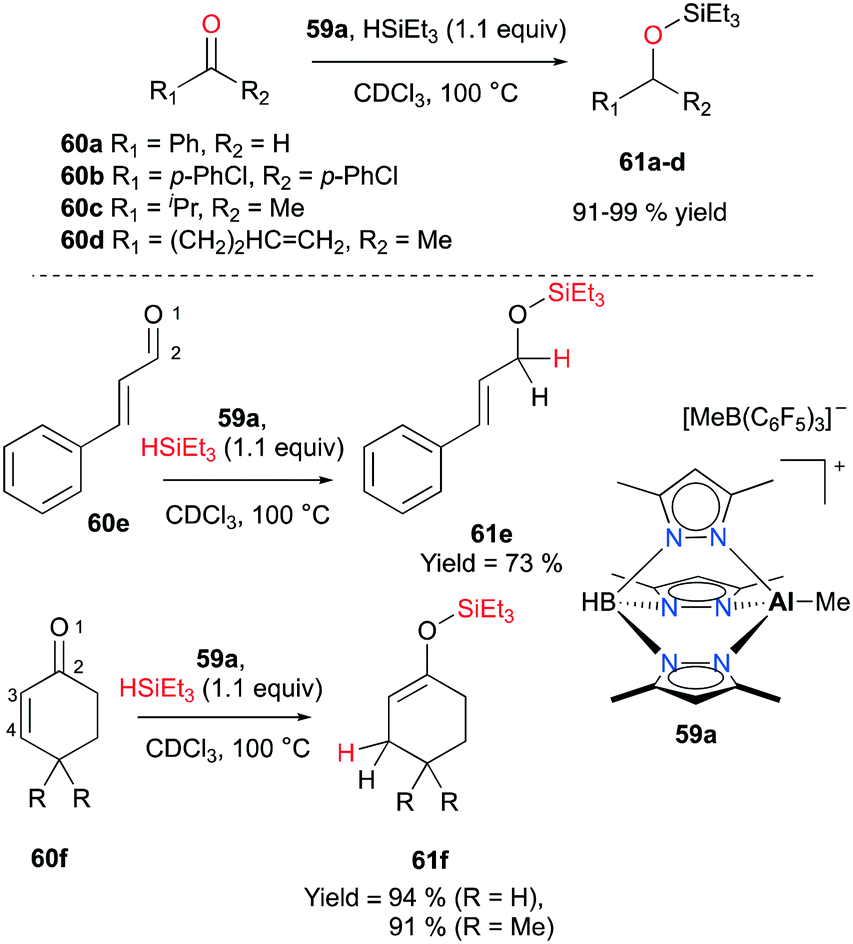
In 2011 Bergman and Koller introduced a well-defined scorpionate cationic complex 59a by methyl abstraction with B(C6F5)3 from its neutral precursor.47 With the optimized catalyst loading (1–2.5 mol%) and temperature (100 °C), complex 59a successfully combined carbonyl compounds 60a–d and HSiEt3 into the corresponding tertiary silyl ethers 61a–d (Fig. 33). Conflicting results have been observed for the addition of a hydride when an α-β-unsaturated aldehyde and ketone were hydrosilylated. The conjugated aldehyde 60e underwent 1,2-addition with HSiEt3, whereas the hydrosilylation of ketones 60f gave rise to 1,4-addition products 61f (Fig. 33).
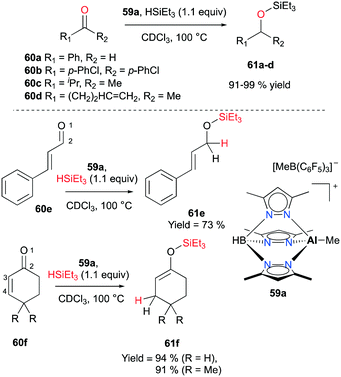 |
| | Fig. 33 Hydrosilylation of aldehydes and ketones 60a–d with 59a (top); 1,2- and 1,4-addition reactions of 60e–f (bottom). | |
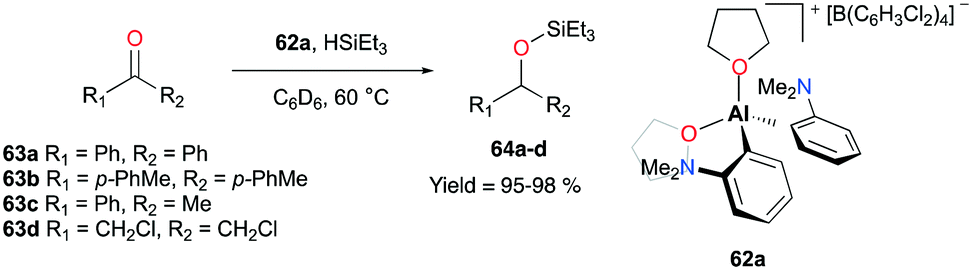
Venugopal and co-workers reported the aryl- and THF-based cationic complex 62a that catalyzed the hydrosilylation of ketones 63a–d to tertiary silylethers in more than 95% yields (Fig. 34).48 Interestingly, 62a exhibited a somewhat opposite behavior towards substrates when compared to the aforementioned system of 59a (Fig. 33). An 1H NMR analysis of the mixture of benzophenone and complex 62a revealed the substitution of two THF ligands by the ketone substrates. On the other hand, the corresponding chemical shift of HSiEt3 did not change in the mixture of 62a. Contrary to these results, Bergman and Koller reported the interactions between 59a and HSiEt3, showing the loss of the 3J coupling of H–Si–CH2 at 55 °C. This finding was further supported by deuterium scrambling between DSiEt3 and H2SiPh2 in the presence of 59a.47
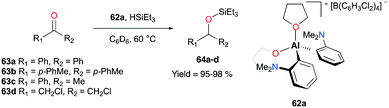 |
| | Fig. 34 Hydrosilylation of ketones with 62a. | |
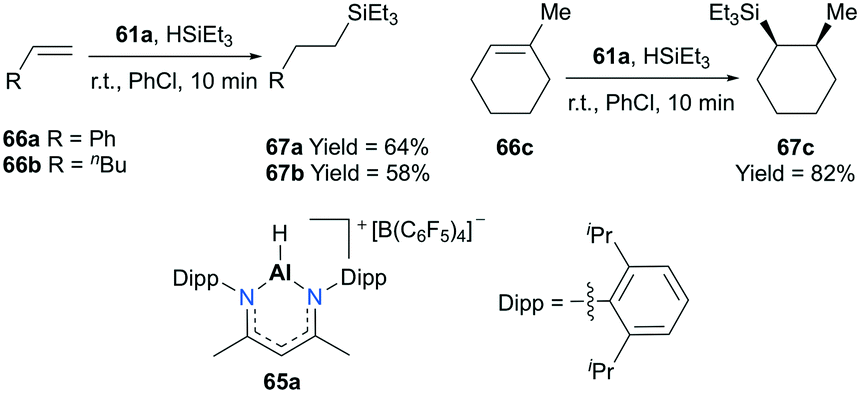
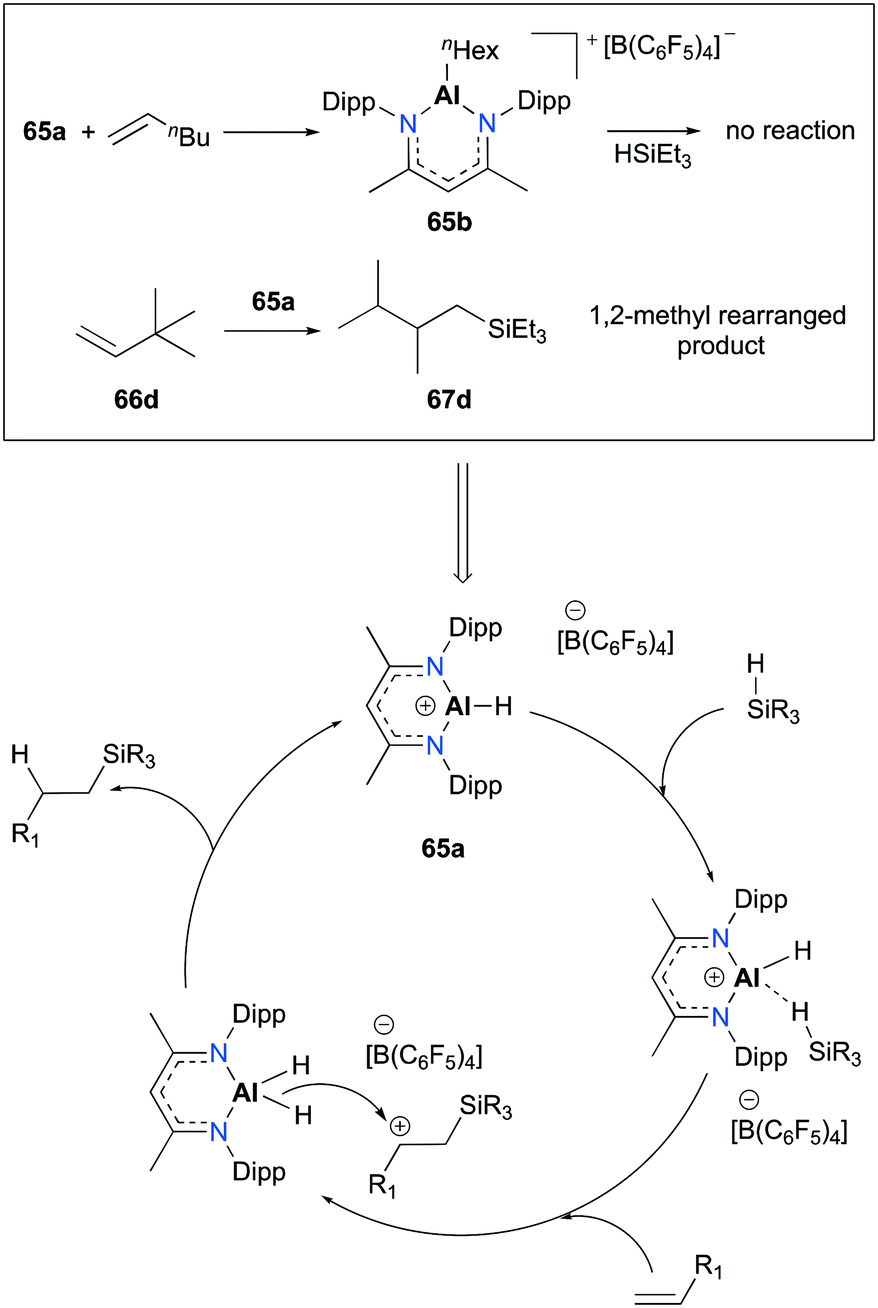
Nikonov and co-workers developed highly active NacNac-based cationic complexes bearing alkyl and hydride moieties for hydrosilylation of alkenes (Fig. 35).49 In particular, acyclic and cyclic alkene substrates were converted to their corresponding silanes (98 to >99% conversion) within a short time span (10 min), using 65a. For the case of substrate 66c, the trans addition product 67c which resulted from the electronic effect of the Si group as well as a stable conformation adopted by the ring was observed.50
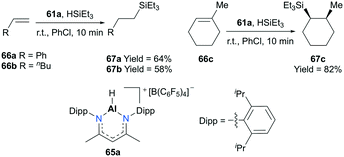 |
| | Fig. 35 Hydrosilylation of alkenes with 65a. | |
Several separate experiments and a notable finding in the transformation of 3,3-dimethyl-1-butene (66d) provided evidence for a plausible mechanism relative to [NacNacAlH][B(C6F5)4] (Fig. 36). The stoichiometric reaction between 65a and 1-hexene successfully afforded complex 65b; however, 65b did not show any reactivity towards HSiEt3. This consequence led to the exclusion of alkene insertion/metathesis catalytic pathways. When 65a was separately treated with the mixture of HSiEt3 and H2SiEt2, mixtures of silanes, such as H4Si, H3SiEt, H2SiEt2, and HSiEt3, were observed. Notably, the hydrosilylation of 66d produced methyl-rearranged silane product 67d. This result supported the formation of a secondary carbocation and a rapid 1,2-methyl migration during the catalytic pathways.
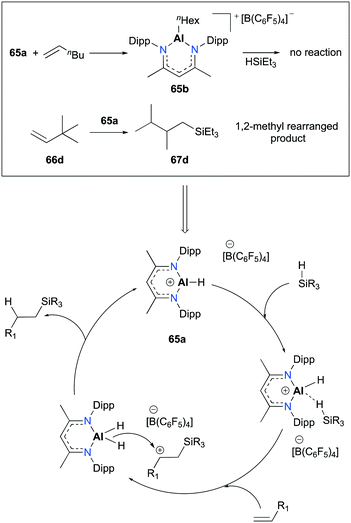 |
| | Fig. 36 Proposed mechanism and evidence for the hydrosilylation of alkenes by 65a. | |
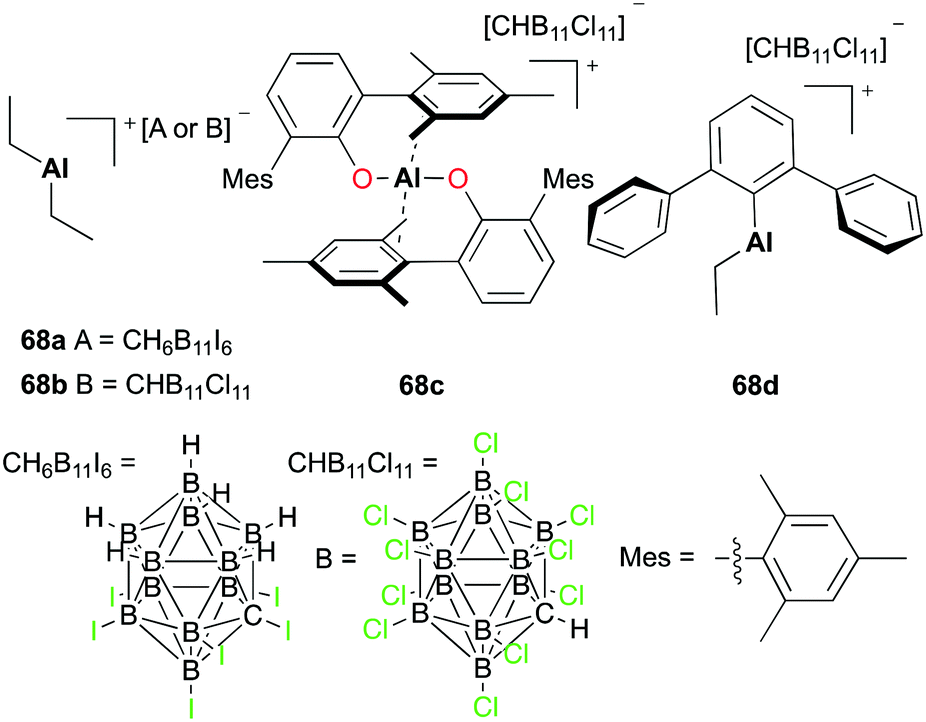
Wehmschulte and co-workers reported the hydrosilylation of CO2 with cationic complexes 68a–d (Fig. 37).51 The catalysis produced multiple products such as methane (CH4), toluene-d5 (C6D5CH3), and diphenylmethane-d10 ((C6D5)2CH2). The distribution of the products was dependent on the catalyst used for the hydrosilylation. Complex 68a catalyzed the reaction between CO2 and Et3SiH, affording methane as a major product. The catalysis of more reactive and robust complex 68c formed C6D5CH3 as a major product. A small amount of DH was generated when 68b–d were used in the hydrosilylation.
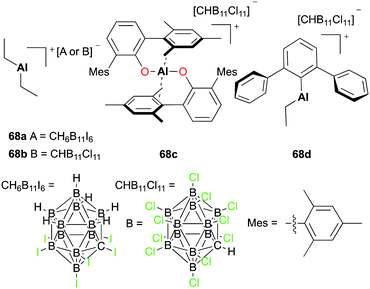 |
| | Fig. 37 Cationic aluminium complexes (68a–d) for CO2 hydrosilylation. | |
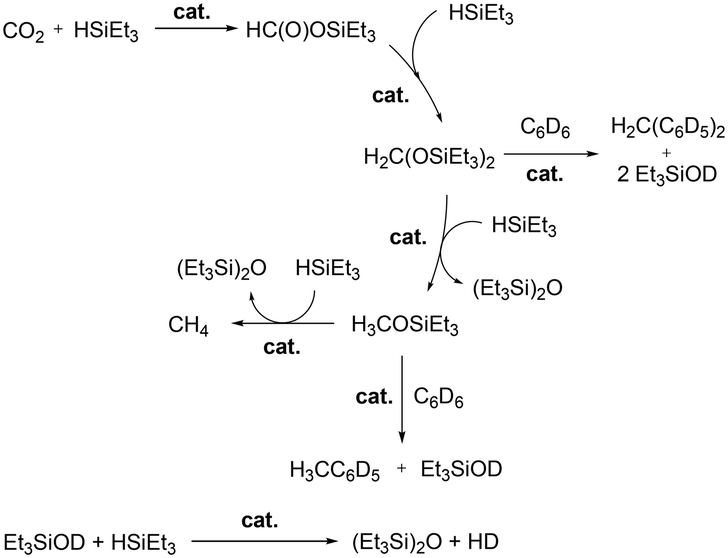
A mechanistic study was carried out by investigating the reaction of potential intermediates HCOOSiEt3 and H3COSiEt3.51a Complex 68a catalyzed the reduction of HCOOSiEt3 in the presence of Et3SiH. The catalysis generated products CH4, C6D5CH3, and (Et3Si)2O as well as intermediate H3COSiEt3. Similarly, the reduction of H3COSiEt3 was catalyzed by 68a and afforded methane and toluene-d5 as the major products. The agreement in the final products as well as the observation of H3COSiEt3 demonstrated the presence of intermediates HCOOSiEt3 and H3COSiEt3 in the hydrosilylation of CO2.
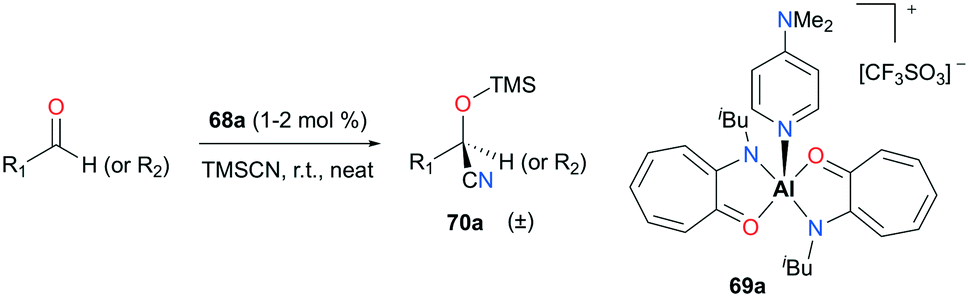
Nagendran and co-workers reported a highly efficient and well-defined cationic aluminum complex (69a) that can catalyze cyanosilylation (Fig. 38).52 The complex was readily prepared from the corresponding neutral complex (AT)2AlOTf and DMAP (4-dimethylaminopyridine). Complex 69a successfully generated a variety of cyclic and acyclic cyanosilyl ether products 70a, using trimethylsilylcyanide (TMSCN) as a reducing reagent. Although the reaction conditions were mild (room temperature and 1–2% cat. loading), high yields (>93%) were observed for a broad range of substrates.
