
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous enzymatic stirred tank reactor cascade with unconventional medium yielding high concentrations of (S)-2-hydroxyphenyl propanone and its derivatives†
Reinhard
Oeggl
ab,
Juliane
Glaser
ac,
Eric
von Lieres
 a and
Dörte
Rother
*ab
a and
Dörte
Rother
*ab
aIBG-1: Biotechnology, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de
bAachen Biology and Biotechnology (ABBt), RWTH Aachen University, 52074 Aachen, Germany
cDigital Integration & Predictive Technologies (DIPT), Amgen Research (Munich) GmbH, Staffelseestr. 2, 81477 Munich, Germany
First published on 8th October 2021
Abstract
The implementation of biocatalysis in flow chemistry offers synergistic synthesis advantages in line with green chemistry principles. Yet, the conversion of high substrate concentrations is in many cases hindered by insolubility issues or substrate toxicity. Here, the continuous synthesis of (S)-2-hydroxyphenyl propanone (2-HPP) from inexpensive benzaldehyde and acetaldehyde in a methyl tert-butyl ether based organic reaction environment, namely micro-aqueous reaction system, has been established. Kinetic parameters of the applied whole cell catalyst were identified to design a continuous process for (S)-2-HPP synthesis. This revealed a necessity to distribute acetaldehyde over a spatial coordinate to remain below a toxic concentration threshold. Hence, three continuous stirred tank reactors (cSTR) were conjugated in a technical cascade with an additional influx of acetaldehyde into each unit. The catalytic behaviour of this reaction setup was described based on mass balances and a kinetic model. Enzyme deactivation was described by a novel staged model and compared to a simple generic model. The optimized continuous setup yielded 190 mM (S)-HPP with an ee > 98% over 8 h. The product was easily recovered from the organic reaction environment by crystallization with an isolated yield of 68% and a purity of >99%. Further, the substrate range of the applied catalyst Pseudomonas putida benzoylformate decarboxylase variant L461A was analysed. This revealed numerous halogenated, methoxylated and nitro-derivatives in ortho, meta, and para position, which can in principle be gained by the established process. As an example, the applied cSTR concept was transferred to p-methoxy benzaldehyde with good results even without further optimization.
Introduction
In recent years continuous flow chemistry utilizing biocatalysis has emerged as a promising tool to fulfil the goal of sustainable fine chemical synthesis in the aspects of green chemistry.1,2 A notable difference of flow chemistry with biocatalysts compared to organic synthesis is that predominantly buffered systems are used for catalytic reactions.3 Unfortunately, many substrates leading to valuable compounds suffer from poor substrate solubility and thereby poor overall synthesis efficiency.4,5 Furthermore, also soluble concentrations of substrate may lead to toxic effects on biocatalysts and deactivate them. For instance, the synthesis of (S)-2-hydroxy-1-phenylpropanone ((S)-2-HPP) from acetaldehyde and benzaldehyde in buffered systems is firstly limited by the poor solubility of benzaldehyde in buffered systems, which does not exceed 40 mM. Secondly, the highly reactive substrate acetaldehyde is known to deactivate the biocatalyst catalysing the reaction.6,7Herein, we aim to overcome these challenges and establish a continuous production process for high (S)-2-HPP concentrations in a hydrophobic reaction environment. The process development in the ideal biocatalytic operation window of high substrate concentration while dealing with substrate toxicity will be supported by descriptive reaction models of different complexities. Further, an extended usability of biocatalytic operations in hydrophobic reaction environments is targeted by demonstrating a simple product isolation procedure and a process transfer to a product platform of derivatives.
