
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent organic frameworks as multifunctional materials for chemical detection
Zheng
Meng
 and
Katherine A.
Mirica
*
and
Katherine A.
Mirica
*
Department of Chemistry, Burke Laboratory, 41 College Street, Dartmouth College, Hanover, NH 03755, USA. E-mail: Katherine.A.Mirica@dartmouth.edu
First published on 17th November 2021
Abstract
Sensitive and selective detection of chemical and biological analytes is critical in various scientific and technological fields. As an emerging class of multifunctional materials, covalent organic frameworks (COFs) with their unique properties of chemical modularity, large surface area, high stability, low density, and tunable pore sizes and functionalities, which together define their programmable properties, show promise in advancing chemical detection. This review demonstrates the recent progress in chemical detection where COFs constitute an integral component of the achieved function. This review highlights how the unique properties of COFs can be harnessed to develop different types of chemical detection systems based on the principles of chromism, luminescence, electrical transduction, chromatography, spectrometry, and others to achieve highly sensitive and selective detection of various analytes, ranging from gases, volatiles, ions, to biomolecules. The key parameters of detection performance for target analytes are summarized, compared, and analyzed from the perspective of the detection mechanism and structure–property–performance correlations of COFs. Conclusions summarize the current accomplishments and analyze the challenges and limitations that exist for chemical detection under different mechanisms. Perspectives on how future directions of research can advance the COF-based chemical detection through innovation in novel COF design and synthesis, progress in device fabrication, and exploration of novel modes of detection are also discussed.
 From left to right: Zheng Meng and Katherine A. Mirica | Zheng Meng obtained his BS from the University of Science and Technology Beijing in 2011. He received his PhD from the Institute of Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Chuan-Feng Chen in 2016. He then moved to Dartmouth College to work as a postdoctoral researcher with Prof. Mirica. His current interests focus on the development of new two-dimensional conductive framework materials and their applications in electronics, sensing, and catalysis. Dr. Katherine Mirica is an Associate Professor of Chemistry at Dartmouth College. She received her BS in Chemistry from Boston College, where she worked in the laboratory of Prof. Lawrence T. Scott. Katherine earned her PhD in Chemistry from Harvard University under the supervision of Prof. George M. Whitesides and completed her postdoctoral training with Prof. Timothy Swager at the Massachusetts Institute of Technology. Dr Mirica began her independent career at Dartmouth College in 2015 and was promoted to Associate Professor in 2021. Her current research program aims to design, synthesize, and gain fundamental insight into structure–function relationships of molecularly precise multifunctional materials with potential applicability for deployment as portable chemical sensors, personal protective equipment, and energy-efficient devices. |
1. Introduction
1.1 Development of materials for the advancement of chemical detection
Sensitive and selective detection of chemical and biological analytes is an important branch of analytical science for identifying targets and determining their concentrations. Sensing has been widely applied in various scientific and technological fields ranging from analytical chemistry, life science, materials science, biomedical diagnostics to drug discovery, food security, personal care, environmental monitoring, and the Internet of Things.1–4 Traditionally, advances in chemical detection have largely benefited from the development of various instrumental methods, such as spectroscopic techniques,5,6 detectors coupled to chromatography,7,8 mass spectrometry,9 electrochemical methods,10 and others,11 that have achieved ultrasensitive detection of chemical and biological analytes. Although instrumental analytical approaches continue to evolve and yield capacities on breathtaking scales of complexity and sensitivity, with the increasingly diversified purposes of chemical detection associated with urgent concerns over health and environment, the development of low-cost, convenient, real-time, and highly efficient methods based on portable, wearable, and miniaturized devices for chemical analysis is greatly demanded.12–17Materials chemistry has historically played an indispensable role in analytical chemistry.18–20 The discovery of new materials has triggered the development of new analytical methods and instrumentation capable of detecting analytes at the single-molecule level.21 The ability to measure subtle chemical and physical changes in a material has led to the development of materials that can induce sensing responses and transduction of electrical or spectroscopic signals by changing their optical or electronic properties, such as changes in impedance, color, fluorescence, Raman scattering, and surface plasmon resonance.22 Stimuli-responsive materials have been demonstrated to be useful for detecting a diverse set of analytes, including gases, pH, ATP, microRNA, pesticides, proteins, and bacteria. In addition to serving as analyte-responsive and signal transduction components,17,22,23 materials can also serve as key components that aid chemical detection, such as an auxiliary selective layer, adsorption module, or scaffold, to couple with classical instrumental techniques for the improvement of the detection performance. For example, materials have been used as conventional sorbents in the solid-phase extraction technique for the pretreatment of the samples or as stationary phases in gas and liquid chromatography or electrochromatography for highly efficient target separations.24–26
Progress in chemistry and materials science over the past century has given rise to several distinct classes of materials, such as zeolites,27 conjugated polymers,28 graphene,29 carbon nanotubes,30 and metal–organic frameworks (MOFs),31–33 that have significantly propelled the advancement of chemical detection. The unique optical, electronic, and catalytic properties of these materials have been harnessed to create new detection platforms for extensive and successful applications in chemical detection. For example, graphene has been applied for the fabrication of highly sensitive, wearable, self-powered sensors for the detection of various ions and biological molecules, owing to its extraordinary electrical, chemical, optical, and mechanical properties.34–36 Conjugated polymers have been developed as reliable materials for the detection of explosives.37–40 The utilization of carbon nanotubes has enabled their integration into smart and mobile devices toward portable, wireless, non-line-of-sight gas sensing.41–44 The development of new chemically precise materials that can retain the useful attributes of these established materials while integrating new features, such as porous scaffolds that provide high surface area for analyte uptake and modular accessibility through bottom-up self-assembly, is poised to provide new opportunities for advancing the field of chemical and biological detection.
1.2 COFs as emergent multifunctional materials for chemical detection
COFs constitute a class of extended and crystalline porous materials comprising lightweight elements linked together by covalent bonds, and feature precise periodicity in the skeleton with pre-designable pore parameters. Since the landmark synthesis of COFs by Yaghi and coworkers in 2005,45 the field of COFs has grown tremendously and has attracted intense attention from researchers with diverse expertise.46–48 As a new generation of porous materials, COFs possess several unique features of chemical modularity, accessibility through bottom-up synthesis from earth-abundant elements, large surface area, high chemical stability with robust chemical linkages, low density, tunable pore sizes, and other modular optical and electronic features. These desirable properties provide new opportunities for a wide range of potential applications in gas adsorption and storage,49–53 separation,54–59 catalysis,60–65 sensing,66–69 drug delivery,70 optoelectronics,71–73 and energy storage.46,74–79 With the progress in the design and synthesis of COFs and the discovery of their physical and properties, COF-based materials exhibit great potential in analytical chemistry.COFs have several distinct features and advantages that make them an intriguing group of materials for a new approach to chemical detection. First, COFs have great tunability that allows atomically precise integration of functional organic units into extended and ordered frameworks in a modular and bottom-up manner. The installation of functional and chemically responsive units, such as optically active fragments, Lewis acidic or basic sites, redox-active centers, and catalytic units, can proceed by the judicious selection of building units with control over the linkage of those building blocks to the desired properties of the target material in the context of chemical detection. The incorporation of specific recognition sites into building blocks is an effective way of generating COFs with selective detection capabilities. The functional groups that are introduced into COFs during their synthesis can be further utilized for post-synthetic modification to render different capabilities, such as improved sensitive and selective recognition of analytes.80 Fine-tuning of the optical and electronic properties for modulating the resulting detection performance can be achieved by a systematic change of the constituent building blocks among a series of structurally related COFs. This modular control, together with the crystalline nature of COFs, makes them perfect systems for investigating structure–property–performance correlations, assisting in the evolution of the analytical performance.
Second, the intrinsic porosity of COFs with large surface-to-volume ratios is also advantageous in chemical detection. The Brunauer–Emmett–Teller (BET) surface areas of two-dimensional (2D) and three-dimensional (3D) COFs have exceeded 300081 and 5000 m2 g−1,82 respectively. These large surface-to-volume ratios of COFs ensure a tremendous accessible surface area for material–analyte interactions, which are beneficial for realizing high sensitivity, even at extremely low concentrations of the analyte. The porous molecular scaffold with the functional sites can provide a platform for the immobilization of additional recognition components and other supplementary attachments (e.g., metallic nanoparticles, metal oxides, and enzymes), which can further improve the sensing performance. Additionally, the intrinsic porous structure allows efficient uptake of analyte molecules, which is beneficial for the preconcentration of the analyte to enhance the detection sensitivity. Selectivity for the uptake of guest molecules can also be further optimized through the engineering of pore parameters.55
Third, certain classes of COFs can be constructed using robust covalent bonds and thereby can show enhanced chemical and thermal stability when compared with their counterparts, MOFs, which are usually connected by relatively weak coordination bonds.83,84 The reinforced stability of COFs can be obtained by providing hydrogen bonding interactions,85 weakening the polarity of the amine bond,86,87 introducing enol–keto tautomerizations,88 utilizing a Michael addition–elimination/benzoxazole pathway,89 and constructing polyarylether90 and pyrazine linkages.91–93 The state-of-the-art approaches to stabilize COFs have resulted in materials that are highly stable to hydrolysis, a wide range of pH, and reductive and oxidative environments.79,94,95 These advances can ensure the applicability of COFs for chemical detection in harsh chemical or physical conditions, such as high ionic strength, high acidity, high basicity, or high operating temperatures.
Overall, chemical detection can take full advantage of the crystalline nature, diverse tunable structure, porous and stable skeleton, and a wide range of other properties of COFs to develop detection platforms. Because of these diversified structural features and properties, COFs have been utilized as multifunctional materials for chemical detection. Different types of chemical detection modes using COFs have been established, including chromism, luminescence, electrical transduction (including electrochemical and chemiresistive detection), chromatographic and spectrometric detection, surface-enhanced Raman spectroscopy (SERS), and quartz crystal microbalance (QCM) (Section 2). Among these methods, detection based on chromism, luminescence, electrochemistry, chromatography, and spectrometry has now been well established. Examples based on SERS, QCM, and chemiresistive detection, although still limited at this time, constitute emerging areas with room for future development.
COFs play multiple roles in analytical detection mechanisms (Fig. 1). In the detection using chromism, luminescence, and electrical transduction, COFs can serve as both receptor and transducer, where they recognize analytes and produce a readable signal attributed to the changes in their photophysical and electronic properties. In some of the electrochemistry-based, SERS, and QCM detection modes, COF-based materials only function as receptors for selective binding of analytes. In chromatographic and spectrometric detection, COFs play an important role as an adsorbent in the selective extraction of an analyte from a complex sample containing trace amounts of the target. COFs also provide porous scaffolds for loading other active materials in building electrochemical detection platforms or serve as an ionization matrix in MS for small molecule detection. By reviewing these detection schemes, we summarize key accomplishments for COF-based chemical detection, and numerous opportunities that exist for both fundamental and applied research in this growing field.
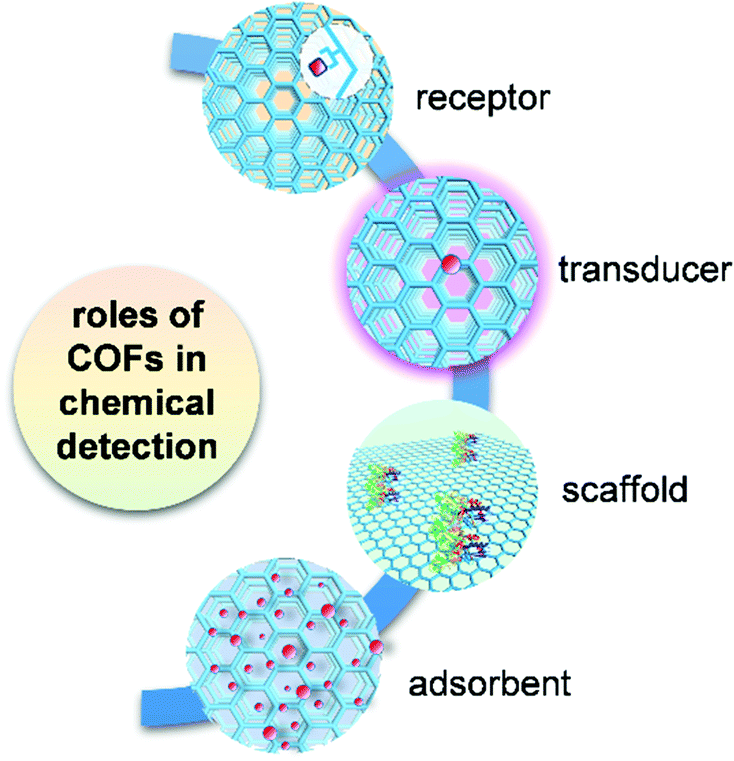 | ||
| Fig. 1 Multiple roles of COFs in the application of chemical detection. | ||
1.3 Scope of the review
In this review, we focus on the recent progress in chemical detection that uses COFs as an integral part of the analytical system. We demonstrate in detail the application of COFs in chemical detection based on different mechanisms categorized into four groups: (1) chromism, (2) luminescence, (3) electrical transduction, and (4) chromatographic and spectrometric detection. We also include the discussion of detection mechanisms that are relatively less established, but different from those mentioned above, including SERS, laser desorption/ionization time-of-flight mass spectrometry, and QCM. We provide a brief tutorial on the basic operation principles of each detection mode to establish a clear connection between the detection mechanism and the unique structural characteristics and properties of COFs. In the discussion of each detection mode, the advances in chemical detection using COFs are highlighted chronologically and by dividing analytes into three major groups: gases and volatiles, ions, and biomolecules, whenever possible. The key parameters of detection performance for target analytes are summarized, compared, and analyzed in the context of the structure–property–performance correlations of COFs. We conclude by summarizing the current accomplishments and analyzing the challenges and limitations that exist for chemical detection under each mechanism. We give perspectives on the future trends that will drive COF-based chemical detection to a new stage through innovation in novel COF design and synthesis, advances in device fabrication, and exploration of novel modes for detection.With the rapid development in the synthesis and discovery of the properties of COFs, as well as the advances in the fabrication techniques of COFs, their applications in chemical detection have burgeoned into a new research field. Only a few reviews have focused on the summary of the potential of COFs in chemical sensing,68,69 and the systematic discussion of the progress and challenges of COFs in the broader context of chemical detection and from the perspective of different detection mechanisms is still lacking. One of the important aims in the organization of this review is to help the readers appreciate how the unique and diversified properties of COFs can be harnessed in multiple ways to develop chemical detection systems based on different detection principles within a field that is in a period of rapid growth.
The synthesis of COFs can be achieved through bottom-up connections of organic building units with well-defined geometry by various covalent linkages, such as boronic ester, imines, hydrazones, azines, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C coupling, and dioxines, yielding extended, crystalline, porous, solid-state materials.46,47,79,96–100 The applications of COFs often depend on the embedded chemical functionality. Functionality that cannot be introduced into COFs during direct synthesis can be accessed through post-synthetic modification using metal complexation, covalent attachment with existing pendant groups, and chemical conversion of linkages.80 The synthesis, modification, and fabrication strategies of COFs have already been well documented,46–48,79,80,96–100 and, therefore, are omitted in this review, though chemical detection methods using COFs build upon on this groundwork. Fundamental understanding of the structure–property relationships of COFs can guide the design of new COFs for different purposes, such as gas capture, catalysis, and energy storage, to name just a few. For the discussions of structure–property relationships in a general sense, the readers are encouraged to refer to existing information on this topic.46,47,96 Chemical detection using amorphous porous covalent organic polymers40,101–106 is not discussed in this review due to the amorphous nature and lack of an atomically precise structure of these materials.
C coupling, and dioxines, yielding extended, crystalline, porous, solid-state materials.46,47,79,96–100 The applications of COFs often depend on the embedded chemical functionality. Functionality that cannot be introduced into COFs during direct synthesis can be accessed through post-synthetic modification using metal complexation, covalent attachment with existing pendant groups, and chemical conversion of linkages.80 The synthesis, modification, and fabrication strategies of COFs have already been well documented,46–48,79,80,96–100 and, therefore, are omitted in this review, though chemical detection methods using COFs build upon on this groundwork. Fundamental understanding of the structure–property relationships of COFs can guide the design of new COFs for different purposes, such as gas capture, catalysis, and energy storage, to name just a few. For the discussions of structure–property relationships in a general sense, the readers are encouraged to refer to existing information on this topic.46,47,96 Chemical detection using amorphous porous covalent organic polymers40,101–106 is not discussed in this review due to the amorphous nature and lack of an atomically precise structure of these materials.
MOFs have been historically recognized as a group of structurally related materials of COFs, which share several key features with COFs, including high surface area, permanent porosity, extended crystalline structure, and modular and tunable accessibility. The applications of MOFs in chemical sensing have already been well summarized in multiple reviews.107–114 These reviews are helpful for the understanding of the unique role of COFs in chemical detection, in contrast to their MOF counterparts. Compared with MOFs, COFs possess several distinct features that make them unique in the context of chemical detection. Different from the relatively labile coordination linkages in MOFs, several types of strong covalent linkages have been exploited to obtain COFs with extraordinary thermal and chemical stability.46,96 This feature is essential for chemical detection under harsh conditions. Metal ions incorporated in MOFs usually have nontrivial effects on the optical, electrical, and electrochemical properties of the frameworks resulting from the intrinsic properties of metal ions or complex metal-to-metal and/or metal-to-ligand interactions. Since there are usually no metals embedded in COF structures, the engineering of targeted properties of COFs for chemical detection can be more straightforward, as compared to MOFs.108,115 The relatively low-density feature of COFs ascribed to their composition of light elements may also be beneficial for the development of wearable and large-area chemical sensing devices. Finally, reliance primarily on earth-abundant elements of C, H, O, and N for COF construction can be beneficial from the perspective of sustainability and cost. While the use COFs as functional materials in chemical detection is still relatively understudied compared with MOFs, Table 1 summarizes the unique features of COFs as an important candidate for chemical detection, in comparison with other relatively established materials, including carbon nanotubes,41,116 graphene,34,117 metal oxides,118,119 conjugated polymers,120 and MOFs.107–113
| Example | Advantages | Disadvantages | Detection modes |
|---|---|---|---|

|
• High surface-to-volume ratio | • Requirement of chemical modification to enhance selectivity | • Electrochemical |
| • High aspect ratio | • Difficulty in establishing reliable electrical contacts | • Chemiresistive | |
| • Excellent stability | • Difficulty in purification | • Optical detection | |
| • High density of reactive sites | • Limited structure and precision control | ||
| • Good thermal stability | |||
| • Compatible with device miniaturization | |||
|
• Large surface-to-volume ratio | • Zero bandgap | • Electrochemical |
| • Good optical transparency | • Lack of effective analyte binding sites in its pure form | • Chemiresistive | |
| • Excellent mechanical flexibility | |||
| • Good functionalization ability | |||
| • Potential for good processability | |||
| • Compatible with ultra-thin silicon channel technology | |||

|
• Strong analyte binding | • Low surface area | • Chemiresistive |
| • Good mechanical strength | • Difficulty with miniaturization | • Electrochemical | |
| • Good thermal stability | • Slow dynamics of analyte transport | ||
| • Easy to interface with solid-state devices | |||
| • Good designability to improve selectivity | |||
|
• Feasibility of introducing an analyte receptor | • Possibility of swelling during measurement | • Fluorescence |
| • Collective (molecular wire) effect for amplified sensitivity | • Limited solubility in aqueous solution | • Conductometric | |
| • Wide tunability of conductivity | • Dependence on molecular design for high selectivity | • Potentiometric | |
| • Good processability | • Colorimetric | ||
|
• Large surface area and porous structure | • Stability in harsh environment | • Fluorescence |
| • Atomically precise structure | • Challenge in the fabrication of materials into devices | • Electrochemical | |
| • Abundant active sites | • High cost of large-scale production | • Chemiresistive | |
| • Tunable pore geometry, surface chemistry, and physical properties | • Electromechanical | ||
|
• Large surface area and porous structure | • Limited processability | • Fluorescence |
| • Potential for atomically precise structure | • Challenge in the synthesis of material with high crystallinity and controlled morphology | • Colorimetric | |
| • Abundant active sites | • High cost of large-scale production | • Electrochemical | |
| • Tunable pore geometry, surface chemistry, and physical properties | • Chemiresistive | ||
| • Generally contains light and earth-abundant elements | |||
| • Potential to be highly stable | |||
2. COFs for different types of chemical detection
2.1 Detection based on chromism
COFs have been used in two different modes for chromism-based detection: (1) the chromism phenomenon directly arising from the changes in the absorption properties of COFs; (2) the chromism phenomenon not arising directly from COFs, and instead arising from changes in the color of external dye molecules. The first chromism mode can be further detailed by two situations. First, the material–analyte interaction perturbs the electronic transition responsible for the coloration and results in a change in the absorption of the chromophoric component of the COF.122 For example, in solvatochromism, one type of commonly seen chromism induced by solvent, solvent molecules with different polarities can stabilize the excited or ground state of a chromophoric component of the COFs by lowering the LUMO or HOMO level of the chromophore, and respectively causing a red or blue shift. Second, the material–analyte interaction triggers the isomerization of the chromophoric component of COFs, which subsequently changes the electronic transition responsible for the coloration.123,124 For example, the H-bonding interaction between the analyte and COFs can result in iminol-to-ketoenamine tautomerism of the COF fragment, causing a change in the absorption spectrum.124 In most COF structures, the extended conjugated skeleton or aromatic units can serve as chromophores that show characteristic and intensive absorption covering ultraviolet to visible, or even extending to the near-infrared range.125 The interactions between analyte molecules and COFs, including hydrogen bonding123,124,126,127 and protonation,128,129 can initiate an absorption change of these chromophores through either of the two situations described above, which has been utilized for the detection of volatile molecules, including humidity,123,126,127 organic solvent molecules,126 and acids.128,129
In the second mode, absorption-based detection using COFs is realized by the chromism of external dye molecules. Here, instead of exhibiting chromism, COFs serve as enzyme mimics to catalyze/mediate redox reactions of dye molecules, leading to a color change of the dyes. When analytes function as oxidants or reductants, the concentration of analytes is proportional to the change of the intensity of the absorption spectrum of the dye.130,131 When an analyte forms a COF–analyte conjugate or complex that has improved catalytic ability for the redox reaction, the concentration of the analyte can be related to the adsorption change of the dye under a given amount of time.132–135 The ability of some COFs to mediate coloration reactions of external dyes has been mainly utilized for the detection of some metal ions132–135 and several biomolecules.130,131,133
Volatiles. A suitably designed COF treated as a periodic combination of molecular dyes can provide the advantages of insolubility and chemical and photochemical stability over small molecule dyes for detecting target molecules in a stream of gas or liquid.85 Auras and coworkers developed the first solvatochromic Py–TT COF composed of pyrene (Py) and thieno-[3,2-b]thiophene (TT) units, which showed strong color shifts when exposed to water or solvent vapors (Fig. 2a).126 Exposing the Py–TT COF film to a humidified N2 stream produced a color change from orange to dark red. Transmission ultraviolet-visible (UV-vis) spectra recorded at different relative pressures of H2O revealed the appearance of an absorption band in the 520–640 nm region and a simultaneous decrease in the 440–500 nm and 280–380 nm spectral regions (Fig. 2b). The COF film exhibited the highest sensitivity towards humidity changes between relative pressures of H2O in the range of 0.64–0.79 (Fig. 2c). The response to changes in the surrounding atmosphere occurred within milliseconds, outperforming commercially available humidity sensors by more than an order of magnitude (Fig. 2d). In addition to this rapid response, the COF film displayed excellent reversibility and stability over at least 4000 switching cycles of humidity and solvent vapor. The Py–TT COF showed positive solvatochromism, in which the absorption was red-shifted with increasing polarity (Fig. 2e); in this case, the increasing polarity of the surrounding medium provided preferential stabilization of the excited state over the ground state, thus lowering the energy required for photoexcitation. Experimental data and time-dependent density-functional theory (TD-DFT) calculations further suggested that the solvatochromism possessed a pronounced charge-transfer character, which involved a reduction of the electron density on the pyrene moieties and a gain in electron density of the thienothiophene bridges (Fig. 2f). The absence of structural or chemical changes of the COF during the solvatochromism may explain the rapid response to humidity. The observation that electronic transitions in this material can be manipulated reversibly and that intramolecular charge transfer can be facilitated via the inclusion of chemically inert guest molecules has the potential to impact the development of stimuli-responsive organic electronics.126
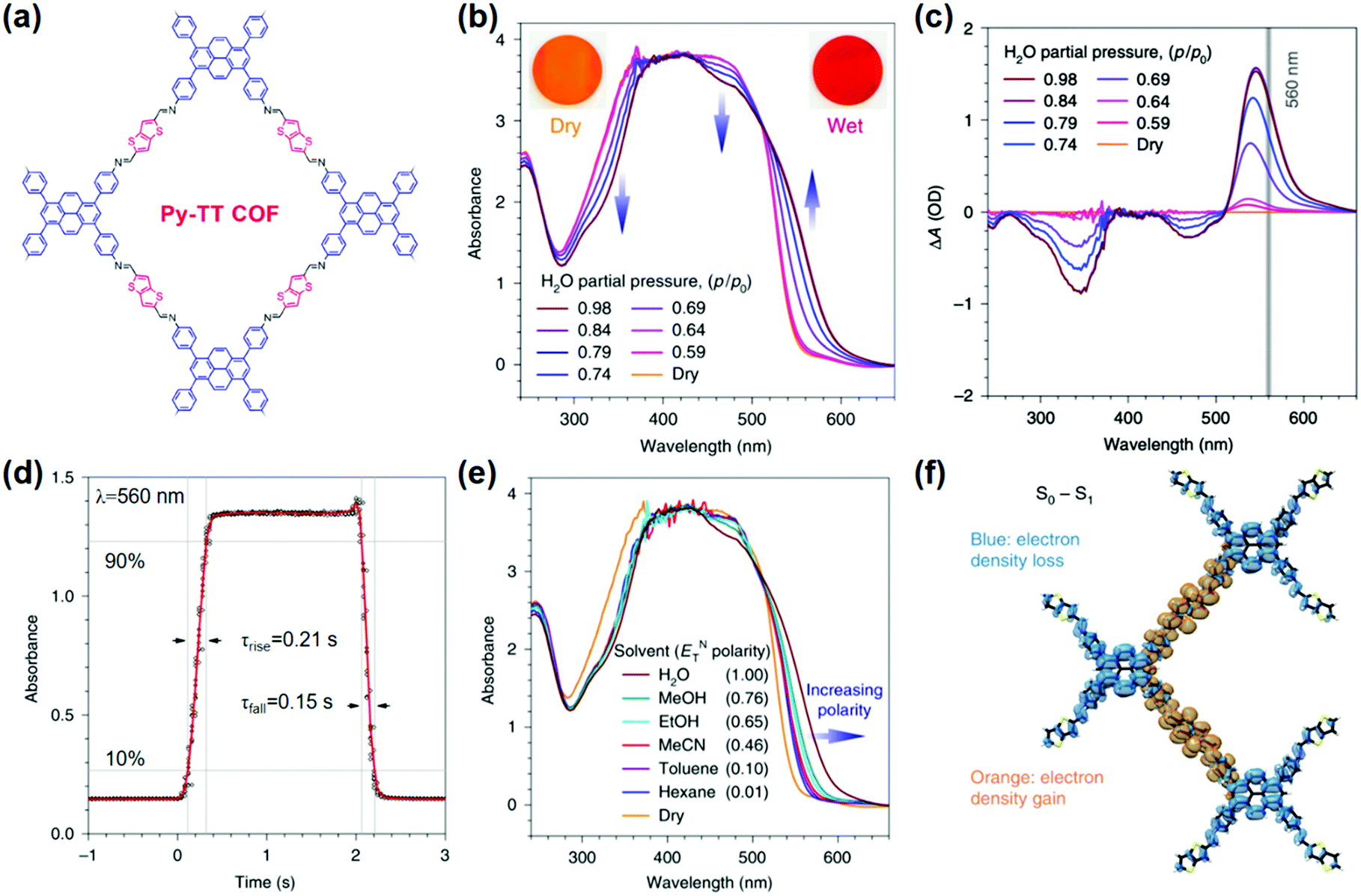 | ||
| Fig. 2 (a) Chemical structure of Py-TT COF. (b) UV-vis absorption spectra of the Py–TT COF film recorded at different relative pressures of H2O in N2. (c) Plots of the humidity-induced absorbance changes at different H2O relative pressures. The grey line indicates the wavelength used for the response time measurements. (d) The solvatochromic response of the Py–TT COF film towards step changes between dry and H2O-saturated N2 streams. (e) UV-vis spectra of the same COF film in saturated atmospheres of various solvents. (f) TD-DFT calculated electron density difference upon one-electron excitation from the ground state (S0) to the first singlet excited state (S1).126 Adapted with permission from ref. 126. Copyright 2018, Springer Nature. | ||
Liu and coworkers reported another crystalline DHNDA-TAPP COF made of 2,6-dihydroxynaphthalene-1,5-dicarbaldehyde (DHNDA) and 2,4,6-tris(4-aminophenyl)-pyridine (TAPP) for humidity detection. Nanofibers of COF DHNDA-TAPP were epitaxially grown on the aramid microfiber surface to form a functional nanocomposite,123 which showed a reversible humidity response with increasing RH from 20% to 100% through a color change from light yellow to dark red detectable by an unaided eye.123 The authors suggested that the water-promoted transformation of isomers between enol form and keto form was likely responsible for the color change.136 The change in the absorption spectrum induced by water-triggered enol to keto tautomerism was further supported and systematically studied by Loh and coworkers.127 This study found that the enol to keto tautomerism significantly affected the charge transfer between the electron donor and acceptor components within the COFs containing triphenylamine or salicylideneaniline building units, and thus altered the optical bandgap.
Based on the iminol-to-ketoenamine tautomerism, in 2020 Dichtel and coworkers demonstrated the chromism of the TAPB-PDA-OH COF made of 1,3,5-tris(4-aminophenyl)benzene (TAPB) and terephthaldehyde (PDA) for colorimetric humidity detection (Fig. 3a).124 The TAPB-PDA-OH COF changed color upon solvation, with a preferred response to high-polarity solvents, such as ethylene glycol and water (Fig. 3b). The absorption of the device made of TAPB-PDA-OH film dramatically increased with the increase of humidity from 0% to 100% (Fig. 3c). The COF film showed a fast and highly reversible response and recovery when switching between humid and dry air within 1 s and 9 s, respectively (Fig. 3d). The response was indistinguishable from that of the first sensing even after more than a month of ambient storage, indicating good stability of this sensor (Fig. 3e). Spectroscopic studies indicated that the chromism responsible for the humidity sensing was attributed to the water-mediated tautomerism in the TAPB-PDA-OH COF. DFT calculations on a related model compound indicated that the diiminol tautomer was strongly preferred in the gas phase; however, in the presence of two water molecules, the diiminol and cis-iminol/ketoenamine were nearly isoenergetic, indicating that both tautomers can be present in significant quantities in a humid environment (Fig. 3f). The authors concluded that the stronger acceptor nature of the central ring of the iminol/ketoenamine tautomer compared to the diiminol resulted in a charge-transfer-type excited state, leading to a new absorption feature.124
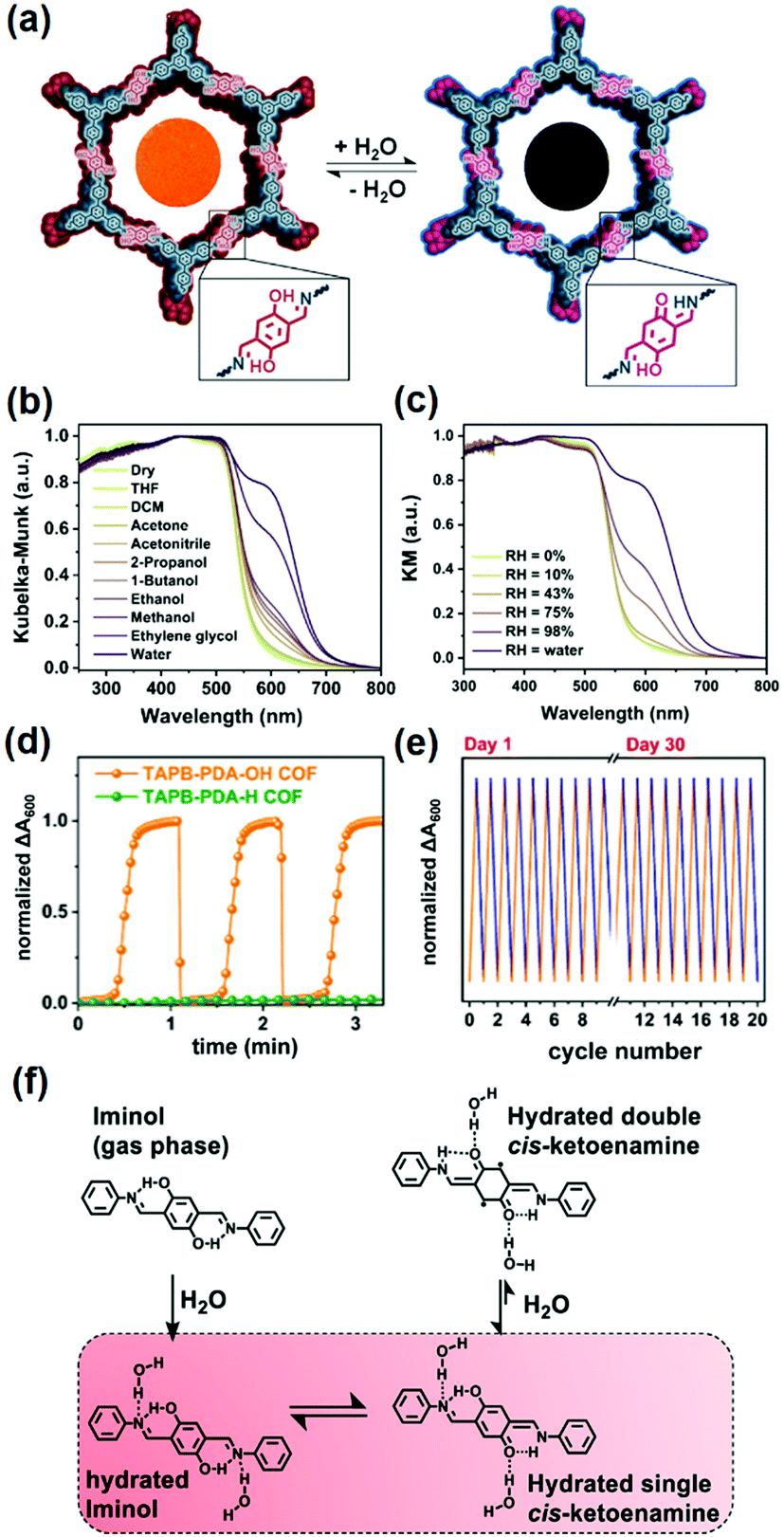 | ||
| Fig. 3 (a) COF TAPB-PDA-OH as a colorimetric humidity sensing device based on the iminol-to-ketoenamine tautomerism. Diffuse reflectance spectroscopy plots for the TAPB-PDA-OH COF powder exposed to (b) different solvents and (c) different levels of humidity. (d) Humidity sensing using TAPB-PDA-OH COF films using the change in absorbance at 600 nm. (e) Humidity sensing using TAPB-PDA-OH films over multiple cycles. (f) Chemical structures of the different tautomers of the small-molecule model compound.124 Reprinted with permission from ref. 124. Copyright 2020, American Chemical Society. | ||
The above examples indicated that water-promoted isomerization of COFs is the main mechanism used for the detection of humidity. In contrast, the detection of acids has been mainly based on Lewis acid–base reactions between the protons and the nitrogen-containing sites (e.g., CN) present in COFs. For example, Kulkarni et al. reported a COF PBHP-TAPT made of (1,4-phenylene)bis(3-hydroxyprop-2-en-1-one) (PBHP) and 1,3,5-tris-(4-aminophenyl)triazine (TAPT), in which triazine was the Lewis-basic moiety for HCl detection.128 The COF powder changed its color from orange to red (525 nm to 630 nm) within seconds after exposure to HCl vapor. This color change was fully reversible when the HCl-treated sample was exposed to NH3 vapor under the same conditions. The PBHP-TAPT COF showed high sensitivity to low concentrations of HCl gas down to 20–50 ppm, with a high threshold at around 3000 ppm. The rapid color change offered an advantage over other optical sensors for corrosive gases, where good reversibility and real-time response can be challenging to achieve.137
In addition to the triazine group mentioned in the above example,137 Auras and coworkers demonstrated the utilization of imine linkages as the functional moiety in COFs for acid vapor sensing.129 A series of dual-pore perylene (Per)-based COFs, including Per-1P, Per-N, and Per-Py, featuring phenylene–NCH–bridge–HCN–phenylene units were investigated (Fig. 4a). The protonation of the nitrogen lone pair of imines typically leads to their decomposition if small amounts of water are present;138 however, the authors found that the close-packed structures of 2D COFs provided sufficient stabilization against hydrolysis and enable fully reversible protonation and deprotonation (Fig. 4b).129 The lowest-energy optical transitions of these imine-linked COFs were characterized by charge-transfer with the first singlet excited state localized on the imines. Since the protonated imines are stronger electron acceptors than their free-base counterparts, the energies of these transitions are lowered, leading to red-shifted absorption and emission spectra. Trifluoroacetic acid at a low concentration of 35 μg L−1 led to a measurable change in the absorption spectrum with protonation-induced absorption and bleach bands extending from 440 to 800 nm, which became more prominent with increasing acid vapor concentration, without any saturation effects (Fig. 4c). Protonation of the phenylene–NCH–bridge–HCN–phenylene units proceeded in a two-step process, with the second protonation requiring significantly stronger acids, since the monoprotonation reduced the basicity of the neighboring imine. The mono- and the bisprotonated species featured distinct absorption features in the red and near-infrared spectral regions (Fig. 4d). These features may enable these COFs to act as colorimetric sensors for simultaneous determination of the acid strength and concentration in nonaqueous solutions, where traditional pH electrodes are not applicable.
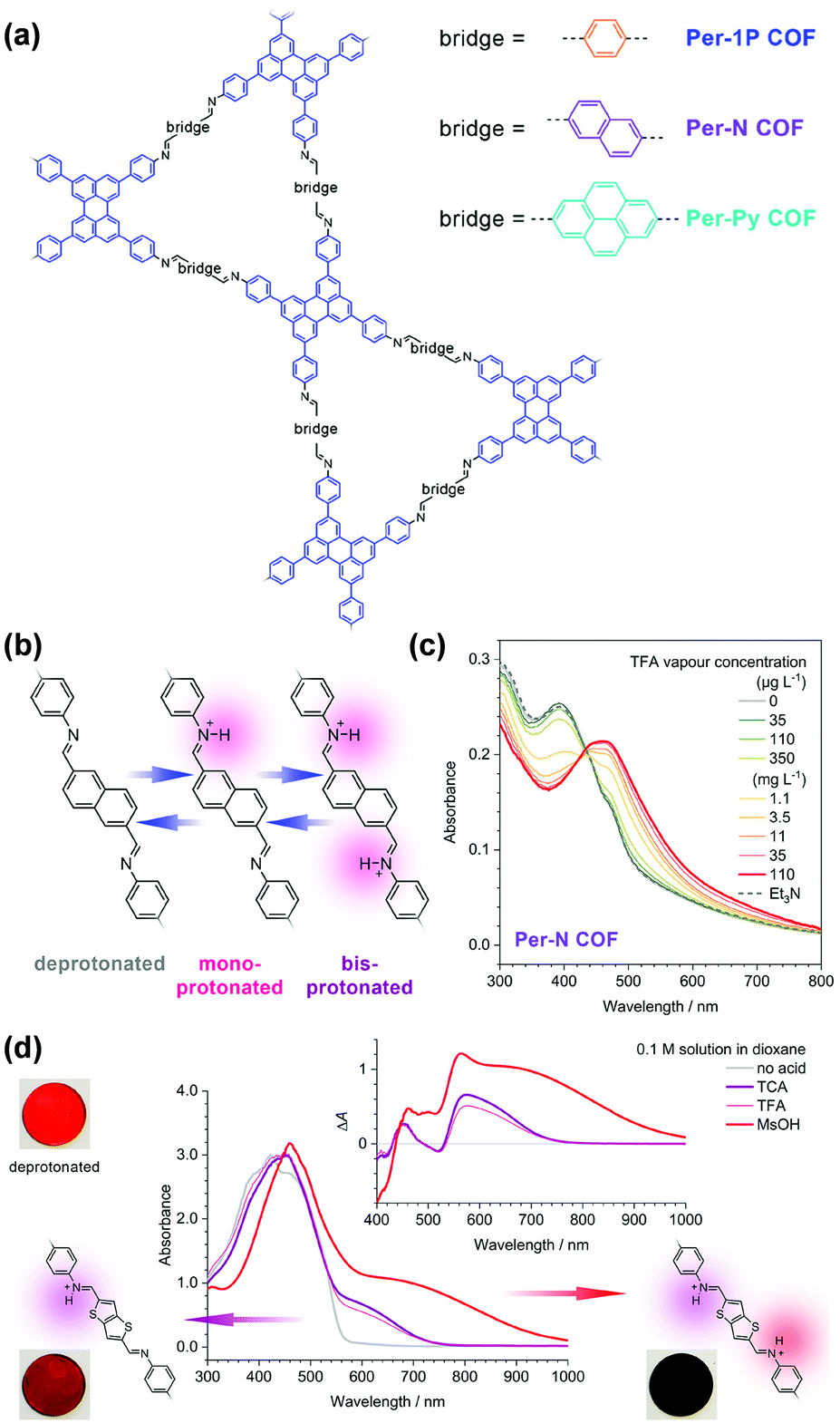 | ||
| Fig. 4 (a) Structure of the perylene-based COFs. (b) The imine-linked bridges of the Per-N COF can be protonated stepwise. (c) Absorption spectra of the Per-N COF film upon protonation in TFA vapor of increasing concentration (green to red lines). (d) Protonation of the Py-TT COF with different acid solutions. TFA and TCA solutions produced selectively the monoprotonated bridge unit with a characteristic absorption band between 550 and 750 nm. The considerably stronger MsOH generated a much stronger absorption band which extends further into the IR. TCA, trichloroacetic acid; TFA, trifluoroacetic acid; MsOH, methanesulfonic acid.129 Reprinted with permission from ref. 129. Copyright 2019, American Chemical Society. | ||
The above examples demonstrate chromism-based detection of volatile analytes based on the intrinsic optical properties of COFs themselves. For chromism-based detection of ions and biomolecules, researchers have successfully harnessed the catalytic properties of COFs. The most exploited system so far has been based on mimicking peroxidases using COFs, in which COFs catalyze a redox reaction and change the adsorption of external dyes. The characteristic features of COFs, such as high porosity, high surface area, abundant active sites, and good thermal and chemical stability coupled with π-electronic structures that can provide excellent electron transfer properties, are highly desirable for designing mimics of peroxidases to mediate the redox reaction for chromism-based detection.
