
Identification of electron-rich mononuclear Ni atoms on TiO2-A distinguished from Ni particles on TiO2-R in guaiacol hydrodeoxygenation pathways†
Xiaoqiang
Zhang
ab,
Peifang
Yan
a,
Bin
Zhao
c and
Z. Conrad
Zhang
 *a
*a
aState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: zczhang@yahoo.com
bUniversity of Chinese Academy of Sciences, Beijing 10049, P. R. China
cState Key Laboratory of Fine Chemicals, PSU-DUT Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China
First published on 31st October 2020
Abstract
The electronic and structural differences of anatase TiO2 (TiO2-A) and rutile TiO2 (TiO2-R) supported Ni catalysts are studied to understand the distinctively different catalytic pathways in guaiacol hydrodeoxygenation over Ni/TiO2-A and Ni/TiO2-R catalysts. Temperature programmed reduction profiles of NiO/TiO2-A and NiO/TiO2-R reveal two types of metal–support interactions. NiO on TiO2-R requires much higher temperature to reduce than NiO on TiO2-A by hydrogen reduction. Surface TiO2-A is partially reduced together with NiO at low temperature driven by strong interaction between reduced Ni and the reducible TiO2-A surface. In situ X-ray photoelectron spectroscopy (XPS), high resolution TEM (HRTEM), and diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy of adsorbed CO reveal two types of reduced Ni species on Ni/TiO2-A: Ni0 particles and atomically dispersed electron-rich Niδ− atoms. While Ni0 particles are formed only at high Ni loadings, they are fully encapsulated by an overlayer of partially reduced TiOx, making the surface of Ni particles totally inaccessible to CO and to reactants. The electron-rich Niδ− atoms are favorably formed at all Ni loadings on the TiO2-A surface and show only linearly adsorbed CO in the DRIFT spectra. Electron transfer from Ti and O on the TiO2-A surface to atomically dispersed Ni is indicated by the observation of dominant electron-rich Niδ− species in conjunction with Ti(4+δ)+ and O(2−δ)− on Ni/TiO2-A following H2 reduction. In contrast, the chemical states of metallic Ni0 and Ti4+ remain unchanged on Ni/TiO2-R after H2 reduction. The atomically dispersed Niδ− species, formed only on the Ni/TiO2-A surface, is proposed as the active site responsible for the selective hydrodeoxygenation of guaiacol to phenolics. In contrast, the metallic Ni0 particles formed on the Ni/TiO2-R surface catalyze the typical catalytic hydrogenation, preferentially saturating the aromatic ring.
1. Introduction
Strong metal–support interaction (SMSI) generally exists between reduced metal catalysts supported on partially reducible oxide supports.1–5 SMSI effects are manifested by metal particle encapsulation4,6 and stabilization7 by partially reduced supports.5,8,9 For supported metallic hydrogenation catalysts, the surfaces of metal particles are the active sites in dissociating molecular H2 into H atoms, thereby lowering the activation energy of hydrogenation reactions. Most group VIII metals such as Pt and Ni are well known hydrogenation catalysts.10,11 These metals typically catalyze the full hydrogenative saturation of aromatic rings. Forming alloys with inert or less active metals such as Au,12 Cu (ref. 13) and Fe (ref. 14) may alter the hydrogenation selectivity by changing the ensemble structure of the active metal surface. However, this strategy often causes reduced dispersion of active metals. The SMSI effect is found to cause the formation of a reducible oxide support overlayer on the reduced metal particle surface, resulting in altered catalytic selectivity for hydrogenation reactions.4,15Among various supports, TiO2 has been found to exhibit the SMSI effect for improved partial hydrogenation selectivity of supported metal catalysts in reactions such as acetylene hydrogenation to ethylene,9,16,17 CO2 hydrogenation to methanol,18 and α,β-unsaturated aldehyde hydrogenation to α,β-unsaturated alcohols.19,20 On a Pd/anatase TiO2 (TiO2-A) catalyst, the generation of Ti3+ defect sites was proposed to favor the desorption of ethylene in selective acetylene hydrogenation, while a Pd/rutile TiO2 (TiO2-R) catalyst resulted in full hydrogenation of acetylene to ethane due to the lack of the SMSI effect.9,16 Recent studies have shown that the SMSI effect can alter the metal electronic properties via charge transfer between the metal and support in TiO2 supported metal catalysts.6,21–25 Negatively charged Au species (Auδ−) were found in Au/TiO2-A as a result of migration of TiO2 onto the metal surface due to the SMSI effect.6,22 The Auδ− species capable of CO chemisorption was attributed to largely enhanced catalytic activity in the water gas shift reaction.22 It has also been proposed that the charge transfer at the perimeter sites of the supported metal and the supporting metal oxide enhanced the activity.24,25 While TiO2-A has been widely reported to exhibit intriguing functions to alter the electronic properties of the supported metal particles for promoting catalytic performances, there remains a lack of full understanding on the origins of the electronic effects on the metal relating to the surface structure of the support.
Guaiacol (2-methoxyphenol) is a representative product from lignin-derived bio-oils. Catalytic hydrodeoxygenation of guaiacol to sustainable fuels and chemicals has been widely studied.26,27 Guaiacol comprises three C–O bonds, Caryl–OH, Caryl–OCH3, and CarylO–CH3, as well as an aromatic ring. Hydrogenative saturation of the aromatic ring readily takes place on unmodified metal particle surfaces, whereas the various types of C–O bonds may be cleaved at higher temperatures by hydrogenolysis or simply dehydration following the saturation of the aromatic ring in phenolics. For example, with a Ni catalyst supported on non-reducible metal oxides, saturated hydrocarbons were typically the main hydrogenation products.28–31 Zhang et al.28 showed that, with Ni supported on ZrO2 and SiO2–ZrO2, cyclohexane was the main product in 80% selectivity at a guaiacol conversion of 40%. Recently, TiO2-A supported Ni was found to exhibit high selectivity to phenolics in hydrodeoxygenation of guaiacol,32,33 in contrast to the typically known catalytic characteristics of metallic Ni catalysts in saturating aromatic rings. Modification with Mo as in a Ni–Mo/TiO2-A catalyst resulted in partial suppression of the aromatic ring saturation with 60% selectivity to phenolics in guaiacol conversion.32
In a recent study, we identified a phenomenon of cross migration between Ni from large Ni particles and partially reduced TiOx in a physical mixture of the Ni particles and TiO2-A under H2.33 It was verified that the migration of partially reduced TiOx fully encapsulated the Ni particles due to the SMSI effect, causing the Ni particles to become totally inaccessible to reactants including H2 and therefore totally inactive in hydrogenation, while the migration of Ni species over to the TiO2-A support surface produced active and selective catalytic sites for producing phenolics in guaiacol hydrogenation. These observations helped to establish the passivated nature of the supported Ni particles formed on catalysts prepared by the conventional impregnation method with high Ni loadings on a TiO2-A support. Although the active Ni sites for selective hydrodeoxygenation of guaiacol to phenolics were recognized as highly dispersed Ni species invisible under high resolution transmission electron microscopy (HRTEM), the electronic state and the structure of the Ni species remain to be unveiled. In comparison, TiO2-R supported Ni catalysts did not show similar results. Therefore, the aim of this work is to study the electronic and structural properties of the active sites on the Ni/TiO2-A catalyst and further to understand the fundamental differences of the supported Ni on the surfaces of TiO2-A and TiO2-R. Consequently, a correlation between the active sites and the mechanistic pathways associated with each of the catalyst in guaiacol conversions is established.
2. Experimental
2.1. Catalyst preparation
Commercial 40 nm anatase TiO2 (TiO2-A) and 40 nm rutile TiO2 (TiO2-R) with a purity of 99.9% were obtained from Aladdin. Ni/TiO2-A and Ni/TiO2-R were prepared by the incipient wetness impregnation method using an aqueous solution of Ni(NO3)2 (Aladdin, 99.9%). The samples were dried overnight at 120 °C, then calcined by heating to 400 °C at a heating rate of 10 °C min−1 and maintained at 400 °C for 4 h in dry air. The calcined samples were further reduced in 10 vol% H2/Ar for 1 h at specified temperatures. For example, when Ni/TiO2-A was reduced in hydrogen for 1 h at 400 °C, the sample was denoted as Ni/TiO2-A 400/1 h.2.2. Catalyst characterization
The Ni content of the samples was analyzed by using a PerkinElmer Optima 8000 inductively coupled plasma-optical emission spectrometer (ICP-OES). The samples were dissolved in mixed nitric acid and hydrofluoric acid under ultrasound before analysis.The X-ray diffraction (XRD) patterns of the samples were measured on a PANalytical X'Pert powder X-ray diffractometer equipped with a graphite monochromator and Cu Kα radiation source. The measurements were conducted using a PIXcel 1D detector and operated at 40 kV. The materials were identified by comparing the diffraction data with reference patterns in the database (PDF2-2004).
Temperature-programmed reduction in hydrogen (H2-TPR) was performed on a Micromeritics AutoChem II 2920 chemisorption analyzer. For the H2-TPR experiment, approximately 80 mg of sample was placed in a tubular quartz reactor. The sample was reduced in a 10% H2/Ar stream by heating from ambient temperature to 750 °C at a heating rate of 10 °C min−1. The consumed hydrogen was detected by using a thermal conductivity detector (TCD) and quantified by using a calibration plot based on quantitative measurement of Ag2O TPR profiles using a similar protocol.
In situ X-ray photoelectron spectroscopy (in situ XPS) patterns were recorded on a ThermoFisher ESCALAB 250Xi equipped with a monochromated Al Kα (1486.6 eV) radiation source. Prior to the analysis, the samples were pressed into pellets which were in situ reduced at 400 °C for 1 h after reaching 400 °C from ambient temperature with a heating rate of 10 °C min−1 in 10% H2/Ar. The C1s peak was used as the reference at 284.6 eV with a precision of 0.02 eV.
High-resolution transmission electron microscopy (HRTEM) images of the samples were obtained on a JEM-2100 microscope operated at 200 kV. The samples were suspended in ethanol. A few drops of the suspension were added to the TEM grid for TEM measurements. The distributions of Ni particle sizes were calculated by measuring more than 100 particles in the TEM image using Nano Measure 1.2.
Diffuse reflectance infrared Fourier transform (DRIFT) spectra of adsorbed CO were collected in an in situ reaction cell on a Thermo Scientific Nicolet iS50 equipped with an MCT detector. Prior to analysis, the sample was loaded into the reaction cell and reduced in 10% H2/Ar with a temperature ramp of 10 °C min−1 to a specified temperature for 1 h and was then purged in Ar and cooled to 30 °C. DRIFT spectra were recorded after CO chemisorption (5% CO/Ar) at 30 °C for 10 minutes and subsequently Ar was used to remove gaseous CO at 50 mL min−1. The background and catalyst sample spectra were scanned 64 times at one setting with a 4 cm−1 resolution. CAUTION: toxic gaseous Ni carbonyl could be formed (and was indeed formed) and the use of carbonyl traps for DRIFT experiments is a necessary safety requirement.
