
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Static quenching upon adduct formation: a treatment without shortcuts and approximations
Damiano
Genovese
 ,
Matteo
Cingolani
,
Enrico
Rampazzo
,
Luca
Prodi
* and
Nelsi
Zaccheroni
,
Matteo
Cingolani
,
Enrico
Rampazzo
,
Luca
Prodi
* and
Nelsi
Zaccheroni
Department of Chemistry “Giacomo Ciamician”, Università degli Studi di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: luca.prodi@unibo.it
First published on 18th June 2021
Abstract
Luminescence quenching is a process exploited in transversal applications in science and technology and it has been studied for a long time. The luminescence quenching mechanisms are typically distinguished in dynamic (collisional) and static, which can require different quantitative treatments. This is particularly important – and finds broad and interdisciplinary application – when the static quenching is caused by the formation of an adduct between the luminophore – at the ground state – and the quencher. Due to its nature, this case should be treated starting from the well-known law of mass action although, in specific conditions, general equations can be conveniently reduced to simpler ones. A proper application of simplified equations, though, can be tricky, with frequent oversimplifications taking to severe errors in the interpretation of the photophysical data. This tutorial review aims to (i) identify the precise working conditions for the application of the simplified equations of static quenching and to (ii) provide general equations for broadest versatility and applicability. The latter equations can be used even beyond the sole case of pure quenching, i.e., in the cases of partial quenching and even luminescence turn-on. Finally, we illustrate different applications of the equations via a critical discussion of examples in the field of sensing, supramolecular chemistry and nanotechnology.
 Damiano Genovese | Damiano Genovese received his PhD in Chemistry at University of Bologna in 2011, and two grants to fund research stays at Ecole Normale Superieure (Paris, France) and Harvard University (USA). He was an Alexander von Humboldt Fellow in Karlsruhe Institute of Technology (Germany). He was awarded the ENI prize 2013 for his “Debut in Research” and the GIF Young Investigator Award 2018. He is now in a tenure-track for associate professorship (RTD-b) at Università di Bologna. His research spans from colloids physics to surface and polymer chemistry, from self-assembling systems to nanostructured photoactive materials for optoelectronics, sensing, and biomedicine. |
 Matteo Cingolani | He received a Master's degree in Photochemistry and Molecular Materials in 2018. In the same year he was granted a research fellowship on the design and characterization of light activated nanostructures based on silica doped nanoparticles for medical application. Since 2019 Matteo is a PhD student in Nanoscience and his research focuses on the study of interactions of fluorescently labelled probes with different nano- and micro-structures through fluorescence microscopy techniques. His research is directed towards biological, medical and environmental applications. |
 Enrico Rampazzo | Born in Verona in 1973. He completed his PhD in chemistry at the University of Padua under the supervision of prof. Umberto Tonellato and prof. Fabrizio Mancin. He was a postdoctoral fellow under the supervision of prof. Luca Prodi and prof. Marco Montalti, and then fix-term researcher at the Photochemical Nanosciences Laboratory of the University of Bologna (Italy). He is now associate professor of general and inorganic chemistry at the University of Bologna. His research focusses on the development of luminescent dyes, sensors and (electro)luminescent systems based on supramolecular systems, dye doped silica nanoparticles and polymers. |
 Luca Prodi | Luca Prodi received his PhD in 1992. In 2006 he become a full Professor of General and Inorganic Chemistry at the University of Bologna where has been Head of the “Giacomo Ciamician” Department of Chemistry from 2015 to 2018. Since his graduation he dealt with the applications of photochemistry, focussing his interest – in the recent years – on the design of photoactive silica nanoparticles for nanomedicine. He is an inventor of several international patents and a co-founder of two spin-off companies. |
 Nelsi Zaccheroni | Nelsi Zaccheroni studied and obtained her PhD in Chemical Sciences (1997) at the University of Bologna (Italy), where she is now Associate Professor in General and Inorganic Chemistry since 2014. She was postdoctoral fellow within a TMR-CEE project at the University College of Dublin (Ireland) and then visiting professor in Australia and Canada, she is also co-inventor of a few patents. Her research interests are mainly focused on luminescent systems for imaging and sensing, spanning from molecular to nanostructured materials (including responsive polymers), for biomedical and environmental applications. |
Key learning points(1) Static quenching is a different mechanism than dynamic, therefore the linear “pseudo Stern–Volmer” relationship and other simplified approaches only apply in limited conditions.(2) The general equations for static quenching when this is caused by adduct formation are provided and compared with the systematic deviations of the pseudo Stern–Volmer approach. (3) The general equations allow error-free analysis of quenching data, provided that they are correctly acquired (or corrected post-acquisition). (4) The general equation allows broader application, also to cases of (i) partial quenching and (ii) luminescence turn-on. (5) Critical discussion of static quenching applications in sensing, supramolecular chemistry and nanotechnology facilitate transfer of knowledge to real cases. |
1. Introduction
All processes inducing the decrease of the luminescence intensity of a certain sample are termed luminescence quenching processes. Their importance in many fields, including chemistry and biology, is enormous. Luminescence quenching can be considered as a variation of an output signal (luminescence intensity) due to the nature and concentration of an input (the quencher), and thus can fit the definition of a sensing process. The application of chemical sensors is transversal to many fields of science and technology1–3 from the study of health risks or performance of medical therapies, to environmental risks, to agriculture and technological production monitoring. Chemical sensors are a thematic of current great scientific interest, and a significant part of them deals with luminescent chemosensors with transduction mechanisms based on quenching.4 Since the efficiency of the quenching process can also depend on the distance between the luminescent unity and the quencher, this process can be also used as a “molecular ruler”, to study, for example, the occurrence of chemical or biological reactions or conformational changes of large systems such as proteins.5The cross-cutting interest for luminescence quenching arises, therefore, from the concurrence of its many advantages: (i) experimental easiness, repeatability and reproducibility; (ii) cost-effectiveness, from purchase to maintenance of instrumentation; (iii) high sensitivity, that can lead to low detection limits and (iv) high spatial and temporal resolution, even in real-time. Added values are that quenching processes are often non-destructive and require small amounts of sample, a particularly beneficial feature in fields like biology, biochemistry and medicine where samples can be – by their nature – very small in size.
The definition of luminescence quenching is simple and straightforward, but the chemical processes and related mechanisms leading to quenching can vary and require different quantitative treatments. First of all, luminescence quenching is typically distinguished in dynamic (collisional) or static quenching. It is important to underline that both dynamic and static quenching are characterized by intimate mechanisms of deactivation of the excited state including, for example, energy (Förster and Dexter) or electron transfer, formation of excimer or exciplex, intersystem crossing or internal conversion. Unraveling the actual intimate mechanism leading to luminescence quenching is a complex task, that often requires information stemming from multiple analytical techniques, and that is beyond the scope of this tutorial review, which instead focusses on the correct recognition of static quenching upon chemical association, regardless of the specific intimate mechanism.
Both dynamic and static quenching have been known and studied for a long time. The first to be quantitatively discussed has been the dynamic one,6 that can be defined as a bimolecular process, occurring via diffusion-controlled collision events between the luminophore – at the excited state – and the quencher. Only after a few decades, experimental pieces of evidence took different scientists to introduce and investigate the static quenching mechanism.7 In particular, photophysical measurements of intensity variations in the presence of a quencher that deviated from the linear trend typical of dynamic quenching (see infra) were used to prove and study the formation and the stability of a fluorophore–quencher complex. The evidence and theorization of static quenching – as named by Gregorio Weber, its first theorist7 – had and has a great impact on the interpretation of processes occurring within complex matrixes such as the biological ones. As we will discuss later, many are the processes that can be categorized as static quenching, and that have been treated in the literature even with highly sophisticated mathematical and physical approaches.