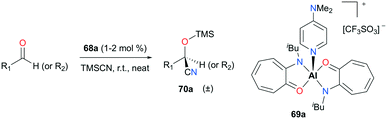 |
| | Fig. 38 Cyanosilylation of ketones and aldehydes with 69a. | |
2.8. Epoxidation and epoxide deoxygenation
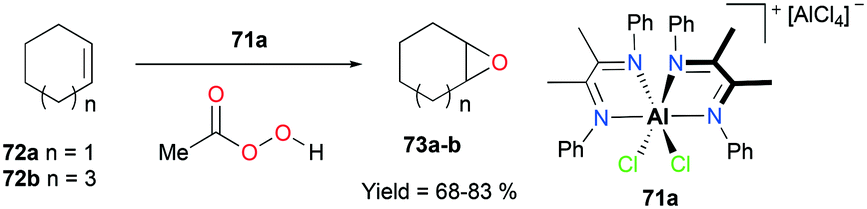
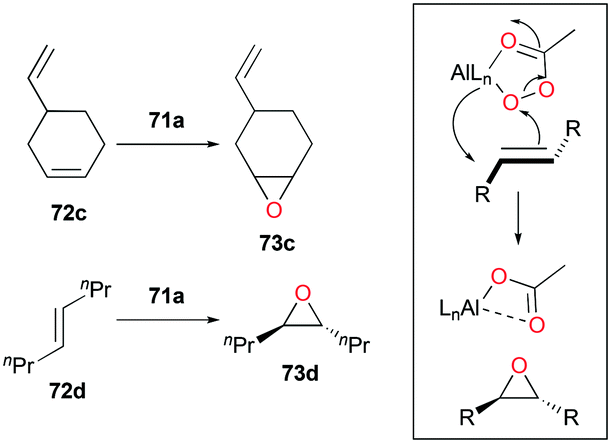
Alpha diimine-based cationic complex 71a, reported by Goldsmith and co-workers, catalyzed the epoxidation of olefins (Fig. 39).53 Complex 71a was synthesized from the reaction between equimolar amounts of neutral α-diimines and AlCl3. The reaction of internal alkenes 72a–d with peracetic acid (PPA) as an oxidizing reagent generated moderate to good yields of epoxides 73a–d. The exclusive epoxidation at the internal alkene of 4-vinylcyclohexene 72c clearly showed that 71a preferentially oxidized the more electron-rich alkene, rather than the sterically less encumbered terminal alkene (Fig. 40). The exclusive product of trans-2,3-dipropyloxirane (73d) supported the concerted process of an oxygen atom transfer to the corresponding alkene.54
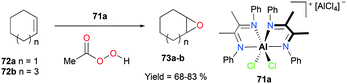 |
| | Fig. 39 Epoxidation of internal alkenes using 71a and PA. | |
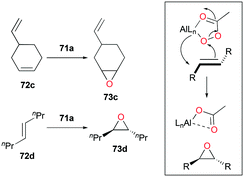 |
| | Fig. 40 Selectivity of 71a towards alkene regions and the proposed concerted process. | |
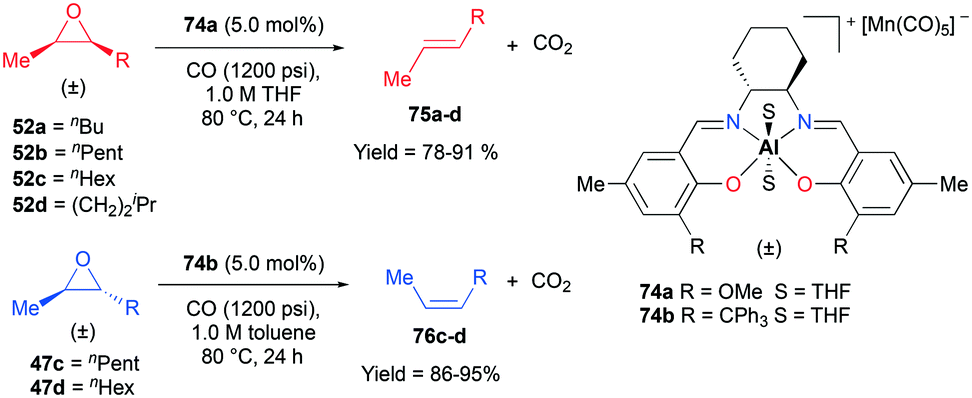
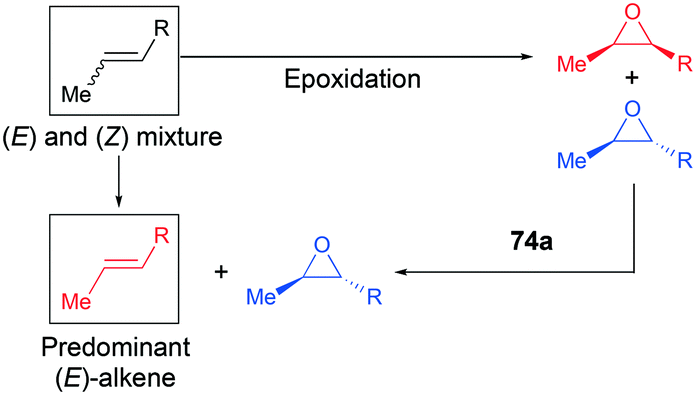
Recently, a remarkable catalytic system exhibiting de-epoxidation was developed by Coates and co-workers (Fig. 41).55 Using salen-based cationic complexes 74a and 74b as well as appropriate solvents, substrates, and a counter anion, 2,3-cis- and trans-epoxides 52a–d and 47c–d were reduced to alkenes 75a–d and 76c–d with inversion of the stereochemistry of epoxides. In addition to the good to high yields, it was shown that this catalytic system is a great strategy to isolate a single geometric isomer from a mixture of alkenes, exploiting the faster reaction rate of 74a towards cis epoxides. (Fig. 42).
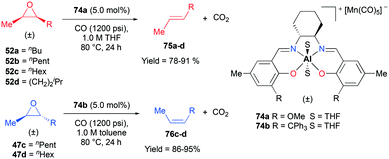 |
| | Fig. 41 Deoxygenation of 2,3-cis- and trans-epoxides with 74a and 74b. | |
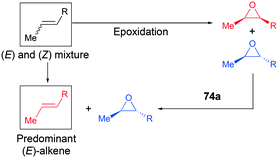 |
| | Fig. 42 A strategy for the isolation of one geometrical isomer of alkenes. | |
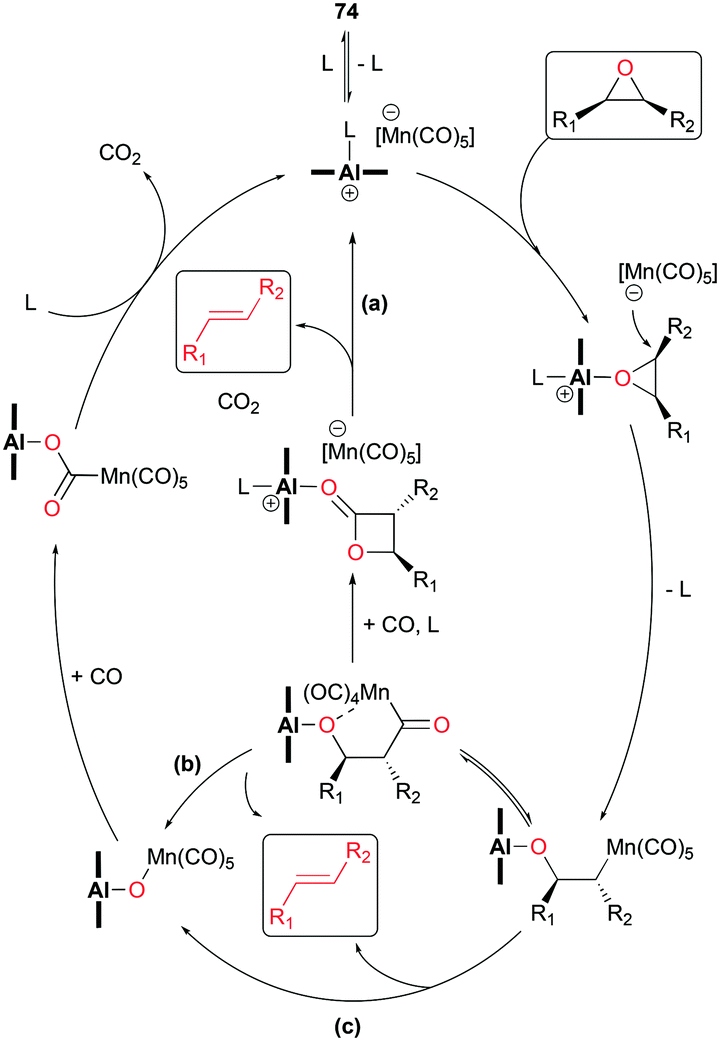
In light of the inverted stereochemistry of products, it was suggested that the deoxygenation mechanism is related to the initial steps in epoxide carbonylation (Fig. 23). In addition, it was proposed that the resulting alkenes are ultimately generated from one of the three plausible pathways such as: (a) ring closure followed by Lewis acid-mediated decarboxylation, (b) formation of acyl manganese followed by alkene generation, and (c) β-oxygen elimination from manganese alkyl (Fig. 43).
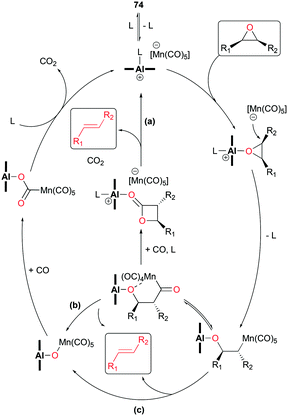 |
| | Fig. 43 Mechanism of deoxygenation of epoxides. | |
2.9. Coupling and dimerization of substrates
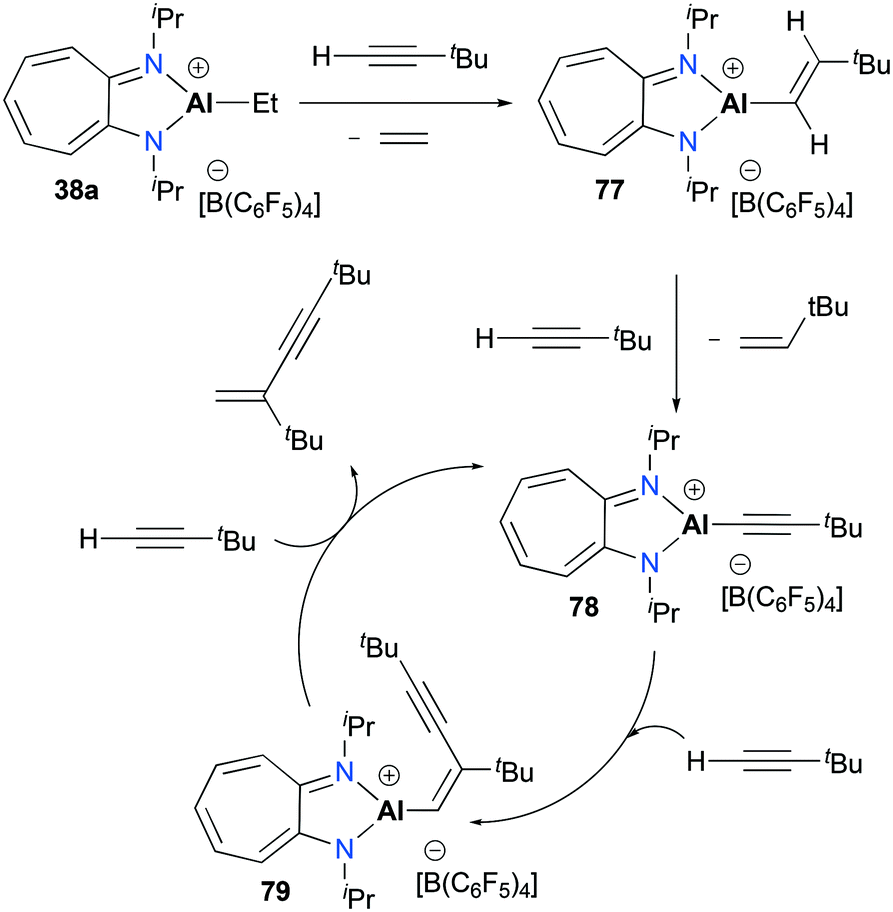
Jordan and co-workers reported the dimerization of tert-butyl acetylene with [(iPr2-ATI)AlEt][B(C6F5)3] 38a which was discussed above (Fig. 17).22,32 The individual catalytic pathways were studied by characterizing intermediates occurring in a catalytic cycle (Fig. 44). The stoichiometric reaction between 38a and tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH produced ethylene and the vinyl aluminum precatalyst 77 quantitatively. When another 1 equivalent of the acetylene was treated with 77, the reaction allowed a mixture of 78, 79, and 77 with a ratio of 4:1:1. Under catalytic conditions, NMR spectra revealed 79 as the only intermediate, indicating that 79 is most likely to be the resting state of the catalytic cycle.
CH produced ethylene and the vinyl aluminum precatalyst 77 quantitatively. When another 1 equivalent of the acetylene was treated with 77, the reaction allowed a mixture of 78, 79, and 77 with a ratio of 4:1:1. Under catalytic conditions, NMR spectra revealed 79 as the only intermediate, indicating that 79 is most likely to be the resting state of the catalytic cycle.
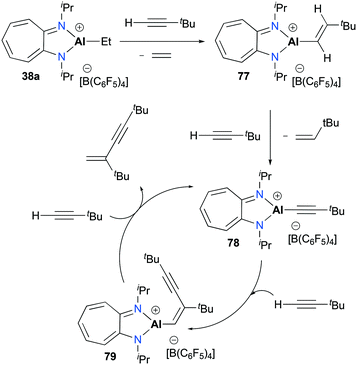 |
| | Fig. 44 Mechanism of tert-butyl-acetylene dimerization with 38a. | |
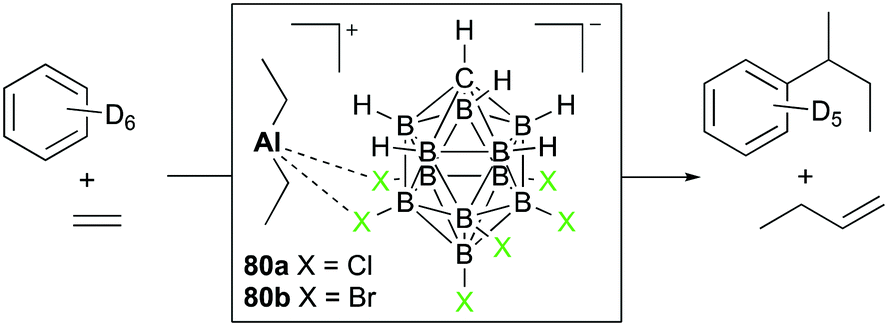
Reed and Sen introduced diethyl aluminum complexes 80a and 80b having tight ion-pairings with halogenated carboranes (Fig. 45).56 The reactivity towards substrates was dominated by the Lewis acidity of aluminum centers, keeping aluminum–Et bonds intact. With 80a dissolved in benzene-d6, the reaction with ethylene (150 psi) generated 1-butene and sec-butylbenzene as the major products. Based on the observed products, sequential steps including coordination of ethylene, generation of the carbocation, and attack of benzene at the carbocation were envisioned as the major mechanistic pathways.
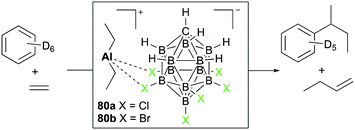 |
| | Fig. 45 Coupling of ethylene and benzene with 80a. | |
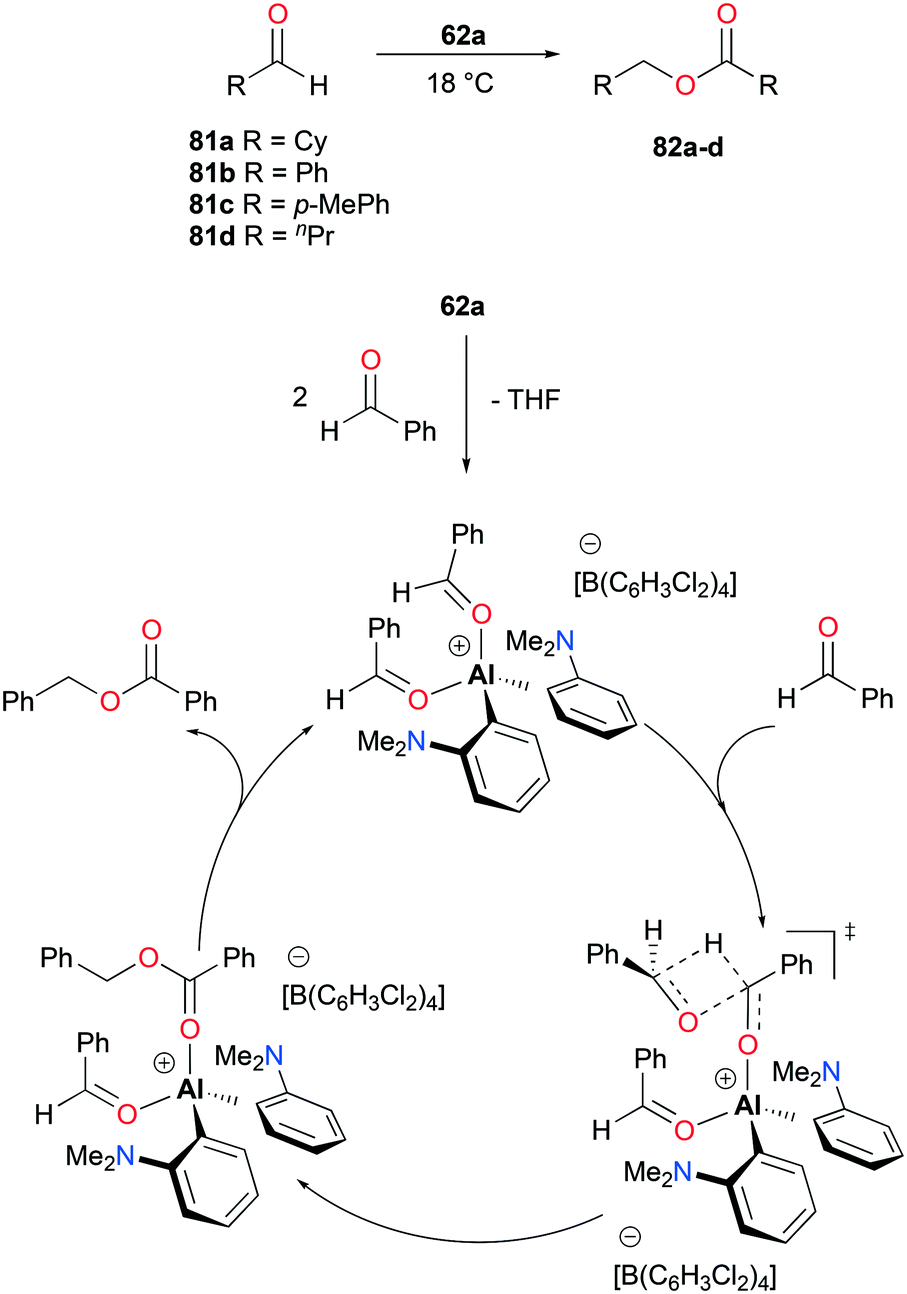
The aforementioned complex 62a (Fig. 34), introduced by Venugopal and co-workers, catalyzed the Tishchenko reaction – the formation of esters from the dimerization of aldehydes (Fig. 46).48 The catalysis with aldehydes 81a–d generated alkyl and aryl esters 82a–d at ambient temperature. The reaction pathways, studied by kinetic analysis and DFT calculations, include four major steps with a notable transition state such that an incoming benzaldehyde formed a four-membered ring transition state with one of the coordinating aldehydes, overcoming an activation barrier of 25.6 kcal mol−1.
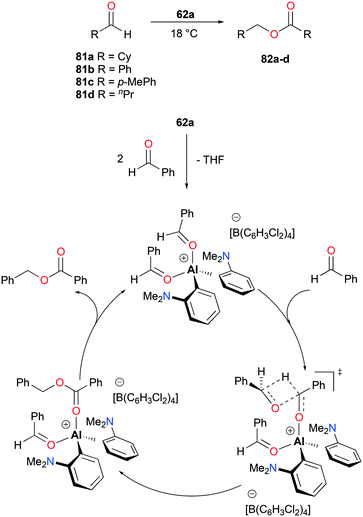 |
| | Fig. 46 Tishchenko reaction with 62a and its mechanism. | |
3. Cationic gallium complexes
Gallium is primarily found in its +3 oxidation state. Ga(I) species are rare and are mainly used as reagents for stoichiometric reactions as they tend to be unstable towards disproportionation.57 Neutral gallium(III) complexes have been used as versatile catalysts in a variety of reactions.58 Yet, their hygroscopic nature limits their ability requiring a large excess of catalyst to reach the desired conversion and efficiency.59 In 1992, Cowley and co-workers proposed the first definitive structural evidence for a solvated dimethylgallium cation complex.60 With the development and characterization of cationic gallium complexes, structural information of many cationic gallium complexes has been reported to date.60,61
3.1. Ring opening polymerization (ROP) of cyclic ethers
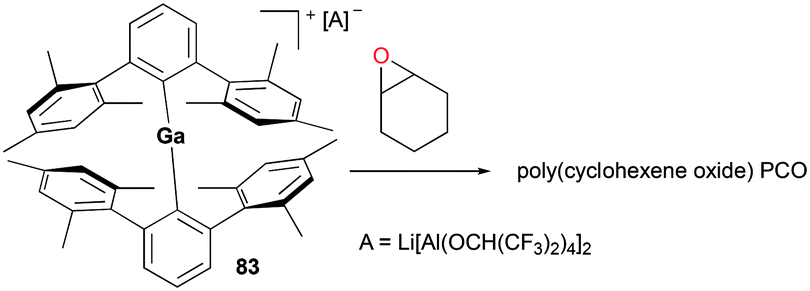
A two-coordinate cationic gallium complex [(2,6-Mes2C6H3)2Ga][Li[Al(OCH(CF3)2)4]2] (Mes = 2,4,6-Me3C6H2) 83 incorporating a bulky m-terphenyl ligand was reported by Wehmschulte and co-workers (Fig. 47).62 Complex 83 was generated through salt metathesis between [2,6-Mes2C6H3]2GaCl and 2 equivalents of Li[Al(OCH(CF3)2)4]. Despite being unreactive towards 1-octene, 83 immediately polymerized 173 equivalents of cyclohexene oxide in C6H5Cl in an exothermic reaction to obtain poly(cyclohexeneoxide) (PCO).