Ions. Xiong and coworkers developed a Cu2+ detection method by mimicking the peroxidase with a covalent triazine framework (CTF).132 The peroxidase-like catalytic activity of the CTF can be modulated by Cu2+ ions. In the presence of Cu2+ ions, the catalytic activity of the CTF was significantly enhanced, which enabled oxidation of the colorless substrate 3,3′,5,5′-tetramethylbenzidine (TMB) to bright blue oxidized TMB (oxTMB) in the presence of H2O2. Based on this colorimetric method, a good linear relationship between the adsorption of oxTMB and concentration of Cu2+ ions at 1.0 to 80.0 μg L−1 could be obtained with a limit of detection (LOD) at 0.05 μg L−1. The high specificity of Cu2+–N chelation resulted in good selectivity for Cu2+ over other competing ions, such as Na+, K+, Ba2+, Zn2+, Al3+, and Ce3+. This strategy was further applied for the detection of Cu2+ ions in real food samples with recoveries of 96–105%, constituting a promising approach for the rapid, selective, sensitive, and low-cost detection of Cu2+ ions.132 Considering the slow analyte diffusion resulting from the 3D nature of the bulk CTF, this method was optimized by using an iron-modified two-dimensional covalent triazine framework (2D Fe-CTF) for F− detection.133 The 2D Fe-CTF sheets with favorable dispersibility, large surface areas, and abundant accessible active sites allowed fast analyte diffusion, enabling detection of F− ions in the 50–700 nM range with a low limit of detection (signal/noise = 3) of 5 nM and good selectivity over various interfering ions, including Cl−, Br−, SO42−, NO3−, HCO3−, HCOO−, CO32−, PO43−, and CH3COO−.133
Recently, Qiu and coworkers used bipyridine-containing COF (Tp-Bpy) nanosheets (NSs) for highly sensitive mercury sensing.134 The COF synthesized from 2,4,6-triformylphloroglucinol (Tp) and 5,5′-diamino-2,2′-bipyridine (Bpy) had regular pore structures and abundant nitrogen-containing functional groups, which served as active sites for the in situ generation of gold nanoparticles (Au NPs) to create a functionalized composite (Fig. 5a). The presence of Hg2+ significantly improved the catalytic activity of the Au NPs@Tp-Bpy nanocomposite in the oxidation of colorless TMB to bright blue oxTMB, accompanied by a significant colorimetric response (Fig. 5b). At the same time, the anchoring of Au NPs onto Tp-Bpy NSs through coordination bonds can enhance the dispersibility and stability of the Au NPs, which otherwise can easily aggregate, leading to an unexpected decrease of their catalytic activity. This synergistic effect of the increased mimetic activity of the Au amalgam and the higher access probability of Hg2+ provided by Tp-Bpy nanosheets rendered the Au NPs@Tp-Bpy nanocomposite with a high sensitivity for the detection of Hg2+ with an ultralow detection limit of 0.33 nM (Fig. 5c). Other metal ions, including Cr3+, Fe3+, Cu2+, Zn2+, and Mg2+, had minimal effects on Hg2+ detection (Fig. 5d).134 The strategy based on the color change of TMB catalyzed by Au NPs@COF was also utilized by Yan and coworkers for mercury detection by using a novel COF employing tris-(4-formyl-phenyl)triazine and 4,4′-azodianiline as the monomers for the determination of the mercury content in water.135
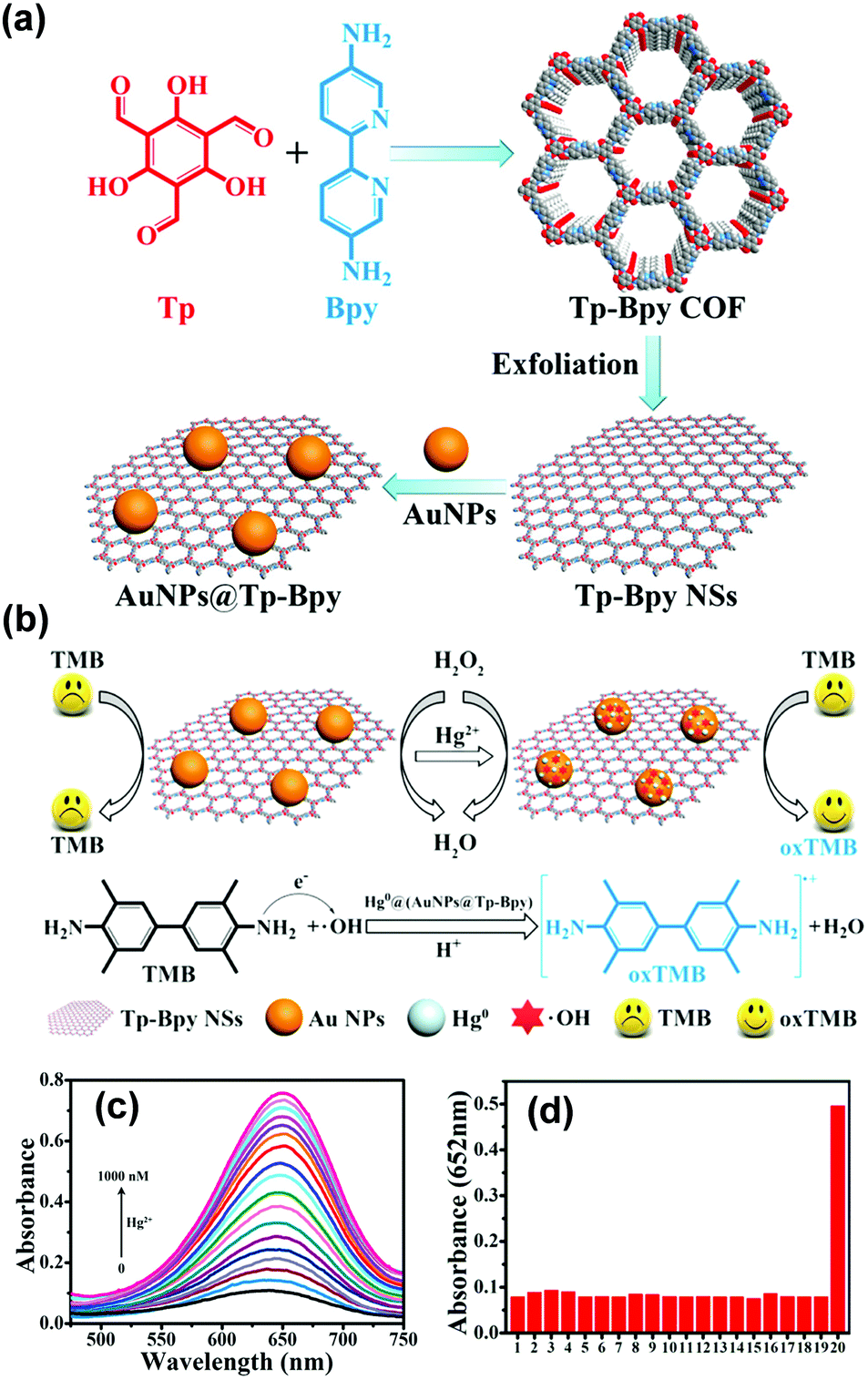 | ||
| Fig. 5 (a) The synthesis of the Au NPs@Tp-Bpy nanocomposite. (b) Mechanism of colorimetric detection of mercury ions using Au NPs@Tp-Bpy. (c) UV-vis spectra after addition of Hg2+ to 1000 nM. (d) Catalytic activity of the Au NPs@Tp-Bpy nanocomposite stimulated by different metal ions (1, blank; 2, Cr3+; 3, Fe3+; 4, Cu2+; 5, Zn2+; 6, Mg2+; 7, Co2+; 8, Ag+; 9, Pb2+; 10, Ni2+; 11, Na+; 12, K+; 13, Ca2+; 14, Al3+; 15, Ba2+; 16, Mn2+; 17, NH4+; 18, Cd2+; 19, Fe2+; and 20, Hg2+).134 Adapted with permission from ref. 134. Copyright 2019, American Chemical Society. | ||
Biomolecules. Hou and coworkers found that the triazine-based COF CTF-1 possessed both intrinsic peroxidase-like activity and oxidase-like activity because it was capable of catalyzing the oxidation of chromogenic substrates, such as TMB, with or without H2O2 with high efficiency. The peroxidase-like activity of CTF-1 may be derived from the formation of hydroxyl radicals (˙OH), while the oxidase-like activity of CTF-1 mostly stemmed from the generation of O2− and 1O2 derived from the dissolved O2. Utilizing this design, the authors developed a colorimetric detection platform composed of TMB and CTF-1 for the detection of biothiols.130 The absorbance at 652 nm decreased linearly with the increasing concentration of biothiols. This platform was used to detect three biothiols, including glutathione (GSH), cysteine (Cys), and homocysteine (Hcy), with a limit of detection at 0.6 μM.130 Compared with other sensing systems with similar dynamic ranges and LODs,139 the method demonstrated simplicity, low cost, and did not require extra light sources and additional chemicals, especially H2O2, making it a promising enzyme-mimetic alternative for biosensing. Analogous strategies harnessing the catalytic performance of COFs as enzymatic mimics to mediate the coloration of chromogenic substrates have also been developed for the detection of sarcosine and ochratoxin A at sub-μM concentrations133 and the detection of glucose with LOD at 1.0 μM.131
Chromism-based detection has been used to realize the analysis of volatiles, ions, and biomolecules (Table 2). Taking advantage of the solid-state nature, porous skeleton, chemically stable structure, pre-designable active sites, and tunable optical properties of COFs, this detection approach can generally provide the advantages of signal visibility, fast response, good sensitivity, and suitability for on-site detection.140 Although in a few cases COFs can be used directly in colorimetric detection and their response can be qualitatively visualized by the unaided eyes, the accurate determination of the analyte concentration relies on the monitoring of the absorption spectrum of the chromic materials. Because of their polymeric nature, COFs cannot be directly dissolved in solvents for the absorption spectrum measurement; instead, suspensions of COFs have to be employed. For detection where COFs serve as catalysts to mediate the color change of external dyes, monitoring of the absorption spectrum can be routine and straightforward. However, for the detection based on chromism of COFs, the aggregation state needs to be controlled to ensure the reproducibility and long-term stability of COF suspensions, because both the intensity and the range of the absorption can be a function of the packing states of chromophores in COFs, especially for COFs that have a layered structure.
| Analyte | COF | Detection architecture | LOD | Detection range | Other detection parameters | Ref. |
|---|---|---|---|---|---|---|
| H2O | DHNDA-TAPP | DHNDA-TAPP/aramid microfiber | N/A | 20–100% RH | Reversible and repeatable | 123 |
| H2O | TAPB-PDA-OH | TAPB-PDA-OH film | N/A | 10–100% RH | In gas phase, reversible, response time 9 s, recovery time 1 s | 124 |
| H2O | Py–TT | Py–TT film | N/A | 0.59–0.98 (p/p0) | In gas phase, reversible, response time 0.21 s, recovery time 0.15 s | 126 |
| HCl | PBHP-TAPT | PBHP-TAPT powder | <20 ppm | 20–3000 ppm | In gas phase, response time < 2 s | 128 |
| HCl | Per-N | Per-N film | 35 μg L−1 | 35–110 μg L−1 | In THF solution, reversible when treated with Et3N | 129 |
| HCl | COF-ETBA-DAB | COF-ETBA-DAB suspension | N/A | 0.13–91 mM | In 1,4-dioxane suspension, reversible | 141 |
| Cu2+ | CTF | CTF/TMB/H2O2 | 0.08 μg L−1 | 1.0–80 μg L−1 | In water, pH = 4.0, selectivity over Na+, K+, Ba2+, Zn2+, Al3+, Ce3+, Co2+, Ni2+, Fe3+, Mn2+, and Cr3+; vegetable and water sample | 132 |
| Hg2+ | Tp-Bpy | Au NPs@Tp-Bpy/TMB/H2O2 | 0.33 nM | 0–1000 nM | 200 mM H2O2, pH = 4.0; selectivity over Cr3+, Fe3+, Cu2+, Zn2+, Mg2+, Co2+, Ag+, Pb2+, Ni2+, Na+, K+, Ca2+, Al3+, Ba2+, Mn2+, NH4+, Cd2+, and Fe2+ | 134 |
| Hg2+ | PTAzo | PTAzo/TMB/H2O2 | 0.75 nM | 5–300 nM | In acetic acid, pH = 4.0; selectivity over Ca2+, Mn2+, Cr3+, Pb2+, Ba2+, K+, Na+, Fe3+, Mg2+ and Cu2+ | 135 |
| F− | Fe-CTF | Fe-CTF/TMB/H2O2 | 5 nM | 50–700 nM | Selectivity over Cl−, Br−, SO42−, NO3−, HCO3−, HCOO−, CO32−, PO43−, and CH3COO− | 133 |
| GSH | CTF-1 | CTF-1/TMB/H2O2 | 0.65 μM | 1–100 μM | In 0.1 M NaAc buffer, pH = 4.0 | 130 |
| Cys | CTF-1 | CTF-1/TMB/H2O2 | 0.62 μM | 1–100 μM | In 0.1 M NaAc buffer, pH = 4.0 | 130 |
| Hcy | CTF-1 | CTF-1/TMB/H2O2 | 0.68 μM | 1–140 μM | In 0.1 M NaAc buffer, pH = 4.0 | 130 |
| Glucose | Fe-COF | Fe-COF + TMB + H2O2 | 1.1 μM | 30–500 μM | In 0.2 M NaAc buffer, pH = 5, selectivity over sucrose, maltose, and lactose | 131 |
| Sarcosine | Fe-CTF | Fe-CTF/TMB/H2O2 | 0.56 μM | 10–100 μM | In 0.2 M PBS buffer, pH = 8.0 | 133 |
| Ochratoxin A | Fe-CTF | Fe-CTF/TMB/H2O2 | N/A | 0.2–0.8 μM | In 50 mM NaAc buffer, pH = 4.0 | 133 |
| H2O2 | Cu2+-CTF | Cu2+-CTF/TMB/H2O2 | N/A | 2.5–140 μM | In 0.1 M NaAc buffer, pH 4.0 | 142 |
2.2 Detection based on luminescence
General principles of luminescence-based detection. Luminescence is the emission of light upon absorption of energy from various sources, such as chemical energy, electromagnetic radiation, ionizing radiation, heat, pressure, and others. Fluorescence and phosphorescence are two important types of luminescence resulting from the absorption of photons.143 Fluorescence is a spin-allowed radiative transition from the lowest singlet excited state S1 of the fluorophore to its singlet ground state S0. Phosphorescence refers to the spin-forbidden radiative transition from the triplet state T1 to ground state S0 (Fig. 6).115,144 So, fluorescence occurs immediately upon excitation with a short decay time (t < 10 ms). Phosphorescence, in contrast, emits continuously for a certain period after the excitation is switched off (t > 0.1 s).145
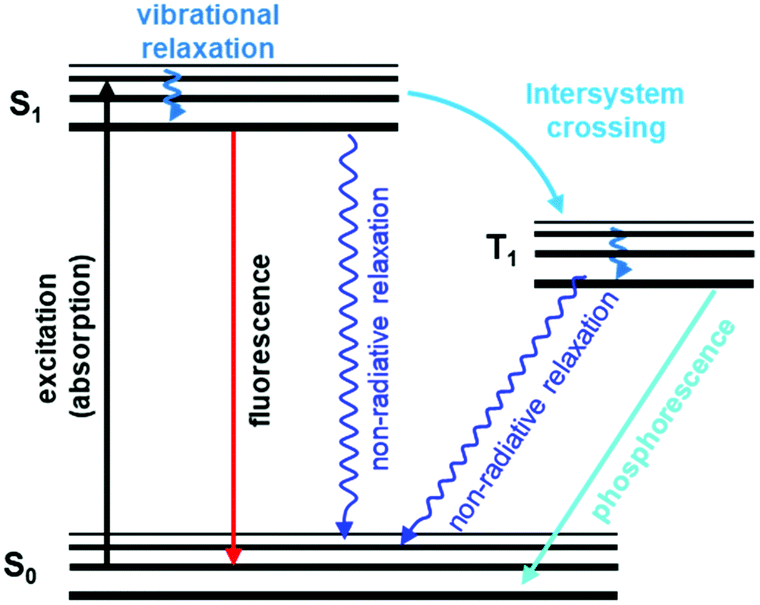 | ||
| Fig. 6 A typical Jablonski diagram displaying schematically electronic states involved in luminescence phenomena.146 Adapted with permission from ref. 146. Copyright 2020, Royal Society of Chemistry. | ||
There are several parameters of fluorescence emission that can be used to characterize a fluorescent material. First, fluorescence intensity (I) can be measured at the given wavelength of excitation and emission (usually, band maxima), whose dependence on emission wavelength gives the fluorescence emission spectrum. Second, emission anisotropy is a function of the fluorescence intensities obtained at two different polarizations, vertical and horizontal. Third, emission can also be characterized by fluorescence lifetime (τ), which is the measurement of the time a fluorophore spends in the excited state before returning to the ground state by emitting a photon.
Changes in fluorescence emission properties, such as quenching and enhancement of the intensity, shifts of the fluorescence spectrum, and changes in fluorescence lifetime because of interactions of analytes with fluorophores, can be used to realize sensitive and cost-effective chemical detection.147–150 Based on different photophysical processes, general fluorescence detection mechanisms mainly include (1) various charge transfer (CT) processes, including photo-induced electron transfer (PET), intramolecular charge transfer (ICT), metal–ligand charge transfer (MLCT), and twisted intramolecular charge transfer (TICT), (2) energy transfer (ET) processes, including electronic energy transfer (EET) and fluorescence resonance energy transfer (FRET), (3) formation of an exciplex, (4) aggregation-induced emission, and (5) excited-state intramolecular proton transfer (ESIPT).151
CT is the most widely applied mechanism employed in fluorescence-based detection.152 By interacting with analytes, fluorescence of a fluorophore can be quenched due to the PET process. The inhibition of the PET process can recover the fluorescence. For a fluorescent material based on intramolecular charge transfer, enhancement or suppression of the ICT process will lead to a red or blue shift in its emission spectrum. A ratiometric signal can be obtained, which can eliminate ambiguities by self-calibration of two emission bands and allow quantitative determination.153 MLCT is the charge transfer taking place between a ligand and a transition metal cation and is commonly observed in processes involving transition metal binding.154 TICT occurs upon photoexcitation in molecules that usually consist of a donor and acceptor linked by a single bond.155 Following intramolecular twisting, the TICT state returns to the ground state either through a red-shifted emission or by nonradiative relaxation. Since intramolecular rotation is very sensitive to polarity and/or steric hindrance, TICT can be used for the detection of solvents, (micro)viscosity, and chemical species.
The mechanism of ET involves a donor fluorophore, which may transfer its excitation energy to a nearby acceptor chromophore in a non-radiative fashion through long-range dipole–dipole interactions.156 Based on the distance of interaction between the energy states of donor and acceptor, ET can be classified as electronic energy transfer or fluorescence resonance energy transfer. EET requires a distance between donor and acceptor within 10 Å, while FRET requires a certain degree of spectral overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor. The distance between the donor and acceptor should be from 10 to 100 Å for efficient FRET to occur.151 For this reason, fluorescence detection based on the ET process is distance-dependent.
In addition to CT and ET processes, the formation of an exciplex is another important mechanism for fluorescence detection. An exciplex is a short-lived heterodimeric molecule formed by the interaction of a fluorophore in the excited state with an analyte molecule. The emission spectrum of an exciplex is red-shifted compared with that of the sole fluorophore, and a dual emission from the monomeric fluorophore and exciplex is often observed simultaneously.157 Exciplex formation or deformation upon interaction with a guest species allows the detection by simply monitoring the exciplex band.158
The AIE phenomenon utilizes the restriction of intramolecular rotation in aggregates to enhance the fluorescence emission.71,159,160 Unhindered intramolecular rotation in molecules in the free state can lead to nonradiative decay of the corresponding excited states, making fluorophores nonemissive. Upon aggregation in a suitable environment, intramolecular rotation is restricted, and the emission is thus greatly enhanced. The AIE effect can be tuned by the analyte molecule through electrostatic interaction, coordination interaction, hydrophobic interaction, steric hindrance, or the influence of polarity and viscosity to result in the characteristic change of the fluorescence emission.151,161,162
In ESIPT systems, photoexcited molecules relax their energy through tautomerization by the transfer of protons.163,164 ESIPT is generally a fast process with reported values ranging from fractions of picoseconds to tens of picoseconds. It is easy to recognize in steady-state spectra because the absorbance is generally similar to that of the parent fluorophore but the fluorescence is significantly different. Since ESIPT is much faster than the emission process (radiative decay), the fluorescence observed for ESIPT chromophores is often from the tautomer triggered by the proton dislocation. The accompanied geometry relaxation can result in a large Stokes shift where spectral overlap between absorption and emission is minimized.165 The ESIPT process can be perturbed by many intermolecular interactions, such as removal of the proton involved in the ESIPT process, and thus can be used for detecting analytes.151
Molecular design of fluorescent COFs. The molecular design of fluorescent COFs has harnessed the fundamental principles of molecular engineering of other fluorescent materials, including the introduction of well-established fluorescent chromophores, the tuning of the fluorescence properties with modification groups, and the elimination of aggregation-induced fluorescence quenching. The utilization of aromatic and conjugated molecular entities with high probabilities for S0 → S1 transition and π* → π relaxation can give rise to optical emission or photoluminescence when subject to excitation.71,166–168 Consequently, fluorescent COFs contain large π-conjugated building elements—phenyl, triazine, pyrene, triphenylene, and triphyenylbenzene—many of which are well known fluorescent chromophores.68,169,170 We will see quite a few examples in the following discussions in Sections 2.2.2–2.2.4, where fluorescent COFs used for detection are built upon by directly incorporating fluorescent chromophores. The emission property of these chromophores in COFs can be further modulated to different degrees based on the nature of the linkages. The boronate ester linkages have been popular for the construction of COFs for their reversible feature; however, this type of linkages typically doesn't allow for efficient π-delocalization with the constituent building blocks.47,125 Thus, for many of the COFs created using boronate ester linkages, the fluorescence properties of the COF mainly originate from the building units. For COFs linked by conjugated linkages, such as imine, triazine, phenazine, or olefins, their optical properties including the fluorescence emissions, can be significantly different from that of the constituent building blocks. The position of the emission peak, luminescence quantum yield, and fluorescence intensity of chromophores in COFs can be modulated by the electronic effects from other components.
The utilization of fluorescent chromophores is a straightforward approach for attaining fluorescence property; this approach also offers unique properties in COFs that are desirable for optoelectronic and photovoltaic device applications.125 However, the presence of large conjugated fragments can lead to the packing of periodic columnar π-arrays in COFs, which can trigger the thermal decay of photoexcited states and result in reduced or nonemissive characteristics.71,159,160 This aggregation-caused quenching (ACQ) mechanism is ubiquitous for COFs and is, perhaps, the reason why highly luminescent COFs are rarely achieved even though various luminescent π-units have been employed for the synthesis of COFs.171–173 To overcome the ACQ effect, two effective strategies have been employed. The first strategy relies on the spatial separation and spacing of the π-conjugated fluorescent chromophores. This concept had previously been utilized for the development of highly fluorescent conjugated polymers through the introduction of structurally rigid and bulky units into chains of polymers to minimize polymer aggregation and self-quenching.120,174 As for the design of fluorescent COFs with this strategy, both the incorporation of bulky groups175,176 and the construction of a 3D topology177 have been demonstrated, in which the fluorescent chromophores are spatially separated to avoid aggregation. In the second strategy, the concept of aggregation-induced emission (AIE)178,179 is utilized to obtain highly luminescent COFs, which is in sharp contrast with the first strategy where the aggregation is designed to be minimized. This strategy is implemented by introducing AIE active chromophores, which usually have freely rotating groups that enable the energy of the excited state to be released in the form of thermal decay by molecular rotations rather than in the form of fluorescence emission. When these AIE chromophores aggregate, restrictions of molecular rotations restore the release of energy during electron transition in the way of fluorescence emission. In the construction of COFs using the AIE concept, the intralayer covalent bonding and interlayer noncovalent π-interactions can work synergistically to reduce the rotation-induced thermal decay of the photoexcited state to afford high fluorescence emission.173
In short, the direct incorporation of well-established fluorescent chromophores is a straightforward way to construct fluorescent COFs. The fluorescence emissions will be mainly determined by the choice of the chromophores, but will also be affected by the type of linkage. However, the aggregation-induced quenching mechanism can make this method ineffective in getting highly emissive COFs. Subsequently, spatial separation of the π-conjugated fluorescent chromophores to avoid aggregation and the use of the AIE concept have become new strategies for achieving highly emissive properties. Taken together, these strategies also illustrate that the fluorescence properties of COFs are dominated by the choice of organic building units. In comparison, in MOFs, luminescence can arise from a few comparable contributors, including direct excitation of organic ligands (particularly from the highly conjugated ligands), metal-centered emission, and charge transfer (such as ligand-to-metal charge transfer and metal-to-ligand charge transfer).115 Therefore, the mechanism of luminescence of COFs has the potential to be more straightforward and relatively easy to engineer and exploit compared to MOFs.
Advantages of COFs for fluorescence-based chemical detection. Fluorescence-based chemical detection can rely on the analyte–COF interaction that triggers changes in emission property, including wavelength and intensity of emission, or the rise of a new emission band, through multiple feasible mechanisms.180–182 Fluorescence detection of most of the volatiles, gases, and ions discussed in the following subsections is based on CT between COFs and analytes which usually results in quenching of the luminescence of COFs. These analytes usually possess appropriate receptor energy levels, for example, the LUMO of electron-deficient nitroaromatics183 and unoccupied d-orbitals of transition metal ions.176 The quenching can thus be ascribed to nonradiative charge transfer from COF donors to analyte receptors. When the emission band of COFs overlaps with the absorption band of receptor analytes to some degree, the interaction of an excited state fluorophore in COFs with analytes results in radiationless deactivation of the fluorophore to the ground state through the ET process.184 Interactions between the fluorophore of COFs and the analyte species, such as hydrogen bonding160,185–187 and coordination bonding, can also reduce π-electron delocalization or structural coplanarity, which may also quench the fluorescence of the system. Moreover, the solvent polarity and the local environment also have profound effects on the emission spectral properties through the aggregation-caused quenching or aggregation-induced emission enhancement effect.178
In addition to the feasibility of changing the fluorescence properties via the diverse mechanisms mentioned above, three features make COFs excellent candidates for fluorescence-based detection. First, COFs have large surface-to-volume ratios, which can provide abundant active sites for effective material–analyte interaction to enhance sensitivity. Their intrinsic nanoporous structure is beneficial for the preconcentration of analytes to strengthen the sensing response. Second, the conjugated structure of many COFs is capable of amplifying the fluorescence response from an individual COF–analyte interaction to create a bulk response of the entire COF scaffold through electron delocalization.188 This effect resembles the “molecular wire effect” coined by Swager39,120,148 for the sensing using conjugated organic polymers (COPs), in which the donor ability of COPs is enhanced in the excited state due to the delocalized π* state, offering a platform for the easy migration of excitons to interact with analytes (acceptor). Critical to the implementation of this effect is the good dispersion of chromophores within bulk materials to avoid self-quenching.148 While this design criterion has been achieved by using bulky groups in COPs, COFs can potentially offer a unique opportunity for strategic positioning of fluorescent subunits with atomic precision within porous scaffolds through reticular molecular engineering. Third, both the “bottom-up” and “post-synthetic” strategies can be applied to achieve specific and effective material–analyte interactions to enhance the selectivity and sensitivity of COF-based sensors.
So far, efforts in the use of COFs for chemical detection have been centered on the fluorescence quenching or enhancement triggered by analyte molecules. These investigations have revealed the potential of COFs in the quantitative detection of explosives based on nitroaromatics, heavy metal ions, small molecular compounds, and biologically relevant analytes. This section provides a detailed account of COFs in fluorescence detection of analytes by dividing the analytes into three groups: volatiles and gases, ions, and biology-related molecules.
Nitroaromatics and other explosives. Nitroaromatics, including 2,4,6-trinitrotoluene (TNT), dinitrotoluene (DNT), and 2,4,6-trinitrophenol (TNP), constitute an important group of explosives that possess high explosive efficacy. Some nitroaromatics, like TNP, also have mutagenic activities to cause acute health problems, such as headache, liver injury, and diarrhea. Nitroaromatics are also a major source of environmental pollution. Hence, the highly selective and sensitive detection of nitroaromatics has attracted research concern.
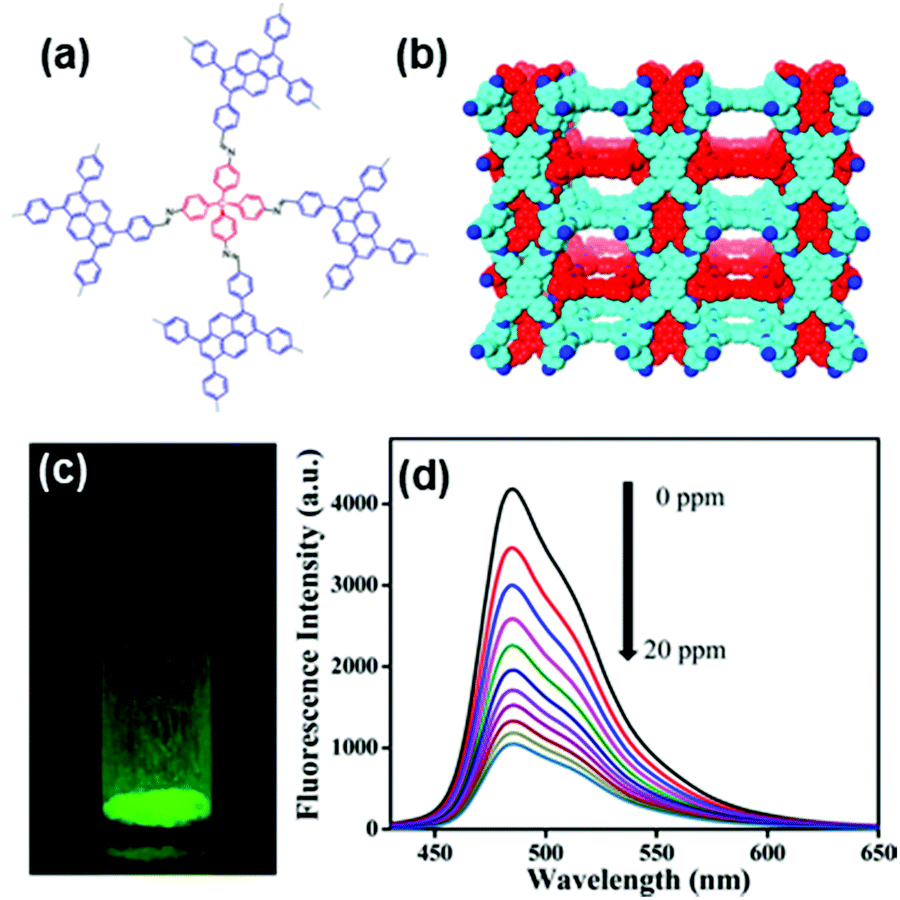
Zhu and coworkers synthesized a 3D crystalline porous aromatic framework (PAF-15) with high fluorescence quantum yield by assembling luminescent building blocks of tetra(4-dihydroxyborylphenyl)germanium (TBPGe) and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP).188 PAF-15 exhibited a strong fluorescence with an absolute quantum yield of 14% as a suspension in CHCl3. A distinct quenching effect was observed upon the addition of nitroaromatics such as nitrobenzene, 2,4-DNT, and TNT, which could result from the electrostatic interaction between PAF-15 and electron-deficient compounds.189 The addition of other common aromatic compounds such as benzene, toluene, chlorobenzene, bromobenzene, phenol, and aniline did not affect the fluorescence intensity of PAF-15. Zhang et al. prepared nanoparticles of a new type of melamine-based Schiff base network SNW-1 by microwave-assisted synthesis for the detection of nitroaromatic explosives.183 The as-synthesized SNW-1 nanoparticles exhibited high sensitivity and selectivity, as well as fast response to TNT, 2,4,6-trinitrophenylmethylnitramine, and TNP without interference by common organic solvents (Fig. 7). Time-dependent fluorescence quenching by DNT vapor showed a sharp increase in only 10 s. The detection limit for DNT vapor was 9.8 ppb, comparable with porous silicon films. The authors ascribed the observed sensing performance for these nitroaromatic explosives to the nanoscale size and unique hierarchical porosity of such fluorescence-based sensing material.183
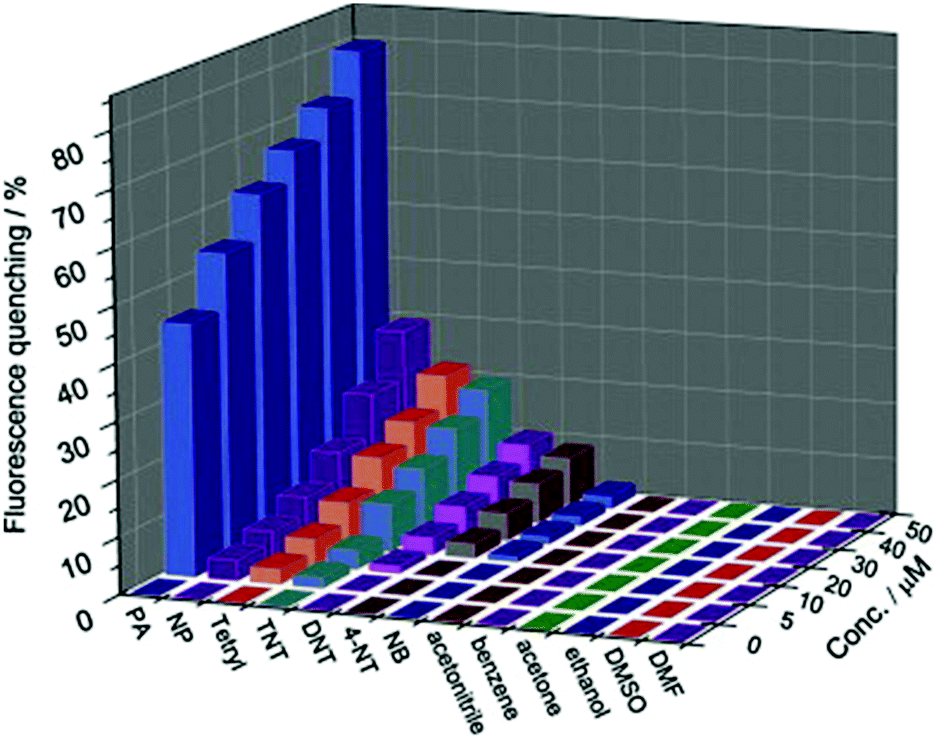 | ||
Fig. 7 Fluorescence quenching results of nitroaromatic explosives and interferents to the emission of the SNW-1 nanoparticles at various concentrations in a THF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v) mixture.183 Adapted with permission from ref. 183. Copyright 2012, Elsevier. :9, v/v) mixture.183 Adapted with permission from ref. 183. Copyright 2012, Elsevier. | ||
Although these earlier studies demonstrate promising application of COFs for the detection of electron-deficient nitroaromatics over other chemical species,183,188 selective detection of certain nitroaromatics is still challenging because of the limited selectivity of the interaction between the analyte and COFs. To improve the sensitivity and selectivity for nitroaromatics, two main approaches have been adopted to engineer the chemical structures of COFs: (1) the introduction of active binding sites160,190–192 and (2) the adjustment of the electronic feature of the fragments in COFs through the choice of building blocks.193,194 Both these methods can tune the material–analyte interactions electronically.
For example, Jiang and coworkers reported the development of an azine-linked COF Py-Azine with high crystallinity, porosity, and robust chemical stability for the selective detection of TNP.160 In the Py-Azine COF, the pyrene units at the vertices and the azine linkers on the edges of 2D polygon sheets stack to form periodic pyrene columns and one-dimensional channels with exposed azine units on the pore walls (Fig. 8a). The authors found that these azine units can provide open docking sites for forming hydrogen-bonding interactions with guest molecules,195 resulting in fluorescence quenching and achieving selective detection of guests. When the suspension of the Py-Azine COF was exposed to the TNP vapor, the COF fluorescence was quickly quenched. The fluorescence quenching reached 69% in response to a concentration of TNP in acetonitrile that was as low as 70 ppm (Fig. 8b). When other nitrobenzene compounds were tested under the same condition, fluorescence quenching of the COF was only 13%, 5%, 3%, and 3% for DNP, DNT, NP, and NT, respectively (Fig. 8c). The authors proposed that the selective detection of TNP among other nitrobenzene compounds may result from the hydroxy unit and the three nitro groups of TNP that form hydrogen-bonding interactions with the open nitrogen atoms in the azine units on the pore walls and provides the most deficient π-system for driving the fluorescence quenching.160
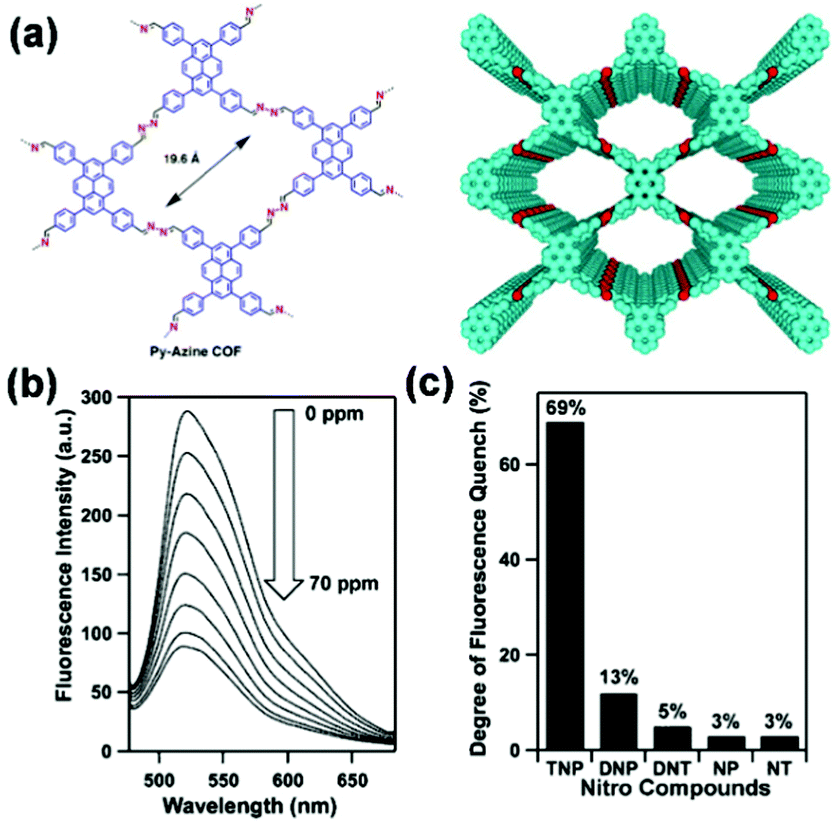 | ||
| Fig. 8 (a) Chemical structure and model of COF Py-Azine. (b) Fluorescence quenching of the Py-Azine COF upon the addition of TNP in acetonitrile. (c) Degree of fluorescence quenching upon addition of the nitro compounds (70 ppm).160 Adapted with permission from ref. 160. Copyright 2013, American Chemical Society. | ||
Since the aggregated layers in the bulk form may mask active sites, bulk COFs can exhibit ineffective interaction with analytes, leading to moderate chemical sensing ability. To address this shortcoming, Banerjee and coworkers exfoliated the aggregated π-stacked COF layers using the liquid phase exfoliation method to produce covalent organic nanosheets (CONs) from imide-based COFs (TpBDH and TfpBDH, Tp, 1,3,5-triformylphloroglucinol, Tfp, 1,3,5-tris(4-formylphenyl)benzene, BDH, pyromellitic-N,N′-bisaminoimide, Fig. 9a).190 TfpBDH-CONs showed up to 10 times increased luminescence intensity in both isopropyl alcohol solution and in solid-state (deposited on paper strips) compared to their bulk counterparts. TfpBDH-CONs exhibited maximum ca. 63% quenching efficiency towards TNP at 5.4 × 10−5 M concentration in IPA over other nitroaromatic explosives (TNT: 31%, DNT: 3%, DNP: 23%, and NP: 4%), which can be attributed to the electronic charge transfer from the HOMO of the picrate anion (TNP−) to the LOMO of the protonated TfpBDH-CONs (Fig. 9b and c). The fluorescence lifetime reduced sharply with the increase of TNP concentration, suggesting that the quenching was dynamic. On the other hand, in the solid state, a “turn-on” type detection capability for TNP was observed. This “turn-on” sensing behavior in the solid state could be due to the proton transfer from TNP to the basic nitrogen atom of the imine (–CN–) bond. The PL emission maxima in the solid state were red-shifted and enhanced by 10 times under 10−3 M TNP (Fig. 9d). Other nitroaromatics did not show significant effects on TfpBDH-CONs in the solid state (Fig. 9e). These findings described a new approach towards developing efficient fluorescence chemosensor materials for both visual and spectroscopic detection of nitroaromatic compounds.
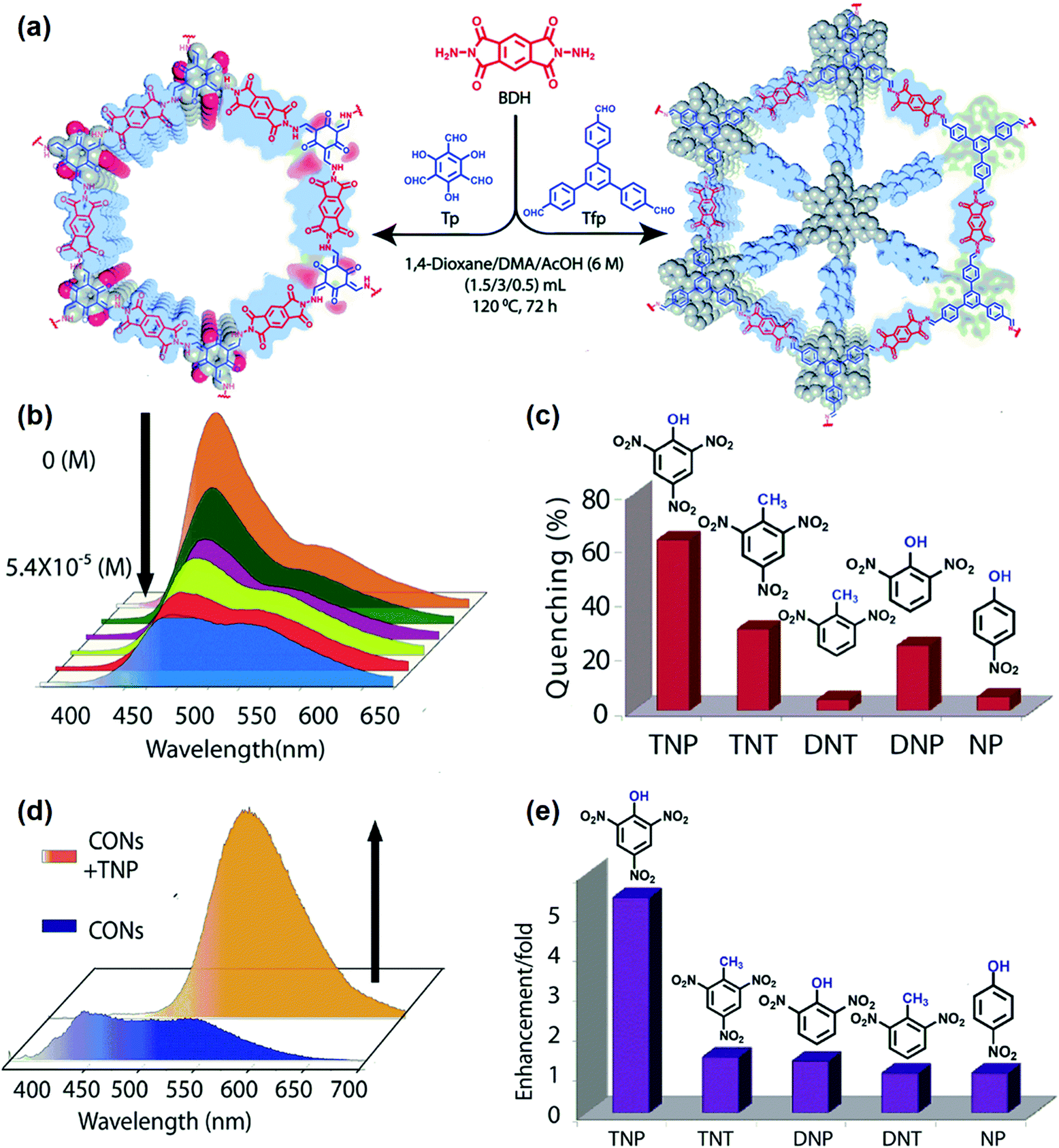 | ||
| Fig. 9 (a) Schematic of the synthesis of TpBDH-COFs and TfpBDH-COFs. (b) Fluorescence “turn-off” sensing using TfpBDH-CONs in an isopropyl alcohol solution. (c) The selectivity of TfpBDH-CONs towards TNP based on fluorescence quenching. (d) Fluorescence enhancement of “turn-on” sensing in the solid phase. (e) The selectivity of TfpBDH-CONs towards TNP based on fluorescence enhancement.190 Adapted with permission from ref. 190. Copyright 2015, Royal Society of Chemistry. | ||
COFs in the form of nanosheets were also recently made by Xian and coworkers for sensitive detection of TNP.191 Few-layered polyimide CONs (PI-CONs) were exfoliated from bulk PI-COF made of tetra(4-aminophenyl) porphyrin and perylenetetracarboxylic dianhydride, which showed enhanced activity relative to that of PI-COF. The fluorescence intensity of PI-CONs gradually reduced with the increasing concentration of TNP. This fluorescence quenching can be attributed to a combination of ground-state electron transfer from TNP− to PI-CONs and the inner filter effect (IFE) in which the excitation and/or emission light of PI-CONs was partly absorbed by TNP. This approach provided a detection limit of 0.25 μM. Other interferents, including nitrobenzene (NB), nitrotoluene (NT), DNT, TNT, 4-chlorophenol (MCP), and hydroquinone (HQ), had negligible impacts on the detection of TNP because of the less efficient charge transfer and IFE between these interferents and PI-CONs. More recently, Zamora and coworkers reported the application of exfoliated nanosheets of pyrene-IMDEA-COF for the sensing of aromatic pollutants in water.196 The colloidal IMDEA-COF-1 nano-layers showed remarkable fluorescence towards a variety of organic pollutants and dyes, including NB, dinitrobenzene (DNB), and methylene blue (MB).
Although being widely used,190,191 COFs with conventional Schiff base structures can have limited hydrolytic stability in acid and base, or even in the presence of water, which is a concern for their long-term stability in detection under harsh conditions. H-bonded keto–enamine type COFs, instead, can give highly stable and crystalline structures compared with conventional Schiff base structures, thus providing an effective solution for the stability issues.85,88 Toward this end, Bhaumik and coworkers reported a new triazine functionalized keto–enamine based COF TRIPTA that introduced high thermal and chemical stability.192 TRIPTA was found to be highly luminescent when suspended in polar solvents upon irradiation and was able to detect various nitroaromatic compounds with good sensitivity by fluorescence quenching at concentrations as low as 10−8 M. The maximum fluorescence quenching was observed for TNP (61.7% at 5.46 × 10−7 M) with a Stern–Volmer constant of 2.7 × 106 M−1.
Since nitroaromatics possess electron-deficient rings, embedding fragments with strategically chosen electronic features into COFs represents another effective way to facilitate charge transfer interaction for the improvement of sensitivity, as well as selectivity over other non-aromatic interferents. Murugavel and coworkers found that electron-rich amine substituted triphenylbenzenes (TAPB) can interact with nitroaromatics in the solid-state and can serve as effective fluorescence sensors for nitroaromatic analytes.197–199 Inspired by those studies, the authors integrated the TAPB fluorophore into imine and β-ketoenamine based COFs for the detection of nitroaromatics.193 The fluorescence of those COFs was effectively quenched in the presence of different nitroaromatic compounds, such as TNP, DNT, p-DNB, and mDNB, among which TNP was the most efficient quencher, probably because of the proton transfer from TNP to the nitrogen atoms of the COFs. The aromatic electron-rich triphenylamine group was also incorporated into COF-BABD-BZ (BABD, 1,4′-(bis(4-formylphenyl)amino)-[1,10-biphenyl]-3,5-dicarbaldehyde, BZ, benzidine) by Dai and coworkers for the detection of TNP.200 The COF showed a quick color change in its fluorescence from golden yellow to dark brown upon the addition of TNP and exhibited good selectivity towards TNP compared to other nitroaromatics.