2.3. Reaction study
Guaiacol conversion under H2 pressure was performed in a sealed 50 mL stainless steel batch reactor (Beijing Century Senlong Experimental Apparatus Co. Ltd.). In a typical run, the reactor was charged with a mixture of 1.2 g guaiacol in 25 mL decane and 0.25 g of catalyst. The reactor was purged with N2 for 10 min and pressurized with 4 MPa of H2. To allow comparison among the different catalysts, the same reaction conditions were used for all catalysts. After reaction, 0.2394 g n-tetradecane as an internal standard and 30 mL ethanol were added into the reactor. The liquid products were identified by GC–MS (Agilent 7890A-5975C, HP-5MS) and quantified by gas chromatography (Agilent 7890A) using a flame ionization detector (FID) and an HP-5 column (30 m × 0.32 mm × 0.25 μm). The gas product was analyzed by gas chromatography (Agilent 7890A) using a FID and an HP-AL/S (25 m × 0.32 mm × 8 μm).The conversion of guaiacol (Xguaiacol, %), the product selectivity (Sproduct i, %), and the product yield (Yproduct i, %) were calculated according to eqn (1)–(3) below. In these equations, ninitial guaiacol is the initial amount of guaiacol (mol), nfinal guaiacol is the final amount of guaiacol (mol), nproduct i is the amount of product i in the reaction mixture and nreacted guaiacol is the amount of guaiacol that reacted.
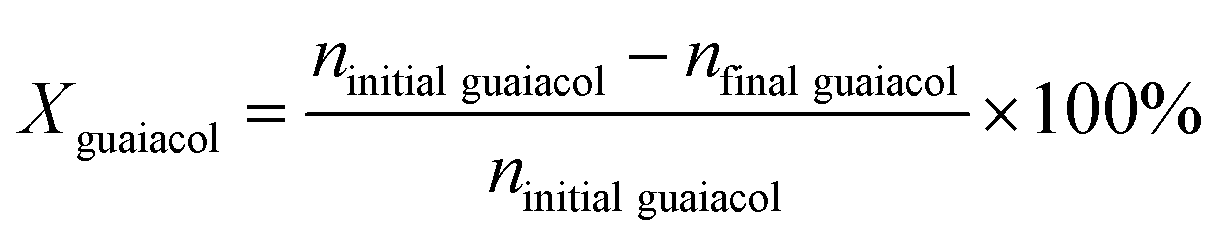 | (1) |
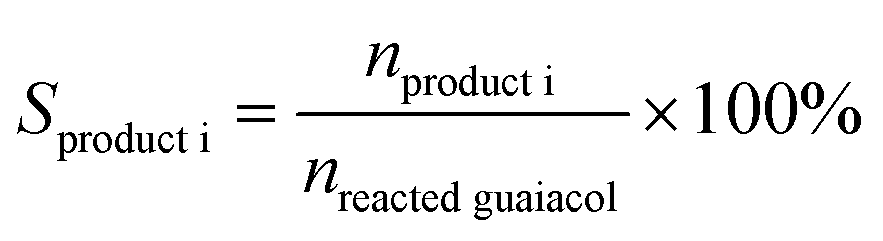 | (2) |
 | (3) |
3. Results and discussion
3.1. Catalyst characterization results
In the first phase of this study, we prepared catalysts with nearly the same Ni loading on TiO2-A and TiO2-R supports (Table 1). A nominal Ni loading of 3 wt% on TiO2-A and TiO2-R was chosen to ensure that metallic Ni particles were formed on both supports after H2 reduction. A threshold value of 1 wt% Ni loading was found in a previous study for the formation of Ni particles on TiO2-A, which became clearly observed in HRTEM when the Ni loading was above 2 wt%.33 A Ni loading of 3 wt% was an appropriate choice because higher Ni loadings above 2 wt% up to 8 wt% was found to have little contribution to the catalytic hydrodeoxygenation performance on the TiO2-A support. Ni loadings of 0.5 wt% and below were also systematically studied to characterize the properties of the catalytically responsible but invisible Ni species on TiO2-A under HRTEM.33The accurate Ni loading on the nominal 3% Ni/TiO2-A sample was 2.85 wt% based on ICP-OES elemental measurements, and that on the nominal 3% Ni/TiO2-R was 2.91 wt% (Table 1). For the samples reduced at 400 °C in H2 for 1 h, the samples 3% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-R 400/1 h are named for simplicity of presentation.
The X-ray diffractograms of the supports and catalysts are shown in Fig. 1. The XRD patterns of the 3% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts are identical to that of TiO2-A and TiO2-R,34 respectively. There is no visible bulk phase change in the XRD patterns, and no peak is attributable to Ni and NiO,30 indicating that no big Ni and NiO particles are formed from the reduction of supported Ni species.
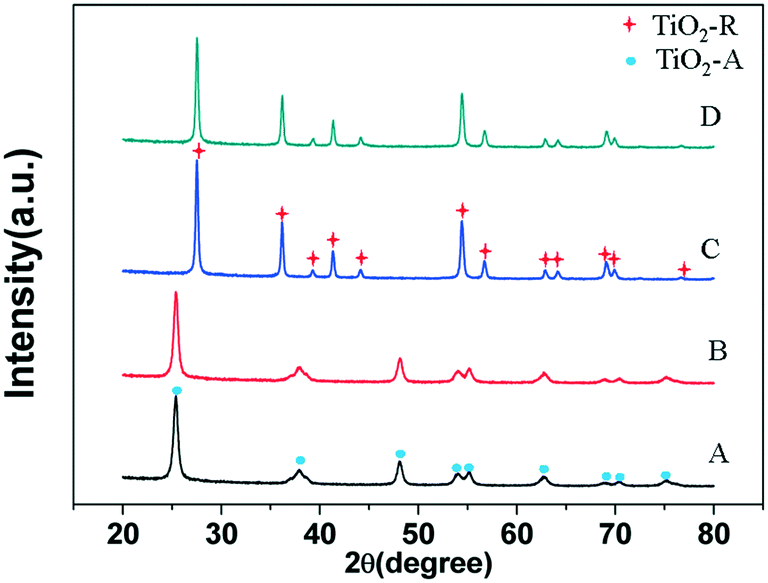 | ||
| Fig. 1 X-ray diffractograms of (A) TiO2-A; (B) 3% Ni/TiO2-A 400/1 h; (C) TiO2-R and (D) 3% Ni/TiO2-R 400/1 h. | ||
The HRTEM images of the supports, and the 3% Ni/TiO2-R 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts are shown in Fig. 2. Ni nanoparticles dominantly in the size range of 1–2 nm were formed for both the 3% Ni/TiO2-R 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts (Fig. 2E and F). The observation of the Ni particles on these two TiO2 supports are expected at Ni loadings above 2 wt% on TiO2-A,33 but Ni particles were formed at low Ni loadings (e.g. 0.5 wt%) only on TiO2-R (will be discussed below).
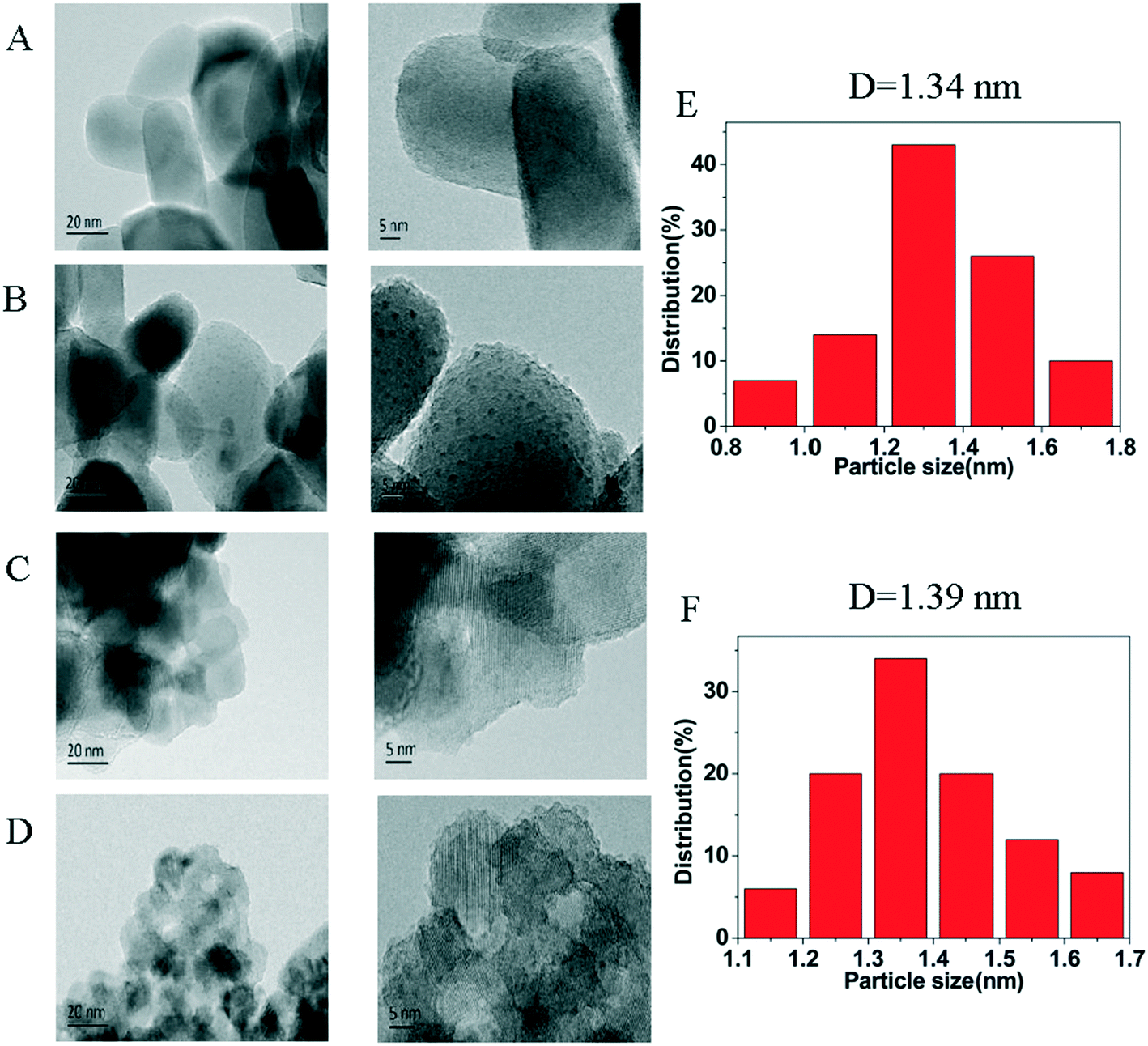 | ||
| Fig. 2 HRTEM images of (A) TiO2-R; (B) 3% Ni/TiO2-R 400/1 h; (C) TiO2-A and (D) 3% Ni/TiO2-A 400/1 h; the histograms of the size distributions of Ni nanoparticles for 3% Ni/TiO2-R 400/1 h (E) and for 3% Ni/TiO2-A 400/1 h (F). | ||
Fig. 3 shows the temperature programmed reduction (TPR) profiles of the supports and the catalysts in 10% H2/Ar. The peaks are a measure of H2 consumption in the reduction of catalyst components at a temperature up to 750 °C. Table 2 shows the H2 consumption profiles of the 3% Ni/TiO2-R and 3% Ni/TiO2-A catalysts. While the H2/Ni ratio was 0.92 (Table 2) for the 3% Ni/TiO2-R catalyst, in contrast, a much higher H2/Ni ratio of 1.77 was determined for the 3% Ni/TiO2-A catalyst at a temperature above 450 °C. Although significantly more H2 was consumed compared to that required by the stoichiometry for the full reduction of Ni2+ species in the reduction of the 3% Ni/TiO2-A catalyst, only one intense TPR peak appeared at 260 °C on this catalyst. The large excess consumption of H2 in the TPR indicated a substantially enhanced reduction of surface TiO2-A at the temperature NiO was reduced,20 although not all Ni2+ was reduced (according to the XPS results, to be discussed below). The concomitant reduction of both NiO and the reducible TiO2-A together at a low temperature is indicative of a thermodynamically favorable interaction between the reduced Ni and the partially reduced TiO2-A. In comparison, the reduction of NiO on TiO2-R was much more energetically demanding as evidenced by the TPR profile of the 3% Ni/TiO2-R catalyst extending up to 550 °C. The high NiO reduction temperature on the 3% Ni/TiO2-R catalyst suggests a strong interaction between NiO and the non-reducible TiO2-R (Fig. 3). Therefore, the difference in the TPR profiles between the 3% Ni/TiO2-R and the 3% Ni/TiO2-A catalysts may be attributed to the different types of interactions between NiO and reduced Ni with titania of different crystalline structures.