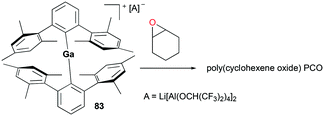 |
| | Fig. 47 Oligomerization of cyclohexene oxide with complex 83. | |
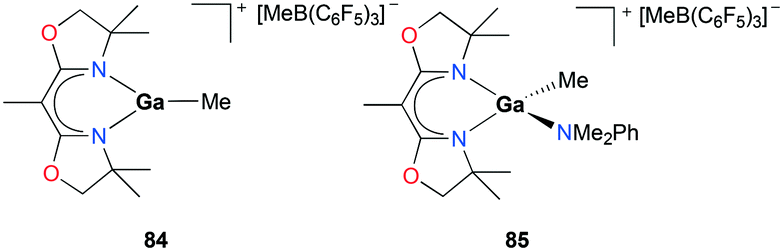
Polymerization of cyclic ethers was further studied in-depth with cationic gallium complexes supported by bis(oxazolinato) ligands by Dagorne, Bellemin-Laponnaz, and co-workers (Fig. 48).63 The cationic alkylgallium complex [(BOX-Me2)GaMe][MeB(C6F5)3] (BOX = bis(oxazolinato)) 84 was synthesized through methyl abstraction from [(BOX)GaMe2] with 1 equivalent of B(C6F5)3. The resulting cation was more stable compared to its aluminum counterparts, as expected of the less Lewis acidic gallium center compared to aluminum.64 To overcome the lack of stability of 84, the methylgallium complex [(BOX-Me2)Ga(Me)(NMe2Ph)][MeB(C6D5)3] 85 was prepared through the reaction of [(BOX-Me2)GaMe2] with 1 equivalent of B(C6F5)3 in the presence of 1 equivalent of NMe2Ph. Complex 85 was stable and showed a fast exchange of NMe2Ph on the NMR timescale. In the light of the nonlabile behavior of aluminum analogues under the same conditions, this lability of NMe2Ph to gallium showed the lower Lewis acidity of the cationic gallium center compared to that of the aluminum center.64
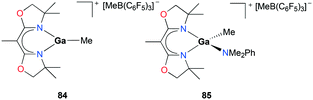 |
| | Fig. 48 Bis(oxazolinato) ligated cationic gallium complexes (84–85) studied for propylene oxide oligomerization. | |
Complexes 84 and 85 were investigated for their ability to oligomerize propylene oxide (PO) (Fig. 48). Complex 84 catalyzed the oligomerization of PO, but increasing the reaction time did not lead to conversion higher than 40% attributed to the fast decomposition of 84 into inactive species in the presence of excess PO within 5 min. Complex 85 also successfully oligomerized PO reaching a higher conversion of 85% compared to that of 84 reflecting its enhanced stability. Size exclusion chromatographic data showed clear multimodal characteristics of the oligomerization process suggesting the presence of more than one active species during the reaction.
3.2. Polymerization of cyclic esters
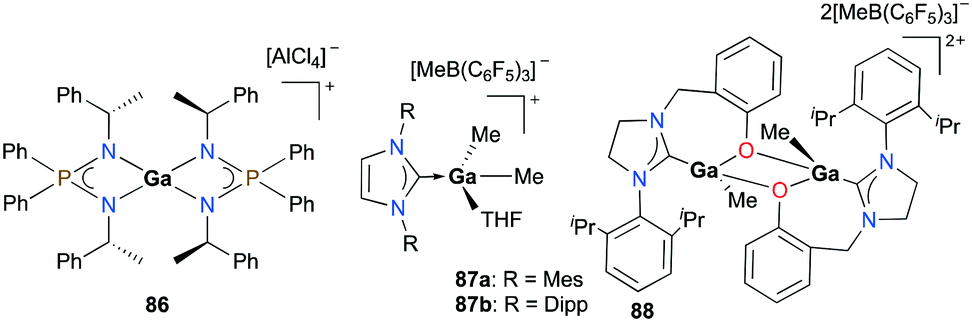
Cationic gallium complexes with iminophosphoamide or NHC-derived ligands showed poor or no reactivity towards the polymerization of cyclic esters. Roesky and co-workers reported the first example of an enantiopure gallium complex coordinated by chiral iminophosphoamides [L2Ga][AlCl4] (L = [Ph2P(N(R)CH(CH3)Ph)2]) 86 (Fig. 49).65 Complex 86 was prepared through stoichiometric chloride abstraction of L2GaCl by AlCl3 in THF. However, complex 86 was catalytically inactive for the ROP of racemic lactide (rac-LA). Cationic gallium complexes stabilized by NHCs, 87a–b, reported by Dagorne and co-workers were also inactive for lactide ROP (Fig. 49).28 They showed no reactivity in ROP of lactide in the presence of BnOH as the chain transfer agent, which may be attributed to their lower Lewis acidity compared to aluminum analogues reported in the same study. Interestingly, it was suggested that this catalytic activity may occur by the action of the free NHC ligand after its decoordination from the gallium center.66
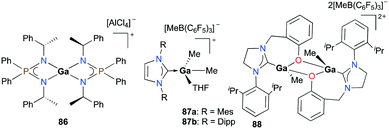 |
| | Fig. 49 Cationic gallium complexes studied for lactide polymerization. | |
Horeglad and co-workers reported a dinuclear alkylgallium aryloxide cation supported by NHC, 88 (Fig. 49).67 Complex 88 showed poor reactivity for the ROP of rac-LA in the presence and absence of iPrOH. The reaction without iPrOH had significant amounts of unreacted rac-LA, highlighting the importance of iPrOH. Moreover, 88 catalyzed the polymerization of ε-CL to produce cyclic PCL, indicated by the loss of end groups in the 1H NMR spectrum.
3.3. Polymerization of olefins
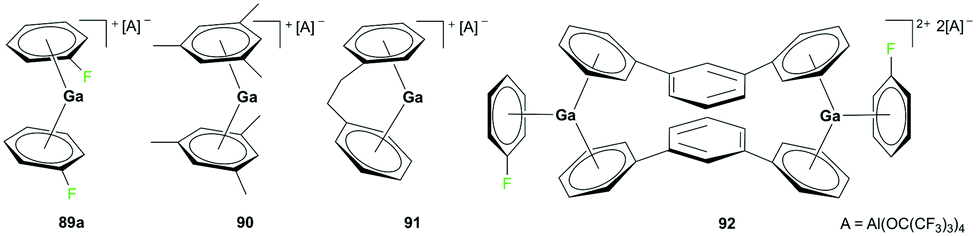
Although the cationic gallium complex with an NHC ligand 88 (Fig. 49) and the analogous species with a methyl substituent showed no sign of polymerization of ethylene (−5 bar, 50 °C, 3 h), the cationic gallium arene complexes 89–93 showed catalytic reactivity for olefin polymerization (Fig. 50 and 51).
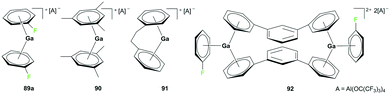 |
| | Fig. 50 Cationic gallium catalysts for the polymerization of isobutylene. | |
Krossing and co-workers reported the oxidation of elemental gallium with Ag[Al(ORF)4] (RF = C(CF3)3) in C6H5F using ultrasonic activation to form complex 89a (Fig. 50).68 Complex 89a was a versatile catalyst for the polymerization of isobutylene (IB) into highly reactive polyisobutylene (HR-PIB) as well as 2,4,4-trimethyl-1-pentene. Complex 90 was generated using ligand exchange between 89a and 2 equivalents of 1,3,5-Me3C6H3 in C6H5F, highlighting the significance of C6H5F for stabilizing gallium in its univalent oxidation state. Derivatives of 89a were synthesized through ligand exchange of 89a and PhC2H4Ph or 1,3-Ph2C6H4 in C6H5F at room temperature to yield 91 and 92, respectively.69
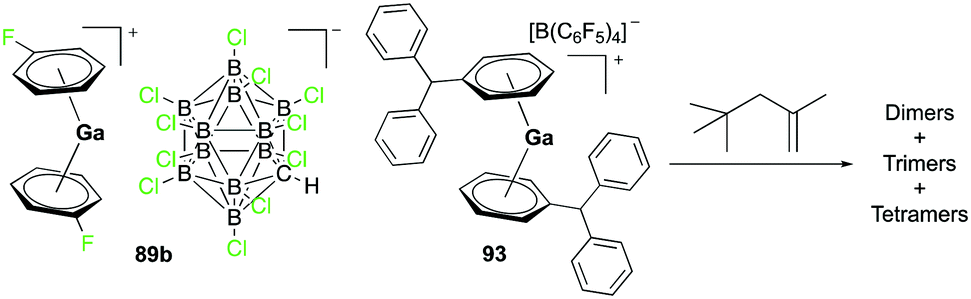
Complex 89b, the analogue of 89a with the [CHB11Cl11]− anion, was reported by Wehmschulte and co-workers to be obtained by reductive elimination of H2 from the intermediate [H2Ga]+ species in the presence of [Et3Si][CHB11Cl11] and C6H5F at room temperature (Fig. 51).70 Complex 93 was generated similarly using [Ph3C][B(C6F5)4] and (H2GaCl)2 in the presence of excess Et3SiH. Complexes 89b and 93 were catalysts for the oligomerization of 2,4,4-trimethyl-1-pentene, yielding a mixture of dimers as the major product along with trimers and tetramers.
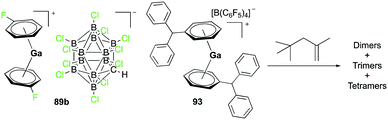 |
| | Fig. 51 Cationic gallium catalysts for the polymerization of 2,4,4-trimethyl-1-pentene. | |
3.4. Epoxidation of alkenes
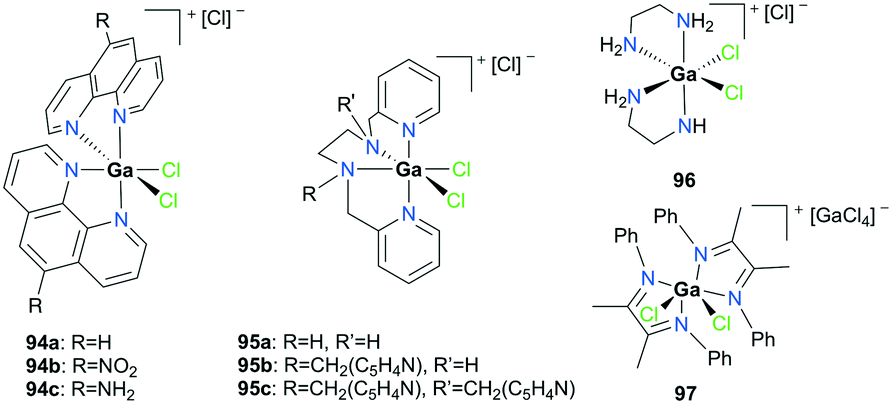
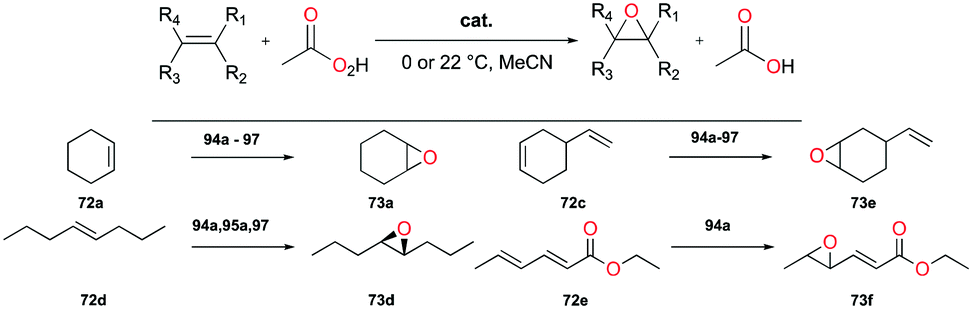
Goldsmith and co-workers, for the first time, reported cationic gallium complexes supported by phenanthrolines (Fig. 52).71 In particular, complex 94a catalyzed the epoxidation of a variety of alkenes including 72a, 72c–72e (Fig. 53). Complex 94a could not catalyze the epoxidation of alkenes in the presence of H2O2 likely due to lower affinity of H2O2 binding to Ga(III) compared to Al(III).72 Therefore, peracetic acid (PAA) and custom-made analogues (PAAR) with lower acidity compared to that of commercially available PAA73 were used. For the species containing more than one alkene group like 72e, the more electron-rich site was oxidized, which was also shown in Mn(II)-catalyzed alkene epoxidation.73 The authors hypothesized that the reactivity stems from the electron-withdrawing nature of the phenanthroline ligand which amplified the Lewis acidity of the Ga(III) center. They observed that with the dissociation of the phenanthroline ligand, the catalyst loses its activity, and this was overcome by adding a free phenanthroline ligand to the initial reaction mixture.
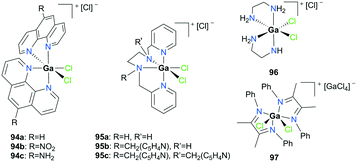 |
| | Fig. 52 Cationic gallium complexes for alkene epoxidation. | |
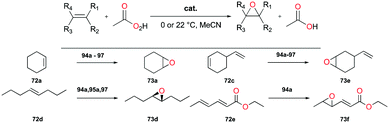 |
| | Fig. 53 Selected cationic gallium-catalyzed epoxidation of alkenes. | |
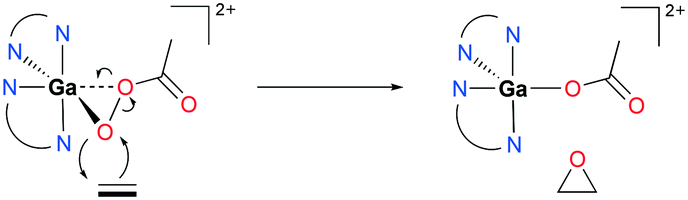
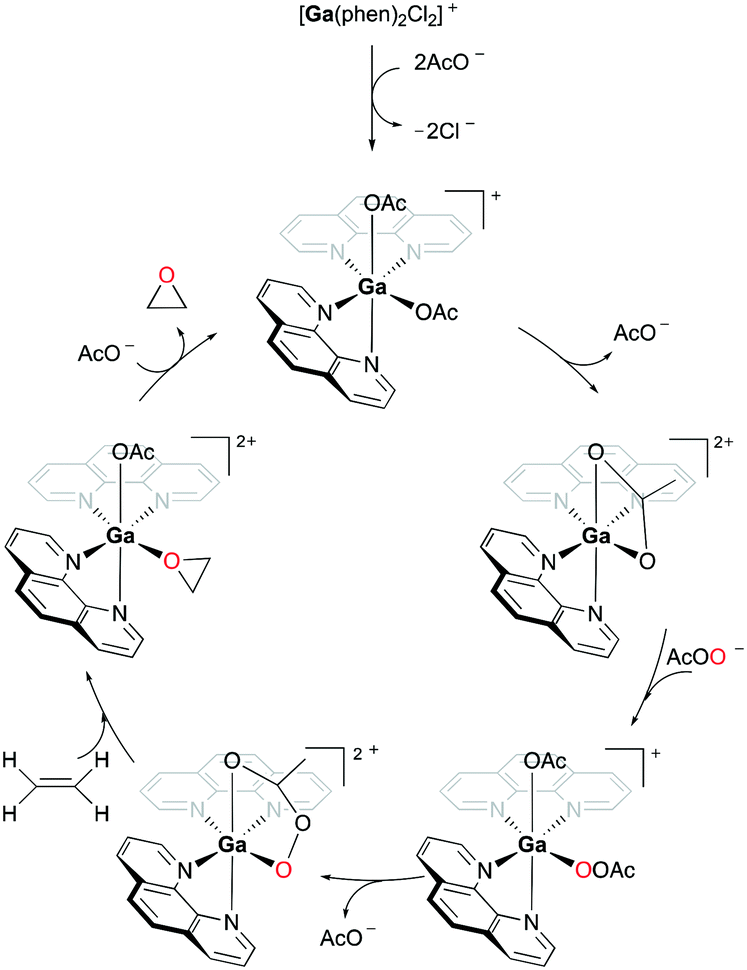
When 94a is used in 1% catalysis loading, epoxides are produced as the exclusive products. However, at catalyst concentration less than 1%, the epoxidation of 72a produces multiple products including 3-cyclohexenol and 1,2-cyclohexanediol (39%, 51%, respectively). As epoxides do not react with Ga(III) and PAA in the reaction, it is speculated that these two products are not derived from the epoxide products. This supported the mechanism of olefin epoxidation by the catalyst which follows a Sharpless-type mechanism at a high PAA/Ga(III) ratio, rather than overoxidation (Fig. 54).74 Moreover, the oxygen-transferring agent was predicted to be a [Ga(phen)2(PI)]2+ (PI = peracetate) species, calculated through subsequent DFT studies.54 The complex forms a stable five-membered metallacycle with bidentate PI (Fig. 55).75
 |
| | Fig. 54 Proposed Sharpless-type epoxidation mechanism. | |
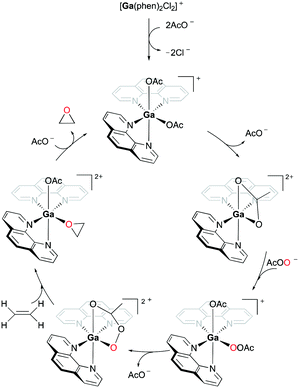 |
| | Fig. 55 Mechanism of cationic gallium-catalyzed alkene epoxidation from a DFT study.54,76 | |
3.5. Hydroarylation and transfer hydrogenation
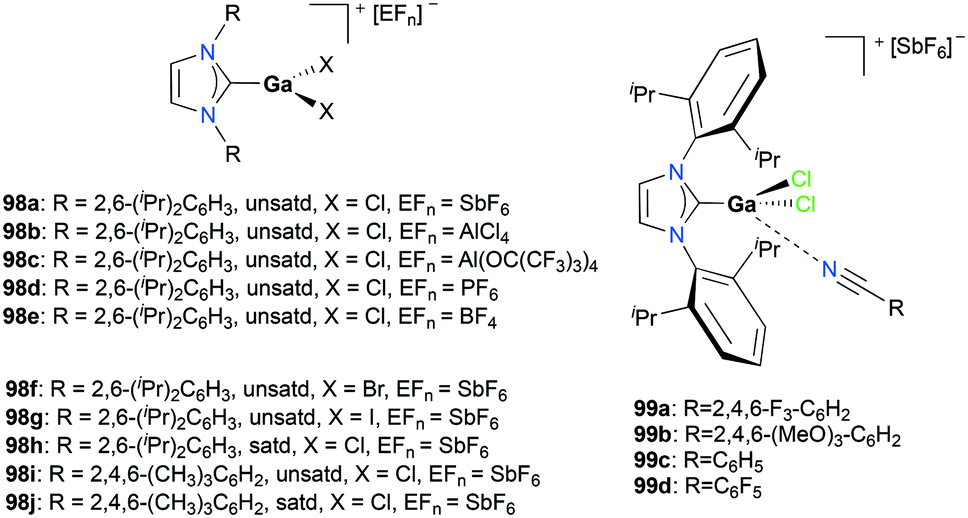
Gandon and co-workers reported a series of cationic Ga(III) halide catalysts incorporating NHC ligands, 98a, 98d–j and 99a–d used as catalysts for one-pot hydroarylation of 100a (Fig. 56).59,77 They showed excellent reactivity for hydroarylation, transfer hydrogenation, and cyclizations of alkynes and alkenes (Fig. 57). For [IPrGaCl2][SbF6] 98a, an excess of silver with respect to gallium increased the reaction rate, which can be due to the lower number of di- or tricationic gallium-centered complexes generated, which was significant for the case with 1 mol% (IPr)GaCl3 (IPr:R = 2,6-(iPr)2C6H3, unsatd).78 Bromide and iodide adducts, 98f and 98g, and complexes with other NHC substituents, 98h–98j also catalyzed the reaction.