The stereo- and chemoselective synthesis of (S)-2-HPP from benzaldehyde and acetaldehyde is so far only described by the Pseudomonas putida benzoylformate decarboxylase (PpBFD), a thiamine-dependent carboligase.8 An advantage is that the most selective enantioselective PpBFD variant L461A meets the enantiopurity required for fine chemicals by achieving a remarkable ee of 98% for (S)-2-HPP (Fig. 1).8,9 Nonetheless, this variant, as well as many ligases applied for 2-hydroxy ketone synthesis, suffer from acetaldehyde sensitivity and poor benzaldehyde solubility.10–17
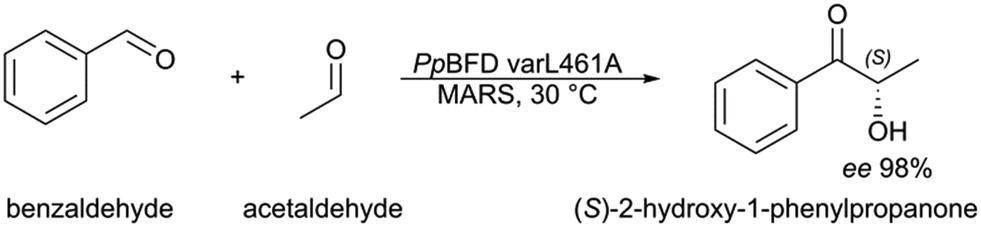 | ||
| Fig. 1 Synthesis of (S)-2-hydroxy-1-phenylpropanone. Benzaldehyde and acetaldehyde are ligated to the chiral (S)-2-hydroxy ketone by Pseudomonas putida benzoylformate decarboxylase variant L461A in a micro-aqueous reaction system (MARS). | ||
The challenge of benzaldehyde solubility was recently mastered in batch applications by applying a moisture controlled micro-aqueous reaction system (MARS).6,7,17–20 MARS provides solvent sensitive enzymes with a stable monophasic environment to react in hydrophobic solvent solutions. This system is obtained by suspending a lyophilized whole cell (LWC) catalyst in pure substrate solutions or substrate/solvent mixtures and adding a minor buffer fraction.18,19 The buffer fraction should not exceed the solubility limit. The expression levels of recombinant PpBFD varL461A in the LWCs are elevated to such an extent that background reactions are rendered negligible.6,7,17,19,21 The potential of this unconventional reaction medium in batch synthesis for (S)-2-HPP production was successfully demonstrated by Wachtmeister et al., by obtaining 340 mM product with an ee of 98%.17 The promising results of other MARS batch applications further encourage an exploration of its potential in continuous operation to achieve synthesis at high substrate concentrations.6,7,19
While a MARS elevates the limits of benzaldehyde solubility, the challenge of enzyme deactivation by acetaldehyde persists. To utilize the high concentrations possible in MARS, acetaldehyde has to be added in smaller portions. In classic continuous syntheses this is realized with a continuous cross flow reactor (Fig. 2A), which allows the distribution of acetaldehyde over the whole column length. For practical reasons in the herein performed process development, this setup is emulated by setting up a continuous stirred tank reactor cascade (cSTR-c; Fig. 2B). This allows a distribution of the total acetaldehyde amount over the respective cSTR-c units and thereby keeping local substrate concentration below a level that would trigger an accelerated enzyme deactivation.
 | ||
| Fig. 2 Methods to minimize effects of substrate toxicity in continuous processes. (A) A continuous cross flow reactor is a suitable method to distribute the total substrate amount, in this case acetaldehyde, over the length of the column avoiding toxification by substrates. In lab-scale such a setup is emulated by (B) a continuous stirred tank reactor cascade (cSTR-cascade). In this manner high concentration of continuously synthesized (S)-2-HPP can be gained, while reducing effects of substrate toxicity to a minimum. This is done by controlling influx flow rates Fi [m3 s−1] and the addition of acetaldehyde ci [mM] (i ∈ {2, 3}) in the defined constant reactor volume V [m3]. | ||
The molecular reactions inside the cSTR-c with biocatalytic synthesis and simultaneous enzyme deactivation are poorly understood. Mechanistic models are a powerful tool to gain a better understanding already from small data sets. Here, a simplified mechanistic model is developed for process analysis and optimization. The reactor model relates the concentration changes of reactants upon enzymatic conversion in the respective unit of the cSTR-cascade to a mass balance of fluxes into and out of that cSTR-c unit. The catalyst is modelled by a 2nd order Michaelis–Menten kinetic with reversible inhibition by acetaldehyde and additional acetaldehyde concentration-dependent enzyme deactivation. The model is calibrated using measurement data in order to predict substrate turnover rates, (S)-2-HPP conversion, and stable operation time to achieve a stable product output in the cSTR-cascade.
The gained product is easily isolated by utilizing the benefits of MARS.6,7,17–19 The recently demonstrated direct product crystallization from the reaction solution, which is facilitated by the well distinguished physicochemical properties of all reactants in MARS, is intruguing.6
To extend the applicability of the presented process, the substrate range of PpBFD varL461A is analysed with respect to halogenated, methoxylated and nitro-benzaldehyde derivatives. As proof of concept, the p-methoxy benzaldehyde derivative is used without further parameter optimization in the cSTR-cascade to prove the general applicability for the synthesis of (S)-2-HPP derivatives in the outlined manner.
Methods and materials
Materials
Methods
In the following the term conversion is defined as the percentage share of benzaldehyde or benzaldehyde derivative that has been carboligated with acetaldehyde to the desired 2-HPP product by mass balancing the measured aromats benzaldehyde and (S)-2-HPP.The obtained orthogonal data array of specific initial activities was utilized to determine kinetic reaction parameters by inverse fitting with fmincon MATLAB (MathWorks 2017b, USA; parameter constrains see ESI† Table S1).22 As reaction kinetic formula a Michaelis–Menten kinetic of the 2nd order (eqn (1)) with a substrate inhibition term for acetaldehyde was applied.15,23 For benzaldehyde no inhibition was detected in the applied concentration range. Hence a benzaldehyde inhibition term is neglected.
 | (1) |
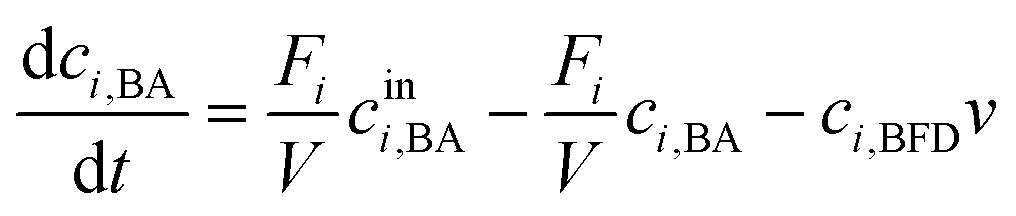 | (2a) |
 | (2b) |
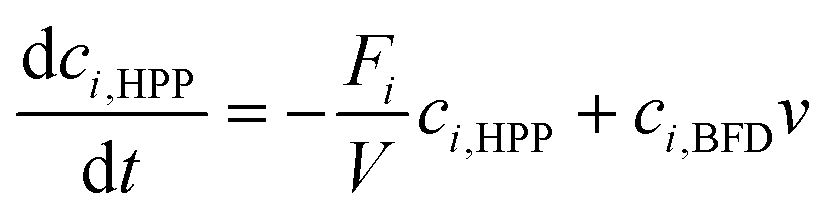 | (2c) |
The resulting differential equation system was solved using the ode15s solver in MATLAB 2017b. For initial conditions inside the reactor, the corresponding inlet concentrations cini,XX were used. Since LWC instead of purified enzymes are used, the amount of PpBFD varL461 is estimated by assuming that one dry E. coli cell weighs 280 fg, of which 155 fg accounts for the total protein.29,30 Of this total protein content 58% are assumed to be PpBFD varL461. This accounts for a total PpBFD varL461 concentration of 15.8 mmol g−1LWC based on a molecular enzyme weight of 57 kDa.31,33
Modelling of a continuous stirred tank reactor cascade with 3 units. A model describing the cSTR-cascade in Fig. 2B was established by connecting the outlet of the first unit with the inlet of the second and the outlet of the second unit with the inlet of the third. The flow rates of the second and third units are increased by the feed rates from the added stock solutions according to eqn (3a) and (b).
| Fi+1 = Fi + Fstocki+1 i ∈ {1, 2} | (3a) |
 | (3b) |
For continuous synthesis of p-methoxy benzaldehyde, three cSTR units were aligned in series as depicted in Fig. 2B. The concentration cini,XX contained 250 mM p-methoxy benzaldehyde and 100 mM acetaldehyde in MTBE. The respective acetaldehyde stock solutions cstock2 and cstock3 contained 3.5 M acetaldehyde dissolved in MTBE. Pumping velocities in the setup were adjusted accordingly to F1 100 μL min−1, Fstock2 4 μL min−1, and Fstock3 3 μL min−1. Samples were collected manually at the outlet of the third cSTR unit. Sample preparation and analysis are described below.
Analytics
Results and discussion
Optimal substrate concentrations in a MARS (batch determination)
First the optimal paring of substrate concentrations in MARS was investigated in batch experiments. With regard to high reaction rates an initial activity screening was performed with acetaldehyde ranges from 25 mM to 200 mM paired with benzaldehyde concentrations from 50 mM to 300 mM.The screening revealed an acetaldehyde optimum between 60 to 100 mM in terms of initial activity, determined within up to 5 min reaction time (Fig. 3). Higher concentrations resulted in an activity decline, which is assumed to be due to substrate inhibition. These findings are consistent with batch observations for PpBFD varL461A in MARS by others.17 Compared to PpBFD reactions in buffer environment with an acetaldehyde optimum at 400 mM to 500 mM, the reaction in MARS presented here shows its acetaldehyde optimum at four times lower concentrations.9,12,13 At first glance, this finding was unexpected. A possible explanation can be the polar nature of acetaldehyde, which would favour an inhomogeneous distribution in MARS, meaning it locally accumulates in the hydrate shell of the biocatalyst. Therefore, the local concentrations of acetaldehyde could hypothetically be higher and correspond more to the optimum in the buffer.