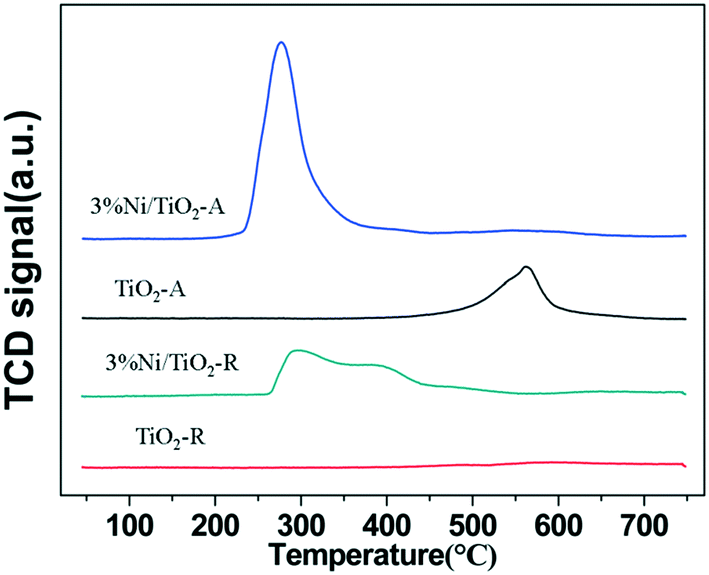 | ||
| Fig. 3 H2-TPR profiles of the supports and supported Ni catalysts. | ||
As first-row transition metals, the atomic numbers of Ni and Ti are close in the periodic table, making it difficult to obtain sufficient contrast to distinguish between Ni and Ti atoms by HRTEM. Similarly, Ni with a lower X-ray absorption energy of 8332.8 eV at its K-edge, supported on TiO2 with a Ti K-edge absorption energy of 4966.4 eV,35 and the strong TiO2 X-ray absorption background would make it difficult to obtain conclusive EXAFS data on Ni atoms, particularly with poor sensitivity at low Ni loadings (<0.5 wt%) below the threshold loading of 1 wt%, which is essential to ensure the absence of large Ni particles.33 Therefore, in this work, we resort to XPS and DRIFT spectroscopy for the characterization of the Ni species.
X-ray photoelectron spectroscopy (XPS) was performed to study the electronic state of Ni, as a result of the interaction of Ni with titania after in situ reduction at 400 °C (Fig. 4). The results of quantitative analyses of Ni, Ti, and O species in various chemical states based on standard curve fitting for the 3% Ni/TiO2-R 400/1 h, 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts are summarized in Table 3. All unreduced samples of the 3% Ni/TiO2-R, 3% Ni/TiO2-A and 0.5% Ni/TiO2-A catalysts showed signatures of Ni2+ with the Ni 2p binding energy at 855.6 eV, and an associated satellite peak at 861.2 eV (Fig. S1A†). The peaks at 852.5, 855.6, and 861.2 eV on 3% Ni/TiO2-R after in situ reduction correspond to the Ni 2p binding energies of Ni0, Ni2+ and the satellite, respectively (Fig. 4A1).5,36 In contrast to the 3% Ni/TiO2-R 400/1 h catalyst which showed only reduced Ni0 species at 852.5 eV, the fitting of the XPS spectrum of the 3% Ni/TiO2-A 400/1 h catalyst revealed two reduced Ni species, one at 852.5 eV (Ni0), and another at 851.9 eV (Fig. 4A2). For the 0.5% Ni/TiO2-A 400/1 h catalyst, only one reduced Ni species at a Ni 2p binding energy of 851.9 eV was observed (Fig. 4A3). Previously, electron-rich Ni species were reported for Ni@TiO2−x (ref. 5) and Ni/ZnO (ref. 37) catalysts with a Ni 2p binding energy of 851.0 eV and 851.8 eV, respectively. Similarly, the Ni species with a binding energy of 851.9 eV found only on the TiO2-A supported Ni catalysts in this work is attributed to electron-rich Ni (Niδ−) species. As will be discussed below (Fig. 6), DRIFT spectroscopy of adsorbed CO further supports the presence of the Niδ− species. On the 3% Ni/TiO2-R 400/1 h catalyst, the fraction of the Ni0 species (852.5 eV) was 16.9% (Table 3). In comparison, the fraction of the Ni0 species was 37.4%, in addition to 9.9% being the Niδ− species on the 3% Ni/TiO2-A 400/1 h catalyst. The total amount of reduced Ni on the 3% Ni/TiO2-A 400/1 h catalyst was nearly 3 times that on the 3% Ni/TiO2-R 400/1 h catalyst. The much lower fraction of the reduced Ni species on the 3% Ni/TiO2-R 400/1 h catalyst is consistent with the difficulty in reducing the NiO on TiO2-R as shown by TPR (Fig. 3). The multiple peaks spanning over a wide temperature range in the TPR profiles for the 3% Ni/TiO2-R 400/1 h catalyst (Fig. 3) may suggest some, although limited, reduction of TiO2 that consumed a fraction of the measured H2 uptake. The relatively low H2 consumption corresponding to a H2/Ni ratio of 0.92 together with the poor in situ reducibility of NiO on the 3% Ni/TiO2-R 400/1 h catalyst prior to XPS measurements further supports the strong interaction of NiO with the dominant TiO2-R crystalline surfaces.
 | ||
| Fig. 4 (A) In situ XPS Ni 2p spectra of (A1) 3% Ni/TiO2-R 400/1 h, (A2) 3% Ni/TiO2-A 400/1 h, and (A3) 0.5% Ni/TiO2-A 400/1 h; (B) in situ XPS Ti 2p spectra of (B1) 3% Ni/TiO2-R 400/1 h, (B2) 3% Ni/TiO2-A 400/1 h, and (B3) 0.5% Ni/TiO2-A 400/1 h; (C) in situ XPS O 1s spectra of (C1) 3% Ni/TiO2-R 400/1 h, (C2) 3% Ni/TiO2-A 400/1 h, and (C3) 0.5% Ni/TiO2-A 400/1 h. | ||
| Sample | Ni species (%) | Ti species (%) | O species (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Niδ− | Ni0 | Ni2+ | Ti4+ | Ti(4+δ)+ | O2− | O(2−δ)− | OH− | |
| n.d.: not detected. | ||||||||
| 3% Ni/TiO2-R 400/1 h | n.d. | 16.9 | 83.1 | 100.0 | n.d. | 88.9 | n.d. | 11.1 |
| 3% Ni/TiO2-A 400/1 h | 9.9 | 37.4 | 52.7 | 22.5 | 77.5 | 31.9 | 58.4 | 9.7 |
| 0.5% Ni/TiO2-A 400/1 h | 38.1 | n.d. | 62.9 | 34.5 | 65.5 | 33.8 | 47.5 | 18.7 |
For the 0.5% Ni/TiO2-A 400/1 h catalyst, the fraction of the Niδ− species was as high as 38.1% with the absence of Ni0 species. No Ni particles were observed under HRTEM at this low Ni loading on the support.33 For 3% Ni/TiO2-A 400/1 h, Ni0 species appeared in XPS and Ni nanoparticles were unambiguously observed (Fig. 2). We calculated the amount of Niδ− species on both the 3% Ni/TiO2-A 400/1 h and the 0.5% Ni/TiO2-A 400/1 h catalysts. The 9.9% being the Niδ− species on the 3% Ni/TiO2-A 400/1 h catalyst corresponded to 47.9 μmol Niδ− species per gram of catalyst (g-catalyst). And on the 0.5% Ni/TiO2-A 400/1 h catalyst, the amount of the Niδ− species corresponded to 30.5 μmol g-catalyst−1. Therefore, the reduction of 0.5 wt% Ni on TiO2-A did not form sufficient Niδ− species to cover the surface in view of the increased Niδ− species on the 3% Ni/TiO2-A 400/1 h catalyst and the absence of Ni0 species on the 0.5% Ni/TiO2-A 400/1 h catalyst. These results suggest that formation of Niδ− species as a result of SMSI between the reduced Ni atoms and the partially reduced surface TiO2-A is thermodynamically favored compared to the formation of Ni0 metal clusters. As the formation of Ni nanoparticles with increasing Ni loading above 2 wt% on the TiO2-A support did not increase guaiacol conversions due to encapsulation by an overlayer of TiOx,33 we consider that the amount of Niδ− species on 3% Ni/TiO2-A 400/1 h represents the maximum, which is further demonstrated by DRIFT spectroscopy of adsorbed CO below (Fig. 6C, to be discussed below). Accordingly, the surface density of the Niδ− species on 3% Ni/TiO2-A 400/1 h, which has a BET surface area of 98.1 m2 g−1, is estimated at 3.2 × 10−3 Niδ− per nm2. This small value suggests that the Niδ− species are likely limited to the surface of the reducible TiO2-A, although oxygen vacancies may be also present in the bulk of the support.
The Ti 2p XPS spectra show the Ti state of in situ reduced samples (Fig. 4B) and unreduced samples (Fig. S1B†). The binding energy at 458.3 eV corresponds to the typical Ti4+ species, according to a previous assignment.5,6 However, a binding energy at 458.9 eV, 0.6 eV higher than that of the Ti4+ species at 458.3 eV, for the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts (Fig. 4B2 and B3, respectively) may be attributed to more electron-deficient Ti(4+δ)+ species.38,39 Charge transfer may have taken place, particularly in view of the Niδ− species generated on the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts. In a previous study with Ni dispersed on a TiO2-A surface after reduction at 500 °C, electron transfer from TiO2 to Ni was proposed and ascribed to lower Fermi levels of Ni instead of TiO2.40 Hence, the most probable explanation for the formation of the Ti(4+δ)+ species, concomitant to the formation of the Niδ− species only on the TiO2-A surface, may be similarly attributed to electron transfer favoring atomically dispersed Niδ− species. The co-existence of the Ti(4+δ)+ and Niδ− species only after H2 reduction on the Ni/TiO2-A surface was further verified by oxidation of the reduced sample in air, which caused the simultaneous disappearance of the Niδ− and Ti(4+δ)+ species in XPS measurements. In addition, the Niδ− and Ti(4+δ)+ species were absent in the XPS spectrum of the reduced 3% Ni/TiO2-R 400/1 h sample which only showed metallic Ni0.