Incorporating both π-electron rich and π-electron deficient cores into COFs can lead to an enhancement in the strength and selectivity of material–analyte interactions, thus enabling the recognition of aromatics from other volatiles. Das and Mandal reported the differentiation of π-electron rich and π-electron deficient aromatics with similar structures and physical properties using COF 1. This COF bears a π-electron deficient triazine core and a π-electron rich naphthalene core connected with –O– and N– donor Lewis basic sites (Fig. 10a).194 The recognition was based on a dual readout constructed from the lifetime or emission intensity and quantum yield of COF 1, which was highly dependent on the interaction with aromatics.194 The electron-deficient triazine core in COF 1 allowed the selective uptake of a π-electron rich unsaturated moiety (benzene) over its saturated congener (cyclohexane); in turn, the electron-rich core was very efficient for the selective and ultra-low detection of highly explosive TNP in water down to 68 ppb (Fig. 10b). The rapid real-time detection of nitroaromatic compounds can be realized through both contact mode and instant spot measurement mode by immobilizing COF 1 on paper strips (Fig. 10c).
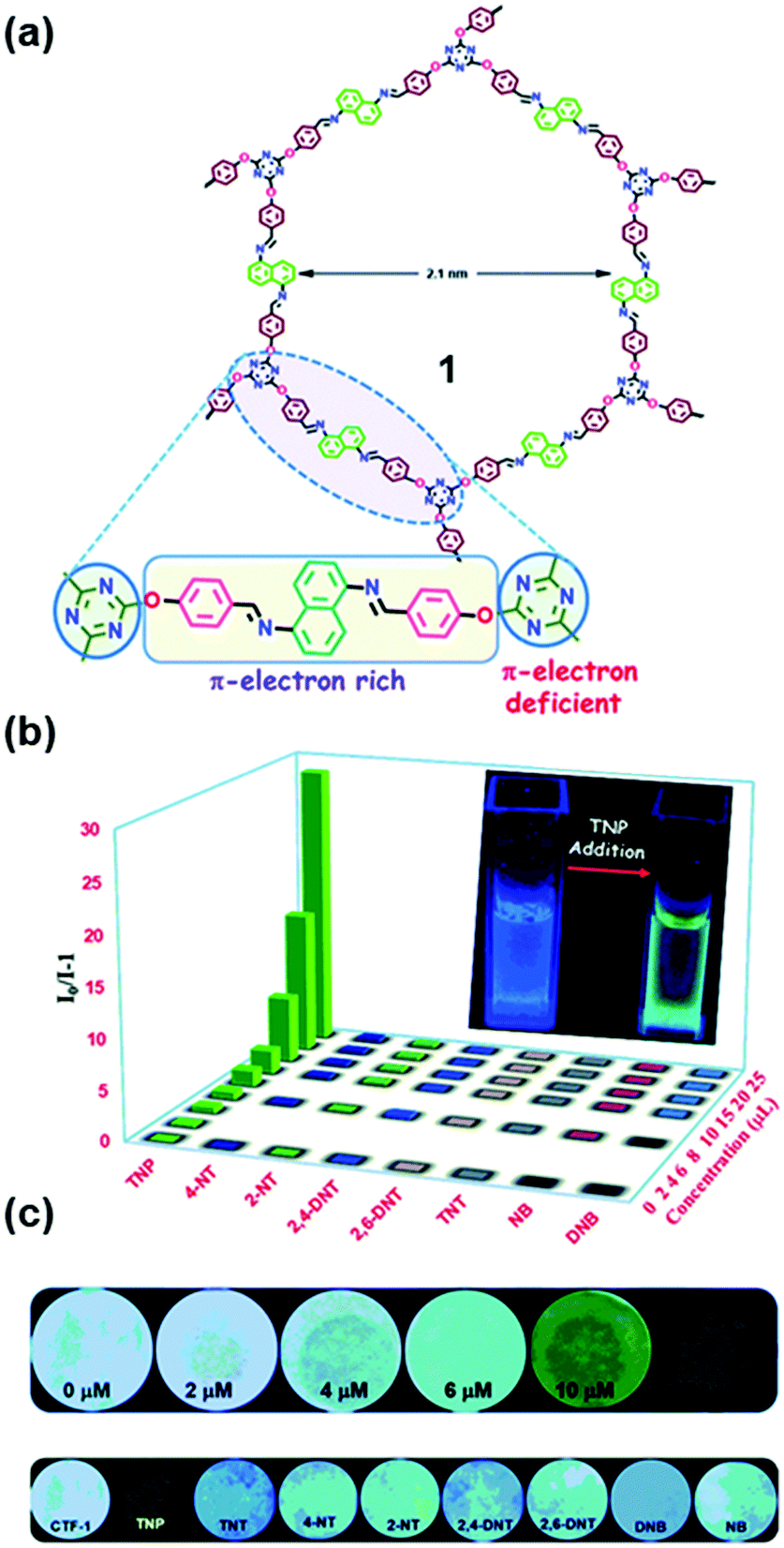 | ||
| Fig. 10 (a) Chemical structure of COF 1 containing both electron rich and electron deficient units. (b) Stern–Volmer plot of all of the analytes in water showing high selectivity for TNP. (c) Photograph of Whatman filter paper strips coated with 1 and different concentrations of TNP (upper row) and paper strips coated with 1 and 20 μM concentration of different nitroaromatics (lower row).194 Adapted with permission from ref. 194. Copyright 2018, Royal Society of Chemistry. | ||
Incorporating aromatic units is a straightforward way to construct fluorescent COFs; however, the majority of 2D COFs, which have large periodic π–π stacked structures, exhibit limited emissive characteristics due to aggregation-caused quenching (ACQ). Highly fluorescent COFs are still in great demand for chemical detection. To circumvent the ACQ effect, the introduction of an AIE moiety has been proven to be an effective strategy to afford highly emissive 2D COFs, because the intralayer covalent linkages and interlayer π-interaction can work synergistically to impede the rotation-induced energy dissipation (see also Section 2.2.1 discussed earlier).173 Recently, Loh and coworkers developed highly luminescent (quantum yield up to 21.1%) imine-based Py-TPE-COF by the integration of a non-planar tetraphenylethene (TPE) building unit with a pyrene unit.201 The photoluminescence intensity decreased monotonically with the addition of TNP, and the quenching percentage increased to 95.5% at 10 ppm TNP. Upon exposure to other non-explosive molecular analogs with similar structures, including DNP, DNT, NT, and 2-nitrophenol (NP), Py-TPE-COF showed only a small to marginal decrease (<15.6%) in the PL intensity.
In most of the examples demonstrated above, one of the important features of COFs being utilized is the face-to-face stacking modes of their 2D sheets. Unlike the layered structures in 2D COFs, 3D COFs can possess more open sites that can be beneficial for the material–analyte interaction. In addition, as the building blocks are isolated in the 3D network, the incorporation of fluorescent units into 3D COF can result in materials with interesting emissive properties that may be different from those of 2D analogs. Through the selective choice of the geometry of the precursors and the connection patterns, Wang and coworkers demonstrated the synthesis of a novel 3D pyrene-based COF 3D-Py-COF (Fig. 11a and b) and its potential in explosive detection.177 3D-Py-COF adopted a two-fold interpenetrated pts topology and showed an intense yellow-green luminescence with a band centered at 484 nm (Fig. 11c). The gradual addition of TNP quenched the fluorescence of the 3D-Py-COF suspension (Fig. 11d). When the concentration of TNP was 20 ppm, a fluorescence quenching degree of 75% was reached with a Stern–Volmer curve quenching constant (Ksv) of 3.1 × 104 M−1, indicating that 3D-Py-COF was sensitive to TNP.
 | ||
| Fig. 11 (a) Chemical structure and (b) the space-filling model of 3D-Py-COF with a two-fold interpenetrated pts network. (c) Photography of the 3D-Py-COF powders under UV light irradiation. (d) Fluorescence quenching upon addition of PA in DMF.177 Adapted with permission from ref. 177. Copyright 2016, American Chemical Society. | ||
Acid/base and other vapors. Jiang and coworkers developed a highly luminescent COF (Fig. 12a) TPE-Ph by introducing an AIE-active tetraphenylethene (TPE) unit which showed sensitive response to NH3 gas.173 The TPE-Ph COF exhibited a high luminescence quantum yield of up to 32%, which was 2- to 3-fold that of the tetraphenylethene molecule. This enhancement suggested that the π–π interactions between layers of COFs, which were absent in the molecular tetraphenylethene material, additionally contribute to the higher fluorescence quantum yield (Fig. 12b). Because the boronate linkages of the TPE-Ph COF can form a Lewis acid–base pair with ammonia, the TPE-Ph COF suspension exhibited rapid luminescence quenching upon ammonia exposure (Fig. 12c). Ammonia at the concentration of 1 ppm could lead to a 30% decrease of fluorescence intensity, demonstrating that the TPE-Ph COF was a highly sensitive ammonia detector at a sub-ppm level. The Stern–Volmer plot revealed an almost linear curve, whereas the fluorescence quenching rate constant was evaluated to be as high as 4.1 × 1014 M−1 s−1 (Fig. 12d). This work demonstrated that the utilization of the concept of AIE could lead to a new generation of highly luminescent COF materials that retain high quantum yields in both solid and solutions and function as highly sensitive sensors to detect specific chemicals.
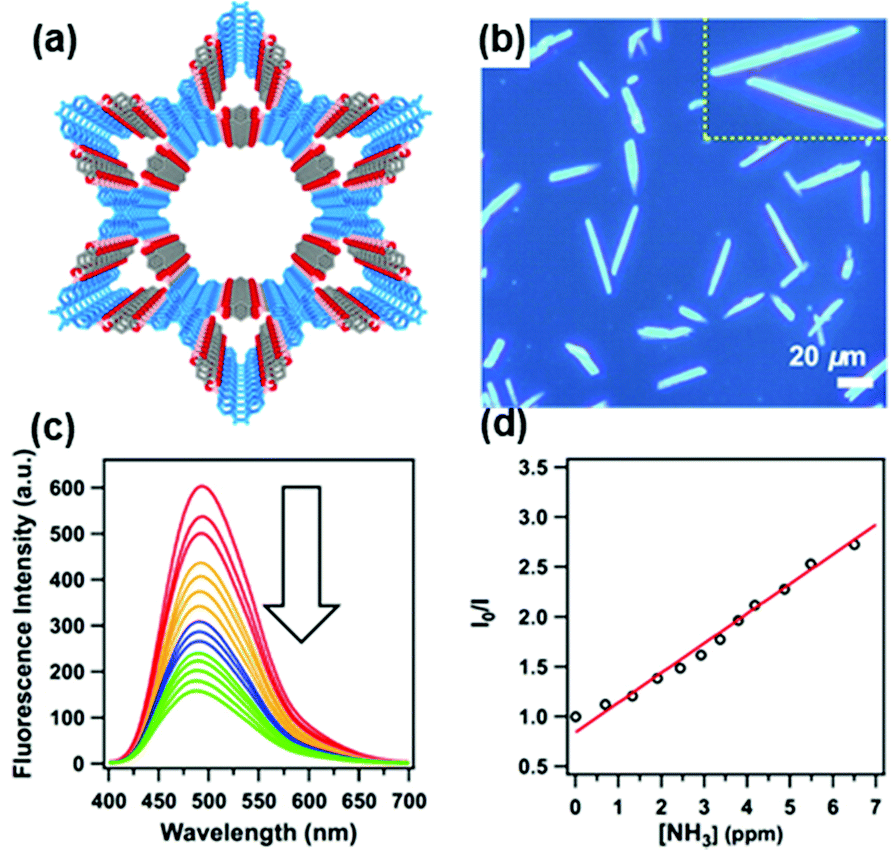 | ||
| Fig. 12 (a) Structural representation of the TPE-Ph COF. (b) Fluorescence microscopy images of TPE-Ph COF samples. (c) Fluorescence spectral change of the TPE-Ph COF upon the addition of ammonia. (d) Stern–Volmer plot of fluorescence quenching by ammonia.173 Adapted with permission from ref. 173. Copyright 2016, American Chemical Society. | ||
Using the same type of fluorophore, Zhao and coworkers reported the detection of HCl gas with a TPE-based 2D COF COF-ETBA-DAB made of 4,4′,4′′,4′′′-(ethane-1,1,2,2-tetrayl)tetra benzaldehyde (ETBA) and 1,4-diaminobenzene (DAB).141 Upon exposure to HCl gas, COF-ETBA-DAB displayed a new emission band with a very strong intensity centered at 670 nm. Meanwhile, the color of the COF crystallites exhibited an obvious change from yellow to red upon less than 1 s HCl exposure, faster than most HCl gas sensors previously reported.202–204 The addition of trace HCl with a concentration of 4.7 ppm could induce a significant change in the fluorescence emission of COF-ETBA-DAB, suggesting high sensitivity. The red color of the HCl-treated COF could be recovered to yellow upon exposure to ammonia vapor. Spectroscopic study indicated that the imine nitrogen atoms in COF-ETBA-DAB may undergo a quick protonation process in the atmosphere of HCl gas, which imparted an influence on the conjugated structure of the TPE skeleton, resulting in changes in the fluorescence emission as well as the color of the COF.141
From the perspective of their response mode, most fluorescent sensors rely on the measurement of a single intensity-varying fluorescence signal, which is prone to interference by the intensity of the excitation, collection efficiency of the emission signal, and inhomogeneous distribution of the probe. Instead, the ratiometric sensor has self-built-in corrections of two signal peaks and thus affords more accurate measurement over a single-output fluorescence sensor.205 Yan and coworkers reported a rationally designed COF with dual fluorescence emissions for the ratiometric sensing of trace water in organic solvents.206 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (Tz) and 2,5-dihydroxyterephthalaldehyde (Da) were chosen as the ligands to synthesize COF TzDa, where the Tz moiety can offer the docking sites and the Da moiety can provide the hydrogen bonds (Fig. 13a). The Tz moiety endowed the TzDa COF with an ICT effect, while the hydrogen-bonding interactions between Da and imine resulted in an excited-state intramolecular proton transfer effect. As a result, TzDa gave two main fluorescence emissions around 500 and 590 nm (Fig. 13b). The ratio of the two fluorescence emissions of TzDa linearly increased with the water content in organic solvents, such as ethyl acetate (Fig. 13c). Hydrogen bonding interactions between water and the Tz moiety blocked the ICT process from triazine to the phenyl group, increasing the possibility of nonradiative relaxation of TzDa, which consequently led to the decrease of fluorescence emission intensity at 500 nm (Fig. 13d). The TzDa COF can be used to detect trace amounts of water (0.006–0.085%) in various organic solvents, including isopropyl alcohol, acetone, tetrahydrofuran, ethyl acetate, and ethanol. Comparison experiments showed that the results obtained by using TzDa showed comparable accuracy with those obtained by gas chromatography in determining the water content in six commercial organic solvents.206
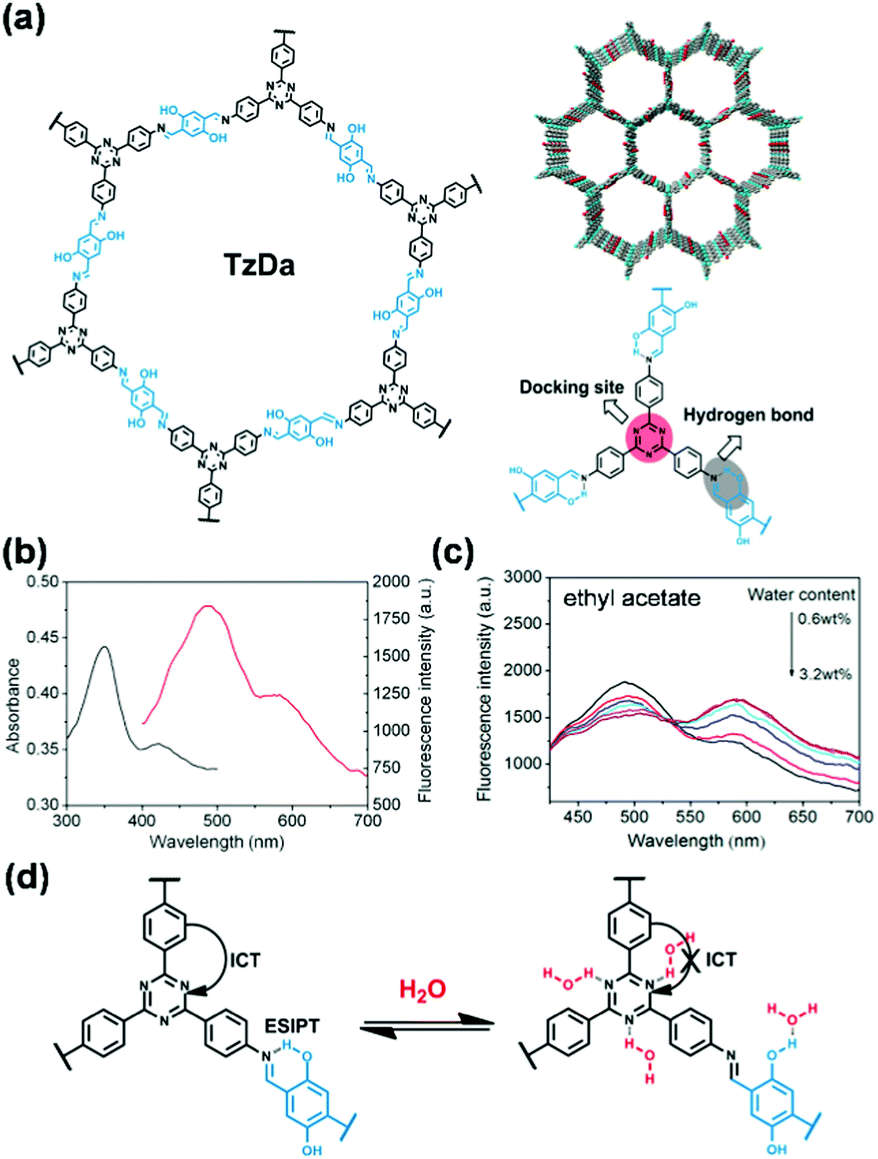 | ||
| Fig. 13 (a) Chemical structure and model of TzDa and the illustration of the docking sites and hydrogen bond in TzDa. (b) UV-vis absorption (black) and fluorescence (red) spectra of TzDa in pure ethyl acetate. (c) Fluorescence spectra of TzDa in ethyl acetate with different water content. (d) H2O-sensing mechanism of TzDa.206 Adapted with permission from ref. 206. Copyright 2017, American Chemical Society. | ||
Others. Qin and coworkers demonstrated the use of mechanochromic luminescent COF systems for highly selective detection of hydroxyl radicals (˙OH).207 The fluorescence intensity of COF-TpMA exhibited linear fluorescence quenching with the ˙OH concentration (0–10 μM), and was mostly unperturbed by the other reactive oxygen species. The mechanistic study indicated that fluorescence quenching was likely a static quenching attributed to the electron transfer of hydroxyl radicals to the π-conjugated framework that induced aggregation.176 COF-TpMA was also applied for the detection of exogenous ˙OH in living cells.
The use of fluorescent COFs has enabled the detection of various metal ions, including Cr3+, Hg2+, Cu2+, Fe3+, Au+ and Al3+, protons (pH), as well as F− anions. The detection of metal ions is mostly based on fluorescence switching due to the charge transfer between metal ions and excited states of COFs, whereas the detection of protons and F− is based on fluorescence change resulting from the formation of the protonated and deprotonated complex of COFs in the presence of protons and F−, respectively.
Metal ions. Oxygen-containing and nitrogen-containing groups, such as –C
N–, –NH2, CO, and –OH, are ubiquitous in a lot of COF structures. These groups can be formed during COF formation, embedded in the building blocks, or present at the edges and defect sites of COF crystallites. These groups generally have binding affinities to various metal ions because of the lone pair electrons in these moieties. This feature of COFs has been harnessed to achieve the detection of metal ions, though the design rules for selectivity have not always been deliberate.
For example, Qiu and coworkers prepared nanosheets of a luminescent crystalline polyimide (PI) for Cr3+ sensing.213 The PI nanosheets were examined for luminescence detection toward a series of metal ions. It was found that PI nanosheets were highly sensitive and selective in the detection of Cr3+ over the interference of other metal ions, such as K+, Ca2+, Cd2+, Pb2+, Mn2+, Hg2+, Mg2+, Co2+, Cu2+, and Zn2+. The selectivity for Cr3+ was attributed to differences in coordination bonding (coordination mode or bonding strength) between various metal ions and nitrogen/oxygen atoms in the PI framework. The authors hypothesized that binding of the PI framework to Cr3+ may reduce the antenna efficiency of the PI frameworks, and result in quenching of the luminescence. In another example, Fan and coworkers utilized the triazine units and edge nitrogenous groups of quantum dots made from CTF-1 to detect Hg2+.214 The purpose of employment of quantum dots was the more abundant edge groups in the quantum dot form of COF CTF-1 than in the bulk form.
Strategic improvement of selectivity for specific ions can be achieved by introducing active sites that exhibit selective recognition ability for the analyte of interest. Examples can be seen in the selective detection of Hg2+175 and Au+.215 Generally, to achieve high sensitivity, unique selectivity, and simple handling, fluorescent organosulfur compounds are used as effective Hg2+ chemosensors.216–218 Inspired by these precedents, Wang and coworkers reported the first application of fluorescent COFs for detecting and removing Hg2+ ions by introducing evenly and densely distributed thioether side chains into COF-LZU8.175 This bottom-up construction of COF-LZU8 not only generated a rigid and extended π-conjugation framework as the signal transducer but also embedded the thioether side chains as the cation receptor for Hg2+ (Fig. 14a). The contorted structure of COF-LZU8 enabled efficient avoidance of the ACQ effect,178 thereby, imparting this COF with high fluorescence both in the solid state and in the dispersion form, in contrast to its building blocks. Upon the addition of Hg2+, COF-LZU8 showed efficient fluorescence quenching (Fig. 14b). Comparison experiments using a structurally related COF without thioether side chains suggested that the quenched fluorescence originated from the interaction between the thioether groups of the COF and Hg2+, which resulted in electron transfer from the COF to the LUMO of Hg2+. Among various metal ions examined, only Hg2+ caused a significant fluorescence quenching of COF-LZU8 (Fig. 14c and d).175 The detection limit for Hg2+ was 25.0 ppb, superior to many thioether-functionalized chemosensors.219–221 Besides the excellent selectivity and sensitivity for detecting Hg2+, COF-LZU8 was also able to effectively remove >98% Hg2+ in water with residual Hg2+ less than 0.2 ppm. This COF material may serve as a new candidate with intrinsic multipurpose functionalities for simultaneous detection and removal of toxic metal ions.175
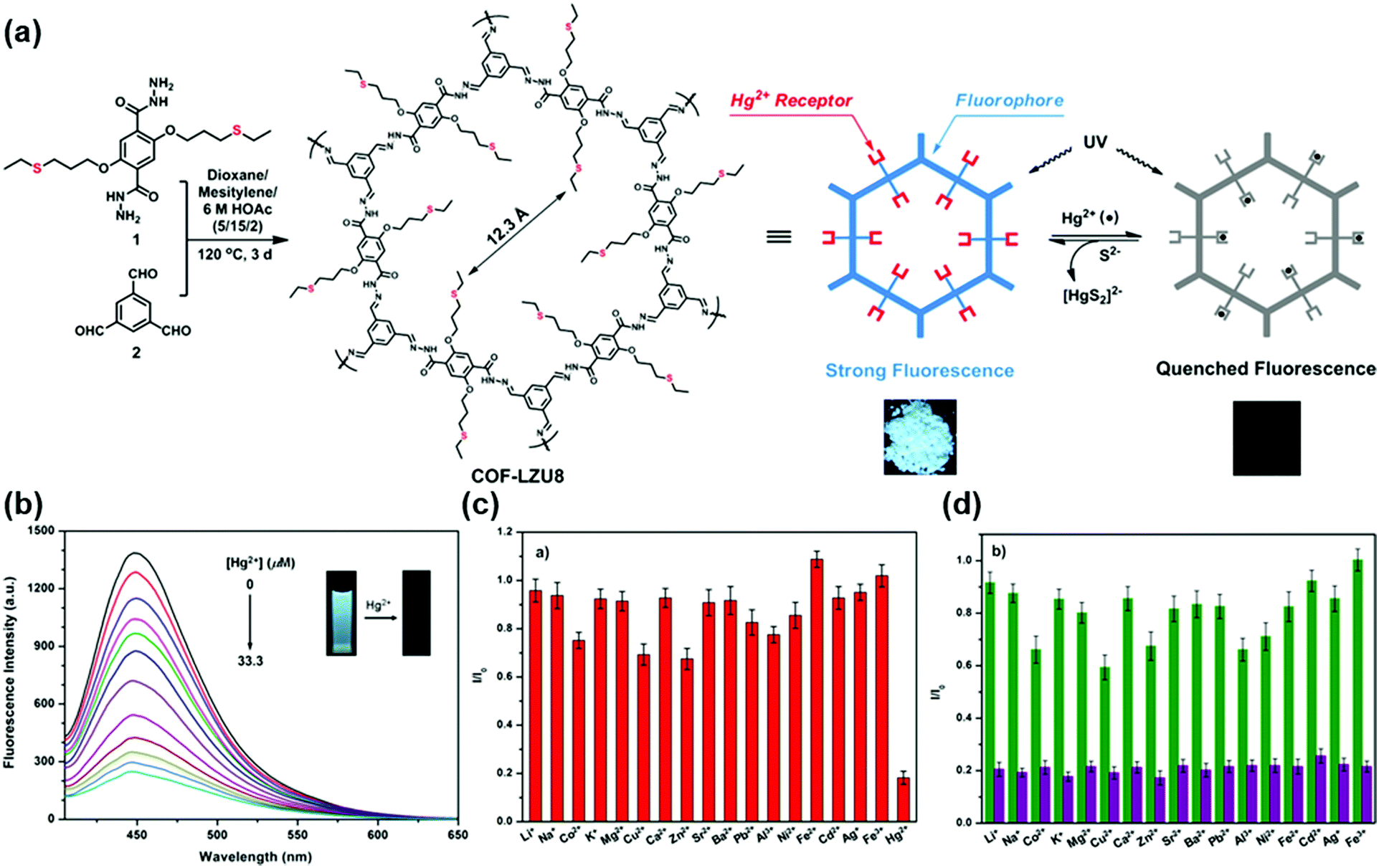 | ||
| Fig. 14 (a) Synthesis and structure of COF-LZU8. (b) Fluorescence titration of COF-LZU8 dispersed in acetonitrile upon the gradual addition of Hg2+ (λex = 390 nm). Inset: Fluorescence titration of COF-LZU8 dispersed in acetonitrile upon the gradual addition of Hg2+. Selectivity of COF-LZU8 toward Hg2+ detection: (c) fluorescence response of COF-LZU8 in the presence of different metal ions (33.3 μM) in acetonitrile and (d) competition experiments. The green bars in (d) represent the emission intensity of COF-LZU8 in the presence of the competing ions (2.0 eq.), and the purple bars represent the emission intensity upon the addition of Hg2+ (1.0 eq.) to the above solutions.175 Adapted with permission from ref. 175. Copyright 2016, American Chemical Society. | ||
Although the fluorescence quenching effect can provide straightforward luminescence change that often can be directly visualized, the sensitivity of the detection depends on the magnitude of the fluorescence change. Therefore, the use of highly fluorescent COFs is one applicable way to enhance sensitivity. For example, Liu and coworkers introduced bulky tert-butyl groups into COF-JLU3 to adjust the π–π interaction between the layers to increase the luminescence efficiency for the detection of Cu2+.176 COF-JLU3 exhibited high luminescence efficiency with an absolute fluorescence quantum yield of 9.91%. The fluorescence emission of COF-JLU3 showed a proportional decrease with the concentration of Cu2+ ions in the range from 0 to 0.4 mM with a LOD determined at 0.31 mM and good selectivity over other interfering ions, such as Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Zn2+, Cd2+, Ni2+, Pb2+, etc. Alternatively, using a strategy that resembles the AIE concept, Jiang and coworkers developed a series of highly emissive carbon-conjugated frameworks (sp2c-COFs) constructed by CC linkages, which showed selectivity for Cu2+.222 The backbone between two proximate pyrene knots consisted of a cyano-substituted phenylenevinylene adopting twisted conformation, which served as an AIE-like chromophore to avoid the aggregation-caused fluorescence quenching (Fig. 15a).223 The highly emissive property of sp2c-COFs compared with the constituent monomer 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy, Fig. 15b), as well as the cyano side groups in the frameworks that can serve as a ligand for triggering supramolecular interactions with specific metal species, renders sp2c-COFs the ability to detect specific guest species. The addition of Cu2+ quenched the fluorescence intensity of sp2c-COF in THF (Fig. 15c). By contrast, the addition of Zn2+ which cannot ligate with the –CN groups could not quench the emission (Fig. 15d). These results suggested that interaction with cyano groups was essential for triggering fluorescence sensing. sp2c-COF was highly sensitive to the Cu2+ ion and achieved a LOD of 88 ppb.222
 | ||
| Fig. 15 (a) Chemical structure of sp2c-COF. (b) Images of sp2c-COF and the TFPPy monomer dispersed in water under a UV lamp. Fluorescence spectral change of sp2c-COF upon addition of (c) Cu2+ and (d) Zn2+ ions.222 Adapted with permission from ref. 222. Copyright 2018, Springer Nature. | ||
In addition to detecting Cr3+, Hg2+, Au+, and Cu2+ ions mentioned above, fluorescent COFs were also applied for the sensing of Fe3+,184,224–227 Al3+,228 and UO22+.229 The introduction of cites with strong metal binding affinity can enhance the sensitivity of detection for metal ions. For example, Chen and coworkers reported the TaDAP COF constructed by the condensation between 1,3,5-tris-(4-aminophenyl)triazine and 2,6-diformylpyridine, which was able to detect Fe3+ in the range of 0.02–0.2 mM.225 However, only a moderate sensitivity with a LOD of 18 μM can be achieved, probably because Fe3+ binding provided by the unidentate pyridine or triazine in the TaDAP COF was relatively weak.225 By utilizing a strong chelating effect, Zhang and coworkers designed and synthesized a hydrazone-linked COF Bth-Dma, which showed improved sensitivity for the detection of Fe3+ ions in aqueous solution.226 The chelation between Fe3+ and the COF was realized by the predesigned O,N,O′-chelating sites in the pore wall of the Bth-Dma COF,227 which can affect fluorescence quenching through energy or electron transfer between the excited states of the COF and the d-orbital of Fe3+.230 As a “turn-off” type fluorescence sensor, the Bth-Dma COF could achieve a low LOD of 0.17 μM in a wide dynamic response range of 0.5–100 μM Fe3+.
In the examples mentioned above, the fluorescence quenching mechanism was used in the detection process. The dissipation of excitation energy by photo-induced electron transfer upon metal ion binding is usually responsible for this “turn-off” type response. Compared with the turn-off mechanism, the advantage of the fluorescence turn-on mechanism in detection is that if a completely dark background (completely quenched state) can be achieved, even a weak turn-on signal can be readily measured and thereby can allow trace detection.231,232 The construction of turn-on type fluorescence detection relies on the mediation of the luminescence or quenching mechanism by utilizing an appropriate molecular design.
In some COFs, the luminescence properties can be primarily dependent on the linkers. When the photoinduced electron transfer process occurs in the linkers, it can cause excitation energy dissipation and lead to weak photoluminescence. In this case, preventing the fluorescence quenching process by coordinating the COF with metal ions can relieve the PET process, providing a promising approach to achieve “turn-on” type fluorescence detection. Using this strategy, Qiu and coworkers reported the first “turn-on” fluorescence sensor used for highly sensitive and selective detection of Al3+ based on COF nanosheets Bpy-NSs that incorporated bipyridine linkers.228 The coordination of bipyridine sites in Bpy-NSs with Al3+ can eliminate the fluorescence quenching process caused by photoinduced electron transfer to result in a fluorescence “turn-on” effect (Fig. 16a). The fluorescence linearly increased with Al3+ concentration and achieved a 15.7-fold improvement with absolute fluorescence quantum yield increased from 0.15% to 1.74% (Fig. 16b). Although bipyridine has been reported to have coordination affinities to a few transition metal ions (e.g., Cu2+, Fe3+, and Co2+),233 metal ions, including Zn2+, Cd2+, Mn2+, Cr3+, Pb2+, Mg2+, etc., had little effect on the fluorescence of Bpy-NSs (Fig. 16c). The authors ascribed this selectivity to the higher energy levels of unoccupied d orbitals of the interfering ions than the 3s and 3p orbitals of Al3+; therefore, PET could not be effectively eliminated by these interfering ions.228
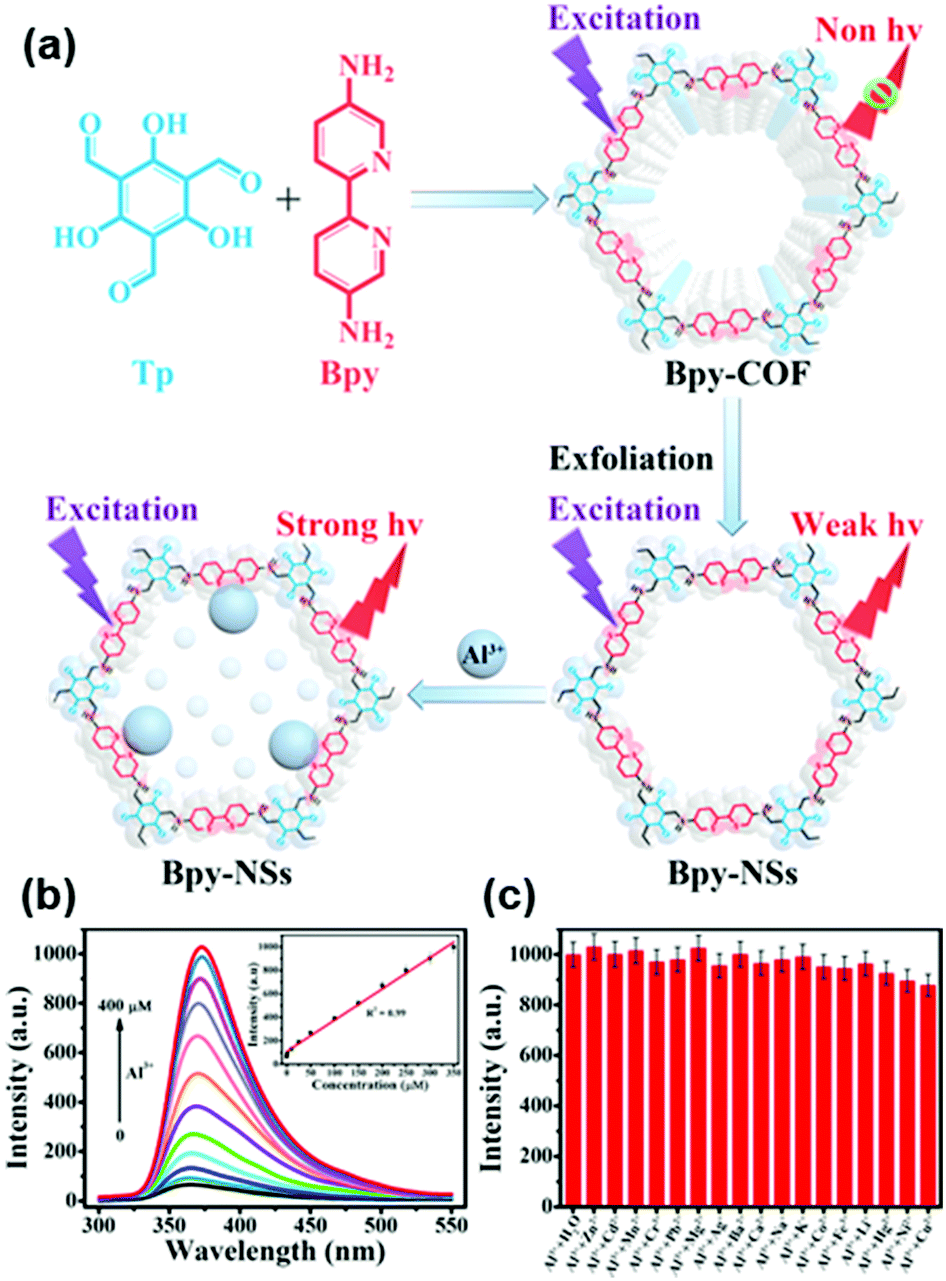 | ||
| Fig. 16 (a) The schematic diagram for preparation of Bpy-NSs and detection of Al3+ in which Al3+ binding at the bipyridine sites inhibits the photoinduced electron transfer to result in a fluorescence “turn-on” effect. (b) Fluorescence spectral changes of Bpy-NSs after the addition of different concentrations of Al3+. The inset is a linear calibration plot for Al3+ detection. (c) Fluorescence intensity changes of Bpy-NSs at 370 nm in the presence of Al3+ and other metal ions (350 μM).228 Adapted with permission from ref. 228. Copyright 2019, American Chemical Society. | ||
In addition to metal ions, fluorescent COFs were also used for pH detection in aqueous media.234,235 pH is fundamental to biological function and its measurement is therefore crucial across all biosciences. Although fluorescent COFs show great potential in the detection of organic volatiles, the weak stability and hydrophobicity of general Schiff base-based skeletons in aqueous solution can restrict their potential application in pH sensing. For developing pH-sensing systems, Liu and coworkers developed a β-ketoenamine based 2D COF (COF-JLU4), which had outstanding hydrolytic stability because of the stable and hydrophilic ketoenamine linkages.234 The fluorescence intensity of COF-JLU4 decreased gradually with the increase of pH from 9.0 to 13.0 (Fig. 17a), which was attributed to the deprotonation process of the nitrogen in the framework.236 When the pH was below 4.5, the luminescence emission at 428 nm increased dramatically with the decrease of pH. The fluorescence enhancement and blue-shift in acidic solution may be raised from the protonation of nitrogens (Fig. 17b). This pH-dependent fluorescence change could be recycled multiple times with similar degrees of fluorescence quenching (Fig. 17c and d).234
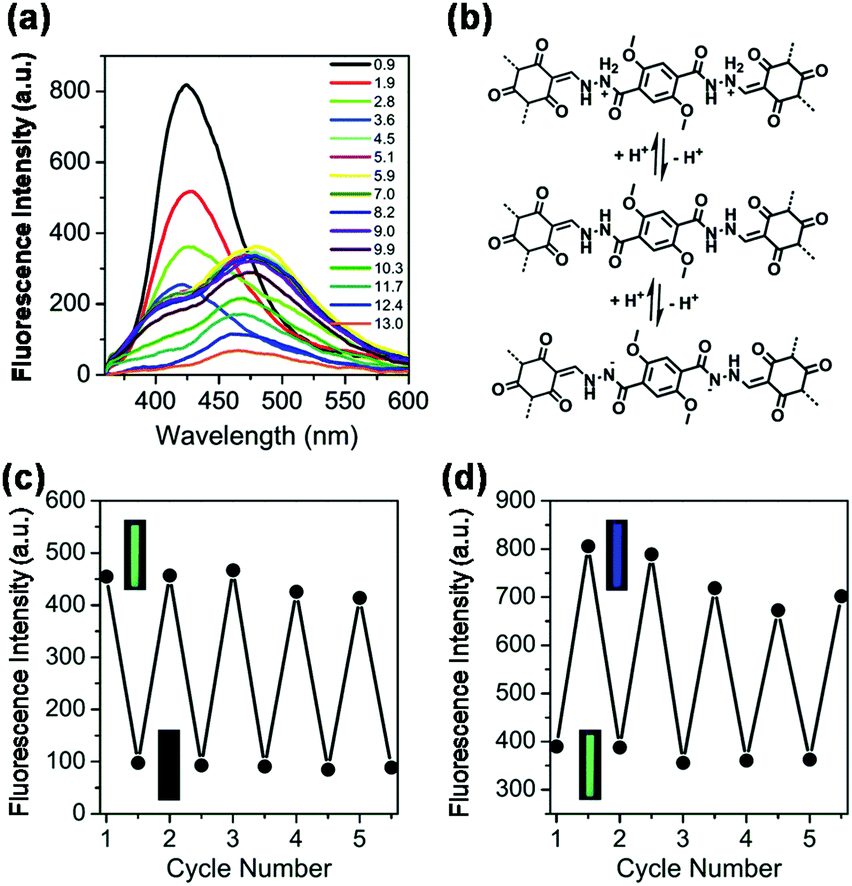 | ||
| Fig. 17 (a) pH-Dependent fluorescence of COF-JLU4 in aqueous solutions with pH ranging from 0.9 to 13.0 measured under excitation at 334 nm. (b) Deprotonation and protonation processes of the COF-JLU4 framework in acidic and basic media. Reversible optical responses of COF-JLU4 (c) between the neutral (pH = 7.0) and basic conditions (pH = 13.0) and (d) between the neutral (pH = 7.0) and acidic conditions (pH = 1.0).234 Adapted with permission from ref. 234. Copyright 2016, Royal Society of Chemistry. | ||
Compared with the detection of cations, the fluorescence detection of anions using COFs is currently less explored. This situation is probably because the interactions between COFs and anions are still less explored and not well established. So far, the only report for the detection of anions is for F−, which was achieved by Jiang and coworkers using TFPPy–DETHz-COF synthesized by condensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy) and 2,5-diethoxyterephthalohydrazide (DETHz).237 The key to realizing the detection of F− was the so-called pinpoint surgery on the nitrogen site of the hydrazone linkage by F−, which scissored the N–H bond to give an N− anion. This reaction eliminated the fluorescence quenching pathway and enabled an improvement of the light-emitting activity of the COF (Fig. 18a and b).237 This COF emitted a weak green-yellow luminescence at 540 nm in THF with an absolute fluorescence quantum yield of only 4.5%. The addition of tetrabutylammonium fluoride to the THF dispersion of TFPPy–DETHz-COF increased the absolute fluorescence quantum yield to 17%, 3.8 fold that of the original one (Fig. 18c); on the other hand, other anions, including Cl−, Br−, I−, and NO3−, did not change the luminescence under otherwise the same conditions (Fig. 18d and e). Time-resolved fluorescence spectroscopy suggested that the deprotonation of –NH groups by F− ions suppressed the electron transfer from the linkage to the pyrene skeleton. Other halogen anions were inert because of their incapability to initiate the deprotonation of the –NH group. TFPPy–DETHz-COF was not only a switch-on type sensor but also showed high sensitivity to F− with a detection limit down to 50.5 ppb (Fig. 18f), which was ranked the best among F− sensors.238
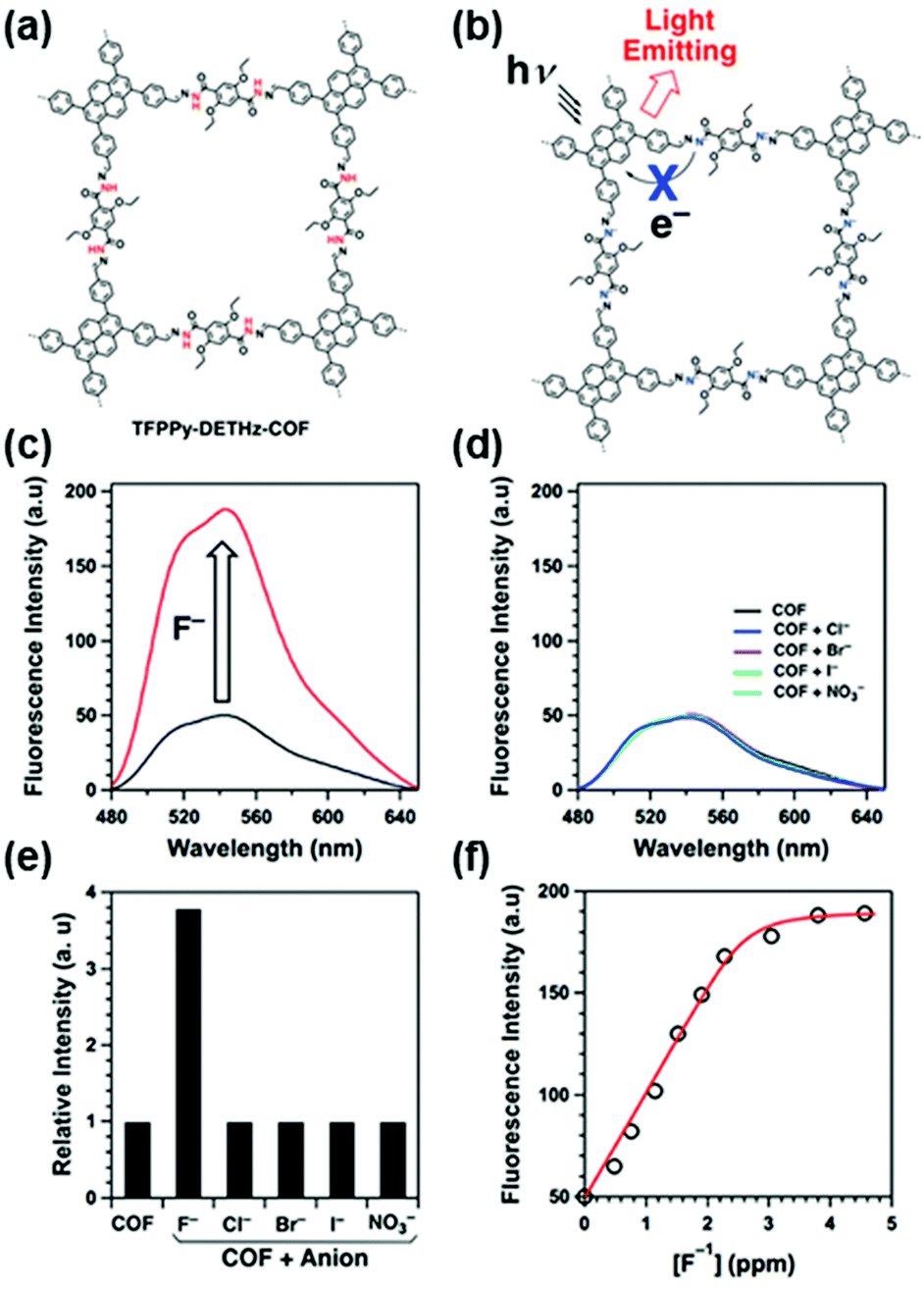 | ||
| Fig. 18 (a) Chemical structure of hydrazone-linked TFPPy–DETHz-COF. (b) The fluorescence enhancement mechanism for F− detection. Florescence spectral change of TFPPy–DETHz-COF upon addition of (c) F− and (d) Cl−, Br−, I−, and NO3−. (e) Relative fluorescence intensity of TFPPy–DETHz-COF upon the addition of anions. (f) The plot of fluorescence intensity versus [F−].237 Adapted with permission from ref. 237. Copyright 2018, American Chemical Society. | ||
So far, two different strategies have been used for the construction of COF based fluorescence detection platforms for biomolecules. In the first strategy, COFs are used as scaffolds or host matrices to support fluorescent probes (e.g., quantum dots, fluorescent metal ions) and receptors (e.g., molecularly imprinted polymers, bioreceptors) to form composite materials for detection, taking advantage of the porous structure and large surface-to-volume ratio of COFs. This strategy has been applied for the detection of DNA,240,241 proteins,242 natural antioxidants,243 antibiotics,244 and others.245–247 Since this strategy relies on the modification of COFs to attain fluorescence property and specific binding ability with analytes, we can see in the following discussions that the sensitivity and selectivity of these detection platforms highly depend on the type of auxiliary receptors. In the second strategy, prudent molecular design is used to construct COFs with intrinsic luminescence property and analyte binding capability. In this case, COFs are used as both responsive materials and transducers to yield detectable fluorescence signals that can be correlated to the identity and concentration of analytes. Recent efforts using this strategy have been devoted to the detection of DNA,248 amino acids,249 and small biology-related molecules.250,251
In 2017, Zhang and coworkers demonstrated a sensing platform for DNA detection by using ultrathin COF nanosheets generated from solvent-assisted liquid sonication from a structurally flexible TPA-COF (Fig. 19a and b).240 The proposed detection mechanism is shown in Fig. 19c. Two hairpin DNA probes, H1 and H2, were applied, in which H1 was labeled with a fluorescent dye. Both of them can be adsorbed on the surface of TPA-COF NSs by π–π stacking interactions, resulting in the fluorescence quenching of the dye. When the target DNA is present, it can specifically trigger a hybridization chain reaction (HCR) between H1 and H2 to yield long double-stranded DNA (dsDNA), which has a very weak interaction with TPA-COF NSs. Therefore, the HCR generated long dsDNA will move away from the surface of TPA-COF NSs and result in the recovery of fluorescence of the dye, providing a quantitative detection of target DNA (Fig. 19d). Compared to the bulk TPA-COF material, TPA-COF NSs exhibited better fluorescence quenching ability (Fig. 19e), probably attributed to the increased surface area of TPA-COF NSs. The fluorescence intensity increased with the concentration of thymine (T) and exhibited a linear relationship in the range of 0–1 nM with a LOD of 20 pM (Fig. 19f), which was comparable with or even better than that of most 2D nanomaterial-based fluorescence DNA sensors.240 Using a similar detection mechanism, Yan and coworkers also developed a fluorescence turn-on platform for sensing DNA using COF TpTta.241 Under optimal conditions, a LOD (S/N = 3) of 3.7 nM was achieved. This TpTta COF-based sensing platform is also extendable for fluorescence turn-on detection of other biomolecules, such as ATP, by simply replacing the probe molecule.241
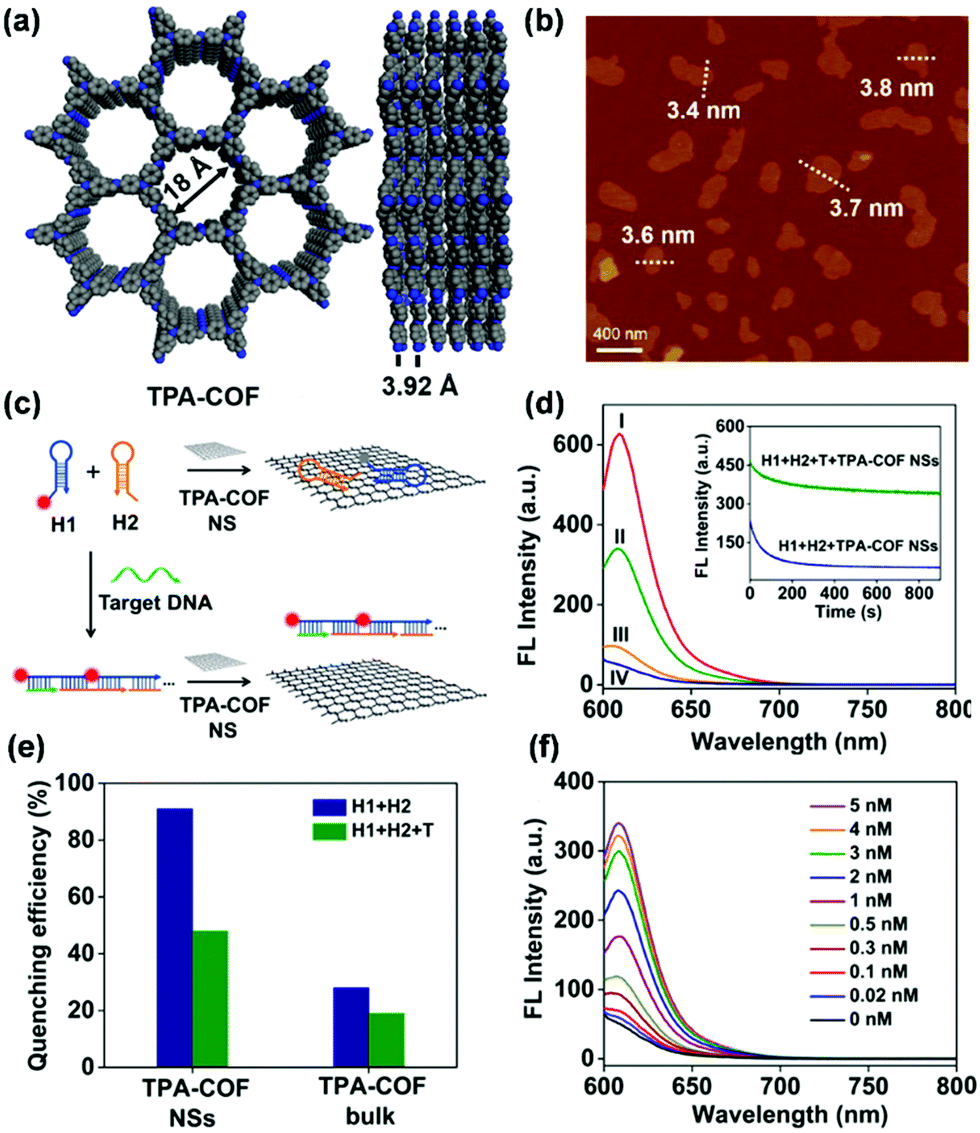 | ||
| Fig. 19 (a) Front and side view of the crystal structure model of TPA-COF. (b) AFM image of TPA-COF NSs. (c) Schematic illustration of a TPA-COF NS-based fluorescence sensor for the detection of DNA. (d) Fluorescence spectra under different experimental conditions: (I) H1 + H2; (II) H1 + H2 + T + TPA-COF NSs; (III) H1 + T + TPA-COF NSs; and (IV) H1 + H2 + TPA-COF NSs. Inset: Kinetic study on the fluorescence change of H1 + H2 and H1 + H2 + T in the presence of TPA-COF NSs. (e) Fluorescence quenching efficiency of TPA-COF NSs and bulk TPA-COF. (f) Fluorescence spectra of the proposed sensing platform in the presence of different concentrations of thymine.240 Adapted with permission from ref. 240. Copyright 2017, American Chemical Society. | ||
In addition to bioreceptors,240 incorporation of molecularly imprinted polymers (MIPs) is another effective strategy to encode extrinsic material–analyte interaction onto COFs. MIPs are polymers with highly selective and effective recognition sites that are spatially and chemically complementary to the template used in the polymerization process.252,253 Liu and coworkers developed a protocol for sensing bovine hemoglobin (BHb) using CdSe/ZnS quantum dot (QD)-grafted COF TpPa coupled with surface molecular imprinting technology.242 TpPa was treated as a support for BHb-imprinted polymer-coated QDs whose amino group can non-covalently interact with protein targets selectively and sensitively to trigger detectable fluorescence signals. The QD-grafted COF could detect BHb concentrations over the range of 0.002–0.2 mg mL−1 with a LOD of 5.4 × 10−4 mg mL−1. Various metal ions, such as Ca2+, Fe2+, Cu2+, Fe3+, Na+, and Zn2+, did not affect BHb detection. The strategy of adopting MIP-coated quantum dot-grafted COFs was also utilized for the detection of ferulic acid,243 amino acid tyramine,245,246 and a metabolite of carbadox in tissues, namely, quinoxaline-2-carboxylic acid.247
Recently, Yan et al. developed a lanthanide-functionalized COF-based hybrid material Eu@TpPa-1, which can act as a “turn-on” fluorescence sensor toward levofloxacin.244 In the detection system, Eu3+ played a bridge role to exert its characteristic emission in different concentrations of levofloxacin; TpPa-1 served as a host matrix to protect the introduced Eu3+. Eu@TpPa-1 showed strong pink-white and yellow emissions toward the high and low concentrations of levofloxacin, respectively, giving a linear range of 10−6–10−2 M and a LOD at 0.2 μM. Eu@TpPa-1 was successfully applied for sensing levofloxacin in the actual serum and urine samples with LODs at 0.2 and 0.6 μM, respectively.