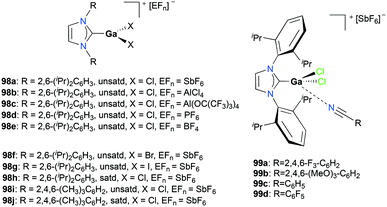 |
| | Fig. 56 Cationic gallium complexes for hydroarylation/transfer hydrogenation. | |
 |
| | Fig. 57 Cationic gallium-catalyzed hydroarylation of alkynes. | |
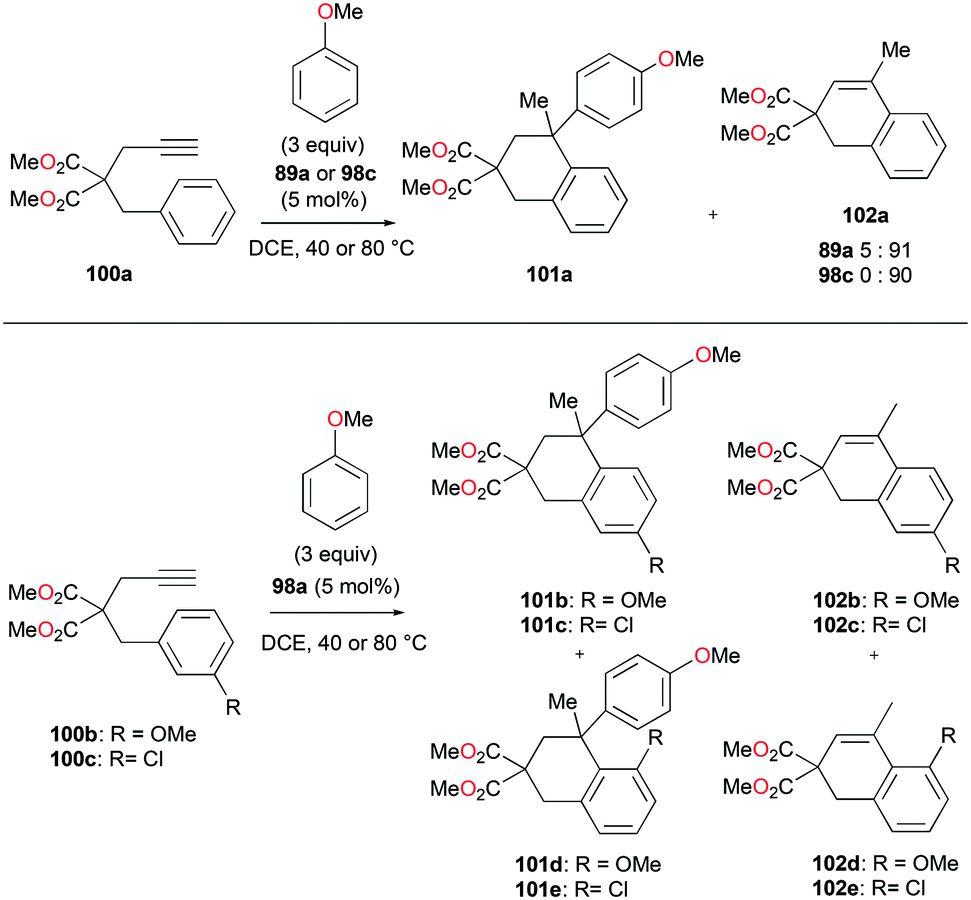
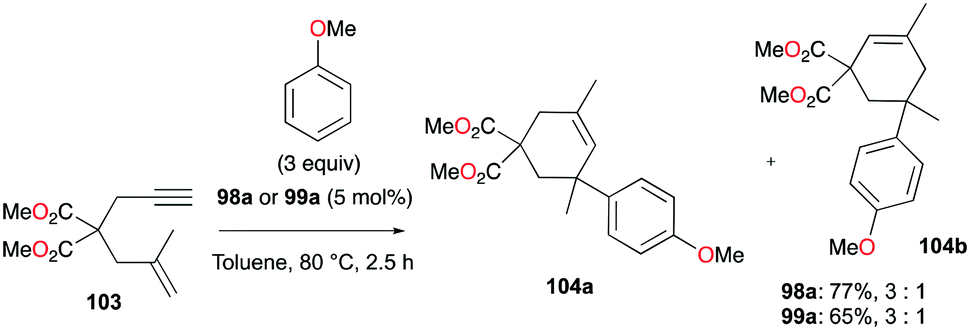
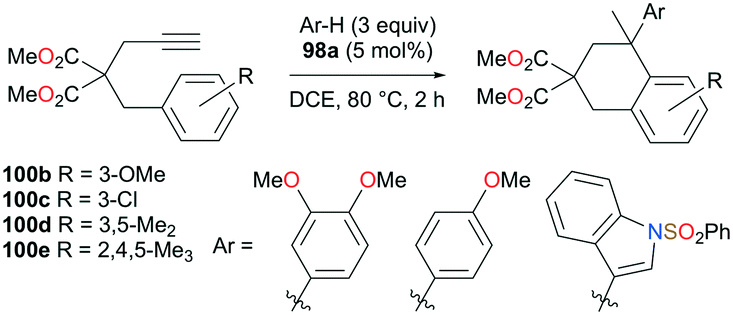
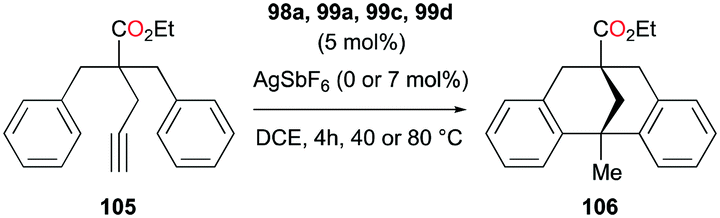
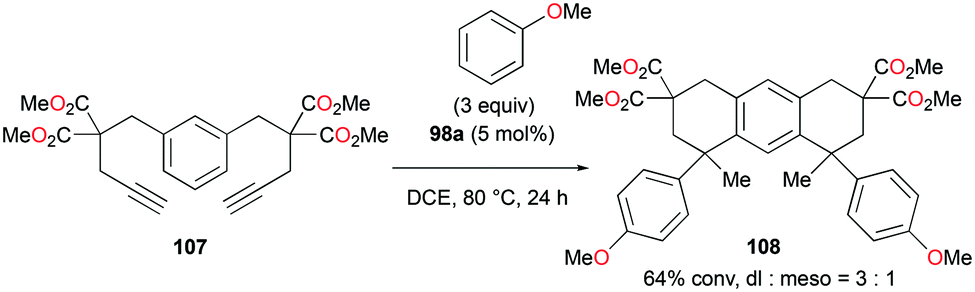
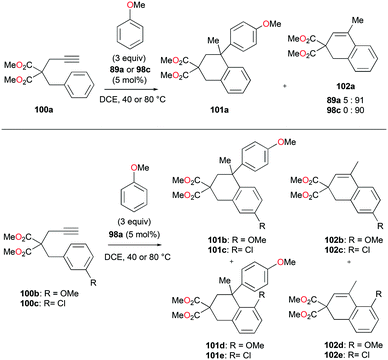
The reversibility of the hydroarylation was investigated with 100b and 103 by Bour, Gandon, and co-workers (Fig. 57 and 58).79 For the reaction of 100b, both para and ortho isomers were produced, but with increasing reaction time, the ratio of 101b:101d increased from 14:14 to 60:22 at 40 °C. This rearrangement can be attributed to the molecule preferring the orientation with less steric hindrance and gave a hint of the reversibility of C–C bond formation of the reaction. The reaction was also successful with indole derivatives and 1,2-dimethoxybenzene, being able to substitute the anisole group (Fig. 59). Intramolecular hydroarylation of 105 to produce 106 was also successful with the four complexes (Fig. 60). Among the four complexes, 98a showed the best reactivity with significantly higher yield compared to the other complexes. Furthermore, 98a catalyzed the double hydroarylation of substrates with more than one alkyne functionality 107 with 64% conversion (Fig. 61).
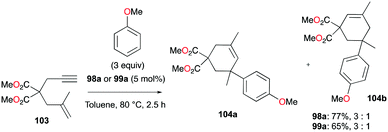 |
| | Fig. 58 Hydroarylation of an enyne with 98a or 99a.59 | |
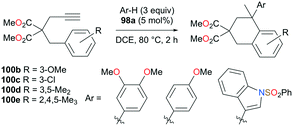 |
| | Fig. 59 Hydroarylation catalyzed by complex 98a. | |
 |
| | Fig. 60 Intramolecular hydroarylation of an alkyne with cationic gallium catalysts. | |
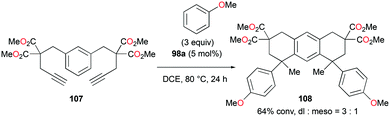 |
| | Fig. 61 Double hydroarylation of an alkyne.80 | |
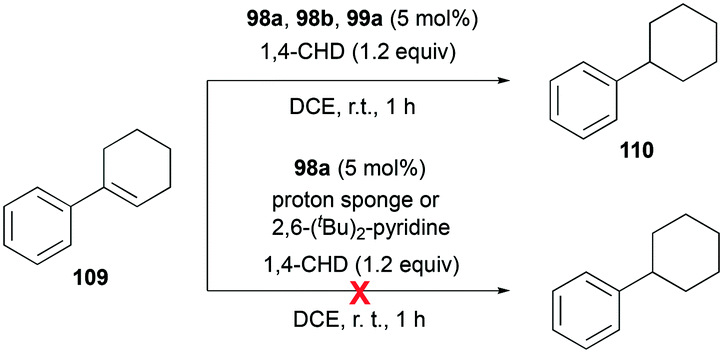
Gandon and co-workers investigated the catalytic activity of 98a (Fig. 56) for transfer hydrogenation of cyclohexenylbenzene 109 to produce 110, using 1,4-cyclohexadiene (1,4-CHD) (Fig. 62).80 The analogous complexes 98b and 99a also showed high reactivity, both reaching 87% conversion for an identical reaction. The presence of bases significantly influenced the reaction, decreasing the yield down to 0%.
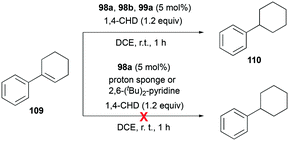 |
| | Fig. 62 Transfer hydrogenation of cyclohexenylbenzene. | |
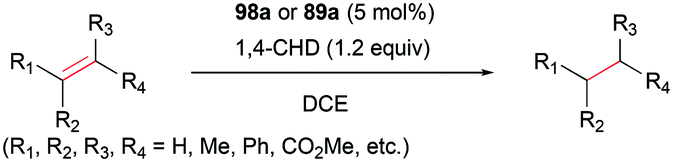
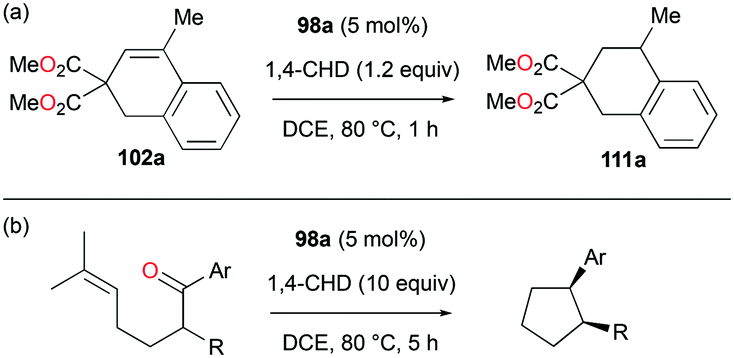
Alkenes with different substituents were tested for transfer hydrogenation by complex 98a with 1,4-CHD (Fig. 63). Among these, 1,1-diphenylethylene was fully converted into 1,1-diphenylethane, while tetraphenylethene was not converted. 89a was also studied for its reactivity for transfer hydrogenation of alkenes with 1,4-CHD.81 All acyclic or cyclic alkenes were hydrogenated in moderate to high yields from 43% to 99%. The functional groups on alkenes, methoxy and nitro groups were not affected by these conditions. 98a and 89a could also effectively catalyze the transfer hydrogenation of cycloalkenes such as the conversion of 102a to 111a (Fig. 64). In the presence of 1,4-CHD in DCE, stereoselective tandem carbonyl-olefin metathesis/transfer hydrogenation of an aryl ketone bearing a dimethyl substituted olefin terminus was also catalyzed by 98a (Fig. 64).82 The reaction yielded 1,2-cis-disubstituted cyclopentanes and cyclohexanes with moderate to high yields using different substrates. Computational studies supported the proposed role of Ga(III) homodimers in the two-step mechanism and revealed the unexpected activation of 1,4-CHD.
 |
| | Fig. 63 Transfer hydrogenation of a series of alkenes by 98a and 89a. | |
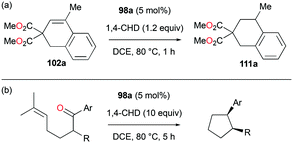 |
| | Fig. 64 Transfer hydrogenation of cycloalkenes (a) and carbonyl–olefin metathesis/transfer hydrogenation tandem reaction (b) by 98a. | |
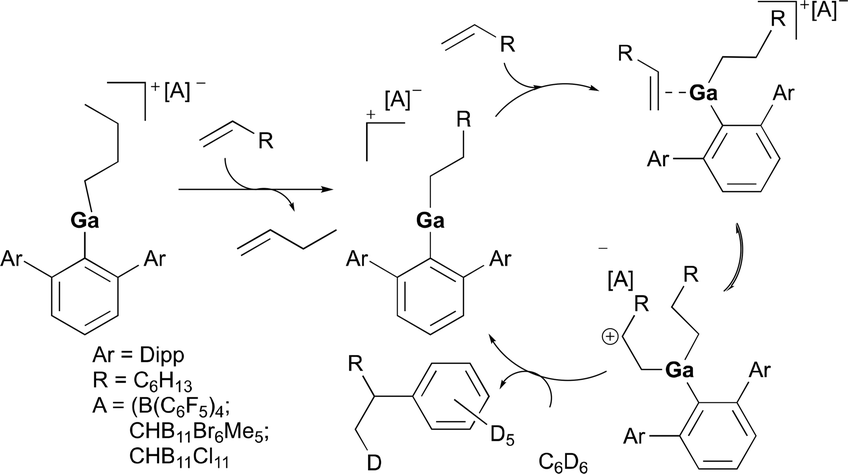
Cationic gallium complexes, 112a and 112b, were reported as catalysts for the hydroarylation of olefins by Wehmschulte and co-workers (Fig. 65).83 The cationic butyl(terphenyl)gallium compounds 112a–b were synthesized through butanide abstraction with the trityl salts of three different weakly coordinating anions [B(C6F5)4]−, [CHB11Br6Me5]−, and [CHB11Cl11]−. Both 112a and 112b catalyzed the arylation of 1-butene at a slow rate. The more thermally stable complex 112b was further studied for its reactivity towards 1-octene (Fig. 66). Interestingly, after 15 minutes from the addition of 1-octene into the solution of 112b in 6:1 C6D6 and C6D5Cl, both 1-octene and 1-butene were present indicating the dissociation of the butyl ligand, in which both signals disappeared after 48 hours. After 21 days at room temperature, no olefin signals were detected, and octyl signals were predominant which include [(2,6-Dipp2C6H3)-Ga(octyl)]+ and various octylbenzenes, indicating successful olefin/alkyl exchange followed by slow octene isomerization and benzene alkylation.
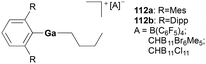 |
| | Fig. 65 Cationic gallium catalyst for benzyl alkylation. | |
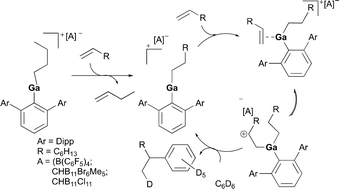 |
| | Fig. 66 Proposed mechanism of benzyl alkylation by cationic gallium complexes. | |
3.6. Cyclization of alkenes and alkynes
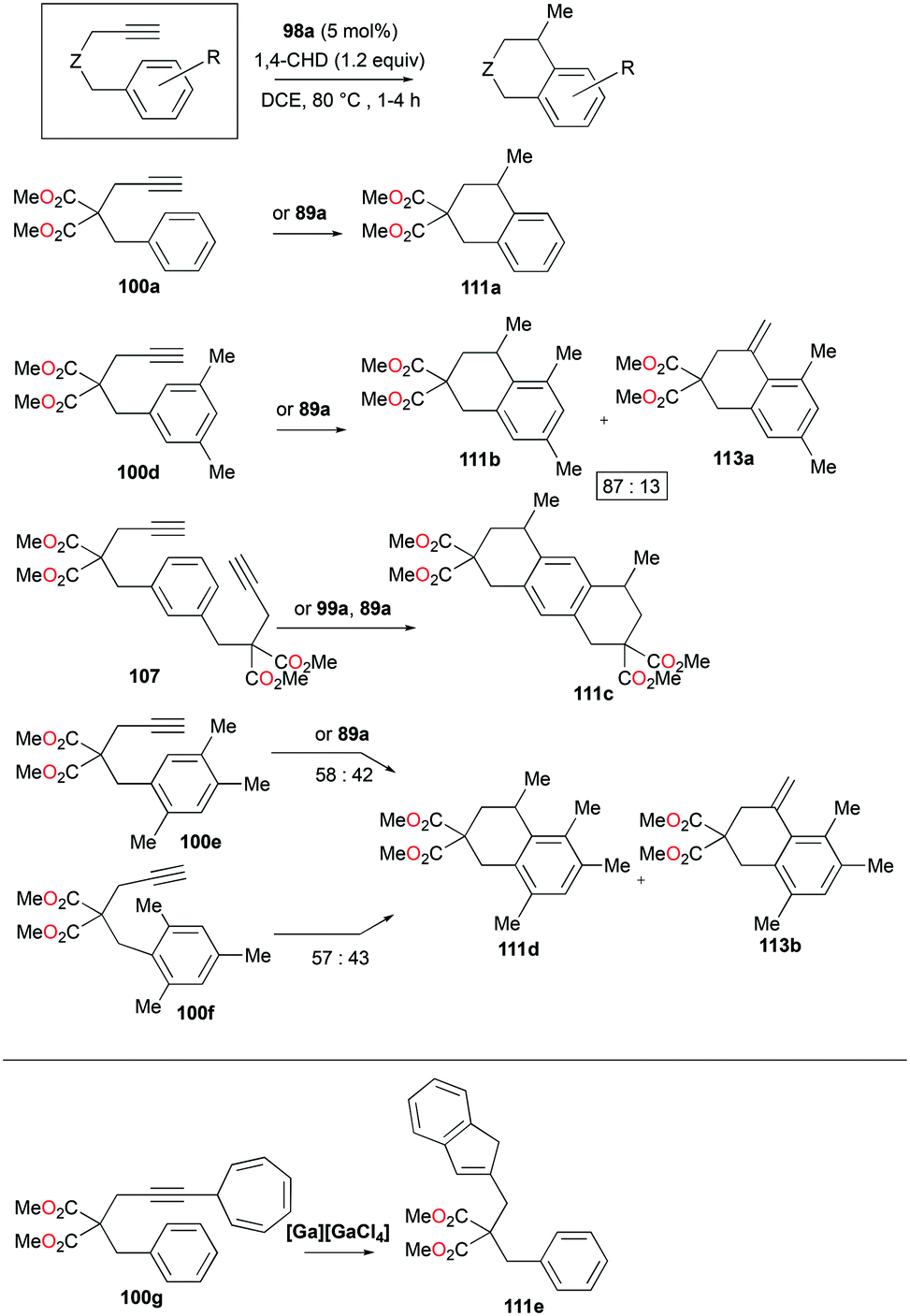
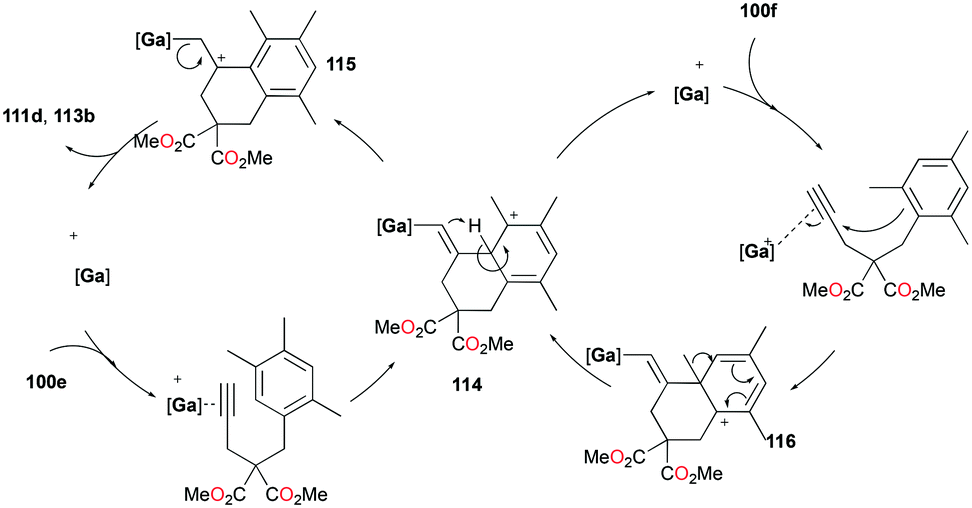
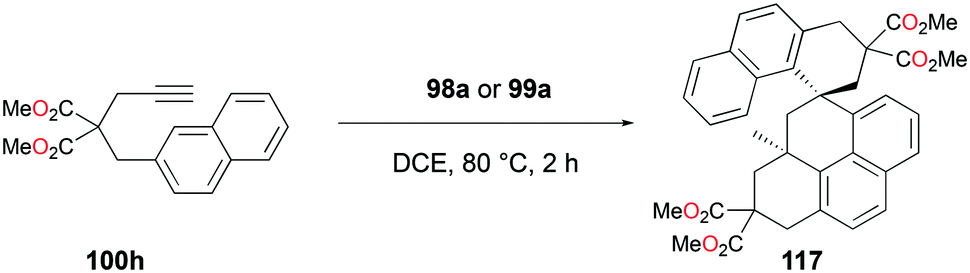
The conditions for transfer hydrogenation of alkenes can be used directly to catalyze the cyclization of arenynes. NHC-ligated cationic gallium catalysts such as 98a (Fig. 56) were also active catalysts for bimolecular or trimolecular cyclization of alkynes (Fig. 67).80 Tandem reactions for species with two alkyne groups 107 were also successful. Catalyst 89a showed higher reactivity for hydrogenative cyclizations.81 The reaction with methyl-substituted arenynes yielded regioisomers (Fig. 67).79 Surprisingly, in the reaction of 100e and 100f, the same products 111d and 113b were obtained. The reaction mechanisms were studied through DFT studies with GaCl3 as a model species (Fig. 68) and showed that the ester group in the backbone of substrates can influence the mechanism of metal-catalyzed carbocyclization reactions by Gandon and co-workers.84 In the case of 100e, the reaction involves Wheland intermediate 114 undergoing a 1,3-proton shift to give 115 and for 100f, Wheland-type complex 116 is produced after ipso attack and undergoes a 1,2-methyl shift to give 114. This mechanism suggests that the dimerization of naphthalene-derived arenyne 100h by 98a or 99a originates from the reaction of the last complex of the cycle with the product (Fig. 69).79
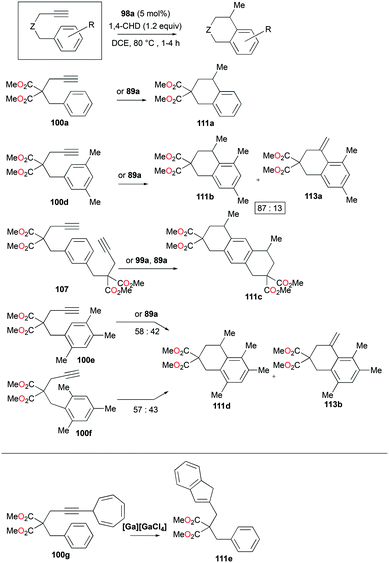 |
| | Fig. 67 Cyclization of arenynes by cationic gallium complexes. | |
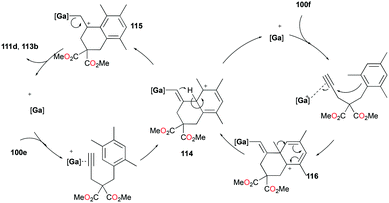 |
| | Fig. 68 Mechanism of carbocyclization of an arenyne by a cationic gallium complex. | |
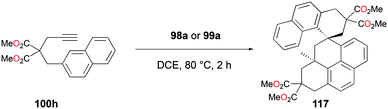 |
| | Fig. 69 Dimerization of an arenyne by cationic gallium catalysts. | |
Bour, Gandon, and co-workers reported the Ga(I)-catalyzed isomerization of 100g and their derivatives, highlighting that such a reaction can be catalyzed by the soft π-acidic behaviour of [Ga][GaCl4].85 Their subsequent study on skeletal reorganization of 7-alkynylcycloheptatriene with a 2,2-dimethyl-1,3-dioxane tether showed different ratios of product species with respect to the mol% of 89a as the catalyst.85b In contrast, complex 98a and [Ga][GaCl4] showed either degradation or no catalytic reactivity toward the same reaction.
Gandon, Bour, and co-workers tested 89a (Fig. 50) for the cycloisomerization of enyne 118 into 119 (Fig. 70).81 This reaction is known to be difficult with typical Ga(III) species as they tend to polymerize the substrate, which results in low yields of 17% and 32% for GaCl3 and 98c (Fig. 56), respectively. Cationic Ga(I) complex 89a had a yield of 45%, with a preference for producing the cycloisomerization product. Novikov, Tomilov, and co-workers reported the cycloaddition/annulation of styrylmalonates with aromatic aldehydes and styrene, using cationic gallium complexes with various counter-anions, [tBu4PcGa][A] (A = SbF6, Sb2F11, AsF6, B(ArF)4).86 In the [2 + 3] annulation process of styrylmalonate with benzaldehyde, [tBu4PcGa][Sb2F11] showed remarkably the best catalytic reactivity, with 80% yield, compared to the analogous species with other counter-anions or GaCl3. It also efficiently catalyzed the reaction with styrene with 60% yield (dr ∼4/1, 80 °C).
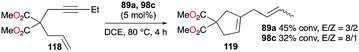 |
| | Fig. 70 Cycloisomerization of an enyne. | |
3.7. C–O bond formation
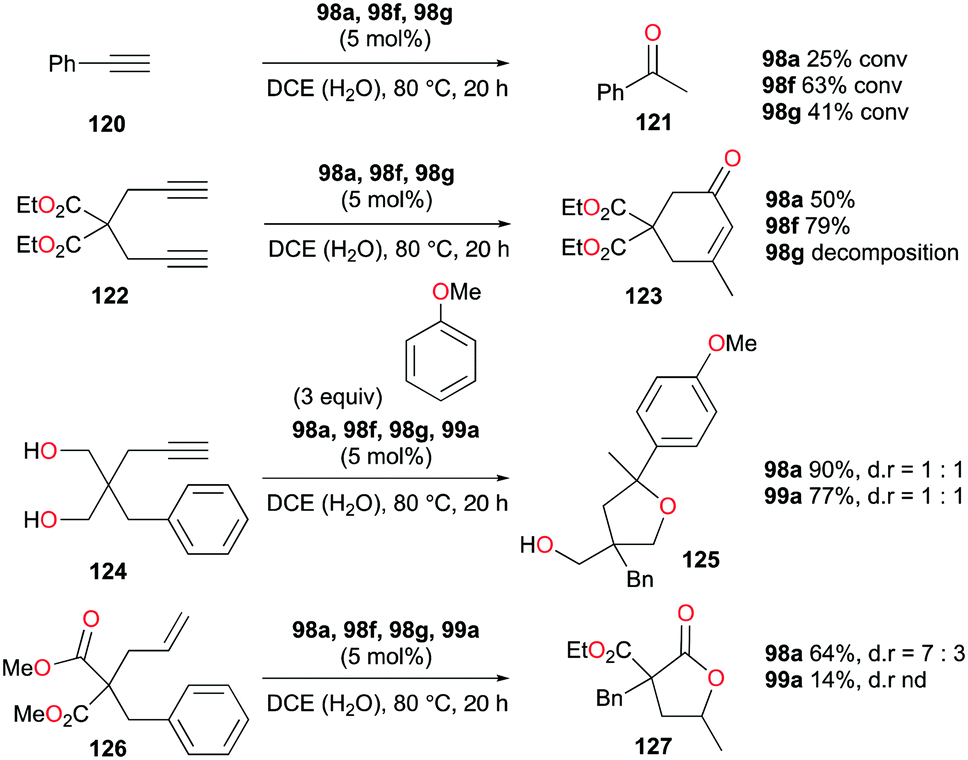
The formation of C–O bonds by activation of alkynes and alkenes by 98a (Fig. 56) was investigated by Bour, Gandon, and co-workers (Fig. 71).79 The hydration of phenylacetylene 120 to produce acetophenone 121 was catalyzed by 98a in wet DCE. The nature of the halide on the catalyst affected the catalysis significantly.87 Among the three catalysts, 98f with the bromide ligand resulted in the highest yields. The conversion of 124 into 125 and the dealkylative lactonization of complex 126 into lactone 127 through ketal species in wet DCE were also successful. The conversion of 126 into 127 was reported as a side reaction of Lewis acid-catalyzed cycloisomerization.88
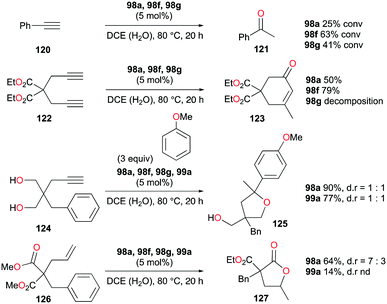 |
| | Fig. 71 Catalytic C–O bond formation by cationic NHC gallium complexes. | |
3.8. Alcohol activation
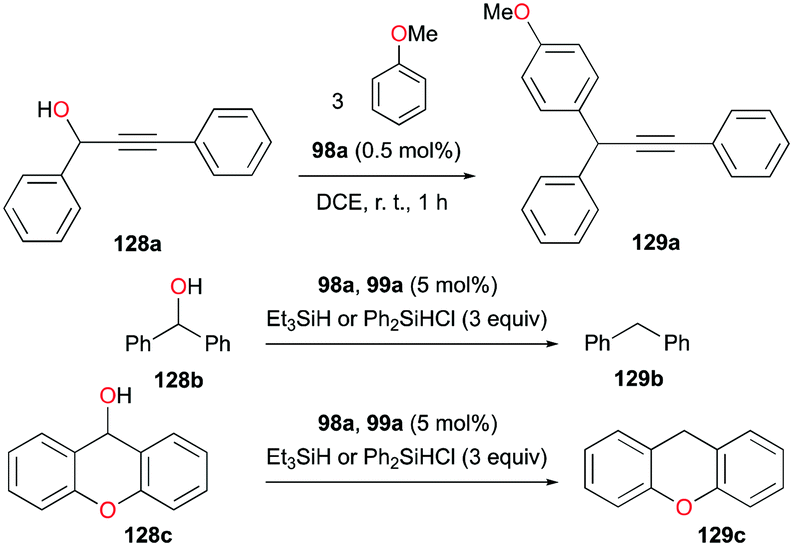
In the same study by Bour, Gandon, and co-workers, 98a (Fig. 56) catalyzed alcohol activation through C–C or C–H formation reactions (Fig. 72).79 The reaction of 128a with anisole catalyzed by 98a regioselectively yielded the Friedel–Crafts product 129a with substitution at the para position.89 In the presence of silanes Et3SiH and Ph2SiHCl, 98a and 99a promote the efficient catalytic deoxygenation of alcohols.90 The silanes used in the reaction had a significant influence on the reaction of 128b, where Ph2SiHCl was more efficient than Et3SiH. Moreover, although the reduction of other alcohols was successful, reduction of podophyllotoxin and 1-adamantanol required a longer reaction time and higher temperature, respectively.
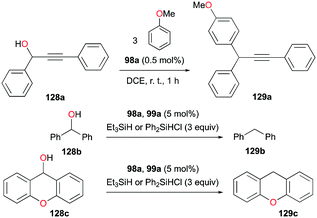 |
| | Fig. 72 Selected alcohol activation via C–C and C–H bond formation. | |
3.9. Hydrosilylation
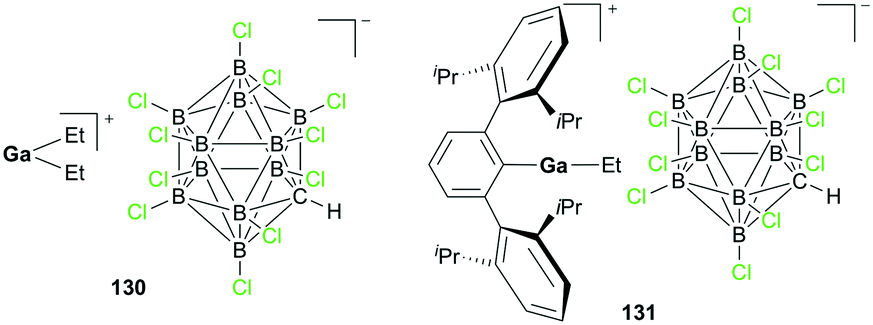
Wehmschulte and co-workers reported cationic gallium complexes 130 and 131 as catalysts for CO2 hydrosilylation (Fig. 73).91 Complexes 130 and 131 were prepared through ethide abstraction of GaEt3 and (2,6-Dipp2C6H3)–GaEt2 by silylium salt [Et3Si][CHB11Cl11], respectively. The catalytic activities of the two compounds for the reduction of CO2 were studied on the NMR scale in C6D6 or C6D5CD3. The reaction of the catalysts and 20 equiv. of Et3SiH in the presence of CO2 (ca. 1.3 atm) proceeds through multiple steps to yield the product (Et3Si)2O (Fig. 74).51b,92 C6D5CH3 and (C6D5)2CH2 were formed by the side reaction of Lewis acid-catalyzed Friedel–Crafts alkylation of C6D6 with Et3SiOCH3 and (Et3SiO)2CH2, respectively. Both successfully catalyzed the reaction, yet 130 reached only up to 82.6% conversion leaving unreacted Et3SiH even after 1 week at 80 °C, whereas 131 reached full conversion after 8 hours. While 130 catalyzed the hydrosilylation with low activity, attributed to its lower Lewis acidity and the tendency of 130 to form a polymeric structure, complex 131 was very reactive towards the formation of Et3SiOCH3.
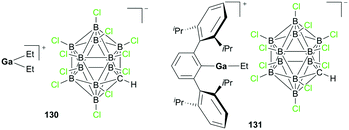 |
| | Fig. 73 Cationic gallium complexes for hydrosilylation. | |
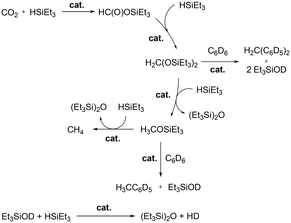 |
| | Fig. 74 Important steps and side reactions of CO2 reduction by Et3SiH catalyzed by Lewis acidic cationic gallium complexes.91 | |
Complexes 89b and 93 (Fig. 50–51) by Wehmschulte and co-workers were also investigated for their ability to catalyze the hydrosilylation of three species; benzophenone, 1-hexane, and CO2.70a Both complexes were active towards the hydrosilylation of benzophenone and 1-hexane. Benzophenone was rapidly reduced into diphenylmethane with Et3SiH; Ph2C(H)OSiEt3 was not detected as a byproduct. Complex 93 was more active for the hydrosilylation of benzophenone. For the hydrosilylation of 1-hexene with Et3SiH with 1% catalyst, the anti-Markovnikov product was obtained after several hours at 70 to 90 °C.
4. Cationic indium complexes
Compared to the lighter group 13 element gallium, simple indium halide salts as starting materials in the +1 oxidation state can be readily prepared and they are commercially available; however, they are insoluble in most common organic solvents or decompose in the presence of water or other bases.93 Although Macdonald and co-workers reported the synthesis of [In][SO3CF3] as a new source of monovalent indium which is considerably soluble and stable in various organic solvents,94 there have been a handful of both cationic In(I) and In(III) complexes with only structural information,61e,95 and the catalytic reactivity of cationic indium complexes has been rarely reported to date.
4.1. Polymerization of cyclic ethers
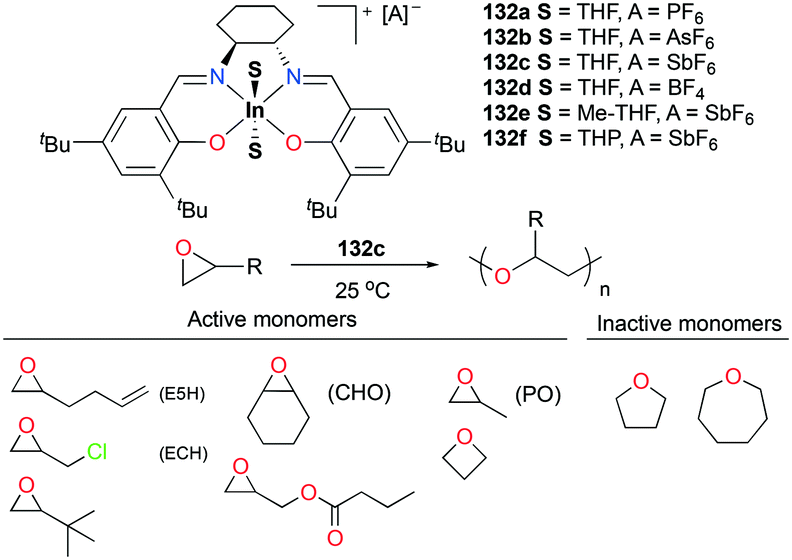
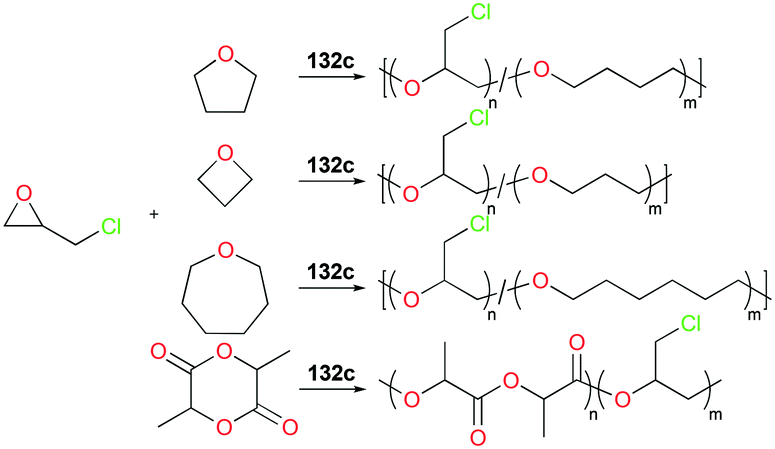
As discussed above, polymerization of epoxides such as propylene oxide (PO) has been dominated by cationic aluminum complexes among other group 13 metal cations. Mehrkhodavandi and co-workers reported the synthesis of a series of discrete cationic indium complexes (±)-[(ONNO)In(S)2][A] (S = THF and A = PF6− (132a), AsF6− (132b), SbF6− (132c), BF4− (132d); A = SbF6− and S = Me-THF, 2-methyltetrahydrofuran (132e); THP, tetrahydropyran (132f)) by salt metathesis with a variety of silver salts in corresponding solvents (Fig. 75).96 Complex 132c polymerized various functionalized epoxides to produce the respective polyethers with high conversions. Although complex 132c polymerized 200 equiv. of neat PO at 25 °C to high molecular weights PPO (Mn = 11900 g mol−1, Đ = 1.36), the polymerization reaction afforded stereo- and regio-irregular PPO, which is one of the characteristics of the cationic ring-opening mechanism. For larger cyclic ethers, 100 equiv. of oxetane was polymerized by 132c (25 °C, CH2Cl2, 24 h, Mn = 12200 g mol−1, Đ = 1.56), whereas THF and oxepane were not homopolymerizable under the same reaction conditions.
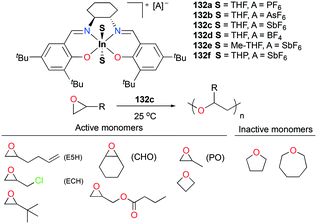 |
| | Fig. 75 A series of cationic indium salen complexes 132a–f and homopolymerization of different cyclic ethers by 132c. | |
Copolymerization of equimolar mixtures of epichlorohydrin (ECH) with THF, oxetane or oxepane with 132c at 25 °C produced the corresponding high molecular weight copolymers with low dispersities (Fig. 76). While racemic lactide (rac-LA) was not homopolymerizable by 132c, a copolymer of ECH and rac-LA was obtained by copolymerization of 200 equiv. of ECH and 653 equiv. of rac-LA in the melt, implying that rac-LA was polymerized by generated indium alkoxide species after initiation via a coordination and insertion mechanism. A counter ion effect was observed: only 132c achieved full conversion of epoxides in 24 h, whereas complexes 132a and 132b were deactivated in 100 min with conversions of 54 and 67%, respectively. The initiation activity of the complexes (132c, 132e, and 132f) for ROP of E5H varied depending on coordinated solvent molecules. The trend of initiation activity (132e > 132c > 132f) showed an inverse correlation with the Gutmann donor number of each solvent donor (THP > THF > Me-THF).97
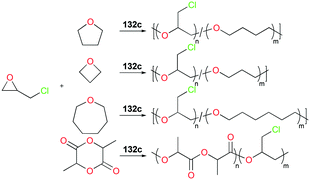 |
| | Fig. 76 Copolymerization of epichlorohydrin with different cyclic ethers or rac-LA catalyzed by 132c. | |
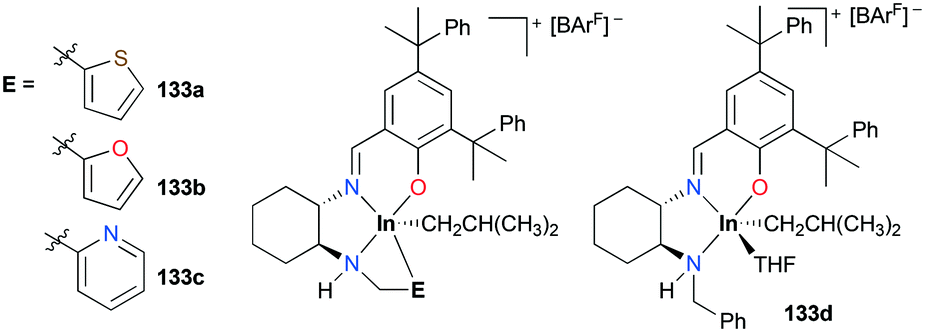
Mehrkhodavandi and co-workers reported catalytically active cationic alkyl indium complexes supported by hemi-salen type ligands bearing hemilabile pendant donor groups to show the impact of the hemilability on the stability and reactivity of the complexes towards polymerization of ECH and cyclohexene oxide (CHO) (Fig. 77).98 Cationic alkyl indium complexes 133a–c were prepared by protonation of the corresponding neutral dialkyl analogues with [HNMe2Ph][BArF] at room temperature in either coordinating or non-coordinating solvents. However, complex 133d was prepared only at −30 °C in THF due to the absence of a donor arm (Fig. 77). Although the first cationic indium complex supported by a hemilabile ligand was investigated by Mountford and coworkers,95g their complex exhibited no catalytic reactivity.
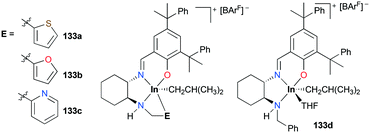 |
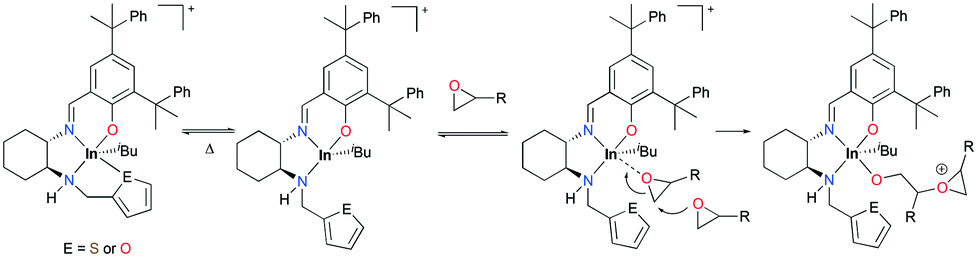
| | Fig. 77 A series of cationic indium complexes bearing a hemilabile pendant donor 133a–c and a non-hemilabile pendant donor 133d. | |
The trend of ROP of ECH via a cationic ring-opening mechanism (133d > 133a > 133b > 133c) showed an inverse correlation with the donor ability of the pendant groups (133d < 133a < 133b < 133c). Complex 133d polymerized 300 equiv. of ECH with an initiation efficiency of 99% at 25 °C (C6D6, 24 h, Mn = 20500 g mol−1, Đ = 1.50), while complex 133c did not polymerize ECH at either 25 or 80 °C. Although complexes 133a and 133b catalyzed the ROP of 300 equiv. of ECH at 25 °C, their initiation efficiencies were 61 and 39%, respectively. The dissociation of the hemilabile donor is required to initiate the ROP of ECH; therefore the initiation efficiency of each complex shows that the dissociation of the pendent arm for 133a and 133b is reversible, whereas that for 133c is not, depending on the strength of the donor (Fig. 78). However, for a highly reactive monomer, CHO was polymerized in a relatively controlled fashion at 25 °C by 133c (C6D6, 24 h, Mn = 108600 g mol−1, Đ = 1.47), whereas 133a and 133b formed polyethers with high dispersity under the same reaction conditions.
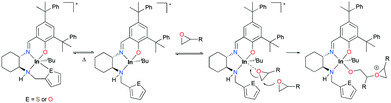 |
| | Fig. 78 Proposed mechanism for the polymerization of epoxides by 133a–b. | |
4.2. Polymerization of cyclic esters
Since the first attempt to polymerize rac-LA using cationic aluminum complexes reported by Bertrand and co-workers in 1998,21 there have been some contributions to the development of cationic aluminum and gallium complexes but cationic indium complexes have not been studied for the ring-opening polymerization of cyclic esters until recently. The first example of a cationic indium complex capable of ROP of lactide via a coordination and insertion mechanism is 133c (Fig. 77).98 Complex 133c has an isobutyl group as a nucleophilic group reactive towards the incoming rac-LA and the analysis of the polymer end-groups by MALDI-TOF mass spectrometry confirms that 133c initiated the ROP of rac-LA without an external initiator. While 250 equiv. of rac-LA was polymerized by 133c (100 °C, toluene, 24 h, Mn = 130000 g mol−1, Đ = 1.32), 133a was unreactive towards rac-LA and 133b showed only 20% conversion of rac-LA under the same reaction conditions, suggesting that the pyridyl pendant donor group decreased the electrophilicity of the indium center, making the In–C bond polarized enough to initiate the polymerization.
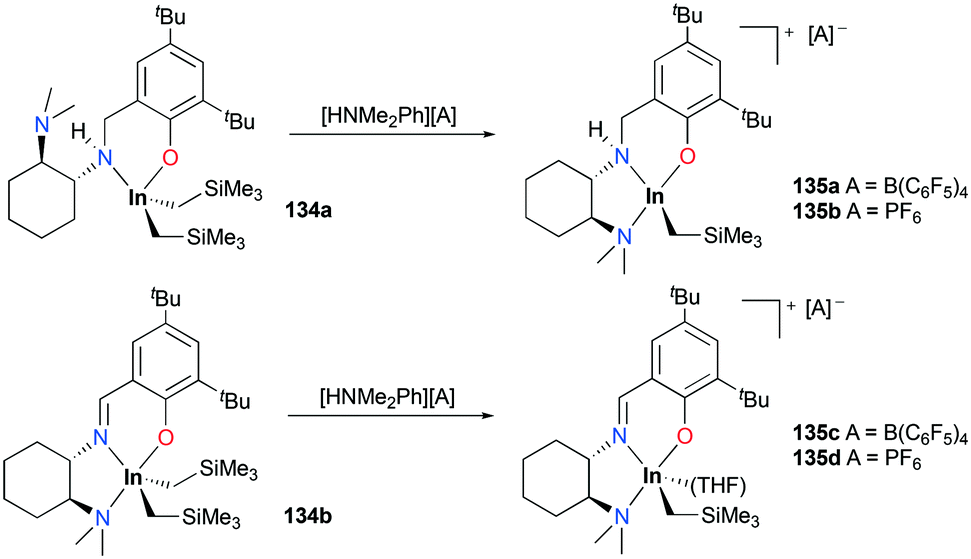
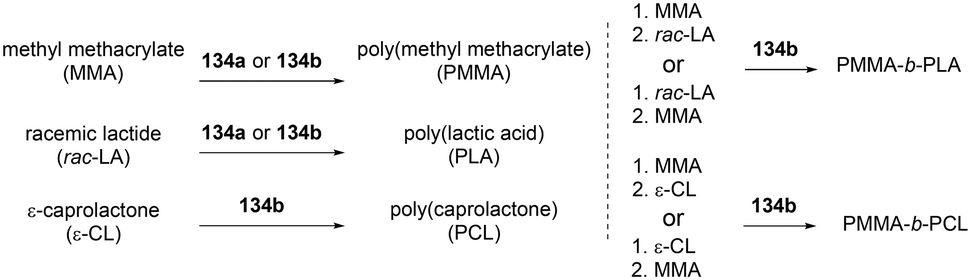
A series of cationic alkyl indium complexes supported by tridentate Schiff base ligands were reported by Mehrkhodavandi and co-workers (Fig. 79).99 The cationic alkyl indium complexes 135a–d were generated by protonation of dialkyl indium complexes 134a–b with anilinium salts [HNMe2Ph][A] (A = B(C6F5)4, PF6) at room temperature in THF.
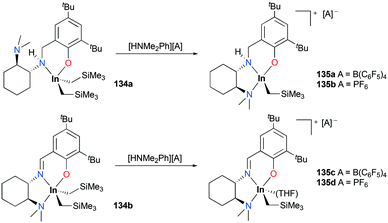 |
| | Fig. 79 Synthesis of cationic indium complexes 135a–d. | |
Both of these neutral and cationic alkyl indium complexes were investigated as catalysts for the formation of the poly(methyl methacrylate) poly(lactide) block copolymer (PMMA-b-PLA) through simple sequential addition of monomers (Fig. 80). Prior to the investigation of the block copolymerization, the neutral and cationic alkyl complexes showed different reactivities in the homopolymerization of MMA and rac-LA. Cationic complexes 135a–135c were unreactive towards methyl methacrylate (MMA) polymerization, whereas neutral complexes 134a and 134b catalyzed the polymerization of MMA (200 equiv., 60 °C, toluene, 48 h, Mn = 149000 g mol−1, Đ = 1.78; Mn = 156200 g mol−1, Đ = 1.52, respectively). While polymerization of rac-LA catalyzed by neutral complex 134a and 134b reached 97% conversion, cationic complexes 135a and 135c produced a modest amount of PLA, respectively, with 84 and 39% conversion at 100 °C in 24 h under the same conditions.
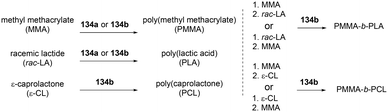 |
| | Fig. 80 Homo- and copolymerization of MMA with 134b. | |
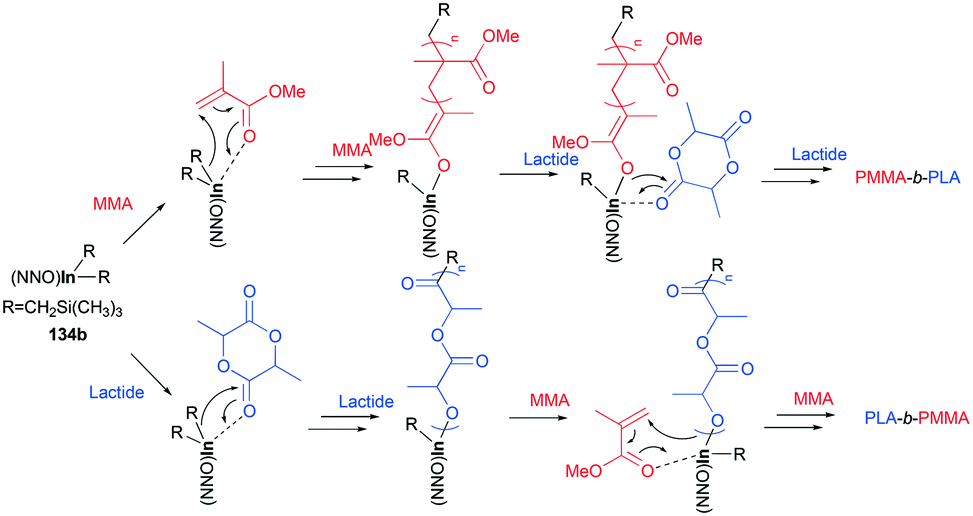
The cationic complexes did not polymerize MMA due to the insufficient nucleophilicity of –CH2Si(CH3)3; thus the cationic complexes were not effective catalysts for block copolymerization of rac-LA/ε-CL and MMA. Only the neutral complexes were found to catalyze the sequential block copolymerization of rac-LA/ε-CL and MMA via an alkyl- (–CH2Si(CH3)3) or alkoxide-(generated in situ) initiated coordination–insertion mechanism for cyclic esters, and an alkyl- or alkoxide mediated conjugate Michael addition for MMA (Fig. 81).
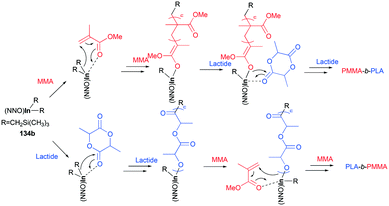 |
| | Fig. 81 Proposed mechanism for the synthesis of a block copolymer by 134b. | |
4.3. Coupling of epoxides and lactones
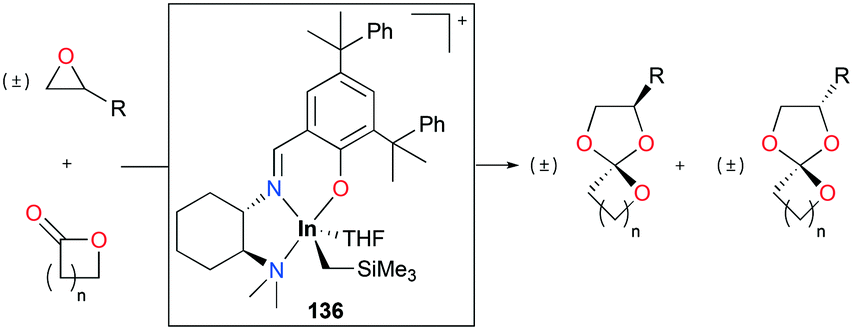
Spiro orthoesters (SOEs) are a family of expanding monomers which can overcome unfavorable volume shrinkage with zero shrinkage or even volume expansion during polymerization. SOEs have been synthesized mostly by acid-mediated coupling of epoxides and lactones. However, the poor selectivity of SOE formation, low yields, and limited scope of functionality in SOEs lead to the need for the development of new catalysts.
Mehrkhodavandi and co-workers reported cationic alkyl indium catalyst 136 for the coupling of functionalized epoxides with lactones to form spiro orthoesters (SOEs) quantitatively (Fig. 82).100 As a representative reaction, the reaction of an equimolar mixture of 1,2-epoxy-7-octene (EOE) with ε-caprolactone (ε-CL) at 60 °C in C6H6 with a 2.5% loading of 136 converted both EOE and ε-CL quantitatively to 2-(hex-5-en-1-yl)-1,4,6-trioxaspiro[4.6]undecane (SOE1) as a racemic mixture of diastereomers in 24 h. The scope of the reactivity of 136 was expanded to other epoxides bearing different functional groups and lactones with different ring sizes.
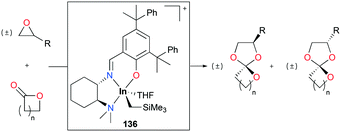 |
| | Fig. 82 Coupling of epoxides and lactones by 136. | |
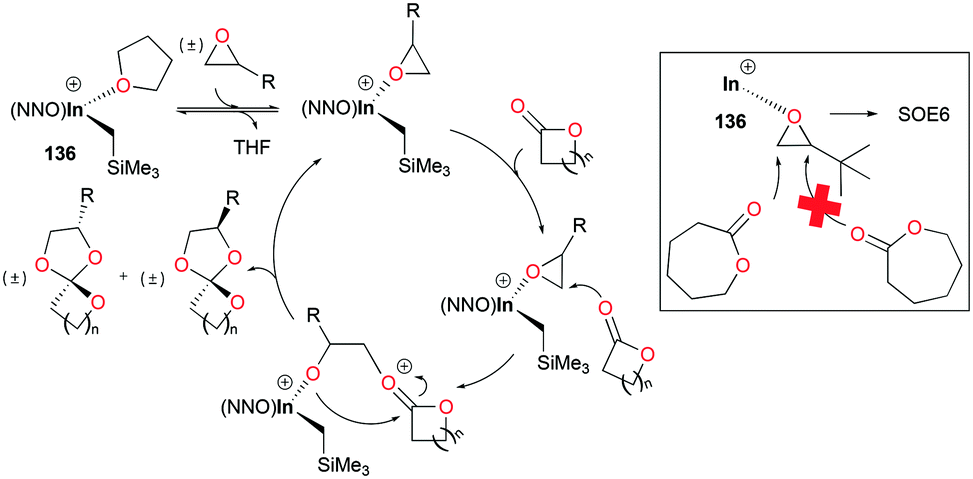
The major competing reactions in this system are the homopolymerization of epoxides and lactones. Control experiments showed that 136 did not polymerize epoxides or lactones under the reaction conditions. However, in SOE formation reactions, both epoxides and lactones were fully converted with high yields of SOEs and none of the side products were observable, suggesting the excellent selectivity of this catalytic system for SOE formation (Fig. 83). Given the general method for the synthesis of SOEs showing low yields (<50%) and by-products,101 this catalytic system shows the highest yields of SOEs (up to 90% isolated yield) and selectivity for SOE formation.
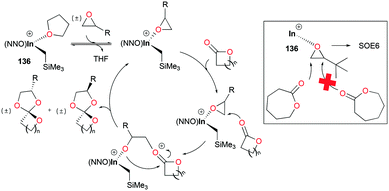 |
| | Fig. 83 Proposed mechanism for the synthesis of SOEs by 136. | |
4.4. Hydroarylation of alkynes and transfer hydrogenation of alkenes
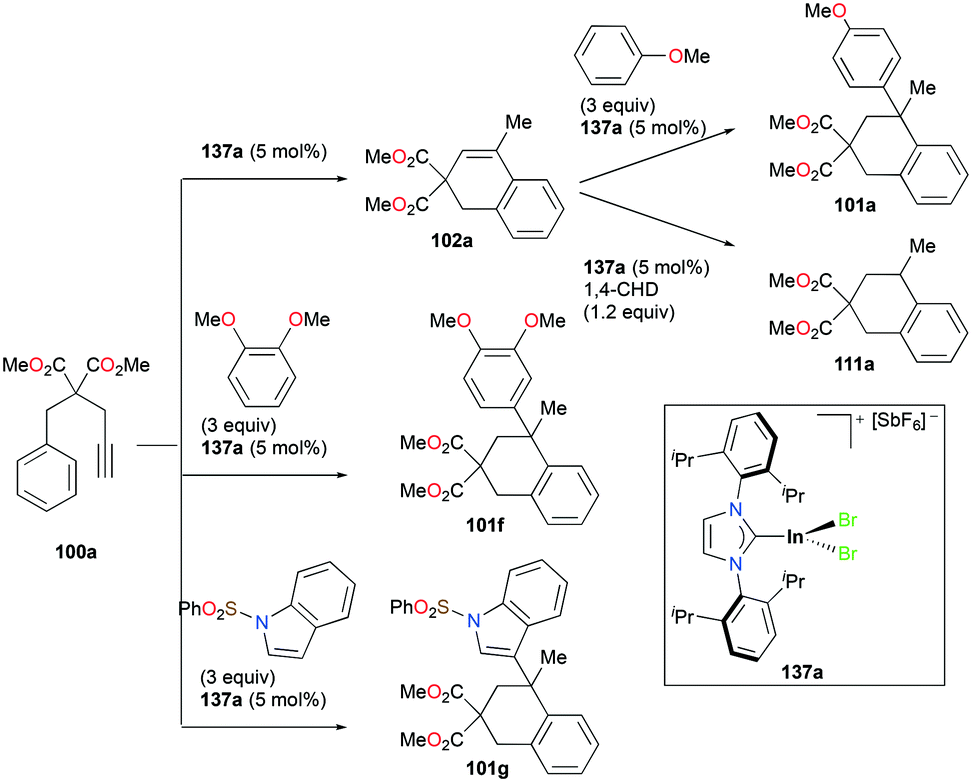
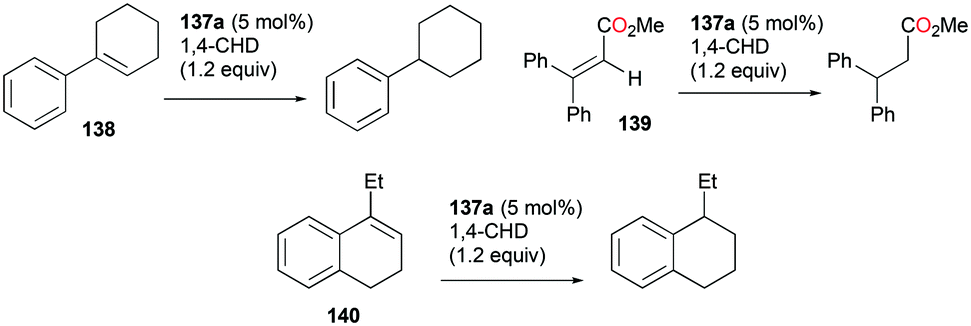
Gandon and co-workers have reported cationic indium complex 137a for the dihydroarylation of arenyne 100a (Fig. 84).102 The in situ generated complex 137a reacts with arenes to give the respective products 102a and 101a, f–g. The intramolecular hydroarylation of 100a formed minor product 102a, which underwent either a secondary intermolecular hydroarylation with anisole to form major product 101a with 62% isolated yield or transfer hydrogenation in the presence of hydrogen transfer agent 1,4-cyclohexadiene (1,4-CHD) to form product 111a. When 1,2-dimethoxybenzene or 1-(phenylsulfonyl)indole was used for dihydroarylation of 100a, full conversion was achieved faster and the isolated yields improved.
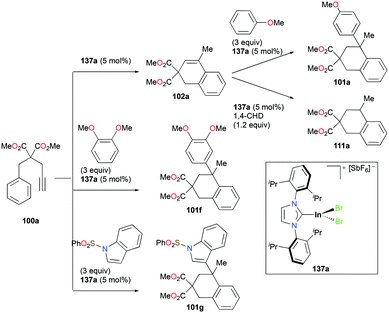 |
| | Fig. 84 Dihydroarylation of arenyne 100a by 137a. | |
In addition to dihydroarylation, complex 137a catalyzed the transfer hydrogenation of other alkenes (138–140) in the presence of 1,4-CHD (Fig. 85). Cyclohexenylbenzene 138 was fully reduced in 2 h at 20 °C. Although the hydrogenation of the acyclic substrate 139 required a higher temperature (80 °C) and longer reaction time (20 h), the yield of the corresponding product was 58%, whereas the hydrogenation of the cyclic alkene 140 took place at 20 °C with a higher yield (80%) in 1 h. The neutral indium complex (IPr)InBr3 catalyzed none of the dihydroarylation and transfer hydrogenation reactions.
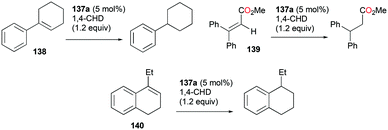 |
| | Fig. 85 Transfer hydrogenation of alkenes 138–140 by 137a. | |
4.5. Cyclization of alkenes and alkynes
The enyne 141 was cycloisomerized with cationic indium complex 137a (Fig. 86). Although the reaction achieved full conversion of 141, the products were a mixture of unidentified products containing low amounts of the desired product. When the anion was changed and [(IPr)InBr2][Al(OC(CF3)3)4] (137b) was used instead of 137a, the same reaction in DCE at 80 °C cleanly gave the desired product in 2 h with 56% isolated yield (E:Z = 6:1). This result showed a counterion effect on the reactivity of cationic NHC indium complexes.
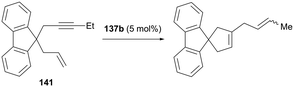 |
| | Fig. 86 Cycloisomerization of enyne 141 by 137b. | |
Allene, one of the fundamental synthetic building blocks particularly for cyclization transformation, formed six-membered heterocycles with α,β-unsaturated conjugated compounds via [4 + 2] cycloaddition. Such cyclization has been mainly achieved by chiral Lewis base catalysts with activated allenes, and there have been no reports using non-activated allenes. However, regio- and enantioselective [4 + 2] annulation of β,γ-unsaturated α-keto esters and non-activated allenes by a chiral cationic indium phosphate complex was reported by Luo and co-workers (Fig. 87).103 The in situ generated cationic indium complex [In(OP*)2][BArF] (OP* = chiral phosphate, BArF = B[3,5-(CF3)2C6H3]4) catalyzed the representative reaction of allene 143 and keto ester 144 in CH2Cl2 at room temperature, resulting in the formation of the regioisomeric adducts 145a and 145b, which are products of terminal or internal cycloaddition to the allene, respectively. The cationic indium complex generated from silver phosphate (142) gave superior results. The isolated regioisomeric products consisted of 70% desired adduct 145a (Z/E = 6:1) with 94% ee and 30% 145b (Z/E = 9:1) with 73% ee. The regio- and enantioselectivity were improved (145a/145b = 85:15; 98 and 92% ee for 145a and 145b, respectively) when the same reaction was conducted in CHCl3 instead of CH2Cl2. Control experiments revealed that none of InBr3, phosphoric acid, and AgBArF solely catalyzed the reaction, and the combination of InBr3 and phosphoric acid was also inactive towards the reaction, indicating that the cationic indium is the active center.
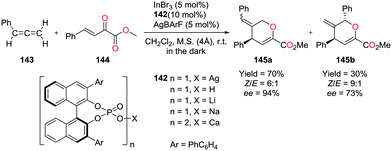 |
| | Fig. 87 Representative [4 + 2] annulation reaction of allene 143 and keto ester 144. | |
The scope of both substrates was expanded to a variety of allenes and keto esters (Fig. 88). The [4 + 2] annulation reaction of allene 143 with various β,γ-unsaturated α-keto esters or various allenes with keto ester 144 showed similar reactivities under the same reaction conditions. The desired cycloadducts were obtained in good yields with up to 99% ee and an 11:1 Z/E ratio. Overall, this suggested the marginal substituent effects of allenes and keto esters on the regioselectivity and enantioselectivity of this system.
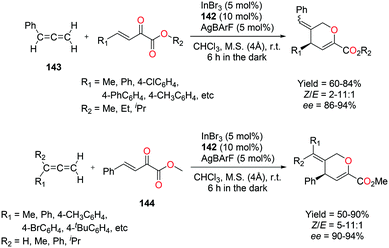 |
| | Fig. 88 [4 + 2] annulation reaction of various allenes and keto esters. | |
Corey and co-workers demonstrated the enantioselective InBr3- or InI3-catalyzed polycyclization of acetylenic substrates, leading to bi-, tri- and tetracyclic products (Fig. 89).104 The unusually high affinity between In(III) halide and carbon–carbon triple bonds activated the CC bond to initiate the enantioselective cationic polycyclization. On the basis of this study, the same group reported a catalytic system, using InI2+.105 The ligandless cation InI2+ salts [InI2][A] (A = SbF6− (146a), B[3,5-(CF3)2C6H3]4− (146b)) were prepared by salt metathesis with the corresponding silver salts. Due to their enhanced affinity for the acetylenic π-system cations, 146a–b required lower catalyst loading and were more reactive compared to InBr3 or InI3.
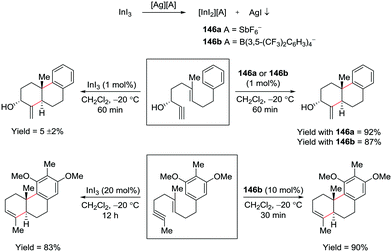 |
| | Fig. 89 Relative reactivities of InI3, 146a, and 146b in the polycyclization reactions. | |
The scope of the activation of the CC bond by InI2+ was expanded to the synthesis of spiro rings and acetylenic ketalization (Fig. 90). Each reaction formed the respective products in good yields with 10 mol% catalyst loading. It is likely that the selective activation of the CC bond in the presence of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond can be explained by an interaction between each vacant p-orbital of indium and one of the π-orbitals of the CC bond.
C bond can be explained by an interaction between each vacant p-orbital of indium and one of the π-orbitals of the CC bond.
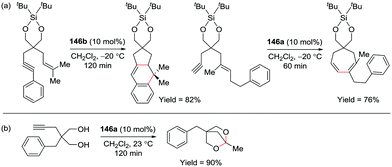 |
| | Fig. 90 (a) Synthesis of spiro rings and (b) acetylenic ketalization by 146a–b. | |
A rare example of a cationic indium complex with a lower oxidation state was reported by Bour, Gandon and co-workers. The cationic indium(I) complex, [In(PhF)2][Al(OC(CF3)3)4], catalyzed the cyclization of 7-alkynyl cycloheptatriene (100g) into an indene (111e), which can also be achieved by cationic gallium(I) complex (Fig. 67).85a While neutral indium(I) species such as InCl or InOTf were inactive towards the same reaction, the cationic indium(I) complex selectively converted 100g into 111e at 80 °C in DCE or toluene.
4.6. Carbonyl–ene reaction
Loh and co-workers reported a neutral indium complex supported by a chiral pyridine-bis(oxazoline) (pybox) ligand, which was generated in situ and catalyzed the asymmetric carbonyl–ene reactions of glyoxylates with 1,1-disubstituted or 1,1,2-trisubstituted olefins with high enantioselectivity.106 However, this system required 4–6 days to complete the reaction and a low reaction temperature (0 °C) to gain enantioselectivity. Therefore, the same group reported more reactive cationic indium–pybox complexes for the highly enantioselective carbonyl–ene reactions of ethyl glyoxylate with various olefins (Fig. 91).107 The cationic indium–pybox complexes were prepared in situ from InCl3, pybox (147a) and different silver salts (AgSbF6, AgBF4, AgPF6 and AgClO4) in 1,2-dichloroethane (DCE) at room temperature. Among those, the complex generated with AgSbF6 provided the best results. The representative reaction of ethyl glyoxylate (148) and α-methylstyrene (149) catalyzed by the cationic indium complex required a significantly shortened reaction time at room temperature and showed high enantioselectivity.
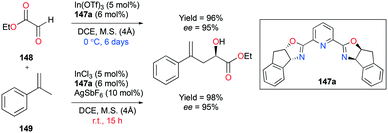 |
| | Fig. 91 Asymmetric carbonyl–ene reactions of ethyl glyoxylate (148) and α-methylstyrene (149) by the neutral or cationic indium pybox complexes. | |
The scope of the reaction was expanded to other 1,1-disubstituted or 1,1,2-trisubstituted olefins (Fig. 92). In most cases, the cationic indium complex completed the reactions in 15–20 h at room temperature with better enantioselectivities (up to 96% ee) and yields (up to 98%) of the respective homoallylic alcohols compared to the neutral analogue. In contrast to the neutral species, high enantioselectivities and yields were obtained by the cationic indium complex regardless of the presence of electron-withdrawing or donating groups in olefins.
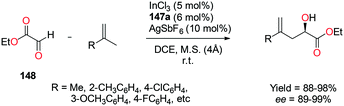 |
| | Fig. 92 Selected asymmetric carbonyl–ene reactions of ethyl glyoxylate (148) and various olefins by the cationic indium pybox complex. | |
In a subsequent effort, the same group reported the highly anti-diastereoselective and enantioselective carbonyl–ene reaction of glyoxylates and trisubstituted olefins resulting in the formation of two stereogenic centers in homoallylic alcohols by using the same cationic indium complex (Fig. 93).108 Other ligands (147b–d) were applied in this reaction; however, 147a provided the best results. The representative reaction of ethyl glyoxylate (148) and (E)-but-2-en-2-ylbenzene (150) in DCE at room temperature afforded the desired product in 48 h with 90% yield, a 94:6 diastereomeric ratio (dr) and 95% ee.
 |
| | Fig. 93 Enantioselective and anti-diastereoselective carbonyl–ene reactions of ethyl glyoxylate (148) and (E)-but-2-en-2-ylbenzene (150) by the cationic indium pybox complexes. | |
Reactions of other glyoxylates with different ester substituents and 150 were conducted to examine the ester substituent effect under the same reaction conditions. While the reactions with glyoxylates possessing linear ester substituents such as methyl, ethyl and n-butyl groups showed consistently high diastereoselectivities (92:8–94:6 dr), enantioselectivities (89–95% ee) and yields (84–90%), the reaction with bulky isopropyl glyoxylate obtained a decreased diastereoselectivity (85:15 dr) and yield (75%).
The scope of trisubstituted alkenes was expanded with ethyl glyoxylate for this asymmetric glyoxylate–ene reaction (Fig. 94). In most cases, the cationic indium complex catalyzed the formation of desired homoallylic alcohols in good yields (up to 90%) with high diastereoselectivities (up to 99:1 dr) and enantioselectivities (up to 99%). As in previous work, a tolerance towards the electron-donating and withdrawing groups in alkenes was observed. Furthermore, the investigation of the steric effect of olefins by changing R2 to a propyl group or R3 to an ethyl group showed still high selectivities (99:1 dr, 96% ee; 91:9 dr, 92% ee, respectively). However, when cyclopentenylbenzene was employed with ethyl glyoxylate, the resulting product was obtained in 75% yield with a lowered dr (88:12) and high enantioselectivity (99% ee).
 |
| | Fig. 94 Selected enantioselective and anti-diastereoselective carbonyl–ene reactions of ethyl glyoxylate (148) and various trisubstituted alkenes by the cationic indium pybox complex. | |
The effect of the steric configuration of olefins was also investigated using the (Z) isomer of 150. While the reaction of the (E) isomer of 150 and 148 afforded the corresponding product in 90% yield with a 94:6 dr and 95% ee, the use of the (Z) isomer of 150 in the same reaction gave the product in only 17% yield with the same selectivities, suggesting the need for a proton in the β-cis substituent for this system (Fig. 95).
 |
| | Fig. 95 Investigation of the stereo effect of trisubstituted alkenes for carbonyl–ene reactions. | |
4.7. Other cationic indium complexes in catalysis
Jordan and co-workers reported a series of cationic indium complexes based on [(iPr2-ATI)In(Me)][A] (iPr2-ATI = N,N′-diisopropylaminotroponiminate; A = B(C6F5)4− (153a), MeB(C6F5)3− (153b))109 and compared the catalytic reactivities of 153a with those of cationic aluminum analogues (Fig. 96).22,32,61c Complex 153a was prepared by the reaction of 151 with [Ph3C][B(C6F5)4] followed by thermolysis of 152. Complex 153a was formed via electrophilic addition of CPh3+ to 151 prior to methyl abstraction by dissociation of CPh3+ from 152, whereas 153b was formed directly via methyl abstraction of 151 by B(C6F5)3. The addition of an appropriate Lewis base to 153a and 153b yielded [(iPr2-ATI)In(Me)(L)][B(C6F5)4] (L = NMe2Ph (154a), CH3CN (154b), Me2CO (154c), PMe3 (154d)) and [(iPr2-ATI)In(Me)(L)][MeB(C6F5)3] (L = NMe2Ph (154e), PMe3 (154f)), respectively.
 |
| | Fig. 96 Synthesis of cationic indium complexes 152–154f. | |
The catalytic reactivity of 153a, in particular, was investigated. However, 153a was unreactive towards ethylene, tBuCCH, H2 and CO, while analogous cationic aluminum complexes [(iPr2-ATI)Al(R)][B(C6F5)4] (R = Et, iBu) catalyzed the dimerization of tBuCCH by an insertion/s-bond metathesis mechanism (C6D5Cl, 23 °C, ca. 4 t.o./h, >90% selectivity for the product) (Fig. 44) and the polymerization of isobutylene and propylene oxide was initiated by these cationic aluminum complexes, respectively. Only trace activity for isobutylene polymerization by 153a was observable. Details of the polymerization with 153a were not reported. These differences in the reactivity illustrate that alkyl indium complexes are generally less reactive than alkyl aluminum analogues due to the lower Lewis acidity and In–C bond polarity of indium complexes.
Wehmschulte and co-workers reported the arene-solvated indium(I) species [In(C7H8)3][CHB11Cl11] (155a) and [In(C6H5Br)1.5][CHB11Cl11] (155b) obtained by a redox reaction of indium powder and the silver salt Ag(CHB11Cl11) at 80 °C in toluene or bromobenzene (Fig. 97(a)).95p The structure of 155a consisted of an In(I) cation coordinated to three toluene molecules and weakly to one Cl of the anion. Contrarily, the structure of 155b comprised only 1.5 molecules of bromobenzene per In(I) cation on average, and four distinct In(I) coordination environments were observed. One of them is depicted in Fig. 97(a).
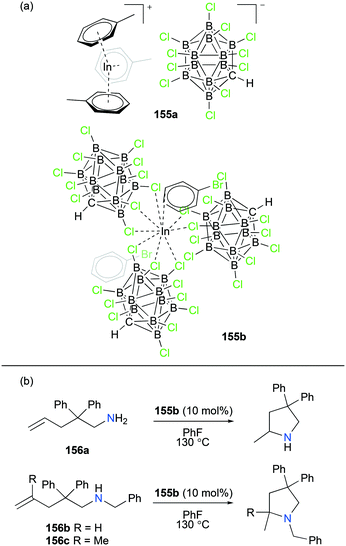 |
| | Fig. 97 (a) The arene-solvated indium(I) species (155a–b) and (b) intramolecular hydroamination of primary and secondary aminopentenes 156a–c with 155b. | |
In particular, the catalytic reactivity of the cationic indium(I) species 155b was investigated for the intramolecular hydroamination of aminopentenes (156a–c) in fluorobenzene at 130 °C (Fig. 97(b)). The primary amine 156a was converted to its corresponding heterocyclic product with 95% conversion in 25 hours. However, the activity of 155b was significantly slower for the secondary amines, for example, 97% conversion of 156c was achieved in 94 hours.
Conclusions
A number of discrete cationic group 13 complexes have been generated via various synthetic routes and their reactivity has been explored. An increasing interest in main group metals in catalysis has recently led to the development of discrete cationic group 13 complexes supported by deliberately designed ligands and the investigation of their catalytic reactivities.
Although cationic species show superior reactivities compared to neutral analogues in most cases and their enhanced Lewis acidity allows them to be potentially excellent catalysts in Lewis acid-mediated transformations as shown in the present review, the scope of reactivity is still narrow. There are well-studied applications, for instance, the reactivity and mechanism study of carbonylation of epoxides by Coates' cationic aluminum complexes showing excellent selectivity; however, more efforts should be made to investigate and expand the reactivities of cationic group 13 complexes for the economic production of useful molecules and delicately designed polymers for complex applications such as the transformation of CO2 and synthesis of functionalized copolymers. In this respect, appropriately designed ligands will play a role in the selectivity of catalysts and influence the catalytic activity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge support from the Natural Sciences and Engineering Research Council of Canada.
Notes and references
-
(a) N. Chatani, H. Inoue, T. Kotsuma and S. Murai, J. Am. Chem. Soc., 2002, 124, 10294–10295 CrossRef CAS;
(b) H. Inoue, N. Chatani and S. Murai, J. Org. Chem., 2002, 67, 1414–1417 CrossRef CAS;
(c) V. Mamane, P. Hannen and A. Furstner, Chem. – Eur. J., 2004, 10, 4556–4575 CrossRef CAS;
(d) S. M. Kim, S. I. Lee and Y. K. Chung, Org. Lett., 2006, 8, 5425–5427 CrossRef CAS;
(e) S. I. Lee, S. H. Sim, S. M. Kim, K. Kim and Y. K. Chung, J. Org. Chem., 2006, 71, 7120–7123 CrossRef CAS;
(f) E. M. Simmons and R. Sarpong, Org. Lett., 2006, 8, 2883–2886 CrossRef CAS;
(g) H. J. Li, R. Guillot and V. Gandon, J. Org. Chem., 2010, 75, 8435–8449 CrossRef CAS.
-
S. Dagorne and S. Bellemin-Laponnaz, in The Group 13 Metals Aluminium, Gallium, Indium, and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, Wiley, West Sussex, UK, 2011, p. 654 Search PubMed.
-
(a) K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS;
(b) I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS;
(c) D. A. Evans, J. M. Janey, N. Magomedov and J. S. Tedrow, Angew. Chem., Int. Ed., 2001, 40, 1884–1888 CrossRef CAS.
-
(a) S. Dagorne, M. Normand, E. Kirillov and J. F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS;
(b) A. Gualandi, F. Calogero, S. Potenti and P. G. Cozzi, Molecules, 2019, 24, 1716 CrossRef CAS.
-
(a) P. Cintas, Synlett, 1995, 1087–1096 CrossRef CAS;
(b) J. Auge, N. Lubin-Germain and J. Uziel, Synthesis, 2007, 1739–1764 CrossRef CAS;
(c) Z.-L. Shen, S.-Y. Wang, Y.-K. Chok, Y.-H. Xu and T.-P. Loh, Chem. Rev., 2013, 113, 271–401 CrossRef CAS;
(d) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS;
(e) I. Yu, T. Ebrahimi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Dalton Trans., 2015, 44, 14248–14254 RSC;
(f) T. Ebrahimi, D. C. Aluthge, B. O. Patrick, S. G. Hatzikiriakos and P. Mehrkhodavandi, ACS Catal., 2017, 7, 6413–6418 CrossRef CAS.
-
(a) U. Schneider and S. Kobayashi, Acc. Chem. Res., 2012, 45, 1331–1344 CrossRef CAS;
(b) C. Bour and V. Gandon, Coord. Chem. Rev., 2014, 279, 43–57 CrossRef CAS;
(c) C. Fliedel, G. Schnee, T. Aviles and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63–86 CrossRef CAS;
(d) L. C. Wilkins and R. L. Melen, Coord. Chem. Rev., 2016, 324, 123–139 CrossRef CAS;
(e) W. L. Li, X. L. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017, 350, 14–29 CrossRef CAS;
(f) C. R. Goldsmith, Coord. Chem. Rev., 2018, 377, 209–224 CrossRef CAS;
(g) A. Dogra and N. Gupta, ChemistrySelect, 2019, 4, 10452–10465 CrossRef CAS.
- D. A. Atwood, Coord. Chem. Rev., 1998, 176, 407–430 CrossRef CAS.
-
(a) S. H. Strauss, Chem. Rev., 1993, 93, 927–942 CrossRef CAS;
(b) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS;
(c) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC.
- S. Dagorne and D. A. Atwood, Chem. Rev., 2008, 108, 4037–4071 CrossRef CAS.
- S. Dagorne and R. Wehmschulte, ChemCatChem, 2018, 10, 2509–2520 CrossRef CAS.
-
(a) D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37–52 CrossRef CAS;
(b) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC;
(c) S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC;
(d) A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS;
(e) J. P. Sestelo, L. A. Sarandeses, M. M. Martinez and L. Alonso-Maranon, Org. Biomol. Chem., 2018, 16, 5733–5747 RSC.
- D. A. Atwood, J. A. Jegier and D. Rutherford, J. Am. Chem. Soc., 1995, 117, 6779–6780 CrossRef CAS.
-
(a) D. A. Atwood, J. A. Jegier and D. Rutherford, Inorg. Chem., 1996, 35, 63–70 CrossRef CAS;
(b) J. A. Jegier and D. A. Atwood, Inorg. Chem., 1997, 36, 2034–2039 CrossRef CAS;
(c) J. A. Jegier, M.-Á. Muñoz-Hernández and D. A. Atwood, J. Chem. Soc., Dalton Trans., 1999, 2583–2588 RSC;
(d) S. Liu, M.-A. Munoz-Hernandez and D. A. Atwood, J. Organomet. Chem., 2000, 596, 109–114 CrossRef CAS;
(e) M.-A. Munoz-Hernandez, M. L. McKee, T. S. Keizer, B. C. Yearwood and D. A. Atwood, J. Chem. Soc., Dalton Trans., 2002, 410–414 RSC.
-
(a) S. Dagorne, L. Lavanant, R. Welter, C. Chassenieux, P. Haquette and G. Jaouen, Organometallics, 2003, 22, 3732–3741 CrossRef CAS;
(b) S. Dagorne, C. R. Chim., 2006, 9, 1143–1150 CrossRef CAS.
- S. Dagorne, I. Janowska, R. Welter, J. Zakrzewski and G. Jaouen, Organometallics, 2004, 23, 4706–4710 CrossRef CAS.
- J.-T. Issenhuth, J. Pluvinage, R. Welter, S. Bellemin-Laponnaz and S. Dagorne, Eur. J. Inorg. Chem., 2009, 31, 4701–4709 CrossRef.
- M. Haddad, M. Laghzaoui, R. Welter and S. Dagorne, Organometallics, 2009, 28, 4584–4592 CrossRef CAS.
- L. S. Baugh and J. A. Sissano, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1633–1651 CrossRef CAS.
- N. Nakata, Y. Saito and A. Ishii, Organometallics, 2014, 33, 1840–1844 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- N. Emig, H. Nguyen, H. Krautscheid, R. Réau, J.-B. Cazaux and G. Bertrand, Organometallics, 1998, 17, 3599–3608 CrossRef CAS.
- A. V. Korolev, I. A. Guzei and R. F. Jordan, J. Am. Chem. Soc., 1999, 121, 11605–11606 CrossRef CAS.
- J. Lewiński, P. Horeglad, M. Dranka and I. Justyniak, Inorg. Chem., 2004, 43, 5789–5791 CrossRef.
- S. Milione, F. Grisi, R. Centore and A. Tuzi, Organometallics, 2006, 25, 266–274 CrossRef CAS.
- S. Dagorne, F. Le Bideau, R. Welter, S. Bellemin-Laponnaz and A. Maisse-François, Chem. – Eur. J., 2007, 13, 3202–3217 CrossRef CAS.
- A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, C. Alonso-Moreno, J. A. Castro-Osma, I. Márquez-Segovia, L. F. Sánchez-Barba, A. M. Rodríguez and J. C. Garcia-Martinez, Organometallics, 2011, 30, 1507–1522 CrossRef CAS.
- M. Lamberti, I. D'Auria, M. Mazzeo, S. Milione, V. Bertolasi and D. Pappalardo, Organometallics, 2012, 31, 5551–5560 CrossRef CAS.
- G. Schnee, A. Bolley, C. Gourlaouen, R. Welter and S. Dagorne, J. Organomet. Chem., 2016, 820, 8–13 CrossRef CAS.
- J. Kiriratnikom, S. Chotchatchawankul, S. Haesuwannakij, S. Kiatisevi and K. Phomphrai, New J. Chem., 2018, 42, 8374–8383 RSC.
- H. Plommer, J. N. Murphy, L. N. Dawe and F. M. Kerton, Inorg. Chem., 2019, 58, 5253–5264 CrossRef CAS.
- M. Bochmann and D. M. Dawson, Angew. Chem., Int. Ed. Engl., 1996, 35, 2226–2228 CrossRef CAS.
-
(a) E. Ihara, V. G. Young and R. F. Jordan, J. Am. Chem. Soc., 1998, 120, 8277–8278 CrossRef CAS;
(b) A. V. Korolev, E. Ihara, I. A. Guzei, V. G. Young and R. F. Jordan, J. Am. Chem. Soc., 2001, 123, 8291–8309 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- M. Bruce, V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 2523–2524 RSC.
- P. A. Cameron, V. C. Gibson, C. Redshaw, J. A. Segal, M. D. Bruce, A. J. P. White and D. J. Williams, Chem. Commun., 1999, 1883–1884 RSC.
- B. Lian, H. Ma, T. P. Spaniol and J. Okuda, Dalton Trans., 2009, 9033–9042 RSC.
- S. Dagorne, M. Bouyahyi, J. Vergnaud and J.-F. Carpentier, Organometallics, 2010, 29, 1865–1868 CrossRef CAS.
-
(a) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS;
(b) Y. D. Y. L. Getzler, V. Kundnani, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 6842–6843 CrossRef CAS;
(c) Y. D. Y. L. Getzler, M. Viswanath, E. B. Lobkovsky and G. W. Coates, Pure Appl. Chem., 2004, 76, 557–564 CAS;
(d) T. L. Church, Y. D. Y. L. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125–10133 CrossRef CAS;
(e) T. L. Church, C. M. Byrne, E.
B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 8156–8162 CrossRef CAS;
(f) J. M. Rowley, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 4948–4960 CrossRef CAS;
(g) M. Mulzer, B. T. Whiting and G. W. Coates, J. Am. Chem. Soc., 2013, 135, 10930–10933 CrossRef CAS;
(h) M. Mulzer, J. R. Lamb, Z. Nelson and G. W. Coates, Chem. Commun., 2014, 50, 9842–9845 RSC;
(i) M. Mulzer and G. W. Coates, J. Org. Chem., 2014, 79, 11851–11862 CrossRef CAS;
(j) M. Mulzer, B. J. Tiegs, Y. Wang, G. W. Coates and G. A. O'Doherty, J. Am. Chem. Soc., 2014, 136, 10814–10820 CrossRef CAS;
(k) M. Mulzer, W. C. Ellis, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 1928–1933 RSC;
(l) A. K. Hubbell, A. M. LaPointe, J. R. Lamb and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 2474–2480 CrossRef CAS;
(m) E. W. Dunn, J. R. Lamb, A. M. LaPointe and G. W. Coates, ACS Catal., 2016, 6, 8219–8223 CrossRef CAS.
- Q. Chen, M. Mulzer, P. Shi, P. J. Beuning, G. W. Coates and G. A. O'Doherty, Org. Lett., 2011, 13, 6592–6595 CrossRef CAS.
- V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2781–2784 CrossRef CAS.
- M.-A. Muñoz-Hernandez, S. Parkin, B. Yearwood, P. Wei and D. A. Atwood, J. Chem. Crystallogr., 2000, 30, 215–218 CrossRef.
- N. Funken, Y.-Q. Zhang and A. Gansäuer, Chem. – Eur. J., 2017, 23, 19–32 CrossRef CAS.
-
(a) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1994, 59, 7195–7196 CrossRef CAS;
(b) J. Prandi, J. L. Namy, G. Menoret and H. B. Kagan, J. Organomet. Chem., 1985, 285, 449–460 CrossRef CAS;
(c) C.-L. Chang, M. P. Kumar and R.-S. Liu, J. Org. Chem., 2004, 69, 2793–2796 CrossRef CAS.
- J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 15049–15054 CrossRef CAS.
- J. R. Lamb, Y. Jung and G. W. Coates, Org. Chem. Front., 2015, 2, 346–349 RSC.
- E. T. McBee, W. L. Dilling and H. P. Braendlin, J. Org. Chem., 1962, 27, 2704–2710 CrossRef CAS.
- J. Koller and R. G. Bergman, Organometallics, 2012, 31, 2530–2533 CrossRef CAS.
- R. Kannan, R. Chambenahalli, S. Kumar, A. Krishna, A. P. Andrews, E. D. Jemmis and A. Venugopal, Chem. Commun., 2019, 55, 14629–14632 RSC.
- K. Jakobsson, T. Chu and G. I. Nikonov, ACS Catal., 2016, 6, 7350–7356 CrossRef CAS.
- K. Yamamoto and M. Takemae, Synlett, 1990, 5, 259–260 CrossRef.
-
(a) M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS;
(b) R. J. Wehmschulte, M. Saleh and D. R. Powell, Organometallics, 2013, 32, 6812–6819 CrossRef CAS;
(c) M. Saleh, D. R. Powell and R. J. Wehmschulte, Organometallics, 2017, 36, 4810–4815 CrossRef CAS.
- M. K. Sharma, S. Sinhababu, G. Mukherjee, G. Rajaraman and S. Nagendran, Dalton Trans., 2017, 46, 7672–7676 RSC.
- C. A. Koellner, N. A. Piro, W. S. Kassel, C. R. Goldsmith and C. R. Graves, Inorg. Chem., 2015, 54, 7139–7141 CrossRef CAS.
- M. L. McKee and C. R. Goldsmith, Inorg. Chem., 2014, 53, 318–326 CrossRef CAS.
- J. R. Lamb, A. K. Hubbell, S. N. MacMillan and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 8029–8035 CrossRef CAS.
- K.-C. Kim, C. A. Reed, G. S. Long and A. Sen, J. Am. Chem. Soc., 2002, 124, 7662–7663 CrossRef CAS.
-
(a) Y. Kuniya and M. Hosaka, J. Cryst. Growth, 1975, 28, 385–391 CrossRef CAS;
(b) C. Dohmeier, D. Loos and H. Schnockel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129–149 CrossRef CAS.
- S. Ghosh, R. R. Gowda, R. Jagan and D. Chakraborty, Dalton Trans., 2015, 44, 10410–10422 RSC.
- S. Tang, J. Monot, A. El-Hellani, B. Michelet, R. Guillot, C. Bour and V. Gandon, Chem. – Eur. J., 2012, 18, 10239–10243 CrossRef CAS.
- D. A. Atwood, R. A. Jones, A. H. Cowley, S. G. Bott and J. L. Atwood, J. Organomet. Chem., 1992, 425, C1–C3 CrossRef CAS.
-
(a) J. A. C. Clyburne, R. D. Culp, S. Kamepalli, A. H. Cowley and A. Decken, Inorg. Chem., 1996, 35, 6651–6655 CrossRef CAS;
(b) S. Dagorne, I. A. Guzei, M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 2000, 122, 274–289 CrossRef CAS;
(c) A. V. Korolev, F. Delpech, S. Dagorne, I. A. Guzei and R. F. Jordan, Organometallics, 2001, 20, 3367–3369 CrossRef CAS;
(d) X. Sava, M. Melaimi, N. Mezailles, L. Ricard, F. O. Mathey and P. Le Floch, New J. Chem., 2002, 26, 1378–1383 RSC;
(e) N. R. Bunn, S. Aldridge, D. L. Kays, N. D. Coombs, A. Rossin, D. J. Willock, J. K. Day, C. Jones and L.-L. Ooi, Organometallics, 2005, 24, 5891–5900 CrossRef CAS;
(f) B. Buchin, C. Gemel, T. Cadenbach, R. Schmid and R. A. Fischer, Angew. Chem., Int. Ed., 2006, 45, 1074–1076 CrossRef CAS;
(g) A. M. Felix, D. A. Dickie, I. S. Horne, G. Page and R. A. Kemp, Inorg. Chem., 2012, 51, 4650–4662 CrossRef CAS;
(h) A. El-Hellani, J. Monot, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 506–514 CrossRef CAS;
(i) K. Glootz, D. Kratzert and I. Krossing, Z. Anorg. Allg. Chem., 2020, 646, 523–525 CrossRef CAS;
(j) I. A. Guzei, S. Dagorne and R. F. Jordan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 143–144 CrossRef CAS;
(k) K. Glootz, D. Kratzert, D. Himmel, A. Kastro, Z. Yassine, T. Findeisen and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 14203–14206 CrossRef CAS.
- R. J. Wehmschulte, J. M. Steele, J. D. Young and M. A. Khan, J. Am. Chem. Soc., 2003, 125, 1470–1471 CrossRef CAS.
- S. Dagorne, S. Bellemin-Laponnaz, A. Maisse-François, M. N. Rager, L. Jugé and R. Welter, Eur. J. Inorg. Chem., 2005, 20, 4206–4214 CrossRef.
- S. Dagorne, S. Bellemin-Laponnaz and R. Welter, Organometallics, 2004, 23, 3053–3061 CrossRef CAS.
- B. Goswami, R. Yadav, C. Schoo and P. W. Roesky, Dalton Trans., 2020, 49, 675–681 RSC.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS.
- A. M. Dąbrowska, A. Hurko, M. Dranka, V. Varga, M. Urbańczyk and P. Horeglad, J. Organomet. Chem., 2017, 840, 63–69 CrossRef.
-
(a) M. R. Lichtenthaler, A. Higelin, A. Kraft, S. Hughes, A. Steffani, D. A. Plattner, J. M. Slattery and I. Krossing, Organometallics, 2013, 32, 6725–6735 CrossRef CAS;
(b) J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228–3231 CrossRef CAS.
- M. R. Lichtenthaler, S. Maurer, R. J. Mangan, F. Stahl, F. Mönkemeyer, J. Hamann and I. Krossing, Chem. – Eur. J., 2015, 21, 157–165 CrossRef CAS.
-
(a) R. J. Wehmschulte, R. Peverati and D. R. Powell, Inorg. Chem., 2019, 58, 12441–12445 CrossRef CAS;
(b) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS.
-
(a) A. J. Carty, K. R. Dymock and P. M. Boorman, Can. J. Chem., 1970, 48, 3524–3529 CrossRef CAS;
(b) A. T. McPhail, R. W. Miller, C. G. Pitt, G. Gupta and S. C. Srivastava, J. Chem. Soc., Dalton Trans., 1976, 1657–1661 RSC;
(c) W. Jiang, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2012, 51, 2725–2727 CrossRef CAS.
- A. G. DiPasquale and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 1812–1813 CrossRef CAS.
-
(a) A. Murphy, A. Pace and T. D. P. Stack, Org. Lett., 2004, 6, 3119–3122 CrossRef CAS;
(b) A. Murphy and T. D. P. Stack, J. Mol. Catal. A: Chem., 2006, 251, 78–88 CrossRef CAS.
-
(a) M. L. Kuznetsov, Y. N. Kozlov, D. Mandelli, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2011, 50, 3996–4005 CrossRef CAS;
(b) K. Sharpless, J. Townsend and D. Williams, J. Am. Chem. Soc., 1972, 94, 295–296 CrossRef CAS.
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- C. Bour and V. Gandon, Coord. Chem. Rev., 2014, 279, 43–57 CrossRef CAS.
- C. Bour, J. Monot, S. Tang, R. Guillot, J. Farjon and V. Gandon, Organometallics, 2014, 33, 594–599 CrossRef CAS.
- D. Vidovic, M. Findlater, G. Reeske and A. H. Cowley, J. Organomet. Chem., 2007, 692, 5683–5686 CrossRef CAS.
- B. Michelet, S. Tang, G. Thiery, J. Monot, H. Li, R. Guillot, C. Bour and V. Gandon, Org. Chem. Front., 2016, 3, 1603–1613 RSC.
- B. Michelet, C. Bour and V. Gandon, Chem. – Eur. J., 2014, 20, 14488–14492 CrossRef CAS.
- Z. Li, G. Thiery, M. R. Lichtenthaler, R. Guillot, I. Krossing, V. Gandon and C. Bour, Adv. Synth. Catal., 2018, 360, 544–549 CrossRef CAS.
- A. Djuroyic, M. Vayer, Z. L. Li, R. Guillot, J. P. Baltaze, V. Gandon and C. Bour, Org. Lett., 2019, 21, 8132–8137 CrossRef.
- J. D. Young, M. A. Khan, D. R. Powell and R. J. Wehmschulte, Eur. J. Inorg. Chem., 2007, 12, 1671–1681 CrossRef.
- B. Michelet, G. Thiery, C. Bour and V. Gandon, J. Org. Chem., 2015, 80, 10925–10938 CrossRef CAS.
-
(a) M. Vayer, R. Guillot, C. Bour and V. Gandon, Chem. – Eur. J., 2017, 23, 13901–13905 CrossRef CAS;
(b) M. Vayer, C. Bour and V. Gandon, Eur. J. Org. Chem., 2020, 33, 5350–5357 Search PubMed.
- R. A. Novikov, A. A. Levina, D. D. Borisov, A. D. Volodin, A. A. Korlyukov, Y. V. Tkachev, Y. B. Platonova, L. G. Tomilova and Y. V. Tomilov, Organometallics, 2020, 39, 2580–2593 CrossRef CAS.
-
(a) C. Zhang, D.-M. Cui, L.-Y. Yao, B.-S. Wang, Y.-Z. Hu and T. Hayashi, J. Org. Chem., 2008, 73, 7811–7813 CrossRef CAS;
(b) M. Presset, B. Michelet, R. Guillot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Chem. Commun., 2015, 51, 5318–5321 RSC.
-
(a) I. J. S. Fairlamb, S. Grant, S. Tommasi, J. M. Lynam, M. Bandini, H. Dong, Z. Lin and A. C. Whitwood, Adv. Synth. Catal., 2006, 348, 2515–2530 CrossRef CAS;
(b) F. Grau, A. Heumann and E. Duñach, Angew. Chem., Int. Ed., 2006, 45, 7285–7289 CrossRef CAS;
(c) B. Cacciuttolo, S. Poulain-Martini and E. Duñach, Eur. J. Org. Chem., 2011, 20, 3710–3714 CrossRef.
- C.-F. Xu, M. Xu, L.-Q. Yang and C.-Y. Li, J. Org. Chem., 2012, 77, 3010–3016 CrossRef CAS.
-
(a) T. Miyai, M. Ueba and A. Baba, Synlett, 1999, 2, 182–184 CrossRef;
(b) M. Yasuda, Y. Onishi, M. Ueba, T. Miyai and A. Baba, J. Org. Chem., 2001, 66, 7741–7744 CrossRef CAS;
(c) A. Baba, M. Yasuda, Y. Nishimoto, T. Saito and Y. Onishi, Pure Appl. Chem., 2008, 80, 845–854 CAS;
(d) V. J. Meyer and M. Niggemann, Chem. – Eur. J., 2012, 18, 4687–4691 CrossRef CAS;
(e) G. N. Kumar and K. K. Laali, Org. Biomol. Chem., 2012, 10, 7347–7355 RSC;
(f) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS.
- M. Saleh, D. R. Powell and R. J. Wehmschulte, Organometallics, 2017, 36, 4810–4815 CrossRef CAS.
-
(a) M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS;
(b) J. Chen, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2016, 138, 5321–5333 CrossRef CAS.
-
(a) D. G. Tuck, Chem. Soc. Rev., 1993, 22, 269–276 RSC;
(b) J. A. J. Pardoe and A. J. Downs, Chem. Rev., 2007, 107, 2–45 CrossRef CAS.
-
(a) C. L. B. Macdonald, A. M. Corrente, C. G. Andrews, A. Taylor and B. D. Ellis, Chem. Commun., 2004, 250–251 RSC;
(b) B. F. T. Cooper and C. L. B. Macdonald, New J. Chem., 2010, 34, 1551–1555 RSC.
-
(a) F. W. B. Einstein and D. G. Tuck, J. Chem. Soc. D, 1970, 1182–1183 RSC;
(b) H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1985, 24, 893–904 CrossRef;
(c) W. T. Robinson, C. J. Wilkins and Z. Zeying, J. Chem. Soc., Dalton Trans., 1990, 219–227 RSC;
(d) M. Sigl, A. Schier and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1998, 53, 1301–1306 CAS;
(e) M. Sigl, A. Schier and H. Schmidbaur, Eur. J. Inorg. Chem., 1998, 2, 203–210 CrossRef;
(f) I. A. Guzei, F. Delpech and R. F. Jordan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, e327–e328 CrossRef;
(g) D. A. Robson, S. Y. Bylikin, M. Cantuel, N. A. H. Male, L. H. Rees, P. Mountford and M. Schröder, J. Chem. Soc., Dalton Trans., 2001, 157–169 RSC;
(h) A. H. Cowley, C. L. Macdonald, J. S. Silverman, J. D. Gorden and A. Voigt, Chem. Commun., 2001, 175–176 RSC;
(i) J. N. Jones, C. L. B. Macdonald, J. D. Gorden and A. H. Cowley, J. Organomet. Chem., 2003, 666, 3–5 CrossRef CAS;
(j) W. Uhl, C. Emden, G. Geiseler and K. Harms, Z. Anorg. Allg. Chem., 2003, 629, 2157–2167 CrossRef CAS;
(k) C. G. Andrews and C. L. B. Macdonald, Angew. Chem., Int. Ed., 2005, 44, 7453–7456 CrossRef CAS;
(l) N. N. Chamazi, M. M. Heravi, T. Breyhan and B. Neumüller, Z. Anorg. Allg. Chem., 2007, 633, 1243–1245 CrossRef CAS;
(m) I. Peckermann, D. Robert, U. Englert, T. P. Spaniol and J. Okuda, Organometallics, 2008, 27, 4817–4820 CrossRef CAS;
(n) T. Jurca, J. Lummiss, T. J. Burchell, S. I. Gorelsky and D. S. Richeson, J. Am. Chem. Soc., 2009, 131, 4608–4609 CrossRef CAS;
(o) I. Peckermann, T. S. Dols, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2010, 695, 2325–2328 CrossRef CAS;
(p) K. M. Osman, D. R. Powell and R. J. Wehmschulte, Inorg. Chem., 2015, 54, 9195–9200 CrossRef CAS;
(q) D. Mukherjee, T. P. Spaniol and J. Okuda, Dalton Trans., 2017, 46, 651–655 RSC.
-
(a) D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC;
(b) D. C. Aluthge, E. X. Yan, J. M. Ahn and P. Mehrkhodavandi, Inorg. Chem., 2014, 53, 6828–6836 CrossRef CAS;
(c) D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284–5292 RSC;
(d) A. B. Kremer, R. J. Andrews, M. J. Milner, X. R. Zhang, T. Ebrahimi, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, Inorg. Chem., 2017, 56, 1375–1385 CrossRef CAS;
(e) C. Diaz, T. Ebrahimi and P. Mehrkhodavandi, Chem. Commun., 2019, 55, 3347–3350 RSC.
- D. J. Darensbourg and W.-C. Chung, Polyhedron, 2013, 58, 139–143 CrossRef CAS.
- C. Goonesinghe, H. Roshandel, C. Diaz, H.-J. Jung, K. Nyamayaro, M. Ezhova and P. Mehrkhodavandi, Chem. Sci., 2020, 11, 6485–6491 RSC.
- H.-J. Jung, I. Yu, K. Nyamayaro and P. Mehrkhodavandi, ACS Catal., 2020, 10, 6488–6496 CrossRef CAS.
- H.-J. Jung, C. Chang, I. Yu, D. C. Aluthge, T. Ebrahimi and P. Mehrkhodavandi, ChemCatChem, 2018, 10, 3219–3222 CrossRef CAS.
- H. Nishida, F. Sanda, T. Endo, T. Nakahara, T. Ogata and K. Kusumoto, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 68–73 CrossRef CAS.
- B. Michelet, J.-R. Colard-Itté, G. Thiery, R. Guillot, C. Bour and V. Gandon, Chem. Commun., 2015, 51, 7401–7404 RSC.
- L. Wang, J. Lv, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2017, 56, 10867–10871 CrossRef CAS.
-
(a) K. Surendra, W. Qiu and E. J. Corey, J. Am. Chem. Soc., 2011, 133, 9724–9726 CrossRef CAS;
(b) W.-W. Qiu, K. Surendra, L. Yin and E. J. Corey, Org. Lett., 2011, 13, 5893–5895 CrossRef CAS.
- K. Surendra and E. J. Corey, J. Am. Chem. Soc., 2014, 136, 10918–10920 CrossRef CAS.
- J.-F. Zhao, H.-Y. Tsui, P.-J. Wu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2008, 130, 16492–16493 CrossRef CAS.
- J.-F. Zhao, T.-B. W. Tjan and T.-P. Loh, Tetrahedron Lett., 2010, 51, 5649–5652 CrossRef CAS.
- X. Zhang, M. Wang, R. Ding, Y.-H. Xu and T.-P. Loh, Org. Lett., 2015, 17, 2736–2739 CrossRef CAS.
- F. Delpech, I. A. Guzei and R. F. Jordan, Organometallics, 2002, 21, 1167–1176 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*