The most common case of static quenching is however represented by the emission intensity decrease due to the formation of an adduct between the luminophore – at the ground state – and the quencher, in which the photophysical properties of the luminophore are profoundly altered. This is an equilibrium process; therefore, the concentrations in solution of the three different species – luminophore, quencher and adduct – is governed by the equilibrium association constant.
In the ‘60s and ‘70s, this kind of static quenching played a key role in the development of supramolecular chemistry, which had its birth with the pioneering work of Lehn,8 Pedersen9 and Cram10 who were awarded with the Nobel prize in 1987. This research field is still in continuous expansion and recognized as one of the topics of modern chemistry and is based on intermolecular interactions among suitable moieties that yield organized systems with emergent features. During years quenching has become, also in this field, a key diagnostic tool to quantitatively monitor such interactions with high time and space resolution and high sensitivity.11
However, even if quenching processes involve easy to perform reactions that are relatively easy to follow by luminescence intensities variations, their quantitative treatment and interpretation has been often misleading. Simplified equations have been used in place of the most general ones, regardless of the specific conditions, compatible or not with the operated simplifications. This tutorial review aims to help researchers to identify the precise working conditions for the application of the different equations of static quenching in case of chemical association, and, in addition, to provide the most general equations, also showing their superior versatility and applicability, which extends even beyond the sole case of quenching.
2. Dynamic quenching
To start this tutorial review, tracing back to the first papers quantitatively treating dynamic and static quenching will help reconstructing some fundamental hints that got lost or ambiguously reported in a sort of “telephone game” over the years.Luminescence quenching resulting from the collision between the emitting moiety and the quencher is called, collisional or – more commonly – dynamic quenching. The kinetics of this process was studied in early 1900s by Otto Stern and Max Volmer6 that proposed the well-known Stern–Vomer equation quantitatively describing this process and evidencing a linear dependence of Φ0/Φ from the concentration of the quencher in homogeneous solvents:
| Φ0/Φ = τ0/τ = 1 + kqτ0[Q] = 1 + KSV[Q] | (1) |
Plotting I0/I versus [Q] a linear dependence is thus expected, and the interpolation of the data gives a straight line with a slope equal to KSV and intercepting the y-axis at the value of one. It is worth noting that, because of the nature of this process, kq is typically not higher than the diffusion rate constant, kdiff, which depends on the temperature and the viscosity of the solvent. At room temperature for the most common solvents kdiff is lower than 4 × 1010 L mol−1 s−1 (in water at 25 °C kdiff = 7.4 × 109 L mol−1 s−1).13
Dynamic quenching is a deactivation process that depopulates the excited state competing with emission; consequently, the decrease of the excited state lifetime must be proportional to the decrease of the luminescence quantum yield. Thus, a characteristic feature of the dynamic quenching is the equivalence τ0/τ = I0/I.14,15
A detailed discussion of the derivation, application and interpretation of the Stern–Volmer equation is a wide and extremely interesting topic but it is beyond the goals of this tutorial review. We want to focus our attention, in fact, on the static quenching that rises, in our opinion, more shadows in its treatment. We will start with a brief introductory discussion on the first studies on this topic in the following section.
3. First approaches to static quenching
Early on, experimental data on the application of the Stern–Volmer equation to different systems, above all biological ones, showed deviations from the linear behaviour that were explained, initially by Frank and Wawilov, with the formation of short-living complexes.16 They proposed that, besides dynamic quenching, an additional mechanism could contribute to the decrease of the luminescence. During excitation, one or more quencher molecules could be statistically located inside an interaction sphere around the luminescent species, at a suitable interaction distance to form a dark complex, leading to its instantaneous quenching, while outside this sphere the quencher does not affect fluorescence. To take into account this additional contribution to the luminescence decrease, a new factor was introduced into the Stern–Volmer equation, i.e., the exponential factor eV[Q], where the term V represents an active volume surrounding the excited fluorophore.| I0/I = (1 + KSV[Q])eV[Q] | (2) |
After the observations of H. Weil-Malherbe on the quenching effects of purines on the emission intensity of 3,4-benzopyrene in acidic solutions, where the active quenching agent is the caffeine ion via the instauration of specific intermolecular forces,17 the first papers deeply investigating quenching as pure complex formation were published starting from the end of the 1940s by Gregorio Weber.7 His research work dealt with fluorescence quenching in solution by dark complex formation (riboflavin and caffeine) and he used this process to determine the association constant of the complex itself.
The equations proposed for the quantitative treatment of the experimental data were derived in the precise case in which: (i) the complex does not contribute to the emission (i.e., it is completely quenched or its residual emission is negligible) and, very importantly, (ii) the concentration of the fluorophore is negligible in comparison with the quencher one. In these well-defined conditions, Weber found again a linear dependence of I0/I on the concentration of the quencher deriving an equation completely analogous to the Stern–Volmer one:
| I0/I = 1 + Ka[Q] | (3) |
In the same year, Boaz and Rollefson18 tried to fit the curvatures deviating from the Stern–Volmer law with equations that resulted in a quadratic dependence from the quencher concentration. Basing their discussion on the model proposed by Frank and Wawilov,16 but specifically detailing the complex formation in the active volume, they explained the quadratic dependence with the following equation that considered the contributions from both quenching mechanisms:
| I0/I = (1 + KSV[Q])(1 + Ka[Q]) | (4) |
 | ||
| Scheme 1 Association equilibria between F and Q at the ground and excited states, where Ka is the association constant, k+* and k−* are the association and dissociation kinetic constant at the excited state, hν, knr and kr indicate the energy of the absorbed photon, the non-radiative and radiative kinetic constants, for F and A respectively without and with apostrophe. Adapted from ref. 19 with permission of the American Chemical Society, copyright 1970. | ||
This scheme tries to include all possible interactions between the fluorophore F and the quencher and is also suitable to explain complexes that induce an activation of the fluorescence of the emitting species and not only a quenching. In this hectic framework, many groups started to use these strategies to probe structural dispositions and fluctuations in macromolecules exploiting different quenchers20 and the compartmentalization of components in micelles and membranes.21
All these studies took advantage of the equations reported above that are still extensively applied but, over the years, a fundamental point has been often underestimated or even forgotten: eqn (3) and (4) have been derived in specific conditions, where the complex A is quenched and, above all, where [F] is negligible compared to [Q].