Fig. 4C1 shows the fitted O 1s binding energies corresponding to the titania lattice (529.7 eV) and OH− (531.0 eV).41 The binding energy at 530.3 eV on the 3% Ni/TiO2-A 400/1 h (Fig. 4C2) and 0.5% Ni/TiO2-A 400/1 h catalysts (Fig. 4C3) was 0.6 eV higher than that of the O2− species (529.7 eV) on the 3% Ni/TiO2-R 400/1 h catalyst. A previous study reported that oxygen vacancies are introduced on TiO2 by co-milling of TiO2 nanoparticles with poly(tetrafluoroethylene) (PTFE) powder. A shift of the O2− species to a higher binding energy was observed and attributed to electron transfer to atoms neighboring the oxygen vacancies as a result of the co-milling.42 In Fig. 4C2 and C3, the binding energy shift from the typical O2− species at 529.7 eV to O(2−δ)− species at 530.3 eV on the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts occurred concomitantly with the binding energy shift of the Ti4+ species at 458.3 eV to the Ti(4+δ)+ species at 458.9 eV. Taking all the XPS results together, it may be suggested that electrons bound to oxygen and titanium ions shift toward Ni atoms occupying the oxygen vacancies. In comparison to the unchanged 2p binding energies of Ni0 and Ti4+ (Fig. 4A1 and B1) on the 3% Ni/TiO2-R 400/1 h catalyst after H2 reduction, the distinctive presence of the Niδ−, Ti(4+δ)+ and O(2−δ)− species on TiO2-A (at both 3 wt% and 0.5 wt% Ni loadings) revealed the dramatic difference in the metal–support interactions between Ni on TiO2-A and Ni on TiO2-R. As shown in Table 3, Ti(4+δ)+ and O(2−δ)− species dominated on the surface of the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts. However, the amount of Niδ− species on these catalysts was not sufficient to account for the oxygen vacancies affiliated with the Ti(4+δ)+ species on the surface. We must take into consideration the large excess of H2 consumed in the reduction step compared to that needed for the full reduction of NiO to Ni0 and Niδ− species based on quantitative analysis of the XPS profiles.
To further investigate the cause for excess H2 consumption, we prepared and characterized a series of Ni/TiO2-A catalysts of low Ni loadings by impregnation. The accurate Ni loadings on Ni/TiO2-A are shown in Table S1.†Fig. 5 shows the H2-TPR profiles of Ni/TiO2-A with Ni loadings between 0.1 and 0.5 wt%. The TiO2-A reduction peak was shifted to a lower temperature even at a Ni loading as low as 0.1 wt% on TiO2-A (Fig. 5). With increasing Ni loading in this range, the reduction temperature of TiO2-A was further decreased. The H2 consumptions for the reduction of TiO2-A were substantially higher than that for reduction of NiO as revealed by the rather high H2/Ni ratios (Table 4). The excess H2 consumption may be explained by hydrogen spillover that also led to the decreased reduction temperature of TiO2-A. Hydrogen atoms, created by dissociative chemisorption of H2 molecules on reduced Ni sites, spill over the reducible TiO2-A surface and are consumed for the reduction of TiO2-A surface sites. We attribute the excess H2 consumptions to the formation of TiO–H species,43,44 the creation of oxygen vacancies by H2O formation,45,46 and the formation of Ti–H (hydride) species.47
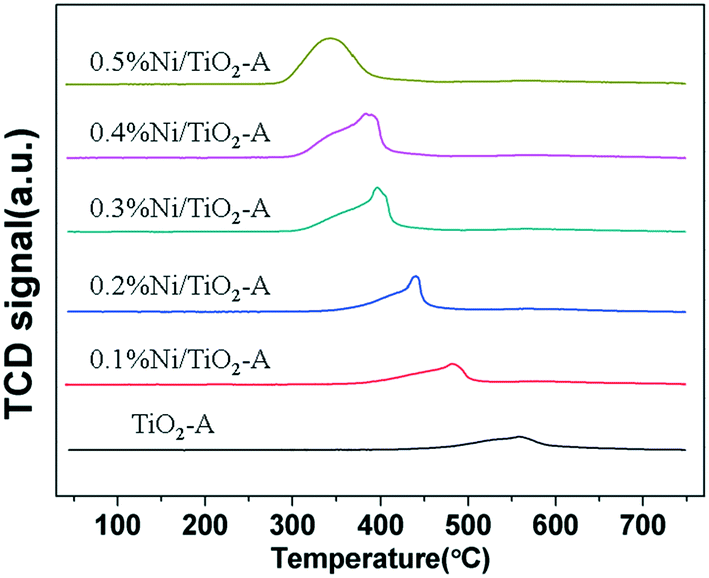 | ||
| Fig. 5 H2-TPR profiles of the TiO2-A supported Ni catalysts with low Ni loadings. | ||
Although a large excess of H2 was consumed in the reduction step, there was no Ti3+ observed in the XPS results. In a recent study on pre-reduced Ni/TiO2-A, we observed a large amount of H2 that was desorbed at 90 °C during temperature programmed desorption in Ar corresponding to a H/Ni ratio of 4.33 The significantly higher H/Ni ratio over that of a stoichiometric ratio of H/Ni of 1 typically used to measure Ni dispersion on a support indicates that adsorbed H on the surface of TiO2-A formed TiO–H species and Ti–H species. The apparent absence of a Ti3+ signal in XPS (Fig. 4B2 and B3) may be explained by hydride filling the oxygen vacancies to form Ti–H species much like the formation of the Niδ− species on the Ni/TiO2-A catalyst. The H− or Niδ− species occupying an oxygen vacancy in the tetra-coordinated [TiO4] surface is proposed to account for the transferred electrons corresponding to the change from Ti4+/O2− on unreduced Ni/TiO2-A to Ti(4+δ)+/O(2−δ)− on reduced Ni/TiO2-A.
In previous studies, Niδ− species were observed on TiO2 and ZnO supports under reducing conditions.5,37 The formation of Niδ− species was rationalized by electron transfer from TiO2 (ref. 5) or ZnO (ref. 37) due to strong metal–support interaction (SMSI). In the present work, Ni exhibited two reduced chemical states, Ni0 and Niδ−, on 3% Ni/TiO2-A 400/1 h. At the TiOx encapsulated Ni particle surface, Niδ− species could form as the outmost layer of Ni atoms that have direct access to the oxygen vacancies of an oxide overlayer, while the bulk of Ni in the inner part of the particle could remain as Ni0 species. However, this scenario where Ni0 and Niδ− species coexisted on the same Ni particle may be less likely since metallic Ni particles are electron conductors. While the XPS results showed the presence of both Ni0 and Niδ− species on the 3% Ni/TiO2-A 400/1 h catalyst (Fig. 4A2), HRTEM images of the 3% Ni/TiO2-A 400/1 h catalyst showed Ni particles of 1–2 nm on the TiO2-A surface (Fig. 2D and F). To reveal if the Ni0 and Niδ− species are associated, we turn to the characterization results of the 0.5% Ni/TiO2-A 400/1 h catalyst. Although the XPS analysis showed that the Niδ− species were the only reduced Ni species on the 0.5% Ni/TiO2-A 400/1 h catalyst, the HRTEM images of this catalyst did not show detectable Ni particles on the TiO2-A surface.33 Therefore, we propose that the Niδ− species on Ni/TiO2-A are located at the oxygen vacancies of the reducible TiO2-A surface lattice due to the SMSI between the reduced Ni atoms and the partially reduced surface TiO2-A.
The oxygen vacancies have been proposed as stabilizing sites in metal oxides to prevent the leaching and aggregation of isolated metal atoms.48–50 Oxygen vacancies on P25, as a support having mixed anatase and rutile TiO2 structures, were found to stabilize atomically dispersed Pd atoms.50 TiO2-A is a reducible oxide and the creation of oxygen vacancies on its surface resulted in enhanced electron transfer between the TiO2 and active metals.51 Sanchez et al.52 suggested that atoms or small metallic clusters were able to “drill a hole” into the support with the increase of concentration of oxygen vacancies during reduction. Studies demonstrated that atomically dispersed Pt and Rh species interacted with two O atoms from OH groups on the TiO2-A surface and thus formed Pt2+ and Rh2+ species.53–55 The metals in the ionic state were stable in oxidative treatment. In this work, however, the atomic Niδ− species is electron-rich Niδ− generated by H2 reduction of both NiO and the TiO2-A surface. This Niδ− species and the Ti(4+δ)+/O(2−δ)− species were found to disappear in subsequent oxidative treatment. Thus, the Niδ− species associated only with the reducible TiO2-A surface and absent on the TiO2-R surface indicate that the Niδ− species are not associated with two O atoms on the TiO2-A surface as for the Pt2+ and Rh2+ species. Therefore, we propose that the H− and Niδ− species are located at the oxygen vacancies as results of the electron transfer from Ti4+/O2−. The DRIFT spectroscopy of adsorbed CO below provides further evidence on the atomically isolated Niδ− species on TiO2-A. The unreduced Ni2+ and a fraction of spillover H species may be associated with the surface O2− atoms, similar to the Pt2+ and Rh2+ species.53–55 However, such unreduced Ni2+ species are not catalytically relevant in the hydrodeoxygenation of guaiacol.
To further differentiate the nature of Ni species generated on TiO2-A and TiO2-R after reduction in H2, DRIFT spectroscopy of chemisorbed CO was used to probe the electronic properties of Ni species on the supports. The DRIFT spectrum of the 3% Ni/TiO2-R 400/1 h catalyst in Fig. 6A1 shows the presence of an intense linearly adsorbed CO peak at 2090 cm−1 and bridging CO peaks at 1950 and 1834 cm−1 on reduced Ni. When the Ni loading was decreased to 0.5 wt%, the linearly adsorbed CO band was shifted to 2060 cm−1 with a decreased intensity due to the lower Ni loading. An intense bridging CO band at 1824 cm−1 was observed on the 0.5% Ni/TiO2-R 400/1 h catalyst (Fig. 6A2), again indicative of the formation of metallic Ni particles even at this low Ni loading on TiO2-R. The frequency of the linear CO peak varied with Ni loading on TiO2-R. The shift to a lower frequency at 2060 cm−1 (Fig. 6A2) has been attributed to, in accordance with the results of CO adsorption on single-crystal nickel planes,56 linear-CO adsorption on planar sites typical of the (100) or (110) planes. However, the assignment of high frequency at 2090 cm−1 is less straightforward. The widely accepted assignment of the peak at 2090 cm−1 is more than one CO molecule linearly bonded to one Ni atom with high-coordination (sub-carbonyl species).57,58 The ratio of the intensities of the bridge-CO to the linear-CO on 0.5% Ni/TiO2-R 400/1 h was higher than that on 3% Ni/TiO2-R 400/1 h. Generally, the ratio is increased with decreasing Ni particle size because smaller particles generally contain a higher concentration of defect sites than larger particles.56 The DRIFT spectrum of bridging CO (Fig. 6A2) is consistent with the formation of reduced Ni particles on 0.5% Ni/TiO2-R 400/1 h, providing additional evidence of the weak interaction of reduced Ni with the surface of TiO2-R.