 | ||
| Fig. 3 Initial activity measurements. The initial catalytic velocity of PpBFD varL461A for (S)-HPP synthesis at substrate concentrations of 25 to 200 mM acetaldehyde and 50 to 300 mM benzaldehyde was measured; 30 °C, 1000 rpm, ntechnical = 4. | ||
In case of benzaldehyde, the initial activities obtained a local high at 300 mM (Fig. 3). To identify, whether this local benzaldehyde optimum also corresponded to a global maximum, the screening was extended to benzaldehyde concentrations of up to 5 M (ESI† Fig. S3). Since no synergistic effects were observed between the ratio of acetaldehyde and benzaldehyde, this extended screening was performed with only one acetaldehyde concentration of 100 mM. The extended screening showed that in fact concentrations of up to 500 mM lead to an increased initial activity before it declines again (ESI† Fig. S3). Hence, MARS can be a suitable reaction environment for high aromatic substrate concentrations, which can circumvent long term enzyme deactivation due to substrate toxicity.
Calculation of apparent kinetic parameters and substrate inhibition in a MARS (batch determination)
Next, kinetic parameters for the carboligation towards (S)-2-HPP by PpBFD varL461A in a lyophilised whole cell formulation in MARS were obtained by fitting the initial activity screening results (Fig. 3) with the 2nd order Michaelis–Menten (eqn (1)). This results in a reaction-kinetic model, which is designed to be later coupled to the mechanistic cSTR-c model. It is possible that the substrate and product concentrations within the lyophilized whole cells (LWCs) and the liquid phase are different. As the concentrations in the cells cannot be detected, an even distribution was assumed for the calculations. Consequently, only apparent parameters could be calculated.This first fit resulted in a VMAX of 9.2 U mg−1 for LWC catalyst formulations, a KM,BA of 278.3 mmol L−1, a KM,AA of 1693.4 mmol L−1, and an inhibitory constant Kinh,AA of 1.8 mmol L−1. When evaluating these results, it has to be stressed that they are numerical results obtained with a Mathlab solver within defined limits (Table S1†). While the values themselves may not reflect real values, otherwise a reaction would hardly take place, they are the best obtained fit based on the orthogonal initial activity matrix and model within the defined limits. Probably, other presently unknown variables of the non-natural reaction environment besides acetaldehyde have an influence on the catalyst performance.
For a subsequent cSTR-c application, a setup of three cSTR-c units in series were investigated. Based on the restriction to three cSTR-c units it was estimated that 250 mM benzaldehyde can be converted with acetaldehyde amounts being added slightly above the equimolar amount.
Transfer of operation parameter into one cSTR-cascade unit (continuous)
Determination of acetaldehyde dependent deactivation
During kinetic parameter investigation, the influence of different acetaldehyde concentrations on initial rate activity was determined. Now, the acetaldehyde dependent deactivation term kD of PpBFD varL461A was examined. Therefore, acetaldehyde concentrations in a range of 60 mM to 250 mM were investigated over time in one cSTR unit (technical setup scheme cf. ESI† Fig. S2A). The determination of kD is essential to find optimal acetaldehyde concentrations and therewith an acetaldehyde feed for the three cSTR-c units. The experimental results verified a proportional correlation of acetaldehyde concentration and enzyme deactivation (Fig. 4). Enzyme deactivation is here derived from a decline in (S)-2-HPP formation. | ||
| Fig. 4 Continuous operation of a cSTR-cascade consisting of 3 units. The staged deactivation model was used, and the Michaelis–Menten parameters were estimated from process data of a single cSTR unit. Displayed is the conversion over the whole cSTR-cascade. At the beginning, the first cSTR unit is filled (star) and then eflux flows into the second unit (triangle) and then in the third (dots). A measurement in the 1st and 2nd unit is omitted by the technical setup once they are connected to the subsequent unit, as measurement is conducted at the efflux position. Here, the simulated data projects the conversion over time in the respective units. | ||
The process model (eqn (3a) and (b)) was fitted to the experimentally retrieved data to determine the enzyme deactivation term kD to quantitatively describe the long-term process. The obtained model was able to describe measured benzaldehyde conversions in a single cSTR unit in combination with acetaldehyde concentrations of up to 120 mM very well (ESI† Fig. S5C). For higher acetaldehyde concentrations, the long-term conversion profile deviated considerably from measured data. Hence, the Michaelis–Menten parameters were re-estimated together with kD using a genetic algorithm and gradient search in MATLAB (ESI† Fig. S5). The re-estimation revealed that Michaelis–Menten kinetic parameters under process conditions in a single cSTR unit have a marginally (by 0.1 U mg−1) different VMAX of 9.3 U mg−1, but a 5-fold increased Kinh,AA of 9.9 mM, while KM values for acetaldehyde and benzaldehyde remained unaffected. The re-estimated kD was 2.1 × 10−6 s−1. These process Michaelis–Menten parameters were found to describe the conversion of benzaldehyde to (S)-2-HPP in one cSTR-cascade unit more adequately than the previously determined parameters from initial activity measurements (Fig. 4). The kinetic differences are likely caused by altered mass transfer in the 9 mL cSTR-c unit with internal stirring as compared to the 1 mL batch experiments with external stirring in which the initial activities were recorded. Importantly, the process kinetic parameters together with kD described an ideal acetaldehyde concentration of 100 mM. This finding aligns with the experimental data gained in initial activity screenings in batch. Conclusively, 100 mM acetaldehyde was applied in further setups as this concentration yields the best compromise between fast catalytic velocity due to sufficiently high amounts of acetaldehyde, low acetaldehyde initial rate inhibition, and a relatively low enzyme deactivation rate over time (ESI† Fig. S5).
| Process operation | Catalyst | Conversion [%] | Final (S)-HPP concentration [M] | ee [%] | Retention time [h] | Catalyst load [g L−1] | sSTY [g L−1 d−1 g−1catalyst] |
|---|---|---|---|---|---|---|---|
| cSTR-c | LWC | 76 | 0.19 | 98 | 0.5 | 100 | 0.190 |
| Fed-batch | LWC | 68 | 0.34 | 98 | 6 | 100 | 0.085 |
Product isolation
In a next step, the synthesized (S)-2-HPP was purified with the aim to gain an in situ product crystallization. Thus, the physicochemical parameters of all compounds were collected to deduce a suitable downstream process (ESI† chapter V, Table S3). In a step-wise distillation at mild vacuum both, acetaldehyde and MTBE, were successfully stripped from the product solution (Fig. 5). The remaining product mixture contained benzaldehyde and (S)-2-HPP. Unfortunately, benzaldehyde could not be stripped from the solution at 50 °C and full vacuum. Following the ideal gas law, the temperature could in theory be increased to strip benzaldehyde. This strategy was not followed, as a temperature increase might reduce the stereoselectivity and regioselectivity of the product, as (S)-2-HPP is prone to tautomerise at higher temperatures. Hence, a separation based on the distinguished melting points of both chemicals was performed by cooling crystallisation at 4 °C. Under these conditions (S)-2-HPP crystals grew within a few hours. Upon filtration and washing with cooled petrol ether, an isolated yield of 68% with a crystal purity of 99% was achieved. This simple technique, moderate vacuum combined with a cooling crystallization, underscores the advantages of MARS in terms of product isolation. In addition, other products are reported to have been isolated from MARS by crystallization.6 Combined, the presented results demonstrate that MARS is an easily applicable reaction system enabling facile product isolation by crystallization.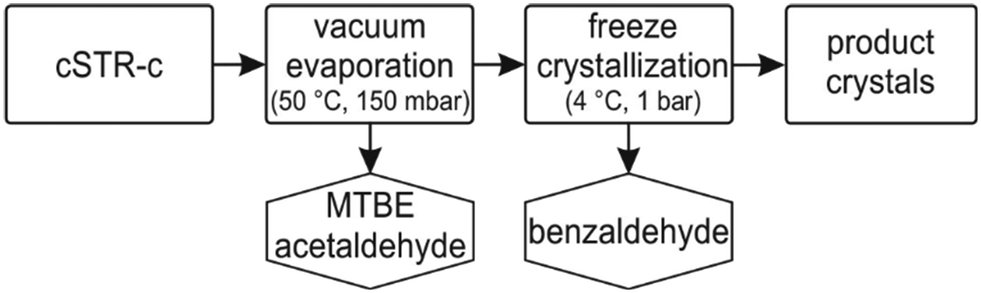 | ||
| Fig. 5 Block flow diagram for (S)-HPP isolation. | ||
Substrate range screening to broaden product platform
Having established a robust synthesis process for (S)-2-HPP with an easy and fast product isolation, it would be desirable to use the process to gain access to a whole (S)-2-HPP derivative platform, which can be used for further chemical syntheses. Hence, nitro-, halo-, and methoxy-derivatives of benzaldehyde substituted at the ortho-, meta-, and para-position were screened for mixed carboligation products with acetaldehyde with the enzyme PpBFD varL461A. The outcome of this substrate range screening in MARS revealed, that all (S)-2-HPP derivatives except for the meta-nitro derivative can be produced (Table 2). Even more convincing is the exceptional ee of >99%, which was obtained for almost all derivatives when using PpBFD varL461A. Also, chemoselectivity was excellent, as no phenylacetylcarbinol (IUPAC: 1-hydroxy-1-phenyl-propan-2-one) derivatives were detected.|
|
|
|
||||
|---|---|---|---|---|---|---|
| Conv. [%] | ee [%] | Conv. [%] | ee [%] | Conv. [%] | ee [%] | |
| R = F | 82 | 84 | 95 | 99 | 58 | 99 |
| R = Cl | 22 | 99 | 87 | 96 | 48 | 99 |
| R = Br | 8 | 99 | 81 | 99 | 45 | 99 |
| R = MeO | 19 | 99 | 85 | 96 | 56 | 95 |
| R = NO2 | 33 | 99 | 1 | 69 | 99 | 99 |


Transfer of cSTR-c to an a para-methoxylated (S)-2-HPP derivative
As an example, para-methoxy-benzaldehyde was selected to proof transferability of the cSTR-c to (S)-2-HPP synthesis to other products. In a quick proof-of-principle without determination of kinetic parameters, already 41% conversion with an ee of 99% could be obtained (ESI† Fig. S9, for 1H-NMR determination see S11) without any product specific optimization. This is almost equally as good as a recently presented fed-batch process optimized in MARS, which achieved a 46% conversion of para-methoxy benzaldehyde to the target (S)-2-HPP derivative.6 A determination of kinetic parameters would certainly help to further improve reaction conditions in the cSTR and thus lift it to levels of the fed-batch process or even beyond. But this resource- and time-saving rapid transferability test already shows that the presented cSTR-cascade is a stable synthesis system that can even be transferred to other substrates to obtain a product platform of (S)-2-HPP derivatives.Summary and conclusion
The herein presented work combines the strengths of a MARS environment, continuous process operation, and model-driven process optimization to selectively obtain 190 mM (S)-2-HPP with an ee > 98% over 8 h.First, kinetic behaviour of applied PpBFD varL461A LWCs in MARS was analysed to determine ideal starting concentrations of the carboligation substrates benzaldehyde and acetaldehyde. It was shown that benzaldehyde concentrations of up to 500 mM and acetaldehyde up to 100 mM can be applied before substrate inhibition sets in. The collected data was used subsequently to deduce apparent Michaelis–Menten parameters. Based on the kinetic data, a continuous process in a cSTR-cascade was selected to minimize effects of substrate inhibition and enzyme deactivation (Fig. 2). Second, options for enhancing catalyst stability were explored to prolong continuous operation. This revealed the supplementation of 5 mM ThDP with 25 mM Mg2+ in the 22% (v v−1) MARS buffer fraction as beneficial for enhanced stability. Moreover, the distribution of acetaldehyde over the cSTR-cascade was found to be vital, as enzyme deactivation was observed to be caused by elevated acetaldehyde concentrations. For quantifying this, a novel enzyme deactivation kinetic was proposed that describes the deactivation of the tetrameric PpBFD varL461A as combination of the independent deactivation of its monomers. The final reaction model determined concentrations of 120 mM acetaldehyde as threshold level, beyond which enzyme deactivation accelerates disproportionately fast. This was accounted for by distributing the total acetaldehyde amount which is required to convert 250 mM benzaldehyde over three cSTR-units. The model was also used for computing acetaldehyde stock solution flow rates Fi,stock (Fig. 2B) that prevent an exceeding of 100 mM acetaldehyde in the respective cSTR-cascade units. Third, all determined process parameters were successfully applied for design a cSTR-cascade with a productivity of 4.56 g g−1LWC (S)-2-HPP. The whole synthesis process was supported by a mechanistic model with staged deactivation, which illustrated the kinetic reaction behaviour in this continuous reaction setup in an unconventional reaction environment. Although the continuous (S)-2-HPP synthesis established here delivers lower product titres compared to an established fed-batch process, a doubled sSTY could be achieved due to catalyst retention and reuse. Hence, the economic feasibility of biocatalytic processes is significantly increased by continuous process operation. In a subsequent downstream processing the organic reaction environment MARS facilitated product isolation and permitted direct product crystallization with an isolated yield of 68% and a crystal purity of 99%. Ultimately, a substrate screening with PpBFD varL461A was performed to identify 2-hydroxy ketone derivatives which are also accessible via this process setup. Among all tested bromo-, chloro-, fluoro-, methoxy-, and nitro-derivatives at the ortho, meta, and para position, only meta-nitro-benzaldehyde could not be converted. All others exhibited excellent enantio- and regioselectivity. Among them para-methoxy-benzaldehyde was transferred in a proof of principle into the setup cascade and yielded in an unoptimized state already 41%. This verifies the potential of this technical setup in combination with the enzyme to be applicable for the production of a product platform.
Conclusively, this biocatalytic reaction system appeals with its continuous output of high product concentrations and an microaqueous reaction environment granted by MARS. This quite possibly could facilitate a vast implementation of biocatalysis in complex continuous flow synthesis (e.g. unconventional reaction environment) in the near future.
Abbreviations
| c | Concentration [mM] |
| cSTR | Continuous stirred tank reactor |
| cSTR-c | Continuous stirred tank reactor cascade |
| F | Flow velocity [m3 s−1] |
| 2-HPP | 2-Hydroxy-1-phenylpropanone |
| LWC | Lyophilized whole cells |
| MARS | Micro-aqueous reaction system |
| MgSO4 | Magnesium sulfate |
| PpBFD | Pseudomonas putida benzoylformate decarboxylase |
| MTBE | Methyl tert-butyl ether |
| τ | Retention time |
| TEA | Triethylamine |
| ThDP | Thiamine diphosphate |
| V | Reactor volume [m3] |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Helmholtz-Association within the framework of the young investigator's group “synthetic enzyme cascades” for funding this project. Further we thank the company Alfa Laval for their valuable input on membrane selection. Also, we thank Christoph Westerwalbesloh and Martina Pohl for fruitful discussions on kinetics and process modelling. Many thanks go to Lilia Arnold for her great investment in the practical work of this academic piece.Notes and references
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211–230 CAS.
- U. T. Bornscheuer, Philos. Trans. R. Soc., A, 2018, 376, 1–5 CrossRef PubMed.
- R. León, P. Fernandes, H. M. Pinheiro and J. M. S. Cabral, Enzyme Microb. Technol., 1998, 23, 483–500 CrossRef.
- H. Meyer, E. Eichhorn, S. Hanlon, S. Lütz, M. Schürmann, R. Wohlgemuth and R. Coppolecchia, Catal. Sci. Technol., 2013, 3, 29–40 RSC.
- R. Oeggl, T. Maßmann, A. Jupke and D. Rother, ACS Sustainable Chem. Eng., 2018, 6, 11819–11826 CrossRef CAS.
- A. Jakoblinnert and D. Rother, Green Chem., 2014, 16, 3472–3482 RSC.
- D. Gocke, L. Walter, E. Gauchenova, G. Kolter, M. Knoll, C. L. Berthold, G. Schneider, J. Pleiss, M. Müller and M. Pohl, ChemBioChem, 2008, 9, 406–412 CrossRef CAS.
- W. H. De Camp, J. Pharm. Biomed. Anal., 1993, 11, 1167–1172 CrossRef CAS PubMed.
- T. Dünnwald, A. S. Demir, P. Siegert, M. Pohl and M. Müller, Eur. J. Org. Chem., 2000, 2000, 2161–2170 CrossRef.
- H. Iding, T. Dünnwald, L. Greiner, A. Liese, M. Müller, P. Siegert, J. Grötzinger, A. S. Demir and M. Pohl, Chem. – Eur. J., 2000, 6, 1483–1495 CrossRef CAS.
- B. Lingen, J. Grötzinger, D. Kolter, M.-R. Kula and M. Pohl, Protein Eng., 2002, 15, 585–593 CrossRef CAS PubMed.
- P. Domínguez de María, T. Stillger, M. Pohl, M. Kiesel, A. Liese, H. Gröger and H. Trauthwein, Adv. Synth. Catal., 2008, 350, 165–173 CrossRef.
- M. Berheide, S. Kara and A. Liese, Catal. Sci. Technol., 2015, 5, 2418–2426 RSC.
- A. V. Presečki, L. Pintarić, A. Švarc and Đ. Vasić-Rački, Bioprocess Biosyst. Eng., 2018, 41, 793–802 CrossRef PubMed.
- O. P. Ward and A. Singh, Curr. Opin. Biotechnol., 2000, 11, 520–526 CrossRef CAS.
- J. Wachtmeister, A. Jakoblinnert and D. Rother, Org. Process Res. Dev., 2016, 20, 1744–1753 CrossRef CAS.
- T. Yamane, Biocatalysis, 1988, 2, 1–9 CrossRef CAS.
- V. Erdmann, U. Mackfeld, D. Rother and A. Jakoblinnert, J. Biotechnol., 2014, 191, 106–112 CrossRef CAS PubMed.
- A. Jakoblinnert, R. Mladenov, A. Paul, F. Sibilla, U. Schwaneberg, M. B. Ansorge-Schumacher and P. Domínguez de María, Chem. Commun., 2011, 47, 12230 RSC.
- J. Wachtmeister and D. Rother, Curr. Opin. Biotechnol., 2016, 42, 169–177 CrossRef CAS PubMed.
- G. Goetz, P. Iwan, B. Hauer, M. Breuer and M. Pohl, Biotechnol. Bioeng., 2001, 74, 317–325 CrossRef CAS PubMed.
- H. Bisswanger, Enzyme Kinetics, WHILEY-VCH Verlag GmbH, Weinheim, 1st edn, 2002 Search PubMed.
- B. U. Kragl, D. Gygax, O. Ghisalba and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1991, 101, 827–828 CrossRef.
- J. Wachtmeister, P. Mennicken, A. Hunold and D. Rother, ChemCatChem, 2016, 8, 607–614 CrossRef CAS.
- M. Zavrel, T. Schmidt, C. Michalik, M. Ansorge-Schumacher, W. Marquardt, J. Büchs and A. C. Spiess, Biotechnol. Bioeng., 2008, 101(1), 27–38 CrossRef CAS PubMed.
- E. S. Polovnikova, M. J. McLeish, E. A. Sergienko, J. T. Burgner, N. L. Anderson, A. K. Bera, F. Jordan, G. L. Kenyon and M. S. Hasson, Biochemistry, 2003, 42, 1820–1830 CrossRef CAS PubMed.
- R. W. Lencki, J. Arul and R. J. Neufeld, Biotechnol. Bioeng., 1992, 40, 1421–1426 CrossRef CAS PubMed.
- F. C. Neidhardt, K. Tummler and R. Milo, Cell biology by the numbers, Taylor & Francis, New York, 1990 Search PubMed.
- R. Milo, Overall macromolecular composition of E. coli cell.
- M. S. Hasson, A. Muscate, M. J. McLeish, L. S. Polovnikova, J. A. Gerlt, G. L. Kenyon, G. A. Petsko and D. Ringe, Biochemistry, 1998, 37, 9918–9930 CrossRef CAS.
- J. Döbber, T. Gerlach, H. Offermann, D. Rother and M. Pohl, Green Chem., 2018, 20, 544–552 RSC.
- F. H. Andrews and M. J. McLeish, FEBS J., 2013, 280, 6395–6411 CrossRef CAS PubMed.
- M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed.
- B. Mojsoska, G. Carretero, S. Larsen, R. V. Mateiu and H. Jenssen, Sci. Rep., 2017, 7, 42332 CrossRef CAS PubMed.
- A. F. Makarchikov, B. Lakaye, I. E. Gulyai, J. Czerniecki, B. Coumans, P. Wins, T. Grisar and L. Bettendorff, Cell. Mol. Life Sci., 2003, 60, 1477–1488 CrossRef CAS PubMed.
- M. Zampieri, K. Sekar, N. Zamboni and U. Sauer, Curr. Opin. Chem. Biol., 2017, 36, 15–23 CrossRef CAS PubMed.
- M. Oldiges, S. Noack and N. Paczia, Metabolomics in Biotechnology (Microbial Metabolomics), Metabolomics in Practice, 2013, pp. 379–391 Search PubMed.
- N. N. Rao, S. Lütz, K. Würges and D. Minör, Org. Process Res. Dev., 2009, 13, 607–616 CrossRef CAS.
- A. Liese, K. Seelbach and C. Wandrey, Industrial Biotransformation, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2006 Search PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01666g |
| ‡ Concentrations in the buffered stock solution were up to 5 mM ThDP and 25 mM MgSO4. |
| § Notably, the low density of acetaldehyde made it technically impossible to pump pure acetaldehyde and required a concentration below 4 M in MTBE to obtain reliable pumping. |
| This journal is © The Royal Society of Chemistry 2021 |