4. Static quenching when complexity arises
The situation, however, can soon become quite complex. The distance at which the intimate mechanism operates is crucial to determine which factors must be conveniently considered and, thus, which equation should be applied. In particular, in case of short-range interactions, such as electron transfer, quenching can typically occur only if a collisional complex – and in this case, eqn (1) should be applied – or a preformed stable chemical adduct (for which the possible equations will be discussed in the next section) are formed. Yet, quenching through long-range interactions has not a common solution for all possible conditions. A common example is represented by the energy transfer process according to the Förster mechanism, that can be efficient even at distances longer than 5 nm. In low viscosity solutions, deviations from the Stern–Volmer relationship can occur even when the quenching of F is mainly governed by diffusional events. As long as![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) , i.e., the reciprocal mean molecular diffusion distance during the excited state lifetime τ of F (that can be expressed by
, i.e., the reciprocal mean molecular diffusion distance during the excited state lifetime τ of F (that can be expressed by  , D being the sum of the diffusion coefficient of F and Q) is >3R0 (R0 being the Förster distance), at low-moderate concentrations of Q, the quenching follows eqn (1). However, since a close contact between the two partners is not required, in this case the observed quenching constant can exceed the diffusion rate constant kdiff.22 However, when ≤ 3R0 and at high concentrations, more complicated treatments have to be applied.22 Long-range mechanisms, including the Förster mechanism, are active also in highly viscous solvents and rigid matrices. In these situations, diffusional processes can be ignored, and the mutual distance between the quencher Q and F* can be considered constant during the excited state lifetime, representing a possible case of static quenching in absence of chemical association. In this case, defining the critical concentration of the quencher Q, c0Q, as:
, D being the sum of the diffusion coefficient of F and Q) is >3R0 (R0 being the Förster distance), at low-moderate concentrations of Q, the quenching follows eqn (1). However, since a close contact between the two partners is not required, in this case the observed quenching constant can exceed the diffusion rate constant kdiff.22 However, when ≤ 3R0 and at high concentrations, more complicated treatments have to be applied.22 Long-range mechanisms, including the Förster mechanism, are active also in highly viscous solvents and rigid matrices. In these situations, diffusional processes can be ignored, and the mutual distance between the quencher Q and F* can be considered constant during the excited state lifetime, representing a possible case of static quenching in absence of chemical association. In this case, defining the critical concentration of the quencher Q, c0Q, as:and assuming a statistical distribution of their distances, the following equation applies:

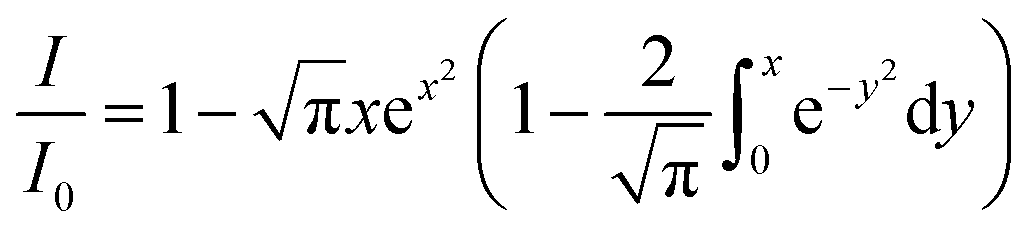 | (5) |
For the sake of completeness, it has to be underlined that the factors responsible for the deviations from the linear relationships expressed by eqn (1) and (3) are manyfold and include, together with Förster energy transfer, also chemical association with high affinity, reversible quenching in excimer and exciplex formation, energy migration as in conjugated polymers, and the confinement of species in different environments, such as in micelles and nanoparticles. In addition, pulsed excitation of luminescence opens up an additional dimension in the study of its quenching.23 These last situations require always a complex mathematical treatment, that can be found in specific review articles.23
In this context, it is quite surprising that in the most common and important case, and for which the mathematical treatment is relatively simple, i.e., for the case of static quenching upon formation of an adduct between the luminophore and the quencher, only the approximate eqn (3) is typically considered, often overlooking the conditions that make it valid.
5. Static quenching upon adduct formation: different equations for different conditions
In the case of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, the association of the fluorophore F and the quencher Q leads to the formation of an adduct that can be generically indicated as A according to the following equilibrium:
:1 stoichiometry, the association of the fluorophore F and the quencher Q leads to the formation of an adduct that can be generically indicated as A according to the following equilibrium:| F + Q ⇌ A | (6) |
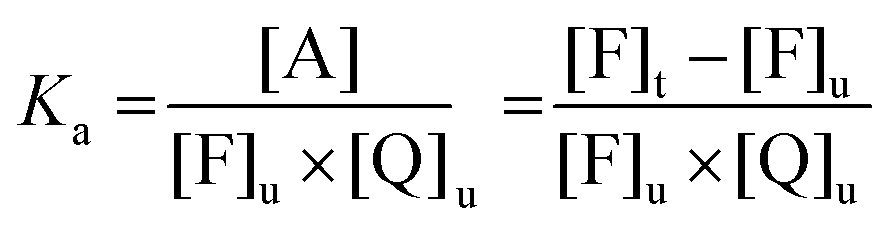 | (7) |
Eqn (7) can be then rewritten as:
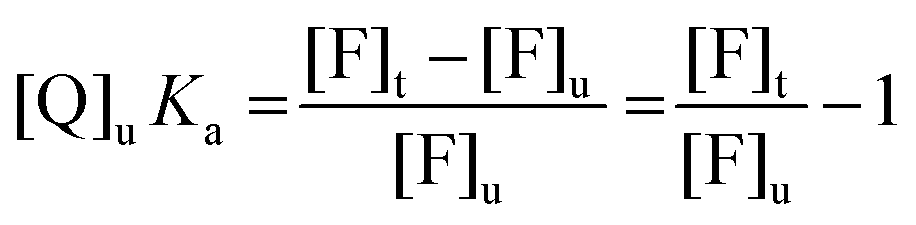 | (8) |
 | (9) |
Combining eqn (8) and (9), eqn (10) can be obtained:
 | (10) |
 | (11) |
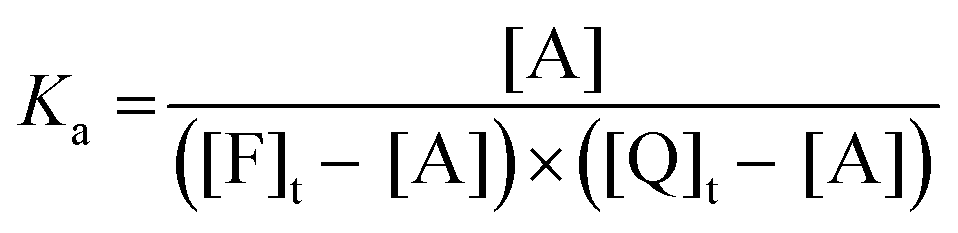 | (12) |
 | (13) |
 | (14) |
 | ||
| Fig. 1 Plots of the I0/I ratio upon increasing [Q]t according to eqn (11) (“pseudo Stern–Volmer”, red lines) and eqn (14) (non-approximate equation, black lines) for different magnitudes of association constants (made absolute using the parameter Ka × [F]t). The dashed cyan lines indicate possible attempts to fit the I0/I trends with a linear fit considering only low [Q]t regimes. | ||
 | ||
| Fig. 2 Trends of the I0/I ratio upon increasing [Q]t according to eqn (14) (a) and eqn (24) (c and e) and their inverse plots (emission variation I/I0, plots b, d and f). Different magnitudes of association constants are shown in each plot (Ka × [F]t = 0.05, 0.5, 1, 5 and 50 as represented by black, red, green, yellow and blue lines, respectively). Plots represent the different cases of complete quenching (ΦA = 0, plots a and b), of partial quenching (ΦA = 0.2 relative to ΦF, i.e. ΦA is 20% of ΦF, plots c and d), and of luminescence turn-on (ΦA = 5 relative to ΦF, i.e. ΦA is 5 times larger than ΦF, plots e and f). | ||
It is now very important to analize the trend of lifetimes in case of static quenching: whatever is Ka, contrary to what is observed for a dynamic quenching τ0/τ does not follow the trend shown by I0/I, since the only emitting species is [F]u, if A is not florescent, and only τ0 can be observed. The observation of only one lifetime, independent of the concentration of the quencher, can thus be an important additional support to distinguish between dynamic and static quenching. Some authors suggest, as a further possibility to distinguish between these two mechanisms, to observe the temperature dependence of the Stern–Volmer plot. This suggestion is based on the supposition that – upon an increase of the temperature – it should be observed an increase of the slope in case of the former (because of an increase of the diffusional quenching rate constant kq) and a decrease of the slope in the case of static quenching (because of a supposed decrease of Ka).26 We would like to suggest to use this criterion with some caution since, while it is always true that kq, being a kinetic rate constant, increases with temperature (and, in general, a temperature increase induces a decrease of the solvent viscosity), how the thermodynamic association constant Ka varies upon changing the temperature depends on the enthalpy of the process; for endothermic reactions, in fact, also Ka increases when the temperature increases, as it has been also found in some cases.27,28
Finally, from the experimental point of view, it is important to note that all suitable corrections must always be applied to take into account possible inner filter effects before drawing any conclusion about quenching in solution; indeed, if the added species presents a not negligible absorbance (i.e., higher than 0.05) at the excitation and/or at the emission wavelengths, a decrease of the emission intensity of F could be observed even in the absence of dynamic or static quenching. In non-ideal conditions of varying absorbance at excitation and/or emission wavelengths, we recommend using accurate corrections,12,13 while very approximate approaches, such as the one obtained using the expression Fcorr = Fobse(Aex+Aem)/2,24 should be avoided.
In addition, when the I0/I ratio becomes very high – i.e., when the fluorescence is highly quenched (in case of a high value of Ka this can occur even at a low concentration of Q) – then a high-sensitivity, low-background fluorometer is required to record reliable data.
As a final remark, plotting I/I0 rather than I0/I can help to reveal the occurrence of a non-linear association, in particular upon addition of the first few equivalents of quencher, as shown in Fig. 2b.
6. The most general approach to an intensity change caused by association
The general eqn (14), as already mentioned, is valid in all conditions provided that the formation of the adduct follows a 1:1 stoichiometry, and the formed adduct is not fluorescent, i.e., in case of complete quenching. However, this is not always the case; many processes, in fact, can induce only a partial quenching or even take to an increase in the luminescence intensity. For this reason, non-approximate equations suitable for these events are of utmost importance for relevant fields such as luminescent chemosensors, luminescent supramolecular adducts, including molecular machines, and luminescent nanostructures.
Pursuing the most general approach (while keeping the assumption of a 1:1 stoichiometry), it is important to bear in mind that a change in the luminescence intensity is produced if the luminescence quantum yield and/or the absorption coefficient of F changes upon complexation with Q forming the adduct A. In this case, the observed intensity can be described by the following equation:
| I = εFφF[F]u + εAφA[A] | (15) |
Similarly, the corrected intensity in absence of the quencher, I0, can be described by the equation:
| I0 = εFφF[F]t | (16) |
| I = εFφF([F]t − [A]) + εAφA[A] = εFφF[F]t + (εAφA − εFφF)[A] | (17) |
 | (18) |
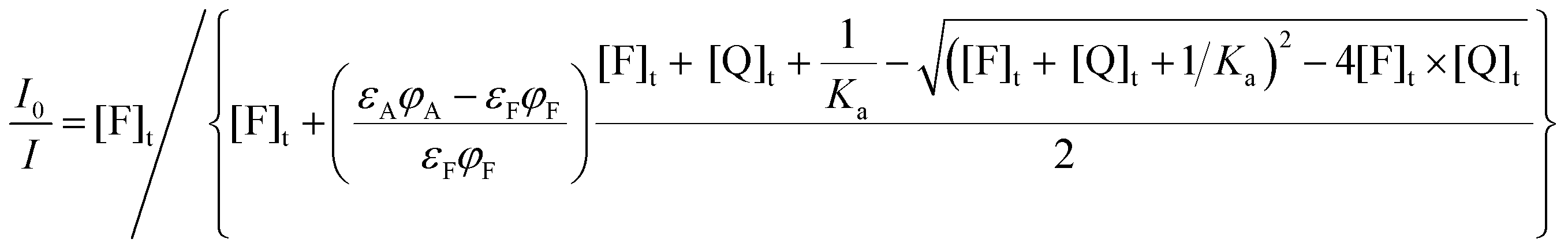 | (19) |
:1 stoichiometry as in the process 6 and (ii) accurate corrections considering inner filter effects have been performed.
In a simpler, but more common case, in which the absorption coefficient can be considered constant and only Φ changes upon association, eqn (15)–(19) can be simplified to eqn (20)–(24) respectively:
| I = φF[F]u + φA[A] | (20) |
| I0 = φF[F]t | (21) |
| I = φF([F]t − [A]) + φA[A] = φF[F]t + (φA − φF)[A] | (22) |
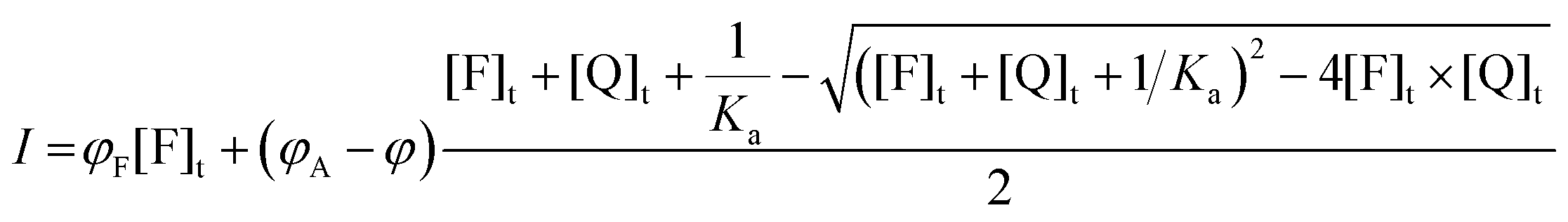 | (23) |
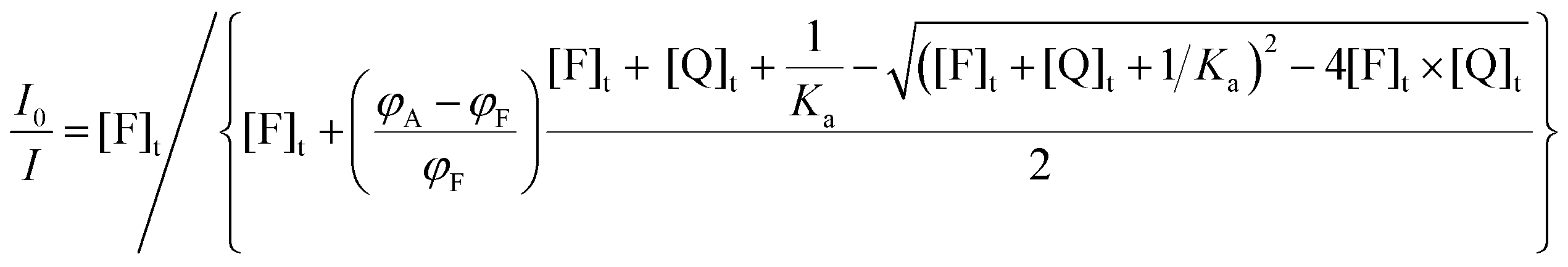 | (24) |
If a decrease of the corrected luminescence is observed, in eqn (15)εFφF > εAφA (or φF > φA in eqn (20)); the I0/I and I/I0 plots relative to different Ka × [F]t values in the case of 20% of residual emission of the adduct are shown as an example in Fig. 2c and d, respectively. As it can be seen, in this case the I0/I versus [Q]t plot tends to plateau, characterized by I0/I > 1, which is determined by the ratio of the intensity of the free and complexed fluorophore, respectively. In this case two different lifetimes should be measured, i.e., the ones of the free and complexed fluorophore with different proportions (and thus, pre-exponential terms)24 depending on the concentration of Q.