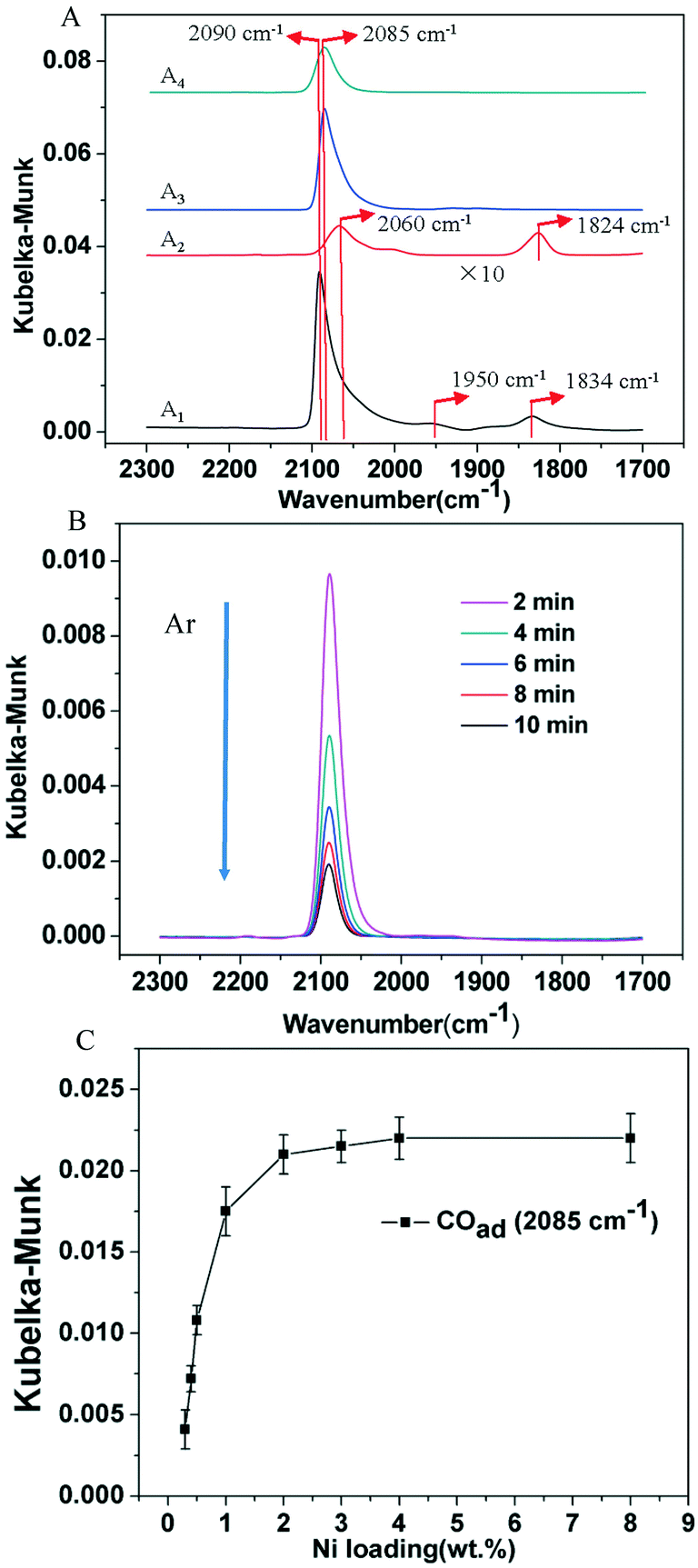 | ||
| Fig. 6 (A) In situ DRIFT spectra of CO on (A1) 3% Ni/TiO2-R 400/1 h, (A2) 0.5% Ni/TiO2-R 400/1 h, (A3) 3% Ni/TiO2-A 400/1 h and (A4) 0.5% Ni/TiO2-A 400/1 h upon CO chemisorption at 30 °C; (B) in situ DRIFT spectra of CO on the 0.5% Ni/TiO2-A 400/1 h catalyst during Ar flushing after saturated CO coverage at 30 °C; (C) Kubelka–Munk intensity of in situ DRIFT spectra of CO on Ni/TiO2-A 400/1 h with different Ni loadings. | ||
In contrast, on the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts (Fig. 6A3 and A4), only one linear CO peak at 2085 cm−1 was observed. The same frequency of linear CO peak on the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts indicates that CO adsorbed on the same Ni species in spite of the presence of Ni nanoparticles as shown by HRTEM (Fig. 2), which is consistent with Ni0 as shown by the XPS results (Fig. 4A2). Here, a red shift from 2090 cm−1 on the 3% Ni/TiO2-R 400/1 h catalyst to 2085 cm−1 on the 3% Ni/TiO2-A 400/h and 0.5% Ni/TiO2-A 400/1 h catalysts is noted. For CO adsorbed on the Ni surface, the IR absorption wavenumber of linearly adsorbed CO reflects the Ni electronic state based on the d–π feedback principle. The reproducible red shift of the absorption peak of linearly adsorbed CO on both the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts in comparison to that on TiO2-R supported metallic Ni is attributed to the Niδ− species on TiO2-A, which is in good agreement with the results of XPS. The absence of bridging CO peaks on the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts indicates the absence of accessible or available Ni particle surfaces on the catalysts.
Fig. 6B shows DRIFT spectra of 0.5% Ni/TiO2-A 400/1 h collected following the saturation of Niδ− sites by CO at 30 °C, at various purging times in Ar. It was observed that the CO stretch band remained symmetrical throughout the desorption process, and the full width at half maximum (FWHM) of the band remained constant at 25 cm−1. Moreover, the vibrational frequency remained constant as CO was desorbed. It has been proposed that the constant vibrational frequency and the FWHM of the CO band can be used to qualitatively address the question of the uniformity of metal sites distributed on a support.59,60 The isolated Pt atoms on TiO2-A exhibited narrow and constant CO stretches, which was attributed to the uniform coordination environments on the support.59 The vibration frequency of CO adsorbed on the Niδ− sites is independent of the coverage, indicating that there is no interaction between the adsorbed CO molecules. Therefore, CO must be spatially isolated on atomically dispersed Niδ− atoms. Lastly, the retained symmetry and FWHM of the vibrational band of CO on Niδ− sites during CO desorption is another strong evidence that the chemical nature of Ni–CO species is uniform, most likely located at a common Niδ− adsorption site on the TiO2-A.
The DRIFT spectra of adsorbed CO on Ni/TiO2-A 400/1 h with Ni loadings covering the range of 0.3 to 8 wt% are shown in Fig. S3.† It is important to note that the linearly adsorbed CO with the absorption peak at 2085 cm−1 was not shifted and no CO absorption peak appeared to indicate bridging CO adsorption on Ni particles over this wide range of Ni loadings, although Ni particles became visible at and above 2 wt%. The plot of the Kubelka–Munk intensity of in situ DRIFT spectra of CO on Ni/TiO2-A 400/1 h with Ni loadings of 0.3–8 wt% as shown in Fig. 6C therefore shows the dependence of Niδ− sites on Ni loading on the TiO2-A support. This plot demonstrates the linear increase of the Niδ− species with Ni loadings below 1 wt% on TiO2-A, and the maximum amount of Niδ− species on the TiO2-A surface was reached at and above 2 wt% Ni loading.
3.2. Distinguished catalytic pathways of the Ni/TiO2-R 400/1 h and Ni/TiO2-A 400/1 h catalysts in hydrodeoxygenation of guaiacol
The guaiacol conversions and product selectivities over the Ni/TiO2-A 400/1 h and Ni/TiO2-R 400/1 h catalysts shown in Table 5 are in full conformity with our recent work,33 showing clearly distinguishable reaction pathways. The major characteristic differences between the two types of catalysts in guaiacol conversions based on the results of this work (Table 5) are summarized below:| Catalystb | RTc/°C | Conv.% | C6-Ring product sel.% | Othersd | C1 sel.% | TONe | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
CH4 | CH3OH | |||||
a Reaction conditions: guaiacol 1.20 g, decane 25 mL, P (H2) = 4 MPa, 500 rpm, 2 h.
b All the catalysts were reduced in 10% H2/Ar at 400 °C for 1 h before reaction.
c RT represents reaction temperature.
d Others corresponding to unidentified C8+ heavier products are calculated based on mass balance.
e TON values of 3% Ni/TiO2-R 400/1 h are based on the metal dispersions by particle size estimated by HRTEM. The metal dispersion is calculated by the following equation:  , where MNi, NA, σ, and ρNi are the molecular weight of Ni (58.7 g mol−1), the Avogadro's number (6.02 × 1023 mol−1), the atomic cross-sectional area (0.0649 nm2), and the metal (Ni) density (8.9 g cm−3), respectively. S is the superficial area of a Ni nanoparticle and V is the volume of a Ni nanoparticle. TON values of 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h are based on the amount of Niδ− species measured by in situ XPS.
f n.d.: not detected. , where MNi, NA, σ, and ρNi are the molecular weight of Ni (58.7 g mol−1), the Avogadro's number (6.02 × 1023 mol−1), the atomic cross-sectional area (0.0649 nm2), and the metal (Ni) density (8.9 g cm−3), respectively. S is the superficial area of a Ni nanoparticle and V is the volume of a Ni nanoparticle. TON values of 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h are based on the amount of Niδ− species measured by in situ XPS.
f n.d.: not detected.
|
|||||||||||
| 3% Ni/TiO2-A | 200 | 3.5 | 93.7 | 0 | 0 | 0 | 0 | 6.3 | 100 | n.d.f | 28.2 |
| 250 | 19.7 | 84.2 | 9.1 | 0 | 0 | 0 | 6.7 | 82.2 | 17.8 | 159.2 | |
| 300 | 48.4 | 82.7 | 11.4 | 0 | 0 | 0 | 5.9 | 82.4 | 17.6 | 391.0 | |
| 0.5% Ni/TiO2-A | 200 | 2.1 | 91.3 | 0 | 0 | 0 | 0 | 8.7 | 100 | n.d. | 26.6 |
| 250 | 13.2 | 82.5 | 8.9 | 0 | 0 | 0 | 8.6 | 78.5 | 21.5 | 167.4 | |
| 300 | 31.6 | 80.9 | 9.7 | 0 | 0 | 0 | 9.4 | 85.8 | 14.2 | 401.0 | |
| 3% Ni/TiO2-R | 200 | 12.8 | 0 | 0 | 0 | 5.9 | 89.5 | 4.6 | n.d. | n.d. | 26.4 |
| 250 | 43.7 | 0 | 0 | 0 | 4.8 | 91.8 | 3.4 | n.d. | n.d. | 90.3 | |
| 300 | 89.1 | 0 | 0 | 3.5 | 33.8 | 57.9 | 4.8 | 21.3 | 78.7 | 184.1 | |

(1) At low guaiacol conversion at 200 °C, the primary product was phenol on both 0.5% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts. At higher guaiacol conversions by increasing the reaction temperature to 250 °C and further to 300 °C, cresol formed but only as a minor product on both catalysts. Cresol was reported to form as a product of intermolecular methyl transfer from guaiacol to phenol on a Au/TiO2-A catalyst.61 The formation of cresol on the 0.5% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts appears consistent with the bimolecular methyl transfer pathway as it occurred only after sufficient phenol was produced. Only hydrogenolysis of C–O bonds occurred on these two Ni/TiO2-A 400/1 h catalysts.
(2) In contrast, hydrogenation of aromatic rings prevailed on the 3% Ni/TiO2-R 400/1 h catalyst. The primary product was methoxycyclohexanol at low guaiacol conversions due to full aromatic ring saturation, even at low Ni loading (0.5 wt%) on Ni/TiO2-R 400/1 h (Fig. S2†). There were no detectable phenolic products at various guaiacol conversion levels on the Ni/TiO2-R 400/1 h catalysts. CH4 and CH3OH were detected only at high guaiacol conversions at 300 °C, suggesting that the primary product, methoxycyclohexanol, was stable under the reaction temperature of 250 °C or below. This observation clearly indicates that CH4 and CH3OH are the consecutive reaction products of methoxycyclohexanol. There are two possible pathways for the demethoxylation of methoxycyclohexanol to cyclohexanol: (a) surface acid site catalyzed demethoxylation with the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond which can be rapidly saturated by hydrogenation over the Ni particles; (b) hydrogenolysis of C–OCH3 over the Ni nanoparticles. The very low cyclohexane selectivity at high guaiacol conversion may suggest that the surface acidity is rather weak. The presence of CH4 may suggest possible C–O hydrogenolysis, which could take place at the interface of the Ni particles and TiO2-R surface.
C double bond which can be rapidly saturated by hydrogenation over the Ni particles; (b) hydrogenolysis of C–OCH3 over the Ni nanoparticles. The very low cyclohexane selectivity at high guaiacol conversion may suggest that the surface acidity is rather weak. The presence of CH4 may suggest possible C–O hydrogenolysis, which could take place at the interface of the Ni particles and TiO2-R surface.
(3) The rate of aromatic ring hydrogenation over the Ni/TiO2-R 400/1 h catalyst was much higher than that of CarylO–CH3 hydrogenolysis over the Ni/TiO2-A 400/1 h catalyst based on the guaiacol conversions. The TON values of the 3% Ni/TiO2-R 400/1 h catalyst, defined as the number of guaiacol molecules converted per surface Ni atom in the duration of the reaction (2 h), reached 184.1 at 300 °C. The TON values in hydrodeoxygenation of guaiacol based on the determined Niδ− sites over the 0.5% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts are 401 and 391, respectively, at 300 °C, which are more than double that on the 3% Ni/TiO2-R 400/1 h catalyst. For the 3% Ni/TiO2-R 400/1 h catalyst, the principal products were ring-saturated cyclohexanol and methoxycyclohexanol, which contrasted sharply with the high selectivity to phenol with the TiO2-A supported Ni catalysts. The hydrogenative aromatic ring saturation is an exothermic reaction, and the C–O bond hydrogenolysis is typically an endothermic reaction. The different TON values and products over the Ni/TiO2-R 400/1 h and Ni/TiO2-A 400/1 h catalysts suggested the different active sites for guaiacol conversion, as the former was metallic Ni catalyzed aromatic ring saturation and the latter only catalyzed C–O bond hydrogenolysis. But for the 0.5% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts, the TON values of the two Ni/TiO2-A 400/1 h catalysts are also very close at 200 °C and at 250 °C (Table 5). The closely matched TON values under the same reaction conditions for the two TiO2-A supported catalysts at Ni loadings of 0.5 wt% and 3 wt% imply that a similar type of active site was responsible for guaiacol conversion across the two Ni/TiO2-A 400/1 h catalysts. We attribute this site to Niδ− species.
Supported Ni particles are known to catalyze saturated hydrogenation of aromatic rings. The reported monometallic nickel catalysts typically showed high hydrogenation activity in aromatic ring saturation forming methoxycyclohexanol, cyclohexanol and cyclohexane as the major products (Table 6).28,29,31,33,62–66 The Ni/TiO2-R 400/1 h catalyst in this work showed similar catalytic performance. By forming Ni–Cu and Ni–Fe bimetallic catalysts, the selectivity to phenol was increased.63,66 In our work, monometallic Ni supported on TiO2-A showed 100% selectivity to phenolics with drastically different performance from monometallic Ni catalysts on other supports.
| Catalyst | Support | Reaction condition | Major product | Ref. | |
|---|---|---|---|---|---|
| T (°C) | P (MPa) | ||||
| Ni | SiO2 | 320 | 17 | Cyclohexane, benzene | 62 |
| Ni | C | 400 | 3 | Cyclohexane, cyclohexanol | 63 |
| Ni | Al2O3 | 200 | 5 | Methoxycyclohexanol, cyclohexanol | 64 |
| Ni | ZrO2 | 300 | 5 | Cyclohexane, toluene | 28 |
| Ni | HZSM-5 | 200 | 3 | Phenol, cyclohexanol | 31 |
| Ni | Beta | 230 | 4 | Cyclohexane | 29 |
| Ni | MCM-41 | 250 | 5 | Cyclohexane, methoxycyclohexanol | 65 |
| Ni | CeO2 | 300 | 4 | Cyclohexane, cyclohexanol | 33 |
| Ni | SiO2–ZrO2 | 300 | 5 | Cyclohexane, cyclohexene | 66 |
| Ni–Cu | SiO2 | 320 | 17 | Cyclohexanol, methoxycyclohexanol | 62 |
| Al2O3 | Cyclohexanol, cyclohexanone | ||||
| CeO2–ZrO2 | Methoxycyclohexanol, cyclohexanone | ||||
| Ni–Cu | ZrO2 | 300 | 5 | Toluene, cyclohexane | 66 |
| SiO2–ZrO2 | Cyclohexane, methylcyclohexane | ||||
| Ni–Cu | C | 300 | 5 | Methoxycyclohexanol, cyclohexanol | 13 |
| Ni–Fe | C | 400 | 3 | Phenol, cyclohexanol | 63 |
| Ni–Mo | SiO2 | 410 | 0.1 | Benzene, toluene | 67 |
| Ni–Co | ZSM-5 | 200 | 5 | Methoxycyclohexanol, cyclohexanol | 64 |
| Al2O3 | Methoxycyclohexanol, cyclohexanol | ||||
| Ni | TiO2-A | 300 | 4 | Phenol, cresol | This work |
| Ni | TiO2-R | 300 | 4 | Methoxycyclohexanol, cyclohexanol | This work |
The DRIFT spectra showing the two distinct bridging CO absorption bands, centered at 1834 cm−1 and 1824 cm−1 over the 3% Ni/TiO2-R 400/1 h (Fig. 6A1) and 0.5% Ni/TiO2-R 400/1 h (Fig. 6A2) catalysts, respectively,68,69 indicate that the surface of the Ni particles on the TiO2-R is accessible to guaiacol and H2 due to lack of strong interaction between Ni particles and the poorly reducible TiO2-R surface. As an expected result, hydrogenative aromatic ring saturation prevailed in guaiacol hydrogenation over the Ni/TiO2-R catalysts irrespective of the Ni loading (Table 5 and Fig. S2†). In contrast, the observation of only linearly adsorbed CO on Niδ− species at 2085 cm−1 by DRIFT spectroscopy, irrespective of Ni loading and the presence of Ni particles on the TiO2-A support, establishes the dominant Niδ− sites due to full encapsulation of Ni particles by a partially reduced amorphous TiOx overlayer.33 Therefore, for both the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts, the same catalytic selectivity to dominantly phenol and the similar TON values further verify that the atomically dispersed Niδ− species on the TiO2-A surface are the active sites responsible for the hydrodeoxygenation performance. The demonstrated corresponding linear increase of both the guaiacol conversion and the Kubelka–Munk intensity of in situ DRIFT spectra of CO with Ni loading on Ni/TiO2-A 400/1 h further support the identification of the Niδ− species in this work as the active site for the selective hydrodeoxygenation of guaiacol to phenolics (Table 5).
To verify that the high phenolic selectivity over Ni/TiO2-A was indeed due to poor ring hydrogenation by this catalyst, we also compared the product selectivity of 3% Ni/TiO2-A 400/1 h at 100% guaiacol conversion by prolonging the reaction time. The 100% phenolic selectivity at 100% guaiacol conversion in Fig. 7A confirmed that the selectivity difference between the Ni/TiO2-R 400/1 h and Ni/TiO2-A 400/1 h catalysts was not due to different conversions, but rather due to the inherent selectivity of the Ni/TiO2-A 400/1 h catalyst for hydrogenolysis of C–O bonds. The stability of the 0.5% Ni/TiO2-A 400/1 h catalyst was evaluated by using the catalyst in four consecutive tests. There was no further treatment of the catalyst after recovering it by filtration. The results are reproducible for the guaiacol conversion and product yields in the recycling tests except for an insignificant decrease likely due to a small loss from handling (Fig. 7B). Furthermore, the DRIFT spectra of the used 0.5% Ni/TiO2-A 400/1 h catalyst showed only one identical peak at 2085 cm−1 compared to that of the fresh 0.5% Ni/TiO2-A 400/1 h catalyst (Fig. 7C). The retained wavenumber of the peak suggested that the Niδ− species remained unchanged after repeated guaiacol conversions. Thus, the 0.5% Ni/TiO2-A 400/1 h catalyst was verified to be stable under the reaction conditions.
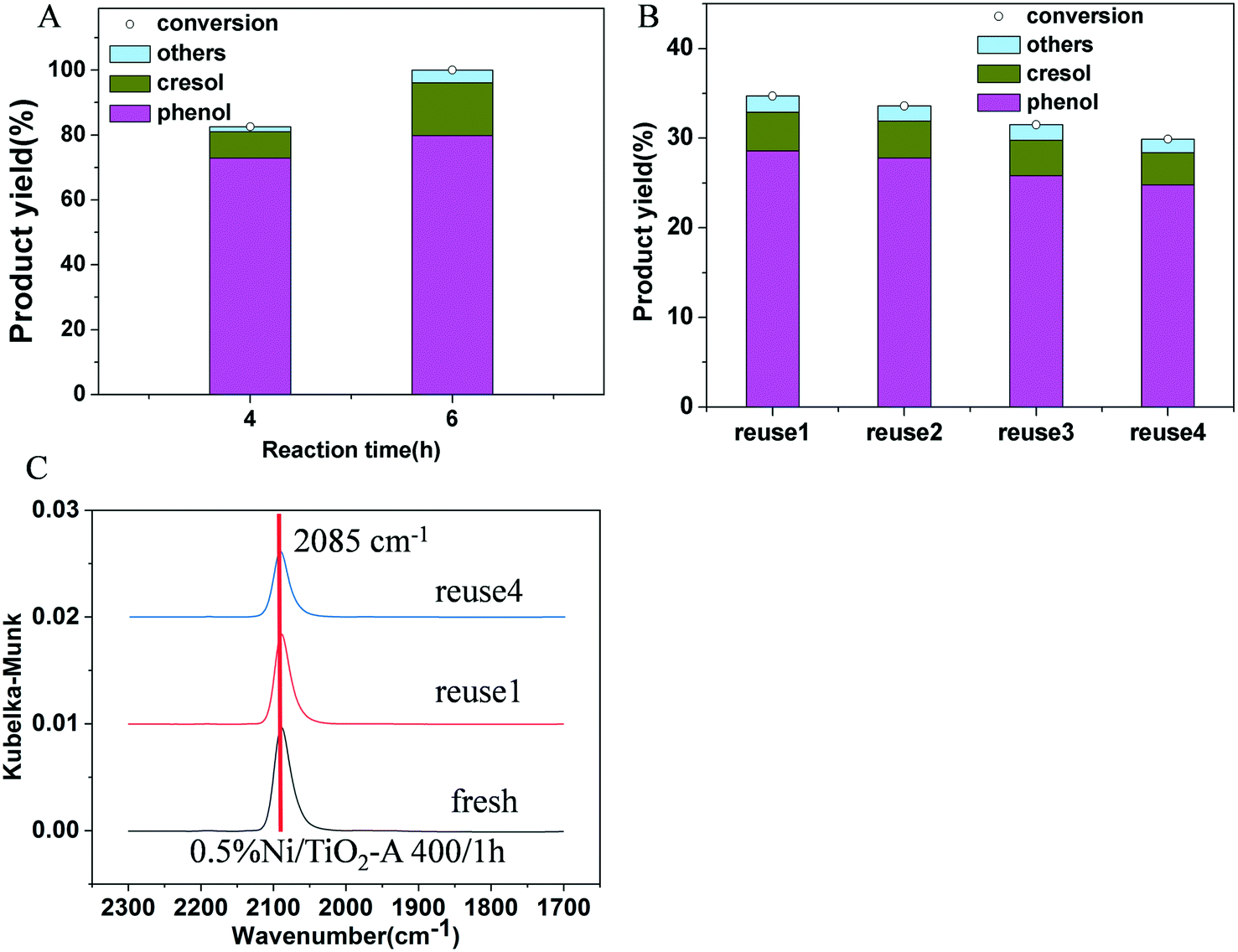 | ||
| Fig. 7 (A) Effect of reaction time on product selectivity of the 3% Ni/TiO2-A 400/1 h catalyst; (B) recycling tests of 0.5% Ni/TiO2-A 400/1 h. The recycled catalyst was used without further pretreatment after separation from the reaction mixture in the previous test; “Others” corresponding to unidentified C8+ heavier products are calculated based on mass balance. Reaction conditions: guaiacol 1.20 g, decane 25 mL, P (H2) = 4 MPa, 500 rpm, 2 h except that indicated in part A; (C) in situ DRIFT spectra of the 0.5% Ni/TiO2-A 400/1 h catalyst after guaiacol reaction upon CO chemisorption at 30 °C. | ||
The difference in the catalytic pathways between the Ni/TiO2-A and Ni/TiO2-R catalysts in guaiacol hydrogenations can now be rationalized by the clearly distinguished active Ni sites in this work. The structure of guaiacol contains three types of C–O bonds, i.e., Caryl–OH, Caryl–OCH3, and CarylO–CH3, with the respective bond dissociation energy of 466, 409–421 and 262–276 kJ mol−1.31 In view of the bond dissociation energy, the energy barrier of CarylO–CH3 bond scission is the lowest among the three. Therefore, hydro-demethylation via CarylO–CH3 bond scission is expected to be more favorable compared with hydro-demethoxylation via cleavage of the Caryl–OCH3 bond in terms of bond strength. Sulfide CoMo and NiMo catalysts have been reported to catalyze hydrogenolysis of guaiacol through hydro-demethylation, forming catechol.70 On the 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts, the primary product being phenol with only CH4 detected as a by-product at low guaiacol conversions (Table 5) is in good agreement with the prediction based on the bond dissociation energy. Hydro-demethoxylation only occurred at higher temperature, as detected at 300 °C. The results suggest that hydro-demethylation is the preferred reaction pathway in guaiacol conversion on the Niδ− site of both 3% Ni/TiO2-A 400/1 h and 0.5% Ni/TiO2-A 400/1 h catalysts.
Lignin-derived model compounds (phenol, anisole and catechol) were also investigated over the 3% Ni/TiO2-A 400/1 h catalyst to study the potential of Niδ− species for hydrogenolysis of their C–O bonds (Fig. S4†). As shown, no phenol conversion on the 3% Ni/TiO2-A 400/1 h catalyst indicated that the Niδ− species was not active for hydrogenolysis of the C–O bond on phenol. For anisole, phenol in 100% selectivity was formed at 12.1% anisole conversion. It is noted that the anisole conversion was lower compared to guaiacol conversion under the same conditions. Catechol showed 100% conversion on the 3% Ni/TiO2-A 400/1 h catalyst, in agreement with the reported literature that hydrodeoxygenation of catechol to phenol is a facile kinetic step.61 These results further demonstrate that the Niδ− species on the 3% Ni/TiO2-A 400/1 h catalyst has very good phenol selectivity.
The H2 pressure dependencies for the 0.5% Ni/TiO2-A 400/1 h and 3% Ni/TiO2-A 400/1 h catalysts were measured at 300 °C with a constant guaiacol concentration over a H2 pressure range from 2 to 6 MPa. The plots shown in Fig. 8A give fits for hydrogen reaction orders of 0.500 for the 0.5% Ni/TiO2-A 400/1 h catalyst and 0.699 for the 3% Ni/TiO2-A 400/1 h catalyst. We failed to find direct information about the reaction order with respect to hydrogen in the hydrodeoxygenation process though some literature studies assumed that the hydrogen reaction order is zero order or first order.71,72
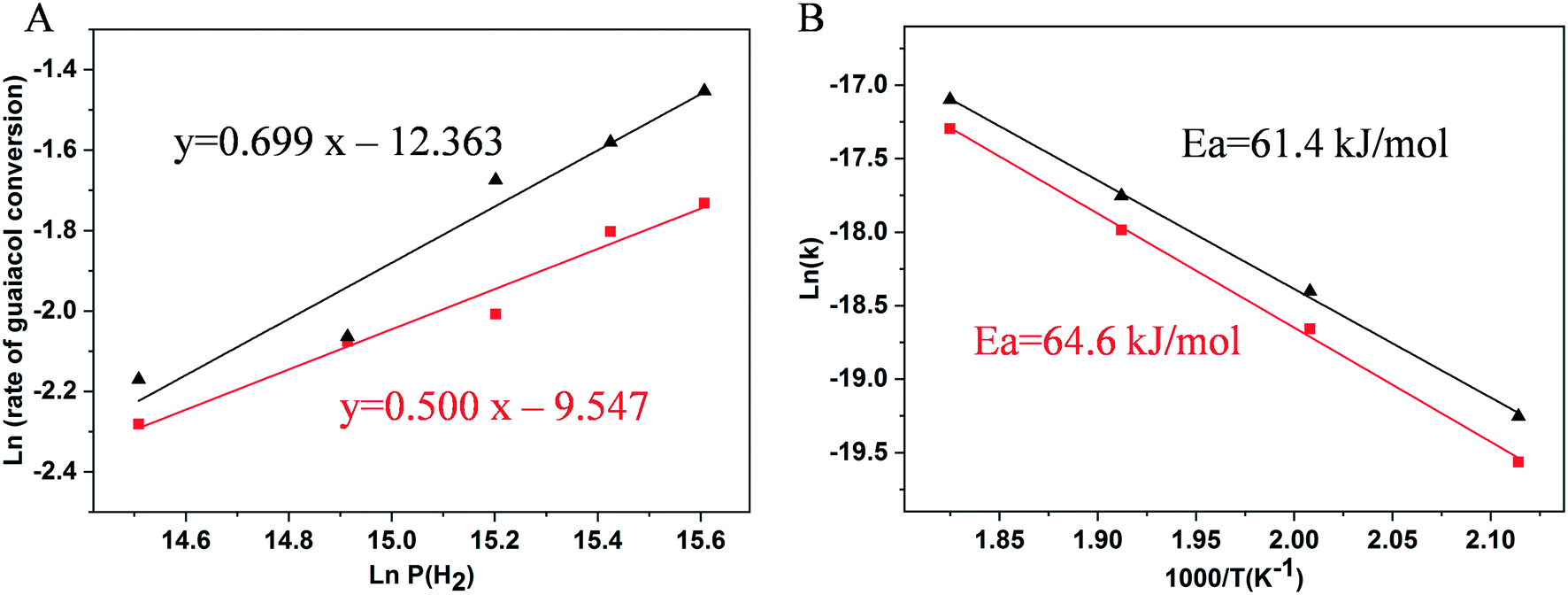 | ||
| Fig. 8 (A) The H2 pressure dependencies for the 0.5% Ni/TiO2-A 400/1 h (red line) and 3% Ni/TiO2-A 400/1 h (black line) catalysts at 300 °C; (B) Arrhenius plots for the temperature dependencies of guaiacol conversion over the 0.5% Ni/TiO2-A 400/1 h (red line) and 3% Ni/TiO2-A 400/1 h (black line) catalysts. | ||
As presented by eqn (4), the rate of reaction, r, is proportional to the guaiacol concentration C and hydrogen pressure P:
| r = k × C × Pm | (4) |
Based on the new understanding of the electronic and structural differences between the active Ni species on TiO2-A and on TiO2-R, possible pathways of guaiacol conversion over the Ni/TiO2-A 400/1 h and the Ni/TiO2-R 400/1 h catalysts are proposed in Scheme 1. With the Ni/TiO2-A 400/1 h catalyst, the Niδ− species catalyzed the energetically favorable hydrogenolysis of the CarylO–CH3 bond to form catechol in the first step, producing CH4 as the dominant C1 product (Table 5). Little catechol was detected in the reaction, likely due to the rapid hydrodeoxygenation of catechol to phenol. It has been reported that hydrodeoxygenation of catechol to phenol is a facile kinetic step.61 In addition, demethoxylation of guaiacol may proceed at a slower rate, thus producing phenol directly with methanol as the minor side product only at higher temperatures. In contrast, over the Ni/TiO2-R 400/1 h catalyst (Scheme 1), the favorable pathway was the hydrogenation of aromatic rings on metallic Ni particles, with little C–O bond hydrogenolysis, producing methoxycyclohexanol as the primary product. The consecutive reactions producing CH3OH and CH4 from hydrogenation of methoxycyclohexanol are much less favorable because CH3OH and CH4 were not detected at lower temperatures (200 °C and 250 °C), in agreement with the low cyclohexanol selectivity (Table 5). The acidity of the Ni/TiO2-R catalyst must be fairly weak to account for the low cyclohexane selectivity at 300 °C, at which the primary product methoxycyclohexanol remained as the major product. Demethylation and demethoxylation are slow reaction steps on the Ni/TiO2-R catalysts (Scheme 1).
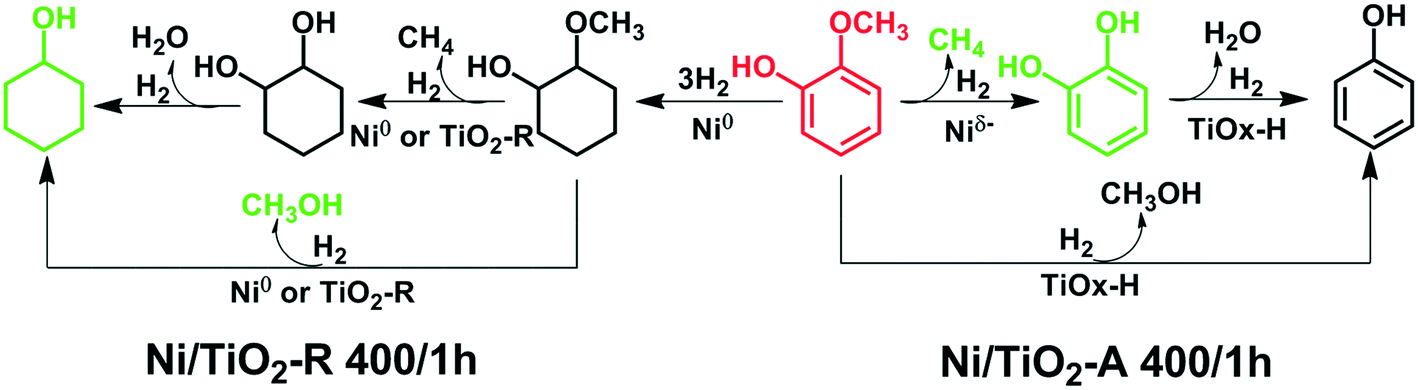 | ||
| Scheme 1 Reactions in guaiacol hydrogenation over the supported Ni catalysts. | ||
4. Conclusion
The differences in the chemical states and surface locations of Ni species on Ni/TiO2-A and Ni/TiO2-R catalysts are revealed through this work. Two distinctively different types of strong interactions between reduced Ni and the reducible TiO2-A surface and between NiO and the poorly reducible TiO2-R surface are demonstrated. The strong interaction between reduced Ni and the reducible TiO2-A surface on the Ni/TiO2-A 400/1 h catalyst led to enhanced reduction of NiO together with a fraction of surface TiO2-A at a low temperature. The absence of metallic Ni particles by HRTEM and the identified atomically dispersed Niδ− species without Ni0 species on the 0.5% Ni/TiO2-A 400/1 h catalyst indicate that the formation of Niδ− species is more energetically favorable than the formation of Ni0 species at low Ni loading. The results of in situ XPS and DRIFT spectroscopy of adsorbed CO show that the atomically dispersed Niδ− species may be located at the oxygen vacancies of the reducible TiO2-A surface lattice. In contrast, this interaction was not found on the Ni/TiO2-R 400/1 h catalyst. The Ni particles were formed on the Ni/TiO2-R 400/1 h catalyst irrespective of Ni loading and were accessible to reactants, as verified using DRIFT spectra of adsorbed CO. The Ni particles on the Ni/TiO2-R 400/1 h catalyst preferentially catalyzed the hydrogenative saturation of the guaiacol aromatic ring. While Ni particles were formed on Ni/TiO2-A 400/1 h only at higher Ni loading, the surface of Ni particles was inaccessible to CO due to full encapsulation by a TiOx overlayer, and was incapable of hydrogenating the aromatic ring in guaiacol. The Ni/TiO2-A 400/1 h catalyst only selectively catalyzed the hydrogenolysis of C–O bonds by the atomically dispersed Niδ− species.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Professor Harold H. Kung of Northwestern University for his suggestions in the revision of this paper. This work was supported by the National Natural Science Foundation of China (Grants 21690084, 21721004 and 21932005) and the Innovation Foundation of DICP (Grant DICP I201936).References
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- J. Van De Loosdrecht, A. M. Van Der Kraan, A. J. Van Dillen and J. W. Geus, J. Catal., 1997, 170, 217–226 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS.
- M. Xu, S. He, H. Chen, G. Cui, L. Zheng, B. Wang and M. Wei, ACS Catal., 2017, 7, 7600–7609 CrossRef CAS.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef.
- L. Wang, J. Zhang, Y. Zhu, S. Xu, C. Wang, C. Bian, X. Meng and F. S. Xiao, ACS Catal., 2017, 7, 7461–7465 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS.
- J. Panpranot, K. Kontapakdee and P. Praserthdam, Appl. Catal., A, 2006, 314, 128–133 CAS.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS.
- J. Zhang, G. Chen, D. Guay, M. Chaker and D. Ma, Nanoscale, 2014, 6, 2125–2130 RSC.
- A. B. Dongil, B. Bachiller-Baeza, I. Rodríguez-Ramos, J. L. G. Fierro and N. Escalona, RSC Adv., 2016, 6, 26658–26667 RSC.
- R. M. Palomino, J. W. Magee, J. Llorca, S. D. Senanayake and M. G. White, J. Catal., 2015, 329, 87–94 CrossRef CAS.
- R. M. Kennedy, L. A. Crosby, K. Ding, C. P. Canlas, A. Gulec, L. D. Marks, J. W. Elam, C. L. Marshall, K. R. Poeppelmeier and P. C. Stair, Catal. Lett., 2018, 148, 2223–2232 CrossRef CAS.
- S. Riyapan, Y. Boonyongmaneerat, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Today, 2015, 245, 134–138 CrossRef CAS.
- A. J. McCue, F. M. McKenna and J. A. Anderson, Catal. Sci. Technol., 2015, 5, 2449–2459 CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS.
- R. Zanella, C. Louis, S. Giorgio and R. Touroude, J. Catal., 2004, 223, 328–339 CrossRef CAS.
- W. Lin, H. Cheng, L. He, Y. Yu and F. Zhao, J. Catal., 2013, 303, 110–116 CrossRef CAS.
- M. Xu, S. Yao, D. Rao, Y. Niu, N. Liu, M. Peng, P. Zhai, Y. Man, L. Zheng, B. Wang, B. Zhang, D. Ma and M. Wei, J. Am. Chem. Soc., 2018, 140, 11241–11251 CrossRef CAS.
- N. Liu, M. Xu, Y. Yang, S. Zhang, J. Zhang, W. Wang, L. Zheng, S. Hong and M. Wei, ACS Catal., 2019, 9, 2707–2717 CrossRef CAS.
- C. J. Pan, M. C. Tsai, W. N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S. Y. Cheng and B. J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS.
- S. Bonanni, K. Aït-Mansour, H. Brune and W. Harbich, ACS Catal., 2011, 1, 385–389 CrossRef CAS.
- K. V. Kovtunov, D. A. Barskiy, O. G. Salnikov, D. B. Burueva, A. K. Khudorozhkov, A. V. Bukhtiyarov, I. P. Prosvirin, E. Y. Gerasimov, V. I. Bukhtiyarov and I. V. Koptyug, ChemCatChem, 2015, 7, 2581–2584 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- Q. Lai, C. Zhang and J. H. Holles, Catal. Sci. Technol., 2017, 7, 3220–3233 RSC.
- X. Zhang, T. Wang, L. Ma, Q. Zhang, Y. Yu and Q. Liu, Catal. Commun., 2013, 33, 15–19 CrossRef CAS.
- P. Yan, M. M. J. Li, E. Kennedy, A. Adesina, G. Zhao, A. Setiawan and M. Stockenhuber, Catal. Sci. Technol., 2020, 10, 810–825 RSC.
- P. M. Mortensen, D. Gardini, H. W. P. De Carvalho, C. D. Damsgaard, J. D. Grunwaldt, P. A. Jensen, J. B. Wagner and A. D. Jensen, Catal. Sci. Technol., 2014, 4, 3672–3686 RSC.
- W. Song, Y. Liu, E. Baráth, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC.
- A. Aqsha, L. Katta and N. Mahinpey, Catal. Lett., 2015, 145, 1351–1363 CrossRef CAS.
- X. Zhang, P. Yan, B. Zhao, K. Liu, M. C. Kung, H. H. Kung, S. Chen and Z. C. Zhang, ACS Catal., 2019, 9, 3551–3563 CrossRef CAS.
- K. J. A. Raj, M. G. Prakash, R. Mahalakshmy, T. Elangovan and B. Viswanathan, Catal. Sci. Technol., 2012, 2, 1429–1436 RSC.
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125–142 CrossRef CAS.
- B. Seemala, C. M. Cai, C. E. Wyman and P. Christopher, ACS Catal., 2017, 7, 4070–4082 CrossRef CAS.
- W. Wang, X. Li, Y. Zhang, R. Zhang, H. Ge, J. Bi and M. Tang, Catal. Sci. Technol., 2017, 7, 4413–4421 RSC.
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-vante, J. Phys. Chem. C, 2011, 115, 20153–20159 CrossRef CAS.
- B. Xin, P. Wang, D. Ding, J. Liu, Z. Ren and H. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS.
- J. M. Herrmann, J. Catal., 1984, 89, 404–412 CrossRef CAS.
- J. L. Rodríguez, T. Poznyak, M. A. Valenzuela, H. Tiznado and I. Chairez, Chem. Eng. J., 2013, 222, 426–434 CrossRef.
- J. Shi, B. Bauer, A. Feldhoff, M. Senna, V. Sepela, V. Laporte and K. Becker, J. Solid State Chem., 2012, 187, 51–57 CrossRef.
- M. M. Islam, M. Calatayud and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 6809–6814 CrossRef CAS.
- W. Wan, X. Nie, M. J. Janik, C. Song and X. Guo, J. Phys. Chem. C, 2018, 122, 17895–17916 CrossRef CAS.
- T. O. Omotoso, B. Baek, L. C. Grabow and S. P. Crossley, ChemCatChem, 2017, 9, 2642–2651 CrossRef CAS.
- K. Liu, P. Yan, H. Jiang, Z. Xia, Z. Xu, S. Bai and Z. C. Zhang, J. Catal., 2019, 369, 396–404 CrossRef CAS.
- G. Hu, Z. Wu and D. Jiang, J. Phys. Chem. C, 2018, 122, 20323–20328 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- S. Zhang, Z. Xia, T. Ni, Z. Zhang, Y. Ma and Y. Qu, J. Catal., 2018, 359, 101–111 CrossRef CAS.
- J. Szanyi, N. C. Nelson, L. Chen, D. Motta and L. Kovarik, Angew. Chem., Int. Ed., 2020, 2–9 Search PubMed.
- Y. Tang, S. Zhao, B. Long, J. Liu and J. Li, J. Phys. Chem. C, 2016, 120, 17514–17526 CrossRef CAS.
- M. G. Sanchez and J. L. Gazquez, J. Catal., 1987, 104, 120–135 CrossRef CAS.
- H. Viet, G. Pacchioni, L. Derita and P. Christopher, J. Catal., 2018, 367, 104–114 CrossRef.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Nat. Commun., 2019, 10, 4488 CrossRef.
- L. Derita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS.
- D. G. Blackmond and E. I. Ko, J. Catal., 1985, 96, 210–221 CrossRef CAS.
- M. Primet, J. A. Dalmon and G. A. Martin, J. Catal., 1977, 46, 25–36 CrossRef CAS.
- X. Zhu, Y. Zhang and C. Liu, Catal. Lett., 2007, 118, 306–312 CrossRef CAS.
- L. Derita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS.
- C. Asokan, H. V. Thang, G. Pacchioni and P. Christopher, Catal. Sci. Technol., 2020, 10, 1597–1601 RSC.
- J. Mao, J. Zhou, Z. Xia, Z. Wang, Z. Xu, W. Xu, P. Yan, K. Liu, X. Guo and Z. C. Zhang, ACS Catal., 2017, 7, 695–705 CrossRef CAS.
- M. V. Bykova, D. Y. Ermakov, V. V. Kaichev, O. A. Bulavchenko, A. A. Saraev, M. Y. Lebedev and V. Yakovlev, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS.
- H. Fang, J. Zheng, X. Luo, J. Du, A. Roldan, S. Leoni and Y. Yuan, Appl. Catal., A, 2017, 529, 20–31 CrossRef CAS.
- M. Zhou, J. Ye, P. Liu, J. Xu and J. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 8824–8835 CrossRef CAS.
- S. Qiu, Y. Xu, Y. Weng, L. Ma and T. Wang, Catalysts, 2016, 6, 134–149 CrossRef.
- X. Zhang, Q. Zhang, T. Wang, L. Ma, Y. Yu and L. Chen, Bioresour. Technol., 2013, 134, 73–80 CrossRef.
- T. He, X. Liu, Y. Ge, D. Han, J. Li, Z. Wang and J. Wu, Catal. Commun., 2017, 102, 127–130 CrossRef CAS.
- M. Mihaylov, K. Hadjiivanov and H. Knözinger, Catal. Lett., 2001, 76, 59–63 CrossRef CAS.
- K. Hadjiivanov, H. Knözinger and M. Mihaylov, J. Phys. Chem. B, 2002, 106, 2618–2624 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- Y. Xiao, R. Lagare, L. Blanshan, E. N. Martinez and A. Varma, AIChE J., 2020, 66, 1–8 CrossRef.
- M. V. Bykova, S. G. Zavarukhin, L. I. Trusov and V. A. Yakovlev, Kinet. Catal., 2013, 54, 40–48 CrossRef CAS.
- D. Gao, Y. Xiao and A. Varma, Ind. Eng. Chem. Res., 2015, 54, 10638–10644 CrossRef CAS.
- E. Laurent and B. Delmon, Appl. Catal., A, 1994, 109, 97–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01720e |
| This journal is © The Royal Society of Chemistry 2021 |