Although proven to be effective, complex fabrication processes are usually required for the modification of the COFs to execute detection in the above examples. Detection systems with concise configurations and good performance can be realized by constructing intrinsically fluorescent and responsive COFs. To introduce inherent analyte binding ability into COFs, Ajayaghosh and coworkers incorporated cationic fragment ethidium bromide into COF EB-TFP for the selective detection of double-stranded DNA (dsDNA) (EB-TFP).248 In an aqueous medium, the COF EB-TFP self-exfoliated into ionic nanosheets (EB-TFP-iCONs). DNA with a long and periodic phosphate backbone can interact electrostatically with positively charged EB-TFP-iCONs, leading to the formation of hybrid EB-TFP-iCONs-DNA crystalline nanosheets (Fig. 20a and b). The EB-TFP-iCONs in the presence of dsDNA showed enhanced fluorescence at 600 nm and decreased intensity of the green emission at 510 nm (Fig. 20c). The authors hypothesized that this DNA-assisted reassembly of EBTFP-iCONs may create a hydrophobic environment over the EB fragment of the iCONs, thereby preventing the excited-state proton transfer process to water and enhancing the emission. The addition of other phosphate-containing molecules such as monobasic and dibasic phosphates (MBP and DBP) and adenosine mono-, di-, and triphosphates (AMP, ADP, and ATP) to EB-TFP-iCONs did not result in any change in emission (Fig. 20d), indicating the selective interaction of double-stranded calf thymus DNA (ctDNA). Steady-state and time-resolved emission studies revealed that the reassembly phenomenon was highly selective for dsDNA when compared to single-stranded DNA (ssDNA), showing the potential of using EB-TFP-iCONs as a 2D fluorescent platform for the label-free detection of complementary DNA strands.248
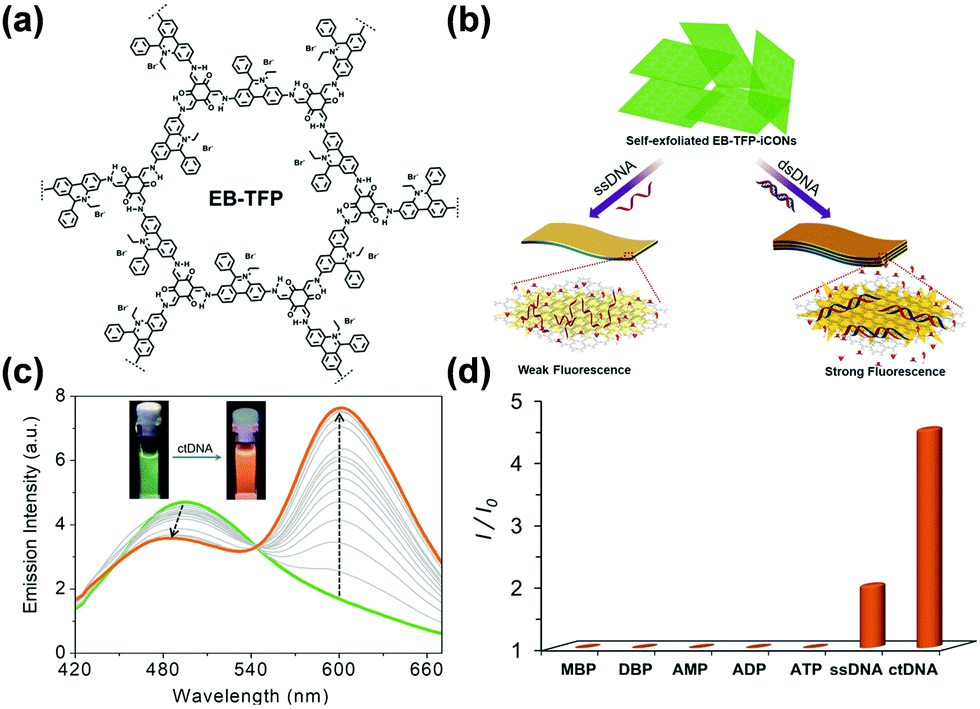 | ||
| Fig. 20 (a) Structure of EB-TFP-iCOF. (b) Schematic of EB-TFP-iCONs as a fluorescent platform for label-free detection of dsDNA. (c) Emission (λex = 350 nm) spectral changes of EB-TFP-iCONs upon the addition of ctDNA (0–32 μm). Inset: Corresponding changes in emission color upon illuminating with 365 nm UV light. (d) Selective fluorescence enhancement (λem = 600 nm) of EB-TFP-iCONs upon adding MBP, DBP, AMP, ADP, ATP, ssDNA (20-mer), and ctDNA.248 Adapted with permission from ref. 248. Copyright 2018, Wiley-VCH. | ||
Utilizing azine moieties as potential recognition sites, Zhao and coworkers demonstrated a signal amplification effect for amino acids and small pharmaceutical molecules using the ultrathin nanosheets of azine containing 2D COFs (NUS-30, NUS-31, and NUS-32) with AIE active tetraphenylethylene (TPE) moieties.249 The azine moieties played a key role in providing binding affinity toward amino acids by hydrogen bonds. Because of hydrogen-bond induced electron or energy transfer, a series of amino acids (L-alanine, L-threonine, and L-tryptophan) and pharmaceutical molecules (L-dopa) can trigger effective fluorescence quenching of COF nanosheets. The binding affinity of the COFs toward these amino acids increased with the total number of azine moieties within the COF structures. Compared with bulk COFs, COFs in nanosheet forms exhibited enhanced sensing sensitivity, indicating that the large exposed surface in the 2D sheets can allow sufficient contact and interaction with amino acids. The COF nanosheets were further integrated into membranes by electrospinning for detecting amino acids, though extended response time was needed compared with solution-based detection. Nevertheless, these results suggested that 2D COF nanosheets are promising material candidates for the fabrication of composite materials or devices for practical biosensing applications.249
Many biologically active molecules are chiral, including the naturally occurring amino acids, carbohydrates, and others. The discrimination of chirality from enantiomeric mixtures is of great importance because the physiological activities and biological functions are distinct for different enantiomers.254 Although progress has also been made in incorporating chiral hydroxyl functionalities into COF structures for chromatographic separation of enantiomers,57,255 it remains a challenge to introduce functional groups in chiral COFs for chiral optics and sensing. Through the incorporation of chiral groups into fluorescent COFs, detection of enantiomeric biomolecules can be possible. Cui and coworkers demonstrated fluorescent chiral COFs for chirality sensing of terpene flavor molecules,250 which are a large and diverse class of naturally occurring organic chemicals with broad use and industrial relevance.256 Two imine-linked chiral fluorescent COFs (CCOF 7 and CCOF 8) were elaborately designed and prepared by linking an enantiopure 1,1′-bi-2-naphthol (BINOL)-based linear dialdehyde with a tris(4-aminophenyl)benzene derivative or tetrakis(4-aminophenyl)ethene as building blocks (Fig. 21a and b). The COF can be readily exfoliated into ultrathin 2D nanosheets (Fig. 21c and d) and electrospun to make free-standing nanofiber membranes. In both the solution and membrane form, the fluorescence of COF nanosheets can be effectively quenched by chiral odor vapors via supramolecular interactions with the immobilized BINOL moieties. When nanosheets of CCOF 7 were treated with the two enantiomers of (α)-pinene, the rate of quenching caused by the (−)-enantiomer was faster than that with the (+)-enantiomer with a quenching ratio [QR = Ksv(−)/Ksv(+)] of 3.41, showing enantioselectivity in the fluorescence recognition (Fig. 21e). Control experiments showed that the bulk sample of CCOF 7 exhibited much lower enantioselective performance than 7-NS (Fig. 21f). This feature may be attributed to the fact that the NSs have a more exposed external surface and accessible active binding sites, allowing sufficient contact and interaction with the analyte.162,240 Similar selectivity was also observed using the nanofiber membrane of CCOF 7 and for other terpenes, including limonene, fenchone, terpinen-4-ol, and carvone, with the QR values ranging from 1.20 to 3.41 (Fig. 21g and h). Compared to the BINOL-based homogeneous and membrane systems, the COF nanosheets exhibited greatly enhanced sensitivity and enantioselectivity owing to the confinement effect and the conformational rigidity of the sensing BINOL groups in the framework.250 Integrating chiral auxiliary inside the open channels of COFs capable of amplifying chiral discrimination of analytes provided a feasible strategy toward the rational design of porous materials for chiral sensing of other biologically important molecules.
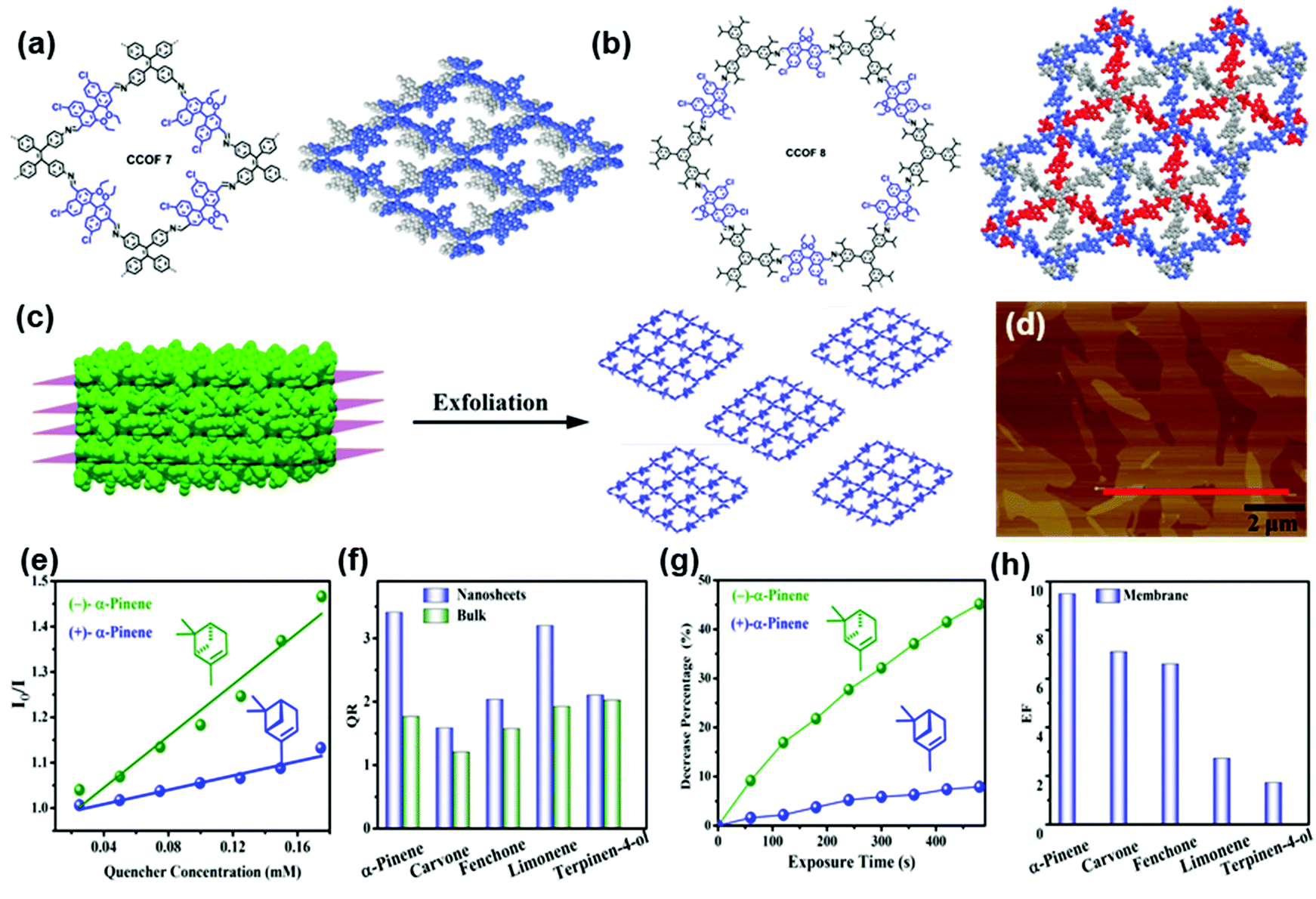 | ||
| Fig. 21 (a) Chemical structure and refined 2D crystal model of CCOF 7. (b) Chemical structure and refined 2D crystal model of CCOF 8. (c) Schematic diagram of exfoliation of COFs into ultrathin 2D NSs. (d) AFM image of the exfoliated nanosheet of CCOF-7. (e) Stern–Völmer plots of CCOF7-NS upon titration of α-pinene in MeCN. (f) Enantioselective quenching ratio for several terpenes. (g) Decrease of fluorescence of the membrane made of CCOF7-NS upon exposure to α-pinene. (h) Enantioselective fluorescence decrease ratio of the membrane for several vapors.250 Adapted with permission from ref. 250. Copyright 2019, American Chemical Society. | ||
The fluorescence property of COFs has also been utilized in the detection of intracellular hydrogen sulfide. Small-molecule fluorescent probes have been widely applied for the detection and imaging of various biological targets; however, the biosensing performance of a small-molecule fluorescent probe can be susceptible to the complex biological environment. For example, intricate intracellular enzymes may affect the selectivity and sensitivity of the designed probe.257 Zhang and coworkers envisaged that COFs can circumvent the problems associated with the interference by enzymes as encountered by small-molecule fluorescent probes, considering that the nanoscale pores of COFs can filter biomacromolecules (Fig. 22a).251 To realize this concept, the authors designed a hybrid fluorescent probe TpASH–NPHS comprised of an imine-linked COF TpASH and a two-photon fluorescent probe 4-amino-1,8-naphthalimide derivative (NPHS) anchored to the COF through alkylation of the phenolic hydroxyl group of TpASH. The azide units on NPHS can be reduced to amino groups in the presence of H2S, which will enhance the fluorescence emission of NPHS. The hybrid fluorescent probe TpASH–NPHS was able to detect the concentration of NaHS in the range of 0–20 μM with a LOD of 0.11 μM (Fig. 22b and c). The fluorescence of TpASH–NPHS was insensitive to various interferents, including common anions, biothiols, cysteine, homocysteine, and reactive oxygen species under normoxic and hypoxic conditions (Fig. 22d). The fluorescence emission of TpASH–NPHS in the lysates of HepG2 cells was not affected but the small molecule fluorescent probe CNPHS showed an emission increase (Fig. 22e), indicating that this COF nanoprobe was able to abate the biomacromolecular interference. Because of the interference-resistant ability, TpASH–NPHS was used to image endogenous H2S and to monitor the expression levels of the H2S-generating enzyme cystathionine g-lyase in a cirrhotic liver model.251 This TpASH–NPHS system provided an example for the exploitation of COFs for interference-resistant and selective biosensing by fully utilizing their predesigned topology and pore structure.
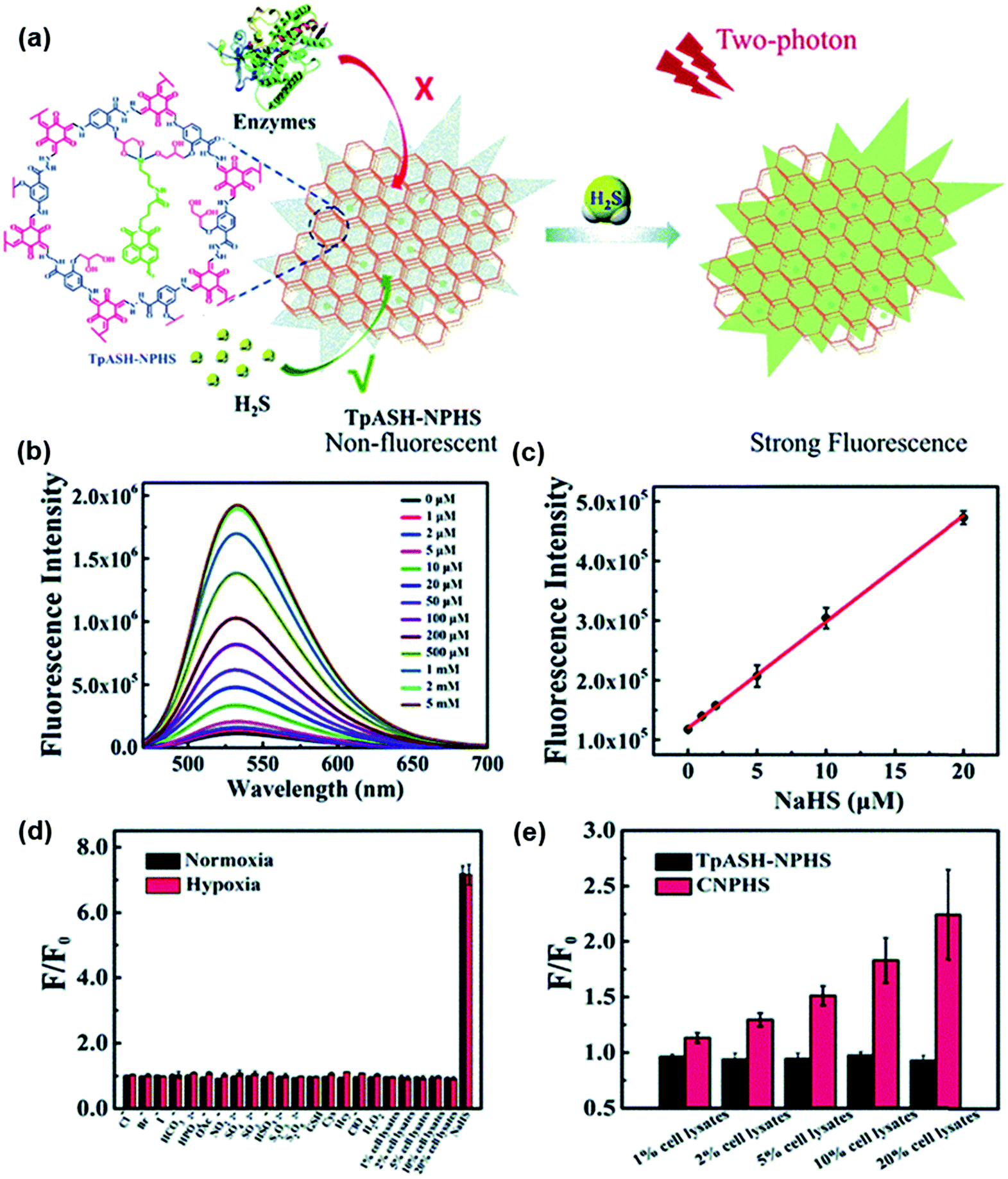 | ||
| Fig. 22 (a) Constitution and mode of action of the COF-based nanoprobe TpASH–NPHS. (b) The fluorescence emission spectra of TpASH–NPHS in the presence of different concentrations of NaHS. (c) Linear response at low NaHS concentrations. (d) Fluorescence response of TpASH–NPHS to various substances under normoxic (21% O2) and hypoxic (<0.1% O2) conditions. (e) The comparison of fluorescence response between TpASH–NPHS and a small-molecule probe CNPHS to different concentrations of HepG2 cell lysates under hypoxia.251 Adapted with permission from ref. 251. Copyright 2018, Royal Society of Chemistry. | ||
The use of COFs for fluorescence-based chemical detection is a rapidly developing field. In recent years, COFs as key components in fluorescence detection have been successfully applied for the analysis of various volatiles, gases, ions, and biological analytes (Table 3). Although encouraging developments have been made, there are still a few challenges remaining. Firstly, because of the insolubility of COFs, most of the fluorescence detection using COFs is carried out in solution, in which a suspension or colloid of COFs has to be made for the measurement of the emission spectrum. However, the aggregation of COF particles can cause decay of fluorescence, which can eventually affect the long-term stability of the detection system. The use of the AIE strategy can provide a promising solution for constructing highly fluorescent COFs. In addition, the exploration of making homogeneous and monodispersed nanoparticles (e.g. QDs) and nanosheets of COFs may also provide stable detection systems meanwhile enhancing the sensitivity due to abundant exposed surface groups of nanoparticles and nanosheets.258 Secondly, in the current sensing platforms, the fluorescence turn-off mechanism based on metal–ligand coordination or donor–acceptor energy and charge transfer is much more frequently used than the fluorescence turn-on mechanism.175,176,190,191 Compared to fluorescence turn-on sensors, fluorescence turn-off sensors can suffer from the limitations of higher background fluorescence and lower sensitivity, raising a great demand for developing highly luminescent COFs.259,260 Only a few turn-on types of platforms have been developed for the detection of ions228,237 and biology-related molecules,240,241,244,251 and detection based on the switching of intrinsic fluorescence of COFs remains rare.228,237 The challenges for developing fluorescence turn-on type detection systems may lie in the limited understanding and control of the fluorescence emission mechanism of COFs under the participation of the analyte molecules, especially in a complex detection environment. Learning from the well-established small molecule-based fluorescence detection systems and introducing them into framework materials may be one of the feasible solutions. Thirdly, although satisfactory selectivity for certain analytes has been demonstrated, the selectivity of most COF-based sensing strategies is limited due to the lack of a specific recognition group. Strategic molecular design is needed to install sophisticated recognition elements in COFs. Post-synthetic modification strategies have played a key role in the development and construction of functionalized COFs, which can endow COFs with effective and selective active sites for analyte binding. However, current post-synthetic modifications of COFs are limited to channel wall functionalization. Precise control of the quantity and distribution of functional groups within COFs is still challenging. Therefore, directly embedding the active sites in the building blocks for the reticular synthesis can be another alternative, though the compatibility between active sites and reticular reactions for COF synthesis needs to be carefully considered.
| Analyte(s) | Type of response | COF | Detection architecture | LOD | Detection range | Other detection parameters | Ref. |
|---|---|---|---|---|---|---|---|
| TNP | Turn-off | Py-Azine | Py-Azine suspension | N/A | 0–70 ppm | In acetonitrile; selectivity over DNP, DNT, NP, and NT | 160 |
| TNP | Turn-off | 3D-Py-COF | 3D-Py-COF suspension | N/A | 0–20 ppm | In DMF | 177 |
| TNP | Turn-off | SNW-1 | SNW-1 suspension | 0.05 μM | 0.2–52.4 μM | In 10% THF aqueous solution; selectivity over NB, 4-NT, DNT, TNT, tetryl, NP, DMF, DMSO, ethanol, acetone, acetonitrile, and benzene | 183 |
| TNP | Turn-off | TfpBDH | TfpBDH-CONs suspension | N/A | 0–5.4 × 10−5 M | In isopropyl alcohol; selectivity over TNT, DNP, DNT and NP | 190 |
| TNP | Turn-on | TfpBDH | Solid state TfpBDH-CONs | 10−3 M | 5.4 × 10−3 M | In gas phase; selectivity over TNT, DNP, DNT and NP | 190 |
| TNP | Turn-off | PI-COF | PI-CONs dispersion | 0.25 μM | 0.5–21 μM | In ethanol; selectivity over NB, NT, DNT, TNT, HQ, AN, PPD, NP, and DNP | 191 |
| TNP | Turn-off | TRIPTA | TRIPTA suspension | N/A | 5.2 × 10−8–5.46 × 10−7 M | In acetonitrile | 192 |
| TNP | Turn-off | TAPB-TFPB | TAPB-TFPB dispersion | N/A | 0–13 ppm | In acetonitrile; selectivity over DNT, p-DNB and mDNB | 193 |
| TNP | Turn-off | iPrTAPB-TFPB | iPrTAPB-TFPB dispersion | N/A | 0–22 ppm | In acetonitrile; selectivity over DNT, p-DNB and mDNB | 193 |
| TNP | Turn-off | TAPB-TFP | TAPB-TFP dispersion | N/A | 0–15 ppm | In acetonitrile; selectivity over DNT, p-DNB and mDNB | 193 |
| TNP | Turn-off | iPrTAPB-TFP | iPrTAPB-TFP dispersion | N/A | 0–23 ppm | In acetonitrile; selectivity over DNT, p-DNB and mDNB | 193 |
| TNP | Turn-off | 1 | 1 dispersion | N/A | 2–25 μM | In water; selectivity over 4-NT, 2-NT, 2,6-DNT, NB, 2,4-DNT, and DNB | 194 |
| TNP | Turn-off | COF-BABD-DB | COF-BABD-DB suspension | N/A | 1–12.5 μM | In acetonitrile; selectivity over DNP, Np, NT, NB, DNB, and DNT | 200 |
| TNP | Turn-off | COFBABD-BZ | COFBABD-BZ suspension | N/A | 1–12.5 μM | In acetonitrile; selectivity over DNP, DNB, NP, NT, NB, and DNT | 200 |
| TNP | Turn-off | TPE-Ph | TPE-Ph dispersion | N/A | 1–25 ppm | In acetone; selectivity over DNP, DNT, NT, and NP | 201 |
| TNP | Turn-off | iPrTAPB-Azo-COP | iPrTAPB-Azo-COP dispersion | N/A | 0–13 ppm | In acetonitrile; selectivity over DNT and m-DNB | 261 |
| DNT | Turn-off | PAF-15 | PAF-15 dispersion | N/A | 1.5–150 ppm | In CHCl3; selectivity over benzene, toluene, chlorobenzene, bromobenzene, phenol, and aniline | 188 |
| TNT | Turn-off | PAF-15 | PAF-15 dispersion | N/A | 1.5–150 ppm | In CHCl3; selectivity over benzene, toluene, chlorobenzene, bromobenzene, phenol, and aniline | 188 |
| NB | Turn-off | PAF-15 | PAF-15 dispersion | N/A | 1.5–150 ppm | In CHCl3; benzene, toluene, chlorobenzene, bromobenzene, phenol, and aniline | 188 |
| DNP | Turn-off | TRIPTA | TRIPTA dispersion | N/A | 5.2 × 10−8–5.46 × 10−7 M | In acetonitrile | 192 |
| DNT | Turn-off | TRIPTA | TRIPTA dispersion | N/A | 5.2 × 10−8–5.46 × 10−7 M | In acetonitrile | 192 |
| NP | Turn-off | TRIPTA | TRIPTA dispersion | N/A | 5.2 × 10−8–5.46 × 10−7 M | In acetonitrile | 192 |
| p-DNB | Turn-off | iPrTAPB-Azo-COP | iPrTAPB-azo-COP dispersion | N/A | 0–329 ppm | In acetonitrile; selectivity over DNT and m-DNB | 261 |
| TATP | Turn-off | 3′PD | 3′PD dispersion | ∼1 μM | 5–100 μM | In DCM | 262 |
| Janus Green | Turn-off | IMDEA-COF-1 | IMDEA-COF-1 suspension | N/A | 10–40 μM | In aqueous solution; selectivity over methylene blue, malachite green, bromophenol blue, crystal violet, and thionin | 196 |
| H2O | Ratiometric | TzDa | TzDa dispersion | 0.085, 0.022, 0.026, 0.006, and 0.034 wt% | 1.1–7.7, 0.1–5.1, 0.5–5.1, 0.6–3.2, and 0.4–2.5 wt% | In IPA, acetone, THF, EtOAc, and ethanol | 206 |
| NH3 | Turn-off | TPE-Ph | TPE-Ph dispersion | <0.3 ppm | 0.3–9.7 ppm | In toluene | 175 |
| NH3 | Turn-on | COP-1 | COP-1 dispersion | 0.059% (v/v) | 1.16–5.80% (v/v) | In gas phase; selectivity over benzene, methylbenzene, dimethylbenzene and trichloromethane; reversible | 137 |
| HCl | Turn-off | COP-1 | COP-1 solid film | 0.011% (v/v) | 1.16–5.80% (v/v) | In gas phase; selectivity over CO2, SO2, H2S and Cl2; reversible | 137 |
| HCl | N/A | COF-ETBA-DAB | COF-ETBA-DAB dispersion | <0.13 mM | 0.13–91 mM | In 1,4-dioxane; response time <1 s | 141 |
| H2S | Turn-on | TpASH | TpASH–NPHS dispersion | 0.12 μM | 1 μM–5 mM | In PBS, pH = 7.4; selectivity over Cl−, Br−, I−, HCO3−, HPO42−, OAc−, NO2−, SO42−, SO32−, HSO3−, S2O32−, S2O52−, GSH, Cys, Hcy, ClO− and H2O2; imaging in live cells, tumor tissues, cirrhotic liver model | 251 |
| Cr3+ | Turn-off | PI | PI nanosheet suspension | N/A | 1 × 10−4–1 × 10−2 M | In water; K+, Ca2+, Cd2+, Pb2+, Mn2+, Hg2+, Mg2+, Co2+, Cu2+, and Zn2+ | 213 |
| Au+ | Turn-off | TTB-COF | TTB-COF dispersion | 0.87 μM | 1.0–10.0 μM | In acetonitrile; Fe3+, Ni2+, Co2+, Zn2+, and Cd2+ | 215 |
| Cu2+ | Turn-off | COF-JLU3 | COF-JLU3 dispersion | 0.31 μM | 0–200 μM | In THF; selectivity over Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Zn2+, Cd2+, Ni2+, Pb2+, Co2+, Ag+, Fe3+ and Al3+ | 176 |
| Cu2+ | Turn-off | sp2c-COF | sp2c-COF dispersion | 88 ppb | 0–0.3 × 10−4 M | In THF; selectivity over Zn2+ | 222 |
| Cu2+ | Turn-off | N/A | Dispersion of carbon quantum dot scaffolded COFs | 0.50 nM; 2.4 nM | 0.0010–10.0 μm; 0.0032–32.0 μm | pH = 7.0; selectivity over Pb2+, Mg2+, Hg2+, Zn2+, Fe2+, Fe3+, Co2+, Cd2+, Cr2+, Ni2+, K+, Ca2+, Na+, Gly, Cys, and HSA; blood, wastewater | 263 |
| Fe3+ | Turn-off | DhaTab | DhaTab dispersion | 0.12 μM | 5–500 μM | In DMF/H2O (9:1, v/v); selectivity over Na+, Mg2+, Al3+, K+, Ca2+, Cu2+, Zn2+, Pb2+, Ag+, and Cd2+ |
184 |
| Fe3+ | Turn-off | PI-COF 201 | PI-COF 201 suspension | 0.13 μM | 0–1 mM | In DMF; selectivity over Na+, K+, Co2+, Pb2+, Sr2+, Mg2+, Ca2+, Ag+, La3+, Ce3+ and Cu2+ | 224 |
| Fe3+ | Turn-off | PI-COF 202 | PI-COF 202 suspension | 0.13 μM | 0–1 mM | In DMF; selectivity over Na+, K+, Co2+, Pb2+, Sr2+, Mg2+, Ca2+, Ag+, La3+, Ce3+ and Cu2+ | 224 |
| Fe3+ | Turn-off | TaDAP | TaDAP dispersion | 18 mM | 0.02–0.86 mM | In DMF/H2O (9:1, v/v); selectivity over Na+, Ag+, K+, Ca2+, Cu2+, Co2+, Mn2+, Ba2+, Co2+, Zn2+, Pb2+, Mg2+, Cr3+, and Al3+ |
225 |
| Fe3+ | Turn-off | Bth-Dma | Bth-Dma dispersion | 0.17 μM | 0.5–100 μM | In water; selectivity over Na+, K+, Mg2+, Ba2+, Mn2+, Ca2+, Gd3+, Zn2+, Ni2+, Co2+, La3+, Nd3+, Fe2+, or Cu2+ | 226 |
| Al3+ | Turn-on | Bpy-COF | Bpy-COF NS dispersion | N/A | 0–400 μM | In water; selectivity over Zn2+, Cd2+, Mn2+, Cr3+, Pb2+, Mg2+, Ag+, Ba2+, Ca2+, Na+, K+, Co2+, Fe3+, Li+, Hg2+, Ni2+, and Cu2+ | 228 |
| UO22+ | Turn-off | TFPT-BTAN-AO | TFPT-BTAN-AO dispersion | 6.7 nM | 0–20 μM | In H2O; selectivity over Co2+, Ca2+, Cd2+, Cs+, Cr3+, Cs+, Mn2+, Fe3+, Cu2+, Ni2+, Al3+, Zn2+, Mg2+, Pd2+, Ba2+, Ce3+, Nd3+, Sm3+, and La3+ | 229 |
| UO22+ | Turn-off | NS-COF | NS-COF NS dispersion | N/A | 0.8–36 μM | In DMF; selectivity over Nd3+, Sm3+, La3+, Ce3+, Mn2+, Sr2+, Ba2+, Ni2+, Co2+, and Zn2+ | 264 |
| F− | Turn-on | TFPPy–DETHz-COF | TFPPy–DETHz-COF dispersion | N/A | 0–4 ppm | In THF; selectivity over Cl−, Br−, I−, and NO3− | 237 |
| H+ | Turn-on | COF-JLU4 | COF-JLU4 dispersion | N/A | pH 0.9–13 | In aqueous solution; reversible | 234 |
| H+ | Turn-off | COF-HQ | COF-HQ suspension | N/A | pH 1–5 | In aqueous solution | 235 |
| DNA | Turn-on | TPA-COF | Suspension of TPA-COF NSs/H1 + H2 | 20 pM | 0.02–5 nm | 50 mM Na2HPO4 and 0.75 M NaCl; pH = 7.4; selectivity over T, single-base mismatch DNA, random DNA | 240 |
| DNA | Turn-on | TpTta | FAM-labeled prob/TpTta suspension | 3.7 nm | 10–100 nm | 10 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2, pH = 7.4 | 241 |
| DNA | Ratiometric | EB-TFP | EB-TFP suspension | N/A | 0–32 μM | 20 mM phosphate buffer, 2 mM NaCl, pH = 7.2; selectivity over MBP, DBP, AMP, ADP, ATP, and ssDNA | 248 |
| ATP | Turn-on | TpTta | FAM-labeled ATP aptamer/TpTta suspension | N/A | 25–200 mM | 20 mM Tris–HCl, 140 mM NaCl, 5 mM KCl, pH = 7.4 | 241 |
| BHb | Turn-off | TpPa | CdSe/ZnS QDs/TpPa dispersion | 5.4 × 10−4 mg mL−1 | 0.002–0.2 mg mL−1 | PBS buffer, pH = 6.7; Ca2+, Fe2+, Cu2+, Fe3+, Na+, Zn2+, K+, and Mg2+ | 242 |
| Ferulic acid | Turn-off | TpPa | QDs-grafted TpPa/MIP dispersion | 5 μg kg−1 | 0.03 to 60 mg kg−1 | In ethanol; selectivity over cinnamic acid, syringic acid, and caffeic acid | 243 |
| Levofloxacin | Turn-on | TpPa-1 | Eu@TpPa-1 suspension | 0.2 μM | 10−6–10−2 M | In water; selectivity over urea, L-proline, NaHCO3, NaCl, KCl, glucose, MgCl2, CaCl2, creatinine, Na2SO4, creatine, NH4Cl; serum, urine | 244 |
| Tyramine | Turn-off | TpPa | CdSe–ZnS QD grafted TpPa/MIOP dispersion | 7.0 μg kg−1 | 35–35000 μg kg−1 |
In ethanol; selectivity over HIS and PEA; meat sample | 245 |
| Tryptamine | Turn-off | TpPa | CNDs-embedded TpPa/MIPs dispersion | 7.0 μg kg−1 | 0.025–0.4 mg kg−1 | In ethanol; selectivity over HIS | 246 |
| Quinoxaline-2-carboxylic acid | Turn-off | TpPa | CdSe–ZnS QD grafted TpPa/MIPs dispersion | 0.85 μmol L−1 | 1–50 μmol L−1 | In acetonitrile/methanol, PBS, pH = 7.5; CBX, MEQ, MQCA, and OLA; meat samples | 247 |
| L-Phenylalanine | Turn-off | NUS-30 | NUS-30 NS dispersion | N/A | N/A | In acetonitrile | 249 |
| L-Dopa | Turn-off | NUS-30 | NUS-30 NSs/PVDF membrane | N/A | N/A | N/A | 249 |
| (−)-α-Pinene | Turn-off | CCOF 7 | CCOF 7 NS suspension | N/A | 10–170 μM | In acetonitrile; selectivity over (+)-α-pinene | 250 |
| (−)-α-Pinene | Turn-off | CCOF 7 | CCOF 7@PVDF | N/A | 0–150 μM | In acetonitrile; selectivity over (+)-α-pinene | 250 |
| 4-Ethylguaiacol | Turn-off | TAPA | QD-grafted COFs@MIPs dispersion | 17 ng mL−1 | 0.025–1 μg mL−1 | In ethanol; selectivity over maltol, 4-vinylguaiacol, and p-cresol; response time 15 min; wine sample | 265 |
| ˙OH | Turn-off | COF-TpMA | COF-TpMA suspension | N/A | 0.1–10 μg mL−1 | In DMSO/PBS buffer (1:9, v/v, 20 mM), pH = 7.4; response time 2 min; cells |
207 |
2.3 Detection based on electrically transduced methods
Electrochemical detection technique. Electrochemical detection is one of the largest groups of analytical methods that have been widely applied for chemical detection. It is recognized to be a promising technology for specific and sensitive detection of various types of analytes in the solution phase due to its capability for fast, simple, direct, and sensitive analysis even in a complex sample matrix and due to the great potential in device miniaturization, integration, and commercialization.266 In electrochemical detection, the responsive material can interact with the analyte of interest through chemical or biological interaction, such that the charge transport characteristic of the tested system can be transduced into a measurable electrical signal in the form of potential, current, charge accumulation, or conductance/impedance change. Based on the way of acquiring and monitoring electrical signals, analytical techniques of electrochemistry can be further divided into three main categories, namely, potentiometry, amperometry, and conductometry, which measure the potential, current, and conductance/resistance of the detection system, respectively.
In potentiometric measurements, the difference in the electrical potential of the working electrodes relative to the potential of a reference electrode is measured, usually by using a two-electrode galvanic cell under ideally zero current conditions. A local equilibrium is built at the electrode–sample interface as a result of analyte partitioning between the working electrode and the sample solution. The measured potential is used to determine the analytical quantity of interest in the solution. This potential can be described by the Nernst equation, which reveals a linear dependence of the response, E, on the logarithm of a function of the activity of the ion in solution.
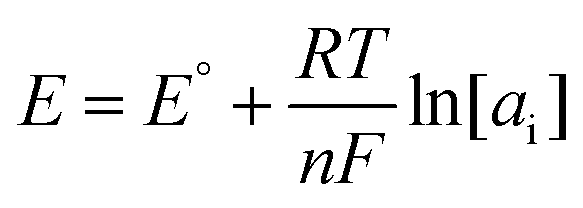
In amperometric detection, the current is measured over time with a fixed potential in a three-electrode set-up comprising a working electrode, counter electrode, and reference electrode. During amperometric detection, electrochemical reactions of analytes occur at the electrode surface. The essential operational feature of amperometric detection is the electron transfer between the working electrode and the analyte. When the working electrode is placed in a stirred solution (batch mode) or a flowing stream (continuous mode), and with the working potential fixed at a value corresponding to the limiting current plateau region, the current, iL, is a function of the concentration c of one redox species:
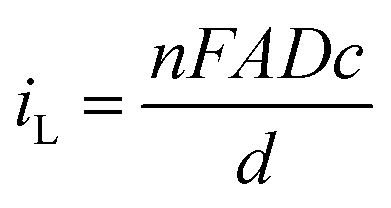
Voltammetric detection shares a similar set-up with amperometric detection. The difference is that the potential applied in voltammetric detection is continuously changed following a well-defined ramp. Typical potential ramps can be linear, and the corresponding technique is called linear sweep voltammetry (LSV). Alternatively, if the potential is cycled between two limiting values with a defined rate, this technique is called cyclic voltammetry (CV). The potential also can be pulsed with a certain amplitude and frequency, like in the case of differential pulse voltammetry (DPV) or square wave voltammetry (SWV). All these techniques are characterized by a signal that depends on the capability of the analytes to be either oxidized and/or reduced at the working electrode. The position and intensity of the corresponding current–potential wave can be respectively used as a fingerprint for identifying an analyte and as a reference signal for quantifying its concentration.
Conductometric detection is carried out by measuring the impedance or conductance change of a thin film or bulk solution in the presence of analyte species. The most used conductometric technique is electrochemical impedance spectroscopy (EIS), which has been used to identify and separate different contributions to the electric and dielectric responses of a material. Similar to voltammetric/amperometric techniques, EIS predominantly utilizes a three-electrode configuration for electroanalytical measurements. A sinusoidal potential excitation with a small magnitude is imposed.267 The current response is a sinusoidal wave with the same frequency of perturbation potential, but with different amplitude and phase.268,269 The magnitude of the recorded phase shift depends on the nature of the electrolyte, diffusion process, electrode kinetics, and chemical reactions that take place in the electrochemical cell.10 For instance, a change in the impedance of the electrochemical cell can be used to follow reactions that produce a change in the number, charge, or mobility of analytes; a change in capacitance is indicative of the change in the dielectric constant of the recognition layer at the electrode surface due to the presence of analytes.270
Chemiresistive detection. Chemiresistive detection is one of the widely applied methods for electrically transduced sensing of gaseous and volatile analytes. A chemiresistive device, called chemiresistor, usually consists of two electrodes connected with a chemiresistive material deposited onto an insulating support. With the aid of amperometric detection, by monitoring the current flowing through the device under a fixed potential, the conductivity/resistance change of responsive materials upon their interactions with analyte molecules is analyzed. Compared with optical detection, chemiresistive detection offers the advantages of simplicity, compatibility with conventional direct current circuits, low cost, and the ease of high precision measurement. Materials with suitable conductivity are required to ensure detectable electrical signals for the implementation of chemiresistive detection. Various conductive materials, like graphene,34,117 metal oxides,118,119,271 and carbon nanotubes,116 have been extensively applied for chemiresistive detection.
Potential advantages of COFs for electrically transduced chemical detection. Considering that there are multiple analytical configurations available, a material can be utilized in different ways for electrically transduced detection. Firstly, for selective recognition of a species of interest, a recognition element is required for the development of electrochemical detection. For example, in potentiometric detection, the perm-selective membrane is a key component that selectively binds to the target ion. In amperometric or voltammetric detection, the recognition element should selectively allow diffusion of the electrochemically active species to the electrode surface. Secondly, materials with good electron transport properties are critically important for their utilization as electrode materials for electrically transduced detection. The charge transfer interactions at the interface between an electrode and analytes in a liquid or gas phase determine the intensity of electrical signals being transduced. Sufficient electrical conductance is needed for the recognition layer of the working electrode in amperometric/voltammetric and chemiresistive detection to enhance the electron transfer.
COFs have porous and stable structures, highly accessible and programmable active sites, and tunable electrical properties. This multifunctionality makes COFs promising and attractive materials in electrically transduced detection. First, COFs can be constructed with abundant accessible analyte binding sites through reticular synthesis or post-modification. Thus, COFs can be used as independent selective layers to modify the working electrodes to endow selectivity for a working electrode.272,273 Second, COFs can be designed to have high surface areas, chemically stable skeletons, and good compatibility with other nanomaterials. Therefore, COFs can serve as excellent scaffolds to support different kinds of receptors, as electron mediators, as a conductive matrix, and/or as an electrochemically active material that can provide selective recognition ability or enhance electrochemical performance for the detection system.274–282 Thirdly, through the choice of building blocks, the method for reticular synthesis, and post-functionalization, COFs can be built with inherent electrochemical activity and conductivity. In contrast to the first two situations, COFs themselves can act as active materials to mediate redox reactions of specific analytes for electrochemical detection283 or as transducers in chemiresistive detection.91,128 Among these three aspects, using COF as scaffolds for the modification of working electrodes in an electrochemical platform is currently the most applied strategy for electrically transduced detection.
In 2014, Wang et al. reported the first example of using COF (COF-LZU1) for electrochemical sensing of biomolecules.272 The imine-linked COF-LZU1 was grown on an amino-functionalized silicon substrate, leaving a large number of edge amino groups on the surface of COF films (Fig. 23). These exposed amino groups are very helpful for adhesion of biomolecules, such as proteins or DNA, through strong electrostatic interactions. In aqueous solutions, the functional groups on biomolecules can be electrolyzed, which can promote the electrostatic interactions between biomolecules and COF films and improve the electronic charges flowing at the interfaces. The charged biomolecular species could also diffuse into the pores of COFs, resulting in a further enhancement of the electrochemical property. Consequently, EIS results demonstrated that the charge-transfer resistance (Rct) of the composite electrodes declined upon bovine serum albumin (BSA) adsorption and probe DNA immobilization, indicating that the adsorption of biomolecules onto COF films strengthened the electrochemical activity of the functional film.289,290 This study showed that COFs could be alternative materials for biosensors by taking advantage of their functional groups on surfaces and porous structures.
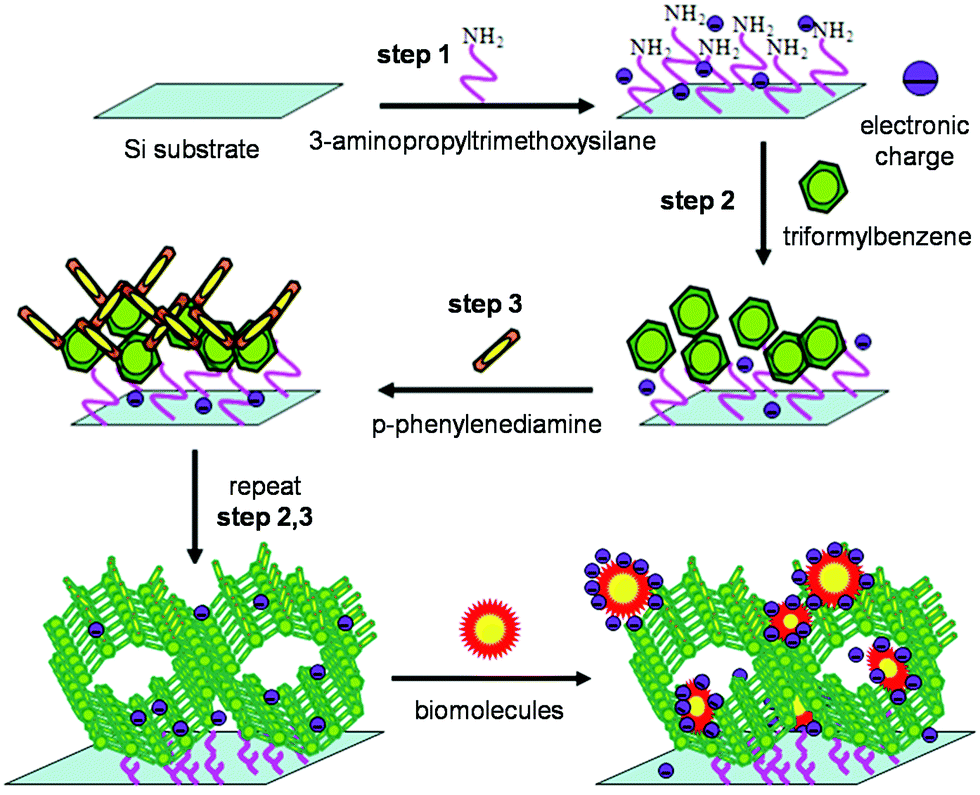 | ||
| Fig. 23 Construction of an imine-linked COF on the surface for bio-molecular detection.272 Reprinted with permission from ref. 272. Copyright 2014, John Wiley and Sons. | ||
COFs with an ordered structure, high porosity, low density, good biocompatibility, and outstanding stability291,292 can act as platforms for anchoring selectors, such as aptamer strands275,278,285 and antibodies,274,284 for detecting other biomolecules. Aptamers are single-stranded DNA or RNA oligonucleotides that can show specific affinity toward target molecules. Zhou and coworkers reported a novel multiple electrochemical aptasensor based on COF TBAPy-MA-COF-COOH for sensitive and simultaneous detection of miRNA 155 and miRNA 122.275 The nanowires of TBAPy-MA-COF-COOH were used as a platform for anchoring single-stranded DNA (ssDNA), which was further hybridized with the complementary aptamer (cApt) probes of miRNA 155 and miRNA 122. Shell-encoded Au NPs with Cu2O were used as signal labels to connect to aptamer cApts. When contacting miRNAs, the signal labels immobilized with cApts were released from the hybridized DNA complex, which resulted in a substantial decrease of the DPV peak current of the signal labels. This TBAPy-MA-COF-COOH-based aptasensor can detect miRNA 155 and miRNA 122, with ultrasensitive low detection limits of 6.7 and 1.5 fM (S/N = 3), respectively, in a wide linear range of 0.01–1000 pM.
By anchoring antibodies and aptamers as selectors to COFs, several platforms for the detection of C-reactive protein (CRP) were developed.274,276,284,285 For example, Yang and coworkers constructed a label-free CRP immunosensor using Pd(II)-coordinated COF-LZU1 (Pd/COF-LZU1)60 immobilized with anti-CRP on the surface of a glassy carbon electrode (GCE).274 When anti-CRP binds with CRP, the formed immune complex obstructs the electric signal of Fe(CN)64−/3−, which can be used to realize label-free detection of CRP by EIS and DPV. Under optimal experimental conditions, a linear range of 5–180 ng mL−1 with an LOD of 1.66 ng mL−1 was achieved.274 An ultrasensitive electrochemical immunoassay for the detection of CRP was later demonstrated by the same group by using COF-LZU8 and the Au NPs-CoS/graphene oxide (GO) nanocomposite.284 COF-LZU8 was functionalized by Au NPs and thionine (Thi) to serve as the electron transfer mediator, while the Au NPs-CoS/graphene oxide (GO) nanocomposite was utilized to immobilize the CRP antibody. Due to the high surface area of COF-LZU8, a great quantity of Thi could be adsorbed on the surface of the COF to amplify the PDV signal. Under optimum conditions, the system achieved a linear range of CRP detection of 0.05–150 ng mL−1 with an LOD of 0.016 ng mL−1. The same group also developed a sandwich CPR electrochemical immunosensor comprising Au NP functionalized COF (Au NPs@COF-TpPa-1) as the matrix to immobilize the CRP antibody and nano-mimetic enzyme Co3O4 NPs as tags to label the secondary CRP antibody.276 This immunosensor can achieve a LOD of 0.017 ng mL−1 for CRP and with good selectivity over other interferents, such as carcinoembryonic antigen (CEA), human chorionic gonadotropin (HCG), BSA, and prostate specific antigen (PSA).276 A photoelectrochemical aptasensor for the label-free detection of CRP based on a 2D porphyrinic COF (p-COF) was developed by Zhang et al.285 p-COF and aptamer modified silver NPs were used to generate a functionalized ITO/NiS electrode. CRP promoted the electron transfer between the active molecules H2O2 and the electrode, diminishing the amplification effect of the photocurrent induced by H2O2, and leading to a decrease in the photocurrent response of the modified ITO/NiS electrode. The developed aptasensor exhibited a wide linear range of 0.5–100 ng mL−1 and a LOD of 0.1 ng mL−1.
By introducing aptamers into a porphyrin-based COF (p-COF), Yan et al. developed an aptasensor that can selectively and sensitively bind epidermal growth factor receptor (EGFR) and detect human breast carcinoma MCF-7 cells.278 The label-free EGFR-targeting aptamer was immobilized onto p-COF via electrostatic interactions, π–π stacking, and hydrogen bonding (Fig. 24a). When aptamer/p-COF was exposed to EGFR solution or living MCF-7 cells, interaction via biorecognition between these strands and EGFR or cells lowered the access of the redox probe ([Fe(CN)6]3−/4−) to the surface of the modified working electrode, leading to a decrease in electrochemical signals. The decrease in electrochemical responses was successively determined by CV (Fig. 24b), DPV, and EIS (Fig. 24c). The p-COF-based aptasensor exhibited extremely low LODs of 5.64 and 7.54 fg mL−1 obtained from DPV and EIS, respectively, with a broad linear detection range of 0.05–100 pg mL−1 EGFR. Interferents, including CEA, PSA, MUC1, HER2, VEGF, IgG, PDGF-BB, and BSA, had negligible effects on EGFR detection (Fig. 24d). When detecting living MCF-7 cells, the p-COF-based aptasensor showed a LOD of 61 cells mL−1 with a linear detection range of 500–105 cells mL−1. The authors attributed the sensing performance of the p-COF aptasensor, including high selectivity, good stability, reproducibility, acceptable recyclability, and favorable applicability in human serum samples, to a few structural features of the COF: (1) the two-dimensional nanosheet structure of p-COF can produce abundant binding sites for aptamer strands or biomolecules;293 (2) the highly conjugated structure of p-COF can enhance the interaction with biomolecules, as well as improve the electrochemical activity; (3) the large channels of p-COF can facilitate aptamer immobilization and increase the amount of the probe adsorbed, further enhancing the detection sensitivity toward analytes.
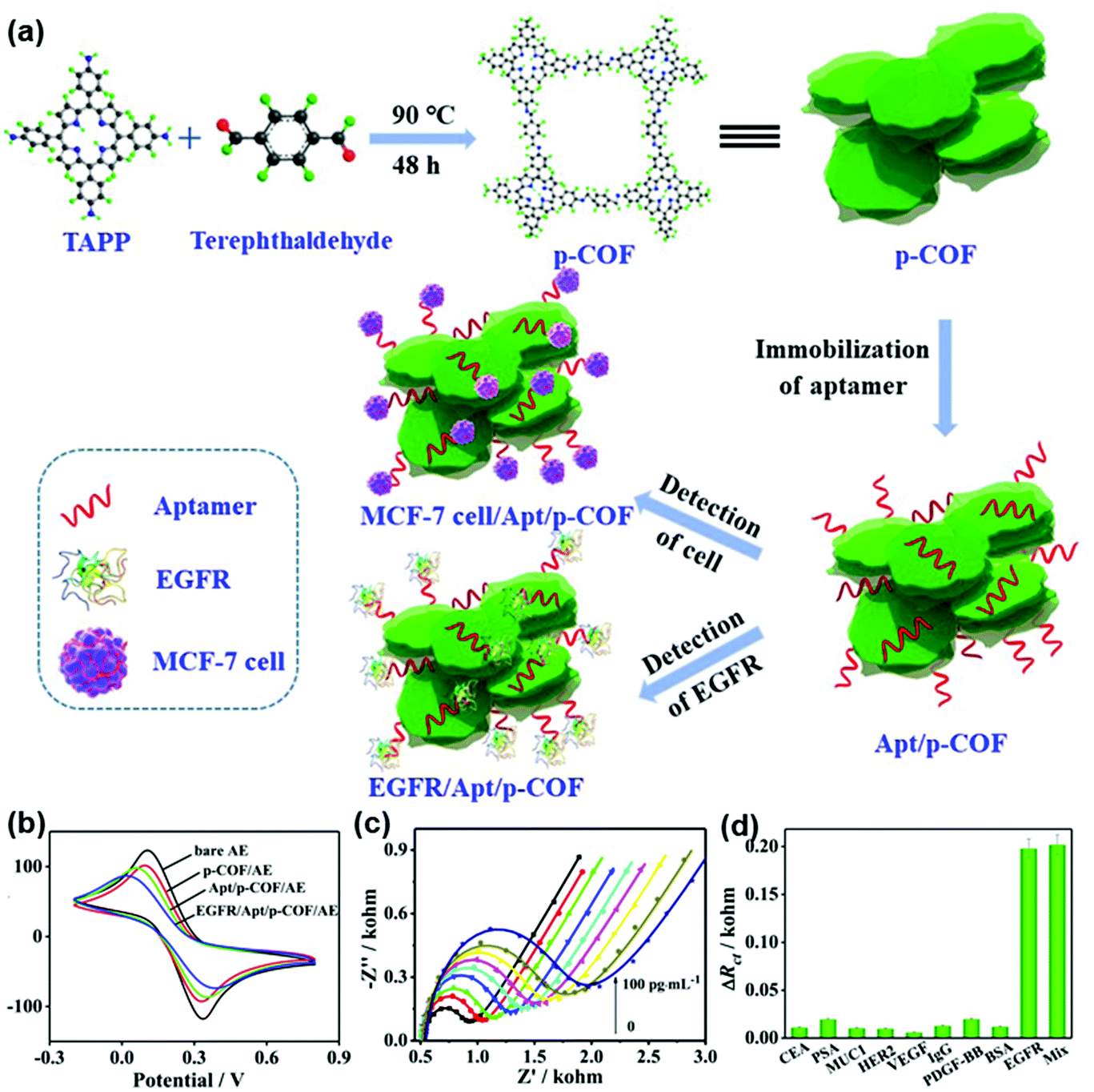 | ||
| Fig. 24 (a) Schematic diagram of the p-COF-based aptasensor for detecting EGFR or MCF-7 cells. (b) CV curves of the p-COF-based aptasensor for detection of EGFR using the Au electrode (AE), p-COF/AE, Apt/p-COF/AE, and EGFR/Apt/p-COF/AE. (c) EIS responses of Apt/p-COF/AE with different EGFR concentrations of 0, 0.05, 0.1, 0.5, 1, 5, 50, and 100 pg mL−1. (d) Change of the charge transfer impedance (ΔRct) values of the p-COF-based electrochemical aptasensor by separately adding the interferents CEA, PSA, MUC1, HER2, VEGF, IgG, PDGF-BB, and BSA with a concentration of 0.1 ng mL−1, EGFR (0.1 pg mL−1), and their mixture.278 Adapted with permission from ref. 278. Copyright 2019, Elsevier. | ||
A sandwich-type electrochemical immunosensor was constructed by Ren and coworkers for the ultrasensitive detection of cardiac troponin I (cTnI),277 the accurate and rapid detection of which can help prevent acute myocardial infarction. In this immunosensor, TiO2 NPs modified with conductive polypyrrole (TiO2-PPy) were used as the substrate material for the immobilization of the primary antibody Ab1-cTnI. Au NP doped COFs and the electron mediator toluidine blue (TB-Au-COFs) behaved as labels for achieving signal amplification (Fig. 25a). Meanwhile, TB-Au-COFs also provided active sites for the immobilization of the secondary antibody Ab2-cTnI. Square wave voltammetry (SWV) showed a linear increase of current with the increase of cTnI concentrations from 0.5pgmL−1 to 10.0ngmL−1 (Fig. 25b and c), affording a low LOD of 0.17 pg mL−1. When this immunosensor was used to test cTnI in human serum samples, good recoveries (96.0–102%) and low relative standard deviations (1.8–3.9%) were achieved.277
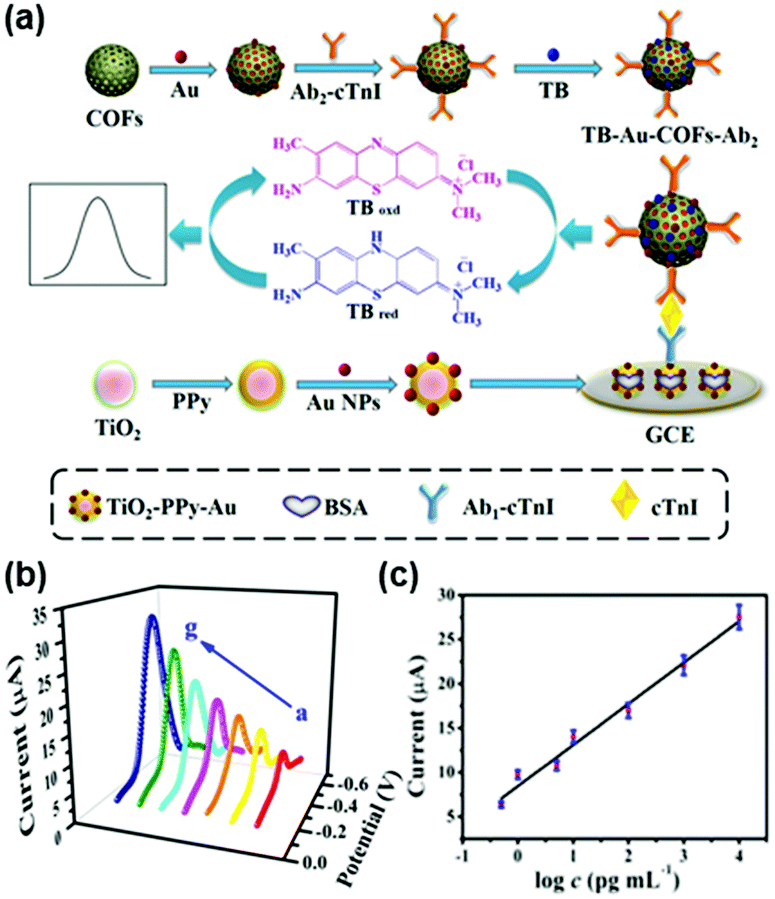 | ||
| Fig. 25 (a) Schematic illustration of the sandwich-type electrochemical immunosensor. (b) SWV current responses with increasing cTnI concentrations from 0.5pgmL−1 to 10.0ngmL−1. (c) Calibration curve of the immunosensor for the detection of different concentrations of cTnI.277 Adapted with permission from ref. 277. Copyright 2018, Elsevier. | ||
Besides the detection of DNA,272 RNA,275 and proteins274,277,278,284,285 demonstrated above, COFs have also been investigated for the detection of antibiotics279,280 and drug molecules.281,282,286 In those studies, without exceptions, COFs have been carefully modified with various functional nanomaterials including MOFs,279,286 Au NPs,281 and MWCNTs,282 and/or connected with selectors, such as aptamer280,286 and MIP,282 to form a compositional material. Usually, the large surface area and abundant functional groups of COFs, the electrochemical activities or catalytic abilities of the nanoparticles, and/or selective binding ability from the selectors can function conjointly to result in a synergistic effect to improve the detection performance of the compositional material which the components in isolation are not able to achieve. For example, Au NPs are one kind of widely used nanomaterials in electrochemical sensors due to their unique catalytic activity; however, free Au NPs tend to agglomerate which is detrimental to their catalytic performance. TAPB-DMTP-COFs can act as the host matrix to support Au NPs with the unsaturated amine group present on TAPB-DMTP-COF, which can effectively avoid the agglomeration of Au NPs.281 The electrochemical detection using the Au NP modified COF composite (TAPB-DMTP-COF/Au NPs) exhibited a good electrocatalytic activity toward the oxidation of chlorogenic acid (CGA)294,295 and displayed a wide linear range of 1.0 × 10−8–4.0 × 10−5 mol L−1, a low LOD of 9.5 × 10−9 mol L−1 as well as a good repeatability of 4.1% in 2.0 × 10−5 mol L−1 CGA.281 Compared with Au NPs, the improved stability of the system resulted in excellent durability with a cycling time of more than 100.281
COFs developed thus far experienced limited utility as electrode materials in electrochemical analysis, due to their low conductivity resulting in low current output. Combining COFs with a conductive matrix296,297 provides a practical strategy to overcome the conductivity issues of COFs to improve their electrochemical performance.298 Xu and coworkers used a composite comprising conductive amino-functionalized carbon nanotubes and COFTTA–DHTA (NH2-MWCNT@COFTTA–DHTA), as well as molybdenum disulfide (MoS2) to modify the GCE as a novel electrochemical sensor for selective recognition of sulfamerazine (SMR).282 A molecularly imprinted polymer (MIP) membrane was anchored on the surface of this modified GCE further to achieve selective recognition of SMR. The electrochemical performance of the assembled electrochemical sensor was investigated with DPV and CV. This sensor displayed excellent selectivity for SMR and good reproducibility. Under optimal conditions, the sensor offered a wide current response for SMR from 3.0 × 10−7 to 2.0 × 10−4 mol L−1, with a LOD of 1.1 × 10−7 mol L−1. Furthermore, this developed sensor was successfully applied for the determination of SMR in pork and chicken samples with recoveries of 86.0–102.0%; the results were comparable to those obtained by high-performance liquid chromatography.282
COF-based conductive composites can be fabricated into electrodes for electrochemical detection of metal ions. For instance, Wang and coworkers developed an electrochemical sensor for the sensitive and selective determination of Pd2+ in an aqueous medium using a carbon paste electrode by mixing TAPB-DMTP-COF with graphite.299 Pd2+ was detected by differential pulse anodic stripping voltammetry. Under optimum conditions, the method showed excellent linearity to the concentration of Pd2+ in the range of 0.0050 to 2.0 μmol L−1 with a LOD of 1.9 nM. The extraordinary performance was ascribed to the high specific surface of TAPB-DMTP-COF that provided a high effective electroactive surface area and the abundant terminal amino groups at the edges of the COF that had coordination capability to Pd2+. Sulfhydryl functionalized COF modified Au electrodes were fabricated by Wang et al. for the selective detection of Pd2+ in water samples.287 The sulfhydryl groups were introduced by post-synthetic modification to enhance the selectivity for Pd2+. A LOD of 0.015 ng mL−1 was obtained, lower than the level of Pd2+ in drinking water permitted by the WHO (10 ng mL−1). Metal ions, such as Mg2+, Ni2+, Zn2+, Cr2+, Fe2+, Cu2+, Cd2+, Hg2+, and Ca2+, have a negligible influence on Pd2+ detection.287
Utilizing the affinity of the boron ester linkage towards water molecules, Pal and coworkers developed a truxene-based boron ester COF (COF-TXDBA) for the measurement of relative humidity (RH) using EIS.273 The condensation reaction between the truxene based ligand and 1,4-phenylenediboronic acid (DBA) gave a crystalline COF with boron ester linkages. The powder X-ray diffraction experiment indicated that boron ester linkages interacted with water molecules in the planar sheets of COF backbones with the empty orbitals of the boron atom interacting with the electron lone pair of the oxygen atom in the water molecule. Under low RH, water molecules were adsorbed on the surface of COF-TXDBA to form the first chemisorbed layers, which were discontinuous under low RH, thus resulting in high impedance. However, under high RH, a multilayer of water molecules can form, which favors the charge transportation process by the Grotthuss chain reaction (H2O + H3O+ → H3O+ + H2O), and results in a sharp increase in the proton conductivity of the sensor.300 This COF-TXDBA based RH sensor exhibited a reversible change of 3 orders of magnitude in impedance in the RH range of 11–98% with a response and recovery time of 37 s and 42 s, respectively.
Redox-active materials can usually serve as redox mediators to couple with the redox reactions of electrochemically active analytes. The redox potential of analyte species can then be brought to moderate or low values, which will be easier to access using normal amperometric and voltammetric testing conditions. The possible signals from redox-active interference species or electrolytes can also be minimized. To serve as an electron mediator in electrochemical detection, COFs have largely relied on the external electrochemically active components, such as redox-active organic molecules277,284 and Au NPs.281 The intrinsic redox activity of COFs has been less utilized. However, incorporating redox-active ligands can be used as a strategy to generate COFs with multiple redox processes,74,301–307 which could be used as active materials to catalyze specific substances and to construct ratiometric electrochemical sensors. Wang and coworkers reported a COF (COFDHTA-TTA) with electroactive monomers 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and 2,5-dihydroxyterethaldehyde (DHTA) as multiple redox-active sites as a novel electrochemical sensing and biosensing platform (Fig. 26a).283 COFDHTA-TTA itself can be directly used as an active material in a ratiometric electrochemical sensor for the determination of hydrogen peroxide and pH level based on current and potential signals. The ratio of current at −0.5 and 0.3 V linearly increased with H2O2 concentration from 8.06 to 400 μM, and the LOD was estimated to be 2.42 μM (Fig. 26b). The DPV response of COFDHTA-TTA/GCE in the presence of interfering chemicals, including glucose, galactose, mannose, fructose, Cl−, K+, UA, and AA, had only slight effects on H2O2 detection (Fig. 26c). As the electron transport process of COFDHTA-TTA/GCE was accompanied by H+ transfer, pH could be also determined. The peak potential at around −0.15 V linearly shifted as the pH of PBS (phosphate buffered saline) solution varied from 11.0 to 3.0 with a high sensitivity of 64.2 mV pH−1 (Fig. 26d). Because COFDHTA-TTA had a good catalytic activity for oxygen reduction reaction in PBS (pH = 7.0), glucose oxidase was further loaded on COFDHTA-TTAvia its nanosized pores and abundant active sites from the N-rich skeleton for glucose sensing. The glucose oxidase-COFDHTA-TTA system exhibited linear responses to glucose from 1.26 μM to 6.0 mM at −0.30 V, as well as from 0.60 μM to 6.0 mM at −0.53 V (Fig. 26e). This work presented an example to construct electrochemical sensors by directly using redox-active COFs as electroactive materials.283
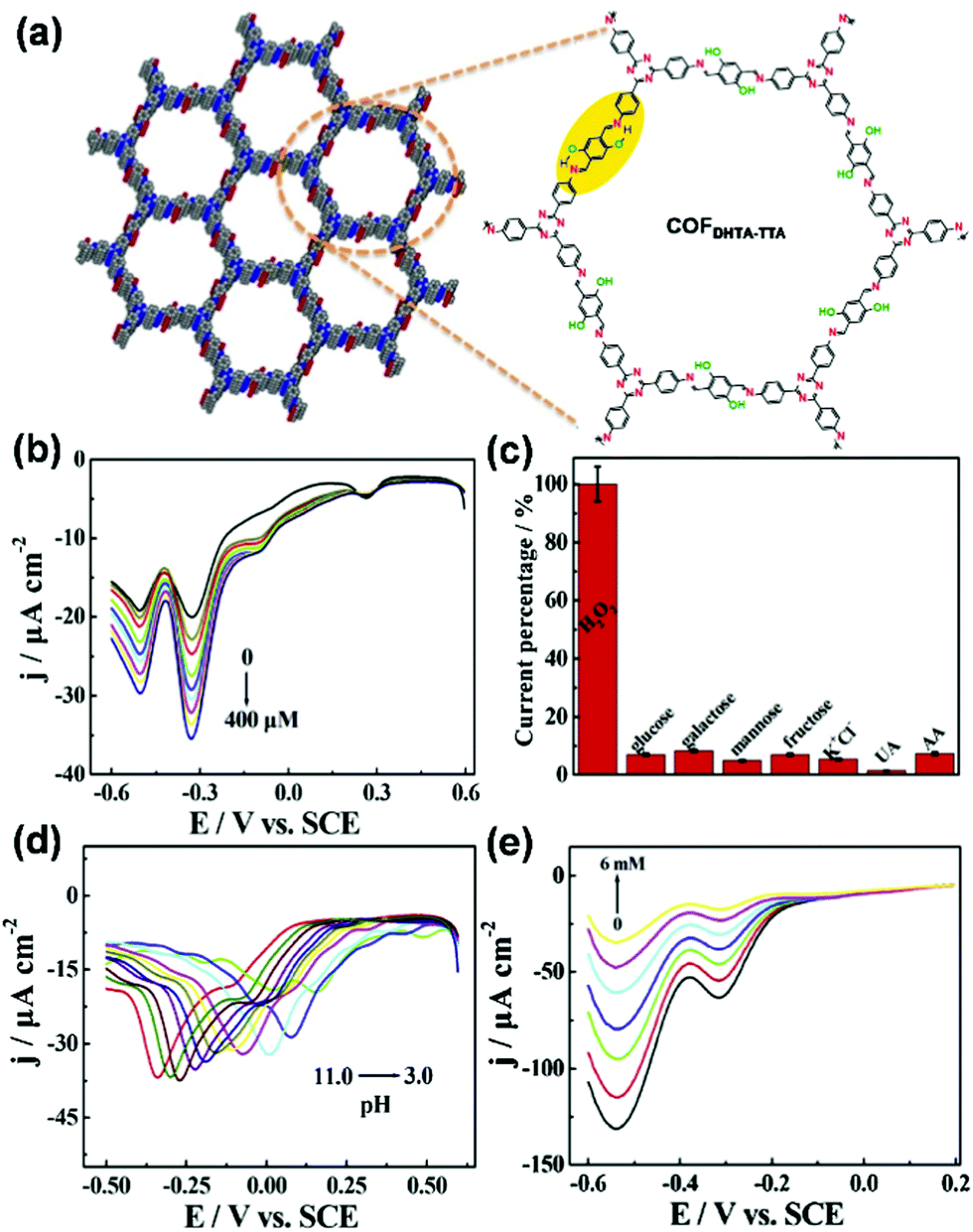 | ||
| Fig. 26 (a) Chemical model and structure of COFDHTA-TTA. (b) DPV responses of COFDHTA-TTA/GCE in 0.2M N2-statured PBS (pH = 7.0) toward different H2O2 concentrations. (c) DPV response of COFDHTA-TTA/GCE toward glucose, galactose, mannose, fructose, Cl−, K+, UA, and AA in 0.2M N2-saturated PBS (pH = 7.0). (d) DPV response of COFDHTA-TTA/GCE in 0.2M N2-saturated PBS with different pH. (e) DPV responses of GOD/COFDHTA-TTA/GCE in 0.2M O2-statured PBS (pH = 7.0) with constant addition of glucose.283 Adapted with permission from ref. 283. Copyright 2019, Elsevier. | ||
As demonstrated in the previous sections, COFs are appealing candidates for the fabrication of devices for chemical detection in the context of their large surface-to-volume ratio, chemically robust skeleton, and programmable functional active sites for analytes. However, significant challenges arise when utilizing them directly in electrically transduced detection as stimuli-responsive electrodes because most COFs have insufficient electrical conductivity to be integrated into electronic devices. Several conductive COFs, however, have been recently developed91–93,308–311 by taking inspiration from the development of other classes of conductive materials including conductive organic polymers,312,313 conductive organic molecular solids,314–316 and conductive metal–organic frameworks.317–319 The construction of conductive COFs usually follows the basic design principles of maximizing the through-bond and through-space charge transport by employing electroactive building units, conjugated linkages, stacked structures, and chemical doping.91–93,125,262,308–311,320–322 These developments of COFs with moderate to good electrical conductivity78,91,92,125,309–311,320,322,323 provide new opportunities for electrically transduced detection.
In 2019, our research team constructed a conductive COF (COF-DC-8) through the aromatic annulation of 2,3,9,10,16,17,23,24-octa-aminophthalocyanine nickel(II) and pyrene-4,5,9,10-tetraone and demonstrated the first implementation of a conductive COF as the active material for chemiresistive detection (Fig. 27a).91 The intrinsic bulk conductivity of the COF material reached 2.51 × 10−3 S m−1. This good electrical conductivity, together with the ability of the built-in nickelphthalocyanine units to interact with a wide range of analytes, made this COF a promising candidate for chemiresistive gas detection. Upon integration into chemiresistive devices, COF-DC-8 showed excellent responses to various reducing and oxidizing gases, including NH3, H2S, NO, and NO2 (Fig. 27b–e). COF-DC-8 was found to be especially sensitive toward oxidizing gases, with pronounced responses observed for NO and NO2, which was consistent with the p-type characteristic of the COF. The LODs for these gases reached the parts-per-billion (ppb) level, which was 70 ppb for NH3, 204 ppb for H2S, 5 ppb for NO, and 16 ppb for NO2, based on response values at 1.5 min exposure. Electron paramagnetic resonance spectroscopy and X-ray photoelectron spectroscopy studies suggested that the chemiresistive response of COF-DC-8 involved charge-transfer interactions between the analyte and the nickelphthalocyanine component of the framework.91 This study demonstrated the high potential of using conductive COFs in chemiresistive detection.
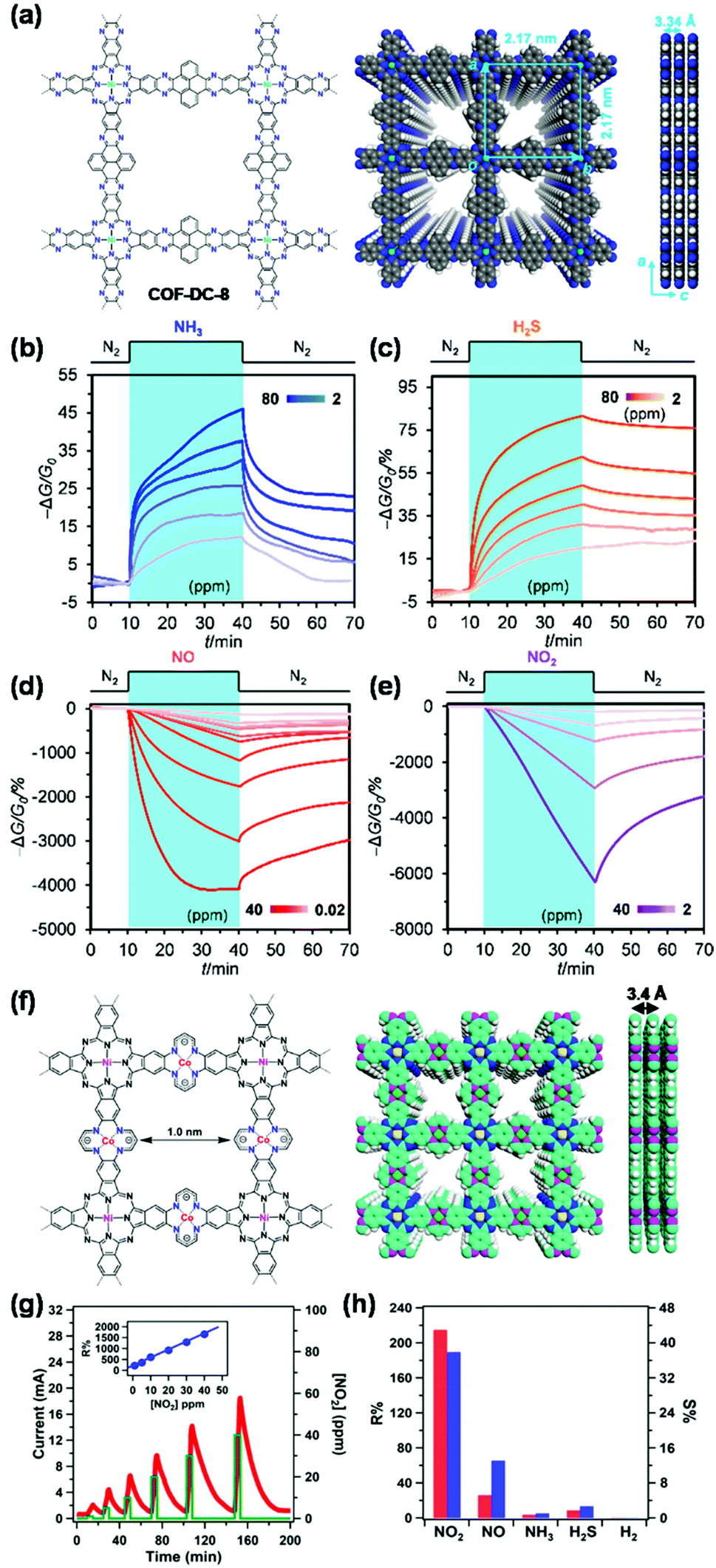 | ||
| Fig. 27 (a) Chemical structure and model of COF-DC-8. Chemiresistive responses of devices with integrated COF-DC-8 to (b) NH3, (c) H2S, (d) NO, and (e) NO2 under an applied voltage of 1.0 V and in an atmosphere of dry nitrogen.91 Adapted with permission from ref. 91. Copyright 2019, American Chemical Society. (f) Chemical structure and model of NiPc-CoTAA. (g) Time-dependent response–recovery current plot of the NiPc-CoTAA film at NO2 concentrations from 1 to 40 ppm. The inset shows the linear relation between R% and NO2 concentration. (h) Responsivity and sensitivity of the NiPc-CoTAA film towards different gases NO2, NO, NH3, H2S, and H2 at 1 ppm (red bar, R%; blue bar, S%).288 Adapted with permission from ref. 288. Copyright 2021, John Wiley and Sons. | ||
Recently, Huang and coworkers developed a new kind of conductive phthalocyanine-based framework NiPc-CoTAA for NO2 detection (Fig. 27f).288 NiPc-CoTAA featured a conjugated tetraaza[14]annulene linkage and densely stacked π-units, which rendered this material with a low band gap of 0.86 eV and high electrical conductivity up to 8.16 × 10−3 Sm−1. The thin film device of NiPc-CoTAA made by the steam-assisted conversion method exhibited significant sensitivity towards several redox gases, including NO2, NO, NH3, H2S, and H2. Upon exposure to NO2 gas at concentrations of 1–40 ppm at 298 K, the NiPc-CoTAA thin-film device exhibited a high response up to 1658% with good linearity (Fig. 27g). NiPc-CoTAA exhibited a superior NO2 sensing selectivity to that of NO, NH3, H2S, and H2 at 298 K (Fig. 27h). These results demonstrated that the NiPc-CoTAA film not only exhibited high sensitivity, but also good selectivity as a chemiresistive sensor.288
As the analyte–material interaction can result in changes of both optical and electrical properties of COFs, it is possible to realize different types of transduction modes by using the same COF detection system. Bojdys and coworkers reported a 2D COF PBHP-TAPT made up of chemoresistant β-amino enone bridges (PBHP) and Lewis-basic triazine moieties (TAPT), which was capable of real-time, reversible sensing of volatile acids and bases through both the optical and conductivity changes of the material.128 The study showed that a single-site protonation occurred at the triazine moiety upon the exposure to acid. This protonation gave the triazine moieties a localized positive charge, drawing π-electron density from the neighboring functional groups of triazine units, which caused a fluorescence quenching of the COF accompanied by a color change from yellow to red. Interestingly, besides optical changes, the acid–base exposure cycle led to a reversible conductivity change of the COF. PBHP-TAPT had a normalized conductivity of 1.32 × 10−8 S m−1, which upon protonation was increased by 170-fold to 2.18 × 10−6 S m−1. The conductivity dropped close to the original value when the sample was regenerated with NH3 vapors. Solid-state UV-vis diffuse reflectance spectroscopy showed that the bandgap of the PBHP-TAPT COF decreased by ∼0.3 eV upon protonation. This study illustrated that both the optical and electronic property change of a COF can be used for chemical detection. Taking full advantage of the porous structure and the fully aromatic donor–acceptor conjugation of COFs is a powerful approach to design practical sensors and switches.128
COFs have shown good potential in electrically transduced detection in various biology-related analytes, metal ions, humidity, pH, and gases (Table 4). Several platforms have been utilized for real sample detection, showing excellent sensitivity and selectivity.276–278,282,287 Those achievements capitalize on the intriguing features of large surface area, abundant active binding sites, and feasible functionalization ability. Despite the encouraging progress, the use of COFs for electrically transduced detection is still at a very early stage and there are still several impending challenges in making use of the full potential of COFs in electrically transduced detection. It is worth noting that most of the current detection systems relied heavily on the construction of composite materials for the improvement of sensing performance. The fabrication of these composites usually needs tedious engineering procedures that involve multiple components. The resulting composites may be challenged in long-term stability that can be detrimental to the detection performance.324 To date, there is still no report on which COFs are used solely as electrode materials for electrochemical detection. One major reason for this is that many COFs have low electrical conductivity. The combination with conductive additives provides a solution for this issue;282,299 however, it may be at the expense of device simplicity and stability. Using composite materials may also result in the reduction of surface area, the shielding of analyte binding sites, as well as the interference of redox-active components embedded within the COF, leading to unpredictable changes in the detection process.319 Compared with the use of COF composites, the epitaxial growth of COFs on conductive substrates may provide a better alternative for obtaining stable electrode materials with improved electrochemical performance.298,302,325,326 Last, but not least, the development of COFs that have an elegant combination of high electrical conductivity, specific active binding sites for the target, as well as redox-active units is expected to lead to advanced applications of this group of multifunctional materials in electrically transduced detection.