On the contrary, if an increase of the corrected luminescence is observed, in eqn (15)εexcφem < εexc′φem′ (or φem < φem′ in eqn (20)); the I0/I and I/I0 plots relative to different Ka × [F]t values in the case of a 5-fold enhancement of the emission are shown as an example in Fig. 2e and f, respectively. Again, the curves tend to plateau but in this case defined by a I0/I < 1.
Importantly, we think that when proper titration (and corrections) are performed, the interpolation of the experimental points of I0/I according to eqn (14), eqn (19) or eqn (24) (or I according to eqn (18) or eqn (23)), depending on the final effect of the quencher, can yield quite precise values of Ka – in case of a 1:1 adduct as indicated in the process 6 – without any hidden approximation. This could allow avoiding even severe errors that can arise plotting the data with non-general equations such as the widely adopted Benesi–Hildebrand one.29 Very often, in fact, also this equation is applied without explicitly addressing its typical limitations: it strictly describes a 1:1 stoichiometry (although some changes can be done to consider different stoichiometries) and, even more importantly, it is valid only in cases where the concentration of the quencher Q, or guest in a host–guest system, is much higher than the concentration of the complex.4,29 The underestimation of the conditions of application often takes to force the linear fitting of data sets that are instead not linear.
7. Application in sensing, diagnostics and nanotechnology
While fluorescence quenching is ubiquitous in science and technology, the distinction of dynamic and static quenching can be an overriding priority – not always straightforward – to obtain correct and quantitative information. In this section, we critically discuss some examples, gathered in different categories (a list that cannot be exhaustive, since fluorescence quenching is exploited in many other field, including the one related to solar cells30), aimed at a better understanding of the concepts discussed in this tutorial review.In order to provide the reader with a tool to favour an easy comparison of any reported example with the treatment proposed in this review, we have summarized in Scheme 2 all steps – necessary or to be avoided – from data collection to analysis, for a correct quantitative interpretation of quenching phenomena.
 | ||
| Scheme 2 Steps that should be performed – or avoided – to correctly interpret quenching phenomena. | ||
7.1 Quenching of the intrinsic fluorescence of proteins
The decrease of the intrinsic fluorescence of protein (and in particular of human and bovine serum albumin, HSA and BSA, respectively) by a potential guest is one of the most common examples of the possibility offered by static quenching, with potential applications that include pharmaceutical sciences and food technology. In many manuscripts it is possible to find an explicit statement asserting that both dynamic and static quenching are expected to give a linear dependence of the Stern–Volmer plot, the only reason for a deviation being a combination of these two mechanisms, as from eqn (4).A linear Stern–Volmer plot has been effectively observed in the case of the quenching of the HSA luminescence upon addition of tolperisone hydrochloride,31 with a concentration range that is in perfect agreement with what suggested at point 1 of Scheme 2. From the analysis of the plot, in the case of dynamic quenching a kd > 5 × 1012 M−1 s−1 would be obtained, a value significantly higher than its possible upper limit, thus excluding this mechanism. The authors provided other spectroscopic and calorimetric evidence of static associations. In agreement with a negative enthalpy of the process, a decrease of the Stern–Volmer constant with temperature was also observed.
A critical analysis shows that an almost linear relationship could be effectively expected in these experimental conditions since the Ka (2.3 × 104 M−1 at 25 °C) and the HSA concentration (2 × 10−6 M) are similar to those represented by the black curve of Fig. 1 (Ka × [F]t = 0.05). Even the quenching of BSA fluorescence by 2-amino-6-hydroxy-4-(4-N,N-dimethylaminophenyl)-pyrimidine-5-carbonitrile, an anti-bacterial agent,28 was explained in terms of an association process also because of the linear dependence of the I0/I plot vs. the concentration of Q. In agreement with this attribution, also in this case, the calculated diffusional rate constant (1013 M−1 s−1) would be too high to support a dynamic quenching. Specifically, the concentration of the fluorophore (2 × 10−5 M−1) and the calculated KSV (ca. 1.5 × 105 M−1) would lead to obtain a non-linear behaviour that is intermediate between the green and yellow curves shown in Fig. 2a. It has however to be noted that the Stern–Volmer plot shown by the authors has been obtained in a concentration range of the quencher up to 7 × 10−7 M, i.e., up to 0.35 equivalents of the fluorophore, much lower than the one required for a correct interpretation (point 1 of Scheme 2; see also the caution indicated in the last row of the scheme). In this range, linearity can be assumed, but it would be much more informative a plot obtained extending the concentration of the quencher up to few equivalents. Furthermore, an increase of the association constant has been observed upon increasing temperature, leading to the calculation of a positive enthalpy for the association process.
A similar behaviour has been also observed for the association between rosmarinic acid (RA) and HSA.27 In the latter instance, the authors observed a positive deviation trend (upward curvature) of the Stern–Volmer plot (see Fig. 3), which, using the authors’ words, could be either the result of a combination of static and dynamic quenching processes or due to the higher concentrations of the ligands around the fluorophore.
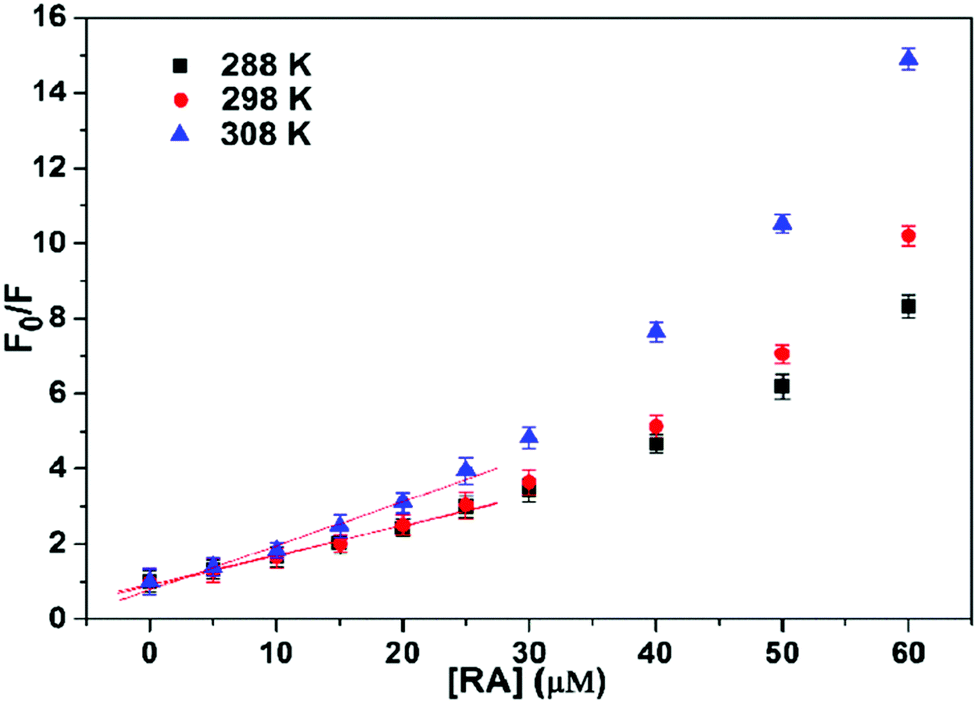 | ||
| Fig. 3 Stern–Volmer plots for the quenching of HSA fluorescence by RA at different temperatures. CHSA = 5.0 × 10−6 M. Reproduced from ref. 27 with permission from Elsevier, copyright 2016. | ||
On the contrary, as already discussed in Section 4 and depicted in Scheme 2, this was exactly what it should be expected in case of pure static quenching, and the analysis of the whole plot, and not only of the first linear portion, would have led to a more precise treatment.
As a final comment for this kind of study, the simplified version of the inner filter effect is generally applied, also in the examples discussed above, while a more accurate correction should be taken into account to avoid misinterpretations of the emission data, as indicated at point 3 of Scheme 2. For these experimental conditions, in fact, with excitation at wavelengths shorter than 300 nm and emission between 300 and 400 nm, inner filter effects play an important role, since absorption of the quencher is often expected in the same range.
7.2 Luminescent molecular chemosensors
Besides proteins, it is very important to underline that sensing, in general, is the widest application field of fluorescence quenching. The variation of an analytical signal (fluorescence) in response to the presence and concentration of a chemical species (the quencher) describes indeed the design of a chemosensor. The dynamic range of a chemosensor depends on the quenching mechanism: in dynamic quenching the intensity behaviour is governed by the Stern–Volmer relationship (eqn (1)), and in general offers a wide linear range, with analytical parameter governed by the luminescence quantum yield of the luminophore and kq. Conversely, in static quenching the dynamic range is determined by the ratio between association constant and concentration: the same chemical species can be detected in different concentration ranges depending upon their association constant with the fluorescent species.32 This has been widely exploited in the sensing of species of high biological and environmental relevance. In our experience, in association induced luminescence quenching or enhancement processes, the association constant can be efficiently determined fitting the luminescence intensity – once suitably corrected for inner filter effect12,33,34 – according to eqn (18) or to eqn (24) when the absorption spectrum of the luminophore is significantly altered.In general, when the chemosensor is operating in its optimal dynamic range,32 the approximation that could lead to a linear I0/I plot as described by eqn (11) does not apply, and curvature should be expected. It has to be mentioned here that this awareness is not very widespread as, on the contrary, the belief of an always linear relationship between I0/I and the concentration of the quencher; this leads to wrong assumptions and data discussion in many cases. For example, looking at the quenching of an azine-based “turn-off” system by Fe3+ ions (4,4-((ethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(methanylylidene) bis(methanylylidene)) bis(2-methoxyphenol) (EBHMM),35 the fluorescence intensity of the chemosensor has been linearly interpolated although an upward curvature could be observed (Fig. 4), in contrast with what reported in Scheme 2.
 | ||
| Fig. 4 Stern–Volmer plots for the quenching of EBHMM by Fe3+, as described in ref. 35. [EBHMM]= 5.0 × 10−6 M. Reproduced from ref. 35 with permission from Elsevier, copyright 2020. | ||
Discussing the Stern–Volmer plot, the possible occurrence of a dynamic quenching was also mentioned, although at the concentrations reported in Fig. 4 this would require an EBHMM excited-state lifetime of several microseconds, an unlikely value for the fluorescence of an organic species in an aerated solution. Moreover, the same intensity data were plotted using the Benesi–Hildebrand equation, which is intended to give association constants and thus cannot be applied in any case involving even a partial dynamic quenching contribution. Unfortunately, this contradiction is not rare in literature. Finally, the calculated Ka (1.0034 M−1) cannot explain a significant quenching in the experimental conditions used.
Another example of questionable assumptions based on the expectation of a linear I0/I plot for static quenching can be represented by the quenching of the fluorescence of triaryl imidazoles by trinitrophenol (TNP),36 also known as picric acid. Fluorescence quenching is quite often proposed for the detection of explosives. In this case, in the presence of curvature at high concentrations, ruling out – because of the constancy of the excited lifetime – the possibility of the concomitant occurrence of static and dynamic quenching, the authors invoked the occurrence of a “super amplified quenching effect”.
It is to note at this point that many ON–OFF chemosensors have been reported in the literature for Fe3+ and TNP. They can indeed be good candidates to quench luminophores having suitable reducing properties at their excited states; however, they show significant absorbance up to 400 nm and beyond, and for this reason a specific attention should be used in these cases in obtaining corrected intensity data, due to possible changes in absorbance at the excitation wavelength, before drawing any conclusion, as reported in Scheme 2, point 3.
7.3 Luminescence changes in nanomaterials
Chemosensors have been prepared also engineering a variety of nanostructures,37 whose versatility has been proved to be useful to optimize biocompatibility and emission wavelength. In particular, the NIR region is the emission spectral range of choice for best applicability in vivo, due to the transparency window of biological tissues falling in this region. Recently, a chemosensor based on carbon nanotubes for hydrogen peroxide was used to quantitatively monitor plant health in response to various stresses, via a multistep mechanism combining high-affinity association, diffusion of H2O2, and a Fenton-type chemical reaction producing OH˙ radicals.38 In this type of nanomaterials, showing luminescence quenching with complex multistep mechanisms, the simple cases of static or dynamic quenching do not readily apply, and quantitative information can only be obtained with dedicated calibration curves.Fluorescence quenching and further photophysical phenomena such as excimer formation or donor–acceptor FRET have also been employed to monitor and verify the hierarchical organization of organic–inorganic nanoparticles. Also in this field, the distinction between static and dynamic quenching is not trivial, since the formation of compartmentalized environments with different viscosity and rigidity mixes the concepts of pure chemical association and pure diffusion. For example, the inclusion of a quencher in a rigid compartment in close proximity to another compartment containing fluorescent moieties can lead to quenching of their fluorescence, even though there is no diffusion, nor chemical association or affinity between the two molecular partners. In these cases, a possible approach could be the one illustrated discussing the Förster energy transfer in high viscous media. Yet, the observed dependence of fluorescence intensity and lifetime on quencher concentration results similar to the case of static quenching, pointing to a more fruitful description of this non-ideal case as a static-like quenching, with a discussion of association constants and affinities not between fluorophore and quencher, but rather between these two moieties and the compartmentalized environments.39,40 Fluorescence quenching provided insights on the growing mechanism of complex nanoarchitectures obtained, among other methods, via (i) self-assembly of surfactants and organic–inorganic precursors in core–shell structures41 or by (ii) shell-by-shell assembly of organic layers around NP cores.42 These highly order organic–inorganic hybrid architectures represent an emerging field at the interface of synthetic chemistry, nanotechnology, and materials science, with emerging properties such as controlled dispersibility and stability in various solvents, trapping of guest molecules (for drug delivery or water cleaning) and compartmentalization of photoactive components for advanced functionalities, such as cascade FRET or photoswitchable fluorescence.43
7.4 Fluorescence changes in molecular machines
In the broad field of nanotechnology, static quenching is widely used also in the field of molecular machines, where it is often the diagnostic signal of assembly and disassembly steps of machinery components, offering also high temporal control.44,45 In this context, a typical example is represented by the possibility to monitor the formation of psedurotaxanes, determining also the association constant among the different components. For example, in the design of a molecular-level plug/socket system, it has been observed a strong quenching of a binaphthyl unit incorporated into a crown ether and a protonated 9-methyl(aminomethyl)anthracene thorough an efficient energy transfer process.46 In this case, the authors noted that the inner filter effects, in particular due to the absorption by the anthracenyl moiety of the unquenched emission of the binaphthyl unit, could lead to large experimental errors. Despite this, a correct treatment (point 3, Scheme 2) led them to assign a 1:1 stoichiometry to the formed adduct and to estimate the lower limit of the association constant (>1 × 105 M−1). In a more recent example, Credi and coworkers reported the formation of two pseudorotaxane complexes between two calix[6]arenes bearing naphthyl units and dioctylviologen ditosylate (DOV × 2TsO, see Fig. 5).47
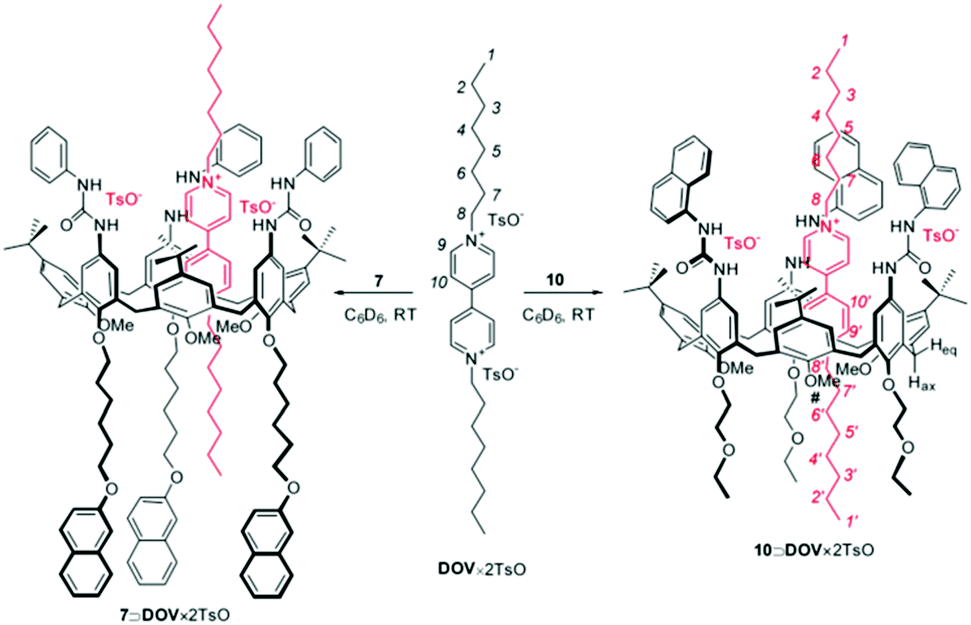 | ||
| Fig. 5 Schematic representation of the pseudorotaxanes between two calix[6]arenes and dioctylviologen ditosylate (DOV × 2TsO). Reproduced from ref. 47 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2016. | ||
In both cases the formation of the adduct caused a not complete quenching of the naphthyl units appended to the host, revealing a 1:1 stoichiometry. In the case of calix[6]arene 7 (Fig. 6) the fitting of the plot according to eqn (23) yielded a value of the association constant logK = 7.0 ± 0.2, while in the case of calix[6]arene 10 only a lower limiting value of the association constant (logK > 7.5) was obtained, a situation that is common when Ka × [F]t > 50.
 | ||
| Fig. 6 Luminescence spectral changes (λexc = 315 nm) upon addition of increasing amounts of DOV × 2TsO to a 3.0 × 10−5 M solution of 7. The inset shows the titration curve obtained by plotting the emission intensity at 350 nm as a function of the DOV × 2TsO equivalents; the full line is the data fitting corresponding to a 1:1 binding model, according to eqn (23). Conditions: air-equilibrated CH2Cl2, room temperature. Reproduced from ref. 47 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2016. | ||
In both cases, as indicated in Scheme 2, from the residual intensities the authors were able to determine the unimolecular rate constants (that should not be confused with the bimolecular rate constant relative to the dynamic quenching) for the quenching constant inside the adduct, providing additional information that could be used for the design of even more performing systems.
7.5 Fluorescence quenching applied to DNA nanotechnology
Quenching is a primary tool also in nucleic acids nanotechnology. In this case, quenching is primarily static because it is based on the affinity between complementary strands, even though the amount of quenching in the OFF and ON states may depend on the conformational freedom of the strands.32 A typical example is represented by the so-called molecular beacons. They are fluorescent biosensors made of single-strand DNA, with the peculiarity, respect to the systems described so far, that both the fluorophore and the quencher are covalently linked to the probe. Molecular beacons exist in two conformations: a closed one, in which the fluorophore is sufficiently close to the quencher to become almost not luminescent, and an open one, suitable to bind the target analyte, in which the distance between the two units is much larger, making this conformation more fluorescent. The presence of the analyte thus shifts the equilibrium towards the open form leading to a fluorescence increase. With proper strategies including allostery and cooperativity, Ricci demonstrated the possibility to design biosensors – working in the optimal dynamic range – for many kinds of analytes, not limited to nucleic acids but including small molecules such as cocaine, antibodies40 and proteins such as trombin.48 Looking at fluorescence intensity it is possible to measure the association constant between the probe and the analyte and the concentration of the latter. Although this is not always reported, it could be possible also to study the equilibrium between the two conformations of the molecular beacon in absence of the analyte, an important parameter for the evaluation of its analytical performance and its dynamic range.Apart from the molecular beacon approach, fluorescent reporters have been linked to nucleic acid strands to monitor their conformation and folding dynamics and their response upon interaction with a variety of substrates and surfaces. By the way, an association changing the conformation of the probe, for example leading to the formation/disruption of H-aggregates, and thus switching its fluorescence is commonly used in biosensing.49
Interfacing DNA with nanomaterials has emerged as a prosperous research field owing to applications in biosensing, gene and drug delivery, and directed assembly of nanomaterials.48 Interactions with graphene oxide nanosheets have been investigated via fluorescence quenching of FAM-functionalized DNA,50 while the integration of oligonucleotides with AuNPs and anti-AFB1 monoclonal antibodies yielded a new fluorescence turn-on sensor for homogeneous detection of aflatoxin B1 (AFB1), a potent low molecular weight mycotoxin.51 Remaining in the field of sensing, Willner et al. introduced a DNA tetrahedron aimed at the parallel and multiplexed analysis of different targets: miRNAs, DNA-cleaving enzymes, and aptamer–ligands. Three fluorophores were integrated as the transducers of the parallel and multiplexed analysis of the different analytes.52 Another DNA tetrahedron based multicolour nanoprobe was used for simultaneous imaging of three tumour-related mRNAs in living cells. Here, the fluorescence quenching served as the initial step to guarantee a dark background and a sufficient contrast to observe fluorescence restoration caused by competitive chain replacement reaction in cancerous cells.53 The quenching of fluorescent nucleic acids is also employed to generate molecular logic devices (MLDs), promising for applications in bioanalysis, computing, and others requiring Boolean logic.54 Finally, the quenching of fluorescent DNA was employed to improve the contrast of a super-resolution fluorescence imaging technique, DNA-PAINT. The insertion of a quencher resulted in the Quencher-Exchange-PAINT technique, which promises enhanced efficiency for multiplexed imaging of complex nanostructures, in thick tissues, and without the need for washing steps.55
8. Conclusions
The possibility for a given chemical species to modify the photophysical properties of a luminophore, including the complete quenching of its intensity, is widely used far beyond the field of chemistry. However, when this event is caused by the formation of an adduct, many researchers still approach its treatment with the belief that the I0/I plot vs. the concentration of the quencher should always have linear behaviour, a belief that is explicitly stated in many articles. A linear plot, though, can be expected only for complete quenching and when the association constant is relatively low, i.e., for an association process with a 1:1 stoichiometry, when the product of the association constant and the concentration of the fluorophore is equal or lower than 0.05.
In all the other conditions, an upward curvature should be expected, and this tutorial review provides the tools to approach them all. This general fitting approach can provide additional information, such as the association constant of the process and the photoluminescence quantum yield of the complex, avoiding misinterpretations of the experimental results. We would like also to remind that, to avoid artifacts it is essential to suitably consider, before any interpretation, the possible occurrence of inner filter effects.
To conclude, this review is intended to offer a useful tool to support the widest researcher audience in different fields to rigorously plan and analyse photophysical data – even beyond the sole case of quenching – without errors due to oversimplifications or improper application of simplified equations. We believe that it is now time for the ready and fruitful use of the most general equations that – even if lengthy – are easy to use with any modern fitting software. The great advantage of turning to the use of the general equations is the broadest versatility and applicability, in all regimes of Ka × [F]t, and even in cases of partial quenching or luminescence turn-on processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Italian Ministero dell’Istruzione, Università e Ricerca, MIUR (Project PRIN 2017EKCS35). The paper is published with the contribution of the Department of Excellence program financed by MIUR (MIUR, L. 232 del 01/12/2016).Notes and references
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 Search PubMed.
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 Search PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 Search PubMed.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 Search PubMed.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 Search PubMed.
- O. Stern and M. Volmer, Z. Phys., 1919, 20, 183–188 Search PubMed.
- G. Weber, Trans. Faraday Soc., 1948, 44, 185–189 Search PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 Search PubMed.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 Search PubMed.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1982, 21, 155–173 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 Search PubMed.
- A. Credi and L. Prodi, J. Mol. Struct., 2014, 1077, 30–39 Search PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis Group, 3rd edn, 2006 Search PubMed.
- F. Perrin, Ann. Phys., 1929, 10, 169–275 Search PubMed.
- S. I. Wawilow, Acta Phys. Pol., 1936, 5, 417 Search PubMed.
- I. M. Frank and S. I. Wawilov, Z. Phys., 1931, 69, 100 Search PubMed.
- H. Weil-Malherbe, Biochem. J., 1946, 40, 351–363 Search PubMed.
- H. Boaz and G. K. Rollefson, J. Am. Chem. Soc., 1950, 72, 3435–3443 Search PubMed.
- W. M. Vaughan and G. Weber, Biochemistry, 1970, 9, 464–473 Search PubMed.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4171–4179 Search PubMed.
- E. Blatr, R. C. Chatelier and W. H. Sawyer, Biophys. J., 1986, 50, 349–356 Search PubMed.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, 2009 Search PubMed.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Nature, 3rd edn, 2006 Search PubMed.
- R. C. Evans, P. Douglas and H. D. Burrows, Applied Photochemistry, Springer, 1st edn, 2013 Search PubMed.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R60 Search PubMed.
- X. Peng, X. Wang, W. Qi, R. Su and Z. He, Food Chem., 2016, 192, 178–187 Search PubMed.
- V. D. Suryawanshi, L. S. Walekar, A. H. Gore and P. V. Anbhule, J. Pharm. Anal., 2016, 6, 56–63 Search PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 Search PubMed.
- M. Gerhard, B. Louis, R. Camacho, A. Merdasa, J. Li, A. Kiligaridis, A. Dobrovolsky, J. Hofkens and I. G. Scheblykin, Nat. Commun., 2019, 10, 1–12 Search PubMed.
- G. Rabbani, E. J. Lee, K. Ahmad, M. H. Baig and I. Choi, Mol. Pharmaceutics, 2018, 15, 1445–1456 Search PubMed.
- F. Ricci, A. Valle, A. J. Simon, A. Porchetta and K. W. Plaxco, Acc. Chem. Res., 2016, 49, 1884–1892 Search PubMed.
- A. V. Fonin, A. I. Sulatskaya, I. M. Kuznetsova and K. K. Turoverov, PLoS One, 2014, 9, e103878 Search PubMed.
- J. Kimball, J. Chavez, L. Ceresa, E. Kitchner, Z. Nurekeyev, H. Doan, M. Szabelski, J. Borejdo, I. Gryczynski, Z. Gryczynski and Z. Gryczynski, Methods Appl. Fluoresc., 2020, 8, 033002 Search PubMed.
- S. Manigandan, A. Muthusamy, R. Nandhakumar and C. Immanuel, J. Mol. Struct., 2020, 1208, 127834 Search PubMed.
- Jigyasa and J. K. Rajput, Sens. Actuators, B, 2018, 259, 990–1005 Search PubMed.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 Search PubMed.
- H. Wu, R. Nißler, V. Morris, N. Herrmann, P. Hu, S.-J. Jeon, S. Kruss and J. P. Giraldo, Nano Lett., 2020, 20, 2432–2442 Search PubMed.
- V. N. Morozov, M. A. Kolyvanova, O. V. Dement, V. M. Rudoy and V. A. Kuzmin, J. Lumin., 2020, 219, 116898 Search PubMed.
- A. Porchetta, R. Ippodrino, B. Marini, A. Caruso, F. Caccuri and F. Ricci, J. Am. Chem. Soc., 2018, 140, 947–953 Search PubMed.
- D. Genovese, E. Rampazzo, S. Bonacchi, M. Montalti, N. Zaccheroni and L. Prodi, Nanoscale, 2014, 6, 3022–3036 Search PubMed.
- L. M. S. Stiegler, T. Luchs and A. Hirsch, Chem. – Eur. J., 2020, 26, 8483–8498 Search PubMed.
- S. Cheung and D. F. O’Shea, Nat. Commun., 2017, 8, 1885 Search PubMed.
- Q.-H. Guo, Y. Qiu, X. Kuang, J. Liang, Y. Feng, L. Zhang, Y. Jiao, D. Shen, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 14443–14449 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 Search PubMed.
- E. Ishow, A. Credi, V. Balzani, F. Spadola and L. Mandolini, Chem. – Eur. J., 1999, 5, 984–989 Search PubMed.
- G. Orlandini, G. Ragazzon, V. Zanichelli, L. Degli Esposti, M. Baroncini, S. Silvi, M. Venturi, A. Credi, A. Secchi and A. Arduini, ChemistryOpen, 2017, 6, 64–72 Search PubMed.
- Y. Hu, A. Cecconello, A. Idili, F. Ricci and I. Willner, Angew. Chem., Int. Ed., 2017, 56, 15210–15233 Search PubMed.
- I. A. Karpenko, M. Collot, L. Richert, C. Valencia, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 Search PubMed.
- A. Lopez, B. Liu, Z. Huang, F. Zhang and J. Liu, Langmuir, 2019, 35, 11932–11939 Search PubMed.
- L. Xu, H. Zhang, X. Yan, H. Peng, Z. Wang, Q. Zhang, P. Li, Z. Zhang and X. C. Le, ACS Sens., 2018, 3, 2590–2596 Search PubMed.
- Z. Zhou, Y. S. Sohn, R. Nechushtai and I. Willner, ACS Nano, 2020, 14, 9021–9031 Search PubMed.
- S. Wang, M. Xia, J. Liu, S. Zhang and X. Zhang, ACS Sens., 2017, 2, 735–739 Search PubMed.
- M. Massey, I. L. Medintz, M. G. Ancona and W. R. Algar, ACS Sens., 2017, 2, 1205–1214 Search PubMed.
- T. Lutz, A. H. Clowsley, R. Lin, S. Pagliara, L. Di Michele and C. Soeller, Nano Res., 2018, 11, 6141–6154 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |