
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselective heterogeneous iridium catalyzed hydrogenation of cinnamalaniline†
Risto
Savela
 *a,
Nataliya D.
Shcherban
b,
Marko M.
Melander
c,
Igor
Bezverkhyy
d,
Irina L.
Simakova
e,
Otto
Långvik
a,
Ekaterina
Kholkina
f,
Tamara
Schindler
a,
Annabelle
Krauβ
a,
Karoliina
Honkala
c,
Dmitry Yu.
Murzin
f and
Reko
Leino
a
*a,
Nataliya D.
Shcherban
b,
Marko M.
Melander
c,
Igor
Bezverkhyy
d,
Irina L.
Simakova
e,
Otto
Långvik
a,
Ekaterina
Kholkina
f,
Tamara
Schindler
a,
Annabelle
Krauβ
a,
Karoliina
Honkala
c,
Dmitry Yu.
Murzin
f and
Reko
Leino
a
aJohan Gadolin Process Chemistry Centre, Laboratory of Molecular Science and Technology, Organic Chemistry Research Group, Åbo Akademi University, FI-20500 Turku, Finland. E-mail: risto.savela@abo.fi
bL.V. Pisarzhevsky Institute of Physical Chemistry, National Academy of Sciences of Ukraine, 31 pr. Nauky, Kiev, 03028, Ukraine
cDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland
dLaboratoire Interdisciplinaire Carnot de Bourgogne, UMR 6303 CNRS-Université de Bourgogne-Franche Comté, 9 Av. A. Savary, BP 47870, 21078, Dijon Cedex, France
eBoreskov Institute of Catalysis, pr. Ak. Lavrentieva 5, 630090, Novosibirsk, Russia
fJohan Gadolin Process Chemistry Centre, Laboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Turku, Finland
First published on 9th December 2020
Abstract
Selective hydrogenation of unsaturated imines over heterogeneous catalysts is an ecologically feasible and effective way to produce commercially valuable saturated imines and unsaturated amines under mild conditions, avoiding the utilization of toxic halides. The liquid-phase hydrogenation of a model imine, cinnamalaniline, over Ir, Ru, Pd and Au catalysts was studied in polar protic (methanol, 2-propanol), polar aprotic (methyl tert-butyl ether) and non-polar aprotic (toluene) solvents at 40–80 °C under atmospheric hydrogen pressure. Different metal oxides (Al2O3, ZrO2, SiO2) and carbon composites based on carbon nitrides synthesized by pyrolysis of ethylenediamine or melamine modified by KOH, HNO3, NH4Cl or TEOS (including template KIT-6), porous carbon (samples prepared by pyrolysis of sucrose, including template KIT-6) and mesoporous carbon Sibunit were applied to study the effect of the support. Among the tested metals, iridium exhibited the most promising catalytic performance in terms of hydrogenation activity and chemoselectivity towards unsaturated amine. The use of Ir on carbon nitrides (C3N4–NH4Cl, CxNy-KIT-6) and amphoteric metal oxides (ZrO2, Al2O3) in nonpolar aprotic toluene solvent provided the best selectivity towards unsaturated amine by minimizing oligomerization. Computational studies indicate that the selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group on Ir results from a favorable cinnamalaniline adsorption geometry at high surface coverage. Comparable heterogeneously catalyzed highly chemoselective hydrogenation of unsaturated imine into unsaturated amine under atmospheric hydrogen pressure and low temperatures has not been reported previously.
N group on Ir results from a favorable cinnamalaniline adsorption geometry at high surface coverage. Comparable heterogeneously catalyzed highly chemoselective hydrogenation of unsaturated imine into unsaturated amine under atmospheric hydrogen pressure and low temperatures has not been reported previously.
Introduction
While several classical and homogeneously catalyzed synthetic methods exist to form carbon nitrogen bonds to produce alkyl or aryl substituted amines, the selective heterogeneously catalyzed methods for unsaturated amine synthesis are scarce. One of the most utilized classical methods is the nucleophilic substitution reaction using alkyl halides in combination with a suitable amine. Disadvantages of this generally efficient and simple-to-operate method include the use of toxic and/or expensive organic halides and the discharge of corroding organic salt waste.1 A number of undesired side reactions may also take place, resulting in the formation of higher amines. Alternatives to nucleophilic substitution include e.g. reductive amination,2 hydroamination3 and alcohol amination via hydrogen borrowing.4 Mechanistically, the final step in the reductive amination and alcohol amination via hydrogen borrowing involves reduction of the imine or enamine moieties formed, which in the presence of other unsaturated bonds may result in lower selectivity. Unsaturated amines are important building blocks for production of several valuable compound classes.2c,5 In particular, unsaturated terpene amines, readily prepared from terpenes and amines by reductive amination, have been used as intermediates of potential drugs for neurological diseases.6Selective reduction of imines in the presence of olefins can be achieved by stoichiometric amounts of a suitable hydride source, such as sodium borohydride. While such hydride reducing agents are convenient at a smaller scale, they suffer from limited shelf life and excessive waste formation during work up and purification. Thus, selective heterogeneous hydrogenation methods would simplify and limit the amount of waste formed in scaling up of the corresponding reduction reactions.1 Various homogeneously7,8 and heterogeneously9 catalyzed hydrogenation methods for imine reduction have been developed recently, with particular focus on enantioselectivity.10 Yet, only a few of the homogeneously catalyzed imine hydrogenation11 and hydrosilylation12 methods developed to date, tolerate olefinic moieties in the substrate. Similarly, selective heterogeneous hydrogenation13 and hydrosilylation14 methods are scarce. Earlier, a series of studies on selective synthesis of unsaturated amines through one-pot monoterpene amination and controlled hydrogenation of competitive CC and CN groups in the presence of gold catalysts at 100–180 °C have been reported.15
While homogeneously catalyzed selective imine hydrogenation reactions are plentiful, the main objective of the present work was to develop a heterogeneous catalyst system for chemoselective hydrogenation of imine CN bonds in the presence of allylic CC bonds, at temperatures below 100 °C and at atmospheric hydrogen pressure. Optimally, this would allow removal of the catalyst by filtration followed by simple purification procedure, decreasing both the time and costs associated with chemical transformations. Cinnamalaniline (1) (Scheme 1) was selected as a model compound, being readily prepared in significant quantities by condensation of cinnamaldehyde and aniline in the presence of acid catalyst. A variety of aniline and cinnamaldehyde derivatives are also commercially available, that could be easily utilized to synthesize different allylic imines, similar to compound 1, followed by hydrogenation to the corresponding derivatives of compound 2. These types of compounds could be further used in the synthesis of various biologically active compounds, such as herbicides or fungicides,16 or precursors for compounds with antiviral properties.17 Also, the reactivity of unsaturated bonds in compound 1 can mimic benzodiazepine derivative precursors.11e Hydrogenation of cinnamalaniline may proceed either via initial CN bond hydrogenation to provide the unsaturated amine 2 or by –CC– bond hydrogenation to saturated imine 3, followed by further hydrogenation to the fully saturated amine 4, as illustrated in Scheme 1.
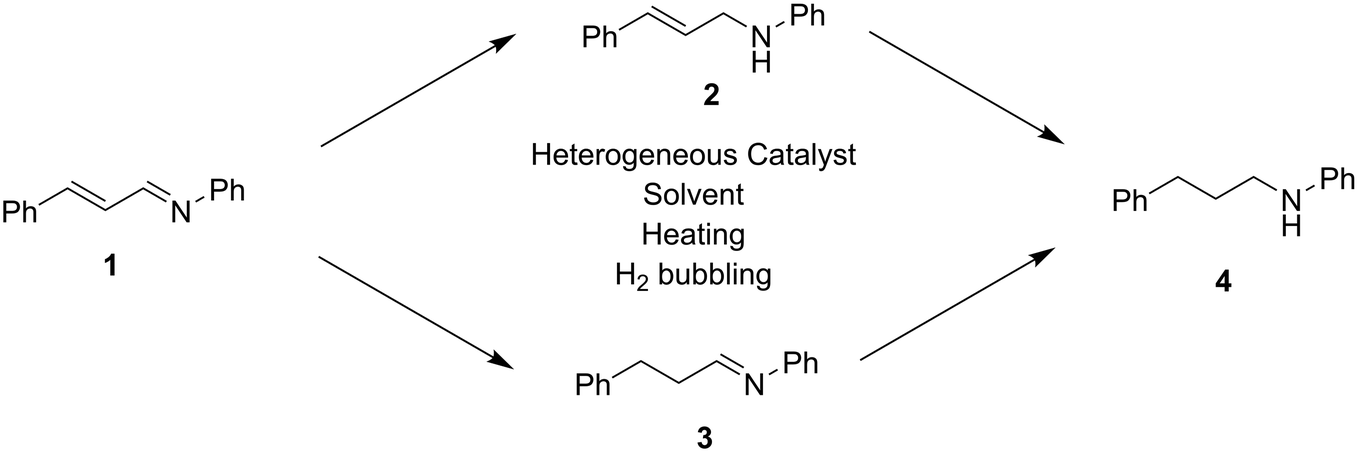 | ||
| Scheme 1 The reaction scheme of cinnamalaniline hydrogenation. | ||
In this work, heterogeneous noble metal catalysts on different support materials were investigated for selective hydrogenation of an allylic imine. Based on the previous reports on homogeneously and heterogeneously catalyzed hydrogenation and hydrosilylation of imines,11–15 monometallic iridium, palladium, ruthenium and gold catalysts were selected. Heterogeneous platinum catalysts were excluded due to their earlier observed rapid deactivation and poor recycling characteristics in the hydrogenation of imines.18 Following the initial screening of the metal catalysts, the influence of support and kinetics in heterogeneously catalyzed hydrogenation of cinnamalaniline was explored. Furthermore, DFT studies were utilized to examine the hydrogenation mechanism and to explain the experimentally observed regioselectivity towards unsaturated amine 2 displayed by iridium-based heterogeneous catalysts.
Results and discussion
Catalyst characterization
The support structures were studied using XRD and the results from XRD study of the phase composition of the supports are presented in Fig. 1. All the synthesized carbon nitride supports exhibit one main reflex (002) at 2θ ∼ 27.3–27.6° (Fig. 1 left), which is characteristic of the interlayer packaging of conjugated aromatic systems. These high-angle reflexes correspond to an average interplanar distance d = 0.327–0.322 nm, similar to the (002) plane of graphitic carbon nitride. The average crystallite sizes calculated from the (002) broadening of the reflex by the Scherrer formula are ca. 10 nm. Carbon supports C-KIT-6, CxNy-KIT-6 and C-micro are X-ray amorphous due to their turbostratic structures, which are characterized by a varying ordering degree of the packing of graphite layers.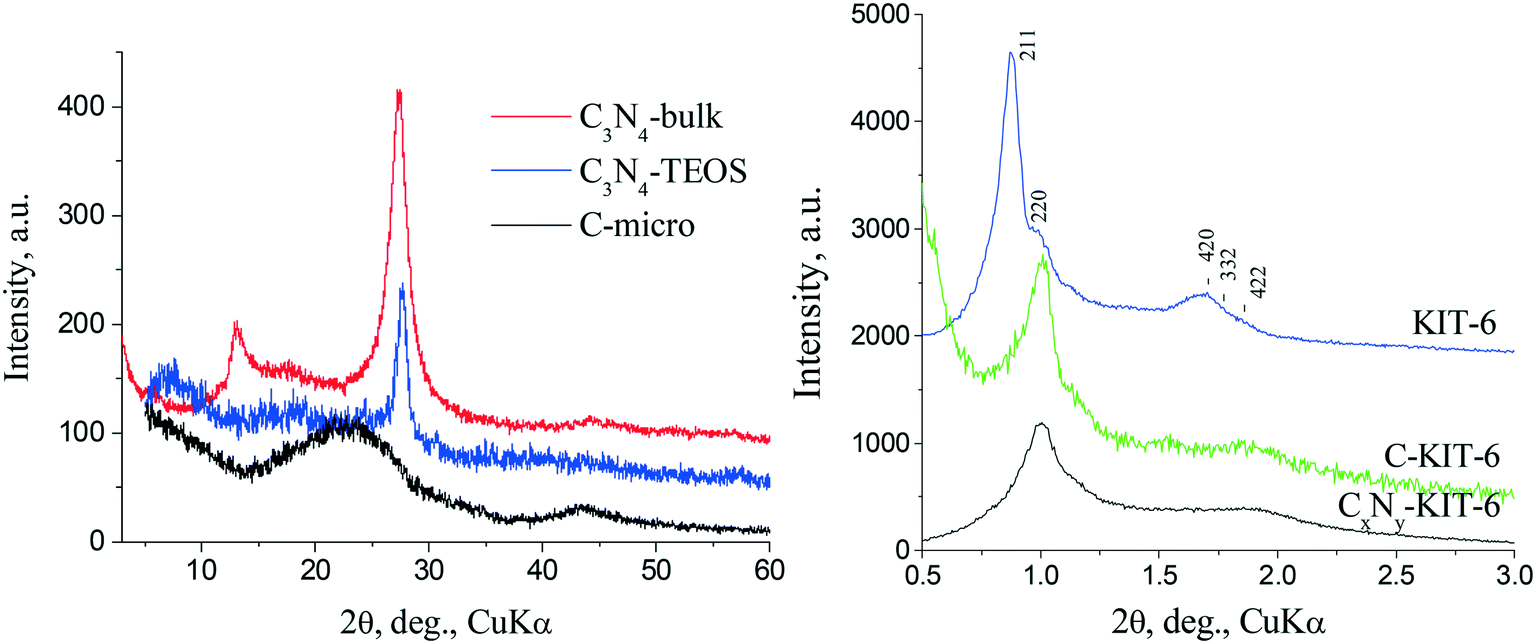 | ||
| Fig. 1 X-ray diffraction patterns of the prepared supports: left: carbon nitride and microporous carbon; right: initial silica MMS KIT-6, CxNy-KIT-6 and C-KIT-6. | ||
The resulting carbons have a spatially ordered mesostructure corresponding to the initial mesoporous molecular sieve (MMS). Thus, the XRD pattern (Fig. 1 right) of the initial hard template KIT-6 is characterized by well-separated hkl reflexes, which define a highly ordered cubic structure with Ia3d symmetry type. Due to decrease of the interplanar distances in carbon nitride obtained in KIT-6 (CxNy-KIT-6), the peaks in the diffraction pattern are shifted towards larger angles, compared to the initial MMS. The decrease in the intensity of the reflexes in the diffraction pattern of CxNy-KIT-6 indicates thickening of the walls in the product compared to the KIT-6. Therefore, the samples obtained after removal of the hard template replicate to a certain extent the corresponding MMS.
Impregnation of carbon nitrides, carbon supports, as well as metal oxides with H2IrCl6 followed by reduction with hydrogen led to the formation of iridium nanoparticles with an average diameter within the range of 1.3–1.7 nm (Fig. 2). On alumina support, the average iridium nanoparticle size was ca. 1 nm. The obtained Ir nanoparticles are mainly uniformly distributed on the support surface.
 | ||
| Fig. 2 TEM images of the prepared supported Ir catalysts: from left to right. Above: Ir/C3N4–HNO3, Ir/C3N4–NH4Cl, Ir/CxNy-KIT-6; below: 3% Ir/ZrO2, 4% Ir/Al2O3 and histogram of 4% Ir/Al2O3. | ||
Nitrogen physisorption was used to determine the microtextural characteristics of carbon nitride and carbon supports. The surface areas and pore sizes of all carbonaceous materials are presented in Table 1. Catalyst supports C3N4–NH4Cl (84 m2 g−1), C-KIT-6 (1570 m2 g−1), CxNy-KIT-6 (585 m2 g−1), mesoporous C (Sibunit) (354 m2 g−1) and sucrose-derived C-micro (744 m2 g−1) exhibit higher specific surface area values compared to C3N4-bulk (10 m2 g−1) and C3N4–TEOS (28 m2 g−1). In case of the template based samples, C-KIT-6 featured a three-fold higher specific surface area compared to CxNy-KIT-6, with both KIT derived samples displaying well-developed pore structures. Similar to the KIT family, C3N4–NH4Cl material and mesoporous carbon Sibunit exhibited a rather large mesopore volume with an average pore diameter ranging from 11 to 20 nm, while C-micro material contains only micropores with an average diameter of 2 nm that could hamper the accessibility of the metal active sites for bulky substrate molecules.
| Sample | V micro, cm3 g−1 | D micro, nm | V meso, cm3 g−1 | D meso, nm | S meso, m2 g−1 | S BET, m2 g−1 | V sum, cm3 g−1 |
|---|---|---|---|---|---|---|---|
| C3N4-bulk | 0 | — | 0.06 | ∼50 | 10 | 10 | 0.06 |
| C3N4–TEOS | 0 | — | 0.14 | 30 ± 12 | 25 | 28 | 0.14 |
| C3N4–NH4Cl | 0 | — | 0.42 | ∼20 | 82 | 84 | 0.42 |
| C3N4–HNO3 | 0 | — | 0.10 | ∼30 | 20 | 22 | 0.10 |
| C3N4-KOH | 0 | — | 0.12 | ∼24 | 23 | 25 | 0.12 |
| C-KIT-6 | 0.07 | 0.55 | 1.86 | ∼11 | 1300 | 1570 | 1.93 |
| CxNy-KIT-6 | 0.06 | 0.52 | 0.58 | ∼14 | 440 | 585 | 0.7 |
| C-micro | 0.29 | 0.46 | 0 | — | 2 | 744 | 0.29 |
Details of the microtextural characteristics of both the fresh and spent metal oxide supported iridium catalysts are given in Table 2. Based on the measured pore volumes the materials mainly exhibit mesoporous characteristics. Deposition of the metal substantially influenced the surface area of the support, which can be explained by blocking of micropores.
| Sample | V meso, cm3 g−1 | D meso, nm | S t-plot, m2 g−1 | S BET, m2 g−1 | V Σ, cm3 g−1 | |
|---|---|---|---|---|---|---|
| a In parenthesis surface area of the supports. | ||||||
| Ir/ZrO2 | Fresh | 0.18 | 14 | 22 | 44 (106)a | 0.18 |
| Spent | 0.14 | 12 | 15 | 34 | 0.14 | |
| Ir/Al2O3 | Fresh | 0.14 | 13 | 27 | 54 (140)a | 0.14 |
| Spent | 0.10 | 13 | 13 | 36 | 0.10 | |
| Ir/C | Fresh | 0.29 | 2–4 | 200 | 221 (350)a | 0.29 |
| Ir/SiO2 | Fresh | 0.49 | 10 | 203 | 242 (250)a | 0.13 |
| Spent | 0.10 | 10 | 13 | 36 | 0.10 | |
The basicity of carbon- and oxide-based supports was measured using temperature-programmed desorption of CO2 (TPD CO2). In a typical TPD curve of carbon dioxide of the synthesized C3N4 samples (Fig. 3), only one broad peak in the TPD profiles can be found independent of the modification type. The concentration of basic sites on similar materials was reported to be ca. 40–60 μmol g−1.19 The peak temperatures are all in the range of ca. 125–135 °C showing that the base sites are of weak strength. The temperature peak of carbon dioxide desorption at ca. 90–100 °C can be attributed to CO2 physisorption.20 Data on the basicity of Ir/ZrO2 and Ir/Al2O3 are provided in Table 3 illustrating that the materials are mildly basic with the predominance of weak basic sites.
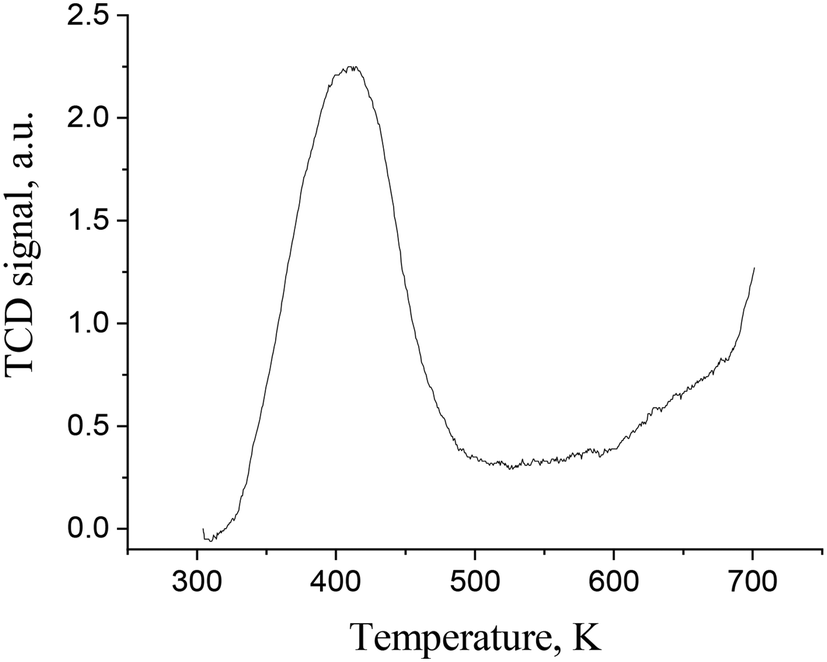 | ||
| Fig. 3 CO2-TPD profile of the prepared carbon nitride (C3N4-bulk). | ||
| Catalyst | Concentration of basic sitesa μmol g−1 | |||
|---|---|---|---|---|
| Weak | Medium | Strong | Total | |
| a The basic sites were divided into three groups – weak (<750 K), medium (750–1050 K) and strong (>1050 K) based on the ability to desorb the probe molecule (CO2) from the surface of active sites. | ||||
| Ir/Al2O3 | 14.3 | 10.8 | 1.4 | 26.5 |
| Ir/ZrO2 | 12.6 | 6.2 | 0.8 | 19.6 |
Catalyst screening
Initially, alumina supported Ir, Pd, Au and Ru catalysts were investigated for their catalytic activity and selectivity in the cinnamalaniline (1) hydrogenation, followed by screening of several support materials, such as different carbon and carbon nitride materials, Al2O3, ZrO2, and SiO2. The initial screening was performed at 40 °C and atmospheric hydrogen pressure in methanol, which is a readily available, cheap and often utilized solvent in hydrogenation reactions. The results from catalyst screening are collected in Table 4.| Catalyst | Conversion of 1 (%) | Selectivity to 2a (%) | GC-based yield | Mass balance | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| Compound 1 250 mg (1.21 mmol), methanol 59.5 mL, tetradecane 68 μL (0.26 mmol; introduced in 0.5 mL of toluene) and the corresponding catalyst (1 mol%, for Ir and Au and 2 mol% for Ru and Pd) at 40 °C for 5 hours.a Selectivity (%) = [2]/([1]0 − [1]6h) × 100. | ||||||
| Pd/Al2O3 | >99 | 0 | — | 32 | 50 | 82 |
| Ru/Al2O3 | 15 | 0 | — | — | — | 75 |
| Ru/C | 97 | 0 | — | 6 | 5 | 14 |
| Au/Al2O3 | 15 | 20 | 3 | 2 | — | 87 |
| Ir/Al2O3 | >99 | 44 | 44 | — | 16 | 60 |
| Ir/ZrO2 | >99 | 48 | 48 | — | 12 | 60 |
| Ir/SiO2 | 0 | — | — | — | — | — |
| Ir/C | 88 | 13 | 11 | 2 | — | 25 |
| Ir/C3N4-bulk | 50 | 42 | 21 | — | 3 | 74 |
| Ir/C3N4–TEOS | 44 | 41 | 18 | — | 3 | 77 |
| Ir/C3N4–HNO3 | 17 | 35 | 6 | — | 2 | 91 |
| Ir/C3N4–KOH | 39 | 41 | 16 | — | 4 | 81 |
| Ir/C3N4–NH4Cl | 88 | 51 | 45 | 4 | 10 | 70 |
| Ir/C-KIT-6 | 94 | 30 | 28 | 5 | 5 | 44 |
| Ir/CxNy-KIT-6 | >99 | 39 | 39 | — | 18 | 57 |
| Ir/C-micro | 14 | 0 | — | — | — | 86 |
According to GC analysis, and verification against separately prepared compounds, the main products included: the unsaturated amine, 1-aniline-3-phenyl-3-propene (2), from CN bond hydrogenation; N-(3-phenylpropylidene)benzamine (3), from CC bond hydrogenation; and N-(3-phenylpropyl)aniline (4) from hydrogenation of both CC and CN bonds. In addition, on the basis of the incomplete mass balance of compounds 1–4, unknown high molecular mass oligomerization products were formed, which could not be detected by GC-FID and GC-MS methods.
Both alumina supported ruthenium and gold catalysts exhibited poor conversion of compound 1 with barely detectable amounts of the hydrogenation products 2–4 (Table 4). In contrast, the carbon supported ruthenium catalyst reached a high conversion of 1, yielding mostly unknown by-products and, consequently, was not investigated further. Palladium on alumina showed a high conversion of compound 1 and was initially selective towards olefin bond hydrogenation yielding compound 3, followed by further hydrogenation to the saturated amine 4. Similar to the palladium catalyst, the alumina supported iridium catalyst exhibited high conversion of compound 1 with high selectivity towards the imine bond hydrogenation. It was found that, for alumina supported metals, conversion of cinnamalaniline increases in the series: Ru ∼ Au ≪ Pd ∼ Ir, whereas selectivity towards the unsaturated amine 2 adheres to the following order: Ru ∼ Pd < Au ≪ Ir.
Preliminary metal screening showed that Ru, Pd and Au catalysts were not effective for chemoselective hydrogenation of the CN bond in compound 1 under the investigated conditions, while Ir exhibited high catalytic hydrogenation activity and chemoselectivity towards the unsaturated amine 2. Similar chemoselectivity has been observed for Ir catalysts in the hydrogenation of multifunctional oxygenates, such as citral21 and crotonaldehyde.22
Catalyst support screening – metal and silicon oxides
Next, the influence of support on the catalytic activity in the chemoselective cinnamalaniline hydrogenation was investigated for silica, zirconia and carbon-based Ir-catalysts with the results summarized in Table 4. It was demonstrated that all active Ir-catalysts are regioselective towards compound 2, which can be explained by favorable adsorption of the CN bond on pure iridium surface as shown by computational studies below. Silica-supported iridium was found to be completely inactive, while both alumina and zirconia supported iridium catalysts exhibited high conversions of cinnamalaniline to product 2 with overall yields up to ∼50% based on GC-analysis. The small differences in selectivity towards unsaturated amine 2 between alumina and zirconia supported catalysts can be correlated with the metal oxide base strength, which in terms of proton affinity was estimated to be similar for ZrO2 (900 kJ mol−1) and Al2O3 (935 kJ mol−1).23 For iridium supported on silica, alumina and zirconia, the conversion of cinnamalaniline increases in the series: SiO2 ⋘ ZrO2 ∼ Al2O3, for the last two supports the yields of the unsaturated amine 2 and undetectable side products were also comparable.
Catalyst support screening – carbon based materials
Iridium supported on microporous carbon (Ir/C-micro) displayed negligible activity, which is tentatively explained by low accessibility of the bulky reagents to the active iridium sites in the micropores. In comparison, the iridium on carbon with a mesoporous support structure (Ir/C-KIT-6) demonstrated 30% selectivity towards compound 2 and 5% selectivity towards compound 3 at 94% conversion in 5 hours. Depositing of the iridium particles on similarly prepared carbon nitride-based supports resulted in improved selectivity towards compound 2. For example, iridium supported on CxNy-KIT-6 achieved ca. 39% selectivity to compound 2 at full conversion within the same reaction time. More prominent nitrogen doping of the support further improved the selectivity at the expense of activity, affording 42% selectivity towards compound 2 at 50% conversion in the case of Ir/C3N4-bulk. The highest catalytic activity for carbon nitride-based supports was achieved with Ir/C3N4–NH4Cl, reaching up to 51% selectivity to compound 2 with 88% conversion, comparable to that of Ir/Al2O3 and Ir/ZrO2. The improvements in selectivity can be attributed to the highly developed porous structure of C3N4–NH4Cl compared to other C3N4 materials. Increase in the catalytic activity of carbon nitride supported Ir catalysts (Ir/C3N4–NH4Cl and Ir/CxNy-KIT-6), in comparison with Ir on nitrogen-free carbon supports, results from their higher basicity, as confirmed by CO2-TPD data (Fig. 3). Aside from the textural and chemical properties, the role of different supports in fine-tuning the selectivity to the compound 2 can be attributed to the acid–base properties of the utilized support, i.e., supports with mainly weak basic sites (Ir/C3N4–NH4Cl and Ir/CxNy-KIT-6) can yield similar activity and selectivity as amphoteric Ir/ZrO2 and Ir/Al2O3. At the same time, the overall mass balance seems to be improved by limiting side reactions due to narrow range of basic sites exhibited by the carbon nitrides (Ir/C3N4–NH4Cl).Effects of the support characteristics on hydrogenation selectivity
Based on the relative hydrogenation selectivity, compound 2 is the predominant hydrogenation product independent of the iridium catalyst utilized (Fig. 4, middle). By also considering the undesired side reactions, it can be seen that application of the Ir/C3N4–NH4Cl catalyst reaches the highest selectivity towards 2 at a moderate conversion level of compound 1 but with a loss of selectivity as the reaction progresses. In comparison, Ir/CxNy-KIT-6 catalyst was less selective, yet maintaining the same level of selectivity until full conversion of cinnamalaniline was achieved. Ir/C3N4-bulk catalyst seems to exhibit very high selectivity (approx. 90%) towards 2 at a very low conversion of cinnamalaniline, which can be attributed at least partially to the detection/integration limits of the instrumentation used to estimate the yield.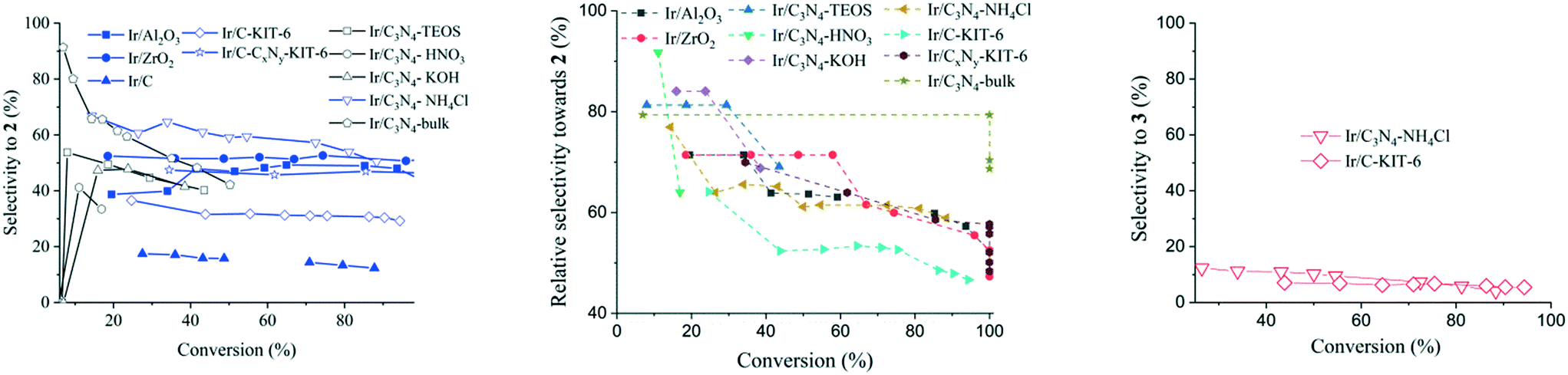 | ||
| Fig. 4 Dependence of (left) selectivity and (middle) relative selectivity to the unsaturated amine 2 and (right) selectivity to the saturated imine 3 as a function on cinnamalaniline 1 conversion. Reaction conditions: compound 1 250 mg (1.21 mmol), methanol 59.5 mL, tetradecane 68 μL (0.26 mmol; introduced in 0.5 mL of toluene), catalyst loading (1 mol%) at 40 °C. | ||
Hydrogenation selectivity profiles obtained with iridium on strong alkali or silicate modified carbon nitride supports, (Fig. 4, Ir/C3N4–KOH and Ir/C3N4–TEOS respectively) were comparable to each other, but significantly below the selectivity obtained with Ir/C3N4–NH4Cl. This suggests that strongly basic supports can hinder the adsorption of the imine bond (CN) on the iridium surface, whereas moderately basic supports provide better adsorption of the imine bond on the iridium surface leading to increased selectivity towards compound 2.
Interestingly, for all iridium catalysts on modified carbon nitride supports, the hydrogenation selectivity towards compound 2 decreases with conversion, especially in the case of Ir/C3N4–HNO3. The use of acidic supports (e.g., C3N4–HNO3) leads to a significant decrease of selectivity towards the unsaturated amine, which can tentatively be associated with the subsequent olefin oligomerization over acid sites of the support. Moreover, in comparison to iridium on alumina or zirconia, the hydrogenation selectivity towards compound 2 was further decreased by faster subsequent –CC– bond hydrogenation to yield the fully saturated compound 4. It is worth to point out that the cinnamalaniline hydrogenation in methanol over iridium catalysts seems to take place exclusively via compound 2, excluding Ir/C3N4–NH4Cl and Ir/C-KIT-6 catalysts (Fig. 4).
Analysis of the basicity of C3N4 supports modified with KOH, TEOS, HNO3 and NH4Cl by CO2-TPD (Fig. 3) did not result in any quantitative correlations between the support basicity and selectivity towards unsaturated amine 2. Nevertheless, a clearly different catalytic behavior in terms of selectivity towards compound 2 was observed for iridium on nitrogen-free carbon supports C and C-KIT, in comparison to that of Ir/C3N4–NH4Cl, Ir/CxNy-KIT-6, Ir/ZrO2 and Ir/Al2O3 (Fig. 4). This indicates that the chemical nature of the support is probably less important than the availability of basic sites with an optimal strength on these supports.
Influence of the solvent and temperature
The selectivity towards hydrogenation of polyunsaturated compounds containing polar unsaturated bonds generally depends on the solvent polarity.24 Thus, the influence of the solvent on hydrogenation of the CN bond vs. CC in cinnamalaniline was investigated with two polar protic solvents, methanol and 2-propanol, polar aprotic methyl tert-butyl ether (MTBE) and one aprotic solvent toluene. Both Ir/ZrO2 and Ir/Al2O3 catalysts were selected for this study, as they displayed high conversion of cinnamalaniline and very good selectivity towards the desired product 2. The results are presented in Table 5. In protic solvents, both catalysts exhibited very similar selectivities and overall mass balance, while the conversion of cinnamalaniline in methanol was somewhat higher than in 2-propanol. Application of the polar aprotic MTBE resulted in a significantly lower conversion of cinnamalaniline albeit at higher mass balance indicating a need of proticity for polar solvents in order to obtain high conversion and selectivity towards 2. The use of toluene as a solvent significantly improved the mass balance at a slight expense of reactivity. The solvent choice significantly influences the rate of cinnamalaniline hydrogenation to compound 2, followed by hydrogenation to compound 4. In toluene, the hydrogenation rate is substantially slower than in alcohol, as illustrated in Fig. 5 and 6 for Ir/Al2O3, due to probably competitive adsorption of toluene and the substrate. Generally, the influence of solvent nature on conversion and selectivity could be very complex, including hydrogen solubility, competitive adsorption, solvation of the transition state and donation of protons.25 A detailed investigation of the solvent effect is beyond the scope of the current study.
| Catalyst | Solvent | Conversion of 1 (%) | GC-based yield | Mass balance | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| Compound 1 250 mg (1.21 mmol), solvent 59.5 mL, tetradecane (68 μL, 0.26 mmol; introduced in 0.5 mL of toluene) and catalyst (1 mol%) at 40 °C for 5 h. | ||||||
| Ir/ZrO2 | Methanol | >99 | 48 | — | 12 | 60 |
| Toluene | 81 | 48 | 4 | 13 | 84 | |
| 2-Propanol | 94 | 47 | — | 13 | 66 | |
| MTBE | 44 | 19 | 2 | 3 | 81 | |
| Ir/Al2O3 | Methanol | >99 | 44 | — | 16 | 60 |
| Toluene | 76 | 47 | 4 | 17 | 92 | |
| 2-Propanol | 78 | 37 | — | 17 | 74 | |
| MTBE | 24 | 7 | — | 1 | 85 | |
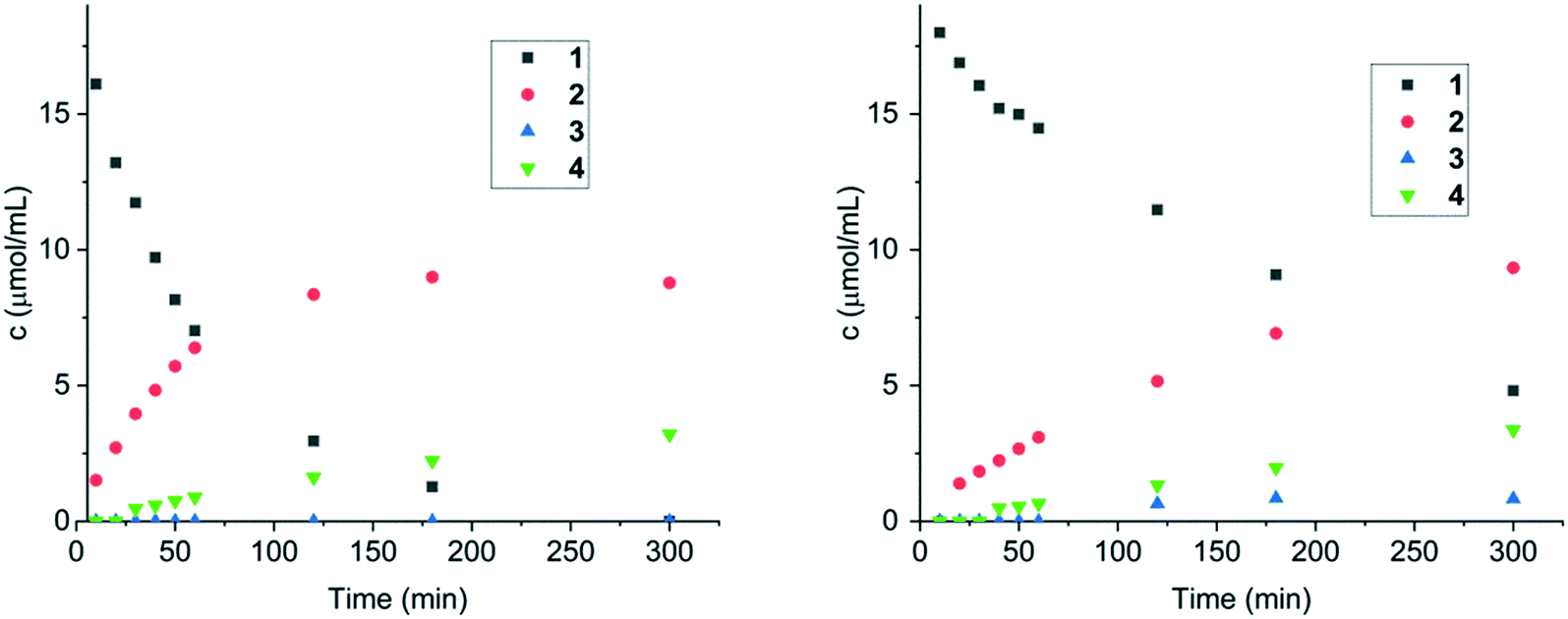 | ||
| Fig. 5 Concentrations of compounds 1–4vs. reaction time over 1 mol% Ir/Al2O3 catalyst in cinnamalaniline 1 hydrogenation using methanol (left) and toluene (right) at 40 °C. Reaction conditions: compound 1 250 mg (1.21 mmol), corresponding solvent 59.5 mL, tetradecane (68 μL, 0.26 mmol; introduced in 0.5 mL of toluene) and the corresponding catalyst (1 mol%) at 40 °C for 5 h. | ||
 | ||
| Fig. 6 Selectivity towards compounds 2–4vs. conversion of cinnamalaniline 1 over 1 mol% Ir/Al2O3 catalyst using methanol (left) and toluene (right) at 40 °C. Reaction conditions: compound 1 250 mg (1.21 mmol), corresponding solvent 59.5 mL, tetradecane (68 μL, 0.26 mmol; introduced in 0.5 mL of toluene) and the corresponding catalyst (1 mol%) at 40 °C for 5 h. | ||
Next, influence of the reaction temperature on the hydrogenation of cinnamalaniline was investigated. Based on solvent screening, toluene was selected as the solvent and the performance of both Ir/Al2O3 and Ir/ZrO2 catalysts was evaluated at three different reaction temperatures (40, 60 and 80 °C). The temperature elevation resulted in similar increase in hydrogenation rates for both catalysts (Table 6). The concentration profiles for Ir/Al2O3 and Ir/ZrO2 (see ESI†) at different temperatures suggest that an increase in the reaction temperature did not have a significant effect on the selectivity towards 2 (Fig. 6 and 7). Notably, Ir/Al2O3 maintained a better overall mass balance than Ir/ZrO2 for compounds 1–4 (Table 6). While the hydrogenation rate of compound 2 is slightly higher at 80 °C compared to 40 and 60 °C, it appears that compound 4 is mainly formed by hydrogenation of the imine bond of compound 3 (Fig. 7). The activation energies for hydrogenation of cinnamalaniline calculated from the data obtained from experiments at different temperatures are 18.3 kJ mol−1 and 14.2 kJ mol−1 for Ir/Al2O3 and Ir/Zr2O respectively, which are lower than the values typically reported for hydrogenation reactions.26
| Catalyst | Temperature (°C) | Conversion of 1 (%) | GC-based yield | Mass balance | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| Compound 1 250 mg (1.21 mmol) was dissolved in toluene (60 mL containing tetradecane 68 μL, 0.26 mmol) and catalyst (1 mol%) at 40–80 °C for 5 hours. | ||||||
| Ir/ZrO2 | 40 | 81 | 48 | 4 | 13 | 84 |
| 60 | 93 | 47 | 2 | 21 | 77 | |
| 80 | >99 | 53 | — | 27 | 80 | |
| Ir/Al2O3 | 40 | 76 | 47 | 4 | 17 | 92 |
| 60 | 92 | 56 | 3 | 27 | 91 | |
| 80 | >99 | 55 | — | 35 | 90 | |
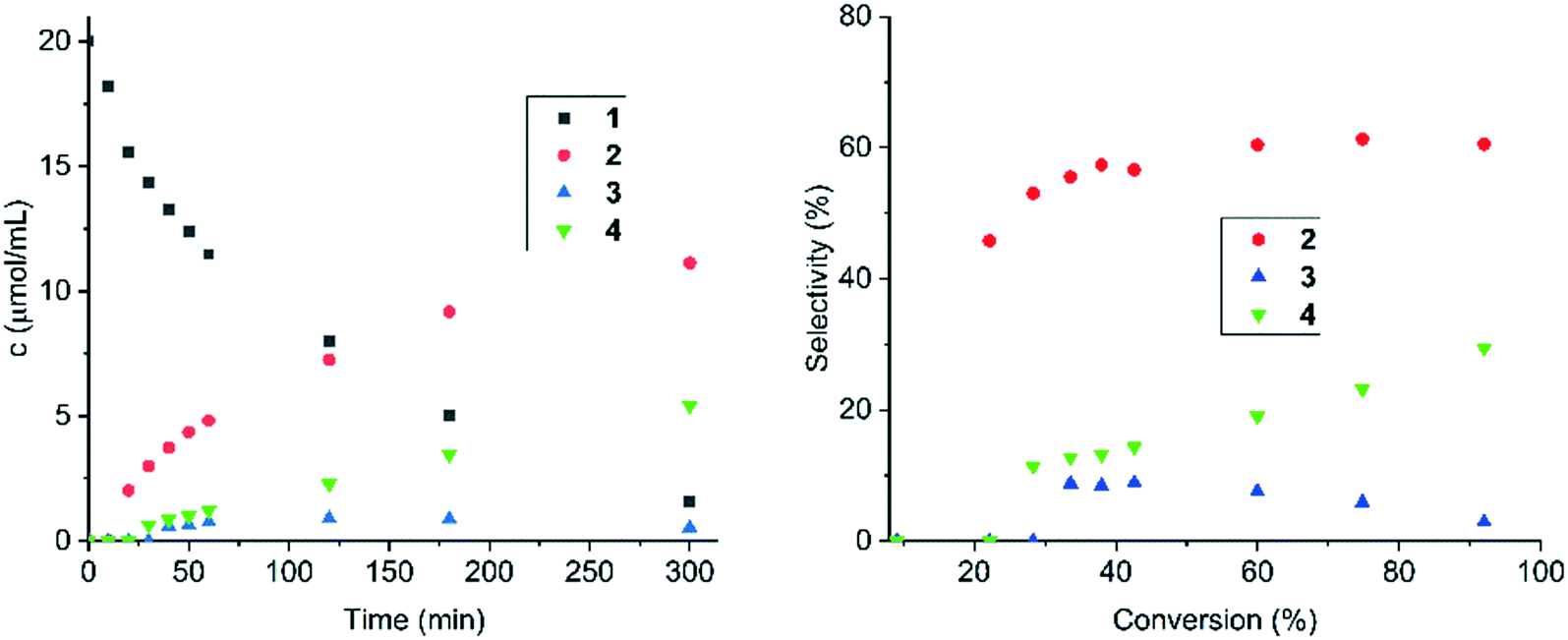 | ||
| Fig. 7 Right: Concentrations of compounds 1–4vs. time in cinnamalaniline hydrogenation over Ir/Al2O3at 60 °C; and left: selectivity towards compounds 2–4vs. conversion of cinnamalaniline 1 at 60 °C. Reaction conditions: compound 1 250 mg (1.21 mmol), toluene (60 mL containing tetradecane 68 μL, 0.26 mmol), catalyst loading (1 mol%), 5 h. | ||
On carbon composite supported catalysts, Ir/C3N4–NH4Cl and Ir/CxNy-KIT-6, the hydrogenation of cinnamalaniline in toluene at 60 °C resulted in similar improvements in the selectivity and mass balance closure as observed for Ir/ZrO2 and Ir/Al2O3 catalysts. While in methanol these carbon nitrides supported catalysts exhibited somewhat better characteristics, in toluene the amphoteric Ir/Al2O3 catalyst showed better characteristics for selectivity and mass balance closure. Comparison of the four selected catalysts is presented in Table 7 with the concentration and selectivity profiles collected into ESI.† The results of this work clearly demonstrate that when searching for optimal cinnamalaniline hydrogenation catalysts with high stability and selectivity, in addition to the metal, the support and the solvent have a crucial role in fine-tuning of the catalytic system.
| Catalyst | Conversion of 1 (%) | GC-based yield | Mass balance | ||
|---|---|---|---|---|---|
| 2 | 3 | 4 | |||
| Compound 1 250 mg (1.21 mmol), toluene (60 mL containing tetradecane 68 μL, 0.26 mmol) and catalyst (1 mol%) at 60 °C for 5 hours.a After 3 hours. | |||||
| Ir/ZrO2 | 93 | 47 | 2 | 21 | 77 |
| Ir/Al2O3 | 92 | 56 | 3 | 27 | 91 |
| Ir/C3N4–NH4Cl | 77 | 36 | 5 | 22 | 86 |
| Ir/CxNy-KIT-6a | >99 | 50 | — | 29 | 79 |
Catalyst deactivation and side products
A brief investigation of deactivation of the catalyst and the side reactions was carried out. Hot filtration of the catalyst from the reaction mixture stopped the reaction (see ESI†). This verifies that the hydrogenation proceeds on the supported iridium particles which do not leach from the support during the reaction. Recycling of the spent Ir/Al2O3 catalyst was also tested. While the cinnamalaniline hydrogenation rate did not significantly decrease, selectivity towards compound 2 was clearly influenced and the mass balance of compounds 1–4 decreased significantly (see ESI†). This may be due to deposition of oligomers into the pores of the utilized catalysts in line with the decrease in the surface area of the spent catalysts (Table 2). Most likely, the balance between the acidic/basic sites and activity of iridium per se is crucial to achieve high hydrogenation selectivity. This type of behavior has also been previously reported in the case of heterogeneous cinnamaldehyde hydrogenation.27 Based on the 1H-NMR-analysis, exposing cinnamalaniline to the fresh catalyst in the absence of hydrogen leads into barely detectable isomerization/oligomerization under the utilized reaction conditions. In addition, hydrogenation of compound 2 to 4 proceeds without any side reactions. It is likely that compound 3 is the main intermediate for the formation of oligomeric compounds via the aldol condensation-type reactions, via e.g. transimination with either compound 2 or 4, as hydrocinnamaldehyde (hydrolysed from compound 3) has been shown to dimerize in the presence of metal oxides and primary and secondary amines.28 A possible role of compound 2 (and 4) was verified by mixing cinnamalaniline, hydrocinnamaldehyde and 10 mol% of compound 2 for 4 hours at 40 °C followed by HPLC-SEC and 1H-NMR analysis. The results obtained from size-exclusion chromatography are similar to those obtained in solvent screening experiments, i.e. protic polar solvents resulting in faster oligomerization than aprotic solvents (see ESI† for the HPLC-SEC chromatograms). Based on 1H-NMR analysis, oligomeric structures are mainly generated from hydrocinnamaldehyde, while extended heating (at least for 16 hours) can incorporate cinnamalaniline to the oligomeric/polymeric structures.Computational studies on the regioselective hydrogenation of the conjugated imine on iridium
The experimental results indicate that regioselective hydrogenation of 1 is dictated by the nature of metal and, to a minor degree, also by the catalyst support and the solvent. Given that iridium was identified as the most regioselective among the studied metals, our goal is to understand the origin of Ir′s regioselectivity. For this, we performed density functional theoretical calculations to elucidate the mechanistic details controlling the selective hydrogenation of 1 on the most abundant Ir(111) surface. To reduce the computational burden, the phenyl substituents were replaced by vinyls, the smallest conjugated moieties and this part of a reactant is not considered amenable towards hydrogenation.Gas-phase reaction thermodynamics
First, we considered the thermodynamics for the formation of the two main products, 2 and 3 and computed the corresponding gas-phase reaction energies, which are ΔEr2 = −58 kJ mol−1 and ΔEr3 = −69 kJ mol−1, respectively. This means that formation of product 3 is thermodynamically more favorable and a selective catalyst is required to form product 2.Adsorption energies
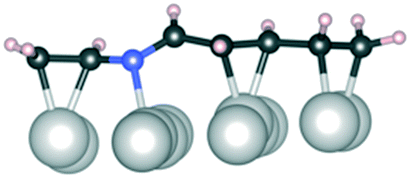
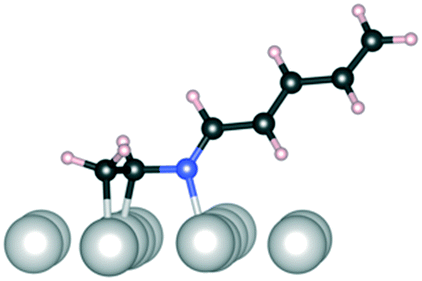
Initially, several adsorption geometries were studied for reactant 1 and products 2 and 3 on Ir(111) and the most stable optimized adsorption geometries together with the corresponding adsorption energies are presented in Table 8. To guarantee a sufficiently unbiased probing of the adsorption geometries, a thorough semi-global structure search was performed using the minima-hopping algorithm as detailed in the Computational details section. The reactant 1 was found to bind equally strongly from the one CN bond or simultaneously from both CN/CC bonds. The most stable adsorption geometry of 2 is bound to Ir(111) from both the now hydrogenated C–N and the conjugated CC bonds lying flat on the surface; all geometries with the C–N tail detached from the surface are highly unstable (over 50 kJ mol−1 less stable) compared to the flat-lying geometry. The flat-lying geometry is prone to further hydrogenation, which is expected to be even easier than the first hydrogenation of the CN double bond as the once hydrogenated molecule is less conjugated than 1. We note that Ir can also catalyze full hydrogenation to 4 as shown in Tables 5–7 but only minor amounts of the CC hydrogenated 3 are observed. However, from the CC/CN bound geometry one would expect the product distribution to be a mixture of 2, 3, and 4 as hydrogenation of the CN and CC bonds are kinetically and thermodynamically similar as shown in Fig. 8. Therefore, we need to identify a pathway leading preferentially to 2 and possibly 4 without forming 3 at any point.
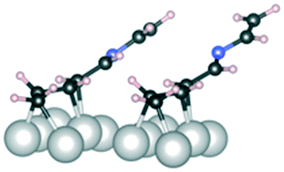
C and CN refer to bonding to the Ir(111) surface from the CC and CN double bonds, respectively. Dark grey: carbon, blue: nitrogen, light grey: iridium, and pale pink: hydrogen
| Conformation | 1 | 1 | 1 | 2 |
|---|---|---|---|---|
| CC and CN |
CN |
CC |
C–N and CC |
|
| Side view |
|
|
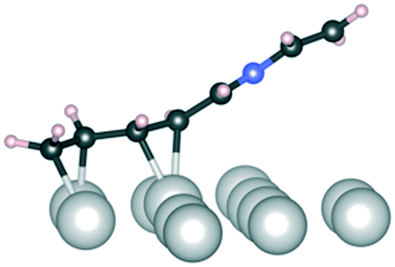
|
|
| E ads (kJ mol−1) | −150 | −160 | −60 | −190 |
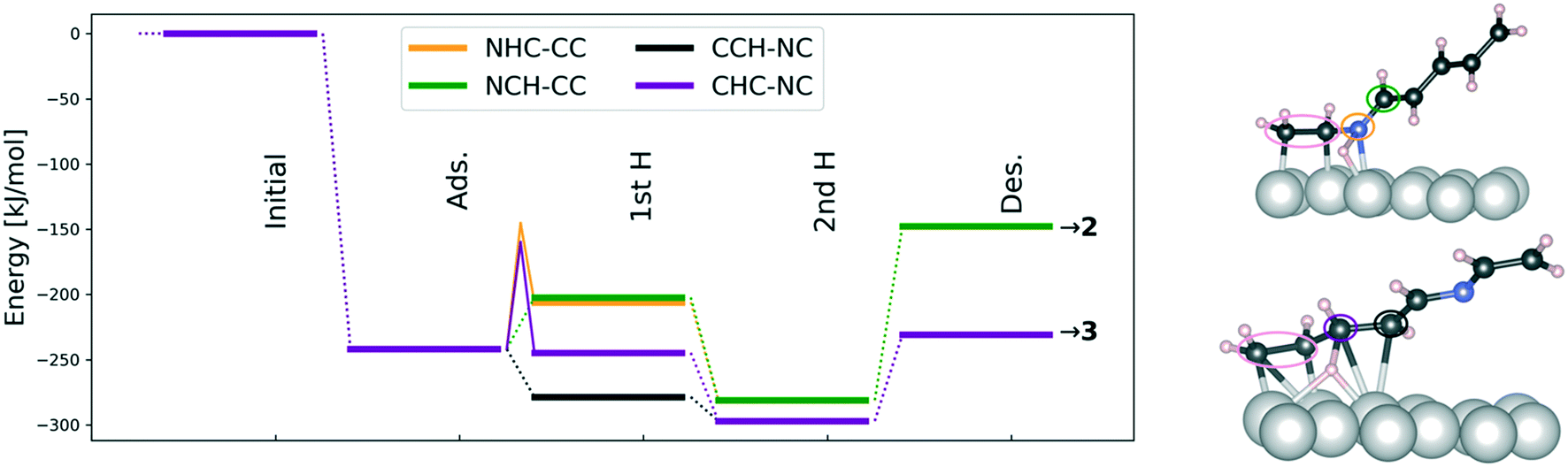 | ||
| Fig. 8 Left: Potential energy profile for the hydrogenation starting from the CC/NC geometry using gas-phase hydrogen as the reference. In NHC–CC the N is hydrogenated first and in NCH–CC the C is hydrogenated first with the CC bond being away from the surface leading to product 2. In CHC–NC and CCH–NC geometries the CC double bond is hydrogenated in different order with CN being away from the surface and the pathways lead to product 3. Ads denotes adsorbed reactant 1 and Des refers to desorbed, gas-phase products while 1st H (2nd H) stands for the number of added hydrogen. The dotted and thin solid lines indicate thermodynamic changes and barriers, respectively. Right: Transition state structures for first hydrogenation along the NHC–CC (top) and CHC–NC (bottom) pathways. The reactive N and C are circulated with colors corresponding to the energy profiles; the pink oval marks the vinyl group acting as a proxy for benzyl. | ||
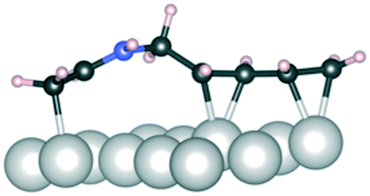
It has been previously shown that surface coverages are crucial for determining the selectivity of metal-catalyzed hydrogenation of conjugated organic molecules.29 This together with the fact that we are able to find strongly exothermic adsorption geometries for reactant 1 indicates that the Ir catalyst surface has relatively high concentration of adsorbed species and therefore the surface coverage effects are considered. Table 9 summarizes the adsorption energies and structures for the reactant at higher surface coverage. The results show that adsorption of the reactant 1 is energetically highly favorable even in the presence of another reactant in its vicinity. Crucially, the CN group is bound much stronger than the CC group and in all the high coverage geometries the one or the other end of 1 spontaneously lifts up from the surface during structure optimization. This makes the initial high-coverage geometry susceptible to selective hydrogenation either from the CC or the CN group. However, the geometry where 1 is bound exclusively via the CN-bond (1, 2CN geometry in Table 8) is significantly more stable than mixed binding with both CC and CN (1, CC/CN) or exclusively CC-bound (1, 2CC) geometries. This indicates that at high surface coverage the CN-bound geometry is clearly the most abundant adsorbed reactant conformation as shown in Table 9 – a pathway starting from this adsorption geometry is likely to lead to 2 and even 4 while formation of 3 is avoided.
C and 2CN, both reactant molecules are bound either from two CC bonds or two CN bonds, respectively. In the CC/CN configuration, one reactant molecule is bound from the CC bond and the other one from the CN bond. The surface abundance is computed as the Boltzmann weight at 40 °C
| Above, 1 CC/CN |
1, 2CC |
1, 2CN |
1, CC/CN |
|
|---|---|---|---|---|
| Geometry |
|
|
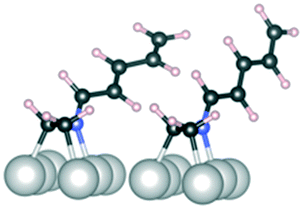
|
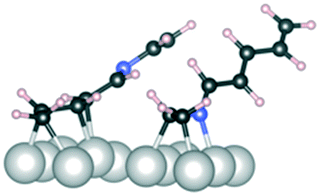
|
| E ads (kJ mol−1) | −80 | −160 | −120 | |
| Abundance | 1 × 10−14 | 1 | 1 ×10−7 |
Hydrogenation thermodynamics and barriers
After considering the adsorption characteristics of the reactant 1, the elementary kinetics and thermodynamics of forming either 2 or 3 at high surface coverage were addressed. Fig. 8 presents the thermodynamic analysis and reveals that cinnamalaniline is preferentially hydrogenated at the CC double bond (CCH and CHC in Fig. 8) leading to formation of 3. The CN hydrogenation is also thermodynamically favorable but desorption of the product 2 is more endothermic than that of product 3. Hence, formation of the minor product 3 is more thermodynamically favored also on the Ir surface.
Given the complexity of the system, entropy effects were only qualitatively addressed for the adsorption and desorption steps as detailed in the Materials and methods section. Including the decrease (increase) in rotational and translational degree of freedom during adsorption (desorption) makes the adsorption energies more endothermic by ∼66 kJ mol−1 and decreases the thermodynamic desorption barriers by ∼65 kJ mol−1. Hence, the adsorption is still highly exergonic but accounting for the major entropic contributions enhances desorption while the overall thermodynamics remain rather unchanged. Entropy changes in elementary steps taking place solely on the surface are considered minor.
As the elementary thermodynamic analysis is unable in explaining the regioselective formation of the experimentally observed major product on Ir, it is necessary to address the reaction kinetics. In the high-coverage adsorption geometry leading to product 2 (green and orange color in Fig. 8), there are no reactive carbon atoms close to the surface as shown in Fig. 8 and Table 9. Our calculations indicate that the reactive carbon atom can only approach the surface after N has been hydrogenated. Therefore, the hydrogenation mechanism of the complex imine 1 is different from the mechanism of methylene imine (H2CNH) hydrogenation presented by Pera-Titus and Sautet,30 where the C is hydrogenated before N. For these reasons, we computed the first hydrogenation barriers for N (NHC–CC in Fig. 8) and C (CHC–NC in Fig. 8), which may determine product selectivity and lead to product molecules 2 and 3, respectively. The barriers for these competing hydrogenation steps are slightly different being ΔE‡NHC–CC = 100 kJ mol−1 and ΔE‡CHC–NC = 85 kJ mol−1 indicating a slight kinetic preference towards forming the minor product 3 over the major product 2 on an Ir catalyst but suggesting that both hydrogenations are feasible under reaction conditions. This in turn means that if the reactant favors an adsorption geometry, where both CN and CC double bonds are sufficiently close to the Ir catalyst surface, the product distribution includes both 2 and 3 species and very likely also fully hydrogenated imine in similar proportions.
As the experiments clearly demonstrate selectivity towards 2, the regioselectivity must be controlled by the reactant adsorption energy and conformation at high-surface coverages as has been shown previously for other selective metal-catalyzed hydrogenations.29 This is indeed the case here because the concentration of the CC bound reactant configurations, giving product 3 on Ir(111), is very low due to the substantial binding energy difference between NC and CC bound geometries. This leads to very different abundances (see Table 9) of reactant adsorption geometries. We therefore ascribe the experimentally observed regioselective formation of 2 on Ir to the high surface coverage of CN-bound reactant geometry despite the fact that thermodynamically and kinetically the formation of 3 is slightly more favorable.
Conclusions
Liquid-phase hydrogenation of cinnamalaniline in polar (methanol, 2-propanol) and non-polar (toluene) solvents over monometallic iridium, ruthenium, palladium and gold catalysts with different carbon-based and metal oxide supports at 40–80 °C and atmospheric hydrogen pressure was studied. In addition to zirconia, silica, alumina and microporous carbon supports, templated (silica KIT-6) mesoporous carbon, Sibunit supports and carbon nitride supports were investigated. The carbon nitrides were prepared both as bulk and chemically modified materials, using different pretreatments and templates followed by iridium impregnation.It was found that for alumina supported metals iridium proved superior in terms of selectivity towards CN bond hydrogenation. Both carbon nitride (Ir/C3N4–NH4Cl, Ir/CxNy-KIT-6) and amphoteric metal oxide (Ir/ZrO2, Ir/Al2O3) based catalysts provided the best regioselectivities towards hydrogenation of the CN group, leading to the formation of the unsaturated amine. A non-polar aprotic solvent significantly improved the mass balance closure at the expense of reactivity, while an increase of the reaction temperature did not affect selectivity to the unsaturated amine. Based on DFT calculations, the regioselectivity of an Ir catalyst is dictated by the reactant adsorption geometry and substantially more favorable adsorption via the CN bond leading to the experimentally observed product.
Experimental
Chemicals (reagent grade) and solvents (HPLC-grade) utilized in hydrogenation experiments were bought from commercial sources and utilized as is. Argon (99.996%) and hydrogen (99.999%) gases (Linde Gas-AGA) were utilized. Cinnamalaniline was prepared according to a literature procedure31 and verified by NMR spectra measured on an 500 MHz NMR spectrometer. The measured NMR spectra were calibrated using residual solvent signal as internal standard.32 The molar weight profile was determined by high pressure size-exclusion chromatography (HP-SEC) using an Agilent 1100 series HPLC instrument equipped with a G1315B DAD-detector, 2× Jordi Gel DVB 500A (300 mm × 7.8 mm) columns and a 50 mm × 7.8 mm guard column. This setup allows selective analyses of aromatic compounds. The samples were analyzed in THF with one percent AcOH as eluent at a flow rate of 0.8 mL min−1 with 35 min analysis time/sample. The samples were dissolved in the eluent solution to yield a concentration of 1 mg mL−1.
Catalyst preparation
Carbon nitride denoted as C3N4-bulk was prepared by bulk pyrolysis of melamine. Melamine (5 g) was heated in air at 525 °C for 2 h to provide a yellow carbon nitride powder. For synthesis of the C3N4–TEOS support, a mixture containing melamine and the products of TEOS hydrolysis with the melamine: TEOS molar ratio of 1.5 was subjected to thermal treatment. To prepare this mixture, the desired amount of melamine was dissolved in distilled water under heating, followed by addition of the corresponding amount of TEOS, stirring for 30 min and drying at 100 °C until complete evaporation of the solvent. The C3N4–HNO3 material was prepared from melamine precipitated from ethylene glycol with 0.6 M nitric acid solution (HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :melamine molar ratio = 6). For synthesis of C3N4–KOH, melamine treated with potassium hydroxide was used. For this purpose, a solution of melamine (1.6 g) and KOH (0.02 g) in distilled water (200 ml) was stirred for 1 h at 90 °C and then dried at 100 °C until complete evaporation of the solvent. The C3N4–NH4Cl material was obtained by heat treatment of a mixture of melamine (1 g) with ammonium chloride (6.37 g). All melamine derived samples were heated under similar conditions (525 °C, 2 h). The obtained materials were washed with distilled water, and in the case of C3N4–TEOS, the silica component was removed by treating the resulting powder with 15% HF solution.
:melamine molar ratio = 6). For synthesis of C3N4–KOH, melamine treated with potassium hydroxide was used. For this purpose, a solution of melamine (1.6 g) and KOH (0.02 g) in distilled water (200 ml) was stirred for 1 h at 90 °C and then dried at 100 °C until complete evaporation of the solvent. The C3N4–NH4Cl material was obtained by heat treatment of a mixture of melamine (1 g) with ammonium chloride (6.37 g). All melamine derived samples were heated under similar conditions (525 °C, 2 h). The obtained materials were washed with distilled water, and in the case of C3N4–TEOS, the silica component was removed by treating the resulting powder with 15% HF solution.
To synthesize the C-KIT-6 support, a desired amount of the initial silica mesoporous molecular sieve (MMS) KIT-6 (cubic symmetry) was mixed with an aqueous solution containing sucrose and concentrated sulfuric acid (i.e., 1.25 g and 0.14 g, respectively, per 1 g of MMS with the pore volume of 1.3 cm3 g−1). The resulting suspension was first dried at 100 °C for 6 h, and then for additional 6 h at 160 °C. This was followed by mixing again with an aqueous solution of sucrose and sulfuric acid (0.8 g and 0.08 g, respectively, per 1 g of MMS with the same pore volume). After repeated treatment under identical conditions (100 and 160 °C), the obtained dark brown powder was heated (heating rate 5 °C min−1) under argon to 900 °C and retained for 2.5 h. The silica component was removed by treatment of the obtained powder with 15% HF solution. The product was filtered, washed with ethanol and dried at 100 °C.
For synthesis of CxNy-KIT-6, the initial silica KIT-6 (0.5 g) was added to a mixture of 70% aqueous solution of ethylenediamine (2 ml) and carbon tetrachloride (1.5 ml) and heated at 90 °C for 6 h under reflux. The resulting dark brown mass was dried at 60 °C for 12 h and mixed again with a mixture of 70% ethylenediamine solution (2 ml) and carbon tetrachloride (1.5 ml). The mixture was reheated at 90 °C for 6 h under reflux and dried. The obtained powder was then heated under inert (argon) atmosphere to 600 °C (heating rate 3 °C min−1) and kept at that temperature for 5 h. The silica hard template KIT-6 was removed by treatment in HF solution, the obtained product was then washed several times with distilled water and ethanol, and finally dried at 100 °C.
For synthesis of the C-micro support, sucrose was kept in an argon flow at 900 °C for 2.5 h.
Zirconia and alumina were precalcined in air at 500 °C for 2 hours before catalyst preparation.
To prepare Ir catalysts over C-micro, C3N4 family, KIT-6, CxNy-KIT-6 and C-KIT-6 with 3 wt% metal loading a weighted amount of the support was mixed with 0.5 M H2IrCl6 aqueous solution, stirred at room temperature overnight, dried at 100 °C followed by reduction in flowing hydrogen at 300 °C for 2 h.
Incipient wetness impregnation with an aqueous solution of IrCl3 hydrate was used for preparation of 3 wt% Ir/ZrO2, 3 wt% Ir/SiO2, 3 wt% Ir/C and 4 wt% Ir/Al2O3 followed by drying at 110 °C for 17 h and reduction by molecular hydrogen from room temperature up to 400 °C with a ramp rate of 3 °C min−1, thereafter holding in hydrogen atmosphere for 3 h to fully reduce IrCl3.
Au/Al2O3 (1 wt%) was prepared by deposition–precipitation on alumina prepared by a sol–gel method33 using urea as the precipitation agent. First, 4 g of the prepared alumina support were added to 400 ml of aqueous solution of HAuCl4 (4.2 × 10−3 M) and urea (0.42 M). The initial pH of solution was ca. 2. After vigorous stirring at 80 °C for 4 h, the suspension was filtered and washed with ammonium hydroxide (25 M) for 30 min. The pH of the solution after stirring with ammonium hydroxide was ca. 10. Finally, the sample was washed in water until pH 7, filtered and dried at room temperature for 24 h.
Catalyst characterization
The phase composition of the catalysts was analyzed using X-ray diffractometer D8 Advance (Bruker AXS) with CuKα-radiation.The porous structure characterization of carbon nitride and other carbon based supports was performed by N2 physical adsorption at 77 K using Sorptomatic 1990, after outgassing the samples at 473 K under vacuum for 4 h. The total surface area, SBET, was calculated by the BET equation.34 The mesopore size was determined using the Barrett–Joyner–Halenda method from the desorption branch of the isotherm.35 The micropore size was calculated according to the Horvath–Kawazoe equation.36
Measurements of the surface area and pore volume of the supported Ir catalysts were performed by nitrogen physisorption using MicroActive 3FlexTM 3500 (Micromeritics®). BET (Brunauer–Emmett–Teller) method was applied for determination of the specific surface area. External surface area was achieved using t-plot method. The mesopore volumes were obtained using the BJH (Barrett–Joyner–Halenda) method. Micropore volume was evaluated by Dubinin–Radushkevich method. Catalysts were pretreated under vacuum (0.05 mbar) and heated to 180 °C for at least 7 h for moisture removal prior to measurements performed at 77 K. An exception was Ir/C, which pretreatment was done at 120 °C.
TEM images were obtained using the field emission TEM JEM-2100F (JEOL) and JEM-2100 (JEOL) instruments with an accelerating voltage of 200 kV. The samples were dispersed in ethanol in an ultrasonic bath for 5 min, followed by deposition of the suspension on a copper grid coated with a carbon film.
Basicity of the supports was determined by temperature programmed desorption of CO2 using a Micromeritics Autochem 2910 equipment. Prior to adsorption of CO2, the materials were heated to 400 °C (10 °C min−1) in a 10 ml min−1 flow of helium and kept at this temperature for 60 min. Thereafter, the samples were cooled to ambient temperature and CO2 was adsorbed for 30 min with a 50 ml min−1 flow. After CO2 adsorption, the samples were flushed with helium (20 ml min−1) for 30 min to remove physisorbed CO2. The temperature programmed desorption was carried out with a 10 °C min−1 heating rate until 600 °C recording the TCD signal every second.
Hydrogenation experiments of cinnamalaniline
All catalysts employed were heated to 150 °C under H2 atmosphere for 1 h before hydrogenation experiments. A four necked glass reactor was purged with argon followed by addition of cinnamalaniline (250 mg, 1.21 mmol) and solvent (toluene, 2-propanol or methanol; 60 mL, containing tetradecane 68 μL, 0.26 mmol; in the case of methanol, the solution contained 0.5 mL of toluene in order to solve tetradecane). A gas bubbler was introduced into the reactor and the solution was purged with hydrogen for ca. 10 minutes. To minimize any possibilities of detonation due to static discharge the hydrogen atmosphere was switched to argon prior the addition of the catalyst. The mixtures were heated to either 40 °C, 60 °C or 80 °C and hydrogen bubbling was continued maintaining the H2 pressure of ca. 1 bar. The reaction was followed by gas chromatograph (GC) with flame ionization detector (FID), equipped with HP-5 column (30 m × 320 μm × 0.25 μm), and He as the carrier gas, using the following temperature program: injector 220 °C, oven T initial = 50 °C (4 min), rate 20 °C min−1, T final = 300 °C, hold 5 min.Selectivity towards compounds 2–4 was calculated by dividing the GC-based yield of 2, 3 or 4 by converted cinnamalaniline 1; while for calculation of relative selectivity: the GC-based yield of 2 was divided by the combined GC-based yields of the known products 2–4.
Oligomerization experiments
Into 3 mL vial with magnetic stirred was added hydrocinnamaldehyde (34 mg, 0.25 mmol), cinnamalaniline (52 mg, 0.25 mmol), N-(3-phenyl-2-propen-1-yl)-benzenamine (3 mg, 0.013 mmol) and solvent (3 mL). The mixture was heated to 40 °C and stirred for 4 hours, followed by analysis of the reaction mixture with HP-SEC.Computational methods
All calculations were performed using the GPAW-1.5.1 software37 and ASE.37 The van der Waals corrected, surface-specific BEEF-vdW functional38 was used. The grid spacing was set to a standard value of 0.18 Å. The computed bulk lattice constant is 3.91 Å which is in good agreement with the experimental value of 3.84 Å. For the surface calculations a 3 × 4 × 4 slab was used with the bottom two layers fixed and a 3 × 2 × 1 k-point sampling was used. The geometry optimizations were considered converged when the residual force was below 0.05 eV Å−1. To simulate the liquid environment, the SCMVD implicit solvent method39 with water parameters was adopted to mimic the methanol environment. To locate transition states the nudged-elastic band40 with 5 intermediate images was utilized. For semi-global optimization the minima hopping method41 was applied. In this approach short molecular dynamics simulations are run at a high temperature (1200 K) to escape local minima. Subsequently local geometry optimization is performed. With this approach at least 20 minima around the geometries 1, 2, and 3 were studied resulting in a wide search space for the conformers.Given the complexity of the considered molecule the entropic effects were not considered as this would require extensive sampling of the vibrational and torsional modes as well as 2D translation/rotational movement of the adsorbed complexes for reliable results and is not within the scope of the present study. The entropic effects would be most significant for the adsorption and desorption. These contributions can be approximated by removing/adding one translational degree of freedom during adsorption/desorption. For this, we computed the translational and rotational energies and entropies within the 3D ideal gas approximation for the solution phase molecules. For adsorbed species, the 2D hindered rotor/translator approximation with 50 kJ mol−1 barriers for both translation and rotation were used to approximate energies and entropies of the corresponding degrees of freedom. The experimental temperature of 60 °C and 1 bar. With these corrections, the adsorption free energies become weaker by 66 kJ mol−1 and the desorption more facile by 65 kJ mol−1.
Author contributions
The work was originally conceptualized by RS, DYM, RL and OL. Non-commercial catalysts were synthesized and characterized by NDS, IB, EK and IS. RS and OL supervised the hydrogenations experiments of Erasmus-interns TS and AK. The hydrogenation experiments were finalized by RS. MMM and KH devised the computational approach and analyzed the results. MMM performed the calculations. All authors contributed in different extent in writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MMM and KH acknowledge funding by the Academy of Finland through projects 307853 and 307623. The computational studies were made possible by the Finnish Grid and Cloud Infrastructure – FGCI. RS acknowledges funding by the Academy of Finland through project 295364. Synthesis of some catalysts (Ir on alumina, zirconia, silica, Sibunit) was supported by RFBR Grant 18-53-45013 IND_a, characterization (TEM, N2 physisorption, XRF) was supported by Ministry of Science and Higher Education of the Russian Federation. NDS acknowledges the support of the National Academy of Sciences of Ukraine to the project “New effective nanoscale catalysts for the production of valuable organic compounds from bio raw materials and products of its conversion” (no. 19/02-2020). Help of Lucas Lagerquist with HP-SEC analysis is gratefully acknowledged.Notes and references
- (a) G. W. Gribble, Chem. Soc. Rev., 1998, 27, 395–404 RSC; (b) M. Periasamy and M. Thirumalaikumar, J. Organomet. Chem., 2000, 609, 137–151 CrossRef CAS; (c) M. V. N. de Souza and T. R. A. Vasconcelos, Appl. Organomet. Chem., 2006, 20, 798–810 CrossRef CAS.
- (a) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS; (b) A. A. Pletnev, Amines, in Comprehensive Organic Transformations, ed. R. C. Larock, John Wiley & Sons, Hoboken NJ, 3rd edn, 2018, vol. 2, pp. 1479–1716 Search PubMed; (c) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS.
- (a) V. I. Isaeva and L. M. Kustov, Top. Catal., 2016, 59, 1196–1206 CrossRef CAS; (b) C. Lepori and J. Hannedouche, Synthesis, 2017, 49, 1158–1167 CAS; (c) C. Michon, M.-A. Abadie, F. Medina and F. Agbossou-Niedercorn, J. Organomet. Chem., 2017, 847, 13–27 CrossRef CAS; (d) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef CAS; (e) L. F. T. Novaes and J. C. Pastre, Quim. Nova, 2017, 40, 932–945 CAS; (f) J. A. Gurak and K. M. Engle, Synlett, 2017, 28, 2057–2065 CrossRef CAS.
- (a) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef; (b) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC; (c) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS.
- (a) A. Stütz, Angew. Chem., Int. Ed. Engl., 1987, 26, 320–328 CrossRef; (b) A. Shupak, I. Doweck, C. R. Gordon and O. Spitzer, Clin. Pharmacol. Ther., 1994, 55, 670–680 CrossRef CAS.
- (a) A. A. Silver, R. D. Shytle, K. H. Sheehan, D. V. Sheehan, A. Ramos and P. R. Sanberg, J. Am. Acad. Child Adolesc. Psychiatry, 2001, 40, 1103–1110 CrossRef CAS; (b) T. G. Tolstikova, E. A. Morozova, A. V. Pavlova, A. V. Bolkunov, M. P. Dolgikh, E. A. Koneva, K. P. Volcho, N. F. Salakhutdinov and G. A. Tolstikov, Dokl. Chem., 2008, 422, 248–250 CrossRef CAS; (c) D. I. Park, H. G. Kim, W. R. Jung, M. K. Shin and K. L. Kim, Neuropharmacology, 2011, 61, 276–282 CrossRef CAS; (d) I. G. Kapitsa, E. V. Suslov, G. V. Teplov, D. V. Korchagina, N. I. Komarova, K. P. Volcho, T. A. Voronina, A. I. Shevela and N. F. Salakhutdinov, Pharm. Chem. J., 2012, 46, 263–265 CrossRef CAS.
- For Reviews see: (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115(13), 6621–6686 CrossRef CAS; (b) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS; (c) D. Wie and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef.
- For recent Reports see: (a) S. Semwal and J. Choudhurry, ACS Catal., 2016, 6, 2424–2428 CrossRef CAS; (b) S. R. Flynn, O. J. Metters, I. Manners and D. F. Wass, Organometallics, 2016, 35, 847–850 CrossRef CAS; (c) S. Semwal and J. Choudhurry, Angew. Chem., Int. Ed., 2017, 56, 5556–5560 CrossRef CAS; (d) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS; (e) J. Chen, Z. Zhang, B. Li, F. Li, Y. Wang, M. Zhao, I. D. Gridnev, T. Imamoto and W. Zhang, Nat. Commun., 2018, 9, 5000 CrossRef; (f) H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballman, F. D. Proft and S. Haredr, Nat. Catal., 2018, 1, 40–47 CrossRef CAS; (g) Y. Liu, F. Chen, Y.-M. He, C. Li and Q.-H. Fan, Org. Biomol. Chem., 2019, 17, 5099–5105 RSC.
- (a) H. Li, Z. Dong, P. Wang, F. Zhang and J. Ma, React. Kinet., Mech. Catal., 2013, 108, 107–115 CrossRef CAS; (b) M. Nasrollahzadeh, New J. Chem., 2014, 38, 5544–5550 RSC; (c) T. Stemmler, F. A. Westerhaus, A.-E. Surkus, M.-M. Pohl, K. Junge and M. Beller, Green Chem., 2014, 16, 4535–4540 RSC; (d) N. M. Patil and B. M. Bhanage, Catal. Today, 2015, 247, 182–189 CrossRef CAS; (e) M. Bhardwaj, H. Sharma, S. Paul and J. H. Clark, New J. Chem., 2016, 40, 4952–4961 RSC; (f) R. J. Kalbasi and O. Mazaheri, New J. Chem., 2016, 40, 9627–9637 RSC; (g) P. Ji, K. Manna, Z. Lin, A. Urban, F. X. Greene, G. Lan and W. Lin, J. Am. Chem. Soc., 2016, 138, 12234–12242 CrossRef CAS; (h) L. Jiang, P. Zhou, Z. Zhang, Q. Chi and S. Jin, New J. Chem., 2017, 41, 11991–11997 RSC; (i) L. Jiang, P. Zhou, Z. Zhang, S. Jin and Q. Chi, Ind. Eng. Chem. Res., 2017, 56, 12556–12565 CrossRef CAS; (j) H. Sharma, M. Bhardwaj, M. Kour and S. Paul, Mol. Catal., 2017, 435, 58–68 CrossRef CAS; (k) M. N. Shaikh, M. A. Aziz, A. N. Kalanthoden, A. Helal, A. S. Hakeem and M. Bououdina, Catal. Sci. Technol., 2018, 8, 4709–4717 RSC.
- (a) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS; (b) Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769–14827 CrossRef CAS; (c) C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS.
- (a) C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 8952–8965 CrossRef CAS; (b) Y. Misumi, H. Seino and Y. Mizobe, J. Am. Chem. Soc., 2009, 131, 14636–14637 CrossRef CAS; (c) H. Seino, Y. Misumi, Y. Hojo and Y. Mizobe, Dalton Trans., 2010, 39, 3072–3082 RSC; (d) Q. Hu, Y. Hu, Y. Liu, Z. Zhang, Y. Liu and W. Zhang, Chem. – Eur. J., 2017, 23, 1040–1043 CrossRef CAS; (e) Z. Yang, Z. Ding, F. Chen, Y.-M. He, N. Yang and Q.-H. Fan, Eur. J. Org. Chem., 2017, 1973–1977 CrossRef CAS; (f) Y.-N. Duan, X. Du, Z. Cui, Y. Zeng, Y. Liu, T. Yang, J. Wen and X. Zhang, J. Am. Chem. Soc., 2019, 141, 20424–20433 CrossRef CAS.
- (a) P. D. Pham, P. Bertus and S. Legoupy, Chem. Commun., 2009, 6207–6209 RSC; (b) V. Kumar, S. Sharma, U. Sharma, B. Singh and N. Kumar, Green Chem., 2012, 14, 3410–3414 RSC; (c) V. Kumar, U. Sharma, P. K. Verma, N. Kumar and B. Singh, Adv. Synth. Catal., 2012, 354, 870–878 CrossRef CAS; (d) R. Kumar, E. Gravel, A. Hagège, H. Li, D. Verma, I. N. N. Namboothiri and E. Doris, ChemCatChem, 2013, 5, 3571–3575 CrossRef CAS; (e) Y. Corre, W. Iali, M. Hamdaoui, X. Trivelli, J.-P. Djukic, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2015, 5, 1452–1458 RSC; (f) R. J. Maya, S. Poulose, J. John and R. L. Varma, Adv. Synth. Catal., 2017, 359, 1177–1184 CrossRef CAS.
- (a) E. A. Artiukha, A. L. Nuzhdin, G. A. Bukhtiyarova, S. Y. Zaytsev, P. E. Plyusnin, Y. V. Shubin and V. I. Bukhtiyarov, Catal. Sci. Technol., 2015, 5, 4741–4745 RSC; (b) Q. Zhang, S.-S. Li, M.-M. Zhu, Y.-M. Liu, H.-Y. He and Y. Cao, Green Chem., 2016, 18, 2507–2513 RSC.
- (a) B. S. Takale, S. M. Tao, X. Q. Yu, Z. J. Feng, T. Jin, M. Bao and Y. Yamamoto, Org. Lett., 2014, 16, 2558–2561 CrossRef CAS; (b) M. Kumar, V. Bhatt, O. S. Nayal, S. Sharma, V. Kumar, M. S. Thakur, N. Kumar, R. Bal, B. Singh and U. Sharma, Catal. Sci. Technol., 2017, 7, 2857–2864 RSC.
- (a) Y. S. Demidova, I. L. Simakova, M. Estrada, S. Beloshapkin, E. V. Suslov, D. V. Korchagina, K. P. Volcho, N. F. Salakhutdinov, A. V. Simakov and D. Y. Murzin, Appl. Catal., A, 2013, 464–465, 348–356 CrossRef CAS; (b) Y. S. Demidova, E. V. Suslov, I. L. Simakova, D. V. Korchagina, E. S. Mozhajcev, K. P. Volcho, N. F. Salakhutdinov, A. Simakov and D. Y. Murzin, J. Mol. Catal. A: Chem., 2017, 426, 60–67 CrossRef CAS; (c) Y. S. Demidova, E. V. Suslov, I. L. Simakova, K. P. Volcho, N. F. Salakhutdinov, A. Simakov and D. Y. Murzin, Mol. Catal., 2017, 433, 414–419 CrossRef CAS; (d) Y. S. Demidova, E. V. Suslov, I. L. Simakova, D. V. Korchagina, E. S. Mozhajcev, K. P. Volcho, N. F. Salakhutdinov, A. Simakov and D. Y. Murzin, J. Catal., 2018, 360, 127–134 CrossRef CAS.
- (a) A. Stuetz, A. Georgopoulos, W. Granitzer, G. Petranyi and D. Berney, J. Med. Chem., 1986, 29, 112–125 CrossRef CAS; (b) R. K. Sharma, Y. Monga, A. Puri and G. Gaba, Green Chem., 2013, 15, 2800–2809 RSC.
- A. A. Voronov, K. A. Alekseeva, E. A. Ryzhkova, V. V. Zarubaev, A. V. Galochkina, V. P. Zaytsev, M. S. Majik, S. G. Tilve, A. V. Gurbanov and F. I. Zubkov, Tetrahedron Lett., 2018, 59, 1108–1111 CrossRef CAS.
- B. Kovács, R. Savela, K. Honkala, D. Y. Murzin, E. Forró, F. Fülöp and R. Leino, ChemCatChem, 2018, 10, 2893–2899 CrossRef.
- N. D. Shcherban, P. Mäki-Arvela, A. Aho, S. A. Sergiienko, P. S. Yaremov, K. Eränen and D. Y. Murzin, Catal. Sci. Technol., 2018, 8, 2928–2937 RSC.
- J. Wu, W. Hua, Y. Yue and Z. Gao, Acta Phys.-Chim. Sin., 2020, 36, 1904066 Search PubMed (1–7).
- H. Bernas, I. Simakova, P. Mäki-Arvela, I. P. Prosvirin, R. Leino and D. Y. Murzin, Catal. Lett., 2012, 142, 690–697 CrossRef CAS.
- I. L. Simakova, Y. S. Demidova, A. V. Simakov and D. Y. Murzin, J. Sib. Fed. Univ., Chem., 2019, 12, 536–549 Search PubMed.
- V. N. Panchenko, E. A. Paukshtis, D. Y. Murzin and I. L. Simakova, Ind. Eng. Chem. Res., 2017, 56, 13310–13321 CrossRef CAS.
- D. Y. Murzin, Catal. Sci. Technol., 2016, 6, 5700–5713 RSC.
- E. Toukoniitty, P. Mäki-Arvela, J. Kuusisto, V. Nieminen, J. Päivarinta, M. Hotokka, T. Salmi and D. Y. Murzin, J. Mol. Catal. A: Chem., 2003, 192, 135–151 CrossRef CAS.
- R. L. Augustine, Heterogeneous Catalysis for the Synthetic Chemist, Marcel Dekker, New York, 1996 Search PubMed.
- J. Barrault, A. Derouault, G. Courtois, J. M. Maissaint, J. C. Dupin, C. Guimon, H. Martinez and E. Dumitriu, Appl. Catal., A, 2004, 262, 43–51 CrossRef CAS.
- (a) H. Hagiwara, J. Hamaya, T. Hoshi and C. Yokoyama, Tetrahedron Lett., 2005, 46, 393–395 CrossRef CAS; (b) K. A. Ostrowski, D. Lichte, M. Stuck and A. J. Vorholt, Tetrahedron, 2016, 72, 592–598 CrossRef CAS.
- S. Tuokko, P. M. Pihko and K. Honkala, Angew. Chem., Int. Ed., 2016, 55, 1670–1674 CrossRef CAS.
- T. Wang, J. Ibañez, K. Wang, L. Fang, M. Sabbe, C. Michel, S. Paul, M. Pera-Titus and P. Sautet, Nat. Catal., 2019, 2, 773–779 CrossRef CAS.
- H. Ghafuri and M. Roshani, RSC Adv., 2014, 4, 58280–58286 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- G. Perez, S. Fuentes, V. Petranovskii and A. Simakov, Catal. Lett., 2006, 110, 53–60 CrossRef.
- S. G. Gregg and K. S. W. Sing, Adsorption, surface area and porosity, Acad. press, New York, 1982 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- G. Horváth and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef.
- (a) J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B, 2005, 71, 035109 CrossRef; (b) J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dulak, L. Ferrighi, J. Gavnholt, C. Linsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS; (c) A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennigs, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasberg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygensen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef.
- (a) M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS; (b) J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B, 2012, 85, 235149 CrossRef.
- A. Held and M. Walter, J. Chem. Phys., 2014, 141, 174108 CrossRef.
- (a) G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS; (b) G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- A. A. Peterson, Top. Catal., 2014, 57, 40–53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01886d |
| This journal is © The Royal Society of Chemistry 2021 |