| Analyte(s) | Method | COF | Detection architecture | LOD | Detection range | Other detection parameters | Ref. |
|---|---|---|---|---|---|---|---|
| BSA | Electrochemistry: EIS | COF-LZU1 | COF-LZU1/Si | N/A | N/A | 0.1 M KCl | 272 |
| DNA | Electrochemistry: EIS | COF-LZU1 | COF-LZU1/Si | N/A | N/A | 0.1 M KCl | 272 |
| CRP | Electrochemistry: EIS, DPV | COF-LZU1 | Pd-COF-LZU1-anti-CRP/GCE | 1.66 ng mL−1 | 5–180 ng mL−1 | 0.01 M PBS, pH = 7.5; selectivity over BSA, HCG, Gly, Glu | 274 |
| H2O | Electrochemistry: EIS | COF-TXDBA | COF-TXDBA/Ag–Pd electrode | N/A | 11% RH and 98% RH | Response time 37 s, recovery time 42 s, reversible | 273 |
| CRP | Electrochemistry: DPV | COF-LZU8 | Thi-Au NPs-COF-LZU8 | 0.016 ng mL−1 | 0.05–150 ng mL−1 | N/A | 284 |
| CRP | Electrochemistry: EIS, CV | COF-TpPa-1 | Au NPs@COF-TpPa-1/chitosan/GCE | 0.017 ng mL−1 | 0.05–80 ng mL−1 | PBS buffer, pH = 7.5; selectivity over CEA, HCG, BSA, PSA | 276 |
| OTC | Electrochemistry: EIS, CV | MCA | Ce-MOF@MCA/aptamer/AE | 35 fM | 2 × 10−4–1.0 nM | 0.1 M PBS, pH = 7.4; selectivity over NO3−, Cl−, Ca2+, Mg2+, L-alanine, DL-phenylalanine, AMP, bleomycin, doxycycline hyclate, streptomycin sulphate, Kana, UA, and urea; milk, wastewater, and urine samples | 286 |
| CRP | Photoelectrochemical | p-COF | NiS and Ag NPs-p-COF-CRP aptamer/ITO | 0.1 ng mL−1 | 0.5 to 100 ng mL−1 | 0.1 M PBS, pH = 7.4; selectivity over HCG, PSA, MC-LR, BSA, HCG; serum | 285 |
| Chlorogenic acid | Electrochemistry: CV, DPC | TAPB-DMTP-COF | TAPB-DMTP-COF/Au NPs/GCE | 9.5 × 10−9 M |
1.0 × 10−8–4.0 × 10−5molL−1 |
PBS, pH = 7.0; Fe3+, Na+, K+, Ca2+, Cu2+, Zn2+, Mg2+, PO43−, NH4+, Cl−, and SO42−, uric acid, dopamine, guanine, L-dopa, glucose, hydroquinone, creatine, adenine, and catechol, AA, thymol, rutin and quercetin, caffeic acid, gallic acid, and vanillic acid; food and drink samples |
281 |
| Pd2+ | Electrochemistry: CV, DPASV | APB-DMTP-COF | APB-DMTP-COF/CPE | 1.9 nM | 0.0050–2.0 μM | Acetate buffer solution, pH = 4.5; selectivity over Cu2+, Hg2+, Cd2+, Ni2+, Co2+, Mn2+, Fe2+, Zn2+, Cr3+, Sn2+, Ag+, Mg2+, Al3+, K+, Na+, Mg2+, Ca2+, NO3−, F−, Cl−, Br−, SCN−, SO42−, CO32−, PO43−, SiO32−; real water samples | 299 |
| cTnI | Electrochemistry: SWV | PPy | TiO2-PPy-Au/GCE | 0.17pgmL−1 |
0.5 pg mL−1–10.0 ng mL−1 | 0.1 M PBS, pH = 7.4; selectivity over insulin, CEA, PSA, and squamous cell carcinoma antigen; human serum samples | 277 |
| miRNAs | Electrochemistry: DPV | TBAPy-MA-COF | TBAPy-MA-COF NWs/Au NPs/aptamer/GCE | 6.7 fM (miRNA 155) 1.5 fM (miRNA 122) | 0.01–1000 pM | PBS, pH = 7.4; selectivity over miRNA 122, 141, 205, 21; human serum | 275 |
| Dopamine | Electrochemiluminescence | TpBD | MIP/UCNPs/CTpBD-Au/GC | 2 × 10−15 M | 10−14–10−6 M | 0.1 M PBS, pH = 7.4; selectivity over adrenaline, PEA, caffeic acid, UA, AA, Ca2+, NH4+, K+, Na+, SO42−, NO3−, Cl−; human serum | 327 |
| AMP | Electrochemistry: EIS | TPN-COF | apt/Co-MOF@TPN-COF/AE | 0.217fgmL−1 |
0.001–2000pgmL−1 |
0.01M PBS, pH = 7.4; ADR, TOB, RFP, Kana, PSA, BSA, Ca2+, and Na+; human serum, river water and milk |
279 |
| Sulfamerazine | Electrochemistry: DPV | N/A | NH2-MWCNT@COF/GCE | 1.1 × 10−7 M |
3.0 × 10−7–2.0 × 10−4 M |
0.2M Britton–Robinson buffer solution, pH = 5.0; selectivity over glucose, glutamic acid, ascorbic acid; meal samples |
282 |
| ENR | Electrochemistry: EIS | Py-M-COF | AptENR/Py-M-COF/AE | 6.07 fg mL−1 | 0.01pgmL−1–2ngmL−1 |
0.01M PBS, pH = 7.4; selectivity over tetracycline, Kana, TOB, Na+, K+, streptomycin, and OTC |
280 |
| AMP | Electrochemistry: EIS | Py-M-COF | AptAMP/Py-M-COF/AE | 0.04 fg mL−1 | 0.01pgmL−1–2ngmL−1 |
0.01M PBS solution, pH = 7.4; selectivity over tetracycline, Kana, TOB, Na+, K+, streptomycin, and OTC |
280 |
| Pb2+ | Electrochemistry: SWASV | TAPB-BMTTPA-COF | TAPB-BMTTPA-COF/AE | 0.015ng mL−1 |
0.05–20ng mL−1 |
Acetic acid/sodium acetate buffer solutions, pH = 5.0; selectivity over +, Ni2+, Zn2+, Cr2+, Fe3+, Cu2+, Cd2+, Hg2+, Ca2+, Na+ and K+; environmental waters | 287 |
| H2O2 | Electrochemistry: CV, DPV | COFDHTA-TTA | COFDHTA-TTA/GCE | 1.70–2.42 μM | 5.66–400 μM | 0.2 M N2-statured PBS, pH = 7.0; selectivity over galactose, mannose, fructose, Cl−, K+, UA and AA | 283 |
| H+ | Electrochemistry: CV, DPV | COFDHTA-TTA | COFDHTA-TTA/GCE | N/A | pH 3–11 | 0.2 M N2-satruated PBS | 283 |
| Glucose | Electrochemistry: CV, DPV | COFDHTA-TTA | Glucose oxidase GOD/COFDHTA-TTA/GCE | 0.18–0.38 μM | 0.60 μM –6.0 mM | 0.2 M N2-statured PBS, pH = 7.0; selectivity over galactose, mannose, fructose, Cl−, K+, UA and AA | 283 |
| EGFR | Electrochemistry: DPV, EIS | p-COF | apt/p-COF/AE | 5.64 fg mL−1 (DPV) | 0.05–100 pg mL−1 | PBS, pH = 7.4; CEA, PSA, MUC1, HER2, VEGF, IgG, PDGF-BB, and BSA | 278 |
| 7.54 fg mL−1 (EIS) | |||||||
| MCF-7 cells | Electrochemistry: EIS | p-COF | apt/p-COF/AE | 61 cells mL−1 | 500–105 cells mL−1 | 0.1 M PBS, pH = 7.4; selectivity over L929 cells | 278 |
| HCl | Chemiresistor | PBHP-TAPT | PBHP-TAPT pellet | N/A | N/A | Reversible on exposure to NH3 | 128 |
| NH3 | Chemiresistor | COF-DC-8 | COF-DC-8/AE | 56.8–70 ppb | 2–80 ppm | N2 | 91 |
| H2S | Chemiresistor | COF-DC-8 | COF-DC-8/AE | 121 ppb | 2–80 ppm | N2 | 91 |
| NO | Chemiresistor | COF-DC-8 | COF-DC-8/AE | 1–5 ppb | 0.02–40 ppm | N2 | 91 |
| NO2 | Chemiresistor | COF-DC-8 | COF-DC-8/AE | 1–16 ppb | 2–40 ppm | N2 | 91 |
| NO2 | Chemiresistor | NiPc-CoTAA | NiPc-CoTAA thin film/AE | N/A | 1–40 ppm | N2, sensitivity (%) 37.6 ppm−1, selectivity over NO, NH3, H2S, and H2 | 288 |
2.4 Detection based on extraction coupled with chromatography and spectrometry
Therefore, prior to instrumental analysis of trace-level analytes in complex samples, sample preparation is an inevitable part of the extraction and enrichment of target analytes from a sample matrix.330 Extraction techniques are promising tools in sample preparation for separation and enrichment. Solid-phase extraction (SPE), which is based on the partition equilibrium of the analytes between the sample and the solid absorbent,331 can enrich and purify targets simultaneously. It has become the most widely applied extraction technique because of its high extraction efficiency, simple operation, good reproducibility, and low required consumption.24,331 Up to now, a series of SPE methods have been developed, including dispersive microsolid-phase extraction, magnetic solid-phase extraction, and solid-phase microextraction. In microsolid-phase extraction, the suspension of nanomaterial adsorbents is directly mixed into the aqueous sample and dispersed by ultrasonication, and then adsorbents are separated from the sample solution by centrifugation or filtration.332 Magnetic solid-phase extraction (MSPE) is based on the universal dispersion of a magnetizable or magnetic material as the sorbent.333 In MSPE, the solid phase can be easily separated under a magnetic field, which simplifies the preparation steps. Solid-phase microextraction (SPME) involves the use of a fused silica or metal fiber substrate covered with an appropriate sorbent coated thin layer, which can extract different kinds of volatile and non-volatile analytes from the gas or liquid phase.334,335 SPME is a non-exhaustive sample pretreatment and can shorten the time of extraction, improve efficiency and enhance sensitivity due to the integration of sampling, analyte or matrix isolation, and enrichment into a single step.336
In SPE, elimination of backgrounds can be achieved by partitioning the desired analytes from complex samples to the surface of the SPE sorbent. This partition is kinetically or thermodynamically driven by the physiochemical properties of the SPE sorbent. Therefore, the composition and structure of the adsorbent material play an important role in extraction which determines the selectivity and sensitivity of the analysis.337 Polymeric sorbents are often used; however, they may suffer from irreversible sorption, ineffective transportation of analytes, and swelling, which can lead to slow adsorption/desorption kinetics and impair selective adsorption.338–340 Many new coatings have been developed, such as ionic liquid/polymeric ionic liquids,341 molecularly imprinted polymers,342 carbon nanotubes (CNTs),343 graphene,344 and metal/metal oxide nanoparticles.345 However, these materials still have their limitations, such as low adsorption capacity, limited stability in aqueous samples with low or high pH, and low selectivity.
Significant efforts have been devoted to the development of advanced sorbent materials with improved selectivity or specificity towards target analytes, high sorptive capacity, and enhanced chemical and physical-mechanical stability. Currently, there is a trend toward the exploration of crystalline and porous materials for SPE, for example, MOFs, to realize improved sensitivity, selectivity, and repeatability of sample preparation techniques. Still, many MOFs have insufficient chemical stability in solvents, which can result in a decrease of the adsorption capacity after prolonged exposure to the solution of samples, hindering their further applications in SPE.346,347
COFs, as novel porous materials, have shown their advantages in solid-phase extraction. First, covalent organic frameworks have permanent porosity, well-defined pore structures, and high surface areas, which are beneficial in analyte uptake. The programmable pore parameters (size and geometry) can be harnessed to realize the size exclusive effect for selective extraction. Second, the modular synthesis and feasible functionalization ability of COFs allow the introduction of abundant functional groups, which can further enhance the adsorption efficiency and selectivity. Third, COFs can be designed with excellent thermal and chemical stability. Compared with their analogs, such as MOFs,346,347 microporous polymers,348 and hypercrosslinked polymers,349 COFs have been proven to be more stable in water and acidic media. Thus, COFs can be utilized in SPE under harsh conditions but still show good long-term stability. Fourth, COFs are usually made of light elements, such as H, B, C, N, and O, which endow them with processability and feasibility to be integrated with the silica or metal fiber substrate in solid-phase microextraction or to form membranes.350 COFs have been utilized as absorbents in dispersive microsolid-phase extraction, MSPE, and solid-phase microextraction for the detection of industrial chemicals,351–376 pesticides and insecticides,377–382 chemical warfare agents,24 ions,383 drug molecules,384–390 and biomolecules391,392 in safety inspection of water and food, environmental monitoring, and analysis of biological mixtures. In most cases, the detection is performed by coupling solid-phase extraction with HPLC, GC, MS, or chromatography–mass spectrometry combinations to enable quantitative analysis.
Aromatic dyes. Dyes are coloring compounds that are widely used in the fabrication of fibers; however, their residues in food or textiles are strictly regulated. In 2005, Zhang et al. demonstrated the utilization of a crystalline hydrazone linked covalent organic polymer (HL-COP) as an adsorbent in micro-solid-phase extraction for online analysis of trace Sudan dyes in food samples (Fig. 28a).351 This HL-COP adsorbent contained abundant phenyl rings and imine (–C
N) groups with rich π electrons. HL-COP showed remarkable preconcentration ability for six kinds of Sudan dyes with enrichment factors as high as 305–757. The adsorption mechanism may involve π–π affinity, hydrophobic effect, hydrogen bonding, and donor–acceptor interaction, which contributed to its strong recognition affinity to target compounds. The adsorption amounts by HL-COP were 1.0–11.0 fold those by three commonly used commercial adsorbents, including multiwall carbon nanotubes, graphene, and C18 silica gel (Fig. 28b). When HL-COP was applied as an adsorbent of online micro-solid-phase extraction coupled with HPLC for enrichment and analysis of trace Sudan dyes in food samples, low LODs of 0.03–0.15 μg L−1 could be achieved. The method was successfully applied for online analysis of chili powder (Fig. 28c) and sausage samples with recoveries in the range of 75.8–108.2% and 73.8–112.6%, respectively. These results suggested that COFs can serve as promising sample preparation media for sampling, enrichment, and separation purpose in real sample analysis when coupled with the chromatography technique.
 | ||
| Fig. 28 (a) The HL-COP absorbent packed online micro-solid-phase extraction-HPLC system. (b) Extraction performance of HL-COP adsorbents for six Sudan dyes compared with three commonly used commercial adsorbents. (c) Chromatograms for the analysis of the chilli powder: (i) the mixed standard solution of six Sudan dyes (10.0 μg L−1); (ii) direct injection of the extracted solution of chilli powder; (iii) online enrichment of extracted solution of chilli powder; (iv) online enrichment of the extracted solution for the spiked chilli powder sample with 10.0 μg kg−1 of six Sudan dyes. Peaks 1–6 are Sudan Red G, Sudan I, Sudan II, Sudan III, Sudan Red 7B, and Sudan IV, respectively.351 Adapted with permission from ref. 351. Copyright 2015, Elsevier. | ||
Polycyclic aromatic hydrocarbons (PAHs). PAHs are well-known environmental pollutants with strong carcinogenic and endocrine-disrupting properties.393 It is of great importance to monitor and control the amount of PAHs in the environment. A few detection methods that combine SPE355,372 or MSPE353,358,363,365 using COF-LZU1,353 TpBD,358,365,372 COF-SCU1,355 and TpDA363 with HPLC and GC have been established. The high extraction efficiency of these COFs can be usually attributed to strong π stacking and hydrophobic interactions between COFs and polycyclic aromatics. For example, Wang et al. used a COF LZU-1 based magnetic adsorbent for MSPE of PAHs.353 COF LZU-160 was covalently immobilized onto polyethyleneimine-functionalized magnetic nanoparticles (COF-LZU1@PEI@Fe3O4), which allowed rapid separation from the sample solution with the help of a magnetic field. COF-LZU1@PEI@Fe3O4 displayed high extraction efficiency for PAHs such as pyrene, benzo[a]pyrene, fluoranthene, benz[a]anthracene, benzo[a]fluoranthene, and dibenz[a,h]anthracene, attributed to strong π stacking and hydrophobic interaction between COFLZU-1 and polycyclic aromatics. PAHs were then quantified by HPLC after desorption with acetonitrile. The MSPE-HPLC method showed low LODs of 0.2–20 pg mL−1, wide linear ranges, and good reproducibility (relative standard deviations <4.4%). The method was successfully applied to determine PAHs in environmental samples with recoveries ranging from 90.9 to 107.8% for water samples and 85.1 to 105.0% for soil samples.
To realize the efficient and durable separation of the adsorbents and sample solution, Pang et al. reported the use of COF TpBD grown onto stainless steel wire as an SPME fiber, which was used as the solid-phase microextraction fiber to extract PAHs for subsequent GC-MS determination in grilled meat samples.372 COF TpBD was grown on stainless steel wire modified by polydopamine that served as a linker to react with benzidine (Fig. 29a). The TpBD bonded fiber showed good durability, which can stand at least 200 cycles without significant loss of extraction efficiency. The TpBD bonded SPME fiber gave much larger enhancement factors than commercial SPME coatings, including PDMS fibers, PDA fibers, and etched stainless steel wire (Fig. 29b). The developed method could be successfully applied for the determination of trace PAHs in grilled meat samples with recoveries from 85.1% to 102.8%.
 | ||
| Fig. 29 (a) Schematic illustration of fabrication of the TpBD bonded SPME fiber. (b) Comparison of the PDA coated fiber, etched fiber, PDMS coated fiber and TpBD bonded fiber for the extraction of PAHs.372 Adapted with permission from ref. 372. Copyright 2019, Elsevier. | ||
Phenols. Phenols are primarily used to produce polycarbonate plastics and resins or used as deoxidants, and have been implicated in a wide variety of problems, including endocrine disruption, carcinogenicity, and neurotoxicity.394 A few COFs have been utilized as absorbents in magnetic solid-phase extraction,356,359,360,366 solid-phase microextraction,367,369,376 and micro solid-phase extraction362 for the analysis of phenolic species in biological, food, and water samples. For example, Lin and coworkers reported the use of core–shell structured magnetic COF (Fe3O4@COF) nanocomposites as an adsorbent for MSPE of bisphenols (BPs) from human serum samples.356 The Fe3O4@COF-TAPB-PDA nanocomposites with a core–shell structure possessed a high specific surface area (181.36 m2 g−1), uniform mesoporous size (∼3.6 nm), high saturation magnetization (42.7 emu g−1), and good thermal and chemical stability. Using the nanocomposite as an adsorbent, MPSE coupled with HPLC-MS gave good linearity within the concentration range of 0.1–50 μg L−1 of bisphenols. The method was also applicable for the analysis of trace BPs in human serum samples.356 Recently, a method for rapid detection of trace tetrabromobisphenol A (TBBP A) in water media was developed by Yu and coworkers by combining COF-based solid-phase microextraction (SPME) with constant flow desorption ionization mass spectrometry.367 The particles of COF TpBD were directly coated on a disposable glass fiber (Fig. 30a) for the solid-phase microextraction. The porous structure, suitable channel size, secondary amine group, carbonyl group, as well as the benzene/aromatic ring in the framework that allows π–π stacking interaction between TBBPA and TpBD enabled the use of TpBD as an effective adsorbent for organic pollutants. TBBPA was then ionized and transmitted into the linear trap quadrupole-mass spectrometer (Fig. 30b and c). An enrichment factor of 185 was achieved to result in a 3.6 to 7.2-fold increase in the peak area of the TBBPA ion compared to commercial fibers. Moreover, compared with the traditional HPLC or HPLC-MS protocols, the developed method presented significantly reduced analytical time (7 min vs. 30 min) and much lower LOD values (0.92 ng L−1vs. 23–900 ng L−1).
 | ||
| Fig. 30 (a) Schematic illustration of the preparation of the TpBD coated fiber and SPME process. (b) Schematic illustration of SPME coupled with constant flow desorption ionization MS. (c) Photograph of the Taylor cone formed between the end of the fiber and the MS inlet.367 Adapted with permission from ref. 367. Copyright 2019, American Chemical Society. | ||
The selectivity and enrichment ability of COFs could be improved by their functionalization. Zhao and coworkers synthesized a boric acid-functionalized COF (B-COF) by using 2,4,6-trihydroxy-1,3,5-benzenetrialdehyde and benzidine as ligands and 4-aminophenyl-boronic acid as a functionalization linker for the specific enrichment and direct detection of cis-diol-containing compounds by MALDI-TOF MS assay.368 B-COF possessed boric acid groups that have selective affinity to capture cis-diol-containing compounds, including luteolin, riboflavin, and pyrocatechol. High selectivity against target analytes was obtained in the presence of 100 times more anti-interference compounds. The LODs of as low as 0.01–0.50 fg mL−1 were achieved for luteolin, riboflavin, and pyrocatechol with the enrichment of B-COF.368
Solid-phase extraction was also applied for the analysis of phenolic compounds in environmental water,360 drink,366 food samples,359,376 and others362 with good recovery values. In most of the cases, the extraction efficiencies were dominantly determined by the covalent or noncovalent interactions between the analyte and COFs. Different from these examples, a recent study by Zhao and coworkers showed that the size of the pores in COFs may also play a role.369 The authors fabricated a spherical TPB-DMTP-COF with high acid/base stability, thermostability, and large surface area as the coating for solid-phase microextraction.369 They found that the enhancement factors of TPB-DMTP-coated fibers increased with the number of chlorine or methyl substituents. Hence, the strong adsorption affinity could be derived from a size-matching effect with the combination of π–π affinity, H-bonding, and van der Waals forces.
Halogenated aromatics. Halogenated aromatics, usually used as intermediates of industry products, are recognized as a class of highly toxic environmental contaminants.395 The toxicity and bioaccumulation of these compounds make the ability to detect them at extremely low levels a necessity.396 To improve the extraction efficiency and selectivity, Jiang et al. developed an aptamer-functionalized magnetic core–shell material for selective extraction of trace amounts of hydroxylated polychlorinated biphenyls.357 The aptamer bearing amino groups was immobilized on the surface of the magnetic COF-COOH through covalent linkage with carboxyl groups (Fig. 31a). The resulting Fe3O4@CO-Apt material thus combined the advantages of easy separation, porous structure, high surface area, and specific affinity for hydroxylated polychlorinated biphenyls. The extraction procedure of the targets involved the dispersion of Fe3O4@COF-Apt in the sample solution, ultrasonication, and collecting from the solution with a magnet (Fig. 31b). The adsorbed targets on Fe3O4@COF-Apt were eluted with solvent and subjected to subsequent HPLC-MS determination. Compared with the nonmodified COF and Fe3O4, Fe3O4@COF-Apt provided much higher selectivity for hydroxy-2′,3′,4′,5,5′-pentachlorobiphenyl (2-OH-CB 124) (Fig. 31c), indicating that the aptamer played a significant role in improving the selectivity toward 2-OH-CB 124 due to its high binding affinity to the target. The maximum adsorption capacity of Fe3O4@COF-Apt for 2-OH-CB 124 reached 37.17 μg g−1 (Fig. 31d). This aptamer-functionalized COF provided a wide linear range of 0.01–40 ng
mL−1 and a LOD as low as 2.1 pgmL−1 when used for the capture and detection of 2-OH-CB 124 in human serum.
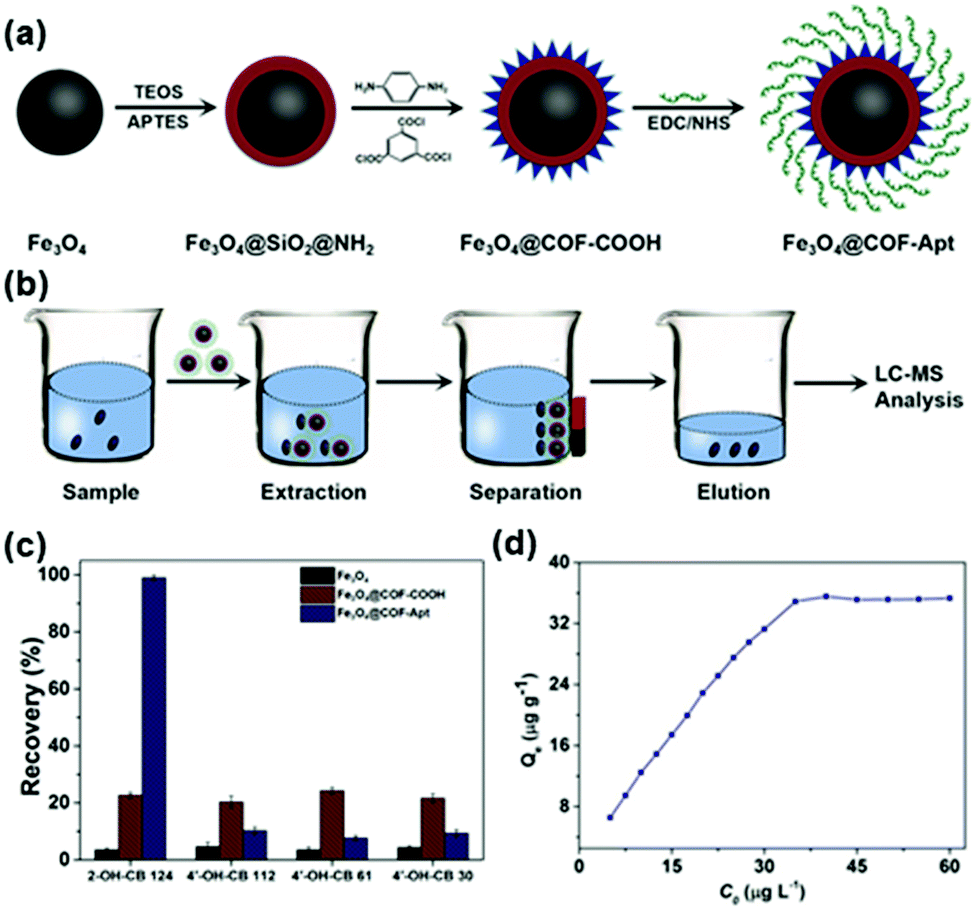 | ||
| Fig. 31 (a) Synthetic scheme of Fe3O4@COF-Aptand and (b) typical process for selective extraction of OH-PCBs with Fe3O4@COF-Apt. (c) Comparison of the extraction efficiency of 2-OH-CB 124, 4′-OH-CB 112, 4′-OH-CB 61, and 4′-OH-CB 30 with Fe3O4, Fe3O4@COF-COOH, and Fe3O4@COF-Apt. (d) Adsorption isotherm of 2-OH-CB 124 with Fe3O4@COF-Apt.357 Adapted with permission from ref. 357. Copyright 2018, John Wiley and Sons. | ||
COFs are normally non-selective to molecules that are smaller than their cavities in the extractions. To develop selective, simple, and rapid extraction pretreatments for the detection of trace polychlorinated biphenyl (PCB) residues in soils, Wang et al. developed a mesoporous titanium dioxide and TPB-TP-COF composite coating (Fe3O4@mTiO2@TPB-TP-COF) onto a Nd-Fe-B magnet for headspace sorptive extraction.364 This new coating exhibited good selectivity over PAHs, like naphthalene and benzopyrene, which was hypothesized as due to a synergistic effect resulting from the unique compositional structure of the Fe3O4@mTiO2@TPB-TP-COF coating.364 The outer COF coating has cavities of uniform size (∼3.9 nm), which may allow PCBs to pass freely while excluding larger-sized macromolecules. The inner mTiO2 coating has a good affinity towards chlorine-containing organic compounds, which can improve the selectivity for PCBs. Under optimal conditions, the LOD was measured to be less than 0.06 ng g−1 with a linear range of 0.01–100 ng g−1. The analysis of brominated370 and fluorinated aromatics373 was also implemented by coupling solid-phase extraction with GC-MS and surface-assisted laser desorption/ionization time-of-flight mass spectrometry, respectively.
Pesticides. Pesticides have been widely applied in agriculture and their residues in the environment and food can cause acute or chronic biological poisoning. Solid-phase extractions using COFs coupled with chromatography and mass spectrometry have been proven to be effective and reliable tools for the analysis of trace pyrethroid and benzoylurea-based pesticide residues in various complex food substrates. The high adsorption capacity and good selectivity are usually from π-stacking and hydrophobic interactions between pesticides and COFs.377–382 For example, Li and coworkers developed an analytical method for the simultaneous determination of pesticides in fruit samples by magnetic solid-phase extraction combined with ultra-high performance liquid chromatography and tandem mass spectrometry.382 A composite made of triazine-imine COF TRI-TER grown on amino-functionalized Fe3O4 was applied as the magnetic sorbent for efficient enrichment and rapid separation of pesticides. LODs ranging from 0.4 to 1.2 ng L−1 were achieved for eight pesticides which can meet the requirement for ultra-trace analysis of pesticides. The established method was successfully applied to actual fruit samples.382
Ions. As discussed in Section 2.2.3, C
N and CO groups present in many COF structures can coordinate to metal ions. In addition, stronger coordination sites could also be strategically introduced into COFs. These features suggest that COFs can be used as sorbents for extraction of metal ions. Wang and coworkers examined two COFs TpBD and CTpBD as sorbents for solid-phase extraction and analysis of trace ions via flow injection followed by inductively coupled plasma mass spectrometry (ICP-MS).383 TpBD showed effective adsorption to only five metal ions, including V4+, Cu2+, As3+, Se4+, and Mo4+, while CTpBD demonstrated SPE performance to up to ten metal ions, such as Cr3+, Mn2+, Co2+, Ni2+, Cd2+, V5+, Cu2+, As3+, Se4+, and Mo6+, probably due to the presence of the COOH functional groups in CTpBD that provided strong coordination with metal ions. Owing to the intrinsic porous structure and metal-binding capability of CTpBD, preconcentration of the target trace elements via the COF-filled on-line SPE column achieved low detection limits of 2.1–21.6 ng L−1 along with a wide linearity range at 0.05–25 μg L−1 for all target ions. This method was successfully utilized for trace element analysis in environmental and food samples.
Biomolecules. Besides the utilization of COFs as absorbents for the detection of aromatics, phenols, pesticides, and ions demonstrated above, their use in the detection of a few biomolecules, including phosphopeptides, glycopeptides, and antibodies, is also reported. The selective enrichment of trace phosphopeptides from complex biological mixtures is indispensable for phosphoproteome studies. In the most common approach for phosphopeptide enrichment, immobilized metal ion affinity chromatography (IMAC) based on the chelation/coordination chemistry is adopted. However, this approach involves tedious synthetic processes due to the use of chelating ligands and solid supports. As an alternative, incorporating metal ions into the COFs with metal coordination capability can provide solid-phase matrices for the selective enrichment of the peptides. Shen and coworkers developed a novel COF-based IMAC material (TpPa-2-Ti4+) by direct immobilization of Ti4+ into the TpPa-2 COF without using extra chelating ligands.385 The permanent porosity, high specific surface areas, well-ordered hexagonal 2D topology, and strong chemical stability endowed TpPa-2-Ti4+ composites with high metal ion loading densities as well as excellent mass transportation and enrichment performance.88 The obtained Ti4+-modified COF demonstrated a low LOD of 4 fmol and satisfactory selectivity for the capture of phosphopeptides from peptide mixtures of tryptic digestion (β-casein
:BSA = 1:100). It was also successfully applied for the enrichment of phosphopeptides from non-fat milk and HeLa cells.385
Post-synthetic modification can offer the advantage of incorporation of on-demand desired functionalities on the surface of COF backbones to improve the extraction capacity and selectivity meanwhile maintaining the structural regularity of COFs. A core–shell structured COF composite (MCNC@COF@Zr4+) with a magnetic colloid nanocrystal cluster (MCNC) as the core and Zr4+ ion-functionalized two-dimensional COFs as the shell was recently reported by Lin and coworkers for efficient and selective enrichment of phosphopeptides.387 A three-step sequential functionalization that involved the conversion of hydroxyl groups to carboxyl groups, the introduction of pamidronic acid (PA) to obtain desired phosphate groups, and the formation of Zr4+-immobilized magnetic COF composites was performed for the post-functionalization of the COF (Fig. 32a). The as-prepared MCNC@COF@Zr4+ can serve as an effective adsorbent for selective enrichment of phosphopeptides attributed to its regular mesoporous structure with high surface area and the high affinity interaction between Zr4+ ions and phosphate groups. Direct analysis (Fig. 32b) or treatment by MCNC@COF composites (Fig. 32c) for tryptic digests of 100 μg mL−1 β-casein did not show obvious signals using MALDI-TOF mass spectra; in contrast, after treating β-casein with the MCNC@COF@Zr4+ composites, three phosphopeptides (β1, β2, and β3) possessing both the phosphorylated parts and dephosphorylated counterparts were identified with enhanced signal intensities (Fig. 32d). A low detection threshold of 10 fmol for phosphopeptides was obtained. Moreover, the unique pore structure endowed a size-exclusion effect of biomacromolecules, such as lysozyme and concanavalin A. This effectiveness of the MCNC@COF@Zr4+ composites in screening biomacromolecules background was advantageous in real complex samples’ analysis, which was further evidenced by selective enrichment of phosphopeptides from the tryptic digests of defatted milk (Fig. 32e) and direct capture of low-abundance 14 endogenous phosphopeptides from 10-fold diluted human serum.387
 | ||
| Fig. 32 (a) Representation of the strategy for preparing Zr4+-immobilized magnetic COFs through sequential post-synthetic modifications. MALDI-TOF mass spectra of the tryptic digests of β-casein from (b) direct analysis, (c) analysis after enrichment with MCNC@COF composites, and (d) analysis after enrichment with MCNC@COF@Zr4+ composites. (e) MALDI-TOF mass spectra of the tryptic digests of defatted milk after enrichment with MCNC@COF@Zr4+ composites.387 Adapted with permission from ref. 387. Copyright 2019, American Chemical Society. | ||
Given the regular pore sizes and controllable chemical affinities by modifications, COFs are also potential candidates for the capture of N-linked glycopeptides, which are a kind of biomarkers for early diagnosis of various diseases.397,398 Gao and coworkers developed a COF-functionalized magnetic graphene biocomposite (MagG@COF-5) as an ultrasensitive hydrophilic matrix for recognizing N-linked glycopeptides.391 Due to the strong π–π interactions between ligands and graphene, layers of COF-5 could be successfully coated on the surface of magnetic graphene via a hydrothermal reaction (Fig. 33a). The coating of the COF on graphene was recognized to possess multipurpose functionalities and could be used as an extraction matrix when coupled with MALDI-TOF-MS for N-linked glycopeptide analysis (Fig. 33b). First, the unique π–π electron structure and strong magnetic properties of magnetic graphene composites provided binding sites for targets and facilitated rapid and convenient separation by a magnet. Second, the coating of COF-5 can be used as a molecular gate to exclude large-sized proteins from complex biological samples through the size-exclusion effect. Third, the high surface area of COF-5 coating can ensure high loading capacity. For these unique characteristics, the MagG@COF-5 biocomposite showed excellent performance in N-linked glycopeptide analysis with a LOD of 0.5 fmol μL−1, an excellent size-exclusion effect (HRP digests/BSA selectivity, 1:600), and good recyclability and reusability. 232 N-linked glycopeptides from 85 glycoproteins were detected by treatment of human serum with this biocomposite, demonstrating the potential of MagG@COF-5 for use in glycoproteome and clinical diagnosis fields.
 | ||
| Fig. 33 (a) Synthesis of the MagG@COF-5 biocomposite. (b) Flow chart of the recognition process for N-linked glycopeptides by using the MagG@COF-5 biocomposite.391 Adapted with permission from ref. 391. Copyright 2017, Royal Society of Chemistry. | ||
The regular arrangement of active sites and porous structure of COFs offer a suitable platform for protein immobilization with consistent orientation, which can serve as a solid matrix for immunoassay. Fang and coworkers developed an immunoaffinity probe, Fe3O4@TpBD-DSS-Ab-MEG, based on magnetic COFs with anchored antibodies for the detection of a cancer biomarker, heat shock protein 90alpha (Hsp90α).392 The fabricated composites with Fe3O4 as the magnetic core and COF TpBD as the shell exhibited several appealing features, including strong magnetic responses (7.96 emu g−1), ordered active groups, a large number of immobilized antibodies (111.7 μg mg−1), and good solvent and thermal stability. Attributed to these features, Fe3O4@TpBD-DSS-Ab-MEG demonstrated a low LOD of 50 pg mL−1 and high selectivity over BSA. Furthermore, 2 peptides of Hsp90α were successfully observed in human plasma. This immunoassay strategy thus offered an ability for accurate analysis of low-abundant protein biomarkers in complicated biological matrices.
As adsorbents for solid-phase extraction,59,399–402 COFs present good adsorption performance towards many analytes, attributed to their high surface area, regular porous channels, abundant functional groups, and stable chemical structures. This sample preparation method, coupled with various chromatographic and spectrometric techniques, has become a powerful tool for the analysis of trace-level analytes in complex samples, ranging from various aromatic compounds, pesticides and insecticides, chemical warfare agents, ions, drug molecules, to biomolecules in environmental, food, and biological samples (Table 5). Even though a lot of reports have shown the appealing detection performance of COFs toward specific analytes in real samples, further development towards the practical use of COF materials for simple, quick, and efficient extraction techniques is still highly desired. Firstly, COFs still possess limited selectivity for analytes with similar structures, which has been seen in the detection of PAHs and ions. Because the interaction between COFs and those analytes is mainly based on weak intermolecular interactions, such as π–π stacking interactions, hydrogen bonding interactions, coordination reactions, and van der Waals force, they usually lack specificity. Combining carefully engineered size-exclusion effect and binding sites introduced through functionalization can provide potential solutions for the selectivity issue. Secondly, most of the reported COFs are highly hydrophobic which can lead to weak adhesion between the sample media and COFs and slow adsorption kinetics. The development of new COFs with well-controlled surface chemistry and channel environment will lead to superior adsorption ability for hydrophilic compounds in aqueous and biological samples. Thirdly, COFs should also be chosen or designed by balancing the feasibility of adsorption and desorption during the SPE process. A COF with a strong affinity to a certain analyte may show higher extraction efficiency, but may lead to incomplete desorption in the subsequent procedure which can affect the accuracy of quantitative analysis. Furthermore, the combination of COFs with other functional materials and substrates may further enhance their adsorption capacity and chemical stability and reduce the cost, and eventually expand the application of COFs as effective adsorbents in sample treatment for analyte detection.
| Analyte(s) | Method | COF | Detection architecture | LOD | Detection range | Other detection parameters | Ref. |
|---|---|---|---|---|---|---|---|
| Sudan dyes | Micro-solid-phase extraction (MSPE)/HPLC | HL-COP | HL-COP/SSF column | 0.03–0.15 μg L−1 | 0.5–100 μg L−1 | pH = 7.0, extraction flow rate of 300 μL min−1, desorption flow rate of 200 μL min−1 and desorption flow volume of 600 μL; chili powder, sausage samples, recoveries 73.8–112.6% | 351 |
| Pyrethroids | SPME/GC | A hydrazone COF | COF/PDA-SSF | 0.11 to 0.23 μg kg−1 | 1–1000 μg kg−1 | Extraction temperature of 50 °C, extraction time of 30 min, and desorption time of 2 min; vegetables and fruits, recoveries 75.6–106.3% | 377 |
| Paclitaxel | Magnetic SPME/HPLC | COF-1 | Fe3O4/COF-1 | 0.02 ng mL−1 | 0.1–200 mL−1 | pH = 9, acetonitrile content of 0.5% (v/v), extraction time of 1 h, sample volume of 20 mL; rat plasma sample, recovery 99.4–103.7% | 384 |
| Phosphopeptides | SPE/MALDI-TOF | TpPa-2 | TpPa-2-Ti4+ | 4 fmol | N/A | 200 μL of buffer (50% extraction-H2O, 1% TFA) for 30 min, eluted with 5% NH4OH for 5 min; β-casein:BSA = 1:100); non-fat milk and HeLa cells |
385 |
| Benzophenones | SPME/HPLC | COF-SH | COF-SH@poly(GMA–EDMA) | 0.4–0.7 ng mL−1 | 1–500 ng mL−1 | pH = 3.0, eluent MeOH flow rate 0.03 mL min−1; urine and serum specimens | 352 |
| N-Linked glycopeptide | Magnetic SPE/LC-MS | COF-5 | Magnetic graphene (MagG)@COF-5 | 0.5 fmol μL−1 | 0.5–100 fmol μL−1 | Extraction solvent H2O/TFA (95:4:1, v/v/v), extraction time 15 min; digests/BSA, 1:600; human serum |
391 |
| PAHs | Magnetic SPE/HPLC | COF-LZU1 | COF-LZU1@PEI@Fe3O4 | 0.2–20 pg mL−1 | 2–2000 pg mL−1 | pH = 9.0, acetonitrile content of 1%, extraction time 30 min, sample volume 20 mL; environmental samples, recoveries 85.1–107.8% | 353 |
| Phthalate esters | Magnetic SPE/GC | CTF | CTFs/Ni | 0.024–0.085 mg kg−1 | 0.08–80 mg kg−1 | pH = 7, NaCl (3%, m/v), desorption solvent acetone, extraction time 5 min, desorption time 5 min; plastic cup and mineral water bottle | 354 |
| Volatile benzene homologues | SPME/GC | COF-SCU1 | COF-SCU1/SSW | 0.03–0.15 ng L−1 | 0.1 to 100 ng L−1 | Extraction time 20 min, temperature 40 °C, desorption temperature 250 °C for 3 min; indoor air samples, recoveries 87.9–103.4% | 355 |
| Bisphenols | Magnetic SPE/LC-MS | TAPB-TPA | Fe3O4@TAPB-TPA | 1.0–78.1 ng L−1 | 0.1–50 ng L−1 | Extraction time 10 min, desorption solvent i-propanol; human serum sample, recoveries 93.0–107.8% | 356 |
| Cyano pyrethroids | SPE-HPLC | TAPB-TP | Molecularly imprinted TAPB-TP | 0.011–0.018 ng g−1 | 0.1–200 ng g−1 | Desorption solvent 4% acetic acid in methanol; fruit, vegetable, and medicine samples, recoveries 4.3–102.7% | 378 |
| OH-PCBs | Magnetic SPE/LC-MS | COF-COOH | Fe3O4@COF-aptamers | 2.1 pg mL−1 | 0.01–40 ng mL−1 | pH = 11, extraction time 30 min, human serum, recoveries > 90% | 357 |
| PAHs | Magnetic SPE/HPLC | TpBD | Fe3O4@TpBD | 0.83–11.7 ng L−1 | 1–100 ng mL−1 | Extraction time 12 min, eluent acetonitrile, desorption time 15 min; food samples, recoveries 84–99% | 358 |
| Endocrine disruptors | Magnetic SPE/HPLC | TpBD | Fe3O4@TpBD | 1.4–8.7 μg L−1 | 10–1000 μg L−1 | Eluent acetonitrile, adsorption time 5 min, desorption time 10 min; food samples, recoveries 89.6–108.9% | 359 |
| Metal ions | SPE/ICP-MS | TpBD | TpBD | 2.1–21.6 ng L−1 | 0.05–25 μg L−1 | pH = 5.0, eluent 0.7 M HNO3; environmental and food samples, recoveries over 85% | 383 |
| Phenolic pollutants | Magnetic SPE/LC | CTF | CTFs/Fe2O3 | 0.09–0.53 ng mL−1 | 25–2000 ng mL−1 | pH = 2, eluent acetonitrile, extraction time 30 min; real water samples, recoveries 85.5–97.7% | 360 |
| Perfluorinated compounds | Magnetic SPE/LC | CTF | CTFs/Fe2O3 | 0.62–1.39 ng L−1 | 5–4000 ng L−1 | Eluent acetone; environmental water samples, recoveries 87–106% | 361 |
| Parabens | Magnetic SPE/HPLC | CTF | Fe3O4/CTF | 0.02 μg L−1 | 0.1–500 μgL–1 | pH = 7.0, eluent methanol-acetonitrile (v/v = 1:1), desorption time 30 s; milk and water samples, recoveries 86–102% |
362 |
| PAHs | Magnetic SPE/HPLC | TpDA | Fe3O4@TpDA | 0.03–0.73 mg L−1 | 5–100 μg L−1 | Extraction solvent acetonitrile, adsorption time 10 min, desorption time 15 min; oil and meat sample, recoveries 85.5–104.2% | 363 |
| Pesticides | SPE/HPLC | DAAQ-TFP | DAAQ-TFP | 0.02–0.08 ng mL−1 | 0.1–160 ng mL−1 | pH = 2–8, 0.2 mL acetonitrile as the eluent with a pre-soaking time of 1 min; water and drink sample, recoveries 85.5–112.7% | 379 |
| Pesticides | SPME/GC | TpPaNO2 | TpPaNO2/stainless steel wire | 0.04–0.25 μg kg−1 | 0.5–225 μg kg−1 | Extraction time 45 min, pH = 6–7, desorption at 260 °C for 6 min; vegetable and fruit samples, recoveries 81.5–111% | 380 |
| Polychlorinated biphenyls | Headspace sorptive extraction/GC-MS | TAPB-PDA | Fe3O4@mTiO2@TAPB-PDA | <0.06ng g−1 |
0.01–100 ng g−1 |
Soil samples, recoveries 94.6–97.2% | 364 |
| Benzoylurea insecticides | SPE/HPLC | TpAzo | TpAzo | 0.1–0.2 ng mL−1 | 1.0–160.0 ng mL−1 | pH = 3–6, eluent acetonitrile; drink and vegetable sample, recoveries 84–108% | 381 |
| Sulfonamide antibiotics | SPE/HPLC | COF made of melamine and phthalaldehyde | COF/polyurethanes | 0.004–1.050μg kg−1 |
0.25–1000ng kg−1 |
pH = 4, eluent methanol; recoveries 82.7–100.8% | 386 |
| Chlorophenols | SPME/GC-MS | TpBD | TpBD/stainless steel wire | 0.3–1.8 μg kg−1 | 3–300 μg kg−1 | pH = 11, extraction temperature 40 °C, extraction time 35 min, desorption temperature of 250 °C; food samples, recoveries 70.2–113% | 376 |
| Fluorescent whitening agents | SPE/HPLC | COP | Fe3O4/COF | 0.1 ng L−1 | 0.5–100 ng L−1 | Extraction time 50 min, eluent acetone, elution time 60 min; cosmetic samples, recoveries 8.2–105% | 365 |
| Phthalate ester | Magnetic SPE/GC-MS | TpBD | Fe3O4@PDA@TbBd | 0.0025–0.01 ng mL−1 | 50–8000 ng mL−1 | pH = 7, extraction time 30 min, eluent acetone, desorption time 10 min; human plasma sample, recoveries 92.3–98.9% | 375 |
| Phenols | SPE/HPLC | TpBD | Fe3O4@TpBD | 0.08–0.21 ng mL−1 | 0.5–1000 ng mL−1 | pH = 6, extraction time, 30 min; drink samples, recoveries 81.3–118.0% | 366 |
| Phosphopeptides | SPE/MS | TfPb-DHBD | Magnetic colloid nanocrystal cluster@TfPb-DHBD@Zr4+ | 10 fmol | N/A | Milk and human serum | 387 |
| Tetrabromobisphenol A | SPME/MS | TpBD | TpBD/glass fiber | 0.92 ng L−1 | 0.01–10 μg L−1 | Water samples, recoveries 83.9–113.1% | 367 |
| cis-Diol-containing compounds | SPE/MALDI-TOF | TpBD | Boric-acid functionalized TpBD | 0.01–0.5 pg mL−1 | 0.1–200 pg mL−1 | pH = 8.0, extraction time 80 min; human serum samples, recoveries 85.3–109% | 368 |
| Nonsteroidal anti-inflammatory drugs | SPE/HPLC | TpBD | TpBD@SiO2 | 0.38–2.92 μg L−1 | 0.002–1.0 μg mL−1 | pH = 2, eluent methanol (1% NH3·H2O), desorption time 10 min; water samples, recoveries 77.3–111.6% | 388 |
| Pesticides | Magnetic SPE/HPLC | TRI-TER | TRI-TER@Fe3O4 | 0.4–1.2 ng L−1 | 5–20000 ng L−1 |
Extraction time 40 min, eluent acetone, desorption time 5 min; fruit samples, recoveries 73–103% | 382 |
| Sulfonamide | Magnetic SPE/HPLC | TpBD | Fe3O4@TpBD | 0.28–1.45 mg L−1 | 1–80 μg L−1 | Eluent acetone; food samples, recoveries 82–94% | 389 |
| Phenols | SPME/GC-MS | PB-DMTP-COF | PB-DMTP-COF-coated fiber | 0.0048–0.015 ng L−1 | 0.05–1000 ng L−1 | Extraction time 60 min, 50 °C, pH = 6, desorption temperature 280 °C; water samples, recoveries 81.3–116.3% | 369 |
| Polybrominated diphenyl ethers | SPME/GC–MS | TpPa-1 | TpPa-1 coated fiber | 0.0058–0.022 ng L−1 | 0.01–100 ng L−1 | Extraction time 40 min, extraction temperature 70 °C, desorption temperature 300 °C; water samples, recoveries 71.9–125.4% | 370 |
| Phthalate esters | Magnetic SPE/GC–MS | TbBd | magG@PDA@TbBd | 0.004–0.02 ng mL−1 | 50–8000 ng mL−1 | pH = 7, extraction time 10 min, eluent dichloromethane, desorption time 10 min; milk samples, recoveries 91.4–108.5% | 371 |
| PAHs | SPME/GC-MS/MS | TpBD | TpBD/stainless steel wire | 0.02–1.66 ng L−1 | 2–200 ng L−1 | Extraction time, 30 min; desorption time, 5 min; extraction temperature, 40 °C; meat sample, recoveries 85.1–102.8% | 372 |
| Chemical warfare agents | SPE/GC-MS and NMR | CTF | CTF | 0.010–0.100 μg mL−1 | 0.040–20.0 μg mL−1 | Eluent DCM, CHCl3, and EtOAc; recoveries 87–100% | 24 |
| Fluorochemicals | SPE/SALDI-TOF MS | COF-LZU1 | COF-LZU1 | 0.016–0.67 ng mL−1 | 10–2000 pg mL−1 | pH = 3; selectivity over humic acid; real water samples, recoveries 77.1–123% | 373 |
| Allergenic disperse dyes | Magnetic SPE/LC-MS/MS | N-Mag-COF | N-Mag-COF | 0.021–0.58 μg kg−1 | 0.5–50 μg kg−1 | Extraction solvent methanol, extraction time 10 min; textile samples, recoveries 72.2–107% | 374 |
| Sulfonamides | SPE/HPLC | SNW-1 | SNW-1@PAN | 1.7–2.7 ng mL−1 | 5–125 ng mL−1 | pH = 5–7, eluent 5% ammonia-methanol; metal samples, recoveries 86–111% | 390 |
| Hsp90α | Magnetic SPE/MS | TpBD | Fe3O4@TpBD-DSS-Ab-ME | 50 pg mL−1 | N/A | pH = 6.0; human plasma samples, selectivity: Hsp90α:BSA = 1:1000 |
392 |
2.5 Detection based on other mechanisms
COFs can be rationally designed to exhibit excellent adsorption capability and high affinity to a series of targets, making them potential candidates for composing hybrid substrates for SERS-based detection. Long and coworkers developed a SERS substrate for the recognition of polycyclic aromatic hydrocarbons with high sensitivity by using COF SNW-1 (Fig. 34a).408 Unlike most SERS substrates made of metal NP decorated hybrids, the Au NPs/SNW-1 nanocomposite with well-controlled particle size and fine-tuned morphology could be fabricated based on rapid physisorption of Au NPs to abundant nitrogen atoms present in SNW-1. Diverse PAHs and their mixtures, including acenaphthene, phenanthrene, anthracene, fluoranthene, pyrene, benzo[a]anthracene, 3,4-benzopyrene, and coronene, could be analyzed by using the developed substrate with high sensitivity and reproducibility. The characteristic Raman signature could be obtained from the PAHs even down to a concentration of tens of nM, and the corresponding LOD for each of the eight target PAHs was down to 10−7 to 10−8 M (Fig. 34b and c). The developed substrate of Au NPs/SNW-1 could be further applied in the SERS analysis of real samples of local tap and pond water with recovery for all the spiked PAHs ranging from 80 to 114%.408
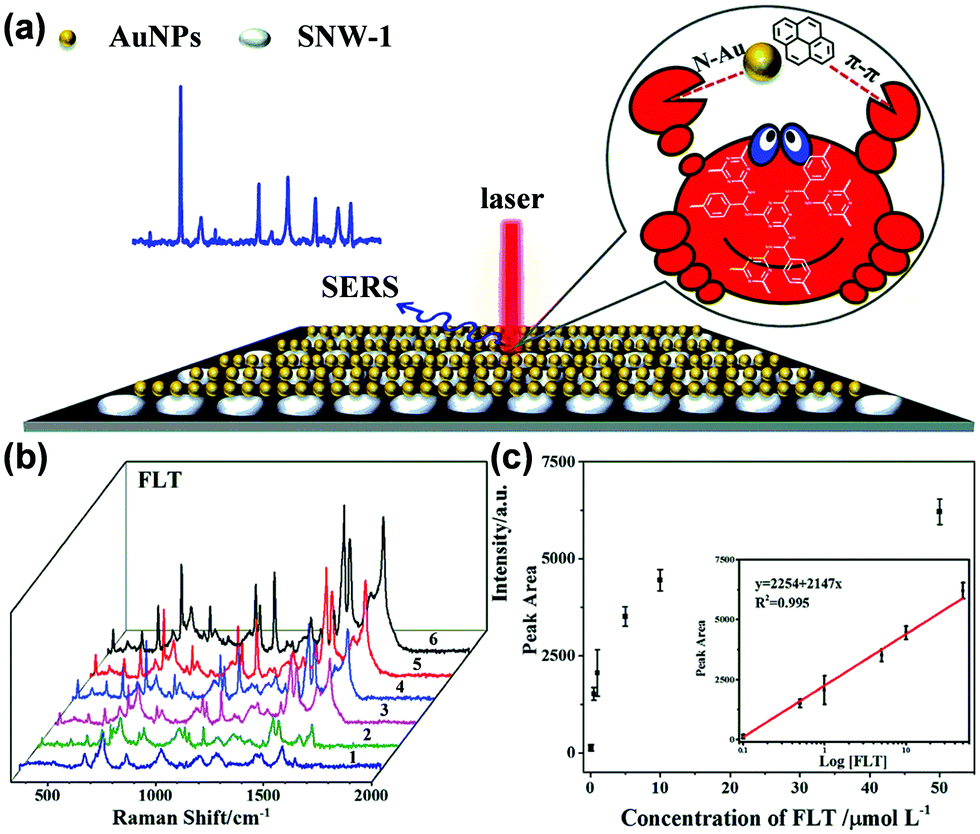 | ||
| Fig. 34 (a) PAHs in close proximity to Au NPs in the nanohybrids of Au NPs/SNW-1. (b) Raman spectra by using Au NPs/SNW-1 as the SERS substrate obtained from FLT at 0.1, 0.5, 1.0, 5.0, 10.0 and 50.0 mM (1–6), respectively. (c) Scatter plots of peak areas of SERS spectra at 1100 cm−1versus the concentrations of FLT. Inset: Calibration curve of the peak areas of Raman spectra versus the logarithm of the concentration of FLT.408 Adapted with permission from ref. 408. Copyright 2017, Royal Society of Chemistry. | ||
Xia and coworkers reported the first example of using a COF as a matrix for LDI-TOF MS,415 in which COF TpBD was used to demonstrate the proof-of-concept.416 Biological molecules, such as amino acids and fatty acids, as well as environmental pollutants, like bisphenol S (BPS) and pyrene, were successfully detected. This method achieved higher desorption/ionization efficiency and less background interference than when using the conventional organic matrices. It exhibited good salt tolerance (500 mM NaCl) and repeatability, and the detection limit of amino acids reached 90 fmol. When this COF matrix was used for amino acid analysis in the honey sample, a recovery of 97 ± 3% was achieved.415 The authors hypothesized that the good performance of COF TpBD as the matrix for LDI-TOF MS originated from its unique structure. The π-conjugated structure may play an important role in assisting the desorption/ionization process because laser adsorption and energy transfer strongly depend on the continuous π-stacking structure. Moreover, the hydroxyl groups of TpBD can enhance the desorption/ionization effects. The conjugated and nanoporous structure of TpBD can also inhibit the production of matrix fragment peaks and prevent target molecules from aggregation, which may effectively accelerate the desorption process from TpBD. Therefore, the unique structure of TpBD resulted in highly efficient transfer of laser energy to small molecules, leading to productive and stable ion intensities.415
To enhance the sensitivity and selectivity of the detection, Zhang and coworkers utilized COF BABE-TFPy which contains abundant and accessible –NH and –OH functionalities in channels (Fig. 35a) as the coating layer of a QCM sensor for various VOCs and a mustard gas simulant, 2-chloroethyl ethyl sulfide (CEES).417 The functional groups in the COF can facilitate the interaction with guest molecules through hydrogen bonding. The frequency changes of the COF coated QCM sensor were linearly related to the concentrations of CEES vapor within the range of 5.6–19.7 ppm, and the sensor could achieve a low LOD of 0.96 ppm (Fig. 35b). The BABE-TFPy coated QCM sensor displayed significantly higher sensitivity to CEES vapor compared to other common VOCs, even when the concentration of the CEES vapor was much lower than that of the other vapors (Fig. 35c). Theoretical calculations further showed that this good selectivity and sensitivity may be ascribed to the formation of dual-hydrogen bonding interactions between the CEES molecule and the backbone of the BABE-TFPy COF (Fig. 35d).417
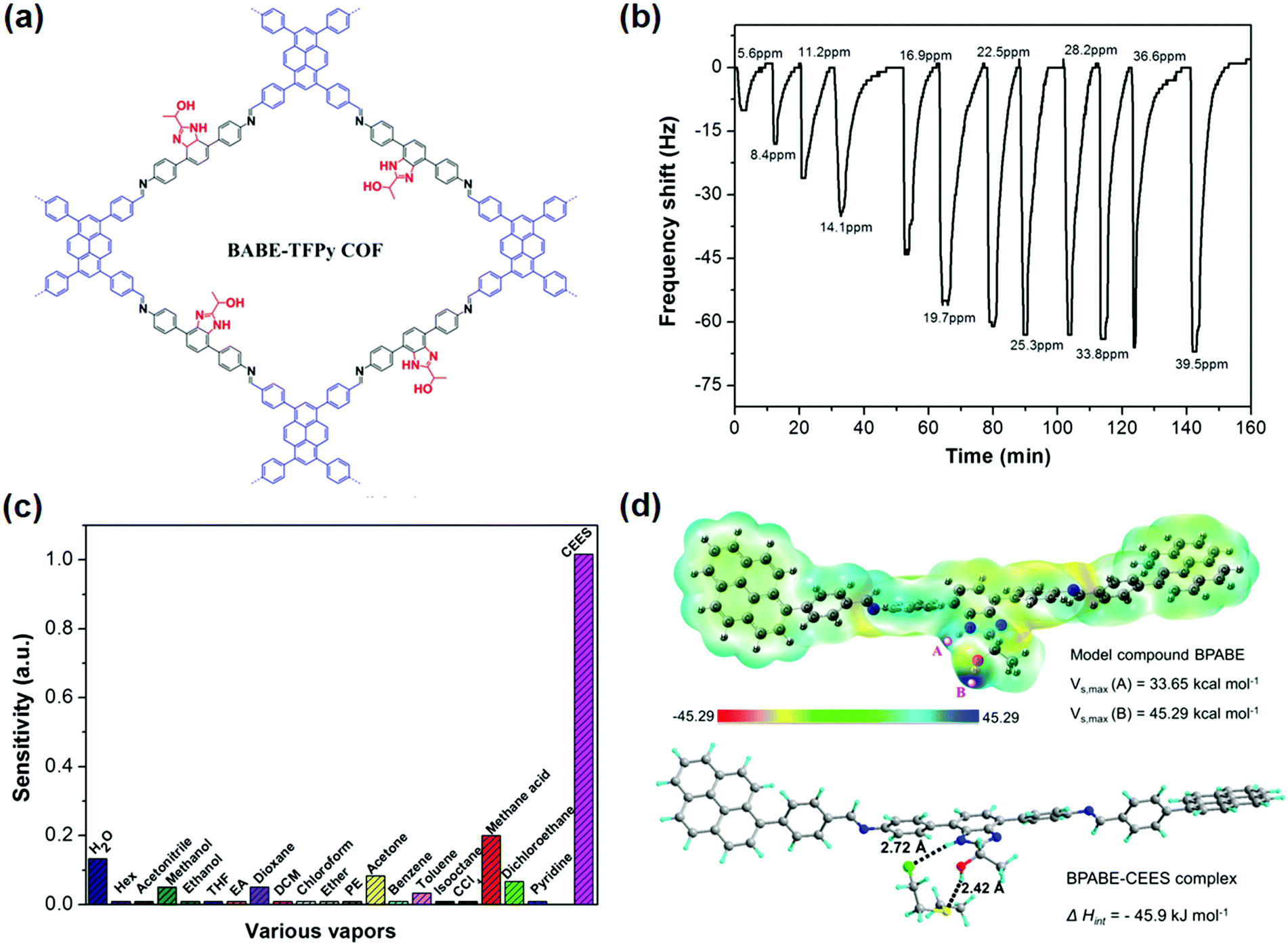 | ||
| Fig. 35 (a) Chemical structure of the BABE-TFPy COF. (b) The dynamic response curve of the BABE-TFPy coated QCM sensor exposed to CEES vapor with increasing concentration at 40 °C. (c) Frequency change of the BABE-TFPy coated QCM sensor in response to various vapors. (d) Electrostatic potential of the model compound BPABE for the BABE-TFPy COF and optimized geometries of the dual-hydrogen bonding between the model compound BPABE and the CEES molecule.417 Adapted with permission from ref. 417. Copyright 2019, American Chemical Society. | ||
Constructing heterostructures with advanced architecture is an effective strategy for enhancing the crystallinity and functional performance of COFs. Xu and coworkers reported MOF/COF heterostructure MOF-GC@COF with ZIF-67 derived graphitic carbon (MOF-GC) as the core and COF-TFP-DMPA as the shell for highly sensitive and selective sensing of formaldehyde vapor.422 The pristine MOF-GC exhibited poor adsorption selectivity toward HCHO vapor. The pristine COF-TFP-DMPA showed high selectivity to HCHO compared to other vapors; however, it suffered from low sensing dynamics. The response dynamics and sensitivity of the heterostructure can be balanced through the tuning of the core/shell ratio. The authors hypothesized that MOF-GC possessing excellent electrical properties and ultrahigh carrier mobility was favorable to promote the free transfer of electrons and for rapid adsorption kinetics, while the COF shell with abundant –NH– functional groups provided selective hydrogen bonding interactions with the aldehyde groups present in HCHO. In addition, the core–shell motif can help prevent the aggregation of MOF-GC cores and promote the formation of hierarchical porosity, which may facilitate effective adsorption and diffusion of gas molecules. Therefore, the MOF-GC@COF heterostructure with the optimized ratio showed a high QCM sensing response toward HCHO, with a pseudo-first-order rate constant of 1.026 min−1, a remarkable detection sensitivity of 151.8 Hz ppm−1, and a superior selectivity over a few other vapors, including formic acid, water, ammonia, etc.422
3. Conclusion and perspectives
Recent efforts dedicated to the design and synthesis of robust, stable, ordered and highly accessible porous COFs, the chemical modifications of COFs,47,80 as well as the fundamental understanding of their physical and chemical properties have paved the way for their applications in chemical detection.68,69,97,125,400,402 COFs as an emerging group of multifunctional materials have been utilized in advancing chemical detection of a relatively broad range of analytes in various media and scenarios. In the demonstrated examples of chemical detection, COFs have played multifaceted roles, including as recognition units for analyte binding, as transducers for transforming the analyte binding event to optical or electrical signals, as scaffolds for supporting other responsive materials, and/or as selective and efficient adsorbents for preconcentration prior to instrumental analysis. These properties are attributed to the unique features of COFs, including an ordered and porous structure, large surface areas, abundant functional groups, a stable skeleton, high structural tunability and functionalization capability, and engineerable optical and electronic properties.In this review, we have summarized the progress in the use of COFs, including COFs with post-synthetic modifications and COF-based composites (e.g., COFs–metal nanoparticles, COFs–polymers, COFs–MOFs, and COFs–enzyme composites) for the detection of volatiles and gases, ions, and biomolecules, the selective and sensitive detection, which is of importance in public safety, health care, disease diagnosis, and environmental monitoring and protection. We have discussed the key advances in the application of COFs in chemical detection based on different analytical mechanisms that have been divided into detection based on chromism, fluorescence, electrochemistry, extraction coupled with chromatography and spectrometry, and other detection modes. We have also described each detection mechanism and correlated the role of COFs in these distinct detection modes with their unique structural features by the analysis and comparison of their structure–property–performance relationships. In doing so, we have aimed to demonstrate how the structural and compositional features of COF-based materials endow them with different optical, electrochemical, electronic, and other chemical and physical properties, and ultimately lead to fascinating and featured applications in the development of chemical detection.
The detection of specific analytes using COFs under different detection methods is summarized in Tables 2–5 by giving the key detection parameters. From these summaries, it is apparent that optical detection, including using chromism and fluorescence change, is the most studied detection mode for volatiles, gases, and ions. Especially, fluorescence-based detection has been proven to be useful in detecting nitroaromatic explosives with reasonable sensitivity. The optical method can usually provide the advantages of signal visibility, fast response, and good sensitivity for on-site detection. Compared with the optical method, the electrochemical method is currently a powerful tool employed in the detection of biologically important analytes. Electrodes modified with COFs and COF-based nanocomposites have achieved sensitivity with LODs down to the level of nmol to fmol L−1, selectivity over a wide range of interferents, and excellent reliability, which is poised for accurate biological analysis and disease diagnostics. SPE coupled with chromatography and spectrometry exhibited capability and robustness for the detection of a broader range of chemicals, ranging from industrial chemicals, ions, drug molecules, to biomolecules. This approach has been successfully applied in real sample analysis, including various food, water, environmental, and biological samples. Some new detection methods, such as SERS, QCM, and chemiresistive detection, have also emerged. The application of these techniques to COF-based analysis brings about the advantages of ultra-low detection limits, high selectivity, and simple and easy detection configurations, and provides on-demand choices for the analysis of specific detection scenarios.
There are several general conceptual and experimental approaches we can learn from these advances that have been proven to be effective for improving the sensitivity, selectivity, response time, and durability of the devices. First, the designability and bottom-up access of COFs can be fully harnessed to introduce specific functional active sites that can interact with target analytes effectively and specifically to realize desired sensitivity and selectivity. As a supplementary strategy, post-synthetic modification has also been proven to be useful in incorporating active sites necessary for host–guest interactions. Second, the engineering of pore size and geometry is an important way of achieving selectivity. Size exclusivity is perhaps the most intuitive selection rule for the recognition of small molecules/ions from biomacromolecular interferents. Thus, the pore size is an important parameter to consider when designing COFs for electrochemical detection or as adsorbents for the preconcentration of analytes. Third, COFs can combine with other nanomaterials (e.g., nanoparticles, SWCNT, and QDs) to form synergistic composites to significantly enhance the analyte recognition, catalytic ability, redox activity, and/or conductivity of the detection system, leading to improved detection capability and performance. Fourth, compared with bulk forms of COFs, thin sheets of COFs have an enhanced surface-to-volume ratio, more accessible surface functional groups, and superior optical and electronic properties. The use of COFs in the form of thin sheets can, therefore, improve the detection performance.
Although some intriguing developments have been made, the application of COFs in the field of chemical detection is still in early stages of development and critical challenges remain to be addressed to implement COFs in analytical applications. Fluorescence-based detection is currently the most investigated method that is based on the fluorescence change of COFs. Even though various fluorescent building blocks can be integrated into the skeletons of COFs, most of the COF-based materials show weak or no emission in their bulk state due to the so-called ACQ effect. Therefore, current approaches to fluorescence detection rely heavily on the measurement of the emission in the solution phase by preparing a COF suspension or colloid, making it very challenging to be used in real-time analysis. This shortcoming limits their use for direct gas and vapor detection in the gaseous environment by using COFs in their solid-state. Utilizing the AIE mechanism to develop COFs with high emissive ability in their solid-state form may provide an opportunity for easy and on-spot detection. In addition, the turn-on type fluorescence detection is still highly desirable for highly sensitive detection. The progress could be built upon a new level of understanding of emission characteristics of COFs and the interaction mechanism between COFs and analytes, which can be prudently assessed with the assistance of fundamental photophysical mechanisms and principles of supramolecular chemistry coupled with computational calculations185,187,423,424 and in situ spectroscopic techniques.108
Electrically transduced detection holds a great opportunity for device miniaturization and integration. COFs have exhibited their potential in the electrically transduced detection of biology-related and gaseous analytes. However, because of limited electrochemical activity and low conductivity, the applications of COFs as sole active electrode materials for electrically transduced detection have been limited. In many cases, COFs are used as scaffolds to combine with metal nanoparticles, conductive matrices, or selectors, to ensure satisfactory detection performance. The dependence on the complex composite structure can bring difficulty in device fabrication and also lead to unpredictable stability issues in the detection process. Fortunately, the great tunability and designability of COFs make it possible to construct COFs that harmoniously integrate the feature of high electrical conductivity, specific active binding sites, and redox-active units, ideally leading to more advanced applications in electrically transduced detection.78,91,125,309–311,320,323 Moreover, the utilization of conductive COFs for chemiresistive detection has emerged,91,128 paving the way for the development of other electronic configurations for electrically transduced detection platforms.
As another important aspect of COFs in chemical detection, COFs have acted as effective adsorbents in solid-phase extraction for complex and trace sample analysis. The experimental parameters for the extraction, including adsorbent dosage, extraction time, pH and ion strength, desorption solvent, and time, can affect the extraction efficiency, and thus need to be examined thoroughly before chromatography and spectrometry analysis. Some of the above procedures can be time-consuming and solvent intensive, and unsuitable for online microfluidic analysis. In this regard, the development of simple, quick, and efficient extraction techniques is urgently needed. Furthermore, the construction of novel COFs with different dimensions, pore geometry, and functional groups could be the key to increase their extraction efficiency and selectivity.
In addition to these challenges for specific detection modes, future directions for chemical detection using COFs also include the development of new strategies to increase the sensitivity and selectivity, the exploration of appropriate methods for the seamless integration of COFs on the substrate or electrode, and the discovery and synthetic access of new COFs with desired properties that can meet the needs of specific detection tasks. The advances of these directions ask for innovations of material design and synthesis, morphological control, and device fabrication by harnessing the useful concepts from reticular chemistry, materials chemistry, supramolecular chemistry, and biochemistry.
Sensitivity and selectivity can be achieved by the combination of the porous structure of COFs and other active components to form stable composite structures. It is known that enzymes have high efficiency and specificity in catalyzing biochemical reactions because of the specific molecular recognition of substrates by enzymes. MOFs have been gaining popularity as platforms for enzyme immobilization for catalysis and sensing,425 due to flexibility in their structural/chemical design and great surface tunability. Likewise, as the counterpart of MOFs, COFs with ordered structure and tunable pore parameters can provide excellent platforms for enzyme immobilization that can retain the integrity and activity of enzymes and enhance their stability and recyclability.425,426 Even though most COFs are microporous with pores not large enough to accommodate enzymes, mesoporous COFs with large pores up to a few nanometers have been reported,427–434 paving the way for the storage of large molecules. Initial investigations on the immobilization of enzymes in mesoporous COFs have revealed excellent enzymatic activity and durability for catalytic reactions.431–433 Transducing these interactions into the changes in physical and electronic properties of COFs may allow the detection of the participating molecules in these interactions. Therefore, we anticipate that the immobilization of enzymes in mesoporous materials is potentially useful for detection, such as sensing of biologically related analytes, by utilizing the greater number of interaction sites for the analyte molecules provided by encapsulated enzymes.
Reliable methods for interfacing COFs with electrodes or incorporating them into the substrate to generate high-quality electronic devices still need to be developed. Studies on relevant MOF-based materials have shown that thin films can offer the features of fast mass and electron transport, maximized exposed surface area, and desired mechanical properties. The exploitation of MOF thin films has realized the fabrication of devices with rapid response, ultrasensitivity, transparency, and flexibility for electronic,435,436 electrochemical, and electromechanical detection.437 Analogously, the growth of thin COF films with control over the film thickness, morphology, and crystallinity is highly desirable for the fabrication of functional, miniaturized, and portable electronic devices for chemical detection in a low-cost and scalable way.98 However, COFs are typically synthesized under solvothermal conditions with monomers and intermediate oligomers partially soluble in the reaction solvent.48 This heterogeneity provides insoluble and unprocessable microcrystalline powder aggregates and usually leads to poor control over the morphology of COFs, as compared to MOFs. Strategic use of the bottom-up principle of COF synthesis and template strategies can offer the opportunity to organize and orient functional π-electron systems into robust periodic structures.100 Pioneering work by Dichtel and co-workers has enabled the growth of oriented 2D layered COF films on substrate-supported single-layer graphene. Recently, the interface synthesis with the aid of surfactants has been utilized to produce ultrathin COF nanosheets with atomic thickness.438,439 In addition to growing thin films, a surface deposition strategy has also been used for integrating COFs with other substrates such as melamine foam440 and capillary columns.57 The terracotta process81 and 3D printing technology441 have also recently been developed for the rapid and facile fabrication of COFs with controlled shape, size, and composition. These advances provide great opportunities for optimizing the procedures from COF synthesis to device fabrication for further exploration in fluorescence, electrochemical, chemiresistive, and chromatography detection.442
Continuous efforts towards making full use of the principles of reticular chemistry to precisely integrate functional building blocks to achieve predesigned compositions, components, and properties of COFs are still highly desired.96 Since the first report of COFs in 2005, the synthesis of the COFs has been rapidly developed and the structural diversity of COFs has greatly expanded, alongside a broadening of the scope of applications of COFs. Nevertheless, control over the growth, defect sites, stacking, and morphology of COFs is still important for establishing precise structure–function correlations. Additionally, computational assessment of COF materials can provide valuable guidance and information for the design, characterization, and property evaluation of COFs.443,444 The development of new COFs with unprecedented optical, electronic, and magnetic properties, as well as stable chemical structures,47,97 is expected to provide opportunities for the advancement of currently existing detection modes and give insights into exploring novel mechanisms for chemical detection.68,69
Abbreviation
| MEG | 2-(2-(2-Methoxyethoxy)ethoxy)ethanamine |
| CEES | 2-Chloroethyl ethyl sulfide |
| Tetryl | 2,4,6-Trinitrophenylmethylnitramine |
| TNP | 2,4,6-Trinitrophenol |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| MQCA | 3-Methylquinoline-2-carboxylic acid |
| PEA | β-Phenylethylamine |
| AF | Aflatoxin |
| ACQ | Aggregation caused quenching |
| AIE | Aggregation induced emission |
| AMP | Ampicillin |
| Ab | Antibody |
| AA | Ascorbic acid |
| AB | Aniline |
| BET | Brunauer–Emmett–Teller |
| BHb | Bovine hemoglobin |
| BSA | Bovine serum albumin |
| CRP | C-Reactive protein |
| CND | Carbon nanodot |
| FAM | Carboxyfluorescein |
| CBX | Carbadox |
| CEA | Carcinoembryonic antigen |
| cTnI | Cardiac troponin I |
| CT | Charge transfer |
| COF | Covalent organic framework |
| CONs | Covalent organic nanosheets |
| COPs | Covalent organic polymers |
| CV | Cyclic voltammetry |
| Cys | Cysteine |
| DON | Deoxynivalenol |
| DCM | Dichloromethane |
| DPV | Differential pulse voltammetry |
| DNP | Dinitrophenol |
| DNB | Dinitrobenzene |
| DNT | Dinitrotoluene |
| ADR | Doxorubicin hydrochloride |
| EIS | Electrochemical impedance spectroscopy |
| EET | Electronic energy transfer |
| ET | Energy transfer |
| ENR | Enrofloxacin |
| EtOAc | Ethyl acetate |
| EGFR | Epidermal growth factor receptor |
| ESIPT | Excited-state intramolecular proton transfer |
| FRET | Fluorescence resonance energy transfer |
| GC | Gas chromatography |
| Glu | Glutamic acid |
| GSH | Glutathione |
| GMA–EDMA | Glycidyl methacrylate–ethylene dimethacrylate |
| Gly | Glycine |
| AE | Gold electrode |
| GO | Graphene oxide |
| Hsp90α | Heat shock protein 90α |
| HIS | Histidine |
| HOMO | Highest occupied molecular orbital |
| HPLC | High performance liquid chromatography |
| Hcy | Homocysteine |
| HCG | Human chorionic gonadotropin |
| HER2 | Human epidermal growth factor receptor 2 |
| HSA | Human serum albumin |
| HCR | Hybridization chain reaction |
| HQ | Hydroquinone |
| OH-PCBs | Hydroxylated polychlorinated biphenyls |
| IgG | Immunoglobulin G |
| ICT | Intramolecular charge transfer |
| IPA | Isopropyl alcohol |
| Kana | Kanamycin |
| LOD | Limit of detection |
| LSV | Linear sweep voltammetry |
| LUMO | Lowest occupied molecular orbital |
| MS | Mass spectrometry |
| MEQ | Mequindox |
| MLCT | Metal–ligand charge transfer |
| MOF | Metal–organic framework |
| MC–LR | Microcystin-LR |
| MSPE | Micro-solid-phase extraction |
| MIP | Molecularly imprinted polymer |
| MUC1 | Mucin 1 |
| DMF | N,N-Dimethylmethanamide |
| NS | Nanosheeet |
| NP | Nanoparticle |
| NB | Nitrobenzene |
| Np | Nitrophenol |
| NT | Nitrotoluene |
| OTA | Ochratoxin A |
| OLA | Olaquindox |
| OTC | Oxytetracycline |
| PBS | Phosphate-buffered saline |
| PET | Photo-induced electron transfer |
| PDGF-BB | Platelet-derived growth factor with two B subunits |
| PPD | p-Phenylenediamine |
| PAHs | Polycyclic aromatic hydrocarbons |
| PDA | Polydopamine |
| PI | Polyimide |
| PSA | Prostate specific antigen |
| QCM | Quartz crystal microbalance |
| NaAc | Sodium acetate |
| SPME | Solid-phase microextraction |
| SWV | Square wave voltammetry |
| SSF | Stainless steel fiber |
| SSW | Stainless steel wire |
| RFP | Streptomycin |
| SERS | Surface enhanced Raman scattering |
| SWCNT | Single walled carbon nanotube |
| DSS | Suberic acid bis(N-hydroxy succinimide ester) |
| TCNQ | Tetracyanoquinodimethane |
| Thi | Thionine |
| 3D | Three-dimensional |
| TOF | Time-of-flight |
| TOB | Tobramycin |
| 2D | Two-dimensional |
| TICT | Twisted intramolecular charge transfer |
| UA | Uric acid |
| VEGF | Vascular endothelial growth factor |
| ZEN | Zearalenone |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from startup funds provided by Dartmouth College, Irving Institute for Energy and Society, Army Research Office Young Investigator Program (W911NF-17-1-0398), Sloan Research Fellowship (FG-2018-10561), Camille Dreyfus Teacher-Scholar Award, NSF CAREER Award (#1945218), Maximizing Investigators’ Research Award from the National Institutes of Health (R35GM138318), 3M Non-Tenured Faculty Award, and US Army Cold Regions Research & Engineering Lab (W913E519C0008), National Science Foundation EPSCoR award (#1757371).Notes and references
- R. A. Potyrailo, Chem. Rev., 2016, 116, 11877 CrossRef CAS PubMed.
- Y. Y. Broza, X. Zhou, M. Yuan, D. Qu, Y. Zheng, R. Vishinkin, M. Khatib, W. Wu and H. Haick, Chem. Rev., 2019, 119, 11761 CrossRef CAS PubMed.
- M. Mayer and A. J. Baeumner, Chem. Rev., 2019, 119, 7996 CrossRef CAS PubMed.
- K. Mitsubayashi, O. Niwa and U. Yuko, Chemical, Gas, and Biosensors for Internet of Things and Related Applications, Elsevier Inc, 2019 Search PubMed.
- W. Zhang, Y. Tang, A. Shi, L. Bao, Y. Shen, R. Shen and Y. Ye, Materials, 2018, 11, 1364 CrossRef PubMed.
- P. R. Haddad, Ion chromatography-principles and applications, Elsevier, 1990, pp. 343–385 DOI:10.1016/s0301-4770(08)61144-6.
- L. A. Ellis and D. J. Roberts, J. Chromatogr. A, 1997, 774, 3 CrossRef CAS.
- G. Guetens, G. De Boeck, M. S. Highley, M. Wood, R. A. A. Maes, A. A. M. Eggermont, A. Hanauske, E. A. de Bruijn and U. R. Tjaden, J. Chromatogr. A, 2002, 976, 239 CrossRef CAS PubMed.
- C. L. Wilkins, Science, 1983, 222, 291 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- D. A. Skoog, F. J. Holler and S. R. Crouch, Principles of Instrumental Analysis, Cengage Learning, 2017 Search PubMed.
- D. Diamond, S. Coyle, S. Scarmagnani and J. Hayes, Chem. Rev., 2008, 108, 652 CrossRef CAS PubMed.
- S. Joo and R. B. Brown, Chem. Rev., 2008, 108, 638 CrossRef CAS PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318 CrossRef CAS PubMed.
- F. Yi, H. Ren, J. Shan, X. Sun, D. Wei and Z. Liu, Chem. Soc. Rev., 2018, 47, 3152 RSC.
- V. A. Aksyuk, Nat. Nanotechnol., 2017, 12, 940 CrossRef CAS PubMed.
- Z. Meng, R. M. Stolz, L. Mendecki and K. A. Mirica, Chem. Rev., 2019, 119, 478 CrossRef CAS PubMed.
- F. Valentini and G. Palleschi, Anal. Lett., 2008, 41, 479 CrossRef CAS.
- R. Murray, Anal. Chem., 2009, 63, 493a CrossRef.
- F. S. Ligler and H. S. White, Anal. Chem., 2013, 85, 11161 CrossRef CAS PubMed.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS PubMed.
- F.-G. Bănică, Chemical Sensors and Biosensors, John Wiley & Sons, Ltd, 2012 Search PubMed.
- J. Janata and A. Bezegh, Anal. Chem., 1988, 60, 62R CrossRef CAS PubMed.
- K. Sinha Roy, D. R. Goud, A. Mazumder, B. Chandra, A. K. Purohit, M. Palit and D. K. Dubey, ACS Appl. Mater. Interfaces, 2019, 11, 16027 CrossRef CAS PubMed.
- N. Negreira, M. Lopez de Alda and D. Barcelo, J. Chromatogr. A, 2013, 1280, 64 CrossRef CAS PubMed.
- J. Zhang and Z. Chen, J. Chromatogr. A, 2017, 1530, 1 CrossRef CAS PubMed.
- D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, Wiley, 1973 Search PubMed.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578, 10.1039/c39770000578.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546 CrossRef CAS.
- M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151 CrossRef CAS.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401 CrossRef CAS.
- F. Yavari and N. Koratkar, J. Phys. Chem. Lett., 2012, 3, 1746 CrossRef CAS PubMed.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, Trends Anal. Chem., 2010, 29, 954 CrossRef CAS.
- E. Singh, M. Meyyappan and H. S. Nalwa, ACS Appl. Mater. Interfaces, 2017, 9, 34544 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed.
- J. Janata and M. Josowicz, Nat. Mater., 2003, 2, 19 CrossRef CAS PubMed.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423 CrossRef CAS.
- J. F. Fennell, Jr., S. F. Liu, J. M. Azzarelli, J. G. Weis, S. Rochat, K. A. Mirica, J. B. Ravnsbaek and T. M. Swager, Angew. Chem., Int. Ed., 2016, 55, 1266 CrossRef PubMed.
- D. R. Kauffman and A. Star, Angew. Chem., Int. Ed., 2008, 47, 6550 CrossRef CAS PubMed.
- K. A. Mirica, J. G. Weis, J. M. Schnorr, B. Esser and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 10740 CrossRef CAS PubMed.
- J. M. Azzarelli, K. A. Mirica, J. B. Ravnsbaek and T. M. Swager, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18162 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- D. Rodriguez-San-Miguel and F. Zamora, Chem. Soc. Rev., 2019, 48, 4375 RSC.
- G. Garberoglio, Langmuir, 2007, 23, 12154 CrossRef CAS PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, 3rd, J. Am. Chem. Soc., 2008, 130, 11580 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed.
- J. L. Mendoza-Cortes, S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, 3rd, J. Phys. Chem. A, 2010, 114, 10824 CrossRef CAS PubMed.
- L. Zhu and Y. B. Zhang, Molecules, 2017, 22, 1149 CrossRef PubMed.
- C. X. Yang, C. Liu, Y. M. Cao and X. P. Yan, Chem. Commun., 2015, 51, 12254 RSC.
- H. Xu, X. Chen, J. Gao, J. Lin, M. Addicoat, S. Irle and D. Jiang, Chem. Commun., 2014, 50, 1292 RSC.
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28, 1277 CrossRef CAS.
- H. L. Qian, C. X. Yang and X. P. Yan, Nat. Commun., 2016, 7, 12104 CrossRef CAS PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665 RSC.
- C. Zhang, B. H. Wu, M. Q. Ma, Z. Wang and Z. K. Xu, Chem. Soc. Rev., 2019, 48, 3811 RSC.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878 CrossRef CAS PubMed.
- R. K. Sharma, P. Yadav, M. Yadav, R. Gupta, P. Rana, A. Srivastava, R. Zbořil, R. S. Varma, M. Antonietti and M. B. Gawande, Mater. Horiz., 2019, 7, 411 RSC.
- T. Yang, Y. N. Cui, H. Y. Chen and W. H. Li, Acta Chim. Sin., 2017, 75, 339 CrossRef CAS.
- T. Wang, R. Xue, Y. L. Wei, M. Y. Wang, H. Guo and W. Yang, Prog. Chem., 2018, 30, 753 Search PubMed.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266 RSC.
- X. Zhang, G. Li, D. Wu, B. Zhang, N. Hu, H. Wang, J. Liu and Y. Wu, Biosens. Bioelectron., 2019, 145, 111699 CrossRef CAS PubMed.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Díaz Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS PubMed.
- M. Dogru and T. Bein, Chem. Commun., 2014, 50, 5531 RSC.
- A. K. Mandal, J. Mahmood and J.-B. Baek, ChemNanoMat, 2017, 3, 373 CrossRef CAS.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258 CrossRef CAS PubMed.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001 RSC.
- S. B. Alahakoon, C. M. Thompson, G. Occhialini and R. A. Smaldone, ChemSusChem, 2017, 10, 2116 CrossRef CAS PubMed.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- Y. Wu, D. Yan, Z. Zhang, M. M. Matsushita and K. Awaga, ACS Appl. Mater. Interfaces, 2019, 11, 7661 CrossRef CAS PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903 RSC.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856 CrossRef CAS PubMed.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed.
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052 CrossRef CAS PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478 CrossRef CAS PubMed.
- D. Stewart, D. Antypov, M. S. Dyer, M. J. Pitcher, A. P. Katsoulidis, P. A. Chater, F. Blanc and M. J. Rosseinsky, Nat. Commun., 2017, 8, 1102 CrossRef PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524 CrossRef CAS PubMed.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587 CrossRef CAS PubMed.
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587 CrossRef CAS PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Cote and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872 CrossRef CAS PubMed.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488 RSC.
- S. Cao, B. Li, R. Zhu and H. Pang, Chem. Eng. J., 2019, 355, 602 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807 CrossRef CAS PubMed.
- X. Deng, Y. S. Fang, S. Lin, Q. Cheng, Q. Y. Liu and X. M. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 3514 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705 CrossRef CAS PubMed.
- D. Gopalakrishnan and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 8357 CrossRef CAS PubMed.
- N. N. Sang, C. X. Zhan and D. P. Cao, J. Mater. Chem. A, 2015, 3, 92 RSC.
- E. Oezdemir, D. Thirion and C. T. Yavuz, RSC Adv., 2015, 5, 69010 RSC.
- L. Guo, X. F. Zeng and D. P. Cao, Sens. Actuators, B, 2016, 226, 273 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- P. Kumar, A. Deep and K.-H. Kim, Trends Anal. Chem., 2015, 73, 39 CrossRef CAS.
- W.-T. Koo, J.-S. Jang and I.-D. Kim, Chem, 2019, 5, 1938 CAS.
- H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364 RSC.
- A. Amini, S. Kazemi and V. Safarifard, Polyhedron, 2020, 177, 114260 CrossRef CAS.
- Ü. Anik, S. Timur and Z. Dursun, Microchim. Acta, 2019, 186, 196 CrossRef PubMed.
- Y. Shu, Q. Ye, T. Dai, Q. Xu and X. Hu, ACS Sens., 2021, 6, 641 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- V. Schroeder, S. Savagatrup, M. He, S. Lin and T. M. Swager, Chem. Rev., 2018, 119, 599 CrossRef PubMed.
- Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283 RSC.
- M. A. Carpenter, S. Mathur and A. Kolmakov, Metal Oxide Nanomaterials for Chemical Sensors, Springer, New York, NY, 2013 Search PubMed.
- C. Wang, L. Yin, L. Zhang, D. Xiang and R. Gao, Sensors, 2010, 10, 2088 CrossRef CAS PubMed.
- S. W. Thomas, 3rd, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed.
- P. Bamfield, Chromic Phenomena, Royal Society of Chemistry, 2010 Search PubMed.
- K. Dimroth, C. Reichardt, T. Siepmann and F. Bohlmann, Justus Liebigs Ann. Chem., 1963, 661, 1 CrossRef CAS.
- W. Huang, Y. Jiang, X. Li, X. J. Li, J. Y. Wang, Q. Wu and X. K. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8845 CrossRef CAS PubMed.
- S. Jhulki, A. M. Evans, X. L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J. L. Bredas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783 CrossRef CAS PubMed.
- H. V. Babu, M. G. M. Bai and M. Rajeswara Rao, ACS Appl. Mater. Interfaces, 2019, 11, 11029 CrossRef CAS PubMed.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Nat. Commun., 2018, 9, 3802 CrossRef PubMed.
- X. Li, Q. Gao, J. Aneesh, H.-S. Xu, Z. Chen, W. Tang, C. Liu, X. Shi, K. V. Adarsh, Y. Lu and K. P. Loh, Chem. Mater., 2018, 30, 5743 CrossRef CAS.
- R. Kulkarni, Y. Noda, D. Kumar Barange, Y. S. Kochergin, P. Lyu, B. Balcarova, P. Nachtigall and M. J. Bojdys, Nat. Commun., 2019, 10, 3228 CrossRef PubMed.
- L. Ascherl, E. W. Evans, J. Gorman, S. Orsborne, D. Bessinger, T. Bein, R. H. Friend and F. Auras, J. Am. Chem. Soc., 2019, 141, 15693 CrossRef CAS PubMed.
- J. He, F. J. Xu, J. Hu, S. L. Wang, X. D. Hou and Z. Long, Microchem. J., 2017, 135, 91 CrossRef CAS.
- J. Wang, X. Yang, T. Wei, J. Bao, Q. Zhu and Z. Dai, ACS Appl. Bio Mater., 2018, 1, 382 CrossRef CAS.
- Y. Xiong, L. Su, X. He, Z. Duan, Z. Zhang, Z. Chen, W. Xie, D. Zhu and Y. Luo, Sens. Actuators, B, 2017, 253, 384 CrossRef CAS.
- L. J. Su, Z. Zhang and Y. H. Xiong, Nanoscale, 2018, 10, 20120 RSC.
- W. R. Cui, C. R. Zhang, W. Jiang, R. P. Liang, S. H. Wen, D. Peng and J. D. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 9408 CrossRef CAS.
- W. Li, Y. Li, H. L. Qian, X. Zhao, C. X. Yang and X. P. Yan, Talanta, 2019, 204, 224 CrossRef CAS PubMed.
- M. Ziolek, J. Kubicki, A. Maciejewski, R. Naskrecki and A. Grabowska, J. Chem. Phys., 2006, 124, 124518 CrossRef PubMed.
- N. Xu, R.-L. Wang, D.-P. Li, Z.-Y. Zhou, T. Zhang, Y.-Z. Xie and Z.-M. Su, New J. Chem., 2018, 42, 13367 RSC.
- N. Popp, T. Homburg, N. Stock and J. Senker, J. Mater. Chem. A, 2015, 3, 18492 RSC.
- R. S. Li, H. Z. Zhang, J. Ling, C. Z. Huang and J. Wang, Appl. Spectrosc. Rev., 2015, 51, 129 CrossRef.
- M. I. Kim, M. S. Kim, M. A. Woo, Y. Ye, K. S. Kang, J. Lee and H. G. Park, Nanoscale, 2014, 6, 1529 RSC.
- F. Z. Cui, J. J. Xie, S. Y. Jiang, S. X. Gan, D. L. Ma, R. R. Liang, G. F. Jiang and X. Zhao, Chem. Commun., 2019, 55, 4550 RSC.
- Y. H. Xiong, Y. M. Qin, L. J. Su and F. G. Ye, Chem. – Eur. J., 2017, 23, 11037 CrossRef CAS PubMed.
- B. Valeur and M. r. N. Berberan-Santos, J. Chem. Educ., 2011, 88, 731 CrossRef CAS.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag Berlin Heidelberg, 1994 Search PubMed.
- G. Feng, G. Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179 RSC.
- J. V. Goodpaster and V. L. McGuffin, Anal. Chem., 2001, 73, 2004 CrossRef CAS PubMed.
- T. M. Swager, Acc. Chem. Res., 1998, 31, 201 CrossRef CAS.
- S. Singh, J. Hazard. Mater., 2007, 144, 15 CrossRef CAS PubMed.
- J. Li, C. E. Kendig and E. E. Nesterov, J. Am. Chem. Soc., 2007, 129, 15911 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
- A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC.
- Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889 CrossRef CAS PubMed.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti, A. Taglietti and D. Sacchi, Chem. – Eur. J., 1996, 2, 75 CrossRef CAS.
- S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731 RSC.
- G. A. Jones and D. S. Bradshaw, Front. Phys., 2019, 7, 100 CrossRef.
- J. Liang, Q. L. Nguyen and S. Matsika, Photochem. Photobiol. Sci., 2013, 12, 1387 CrossRef CAS PubMed.
- J. S. Wu, J. H. Zhou, P. F. Wang, X. H. Zhang and S. K. Wu, Org. Lett., 2005, 7, 2133 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed.
- M. Gao and B. Z. Tang, ACS Sens., 2017, 2, 1382 CrossRef CAS PubMed.
- J. Dong, X. Li, K. Zhang, Y. Di Yuan, Y. Wang, L. Zhai, G. Liu, D. Yuan, J. Jiang and D. Zhao, J. Am. Chem. Soc., 2018, 140, 19816 Search PubMed.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13 CrossRef CAS PubMed.
- S. J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542 CrossRef CAS PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803 RSC.
- J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120 CrossRef CAS PubMed.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416 CrossRef CAS PubMed.
- S. Dalapati, C. Gu and D. Jiang, Small, 2016, 12, 6513 CrossRef CAS PubMed.
- R. Xue, H. Guo, T. Wang, L. Gong, Y. N. Wang, J. B. Ai, D. D. Huang, H. Q. Chen and W. Yang, Anal. Methods, 2017, 9, 3737 RSC.
- D. Y. Chen, C. Liu, J. T. Tang, L. F. Luo and G. P. Yu, Polym. Chem., 2019, 10, 1168 RSC.
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885 CrossRef CAS PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026 CrossRef CAS PubMed.
- S. Dalapati, E. Q. Jin, M. Addicoat, T. Heine and D. L. Jiang, J. Am. Chem. Soc., 2016, 138, 5797 CrossRef CAS PubMed.
- S. Rochat and T. M. Swager, ACS Appl. Mater. Interfaces, 2013, 5, 4488 CrossRef CAS PubMed.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031 CrossRef CAS PubMed.
- Z. P. Li, Y. W. Zhang, H. Xia, Y. Mu and X. M. Liu, Chem. Commun., 2016, 52, 6613 RSC.
- G. Q. Lin, H. M. Ding, D. Q. Yuan, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302 CrossRef CAS PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed.
- M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC.
- Y. Zeng, Z. Fu, H. Chen, C. Liu, S. Liao and J. Dai, Chem. Commun., 2012, 48, 8114 RSC.
- D. Tanaka, S. Horike, S. Kitagawa, M. Ohba, M. Hasegawa, Y. Ozawa and K. Toriumi, Chem. Commun., 2007, 3142, 10.1039/b707947h.
- W. Zhang, L. G. Qiu, Y. P. Yuan, A. J. Xie, Y. H. Shen and J. F. Zhu, J. Hazard. Mater., 2012, 221, 147 CrossRef PubMed.
- L.-L. Wang, C.-X. Yang and X.-P. Yan, Sci. China: Chem., 2018, 61, 1470 CrossRef CAS.
- Y. P. Wang, Z. Y. Zhao, G. L. Li, Y. Yan and C. Hao, J. Mol. Model., 2018, 24, 153 CrossRef PubMed.
- Y. A. Yang, Z. Y. Zhao, Y. Yan, G. L. Li and C. Hao, New J. Chem., 2019, 43, 9274 RSC.
- X. D. Song, Z. Y. Zhao, D. H. Si, X. Wang, F. Y. Zhou, M. R. Zhang, Y. T. Shi and C. Hao, J. Mol. Model., 2019, 25, 248 CrossRef PubMed.
- Y. Yuan, H. Ren, F. Sun, X. Jing, K. Cai, X. Zhao, Y. Wang, Y. Wei and G. Zhu, J. Phys. Chem. C, 2012, 116, 26431 CrossRef CAS.
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137 RSC.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931 RSC.
- C. L. Zhang, S. M. Zhang, Y. H. Yan, F. Xia, A. N. Huang and Y. Z. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415 CrossRef CAS PubMed.
- R. Gomes and A. Bhaumik, RSC Adv., 2016, 6, 28047 RSC.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 10040 RSC.
- P. Das and S. K. Mandal, J. Mater. Chem. A, 2018, 6, 16246 RSC.
- D. Ray, S. Dalapati and N. Guchhait, Spectrochim. Acta, Part A, 2013, 115, 219 CrossRef CAS PubMed.
- P. Albacete, A. Lopez-Moreno, S. Mena-Hernando, A. E. Platero-Prats, E. M. Perez and F. Zamora, Chem. Commun., 2019, 55, 1382 RSC.
- P. Vishnoi, M. G. Walawalkar, S. Sen, A. Datta, G. N. Patwari and R. Murugavel, Phys. Chem. Chem. Phys., 2014, 16, 10651 RSC.
- P. Vishnoi, S. Sen, G. N. Patwari and R. Murugavel, New J. Chem., 2015, 39, 886 RSC.
- P. Vishnoi, M. G. Walawalkar and R. Murugavel, Cryst. Growth Des., 2014, 14, 5668 CrossRef CAS.
- M. W. Zhu, S. Q. Xu, X. Z. Wang, Y. Q. Chen, L. Y. Dai and X. Zhao, Chem. Commun., 2018, 54, 2308 RSC.
- Q. Gao, X. Li, G. H. Ning, K. Leng, B. B. Tian, C. B. Liu, W. Tang, H. S. Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349 RSC.
- M. Hu, W. Kang, B. Cheng, Z. Li, Y. Zhao and L. Li, Microchim. Acta, 2016, 183, 1713 CrossRef CAS.
- M. Hu, W. Kang, Z. Zhong, B. Cheng and W. Xing, Ind. Eng. Chem. Res., 2018, 57, 11668 CrossRef CAS.
- Y.-Y. Lv, J. Wu and Z.-K. Xu, Sens. Actuators, B, 2010, 148, 233 CrossRef CAS.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185 RSC.
- H. L. Qian, C. Dai, C. X. Yang and X. P. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 24999 CrossRef CAS PubMed.
- W. Liu, Y. P. Cao, W. Z. Wang, D. Y. Gong, T. Cao, J. Qian, K. Iqbal, W. W. Qin and H. C. Guo, Chem. Commun., 2019, 55, 167 RSC.
- L. Jarup, Br. Med. Bull., 2003, 68, 167 CrossRef PubMed.
- J. Huff, R. M. Lunn, M. P. Waalkes, L. Tomatis and P. F. Infante, Int. J. Occup. Environ. Health, 2007, 13, 202 CrossRef CAS PubMed.
- P. Bhatti, P. A. Stewart, A. Hutchinson, N. Rothman, M. S. Linet, P. D. Inskip and P. Rajaraman, Cancer Epidemiol., Biomarkers Prev., 2009, 18, 1841 CrossRef CAS PubMed.
- M. Valko, H. Morris and M. T. Cronin, Curr. Med. Chem., 2005, 12, 1161 CrossRef CAS PubMed.
- A. M. El-Sharkawy, O. Sahota, R. J. Maughan and D. N. Lobo, Clin. Nutr., 2014, 33, 6 CrossRef CAS PubMed.
- J.-D. Xiao, L.-G. Qiu, Y.-P. Yuan, X. Jiang, A.-J. Xie and Y.-H. Shen, Inorg. Chem. Commun., 2013, 29, 128 CrossRef CAS.
- Y. Z. Zhu, M. Qiao, W. C. Peng, Y. Li, G. L. Zhang, F. B. Zhang, Y. F. Li and X. B. Fan, J. Mater. Chem. A, 2017, 5, 9272 RSC.
- Z. Zhou, W. Zhong, K. Cui, Z. Zhuang, L. Li, L. Li, J. Bi and Y. Yu, Chem. Commun., 2018, 54, 9977 RSC.
- E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14270 CrossRef CAS PubMed.
- S. Yoon, A. E. Albers, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16030 CrossRef CAS PubMed.
- Y. Zhao and Z. Zhong, J. Am. Chem. Soc., 2006, 128, 9988 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443 CrossRef CAS PubMed.
- A. B. Descalzo, R. Martinez-Manez, R. Radeglia, K. Rurack and J. Soto, J. Am. Chem. Soc., 2003, 125, 3418 CrossRef CAS PubMed.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072 CrossRef CAS PubMed.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- Y. Li, F. Li, H. Zhang, Z. Xie, W. Xie, H. Xu, B. Li, F. Shen, L. Ye, M. Hanif, D. Ma and Y. Ma, Chem. Commun., 2007, 231, 10.1039/b612732k.
- T. Wang, R. Xue, H. Chen, P. Shi, X. Lei, Y. Wei, H. Guo and W. Yang, New J. Chem., 2017, 41, 14272 RSC.
- X. C. Wu, B. W. Wang, Z. Q. Yang and L. G. Chen, J. Mater. Chem. A, 2019, 7, 5650 RSC.
- G. Chen, H. H. Lan, S. L. Cai, B. Sun, X. L. Li, Z. H. He, S. R. Zheng, J. Fan, Y. Liu and W. G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830 CrossRef CAS PubMed.
- A. A. Aruffo, T. B. Murphy, D. K. Johnson, N. J. Rose and V. Schomaker, Inorg. Chim. Acta, 1982, 67, L25 CrossRef CAS.
- W.-R. Cui, C.-R. Zhang, W. Jiang, R.-P. Liang and J.-D. Qiu, ACS Appl. Nano Mater., 2019, 2, 5342 CrossRef CAS.
- W. R. Cui, C. R. Zhang, W. Jiang, F. F. Li, R. P. Liang, J. Liu and J. D. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- M. Zheng, H. Tan, Z. Xie, L. Zhang, X. Jing and Z. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 1078 CrossRef CAS PubMed.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105 RSC.
- B. Esser and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 8872 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790 CrossRef CAS PubMed.
- Y. W. Zhang, X. C. Shen, X. Feng, H. Xia, Y. Mu and X. M. Liu, Chem. Commun., 2016, 52, 11088 RSC.
- C. Long, L. W. He, F. Y. Ma, L. Wei, Y. X. Wang, M. A. Silver, L. H. Chen, Z. Lin, D. X. Gui, D. W. Juan, Z. F. Chai and S. A. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 15364 CrossRef PubMed.
- Q. Lin, X. Zhu, Y.-P. Fu, Y.-M. Zhang and T.-B. Wei, Dyes Pigm., 2015, 112, 280 CrossRef CAS.
- Z. Li, N. Huang, K. H. Lee, Y. Feng, S. Tao, Q. Jiang, Y. Nagao, S. Irle and D. Jiang, J. Am. Chem. Soc., 2018, 140, 12374 CrossRef CAS PubMed.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511 CrossRef CAS PubMed.
- S. Weiss, Science, 1999, 283, 1676 CrossRef CAS PubMed.
- Y. W. Peng, Y. Huang, Y. H. Zhu, B. Chen, L. Y. Wang, Z. C. Lai, Z. C. Zhang, M. T. Zhao, C. L. Tan, N. L. Yang, F. W. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698 CrossRef CAS PubMed.
- W. Li, C. X. Yang and X. P. Yan, Chem. Commun., 2017, 53, 11469 RSC.
- T. H. Ni, D. A. W. Zhang, J. Wang, S. Wang, H. L. Liu and B. G. Sun, Sens. Actuators, B, 2018, 269, 340 CrossRef CAS.
- Y. Wang, Y. Z. Wang and H. L. Liu, Nanomaterials, 2019, 9, 305 CrossRef CAS PubMed.
- J.-M. Wang, X. Lian and B. Yan, Inorg. Chem., 2019, 58, 9956 CrossRef CAS PubMed.
- D. W. Zhang, H. L. Liu, W. T. Geng and Y. P. Wang, Food Chem., 2019, 277, 639 CrossRef CAS PubMed.
- D. W. Zhang, Y. P. Wang, W. T. Geng and H. L. Liu, Sens. Actuators, B, 2019, 285, 546 CrossRef CAS.
- Y. Zhang, D. W. Zhang and H. L. Liu, Polymers, 2019, 11, 708 CrossRef CAS PubMed.
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443 CrossRef CAS PubMed.
- J. Q. Dong, X. Li, S. B. Peh, Y. D. Yuan, Y. X. Wang, D. X. Ji, S. J. Peng, G. L. Liu, S. M. Ying, D. Q. Yuan, J. W. Jiang, S. Ramakrishna and D. Zhao, Chem. Mater., 2019, 31, 146 CrossRef CAS.
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081 CrossRef CAS PubMed.
- P. Wang, F. Zhou, C. Zhang, S. Y. Yin, L. L. Teng, L. L. Chen, X. X. Hu, H. W. Liu, X. Yin and X. B. Zhang, Chem. Sci., 2018, 9, 8402 RSC.
- E. Verheyen, J. P. Schillemans, M. van Wijk, M. A. Demeniex, W. E. Hennink and C. F. van Nostrum, Biomaterials, 2011, 32, 3008 CrossRef CAS PubMed.
- J. E. Lofgreen, I. L. Moudrakovski and G. A. Ozin, ACS Nano, 2011, 5, 2277 CrossRef CAS PubMed.
- L. Friedman and J. G. Miller, Science, 1971, 172, 1044 CrossRef CAS PubMed.
- X. Han, J. Huang, C. Yuan, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 892 CrossRef CAS PubMed.
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408 CrossRef CAS PubMed.
- L. J. O'Connor, I. N. Mistry, S. L. Collins, L. K. Folkes, G. Brown, S. J. Conway and E. M. Hammond, ACS Cent. Sci., 2017, 3, 20 CrossRef PubMed.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58 CrossRef CAS PubMed.
- A. Akthakul, N. Maklakov and J. White, Anal. Chem., 2010, 82, 6487 CrossRef CAS PubMed.
- Y. Wang, J. T. Chen and X. P. Yan, Anal. Chem., 2013, 85, 2529 CrossRef CAS PubMed.
- D. Kaleeswaran and R. Murugavel, J. Chem. Sci., 2018, 130, 1 CrossRef CAS.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421 CrossRef CAS PubMed.
- Y. Y. Cai, Y. Jiang, L. P. Feng, Y. Hua, H. Liu, C. Fan, M. Y. Yin, S. Li, X. X. Lv and H. Wang, Anal. Chim. Acta, 2019, 1057, 88 CAS.
- Y. Li, M. Zhang, X. Guo, R. Wen, X. Li, X. Li, S. Li and L. Ma, Nanoscale Horiz., 2018, 3, 205 RSC.
- H. L. Liu, Y. Zhang, D. W. Zhang, F. P. Zheng, M. Q. Huang, J. Y. Sun, X. T. Sun, H. H. Li, J. Wang and B. G. Sun, Microchim. Acta, 2019, 186, 182 CrossRef PubMed.
- U. Guth, W. Vonau and J. Zosel, Meas. Sci. Technol., 2009, 20, 042002 CrossRef.
- E. Barsoukov and J. R. Macdonald, Impedance Spectroscopy: Theory, Experiment, and Applications, Wiley, 2018 Search PubMed.
- M. E. Orazem and B. Tribollet, Electrochemical Impedance Spectroscopy, Wiley, 2017 Search PubMed.
- A. Lasia, Electrochemical Impedance Spectroscopy and its Applications, Springer, New York, 2014 Search PubMed.
- D. Antuña-Jiménez, G. Díaz-Díaz, M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, Molecularly Imprinted Sensors, 2012, pp. 1–34 DOI:10.1016/b978-0-444-56331-6.00001-3.
- G. Shen, P.-C. Chen, K. Ryu and C. Zhou, J. Mater. Chem., 2009, 19, 828 RSC.
- P. Y. Wang, M. M. Kang, S. M. Sun, Q. Liu, Z. H. Zhang and S. M. Fang, Chin. J. Chem., 2014, 32, 838 CrossRef CAS.
- H. Singh, V. K. Tomer, N. Jena, I. Bala, N. Sharma, D. Nepak, A. De Sarkar, K. Kailasam and S. K. Pal, J. Mater. Chem. A, 2017, 5, 21820 RSC.
- T. Z. Liu, J. R. Xia, Y. Li, W. K. Chen, S. Zhang, Y. Liu, L. Zheng and Y. H. Yang, Chem. J. Chin. Univ., 2015, 36, 1880 CAS.
- Z. Cao, F. Duan, X. Huang, Y. Liu, N. Zhou, L. Xia, Z. Zhang and M. Du, Anal. Chim. Acta, 2019, 1082, 176 CrossRef CAS PubMed.
- Y. Ma, M. Lu, Y. Deng, R. Bai, X. Zhang, D. Li, K. Zhang, R. Hu and Y. Yang, J. Biomed. Nanotechnol., 2018, 14, 1169 CrossRef CAS PubMed.
- T. Zhang, N. Ma, A. Ali, Q. Wei, D. Wu and X. Ren, Biosens. Bioelectron., 2018, 119, 176 CrossRef CAS PubMed.
- X. Yan, Y. P. Song, J. M. Liu, N. Zhou, C. L. Zhang, L. H. He, Z. H. Zhang and Z. Y. Liu, Biosens. Bioelectron., 2019, 126, 734 CrossRef CAS PubMed.
- X. K. Liu, M. Y. Hu, M. H. Wang, Y. P. Song, N. Zhou, L. H. He and Z. H. Zhang, Biosens. Bioelectron., 2019, 123, 59 CrossRef CAS PubMed.
- M. H. Wang, M. Y. Hu, J. M. Liu, C. P. Guo, D. L. Peng, Q. J. Jia, L. H. He, Z. H. Zhang and M. Du, Biosens. Bioelectron., 2019, 132, 8 CrossRef CAS PubMed.
- T. Zhang, Y. L. Chen, W. Huang, Y. Wang and X. Y. Hu, Sens. Actuators, B, 2018, 276, 362 CrossRef CAS.
- Y. F. Sun, L. H. Xu, G. I. N. Waterhouse, M. L. Wang, X. G. Qiao and Z. X. Xu, Sens. Actuators, B, 2019, 281, 107 CrossRef CAS.
- M. Xu, L. Wang, Y. Xie, Y. Song and L. Wang, Sens. Actuators, B, 2019, 281, 1009 CrossRef CAS.
- Y. Deng, X. T. Du, Y. C. Ma, K. L. Zhang, X. Zhang, D. L. Li, R. Y. Bai, R. Hu and Y. H. Yang, Nanosci. Nanotechnol. Lett., 2018, 10, 520 CrossRef.
- X. Zhang, K. N. Chi, D. L. Li, Y. Deng, Y. C. Ma, Q. Q. Xu, R. Hu and Y. H. Yang, Biosens. Bioelectron., 2019, 129, 64 CrossRef CAS PubMed.
- N. Zhou, Y. S. Ma, B. Hu, L. H. He, S. J. Wang, Z. H. Zhang and S. Y. Lu, Biosens. Bioelectron., 2019, 127, 92 CrossRef CAS PubMed.
- R. Y. Wang, W. H. Ji, L. Q. Huang, L. P. Guo and X. Wang, Anal. Lett., 2019, 52, 1757 CrossRef CAS.
- Y. Yue, P. Cai, X. Xu, H. Li, H. Chen, H. C. Zhou and N. Huang, Angew. Chem., Int. Ed., 2021, 60, 10806 CrossRef CAS PubMed.
- A. A. Ansari, A. Kaushik, P. R. Solanki and B. D. Malhotra, Bioelectrochemistry, 2010, 77, 75 CrossRef CAS PubMed.
- Z. Zhang, S. Liu, Y. Shi, Y. Zhang, D. Peacock, F. Yan, P. Wang, L. He, X. Feng and S. Fang, J. Mater. Chem. B, 2014, 2, 1530 RSC.
- L. Bai, S. Z. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128 RSC.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124 CrossRef CAS PubMed.
- Y. Suzuki, M. Endo and H. Sugiyama, Nat. Commun., 2015, 6, 8052 CrossRef CAS PubMed.
- J. Teixeira, A. Gaspar, E. M. Garrido, J. Garrido and F. Borges, BioMed Res. Int., 2013, 2013, 251754 Search PubMed.
- T. W. Crozier, A. Stalmach, M. E. Lean and A. Crozier, Food Funct., 2012, 3, 30 RSC.
- P. Wang, Q. Wu, L. Han, S. Wang, S. Fang, Z. Zhang and S. Sun, RSC Adv., 2015, 5, 27290 RSC.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- B. Sun, J. Liu, A. Cao, W. Song and D. Wang, Chem. Commun., 2017, 53, 6303 RSC.
- T. Zhang, C. W. Gao, W. Huang, Y. L. Chen, Y. Wang and J. M. Wang, Talanta, 2018, 188, 578 CrossRef CAS PubMed.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernandez-Burgos, K. E. Silberstein, G. G. Rodriguez-Calero, R. P. Bisbey, H. D. Abruna and W. R. Dichtel, ACS Nano, 2015, 9, 3178 CrossRef CAS PubMed.
- D.-H. Yang, Z.-Q. Yao, D. Wu, Y.-H. Zhang, Z. Zhou and X.-H. Bu, J. Mater. Chem. A, 2016, 4, 18621 RSC.
- S. Chandra, D. Roy Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074 CrossRef CAS.
- Y. Zhang, S. N. Riduan and J. Wang, Chem. – Eur. J., 2017, 23, 16419 CrossRef CAS PubMed.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16 CrossRef CAS PubMed.
- H. Ding, Y. Li, H. Hu, Y. Sun, J. Wang, C. Wang, C. Wang, G. Zhang, B. Wang, W. Xu and D. Zhang, Chem. – Eur. J., 2014, 20, 14614 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673 CrossRef CAS PubMed.
- S. Kim and H. C. Choi, Commun. Chem., 2019, 2, 60 CrossRef.
- M. Wang, M. Ballabio, M. Wang, H. H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810 CrossRef CAS PubMed.
- H. Yang, S. Zhang, L. Han, Z. Zhang, Z. Xue, J. Gao, Y. Li, C. Huang, Y. Yi, H. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 5366 CrossRef CAS PubMed.
- T. M. Swager, Macromolecules, 2017, 50, 4867 CrossRef CAS.
- M. Kertesz, C. H. Choi and S. Yang, Chem. Rev., 2005, 105, 3448 CrossRef CAS PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948 CrossRef CAS.
- J. L. Petersen, C. S. Schramm, D. R. Stojakovic, B. M. Hoffman and T. J. Marks, J. Am. Chem. Soc., 1977, 99, 286 CrossRef CAS.
- N. S. Hush, Ann. N. Y. Acad. Sci., 2003, 1006, 1 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566 CrossRef CAS PubMed.
- G. Givaja, P. Amo-Ochoa, C. J. Gomez-Garcia and F. Zamora, Chem. Soc. Rev., 2012, 41, 115 RSC.
- M. Ko, L. Mendecki and K. A. Mirica, Chem. Commun., 2018, 54, 7873 RSC.
- S.-L. Cai, Y.-B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S.-R. Zheng, W.-G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693 RSC.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806 CrossRef CAS PubMed.
- V. Lakshmi, C. H. Liu, M. Rajeswara Rao, Y. Chen, Y. Fang, A. Dadvand, E. Hamzehpoor, Y. Sakai-Otsuka, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 2155 CrossRef CAS PubMed.
- L. Wang, C. Zeng, H. Xu, P. Yin, D. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. Ma, Chem. Sci., 2019, 10, 1023 RSC.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230 CrossRef CAS PubMed.
- J. Yoo, S. J. Cho, G. Y. Jung, S. H. Kim, K. H. Choi, J. H. Kim, C. K. Lee, S. K. Kwak and S. Y. Lee, Nano Lett., 2016, 16, 3292 CrossRef CAS PubMed.
- Y. Guo, K. Ruan, X. Yang, T. Ma, J. Kong, N. Wu, J. Zhang, J. Gu and Z. Guo, J. Mater. Chem. C, 2019, 7, 7035 RSC.
- Y. Gu, J. P. Wang, H. P. Shi, M. F. Pan, B. Liu, G. Z. Fang and S. Wang, Biosens. Bioelectron., 2019, 128, 129 CrossRef CAS PubMed.
- D. DeVault, J. Am. Chem. Soc., 1943, 65, 532 CrossRef CAS.
- O. Coskun, North. Clin. Istanb., 2016, 3, 156 Search PubMed.
- S. Mitra, Sample Preparation Techniques in Analytical Chemistry, John Wiley & Sons, Inc., 2003 Search PubMed.
- M.-C. Hennion, J. Chromatogr. A, 1999, 856, 3 CrossRef CAS PubMed.
- A. Chisvert, S. Cárdenas and R. Lucena, Trends Anal. Chem., 2019, 112, 226 CrossRef CAS.
- M. Šafaříková and I. Šafařík, J. Magn. Magn. Mater., 1999, 194, 108 CrossRef.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 2002, 62, 2145 CrossRef.
- I. Vasconcelos and C. Fernandes, Trends Anal. Chem., 2017, 89, 41 CrossRef CAS.
- C.-H. Xu, G.-S. Chen, Z.-H. Xiong, Y.-X. Fan, X.-C. Wang and Y. Liu, Trends Anal. Chem., 2016, 80, 12 CrossRef CAS.
- F. Augusto, L. W. Hantao, N. G. S. Mogollón and S. C. G. N. Braga, Trends Anal. Chem., 2013, 43, 14 CrossRef CAS.
- C. W. Huck and G. K. Bonn, J. Chromatogr. A, 2000, 885, 51 CrossRef CAS PubMed.
- M. Polo, M. Llompart, C. Garcia-Jares and R. Cela, J. Chromatogr. A, 2005, 1072, 63 CrossRef CAS PubMed.
- T. Nema, E. C. Chan and P. C. Ho, Talanta, 2010, 82, 488 CrossRef CAS PubMed.
- H. Yu, T. D. Ho and J. L. Anderson, Trends Anal. Chem., 2013, 45, 219 CrossRef CAS.
- M. M. Sanagi, S. Salleh, W. A. W. Ibrahim, A. A. Naim, D. Hermawan, M. Miskam, I. Hussain and H. Y. Aboul-Enein, J. Food Compos. Anal., 2013, 32, 155 CrossRef CAS.
- L. M. Ravelo-Perez, A. V. Herrera-Herrera, J. Hernandez-Borges and M. A. Rodriguez-Delgado, J. Chromatogr. A, 2010, 1217, 2618 CrossRef CAS PubMed.
- C. Herrero-Latorre, J. Barciela-Garcia, S. Garcia-Martin and R. M. Pena-Crecente, Anal. Chim. Acta, 2018, 1002, 1 CrossRef CAS PubMed.
- M. E. I. Badawy, M. A. M. El-Nouby and A. E. M. Marei, Int. J. Anal. Chem., 2018, 2018, 3640691 Search PubMed.
- J. Ma, Z. Yao, L. Hou, W. Lu, Q. Yang, J. Li and L. Chen, Talanta, 2016, 161, 686 CrossRef CAS PubMed.
- F. Maya, C. Palomino Cabello, R. M. Frizzarin, J. M. Estela, G. Turnes Palomino and V. Cerdà, Trends Anal. Chem., 2017, 90, 142 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708 RSC.
- C. J. Zhang, G. K. Li and Z. M. Zhang, J. Chromatogr. A, 2015, 1419, 1 CrossRef CAS PubMed.
- H. Q. Wang, Z. Li, W. Feng and Q. Jia, New J. Chem., 2017, 41, 13043 RSC.
- R. Wang and Z. L. Chen, Microchim. Acta, 2017, 184, 3867 CrossRef CAS.
- Z. J. Yan, M. He, B. B. Chen, B. Gui, C. Wang and B. Hu, J. Chromatogr. A, 2017, 1525, 32 CrossRef CAS PubMed.
- S. H. Zhang, Q. Yang, Z. Li, W. C. Wang, C. Wang and Z. Wang, Anal. Bioanal. Chem., 2017, 409, 3429 CrossRef CAS PubMed.
- L. Chen, Y. T. He, Z. X. Lei, C. L. Gao, Q. Xie, P. Tong and Z. Lin, Talanta, 2018, 181, 296 CrossRef CAS PubMed.
- D. D. Jiang, T. T. Hu, H. J. Zheng, G. X. Xu and Q. Jia, Chem. – Eur. J., 2018, 24, 10390 CrossRef CAS PubMed.
- N. Li, D. Wu, N. Hu, G. S. Fan, X. T. Li, J. Sun, X. F. Chen, Y. R. Suo, G. L. Li and Y. N. Wu, J. Agric. Food Chem., 2018, 66, 3572 CrossRef CAS PubMed.
- N. Li, D. Wu, J. C. Liu, N. Hu, X. X. Shi, C. J. Dai, Z. W. Sun, Y. R. Suo, G. L. Li and Y. N. Wu, Microchem. J., 2018, 143, 350 CrossRef CAS.
- J. Q. Ma, J. Y. Ren, L. L. Wang, X. Wang, J. M. Lin and R. S. Zhao, J. Sep. Sci., 2018, 41, 3724 CrossRef CAS PubMed.
- J. Y. Ren, X. L. Wang, X. L. Li, M. L. Wang, R. S. Zhao and J. M. Lin, Anal. Bioanal. Chem., 2018, 410, 1657 CrossRef CAS PubMed.
- A. Shahvar, R. Soltani, M. Saraji, M. Dinari and S. Alijani, J. Chromatogr. A, 2018, 1565, 48 CrossRef CAS PubMed.
- X. X. Shi, N. Li, D. Wu, N. Hu, J. Sun, X. X. Zhou, Y. R. Suo, G. L. Li and Y. N. Wu, Anal. Methods, 2018, 10, 5014 RSC.
- Q. Q. Wang, H. H. Wu, F. Y. Lv, Y. T. Cao, Y. Zhou and N. Gan, J. Chromatogr. A, 2018, 1572, 1 CrossRef CAS PubMed.
- L. Xia, X. M. Chen, X. H. Xiao and G. K. Li, J. Sep. Sci., 2018, 41, 3733 CrossRef CAS PubMed.
- Z. H. Deng, X. Wang, X. L. Wang, C. L. Gao, L. Dong, M. L. Wang and R. S. Zhao, Microchim. Acta, 2019, 186, 108 CrossRef PubMed.
- W. Gao, Y. Tian, H. Liu, Y. Q. Cai, A. F. Liu, Y. L. Yu, Z. S. Zhao and G. B. Jiang, Anal. Chem., 2019, 91, 772 CrossRef CAS PubMed.
- K. Hu, Y. X. Lv, F. G. Ye, T. Chen and S. L. Zhao, Anal. Chem., 2019, 91, 6353 CrossRef CAS PubMed.
- L. Liu, W. K. Meng, L. Li, G. J. Xu, X. Wang, L. Z. Chen, M. L. Wang, J. M. Lin and R. S. Zhao, Chem. Eng. J., 2019, 369, 920 CrossRef CAS.
- L. Liu, W. K. Meng, Y. S. Zhou, X. Wang, G. J. Xu, M. L. Wang, J. M. Lin and R. S. Zhao, Chem. Eng. J., 2019, 356, 926 CrossRef CAS.
- Y. J. Lu, B. C. Wang, C. L. Wang, Y. H. Yan, D. P. Wu, H. Z. Liang and K. Q. Tang, Chromatographia, 2019, 82, 1089 CrossRef CAS.
- T. T. Ma, X. F. Shen, C. Yang, H. L. Qian, Y. H. Pang and X. P. Yan, Talanta, 2019, 201, 413 CrossRef CAS PubMed.
- S. H. Wang, H. Y. Niu, D. Cao and Y. Q. Cai, Talanta, 2019, 194, 522 CrossRef CAS PubMed.
- F. F. Wu, Q. Y. Chen, X. J. Ma, T. T. Li, L. F. Wang, J. Hong, Y. H. Sheng, M. L. Ye and Y. Zhu, Anal. Methods, 2019, 11, 3381 RSC.
- Y. H. Yan, Y. J. Lu, B. C. Wang, Y. Q. Gao, L. L. Zhao, H. Z. Liang and D. P. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 26539 CrossRef CAS PubMed.
- T. Wu, X. H. Zang, M. T. Wang, Q. Y. Chang, C. Wang, Q. H. Wu and Z. Wang, J. Agric. Food Chem., 2018, 66, 11158 CrossRef CAS PubMed.
- M. X. Wu, G. Chen, P. Liu, W. H. Zhou and Q. Jia, J. Chromatogr. A, 2016, 1456, 34 CrossRef CAS PubMed.
- W. H. Ji, R. H. Sun, Y. L. Geng, W. Liu and X. Wang, Anal. Chim. Acta, 2018, 1001, 179 CrossRef CAS PubMed.
- Y. H. Song, R. Y. Ma, L. Hao, X. M. Yang, C. Wang, Q. H. Wu and Z. Wang, J. Chromatogr. A, 2018, 1572, 20 CrossRef CAS PubMed.
- M. Wang, X. Zhou, X. Zang, Y. Pang, Q. Chang, C. Wang and Z. Wang, J. Sep. Sci., 2018, 41, 4038 CrossRef CAS PubMed.
- X. L. Wang, R. Y. Ma, L. Hao, Q. H. Wu, C. Wang and Z. Wang, J. Chromatogr. A, 2018, 1551, 1 CrossRef CAS PubMed.
- R. Y. Liang, Y. Peng, Y. L. Hu and G. K. Li, J. Sep. Sci., 2019, 42, 1432 CrossRef CAS PubMed.
- J. M. Liu, X. Z. Wang, C. Y. Zhao, J. L. Hao, G. Z. Fang and S. Wang, J. Hazard. Mater., 2018, 344, 220 CrossRef CAS PubMed.
- Y. L. Chen and Z. L. Chen, Talanta, 2017, 165, 188 CrossRef CAS PubMed.
- H. P. Wang, F. L. Jiao, F. Y. Gao, Y. Y. Lv, Q. Wu, Y. Zhao, Y. H. Shen, Y. J. Zhang and X. H. Qian, Talanta, 2017, 166, 133 CrossRef CAS PubMed.
- X. M. Wang, G. J. Xu, X. L. Wang, X. Wang, M. L. Wang, R. S. Zhao and J. M. Lin, Chin. J. Anal. Chem., 2018, 46, 1990 CAS.
- C. H. Gao, J. Bai, Y. T. He, Q. Zheng, W. D. Ma, Z. X. Lei, M. Y. Zhang, J. Wu, F. F. Fu and Z. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 13735 CrossRef CAS PubMed.
- W. X. Li, N. Chen, Y. Zhu, D. Shou, M. Y. Zhi and X. Q. Zeng, Microchim. Acta, 2019, 186, 76 CrossRef PubMed.
- J. M. Liu, S. W. Lv, X. Y. Yuan, H. L. Liu and S. Wang, RSC Adv., 2019, 9, 14247 RSC.
- Z. M. Yan, B. Q. Hu, Q. L. Li, S. X. Zhang, J. Pang and C. H. Wu, J. Chromatogr. A, 2019, 1584, 33 CrossRef CAS PubMed.
- J. X. Wang, J. Li, M. X. Gao and X. M. Zhang, Nanoscale, 2017, 9, 10750 RSC.
- R. Zhai, X. Gong, J. Xie, Y. Yuan, F. Xu, Y. Jiang, Z. Huang, X. Dai, Y. Zhang, X. Qian and X. Fang, Talanta, 2019, 191, 553 CrossRef CAS PubMed.
- J. Li, L. Yang, S. Luo, B. Chen, J. Li, H. Lin, Q. Cai and S. Yao, Anal. Chem., 2010, 82, 7357 CrossRef CAS PubMed.
- X. Wang, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 2007, 27, 133 CrossRef CAS PubMed.
- C. R. Pearson, Anthropogenic Compounds, 1982, ch. 4, pp. 89–116 DOI:10.1007/978-3-540-47028-1_4.
- P. Liljelind, G. Soderstrom, B. Hedman, S. Karlsson, L. Lundin and S. Marklund, Environ. Sci. Technol., 2003, 37, 3680 CrossRef CAS PubMed.
- P. Perego, L. Gatti and G. L. Beretta, Nat. Rev. Cancer, 2010, 10, 523 CrossRef CAS PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855 CrossRef CAS PubMed.
- N. Li, J. Du, D. Wu, J. Liu, N. Li, Z. Sun, G. Li and Y. Wu, Trends Anal. Chem., 2018, 108, 154 CrossRef CAS.
- J. Wang, J. Li, M. Gao and X. Zhang, Trends Anal. Chem., 2018, 108, 98 CrossRef CAS.
- S. Zhang, Q. Yang, C. Wang, X. Luo, J. Kim, Z. Wang and Y. Yamauchi, Adv. Sci., 2018, 5, 1801116 CrossRef PubMed.
- L. X. Chen, Q. Wu, J. Gao, H. Li, S. Q. Dong, X. F. Shi and L. Zhao, Trends Anal. Chem., 2019, 113, 182 CrossRef CAS.
- X. Xu, H. Li, D. Hasan, R. S. Ruoff, A. X. Wang and D. L. Fan, Adv. Funct. Mater., 2013, 23, 4332 CrossRef CAS.
- E. J. Blackie, E. C. Le Ru and P. G. Etchegoin, J. Am. Chem. Soc., 2009, 131, 14466 CrossRef CAS PubMed.
- E. C. Le, Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794 CrossRef.
- S. Schlucker, Angew. Chem., Int. Ed., 2014, 53, 4756 CrossRef PubMed.
- Y. Yuan, N. Panwar, S. H. K. Yap, Q. Wu, S. Zeng, J. Xu, S. C. Tjin, J. Song, J. Qu and K.-T. Yong, Coord. Chem. Rev., 2017, 337, 1 CrossRef CAS.
- J. He, F. J. Xu, Z. Chen, X. D. Hou, Q. Liu and Z. Long, Chem. Commun., 2017, 53, 11044 RSC.
- A. Sangsuwan, B. Narupai, P. Sae-ung, S. Rodtamai, N. Rodthongkum and V. P. Hoven, Anal. Chem., 2015, 87, 10738 CrossRef CAS PubMed.
- Z. Guo, Q. Zhang, H. Zou, B. Guo and J. Ni, Anal. Chem., 2002, 74, 1637 CrossRef CAS PubMed.
- X. Li, J. Tan, J. Yu, J. Feng, A. Pan, S. Zheng and J. Wu, Anal. Chim. Acta, 2014, 849, 27 CrossRef CAS PubMed.
- A. Nordstrom, J. V. Apon, W. Uritboonthai, E. P. Go and G. Siuzdak, Anal. Chem., 2006, 78, 272 CrossRef PubMed.
- C. Lopez de Laorden, A. Beloqui, L. Yate, J. Calvo, M. Puigivila, J. Llop and N. C. Reichardt, Anal. Chem., 2015, 87, 431 CrossRef CAS PubMed.
- M. Lu, Y. Lai, G. Chen and Z. Cai, Anal. Chem., 2011, 83, 3161 CrossRef CAS PubMed.
- D. Feng and Y. Xia, Anal. Chim. Acta, 2018, 1014, 58 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328 CrossRef CAS PubMed.
- Z. H. He, S. D. Gong, S. L. Cai, Y. L. Yan, G. Chen, X. L. Li, S. R. Zheng, J. Fan and W. G. Zhang, Cryst. Growth Des., 2019, 19, 3543 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
- S. K. Vashist and P. Vashist, J. Sens., 2011, 2011, 1 CrossRef.
- F. Feng, J. Zheng, P. Qin, T. Han and D. Zhao, Talanta, 2017, 167, 94 CrossRef CAS PubMed.
- Y. Gu, Y. N. Wang, X. M. Wu, M. F. Pan, N. Hu, J. P. Wang and S. Wang, Sens. Actuators, B, 2019, 291, 293 CrossRef CAS.
- S. Zhang, Q. Yang, X. Xu, X. Liu, Q. Li, J. Guo, N. L. Torad, S. M. Alshehri, T. Ahamad, M. S. A. Hossain, Y. V. Kaneti and Y. Yamauchi, Nanoscale, 2020, 12, 15611 RSC.
- L.-M. Yang, E. Ganz, S. Wang, X.-J. Li and T. Frauenheim, J. Mater. Chem. C, 2015, 3, 2244 RSC.
- M. Hussain, X. Song, S. Shah and C. Hao, Spectrochim. Acta, Part A, 2019, 224, 117432 CrossRef PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H. C. Zhou, Chem. Soc. Rev., 2017, 46, 3386 RSC.
- Y. Pan, H. Li, J. Farmakes, F. Xiao, B. Chen, S. Ma and Z. Yang, J. Am. Chem. Soc., 2018, 140, 16032 CrossRef PubMed.
- M. Dogru, A. Sonnauer, A. Gavryushin, P. Knochel and T. Bein, Chem. Commun., 2011, 47, 1707 RSC.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975 CrossRef CAS PubMed.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- H. Yang, Y. Du, S. Wan, G. D. Trahan, Y. Jin and W. Zhang, Chem. Sci., 2015, 6, 4049 RSC.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed.
- Q. Sun, C. W. Fu, B. Aguila, J. Perman, S. Wang, H. Y. Huang, F. S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, P. C. Lan and S. Ma, Adv. Mater., 2019, 31, e1900008 CrossRef PubMed.
- S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505 RSC.
- M. S. Yao, X. J. Lv, Z. H. Fu, W. H. Li, W. H. Deng, G. D. Wu and G. Xu, Angew. Chem., Int. Ed., 2017, 56, 16510 CrossRef CAS PubMed.
- J. Wu, J. Chen, C. Wang, Y. Zhou, K. Ba, H. Xu, W. Bao, X. Xu, A. Carlsson, S. Lazar, A. Meingast, Z. Sun and H. Deng, Adv. Sci., 2020, 7, 1903003 CrossRef CAS PubMed.
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Mater. Horizons, 2021, 8, 2387 RSC.
- K. Liu, H. Qi, R. Dong, R. Shivhare, M. Addicoat, T. Zhang, H. Sahabudeen, T. Heine, S. Mannsfeld, U. Kaiser, Z. Zheng and X. Feng, Nat. Chem., 2019, 11, 994 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. A. Perman, T. Butts, F.-S. Xiao and S. Ma, Chem, 2018, 4, 1726 CAS.
- M. Zhang, L. Li, Q. Lin, M. Tang, Y. Wu and C. Ke, J. Am. Chem. Soc., 2019, 141, 5154 CrossRef CAS PubMed.
- T. Zhang, H. Qi, Z. Liao, Y. D. Horev, L. A. Panes-Ruiz, P. S. Petkov, Z. Zhang, R. Shivhare, P. Zhang, K. Liu, V. Bezugly, S. Liu, Z. Zheng, S. Mannsfeld, T. Heine, G. Cuniberti, H. Haick, E. Zschech, U. Kaiser, R. Dong and X. Feng, Nat. Commun., 2019, 10, 4225 CrossRef PubMed.
- E. Tylianakis, E. Klontzas and G. E. Froudakis, Nanoscale, 2011, 3, 856 RSC.
- M. Tong, Y. Lan, Z. Qin and C. Zhong, J. Phys. Chem. C, 2018, 122, 13009 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |