
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dye-sensitized solar cells strike back
Ana Belén
Muñoz-García
 a,
Iacopo
Benesperi
b,
Gerrit
Boschloo
c,
Javier J.
Concepcion
d,
Jared H.
Delcamp
e,
Elizabeth A.
Gibson
b,
Gerald J.
Meyer
f,
Michele
Pavone
g,
Henrik
Pettersson
h,
Anders
Hagfeldt
*ci and
Marina
Freitag
*b
a,
Iacopo
Benesperi
b,
Gerrit
Boschloo
c,
Javier J.
Concepcion
d,
Jared H.
Delcamp
e,
Elizabeth A.
Gibson
b,
Gerald J.
Meyer
f,
Michele
Pavone
g,
Henrik
Pettersson
h,
Anders
Hagfeldt
*ci and
Marina
Freitag
*b
aDepartment of Physics “Ettore Pancini”, University of Naples Federico II, 80126 Naples, Italy
bSchool of Natural and Environmental Science, Newcastle University, Bedson Building, NE1 7RU Newcastle upon Tyne, UK. E-mail: marina.freitag@newcastle.ac.uk
cDepartment of Chemistry, Ångström Laboratory, Uppsala University, P.O. Box 523, 751 20 Uppsala, Sweden. E-mail: anders.hagfeldt@uu.se
dChemistry Division, Brookhaven National Laboratory, Upton, New York 11973, USA
eDepartment of Chemistry and Biochemistry, University of Mississippi, University, MS 38677, USA
fDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA
gDepartment of Chemical Sciences, University of Naples Federico II, 80126 Naples, Italy
hDyenamo AB, Teknikringen 38A, 114 28 Stockholm, Sweden
iUniversity Management and Management Council, Vice Chancellor, Uppsala University, Segerstedthuset, 752 37 Uppsala, Sweden
First published on 30th September 2021
Abstract
Dye-sensitized solar cells (DSCs) are celebrating their 30th birthday and they are attracting a wealth of research efforts aimed at unleashing their full potential. In recent years, DSCs and dye-sensitized photoelectrochemical cells (DSPECs) have experienced a renaissance as the best technology for several niche applications that take advantage of DSCs' unique combination of properties: at low cost, they are composed of non-toxic materials, are colorful, transparent, and very efficient in low light conditions. This review summarizes the advancements in the field over the last decade, encompassing all aspects of the DSC technology: theoretical studies, characterization techniques, materials, applications as solar cells and as drivers for the synthesis of solar fuels, and commercialization efforts from various companies.
 Ana Belen Muñoz García (top left), Iacopo Benesperi (4th column, middle), Gerrit Boschloo (4th column, top), Javier J. Concepcion (2nd column, top), Jared Delcamp (3rd column, top), Libby Gibson (3rd column, bottom), Gerald (Jerry) J. Meyer (bottom left), Michele Pavone (2nd column, bottom), Henrik Pettersson (4th column, bottom), Anders Hagfeldt (top right) and Marina Freitag (bottom right) | Ana Belen Muñoz García (top left) is Associate Professor of Physical Chemistry at the University of Naples Federico II. Dr Iacopo Benesperi (4th column, middle) is a postdoc at Newcastle University. Gerrit Boschloo (4th column, top) is Associate professor in Physical Chemistry at Uppsala University. Dr Javier J. Concepcion (2nd column, top) is the group leader of the Artificial Photosynthesis group at Brookhaven National Laboratory and he serves as BNLs Institutional Coordinator in the solar energy hub Center for Hybrid Approaches in Solar Energy to Liquid Fuels (CHASE). Prof. Jared Delcamp (3rd column, top) is Associate Professor in the Department of Chemistry and Biochemistry at the University of Mississippi. Dr Libby Gibson (3rd column, bottom) is Reader in Energy Materials at Newcastle University. Prof. Gerald (Jerry) J. Meyer (bottom left) is Professor of Chemistry at UNC-Chapel Hill, Vice-Chair for Diversity, and Director of the Center for Hybrid Approaches in Solar Energy to Liquid Fuels (CHASE). Michele Pavone (2nd column, bottom) is Associate Professor of Physical Chemistry at the University of Naples Federico II. Henrik Pettersson (4th column, bottom) is CEO of Dyenamo AB. Professor Anders Hagfeldt (top right) is Vice-Chancellor of Uppsala University and Professor of Physical Chemistry. Dr Marina Freitag (bottom right) is a Royal Society University Research Fellow and Newcastle Academic Track Fellow at Newcastle University. |
1 Introduction
Unprecedented changes in the world's energy production are required to meet with the urgent need to replace fossil fuels to mitigate their effects on climate change, and to keep pace with the ever-increasing global demand for energy. This calls for a rapid shift towards large scale implementation of renewable energy sources, of which sunlight has by far the largest potential. The challenge for scientists is to explore new materials for the creation of devices that can be mass-produced and efficiently convert light energy into electricity or solar fuels at a lower cost with sustainability in mind. Since renewable energy sources currently account for only about 10% of the total energy supply1 (29% of the total electricity supply), there is room for a large increase in energy production from solar cells in the near future.The Sun is the largest source of energy when taking into account both renewable and non-renewable sources, as it supplies the world with 173![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TW of energy each year.2 In other words, more energy from the Sun reaches the Earth in one hour than the human population consumes in a year. Photovoltaic electricity generation has grown at an average rate of more than 34% each year over the last 10 years, making it the world's fastest developing energy technology.3 However, photovoltaic cells contribute only 1% of the global energy production. The International Energy Agency (IEA) predicts a 50% increase in renewable electricity production from 2019 to 2025.4 This fast rise in the capacity of users to produce their own energy offers new possibilities and problems for utilization on a global level. Distributed solar PV systems in residential and commercial buildings as well as in industries are projected to establish a strong market position, and their installed capacity is estimated to almost double to 320 GW by 2025. The Si-based solar technology is presently that most established in manufacturing. Alternative technologies generally offer comparable efficiency to Si (e.g. GaAs or CIGS) in single-junction systems, but they remain expensive owing to manufacturing and material costs. Third-generation photovoltaic devices – hybrid solar cells – use cheap and abundant raw materials with the potential of high efficiencies.4
000 TW of energy each year.2 In other words, more energy from the Sun reaches the Earth in one hour than the human population consumes in a year. Photovoltaic electricity generation has grown at an average rate of more than 34% each year over the last 10 years, making it the world's fastest developing energy technology.3 However, photovoltaic cells contribute only 1% of the global energy production. The International Energy Agency (IEA) predicts a 50% increase in renewable electricity production from 2019 to 2025.4 This fast rise in the capacity of users to produce their own energy offers new possibilities and problems for utilization on a global level. Distributed solar PV systems in residential and commercial buildings as well as in industries are projected to establish a strong market position, and their installed capacity is estimated to almost double to 320 GW by 2025. The Si-based solar technology is presently that most established in manufacturing. Alternative technologies generally offer comparable efficiency to Si (e.g. GaAs or CIGS) in single-junction systems, but they remain expensive owing to manufacturing and material costs. Third-generation photovoltaic devices – hybrid solar cells – use cheap and abundant raw materials with the potential of high efficiencies.4
Exactly 30 years ago, in 1991, Michael Grätzel and his research group realized a new kind of solar cell: the dye-sensitized solar cell, DSC, or Grätzel cell.5 It is a very promising alternative to classical inorganic p–n junction solar cells as it combines molecular systems and nanoparticles to create a device that mimics photosynthesis, with the objective of turning sunlight into a renewable, reliable, and low-cost source of energy closer to existence. The first demonstration of dye injection into a single crystal semiconductor was provided by Gerischer in 1966,6,7 but it was Grätzel's introduction of a mesoporous semiconductor layer that led to the breakthrough in DSC technology. In DSCs, dyes are responsible for light absorption and charge separation and, therefore, for the conversion of photons to electrons. Dyes are bound to mesoporous semiconductors, which are only used to collect the resulting free electrons and transport them to the electrode as current.8 Electrons flow back into the system through a charge transport material, which regenerates the dye molecules, thus closing the circuit.9–11 DSC devices exhibit impressive energy efficiencies of over 13% under full sun illumination.12 Further, they are based on inexpensive starting materials and simple production techniques.13,14 Some concern has been raised about the sealing of liquid junction solar cells.15–18 Therefore, improvements in sealing strategies or the substitution of the liquid electrolyte with a solid charge transfer material will have a large influence on commercialization.19–23
With no clear third generation solar cell technology being dominant for mass production given significant concerns across all technologies, it is expected that DSCs will have years of thriving development ahead toward high efficiency outdoor applications. Additionally, DSCs are exceptional among third generation technologies with regard to specific applications. DSCs can be designed with a high degree of flexibility concerning shape, color, and size, as well as suitability for unique deployment scenarios. DSCs remain a competitive third generation alternative photovoltaic technology for several reasons including: (i) simple preparation methods, which will help to convert solar energy in a sustainable way, (ii) fabrication without the use of toxic materials, and (iii) design flexibility, which allows DSCs to be implemented in many different environments, from transparent smart windows to consumer electronics and indoor applications, which enables the powering of the next digital revolution of widely distributed sensors forming the Internet of Things (IoT).
The research progress during the past ten years in the field of DSCs is marked by important breakthroughs towards their use for a sustainable future. Relentless endeavours made it possible to achieve high efficiencies for DSCs in outdoor and indoor environments. These considerable advances were made by developing new panchromatic rigid-structure dye systems, new redox shuttles and hole transport materials, and by gaining new knowledge about the dyes' and redox shuttles' fundamental behavior. Under full sun illumination (standard AM1.5G), power conversion efficiencies have reached 13% (certified value)12 and 14% (non certified) with co-sensitized organic dyes.24,25 Under artificial light sources, efficiencies were pushed above 34%.12,26 The new redox couples and electrolytes based on cobalt and copper coordination complexes are able to regenerate the dye with less than 0.2 V driving force, which allows for the fabrication of systems with lower thermal losses. Current research and developments are the perquisite to improve efficiencies beyond 20%. Here, this review offers an updated overview of advanced characterization methods and current research trends of this transitioning technology, from the perspectives of device and molecular modelling to state-of-the-art techniques and novel device structures. Every device element, from metal oxides and nanomaterials to new hole transporter materials, dopants, and counter-electrodes, is addressed. Additional applications and constructs are discussed including p-type DSCs, tandem DSCs, and dye-sensitized solar fuel production. Past and current commercialization efforts are also showcased.
1.1 Light and energy
All photovoltaic devices, such as solar cells, convert solar radiation into electricity on the basis of the photovoltaic effect, discovered by the French physicist Alexandre Edmond Becquerel.27 The photovoltaic effect is linked to the photoelectric one, a phenomenon in which electrons are expelled when light shines on a conducting material. For the explanation of this phenomenon, Albert Einstein received the 1921 Nobel Prize in physics, introducing new quantum principles.28 It is described as the appearance of an electric voltage between two electrodes attached to a solid or liquid system when light shines onto it.In space, the solar spectrum resembles that of a black body at a temperature of 5778 K and includes a wide range of wavelengths, from X-rays to radio waves, with the main peak in the visible range (see Fig. 1). While travelling through Earths atmosphere, parts of the spectrum are filtered out (e.g. X-rays) and the solar spectrum reaching the planet surface is different compared to space. The light path through the atmosphere is defined as air mass (AM).29 As the solar spectrum distribution varies during the day and at different locations, a standard reference spectrum was established in order to compare the performance of photovoltaic devices from various manufacturers and research labs. The AM1.5 Global (AM1.5G) spectrum has a combined power intensity of 1000 W m−2 (100 mW cm−2) and is used as standard for the efficiency measurement of solar cells.30,31 The irradiance of sunlight, whose curve is shown in Fig. 1, is defined as the amount of energy of a certain light wavelength shone on a unit area per unit of time, J s−1 m−2 nm−1 (W m−2 nm−1). This spectral irradiance can be integrated over all wavelengths to obtain the overall irradiance in W m−2.
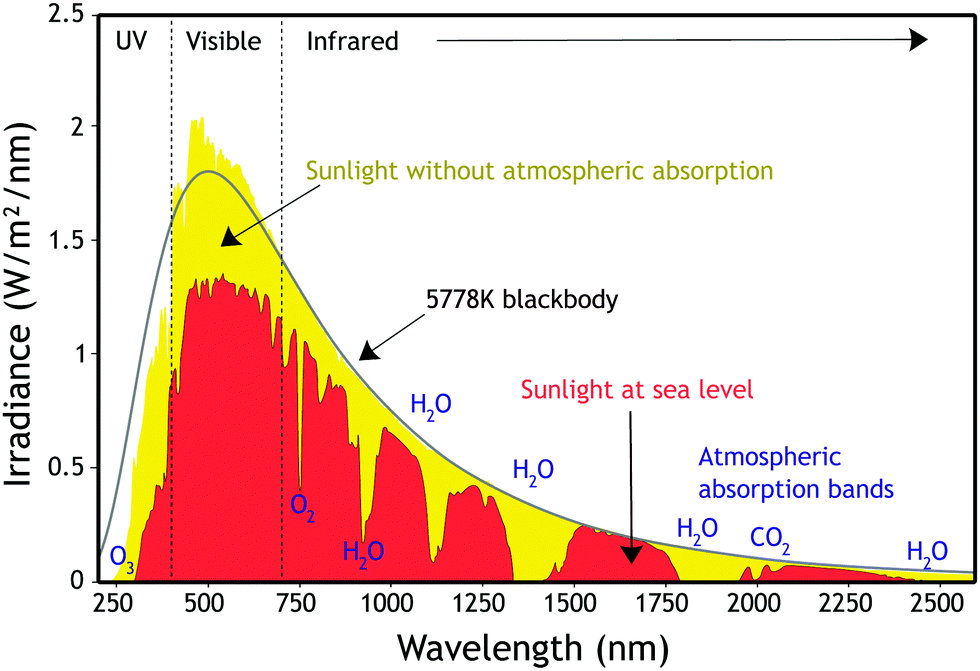 | ||
| Fig. 1 Solar irradiance spectrum. Artwork created by Nick84 and released under Creative Commons BY-SA 3.0 license, ref. 32. | ||
While DSCs perform well under sunlight, since dye light absorption profiles are commonly limited to the visible part of the solar spectrum, they perform even better when illuminated by artificial light sources, whose emission spectrum is similar to the visible range of that of the Sun (Fig. 2).26,33–37 Since any indoor light intensity is orders of magnitude smaller than sunlight and the spectra between the different light sources vary considerably, from an experimental point of view indoor lighting conditions are quite different from the solar irradiance outdoors. The intensity of typical indoor lighting has illuminance values ranging from 200 to 1000 lx (lux, which corresponds to lumen per unit area, lm m−2). For comparison, AM1.5G light has an illuminance value of about 100000 lx. Illuminance is similar to irradiance (measured in W m−2), but it defines light intensity in terms of human eye perception rather than energy. Illuminance cannot be converted to irradiance via a simple mathematical operation and while the latter can be used to quantify solar cell performance directly, the former cannot. At the same illuminance, in fact, different light spectra will produce different irradiance. For example, a light bulb emitting blue light with 1000 lx illuminance will produce more irradiance than a bulb emitting red light with the same illuminance. Only after the lamp spectrum has been determined can the illuminance be obtained from irradiance using eqn (1):
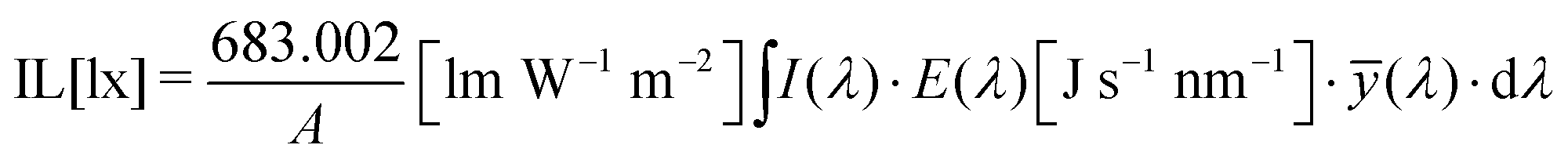 | (1) |
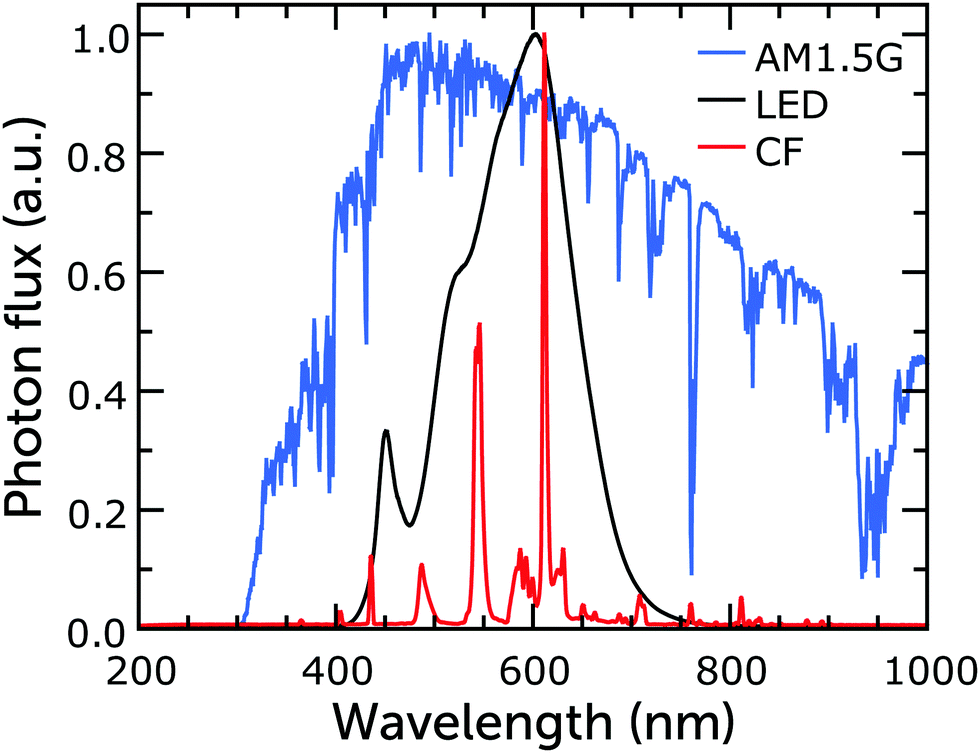 | ||
| Fig. 2 Normalized emission spectra of warm white fluorescent and LED bulbs, and of the AM1.5G standard. Reproduced from ref. 38 with permission from The Royal Society of Chemistry, copyright 2021. | ||
In the case of sunlight measurements there are several guidelines that describe standard experimental conditions, as well as how to test the solar cell, see e.g. ASTM standard E948.39 For indoor measurements, however, no standard has been defined yet.
1.2 Operation principles and structure
The basic components of a dye-sensitized solar cell are the dye-sensitized semiconductor electrode (the working electrode or photoanode), the redox electrolyte and the counter electrode. A monolayer of dye molecules adsorbed on the semiconductor surface is responsible for light absorption in the device. In conventional DSCs, the semiconductor has an n-type character: electrons in the conduction band are responsible for electrical conductivity of the material. Furthermore, the semiconductor has a wide bandgap and does not significantly contribute to solar light absorption. By far, the most applied semiconductor in DSCs is TiO2 with the anatase crystal structure, which has a bandgap of ∼3.2 eV and absorbs only UV light. TiO2 will be assumed as the semiconductor for the remainder of this part, noting here that a large number of semiconductors can actually be used in DSCs.A flat and dense TiO2 electrode with an adsorbed dye monolayer does not absorb enough light to give practically relevant solar-to-electric conversion efficiencies. In order to harvest a large part of the solar spectrum, TiO2 electrodes possessing high-surface areas are used, such as the mesoporous TiO2 electrode. This electrode consists of numerous interconnected nanoparticles that are typically about 20–30 nm in size. The porosity of the electrode is about 50% and its surface area can be several hundred times larger than the projected area. As such, the amount of dye adsorbed is also several hundred times larger than for a flat surface. Dye molecules that are chemically bound to the TiO2 have the best performances in the DSC. These molecules are also in contact with the redox electrolyte that fills the pores of the mesoporous electrode. The redox mediator transports positive charges to the counter electrode, which is typically located in parallel close to the working electrode.
Photoinduced electron transfer from a dye molecule to the conduction band of TiO2 is the first step in the working mechanism of a dye-sensitized solar cell, see Fig. 3. When light is absorbed by the dye (D), an electron is excited to a higher energy level. The excited dye (D*) can subsequently inject an electron into the conduction band of TiO2, which provides a variety of acceptor levels (reaction (1) in Fig. 3). This electron transfer process occurs on the femto- to picosecond time scale.
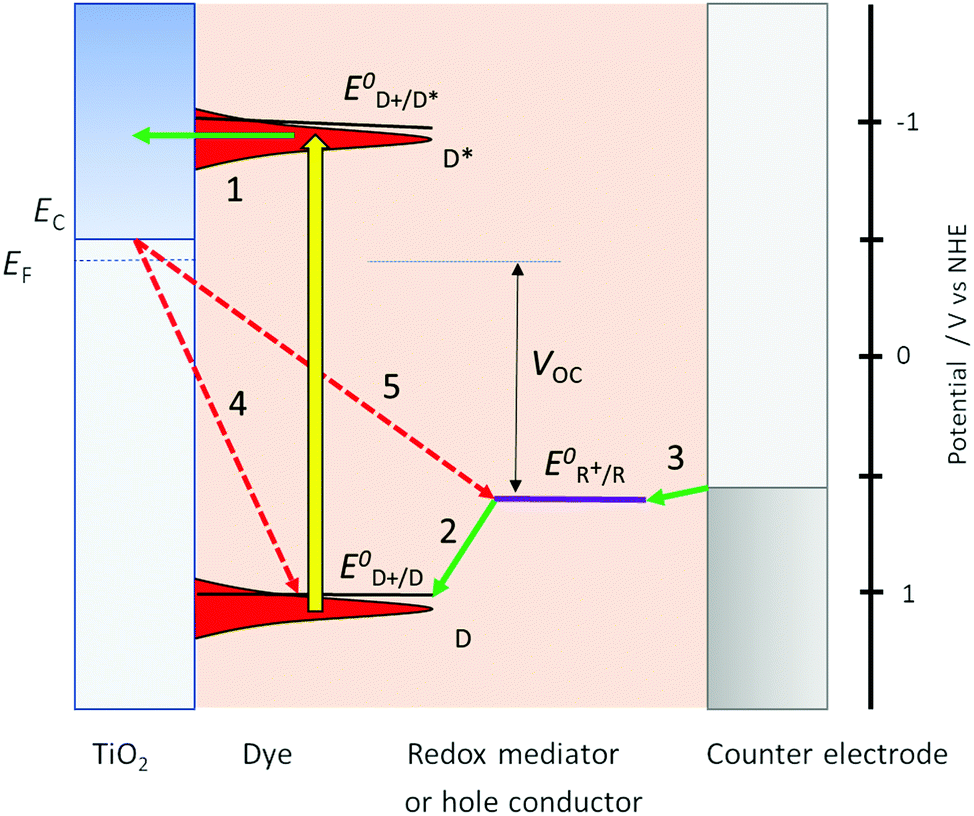 | ||
| Fig. 3 Basic diagram of the dye-sensitized solar cell, displaying working mechanism and energy levels. | ||
Electrons in the mesoporous semiconductor are charge compensated by ions in the surrounding electrolyte, and their transport is driven by electronic drift-diffusion. Electrons are collected at the electrode contact on a millisecond time scale under full sunlight illumination. The slow and light-dependent electron transport is generally explained using a multiple trapping model with an exponential trap distribution below the conduction band,40 however the nature of the traps is still debated. In recent work, it was found that upon electron accumulation into mesoporous TiO2, cations adsorb onto the semiconductor surface.41 This could lead to electrostatic traps for the electrons in mesoporous TiO2 and account for the observation of similar trap distributions for different types of metal oxides.
The sensitized TiO2 is in contact with an electrolyte containing a redox mediator (R+/R) that regenerates the dye (i.e. reduction of the oxidized dye D+, reaction (2) in Fig. 3), and also transfers positive charges from the working to the counter electrode, by means of diffusion of R+. At the counter electrode R+ is reduced to R (reaction (3)). The dye regeneration process is typically on the microsecond time scale and must be fast enough to prevent recombination of electrons from the semiconductor to the oxidized dye (reaction (4)). Electrons can also recombine with the oxidized form of the redox mediator (reaction (5)).
Fig. 3 also provides the basic energy level diagram of the DSC. The ground-state energy level of the dye is located just below E0(D+/D), the standard reduction potential of the dye, and is often referred to as the HOMO (highest occupied molecular orbital) level. The energy level of the excited dye D* is obtained by adding the absorbed photon energy. The lowest-lying excited state level is obtained by adding E0–0 (the zero–zero transition energy), which is generally obtained experimentally from the intercept of normalized absorption and fluorescence spectra. This level is often referred to as the LUMO (lowest unoccupied molecular orbital) level.
D* levels should be higher than the conduction band edge EC of the semiconductor to ensure sufficient driving force for efficient photoinduced electron injection. Fluorescence of the dye and non-radiative decay processes are competing with the injection reaction. For optimum DSC performance, D* and EC should possess sufficient electronic overlap, so that a high quantum yield of injection is obtained, while at the same time EC should be as high as possible to obtain a good output voltage in the DSC.
There should also be good matching between the energy levels of dye and redox mediator: sufficient driving force for reduction of the oxidized dye is needed to drive this reaction fast enough to prevent losses through electron/dye recombination. On the other hand, the driving force should not be excessive, as it lowers the voltage output of the DSC.
The voltage output of the DSC is the potential difference between working electrode and counter electrode, see Fig. 3. The potential of the counter electrode is close to that of the redox potential of the electrolyte, and equal to it when no current is flowing, under open-circuit conditions. The potential of the working electrode is equal to the Fermi level of the semiconductor. The Fermi level EF is given by:
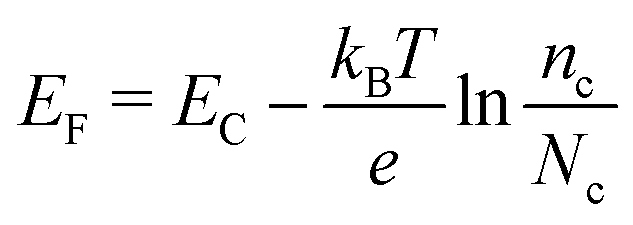 | (2) |
1.3 Device structures
The standard device structure for the DSC is the sandwich cell, in which both working and counter electrodes are based on conducting glass substrates that are placed face-to-face, with a thin layer of the redox electrolyte in between (Fig. 4a). The distance between the electrodes is usually determined by a thermoplastic frame that also acts as the sealing, and it is typically about 25 μm. An even narrower spacing is favorable, as this decreases the resistance due to redox mediator diffusion in the electrolyte.42 Fluorine-doped tin oxide (FTO)-coated glass is most frequently used as conducting glass in DSCs. FTO glass provides a good compromise between high chemical and thermal stability, low sheet resistance and high solar light transmittance. The photoelectrode consists of FTO glass with the mesoporous TiO2 film sintered on top. An optional thin and dense TiO2 layer (the so-called blocking layer), whose function is to decrease electron recombination from the FTO to the redox electrolyte, can be located between the FTO and the mesoporous TiO2. A light-scattering TiO2 layer can be added on top of the mesoporous layer to improve light capture in the device. The counter electrode comprises FTO glass with a catalyst, such as Pt nanoparticles, carbon, or a conducting polymer deposited onto of it. The sandwich structure allows for (semi-)transparent solar cell devices and the possibility for illumination from either side, provided that the counter electrode is transparent.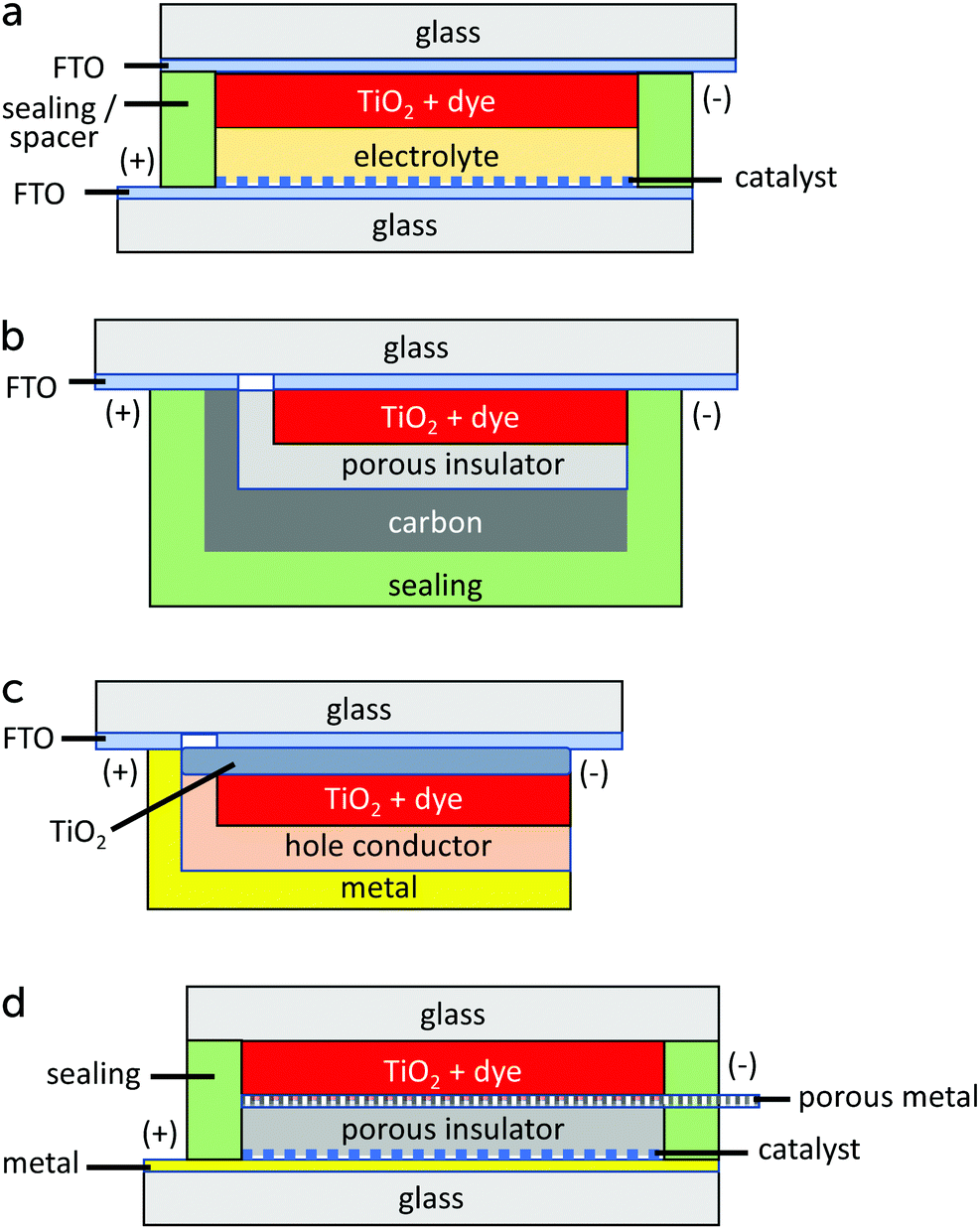 | ||
| Fig. 4 Device structures for dye-sensitized solar cells: (a) sandwich cell, (b) monolithic cell with carbon counter electrode, (c) solid-state DSC (monolithic), and (d) conducting glass-free DSC design. | ||
Monolithic DSC structures have advantages over the sandwich structure from a fabrication and cost point of view. Only one FTO glass substrate is used, onto which the different layers are screen-printed: first the mesoporous TiO2, then a porous insulating layer and finally a porous carbon layer that acts as counter electrode and electrical conductor (Fig. 4b). The redox electrolyte is infiltrated in all three layers, and a back sealing covers the whole device. This device structure is well suited for scaling up to modules with series or parallel interconnections. The highest reported efficiency for a monolithic DSCs with carbon counter electrodes is 7.6%.43 The carbon electrode in the monolithic DSC can be replaced by other conductors. For instance, highly-doped PEDOT films have been used in combination with a porous polyethylene separator film, reaching an efficiency of 7.7%, while also allowing for flexible devices.44 Recently, a Ni metal foil with Cr coating and Pt catalyst was implemented instead of the carbon electrode, and an efficiency of 8.0% was achieved.45
In a solid-state DSC, the liquid redox electrolyte is replaced with a solid hole transporting material (HTM). It is also commonly a monolithic structure, see Fig. 4c.46 A critical step in the fabrication is the infiltration of the hole conductor into the mesoporous TiO2 layer. Solution-based methods do not result in complete pore filling.22 Furthermore, a thin capping HTM layer is needed, onto which the metal contact is evaporated.
It is possible to avoid FTO-coated glass altogether in DSC structures. Several types of back-contact DSC devices have been developed, where the mesoporous TiO2 film is contacted at the back with a porous metal film47 or a metal mesh.48 A suitable metal is titanium, which forms a passivating oxide layer. Alternatively, a stainless steel mesh can be used if it is coated with a thin passivating layer. The counter electrode can also be Ti metal, but it should then be provided with a suitable catalyst. A possible layout of a DSC avoiding conducting glass is shown in Fig. 4d. The advantages of such a DSC are a higher solar light transmittance of the top glass, and a very low sheet resistance of the working and counter electrodes, allowing for much larger area solar cells.
2 Characterization
2.1 Power conversion efficiency and J–V characteristics
The efficiency of a solar cell is its most important performance parameter. We will refer to it as the power conversion efficiency (PCE), in order to clearly distinguish it from quantum efficiencies. The PCE is usually obtained from the current density (current per unit area, J) vs. potential (V) characteristics of the solar cell, recorded under illumination by a solar simulator. The standard measurement condition is illumination with 100 mW cm−2 light with AM1.5G spectral distribution, while the cell is kept at 25 °C.39J–V curves are recorded using a source meter or a potentiostat that can apply a controlled potential to the device and measure the current. Typically, J–V curves are recorded using voltage steps of 5 or 10 mV. After each voltage step some delay time should be applied (more than 100 ms) before the current measurement is done, in order to allow for the current to reach a stable value.49 If the chosen delay time is too short, J–V curves recorded in the forward and reverse direction are not identical: hysteresis is observed. While hysteresis in J–V curves has been widely discussed in the field of perovskite solar cells, it has not attracted much attention in the DSC field. The origin of hysteresis in DSC is attributed to: (i) capacitive currents, caused by (dis)charging of the mesoporous electrode after the potential step,50 and (ii) mass transport in the electrolyte and resulting concentration gradients in the redox couple concentrations.51 Hysteresis becomes very apparent in DSCs with practical electrolytes that are more viscous than the volatile acetonitrile-based electrolytes that are used for record devices.
From the J–V curve several parameters can be determined: JSC, the current density at zero applied potential; and VOC, the open-circuit potential, which is the potential found at zero current. At the maximum power point (MPP) the power output of the device (which is the product of J and V) reaches a maximum, PMPP, see Fig. 5. The fill factor (FF) is the ratio between PMPP and the product of VOC and JSC. A high value of the FF (closer to 1) gives a more square-looking curve and indicates the ability of the solar cell to deliver current and potential at the same time. The PCE is given by eqn (3), where Plight is the power density of the incoming light.
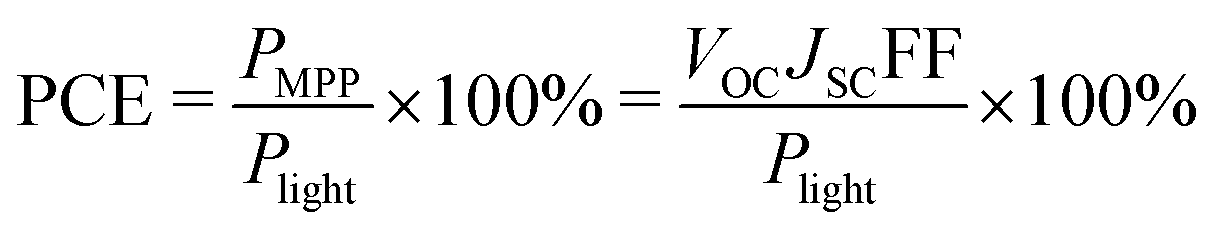 | (3) |
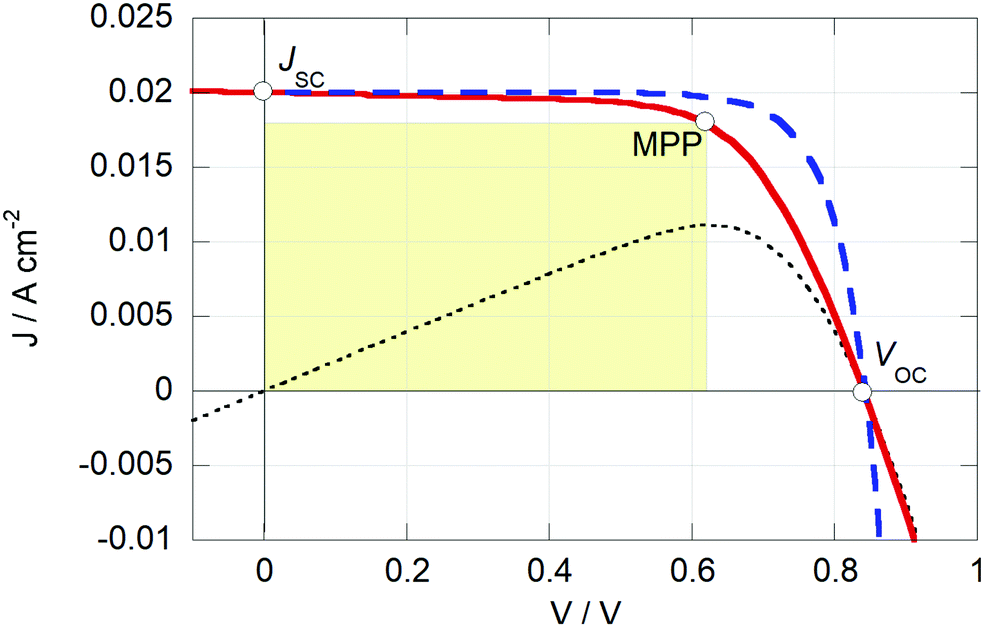 | ||
| Fig. 5 Simulated J–V curves of a solar cell using the Shockley diode model with (red line) and without (blue stripes) series and parallel resistance losses. Rs and Rp are 5 and 1000 Ω cm2, respectively; Js = 1.5 nA cm−2; n = 2. The resistance losses reduce the PCE from 13.1% to 11.2%, due of the reduced fill factor (from 78% to 66%). The black dotted line the is the device's power output with resistance losses. The yellow square represents the device's power output. | ||
In order to correctly calculate the PCE, the active area of the solar cell device needs to be determined accurately. The most reliable method used in the DSC field is to place a black metal mask with an aperture – the area of which is used for the PCE calculation – directly on top of the solar cell. Also, any light entering from the sides should be blocked. This ensures that no light from outside the aperture area is channeled into the solar cell. The aperture area should be either similar to, or smaller than the DSC working electrode.52 If a small aperture is used, part of the DSC is not illuminated. This, however, does not affect the measured PCE much since the non-illuminated areas of the DSC do not contribute much to recombination current in most cases. It is useful to record the J–V curve in the dark as well for further analysis of the solar cell, which should not use the aperture area, but instead the measured working electrode area for correct analysis.
The general shape of the J–V curve of a DSC is well-described by the Shockley diode equation with additional resistive losses, see eqn (4),
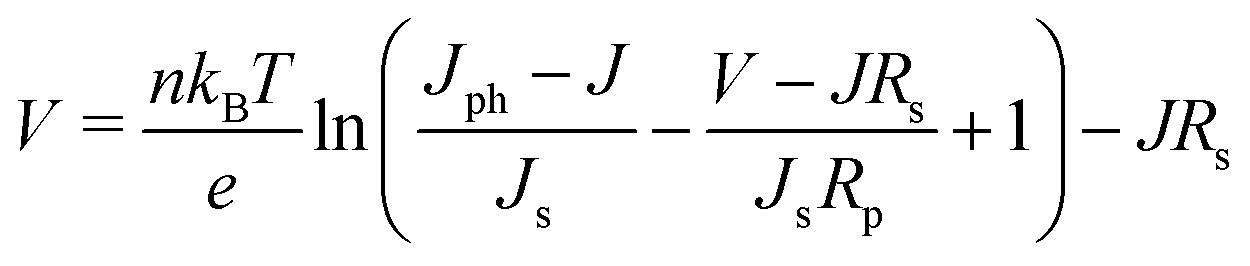 | (4) |
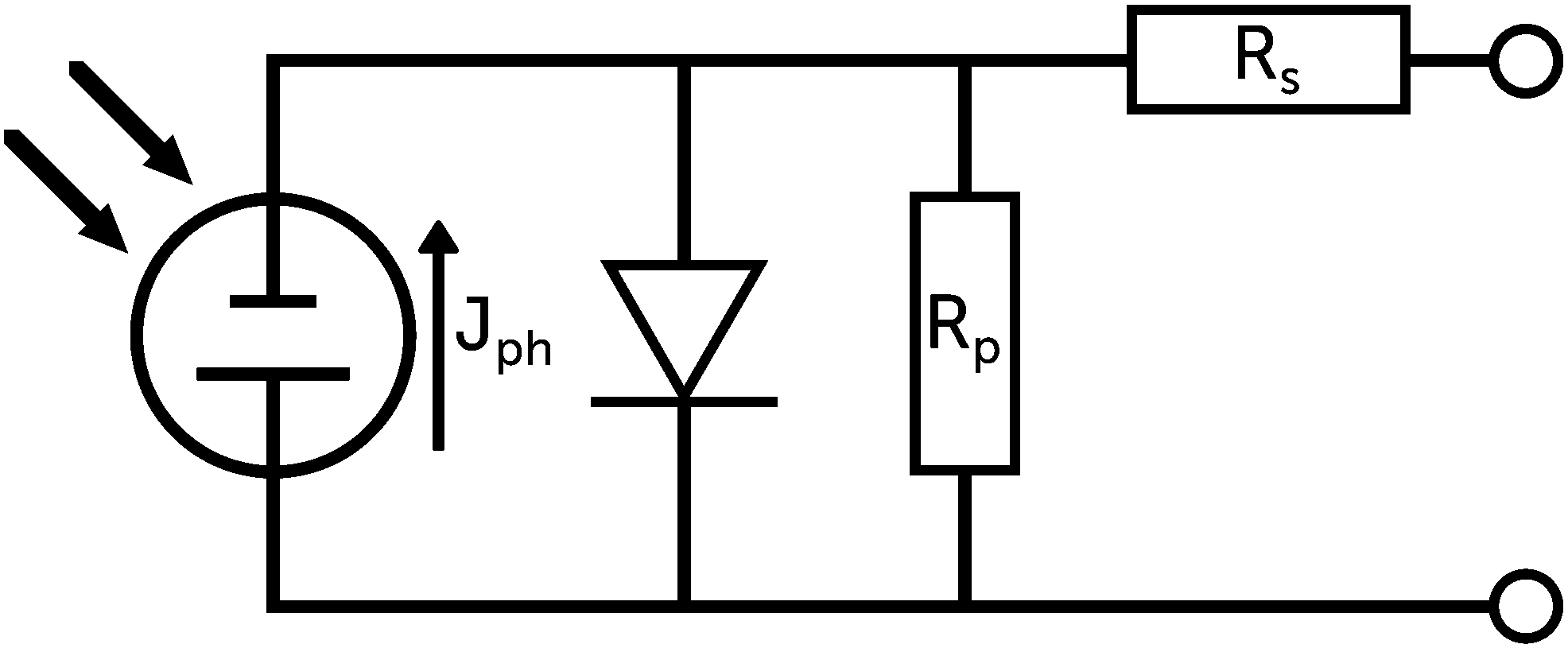 | ||
| Fig. 6 Representation of a solar cell as a schematic circuit. | ||
MPP tracking is an alternative method to obtain the PCE of a solar cell. The perturb-and-observe method is frequently applied where a step-wise change in potential is made and it is checked whether the product of J and V increases or decreases; then, depending on the outcome, the next step is made in either the positive of negative potential direction. MPP tracking is a useful method to prove that the DSC is a stable and regenerative system.
2.2 J–V characterization in ambient light conditions
Although the practicalities of solar cell measurement in ambient light (indoor) conditions are the same as those described above for sunlight simulation, the interpretation of the results is more complex. A brief overview of the challenges and best practices for reporting ambient light J–V measurements is provided here, while a more detailed discussion can be found elsewhere.38,53As detailed in eqn (3), PCE is a function of the power provided by the light source, Plight. In the case of sunlight there is a unique source of light, with well-known characteristics and a constant, standardized value of Plight. Indoor, on the other hand, there is a great variety of different light sources. This leads to the conclusion that, while in simulated sunlight measurements the reported PCE value of a solar cell can always be translated to the device's absolute power output via a simple mathematical operation, the same does not apply to ambient light measurements. In the latter case, in fact, Plight is unknown, and it is the experimentalist's responsibility to measure it accurately for the light source in use. Therefore, when performing and reporting about indoor J–V measurements: (i) extra care should be taken in the determination of Plight for the correct computation of the PCE value, (ii) the make and model of the light source should always be specified, together with its emission spectrum, and (iii) the PMPP value should always be reported alongside the PCE value. This last point is particularly important to facilitate the comparison of results from different laboratories, because a given solar cell configuration may have a very similar PMPP output when illuminated by different light sources, but very different PCE values depending on the overlap between the device absorption and the light source emission spectra.
During practical experiments, in the case of sunlight, the adjustment of the light intensity to the desired value is easily achieved through the use of a reference cell calibrated by a certification authority. However, there cannot be a calibrated reference cell in the case of indoor measurements, unless every laboratory in the world agrees to use the same light bulb. Light intensity determination in ambient light experiments is usually carried out with the use of a lux meter, which provides a value of the illuminance at the measuring spot. However, lux meters are generally bulky tools, and their correct placement inside the testing equipment could be cumbersome. This difficulty arises from one more hurdle that ambient light measurements must overcome compared to simulated sunlight experiments: In the latter case, the intensity of the light source is about two orders of magnitude higher than that present in a common laboratory room. As such, the testing equipment can be easily placed on an open laboratory bench and the eventual contribution to the device photocurrent of the light present in the room will be negligible. In the former case, however, the intensity of the light source is of the same order of magnitude of that present in the laboratory room. Therefore, the testing equipment must be properly encased, so that it is completely isolated from the laboratory environment.
2.3 Incident photon-to-current conversion efficiency (IPCE)
In an IPCE measurement, monochromatic light – typically generated by passing white light through a monochromator – falls onto the solar cell and the short-circuit photocurrent is recorded as a function of the light's wavelength. The IPCE is calculated using eqn (5) and is normally plotted as a function of wavelength, yielding a spectrum that is sometimes referred to as the photocurrent action spectrum.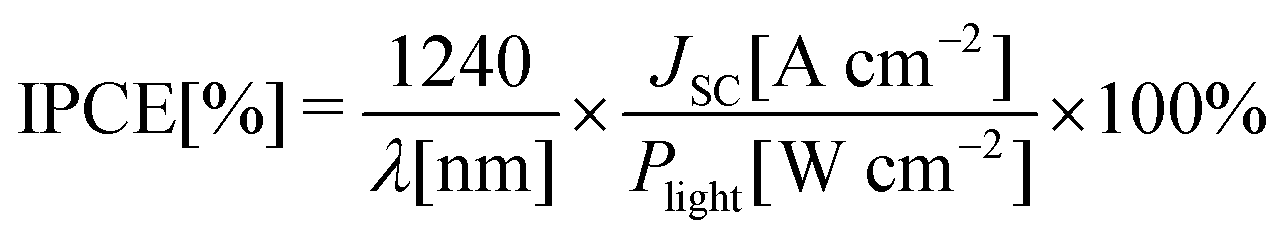 | (5) |
In the equation, λ and Plight are the wavelength and the power density of incident light, respectively. IPCE can be measured using DC or AC methods. In the DC method, only monochromatic light is used, while in the AC method chopped monochromatic light is applied, and a constant white light can be added. The AC photocurrent response is measured using a lock-in amplifier. The two methods should yield the same result, provided that the photocurrent scales linearly with light intensity and that the chopping frequency in the AC mode is sufficiently low.
Integration of the IPCE spectrum with respect to the AM1.5G flux (ϕAM1.5G) gives a calculated value of the JSC,IPCE (eqn (6)):
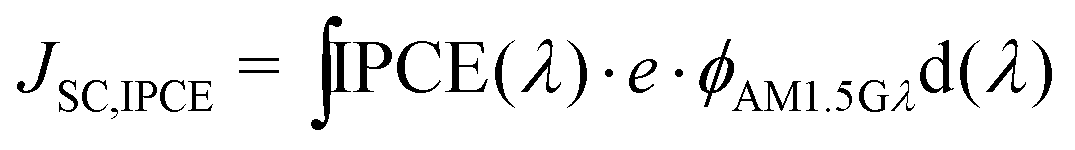 | (6) |
A good match between JSC,IPCE and JSC measured using a solar simulator gives added confidence in the validity of IPCE and JSC measurements. Significant differences can point to calibration errors of the systems.
2.4 Impedance spectroscopy
Small-modulation techniques are particularly useful to study complex systems like the DSC. We can distinguish between electrical modulation techniques, such as electrochemical impedance spectroscopy, and optical modulation techniques, such as transient photovoltage (TPV), discussed below.Electrochemical impedance spectroscopy (EIS) is a widely used general technique in science and technology. A small sinusoidal potential modulation with an amplitude of about 10 mV is superimposed onto a base potential, and the amplitude and phase-shifts of resulting sinusoidal current changes are measured. This is repeated for a large series of frequencies – for DSC typically in the 105–10−1 Hz range – to obtain a complete EIS spectrum. The impedance is given by z = dV/dI and is often represented as a complex number: z = z′+ jz′′, where j is 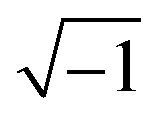 , z′ is the real part of the impedance, and z′′ the imaginary part, which is phase-shifted by 90°. The real part of the impedance reflects resistance, while the imaginary part originates from capacitance and inductance. For a resistor the impedance is independent of frequency, z = R, while for a capacitor z = −(jωC)−1, where C is the capacitance and ω the angular frequency. An equivalent circuit, consisting of electrical elements R, C, L (inductance), CPE (constant phase element, a non-ideal capacitor), and Zd (diffusion impedance or Warburg element) is used to fit the experimental EIS spectrum.
, z′ is the real part of the impedance, and z′′ the imaginary part, which is phase-shifted by 90°. The real part of the impedance reflects resistance, while the imaginary part originates from capacitance and inductance. For a resistor the impedance is independent of frequency, z = R, while for a capacitor z = −(jωC)−1, where C is the capacitance and ω the angular frequency. An equivalent circuit, consisting of electrical elements R, C, L (inductance), CPE (constant phase element, a non-ideal capacitor), and Zd (diffusion impedance or Warburg element) is used to fit the experimental EIS spectrum.
A convenient EIS analysis of DSC is done under illumination at open-circuit conditions. An example is shown in Fig. 7,54 where 3 semicircles can be found, corresponding to three processes in the DSC with significantly differing time constants. The left-hand semicircle, at higher frequencies, is due to the charge transfer resistance at the counter electrode (RCE) and to the double layer capacitance at the counter electrode/electrolyte interface (CCE), giving a time constant τCE = RCE·CCE. At intermediate frequencies, the recombination resistance at the mesoporous TiO2/electrolyte interface, Rrec, and the capacitance of the mesoporous TiO2, CTiO2, form the second semicircle. The electron lifetime in TiO2, τe, is given by τe = Rrec·CTiO2. At the lowest frequencies, the impedance due to diffusion of the redox mediator in the electrolyte, Zd, forms the third semicircle. Zd is given by Zd = Rd·(jω/ωd)−1 tanh(jω/ωd), where Rd is the diffusion resistance and ωd is D/L2, with D the diffusion coefficient and L the effective electrolyte layer thickness.55 The high frequency intercept at the Z′ axis is the series resistance caused largely by the conducting glass RTCO.
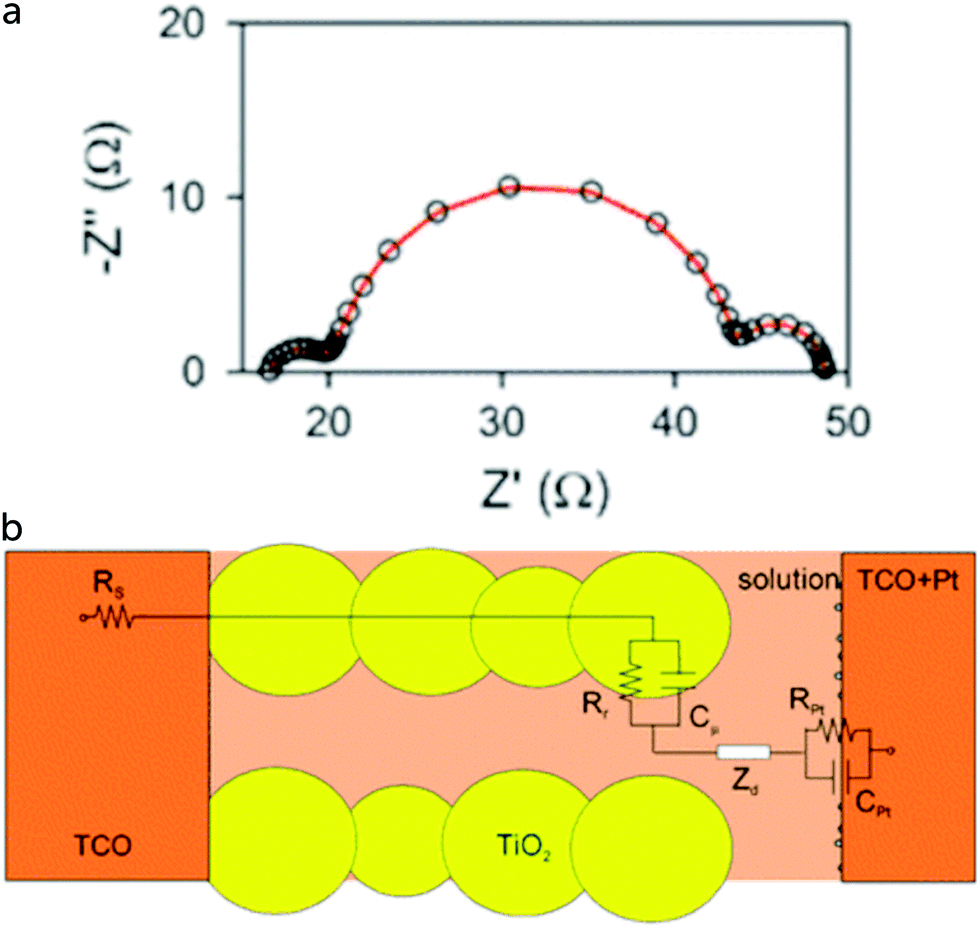 | ||
| Fig. 7 (a) Impedance spectrum (Nyquist plot) of a dye-sensitized solar cell under illumination, recorded at VOC. (b) Schematic model to fit the EIS under these conditions. Adapted from ref. 54 with permission from the PCCP Owner Societies, copyright 2011. | ||
An EIS measurement in the dark at the same applied potential would yield different results: there is for instance no electron recombination to oxidized dye molecules. Furthermore, there could be a rather large current flow in the device, which leads to potential drops and a less well-defined Fermi level in the mesoporous TiO2. The local concentrations of the redox mediator in the device will also be different. However, the advantage of a dark EIS measurement is that it allows for the direct probing of the sensitizer influence on recombination resistance from electrons in TiO2 transferring to the redox shuttle in the absence of increased electrode heat and without competing processes such as recombination to the dye.56
2.5 Opto-electrical transient techniques
Opto-electrical transient measurements and charge extraction methods provide a very useful tool for understanding processes occurring in dye-sensitized solar cells. Detailed description and analysis of such techniques can be found elsewhere.57,58 Opto-electrical transient techniques include photocurrent/voltage transients, that can be performed either as small or large modulation techniques.Light off/on modulation is easy to perform experimentally and can give useful information. Short-circuit photocurrent transients can provide evidence for accumulation or depletion of the redox mediator in different parts of the DSC. For instance, if the concentration of oxidized redox mediator is too low at the counter electrode, a high value of JSC cannot be maintained and electrons in TiO2 will have to recombine with the oxidized dye or redox mediator. Such a situation can occur in viscous electrolytes when the oxidized form of the mediator is present in too low concentration, see Fig. 8.59
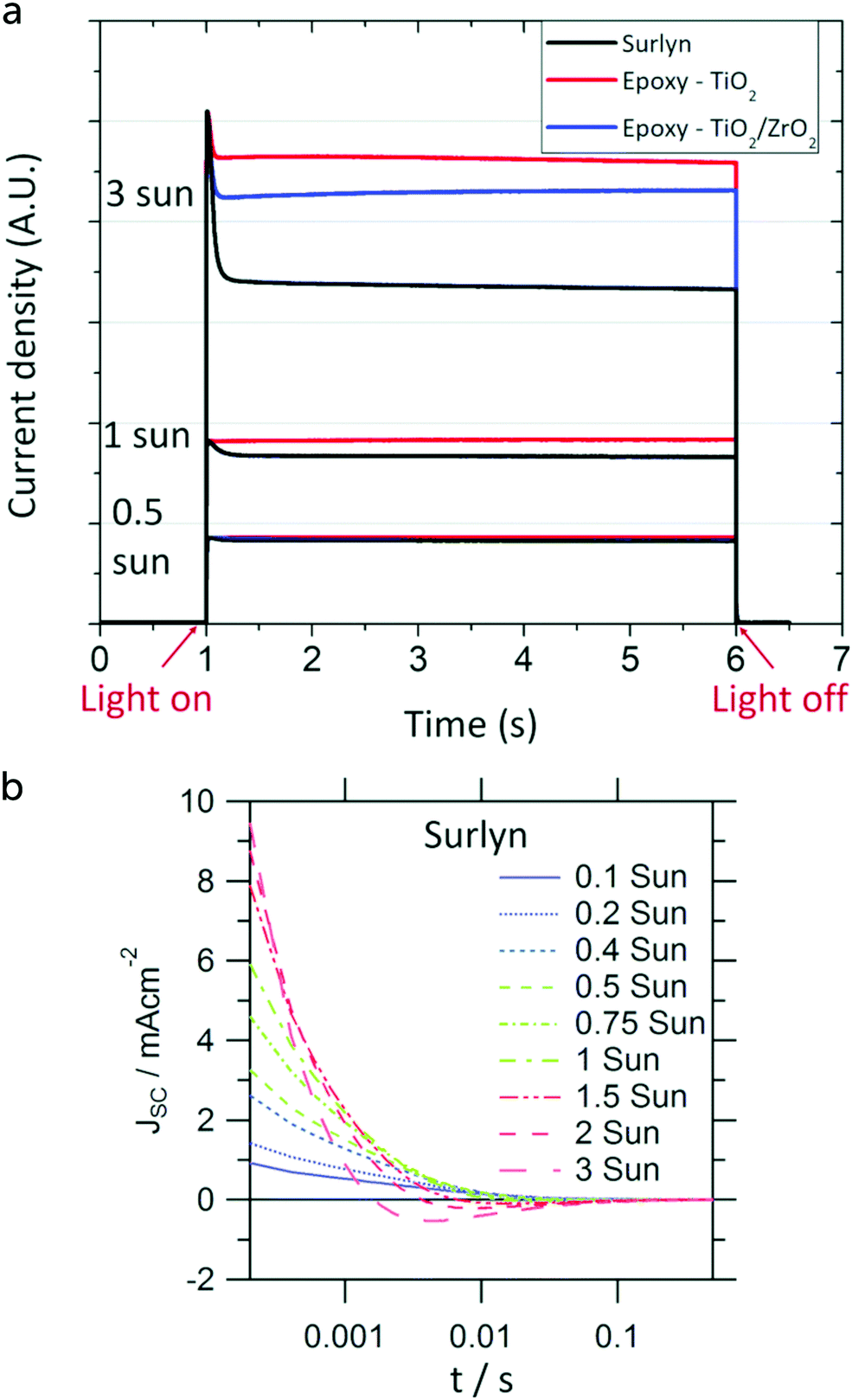 | ||
| Fig. 8 Photocurrent transients of a DSC with a Cu complex-based electrolyte. (a) Under high light intensities and with a relatively thick electrolyte layer (Surlyn: 30 μm) a clear spike is found in the photocurrent onset transient. (b) After switching the light off, a reversal of current can be found in the photocurrent decay transient, due to accumulation of oxidized redox species in the mesoporous electrode, which are reduced by electrons in the TiO2. Adapted from ref. 59 with permission from the PCCP Owner Societies, copyright 2017. | ||
Charge extraction methods provide information about the accumulated electrons in the mesoporous TiO2 electrode as a function of potential and/or light intensity. During the extraction, part of the accumulated electrons may recombine before being collected. The extracted charge should therefore be considered as a lower limit of the actual accumulated charge. Integration of the photocurrent decay transient over time gives a good measure of the accumulated charge in mesoporous TiO2 electrodes under short-circuit illumination conditions. To obtain the charge under open-circuit illumination conditions, a double switch is needed: light is switched off and simultaneously the cell is switched from open-circuit to short-circuit conditions. Plotting the extracted charge as a function of the VOC gives a useful trend that can be used to assess band-edge changes, for instance as a function of the sensitizer or of additives to the electrolyte.
Small optical modulation techniques, namely transient photocurrent (TPC) and photovoltage (TPV), provide information on electron transport in the mesoporous TiO2 and electron recombination, respectively. The modulation can be in the form of a sine wave: the technique is then called IMPS or IMVS (intensity-modulated photocurrent or voltage spectroscopy, respectively), and multiple frequencies are analyzed. Alternatively, the modulation is in the form of a small pulse or of a step, and the response is recorded in the time domain. Similar information can be obtained from EIS measurements, but TPC and TPV in the time domain have the advantage of being a rapid measurement that can be analyzed quickly, since the photocurrent or photovoltage response to a small light modulation has a simple exponential form, where the time constant is the electron transport time (provided that no significant recombination takes place) for photocurrent transients, or the electron lifetime τe for photovoltage transients. Fig. 9 gives an example of charge extraction and photovoltage transient results for different dyes used in co-sensitized DSC devices.60
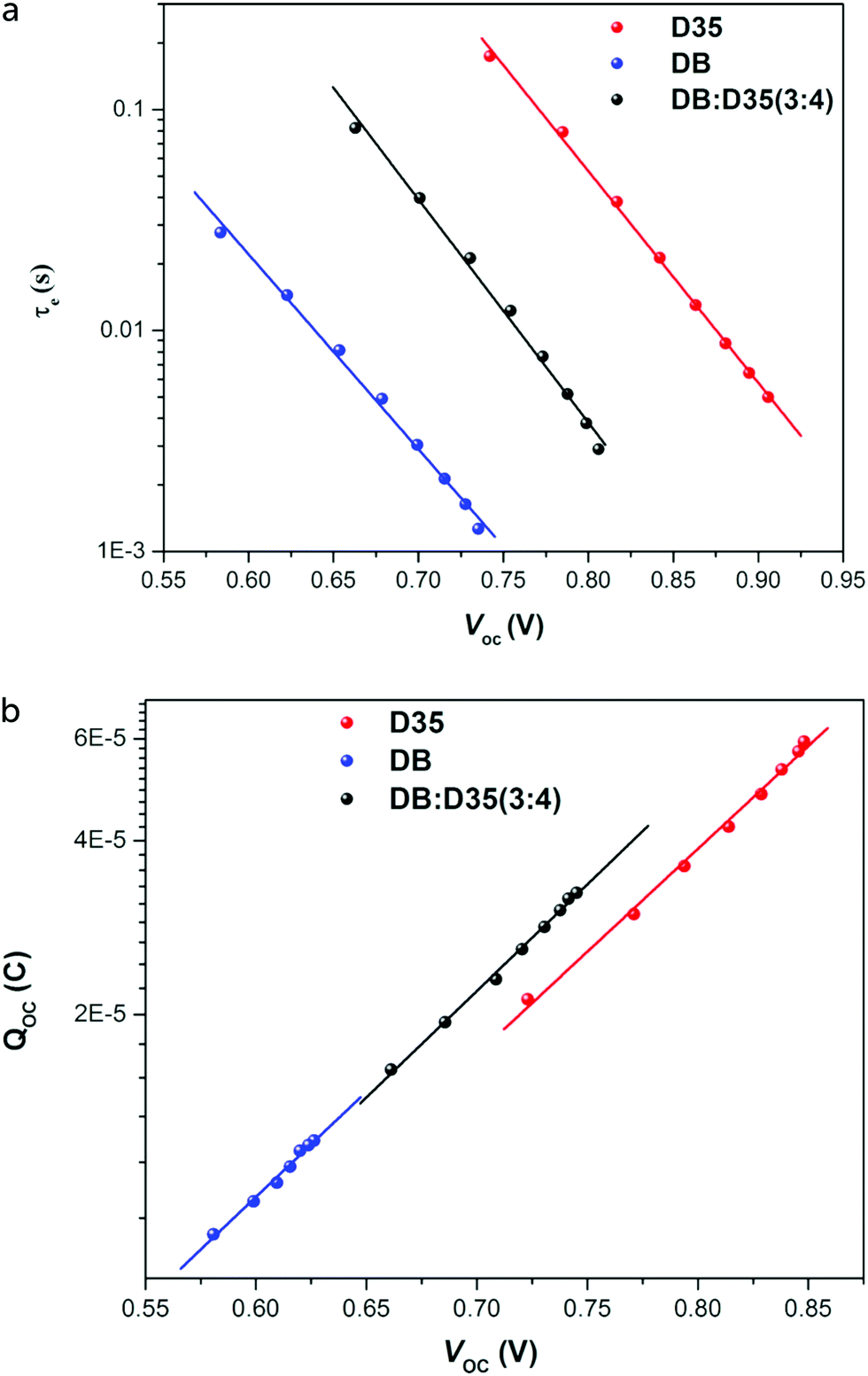 | ||
| Fig. 9 (a) Electron lifetime and (b) accumulated charge as a function of VOC for DSCs with a cobalt-based electrolyte, sensitized with D35, Dyenamo blue (DB), or both. Band-edge shifts of the different dyes are small, however a large difference in electron lifetime is found. Adapted with permission from ref. 60. Copyright 2016 American Chemical Society. | ||
2.6 Spectroscopy
An important attribute of the mesoporous anatase thin films introduced by Grätzel and O'Regan is that they are amenable to spectroscopic characterization from the visible to the terahertz region (400 nm–3 mm) in transmission mode with high signal-to-noise ratios.5 Spectroscopic studies have provided keen insights into the fundamental electron transfer reactions responsible for electrical power generation and recombination reactions that lower efficiency. Such spectroscopic data has also been used to test existing theories of interfacial electron transfer.61 Steady-state spectroelectrochemical measurements provide thermodynamic information on the dye-sensitized interface, while pulsed or modulated light excitation provides access to kinetics. In this section, insights gained over the last ten years from spectroscopic studies of dye-sensitized interfaces are presented. Unless otherwise stated, sensitized anatase TiO2 thin films immersed in organic electrolyte solvents at room temperature can be assumed.Emphasis is placed on the kinetics and mechanisms for photo-induced interfacial charge separation, sensitizer regeneration, and charge recombination. The sensitizer ground and excited state reduction potentials are often taken from measurements in fluid solution and are assumed to remain unchanged upon surface anchoring. However, there is now growing evidence that the physical location of sensitizers within the electric double layer results in behavior very different from that in a fluid solution, a point that is elaborated upon here.62 An interesting observation is that the sensitizer redox chemistry rarely obeys the Nernst equation when anchored to TiO2. Recall that a 59 mV change in the applied potential should result in a factor of ten change in concentration at room temperature, but for sensitizers anchored to TiO2 it typically implies a ∼100 mV potential step. This behavior is typically quantified by the introduction of a “non-ideality” factor (α) in the modified Nernst equation (eqn (7)).
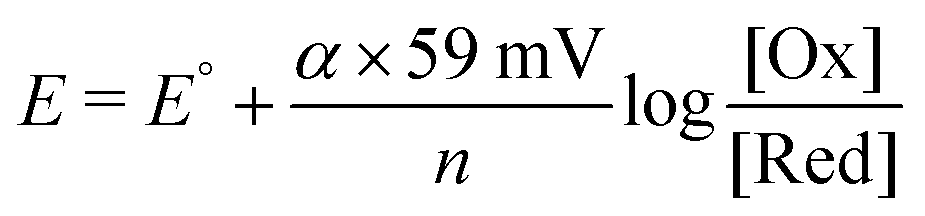 | (7) |
Insights into the origin(s) of this non-ideal equilibrium redox chemistry came from metalloporphyrin sensitizers that had two adjacent quantifiable redox couples when surface anchored, Co(III/II) and Co(II/I).63 The Co(III/II) reduction was nearly ideal yet the Co(II/I) process had a large non-ideality factor of 1.6 ≤ α ≤ 2.5. Such behavior was not easily rationalized with a “Frumkin” model wherein intermolecular interactions influence the redox equilibria. Instead, the data were most consistent with a model wherein a fraction of the electric field was present across the inner Helmholtz plane of the electric double layer. The results indicated that non-ideality was most significant when the TiO2(e−) concentration was high with a percentage potential drop of only ∼15% for the Co(III/II) couple and 45% for Co(II/I).63
Further insights into non-Nernstian redox chemistry were gained from sensitizers where a redox active center closest to the oxide surface showed a higher non-ideality factor α = 1.4 ± 0.2 than a more remote center with α = 1.1 ± 0.1.64 This suggested that proximity to the oxide surface and location within the electric double layer contribute to non-Nernstian behavior. The impact of the electric field on the spectroscopic and the non-exponential kinetics described below remains unknown. More fundamental research is needed to fully elucidate the origin(s) of this intriguing interfacial redox chemistry.
2.6.1.1 Excited-state electron injection. It has been known for some time that electron transfer from a photoexcited sensitizer to TiO2 can occur on ultrafast femtosecond time scales.65 If such excited-state electron injection was quantitative and general, a wide variety of sensitizers and light absorbing materials could be widely employed. Unfortunately, this is not the case. Below, excited-state injection is discussed for inorganic charge transfer excited states and organic sensitizers.
Inorganic charge transfer excited states. A recent advance in excited-state injection was garnered from a kinetic study of [RuII(4,4′-(PO3H2)2-2,2′-bipyridine)(LL)2]2+ sensitizers, where (LL) is an ancillary 2,2′-bipyridine ligand that tuned the excited-state potentials from −0.69 to −1.03 V vs. NHE.68 Excited-state injection showed biphasic kinetics occurring mainly at the 3–30 ps and 30–500 ps range in acidic aqueous solution. The slower process was assigned to injection from the thermally-equilibrated excited state with rate constants that were directly correlated to the excited-state potential E°(RuIII/II*). Strong photoreductants transferred electrons to TiO2 more quickly than did weaker excited state reductants. Electrochemical measurements were used to estimate the TiO2 acceptor state distribution and the overlap with E°(RuIII/II*) was correlated with the injection rate constant. Such behavior is expected based on Gerischer's model for interfacial electron transfer. The faster injection components were not analyzed in detail and were assigned to injection from higher energy unequilibrated excited states. The data indicate that the commonly reported non-exponential kinetics for electron injection can be rationalized by a continuous decrease in the injection rate constants that accompany excited-state relaxation from the initially formed Franck–Condon state to the thermally-equilibrated photoluminescent state (Fig. 10).68
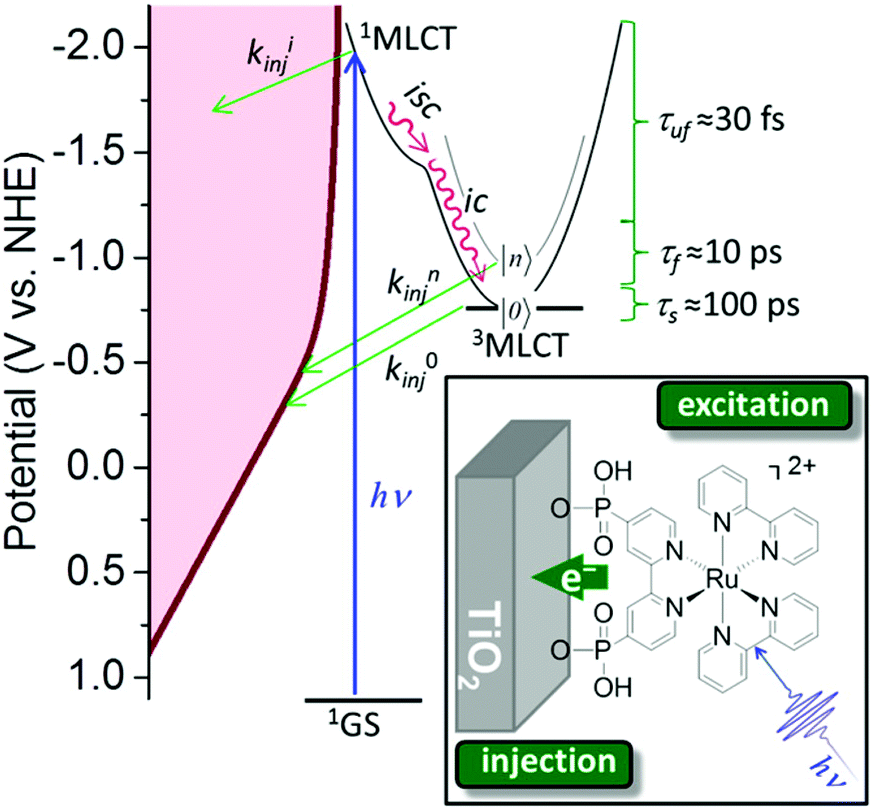 | ||
| Fig. 10 The energetic overlap of the initially-formed Frack-Condon state (1MLCT) and the photoluminescence 3MLCT with the acceptor states in anatase TiO2 at pH 1. Intersystem crossing (isc) and internal conversion (ic) compete kinetically with excited-state injection. Inset shows the structure of a Ru(II) sensitizer undergoing excited-state injection. Adapted with permission from ref. 68. Copyright 2016 American Chemical Society. | ||
Historically, Fe(II) diimine complexes have resulted in very low excited-state injection yields and there is now a detailed theoretical69,70 and experimental71,72 understanding of this. In brief, the charge transfer excited states are rapidly deactivated through low-lying metal-centered states. The exciting discovery of luminescent N-heterocyclic Fe(II) carbene complexes with long-lived excited states has dramatically changed this landscape.73–77 A comprehensive study with electron paramagnetic resonance spectroscopy, transient absorption and terahertz spectroscopies as well as quantum chemical calculations revealed an injection yield of 0.92 from the MLCT excited state.74 Such injection yields were unprecedented for charge transfer excited states based on iron sensitizers. The key to success was the realization of a 18 ± 1 ps charge transfer excited state whose lifetime exceeds that of iron polypyridyl complexes by about a thousand-fold. The nearly quantitative injection yield has motivated many to explore related Fe(II) carbene complexes with ground state Fe(III/II) potentials favorable for regeneration with donors like iodide.75–77 First row transition metal sensitizers based on Cu(I) and Co(I) have also been found to inject electrons efficiently into TiO2 (Fig. 11).78–80
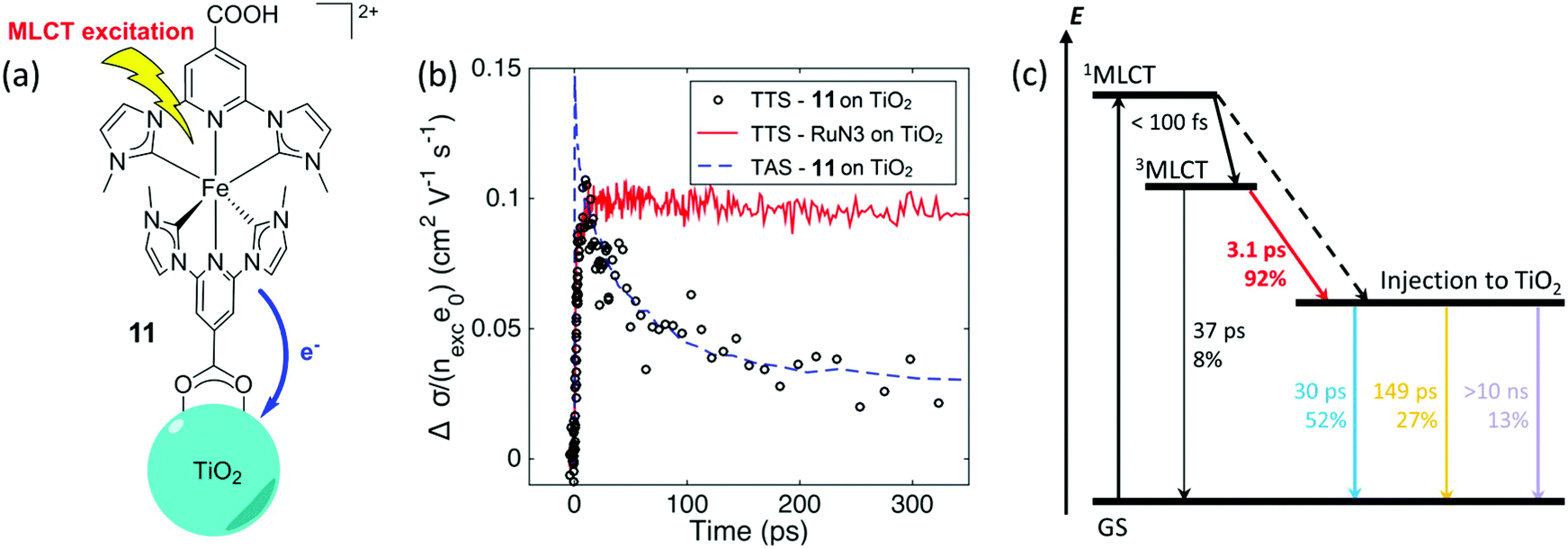 | ||
| Fig. 11 (a) Chemical structure of the N-heterocyclic Fe(II) carbene complex anchored to TiO2. (b) Transient absorption and terahertz kinetic data for the iron carbene complex and for N3. (c) A Jablonski-type diagram. Reprinted with permission from ref. 75. Copyright 2016 American Chemical Society. | ||
Organic excited states. The late Charles Schmuttenmaer reported novel terahertz injection studies of porphyrins and metalloporphyrins anchored to TiO2 and SnO2.79–82 The long-term goal of these studies was dye-sensitized water oxidation, and high potential porphyrins that were weak excited state reductants was the predominant focus. The injection yields were often less than unity on to TiO2 surfaces and were enhanced on SnO2 by virtue of a ∼0.5 eV more positive conduction band edge. On both substrates and similar to the ultrafast injection studies with Ru(II) sensitizers, more rapid injection was observed with porphyrins that were stronger photoreductants in the fluorescent singlet excited state. The THz measurements were made in the absence of an electrolyte. An interesting aspect of the porphyrin sensitizers is the presence of low-lying triplet states whose population was shown to impact the injection yield. The orientation of the porphyrin with respect to the oxide surface was also controlled by functional groups for surface binding on the aromatic porphyrin ring or through axial ligation in metalloporphyrins. It is interesting to note that injection from porphyrins with hydroxamate binding groups was as good as that measured with the more commonly used carboxylate groups.79
Ultrafast excited-state injection studies of porphyrins anchored to TiO2 through well-defined rigid linkers have been reported.83 Application of a time domain vibrational spectroscopy pump degenerate four-wave mixing technique enabled identification of the Raman-active modes triggered by light absorption. The spectral data were assigned to modes based on the linker group and that localized on the porphyrin ring. The data suggested that this four-wave mixing technique can distinguish between vibrational modes generated by light absorption from those generated by excited-state injection.83
In a related study, excited-state injection by (perylene-9-yl)carboxylate into TiO2 was shown to be complete within 12 fs.84 The ultrafast transient absorption data mapped the decay of the singlet excited state and the appearance of the oxidized perylene. Nonadiabatic quantum dynamic simulations indicated that injection was complete within 20 fs, in close agreement with the experimental value. The reorganization energy for electron transfer was estimated to be 220 meV. Non-equilibrium modes in the 1000–1800 cm−1 region were assigned to in-plane asymmetric vibrations of the perylene sensitizers. The agreement between theory and experiment in these studies indicates that these are powerful tools for quantifying vibronic effects at dye-sensitized interfaces.84
The classical iodide/triiodide redox mediators have been the subject of several prior reviews and are only summarized here.85–87 Iodide oxidation yields a metastable species in di-iodide, I2−˙, either through the iodine atom intermediate I˙ + I− → I2−˙ or (possibly) through a concerted pathway. Di-iodide is unstable with respect to disproportionation: 2I2−˙ → I3− + I−. In acetonitrile solutions, the one-electron reduction of I3− by TiO2(e−) is thermodynamically uphill and the equilibrium concentration of I2 is small. These factors allow for efficient transport of the injected electrons with minimal recombination. Iodide oxidation happens on a time scale of hundreds of nanoseconds for most sensitizers. Many researchers concluded that the regeneration by iodide was completely optimized using quantitative Incident Photon-to-Current Efficiency (IPCE) in the short circuit condition. However, at the open-circuit or power point conditions, where the number of electrons in each nanocrystallite is large, there is now clear evidence that regeneration is non-quantitative.88,89 The regeneration quantum yield, Φreg, has been determined spectroscopically by eqn (8), where kreg is the pseudo-first-order regeneration rate constant at molar donor concentration [D].
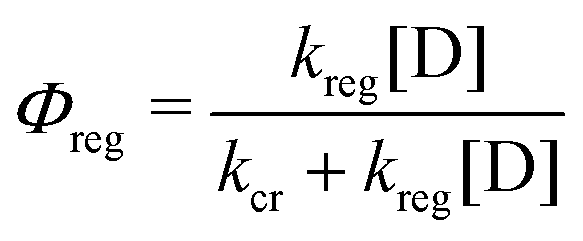 | (8) |
Nanosecond transient absorption kinetic measurements were made with D–π–A sensitizers as a function of the applied potential to simulate conditions along the current–voltage curve. It was found that Φreg decreased from unity to 0.83 at the open-circuit condition with 0.5 M I−. For 0.3 M [Co(bpy)3]2+, the quantum yield decreased to 0.60.88 Irradiance-dependent photoelectrochemical measurements with the classical N3 sensitizer provided the same conclusion: regeneration is quantitative at short-circuit and non-quantitative at the open-circuit and power point conditions.89 For alternative oxides, such as SnO2, regeneration has also been shown to be non-optimal due to the more rapid recombination.90 Realization that regeneration can be better optimized to enhance fill factors and open-circuit photovoltages continues to inspire researchers to design interfaces capable of more rapid regeneration without a significant loss of free energy.
Regeneration kinetics have been enhanced with sensitizers competent of halogen and chalcogen bonding.91–93 Kinetic regeneration studies of organic D–π–A sensitizers where the triphenylamine donor was substituted with halogen atoms were conducted, Fig. 12. In their oxidized form the presence of a σ-hole for halogen bonding was apparent in the sensitizers with Br and I. Transient spectroscopic studies revealed a correlation between the sensitizer halogen bonding ability and the second-order regeneration rate constant by iodide, yet no trend was observed with [Co(bpy)3]2+, which is incapable of halogen bonding. While the power conversion efficiency enhancements were small, these studies provided compelling evidence that halogen bonding can be utilized to enhance regeneration kinetics and yields at dye-sensitized/TiO2 interfaces.
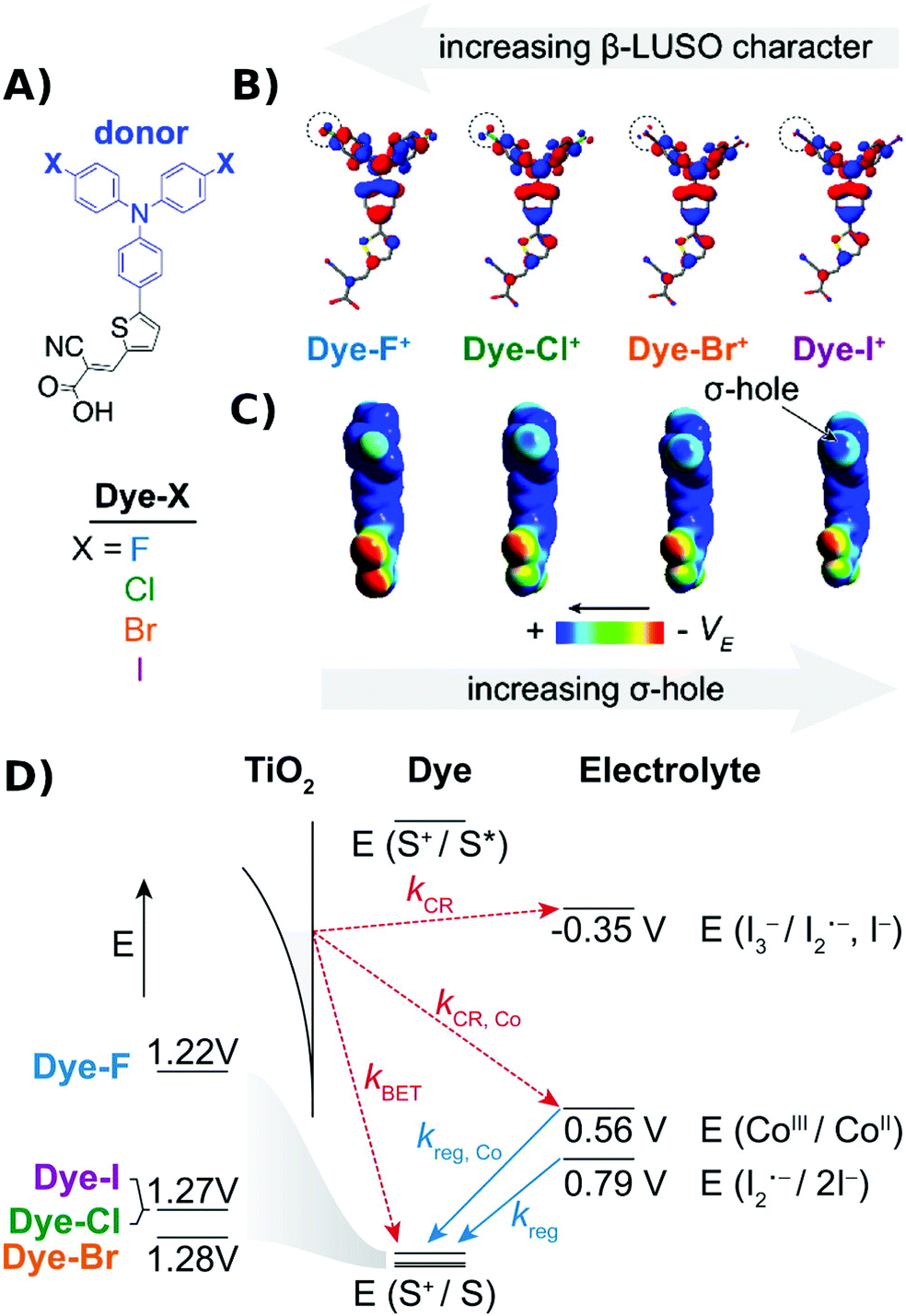 | ||
| Fig. 12 (A) Molecular structures of the Dye-X series. (B and C) DFT models of the singly oxidized forms of Dye-X showing (B) the β-LUSO and (C) the existence of σ-holes on the poles of the terminal halogen substituents for the series, with the exception of Dye-F. (D) Scheme of energy levels and electron transfer processes. Adapted with permission from ref. 92. Copyright 2016 American Chemical Society. | ||
A notably rapid regeneration process was reported for highly cationic Ru(II) sensitizers, [Ru(tmam)2(dcb)]6+, where tmam is the quaternary ammonium derivative, i.e. 4,4′-bis-(trimethylaminomethyl)-2,2′-bipyridine.94 When anchored to TiO2, these sensitizers showed clear evidence of ion pairing with iodide and an anionic cobalt redox mediator (Keq > 104 M−1) in acetonitrile. Injection and regeneration on time scales of less than 10 ns were achieved using Co mediators. Diffusion limitations associated with sensitizer regeneration were improved by ion pairing and the IPCE nearly doubled.94
An interesting aspect of Cu(II/I) bipyridyl mediators is that the two redox states often have very different coordination environments.95–102 The Cu(I) redox state is typically four-coordinate with a pseudo-tetrahedral geometry, while Cu(II) is subject to a Jahn–Teller distortion that is often manifest in five-coordinate complexes with the fifth ligand derived from solvent or counter-ion. In a comprehensive study with three different D–π–A sensitizers, regeneration by the four Cu(I) diimine mediators shown was investigated, Fig. 13.95 These mediators possess methyl groups in the 6,6′ positions of bipyridine and the 4,7 positions of 1,10-phenathroline that prevent planarization of the two ligands in the Cu(II) state, resulting in a significant positive shift in E°(CuII/I). For two of the three sensitizers, the regeneration rates increased with thermodynamic driving force and Φreg ∼ 1 in all cases. Regeneration by [Cu(eto)2]+ was so rapid that in some cases it was unclear experimentally whether injection occurred first or whether a photogalvanic mechanism was operative. Prior work revealed that these Cu diimine complexes were able to quench the sensitizer excited states.96 Density functional theory calculations were used to estimate the reorganization energy – λ – for regeneration in the presence and absence of Lewis-basic 4-tert-butylpyridine (tBP). Interestingly, this analysis indicated that tBP binding to Cu(II) had a dramatic ∼1 eV increase in λ that was predicted to result in charge recombination in the normal region, with Marcus inverted recombination in the absence. The ability to tune redox reactivity with external Lewis bases is a novel aspect of these mediators that may be further optimized for dye-sensitized solar cell applications.95–102
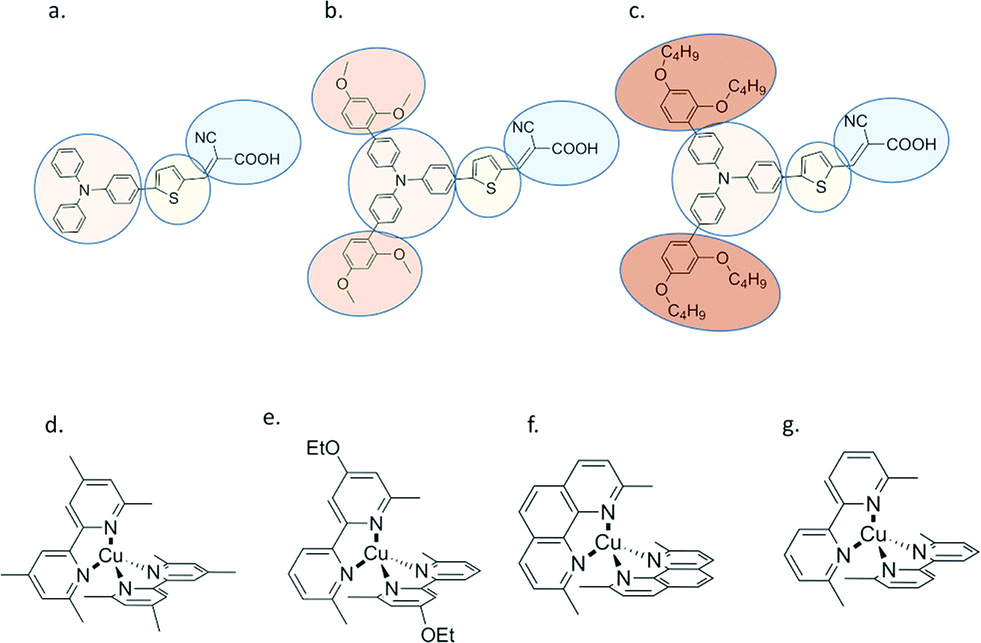 | ||
| Fig. 13 Molecular structures of (a) D5, (b) D45 and (c) D35 dyes, and (d) [Cu(tmby)2]2+/+, (e) [Cu(eto)2]2+/+, (f) [Cu(dmp)2]2+/+ and (g) [Cu(dmby)2]2+/+ complexes. Reprinted with permission from ref. 95. Copyright 2018 American Chemical Society. | ||
A significant advance in regeneration at dye-sensitized p-type NiO was realized with tris(acetylacetonato)iron mediators, abbreviated [FeIII/II(acac)3]0/−.103 The second-order regeneration rate-constant measured spectroscopically was large, 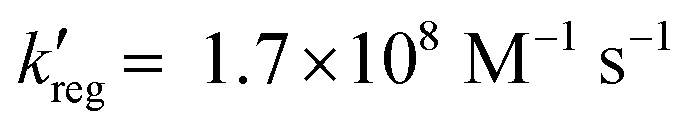 . At the mediator concentrations employed, this rate constant indicated a regeneration yield Φreg = 0.99. This is a particularly notable advance as these iron mediators significantly enhanced the efficiency of dye-sensitized p-type materials.103
. At the mediator concentrations employed, this rate constant indicated a regeneration yield Φreg = 0.99. This is a particularly notable advance as these iron mediators significantly enhanced the efficiency of dye-sensitized p-type materials.103
The discovery that hole-hopping rates were directly correlated with charge recombination kinetics represents an important finding.112 Sensitizers that undergo rapid S + S+→ S+ + S hole-hopping were shown to recombine more rapidly than those that hop more slowly. An example is shown in Fig. 14, where the transient absorption data reports on the charge recombination reaction while the anisotropy reports on hole-hopping. For the D–π–A sensitizer mp13, both hole-hopping and charge recombination responded in a similar fashion to changes in the solvent or external environment.
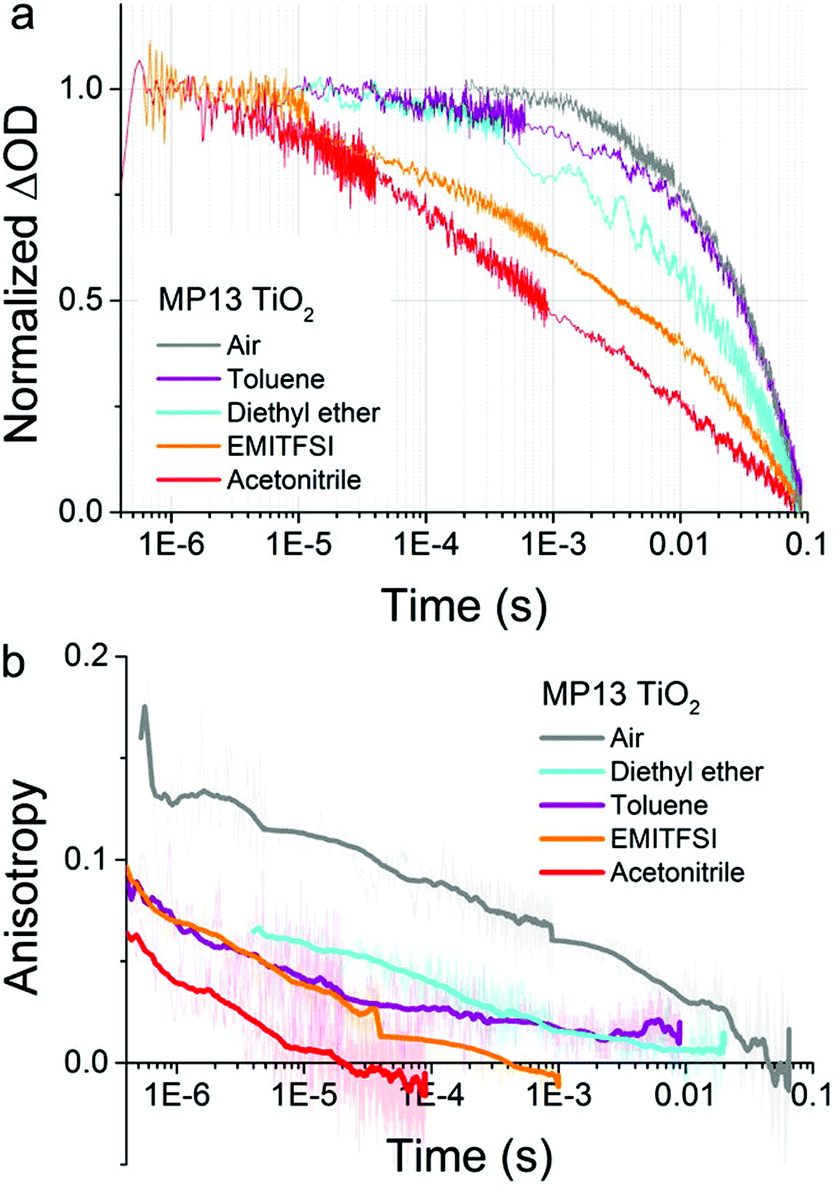 | ||
| Fig. 14 (a) Transient absorption and (b) transient absorption anisotropy spectroscopy on MP13 sensitized TiO2 films on glass immersed in different environments. The films were pumped with pulsed laser excitation at 430 nm while the oxidized dye signal was probed at 770 nm. The solid lines in (b) are obtained by calculating a moving average of the raw data (also displayed in background). Adapted with permission from ref. 112. Copyright 2016 American Chemical Society. | ||
Studies of a homologous series of four sensitizers that maintain the cis-Ru(NCS)2 coordination environment with one surface anchoring group show that they undergo rapid hole-hopping.113,114 The hole-hopping rate constants – khh – measured electrochemically spanned about a factor of seven and followed the same trend as did the charge recombination kinetic data.114 Subsequent temperature and surface coverage-dependent kinetic studies with sensitizers that displayed very different hole-hopping rates also supported the conclusion that rapid hole-hopping promotes charge recombination.115 Interestingly, no correlation between the activation energy for hole-hopping or charge recombination was evident with the solvent dielectric, but both dynamic processes could be tuned by the addition of inert salts to the solvent or by controlling access of electrolyte cations to the oxide surface.116 These results lead to the conclusion that undesired recombination of charges may be reduced by limiting lateral hole-hopping. This implies that hole-hopping may play a greater role in charge recombination than transport of the injected electrons.112 Control of the intermolecular distance between sensitizers and the electrolyte tunes the charge recombination reaction and can favor conditions where the transient spectroscopic data reflects the true interfacial electron transfer event.
Absorption of a photon initiates the formation of one injected electron and one oxidized sensitizer. They are formed in equal numbers and a second-order recombination might be anticipated with the rate law as r = k[S+][TiO2(e−)]. An Ostwald isolation type approach where an applied potential was used to control the number of electrons and oxidized sensitizers identified the rate law as r = k[S+]1[TiO2(e−)]1.117 The Ostwald isolation conditions differ from those encountered in operational solar cells or in transient photovoltage measurements where alternative rate laws have been reported.118 In all cases, the injected electrons reside in spherical nanocrystallites interconnected in a mesoporous thin film, whereas the oxidized dye molecules are restricted to the quasi-two-dimensional oxide surface. Hence, charge recombination is an intriguing process where opposite charges on different sides of an interface come into close proximity before electron transfer occurs.
For fundamental recombination studies, transparent conductive oxide (TCO) materials have some advantages.119–121 They have a metallic character, which permits potentiostatic control of the Fermi level (EF) and, consequently, of the driving force for charge recombination, −ΔG° = nF(E°′ − EF). Quantifying kcr as a function of −ΔG° allows analysis through Marcus-Gerischer theory and access to the total reorganization energy (λ) and to the electronic coupling. Studies with acceptors positioned at variable distances from a TCO surface provided a remarkable result: λ decreases to near zero when the acceptor is most proximate to the oxide surface.121 At distances greater than ∼20 Å in the diffuse part of the electric double layer, λ approximately equals the value expected for homogeneous reactions, λ ≈ 0.9 eV. Thus, dye-sensitization with transparent conductive oxides provides exciting opportunities to test interfacial electron transfer theories and to probe the impact of the electric double layer.
2.6.3.1 Recombination to solution species. It was recently shown that under some conditions electron transfer from TiO2 to acceptors dissolved in fluid solution followed a first-order kinetic model.122,123 Excited-state injection followed by sensitizer regeneration with triphenylamine donors dissolved in solution were used to quantify the reaction TiO2(e−) + TPA+ → TiO2 + TPA. Interestingly, when the thermodynamic driving force for this reaction was large, first-order kinetics were operative, a non-intuitive result that suggests the TPA+ acceptors are electrostatically bound to the oxide surface allowing a uni-molecular-type recombination reaction. When −ΔG° was small, dispersive kinetics were observed and attributed to electron transport to the oxidized TPA. Temperature-dependent studies analyzed through transition state theory indicated that recombination occurs with a highly unfavorable entropy of activation.122 Activation energies were the same (within experimental error) – 12 kJ mol−1 – for all interfacial electron transfer reactions, indicating that the barriers for electron transport and interfacial electron transfer were similar. Eyring analysis indicated a substantial entropy change to the activation barrier.123
The TiO2(e−) + I3− → reaction is known to be kinetically slow on a millisecond time scale, behavior that is typically attributed to an unfavorable positive ΔG°. The identity of Lewis acidic cations present in the electrolyte impacts the reaction kinetics.124–126 Alkaline and alkaline earth cations screen the electric field generated by the injected electrons and also influence charge recombination to organic acceptors.126 Interestingly, the SnO2(e−) + I3− → reaction is much slower than for TiO2 and extends to the seconds time scale, presumably by virtue of the more positive SnO2 donor states.90
2.6.3.2 Sensitizer–bridge–donor (S–B–D) acceptors. A successful approach for inhibiting unwanted charge recombination is to regenerate the oxidized sensitizer by intramolecular electron transfer.127–130 In this approach, electron transfer occurs from a donor D covalently linked through a bridge unit B to the oxidized sensitizer S. An interesting observation was that a relatively small structural change in the planarity of an aromatic bridge altered the electron transfer mechanism from adiabatic to non-adiabatic. Interestingly, recombination to S+ and D+ were the same for adiabatic transfer, while non-adiabatic transfer to D+ was markedly inhibited. The kinetic data revealed that recombination utilized a bridge-orbital pathway.127
In one study, the S+/0 and D+/0 reduction potentials were very similar such that excited state injection created a quasi-equilibrium Keq = k1/k−1 that was quantified over an 80 °C temperature range, TiO2|S+–B–D ⇌ TiO2|S–B–D+. A significant barrier was measured under all conditions indicating that a true redox equilibrium was operative. The magnitude of Keq was closer to unity for the phenyl bridge and hence 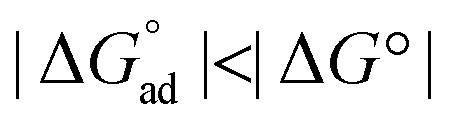 , as had been predicted theoretically. The van't Hoff shown for the adiabatic equilibrium clearly indicates ΔH° = qp = 0, and that the equilibrium constants are determined solely by ΔS°. For the non-adiabatic equilibrium, ΔH°= ± 7.0 kJ mol−1.128 The results show that the magnitude of ΔG° is decreased when adiabatic pathways are operative, a finding that should be considered in the design of S–B–D sensitizers for dye-sensitized solar cell applications.129,130
, as had been predicted theoretically. The van't Hoff shown for the adiabatic equilibrium clearly indicates ΔH° = qp = 0, and that the equilibrium constants are determined solely by ΔS°. For the non-adiabatic equilibrium, ΔH°= ± 7.0 kJ mol−1.128 The results show that the magnitude of ΔG° is decreased when adiabatic pathways are operative, a finding that should be considered in the design of S–B–D sensitizers for dye-sensitized solar cell applications.129,130
3 Theory and computational studies
DSCs offer a unique playground for fundamental studies of complex phenomena concerning sunlight harvesting, charge and mass diffusion across multi-layer heterogeneous interfaces, and electrochemistry. Theory and computation have been key players in providing the scientific foundation to understand and dissect DSC devices, starting from isolated components (e.g. dyes, electrodes) and elementary processes up to electron/ion transport properties at hybrid organic–inorganic and liquid–solid interfaces.131–134 This section presents a brief outline of the state-of-the-art theoretical methods addressing these systems and processes, with a particular focus on cutting-edge studies from the last ten years (Fig. 15).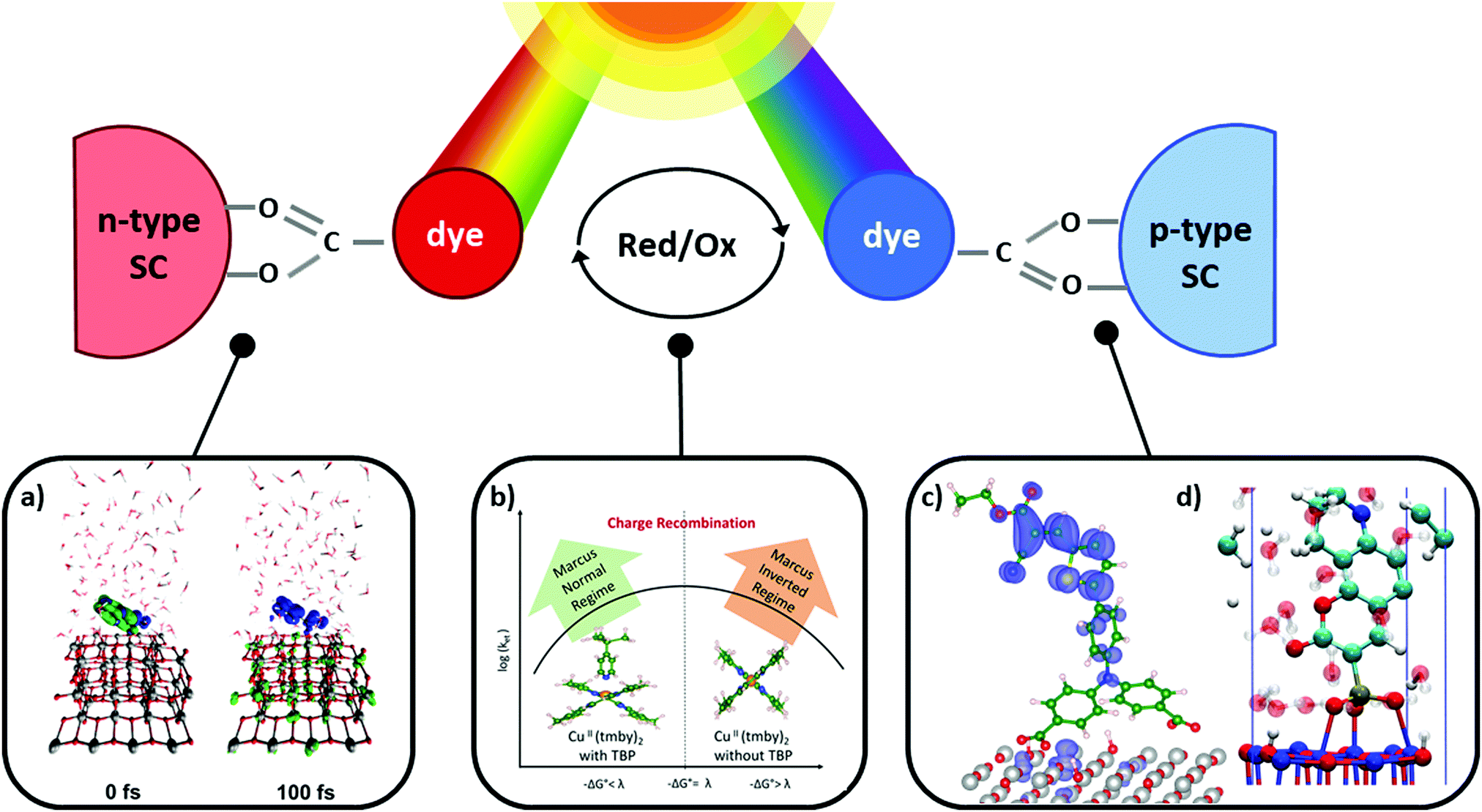 | ||
| Fig. 15 Examples of recent computational studies on DSC components. (a) electron (green) and hole (blue) densities at the beginning of the simulation (t = 0 fs) and upon electron injection (t = 100 fs) for benzohydroxamic acid anchored on TiO2 with full explicit water solvation. Adapted with permission from ref. 135. Copyright 2020 American Chemical Society. (b) Analysis of charge transfer parameters in Cu-based electrolytes. Adapted with permission from ref. 95. Copyright 2018 American Chemical Society. (c) Isosurfaces of band-decomposed charge density of the lowest unoccupied band of the push–pull dye T1/NiO system. Adapted with permission from ref. 136. Copyright 2019 American Chemical Society. (d) Anchoring geometry of C343 as a model dye on NiO during the molecular dynamics simulation in explicit water. Adapted with permission from ref. 137. Copyright 2017 American Chemical Society. | ||
3.1 Theoretical background
Simulation of sunlight conversion to electricity in DSCs calls for the application of several theoretical methods to tackle complex materials and processes that span across several scales of space and time. Light harvesting, dye/electrode charge transfer, electron transport to the charge collector, oxidized dye regeneration, electrolyte diffusion, and reduction at the counter electrode are all processes that occur at different places and with different time frames, from femtoseconds to milliseconds. Therefore, the simulation approach must be multi-scale, starting from the elementary processes at the nano scale and adding step-by-step the effects coming from larger (longer) space (time) scales.Initially, the quantum mechanical (QM) interactions among electromagnetic radiation, electrons, and nuclei need to be properly described. Within this framework, Density Functional Theory (DFT) is the current method of choice for the electronic structure of materials and interfaces,138 and its extension to Time-Dependent DFT (TD-DFT) has also enabled the effective description of excited state properties.139 However, the application of Kohn–Sham DFT and the related TD-DFT still suffers from the approximate nature of the unknown exchange–correlation (XC) density functional.140 This flaw is very relevant for modeling within the context of DSCs as it can jeopardize DFT results reliability in predicting charge transfer processes involving strongly correlated materials (e.g. transition metal oxide-based electrodes) and non-covalent weak interactions (e.g. dispersion forces).141 Recent theoretical advances in XC formulations and other effective approaches have been able to amend most of these drawbacks, but often only on a case-specific base. Moreover, DSC molecular and solid-state components have been traditionally studied within different numerical approximations, with no or little overlap, which has hindered an easy transfer of theoretical advancements from one DSC component to the other. For example, successful TD-DFT approaches for molecular dyes are not numerically feasible for solid-state electrodes. Vice versa, new approaches beyond DFT (e.g. GW142,143 and RPA144) for bulk-extended materials are still not feasible for realistic hybrid interfaces. Thus, the following sections will discuss: (i) the best available approaches for each DSC component, (ii) the relevant physico-chemical properties to be computed, and (iii) how the results from first-principles calculations can be implemented in multi-scale models to predict the overall DSC power conversion efficiency.
3.2 Theoretical description of sensitizers and molecular components
Since the earliest characterization of Ru-based145,146 and organic147 dyes, the computer power and theoretical machinery for modeling excited states of molecular species has considerably grown.148 The advancements in XC functionals (long-range corrected hybrid149 and double hybrid150) and in TD-DFT algorithms (e.g. analytical first derivatives) allowed the molecular design of dyes with specifically tailored properties for application in n-type151,152 and p-type153 photoelectrodes. The combination of long-range corrected density functionals like CAM-B3LYP or ω-B97X and triple-z quality basis sets such as 6-311++G(d,p) and def2_TZVP have provided excellent results even for the challenging cases of intra-molecular charge-transfer excitations.154 When TD-DFT fails, excited-state properties can still be obtained by means of wavefunction-based methods (e.g. CASPT2,155 NEVPT2156 and EOM-CCSD157), whose major limit is the dye size, due to their high computational cost.A key strategy to avoid undesired charge recombination is based on the development of push–pull dyes, where the excited electron is localized close to the electrode (for standard n-type DSCs158) or exposed to the solvent (in photocathodes159). The molecular design of new dyes with such characteristics has been greatly aided by the topological analysis of electron density changes upon photoexcitation, such as the combination of TD-DFT and density-based charge-transfer indexes.160 This approach is based on the analysis of the difference between the charge densities of the excited and the ground states and has been proven to be very effective for molecular dyes,161 including metal-based ones.162,163 Additionally, this approach has been recently updated to account for complex dye structures.164
A significant novel contribution of the DFT-based quantum chemistry approach is related to the new transition metal complexes developed as redox shuttle substitutes to the I−/I3− electrolyte. First-principles approaches have been exploited to assess the molecular parameters related to their redox potential – to be compared with the dye HOMO energy level – in order to evaluate the driving force for dye regeneration,165 as well as to consider the reorganization energies upon oxidation within a diabatic charge transfer scheme based on Marcus theory.166 The results of hybrid DFT on Co and Cu complexes present certain levels of inaccuracy in predicting the redox potentials, with errors usually around 0.2–0.5 eV with respect to experimental data.14 This is due to the approximate nature of the XC density functional when comparing two systems with a different number of electrons. A much better agreement between theory and experiment is achieved in the computation of reorganization energies (λ) and corresponding charge transfer kinetic parameters.95,167
The accuracy in predicting such parameters (photoexcitation, redox potential, reorganization energies) largely depends on the approach used for modeling the chemical environment. A well-known and effective strategy to model the structure and properties of solvated systems is represented by focused models, where the system is partitioned into a chemically interesting core (e.g. the solute in a solution) and the environment, which perturbs the core, modifying its properties. While a level of theory as high as required is retained for the core, the environment is treated in a more approximate way. Two popular alternatives of such approaches are: (i) to consider the environment as a structure-less continuum as in the Polarizable Continuum Model (PCM),168 or (ii) to retain its atomistic resolution within a molecular mechanics (MM) description.169 Both alternative strategies can be effectively coupled to a QM description of the core, and can also be coupled together to overcome their respective limitations.170 In the context of DSC, PCM and hybrid QM/MM approaches have been extensively applied to account for the solvent effects on the physico-chemical properties of dyes and redox shuttles.171
3.3 Simulation of solid-state electrodes and heterogeneous interfaces
The first systematic computational studies on DSCs concerned the main components of the original Grätzel cell, focusing mostly on n-type semiconductor oxides (e.g. TiO2, ZnO, SnO2) and their interfaces with molecular dyes (e.g. dye anchoring groups).131–134 In the last decade, the quest for tandem cells has spurred theoretical studies also on p-type DSC components172 (p-type semiconductors, push–pull dyes, and their interfaces), which were barely studied in the first years of the modern DSC technology. In both cases, studies of electrode and counter electrode materials have relied on the periodic supercell DFT approach, mainly by employing plane-wave basis set and pseudo-potentials replacing core electrons.173–176 Standard local and semi-local XC functionals have been recently replaced mostly by DFT+U177 and hybrid HF-DFT178 for modeling the strong-correlated nature of the transition metal oxides that are commonly employed as electrodes in DSCs. The characterization of band structures with these methods can provide useful hints on the nature of the bandgap and the possible optical properties, as well as on electron/hole mobilities.179 Within this framework, recent studies have explored several possible alternatives to NiO for p-type DSC and tandem cells.180,181 While semi-local DFT (GGA) provides too low of a bandgap, the DFT+U approach strongly depends on the choice of the Hubbard-like U–J parameter. The hybrid HF-DFT approach tends to overestimate the bandgap, and the estimate is also affected by the choice of HF-like exact exchange percentage into the HF-DFT scheme. Methods based on Green function (GW) and on the Random Phase Approximation (RPA), as well as methods based on Bethe-Salpeter equation (BSE) and TD-DFT have the potential of providing results in quantitative agreement with experiments, but their feasibility is hindered by high computational cost.182 Besides these shortcomings, thanks to the relatively good accuracy in predicting bandgap centers by standard DFT and considering the Janak's theorem, it is possible to compute the absolute potentials vs. NHE of the electrode band edges within a surface slab approach.183 In particular, the conduction band (CB) is relevant for photoanodes, and the valence band (VB) is relevant for photocathodes. Comparing these values to the computed HOMO and LUMO energies of the dye provides a powerful tool to assess the quality of a dye/electrode combination. The dye LUMO must be higher in energy than the electrode CB in n-type DSCs and the dye HOMO must be lower than the electrode VB in the p-type counterpart to allow for convenient electron and hole injections, respectively.In the last decade, the availability of more and more powerful computing facilities allowed the study of the dye/electrode interface at the full atomistic scale. From cluster-size electrodes with few atoms,184,185 computational tools now have the capability of simulating the full electrode surfaces with periodic boundary conditions, including the attached dyes186 and, in some cases, also the explicit solvent medium.137 The characterization of dye/electrode interfaces has provided great advancement in the understanding of the complex interfacial electronic processes.187 For both n- and p-type DSCs, it has been possible to assess the strength of the dye-surface anchoring,188–190 the role of dipole moment at the surface in tuning the electrode CB/VB edge potential,191 and the effects of surface polarization192,193 and the electrolyte solution194 on the dyes' electronic structure. The results allowed for a better design of dyes, with specific anchoring groups and with electron-donor/acceptor moieties well distributed into the dye molecular architecture.195
All these studies have paved the route to the recent implementation of real-time TD-DFT simulations of the dye/electrode interface after sunlight absorption and charge separation.196–198 With these approaches, mostly focused on n-type DSCs, it has been possible to dissect the specific mechanism and kinetics of charge transfer between the excited dye and the electrode, as well as of undesired charge recombination events.194 These studies still retain some empiricism, for example in the choice of some parameters that need to be fitted to experiments, but they certainly represent a frontier in the theoretical modeling of DSC interfaces, and we can expect further developments of these tools in the near future.
Last but not least, the importance of using the results from atomistic simulations in macroscopic modelling approaches must be mentioned. For example, the computed charge transfer rates can be implemented in a kinetic Monte Carlo approach for the simulation and interpretation of complex electrochemical measurements (e.g. impedance).199 At the same time, computed parameters derived from the isolated dye, the pristine electrode, and the dye/electrode interface can be conveniently cast in empirical formulae to obtain a realistic estimate of the photo-conversion efficiency.200
3.4 New horizons in modeling DSC devices
The great challenge of finding new materials and interfaces for DSCs requires further advancements in computational techniques. Although the atomistic description of complex materials and interfaces may still benefit from the accuracy and versatility of ab initio methods, new tools are emerging within the ongoing extraordinary revolution in computational sciences that involves Artificial Intelligence (AI) and Data Sciences. DSC development fits in these new approaches at different levels and, indeed, the first AI-based studies on DSC are now reality.201 On one hand, AI under Machine Learning-based approaches has been applied for electrode materials and dyes,202–204 tailoring specific structure–property relationships with deep-learning neural networks rather than first-principles equations. On the other hand, several tools are already available for automated screening and analysis of large datasets,205 compiled from experiments and/or advanced QM calculations, aimed at finding new, unexpected combinations of DSC components that maximize photo-conversion efficiencies, even at different light conditions.206–208 The future of these tools looks bright, together with their further integration within the new promising quantum information technologies.2094 Materials
4.1 Nanostructured metal oxide electrodes
Nanostructured semiconductor electrodes provide a large surface area for dye adsorption, an essential feature for DSCs. The most commonly used type of nanostructured electrode in DSC is the mesoporous electrode, which is composed of 10 to 50 nm-sized nanocrystals and has a porosity of about 50%. Other types of metal oxide nanostructures that have been applied in DSC are nanotubes, nanorods, nanofibers, nanosheets, etc.By far, the most used material for mesoporous electrodes is TiO2 with the anatase crystal structure (Fig. 16). This wide bandgap semiconductor has an indirect bandgap of 3.2 eV. The standard method for the preparation of mesoporous TiO2 electrodes is by screen printing of a suitable paste, followed by annealing in air at high temperature (400–500 °C) to burn out the organic additives required to make a paste with appropriate rheological properties and giving the required porosity. This heat treatment also gives a partial sintering of the TiO2 to make electronic connections between the particles and gives mechanical stability to the film. Depending on the precise composition, the mesoporous TiO2 film can be completely optically transparent, or have a slight white color. Several commercial suppliers offer suitable TiO2 screen printing pastes.
 | ||
| Fig. 16 SEM image of a mesoporous TiO2 film made with the GreatCell Solar 18NR-T paste. | ||
A light scattering layer containing ∼400 nm-sized TiO2 particles is frequently deposited on top of the mesoporous layer. This layer reflects transmitted light back into the active film and usually improves the efficiency for DSC devices that are illuminated through the FTO/glass substrate. Light-scattering particles can also be added to the mesoporous film paste to obtain a similar effect; the latter method is more appropriate for DSC with illumination from the counter electrode side. We refer to ref. 210 for further reading on application of light scattering in DSC.
For best performance, it is common in research papers to apply a TiCl4 treatment: mesoporous TiO2 films are immersed in an aqueous TiCl4 solution, leading to chemical bath deposition of an ultrathin layer of TiO2 (about 1 nm) onto the mesoporous electrode and the underlying conducting glass.211 A further heat treatment is used to crystallize the material and to remove water.212
The porosity and pore size of mesoporous films are particularly important for the use of alternative redox mediators, such as cobalt bipyridine complexes. In this case, a marked improvement of DSC performance was found at one sun illumination, from 1.4% to 4.8%, when the porosity was increased from 52% to 59%.213 Deviations from linearity of photocurrent vs. light intensity plots, as well as photocurrent transients clearly demonstrated the occurrence of mass transport limitations of the redox mediator. Yella et al. demonstrated that best performing DSCs with cobalt bipyridine redox mediator should have a thinner added TiO2 layer deposited by TiCl4 after screen-printing.214
Doping of TiO2 can give some positive effects by adding or removing trap states, changing the band edge levels, improving dye adsorption, and by stabilizing the anatase phase, as recently reviewed by Roose et al.215 For instance, a high VOC of 1.45 V was obtained by Mg doping of TiO2 through an additional MgO/Al2O3 surface treatment and employing a bromide-based redox electrolyte.216 In highly efficient DSCs, however, the state-of-the art mesoporous TiO2 electrodes are not doped.
A large variety of TiO2 nanostructures have been tested in DSCs: one-dimensional structures such as nanotubes and oriented nanorod arrays,217 mesoporous microbeads218 and mesoporous single crystals.219 Templating methods provide a route to ordered mesoporous TiO2 materials, with soft-templating methods using surfactants and hard-templating methods using silica or polystyrene spheres.220 None of these structures, however, outperform standard mesoporous TiO2 electrodes under optimized conditions.
In 1D structures (nanotubes and single crystalline nanorods), faster electron transport is often named as a potential advantage for these structures. In practice, however, the charge collection in mesoporous films is sufficiently high, so that no solar cell improvement can be expected on that basis. Mesoporous TiO2 microbeads are of potential interest for several reasons: first, a high PCE of 10.7% was achieved in a single printed layer;218 second, they can be annealed at high temperature and sensitized before application onto a (flexible) substrate. Furthermore, this and other structures with hierarchical architecture can have an advantage with respect to mass transport in the electrolyte. Mesoporous microbead electrodes outperformed standard mesoporous electrodes when using a more viscous MPN-based cobalt electrolyte at 1 sun light intensity.221 Microbead electrodes were also successfully applied in solid-state DSCs (Fig. 17).222
 | ||
| Fig. 17 SEM micrographs of mesoporous TiO2 microbeads. (a) Adapted with permission from ref. 223. Copyright 2010 American Chemical Society. (b) Adapted from ref. 222 with permission from The Royal Society of Chemistry, copyright 2014. | ||
A disadvantage related to TiO2 as a material for the dye-sensitized solar cell is its photocatalytic activity:224 direct excitation of the semiconductor leads to highly energetic holes that can oxidize organic compounds. This lowers the long-term stability of DSC under illumination. Such degradation can be avoided by adding a UV-filter to the solar cell, but this will lead to additional cost. The UV activity of TiO2 is one reason to look into alternatives.
There are many other metal oxides that can be applied in the working electrode of a DSC. ZnO is the most investigated alternative to TiO2, in a wide variety of nanostructures.225,226 Its electron mobility is much higher than that of TiO2, but its (photo)chemical stability is lower. SnO2 is chemically very stable, has a higher bandgap than TiO2, but a lower conduction band edge energy, leading to a lower photovoltage in DSCs.227 Both ZnO and SnO2 are probably best applied in core–shell structures in DSCs, as discussed below. Table 1 lists alternative n-type semiconductor materials used in DSC that have obtained a PCE of more than 5%.
| Semiconductor | Bandgap (eV) | Nanostructure | Sensitizer – electrolyte | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|
| TiO2 (anatase) | 3.2 | Mesoporous | ADEKA-1/LEG4 – Co(phen)3 | 14.3 | 2015 | 24 |
| TiO2 (rutile) | 3.0 | Nanorod array | N719 – I−/I3− | 11.1 | 2019 | 228 |
| TiO2 (brookite) | 3.2 | Mesoporous | N719 – I−/I3− | 8.2 | 2020 | 229 |
| ZnO | 3.2 | Aggregated nanoparticles | N719 – I−/I3− | 7.5 | 2011 | 230 |
| SnO2 | 3.5 | Nanoparticles/ | N719 – I−/I3− | 6.3 | 2013 | 231 |
| Nb2O5 | 3.6 | Nanorod array | N719 – I−/I3− | 6.0 | 2013 | 231 |
| Nb3O7(OH) | 3.0 | Nanorod array | N719 – I−/I3− | 6.8 | 2013 | 231 |
| Zn2SnO4 | 3.6 | Aggregated nanoparticles | X73 – Co(phen)3 | 8.1 | 2020 | 232 |
| BaSnO3 | 2.9 | Mesoporous | N719 – I−/I3− | 6.6 | 2019 | 233 |
| Ba0.8Sr0.2SnO3 | 3.0 | Mesoporous | N719 – I−/I3− | 7.7 | 2019 | 233 |
Combinations of metal oxides have also been evaluated for DSC in a large number of studies. Scientifically most interesting are so-called core–shell structures, where a nanostructured electrode is covered by an ultra-thin layer of a different material, usually one with a higher bandgap. Deposition is performed by chemical bath deposition (using e.g. TiCl4 for deposition of TiO2) or by atomic layer deposition (ALD). The shell material can be a semiconductor or an insulator such as Al2O3 or SiO2: if sufficiently thin, adsorbed dyes can inject electrons into the core material through tunneling. Typically, rate constants for both electron injection and recombination are significantly reduced. This can lead to an improved solar cell efficiency if the injection efficiency is not significantly decreased. In addition, the shell can lead to added chemical stability (e.g. for Al2O3, SiO2, or TiO2 on ZnO). A few examples of core–shell structures will be given here: in ALD-deposited Al2O3 on mesoporous TiO2, the PCE increased from 6.2% to 8.4% upon 20 ALD cycles. This was partly caused by a higher recombination resistance and partly by a higher dye adsorption of the modified electrode.234 As another example, 3D-bicontinous inverse opal SnO2 structures were synthesized infiltrating a film of monodisperse polystyrene particles with SnCl2 in ethanol, followed by heating, see Fig. 18. A TiO2 shell was formed by chemical bath deposition using TiCl4. The resulting electrodes yielded an efficiency of 8.2% in DSCs, whereas TiO2/TiO2 inverse opal/shell structures yielded 7.2%.235
 | ||
| Fig. 18 (a) Inverse opal SnO2 electrode; (b) after coating with a 170 nm shell of TiO2. Adapted from ref. 235 with permission from The Royal Society of Chemistry, copyright 2016. | ||
4.2 Sensitizers
Photoanodes based on molecular sensitizers at a semiconductor interface for DSCs require that the sensitizer absorbs solar energy and injects electrons into the semiconductor conduction band. Thus, the sensitizer controls the breadth of the solar spectrum used and the quantum yield for electron injection. Additionally, the sensitizer should promote long-lived charge separated states at the interface, and the oxidized sensitizer should rapidly undergo electron transfer from a reducing redox shuttle (RS) to limit the competitive electron back-transfer reaction from electrons in TiO2 to the oxidized dye. The sensitizer is also often tasked with providing insulating groups to protect electrons in TiO2 from recombining with the electrolyte. Recent progress in dye design with respect to these design criteria has fueled much of the increase observed in performance metrics. The atomistic level control with respect to dye design allows for the precise tuning of dye properties. One strategy that has been explored intensely is related to the design of a dye capable of absorbing photons across the visible spectrum and into the near infra-red (NIR) region to maximize the power conversion efficiency from a single photoanode-based device. Estimates of a practical efficiency limit at about 22% PCE are reported if driving forces for electron transfers to a semiconductor and from a redox shuttle to the oxidized dye can be kept to a combined 400 mV or less and the sensitizer can efficiently use photons as low in energy as ∼950 nm.236 Alternatively, an increasingly popular approach is to tailor chromophores to a specific spectral region to be used in co-sensitized or multiple-photoanode-based devices. This second approach increases the complexity of the device, but allows for higher theoretical PCEs. Using similar approximations of 400 mV free energies for electron transfers with the spectrum divided into three equal parts (wide, medium, and narrow optical gaps) from 400–950 nm leads to a practically possible PCE of ∼33%. Thus, significant gains in PCE are possible through research on multiple photoanode systems. Additionally, these materials are attractive for use with existing solar cell technologies as described below. For this strategy to work effectively, the sensitizer (and redox shuttle) needs to be custom tailored to each spectral region for minimal overpotential losses. Both single and multiple photoanode dye design approaches are discussed below with respect to both metal- and organic-based dyes. Notably, the literature with respect to dyes for DSCs is vast and growing rapidly with many exciting findings being reported weekly, which cannot all be highlighted (especially with regard to phthalocyanies, BODIPYs, DPP chromophores, multidonor systems, multiacceptor systems, dual anchor dyes, unique anchoring groups, and non-covalently bound dye–dye and dye-RS systems). The examples below serve to highlight recent select findings on high photocurrent, high photovoltage, deep NIR absorbing dyes, wide optical gap dyes, and high PCE dyes. Select design strategies being used within approximately the last decade are highlighted and should not be viewed as an exhaustive catalogue of dye design approaches. | ||
| Fig. 19 Examples of metal complex-based sensitizers. | ||
| Sensitizer | Electrolyte | Additives | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| N719 | I2, BMII | GuSCN, tBP | 789 | 18.2 | 70.4 | 10.1 | 2008 | 212 |
| CYC-B11 | I2, LiI, DMII | GuSCN, tBP | 743 | 20.05 | 77 | 11.5 | 2009 | 237 |
| Black dye | I2, LiI, DMPII | tBP | 727 | 20.43 | 72.4 | 10.75 | 2012 | 238 |
| TUS-38 | I2, LiI, EMII | tBP | 702 | 23.43 | 72.2 | 11.88 | 2016 | 239 |
| T7 | I2, LiI, DMPII | tBP | 760 | 16.7 | 70 | 8.9 | 2016 | 240 |
| T7 | Co(phen)3 | LiClO4, tBP | 800 | 10.1 | 70 | 5.7 | 2016 | 240 |
| T5 | I2, LiI, DMPII | tBP | 680 | 19.5 | 67 | 8.9 | 2016 | 240 |
| T5 | Co(phen)3 | LiClO4, tBP | 670 | 4.05 | 52 | 1.4 | 2016 | 240 |
| TF-1 | I2, LiI, DMPII | tBP | 670 | 16.7 | 68 | 7.7 | 2016 | 240 |
| TF-1 | Co(phen)3 | LiClO4, tBP | 570 | 6.85 | 39 | 1.5 | 2016 | 240 |
| Ru-1 | Co(phen)3 | LiTFSI, tBP | 837 | 13.2 | 78 | 8.6 | 2013 | 241 |
| Ru-1 | I2, LiI, PMII | GuSCN, tBP | 715 | 16.3 | 75 | 8.7 | 2013 | 241 |
| SA22 | Co(phen)3 | LiTFSI, NOP | 827 | 12.25 | 75.5 | 7.9 | 2016 | 242 |
| SA25 | Co(phen)3 | LiTFSI, NOP | 810 | 10.68 | 77.9 | 6.9 | 2016 | 242 |
| SA246 | Co(phen)3 | LiTFSI, NOP | 845 | 14.55 | 74.7 | 9.4 | 2016 | 242 |
| SA282 | Co(phen)3 | LiTFSI, NOP | 794 | 9.89 | 78.5 | 6.3 | 2016 | 242 |
| SA284 | Co(phen)3 | LiTFSI, NOP | 794 | 11.28 | 76.9 | 7.0 | 2016 | 242 |
| SA285 | Co(phen)3 | LiTFSI, NOP | 807 | 11.85 | 73.6 | 7.2 | 2016 | 242 |
| SA633 | Co(phen)3 | LiTFSI, tBP | 819 | 13.68 | 71.5 | 8.0 | 2017 | 243 |
| SA634 | Co(phen)3 | LiTFSI, tBP | 845 | 13.89 | 70.0 | 8.2 | 2017 | 243 |
| SA635 | Co(phen)3 | LiTFSI, tBP | 809 | 13.03 | 72.1 | 7.6 | 2017 | 243 |
| 51–5ht | Co(bpy)3 | LiTFSI, tBP | 840 | 12.78 | 76.4 | 8.22 | 2016 | 244 |
| 51–5ht | Co(phen)3 | LiTFSI, tBP | 842 | 12.17 | 75.0 | 7.69 | 2016 | 244 |
| 51–5ht | I2, LiI, PMII | tBP | 718 | 15.31 | 74.6 | 8.20 | 2016 | 244 |
| 51–57dht | Co(bpy)3 | LiTFSI, tBP | 844 | 13.56 | 74.2 | 8.49 | 2016 | 244 |
| 51–57dht | Co(phen)3 | LiTFSI, tBP | 898 | 12.32 | 75.4 | 8.34 | 2016 | 244 |
| 51–57dht | I2, LiI, PMII | tBP | 727 | 14.17 | 74.3 | 7.66 | 2016 | 244 |
| 51–57dht.1 | Co(bpy)3 | LiTFSI, tBP | 853 | 13.36 | 75.0 | 8.55 | 2016 | 244 |
| 51–57dht.1 | Co(phen)3 | LiTFSI, tBP | 900 | 13.89 | 76.2 | 9.53 | 2016 | 244 |
| 51–57dht.1 | I2, LiI, PMII | tBP | 740 | 13.53 | 74.9 | 7.50 | 2016 | 244 |
| TFRS-80a | Co(phen)3 | LiTFSI, tBP | 840 | 13.44 | 75.7 | 8.55 | 2014 | 245 |
| TFRS-80a | I2, LiI, DMPII | tBP | 780 | 14.49 | 66.8 | 7.55 | 2014 | 245 |
| TFRS-80a | I2, DMPII | tBP | 890 | 12.93 | 72.7 | 8.37 | 2014 | 245 |
| TFRS-80b | Co(phen)3 | LiTFSI, tBP | 820 | 13.30 | 76.6 | 8.36 | 2014 | 245 |
| TFRS-80b | I2, LiI, DMPII | tBP | 680 | 10.39 | 68.1 | 4.80 | 2014 | 245 |
| TFRS-80b | I2, DMPII | tBP | 780 | 9.81 | 72.5 | 5.55 | 2014 | 245 |
| TFRS-80c | Co(phen)3 | LiTFSI, tBP | 840 | 14.32 | 75.4 | 9.06 | 2014 | 245 |
| TFRS-80c | I2, LiI, DMPII | tBP | 730 | 14.84 | 65.1 | 7.06 | 2014 | 245 |
| TFRS-80c | I2, DMPII | tBP | 880 | 12.41 | 75.6 | 8.26 | 2014 | 245 |
| Ir-1 | Fe(bpy)3 | tBP | 870 | 0.014 | 48 | 0.60 | 2020 | 246 |
| Os-1 | I2, LiI, DMPII | None | 320 | 23.7 | 36 | 2.7 | 2010 | 247 |
| TF-5 | I2, LiI, DMPII | tBP | 640 | 18.0 | 71.6 | 8.25 | 2012 | 248 |
| TF-51 | I2, LiI, DMPII | tBP | 560 | 20.1 | 66.4 | 7.47 | 2012 | 248 |
| TF-52 | I2, LiI, DMPII | tBP | 600 | 23.3 | 63.3 | 8.85 | 2012 | 248 |
| DX3 | I2, LiI, DMPII | tBP | 55 6 | 30.3 | 60.5 | 10.2 | 2015 | 249 |
Wide optical gap sensitizers are important for a number of applications and, within DSC literature, these systems are exceptionally valuable for use in multiple photoanode systems. With respect to these applications, generating a high photovoltage from the high-energy visible photons is critical to avoid thermal free energy losses. The overall PCE of the system is typically not the metric being pursued in these systems since they are often designed with tandem or multiple photoanode systems as the larger goal. Wide optical gap metal-based sensitizers are relatively rarely used in the literature with RSs capable of generating high photovoltages. This may in part be due to the higher photovoltage generating redox shuttles often being 1-electron metal-based RSs. As described above, the design of metal-based dyes that undergo efficient electron transfers with good charge separation lifetimes with metal-based RSs remains a key research direction. However, recently a cyclometalated Ir complex (Ir-1) based on two phenylpyridine ligands and a 4,4′-bis(phosphonomethyl)-2,2′-bipyridine ligand has been used in high photovoltage DSCs with the Fe(bpy)33+/2+ redox shuttle to give 870 mV photovoltage under one-sun and 1.06 V under UV irradiation (Fig. 19).246
Narrow optical gap sensitizers are critical toward the use of lower energy photons in multiple photoanode-based devices (e.g. tandem solar cells). Within this region, the breadth of the IPCE spectrum (and JSC generated) is a key performance metric with the goal being to combine these photoanodes into tandem-type systems. Metal-based sensitizers are exceptional in the >800 nm spectral region within DSC devices. Ru- and Os-based sensitizers specifically have shown exceptional deep NIR photon absorption and conversion properties. The ultrafast electron injection properties of these systems allows for efficient electron transfers prior to excited-state relaxation and therefore enables the efficient harvest of relatively low energy photons with minimal driving force needed for charge injection. Os-1 is a similar structure to N3 which uses two bipyridine-based ligands and a β-diketone in place of the NCS ligands of N3 (Fig. 19).247 Os-1 is broadly absorbing with an IPCE onset near 1100 nm and in excess of 70% across the visible spectrum. A PCE of 2.7% is reported which is low due to a poor VOC (0.32 V) despite the high JSC value of 23.7 mA cm−2. Os dye TF-52 was one of the first sensitizers to reach 1000 nm with a high peak IPCE (∼75%).248 A photocurrent of 23.3 mA cm−2 was reported with an efficiency of 8.85%. Light soaking at 60 °C with TF-52 reveals no significant change in PCE for this device over a 1000 hour measurement. Dye DX3 efficiently uses photons across the visible spectrum with an IPCE onset of ∼1100 nm. The peak IPCE value observed with this system is >80% with the IPCE remaining in excess of 80% from approximately 450 to 900 nm. A JSC in excess of 30 mA cm−2 is observed from DSC devices using this dye. The deep NIR photon use of DX3 leads to the use of a DSC device made from this material in tandem with a perovskite solar cell with the DSC device being used as the narrow bandgap material (21.5% PCE tandem efficiency).249 These dyes are attractive for use in tandem type systems and represent the forefront of high percentage IPCE, broadly absorbing sensitizers. Design of sensitizers that retain high percentage IPCE values throughout the IPCE spectrum and extend IPCE wavelengths to beyond 1100 nm is an intriguing direction for this type of sensitizers that could have significant impact on tandem device designs.
 | ||
| Fig. 20 Contemporary rapid routes to complex organic dyes where X is a halide, M is a transmetallating reagent, and Y is a masked functionality such as a TMS group prior to halide conversion. | ||
| Sensitizer | Electrolyte | Additives | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| D149 | I2, LiI, BMII | tBP | 644 | 19.86 | 69.4 | 8.85 | 2008 | 262 |
| D205 | I2, LiI, BMII | tBP | 710 | 18.68 | 70.7 | 9.40 | 2008 | 262 |
| WS-66 | I2, LiI, DPMII | tBP | 757 | 12.97 | 71 | 7.01 | 2017 | 263 |
| WS-67 | I2, LiI, DPMII | tBP | 711 | 15.91 | 73 | 8.25 | 2017 | 263 |
| WS-68 | I2, LiI, DPMII | tBP | 705 | 17.73 | 67 | 8.42 | 2017 | 263 |
| WS-69 | I2, LiI, DPMII | tBP | 696 | 19.39 | 67 | 9.03 | 2017 | 263 |
| IQ4 | Co(bpy)3 | LiClO4, tBP | 771 | 14.69 | 68.8 | 7.79 | 2014 | 264 |
| IQ4 | I2, LiI, DMII | GuSCN, tBP | 737 | 15.33 | 75.5 | 8.53 | 2014 | 264 |
| YA421 | Co(bpy)3 | LiClO4, tBP | 803 | 15.76 | 71.2 | 9.00 | 2014 | 264 |
| YA421 | I2, LiI, DMII | GuSCN, tBP | 741 | 15.41 | 71.1 | 8.12 | 2014 | 264 |
| YA422 | Co(bpy)3 | LiClO4, tBP | 890 | 16.25 | 73.7 | 10.65 | 2014 | 264 |
| YA422 | I2, LiI, DMII | GuSCN, tBP | 741 | 14.40 | 68.2 | 7.28 | 2014 | 264 |
| DPP13 | I2, LiI, DMII | GuSCN, tBP | 705 | 16.2 | 67 | 7.60 | 2013 | 265 |
| DPP13 | Co(bpy)3 | LiClO4, tBP | 743 | 15.6 | 78 | 8.97 | 2013 | 265 |
| DPP14 | I2, LiI, DMII | GuSCN, tBP | 680 | 16.6 | 68 | 7.73 | 2013 | 265 |
| DPP14 | Co(bpy)3 | LiClO4, tBP | 716 | 15.2 | 76 | 8.23 | 2013 | 265 |
| DPP15 | I2, LiI, DMII | GuSCN, tBP | 684 | 16.9 | 65 | 7.44 | 2013 | 265 |
| DPP15 | Co(bpy)3 | LiClO4, tBP | 745 | 17.6 | 75 | 9.81 | 2013 | 265 |
| DPP17 | I2, LiI, DMII | GuSCN, tBP | 700 | 16.3 | 63 | 7.13 | 2013 | 265 |
| DPP17 | Co(bpy)3 | LiClO4, tBP | 761 | 17.9 | 74 | 10.1 | 2013 | 265 |
| D21L6 | I2, LiI, DMII | GuSCN, tBP | 714 | 13.81 | 72.1 | 7.11 | 2010 | 266 |
| C218 | I2, LiI, DMII | GuSCN, tBP | 768 | 15.84 | 73.5 | 8.95 | 2010 | 266 |
| AP25 | I2, LiI, DMII | GuSCN, tBP | 527 | 19.9 | 65 | 6.8 | 2020 | 267 |
| PB1 | I2, LiI, DMII | GuSCN, tBP | 704 | 12.1 | 75 | 6.50 | 2016 | 268 |
| PB2 | I2, LiI, DMII | GuSCN, tBP | 648 | 12.7 | 75 | 6.24 | 2016 | 268 |
| DP1 | I2, LiI, DMII | GuSCN, tBP | 680 | 10.9 | 75 | 5.61 | 2016 | 268 |
| DP2 | I2, LiI, DMII | GuSCN, tBP | 697 | 13.7 | 76 | 7.41 | 2016 | 268 |
| C268 | I2, DMII, EMII | sulfolane, NBB, GuSCN | 718 | 16.76 | 72.3 | 8.7 | 2018 | 269 |
| D35 | Co(bpy)3 | LiClO4, tBP | 920 | 10.7 | 68 | 6.7 | 2010 | 270 |
| D35 | I2, LiI, TBAI | tBP | 910 | 9.38 | 65 | 5.5 | 2010 | 270 |
| Y123 | I2, LiI, DMII | GuSCN, tBP | 757 | 13.6 | 70 | 7.2 | 2011 | 271 |
| Y123 | Co(bpy)3 | LiClO4, tBP | 855 | 14.6 | 70 | 8.8 | 2011 | 271 |
| Y123 | Co(bpy-pz)2 | LiClO4, tBP | 1020 | 12.54 | 69.4 | 8.87 | 2012 | 272 |
| Y123 | Cu(tmby)2 | LiTFSI, tBP | 1030 | 13.6 | 74 | 10.3 | 2018 | 273 |
| WS-70 | Cu(tmby)2 | LiTFSI, tBP | 1060 | 13.2 | 77 | 11.0 | 2018 | 273 |
| WS-72 | Cu(tmby)2 | LiTFSI, tBP | 1100 | 13.3 | 78 | 11.6 | 2018 | 273 |
| L348 | Cu(tmby)2 | LiTFSI | 1170 | 6.4 | 72.0 | 5.3 | 2018 | 274 |
| L349 | Cu(tmby)2 | LiTFSI | 1160 | 11.0 | 71.7 | 9.2 | 2018 | 274 |
| L350 | Cu(tmby)2 | LiTFSI | 1140 | 13.0 | 76.0 | 11.2 | 2018 | 274 |
| L351 | Cu(tmby)2 | LiTFSI | 1060 | 11.2 | 76.3 | 9.1 | 2018 | 274 |
| NT35 | Cu(tmby)2 | LiTFSI, MBI | 950 | 5.96 | 79.1 | 4.5 | 2021 | 12 |
| XY1b | Cu(tmby)2 | LiTFSI, MBI | 1010 | 15.26 | 76.3 | 11.8 | 2021 | 12 |
| MS4 | Cu(tmby)2 | LiTFSI, MBI | 1170 | 8.86 | 73.0 | 7.6 | 2021 | 12 |
| MS5 | Cu(tmby)2 | LiTFSI, MBI | 1240 | 8.87 | 73.3 | 8.0 | 2021 | 12 |
| SC-1 | Co(bpy)3 | LiTFSI, tBP | 828 | 14.70 | 76.2 | 9.3 | 2017 | 275 |
| SC-2 | Co(bpy)3 | LiTFSI, tBP | 856 | 16.62 | 74.5 | 10.6 | 2017 | 275 |
| SC-3 | Co(bpy)3 | LiTFSI, tBP | 920 | 16.50 | 75.8 | 11.5 | 2017 | 275 |
| C272 | Co(phen)3 | LiTFSI, tBP | 897 | 15.81 | 74.4 | 10.6 | 2015 | 276 |
| C275 | Co(phen)3 | LiTFSI, tBP | 956 | 17.03 | 77.0 | 12.5 | 2015 | 276 |
| R4 | Co(bpy)3 | LiTFSI, tBP | 852 | 17.25 | 75.4 | 11.1 | 2018 | 277 |
| R6 | Co(bpy)3 | LiTFSI, tBP | 850 | 19.69 | 75.4 | 12.6 | 2018 | 277 |
| H1 | Co(bpy)3 | LiTFSI, tBP | 931 | 14.33 | 72.3 | 9.7 | 2019 | 278 |
| H2 | Co(bpy)3 | LiTFSI, tBP | 903 | 15.47 | 74.0 | 10.3 | 2019 | 278 |
| ZL001 | Co(bpy)3 | LiClO4, tBP | 887 | 20.57 | 70.0 | 12.8 | 2019 | 279 |
| ZL003 | Co(bpy)3 | LiClO4, tBP | 956 | 20.73 | 68.5 | 13.6 | 2019 | 279 |
| ADEKA-2 | Co(bpy)3 | LiClO4, tBP | 821 | 15.1 | 75.2 | 9.32 | 2014 | 280 |
| ADEKA-1 | Co(bpy)3 | LiClO4, tBP | 848 | 16.1 | 76.2 | 10.4 | 2014 | 280 |
| ADEKA-1 | Co(Cl-phen)3 | LiClO4, tBP, NaClO4, TBAPF6, TBPPF6, HMIPF6, TMSP, MP | 1036 | 15.6 | 77.4 | 12.5 | 2014 | 280 |
| SFD-5 | Br2, BMIBr, TPABr | GuSCN, tBP | 960 | 6.16 | 53 | 3.1 | 2016 | 216 |
| ADEKA-3 | Br2, BMIBr, TPABr | GuSCN, tBP, TMSP, MP, H2O | 1450 | 4.77 | 56 | 3.9 | 2016 | 216 |
| AP11 | Fe(bpy)3 | LiTFSI, tBP | 1260 | 3.50 | 63 | 2.9 | 2019 | 281 |
| AP14 | Fe(bpy)3 | LiTFSI, tBP | 1320 | 3.40 | 63 | 2.7 | 2019 | 281 |
| AP16 | Fe(bpy)3 | LiTFSI, tBP | 1290 | 3.10 | 65 | 2.6 | 2019 | 281 |
| AP17 | Fe(bpy)3 | LiTFSI, tBP | 1270 | 2.90 | 58 | 2.2 | 2019 | 281 |
| RR9 | Fe(bpy)3 | LiTFSI, tBP | 1420 | 2.8 | 47 | 1.9 | 2018 | 282 |
| YD2 | I2, LiI, DMII | GuSCN, tBP | 770 | 1 8.6 | 7 6.4 | 11 | 2010 | 283 |
| YD2 | Co(bpy)3 | LiClO4, tBP | 825 | 14.9 | 69 | 8.4 | 2011 | 284 |
| YD2-o-C8 | Co(bpy)3 | LiClO4, tBP | 965 | 17.3 | 71 | 11.9 | 2011 | 284 |
| GY21 | Co(bpy)3 | Not specified | 615 | 5.03 | 79.8 | 2.52 | 2014 | 285 |
| GY21 | I2, PMII | LiTFSI, tBP | 552 | 11.50 | 75.1 | 4.84 | 2014 | 285 |
| GY50 | Co(bpy)3 | Not specified | 885 | 18.53 | 77.3 | 12.75 | 2014 | 285 |
| GY50 | I2, PMII | LiTFSI, tBP | 732 | 18.45 | 65.7 | 8.90 | 2014 | 285 |
| SM371 | Co(bpy)3 | LiTFSI, tBP | 960 | 15.9 | 79 | 12.0 | 2014 | 286 |
| SM315 | Co(bpy)3 | LiTFSI, tBP | 910 | 18.1 | 78 | 13.0 | 2014 | 286 |
| SGT-020 | Co(bpy)3 | LiClO4, tBP | 825 | 15.6 | 7 4.7 | 9.6 | 2017 | 287 |
| SGT-021 | Co(bpy)3 | LiClO4, tBP | 819 | 17.9 | 75.4 | 1 1.1 | 2017 | 287 |
| SGT-130 | Co(bpy)3 | LiClO4, tBP | 810 | 16.84 | 72.08 | 9.83 | 2017 | 288 |
| SGT-136 | Co(bpy)3 | LiClO4, tBP | 804 | 18.35 | 74.84 | 11.04 | 2017 | 288 |
| SGT-137 | Co(bpy)3 | LiClO4, tBP | 825 | 19.39 | 73.98 | 11.84 | 2017 | 288 |
| SGT-137 | I2, LiI, DMPII | tBP | 690 | 18.55 | 68.9 | 8.9 | 2020 | 25 |
| SGT-146 | Co(bpy)3 | LiTFSI, tBP | 834 | 16.39 | 74.6 | 10.2 | 2020 | 25 |
| SGT-146 | I2, LiI, DMPII | tBP | 674 | 18.54 | 72.9 | 9.2 | 2020 | 25 |
| SGT-147 | Co(bpy)3 | LiTFSI, tBP | 839 | 17.15 | 73.5 | 10.5 | 2020 | 25 |
| SGT-147 | I2, LiI, DMPII | tBP | 702 | 18.46 | 67.6 | 8.8 | 2020 | 25 |
| SGT-148 | Co(bpy)3 | LiTFSI, tBP | 849 | 17.12 | 72.9 | 10.6 | 2020 | 25 |
| SGT-148 | I2, LiI, DMPII | tBP | 698 | 18.71 | 68.4 | 8.9 | 2020 | 25 |
| SGT-149 | Co(bpy)3 | LiTFSI, tBP | 898 | 17.49 | 72.2 | 11.4 | 2020 | 25 |
| SGT-149 | I2, LiI, DMPII | tBP | 713 | 19.32 | 71.1 | 9.8 | 2020 | 25 |
| SM63 | I2, LiI, DMII | GuSCN, tBP | 700 | 14.43 | 73 | 7.35 | 2016 | 289 |
| LD14-C8 | I2, LiI, DMII | GuSCN, tBP | 730 | 15.72 | 74 | 8.45 | 2016 | 289 |
| WW-3 | Co(bpy)3 | LiTFSI, tBP | 744 | 9.81 | 76.7 | 5.6 | 2014 | 290 |
| WW-4 | Co(bpy)3 | LiTFSI, tBP | 500 | 3.00 | 29.9 | 0.3 | 2014 | 290 |
| WW-5 | Co(bpy)3 | LiTFSI, tBP | 766 | 18.87 | 73.3 | 10.3 | 2014 | 290 |
| WW-6 | Co(bpy)3 | LiTFSI, tBP | 840 | 17.16 | 73.8 | 10.6 | 2016 | 291 |
| WW-7 | Co(bpy)3 | LiTFSI, tBP | 708 | 8.05 | 77.7 | 4.4 | 2016 | 291 |
| WW-8 | Co(bpy)3 | LiTFSI, tBP | 733 | 8.27 | 78.6 | 4.8 | 2016 | 291 |
| WW-9 | Co(bpy)3 | LiTFSI, tBP | 770 | 15.93 | 75.2 | 9.2 | 2016 | 291 |
| YD22 | I2, LiI, PMII | tBP | 700 | 14.92 | 72.43 | 7.56 | 2016 | 292 |
| YD23 | I2, LiI, PMII | tBP | 740 | 17.10 | 71.41 | 9.00 | 2016 | 292 |
| YD24 | I2, LiI, PMII | tBP | 730 | 17.29 | 72.46 | 9.19 | 2016 | 292 |
| YD25 | I2, LiI, PMII | tBP | 720 | 15.22 | 72.66 | 7.93 | 2016 | 292 |
| YD26 | I2, LiI, PMII | tBP | 790 | 15.26 | 73.24 | 8.79 | 2016 | 292 |
| YD27 | I2, LiI, PMII | tBP | 790 | 15.45 | 73.07 | 8.92 | 2016 | 292 |
| YD28 | I2, LiI, PMII | tBP | 760 | 14.07 | 70.60 | 7.58 | 2016 | 292 |
| XW1 | I2, LiI, PMII | tBP | 716 | 14.99 | 66 | 7.13 | 2014 | 293 |
| XW2 | I2, LiI, PMII | tBP | 680 | 15.73 | 64 | 6.84 | 2014 | 293 |
| XW3 | I2, LiI, PMII | tBP | 694 | 15.60 | 68 | 7.32 | 2014 | 293 |
| XW4 | I2, LiI, PMII | tBP | 702 | 16.22 | 70 | 7.94 | 2014 | 293 |
| C1 | I2, LiI, PMII | tBP | 780 | 11.21 | 65 | 5.67 | 2014 | 293 |
| XW9 | I2, LiI, PMII | tBP | 740 | 16.17 | 68.9 | 8.2 | 2015 | 294 |
| XW10 | I2, LiI, PMII | tBP | 739 | 17.51 | 68.0 | 8.8 | 2015 | 294 |
| XW11 | I2, LiI, PMII | tBP | 727 | 18.26 | 70.1 | 9.3 | 2015 | 294 |
| XW14 | I2, LiI, PMII | tBP | 725 | 17.07 | 70 | 8.6 | 2015 | 295 |
| XW15 | I2, LiI, PMII | tBP | 720 | 18.02 | 67 | 8.7 | 2015 | 295 |
| XW16 | I2, LiI, PMII | tBP | 734 | 17.92 | 70 | 9.1 | 2015 | 295 |
| XW17 | I2, LiI, PMII | tBP | 700 | 18.79 | 72 | 9.5 | 2015 | 295 |
| SGT-021 | Co(bpy)3 | LiTFSI, tBP | 848 | 1 6.9 | 7 5.8 | 1 0.8 | 2019 | 296 |
| SGT-023 | Co(bpy)3 | LiTFSI, tBP | 739 | 3.4 | 79.5 | 2.0 | 2019 | 296 |
| SGT-025 | Co(bpy)3 | LiTFSI, tBP | 8 19 | 1 4.1 | 7 8.4 | 9.1 | 2019 | 296 |
| XW26 | I2, LiI, PMII | tBP | 708 | 11.37 | 69.13 | 5.57 | 2017 | 297 |
| XW27 | I2, LiI, PMII | tBP | 710 | 14.08 | 72.26 | 7.17 | 2017 | 297 |
| XW28 | I2, LiI, PMII | tBP | 715 | 19.38 | 72.96 | 10.14 | 2017 | 297 |
| LG1 | I2, LiI, DMII | tBP | 710 | 17.43 | 71 | 8.89 | 2017 | 298 |
| LG2 | I2, LiI, DMII | tBP | 710 | 15.45 | 72 | 7.87 | 2017 | 298 |
| LG3 | I2, LiI, DMII | tBP | 710 | 12.10 | 72 | 6.17 | 2017 | 298 |
| LG4 | I2, LiI, DMII | tBP | 710 | 15.02 | 68 | 7.30 | 2017 | 298 |
| LG5 | I2, LiI, DMII | tBP | 680 | 21.01 | 71 | 10.20 | 2017 | 298 |
| LG6 | I2, LiI, DMII | tBP | 690 | 19.55 | 71 | 9.64 | 2017 | 298 |
| LG7 | I2, LiI, DMII | tBP | 660 | 13.38 | 69 | 6.21 | 2017 | 298 |
| ZZX-N7 | I2, LiI, DMII | GuSCN, tBP | 732 | 15.39 | 63.33 | 7.51 | 2015 | 299 |
| ZZX-N8 | I2, LiI, DMII | GuSCN, tBP | 741 | 14.25 | 69.97 | 7.78 | 2015 | 299 |
| ZZX-N9 | I2, LiI, DMII | GuSCN, tBP | 656 | 15.46 | 70.57 | 7.53 | 2015 | 299 |
| YD2-o-C8T | I2, LiI, DMII | GuSCN, tBP | 730 | 15.6 | 68 | 7.7 | 2015 | 300 |
| YD2-o-C8 | I2, LiI, DMII | GuSCN, tBP | 780 | 17.3 | 65 | 8.8 | 2015 | 300 |
| PZn-HOQ | I2, LiI, DPMII | GuSCN, tBP | 576 | 6.48 | 67.8 | 2.53 | 2014 | 301 |
| DPZn-HOQ | I2, LiI, DPMII | GuSCN, tBP | 595 | 7.81 | 66.4 | 3.09 | 2014 | 301 |
| DPZn-COOH | I2, LiI, DPMII | GuSCN, tBP | 602 | 4.22 | 69.4 | 1.76 | 2014 | 301 |
| mJS1 | Co(bpy)3 | LiTFSI, tBP | 833 | 10.55 | 76.2 | 6.69 | 2021 | 302 |
| mJS2 | Co(bpy)3 | LiTFSI, tBP | 845 | 5.47 | 75.2 | 3.48 | 2021 | 302 |
| mJS3 | Co(bpy)3 | LiTFSI, tBP | 814 | 3.73 | 76.8 | 2.33 | 2021 | 302 |
| bJS1 | Co(bpy)3 | LiTFSI, tBP | 823 | 12.52 | 77.9 | 8.03 | 2021 | 302 |
| bJS2 | Co(bpy)3 | LiTFSI, tBP | 849 | 16.59 | 75.9 | 10.69 | 2021 | 302 |
| bJS3 | Co(bpy)3 | LiTFSI, tBP | 836 | 16.48 | 75.5 | 10.42 | 2021 | 302 |
| LWP12 | Co(bpy)3 | LiTFSI, tBP | 731 | 12.07 | 73.8 | 6.5 | 2016 | 303 |
| LWP13 | Co(bpy)3 | LiTFSI, tBP | 706 | 10.06 | 78.0 | 5.5 | 2016 | 303 |
| LWP14 | Co(bpy)3 | LiTFSI, tBP | 805 | 17.22 | 74.1 | 10.3 | 2016 | 303 |
| SM85 | I2, LiI, DMII | GuSCN, tBP | 578 | 13.4 | 71 | 5.7 | 2019 | 304 |
| H2PE1 | I2, LiI, PMII | tBP | 540 | 5.26 | 73 | 2.06 | 2017 | 305 |
| LS-01 | I2, LiI, PMII | tBP | 530 | 12.58 | 70 | 4.67 | 2017 | 305 |
| LS-11 | I2, LiI, PMII | tBP | 520 | 16.13 | 64 | 5.36 | 2017 | 305 |
| XW40 | I2, LiI, PMII | tBP | 730 | 18.67 | 68.3 | 9.3 | 2019 | 306 |
| XW48 | I2, LiI, PMII | tBP | 755 | 18.34 | 70.2 | 9.7 | 2019 | 306 |
| XW48 | Co(bpy)3 | LiTFSI, tBP | 803 | 15.20 | 73.2 | 8.9 | 2019 | 306 |
| XW49 | I2, LiI, PMII | tBP | 753 | 18.09 | 69.6 | 9.5 | 2019 | 306 |
| XW49 | Co(bpy)3 | LiTFSI, tBP | 837 | 15.60 | 72.9 | 9.5 | 2019 | 306 |
| XW50 | I2, LiI, PMII | tBP | 761 | 18.96 | 70.2 | 10.1 | 2019 | 306 |
| XW50 | Co(bpy)3 | LiTFSI, tBP | 843 | 16.24 | 73.9 | 10.1 | 2019 | 306 |
| XW51 | I2, LiI, PMII | tBP | 781 | 20.07 | 70.2 | 11.1 | 2019 | 306 |
| XW51 | Co(bpy)3 | LiTFSI, tBP | 844 | 15.24 | 75.6 | 9.7 | 2019 | 306 |
| XW41 | I2, LiI, PMII | tBP | 695 | 16.77 | 70.1 | 8.16 | 2019 | 307 |
| XW60 | I2, LiI, PMII | tBP | 715 | 16.77 | 73.1 | 8.8 | 2020 | 308 |
| XW61 | I2, LiI, PMII | tBP | 775 | 21.41 | 74.7 | 12.4 | 2020 | 308 |
| XW62 | I2, LiI, PMII | tBP | 762 | 20.70 | 73.2 | 11.6 | 2020 | 308 |
| XW63 | I2, LiI, PMII | tBP | 763 | 20.63 | 73.7 | 11.6 | 2020 | 308 |
| ISQ1 | Iodolyte Z-50 | 544 | 8.99 | 68.4 | 3.34 | 2018 | 309 | |
| ISQ2 | Iodolyte Z-50 | 558 | 9.62 | 68.7 | 3.68 | 2018 | 309 | |
| ISQ3 | Iodolyte Z-50 | 576 | 10.02 | 72.0 | 4.15 | 2018 | 309 | |
| SQ1 | Iodolyte Z-50 | 579 | 8.33 | 71.1 | 3.43 | 2016 | 310 | |
| sQ2 | Iodolyte Z-50 | 649 | 12.56 | 71.5 | 5.8 | 2016 | 310 | |
| SQ3 | Iodolyte Z-50 | 606 | 9.05 | 69.8 | 3.83 | 2016 | 310 | |
| SQ4 | Iodolyte Z-50 | 622 | 10.10 | 68.7 | 4.31 | 2016 | 310 | |
| SQ5 | Iodolyte Z-50 | 660 | 19.82 | 68.9 | 9.0 | 2016 | 310 | |
| SQ6 | Iodolyte Z-50 | 648 | 14.20 | 68.5 | 6.30 | 2016 | 310 | |
| SQ7 | Iodolyte Z-50 | 646 | 16.67 | 69.9 | 7.53 | 2016 | 310 | |
| YR1 | I2, LiI, DMII | GuSCN, tBP | 524 | 2.88 | 69 | 1.04 | 2013 | 311 |
| YR2 | I2, LiI, DMII | GuSCN, tBP | 563 | 2.77 | 73 | 1.14 | 2013 | 311 |
| YR3 | I2, LiI, DMII | GuSCN, tBP | 604 | 7.26 | 74 | 3.27 | 2013 | 311 |
| YR4 | I2, LiI, DMII | GuSCN, tBP | 613 | 8.53 | 74 | 3.85 | 2013 | 311 |
| YR5 | I2, LiI, DMII | GuSCN, tBP | 605 | 7.80 | 74 | 3.49 | 2013 | 311 |
| YR6 | I2, LiI, DMII | GuSCN, tBP | 642 | 14.8 | 71 | 6.74 | 2013 | 311 |
| TS3 | I2, LiI, DMII | GuSCN, tBP | 622 | 13.1 | 73 | 5.95 | 2013 | 311 |
| JD10 | I2, LiI, DMII | GuSCN, tBP | 635 | 16.4 | 70 | 7.30 | 2013 | 311 |
| T-PA | I2, LiI, DMII | GuSCN, tBP | 644 | 9.6 | 72.2 | 4.6 | 2015 | 312 |
| DTP-PA | I2, LiI, DMII | GuSCN, tBP | 642 | 5.9 | 73.5 | 2.8 | 2015 | 312 |
| DTT-CA | I2, LiI, DMII | GuSCN, tBP | 644 | 13.1 | 71.6 | 6.0 | 2015 | 312 |
| DTT-PA | I2, LiI, DMII | GuSCN, tBP | 621 | 3.7 | 76.3 | 1.8 | 2015 | 312 |
| DTS-CA | I2, LiI, DMII | GuSCN, tBP | 682 | 19.1 | 68.3 | 8.9 | 2015 | 312 |
| DTS-PA | I2, LiI, DMII | GuSCN, tBP | 676 | 10.4 | 70.5 | 5.0 | 2015 | 312 |
| PBut-SC2-T | I2, LiI, DMII | GuSCN, tBP | 650 | 13.4 | 70.4 | 6.1 | 2015 | 313 |
| PBut-SC12-T | I2, LiI, DMII | GuSCN, tBP | 660 | 16.3 | 70.1 | 7.5 | 2015 | 313 |
| PSil-SC12-T | I2, LiI, DMII | GuSCN, tBP | 650 | 15.2 | 71.2 | 7.1 | 2015 | 313 |
| PSil-SC12-DTS | I2, LiI, DMII | GuSCN, tBP | 690 | 16.0 | 69.6 | 7.6 | 2015 | 313 |
| TSQa | I2, LiI, DMPII | None | 450 | 8.05 | 59 | 2.13 | 2013 | 314 |
| TSQb | I2, LiI, DMPII | None | 450 | 8.89 | 61 | 2.43 | 2013 | 314 |
| MSQ | I2, LiI, DMPII | None | 520 | 5.25 | 69 | 1.88 | 2013 | 314 |
| JK-216 | I2, LiI, DMPII | tBP | 610 | 13.93 | 74.0 | 6.29 | 2011 | 315 |
| JK-217 | I2, LiI, DMPII | tBP | 583 | 13.73 | 70.2 | 5.54 | 2011 | 315 |
| WCH-SQ10 | I2, LiI | None | 374 | 9.25 | 51 | 1.77 | 2012 | 316 |
| WCH-SQ11 | I2, LiI | None | 391 | 9.06 | 55 | 1.96 | 2012 | 316 |
| PSQ9 | Iodolyte Z-50 | 577 | 17.07 | 70.35 | 6.93 | 2019 | 317 | |
| PSQ10 | Iodolyte Z-50 | 579 | 16.93 | 69.83 | 6.84 | 2019 | 317 | |
| HSQ2 | I2, LiI, DMPII | None | 584 | 11.55 | 61 | 4.11 | 2014 | 318 |
| HSQ3 | I2, LiI, DMPII | None | 581 | 13.95 | 57 | 4.60 | 2014 | 318 |
| HSQ4 | I2, LiI, DMPII | None | 558 | 15.61 | 65 | 5.66 | 2014 | 318 |
| SPSQ1 | I2, LiI, DMPII | tBP | 627 | 6.51 | 73 | 2.98 | 2016 | 319 |
| SPSQ2 | I2, LiI, DMPII | tBP | 670 | 7.94 | 74 | 3.95 | 2016 | 319 |
| L1 | Cu(tmby)2 | LiTFSI, tBP | 910 | 9.4 | 71 | 6.1 | 2020 | 26 |
| WS-68/WS-5 | I2, LiI, DPMII | tBP | 746 | 14.08 | 67 | 7.67 | 2017 | 263 |
| WS-5/WS-69 | I2, LiI, DPMII | tBP | 753 | 19.56 | 68 | 10.09 | 2017 | 263 |
| AP25/D35 | I2, LiI, DMII | GuSCN, tBP | 551 | 24.5 | 63 | 8.4 | 2020 | 267 |
| C268/SC-4 | I2, DMII, EMII | Sulfolane, NBB, GuSCN | 779 | 18.10 | 71.0 | 10.0 | 2018 | 269 |
| XY1b/Y123 | Cu(tmby)2 | LiTFSI, MBI | 1050 | 15.74 | 79 | 13.1 | 2018 | 320 |
| MS5/XY1b | Cu(tmby)2 | LiTFSI, MBI | 1050 | 15.84 | 81.3 | 13.5 | 2021 | 12 |
| ADEKA-1/LEG4 | Co(phen)3 | LiClO4, NaClO4, TBAPF6, TBPPF6, HMIPF6, tBP, TMSP, MP, CPrBP, CPeBP, COcBP | 1014 | 18.27 | 77.1 | 14.3 | 2015 | 24 |
| ADEKA-1/SFD-5 | Co(phen)3 | LiClO4, NaClO4, TBAPF6, TBPPF6, HMIPF6, tBP, TMSP, MP | 1035 | 16.07 | 77.3 | 12.86 | 2015 | 321 |
| SGT-020/HC-A4 | Co(bpy)3 | LiClO4, tBP | 864 | 15.8 | 76.6 | 10.5 | 2017 | 287 |
| SM315/HC-A4 | Co(bpy)3 | LiClO4, tBP | 893 | 16.4 | 79.4 | 11.6 | 2017 | 287 |
| SGT-021/HC-A4 | Co(bpy)3 | LiClO4, tBP | 910 | 17.5 | 75.3 | 12.0 | 2017 | 287 |
| SGT-137/HC-A1 | Co(bpy)3 | LiClO4, tBP | 884 | 18.37 | 76.7 | 12.45 | 2017 | 288 |
| XW1/C1 | I2, LiI, PMII | tBP | 746 | 17.53 | 71 | 9.24 | 2014 | 293 |
| XW2/C1 | I2, LiI, PMII | tBP | 697 | 18.22 | 70 | 8.96 | 2014 | 293 |
| XW3/C1 | I2, LiI, PMII | tBP | 705 | 18.42 | 70 | 9.05 | 2014 | 293 |
| XW4/C1 | I2, LiI, PMII | tBP | 736 | 20.15 | 71 | 10.45 | 2014 | 293 |
| XW9/C1 | I2, LiI, PMII | tBP | 764 | 17.01 | 71.8 | 9.3 | 2015 | 294 |
| XW10/C1 | I2, LiI, PMII | tBP | 753 | 18.24 | 74.2 | 10.1 | 2015 | 294 |
| XW11/C1 | I2, LiI, PMII | tBP | 746 | 19.52 | 74.0 | 10.6 | 2015 | 294 |
| XW9/WS-5 | I2, LiI, PMII | tBP | 770 | 17.70 | 74.1 | 10.1 | 2015 | 294 |
| XW10/WS-5 | I2, LiI, PMII | tBP | 765 | 19.01 | 76.4 | 11.0 | 2015 | 294 |
| XW11/WS-5 | I2, LiI, PMII | tBP | 760 | 20.33 | 74.4 | 11.5 | 2015 | 294 |
| XW14/WS-5 | I2, LiI, PMII | tBP | 765 | 18.54 | 70 | 9.9 | 2015 | 295 |
| XW15/WS-5 | I2, LiI, PMII | tBP | 763 | 18.88 | 71 | 10.1 | 2015 | 295 |
| XW16/WS-5 | I2, LiI, PMII | tBP | 773 | 19.01 | 72 | 10.4 | 2015 | 295 |
| XW17/WS-5 | I2, LiI, PMII | tBP | 748 | 20.30 | 72 | 10.9 | 2015 | 295 |
| SGT-021/HC-A1 | Co(bpy)3 | LiTFSI, tBP | 849 | 19.2 | 76.8 | 12.6 | 2019 | 296 |
| SGT-023/HC-A1 | Co(bpy)3 | LiTFSI, tBP | 761 | 9.2 | 79.9 | 5.6 | 2019 | 296 |
| SGT-025/HC-A1 | Co(bpy)3 | LiTFSI, tBP | 837 | 17.3 | 76.0 | 11.0 | 2019 | 296 |
| PZn-HOQ/BET | I2, LiI, DPMII | GuSCN, tBP | 573 | 6.87 | 66.8 | 2.63 | 2014 | 301 |
| PZn-HOQ/BET | I2, LiI, DPMII | GuSCN, tBP | 605 | 8.33 | 67.7 | 3.41 | 2014 | 301 |
| XW40/Z1 | I2, LiI, PMII | tBP | 748 | 19.59 | 71.9 | 10.55 | 2019 | 307 |
| XW41/Z1 | I2, LiI, PMII | tBP | 726 | 19.63 | 71.5 | 10.19 | 2019 | 307 |
| XW51/Z2 | I2, LiI, PMII | tBP | 738 | 20.13 | 7 0.5 | 10.5 | 2020 | 308 |
| TSQa/MSQ | I2, LiI, DMPII | None | 440 | 11.57 | 56 | 2.82 | 2013 | 314 |
| SPSQ1/N3 | I2, LiI, DMPII | tBP | 635 | 15.60 | 73 | 7.20 | 2016 | 319 |
| SPSQ2/N3 | I2, LiI, DMPII | tBP | 656 | 17.10 | 73 | 8.20 | 2016 | 319 |
| XY1/L1 | Cu(tmby)2 | LiTFSI, tBP | 1080 | 15.9 | 67 | 11.5 | 2020 | 26 |
| XY1/D35 | Cu(tmby)2 | LiTFSI, tBP | 1070 | 15.3 | 67 | 11.0 | 2020 | 26 |
| D35/Dyenamo blue | Co(bpy)3 | LiClO4, tBP, TPAA | 920 | 15.5 | 73.3 | 10.5 | 2016 | 322 |
| SGT-149/SGT-021 | Co(bpy)3 | LiTFSI, tBP | 912 | 20.86 | 73.2 | 13.9 | 2020 | 25 |
| SGT-149/SGT-021 | I2, LiI, DMPII | tBP | 722 | 22.05 | 70.6 | 11.3 | 2020 | 25 |
The highest performing DSC dyes are typically based on amine donors.323 These groups are tunable in donation strength, offer reversible oxidation potentials, and have multiple positions for addition of insulating groups. Indoline-based donor dyes have been a popular class of materials in the DSC literature. Relatively early success with indoline use in an organic dye was found when D205 demonstrated a PCE of 9.4% as a donor–acceptor (D–A) dye design with a rhodanine acceptor (Fig. 21).262 This PCE value was reported to be the highest observed for an organic dye at the time and fueled wide-spread use of the indoline donor with varied π-bridges and acceptors. WS-69 uses an indoline donor group along benzoxa diazole (BOD), cyclopentadithiophene (CPDT), and phenyl-cyanoacrylic acid (CAA) moieties to generate a device with an IPCE onset nearing 800 nm, which resulted in a JSC of 19.4 mA cm−2 and a PCE of 9% as a single dye device.263 The use of indoline in a donor–π–bridge–acceptor (D–π–A) design allowed expansion of the IPCE onset from 700 nm with D205 to 800 nm with WS-69. A PCE in excess of 10% could be obtained when co-sensitization strategies were employed with WS-69. Increasing the bulk of the indoline donor used with D205 and utilizing a D–A′–π–A design with a quinoxaline auxiliary acceptor gives dye YA422.264 The increased bulk of the donor group led to a dye compatible with a Co-based electrolyte for a PCE of 10.7% without an added co-sensitizer. The use of the same donor on YA422 on a diketopyrrolopyrrole (DPP)-based dye (DPP17) again lead to a >10% PCE device with a bright blue chromophore valuable for aesthetic applications.265
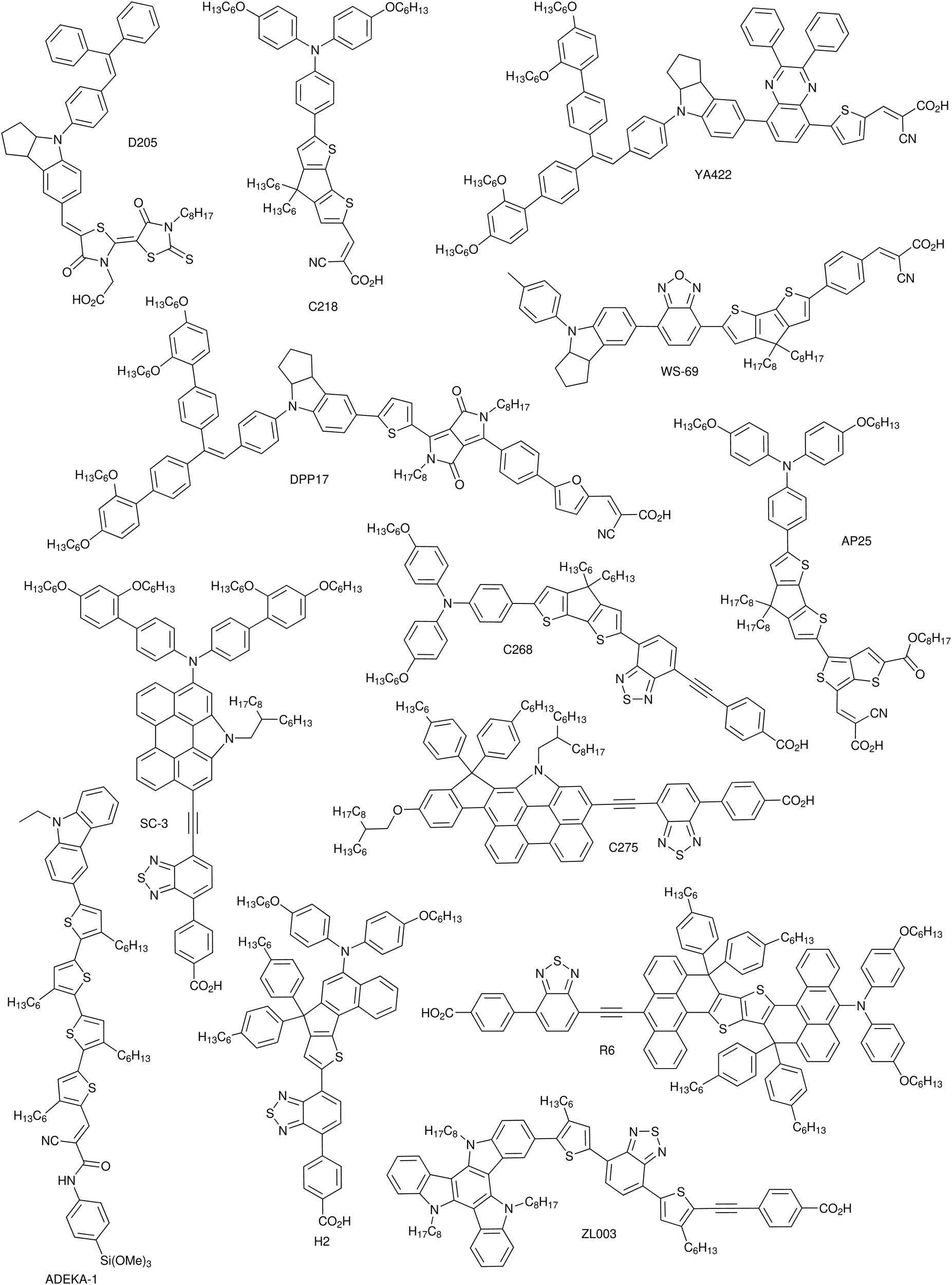 | ||
| Fig. 21 Examples of high-performing organic charge transfer dyes used in DSC devices. | ||
One of the most popular classes of amine donors used in dye design is based on triarylamines (TAAs). TAAs are typically stable and the symmetric aryl groups, before conjugation with the acceptor, allow for ease of incorporation of alkyl chains in multiple dimensions. C218 is a TAA donor-based dye with a CPDT π-bridge and a CAA acceptor which demonstrated a ∼9.0% PCE with an IPCE onset near 700 nm (Fig. 21). In ionic liquid-based devices, exceptional stabilities were noted with nearly no loss in performance under full sun soaking conditions at 60 °C.266 A 3,4-thienothiophene (3,4-TT) group was inserted between the CPDT and CAA groups of C218 to give AP25.267 The 3,4-TT building block is proaromatic by valence bond theory upon ICT, and excited-state aromaticity is observed computationally.268 Proaromatic groups allow for lower energy excitations, which enables the use of lower energy NIR photons. An exceptional photocurrent (JSC = 25 mA cm−2) for an organic dye-based DSC device was reported when AP25 (Fig. 21) was co-sensitized with D35 (Fig. 22). AP25-based DSC devices have an IPCE onset of 900 nm with a peak value of near 90% and the D35-co-sensitized devices showed a PCE of 8.4%. The broad IPCE of the AP25-based DSC device is attractive for use as a narrow optical gap material in tandem and sequential series multijunction (SSM) systems,324–326 yielding DSC devices with PCEs exceeding 10% for both the two and three photoanode devices with an up to 2.1 V open circuit voltage. Replacing the CAA group of C218 with a BTD and a benzoic acid linked with an alkyne group gives C268, which has an IPCE onset red-shifted by 50 nm relative to C218.269 C268 was shown to densely pack on the surface of TiO2 with a co-sensitizer, which enabled the fabrication of possibly the first >10% PCE ionic liquid-based DSC device. Exceptional stability of ionic liquid-based C268 DSC devices is reported during light soaking at 60 °C or at 85 °C when thermally stressed.
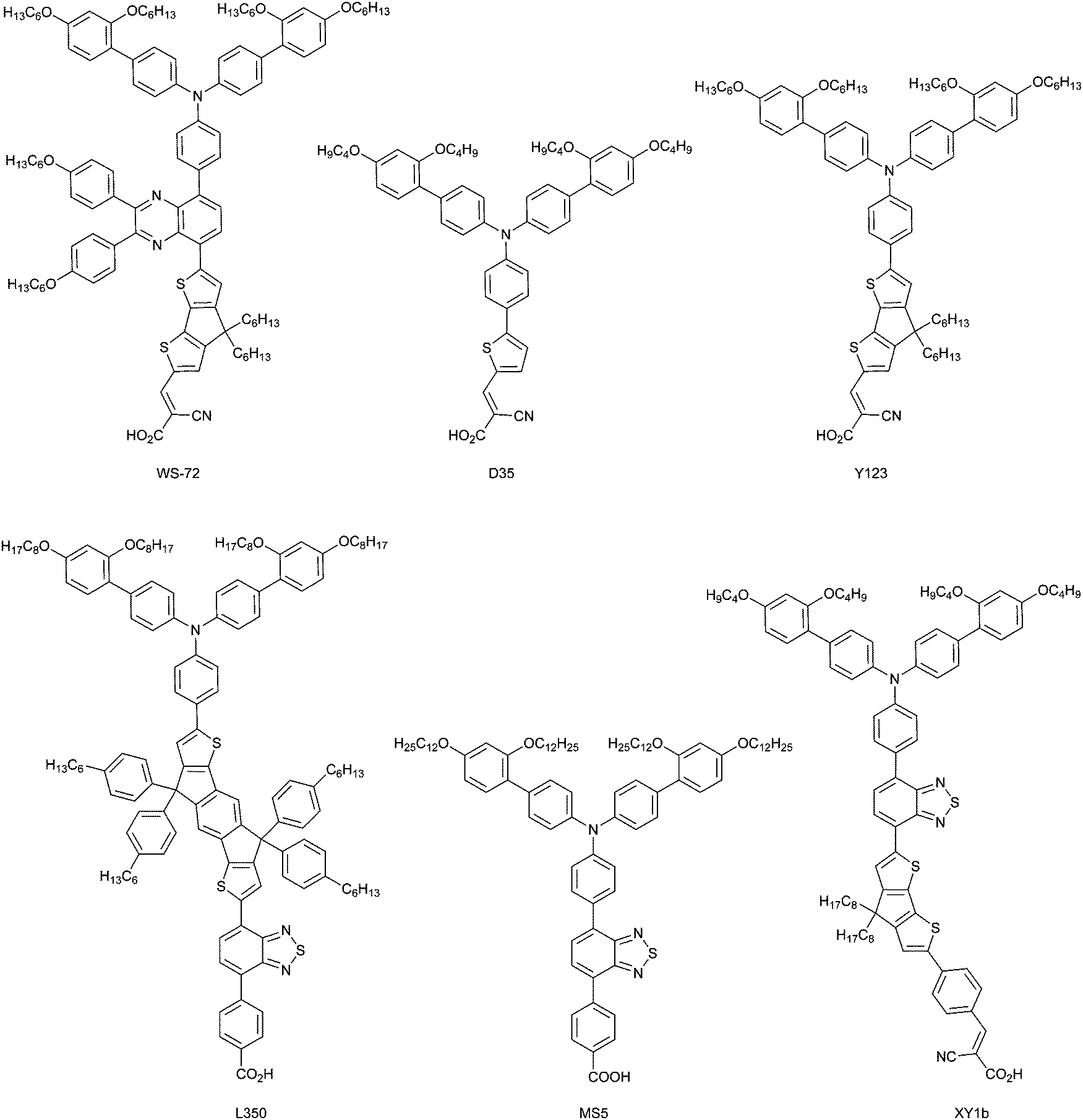 | ||
| Fig. 22 Examples of high-performing organic charge transfer dyes used in DSC devices with “umbrella” type donors. | ||
Amine donor group design has given rise to some of the highest performance DSC devices by enabling the use of 1-electron redox shuttles typically based on Co3+/2+ and Cu2+/+.95,327 For these positively charged 1-electron redox shuttles to facilitate productive electron transfers within the DSC device, exquisite surface protection is needed to slow the recombination reaction of electrons in TiO2 with the oxidized redox shuttle. The most common successful strategy employed with respect to dye design is the use of alkylated donor groups with alkyl chains extending in three dimensions to provide an “umbrella” of insulating groups to protect electrons at the TiO2 surface. One of the first and most widely used materials to demonstrate this concept is the dye D35, which illustrated the benefits of Co3+/2+ redox shuttles relative to I−/I3− (Fig. 22).270 The thiophene π-bridge of D35 was expanded to a CPDT π-bridge to give Y123 with the same CAA acceptor.271,272 The expansion of the π-bridge conjugation length gave a red-shift of the absorption spectrum and allowed for an increase in PCE from 6.7 to 8.8% based on a cobalt redox shuttle. Building from the D35/Y123 D–π–A design, an auxiliary acceptor (A′) strategy was employed with dye WS-72 by insertion of a quinoxaline group between the TAA donor and the CPDT bridge to give a D–A′–π–A design (Fig. 22).273 The D–A′–π–A dye design is reported to enable more favorable electron transfers with extended charge separation durations while red-shifting the absorption spectrum relative to the D–π–A design.328 The D–A′–π–A design often showed not to lower the ground state oxidation potential value significantly despite extending conjugation, which allowed for the continued use of RSs with more positive values in DSC devices for an increase in the theoretical VOC. WS-72 was found to minimize voltage losses when paired with the bis-(4,4′,6,6′-tetramethyl-2,2′-bipyridine)copper(II/I) ([Cu(tmby)2]2+/+) redox shuttle leading to an 11.6% PCE DSC device with a VOC in excess of 1.1 V. The same device and redox shuttle could be solidified to give a solid-state device operating at 11.7% PCE, which is claimed to be the highest known solid-state DSC PCE at the time of the report. L350 uses an indacenodithiophene (IDT) π-bridge with a similar donor group to Y123 and a benzothiadiazole (BTD)-benzoic acid acceptor.274 This design led to a positive ground state oxidation potential (1.04 V vs. NHE) which allowed for the use of the [Cu(tmby)2]2+/+ redox shuttle system to give a 1.14 V open-circuit voltage solar cell for a PCE of 11.2% under full sun conditions. Under low light conditions (1000 lux), an impressive PCE of 28.4% could be obtained. Interestingly, L350 has an optical energy gap of 1.82 eV as estimated from the IPCE onset, which indicates that only 680 mV of total absorbed energy was required to drive both the electron transfer to TiO2 and the regeneration reaction from the redox shuttle. XY1b uses a similar design to that of dye WS-72 with a BTD group in place of the quinoxaline group and a phenyl spacer between the CPDT and CAA groups. Through the use of XY1b, co-sensitizer Y123, redox shuttle [Cu(tmby)2]2+/+, and a direct contact PEDOT counter electrode, a PCE of 13.1% could be obtained under full sun conditions. A 32% PCE at 1000 lux was reported which exceeds the values reported to date with commonly used materials such as silicon and GaAs systems under low light conditions.320 Very recently Zhang et al. have introduced a new dye – MS5 – with a particularly long n-dodecyl “umbrella” alkyl chain and a favorable ground state oxidation potential in respect to the Cu(tmby)2 redox couple, leading to a record device VOC of 1.24 V for a copper redox shuttle-based device.12 The co-sensitization of MS5 with the broader-absorbing XY1b dye resulted in a DSC with a certified PCE of 13.0%, the highest certified efficiency reported to date, while a batch of such devices reached an average 13.5% efficiency when measured in the laboratory. These devices also retained 93% of their initial efficiency after 1000 h of full sun light soaking at 45 °C.
The use of extended π-conjugation systems as donor groups has been an increasing popular strategy for increasing light absorption and improving device PCEs. SC-3 is a perylene-based dye with a bulky diarylamine donor substituted onto a phenanthrocarbazole group (Fig. 21).275 A BTD-benzoic acid acceptor was used with SC-3 to give a dye reported to undergo electron injection from non-relaxed, hot excited states. The fast electron injection coupled with good surface protecting gave the dye 11.5% PCE. Notably, replacing the diarylamine group on SC-3 with an arylether group planarized by a ring fusion strategy led to dye C275, with a higher PCE of 12.5% owing to a high voltage (>950 mV) when using the Co(phen)33+/2+ RS system.276 R6 is designed with a central thienothiophene component fused to two anthracene groups.277 A diarylamine donor and a BTD group with a benzoic acid acceptor complete the conjugated system. Two tetra-substituted sp3-hybridized carbons provide alkyl groups extending above and below the dye conjugated plane to increase solubility and reduce aggregation. R6-based DSC devices have an IPCE onset near 800 nm and give a 12.6% PCE using a Co(bpy)33+/2+-based electrolyte. The devices show a remarkable stability and offer a blue dye for use in aesthetically-driven applications. Dye H2 incorporated a donor group with four alkyl chains with BTD as a π-bridge and benzoic acid as an anchoring group.278 This arrangement led to a high photovoltage (900 mV) when paired with a cobalt redox shuttle, indicating minimal recombination losses due to transfer of an electron from the TiO2 surface to the oxidized redox shuttle. Exceptional stability was observed from a dye analogue during light soaking studies, but ultimately the DSC device PCE was limited by the absorption range of the dye which had an IPCE onset of ∼750 nm. ZL003 was designed with a novel donor group with three alkylated nitrogens, a bisthiophene-substituted benzothiadiazole (BTD), and a benzoic acid anchoring group. This design resulted in exceptional surface protection with minimal recombination losses for a photovoltage loss of only 106 mV based on the theoretical obtainable photovoltage assuming no shift in the TiO2 conduction band taken as −0.5 V versus NHE.279 Notably, ZL003 was found to up-shift the Fermi level of TiO2 by approximately 600–700 mV, which likely contributed to the high photovoltage observed (956 mV) from the ZL003 device with the Co(bpy)33+/2+ RS. The exceptional surface protection, rapid hot electron injection occurring out of locally excited states from the dye to TiO2, and the broad IPCE onset nearing 800 nm led to the highest performing single-dye DSC device reported in the literature at 13.6% PCE.
A large number of anchoring group strategies have been reported in the literature, with strategies often focused on finding strong binding groups which retain facile electron transfer from the photoexcited dye to TiO2. The use of carboxylic acid-based systems is the most popular strategy in the literature owing to their relative ease of preparation and exceptional performance with respect to electron injection. One of the most intriguing motivations for replacing carboxylic acid anchoring groups in DSCs is highlighted with the discovery of ADEKA-1 (Fig. 21).24,280,321 ADEKA-1 features a siloxane-based anchoring group as a tight binding group to TiO2. The siloxane anchoring group enabled the use of a co-sensitizer (LEG4, which is similar to Y123 with OC4H9 rather than OC6H13 alkyl chains on the amine donor, Fig. 22) and a tremendous number of surfaces protecting groups of varied shapes and sizes. This type of extensive co-sensitization is challenging unless a significant difference in anchor binding group strength is present. This strategy has led to the highest performing single DSC device reported in the literature at 14.3% PCE. It is noteworthy that since this discovery, siloxane anchoring groups remain underexplored with respect to incorporation into dye designs which may be due to challenges with identifying the composition of the anchoring group after purification.329
4.2.2.1 Wide optical gap organic sensitizers. A growing body of work is focusing on the design of wide optical gap dyes which have applications in multijunction or tandem DSC devices as the initial photoactive layer and in photoelectrochemical cell systems. For SSM or tandem systems, the photovoltage output from the wide optical gap dye-based DSC is a critical parameter since higher VOC values allow for less free energy waste from high energy visible light (blue) photons. A common objective is to position the dye excited-state energy level near the CB energy of an n-type semiconductor to minimize free energy loss and to position the ground state oxidation potential of the dye positive enough to drive challenging electron transfer reactions. Initial high photovoltage DSCs focused on the use of the Br−/Br3− RS system with wide optical gap dyes. Through the use of Mg-doped TiO2, to shift the CB to a more negative potential, and the Br−/Br3− RS, a theoretical photovoltage of 1.5 V can be obtained.216 A wide optical gap dye with a siloxane-based anchor and a coumarin weak donor (ADEKA-3) was used to give a 1.45 V device at room temperature with 1.5 V observed at 5 °C. A PCE of 3.9% was observed for the room temperature DSC device (Fig. 23). AP14 is designed with an electron deficient thienopyrroledione bridging a benzene with an ether donor and a benzene with a CAA acceptor.281 A 1.73 V versus NHE oxidation potential was measured for AP14 which is positive enough to drive the oxidation of Fe(bpy)32+ in DSC devices to give a 1.32 V device. RR9 is comprised of a BTD π-bridge and a pentaalkylated aryl ether-based weak donor group.282 While the ground state oxidation potential of RR9 is less positive (1.56 V versus NHE) than that reported for AP14, the DSC devices exhibited a higher VOC value of 1.42 V, which was the record high voltage for a room temperature DSC device without the use of TiO2 doping at the time of the report. This device was used in a three active layer SSM DSC device (6-terminal, series wired) as the top layer to give a 3.3 V device where the photovoltage output is >1 V per layer. These systems are inherently limited due to the light absorption of Fe(bpy)33+/2+; however, they provide proof of principle examples of the value of the dye design strategy and indicate the importance of finding a redox shuttle at ≥1.4 V oxidation potential versus NHE that does not absorb visible light for use in SSM or tandem device systems.
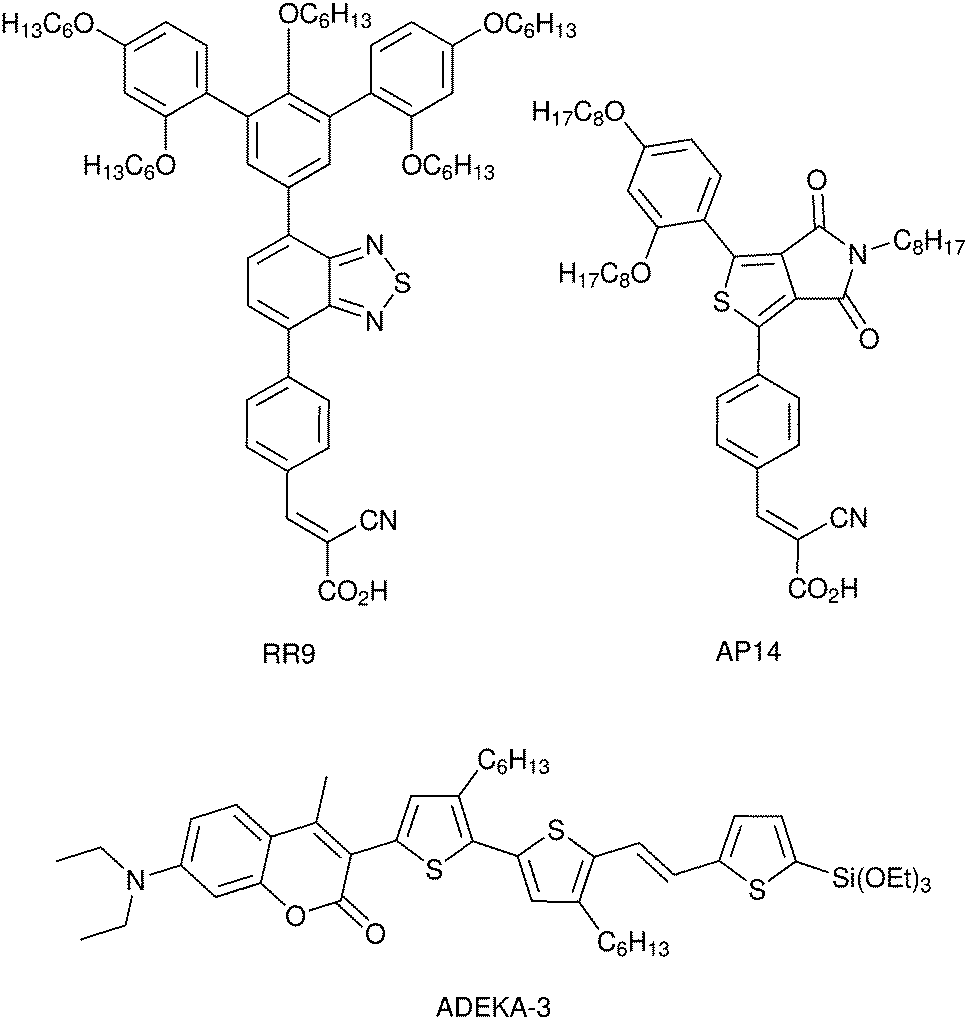 | ||
| Fig. 23 Examples of high voltage dye-designs. | ||
4.2.2.2 Porphyrins. Porphyrins are a primary focus of dye design research due in part to porphyrins being one of the first classes of dyes to show comparable and higher PCEs in DSC devices relative to ruthenium complexes. The donor-porphyrin-acceptor construct is one of the most successful design strategies. In 2010, zinc porphyrin dye YD2 demonstrated an impressive 11% PCE without employing any precious metal, and using a diarylamine donor and benzoic acid acceptor at opposite meso positions of the porphyrin core (Fig. 24).283 Substitution of the remaining two meso positions with de-aggregating tert-butyl-substituted aryls is a key part of this design, although dyes are known with these two meso position being differentiated with high performances.330 YD2-o-C8 is a derivative of YD2 with bis-ortho-substituted alkyl ether substituents on a benzene ring to better disrupt aggregation of the porphyrin dye.284 A complementary organic photosensitizer (Y123, Fig. 22) was used as a co-sensitizer to increase the performance of the YD2-o-C8 device in the 500–650 nm region where porphyrins are relatively weakly absorbing. This co-sensitization gave the highest performing DSC device at the time with a PCE of 12.3%. The landmark PCE was made possible by the use of a 1-electron-based cobalt RS which gave a VOC of nearly 1 V. The introduction of a BTD group near the benzoic acid anchor led to GY50, which better absorbs photons in the 500–650 nm range and eliminated the need for the use of a co-sensitizer.285 A 12.8% PCE was obtained from a single dye DSC device with a JSC of 18.5 mA cm−2 using a cobalt-based electrolyte. This high JSC value was made possible by both red-shifting the Q-band when introducing the BTD group and increasing the absorptivity of the dye throughout the visible spectral region. Comparatively, GY50 with an iodine-based electrolyte system gave a PCE of only 8.9%, which highlights the critical importance of 1-electron-based RSs with regard to high power conversion efficiencies in DSCs. The diarylamine donor group of GY50 was expanded to include an additional aryl group with four total donor-group alkyl chains on SM315 for better TiO2 surface insulation, aimed to slow the recombination of electrons at the TiO2 surface with the cobalt-based electrolyte. This strategy led to a ∼25 mV increase in VOC for SM315 relative to GY50, resulting in the first DSC device reported to reach 13.0% PCE.286 A benzene group on the donor of SM315 was replaced with a fluorene group to give SGT-021.287,296 When benchmarked against SM315, a higher photovoltage (20 mV increase) and photocurrent (1.1 mA cm−2 increase) were obtained. When a non-porphyrin-based organic sensitizer was used as a top cell in a mechanically stacked tandem device, an impressive 14.6% PCE could be obtained.288 Through the incorporation of a D–π–A dye with an exceptionally effective amine donor design to promote favorable charge separation durations, a co-sensitized device with SGT-021 and SGT-149 gave a high PCE of 14.2%.25
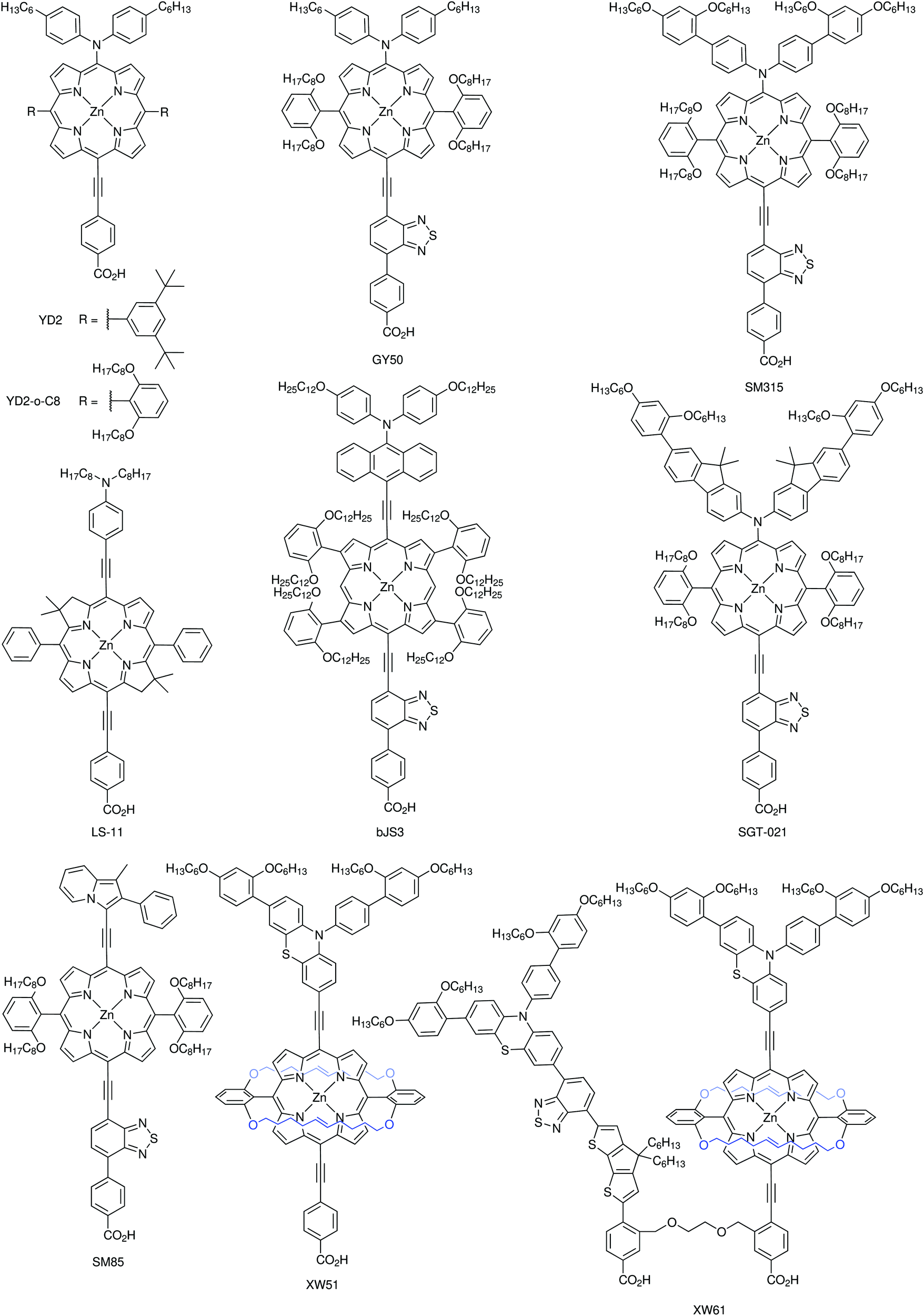 | ||
| Fig. 24 Select porphyrin examples discussed in this review. | ||
To improve further on the exceptional efficiencies described above, the use of lower energy photons (>750 nm) is needed. Numerous strategies have emerged with respect to porphyrin dye design aiming to reduce aggregation through novel constructs, improve spectral response both in the visible and NIR via building block incorporation, co-link of chromophores, and design of supramolecular assembly strategies (tailored aggregation) as referenced and discussed below. With respect to the linear donor-porphyrin-acceptor design with meso-substituted de-aggregating groups, common general methods for extending the absorption range focus on adding donor groups,289–295 fusing non-amine donor groups for π-extended donor groups,331 or adding acceptor groups296–301 as the D and A component to promote lower energy ICT events within the D–porphyrin–A structure. The use of a π-extended donor group has shown promise for improving DSC device performances as well. Specifically, the introduction of an anthracene group between the amine donor and porphyrin (mJS3) resulted in a red shift of both the Soret and Q-band relative to no added anthracene group.302,303 However, the PCE of mJS3 dropped significantly compared to a benchmark YD2-o-C8 DSC cell under identical conditions (2.3 % versus 9.8 %) primarily due to loss of photocurrent with possible aggregation-limited performance for mJS3. De-aggregating groups at the β positions of the porphyrin were explored in the same study and termed a “double fence” porphyrin due to the use of two de-aggregating aryl groups on each side of the porphyrin (see dye bJS3). The double fence strategy shows minimal changes to the dye energetics in solution, and led to a 10.4% PCE cell, which was higher performing than YD2-o-C8 under identical conditions. The massive improvement from 2.3% to 10.4% based on the shift from meso to β-substituted de-aggregative aryls certainly warrants more investigation in this direction. An alternative strategy for red-shifting the porphyrin absorption spectrum has recently been presented which focuses on purposefully inducing aggregation of porphyrin-based dyes with a planarized indolizine donor to allow for an aggregate-induced red-shifting of the absorption spectrum.304 This approach allowed for the shifting of the absorption spectrum substantially on TiO2versus solution (710 nm onset in solution, 875 nm onset on TiO2) and provided an under-explored method of absorbing deeper into the NIR spectral region post-synthesis.
Bacteriochlorins are a class of materials related to porphyrins and are known as a type of hydroporphyrin. These building blocks have been used in DSC dye LS-11 with exceptional NIR photon use until 870 nm in DSC devices.305 LS-11 shows a relatively intense Q-band (112000 M−1 cm−1) compared to many porphyrin-based dyes and multiple absorption features throughout the visible spectral region. However, due to a peak IPCE response of ∼60% and a modest open circuit voltage (0.52 V), the PCE was limited to 5.4%. Further exploration of this class of materials is intriguing given the rare use of NIR photons beyond 800 nm.
Doubly-strapped porphyrins have also shown promise in DSC devices by minimizing aggregate formation thorough the introduction of carbon chains bridging the meso positions such as with dye XW51.306,307 This strategy leads to a high PCE of 11.1% with the I−/I3− RS system. XW51 has demonstrated exceptional stabilities over the course of 1000 hours of ageing.306 XW51 was covalently linked to a “companion” D–A′–π–A organic dye with a complementary absorption spectrum for a 12.4% PCE from an I−/I3− RS-based cell generating 21.4 mA cm−2 of photocurrent with a remarkable photostability to light soaking.308 Significantly diminished performances were reported with a cobalt electrolyte (10.7% PCE), likely due to recombination of electrons in TiO2 with the oxidizing electrolyte. Strategies aimed at complete aggregation mitigation and shifting the absorption spectrum onset of porphyrins to lower energy remain intriguing directions for this class of materials.
4.2.2.3 Squaraines. Squaraine dyes are a popular class of materials in dye-sensitized systems owing to their strong absorption into the NIR spectral region. Squaraine-based dyes have shown some of the deepest NIR photon use in DSC devices known.309 Squaraines typically absorb intensely in the NIR region often between 600–900 nm with molar absorptivities often above 100
000 M−1 cm−1; however, absorption is typically weak in the higher energy spectral region. The literature surrounding this class of materials is expanding dramatically since high performing NIR absorbing chromophores are urgently needed to improve DSC devices. Select examples of squaraine dyes are discussed below (Fig. 25).
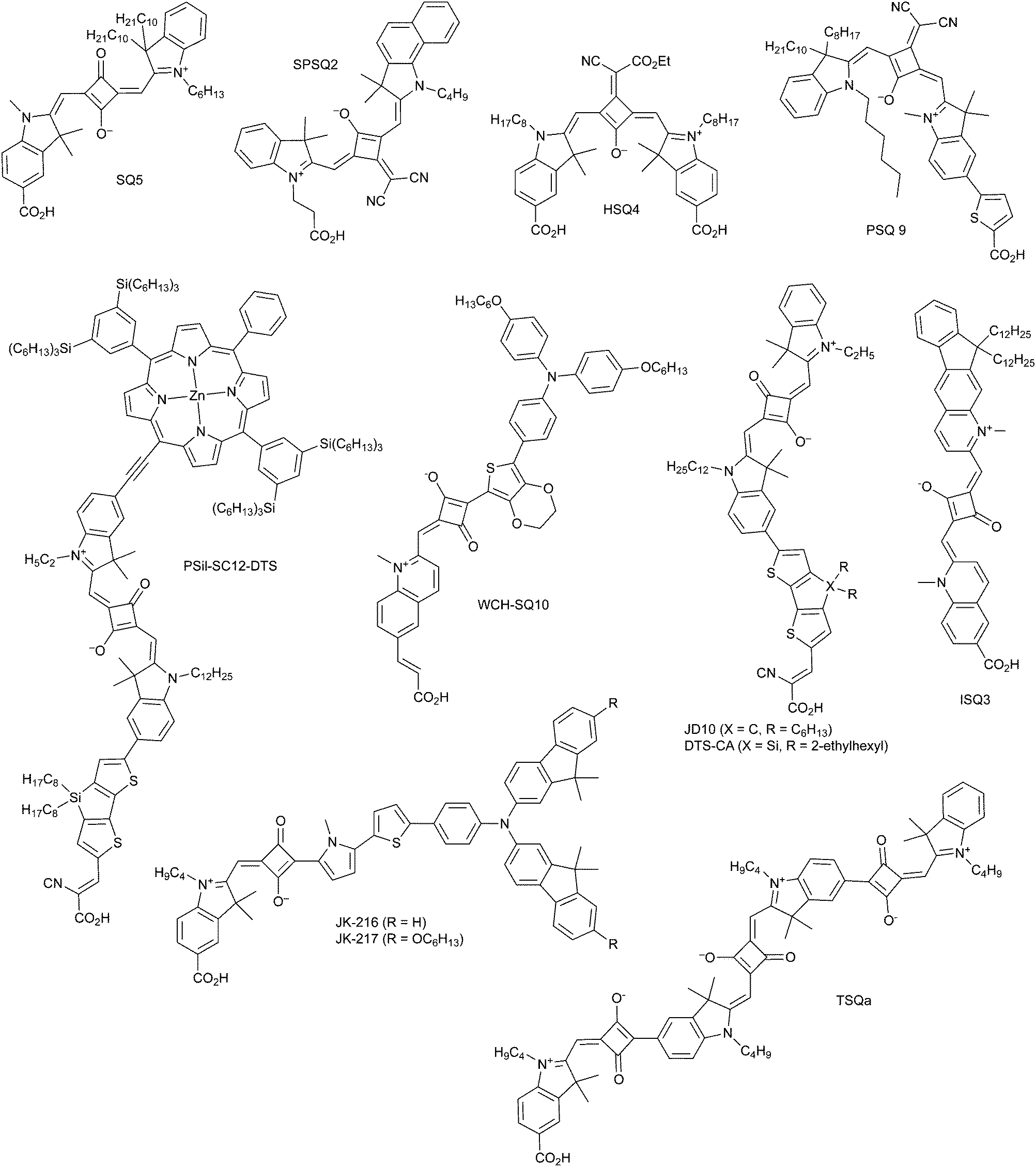 | ||
| Fig. 25 Examples of squaraine-based dyes. | ||
A series of squaraines with systematically varied alkyl groups in- and out-of the π-system plane were evaluated with alkyl group positions both near and far from the TiO2 surface.310 Extending the out-of-plane alkyl groups on the indoline building block furthest from the surface was found to have a dramatic effect on overall DSC device performance. Under identical conditions, the PCE increased from 3.4% with methyl groups in place of long alkyl chains to 7.7% PCE for SQ5 (Fig. 25). Including alkyl chains at the indoline near the TiO2 anchor led to a decrease in PCE to 6.8% which was attributed to lower dye loading. Under fully optimized conditions with reduced chenodeoxycholic acid loadings, SQ5 reached a PCE of 8.9%. These findings are notably recent, and many of the examples discussed below use much shorter alkyl chains on the indoline portion of the dye far from the TiO2 surface. Addition of π-conjugated groups extending from the squaraine chromophore have been used to increase the absorption of dyes in the high energy region and to red-shift the strong NIR absorption further. A series of eight π-bridges were examined with the indoline-based squaraine core showing 4,4-dihexyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT) as the highest efficiency π-bridge studied as part of dye JD10.311 Part of the high performance is attributed to the alkyl chains on CPDT out of the π-system plane leading to reduced aggregation and the introduction of a high energy absorption band upon incorporation of CPDT. Squaraine dyes in general benefit from co-sensitization with visible light-absorbing dyes and when JD10 was co-sensitized with D35 the efficiency could be improved to 7.9% PCE from 7.3% PCE without D35. Upon replacing the alkylated carbon of CPDT with an alkylated silicon atom to give a 4,4-bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b′]dithiophene (DTS) group for dye DTS-CA, the PCE improved to 8.9%.312 DTS-CA was found to have low recombination rates and reduced aggregation, which contributed to the observed high performance. The high energy bands introduced by the CPDT and DTS groups in the 400–550 nm region were modest in intensity but had a strong effect on the IPCE curve in this region. To balance the dye's absorption intensity of the low- and high-energy photons, a porphyrin ring was added to the DTS-CA structure to give PSil-SC12-DTS, which absorbs strongly from 400–550 nm due to the porphyrin core.313 However, despite the balancing of the absorption bands, the peak percent IPCE of the devices with PSil-SC12-DTS dropped from ∼90% with DTS-CA to ∼70%, which was attributed to a lower charge injection efficiency.
DSCs are thought to reach a theoretical maximum practical PCE from a single active layer device near 950 nm.236 Very few dye designs have reached this value. The NIR absorption of squaraine chromophores places them relatively near to this value with IPCE onsets routinely near 800 nm. One approach aimed at a further red-shifting of the squaraine chromophore is based on the use of multiple squaraine building blocks on a single dye such as with TSQa.314 The common bis-indoline-squaraine chromophore has a solution absorption onset of approximately 700 nm. Through the introduction of multiple squaraine building blocks onto the bis-indoline-squaraine chromophore, a solution absorption onset >900 nm could be reached. An IPCE onset of near 1000 nm was obtained with TSQa; however, the peak IPCE was limited to <20%. The addition of multiple squaraine building blocks was found to dramatically lower the dye LUMO energy resulting in a low driving force for electron transfer to TiO2. A second approach to red-shifting squaraine-derived dyes focuses on the de-symmetrization of the commonly used bis-indoline chromophore to allow for the use of a donor–π–bridge group (triarylamine-thiophene-pyrrole based) with a single indoline-squaraine building block as with dyes JK-216 and JK-217.315 An IPCE onset of near 850 nm was obtained with the more red-shifted JK-217. The higher VOC (610 mV) and FF (74%) with JK-216 led to a higher PCE of 6.3% than is observed with JK-217 (VOC = 583 mV, FF = 70%, PCE = 5.5%). Importantly, both dyes were shown to be stable to prolonged light soaking (1000 h at 60 °C) and function well in solid-state devices. WCH-SQ10 is comprised of a triarylamine-3,4-ethylenedioxythiophene donor–π–bridge with a squaraine-quinoline-based structure.316 This design lead to an IPCE onset beyond 1000 nm to give one of the deepest NIR photon accessing organic dyes known. Interestingly, a symmetric core bis-quinoline squaraine dye (ISQ3) shows appreciable light harvesting efficiency on TiO2 reaching 1000 nm, but an IPCE onset near 850 nm.309 This suggests significant influence of the electrolyte on the dye absorbance energy with quinoline-squaraine based materials.
Dicyanomethylene-based squaraine materials show significant red shifts of the absorption spectrum onset relative to the keto squaraine core. Dye PSQ9 has a broad IPCE spectrum reaching ∼850 nm and generating >17 mA cm−2 of photocurrent. Due to a modest photovoltage (577 mV) – as is common in the NIR region with dye sensitized solar cells – the overall power conversion efficiency was limited to 6.9% PCE.317 An ethyl cyanoacetate-derived squaraine dye (HSQ4) with dual anchors was shown to have a substantially increased stability relative to mono-anchored squaraine dyes with no change in PCE after 1000 hours.318 In this same study, the ethyl cyanoacetate group was found to give a dye with a significantly higher excited state oxidation potential than a dicyanomethylene derived dye, which correlated to a higher IPCE peak value (80% versus 70%). Dicyanomethylene squaraines without a conjugated anchoring group have also been shown to function well within co-sensitized DSC devices.319 SPSQ2 was found to increase the performance of N3-based devices by red-shifting the IPCE onset leading to an improved JSC (14.9 mA cm−2 without SPSQ2 and 17.1 mA cm−2 with SPSQ2) and improved PCE (7.1% versus 8.2%).
With substantial recent progress having been shown in co-sensitized DSC devices and in deep NIR photon absorption, continued vigorous research within the area of squaraine dyes is likely and warranted. Notably, the majority of squaraine dye-based DSC devices in the literature rely on the 2-electron I−/I3− RS system, which inherently limits the PCEs of DSC devices. Progressive improvements have been observed with squaraine dyes reaching ∼9% PCE to date with the I−/I3− RS. Similar to the breakthrough performances enabled with porphyrin-based sensitizers, a squaraine dye design that functions well with 1-electron RSs such as Co- and Cu-based systems is needed. This advance in porphyrin designs shifted the PCE from ∼9% to ∼13% when Co RS-compatible dyes were discovered. A similar discovery would greatly benefit squaraine research.
4.2.2.4 Multifunctional DSCs. DSCs have shown exceptional performances as described above in terms of low light intensity use and in tandem of SSM device designs. Additionally, DSCs are intriguing materials for aesthetically important devices owing to the wider range of colors available from the dyes used in these devices. Given the molecular nature of the chromophores being used, photochromic dyes offer a possible strategy for accessing materials with dynamic optical properties and electricity production. DSCs have been shown to operate as photo-chromo-voltaic cells that can be converted from transparent states to visible light absorbing states with the NPI dye (Fig. 26). The use of photochromic dyes is intriguing for building-integrated photovoltaics which can exist in semi-transparent states at night and as visible light absorbing states in the daytime. A key challenge with this approach consists in synthesizing dyes with reasonable power conversion efficiencies in the visible light absorbing state since visible light is competitively used within the devices to both drive electron transfers to the metal oxide semiconductor, and to convert the dye back to the non-visible light absorbing state. The use of diphenyl-naphthopyran has shown exceptional promise in allowing for a PCE >4% with good device stability (50 days tested).199 Interestingly, the diphenyl-naphthopyran building block also allows for thermal conversion or light intensity-based conversion back to a transparent state giving a self-adjusting transmission. Continued research in this area is promising with regard to building integrated photovoltaic markets.
 | ||
| Fig. 26 Photoresponsive NPI in a non-visible light absorbing state (left) and a visible light absorbing state (right). | ||
4.3 Charge transport materials
Although they had been neglected in the early stages of DSC development, charge transport materials (CTMs) are an essential part of this technology and therefore some of the most significant advances in the field of the past decade were made through progress on this component.23,332–338 Research on CTMs branched into the development of materials, the study of their properties and the fundamental understanding of charge transport within the materials and devices. CTMs are responsible for electron transfer between the electrodes and they must be able to regenerate the oxidized dye following light absorption and to be reduced at the counter electrode. Charge transport materials are not only essential for the solar cell efficiency, but they also determine its overall stability. All parameters defining the efficiency of solar cells including the short-circuit photocurrent density (JSC), open-circuit photovoltage (VOC) and the fill factor (FF) are influenced by the properties of charge transport materials and their interface interaction with the electrodes.15,336,339–341 The photocurrent density, even if largely determined by the photon-to-electron conversion abilities of dyes,342,343 is still influenced by the charge transport abilities and recombination pathways of the CTM.344 The VOC depends on the energy alignment between the Fermi level of the TiO2, the ground state of the dye and the overpotential to the CTM.CTMs can be integrated in DSCs in the liquid, quasi-solid and solid state.10 Liquid CTMs or electrolytes in solar cells comprise an organic, aqueous or ionic solvent with a redox couple, for example I−/I3−,345–347 copper14,95,96,346,348–353 or cobalt270,284,286,337,354–356 coordination complexes or organic molecules.357 For DSCs to become commercially viable, significant efforts are being made to develop quasi-solid- and solid-state charge transport materials to ensure sustainability and stability. These CTMs are usually based on organic molecules and polymers333,358,359 or on inorganic and coordination metal complexes. The fundamental differences between the various charge transport materials are the charge mobility and mechanism.10 While in liquid electrolytes there is a prevalence of ionic conductivity, in polymeric and solid-state CTMs the mechanism can be a combination of ionic and electronic transport, or a predominantly electronic process.360
In order to transport charges between the electrodes efficiently, charge transport materials in DSCs must fulfill several requirements:361–363 (i) a redox potential that provides the minimal overpotential, but with a driving force high enough to efficiently regenerate the dye, (ii) low recombination rates with the metal oxide semiconductor and the conductive substrate, (iii) minimal mass transport limitations for fast diffusion through the mesoporous semiconductor towards the counter electrode, (iv) absence or minimization of unwanted chemical and physical interactions with other components of the solar cell to improve overall stability, (v) no or minimal competitive light absorption with respect to the dye.
Currently, there is no ideal electrolyte system that fulfills all requirements, but there are several successful systems that have been discovered, and their advantages and drawbacks will be outlined. Of all the requirements above, the most important characteristics of a redox couple for highly efficient DSCs are fast dye regeneration and slow charge recombination.10Table 4 lists device parameters of DSCs employing various liquid electrolytes referenced in this review, together with the dye used.
| Mediator | Sensitizer | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|
| I−/I3− | N719 | 846 | 17.73 | 75 | 11.18 | 2005 | 145 |
| Br−/Br3− | ADEKA-3 | 1450 | 4.77 | 56 | 3.9 | 2016 | 216 |
| I−/IBr2− | N3 | 790 | 12.8 | 64 | 6.4 | 2007 | 364 |
| I−/I2Br− | N3 | 640 | 9.2 | 41 | 2.4 | 2007 | 364 |
| Co(bpy)3 | D35 | 936 | 12.05 | 69.1 | 7.80 | 2018 | 95 |
| Co(bpy)3 | D45 | 810 | 13.40 | 73.0 | 7.93 | 2018 | 95 |
| Co(bpy)3 | D5 | 713 | 9.45 | 72.8 | 4.91 | 2018 | 95 |
| Co(bpy)3 | N719 | 620 | 3.8 | 76 | 1.8 | 2011 | 365 |
| Co(bpy)3 | Z907 | 744 | 14.0 | 62 | 6.5 | 2011 | 365 |
| Co(bpy)3 | D9L6 | 688 | 10.7 | 72 | 5.32 | 2012 | 165 |
| Co(bpy)3 | D21L6 | 852 | 12.3 | 63 | 6.63 | 2012 | 165 |
| Co(bpy)3 | D25L6 | 854 | 10.8 | 63 | 5.51 | 2012 | 165 |
| Co(bpy)3 | Y123 | 855 | 14.6 | 70 | 8.8 | 2011 | 271 |
| Co(bpy)3 | YD2 | 825 | 14.9 | 69 | 8.4 | 2011 | 284 |
| Co(bpy)3 | YD2-o-C8 | 965 | 17.3 | 71 | 11.9 | 2011 | 284 |
| Co(bpy)3 | SM371 | 960 | 15.9 | 79 | 12.0 | 2014 | 286 |
| Co(bpy)3 | SM315 | 910 | 18.1 | 78 | 13.0 | 2014 | 286 |
| Co(bpy)3 | MK2 | 826 | 13.7 | 69 | 7.8 | 2013 | 366 |
| Co(bpy)3 | LEG1 | 815 | 8.80 | 60 | 4.3 | 2013 | 367 |
| Co(bpy)3 | LEG2 | 830 | 11.2 | 51 | 4.7 | 2013 | 367 |
| Co(bpy)3 | LEG3 | 915 | 8.9 | 68 | 5.5 | 2013 | 367 |
| Co(bpy)3 | LEG4 | 805 | 12.1 | 68 | 6.6 | 2016 | 355 |
| Co(bpy)3 | C218/MKA253 | 810 | 12.2 | 69 | 6.9 | 2016 | 355 |
| Co(phen)3 | D35 | 910 | 7.3 | 62 | 4.2 | 2015 | 368 |
| Co(phen)3 | ADEKA-1/LEG4 | 1014 | 18.27 | 77.1 | 14.3 | 2015 | 24 |
| Co(phen)3 | Z907 | 700 | 3.6 | 56 | 1.4 | 2015 | 368 |
| Co(Me2bpy-pz)2 | D35 | 1020 | 6.1 | 61 | 3.7 | 2013 | 166 |
| Co(bpy-pz)2 | D35 | 1020 | 5.3 | 68 | 3.6 | 2013 | 166 |
| Co(py-pz)3 | D35 | 900 | 2.5 | 66 | 1.5 | 2013 | 166 |
| Co(Mepy-pz)3 | D35 | 880 | 0.78 | 58 | 0.4 | 2013 | 166 |
| SBCC | D35 | 905 | 5.19 | 53.8 | 2.53 | 2014 | 369 |
| Co(phen)3/Co(EtPy)2 | Z907 | 750 | 5.1 | 58 | 2.2 | 2015 | 368 |
| Co(phen)3/Co(EtPy)2 | D35 | 920 | 8.4 | 67 | 5.1 | 2015 | 368 |
| Co(PY5Me2)(tBP) | MK2 | 993 | 8.1 | 76 | 6.1 | 2012 | 337 |
| Co(PY5Me2)(NMBI) | MK2 | 940 | 11.8 | 77 | 8.4 | 2012 | 337 |
| Co(bpyPY4) | MK2 | 757 | 14.7 | 75 | 8.3 | 2013 | 366 |
| Co(ttb) | LEG4 | 810 | 11.6 | 57 | 5.4 | 2016 | 355 |
| Co(ttb) | C218/MKA253 | 805 | 13.0 | 60 | 6.6 | 2016 | 355 |
| Cu(SP)(mnt) | N719 | 660 | 4.4 | 44 | 1.3 | 2005 | 370 |
| Cu(dmp)2 | N719 | 790 | 3.2 | 55 | 1.4 | 2005 | 370 |
| Cu(dmp)2 | C218 | 932 | 11.29 | 66 | 7.0 | 2011 | 346 |
| Cu(dmp)2 | LEG4 | 1020 | 12.6 | 62 | 8.3 | 2016 | 96 |
| Cu(dmp)2 | Y123 | 1060 | 13.61 | 69.2 | 10.3 | 2016 | 14 |
| Cu(dmp)2 | D5 | 1130 | 9.02 | 73.6 | 7.53 | 2018 | 95 |
| Cu(dmp)2 | D45 | 1020 | 9.90 | 74.1 | 7.48 | 2018 | 95 |
| Cu(dmp)2 | D35 | 1140 | 11.40 | 70.6 | 9.22 | 2018 | 95 |
| Cu(dmp)2 | G3 | 860 | 3.8 | 59 | 1.9 | 2016 | 351 |
| Cu(dmp)2 | D | 750 | 4.7 | 36 | 1.3 | 2018 | 371 |
| Cu(phen)2 | N719 | 570 | 0.48 | 43 | 0.12 | 2005 | 370 |
| Cu(bpye)2 | LEG4 | 904 | 13.8 | 71.8 | 9.0 | 2016 | 372 |
| Cu(bpye)2 | Y123 | 627 | 13.2 | 65 | 5.6 | 2020 | 352 |
| Cu(dmby)2 | Y123 | 1070 | 14.15 | 68.7 | 10.0 | 2016 | 14 |
| Cu(dmby)2 | D5 | 1070 | 9.85 | 71.2 | 7.53 | 2018 | 95 |
| Cu(dmby)2 | D45 | 956 | 11.85 | 68.0 | 7.71 | 2018 | 95 |
| Cu(dmby)2 | D35 | 1130 | 11.53 | 60.2 | 7.84 | 2018 | 95 |
| Cu(tmby)2 | Y123 | 1040 | 15.53 | 64.0 | 10.3 | 2016 | 14 |
| Cu(tmby)2 | D5 | 837 | 10.79 | 67.4 | 6.10 | 2018 | 95 |
| Cu(tmby)2 | D45 | 984 | 12.52 | 67.3 | 8.30 | 2018 | 95 |
| Cu(tmby)2 | D35 | 1110 | 12.81 | 66.1 | 9.44 | 2018 | 95 |
| Cu(tmby)2 | L348 | 1170 | 6.4 | 72.0 | 5.3 | 2018 | 274 |
| Cu(tmby)2 | L349 | 1160 | 11.0 | 71.7 | 9.2 | 2018 | 274 |
| Cu(tmby)2 | L350 | 1140 | 13.0 | 76.0 | 11.2 | 2018 | 274 |
| Cu(tmby)2 | L351 | 1060 | 11.2 | 76.3 | 9.1 | 2018 | 274 |
| Cu(tmby)2 | WS-70 | 1060 | 13.2 | 77 | 11.0 | 2018 | 273 |
| Cu(tmby)2 | WS-72 | 1100 | 13.3 | 78 | 11.6 | 2018 | 273 |
| Cu(tmby)2 | D35/XY1 | 1030 | 16.19 | 68 | 11.3 | 2017 | 348 |
| Cu(tmby)2 | Y123/XY1b | 1050 | 13.1 | 79 | 13.1 | 2018 | 320 |
| Cu(tmby)2 | XY1 | 1000 | 13.3 | 67 | 8.9 | 2020 | 26 |
| Cu(tmby)2 | L1 | 910 | 9.4 | 71 | 6.1 | 2020 | 26 |
| Cu(tmby)2 | XY1/L1 | 1080 | 15.9 | 67 | 11.5 | 2020 | 26 |
| Cu(eto)2 | D5 | 828 | 10.12 | 71.5 | 6.00 | 2018 | 95 |
| Cu(eto)2 | D45 | 978 | 12.59 | 66.7 | 8.21 | 2018 | 95 |
| Cu(eto)2 | D35 | 1120 | 11.93 | 66.3 | 8.84 | 2018 | 95 |
| Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2 | G3 | 720 | 9.3 | 66 | 4.4 | 2016 | 351 |
| Cu(2-n-butyl-1,10-phenanthroline)2 | D | 610 | 6.3 | 53 | 2.0 | 2018 | 371 |
| Cu(2-n-butyl-1,10-phenanthroline)2 | G3 | 860 | 10.1 | 66 | 5.7 | 2018 | 373 |
| Cu(2-n-butyl-1,10-phenanthroline)2 | G4 | 780 | 10.1 | 63 | 4.9 | 2018 | 373 |
| Cu(2-mesityl-1,10-phenanthroline)2 | G3 | 830 | 11.4 | 59 | 5.6 | 2018 | 373 |
| Cu(2-mesityl-1,10-phenanthroline)2 | G4 | 840 | 11.7 | 54 | 5.3 | 2018 | 373 |
| Cu(2-tolyl-1,10-phenanthroline)2 | G3 | 870 | 11.1 | 62 | 6.0 | 2018 | 373 |
| Cu(2-tolyl-1,10-phenanthroline)2 | G4 | 870 | 11.1 | 62 | 6.0 | 2018 | 373 |
| Cu(2-phenyl-1,10-phenanthroline)2 | G3 | 880 | 8.0 | 69 | 4.9 | 2018 | 373 |
| Cu(2-phenyl-1,10-phenanthroline)2 | G4 | 810 | 10.2 | 58 | 4.8 | 2018 | 373 |
| Cu(oxabpy) | Y123 | 920 | 9.75 | 69 | 6.2 | 2018 | 353 |
| Cu(1) | Y123 | 689 | 5.7 | 77 | 3.1 | 2020 | 352 |
| Cu(2) | Y123 | 693 | 10.2 | 72 | 4.7 | 2020 | 352 |
| Cu(3) | Y123 | 792 | 7.9 | 75 | 4.3 | 2020 | 352 |
| K4Ni[Fe(CN)6] | N3 | 790 | 8 | 70 | 4 | 2011 | 375 |
| Fe(bpy)3 | RR9 | 1420 | 2.8 | 47 | 1.9 | 2018 | 282 |
| Ferrocene | Carbz-PAHTDTT | 842 | 12.2 | 73 | 7.5 | 2011 | 374 |
| Me10Fc | Carbz-PAHTDTT | 437 | 6.6 | 40 | 1.1 | 2012 | 376 |
| Et2Fc | Carbz-PAHTDTT | 641 | 13.3 | 50 | 4.2 | 2012 | 376 |
| EtFc | Carbz-PAHTDTT | 669 | 12.8 | 56 | 4.8 | 2012 | 376 |
| BrFc | Carbz-PAHTDTT | 671 | 9.3 | 48 | 3.0 | 2012 | 376 |
| Br2Fc | Carbz-PAHTDTT | 599 | 4.4 | 46 | 1.2 | 2012 | 376 |
| Mn(acac)3 | K4 | 765 | 7.8 | 73 | 3.9 | 2014 | 377 |
| Mn(acac)3 | MK2 | 733 | 8.6 | 69 | 4.4 | 2014 | 377 |
| Mn(acac)3 | N719 | 771 | 7.9 | 73 | 4.4 | 2014 | 377 |
| Mn(CF2)3 | MK2 | 800 | 4.95 | 69 | 2.72 | 2016 | 378 |
| VO(salen) | D205/D131 | 740 | 12.3 | 59 | 5.4 | 2013 | 379 |
| VO(hybeb) | N719 | 660 | 5.2 | 58 | 2 | 2015 | 380 |
| T−/T2 | Z907 | 687 | 15.9 | 72 | 7.9 | 2012 | 357 |
| T−/T2 | N719 | 630 | 14.25 | 68 | 6.10 | 2012 | 381 |
| AT−/BAT | N719 | 670 | 13.76 | 68 | 6.27 | 2012 | 381 |
| ET−/BET | N719 | 632 | 9.3 | 71 | 4.2 | 2013 | 382 |
| TEMPO | D-149 | 830 | 9.4 | 70 | 5.4 | 2008 | 383 |
| TEMPO | LEG4 | 965 | 7.74 | 73 | 5.43 | 2015 | 356 |
| TEMPO | D205 | 880 | 9.88 | 75 | 6.5 | 2012 | 384 |
| TEMPO | D205/D131 | 780 | 13.5 | 66 | 7.0 | 2012 | 384 |
| AZA | D205 | 820 | 12.9 | 76 | 8.1 | 2012 | 384 |
| AZA | D205/D131 | 850 | 13.3 | 75 | 8.6 | 2012 | 384 |
| TMTU | D205 | 777 | 16.6 | 49 | 6.32 | 2013 | 385 |
| TMTU | D102 | 770 | 13.8 | 54 | 5.74 | 2013 | 385 |
| TMTU | D131 | 825 | 11.0 | 61 | 5.53 | 2013 | 385 |
| TMTU | N719 | 626 | 10.3 | 50 | 3.22 | 2013 | 385 |
| TMTU | Z907 | 642 | 8.3 | 53 | 2.82 | 2013 | 385 |
| HQ/BQ | N719 | 755 | 10.28 | 66.7 | 5.2 | 2013 | 386 |
| HQ/BQ | CM309 | 755 | 12.10 | 67.8 | 6.2 | 2013 | 386 |
| HQ/BQ | Y123 | 533 | 6.5 | 30 | 1.08 | 2018 | 387 |
| PhHQ/PhBQ | Y123 | 528 | 6.3 | 39 | 1.3 | 2018 | 387 |
| DTHQ/DTBQ | Y123 | 542 | 12.6 | 36 | 2.5 | 2018 | 387 |
| ThymHQ/ThymBQ | Y123 | 455 | 10 | 44 | 2.0 | 2018 | 387 |
4.3.1.1 Halide redox mediators. Initially, successful and efficient DSCs used the iodide/triiodide redox mediator.87,145,388,389 The I−/I3− redox couple shows remarkable performance up to its record PCE of 11.9% (certified, 12.4% non-certified).308,390 The I−/I3− redox couple fulfills several requirements for an ideal electrolyte and it was for several decades the benchmark for research and industry. Advantages of the I−/I3− redox couple include a suitable redox potential for many dyes, small molecular size for high diffusion, good solubility in a wide range of solvents at high concentration for high conductivity, and good stability. However, it also has several drawbacks, which have initiated the search for alternative redox mediators: (i) substantial light absorption of the triiodide and other possible polyiodide species in the 400–500 nm range of the solar spectrum, (ii) corrosivity towards several components of DSCs including the materials used for counter electrodes and sealing, (iii) possible iodine diffusion out of the electrolyte stemming from its high vapor pressure, and especially (iv) the very large driving force of over 0.5 V for dye regeneration due to the two-electron oxidation steps from I− to I3−. Consequently, the VOC attainable from a DSC containing the iodide/triiodide electrolyte is smaller than what is theoretically possible given the choice of dye. Since the overall efficiency of a solar cell scales directly with its VOC, this large driving force constitutes a significant limitation of the I−/I3− redox couple.388
The step towards iodide-free redox mediators begins with bromide/tribromide, which has a more positive potential of an additional 0.35 V, a two-electron transfer, and high solubility in many solvents. Thus, the electrolyte containing the bromide/tribromide redox system can lead to an increased photovoltage, but at the cost of lower JSC values. Hanaya and co-workers successfully implemented the Br−/Br3− electrolyte with the organic dye ADEKA-3 and a Mg2+-doped anatase TiO2 electrode, reaching a photovoltage over 1.4 V and a conversion efficiency close to 4%.216 The development bottleneck for the Br−/Br3−-based electrolyte remains the search for a suitable dye. Bi-Interhalogen redox systems, such as I−/IBr2− and I−/I2Br− were also tested in combination with ruthenium-based sensitizing dyes and reached conversion efficiencies up to 6.4%.362,364
Furthermore, pseudohalogen-based redox couples SCN−/(SCN)2 and SeCN−/(SeCN)2 have been studied with the hope to enhance VOC in DSCs, because their redox potentials are 0.19 and 0.43 V more positive than that of the I−/I3− redox couple, respectively. However, since dye regeneration efficiency with these systems is low, it only resulted in low photocurrents. SeCN− has ambivalent reactivity and can interact with the dye from the Se and N side.391
4.3.1.2 Transition metal coordination complexes. Cobalt-, iron-, copper-, nickel-, manganese- and vanadium-based complexes as one-electron outer-sphere redox couples are currently the most promising and successful candidates to replace the I−/I3− system in DSCs.11 Their characteristics are suitable for the commercialization of DSCs because they have reversible electrochemical properties, structural tunability, and more positive Fermi level values, reduced visible light absorption and superior stability compared to I−/I3−. Metal complexes' electronic properties and redox chemistry can be readily adjusted by altering the central metal cation or, most importantly, the ligands. Marcus theory states that a driving force of 0.2 eV is adequate for outer-sphere single-electron-transfer processes to guarantee a rapid dye regeneration rate, leading to VOC improvements.95,166 The development of novel redox mediators has attracted less interest than that of sensitizing dyes or other materials for different DSC components, but recent developments have renewed the attention to this aspect of DSCs.392
Cobalt coordination complexes. Tridentate (e.g. terpyridines) and bidentate (e.g. bipyridines and phenantrolines) ligands often form octahedral coordination complexes in the most common Co-based redox mediators.166,365,367,393 In 2010, the Hagfeldt group achieved the first successes in high-efficiency DSCs integrating transition metal complexes by combining a novel Co complex-based electrolyte with the organic dye D35. By introducing a succession of complexes with different ligands, the scientists developed a library of redox mediators with a diversity of redox characteristics.270 The initially achieved efficiency of 7% under 1 sun (VOC of 0.92 V and JSC of 10.7 mA cm−2) was reached with the [Co(bpy)3]3+/2+ redox couple (Fig. 27). In 2012, Mosconi et al. were able to show that the formation of an ion pair between the negatively-charged Ru dye and the positively-charged Co complex was responsible for the increase in recombination processes and consequent poor performance of DSCs implementing these systems. This was improved later with addition of larger blocking groups on the Ru dyes.165
 | ||
| Fig. 27 Chemical structures of cobalt coordination complexes-based redox mediators implemented in DSCs. | ||
A follow-up study by Feldt et al. on fundamental aspects of the regeneration and recombination processes of cobalt redox mediators also confirmed that a driving force of 0.25 eV was sufficient to ensure 84% dye regeneration.166,343 The introduction of this new redox mediator system led to a surge in dye development. In 2011 Tsao et al. increased the efficiency with the organic dye Y123, which had a high extinction coefficient thanks to the cyclopentadithiophene (CPDT) π-bridge. DSCs reaching a PCE of 8.8% (VOC = 0.855 V, JSC = 14.6 mA cm−2) under 1 sun were obtained in conjunction with a platinized FTO counter electrode.271 A new family of porphyrin-based dyes was introduced by Yella et al., YD2 and YD2-o-C8, leading to an impressive PCE of 11.9% under full sun (VOC = 0.965 V, JSC = 17.3 mA cm−2).284
The PCE mark of 13% was passed by Mathew et al. with porphyrins improved through a triphenylamine-type hydrophobic donor, leading to dyes SM315 and SM371.286 The highest efficiency reported for DSCs to date is still that obtained with the [Co(phen)3]3+/2+ redox mediator by Kakiage et al., who reached a PCE of 14.3% under full sun (VOC = 1.01 V, JSC = 18.2 mA cm−2) by cosensitizing the ADEKA-1 (MK2 dye variant with an alkoxysilyl binding group) and LEG4 dyes.24 A series of 2,2′-ethylenebis(nitrolomethylidene)diphenol-N,N′-ethylenebis(salicylimine) (salen)-based cobalt complexes was introduced by Nasr-Esfahani et al. in 2014 and reached a PCE of only 2.53% under full sun illumination.369 New complexes were developed by Koussi-Daoud et al. with a cobalt coordination complex Co(EtPy) 2 featuring a terpyridine functionalized with 3,4-ethylenedioxythiophene (EDOT).368 This combination of an electron cascade to the PEDOT counter electrode lead to an enhanced cell efficiency of 5.1% with D35 at 1 sun. The group of U. Bach also introduced new cobalt-based redox mediators with 4-tert-butylpyridine (tBP) and N-methylbenzimidazole (NMBI). The tested complexes [Co(PY5Me2)(tBP)]3+/2+, [Co(PY5Me2)(NMBI)]3+/2+ and [Co(PY5Me2)(MeCN)]3+/2+ reached an efficiency of 8.4% under full sun (VOC = 0.94 V, JSC = 11.8 mA cm−2).337 They further introduced a hexadendate ligand in 2015 to increase the overall stability of cobalt redox mediators. Devices fabricated with this new Co complex, and MK2 or Y123 as dye produced a PCE up to 8.3% under full sun.366,394 In 2016, Freitag et al. introduced the new supramolecular, hemicage cobalt-based mediator [Co(ttb)]3+/2+ with the highly pre-organized hexadentate ligand 5,5′′,5′′′′-((2,4,6-triethyl benzene-1,3,5-triyl) tris(ethane-2,1-diyl))tri-2,2′-bipyridine (ttb) reaching the same performance as with [Co(bpy)3]3+/2+ (bpy = 2,2′-bipyridine) redox mediator and the LEG4 dye.355 Both hexadendate systems exhibit exceptional stability under thermal and light stress.
The addition of aqueous electrolytes aided in the advancement of stabilization and sustainability, and also required the development and use of appropriate hydrophilic dyes. The combination of MK2 and [Co(bpy)3]3+/2+ was utilized by Xiang and colleagues in 2013.395 They eventually achieved aqueous-based devices with a PCE of 5.0% at 1 sun illumination (VOC = 0.687 V, JSC = 9.8 mA cm−2). Dong et al. used the common strategy of introducing surfactants in DSCs and reached a PCE of 5.6% under full sun (VOC = 0.821 V, JSC = 10.17 mA cm−2) with the MK2 dye.396 In 2016, Ellis et al. introduced two complexes with high solubility in water, [Co(bpy)3](NO3)2 and [Co(phen)3]Cl2, and the new dye D51, with a shorter blocking group to allow better wetting in comparison to the organic dye D35. The initial performance reported was 1.4% and 3.4%, respectively, both under 1000 W m−2 illumination.397 In the same study, optimization of [Co(phen)3]Cl3 concentration allowed further performance enhancements to 4.8% and the use of [Co(bpy-pz)3]3]3+/2+ featuring chloride counter ions lead to a 5.5% PCE (VOC = 0.9 V, JSC = 8.1 mA cm−2) under full sun.397
For what concerns DSC operation in ambient light conditions, Venkatesan et al. used the Co(bpy)3 electrolyte in devices sensitized with different dyes.398 The best results were achieved with the Y123 dye, which yielded a PCE of 24.5% at 1000 lx light intensity.
Some disadvantages of cobalt complexes remain. They have a large molecular size leading to slow mass transport and diffusion, large reorganization energies between the oxidation states Co(II) and Co(III) increase the overall energy required to regenerate the dye, and their long-term stability is in question as the complexes in solution will likely undergo ligand exchange, which has to be structurally controlled.
Copper coordination complexes. As alternative redox mediators, Cu2+/+ complexes outperform both iodine- and Co-based electrolytes in combination with various dyes, which was made possible due to lower reorganization energy and minimized overpotential losses.370,399
The significant variations in coordination complex geometries between Cu(I) and Cu(II) species, four-coordinate with tetrahedral geometry vs. four- to six-coordinate (square planar to tetragonal) geometry were anticipated to result in high reorganization energies. However, successful copper coordination complexes used in DSCs were developed by using sterically-hindered ligands to minimize the reorganization energy.
Hattori et al. achieved a maximum PCE of 1.4% for the first time using bis(2,9-dimethyl-1,10-phenantroline)copper(II/I) complexes([Cu(dmp)2]2+/+), Fig. 28.370 This result was later improved by Bai et al.,346 who reached 7% PCE with the C218 organic dye followed by Freitag et al. in 2016, who achieved 8.3% PCE using the D–π–A LEG4 organic dye with a rather high open-circuit voltage of over 1.0 V. Freitag also discovered that the [Cu(dmp)2]2+/+ complex (redox potential of 0.93 V vs. NHE) can achieve good regeneration of the oxidized dye molecules with a driving force as small as 0.14 eV, thus minimizing internal energy losses.96 Cong et al. synthesised a novel Cu mediator – [Cu(bpye)2]2+/+ – featuring the 1,1-bis(2-pyridyl)ethane ligand. A PCE of 9.0% (VOC = 0.90 V, JSC = 14.1 mA cm−2) was achieved, which however declined to 6% after a short light ageing period.372 In 2017, Freitag and co-workers introduced two new redox couples based on Cu bipyridyl complexes, [Cu(dmby)2]2+/+ (0.97 V vs. NHE, dmby = 6,6′-dimethyl-2,2′-bipyridine) and [Cu(tmby)2]2+/+ (0.87 V vs. NHE, tmby = 4,4′,6,6′-tetramethyl-2,2′-bipyridine), which showed efficient organic Y123 dye regeneration at very low driving forces of 0.1 eV.14 The efficiency exceeded 10% under 1000 W m−2 AM1.5G illumination. In their follow-up work Saygili et al. examined the regeneration behavior and recombination processes of [Cu(dmby)2]2+/+, [Cu(tmby)2]2+/+, [Cu(eto)2]2+/+ (eto = 4-ethoxy-6,6′-dimethyl-2,2′-bipyridine), and [Cu(dmp)2]2+/+ in conjunction with organic dyes having various degrees of blocking groups: D5, D35, and D45.95 Their results indicated that DSCs with a combination of D35 and [Cu(dmp)2]2+/+ achieved a very high VOC of 1.14 V without a decrease in JSC. Moreover, with a dye lacking recombination-preventing steric units such as D5, VOC values as high as 1.13 V were possible with [Cu(dmp)2]2+/+ and [Cu(dmby)2]2+/+ electrolytes. Liu et al. introduced a series of indacenodithiophene (IDT)-based D–π–A organic dyes reaching high open-circuit voltage values (>1.1 V) and PCE values of 11.2% at 1 sun.274 Zhang et al. also employed [Cu(tmby)2]2+/+ in conjunction with the novel WS-72 dye, which reduced interfacial electron recombination.
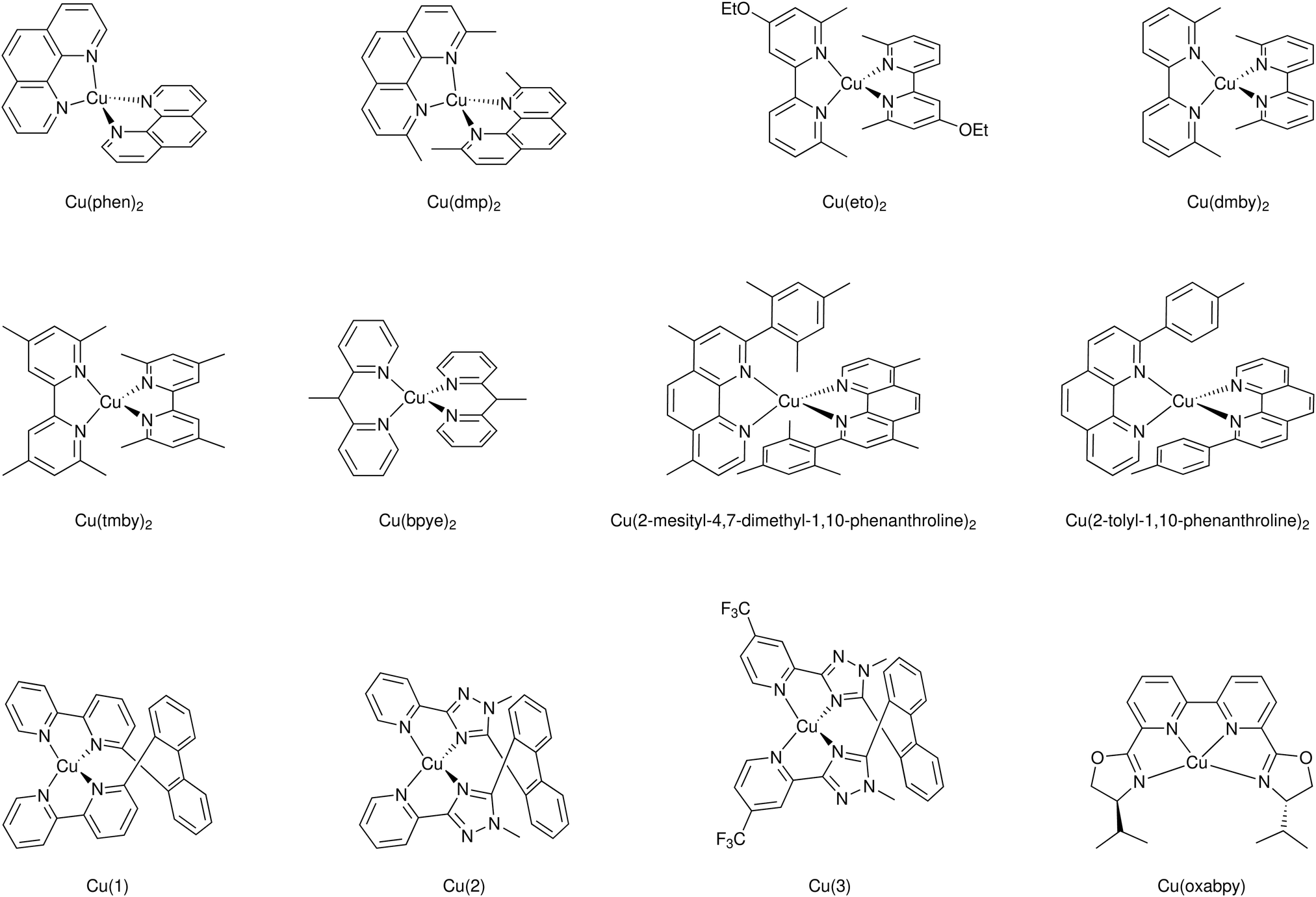 | ||
| Fig. 28 Chemical structures of copper coordination complexes-based redox mediators implemented in DSCs. | ||
Liquid-junction devices generated a notable VOC of 1.1 V together with a PCE of 11.6% under simulated AM1.5G illumination. After drying the liquid electrolyte to create solid-state devices, the PCE increased to 11.7% (JSC = 13.8 mA cm−2, VOC = 1.07 V and FF = 79%).273
In 2017, the field of DSCs experienced a significant push towards indoor applications. Indoor illumination is very different to sun illumination, with an emission spectrum only in the visible and light intensities that are two to three orders of magnitude lower. With high power conversion efficiencies of indoor photovoltaics, the power output obtained under low light illumination is sufficient to power a range of wireless devices belonging to the family of Internet of Things (IoT). Freitag et al. developed a cosensitized DSC with D35 and XY1 dyes employing the [Cu(tmby)2]2+/+ redox couple. The reported PCE was 11.3% at 1 sun and 28.9% at 1000 lx (of a fluorescent light tube).348 A record PCE of 13.1% at full sun (and 32% at 1000 lx) was obtained by Cao et al. using a XY1 and Y123 dye mixture in conjunction with the [Cu(tmby)2]2+/+ redox mediator.320 In 2020, Michaels et al. presented co-sensitized DSCs, where the small organic dye L1 was combined with the XY1 dye to provide VOC and performance values of 910 mV and 34.0%, respectively, at 1000 lx (11.5% at 1 sun). These DSCs were able to power IoT devices capable of machine learning under ambient light.26 The current record of DSC efficiency in ambient light, with a PCE of 34.5% at 1000 lx, belongs to Zhang et al. with devices featuring a MS5/XY1b co-sensitized photoanode and the [Cu(tmby)2]2+/+ redox couple.12
Phenathroline complexes were further developed by Magni et al. They compared [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]2+/+ with [Cu(dmp)2]+ and its oxidized form [Cu(dmp)2Cl]+, which is penta-coordinated. They achieved a maximum 4.4% PCE when coupling these electrolytes with the π-extended benzothiadiazole dye G3. They also analyzed the differences in the steric hindrance effect caused by either the methyl groups in [Cu(dmp)2]+ or the two mesityl rings of [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2](PF6)2, proposing that the latter cause a smaller conformational modification upon oxidation/reduction compared to the former, acting as a “kiss-lock enclosure” that leads to a more negative redox potential.351,400
Colombo et al. developed novel [Cu(2-mesityl-4,7-dimethyl-1,10-phenanthroline)2]PF6 and [Cu(2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline)2]PF6 redox couples with a Fe(II) co-mediator for DSC applications400 and later introduced a series of Cu complexes with different substituents in the α-positions of phenanthroline, with appropriate redox potentials and a distorted tetragonal geometry.401 Dragonetti et al. studied a heteroleptic Cu dye with [Cu(2-n-butyl-1,10-phenanthroline)2]2+/+ and [Cu(dmp)2]2+/+ redox couples. [Cu(dmp)2]2+/+ devices yielded lower photocurrents compared to those based on [Cu(2-n-butyl-1,10-phenanthroline)2]2+/+ due to a higher extinction coefficient of the former, result in agreement with reduced IPCE values at 475 nm when the dmp-based electrolyte was employed.371 [Cu(2-n-butyl-1,10-phenanthroline)2]2+/+ with the new Cu-based dye D achieved the highest PCE of 2% (JSC = 6.3 mA cm−2, VOC = 0.61 V and FF = 0.53). The [Cu(2-mesityl-1,10-phenanthroline)2]2+/+ shuttle produced the best PCE of 3.7% under full sun (JSC = 5.9 mA cm−2, VOC = 0.81 V and FF = 0.77).402 Benazzi et al. developed homoleptic Cu complexes redox couples with low molar absorption coefficient with substituted 1,10-phenanthrolines ([Cu(2-tolyl-1,10-phenanthroline)2]2+/+, [Cu(2-phenyl-1,10-phenanthroline)2]2+/+, and [Cu(2-n-butyl-1,10-phenanthroline)2]2+/+.373
Another polypyridyl complex was presented by Hoffeditz et al., a Cu redox shuttle with the 1,8-bis(2′-pyridyl)-3,6-dithiaoctane (PDTO) ligand. This work showed the ligand exchange processes in the electrolyte upon oxidation from Cu(I) to Cu(II) with the common additive tBP.403 The impact of tBP substitution on Cu(II) species of complexes with bidentate ligands was also studied by Wang et al., who found that tBP replaces the original ligand to form the [Cu(tBP)4]2+ species, which is a poor electron acceptor, leading to high voltages and charge collection efficiencies.404 Heteroleptic Cu(I)-based dyes were investigated by Karpacheva et al. together with homoleptic Cu(II/I) redox couples with a maximum efficiency of 2.06%. The researchers introduced electron-donating methoxy groups in Cu(4,4′-dimethoxy-6,6′-dimethyl-2,2′-bipyridine)2 to decrease the oxidation potential compared to Cu(dmby)2. The performance improvement with the former electrolyte was obtained thanks to a significant JSC increase and despite a decrease in VOC.405
Michaels et al. introduced new copper complexes redox mediators with the tetradentate ligand 6,6′-bis(4-(S)-isopropyl-2-oxazolinyl)-2,2′-bipyridine – [Cu(oxabpy)]2+/+. The ligand allowed to lock the complex in a square-planar geometry, leading to minimized reorganization energies. The gel-like [Cu(oxabpy)]2+/+ complexes showed considerable enhancement of charge transport performance.353 In 2020 Rodrigues et al. introduced a series of three copper redox shuttles with pre-organized tetradentate ligands, which were tested computationally, electrochemically, and in solar cell devices for performance. The rigid tetradentate ligand design achieved a high JSC (14.1 mA cm−2) and more effective electron transfer reactions, which led to an improved VOC value for one of the copper redox shuttle-based devices.352
Iron coordination complexes. An electrolyte based on iron complexes is of high interest as it would represent a sustainable, low cost and non toxic option. In 2012 Daeneke et al. introduced an aqueous hexacyanoferrate electrolyte for DSC. With a high-extinction-coefficient organic dye, MK2, the assembled solar cells reached VOC = 0.761 V, JSC = 7.21 mA cm−2, FF = 75% and PCE = 4.1%.374 Previously, in 2011 Rutkowska et al. successfully implemented a polynuclear electronically/ionically (redox) conducting mixed-valent inorganic material such as nickel(II) hexacyanoferrate(III/II) – ([NiFe(CN)6]2−/− – with a redox potential of approximately 0.84 V vs. NHE, resulting in DSCs of VOC = 0.790 V, JSC = 8 mA cm−2, FF = 70% and PCE = 4%.375
The bipyridine equivalent to cobalt complexes – [Fe(bpy)3]3+/2+ (Fig. 29) – has weaker Fe(II)–N bonds than Co(II)–N, resulting in a high redox potential of 1.37 V. Furthermore, the [Fe(bpy)3]3+/2+ redox couple is well known to be a stable, highly reversible redox system. The RR9 dye, with a low HOMO energy level, was designed to match the redox potential of [Fe(bpy)3]3+/2+ by Delcamp and co-workers. With a driving force of 0.19 eV, the DSCs reached a record VOC of over 1.4 V and a PCE of 1.9%.282
 | ||
| Fig. 29 Chemical structures of iron coordination complexes-based redox mediators implemented in DSCs. | ||
The one-electron, outer-sphere iron-based redox couple ferrocenium/ferrocene (Fc+/Fc) has been extensively investigated in the DSC field thanks to its favourable kinetic properties and to its more positive redox potential, faster electron exchange and lower toxicity in comparison to the iodide/triiodide redox couple. Initial results showed that “plain” Fc+/Fc does not perform well in DSCs, due to high recombination of electrons from both the TiO2 layer and the substrate. Surface passivation, which included spray pyrolysis, atomic layer deposition (ALD), and silane treatment, was used to inhibit recombination. In a subsequent study, Daeneke et al. worked on reducing mass-transport limitations for electrolytes based on the Fc+/Fc redox couple, and addressed recombination issues by depositing thinner layers of TiO2 (18 nm blocking layer, 2.2 μm mesoporous layer and 4.4 μm scattering layer); tBP was also introduced in the electrolyte solution to further passivate the titania surface. Their devices were complemented by the Carbz-PAHTDTT organic dye and by a Pt counter electrode. Such devices performed better (VOC = 0.842 V, JSC = 12.2 mA cm−2, FF = 73%, and PCE = 7.5%) than reference DSCs (VOC = 0.735 V, JSC = 13.3 mA cm−2, FF = 62%, and PCE = 6.1%) and represent the best-performing cells based on the Fc electrolyte to date.374,376
Nickel coordination complexes. Nickel bipyridyl complexes have been tested in battery applications, where they can provide potentials in excess of 2.25 V, with very stable and pseudo-reversible anodic and cathodic half-cell reactions.406,407 For example Ni-bis(dicarbollide), which is comprised of two deboronated (nido-2) o-carborane ligands with η5 coordination, can perform several redox processes with net charges of −2, −1, and 0, corresponding to II–IV oxidation states of the Ni center (Fig. 30). Ni(IV/III) bis(dicarbollide) complexes were used by Li et al. in DSCs, where they provided fast charge transport and a non-corrosive environment. Structural modification of the dicarbollide moiety at the B(9/12) positions with either electron donating or electron withdrawing groups allowed the creation of a class of ligands with different properties. These Ni(IV/III)-dicarbollide mediators however had high reorganization energies during redox processes, which were due to a required cis-to-trans conformational rotation upon electron transfer and lead to low electron exchange rates.408 Spokoyny et al. created a series of redox mediators ranging in redox potentials from 0.37 V to 0.55 V vs. NHE and the highest VOC was obtained for the 3,5-bis(trifluoromethyl)phenyl group, with VOC = 0.850 V; PCEs were in the range between 0.7% and 2%. In DSCs with the N719 photosensitizer, the Ni redox couple with potential 0.77 V vs. NHE rendered a 1.5% efficiency, which was further improved up to JSC = 6.3 mA cm−2 by modifying the photoanode with a nanoparticle-and-aerogel framework possessing a high surface area (13.6 μm thickness), which allowed to reach a PCE of 2.1% (VOC = 0.628 V, JSC = 5.3 mA cm−2, FF = 60%). Further investigations were focused on modification of Ni complexes and the search for an appropriate sensitizer to match this kind of redox couples.406
 | ||
| Fig. 30 Starting with commercially available o-carborane, a five-step, high-yield synthetic strategy is used to create bis(dicarbollide) species from B(9)-functionalized derivatives of the parent carborane. Reprinted with permission from ref. 406. Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Manganese coordination complexes. The search for more sustainable and less toxic redox mediators based on coordination complexes for DSCs was extended to Mn(IV/III) complexes. Manganese can be considered an interesting one-electron outer-sphere redox shuttle candidate because of its variety of accessible redox states (from +2 to +7), low toxicity and abundance. Ideally, the oxidized redox mediator species Mnox, present at the TiO2 surface, should not significantly reduce the lifetime of TiO2 conduction band electrons before Mnox diffuses to the counter electrode. The undesired recombination reaction between electrons at the TiO2 surface and Mnox limits charge collection, as with the ferrocene/ferrocenium couple, and constrains the choice of alternative mediators, which require surface passivation. Some Mn(III) complexes are known to undergo a spin change upon reduction (d4 to d5) that can slow down the undesired recombination.
The first example of application was reported in 2014 by Spiccia et al., who focused on DSCs containing the commercially available [Mn(acac)3]+/0 (acac = acetylacetonate, Fig. 31) with a redox potential of 0.49 V vs. NHE and the MK2 dye, reporting an energy conversion efficiency of 4.4% under AM1.5G, 100 mW cm−2 conditions.377 Carli et al. followed up by developing the derivatives [Mn(CF2)3] (CF2 = 4,4-difluoro-1-phenylbutanate-1,3-dione) and [Mn(DBM)3] (DBM = dibenzoylmethanate).378 This series showed redox potentials in the range between 0.41 V and 0.69 V vs. NHE for [Mn(CF2)3]3+/2+ and [Mn(DBM)3]3+/2+.
 | ||
| Fig. 31 Chemical structures of manganese coordination complexes-based redox mediators implemented in DSCs. | ||
Vanadium coordination complexes. Fundamental electrochemical research on the kinetics and mechanisms of vanadium(V/IV) redox couple reactions in a range of electrolytes, especially for redox flow batteries, is ongoing. For DSCs, in 2013 Nishide and co-workers featured an electrochemically-reversible and fast redox mediator VO(salen) (salen = N,N′-ethylene-bis(salicylideneiminate)), Fig. 32, reaching a conversion efficiency of 5.4% (VOC = 0.74 V and JSC = 12.3 mA cm−2) in a co-sensitized DSC with D205/D131.379 In 2015 Apostolopoulou et al. introduced the oxidovanadium(IV) reversible redox couple [VO(hybeb)]2−/− (where hybeb4− is a tetradentate diaminodiphenolate ligand) with a very low redox potential of −0.047 V vs. NHE. The electrolyte was tested in DSCs with the N719 dye reaching a performance of 2% (VOC = 0.66 V, JSC = 5.2 mA cm−2) under 1 sun illumination.380
 | ||
| Fig. 32 Chemical structures of vanadium coordination complexes-based redox mediators implemented in DSCs. | ||
4.3.1.3 Small organic molecules. Various organic redox active molecules such as TEMPO+/TEMPO, AZA (2-azaadamantan-N-oxyl) Quinone or T−/T2 (T for 1-methyl-1-H-tetrazole-5-thiolate, T2 for the dimer) were tried to circumvent the limitations that still exist with coordination complex redox couples, including inefficient dye regeneration, mass transport limitations of large metal complexes or high electron recombination with the fast outer-sphere redox systems (Fig. 33).
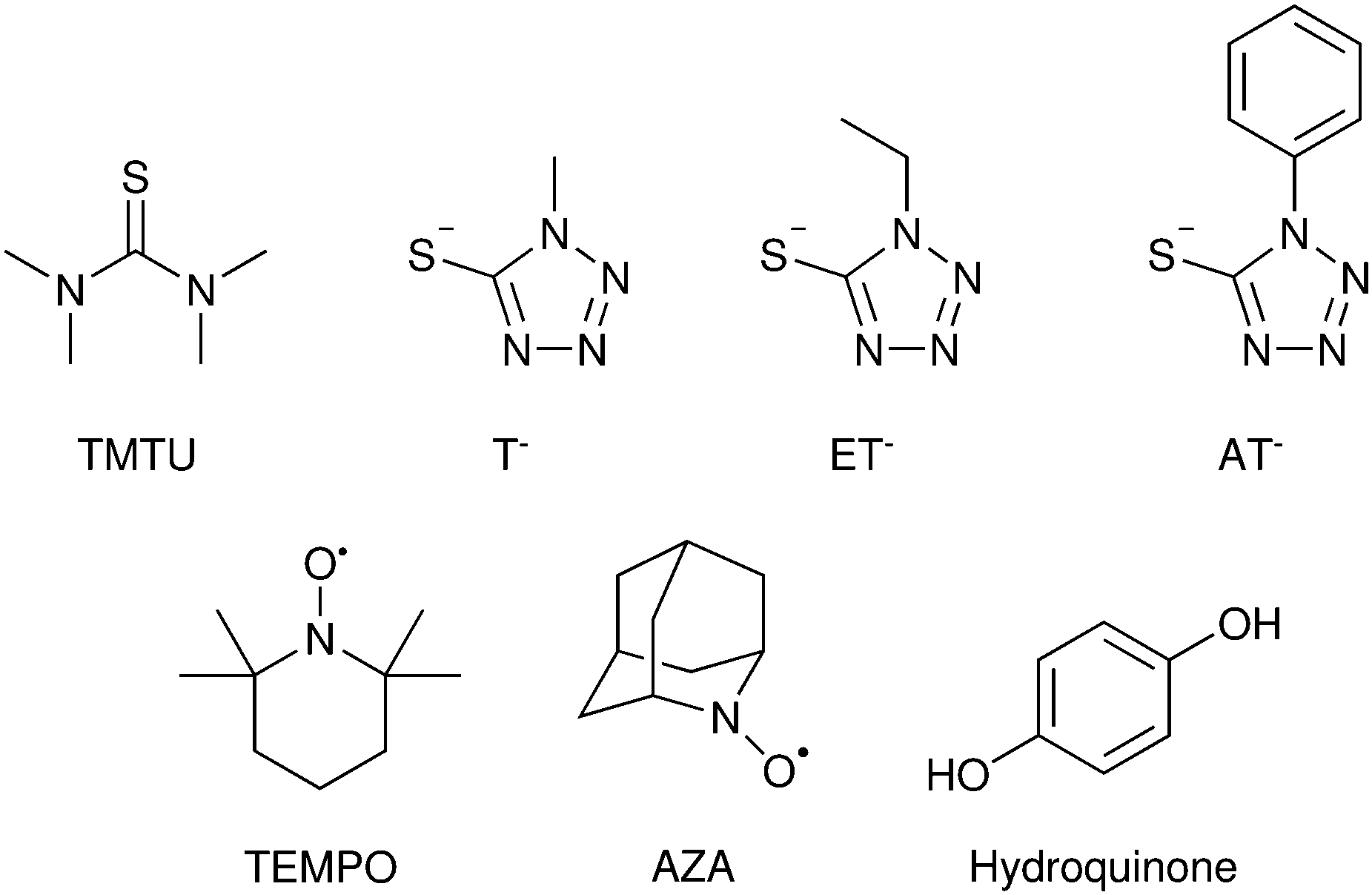 | ||
| Fig. 33 Chemical structures of small organic molecules-based redox mediators implemented in DSCs. | ||
In 2012 Burschka et al. reached a power conversion efficiency of 7.9% with a DSC based on the T−/T2 redox couple together with a PEDOT counter electrode.357 In the same year, Li et al. introduced a new thiolate/disulfide redox couple AT−/BAT,381 an analogue to T−/T2 with more positive redox potential and slower charge recombination reaching promising efficiencies of 6.07%. A year later, supramolecular lithium cation assemblies of crown ether were been used to replace conventional tetraalkylammonium counter-ions in thiolate/disulfide (ET−/BET)-mediated dye-sensitized solar cells, which exhibited high stability and efficiency of 6.61% under 1 sun illumination.382
The redox-active TEMPO was successfully implemented into DSCs as a redox mediator by Grätzel et al. and it improved the VOC over the I−/I3− electrolyte.383 Nitroxide derivatives were also studied as DSC mediators by other groups. However, the VOC was enhanced to the detriment of the cell's short-circuit current density.356,409
Another organic radical – 2-azaadamantan-N-oxyl (AZA) – was used as a stable and highly reactive redox mediator in a DSC. AZA exhibited both an appropriate redox potential and significantly high values of diffusivity, heterogeneous electron-transfer rate, and electron self-exchange reaction rate. These properties gave rise to an enhanced electron-transfer mediation, which lead to a high fill factor and thus excellent photovoltaic performance to achieve a conversion efficiency of 8.6%.384
Liu et al. developed indoline- and ruthenium-based dye-sensitized solar cells with the organic redox couple tetramethylthiourea/tetramethylformaminium disulfide (TMTU/TMFDS2+). This redox couple worked best with the indoline dye D205, reaching a power conversion efficiency of 7.6% under AM1.5G 1 sun illumination. TMTU provided efficient charge collection and injection in all studied devices; however, while regeneration of indoline dyes was also very effective, the regeneration of ruthenium dyes was less so, leading to the decreased performance.385
The hydroquinone/benzoquinone (HQ/BQ) redox pair has increased interest in research as the electron transfer of the redox couple is a thermodynamically reversible process.387 In previous reports, the anionic hydroquinone species (TMAHQ/BQ) was used as a redox mediator in DSCs with the N719 dye as sensitizer and Pt as CE; these systems showed promising photovoltaic characteristics (VOC = 75 5 mV, JSC = 1 0.28 mA cm−2, FF = 66.7%, and PCE = 5.2 %). With the same redox mediator but with PEDOT as counter electrode and the organic dye CM-309, the following parameters were achieved: VOC = 755 mV, JSC = 12.10 mA cm−2, FF 67.8%, and PCE = 6.2%.386
4.3.1.4 Ionic liquids. The use of liquid electrolytes demands perfect sealing of the device to avoid leakage and evaporation of the solvents. To eliminate electrolyte leakage issues in traditional DSCs (i.e. cells with organic solvent-based electrolytes), ionic liquids are used as the electrolyte to improve cell durability. An ionic liquid (IL) is defined as a salt that is liquid at the operational temperature. From a DSC point of view, these molten salts can be described as electrolytes comprised solely of ions.333,410–412 Technically, the difference between ionic liquids and molten salts is given by the melting temperature and some physical characteristics: the former melt below 100 °C and present relatively low viscosity, while the latter melt at high temperatures and are more viscous. When the melting temperature is below 25 °C, we talk about room temperature ionic liquids (RTILs). Ionic liquids (Fig. 34) have found large use as electrolytes in DSCs thanks to the fact that they are chemically and thermally stable, that their viscosity can be adjusted as needed, that they are mostly non-flammable, that they possess high ionic conductivity, and that they are non-reactive in a large range of potentials. From a stability point of view, it is crucial that they have very low vapor pressure, which mitigates evaporation and leaking issues in devices. ILs can play two different roles within DSC electrolytes: they can act as solvents in fully liquid devices, and as organic salts in quasi-solid-state devices. These properties have made ILs a sustainable solution to the problematic use of organic solvents, and ILs with different substituents and ions were prepared and used as redox mediators in dye-sensitized solar cells.
 | ||
| Fig. 34 Examples of cations and anions used in ionic liquids. | ||
Best performances with ILs were reached with imidazolium-based ionic electrolytes. Other IL cations employed are sulfonium, guanidinium, ammonium, pyridinium, or phosphonium, which were also tested as solvent-free electrolytes. The limitations in low diffusion and charge mobility of ILs in comparison to redox mediators in organic solvents remain. Several strategies were employed to improve the mass transport limitations by diluting the ionic liquid with organic solvents, compromising the system with the high volatility of organic solvents. Even in ILs with particularly low viscosity such as imidazolium dicyanamide, the diffusion of the triiodide anion is troublesome at low temperature, while efficiency at high temperature is limited by recombination reactions. An example of low-viscosity electrolytes is represented by the mixture of EMImSCN and PMImI ILs. The diffusion coefficient of triiodide in such electrolyte was 2.95 × 10−7 cm2 s−1, a value 1.6 times higher compared to an electrolyte comprised of PMImI only. DSCs fabricated with this mixed electrolyte in conjunction with the Z907 dye reached a PCE of 7%. ILs' potential advantage over organic solvents remains to be proved, while it is necessary to overcome the main drawbacks of high viscosity and low ion mobility.413
4.3.1.5 Quasi-solid and solid polymer electrolytes. Depending on fabrication strategies, the inclusion of polymers can lead to either quasi-solid (gel) or solid electrolytes. In the former case, the polymer acts as a host matrix for a liquid electrolyte, and it swells to accommodate the liquid inside, forming a gelatinous material that prevents solvent leakage. In the latter case, the redox active components of the charge transport layer are embedded directly within the polymeric structure, without the presence of a solvent.
Gels and quasi-solid polymers. Gel polymer electrolytes (GPEs) are designed to swell and host a liquid electrolyte in the order of tens to hundreds of times their own weight. They can infiltrate and create a contact with the photoanode very effectively in order to ensure fast dye regeneration and, at the same time, possess high conductivity, which leads to quick transport of charges towards the counter electrode.333,338,412,414–420 Polyacrylonitrile (PAN), poly(ethylene oxide) (PEO) derivatives, conducting polymers includ ing polypyrrole (PPy), polyaniline (PAni) and other polymers are the typical host materials (Fig. 35). Dimethyl carbonate (DMC), propylene carbonate (PC) and ethylene carbonate (EC) can be used as organic plasticizers with a large variety of polar solvents, ionic liquids and salts.421,422 A good portion of GPE work in DSCs can be credited to Bella and co-workers, as they showed long-term stability and efficiency of gel electrolytes. The specific approach to create an in situ electrolyte comprises the expansion of a monomer – bisphenol-A-ethoxylate dimethacrylate (BEMA) or poly(ethylene glycol) methyl ether methacrylate (PEGMA) – as well as a photoinitiator into the electrolyte and UV exposure of the assembled solar cell. To prove long-term stability, a DSC fabricated using this method with the LEG4 dye and an electrolyte containing the [Co(bpy)3]3+/2+ redox mediator was first placed in the dark at 60 °C for 1500 h and then subsequently exposed to full sun irradiation for 300 h at 40 °C. At the end of the ageing test the device (initial PCE of 6%) retained 90% of its initial performance.359,423–425 Using polyethylene glycol diacrylate (PEGDA) and PEGMA as copolymers, power conversion efficiencies of up to 4.41% (Table 5) were recorded.426 After inserting fillers based on metal organic frameworks (MOFs) or micro-fibrillated cellulose (MFC) into BEMA or PEGDA and PEGMA polymer blends, a dramatic increase in PCE (up to 7.03%) was observed.340,427,428
 | ||
| Fig. 35 Chemical structures of polymer electrolytes used in DSCs. | ||
| Matrix/polymer | Salt | Sensitizer | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| PPVII | None | N719 | 637 | 13.61 | 71 | 6.18 | 2014 | 412 |
| MPII:SiO2 | I2, NMBI | Z907 | 700 | 13.67 | 73.1 | 7.0 | 2003 | 414 |
| PVP | I2, KI, BMII | N3 | 626 | 15.72 | 55 | 5.41 | 2010 | 415 |
| PVDF-HFP | I2, NMBI, DMPII | Z907 | 730 | 12.5 | 67 | 6.1 | 2003 | 416 |
| BEMA:PEGMA | Co(bpy)3 | LEG4 | 880 | 10.5 | 68 | 6.4 | 2015 | 417 |
| BEMA:PEGMA | I2, NaI | N719 | 580 | 16.0 | 58 | 5.41 | 2013 | 424 |
| PAN-VA | I2, LiI, tBP, DMPII | CYC-B11 | 743 | 18.8 | 76 | 10.58 | 2013 | 418 |
| SGT-626 | I2, LiI, tBP, DMPII | N719 | 764 | 17.55 | 72.53 | 9.72 | 2020 | 419 |
| SGT-612 | I2, LiI, tBP, DMPII | N719 | 782 | 15.27 | 76.6 | 9.1 | 2021 | 420 |
| P(EO-EPI) | I2, LiI | N3 | 670 | 9.1 | 54 | 3.3 | 2008 | 421 |
| Gelator 1 | I2, LiI, DMPII | N719 | 670 | 12.8 | 67 | 5.91 | 2001 | 422 |
| Gelator 2 | I2, LiI, DMPII | N719 | 632 | 11.1 | 65.8 | 4.62 | 2001 | 422 |
| Gelator 3 | I2, LiI, DMPII | N719 | 640 | 11.1 | 63.4 | 4.49 | 2001 | 422 |
| Gelator 4 | I2, LiI, DMPII | N719 | 623 | 11.2 | 66.4 | 4.67 | 2001 | 422 |
| PEO:CMC | I2, NaI, MPII | N719 | 750 | 10.03 | 69 | 5.18 | 2013 | 423 |
| PEGDA:PEGMA | I2, NaI | N719 | 499 | 17.46 | 52 | 4.41 | 2014 | 426 |
| Cellulose | I2, LiI, MPII | N719 | 590 | 8.39 | 67 | 3.33 | 2014 | 340 |
| PEGDA:PEGMA:Mg-MOF | I2, NaI, MPII | N719 | 690 | 12.6 | 55 | 4.80 | 2013 | 427 |
| BEMA:PEGMA:MFC | I2, NaI | N719 | 760 | 15.2 | 61 | 7.03 | 2014 | 428 |
| PMMA | I2, BMII | N719 | 750 | 15.53 | 69 | 8.03 | 2013 | 358 |
| Polystyrene beads | I2, BMII | N719 | 770 | 15.3 | 64 | 7.54 | 2012 | 429 |
| Polyurethane | I2, LiI, BMII | N719 | 740 | 14.97 | 55 | 6.1 | 2011 | 430 |
| PEO:TiO2 | I2, LiI | N3 | 664 | 7.2 | 57.5 | 4.19 | 2002 | 431 |
| HEII | I2, LiI, NMBI, MPII | MK2 | 733 | 14.66 | 69.3 | 7.45 | 2013 | 432 |
| CkC | I2, NaI | N719 | 510 | 7.60 | 53 | 2.06 | 2015 | 433 |
The classic conductivity and diffusivity of the iodine/1-butyl-3-methylimidazolium iodide (BMII) redox system was similar to that of liquid electrolytes and, relative to conventional liquid DSCs, the resulting cells displayed increased stability.358 For devices filled with liquid electrolyte and directed dissolution of polystyrene nanobeads on the counter electrode, resulting in a gel electrolyte, PCEs of 7.54% were registered. The PCE of those devices was close to that of DSCs based on liquid electrolytes (7.59%).429 Finally, when polyurethane was used as gelation matrix, a PCE up to 6.1% was obtained.430
Some research has focused on the use of different nanosized additives, also known as nanofillers (NFs), to enhance charge transport in quasi-solid and solid electrolytes in order to improve solar cell stability and efficiency. Clays, metal oxides, metal nitrides, metal carbides, metal sulphides, and carbonaceous materials may all be used as nanofillers.388,434
Seo et al. used a combination of a PEO-based composite polymer electrolyte with I−/I3− redox mediator and 5 wt% TiO2 nanoparticles, which not only improved the VOC , but also the energy conversion efficiency to 9.2% at 100 mW cm−2 illumination.435 Lee and co-workers have made significant advances since then, including further development of titanium dioxide (TiO2) and titanium carbide (TiC) nanoparticles,436 and, most significantly, graphene oxide sponge (GOS) as nanofillers.437 The conversion efficiency of DSCs with TiO2 nanoparticles as filler was 7.65% in PEO, which is considerably lower than that of the liquid electrolyte reference devices with PCE of 8.34%. The fabricated liquid and quasi-solid DSCs employing TiC nanofillers both obtained a conversion efficiency of 6.3%. By using poly(vinylidene fluoride) PVDF as a co-regulating agent, the quasi-solid solar cells with TiO2 nanofillers achieved an efficiency of 8.32%, comparable to the liquid electrolyte. Furthermore, by including 4 wt% TiO2 nanoparticles as fillers into the printable electrolyte, the PCE was improved to 8.91%. The DSCs remained stable at 50 °C for 1000 h. The GOS nanofillers were added at a concentration of 1.5 wt% in printable electrolytes based on PEO and PVDF for quasi-solid-state dye-sensitized solar cells reaching energy conversion efficiency of 8.88%. Lee et al. also contributed to the development of quasi-solid-state dye-sensitized solar cells for low light conditions,438–440 with the electrolyte specifically optimised with poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP). This was used to prepare polymer gel electrolytes as a gelator of liquid electrolytes with zinc oxide nanofillers resulting in a good performance at 200 lx of 20.11%.441 In addition, Ramesh and co-workers created a gel electrolyte with PVDF-HFP and PEO with SiO2 as nanofiller and the I−/I3− redox pair having a high ionic conductivity of 8.84 mS cm−1 and resulting in DSCs with a PCE of 9.44%.442 Kim and co-workers also presented two types of triblock copolymers prepared by using functionized PEG as macro-RAFT agents: PEG-b-(P(AN-co-BMAAm))2 (SGT-602) and PEG-b-(P(AN-co-DMAAm))2 (SGT-604) with 13–15 wt% TiO2 nanofillers introduced into the gel electrolytes, resulting in efficiencies of 9.30% and 9.39% with SGT-602 and SGT-604, respectively.443
Solid polymers. Polymer electrolytes (PEs) aim at combining the advantages of liquid electrolytes (high ionic conductivity, diffusive transport, and interfacial contact characteristics) with the mechanical benefits of a polymer's resilience and flexibility.333,338,444,445 The majority of inorganic conductors in a host polymer consist of lithium salts (LiI, NaI, LiClO4, LiCF3SO3, LiSCN, NaSCN, NaClO4, LiPF6, etc.).
The selection of polymer hosts for PEs is based on the following characteristics: sufficiently polar and/or groups to form strong cation coordination and low impediment to bond rotation. Poly(ethylene oxide) (PEO) is the host polymer most widely used,410,431 although these systems typically exhibit poor conductivity (10−8 S cm−1),333 which can be increased with the use of blends of various polymers or copolymers and synthetically adapted monomers (Fig. 35).333,446,447
Li et al. introduced a solid-state electrolyte based on an imidazolium iodide compound co-functionalized with hydroxyethyl and ester groups (HEII) and studied the effect that different substituents on the imidazolium ring have on the ionic conductivity of the electrolyte and on the efficiency of solid-state DSCs built with it.432 Bella et al. contributed by constructing biodegradable polymers derived from seaweed as green chemistry-based PE. Carboxymethyl-da-caraageenan (CkC) and NaI/I2-based DSCs display high efficiency of power conversion up to 2.06%.433 Shortcomings of PEs are connected to insufficient pore filling and ionic conduction, which lead to low dye regeneration rates and fast electron recombination at the interfaces between the solid polymer electrolyte and the dye or the metal oxide semiconductor.
New limitations emerge in the manufacturing of dye-sensitized solar cells that arise from the use of solid-state materials, such as poor pore filling of the mesoporous oxide layer. If large molecules with long molecular chains are introduced to mesoporous materials, they are unable to completely penetrate the mesoporous network.450–453 However, in 2011, Burschka et al. presented a ssDSC featuring spiro-OMeTAD with a PCE of 7.2%, thanks to a careful HTM layer optimization with the addition of p-dopants into the precursor solution.454 Given the high performance reached by Burschka, spiro-OMeTAD is often used as a benchmark HTM when presenting new ones, and it has therefore been used in combination with a large number of dyes.455–459 Nevertheless, this material poses many issues and a consensus has been established that affordable, new materials must be sought before ssDSCs' commercial feasibility can be achieved. More in depth, spiro-OMeTAD suffers from poor conductivity and hole mobility unless dopants are used, and it lacks stability over time.458,460,461
4.3.2.1 Organic hole transport materials. Many organic compounds have been investigated as hole transfer materials for ssDSCs. The variety in synthesis helps researchers to develop new materials with the desired properties. New compounds allow the fine-tuning of energy levels, electronic properties, film-forming properties, and solubility in different solvents. Organic hole transport materials have well-defined compositions and molecular weights that ensures consistent properties in different batches. Compared to other compounds, these smaller molecules are better in penetrating the mesoporous layer of the photoanode.462,463
Organic small molecules are the most common class of novel HTMs for ssDSCs. Most of the compounds referenced in this review have a triphenylamine (TPA) donor component in their composition: the nitrogen atom is a strong hole acceptor due to its lone electron pair and it is aided by the presence of three extra phenyl groups. It is possible to tune the energy levels of molecules containing the TPA group by adding substituents – usually the electron-donating group methoxy – to the aromatic rings not connected to the main body of the molecule. The methoxy group, in fact, destabilizes the electronic cloud in the TPA.464 A list of small molecular HTMs is reported along with their related dye and conversion efficiency in Table 6, and their chemical structures are represented in Fig. 36 and 37.
| HTM | Sensitizer | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|
| Spiro-OMeTAD | Y123 | 986 | 9.5 | 76 | 7.2 | 2011 | 454 |
| Spiro-OMeTAD | D102 | 710 | 8.06 | 53 | 3.03 | 2018 | 455 |
| Spiro-OMeTAD | MKA253 | 780 | 12.4 | 63 | 6.1 | 2015 | 456 |
| Spiro-OMeTAD | Z907 | 750 | 7.28 | 64 | 3.5 | 2013 | 457 |
| Spiro-OMeTAD | ID504 | 760 | 9.76 | 64 | 4.8 | 2015 | 458 |
| Spiro-OMeTAD | LEG4 | 900 | 10.10 | 70 | 6.36 | 2016 | 459 |
| 3a | D102 | 860 | 0.32 | 44 | 0.12 | 2014 | 465 |
| 3b | D102 | 680 | 6.32 | 41 | 1.75 | 2014 | 465 |
| X19 | LEG4 | 750 | 9.62 | 62 | 4.5 | 2014 | 466 |
| X51 | LEG4 | 920 | 9.27 | 70 | 6.0 | 2014 | 466 |
| TCz-C3 | D102 | 690 | 6.27 | 51 | 2.21 | 2018 | 455 |
| TCz-C6 | D102 | 590 | 0.86 | 38 | 0.20 | 2018 | 455 |
| TCz-C12 | D102 | 660 | 0.21 | 34 | 0.05 | 2018 | 455 |
| H-DATPA | D102 | 620 | 0.67 | 37 | 0.15 | 2013 | 467 |
| Me-DATPA | D102 | 700 | 1.13 | 43 | 0.34 | 2013 | 467 |
| MeO-DATPA | D102 | 890 | 1.93 | 67 | 1.16 | 2013 | 467 |
| MeO-TPD | LEG4 | 800 | 9.5 | 65 | 4.9 | 2013 | 468 |
| HTM | Z907 | 750 | 8.5 | 51 | 3.3 | 2014 | 469 |
| X1 | MKA253 | 680 | 5.8 | 58 | 2.3 | 2015 | 456 |
| X1 | LEG4 | 880 | 9.44 | 69 | 5.8 | 2017 | 470 |
| X11 | MKA253 | 580 | 4.7 | 62 | 1.7 | 2015 | 456 |
| X11 | LEG4 | 655 | 8.2 | 55 | 3.0 | 2015 | 456 |
| X2 | LEG4 | 810 | 9.79 | 63 | 5.0 | 2015 | 471 |
| X35 | LEG4 | 890 | 9.81 | 63 | 5.5 | 2015 | 471 |
| X3 | LEG4 | 900 | 9.70 | 66 | 5.8 | 2013 | 457 |
| X3 | Z907 | 720 | 8.10 | 63 | 3.7 | 2013 | 457 |
| X14 | LEG4 | 910 | 9.71 | 71 | 6.1 | 2017 | 470 |
| HTM1 | ID504 | 820 | 9.34 | 63 | 4.8 | 2015 | 458 |
| HTM2 | ID504 | 800 | 7.08 | 38 | 2.2 | 2015 | 458 |
| HTM3 | ID504 | 800 | 7.00 | 38 | 2.1 | 2015 | 458 |
| X60 | LEG4 | 890 | 11.38 | 72 | 7.30 | 2016 | 472 |
| PProDOT | N719 | 630 | 10.0 | 56 | 3.5 | 2012 | 473 |
| PEDOP | D35 | 825 | 7.99 | 66 | 4.34 | 2014 | 474 |
| PEDOP | D21L6 | 645 | 7.92 | 59 | 3.05 | 2014 | 474 |
| PEDOP | Z907 | 440 | 1.97 | 53 | 0.46 | 2014 | 474 |
| PEDOT | DPP07 | 770 | 11.13 | 65 | 5.54 | 2016 | 475 |
| PPP-b-P3HT | CYC-B11 | 810 | 8.81 | 65.2 | 4.65 | 2014 | 476 |
| P3HT | CYC-B11 | 750 | 7.71 | 61.1 | 3.53 | 2014 | 476 |
| P3HT | N3 | 628 | 6.29 | 43 | 1.70 | 2014 | 477 |
| P3HT | BzTCA | 880 | 8.22 | 44 | 3.21 | 2014 | 477 |
| P3HT | D102 | 720 | 11.37 | 58 | 4.78 | 2017 | 478 |
 | ||
| Fig. 36 Examples of carbazole-based organic hole conductors. | ||
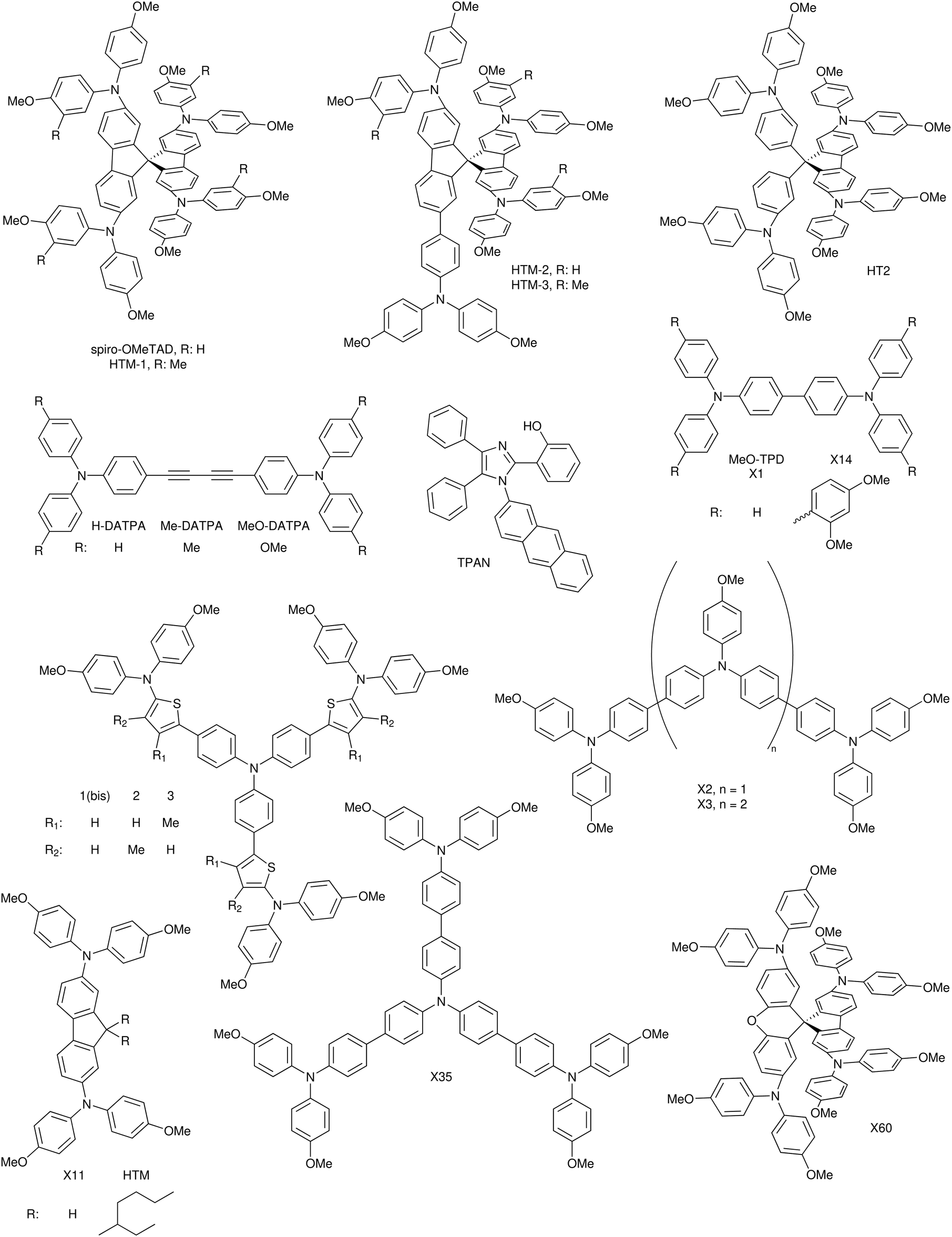 | ||
| Fig. 37 Examples of triphenylamine-based organic hole conductors. | ||
Debia et al. and Xu et al. concurrently developed an HTM (3b465 and X19,466 respectively) based on a carbazole core with a p-methoxyphenyl moiety attached to its nitrogen atom and a di(p-methoxyphenyl)amino group connected in para to each of its phenyl rings. In ssDSCs, 3b was tested with the D102 dye, while X19 with LEG4. These reports provide a good opportunity to highlight the importance of a good dye-HTM combination for what concerns the efficiency of charge transfer. In fact, the best device with 3b-D102 had a PCE of 1.75% (VOC of 680 mV, JSC of 6.32 mA cm−2, FF of 41%), while that with X19-LEG4 had a PCE of 4.5% (VOC of 750 mV, JSC of 9.62 mA cm−2, FF of 62%). The higher current in the latter case can be attributed to different light absorption properties of the two dyes, while the higher VOC and FF are due to a lower series resistance. In a subsequent investigation, Xu et al. presented X51, also based on a carbazole core.466 X19 and X51 are structurally similar, but in the latter case there are two carbazole units that are linked together by a biphenyl moiety bonded to the carbazole nitrogens. As a result, X51's molecular weight almost doubles that of X19. X51 is more conductive than X19, leading to a reduced RS in DSCs, allowing these devices to reach a PCE of 6.0% (VOC of 920 mV, JSC of 9.27 mA cm−2, FF of 70%). Benhattab et al. also connected two carbazoles together, but in this case with alkyl linkers of different lengths (propyl, TCz-C3; hexyl, TCz-C6; and dodecyl, TCz-T12), thus disconnecting electronically the two half molecules. Rather than increasing conjugation as in the case of X51, their efforts were directed to optimize the morphology of the HTM film. The best result was obtained with TCz-C3, with devices reaching a VOC of 690 mV, JSC of 6.27 mA cm−2, FF of 51% and PCE of 2.21%.455
Planells et al. studied four HTMs shaped as rods and comprised of a linear diacetylene core connecting two TPA groups.467 No devices were fabricated with MeS-DATPA (Fig. 37), while cell parameters were VOC = 620 mV, JSC = 0.67 mA cm−2, FF = 37% and PCE = 0.15% for H-DATPA; VOC = 700 mV, JSC = 1.13 mA cm−2, FF = 43% and PCE = 0.34% for Me-DATPA; and VOC = 890 mV, JSC = 1.93 mA cm−2, FF = 67% and PCE = 1.16% for MeO-DATPA. Johansson and co-workers demonstrated that light soaking of full DSCs dramatically improves the efficiency of the solar cell, indicating that ion migration occurs in the solid-state layer. The PCE of their MeO-TPD-based solar cells improved from 1.1% to 4.9% after light soaking.468 Yuan et al. and Liu et al. introduced new HTMs – HTM469 and X11456 – featuring a fluorene center and p-methoxyphenylamino groups connected to each benzene ring. A ssDSC with HTM reached a PCE of 3.3%, while one with X11 reached a PCE of 1.7% with the MKA253 sensitizer and of 3.0% with the LEG4 sensitizer.
Sun and co-workers designed a series of p-methoxy-substituted triphenylamine oligomers, which they used to make X1, X2, X3 and X35.457,471 Optimized devices led to the conclusion that to an increase in number of repeating units corresponded an increase in performance (see Table 6 for champion device details, for X3-based devices VOC was 880 mV, JSC was 9.23 mA cm−2, FF was 62% and PCE was 5.4%). Another effective hole conductor, X14, was created by Sun, Kloo and co-workers. This molecule also presented an expanded aromatic conjugation, since it featured o,p-dimethoxy-substituted phenyl moieties in place of the methoxy groups of X1. The extended conjugation deepened the HOMO level of X14 of about 200 meV compared to X1, while doubling the hole mobility of the former compared to the latter when adding LiTFSI to the HTM layer composition. In the experiments, solar cell efficiency was comparable between the two hole transporting materials. The best X1 samples were the ones that had a PCE of 5.8%, while those with X14 had a PCE of 6.1%. For comparison, the best device based on spiro-OMeTAD displayed a PCE of 5.9%.470 Malinauskas et al. have conducted a study on the long-term stability of spiro-OMeTAD-derived DSCs. They noticed that crystalline domains formed in the originally amorphous spiro-OMeTAD film when the devices were held at 60 °C, which proved the cause of the poor performance of those devices.458 In order to circumvent this limitation they changed spiro-OMeTAD's molecular structure to incorporate asymmetry, reaching high performances with a VOC of 820 mV, JSC of 9.34 mA cm−2, FF of 63% and PCE of 4.8%. HTM-2 and HTM-3, which were more substituted, were also less efficient, with a VOC of 800 mV, JSC of 7.08 mA cm−2, FF of 38% and PCE of 2.2%; and a VOC of 800 mV, JSC of 7.00 mA cm−2, FF of 38% and PCE of 2.1%; respectively.
Xu et al. synthesized X60, the only HTM that could provide comparable results with the benchmark set by Burschka. X60 has a spiro[fluorene-9,9′-xanthene] core linked to p-methoxy substituted diphenylamine side groups, and its spiro moiety costs less than 30 times that of spiro-OMeTAD. They did not have a spiro-OMeTAD-based reference cell, but an X60-based one featured a VOC of 890 mV, JSC of 11.38 mA cm−2, FF of 72% and PCE of 7.30%.472
4.3.2.2 Polymeric hole transporting materials. Using polymers in ssDSCs is more difficult than using small molecules. In practice, for a compound to have excellent electronic properties is not enough. It is also critical to design the device such that the material may permeate the mesosoporous metal oxide and regenerate the dye. Most of the polymers examined here are capable of in situ polymerization; due to this process, monomer molecules can infiltrate the system, and after polymerization, the typically greater conductivity of macromolecules may be utilized. Each article delves into the polymerization process and also refers to the overall structure and characteristics of the monomer itself.
Kim and co-workers introduced a polymer based on a propylenedioxythiophene monomer, ProDOT (Fig. 38).473 PProDOT is similar in structure to PEDOT, but it contains a propylene chain rather than an ethylene one. They employed a solid-state polymerization method in which a dibrominated ProDOT monomer was the starting material. This method is sluggish, but also very inexpensive. A solution of monomers was sprayed onto the photoanode. The solid monomer was put in an oven that was heated at 25 °C and allowed for polymerization to occur with the evaporation of Br2 as a side product. Via coupling with a platinized FTO counter electrode, VOC of 630 mV, JSC of 10.0 mA cm−2, FF of 56% and PCE of 3.5% was reached in terms of photovoltaic performance. Zhang et al. demonstrated the efficiency of PEDOP (poly(ethylenedioxypyrrole)) combined with three separate dyes in suppressing electron recombination, essentially demonstrating the importance of the dye in the system.474 The ssDSCs with D35 dye reached a PCE of 4.34%. D21L6, the second organic dye, performed somewhat worse, with a PCE of 3.05%. However, Zhang et al. demonstrated that the dye DPP07 is as efficient as LEG4 when combined with PEDOT, fabricating a device with a VOC of 770 mV, JSC of 11.13 mA cm−2, FF of 65% and PCE of 5.54%.475
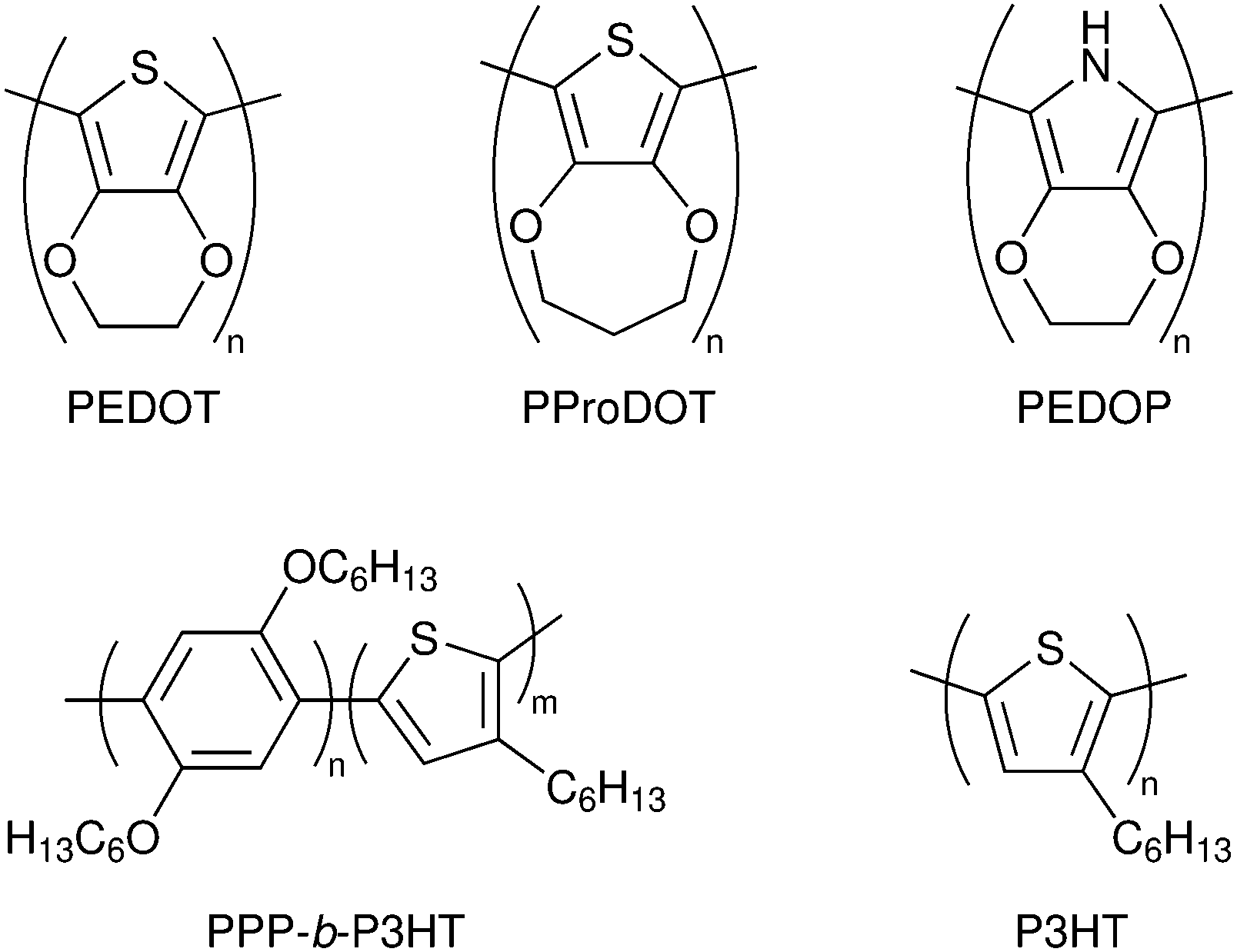 | ||
| Fig. 38 Examples of polymeric hole conductors. | ||
Wang et al. investigated the properties of a pre-polymerized block copolymer of poly(3-hexylthiophene) and poly(2,5-dihexy-p-phenylene), and found that a PPP-b-P3HT-based solar cell achieved a VOC of 810 mV, JSC of 8.81 mA cm−2, FF of 65% and PCE of 4.65%.476
Liu et al. investigated the performance of P3HT with two different dyes. When sensitised with BzTCA, solar cells achieved a VOC of 880 mV, JSC of 8.22 mA cm−2, FF of 44% and PCE of 3.21%, demonstrating that organic dyes are better suited to operate with polymeric HTMs.477 Clément addressed P3HT's usual pore filling problems by creating a highly regioregular polymer with a medium molecular weight and limited dispersion.478 When P3HT with these properties was used in a system with a 2 μm thick TiO2 film, performance improved. Optimized devices had a VOC of 720 mV, JSC of 11.37 mA cm−2, FF of 58% and PCE of 4.78% after HTM deposition and an annealing step at 150 °C to enhance film morphology. In contrast, a device made using spiro-OMeTAD had a PCE of only 3.99%.
4.3.2.3 Inorganic hole transporting materials. Organic HTMs are less stable in water and oxygen than inorganic materials. Generally, inorganic HTMs possess good electronic properties, good conductivity and high temperature stability.479–481 Although these inorganic HTMs already provide good stability in photovoltaic devices, their promise of efficiency remains unfulfilled. Table 7 lists device parameters of DSCs employing various inorganic HTMs referenced in this review, together with the dye used.
| HTM | Sensitizer | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|
| a PC: photonic crystals. | |||||||
| CsSnI3 | N719 | 732 | 19.2 | 72.7 | 10.2 | 2012 | 21 |
| Cs2SnI6 | Z907 | 571 | 13.2 | 61.3 | 4.63 | 2014 | 482 |
| Cs2SnI6 | N719 | 631 | 14.7 | 68.1 | 6.32 | 2014 | 482 |
| Cs2SnI6 | Mix | 623 | 16.9 | 66.1 | 6.94 | 2014 | 482 |
| Cs2SnI6 | Mix + PCa | 618 | 18.6 | 68.0 | 7.80 | 2014 | 482 |
| CuI | N3 | 739 | 14.5 | 69 | 7.40 | 2012 | 483 |
| CuSCN | N719 | 578 | 10.52 | 55.6 | 3.39 | 2012 | 484 |
| Cu(dmp)2 | LEG4 | 1010 | 13.8 | 59 | 8.2 | 2015 | 485 |
| Cu(tmby)2 | Y123 | 1080 | 13.87 | 73.3 | 11.0 | 2017 | 486 |
| Cu(tmby)2 | WS-72 | 1070 | 13.8 | 79 | 11.7 | 2018 | 273 |
| Cu(tmby)2 | XY1:L1 | 1020 | 14.5 | 72 | 10.7 | 2020 | 26 |
| Co(bpyPY4) | Y123 | 768 | 12.12 | 62 | 5.68 | 2016 | 394 |
| Co(bpy)3 | Y123 | 877 | 0.66 | 73 | 0.21 | 2016 | 394 |
Chung et al. used the tin-based perovskite compound CsSnI3 in a N719-sensitized ssDSC.21 With tin fluoride doped into semiconductors, the solar cell developed VOC of 732 mV, JSC of 19.2 mA cm−2, FFs of 72%, and a PCE of 10.2%. To circumvent the volatility of Sn(II)-based perovskites, the Sn(IV)-compound Cs2SnI6 was implemented as hole transport material in solar cells, enabling to harvest holes from different photoanodes with different dyes.482 The PCE of the ssDSC sensitised with Z907 was 4.63%, whereas the PCE of the ssDSC sensitised with N719 was 6.32%. The highest results were obtained using a dye combination of N719, YD2-o-c8, and RLC5. This last system had a VOC of 623 mV, a JSC of 16.9 mA cm−2, a FF of 66%, and a PCE of 6.94%. The output with these dyes was increased even more after including photonic ZnO crystals in the device, reaching a VOC of 618 mV, JSC of 18.6 mA cm−2, FF of 68% with an overall PCE of 7.80% and showing stable output for over 800 hours.
Sakamoto et al. worked on copper iodide, a well-known HTM in the solar cell field. Their analysis discovered how the interface materials affect the formation of CuI layers. The degree of thiocyanate groups in both the dye and counter electrode was crucial for obtaining high efficiency. The variance of the SCN groups in the PEDOT:PSS-based counter electrode resulted in the systems having a greater than two-fold performance compared to those without SCN groups. The successful DSCs showed a VOC of 739 mV, JSC of 14.5 mA cm−2, FF of 69% and PCE of 7.4%.483
Out of the several p-type semiconductors examined for use as hole conductors, the chemical robustness of CuSCN is of particular interest owing to it being a polymeric semiconductor. The solar cells fabricated by Premalal et al. with this HTM included doped p-type copper sulphide nanoparticles and were coated onto a transparent conducting oxide base.484 Triethylamine hydrothiocyanate was used to dope CuSCN and obtain better conductivity; the resulting ssDSC reached a VOC of 578 mV, JSC of 10.52 mA cm−2, FF of 55% and PCE of 3.4%.
4.3.2.4 Metal coordination complex hole transporting materials. Transition metal coordination complexes are a category of materials that incorporates the advantages and disadvantages of both organic and inorganic compounds. As organic compounds they retain an ease-of-processing, but with the high conductivities typical of inorganic compounds, which eliminate the need of p-dopants. The p-dopant is found in the compound itself, and it consists of a complex of the same metal with a higher oxidation state. Energy levels can be varied by modifying the ligand or metal center.485,487–489 Although liquid DSCs have greatly benefited from the implementation of transition metal complexes as electrolytes, as they are far more efficient and less corrosive than iodide/triiodide, only a handful of new compounds of this class have been tested in solid state DSCs so far.284,286,320 Despite this, the best-performing ssDSCs are those employing a metal complex as the hole conductor (see Table 7).
The first researchers to report on ssDSCs based on a metal complex hole conductor were Freitag et al with a phenanthroline-based copper complex with a phenanthroline-based copper complex ([CuII/I(dmp)2]).485 Here, mixed oxidation states of the complex were introduced as solid-state hole transport material. The cell manufacturing technique was identical to the liquid cell construction, but the solvent was allowed to evaporate in air and a fresh injection was repeated until the air gap was filled with solid HTM. They were able to produce a VOC of 1.01 V, JSC of 13.8 mA cm−2, FF of 59% and PCE of 8.2%, surpassing the output of a spiro-OMeTAD-based reference device (5.6%) as well as that of a liquid junction DSC (6.0%).
Further improvements were made by the work of Grätzel and colleagues. Using the copper bipyridyl complex Cu(tmby)2 with the Y123 dye, the authors achieved a VOC of 1080 mV, JSC of 13.87 mA cm−2, FF of 73% and PCE of 11.0%.486
In later research, they developed a new dye for solar cells – WS-72 – able to reduce electron recombination and enhance their efficiency. A solid-state DSC with such dye and Cu(tmby)2 reached a VOC of 1070 mV, JSC of 13.8 mA cm−2, FF of 79% and PCE of 11.7%.273 Most recently, Michaels et al. established a new co-sensitization method using organic dyes XY1 and L1 sensitised solar cells, reporting the first numbers for indoor light conversion with solid-state DSCs of 30% at 1000 lx from a fluorescent lamp (10.7% in full sun).26
Kashif et al. employed a Co(III/II) coordination complex based on a polypyridyl hexadentate ligand: ([Co(bpyPY4)](OTf)2.33) and instead of slow solvent evaporation, the HTM solvent was extracted using vacuum.394 Kashif's top device reached a VOC of 768 mV, JSC of 12.12 mA cm−2, FF of 62% and PCE of 5.68%. For comparison, ssDSCs fabricated with the Co(bpy)3 metal complex, which usually yields excellent efficiencies in liquid DSCs,366 gave an output PCE of only 0.21% because of poor conductivity of the resulting HTM layer with this complex. This demonstrates that only certain metal complexes can be used as hole conductors in ssDSCs.
Additives in solid-state electrolytes and hole transport materials are added to the precursor solution prior to deposition in devices. Some, such as LiTFSI and tBP, are used to alter TiO2 energy levels and passivate its surface as they migrate towards it, allowing for improved charge injection and reduced recombination processes at the TiO2/HTM interface.495–501 In the solid state, they may have the added effect of changing the HTM film morphology. Additionally, certain dopants can directly influence the material. Studies demonstrate that the partial oxidation of the hole conducting substrate leads to increased hole mobility across the layer and, ultimately, conductivity. Oxidizing dopants are necessary for organic compounds and small molecules in particular (see Table 8 for differences in efficiency of DSCs with pristine and doped HTMs), and as an example they must be applied to the spiro-OMeTAD molecule to make it the ideal reference material for ssDSCs.472,502,503 Cappel et al. studied the p-doping properties of LiTFSI in the presence of light and air or N2 atmosphere and Snaith and co-workers continued the work providing a complete description of the doping properties of LiTFSI.461,504 Combined study results showed that oxidation of spiro-OMeTAD by molecular oxygen is activated by LiTFSI regardless of light exposure, while the latter alone is not capable of oxidation. This oxidation process in air has a detrimental side effect, as the redox process consumes Li+ ions, which also serve as additive on the titania surface.
| Dopant | Pristine efficiency (%) | Doped efficiency (%) | Year | Ref. |
|---|---|---|---|---|
| LiTFSI + O2 | 0 | 3 | 2013 | 461 |
| FK102 | 2.3 | 5.6 | 2011 | 454 |
| FK209 | 2.3 | 6.0 | 2013 | 505 |
| FK269 | 2.3 | 6.0 | 2013 | 505 |
| F4TCNQ | 4.55 | 5.44 | 2012 | 506 |
| SnCl4 | 2.52 | 3.40 | 2013 | 507 |
| Spiro(TFSI)2 | 2.34 | 4.89 | 2014 | 460 |
| TeCA | 5.8 | 7.7 | 2015 | 508 |
| TEMPO-Br | 3.99 | 6.83 | 2018 | 509 |
| DDQ | 3.50 | 6.37 | 2018 | 510 |
A Co(III) complex (FK102) has been used as oxidizing dopant in solar cells, which allowed them to attain relatively high efficiencies (Fig. 39).454 The complex oxidized spiro-OMeTAD in solution and the resulting Co(II) species exhibited a low molar extinction coefficient. Upon doping the film's conductivity rose from 4.4 × 10−5 to 5.3 × 10−4 S cm−1, which boosted the overall performance from 2.3 to 5.6%. Two years later Burschka et al. proposed two new Co complexes with better performance, FK209 and FK269.505
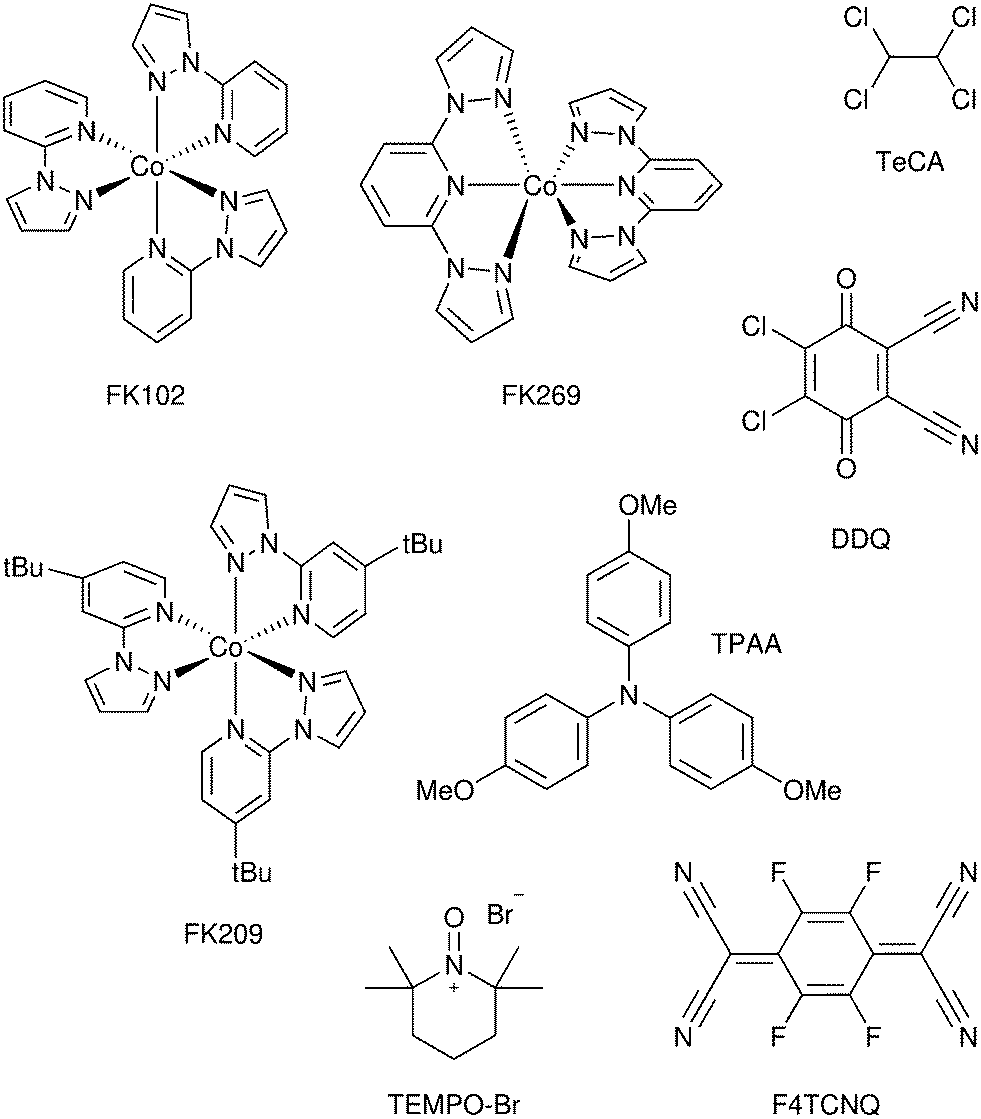 | ||
| Fig. 39 Examples of dopants for hole transporting materials. | ||
Chen et al. oxidized spiro-OMeTAD with the Lewis acid 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ). The use of a dopant produced the spiro-OMeTAD+ species, which was confirmed by a UV-Vis measurement. They used pristine and doped HTMs in ssDSCs, resulting in an increase in efficiency from 0.01 to 0.33%.506 HTM layers with added lithium salt gave efficiencies of 4.55 and 5.44% with and without the presence of F4TCNQ, respectively. Han and colleagues studied a second Lewis acid, SnCl4, which increased conductivity fourfold. The efficiency was 3.4% with a 0.8% doping level of spiro-OMeTAD.507 McGehee and co-workers oxidized the hole conductor itself through the reaction of AgTFSI with Spiro-OMeTAD, removing the need of a p-dopant. Devices built with the pre-oxidized hole conductor demonstrated a significant efficiency increase from 0 to 4.67%.460
Xu et al. reported on 1,1,2,2-tetrachloroethane (TeCA), which they described as a co-solvent. The reason for this is that it is important to keep the TeCA-containing solution under UV light for one minute to allow the spiro-OMeTAD oxidation to take place. System efficiencies increased from 5.8% to 7.7%; for comparison, devices fabricated with FK209 yielded only 6.8% performance.508 TEMPO, previously reviewed among the redox mediators, has also been used as a dopant. Yang et al. reached solar cell efficiency of 6.83% by employing the bromide salt of the oxidised TEMPO.509 A recent study, published by Sun and colleagues, highlighted the effect of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), an oxidant commonly used in chemical synthesis, on ssDSCs. Photovoltaic efficiencies improved from 3.50 to 6.37% when an small quantity of the dopant was introduced.510
4.4 Counter electrodes
The counter electrode (CE) has a major impact on the overall efficiency of DSCs and it performs two main functions: it receives electrons from the external circuit and transmits them into the cell – which necessitates a low resistance – and it acts as a catalyst for the reduction of the oxidized species of the redox mediator. A good CE for DSCs should have the following qualities: high catalytic activity towards the redox mediator, high conductivity, high reflectance, low cost, high surface area, high porosity, low charge-transfer resistance, high exchange current density, chemical resistance to corrosion, energy alignment meeting the potential of the electrolyte's redox couple and good processability for deposition.511,512 For DSCs, a great variety of CE preparation recipes has been demonstrated, including thermal and photo-decomposition,513–516 electrochemical deposition,517–519 chemical vapor deposition,520 and sputter deposition.521–523 The preparation methods greatly affect particle size, surface, morphology, and catalytic and electrochemical characteristics of the electrodes. Smaller particles and larger electrode surface areas provide more catalytic active sites and facilitate improved electrode operation.524Platinum has traditionally been the most common counter electrode active material for DSCs, due to its excellent conductivity and catalytic activity, with PCEs over 12%.284 Nevertheless, Pt still has certain drawbacks to solve, including the high price and rarity of the raw material, poor stability over longer periods, as well as migration towards the photoanode and deposition on the TiO2 layer leading to cell shortage.525–529 Furthermore, due to energy level misalignment, Pt is not very effective in regenerating alternative redox couples such as coordination complexes, T2/T− or polysulfide electrolytes.339 Fortunately, many other materials can be used as CE in DSCs.
Carbon-based materials (Fig. 40)530 are attractive candidates to replace platinum as the CE material in DSCs thanks to advantages such as low cost, abundance, high surface area, high catalytic activity, high electrical conductivity, high thermal stability, corrosion resistance, and high reactivity for redox mediator reduction,388 among other characteristics. An FTO/Au/GNP (graphene nanoplatelets) stack was used as CE to reach a PCE of 14.3%.24,531 The inexpensive and easy preparation, and good stability improve the competitiveness of carbon materials. The key downsides of common CEs based on carbon compounds are an overall worse performance compared to platinized electrodes – in terms of conductivity and catalytic activity – when coupled with the I−/I3− redox couple. Further, poor adhesion to the FTO substrate leads to electrode degradation.532 To mitigate these issues, in recent years researchers from Korea University have doped graphene nanoplatelets with various metals and halogens (Se, Te, Sb, F, I) to improve compatibility of carbon CEs towards the I−/I3− redox couple. These electrodes proved more efficient than those based on Pt, and were also more stable.533–536
 | ||
| Fig. 40 Structures of various carbon allotropes. Reprinted with permission from ref. 530. Copyright 2013 Mineralogical Society of America. | ||
Flexibility, translucency, and facile processing and tuning are all properties of conductive polymers that make them prime candidates as CE materials in DSCs (Fig. 41).173,537 PEDOT (poly(3,4-ethyleneedioxythiophene)), first discovered by the Bayer Lab in the 1980s, is a promising substrate for antistatic and opto-electronic applications due to its high conductivity, outstanding visible light transmittance and extraordinary stability.517 Although PEDOT is an insoluble polymer, it can be easily electrodeposited from its monomer in solution, resulting in excellent conductivity, much higher than that of polyaniline (PAni), polypyrrole (PPy) and polythiophene (PT).537–539 Moreover, the solution to PEDOT's insolubility problem was later solved by co-polymerization with poly(styrene sulfonate) (PSS). PEDOT:PSS is the market pioneer in transparent conductive polymers, it is water-soluble and allows fast manufacturing. Saito et al. investigated for the first time in 2002 PEDOT-based materials – specifically PEDOT:PSS and p-toluenesulfonate (TsO)-doped PEDOT – as CE for DSCs, deposited on FTO via chemical polymerization.540 The PCE of the cell with the PEDOT:TsO CE was almost the same as that with the Pt CE, while in the case of the PEDOT:PSS electrode it was shown that I−/I3− oxidation/reduction processes occurred at higher potentials compared to the other two electrodes, which was attributed to a steric hindrance effect of the PSS component of the polymer.540 By using electrodeposition techniques, PEDOT is now being deployed in the most efficient DSCs, especially due to its high performance in combination with alternative redox mediators and hole transport materials. Tsao et al. showed how electropolymerized PEDOT CEs are much better performing with Co-based redox mediators compared to their Pt counterparts.541 Their best PEDOT-based cell reached a PCE of 10.3%, compared to 7.9% of a Pt-based one. The performance improvement was attributed to a much lower charge transfer resistance of PEDOT towards the Co complex compared to Pt. Freitag et al. achieved a PCE of 11.3% with a copper-mediated DSC featuring a PEDOT CE,348 recently surpassed by Grätzel et al. with a 13.5% PCE cell.12 One more advantage of PEDOT over Pt is that the former is a hole-selecting material. As such, it is possible to fabricate PEDOT-based sandwich-type solar cells without any spacing between the two electrodes without the risk of cell shortage.320,348
 | ||
| Fig. 41 Repeating units of polymers used as counter electrode materials in DSCs. | ||
DSCs incorporating hybrid/mixed CEs outperform devices with single component CEs, thanks to the synergistic effects of the hybrid composite. However, the exact mechanism behind this success is still not fully understood on a fundamental level. Examples of efficient hybrid CEs include platinized PEDOT and a combination of graphene with PEDOT, PAni or Pt.515,519,525,542,543
5 P-type DSCs
5.1 Photocathodes
To increase the efficiency of dye-sensitized solar cells, it has been proposed that the TiO2-based photoanode could be combined in series with a second photoelectrode (i.e. a photocathode) in a tandem device.544,545 In a p–n tandem DSC, the light transmitted by the first photoelectrode can be captured by the second photoelectrode, extending the spectral response to the near IR. The VOC becomes the sum of the two individual (n-type and p-type) devices. Therefore, there is an opportunity to collect more light more efficiently. In principle, tandem DSC devices (p–n DSCs) should overcome the thermodynamic limits of single-junction devices and achieve efficiencies above the Shockley–Queisser limit (theoretically up to 43%).546–548 Unfortunately, the efficiency of p-type DSCs is much lower than that of n-type DSCs, which limits the efficiency of p–n DSCs. For this reason, there has been increasing attention paid to the development of more efficient p-type DSCs in the last 20 years. In these devices the majority charge carriers in the semiconductor are positive holes (h+) and the current flows in the opposite direction to TiO2-based DSCs. Following excitation of the dye with light, electron transfer takes place from the valence band of the semiconductor to reduce the dye, as shown in Fig. 42.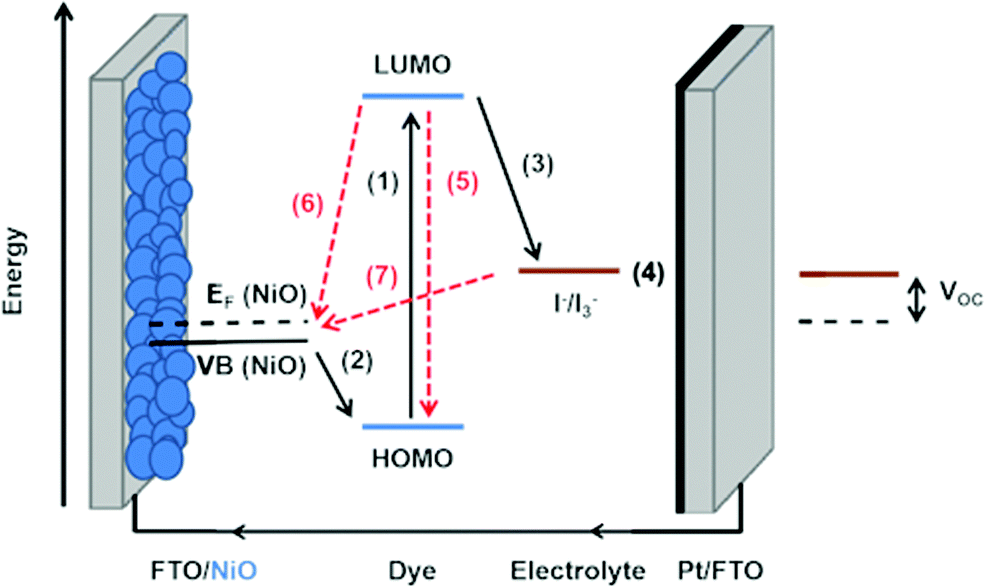 | ||
| Fig. 42 Schematic representation of the charge transfer processes occurring within a NiO-based p-DSC. Recombination processes shown in red. Processes 1–6 defined in the text. Adapted from ref. 544 with permission from The Royal Society of Chemistry, copyright 2019. | ||
Lindquist and coworkers reported the first p-DSC in 1999,549 which used a layer of NiO – a p-type semiconductor – instead of TiO2, and erythrosin B as the photosensitizer. This device had an overall PCE of 0.0076%. By 2010, this had been improved to 0.41% efficiency by improving the quality of the NiO and engineering a dye specifically for NiO.550 However, since the p-DSC efficiency was well below that of n-type devices, the tandem cell efficiency was severely limited (1.91%). Key limitations to the efficiency of p-DSCs include the rapid charge recombination at the dye/NiO and NiO/electrolyte interfaces. Developing photosensitizers that promote charge separation, together with new iodide-free redox mediators can lead to substantial improvements in device efficiencies. Further research into the mechanism, electron transfer dynamics and surface characterisation has enabled further improvements to be made over the following decade, which are summarised in the following sections. By the end of 2020 the highest tandem cell efficiency had reached 4.1%.548
5.2 Semiconductors
NiO is typically chosen as the p-type semiconductor, since it is straightforward to prepare, it has a high-lying valence band edge (0.3 V vs. SCE or 0.54 V vs. NHE at pH 7) and a wide bandgap (3.6–4.0 eV).551–553 There have been extensive articles and reviews on the various synthetic techniques and the challenges of applying NiO in p-DSCs.554–559 A comparison by Gibson et al. found that, based on the P1 dye, the best performance for mesoporous NiO electrodes was reached with a 1–2 μm thick film, with a crystallite size of ca. 20 nm and a specific surface area above 40 m2 g−1.560 The most commonly used synthetic technique is the sol–gel method, due to its simplicity and reproducibility, and pluronic triblock copolymer-templated NiO films satisfy these criteria, giving thicknesses of 1–2 μm and crystal sizes of 15–20 nm.561 Typically, these films are applied in the laboratory by the doctor blade technique, but Jousselme et al. attained promising results (JSC = 3.42 mA cm−2) by inkjet printing a sol–gel precursor.562Despite being straightforward to synthesize and deposit, there are several unfavourable characteristics of NiO. Firstly, whereas TiO2 is non-toxic, NiO is a group 1 carcinogen. The VOC of NiO-based DSCs is limited to 100–200 mV because NiO has a high-lying valence band (0.54 V vs. NHE), which is advantageous in terms of electron transfer to photosensitizers, but leads to a small difference between the Fermi level in the NiO and the redox potential of the electrolyte. NiO also has a low charge diffusion coefficient (∼10−8 cm2 s−1)561,563,564 and the presence of high valence states (e.g. NiIII and NiIV) leads to rapid recombination at the dye/semiconductor and semiconductor/electrolyte interfaces.565,566 This leads to a small diffusion length for holes (2–3 μm), which means thin NiO films must be used.566 Strategies applied over the last 10 years to reduce recombination include applying compact blocking layers on the FTO substrate,567 chemical reduction of the NiO surface,568 surface treatment with an aqueous nickel salt,569 applying a thin, surface layer of Al2O3, B or TiO2,570–572 or adding organic surfactants such as chenodeoxycholic acid.573 Other approaches to improving the electronic properties (either by increasing the hole mobility or lowering the Fermi level) include doping or forming solid solutions with alkali or transition metals such as Li, Co, Mg.574–577 However, a competition between increasing VOC and decreasing JSC is frequently observed, possibly as a result of decreasing the driving force for electron injection if the valence band edge is shifted to more positive potential. The porosity, dye loading and hole transport can be improved by adding graphene or reduced graphene oxide to NiO.578,579 However, despite these modest improvements, the small built-in potential and poor fill factors (typically 30–40%) limit the solar cell efficiency to <1%.
Increasing the solar cell efficiency requires finding a replacement for NiO, ideally with a ca. 0.5 V deeper-lying valence band to match the VOC of TiO2. This is difficult due to the trade-off between conductivity and transparency. Binary or ternary nickel oxides and oxysulfides have been tested in p-type DSCs, but in each case, if the VOC was improved, the current was sacrificed. The potential reasons for this could be physical (insufficient surface area for the dye to adsorb or insufficient porosity for the electrolyte to diffuse), electronic (low dielectric constant or hole mobility) or surface properties such as the presence of high-valence Ni.
K-Doped ZnO thin films, which have high optical transparency (>85%) and a larger hole diffusion coefficient (10−6 cm2 s−1) than NiO, show some promise for p-DSCs (JSC = 0.408 mA cm−2, VOC = 82 mV, and PCE = 0.0012% with C343).580 More encouraging results have been achieved with tin-doped indium oxide (ITO) reaching PCEs of ca. 2%.581,582 Promising results have been obtained with CuO-based DSCs by applying nanoparticles, nanorods or nanowires.583 One-dimensional materials could overcome the shorter transport lifetime for holes in CuO compared to NiO. CuO electrodes are unstable towards I−/I3− , so alternative redox mediators such as cobalt coordination complexes are required.584 An efficiency of 0.19% was reached in combination with zinc phthalocyanine sensitizers and cobalt-based redox mediators.585–587 However, CuO is not optically transparent (Eg = 1.4 eV584). Cu2O is more transparent but less stable than CuO. With C343, a Cu2O device gave a VOC = 0.71 V, a JSC = 1.3 mA cm−2, FF = 46%, and a PCE of 0.42%.588 Cu2O@CuO core–shell structures have been applied to improve the stability, but this has not yet improved the solar cell characteristics (VOC = 315 mV, JSC = 0.14 mA cm−2, PCE = 0.017%).589
Cu-Based delafossites (CuAlO2, CuGaO2, CuFeO2, CuBO2, CuCrO2 and CuCrO2) have been highlighted as potential p-type transparent conductive oxides.590,591 During the last 10 years, attempts have been made to exploit the deeper-lying valence band and high hole mobility of these materials compared to NiO in p-DSCs.584,592–596 Efficiencies of 0.04% have been recorded with CuAlO2, but with delafossites, as with doped NiO, a trade-off between JSC (<1 mA cm−2) and VOC (333 mV) has been found.584,592,597 Better efficiencies of up to 0.18% have been obtained with CuGaO2 in combination with P1 and I−/I3−.598,599 Doping with Mg, Fe and Al improves the specific surface area of CuGaO2 photocathodes and conversion efficiencies comparable with NiO have been reached with Mg:CuGaO2.593,600,601 The best results so far have been with CuCrO2, which reached 0.4% PCE with PMI-6T-TPA and the [Co(en)3]2+/3+ electrolyte, but although the VOC (734 mV) was better than the equivalent NiO device, the JSC (1.23 mA cm−2) was much lower.602 Successful attempts to improve the current include adding plasmonic Au nanoparicles,603 and doping with Mg, Ga and Co, but solar cell efficiencies with delafossites are yet to surpass NiO.604–606
Other proposed alternatives to NiO include mixed chalcogens. LaCuOS has been applied in p-DSCs with PMI-NDI dye but a low PCE (0.002%) was recorded, which the authors attribute to similar valence band edge energies of NiO and LaCuOS, rapid charge recombination and weak binding affinity for the dye on the surface.607 More encouraging results have been reported with spinel cobaltites (MCo2O4; M = Ni, Zn). A NiCo2O4 device with N719 reached a PCE = 0.785% (VOC = 189 mV, JSC = 8.35 mA cm−2, FF = 50%), which is exceptionally high compared to most other p-DSCs fabricated using the standard I−/I3− electrolyte.608,609Table 9 lists the electrochemical properties of the referenced p-type semiconductors, together with the best cell efficiency obtained with them.
| Semiconductor | Bandgap (eV) | Valence band energy (eV vs. vacuum) | Dielectric constant | Max cell efficiency (%) – electrolyte used | Ref. |
|---|---|---|---|---|---|
| NiO | 4.7–4 | −4.94 to −4.7 | 9.7 | 2.51 – Fe(acac)3 | 103,555,571,573,610 |
| K:ZnO | 3.23 | −5.7 | Not reported | 0.012 – I−/I3− | 580 |
| Sn:In2O5 (ITO) | 4.1 | −4.8 | Not reported | 1.96 – Fe(acac)3 | 582,611 |
| CuO | 1.41–1.82 | −4.95 to −5.09 | 18.1 | 0.19 – I−/I3− | 584,587,612 |
| Cu2O | 2.4 | −5.20 | 12 | 0.42 – I−/I3− | 588,613–615 |
| CuAlO2 | 3.5 | −5.68 | 10 | 0.037 – I−/I3− | 590–592,616 |
| CuCrO2 | 3.11 | −5.44 | Not reported | 0.48 – Co(en)3 | 602,603,606 |
| Au@SiO2:CuCrO2 | 3.11 | Not reported | Not reported | 0.31 – T−/T2 | 603 |
| Mg:CuCrO2 | Not reported | Not reported | Not reported | 0.132 – I−/I3− | 604 |
| Ga:CuCrO2 | 3.25–3.30 | −5.39 | Not reported | 0.100 – I−/I3− | 606 |
| AgCrO2 | 3.32 | Not reported | Not reported | 0.0145 – I−/I3− | 595 |
| CuGaO2 | 3.6–3.8 | −5.29 | 0.96 | 0.182 – I−/I3− | 598,599,617 |
| CuFeO2 | 2.03–3.35 | −4.9 to −5.13 | Not reported | 0.0103 – I−/I3− | 597,618 |
| LaOCuS | 3.1 | −4.94 | 4 | 0.002 – Co(dtb-bpy)3 | 607,619,620 |
| NiCo2O4 | 2.06–3.63 | −5.00 | Not reported | 0.785 – I−/I3− | 608,621 |
5.3 Sensitizers
In p-DSCs, the frontier orbitals of the dye must be arranged such that the HOMO lies at more positive potential than the valence band edge of the semiconductor, while the LUMO must be more negative than the redox potential of the electrolyte.622–626 Because the film thickness is limited by the diffusion length in NiO devices (see above), high extinction coefficients are required to capture all incident light. If the photocathode is to be positioned on the bottom of the cell, the dye needs to absorb red-NIR photons. In the first ten years of p-type DSC development commercial dyes were applied, but the first breakthroughs came from developing bespoke “push–pull” systems specifically designed for photocathodes.550,564,627 D–π–A systems, where the electron density is pushed away from the NiO surface on excitation of the dye, improve the charge-separated state lifetime and quantum efficiency. Over the last 10 years, a substantial number of different dye systems have been developed and tested in p-DSCs, typically with NiO.544,610,628–630 Metal complexes such as N719 and N3 generally give poor results in p-DSCs.554 There are a few examples of Ru-based dyes giving promising results with NiO, where there is some charge-transfer character directed away from the semiconductor surface (e.g. an anchoring group is positioned on the electron donating part of the molecule), see Table 10.631–636 Ir complexes (Fig. 43) have also been applied in p-DSCs due to their long lived and strongly oxidizing triplet excited states which favour hole injection into the semiconductor valence band.637–640 The JSC of iridium photosensitizers is generally low due to the narrow absorption spectrum.| Sensitizer | Electrolyte | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | IPCE max (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| K1 | I2, LiI | 96 | 2.91 | 32 | 0.09 | 14 | 2014 | 631 |
| K2 | I2, LiI | 93 | 1.96 | 39 | 0.07 | 9 | 2014 | 631 |
| O3 | I2, LiI | 93 | 3.04 | 35 | 0.099 | ∼20 | 2013 | 632 |
| O13 | I2, LiI | 89 | 2.66 | 31 | 0.074 | ∼19 | 2013 | 632 |
| O17 | I2, LiI | 92 | 2.69 | 34 | 0.085 | ∼16 | 2013 | 632 |
| O8 | I2, LiI | 63 | 0.44 | 36 | 0.009 | 2.02 | 2012 | 633 |
| O11 | I2, LiI | 79 | 1.16 | 36 | 0.033 | 5.49 | 2012 | 633 |
| O12 | I2, LiI | 82 | 1.84 | 34 | 0.051 | 9.08 | 2012 | 633 |
| O18 | I2, LiI | 93 | 3.43 | 33 | 0.10 | Not reported | 2014 | 636 |
| SL1 | I2, DMBII | 104 | 2.25 | 34 | 0.079 | 18 | 2016 | 634 |
| SL2 | I2, DMBII | 77 | 1.5 | 33 | 0.038 | 10 | 2016 | 634 |
| [Ru(bpy)2(H1) | I2, LiI | 95 | 4.06 | 36 | 0.14 | Not reported | 2017 | 635 |
| IrPhen | Co(dtb-bpy)3 | 345 | 0.14 | 44 | 0.021 | ∼4 | 2014 | 637 |
| IrDPQCN2 | Co(dtb-bpy)3 | 508 | 0.25 | 54 | 0.068 | ∼6.2 | 2014 | 637 |
| IrBpystyryl | Co(dtb-bpy)3 | 383 | 0.37 | 44 | 0.061 | ∼10.5 | 2014 | 637 |
| 1 | I2, LiI | 58 | 0.076 | 27 | 0.0012 | 2 | 2017 | 638 |
| AS16 | I2, LiI | 94 | 0.69 | 42 | 0.028 | 17 | 2017 | 638 |
| 2 | I2, LiI | 134 | 0.069 | 40 | 0.0037 | 3 | 2017 | 638 |
| AS17 | I2, LiI | 89 | 0.14 | 42 | 0.0052 | 5 | 2017 | 638 |
| 3 | I2, LiI | 77 | 0.16 | 45 | 0.0056 | 6 | 2017 | 638 |
| AS18 | I2, LiI | 79 | 0.15 | 46 | 0.0055 | 6 | 2017 | 638 |
| AS19 | I2, LiI | 104 | 0.45 | 42 | 0.02 | ∼28 | 2016 | 639 |
| AS9 | I2, LiI | 90 | 0.68 | 36.6 | 0.022 | ∼15 | 2017 | 640 |
| AS10 | I2, LiI | 90 | 0.66 | 37.6 | 0.022 | ∼21.5 | 2017 | 640 |
| AS11 | I2, LiI | 70 | 0.45 | 38.1 | 0.013 | ∼11 | 2017 | 640 |
| AS12 | I2, LiI | 90 | 0.36 | 40.1 | 0.013 | ∼13 | 2017 | 640 |
| AS13 | I2, LiI | 100 | 0.82 | 38.7 | 0.032 | ∼26 | 2017 | 640 |
| AS14 | I2, LiI | 100 | 1.12 | 36.8 | 0.043 | ∼21.5 | 2017 | 640 |
 | ||
| Fig. 43 Examples of metal complex-based sensitizers for p-type DSCs. | ||
Better results have been reported with metal-free systems (see Table 11). The push–pull dye P1 was one of the first organic dyes to achieve a reasonably high JSC. The design was based on the triphenylamine-based dyes used in n-type DSCs and many subsequent dyes for p-DSCs have since been based on this architecture.563,641 Optimised devices with P1 and I−/I3− give IPCE = ca. 63%, and PCE of 0.16%, and P1 has become a benchmark dye for optimising new materials in p-DSCs.550,564,642 In the last decade since these breakthroughs, numerous arylamine-containing molecules have been designed for p-DSCs (Fig. 44), mostly with different acceptor or linker groups,643–649 and a few reports of modified anchoring structure.626,650 Dyes with two acceptor groups per triarylamine unit tend to have a higher absorption coefficient and produce a higher JSC. The highest JSC reported for a p-DSC was produced using CAD3 with two cationic indolium groups as electron acceptors (JSC = 8.21 mA cm−2, λmax = 614 nm, ε = 95000 M−1 cm−1).643,651
| Sensitizer | Electrolyte | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | IPCE max (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | I2, MBII | 153 | 2.06 | 29 | 0.09 | ∼10 | 2010 | 550 |
| 2 | I2, MBII | 176 | 3.40 | 32 | 0.19 | ∼20 | 2010 | 550 |
| 3 | I2, MBII | 218 | 5.35 | 35 | 0.41 | ∼50 | 2010 | 550 |
| P1 | I2, LiI | 89 | 5.37 | 33 | 0.16 | 54 | 2015 | 643 |
| P1 | Co(dtb-bpy)3 | 280 | 1.18 | 30 | 0.10 | ∼20 | 2016 | 652 |
| C343 | I2, LiI | 208 | 0.951 | 32.4 | 0.064 | 7.1 | 2019 | 653 |
| C343 | Co(dtb-bpy)3 | 190 | 0.25 | 32 | 0.015 | ∼2 | 2009 | 627 |
| PI | Co(dtb-bpy)3 | 80 | 0.26 | 26 | 0.006 | ∼3 | 2009 | 627 |
| PINDI | Co(dtb-bpy)3 | 350 | 1.66 | 34 | 0.20 | 31 | 2009 | 627 |
| Eosin B | I2, LiI | 77 | 0.14 | 29 | 0.0032 | Not reported | 2008 | 554 |
| Erythrosin J | I2, LiI | 122 | 0.36 | 26 | 0.011 | Not reported | 2008 | 554 |
| Rhodamine 101 | I2, LiI | 69 | 0.12 | 21 | 0.0022 | Not reported | 2008 | 554 |
| Rhodamine 110 | I2, LiI | 80 | 0.15 | 25 | 0.0031 | Not reported | 2008 | 554 |
| P4 | I2, LiI | 100 | 2.48 | 36 | 0.09 | 44 | 2009 | 641 |
| P2 | I2, LiI | 63 | 3.37 | 31 | 0.07 | 32 | 2010 | 642 |
| P3 | I2, LiI | 55 | 1.36 | 34 | 0.03 | 6 | 2010 | 642 |
| P7 | I2, LiI | 80 | 3.37 | 35 | 0.09 | 26 | 2010 | 642 |
| CAD3 | I2, LiI | 101 | 8.21 | 31 | 0.25 | 50 | 2015 | 643 |
| GS1 | I2, LiI | 106 | 5.87 | 31 | 0.20 | 53 | 2015 | 643 |
| QT-1 | I2, LiI, DMII | 120 | 8.2 | 34 | 0.33 | 60 | 2015 | 644 |
| QT-1 | Co(pz-py)3 | 226 | 6.5 | 34 | 0.50 | Not reported | 2015 | 644 |
| zzx-op1 | I2, LiI | 96 | 5.70 | 38 | 0.21 | 50.1 | 2014 | 645 |
| zzx-op1–2 | I2, LiI | 117 | 7.57 | 40 | 0.35 | 70.2 | 2014 | 645 |
| zzx-op1–3 | I2, LiI | 115 | 6.68 | 40 | 0.31 | ∼57 | 2014 | 645 |
| zzx-op2 | I2, LiI | 111 | 4.00 | 36 | 0.16 | ∼27 | 2014 | 646 |
| zzx-op3 | I2, LiI | 109 | 3.80 | 36 | 0.15 | ∼20 | 2014 | 646 |
| C1 | I2, LiI | 40 | 1.63 | 27 | 0.016 | ∼24 | 2017 | 647 |
| C2 | I2, LiI | 59 | 2.41 | 29 | 0.040 | ∼22 | 2017 | 647 |
| C3 | I2, LiI | 17 | 1.00 | 17 | 0.001 | ∼36 | 2017 | 647 |
| SK2 | I2, LiI | 81 | 0.51 | 33 | 0.014 | ∼14 | 2016 | 648 |
| SK3 | I2, LiI | 82 | 0.54 | 33 | 0.015 | ∼11.5 | 2016 | 648 |
| SK4 | I2, LiI | 134 | 0.43 | 32 | 0.018 | ∼5.6 | 2016 | 648 |
| RBG-174 | I2, LiI | 90 | 2.88 | 36.7 | 0.096 | Not reported | 2018 | 649 |
| COCO | I2, LiI | 91 | 2.45 | 35.9 | 0.080 | Not reported | 2018 | 649 |
| BBTX | I2, LiI | 88 | 4.32 | 33.0 | 0.126 | Not reported | 2018 | 649 |
| COCN | I2, LiI | 77 | 1.53 | 32.3 | 0.038 | Not reported | 2018 | 649 |
| CW1 | I2, LiI | 93 | 3.54 | 35 | 0.114 | ∼36 | 2014 | 626 |
| CW2 | I2, LiI | 118 | 4.05 | 34 | 0.160 | ∼42 | 2014 | 626 |
| 1 | I2, LiI | 50 | 0.83 | 43 | 0.018 | ∼25 | 2019 | 650 |
| 2 | I2, LiI | 103 | 1.6 | 36 | 0.060 | ∼25 | 2019 | 650 |
| 3 | I2, LiI | 49 | 0.87 | 32 | 0.014 | ∼22.5 | 2019 | 650 |
| 4 | I2, LiI | 66 | 0.83 | 33 | 0.018 | ∼25 | 2019 | 650 |
| 5 | I2, LiI | 86 | 1.11 | 37 | 0.036 | ∼25 | 2019 | 650 |
| 6 | I2, LiI | 70 | 0.84 | 23 | 0.014 | ∼21.3 | 2019 | 650 |
| CAD1 | I2, LiI | 87 | 3.32 | 33 | 0.09 | 25 | 2014 | 651 |
| CAD2 | I2, LiI | 96 | 3.25 | 33 | 0.10 | 17 | 2014 | 651 |
| T3 | I2, LiI | 121 | 5.01 | 30.3 | 0.184 | ∼30 | 2015 | 654 |
| T4 | I2, LiI | 119 | 5.31 | 32.9 | 0.208 | ∼32 | 2015 | 654 |
| T5 | I2, LiI | 124 | 4.51 | 33.3 | 0.186 | ∼27 | 2015 | 654 |
| T6 | I2, LiI | 133 | 4.02 | 33.3 | 0.178 | ∼23 | 2015 | 654 |
| T3H | I2, LiI | 133 | 5.56 | 30.5 | 0.226 | ∼32 | 2016 | 655 |
| T4H | I2, LiI | 152 | 6.74 | 31.0 | 0.317 | ∼38 | 2016 | 655 |
| T1 | I2, LiI | 125 | 2.82 | 31 | 0.11 | ∼37 | 2014 | 656 |
| T3 | I2, LiI | 144 | 4.01 | 33 | 0.19 | ∼45 | 2014 | 656 |
| T4 | I2, LiI | 123 | 1.69 | 29 | 0.06 | ∼26 | 2014 | 656 |
| BH2 | I2, DMII | 97 | 4.3 | 31 | 0.13 | Not reported | 2014 | 657 |
| BH4 | I2, DMII | 128 | 7.4 | 30 | 0.28 | Not reported | 2014 | 657 |
| BH6 | I2, DMII | 95 | 4.4 | 31 | 0.13 | Not reported | 2014 | 657 |
| E1 | Co(dtb-bpy)3 | 320 | 0.93 | 44 | 0.13 | ∼13 | 2016 | 652 |
| E2 | Co(dtb-bpy)3 | 320 | 0.78 | 41 | 0.10 | ∼9 | 2016 | 652 |
| O2 | I2, LiI | 94 | 1.43 | 37 | 0.050 | 12.3 | 2011 | 658 |
| O6 | I2, LiI | 97 | 1.04 | 37 | 0.037 | 13.5 | 2011 | 658 |
| O7 | I2, LiI | 90 | 1.74 | 38 | 0.060 | 17.9 | 2011 | 658 |
| QT-1 | I2, LiI, DMII | 120 | 8.2 | 34 | 0.33 | 60 | 2015 | 644 |
| QT-1 | Co(pz-py)3 | 226 | 6.5 | 34 | 0.50 | Not reported | 2015 | 644 |
| EH122 | I2, LiI, DMPII | 134 | 4.39 | 30.3 | 0.178 | ∼28 | 2019 | 659 |
| EH126 | I2, LiI, DMPII | 122 | 3.93 | 30.4 | 0.146 | ∼25.5 | 2019 | 659 |
| EH166 | I2, LiI, DMPII | 131 | 3.47 | 28.4 | 0.129 | ∼20.5 | 2019 | 659 |
| EH162 | I2, LiI, DMPII | 115 | 1.79 | 30.4 | 0.062 | ∼16 | 2019 | 659 |
| EH174 | I2, LiI, DMPII | 137 | 4.84 | 31.2 | 0.207 | ∼28.5 | 2019 | 659 |
| EH170 | I2, LiI, DMPII | 139 | 3.47 | 31.5 | 0.152 | ∼20 | 2019 | 659 |
| BOD1 | I2, LiI | 70 | 0.56 | 38 | 0.015 | Not reported | 2020 | 660 |
| BOD2 | I2, LiI | 40 | 0.48 | 29 | 0.006 | Not reported | 2020 | 660 |
| BOD3 | I2, LiI | 60 | 0.21 | 29 | 0.003 | Not reported | 2020 | 660 |
| 1 | I2, LiI | 79 | 3.15 | 31 | 0.08 | 28 | 2014 | 661 |
| Bodipy-CO2H | I2, LiI | 95 | 1.48 | 36 | 0.05 | 20 | 2015 | 662 |
| 4 | I2, LiI | 97 | 1.60 | 38 | 0.06 | 27 | 2015 | 662 |
| 5 | I2, LiI | 109 | 3.70 | 35 | 0.14 | 44 | 2015 | 662 |
| 6 | I2, LiI | 95 | 1.58 | 35 | 0.05 | 23 | 2015 | 662 |
| 7 | I2, LiI | 106 | 5.87 | 31 | 0.20 | 53 | 2015 | 662 |
| 1 | I2, LiI, BMII | 79 | 0.61 | 25 | 0.012 | 3.2 | 2019 | 663 |
| W1 | I2, LiI | 131 | 2.83 | 34.0 | 0.126 | ∼14 | 2015 | 664 |
| W2 | I2, LiI | 121 | 4.16 | 33.0 | 0.166 | ∼17 | 2015 | 664 |
| W3 | I2, LiI | 134 | 2.32 | 33.1 | 0.103 | ∼9 | 2015 | 664 |
| 1 | I2, LiI | 105 | 1.59 | 35.9 | 0.060 | ∼17 | 2011 | 665 |
| 2 | I2, LiI | 115 | 1.39 | 36.3 | 0.058 | ∼15 | 2011 | 665 |
| 3 | I2, LiI | 113 | 1.38 | 34.0 | 0.053 | ∼14 | 2011 | 665 |
| 4 | I2, LiI | 125 | 2.25 | 33.1 | 0.093 | ∼27.5 | 2011 | 665 |
| 5 | I2, LiI | 122 | 2.18 | 34.6 | 0.092 | ∼17 | 2011 | 665 |
| 6 | I2, LiI | 131 | 2.05 | 32.4 | 0.087 | ∼24 | 2011 | 665 |
| S | I2, LiI | 132 | 2.31 | 33.1 | 0.101 | ∼22.5 | 2011 | 665 |
| p-SQ1 | I2, LiI | 117 | 1.22 | 37.1 | 0.053 | ∼6 | 2012 | 666 |
| p-SQ2 | I2, LiI | 140 | 1.92 | 42.0 | 0.113 | ∼19 | 2012 | 666 |
| BQI | I2, BMII | 140 | 3.00 | 33 | 0.140 | ∼37 | 2017 | 571 |
| BQII | I2, BMII | 137 | 2.17 | 34 | 0.102 | ∼25 | 2017 | 571 |
| I | I2, LiI | 124 | 2.36 | 37 | 0.11 | ∼20 | 2013 | 667 |
| II | I2, LiI | 130 | 2.97 | 35 | 0.14 | ∼29 | 2013 | 667 |
| PMI-CO2H | T−/T2 | 161 | 1.52 | 25.4 | 0.062 | ∼20 | 2020 | 668 |
| PMI-HQ | T−/T2 | 164 | 2.21 | 23.8 | 0.086 | ∼21.5 | 2020 | 668 |
| PMI-DPA | T−/T2 | 168 | 1.33 | 24.6 | 0.055 | ∼26 | 2020 | 668 |
| PMI-acac | T−/T2 | 169 | 2.08 | 27.9 | 0.098 | ∼32 | 2020 | 668 |
| PMI-PO3H2 | T−/T2 | 181 | 1.27 | 17.7 | 0.041 | ∼20 | 2020 | 668 |
| CAD4 | I2, LiI | 84 | 3.96 | 31.6 | 0.105 | Not reported | 2017 | 669 |
| 1 | I2, LiI | 41 | 0.31 | 31 | 0.004 | 10 | 2017 | 670 |
| 2 | I2, LiI | 53 | 0.53 | 30 | 0.009 | 5 | 2017 | 670 |
| 3 | I2, LiI | 61 | 1.17 | 32 | 0.023 | 11 | 2017 | 670 |
| YK-1 | I2, BMII | 102 | 2.33 | 27.9 | 0.064 | ∼13 | 2018 | 671 |
| YK-2 | I2, BMII | 93 | 1.95 | 29.5 | 0.054 | ∼11 | 2018 | 671 |
| JW44 | I2, LiI | 75 | 1.29 | 31 | 0.030 | ∼21 | 2014 | 672 |
| 1 | I2, LiI | 57 | 0.28 | 35 | 0.006 | 5.4 | 2019 | 673 |
| 2 | I2, LiI | 74 | 0.45 | 35 | 0.012 | 8.2 | 2019 | 673 |
| 3 | I2, LiI | 76 | 0.51 | 37 | 0.014 | 9.8 | 2019 | 673 |
| ZnPref | I2, LiI | 98 | 0.19 | 35 | 0.006 | Not reported | 2019 | 673 |
| PP1 | I2, LiI | 132 | 1.45 | 36 | 0.069 | 10 | 2018 | 674 |
| SQ | I2, LiI | 85 | 1.18 | 34 | 0.034 | ∼24 | 2014 | 675 |
| SQ | Co(dtb-bpy)3 | 85 | 0.12 | 30 | 0.0041 | ∼2 | 2014 | 675 |
| PMI-NDI | I2, LiI | 135 | 0.69 | 35 | 0.033 | ∼15 | 2014 | 675 |
| PMI-NDI | Co(dtb-bpy)3 | 315 | 1.06 | 31 | 0.10 | ∼17 | 2014 | 675 |
| SQ-PMI | I2, LiI | 65 | 1.31 | 31 | 0.0026 | ∼24 | 2014 | 675 |
| SQ-PMI | Co(dtb-bpy)3 | 95 | 0.34 | 28 | 0.009 | ∼4 | 2014 | 675 |
| SQ-PMI-NDI | I2, LiI | 95 | 2.73 | 32 | 0.083 | ∼25 | 2014 | 675 |
| SQ-PMI-NDI | Co(dtb-bpy)3 | 175 | 1.17 | 27 | 0.055 | ∼22 | 2014 | 675 |
| 1 | I2, LiI | 100 | 1.89 | 33 | 0.063 | ∼26 | 2016 | 676 |
| 1 | Co(dtb-bpy)3 | 198 | 0.49 | 24 | 0.024 | ∼11 | 2016 | 676 |
| 2 | I2, LiI | 84 | 1.44 | 33 | 0.040 | ∼23 | 2016 | 676 |
| 2 | Co(dtb-bpy)3 | 134 | 0.41 | 24 | 0.013 | ∼7 | 2016 | 676 |
| DPP-Br | I2, LiI | 70 | 0.88 | 33 | 0.020 | ∼21 | 2016 | 676 |
| DPP-Br | Co(dtb-bpy)3 | 103 | 0.26 | 28 | 0.007 | ∼5 | 2016 | 676 |
| 3 | I2, LiI | 90 | 2.03 | 33 | 0.062 | ∼35 | 2016 | 676 |
| 3 | Co(dtb-bpy)3 | 330 | 2.06 | 30 | 0.205 | ∼26 | 2016 | 676 |
| 4 | I2, LiI | 76 | 1.72 | 32 | 0.041 | ∼24 | 2016 | 676 |
| 4 | Co(dtb-bpy)3 | 370 | 1.95 | 29 | 0.21 | ∼25 | 2016 | 676 |
| DPP-NDI | I2, LiI | 81 | 1.79 | 34 | 0.048 | ∼30 | 2016 | 676 |
| DPP-NDI | Co(dtb-bpy)3 | 292 | 1.56 | 29 | 0.13 | ∼28 | 2016 | 676 |
| ISO-Br | I2, LiI | 87 | 0.82 | 34 | 0.025 | ∼5 | 2015 | 677 |
| ISO-Br | Co(dtb-bpy)3 | 182 | 0.80 | 23 | 0.033 | ∼8 | 2015 | 677 |
| ISO-NDI | I2, LiI | 96 | 1.27 | 33 | 0.040 | ∼7 | 2015 | 677 |
| ISO-NDI | Co(dtb-bpy)3 | 260 | 1.54 | 25 | 0.100 | ∼13 | 2015 | 677 |
| ZnPref | I2, LiI, DMBII | 98 | 0.19 | 35 | 0.006 | Not reported | 2016 | 678 |
| ZnP-NDI | I2, LiI, DMBII | 127 | 1.38 | 32 | 0.056 | Not reported | 2016 | 678 |
| ZnP–TPA–NO2 | I2, LiI, DMBII | 107 | 0.29 | 38 | 0.012 | Not reported | 2016 | 678 |
| TCPP | I2, LiI | 128 | 0.8 | 39 | 0.04 | Not reported | 2014 | 679 |
| ZnTCPP | I2, LiI | 158 | 1.5 | 38 | 0.09 | ∼33 | 2014 | 679 |
| ZnP–CO2H–NO2 | I2, LiI, DMBII | 113 | 0.49 | 36 | 0.020 | ∼16 | 2015 | 680 |
| ZnP–eCO2H–NO2 | I2, LiI, DMBII | 114 | 0.48 | 35 | 0.019 | ∼16 | 2015 | 680 |
| ZnP–CO2H–eNO2 | I2, LiI, DMBII | 98 | 0.43 | 32 | 0.013 | ∼14 | 2015 | 680 |
| ZnP–eCO2H–eNO2 | I2, LiI, DMBII | 115 | 0.55 | 34 | 0.022 | ∼10 | 2015 | 680 |
| ZnP–CO2H–eNDI | I2, LiI, DMBII | 127 | 1.38 | 32 | 0.056 | ∼20 | 2015 | 680 |
| ZnP–CO2H–eNDI | Co(dtb-bpy)3 | 195 | 0.5 | 31 | 0.03 | Not reported | 2015 | 680 |
| ZnP–CO2H–BV2+ | I2, LiI, DMBII | 125 | 0.44 | 33 | 0.018 | ∼11.5 | 2015 | 680 |
| 3 | I2, LiI | 134 | 0.956 | 28.9 | 0.037 | 24.3 | 2019 | 653 |
| 3(Ni) | I2, LiI | 206 | 1.199 | 33.2 | 0.082 | 26.0 | 2019 | 653 |
| 4 | I2, LiI | 195 | 1.353 | 33.0 | 0.087 | 23.0 | 2019 | 653 |
| C60trZnPCOOH | I2, LiI | 109 | 1.86 | 37 | 0.076 | Not reported | 2018 | 681 |
| C60trZnPCOOH | Co(dtb-bpy)3 | 244 | 0.63 | 35 | 0.054 | Not reported | 2018 | 681 |
| C60trZnPtrCOOH | I2, LiI | 84 | 1.82 | 33 | 0.050 | Not reported | 2018 | 681 |
| C60trZnPtrCOOH | Co(dtb-bpy)3 | 269 | 0.76 | 36 | 0.074 | Not reported | 2018 | 681 |
| C60ZnPCOOH | I2, LiI | 103 | 1.68 | 37 | 0.063 | Not reported | 2018 | 681 |
| C60ZnPCOOH | Co(dtb-bpy)3 | 175 | 0.71 | 28 | 0.035 | Not reported | 2018 | 681 |
| PhtrZnPCOOH | I2, LiI | 68 | 0.69 | 33 | 0.015 | Not reported | 2018 | 681 |
| PhtrZnPCOOH | Co(dtb-bpy)3 | 48 | 0.22 | 24 | 0.002 | Not reported | 2018 | 681 |
| PMI-6T-TPA | Fe(acac)3 | 568 | 6.4 | 52 | 1.90 | ∼60 | 2018 | 682 |
| ZnP0 | Fe(acac)3 | 327 | 1.9 | 48 | 0.26 | Not reported | 2018 | 682 |
| ZnP1 | Fe(acac)3 | 465 | 4.4 | 45 | 0.92 | ∼43 | 2018 | 682 |
| VG1-C8 | Iodolyte Z-150 | 87 | 0.577 | 37.2 | 0.018 | ∼7 | 2016 | 683 |
| VG10-C8 | Iodolyte Z-150 | 102 | 0.435 | 40.9 | 0.018 | ∼7 | 2016 | 683 |
| VG11-C8 | Iodolyte Z-150 | 93 | 1.160 | 36.1 | 0.043 | ∼10 | 2016 | 683 |
| Erythrosine B | Iodolyte Z-150 | 88 | 1.019 | 36.0 | 0.032 | ∼5.5 | 2016 | 683 |
| BAI–COOH | I2, LiI | 79 | 1.13 | 33 | 0.029 | 7.8 | 2018 | 684 |
| CB5 | EL-HSE | 115 | 1.516 | 34.1 | 0.059 | ∼16 | 2018 | 685 |
| CB6 | EL-HSE | 117 | 1.135 | 31.4 | 0.044 | ∼7 | 2018 | 685 |
| CB7 | EL-HSE | 117 | 2.001 | 32.6 | 0.076 | ∼13 | 2018 | 685 |
| CB8 | EL-HSE | 117 | 1.717 | 32.9 | 0.066 | ∼11 | 2018 | 685 |
 | ||
| Fig. 44 Examples of triphenylamine-based sensitizers for p-type DSCs. | ||
The π-linker (e.g. oligothiophenes, fluorenes) length can also be optimized to maximize the absorption coefficient, the breadth of the spectral response, the energy offset at the interfaces with the semiconductor and electrolyte, the dye loading, the charge-transfer efficiency and recombination rate.652,654–657 PMI-nT-TPA series with oligothiophene bridges of different lengths greatly increased device performances (PCE = 0.09%, 0.19% and 0.41% for n = 1, 2 and 3 respectively) by further extending the charge-separated state lifetime (Fig. 45).550 Other examples include PMI-4T-TPA (JSC = 3.40 mA cm−2),582 T4H (JSC = 6.74 mA cm−2),655 BH4 (JSC = 7.40 mA cm−2),657 PMI-6T-TPA (JSC = 7.0 mA cm−2),686 zzx-op1 (JSC = 4.36 mA cm−2)646 and zzx-op1–2 (JSC = 7.57 mA cm−2).645 Fairly small structural changes to the dye seem to have a big impact, for example comparing O2 (JSC = 1.43 mA cm−2, VOC = 94 mV, FF = 37%, PCE = 0.05%)658 to a thienoquinoidal dye (with a I−/I3− electrolyte: JSC = 8.20 mA cm−2, VOC = 120 mV, FF = 34%, PCE = 0.33%; with a Co(III/II) electrolyte: JSC = 6.5 mA cm−2, VOC = 226 mV, FF = 34%, PCE = 0.50%).644 The EH series of p-type sensitizers with a D–A–π–A framework were prepared containing triphenylamine (TPA) as a donor, an electron-deficient 2,3-diphenylquinoxaline as the auxiliary acceptor, various thiophene derivatives as the π-linkers, methylene malonitrile as the electron acceptor, and carboxylic acid as the anchoring group.659 The p-DSC sensitized by EH174 with a bithiophene π-linker and with one anchoring group performed best (PCE = 0.207%, JSC = 4.84 mA cm−2, VOC = 137 mV, FF = 31.2%) and EH162 with an EDOT π-linker and double anchoring groups performed worst in the series.
 | ||
| Fig. 45 Examples of perylene monoimide- and naphthalene diimide-based sensitizers for p-type DSCs. | ||
The importance of the push–pull structure and the influence of the thiophene π-spacer have been demonstrated with bodipy dyes (Fig. 46). These are relatively straightforward to synthesize and simple modifications to the structure can be made to tune the absorption and emission wavelengths across the visible spectrum.660 The performance of bodipy dyes anchored through benzoic acid at the meso position is quite low, but push–pull bodipy dyes with a triphenylamine donor linked through a thiophene spacer to the bodipy chromophore perform much better (e.g. bodipy-6 PCE = ca. 0.3% and JSC = 3.15 mA cm−2).661 The electronic coupling between the donor and the chromophore is important and bodipy dyes with methyl pyrrole groups give a lower photocurrent compared to the pyrrole analogues (IPCE bodipy-4 = 27%, bodipy-7 = 53%, JSC = 5.87 mA cm−2), which is attributed to better electronic communication with the NiO substrate.662 Kubo et al. reported a NIR-absorbing π-extended dibenzo-bodipy dye applied in p-type DSCs with a I−/I3− electrolyte.663 Despite the push–pull structure – arising from the triphenylamine donor units and nitrothiophene acceptor – and the broad spectral response (up to 850 nm) the performance was still limited by rapid recombination at the dye/NiO interface (VOC = 79 mV, JSC = 0.61 mA cm−2, FF = 25%, PCE = 0.012%).
 | ||
| Fig. 46 Examples of different sensitizers for p-type DSCs. | ||
Generally, having two anchoring groups per triphenylamine unit is less favourable than having two acceptors because the extinction coefficient tends to be higher with two acceptors and the dye loading may be more compact.659 There have been some exceptions, such as the zzx-op series of fluorene-bridged biphenylamine-perylenemonoimide dyes, where the fluorene bridge was directly appended to biphenylamine to ensure good donor/acceptor coupling. W2 with an electron-withdrawing 1,3-benzothiadiazole bridge and an octyl-2-cyanoacrylate acceptor also performed well (JSC = 4.16 mA cm−2, VOC = 121 mV, FF = 33%, PCE = 0.166%).664 In certain cases, such as dye 3 vs. dye 5665 and p-SQ1 vs. p-SQ2,666 a double anchoring group can improve the solar cell performance through enhancing the binding strength between the dye and the semiconductor, thereby facilitating more efficient charge transfer, or by suppressing the dark current.665,666
Typically, carboxylic acid anchoring groups are used; however, until recently, there has been little research into whether or not this is the best choice.687 Alternative anchoring groups have been proposed, including pyridine,571,626,650,667,668 di(carboxylic acid)pyrrole,669,670 hydroxamic acid,671 di(carboxylic acid)triazole,638 catechol,622 carbodithioic acid,622 methyl phosphonic acid,622 acetylacetone (acac),668,672 alkoxysilane,188 coumarin,673 aniline,668 phosphonic acid,668 hydroxyqinoline,668 and dipicolinic acid.668 Phosphonic acid is one of the strongest binding groups and is resistant to both acid and base, but can present some synthetic challenges.668,673 Odobel et al. and Gibson et al. compared the charge-transfer dynamics at the dye/NiO interface for a number of anchoring groups and found that the anchoring group did not significantly influence the rates.668,673 This finding is consistent with the work of Housecroft et al. who compared the benchmark dye P1 with the phosphonic acid derivative PP1.674 The solar cell performance of both dyes was similar, PP1: PCE = 0.054–0.069%, IPCE = 10% at λmax = ∼500 nm; P1: PCE = 0.065–0.079%, IPCE = 13.5% at λmax = 500 nm.
Recombination at the dye/semiconductor surface appears to be a limiting factor to achieving high quantum efficiencies, unlike the analogous TiO2 devices.688,689 Perylene-based donor–acceptor dyads with varying acceptor units (such as either perylene itself coupled to a triarylamine donor, or NDI or C60 appended to a perylene) led to one of the most important breakthroughs in terms of extending the lifetime of the charge-separated state long enough to enable alternative redox mediators to be used (see below).627 The JSC for PMI-6T-TPA and P1 were similar when I−/I3− was used as the electrolyte (JSC = 5.35 vs. 5.48 mA cm−2), but the VOC was larger (218 vs. 84 mV), possibly due to reduced charge recombination at the electrolyte/electrode interface.564 Subsequently, there have been a number of reported dye series showing the benefits of the auxiliary acceptor on reducing charge recombination and, consequently, improving the device performance. These include Warnan et al.'s iodo-squaraines (SQ-PMI-NDI with I−/I3−: JSC = 2.73 mA cm−2, VOC = 95 mV, FF = 32%, PCE = 0.083%; with Co(III/II): JSC = 1.17 mA cm−2, VOC = 175 mV, FF = 27%, PCE = 0.055%),675 and Odobel et al.'s diketopyrrolopyrrole (DPP) and isoindigo series,676,677 which demonstrate the necessity for an appended NDI acceptor group to deliver good solar cell performance. NiO/Th-DPP-NDI produced a JSC of 8.2 mA cm−2, which is comparable to the record dyes CAD3 and QT-1.
Porphyrin dyes have been applied in state-of-the-art n-type DSCs, providing record efficiencies. However, rapid electron–hole recombination has limited their application in p-type DSCs.678,679,690 Odobel et al. attempted to improve their performance by covalently attaching methyl viologen and naphthalene diimide (NDI) acceptors at the meso position (ZnP-NDI dye), but these systems were limited by inefficient regeneration by I−/I3−.680 Chernick et al. developed a series of free-base and nickel asymmetric push–pull porphyrins with alternating meso substituents, electron-withdrawing pentafluorobenzene, electron-donating/coordinating 4-pyridyl ligand, and an electron withdrawing/synthetically modifiable 4-cyanophenyl unit.653 The porphyrins performed similarly to C343 (IPCE = 26%, PCE = 0.082% for the nickel porphyrin). Coordinating an electron acceptor such as C60PPy through the metal center of zinc porphyrins improves the p-DSC performance.673,679 Better p-DSC results were reported by Coutsolelos et al. who applied three covalently-linked donor–acceptor zinc porphyrin-fullerene (ZnP-C60) dyads (C60trZnPCOOH, C60trZnPtrCOOH and C60ZnPCOOH) with a triazole ring spacer between the porphyrin and C60 or anchoring group.681 Long-lived charge-separated states were observed in all three cases, due to a shift in electron density from the chromophore to the acceptor. The lifetime was enhanced by the presence of the triazole spacer for the dyads in solution, but it made only a moderate impact on the rate of charge separation and recombination when the dyads were adsorbed on NiO. However, the triazole ring did improve the photovoltaic performance. The presence of the C60 acceptor improved the solar cell performance compared to the C60-free reference compound PhtrZnPCOOH (with I−/I3− and C60trZnPCOOH: PCE = 0.076%; with Co(III/II) and C60trZnPtrCOOH: PCE = 0.074%). The best performance for a porphyrin photosensitizer in a NiO device so far was reported by Spiccia et al.682 ZnP1 contained a perylenemonoimide (PMI) electron acceptor linked through a fluorene and a Zn(II) porphyrin with alkyl chains as a π-conjugated bridge to a di(p-carboxyphenyl)amine (DCPA) electron donor. The configuration led to a red-shifted absorption onset to the near-IR region (∼800 nm) compared to the PMI-free reference dye ZnP0 (∼650 nm) and the benchmark PMI-6T-TPA (∼700 nm). With the tris(acetylacetonato)iron(III/II) redox mediator, ZnP1 (PCE = 0.92%) outperformed the ZnP0 sensitiser (PCE of 0.29%) but despite the broader spectral response, it did not perform better that the benchmark PMI-6T-TPA dye (2.0% PCE), possibly due to aggregation on the NiO surface.
To complement the state-of-the-art dyes for n-DSCs, red-NIR absorbing dyes have been developed. This is important for tandem devices, where the aim is to increase the spectral response and the VOC. A well-known class of red-NIR absorbing dyes are squaraines such as the VG and p-SQ series.666,683 Indigo is a naturally occurring red-absorbing dye, but its poor solubility makes it challenging to apply in solar cells. A bay-annulated indigo (BAI) was applied in p-DSCs producing a promising photocurrent (JSC = 1.14 mA cm−2), but the performance was limited by aggregation and charge recombination.684 Using a strong electron acceptor to lower the LUMO level in triphenylamine-based push–pull dyes shifts the absorption towards the red.643 Examples are COCO and COCN,649 the pyran-based dyes CB7 and CB8,685 and the CAD series.649,651
5.4 Electrolytes
The I−/I3− liquid electrolyte is most frequently chosen for p-type DSCs for compatibility with n-type DSCs.643 The composition can be optimized for the p-type system by the choice of solvent, typically acetonitrile, and additives, for example using lithium salts to lower the valence band potential, promote charge transport, limit charge recombination and increase the VOC.573,586,628,691,692 Ionic-liquid iodide sources such as 1-butyl-3-methylimidazolium iodide (BMII), 1-ethyl-3-methylimidazolium (EMII) and dimethylpropylimidazolium (DMPII) have also been shown to give good performance.586Drawbacks to the I−/I3− redox mediator include strong light absorption in the blue region, its corrosivity and the small difference between the redox potential of this electrolyte (315 mV vs. NHE) and the Fermi level of NiO, which limits the VOC of these devices to 100–200 mV.628,693 Exchanging I−/I3− for a transparent alternative with a more negative redox potential can increase the VOC of p-type DSCs. For example, the 5,5′-dithiobis(1-phenyl-1H-tetrazole) and sodium 1-phenyl-1H-tetrazole-5-thiolate couple has a redox potential of 245 mV vs. NHE, about 70 mV more negative than that of the iodide electrolyte.668,694 With optimised dyes, this electrolyte improved the VOC compared to I−/I3− and maintained a good JSC.695,696
Coordination complexes have given the most encouraging improvement to device efficiency (see Table 12). Co(III/II) complexes (Fig. 47) offer better optical transparency and tunable redox potentials compared to I−/I3−.697 Slower recombination at the electrolyte/electrode interface and more negative redox potentials than I−/I3− frequently translate to higher VOC (ca. 200–300 mV).698,699 However, a long-lived charge-separated state (dye−/NiO+) is required for dye regeneration with transition metal-based electrolytes to be efficient and in return, not all dyes are suitable. As mentioned above, a secondary electron acceptor, such as PMI or NDI, is required to generate long-lived dye radical anions.697,698 PMI-NDI sensitized NiO and a [Co(dtb-bpy)3]2+/3+ redox electrolyte led to a high VOC of 350 mV and an overall PCE of 0.20%.627 Modification of the peripheral ligands leads to differences in recombination rate and redox potential, leading to efficiencies ranging from 0.04 to 0.24%.697 The first example of a p-type DSC with an efficiency exceeding 1% was with PMI-6T-TPA and Co(III/II) tris(1,2-diaminoethane) ([Co(en)3]2+/3+).698 Interestingly, this redox mediator also performs well in aqueous electrolytes (PCE = 2%, IPCEmax = ∼40% between pH 8–11).700 The device efficiency was raised from 1.3% to 2.51% by substituting Co(en)3 for [Fe(acac)3]0/−.103 This is the highest reported efficiency to date for a p-type DSC.
| Mediator/HTM | Sensitizer | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) | IPCE max (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| Co(dtb-bpy)3 | DPP-NDI | 379 | 1.52 | 29 | 0.17 | Not reported | 2017 | 573 |
| Co(dtb-bpy)3 | PP2-NDI | 342 | 1.72 | 39.7 | 0.31 | ∼21 | 2018 | 696 |
| Co(dtb-bpy)3 | PMI-NDI | 340 | 2.00 | 35 | 0.24 | 33 | 2011 | 697 |
| Co(dtb-bpy)3 | PMI-PhNDI | 210 | 0.78 | 29.3 | 0.048 | ∼14 | 2011 | 699 |
| Co(dtb-bpy)3 | PMI-PhC60 | 180 | 0.58 | 38.8 | 0.040 | ∼23 | 2011 | 699 |
| Co(dtb-bpy)3 | 18 | 85 | 0.342 | 23.6 | 0.007 | Not reported | 2011 | 699 |
| Co(dtb-bpy)3 | 19 | 85 | 0.250 | 28.9 | 0.006 | Not reported | 2011 | 699 |
| Co(dtb-bpy)3 | C343 | 190 | 0.25 | 32 | 0.015 | ∼2 | 2009 | 627 |
| Co(dtb-bpy)3 | PI | 80 | 0.26 | 26 | 0.006 | ∼3 | 2009 | 627 |
| Co(dtb-bpy)3 | PINDI | 350 | 1.66 | 34 | 0.20 | 31 | 2009 | 627 |
| T−/T2 | PMI-CO2H | 161 | 1.52 | 25.4 | 0.062 | ∼20 | 2020 | 668 |
| T−/T2 | PMI-HQ | 164 | 2.21 | 23.8 | 0.086 | ∼21.5 | 2020 | 668 |
| T−/T2 | PMI-DPA | 168 | 1.33 | 24.6 | 0.055 | ∼26 | 2020 | 668 |
| T−/T2 | PMI-acac | 169 | 2.08 | 27.9 | 0.098 | ∼32 | 2020 | 668 |
| T−/T2 | PMI-PO3H2 | 181 | 1.27 | 17.7 | 0.041 | ∼20 | 2020 | 668 |
| T−/T2 | P1 | 304 | 1.73 | 44 | 0.23 | ∼19 | 2013 | 694 |
| T−/T2 | PMI-6T-TPA | 285 | 5.3 | 34 | 0.51 | ∼50 | 2015 | 695 |
| T−/T2 | PP1 | 169 | 1.60 | 30.5 | 0.082 | ∼17 | 2018 | 696 |
| T−/T2 | PP2 | 158 | 1.82 | 31.5 | 0.090 | ∼17 | 2018 | 696 |
| T−/T2 | PP2-NDI | 212 | 4.31 | 33.9 | 0.23 | ∼30 | 2018 | 696 |
| Co(dm-bpy)3 | PMI-NDI | 125 | 2.32 | 29 | 0.08 | 28 | 2011 | 697 |
| Co(dMeO-bpy)3 | PMI-NDI | 200 | 2.42 | 34 | 0.17 | 30 | 2011 | 697 |
| Co(ttb-tpy)2 | PMI-NDI | 240 | 1.61 | 33 | 0.13 | 31 | 2011 | 697 |
| Co(en)3 | PMI-6T-TPA | 654 | 5.23 | 43 | 1.48 | Not reported | 2016 | 700 |
| Fe(acac)3 | PMI-6T-TPA | 645 | 7.65 | 51 | 2.51 | 57 | 2015 | 103 |
| PCBM | DPP-PYRO | 228 | 0.32 | 32 | 0.023 | ∼3 | 2017 | 701 |
| PCBM | DPP-Br | 198 | 0.45 | 32 | 0.028 | ∼4.5 | 2017 | 701 |
| ZnO | BH4 | 480 | 0.346 | 39.4 | 0.07 | ∼3 | 2019 | 702 |
| ZnO | TIP | 535 | 0.855 | 39.8 | 0.18 | ∼5 | 2019 | 702 |
| ZnO | PB6 | 440 | 0.68 | 45 | 0.135 | ∼4 | 2019 | 703 |
| TiO2 | PB6 | 480 | 0.020 | 66 | 0.006 | ∼0.08 | 2018 | 704 |
 | ||
| Fig. 47 Structures of different redox mediators applied in p-DSCs. | ||
In addition to metal complex-based electrolytes, anionic metal oxide clusters known as polyoxometalates (POMs) are versatile and transparent electron reservoirs.709 POMs co-adsorbed on the surface of NiO can slow down the rate of charge-recombination and increase the VOC.710 Lindqvist et al. applied POMs (M6O192−) directly as redox mediators in p-DSCs, giving a four to five-fold increase in VOC compared to I−/I3−.711 Increasing the solubility of POMs could increase the short-circuit current of these cells to deliver competitive efficiencies.
Recently, a few solid-state p-DSCs (p-ssDSC) have been reported.712 Phenyl-C61-butyric acid methyl ester (PCBM) is a well-known solid electron-transfer material used in organic photovoltaics. Tian et al. found that the PCE of their p-ssDSC with P1 and PCBM was low due to slow dye regeneration by the electron transport material.712 Applying molecular dyads such as DPP (diketopyrrolopyrrole)-pyromellitimide (PYRO) can improve the performance.701 However, much improvement is required to deliver an efficient solid-state p-type DSC. Tian et al. followed up their work with organic electron transport mediators by completely removing the electrolyte/organic charge transport component and directly depositing TiO2 or ZnO on the NiO, so that the dye injects electrons directly into the n-type semiconductor and holes directly into the p-type semiconductor.702–704,713 This concept was first introduced by Bandara et al. but incomplete pore filling by the n-type semiconductor limited the cell performance.591,714 Tian et al. ha d optimised the interface between the dye and the semiconductors by engineering the structure of the dye and the deposition of the n-type semiconductor. Solar cells based on the TIP dye, containing an indacenodithieno[3,2-b]thiophene linker, gave PCE = 0.18%, JSC = 0.86 mA cm−2, VOC = 535 mV, FF = 40% and max IPCE of 5%.702
5.5 Photoelectrochemistry and photovoltaic performance
The key charge transfer processes that occur in a p-DSC under operation are summarised in Fig. 42 and the reactions important to photocathodes are:Electron transfer to the excited dye D* from the NiO valence band (“hole injection”):
| D* + NiO → D− + NiO|h+ |
Re-oxidation of the dye by the redox electrolyte (“dye regeneration”):693
| D− + I3− → D + I2−˙ + I˙ |
Diiodide disproportionation to form triiodide and iodide:
| 2I2−˙ → I3− + I− |
Recombination between the reduced dye and a hole in NiO:
| D− + NiO|h+ → D + NiO |
Recombination of a hole in NiO with the reduced species in the electrolyte:
| 2NiO|h + 3I− → 2NiO + I3− |
Over the last ten years, there have been extensive studies into the dynamics of each process. Charge injection is typically a fast process, between 100 femtoseconds to 100 picoseconds according to transient absorption spectroscopy and time-resolved infrared spectroscopy.668,688 The surface electronic states at the interface between NiO and a series of bodipy dyes have been studied by hard and soft XPS and the good overlap between the dye HOMO and semiconductor valence states was consistent with rapid light-induced charge transfer.660 Recombination at the dye−/NiO+ interface, however, is also fast, occurring on a picosecond to nanosecond time scale in simple dye systems such as bodipy and perylene.660,715,716 Regeneration occurs from a nanosecond up to microsecond time scale. Competition between recombination and regeneration is responsible for the poor efficiency for p-type DSCs.642,661,699 Recombination between holes in NiO with the reduced dyes contributes to the low FFs.717,718 A hole-hopping charge transport mechanism has been proposed for NiO, arising from “trap states” such as Ni3+ and Ni4+.568,610,719
The Ni3+ states are important for charge transport and charge recombination at the NiO/electrolyte and NiO/dye interfaces.565,566,577,720 Competition between these processes leads to the short diffusion length and low fill factors observed in NiO-based DSCs.721 Unlike TiO2, the charge carrier lifetime is independent of light intensity or charge density and a charge hopping process, regulated by ions in the electrolyte, takes place at the NiO surface.722 The NiO preparation and deposition route affects both the charge lifetime and transport time.560,564,565 Small amplitude light-modulated transient photocurrent and photovoltage decay measurements and electrochemical impedance spectroscopy (EIS) have also been used to study the effect of doping, of applying an insulating blocking layer and of varying the redox mediator and dye structure on the hole lifetime and transport time.576,697,699,723,724 Application of a NiO blocking layer to suppress charge recombination led to a higher photocurrent and fill factor.725 A Ni(CH3COO)2 treatment to the NiO film was also shown to suppress the hole recombination and led to a 31.3% improvement in the photovoltaic performance.726 Insulating coatings of Al2O3 and TiO2 on the NiO surface increase the recombination resistance and increase the VOC and efficiency of the device.570,571 Chemical treatments such as immersing in NaBH4 or NaOH have also been used to improve the VOC and FF by addressing the Ni3+ surface states and decreasing recombination.568,727,728
Developing new semiconductors, such as alternative metal oxides with better hole mobility compared to NiO or reducing electronic vacancies present above the valence band edge could favour charge transport over recombination.719 Lithium ions ha ve been well-characterized as dopants for NiO and improve the electrical properties of the films, shifting the valence band position to more positive potential, altering the density of states, narrowing the trap energy distribution and increasing the energy barrier for charge recombination.577 Doping NiO with Co has been shown to increase the charge transport lifetime from ∼5 ms for pure NiO to more than two-fold for 2% and 6% Co-doped NiO films. The VOC increased from 122 mV up to a maximum of 158 mV with >6% cobalt doping due to a lowering of the flat-band potential of the NiO by a few tens of mV and also to higher hole lifetimes for the Co-doped cells than those for pure NiO cells.576 Guldi et al. studied the charge transfer processes in CuO photocathodes with I−/I3− using electrochemical impedance spectroscopy.585 They probed the effect of calcination temperature, electrode thickness, and electrolyte ratio on the charge transfer resistance RCT, charge collection efficiency ηcc, diffusion coefficient D and hole lifetime τh and determined that a 300 °C calcination temperature, a film thickness of 5.0 μm and an I−/I3− electrolyte ratio of 2.5:1 gave the optimum balance of dynamics and best device performance. The experiments also revealed less recombination at the electrode/electrolyte interface for CuO compared to NiO.
The dye structure has been shown to affect the charge transfer dynamics. Push–pull donor–acceptor dyes and molecular dyad and triad structures have been developed to extend the charge-separated state lifetimes from tens of picoseconds into the microsecond to millisecond regime.637,666,673,679,699,715,729–731 By extending the linker it is possible to increase the charge-separated state lifetime without decelerating the rate of charge separation.550,657 Varying the coupling between the chromophore and the linker increases the charge-separated state lifetime, but this comes with a sacrifice to the charge injection yield, so a balance must be struck to optimize the performance.634 Adding bulky alkyl chains to the dye, or forming a compact arrangement of dye molecules at the electrode surface inhibit charge recombination at the semiconductor/electrolyte interface, leading to longer charge lifetimes.645,646,725 A surprise came from exploring the charge transfer dynamics of P1 and CAD3,647 which – despite having relatively short charge-separated state lifetimes (ca. <10 ns) – still generate relatively high photocurrents in NiO DSCs. When iodine and lithium iodide were added, the charge-separated state decayed over a one order of magnitude longer time scale compared to the lifetime recorded in the presence of an inert electrolyte. It is possible that there is pre-association of the electron acceptor in the electrolyte with the cationic dyes, or reduction of the high valence states on the surface of NiO by the electron donor in the electrolyte. I− in the electrolyte has been shown to reduce the Ni3+ states, which are thought to be responsible for rapid charge recombination, so a dual effect might be responsible for the increased charge-separated state lifetime in the presence of the redox electrolyte.566,568,691
With electrolytes based on cobalt polypyridyl complexes, the hole lifetimes were shown to be – like with I−/I3− – strongly dependent on light intensity, whereas the hole transport times were largely independent of light intensity. Charge transport times have been found to be almost independent from the structure of the cobalt complexes, but charge lifetimes depend on the steric bulk of the cobalt polypyridyl complex. Most importantly, charge lifetimes were shown to be longer with cobalt complexes (particularly with bulky ligands) compared to I−/I3−.699 Electrolyte additives, such as chenodeoxycholic acid, have also been shown to slow recombination at the electrode/electrolyte interface.573 In these examples, the longer charge lifetimes corresponded with higher open circuit voltage.
5.6 Tandem devices
Tandem DSCs offer an opportunity to increase the solar cell efficiency beyond what can be attained by a single photoelectrode. The top electrode captures the higher energy photons and the transmitted lower energy photons are captured by the bottom electrode. However, the low performance of the photocathodes limits the performance of tandem DSCs. Early studies focused on proving the principle that the VOC of the tandem DSC is the sum of the individual n-type and p-type DSCs, but the devices suffered from very low photocurrents and poor fill factors.547 These first tandem DSCs typically contained I−/I3− as the redox mediator, but substituting it for metal complexes and commercial photosensitizers for dyes designed specifically for photocathodes has led to an improved performance.627 In particular, advances have been made in developing dyes which absorb in the red to NIR region of the solar spectrum to complement state of the art photosensitizers for TiO2 devices. For example, Gibson et al. reported a tandem cell with up to 5.2 mA cm−2 employing the cationic charge-transfer dye CAD3 on NiO and a benchmark charge-transfer dye D35 on TiO2.643 Guldi et al. incorporated Zn(II) phthalocyanines (ZnPc) in photocathodes based on CuO and assembled them in tandem devices with N719 on TiO2, giving a light harvesting range from 300 nm to 800 nm (JSC = 1.28 mA cm−2, VOC = 860 mV, FF = 63%, PCE = 0.69%).732 A more encouraging efficiency of 2.42% was reported by Bach et al. with PMI-6T-TPA as the dye and Fe(acac)3 as the electrolyte.550 Odobel et al. reported a dye-sensitized tandem cell with a diketopyrrolopyrrole (DPP)-based sensitizer at the photocathode (NiO/Th-DPP-NDI) and a TiO2/D35 photoanode. The tandem DSC efficiency was greater than that of the individual p-type and n-type devices (JSC = 6.73 mA cm−2; VOC = 910 mV; PCE = 4.1%).548Deepa et al. reported the most efficient tandem cell to date at 9.76% for a device which included a photocathode with a nickel pthalocyanine dye (NiPcTs) on NiO supported over carbon fabric.733 The photoanode was assembled from conducting core/shell copper@carbon dots anchored to CdS quantum dots on TiO2 and a polysulfide electrolyte was used for compatibility with the CdS. The efficiency of the photocathode half-cell was quite low (0.039%) but when incorporated into the hybrid tandem device it improved the efficiency by almost 3% compared to the photoanode device with carbon fabric alone as the counter electrode (6.69%). Most of the improvement came from the higher photocurrent.
The key issue with tandem devices is that, although great steps have been made in improving the photocurrent density by developing new photosensitizers and improving the photovoltage through developing new redox mediators, the efficiency is still limited by the valence band position of the p-type semiconductor. A semiconductor with a lower valence band than NiO or replacing TiO2 with a material with a higher-lying conduction band is needed to improve the built-in potential of tandem devices. Other than the tandem device by Guldi et al. described above,732 a tandem cell by Kaya et al. assembled from a photocathode of CuCrO2 with a coumarin 6 organic dye, iodide-based redox mediator and N719-sensitized TiO2 photoanode gave a PCE of 2.33% with VOC of 813 mV, JSC of 4.83 mA cm−2, and fill factor of 59%.734 If an alternative p-type transparent semiconductor with a valence band 0.5 V deeper than NiO could be found, an efficiency above 20% would be possible. However, as described above, there is no obvious choice to replace NiO yet.
6 DSCs for solar fuel
The diffused and intermittent nature of solar energy dictates the requirement for energy storage in solar energy conversion strategies. Chemical bonds are arguably the most appealing choice for this goal. For over two billion years, nature's photosynthesis has been converting solar energy into chemical potential, while also sequestering CO2 and producing most of the oxygen in our planet. All fossil fuels we use today are derived from the natural photosynthetic process. Artificial photosynthesis aims to emulate natural photosynthesis to generate solar fuels and commodity chemicals from sunlight using H2O, CO2 and N2 as feedstocks. In the last decade, DSCs have played key roles in one of the fastest-growing artificial photosynthetic approaches, Dye-Sensitized Photoelectrosynthesis Cells (DSPECs). A DSPEC is a modified DSC in which the reduced form of the redox shuttle in the anode compartment is replaced with an oxidation catalyst (e.g. a water oxidation catalyst), while the oxidized form of the redox shuttle in the cathode compartment is replaced with a reducing catalyst (e.g. a proton reduction catalyst). In a DSC the goal is to convert sunlight into electricity to power a device or to charge a battery. In a DSPEC the goal is to convert and store sunlight into chemical bonds, producing O2 or a commodity chemical at the anode and a fuel at the cathode.
Fig. 48 shows a schematic representation of a DSPEC for water splitting. Light-driven water oxidation takes place at the photoanode, composed of a chromophore-catalyst assembly on a mesoporous n-type semiconductor film, and proton/water reduction occurs at a dark Pt cathode. At the photoanode, the chromophore in the chromophore-catalyst assembly is responsible for light absorption and subsequent electron injection from its excited state(s) into the conduction band of the semiconductor. The injected electrons are transported to a transparent conducting oxide (TCO) electrode and delivered to the cathode for proton/water reduction. Electron transfer from the water oxidation catalyst to the oxidized chromophore initiates the activation of the water oxidation catalyst and regenerates the chromophore. This process is repeated four times leading to O2 evolution at the photoanode and H2 evolution at the dark cathode, ideally in a 1:2 O2/H2 ratio, returning the chromophore-catalyst assembly to its initial state.
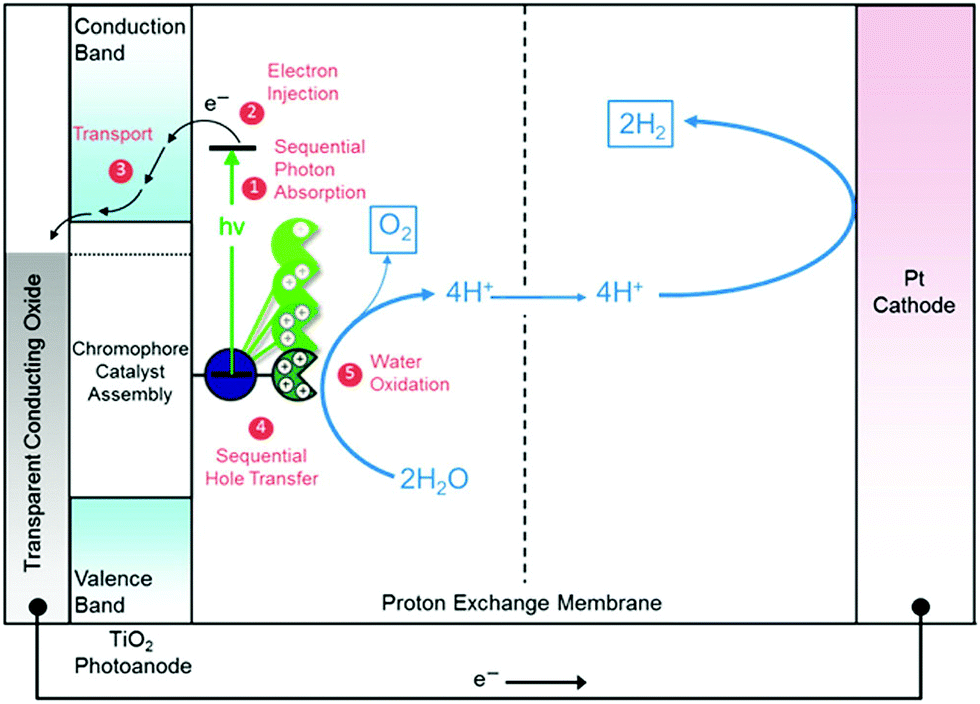 | ||
| Fig. 48 Schematic diagram of a DSPEC for light-driven water splitting with an assembly-derived TiO2 photoanode for water oxidation to O2 and a dark Pt cathode for proton/water reduction to H2. Reprinted with permission from ref. 735. Copyright 2015 American Chemical Society. | ||
Meyer and co-workers reported the first DSPEC in 1999,736 almost a decade after the pioneering DSC work of O'Regan and Grätzel.5 The DSPEC carried out light-driven dehydrogenation of isopropanol to acetone at the photoanode with H2 generation at the dark Pt cathode. It took yet another decade for the development of the first DSPEC for water splitting by Mallouk and co-workers in 2009.737 Nevertheless, the last 12 years have seen an impressive development in this area.735,738–775 The first DSPEC for water splitting reported by Mallouk and co-workers generated a photocurrent of 12.7 μA cm−2 at pH 5.8 under 450 nm light irradiation (7.8 mW cm2) with an internal quantum yield of 0.9% and a faradaic efficiency for O2 generation of 20%.737 Just a decade later, DSPECs are reaching photocurrent densities of ∼2.2 mA cm−2 at pH 7.0 under 1 sun illumination with an incident photon to current efficiency (IPCE) of 29% at 450 nm and faradaic efficiencies for O2 generation over 70%. Correcting for the injection yield of only ∼42% for the chromophore at pH 7.0, the efficiency of the cell, excluding the losses at the core/shell interface, is a remarkable 67%.774
6.1 Photoanodes and photocathodes
In theory, a tandem DSPEC (discussed below in Section 6.5) with both a photoanode and a photocathode could provide significant advantages over a DSPEC with just a photoanode and a dark cathode. Absorption of one photon at the photoanode and one photon at the photocathode by two complementary dyes would emulate the Z-scheme in natural photosynthesis and enable coverage of a wider range of the solar spectrum. In addition, a photocathode would provide additional voltage that could eliminate the need for an applied bias to generate H2 at the photocathode or enable access to fuels from CO2 using catalysts with higher overpotentials than those used to produce H2 as the fuel. Unfortunately, as in the case of DSCs, the development of tandem DSPECs has been hampered by the lack of suitable p-type photocathode materials.In a typical DSC, the photosensitizer or chromophore is anchored to the semiconductor material, while the redox shuttle is free to diffuse from the anode to the cathode and back. In a DSPEC, on the other hand, the oxidation catalyst must be immobilized on the photoanode and it must undergo multiple, successive oxidations to complete one cycle or turnover. For this reason, the position and distance of the oxidation catalyst with respect to the photosensitizer and the semiconductor are key aspects in determining the overall cell performance. This has led to many approaches in the assembly of chromophores and catalysts on the nanoparticles' surfaces of the semiconductor.
The first DSPEC reported used a chromophore-catalyst assembly in which the two were chemically linked through a bridge prior to loading onto the semiconductor surface.736 This design allows precise control of the distance between chromophore and catalyst and positions the catalyst away from the semiconductor surface to inhibit recombination reactions between injected electrons and oxidized catalyst molecules. However, such chromophore-catalyst assembly designs require cumbersome synthetic procedures. The first chromophore-catalyst assembly for water splitting was not suitable for a DSPEC: In the excited state of the chromophore, the excited electron was localized in the bridging ligand and the injection yield into the conduction band of TiO2 was less than 5%.776 Other chromophore-catalyst assembly designs failed to perform in a DSPEC configuration because the oxidized chromophore did not have enough oxidizing power to generate the RuV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O form of the catalyst, a key intermediate for the initial O–O bond formation step.735,777,778
O form of the catalyst, a key intermediate for the initial O–O bond formation step.735,777,778
Introduction of carbene-based water oxidation catalysts in chromophore-catalyst assemblies enabled access to O–O bond formation already at the less-oxidized RuIVO form of the catalyst with additional redox power available from the weakly-coupled Ru(III) chromophore. Water-splitting DSPECs involving a single-site water oxidation catalyst in the chromophore-catalyst assembly were successfully developed.742,749,769
The discovery of the [Ru(bda)(L)2] (bda: 2,2′-bipyridine-6,6′-dicarboxylate; L is a monodentate ligand , Fig. 51) water oxidation catalysts by Sun and co-workers779,780 and their incorporation into chromophore-catalyst assemblies led to significant improvements on DSPEC performance because of their low overpotential and high rates for water oxidation.763,766 This type of catalysts was first used on a DSPEC configuration by loading the catalyst into a Nafion overlayer deposited on top of a Ru(bpy)3-sensitized TiO2 mesoporous film.738 Nevertheless, the first significant DSPEC breakthrough was achieved by co-loading a Ru(bpy)3-type chromophore and a Ru-bda catalyst on TiO2.743 Photocurrent densities up to 1.7 mA cm−2 at pH 6.8 were obtained with a 14% IPCE at 450 nm and 83% faradaic efficiency for O2 generation. This co-loading strategy has been successfully used in DSPEC photoanodes with a variety of chromophore-catalyst combinations.748,753,759,765,768
Mallouk and co-workers introduced a layer-by-layer approach to load chromophores and catalysts on the surface of the semiconductor.737 The authors prepared a Ru(bpy)3-type chromophore containing one phosphonated bipyridine ligand for TiO2-anchoring, and another ligand functionalized with a malonate group that was selective for binding and stabilizing the colloidal IrO2·nH2O water oxidation catalyst nanoparticles. A related layer-by-layer strategy for nanostructured metal oxide films was developed by Meyer and co-workers781 based on previous studies on Si and Au planar electrodes.782,783 This strategy takes advantage of the strong affinity of phosphonate groups for high valent cations such as Zr(IV), and it has been successfully applied in a variety of DSPEC photoanode designs as well as in photocathodes, discussed below.741,745,759,784 In yet another layer-by-layer strategy, a thin film of an oxide (TiO2, Al2O3, etc.) a few nm thick is deposited by atomic layer deposition (ALD) on top of the pre-loaded chromophore. The water oxidation catalyst is then loaded onto this oxide layer using typical metal-oxide anchoring groups. In addition to enabling loading of the catalyst, the ALD overlayer stabilizes and protects the chromophore. The ALD layer-by-layer approach has been extensively used in DSPEC photoanodes.764,767,785
Electropolymerization techniques have also been used to prepare DSPEC photoanodes. In this approach, electropolymerizable groups (e.g. vinyl groups) are introduced in both chromophore and catalysts which end up chemically linked during the electropolymerization process.750,754,761 A variation of this strategy simply electropolymerizes a film of the catalyst on top of a dye-functionalized electrode. The low water solubility of the polymer retains the catalyst molecules on the pores of the mesoporous electrode.752
A recent development for the assembly of chromophores and catalysts on an electrode surface takes advantage of hydrophobic interactions between long alkyl chains to build self-assembled bilayers (SAB, Fig. 49).786 In this approach, a chromophore containing both anchoring groups and long alkyl chains is loaded onto an electrode surface and the resulting chromophore-functionalized electrode is then immersed in a solution of the water oxidation catalyst which has also been functionalized with long alkyl chains. The long alkyl chains in the catalyst molecules self-assemble with the long alkyl chains in the chromophore to create a SAB. This approach allows easy combination of various chromophores and catalysts with the distance between them controlled by the length of the alkyl chains.
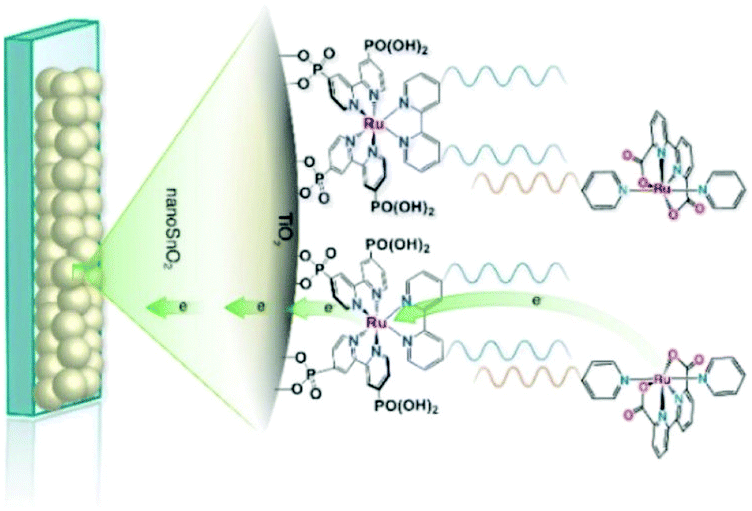 | ||
| Fig. 49 Self-assembled bilayer of a chromophore-catalyst assembly on a metal oxide. Reprinted with permission from ref. 774. Copyright 2019 American Chemical Society. | ||
A water splitting DSPEC built using this strategy reached photocurrent densities of ∼2.2 mA cm−2 under 1 sun illumination at pH 7.0 with an IPCE of 29% at 450 nm and faradaic efficiencies for O2 generation over 70%. Correcting for the injection yield of only ∼42% for the chromophore at pH 7.0, the efficiency of the cell – excluding the losses at the core/shell interface – is a remarkable 67%. At pH 4.7, the cell was operated over a 3 hour period with an 86% faradaic efficiency for O2 generation.774
The first sensitized photocathode for light-driven hydrogen generation was reported by Sun and co-workers.790 It consisted of a cobaloxime molecular catalyst in solution and an organic triphenylamine-type dye anchored on nanostructured NiO. An analogous photocathode, but with the cobaloxime catalyst also anchored to the NiO, was used to prepare an organic dye tandem water splitting DSPEC.753 The cell reached photocurrent densities of −300 μA cm−2 at pH 7 with an IPCE of 25% at 380 nm. Wu and co-workers reported a dye-sensitized photocathode that displayed high stability in strongly acidic solutions.791 As shown in Fig. 50, the organic dye was composed of a triphenylamine (TPA) donor moiety that was linked to two perylenemonoimide (PMI) acceptor groups via oligo-3-hexylthiophene-conjugated π-linker groups on each side of the donor moiety. Carboxylic acid groups on the TPA donors allowed the anchoring on NiO, while the hydrophobic hexyl groups in the thiophene linkers offered protection for both the anchors and the NiO from the very acidic environment in which they were embedded. An acid-stable cubane molybdenum sulphide cluster – [Mo3S4]4+ – was chosen as the proton reduction catalyst. The cell sustained photocurrents beyond −180 μA cm−2 for more than 16 hours at pH 0 in 1.0 M HCl with a 49% faradaic efficiency for H2 generation. Artero and co-workers also reported a NiO-based photocathode using a TPA chromophore covalently linked to a cobaloxime catalyst.792
 | ||
| Fig. 50 Photocathode for hydrogen generation. Reprinted with permission from ref. 791. Copyright 2016 American Chemical Society. | ||
Wasielewski and co-workers used ALD to deposit a thick Al2O3 layer on top of the NiO film with a modified perylene-3,4-dicarboximide chromophore (PMI). In addition to providing protection for the NiO from the aqueous solution, the Al2O3 layer films allowed longer charge separated lifetimes as characterized via femtosecond transient absorption spectroscopy and photoelectrochemical techniques. Light-driven H2 generation was demonstrated with both cobaloxime and Dubois' Ni(L)2-type catalysts (L is a diphosphine).793 Meyer and co-workers also used an ALD layer of Al2O3 on NiO as a bridge between a Ru(bpy)3-type chromophore and a Ni(L)2 proton reduction catalyst, an assembly strategy similar to that reported above for photoanodes.764,767,785
The shortcomings of NiO as a p-type material for photocathodes has prompted scientists to look for new alternatives. Reisner and co-workers have used the delafossite-type material CuCrO2 as a suitable p-type semiconductor for visible light-driven H2 generation.794 The semiconductor was functionalized by co-loading a phosphonated diketopyrrolopyrrole dye with a Ni(L)2 proton reduction catalyst. The hybrid CuCrO2 photocathode displayed a photocurrent of −15 μA cm−2 at 0.0 V vs. RHE in pH 3 aqueous electrolyte solution under UV-filtered simulated solar irradiation. The photocathode displayed good stability and a turnover number of 126 for H2 production was recorded for their Ni(L)2 catalyst during a 2 hour operation. The CuCrO2-based system outperformed a similar photocathode based on NiO, but product generation was limited by the low dye and catalyst loadings. In a follow-up study, macropore architectures of inverse opal CuCrO2 led to a five-fold increase in loading.795
More recently, Meyer and co-workers used boron-doped Si as the p-type material.784 Si nanowires ∼18 μm long were modified by physical vapor deposition of a thin Ti layer (∼10 nm), followed by ALD of a ∼3.0 nm TiO2 layer. The latter protected the p-type Si electrode from photodegradation and allowed anchoring of phosphonate-functionalized perylene-diimide (PDI) chromophores. Ni(L)2 proton reduction catalysts were introduced using the Zr-bridged layer-by-layer approach.781 The integrated photocathode was capable of delivering a photocurrent density of about −1.0 mA cm−2 under zero applied bias (vs. NHE).
Photocathodes for CO2 reduction are even more challenging due to the larger overpotentials of CO2 reduction catalysts compared to proton reduction catalysts. Nevertheless, significant progress has been made on this front in recent years. Ishitani and co-workers reported a photocathode for reduction of CO2 to CO using a NiO electrode functionalized with a Ru(II)-Re(I) supramolecular complex.796 During a 5 hour operation, the photocathode carried out 32 turnovers with a faradaic efficiency of 65% for CO, although the experiments were carried out in a DMF:triethanolamine (5:1) mixture with an applied bias of −1.2 V vs. Ag/AgNO3. The same Ru(II)–Re(I) supramolecular complex on a CuGaO2 p-type semiconductor displayed photoelectrochemical activity for the conversion of CO2 to CO with 68% faradaic efficiency in an aqueous electrolyte solution with an applied bias of −0.7 V vs. Ag/AgCl.797
More recently, Meyer and co-workers developed photocathodes using a novel method based on a binary p–n junction to convert sunlight into electrons with high energy to drive the CO2 reduction reaction to produce formate in an efficient way.798 Such photocathodes featured a semiconductor p–n junction constituted of GaN nanowire arrays on silicon together with surface-bound molecular assemblies to perform light absorption and catalysis. The reduction of CO2 to formate proceeded at a stable photocurrent density of about −1.1 mA cm−2 during 20 h of irradiation, with faradaic efficiencies of up to 64%.
6.2 Photosensitizers
The photosensitizers (or chromophores) used in DSPECs must meet additional demands compared to those used in DSCs. In the photoanode, the oxidized photosensitizer must be capable of oxidizing the water oxidation catalyst through a series of increasingly challenging oxidation states during the water oxidation cycle. In addition to the thermodynamic requirements for such a task, some (or all) of the oxidation steps of the catalysts are proton-coupled in nature and this adds to the kinetic barriers for these oxidations. Because of this, in a DSPEC the photosensitizer remains for longer times in its oxidized form compared to DSCs, which leads to significantly faster decomposition of the photosensitizer. Another important issue is that injection efficiency into the conduction band of the semiconductor is pH-dependent due to the pH dependence of the latter.799,800 In addition, in the aqueous environment where DSPECs operate, long-term stability of the anchoring groups of the photosensitizer remains a challenge. Phosphonic acid groups have been the dominant choice in this regard for both photoanodes and photocathodes, although recent studies include the use of significantly more robust silanes.801–804[Ru(bpy)3]2+-Type chromophores have dominated the DSPEC literature in the photoanode side737,738,741,743,745,748,750,752,754,759,761,765–767,774,784 with a few other examples including zinc porphyrins763 and triphenylamine derivatives.753,762,764 Recent efforts have been made on developing new chromophores with higher oxidation potentials that could enable faster oxidation of the water oxidation catalyst, the use of water oxidation catalysts with higher overpotentials, and DSPEC operation at low pH. Unfortunately, tuning the ground state redox potential of the chromophore commonly also affects their excited state energy levels. Brudvig and co-workers developed a series of CF3-substituted free-base and metalated porphyrins that displayed redox potentials in the 1.25–1.56 V vs. NHE range, higher than the unsubstituted analogues.82 The new porphyrins showed high efficiency for injection into SnO2 but poor injection into TiO2. Meyer and co-workers prepared a series of complexes of the type [Ru(bpy)2(N–N)]2+ (N–N is a polypyridyl ligand with low-lying π* levels). With this approach, the absorption spectra of the new chromophores could be red-shifted up to λmax = 564 nm for the lowest MLCT, compared to 449 nm for the parent [Ru(bpy)3]2+ complex. In addition, the redox potentials for the Ru3+/2+ couples could be enhanced by more than 250 mV. However, these improvements came at the expense of the excited state energy becoming more positive than the conduction band of TiO2, rendering these chromophores unsuitable for excited state electron injection.805 In a follow-up work, introduction of electron-withdrawing groups on the bipyridine ligands enabled a ∼200 mV increase in the Ru3+/2+ couple for surface-bound chromophores. But once again, this improvement resulted in more positive excited state energies and smaller driving forces for electron injection.806 More recently, the introduction of –CF3 and/or –PO3H2 groups on all ligands in tris-homoleptic [Ru(bpy)3]2+-type chromophores resulted in redox potential upshifts of the Ru3+/2+ couple up to 1.6 V vs. NHE while retaining a similar absorption profile and photophysical properties compared to the [Ru(bpy)3]2+ complex.807 These chromophores enabled photochemical water oxidation to be carried out at pH 1 for the first time.
Significant efforts have been also made on developing organic chromophores for both photoanodes and photocathodes. This subject has been recently reviewed by Abbotto and co-workers and it is beyond the scope of this review.808 A recent review on chromophores/sensitizers for photocathodes for both DSCs and DSPECs has been published by Odobel and co-workers.809
6.3 Catalysts
Most studies reported to date in DSPECs have used only a handful of catalysts for both photoanodes and photocathodes. After the first DSPEC for water splitting reported by Mallouk and co-workers737 that used IrOx nanoparticles as water oxidation catalyst in the photoanode, the majority of the reports that followed used either [Ru(tpy)(Mebim-py)(OH2)]2+ (tpy: 2,2′:6′,2′′-terpyridine; Mebim-py: 1-methyl-3-(pyridin-2-yl)-1H-benzo[d]imidazol-3-ium-2-ide)810–813 or [Ru(bda)(L)2], Fig. 51.779,780 | ||
| Fig. 51 Structures of Ru-based water oxidation catalysts. | ||
[Ru(tpy)(Mebim-py)(OH2)]2+ is a single-site water oxidation catalyst and retains its homogeneous catalytic performance when immobilized on the surface of photoanode materials. Nevertheless, its high overpotential and low rates for water oxidation resulted in poor performances for DSPECs using this catalyst. [Ru(bda)(L)2]-type catalysts, on the other hand, follow a bimolecular pathway for water oxidation and do not retain their impressive homogeneous catalytic performance when heterogenized, generating μ-oxo bridged, blue dimer-like structures on the surface of the electrode.814,815 These structures are the true water oxidation catalysts on the surface and their number is only a fraction of all the heterogenized monomeric catalysts that have the proper distance and orientation to generate μ-oxo bridged species. Nevertheless, their high water oxidation activity and low overpotential enable DSPECs using these catalysts to display remarkable performance.
Single-site water oxidation catalysts capable of oxidizing water at high rates and low overpotentials, and which retain their homogeneous catalytic activity when heterogenized could potentially lead to significant improvements in DSPEC performance. Llobet and co-workers have reported single-site water oxidation catalysts with impressive rates although at neutral and basic pH values.816,817 Combining the features of single-site bisphosphonate catalysts ([Ru(bpaH2)(L)2], bpaH2 is 2,2′-bipyridine-6,6′-diphosphonic acid)818 and fast bimolecular [Ru(bda)(L)2]-type catalysts, Concepcion and co-workers have developed hybrid water oxidation catalysts ([Ru(bpHc)(L)2], bpHc is 6′-(hydroxyoxidophosphoryl)-[2,2′-bipyridine]-6-carboxylate) that are faster than the parent catalysts under identical conditions in both chemical and photochemical water oxidation.815,819 Nevertheless, the performance of these catalysts in DSPEC configurations has not been reported to date.
On the photocathode side, catalysts can be separated into two groups: catalysts for proton/water reduction (other than platinum) and catalysts for CO2 reduction. Most studies where a molecular catalyst was used to carry out proton/water reduction at the photocathode used either cobaloxime-type catalysts753,792,793 or the Ni(II) bis(diphosphine) complexes developed by DuBois and co-workers.784,793–795,820–823 A cubane molybdenum-sulfide cluster was also successfully used for proton reduction in extremely acidic (pH 0) conditions and displayed significant stability with up to 16 hours of H2 generation with no degradation.791 However, none of these catalysts have been able to perform at the level of a platinum electrode in a DSPEC. Bias voltages are required to drive H2 evolution even with platinum, with just a few exceptions. Nevertheless, the applied bias is typically due to improper alignment between the conduction band of the photoanode material and the redox potential of the H+/H2 couple rather than overpotential issues related to the proton reduction catalyst. DSPEC studies where water oxidation at the photoanode is accompanied by CO2 reduction at the photocathode are scarce. Ishitani and co-workers have reported CO2 reduction to CO at a CuGaO2 photocathode using a chromophore-catalyst assembly consisting of a [Ru(bpy)3]2+-type chromophore and a [Re(bpy)(CO)3(Br)] catalyst.797 Nevertheless this was not a true DSPEC, because water oxidation was carried out by direct bandgap excitation of the photoanode rather than by sensitization. Meyer and co-workers reported an integrated photocathode based on the [Re(bpy)(CO)3(Cl)] catalyst for CO2 reduction to CO in a CO2-saturated bicarbonate aqueous solution. The integrated photocathode was stable toward CO2 reduction for over 10 h with a faradaic efficiency of ∼65%.802 Meyer and co-workers also reported a series of photocathodes using [Ru(bpy)(CO)2Cl2] as the catalyst for CO2 reduction. The photocathodes reduced CO2 to formate at stable photocurrent densities of around −1.1 mA cm−2 during 20 h of irradiation with faradaic efficiencies of up to 64% in CO2-saturated bicarbonate aqueous solution.798
6.4 Electrode materials
Electrode materials play several key roles in DSPECs. They serve as the solid support for chromophores and catalysts, and in many cases they play a role in chromophore-catalyst integration strategies. In addition, electrode materials are also key in charge separation, and electron collection and/or delivery.The use of SnO2–TiO2 core–shell electrode materials combined with the use of [Ru(bda)(pic)2]-type water oxidation catalysts (Fig. 51) has led to significant developments in DSPECs.754,759,764–767,774,824,825 In the case of SnO2–TiO2 core–shell electrodes, the initial rationale for their better performance compared to bare TiO2 electrodes was based on the difference in the conduction band positions of SnO2 and TiO2. The more positive conduction band of SnO2 should act as a sink from which recombination of injected electrons should be significantly slower. Initial studies by Meyer and co-workers supported this with oxidized chromophores persisting into the millisecond timescale when anchored onto SnO2–TiO2 core–shell surfaces.826 However, follow up studies by the same group discovered that there is actually a new electronic state at the SnO2–TiO2 interface located more positive than both SnO2 and TiO2.800 The success of core–shell electrode materials in DSPECs and other applications is a clear example that finding new materials is not always the only solution. Oftentimes creative solutions with known materials might provide similar or even better outcomes.
Meyer and co-workers used boron-doped Si as the p-type material, protected by a 10 nm Ti layer with an additional 3.0 nm layer of TiO2 for anchoring of chromophores.784 The integrated photocathode was capable of delivering a photocurrent density of about −1.0 mA cm−2 for hydrogen generation under zero applied bias (vs. NHE) using a NiL2 catalyst for proton reduction to H2.
Strategies that creatively combine known materials could prove to be a viable alternative to finding new materials with ideal properties. For example, Meyer and co-workers reported a binary p–n junction strategy to prepare photocathodes that integrate a semiconductor p–n junction (Si/n-GaN) and surface-bound molecular assemblies for light absorption and catalysis. The photocathodes reduce CO2 to formate at stable photocurrent densities of −1.1 mA cm−2 during 20 h of irradiation with faradaic efficiencies of up to 64%.798
6.5 Tandem devices
The net conversion of water and carbon dioxide to oxygen and reduced carbon products in natural photosynthesis is driven by the absorbed energy of two photons for each electron involved in the process (two photosystems in tandem). However, in natural photosynthesis, the two photosystems absorb essentially the same spectral range, which is one of the reasons why this process is relatively inefficient.827,828 A thermodynamic analysis indicates that an approach in which the two photosystems absorb different parts of the light spectrum (tandem junction) is crucial to maximize the capability of converting solar energy into fuels for both natural and artificial photosynthetic systems.827,829,830Fig. 52 shows a schematic diagram of a tandem DSPEC for solar-driven CO2 splitting into CO and O2 by the net reaction 2CO2 + 4hν → 2CO + O2.735 Replacement of the CO2 reduction catalyst in the photocathode with a proton/water reduction catalyst results in a DSPEC for water splitting into O2 and H2. Ideally, the chromophores in the photoanode and photocathode should have complementary spectral absorption profiles. | ||
| Fig. 52 Schematic diagram for a DSPEC for light-driven CO2 splitting into CO and O2 with an assembly-derivatized TiO2 photoanode for water oxidation to O2 and an assembly-derivatized photocathode for CO2 reduction to CO. Reprinted with permission from ref. 735. Copyright 2015 American Chemical Society. | ||
Sun and co-workers reported an organic dye-sensitized tandem DSPEC for light-driven water splitting. The photoanode consisted of a thin film (8 μm) of TiO2 as electrode material, a triphenylamine-based organic dye and a molecular Ru-based catalyst for water oxidation. The photocathode consisted of a thin film (1 μm) of NiO, a triphenylamine-based organic dye and a molecular Co-based catalyst for proton reduction.753 In a 50 mM phosphate buffer at pH 7, the cell reached photocurrent densities of 70 μA cm−2 for water splitting under 100 mW cm−2 irradiation with no applied bias. Meyer and co-workers reported a tandem DSPEC with sustained photocurrents of 250 μA cm−2 over a 2.5 h irradiation time with faradaic efficiencies of 73% and 54% for O2 and H2, respectively.784 The photoanode consisted of a SnO2–TiO2 core–shell electrode with a RuP22+ chromophore and a Ru(bda) water oxidation catalyst assembled using the layer-by-layer approach. The photocathode, described in the previous section, consisted of a boron-doped p-type Si protected with a 10 nm Ti layer with an additional 3.0 nm layer of TiO2 for PDI′ chromophore anchoring. A NiL2 proton reduction catalyst was assembled with the PDI' chromophore via a zirconyl bridge using the layer-by-layer assembly strategy. High energy photons were used at the photoanode for water oxidation and low energy photons were used at the photocathode for proton reduction. The performance of the tandem device was limited by the photoanode. Sherman and co-workers reported an alternative approach to tandem DSPEC devices for water splitting. It combines a typical water splitting DSPEC with a DSC to use more efficiently the solar spectrum and eliminate the need for an applied bias, Fig. 53.760,761
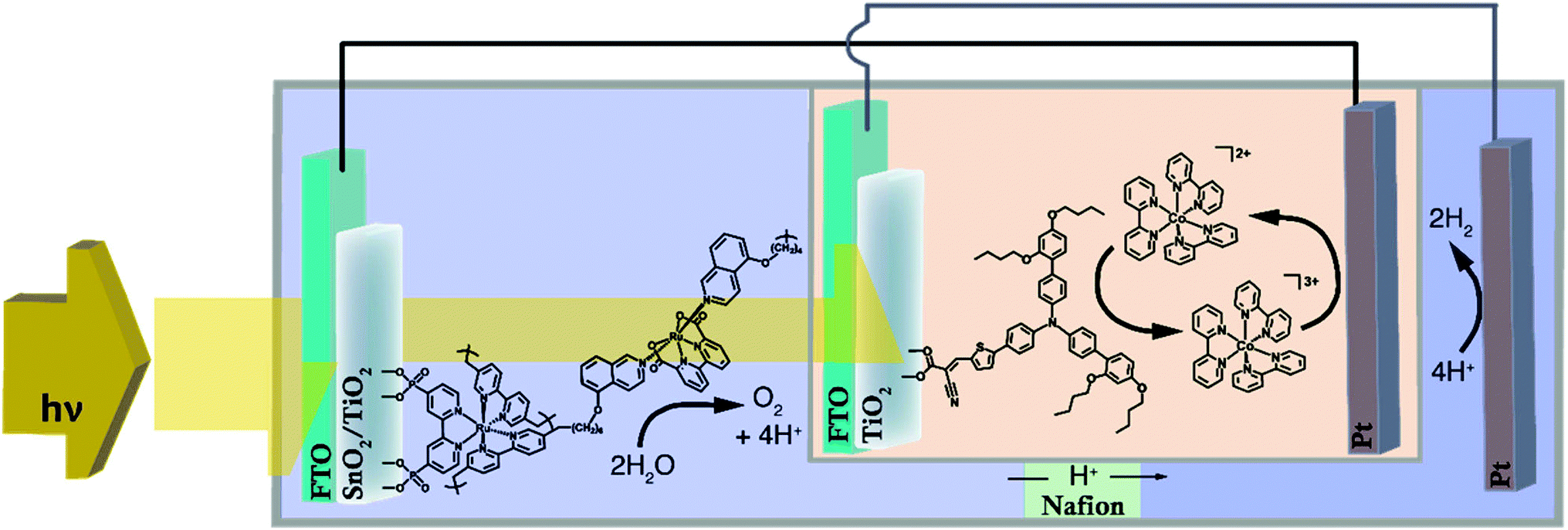 | ||
| Fig. 53 Schematic diagram of a DSPEC wired in series with a DSC. Reprinted with permission from ref. 761. Copyright 2016 American Chemical Society. | ||
The fully assembled tandem cell system consisted of a DSPEC incorporating a SnO2–TiO2 core–shell electrode, a RuP22+ chromophore and a Ru(bda) water oxidation catalyst. The chromophore and catalyst were assembled on the surface of the core–shell electrode via electropolymerization. The photoanode and a dark Pt cathode were wired in series with a DSC employing either the N719 dye and I−/I3− mediator or a D35 dye and the Co(bpy)3 mediator. The tandem cell achieved unbiased photocurrents of 40 μA cm−2 under simulated solar illumination with a solar to hydrogen efficiency of 0.06%.
7 Industrialization and commercialization
The Nature paper by Grätzel and O'Regan5 triggered expectations for a novel low-cost photovoltaic technology with potential to challenge silicon solar cells, which at the time were still forecast to be expensive to manufacture on a large scale. Shortly thereafter, a few pioneering device manufacturing companies initiated DSC development with commercial ambitions, such as Glas Trösch, Leclanché, and Asulab from Switzerland, ABB and INAP in Germany, Ekologisk Energi in Sweden, Solterra in Italy, and Dyesol in Australia. Since then, a range of industrialisation initiatives in different parts of the world have been created. The most intense period was during 2000–2010, when Asian activities were intense, dominated by Japan. An example of the vast Japanese development activities is the fact that >50% of the >2000 novel DSC patent families submitted in the years 2000–2010 had Japanese origin.831 Examples of Japanese companies with strong DSC development during this period are Sharp, Sony, Toyota, Hitachi Maxell, Sanyo, Nippon Oil, Fuji Film, Aisin-Seiki, Fujikura, J-Power Co., Gunze Ltd, Mitsubishi Paper Mills., Sekisui Jushi Corporation, Dai Nippon Printing Company, Nissha Printing, Taiyo Yuden Co., Panasonic Denko, TDK, Spark Plug Co. and Eneos Co Ltd. Equivalent examples from other Asian countries are Dongjin Semichem and Samsung SDI from South Korea and J touch from Taiwan. Further examples of companies with DSC activities during this period are BASF, Bosch, Merck and Tata Steel. Most of these industrial DSC initiatives have been abandoned, whereas some have changed direction during their development, typically from outdoor panels to low-power devices targeting IoT (Internet of Things) applications. In the past ten years, commercial-oriented DSC device activities have been more or less exclusively directed towards see-through aesthetic devices for BIPV applications and small-area devices for low-power applications. Looking at commercialization efforts of the DSC technology throughout the past 30 years, three categories appear: (i) panels to challenge Si, (ii) BIPV via aesthetic devices, (iii) niche products for electronic applications. These diverse efforts are discussed in Section 7.3. Throughout the DSC commercialization, a set of module concepts have been used and thoroughly investigated, each one with their respective strengths and challenges (Section 7.1). In parallel to the device-oriented commercialization activities, there has been supplementary industrialization of required material components, manufacturing equipment and services. However, as the major DSC commercial breakthrough has not taken place yet, these industries still operate at a small scale, with various peaks during the most intense DSC commercialization periods.According to Hagfeldt and co-authors,8 a difficulty of evaluating the performance of DSC modules stems from the fact that various definitions of device efficiency are employed. The efficiency of the active area is used in certain situations, whereas the efficiency of the modules' entire area is used in others. In addition, several module sizes are used, and measurements are performed at varying light intensities. In general, publications dealing with module stability provide lower efficiency figures. The comparison of DSC module findings from various publications should then be evaluated with a grain of salt. Sharp's DSC mini-module, with efficiency of 10.7% from the year 2013, is included in the current table of record solar cell efficiencies.390
7.1 DSC module design
The thorough overview of the five basic DSC module designs presented by Hagfeldt et al. is still relevant.8 This applies to their definition of a DSC module as well, i.e. a device that is considerably larger in both the x and y dimensions compared to a single lab-scale solar cell, and that employs particular solutions to reduce the resistive energy (electron transport) losses. Sandwich and monolithic are still terms used to describe a device construction that has the working and counter electrodes on two separate substrates or on the same one.The bigger size of a DSC module complicates the manufacturing, performance, and stability compared to those of a test cell. Furthermore, the interconnection of cells in a DSC module may create additional efficiency loss routes, such as mismatched performance of linked cells or undesired electrolyte mass transfer between neighbouring cells. The five sandwich and monolithic module concepts, i.e. (i) sandwich Z-interconnection, (ii) sandwich W-interconnection, (iii) sandwich current collection, (iv) monolithic serial connection, and (v) monolithic current collection, have constituted the basis throughout 30 years of DSC device development and commercialization. Their respective advantages and challenges are discussed by Hagfeldt et al.8 Even though there has been an evolution in DSC chemistry, represented by e.g. organic dyes, Cu-based redox mediators and the so-called “zombie cell”,485 the five module designs remain.
One complementary module design deserving attention is the work by Takashima et al. from NGK Spark Club.832 Their so-called ball-grid DSC solution is based on a hybrid copper polyimide flexible substrate covered with a dense carbon counter electrode. The working electrode is contacted to the copper via polymer-cored solder balls. The design efficiently enlarges a DSC cell by combining an efficient current collection grid with a high ratio of active area (95%). In addition, a few interesting novel DSC module design options – driven by simplified production processes – have been presented in the past few years at conferences by representatives of the present DSC industry, such as Exeger in Sweden and Song Textile in South Korea. However, as these designs – to the best of our knowledge – have not been presented in the literature, they are not part of this review. Moreover, Ricoh in Japan have recently launched commercial solid-state DSC products where the device concept has not been found in the literature.
7.2 DSC stability
For any relevant application, good long-term stability of the DSC is crucial. Degradation of the DSC can have various origins:336 (i) dye degradation: dyes can desorb from the TiO2 electrode, a process which is accelerated at higher temperatures. Dyes can also be damaged due to chemical reactions; for instance, they can be unstable in their oxidized state, which is the case for N719. (ii) Electron collection: the TiO2 electrode can change its performance due to loss of electrical contact between neighboring particles or with the FTO substrate. Furthermore, the energy levels of the TiO2 can shift due to changes in the electrolyte. (iii) Redox electrolyte: the redox mediator can undergo chemical changes, such as ligand exchange for cobalt and copper complexes. There can be a loss of the oxidized form of the redox mediator when other species are oxidized due to excitation of TiO2 (e.g., loss of triiodide when holes in TiO2 oxidize solvent molecules). Lastly, evaporation of the solvent can occur. (iv) Counter electrode: the catalyst can be unstable due to the corrosive nature of the redox mediator or it can be poisoned. The stability of Pt-free counter electrodes was reviewed by S. Yun et al.833 (v) Sealing: imperfect sealing can lead to loss of electrolyte and/or introduction of water and oxygen into the system, with detrimental effects. (vi) UV light: direct excitation of TiO2 can lead to damage due to highly oxidizing holes. Typically, a UV filter needs to be included in practical DSC systems for outdoor use for this reason.Best stability data to date is obtained for DSCs based on the iodide/triiodide redox system and ruthenium sensitizers. High-temperature stability of such systems was investigated by Desilvestro and co-workers using electrolytes with different solvents – “HSS” (presumably based on sulfolane), 3-methoxypropionitrile (MPN) and γ-butyrolactone (GBL) – which led, respectively, to final relative PCE values of 83%, 60% and 20% after 1000 h at 85 °C in the dark.834 Sauvage et al. found evidence for solid/electrolyte interphase formation on TiO2 nanoparticles using MPN under such conditions, suggesting that TiO2 acts as a catalyst for electrolyte degradation.835 Mastroianni et al. found that degradation under MPP conditions was much more severe than under open circuit conditions.705 While negligible degradation was found during 3200 h of outdoor testing, significant degradation was found during controlled testing at elevated temperature (1 sun, 85 °C), which was largely attributed to loss of I3− and band edge shifts of the TiO2. The Z907 dye, with hydrophobic tails, was found to be stable upon 1200 h of illumination with iodide-based electrolyte and MPN solvent, even in the presence of large concentrations of water.706 Good stability data for organic sensitizers was reported by Peng Wang et al.269 They used co-sensitized organic dyes C268 and SC-4 in combination with an electrolyte containing DMII and EMII ionic liquids and sulfolane, and recorded just 3% loss of PCE of their solar cells (initial PCE 10.1%) after 1000 h of 1 sun illumination at 60 °C. A 1000 h stability test in the dark at 85 °C led to a 9% loss for the same system (Table 13).
| Redox system – solvent | Sensitizer(s) | Conditions | Initial PCE (%) | Final PCE (relative %) | Year | Ref. |
|---|---|---|---|---|---|---|
| I−/I3− – MPN | N719 | 3200 h, 1 sun, 85 °C, OC | 4.6 | 67 | 2012 | 705 |
| I−/I3− – MPN | N719 | 3200 h, 1 sun, 85 °C, MPP | 4.7 | 28 | 2012 | 705 |
| I−/I3− – MPN | Z907 | 1200 h, 1 sun, 25 °C, OC | 7.0 | 104 | 2019 | 706 |
| I−/I3− – MPN + 20% H2O | Z907 | 1200 h, 1 sun, 25 °C, OC | 5.3 | 123 | 2019 | 706 |
| I−/I3− – DMII, EMII, sulfolane | C268/SC-4 | 1000 h, 1 sun, 60 °C, OC | 10.1 | 97 | 2018 | 269 |
| I−/I3− – DMII, EMII, sulfolane | C268/SC-4 | 1000 h, dark, 85 °C, OC | 10.1 | 91 | 2018 | 269 |
| Co(bpy)3 – MeCN | SM315 | 500 h, 1 sun, 25 °C, MPP | 12.5 | 80 | 2014 | 286 |
| Co(bpy)3 – MPN | Z907 | 2000 h, 1 sun, 25 °C, OC | 4.0 | 91 | 2014 | 707 |
| Co(bpy)3 – MeCN | D35 | 1000 h, 60 °C, OC | 6.4 | 85 | 2014 | 17 |
| Cu(tmby)2 – MeCN | MS5/YX1b | 1000 h, 1 sun, 40 °C, OC | 13.5 | 93 | 2021 | 12 |
| Cu(tmby)2 – MeCN, MPN | Y123 | 432 h, 1 sun, OC | 9.49 | 79 | 2021 | 708 |
| Cu(tme) – MeCN, MPN | Y123 | 432 h, 1 sun, OC | 8.25 | 91 | 2021 | 708 |
The stability of cobalt-based mediators was reviewed by Bella et al. in 2016.836 Mathew et al. performed 500 h light soaking tests under MMP conditions of high-performing porphyrin-sensitized DSCs, after which a loss of 20% was found, partly attributed to dye desorption.286 Jiang et al. investigated long-term stability of Z907-sensitized devices with Co(bpy)3. With MPN as electrolyte solvent, PCE retained 91% of its initial value after 2000 h of continuous 1 sun illumination with cells kept at open circuit.707 1000 h tests for MeCN-based cells under 1 sun and MPP conditions gave no significant degradation for the best cells. Gao et al. performed 1000 h illumination tests at 60 °C for DSC devices with MeCN-based cobalt bipyridine electrolytes and found remarkably good stability for electrolytes with increased concentration of Co2+ and Co3+.17 Boschloo and co-workers investigated the thermal stability of cobalt-based electrolytes with MPN as solvent. They found that addition of bipyridine to the electrolyte could decrease DSC degradation in a 50 days storage test at 70 °C in the dark. With bipyridine and MBI as additives, a 12% loss in PCE was found, compared to a 20% loss with tBP as additive.18 Cobalt complexes with hexadentate ligands were shown to lead to improved stability in DSC illumination tests in comparison to cobalt trisbipyridine, with no degradation after 100 h in 1 sun.355,366
In recent work, Zhang et al. demonstrated good long-term performance for Cu(tmby)2-based electrolytes in a 1000 h light soaking test at 40 °C.12 Ligand exchange with, for instance, tBP could be a problem for long-term stability of these copper complexes.404 Sun and co-workers developed a stable Cu complex with a pentadendate ligand, which did not display facile ligand exchange. PCE remained at 90% of its initial value after 400 h at 1 sun (25 °C), compared to 80% for devices with Cu(tmby)2-based electrolyte.708
For all redox electrolytes, more long-term stability tests under MPP 1 sun illumination conditions are needed to reliably assess the performance of DSCs. Testing under open-circuit conditions will not stress the counter electrode at all. Furthermore, the full redox cycle is not occurring under these conditions, as all electrons in TiO2 will recombine the oxidized dye and redox couple.
In 2010, Hagfeldt et al.8 reviewed the status of DSC module stability up to that year. They highlighted the observation that publications dealing with module stability generally have lower efficiency values than the publications where stability is not mentioned, likely due to more space for encapsulation and/or use of different device chemistry with lower efficiency values. Still, already in 2010, it was evident that long-term stable DSC modules could be realized. A module stability paper that was highlighted was the one from Kato et al.,837 who presented results from 2.5 years of outdoor module tests, resulting in approximately 20% degradation of the initial device performance. By comparing the outdoor module ageing results to accelerated illumination tests on the single cell level, the acceleration factor of the light-soaking test was estimated at 11. Another highlighted paper was the one from Dai et al.,838 who performed one year outdoor testing of their modules resulting in a minor performance decrease, which was not numerically stated in the publication. High temperature storage tests have traditionally been challenging for DSCs. A third highlighted publication was that from Matsui et al.,839 who demonstrated that it is feasible to obtain excellent module stability over 1000 h storage in darkness at 85 °C and 85% relative humidity. An important module stability paper after 2010 is that from Rong et al.840 Monolithic serial-connected devices with a side of 100 cm with solid-state electrolyte passed the following two tests with minor performance decrease: (i) 1000 h at 60 °C, 85% relative humidity (RH) and (ii) 300 temperature cycles between −10 and 60 °C (3 h per cycle). In 2011, Kato et al.841 presented results from 160 days of outdoor tests of DSC modules integrated in solar light devices. They concluded that the JSC gradually increased the first two months before it stabilized, whereas the VOC gradually decreased as the outdoor exposure time proceeded. The overall device efficiency hardly changed. Another publication involving module stability after 2010 is the work from Hinsch et al.842 They present impressive DSC demonstrators with size 60 × 100 cm. However, the stability results (1000 h at 85 °C in darkness) are obtained by a device size of 100 cm2 ( Table 13 ).
It stands clear that the number of publications dealing with DSC module stability in the past 10 years has decreased in relation to the period 2005–2010. We were quite surprised to find a lack of published stability data from the semi-transparent BIPV demonstrators that have been realized around the world (see Section 7.3.2) and the shortage of recent field tests comparing DSC modules with other PV technologies. Likewise, we have not found any recent papers about the stability of low-power DSC modules, likely explained by the fact that this work is carried out by industry where the driving force for publication is low. In addition, Pettersson et al. already in 2001 showed that DSC modules can be very stable under such conditions by demonstrating a mere 4% decrease of the initial performance of a DSC device after half a year of illumination with a fluorescent light (5000 lx).843
7.3 Application categories and commercialization efforts
Despite the different nature of commercialization initiatives performed over the past 30 years, there are few main product categories that can be identified. As a consequence of this, we have divided the targeted applications for DSC into three categories: (i) panels to challenge Si, (ii) BIPV via aesthetic devices, and (iii) niche products for electronic applications. The evolution of each category and their status are discussed below.000 Roof Program in 1999 that dramatically changed the market for photovoltaics. All of this was realized under the assumption that silicon solar cells would face difficulties in reaching manufacturing costs that would make it competitive with conventional energy sources; i.e. there was a need for novel photovoltaic technologies with lower production costs. The leading technologies from this aspect were thin-film PV such as CIS, CIGS and CdTe. Whereas these technologies were targeting high efficiencies and advanced manufacturing processes, characterized by massive investment costs, DSC entered the field from a totally different and unexpected angle, characterized by lower efficiency but basic manufacturing processes and low-cost, scalable raw materials. The investment costs for initiating a DSC production line were foreseen to be a fraction compared to silicon or thin-film technologies. As a result of all of this, DSC attracted many companies that wanted to take on the challenge to commercialize the technology. Moreover, it was a possibility for companies that were not active in the photovoltaic industry to enter the field. As a result of all of this, almost all industrial DSC efforts during 1990–2005 targeted the future global massive PV market. In their 2010 review, Hagfeldt et al. presented a number of DSC device examples from this period that were driven by the target of challenging silicon.8 In the ten-year period 2005–2015, the manufacturing costs of silicon solar cells decreased as a result of the massive Chinese commercialization activities. The previous dream target of manufacturing costs of 1 USD per Wpeak was suddenly dramatically undercut. As a result of this, more emphasis was given to the increase of device efficiency. Consequently, the arguments for DSC as a candidate for future large-scale photovoltaic establishments disappeared, as dramatic efficiency improvements were now required. Even though this coincided with the DSC efficiency breakthroughs from Feldt et al.270 and Yella et al.,284 the entrance of the perovskite technology in 2012 changed the prerequisites for DSCs overnight.844,845 The perovskite technology shared the basic features of DSC, namely cost-efficient scalable manufacturing methods and material components. Even the recent DSC record efficiency of 13.0% in year 202112 is still low compared to those obtained by perovskite solar cells, with a present efficiency record of 25.2%.846 As a result of all this, there are today very few industrial DSC initiatives targeted at challenging silicon PV. In order to change this situation, a significant fundamental scientific breakthrough is required, opening for massive efficiency improvements. Nevertheless, the collective industrial and academic efforts devoted to developing competitive DSC devices for outdoor applications have left important technology testimonies such as module and production technology, proven durability at outdoor conditions, life cycle847 and cost analyses.848 In fact, this collective output has dramatically influenced the development of DSC for BIPV (Section 7.3.2) and low-power applications (Section 7.3.3), as well as the entire perovskite technology.
 | ||
| Fig. 54 (a) The DSC installation at the Conference centre in Lausanne, Switerland, consisting of 1400 W-connected modules of the size 35 × 50 cm2 (in total approx. 150 m2), manufactured by Solaronix in Switzerland. Reproduced with permission from Solaronix S.A., copyright 2021. (b) The DSC installation at the Science Tower in Graz, Austria, consisting of 896 W-connected red DSC devices of 0.6 m2 area each (in total approx. 500 m2), manufactured by H.Glass in Switzerland. Reproduced with permission from H.Glass S.A., copyright 2021. (c) The DSC installation at the Solar Pavillon at Roskilde University in Denmark, consisting of 196 W-connected red DSC panels of area 900 cm2 each (in total approx. 180 m2) made by Dongjin Semichem in South Korea. Architect Jane Ostermann-Petersen. Reproduced with permission from Karina Tengberg, copyright 2021. | ||
All of the aforementioned initiatives were foreseen to represent the commercial breakthrough of aesthetic DSCs for BIPV applications. However, this has not been realized. On the contrary, the industrial activities on see-through aesthetic DSCs seem to have decreased in the past 2–3 years. A tentative explanation for this is that the energy production, i.e. the device efficiencies, were too low to balance the additional cost compared to coloured glass or alternative architectural features, potentially in combination with question marks regarding the product life. However, other similar initiatives are still ongoing, such as the Indian collaboration between Elixir Technologies and CSIR-National Institute for Interdisciplinary Science & Technology (NIIST) (Fig. 55).
 | ||
| Fig. 55 Indian semi-transparent DSC prototypes from Elixir Technologies and CSIR-National Institute for Interdisciplinary Science & Technology (NIIST). Reproduced with permission from the Indian Ministry of Science and Technology, copyright 2021. | ||
All devices in Fig. 54a and b use a module idea based on W-interconnects, i.e. the double-substrate module design contains cells with alternating working and counter electrodes on each substrate. As a consequence, every second cell is irradiated from the counter electrode side, which generally leads to lower current values than irradiation from the working electrode side. A challenge involved is thus to match the current output from adjacent inverted cells. This has commonly been overcome by making the cells illuminated through the counter electrode slightly broader, i.e. a larger active area to compensate for the lower current output. One drawback of this solution is that the ratio of current output from front- and back-side illumination varies with light intensity and illumination angle. Moreover, as semi-transparent devices are illuminated from both sides, the illumination conditions are complicated and unpredictable. Consequently, it is practically impossible to avoid an imbalance in current output between cells. Such imbalance will decrease the overall device performance but it may also result in performance degradation over time. Interestingly enough, we have not found any literature on e.g. the device chemistry and/or the delivered energy values from these installations. This is surprising and unfortunate as these installations would provide highly interesting results and information ranging from device performance to potential degradation modes over time.
Around the beginning of the millennium, activities on flexible DSC were taking off. Companies such as Konarka Technologies, USA, and Sekisui Chemical, Taiyo Yuden Co. and Peccell Technologies, Japan, developed such technologies. The DSC technology of Konarka was a few years later taken over by G24 Innovations (later G24 Power), who initiated a massive effort to commercialize the technology for low-power applications. Their factory in Wales is generally considered as the first large-scale mass production facility for DSC. Various products, such as Logitech keyboards, solar backpacks, solar chargers and solar iBeacons were launched. Whereas G24 targeted large-volume production for broad applications, there were several parallel Japanese initiatives where DSCs were used in solar art demonstrators, e.g. aesthetic devices powering lamps and fans. The lamp charger Hana-Akiri from Sony received a lot of attention, Fig. 56. Similar artistic DSC devices from the same period came from e.g. J Touch Co., Aisin Seiki and Nissha Printing. Retrospectively, it can be concluded that all of these, and many other low-power DSC commercial initiatives in the period 2000–2010, did not trigger a sustainable market demand.
 | ||
| Fig. 56 An example of artistic DSC devices from Sony displayed at the 10th Eco-Products Conference in Tokyo in 2008. Reproduced with permission from Satoshi Uchida, copyright 2021. | ||
The arguments for indoor low-power DSC received novel fuel from the work of Feldt et al.,270 where it stood clear that the combination of organic dyes and one-electron Co-based redox mediators resulted in major performance improvements, with high voltage levels even at low light conditions. In addition, low-power PV became of interest as a result of the increased global activities on IoT applications with forecast billions of small systems requiring low-power supply. As a result, there has been a revival for and a rapid increase in industrial initiatives targeting low-power DSC. The interest for low-power DSC was taken to the next level by the work of Freitag et al.348 By using Cu-based one-electron redox mediators in combination with organic dyes, low-power efficiencies of 28.9% were obtained at 1000 lux. This was followed up by a 32% cell efficiency at 1000 lux by Cao et al.,320 a 34% cell efficiency at 1000 lux by Michaels et al.,26 and a 34.5% cell efficiency at 1000 lux by Zhang et al.12 Interestingly enough, all these pieces of work used the same illumination source (Osram 930 Warm White fluorescent light). However, we highlighted above that characterization of low-power devices is a somewhat confusing part of the PV world since there is no established standard for the illumination and caution should be taken when comparing values (see Section 2.2).53,856 An interesting comparison to low-power perovskite solar cells, however, can be made by the values reported by Meng Li et al.857 They achieved conversion efficiencies up to 35.2% at a device size of 9 mm2 (23.2% at 4 cm2) and 1000 lux using a fluorescent light source (Osram L18W/82). In contrast to the DSC values from Michaels et al.,26 the efficiencies for the perovskite devices were dramatically reduced at lower light intensit ies: 25.7% and 19.5% efficiencies were obtained at 500 and 100 lux, respectively. These perovskite devices include lead, which may be a limitation for commercial exploitation in electronic applications. In addition to DSC and perovskite solar cells, organic solar cells (OPV) represent an additional technology candidate for low-power applications, with confirmed efficiency values up to 28.1% at 1000 lux.858 It is thus a product segment that is becoming crowded by various upcoming technologies. From a strict efficiency point of view, it appears that DSC devices deliver the highest efficiency values at indoor illumination, at least at 500 lux and 100 lux, and at 1000 lux for device size d >1 cm2. This gives companies commercializing low-power DSC the prerequisites to realize the best-performing low-power products. In the commercial race, however, other additional selling points other than indoor efficiency will likely be important, such as price, colour, weight, thickness and flexibility in size and voltage.
The new era of DSC industrialization for niche applications in general, and low-power devices in particular, is confirmed by recent product launches. The DSCs of Fujikura in Japan are already used in wireless multi-sensor device systems such as heatstroke prevention systems and management of large warehouses in Japan, Fig. 57a.859 3GSolar in Israel introduced several DSC options with different transparency and colors to fit many diverse niche applications, including wireless sensor networks, medical and sports devices, security sensors and cameras, agricultural monitors, beacons and electronic signs, computer peripherals, and wearable electronics. Exeger in Sweden has announced that their DSC devices will be used in various consumer electronics devices such as headphones, safe helmets and soft goods. In 2020, Ricoh in Japan launched their solid-state RICOH EH DSC series. These devices are used in applications such as remote controls for projectors and to power IoT sensor systems, Fig. 57b.
 | ||
| Fig. 57 (a) DSC-containing sensor systems from Fujikura in Japan for indoor (left) and outdoor (right) applications, respectively. Reproduced with permission from Fujikura Ltd, copyright 2021. (b) Examples of products from Ricoh containing their solid-state DSC devices: environmental sensors for measuring temperature, humidity, illumination, atmospheric pressure, etc., wireless mouse and remote controls for projectors. Reproduced with permission from Ricoh Company Ltd, copyright 2021. | ||
Out of these DSC products, it is noticeable that Fujikura has different devices for outdoor and indoor use (Fig. 57a). This is likely attributed to the fact that Fujikura worked on outdoor DSC module development before focusing on low-power devices, i.e. they had access to the required chemistry and manufacturing methods for outdoor applications.839 Ricoh appears to be the only producer using solid-state DSCs. Moreover, it is worth noticing that devices from Exeger are marketed as solar cells that are integrated without being seen, Fig. 58, opening for their vision to implement their light harvesting cells on all imaginary surfaces ranging from electronic gadgets to buildings via e.g. blinds, walls, vehicles, bags and furniture.
 | ||
| Fig. 58 Various prototypes including non-visible DSC devices from Exeger in Sweden. Reproduced with permission from Exeger A.B., copyright 2021. | ||
An unexpected side effect of low-power DSC development is the technology Focus-Induced Photoresponse (FIP technique). This technology is based on the discovery that the power output from a DSC is not only dependent on the total flux of incident photons, but also on the size of the area in which they fall. Consequently, when probe light from an object is cast on a detector through a lens, the sensor response depends on how far in or out of focus the object is, i.e. a novel way to measure distances with photodetectors.860 The technology was invented and commercialized by the company Trinamix in Germany, a wholly owned subsidiary of BASF.
8 Outlook: Colourful
Every significant advance over the previous decade in the development of DSCs has been made by the introduction of new principles, techniques, and materials. DSCs are becoming part of the future of electric power generation due to the following characteristics: (i) they are easy to fabricate, (ii) they are manufactured from low-cost materials, (iii) they are environmentally friendly, (iv) they have high conversion efficiencies, and (v) they perform well in diffused light and at high temperatures, conditions in which other technologies cannot compete. Based on creative research work, power conversion efficiencies of up to 20% under sunlight and 45% for ambient light can be anticipated from future DSCs.Detailed understanding of many aspects of the dye-sensitized solar cell is still lacking. Charge recombination is currently the major cause of efficiency loss in DSCs and other solar cells. When one of the components (dye, redox shuttle, or semiconductor) is modified, many processes are impacted, which may boost or lower the overall performance. This needs to be considered at all times when new materials are introduced, and the overall system has to be adapted. DSCs are complex devices and the improvement of only one of their components will not lead to the desired targets in efficiency and stability.
Theory and computation
From the computational perspective, new theoretical tools are needed to push forward our understanding of DSCs beyond the established, successful applications outlined above. Fortunately, thanks to continuously increasing computer power and new computational paradigms, this is the right time for such developments. In silico design and optimization of materials will need to shift from single components to coupled dye/electrode or, ideally, electrode/dye/electrolyte ensembles. New algorithms based on artificial intelligence and machine learning fit this purpose, with training databases obtained from high-throughput computations. Still, the results of such automated discoveries will need to be validated with the magnifying glass of atomistic first-principles calculations, able to dissect electronic and dynamic properties beyond the ideal picture of interfaces considered so far. In particular, we foresee a crucial role of studies addressing defects and additives that can be game changers for reaching desired efficiencies and, regarding processes, charge transfer and recombination events under operating conditions. These advancements in models and methods will bridge the gap between theory and experiments, so that computer and laboratory bench can jointly tackle the design and optimization of new DSCs.Materials
High efficiency and panchromatic organic dye systems have been developed. These are a non toxic, low cost, sustainable, and conveniently accessible option. The next step will be to achieve a fundamental understanding of electron injection from the dye in its excited state into the conduction band of the semiconductor, in order to minimize potential and overall conversion efficiency losses at this interface. The semiconductor requires a modification of the position and of the nature of its conduction band, which can be reached through doping, morphology variation or the use of alternatives to TiO2. The dyes' LUMO level should be tuned to match the potential of the conduction band edge of the semiconductor closely to provide efficient electron injection and minimize energy losses.In a more idealistic direction, DSCs could significantly benefit from the design of a photoinduced molecular rectification strategy built into the chromophore design. The idea of a facile electron transfer to the semiconductor with the cation trapped away from the surface for extended time could ease demands on the rate of dye regeneration by slowing down the competitive back reaction, which could lead to high fill factors thanks to an increase in regeneration efficiency at the maximum power point. The D–π–A dye design is a simple example of this approach that revolutionized the DSC field. If new designs with dramatically higher rectification effects retaining near unity quantum yields for electron injection could be put forward, another revolution within DSCs could be induced, leading to another massive gain in power conversion efficiencies.
Another consideration is the position and packing of molecules on the semiconductor surface, as well as how these factors influence electron transfer kinetics in DSCs. With examples of dyes having exceptionally low recombination losses and exceptionally high conversion efficiencies in devices operating with absorption onsets up to 700 nm in mind, several key directions remain important with regard to DSC dye design. The utilization of photons with >800 nm wavelength with the same efficiency as is observed at 700 nm is another target of the DSC field, with maximal single photoelectrode devices expected to peak at absorption onsets of 950 nm. Additionally, tandem type systems require new chromophores at both high and low energy absorption onsets (high voltage dyes and NIR dyes) paired with appropriate redox shuttles for devices where dye energy levels are well positioned to minimize energy losses. The development of these systems is key for DSCs to exceed the single photoelectrode Shockley–Queisser limit. DSCs have shown exceptional photovoltage outputs from higher energy visible light photons, and the design of dyes maximizing performance in the blue spectral region and of more positive potential redox shuttle systems could be transformative in providing tandem systems to be paired with any smaller-bandgap solar cell technology. The development of one-electron redox shuttles with high performances with transition metal-based sensitizers could provide a needed answer to the lower energy absorption onset challenge, since good sensitizer options already exist but are incompatible with most redox shuttle systems. Furthermore, electron transport in mesoporous semiconductor electrodes is normally described in terms of multiple trapping/detrapping, but the nature of the traps involved is unclear. It has been suggested that the electrostatic interaction between electrons in the semiconductor and ions in the electrolyte could in fact be the origin of such traps.
Future research should further concentrate on electrolyte interactions with electrodes and sensitized dyes, as well as on the impact of these interactions on photoelectrical conversion processes, and on the creation of alternative charge carrier materials to increase charge carriers' transport performance, minimize recombination losses, and improve long-term stability. Another factor to consider in these systems is the replacement of the liquid electrolyte with a solid-state electrolyte or charge transport material to avoid leakage, solvent volatilization, dye photodegradation and desorption, and counter electrode corrosion. This goal has been partially reached thanks to the introduction of metal coordination complexes, but their development is still far behind the efforts made in dye development.
p-type DSCs
Much of the improvement in performance for p-type DSCs has arisen from developments in dyes and new electrolytes. In order to reach efficiencies that compete with thin-film solar cells, the VOC needs to be improved by ca. 0.5 V to match that of typical n-type DSCs. This requires a replacement for NiO which is transparent, conductive, stable and non-toxic. There are very few single materials with all of these properties. Moreover, there are still gaps in our understanding of electron transfer at the interface of p-type metal oxides and dye molecules. Currently, beyond NiO itself, it is not clear what the limitations to p-type DSCs are, but so far, there has been a trade-off between current and voltage that needs to be understood for progress to be made. To realize the potential of p–n tandem DSCs, a concerted effort of materials development combined with state-of-the-art spectroscopy is necessary. Meanwhile, very few examples of solid-state p-type DSCs have been reported and this is a rich area for future development that may overcome some of the challenges associated with liquid cells. Moreover, the factors that limit the performance of solid state DSCs, such as the requirement for thin semiconductor films, may be less limiting in solid-state tandem DSCs.Solar fuels
Most DSPEC studies to date have been carried out at pH values between 4.5 and 8.0, where the injection efficiency of the most commonly used chromophores into the conduction band of wide bandgap semiconductors such as TiO2 is below 50%. In addition, stability of catalysts, chromophores and anchors also decrease as the pH is increased. There are opportunities for significant improvements in DSPEC performance and stability at low pH (e.g. pH 1) where injection efficiencies are close to 100%. Most DSPEC require an applied bias for efficient H2 generation and release. Combining DSPECs and DSCs will eliminate the need for an applied bias and open the door for CO2 reduction photocathodes which typically operate at larger overpotentials than proton reduction photocathodes.Applications
The high sensitivity and efficiency of DSCs in low and ambient light conditions is one of their major benefits. They can be used where diffused solar light prevails over direct solar illumination. For this reason, the essential use of DSCs in building windows is that they operate well not just on the roof, as is the case with direct solar light irradiation in silicon cells. In the light of the global energy report, this advantage of the DSC would also reduce the energy usage represented by buildings. This industry is a major contributor to greenhouse emissions, consuming between 34% and 39% of electricity worldwide. The colors that DSCs can implement are another appealing feature for businesses. DSCs can be used as thin colored and transparent panels, transforming typical walls, skylights, and glass facades into electricity generators.With continued research, it is certain that more interesting features will be revealed that could lead to improved performance of DSCs or to spin-off applications. The aforementioned directions are currently being pursued by researchers and exciting results are expected.
Author contributions
ABMG and MP wrote Section 3. IB wrote Section 2.2, edited the manuscript, compiled tables and drew molecular structures. GB wrote Sections 2.1, 2.3, 2.4, 2.5, 4.1 and 7.2. JJC wrote Section 6. JHD wrote Section 4.2. EAG wrote Section 5. GJM wrote Section 2.6. HP wrote Section 7. AH wrote Section 1. MF wrote Sections 4.3 and 4.4., conceptualized and oversaw the completion of the article. All authors contributed to the writing of the outlook, substantially contributed to the conception and design of the article by interpreting the relevant literature, and contributed to the critical revision and review of the manuscript.Conflicts of interest
GB, HP and AH are co-founders and co-owners of Dyenamo AB.Acknowledgements
The authors would like to thank Dr Leif Häggman for taking the SEM image for Fig. 16. ABMG and Hannes Michaels for proofreading the entire manuscript. MP acknowledges funding from the Italian Ministry of Economic Development in the framework of the Operating Agreement with ENEA for Research on the Electric System. IB acknowledges financial support from the Cloud City government. GB thanks for the support through the STandUP for Energy program. JJC's contribution has been supported by the U.S. Department of Energy, Office of Science, Division of Chemical Sciences, Geosciences, & Biosciences, Office of Basic Energy Sciences under contract DESC0012704 at Brookhaven National Laboratory. JHD acknowledges financial support from the National Science Foundation (NSF-1954922). EAG thanks The North East Centre for Energy Materials EP/R021503/1 and the ERC, starting grant, p-TYPE 715354. GJM gratefully acknowledges support from the Division of Chemical Sciences, Office of Basic Energy Sciences, Office of Energy Research, US Department of Energy (Grant DE-SC0013461). MF acknowledges the support by the Royal Society through the University Research Fellowship (URF\R1\191286) and Research Grant 2021 (RGS\R1\211321).Notes and references
- H. Ritchie, Energy Mix, https://ourworldindata.org/energy-mix.
- D. Archer, Global Warming: Understanding the Forecast, John Wiley & Sons, 2nd edn, 2012 Search PubMed.
- H. Ritchie and M. Roser, Renewable Energy, https://ourworldindata.org/renewable-energy.
- IEA, Renewables 2020, International Energy Agency, 2020 DOI:10.1787/c74616c1-en.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- H. Gerischer, J. Electrochem. Soc., 1966, 113, 1174 CrossRef CAS.
- H. Gerischer, Electrochim. Acta, 1990, 35, 1677–1699 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Stojanović, N. Flores-Diaz, Y. Ren, N. Vlachopoulos, L. Pfeifer, Z. Shen, Y. Liu, S. M. Zakeeruddin, J. V. Milić and A. Hagfeldt, Helv. Chim. Acta, 2021, 104, e2000230 CrossRef.
- I. Benesperi, H. Michaels and M. Freitag, J. Mater. Chem. C, 2018, 6, 11903–11942 RSC.
- M. Freitag and G. Boschloo, Curr. Opin. Electrochem., 2017, 2, 111–119 CrossRef CAS.
- D. Zhang, M. Stojanovic, Y. Ren, Y. Cao, F. T. Eickemeyer, E. Socie, N. Vlachopoulos, J.-E. Moser, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nat. Commun., 2021, 12, 1777 CrossRef CAS PubMed.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Muñoz-García, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CrossRef CAS PubMed.
- R. Harikisun and H. Desilvestro, Sol. Energy, 2011, 85, 1179–1188 CrossRef CAS.
- A. Hinsch, J. M. Kroon, R. Kern, I. Uhlendorf, J. Holzbock, A. Meyer and J. Ferber, Prog. Photovoltaics, 2001, 9, 425–438 CAS.
- J. Gao, M. B. Achari and L. Kloo, Chem. Commun., 2014, 50, 6249–6251 RSC.
- W. Yang, Y. Hao, P. Ghamgosar and G. Boschloo, Electrochim. Acta, 2016, 213, 879–886 CrossRef CAS.
- B. Li, L. Wang, B. Kang, P. Wang and Y. Qiu, Sol. Energy Mater. Sol. Cells, 2006, 90, 549–573 CrossRef CAS.
- J.-H. Yum, P. Chen, M. Grätzel and M. K. Nazeeruddin, ChemSusChem, 2008, 1, 699–707 CrossRef CAS PubMed.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- P. Docampo, A. Hey, S. Guldin, R. Gunning, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2012, 22, 5010–5019 CrossRef CAS.
- J. Zhang, N. Vlachopoulos, M. Jouini, M. B. Johansson, X. Zhang, M. K. Nazeeruddin, G. Boschloo, E. M. J. Johansson and A. Hagfeldt, Nano Energy, 2016, 19, 455–470 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J.-I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- J.-M. Ji, H. Zhou, Y. K. Eom, C. H. Kim and H. K. Kim, Adv. Energy Mater., 2020, 10, 2000124 CrossRef CAS.
- H. Michaels, M. Rinderle, R. Freitag, I. Benesperi, T. Edvinsson, R. Socher, A. Gagliardi and M. Freitag, Chem. Sci., 2020, 11, 2895–2906 RSC.
- E. Becquerel, C. R. Acad. Sci., 1839, 9, 561–567 Search PubMed.
- A. Einstein, Ann. Phys., 1905, 322, 891–921 CrossRef.
- G. Nofuentes, B. García-Domingo, J. V. Muñoz and F. Chenlo, Appl. Energy, 2014, 113, 302–309 CrossRef CAS.
- C. A. Gueymard, D. Myers and K. Emery, Sol. Energy, 2002, 73, 443–467 CrossRef.
- C. A. Gueymard, Sol. Energy, 2004, 76, 423–453 CrossRef.
- Nick84, Spectrum of Solar Radiation (Earth), 14 February 2013. URL: https://commons.wikimedia.org/wiki/File:Solar_spectrum_en.svg.
- J. F. Randall and J. Jacot, Renewable Energy, 2003, 28, 1851–1864 CrossRef CAS.
- N. Tanabe, Fujikura Tech. Rev., 2013, 42, 109–113 Search PubMed.
- F. De Rossi, T. Pontecorvo and T. M. Brown, Appl. Energy, 2015, 156, 413–422 CrossRef.
- I. Mathews, P. J. King, F. Stafford and R. Frizzell, IEEE J. Photovolt., 2016, 6, 230–235 Search PubMed.
- I. Mathews, S. N. Kantareddy, T. Buonassisi and I. M. Peters, Joule, 2019, 3, 1415–1426 CrossRef CAS.
- H. Michaels, I. Benesperi and M. Freitag, Chem. Sci., 2021, 12, 5002–5015 RSC.
- ASTM Standard E948–16, Test Method for Electrical Performance of Photovoltaic Cells Using Reference Cells Under Simulated Sunlight, ASTM International technical report, 2020.
- A. J. Frank, N. Kopidakis and J. van de Lagemaat, Coord. Chem. Rev., 2004, 248, 1165–1179 CrossRef CAS.
- W. Yang, M. Pazoki, A. I. K. Eriksson, Y. Hao and G. Boschloo, Phys. Chem. Chem. Phys., 2015, 17, 16744–16751 RSC.
- L. Han, N. Koide, Y. Chiba, A. Islam, R. Komiya, N. Fuke, A. Fukui and R. Yamanaka, Appl. Phys. Lett., 2005, 86, 213501 CrossRef.
- G. Liu, H. Wang, X. Li, Y. Rong, Z. Ku, M. Xu, L. Liu, M. Hu, Y. Yang, P. Xiang, T. Shu and H. Han, Electrochim. Acta, 2012, 69, 334–339 CrossRef CAS.
- J. Kwon, N.-G. Park, J. Y. Lee, M. J. Ko and J. H. Park, ACS Appl. Mater. Interfaces, 2013, 5, 2070–2074 CrossRef CAS PubMed.
- F. Behrouznejad, N. Taghavinia and N. Ghazyani, Org. Electron., 2018, 57, 194–200 CrossRef CAS.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- Y. Kashiwa, Y. Yoshida and S. Hayase, Appl. Phys. Lett., 2008, 92, 033308 CrossRef.
- Y. Yoshida, S. S. Pandey, K. Uzaki, S. Hayase, M. Kono and Y. Yamaguchi, Appl. Phys. Lett., 2009, 94, 093301 CrossRef.
- K. Takagi, S. Magaino, H. Saito, T. Aoki and D. Aoki, J. Photochem. Photobiol., C, 2013, 14, 1–12 CrossRef CAS.
- N. Koide and L. Han, Rev. Sci. Instrum., 2004, 75, 2828–2831 CrossRef CAS.
- R. Jiang and G. Boschloo, J. Mater. Chem. A, 2018, 6, 10264–10276 RSC.
- H. J. Snaith, Nat. Photonics, 2012, 6, 337–340 CrossRef CAS.
- C.-Y. Chen, Z.-H. Jian, S.-H. Huang, K.-M. Lee, M.-H. Kao, C.-H. Shen, J.-M. Shieh, C.-L. Wang, C.-W. Chang, B.-Z. Lin, C.-Y. Lin, T.-K. Chang, Y. Chi, C.-Y. Chi, W.-T. Wang, Y. Tai, M.-D. Lu, Y.-L. Tung, P.-T. Chou, W.-T. Wu, T. J. Chow, P. Chen, X.-H. Luo, Y.-L. Lee, C.-C. Wu, C.-M. Chen, C.-Y. Yeh, M.-S. Fan, J.-D. Peng, K.-C. Ho, Y.-N. Liu, H.-Y. Lee, C.-Y. Chen, H.-W. Lin, C.-T. Yen, Y.-C. Huang, C.-S. Tsao, Y.-C. Ting, T.-C. Wei and C.-G. Wu, J. Phys. Chem. Lett., 2017, 8, 1824–1830 CrossRef CAS PubMed.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Seró and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
- F. Fabregat-Santiago, J. Bisquert, E. Palomares, L. Otero, D. Kuang, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2007, 111, 6550–6560 CrossRef CAS.
- Q. Wang, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2005, 109, 14945–14953 CrossRef CAS PubMed.
- P. R. F. Barnes, K. Miettunen, X. Li, A. Y. Anderson, T. Bessho, M. Gratzel and B. C. O'Regan, Adv. Mater., 2013, 25, 1881–1922 CrossRef CAS PubMed.
- M. Pazoki, U. B. Cappel, E. M. J. Johansson, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2017, 10, 672–709 RSC.
- R. García-Rodríguez, R. Jiang, E. J. Canto-Aguilar, G. Oskam and G. Boschloo, Phys. Chem. Chem. Phys., 2017, 19, 32132–32142 RSC.
- Y. Hao, Y. Saygili, J. Cong, A. Eriksson, W. Yang, J. Zhang, E. Polanski, K. Nonomura, S. M. Zakeeruddin, M. Grätzel, A. Hagfeldt and G. Boschloo, ACS Appl. Mater. Interfaces, 2016, 8, 32797–32804 CrossRef CAS PubMed.
- H. Tributsch and H. Gerischer, Ber. Bunsen-Ges., 1969, 73, 850–854 CAS.
- A. Zaban, S. Ferrere, J. Sprague and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 55–57 CrossRef CAS.
- S. Ardo, D. Achey, A. J. Morris, M. Abrahamsson and G. J. Meyer, J. Am. Chem. Soc., 2011, 133, 16572–16580 CrossRef CAS PubMed.
- K. Hu, K. C. D. Robson, E. E. Beauvilliers, E. Schott, X. Zarate, R. Arratia-Perez, C. P. Berlinguette and G. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1034–1046 CrossRef CAS PubMed.
- L. Gundlach and F. Willig, ChemPhysChem, 2012, 13, 2877–2881 CrossRef CAS PubMed.
- J. E. Halls and J. D. Wadhawan, Encyclopedia of Applied Electrochemistry, Springer, New York, NY, 2014, pp. 1556–1578 Search PubMed.
- F. Liu, M. Yang and G. J. Meyer, Handbook of Sol-Gel Science and Technology: Processing Characterization and Application; Volume II: Characterization of Sol-Gel Materials and Products, Kluwer Academic Publishers, 2005, pp. 400–428 Search PubMed.
- D. F. Zigler, Z. A. Morseth, L. Wang, D. L. Ashford, M. K. Brennaman, E. M. Grumstrup, E. C. Brigham, M. K. Gish, R. J. Dillon, L. Alibabaei, G. J. Meyer, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2016, 138, 4426–4438 CrossRef CAS PubMed.
- M. C. Carey, S. L. Adelman and J. K. McCusker, Chem. Sci., 2018, 10, 134–144 RSC.
- C. R. Tichnell, J. N. Miller, C. Liu, S. Mukherjee, E. Jakubikova and J. K. McCusker, J. Phys. Chem. C, 2020, 124, 1794–1811 CrossRef CAS.
- E. Jakubikova and D. N. Bowman, Acc. Chem. Res., 2015, 48, 1441–1449 CrossRef CAS PubMed.
- D. N. Bowman, A. Bondarev, S. Mukherjee and E. Jakubikova, Inorg. Chem., 2015, 54, 8786–8793 CrossRef CAS PubMed.
- L. A. Fredin, M. Pápai, E. Rozsályi, G. Vankó, K. Wärnmark, V. Sundström and P. Persson, J. Phys. Chem. Lett., 2014, 5, 2066–2071 CrossRef CAS PubMed.
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed.
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed.
- P. Chábera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef PubMed.
- P. Zimmer, L. Burkhardt, A. Friedrich, J. Steube, A. Neuba, R. Schepper, P. Müller, U. Flörke, M. Huber, S. Lochbrunner and M. Bauer, Inorg. Chem., 2018, 57, 360–373 CrossRef CAS PubMed.
- J. Huang, O. Buyukcakir, M. W. Mara, A. Coskun, N. M. Dimitrijevic, G. Barin, O. Kokhan, A. B. Stickrath, R. Ruppert, D. M. Tiede, J. F. Stoddart, J.-P. Sauvage and L. X. Chen, Angew. Chem., Int. Ed., 2012, 51, 12711–12715 CrossRef CAS PubMed.
- E. C. Brigham, D. Achey and G. J. Meyer, Polyhedron, 2014, 82, 181–190 CrossRef CAS.
- R. L. Milot and C. A. Schmuttenmaer, Acc. Chem. Res., 2015, 48, 1423–1431 CrossRef CAS PubMed.
- S. H. Lee, K. P. Regan, S. Hedström, A. J. Matula, S. Chaudhuri, R. H. Crabtree, V. S. Batista, C. A. Schmuttenmaer and G. W. Brudvig, J. Phys. Chem. C, 2017, 121, 22690–22699 CrossRef CAS.
- J. Jiang, J. R. Swierk, K. L. Materna, S. Hedström, S. H. Lee, R. H. Crabtree, C. A. Schmuttenmaer, V. S. Batista and G. W. Brudvig, J. Phys. Chem. C, 2016, 120, 28971–28982 CrossRef CAS.
- B. Abraham, H. Fan, E. Galoppini and L. Gundlach, J. Phys. Chem. A, 2018, 122, 2039–2045 CrossRef CAS PubMed.
- R. S. Oliboni, H. Yan, H. Fan, B. Abraham, J. P. Avenoso, E. Galoppini, V. S. Batista, L. Gundlach and L. G. C. Rego, J. Phys. Chem. C, 2019, 123, 12599–12607 CrossRef CAS.
- L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119, 4628–4683 CrossRef CAS PubMed.
- J. G. Rowley, B. H. Farnum, S. Ardo and G. J. Meyer, J. Phys. Chem. Lett., 2010, 1, 3132–3140 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- K. C. D. Robson, K. Hu, G. J. Meyer and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1961–1971 CrossRef CAS PubMed.
- F. Li, J. R. Jennings and Q. Wang, ACS Nano, 2013, 7, 8233–8242 CrossRef CAS PubMed.
- B. N. DiMarco, R. N. Sampaio, E. M. James, T. J. Barr, M. T. Bennett and G. J. Meyer, ACS Appl. Mater. Interfaces, 2020, 12, 23923–23930 CrossRef CAS PubMed.
- W. B. Swords, S. J. C. Simon, F. G. L. Parlane, R. K. Dean, C. W. Kellett, K. Hu, G. J. Meyer and C. P. Berlinguette, Angew. Chem., Int. Ed., 2016, 55, 5956–5960 CrossRef CAS PubMed.
- S. J. C. Simon, F. G. L. Parlane, W. B. Swords, C. W. Kellett, C. Du, B. Lam, R. K. Dean, K. Hu, G. J. Meyer and C. P. Berlinguette, J. Am. Chem. Soc., 2016, 138, 10406–10409 CrossRef CAS PubMed.
- F. G. L. Parlane, C. Mustoe, C. W. Kellett, S. J. Simon, W. B. Swords, G. J. Meyer, P. Kennepohl and C. P. Berlinguette, Nat. Commun., 2017, 8, 1761 CrossRef PubMed.
- L. Casarin, W. B. Swords, S. Caramori, C. A. Bignozzi and G. J. Meyer, Inorg. Chem., 2017, 56, 7324–7327 CrossRef CAS PubMed.
- Y. Saygili, M. Stojanovic, H. Michaels, J. Tiepelt, J. Teuscher, A. Massaro, M. Pavone, F. Giordano, S. M. Zakeeruddin, G. Boschloo, J.-E. Moser, M. Grätzel, A. B. Muñoz-García, A. Hagfeldt and M. Freitag, ACS Appl. Energy Mater., 2018, 1, 4950–4962 CrossRef CAS.
- M. Freitag, F. Giordano, W. Yang, M. Pazoki, Y. Hao, B. Zietz, M. Grätzel, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2016, 120, 9595–9603 CrossRef CAS.
- B. Selvaraj, G. Shanmugam, S. Kamaraj, A. Gunasekeran and A. Sambandam, Inorg. Chem., 2021, 60, 1937–1947 CrossRef CAS PubMed.
- Y. Saygili, M. Stojanovic, H.-S. Kim, J. Teuscher, R. Scopelliti, M. Freitag, S. M. Zakeeruddin, J.-E. Moser, M. Grätzel and A. Hagfeldt, J. Phys. Chem. C, 2020, 124, 7071–7081 CrossRef CAS.
- A. Glinka, M. Gierszewski, B. Gierczyk, G. Burdziński, H. Michaels, M. Freitag and M. Ziółek, J. Phys. Chem. C, 2020, 124, 2895–2906 CrossRef CAS.
- K. Kannankutty, C.-C. Chen, V. S. Nguyen, Y.-C. Lin, H.-H. Chou, C.-Y. Yeh and T.-C. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 5812–5819 CrossRef CAS PubMed.
- T. Higashino, H. Iiyama, S. Nimura, Y. Kurumisawa and H. Imahori, Inorg. Chem., 2020, 59, 452–459 CrossRef CAS PubMed.
- S. O. Fürer, R. A. Milhuisen, M. K. Kashif, S. R. Raga, S. S. Acharya, C. Forsyth, M. Liu, L. Frazer, N. W. Duffy, C. A. Ohlin, A. M. Funston, Y. Tachibana and U. Bach, Adv. Energy Mater., 2020, 10, 2002067 CrossRef.
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed., 2015, 54, 3758–3762 CrossRef CAS PubMed.
- H. Imahori, S. Kang, H. Hayashi, M. Haruta, H. Kurata, S. Isoda, S. E. Canton, Y. Infahsaeng, A. Kathiravan, T. Pascher, P. Chábera, A. P. Yartsev and V. Sundström, J. Phys. Chem. A, 2011, 115, 3679–3690 CrossRef CAS PubMed.
- S. Ye, A. Kathiravan, H. Hayashi, Y. Tong, Y. Infahsaeng, P. Chabera, T. Pascher, A. P. Yartsev, S. Isoda, H. Imahori and V. Sundström, J. Phys. Chem. C, 2013, 117, 6066–6080 CrossRef CAS.
- J. Nelson, S. A. Haque, D. R. Klug and J. R. Durrant, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205321 CrossRef.
- J. Nelson and R. E. Chandler, Coord. Chem. Rev., 2004, 248, 1181–1194 CrossRef CAS.
- J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15374–15380 CrossRef CAS.
- S. Ardo and G. J. Meyer, J. Am. Chem. Soc., 2010, 132, 9283–9285 CrossRef CAS PubMed.
- S. Ardo and G. J. Meyer, J. Am. Chem. Soc., 2011, 133, 15384–15396 CrossRef CAS PubMed.
- M. D. Brady, L. Troian-Gautier, T. C. Motley, M. D. Turlington and G. J. Meyer, ACS Appl. Mater. Interfaces, 2019, 11, 27453–27463 CrossRef CAS PubMed.
- D. Moia, A. Szumska, V. Vaissier, M. Planells, N. Robertson, B. C. O'Regan, J. Nelson and P. R. F. Barnes, J. Am. Chem. Soc., 2016, 138, 13197–13206 CrossRef CAS PubMed.
- A. V. Müller, K. T. de Oliveira, G. J. Meyer and A. S. Polo, ACS Appl. Mater. Interfaces, 2019, 11, 43223–43234 CrossRef PubMed.
- R. N. Sampaio, A. V. Müller, A. S. Polo and G. J. Meyer, ACS Appl. Mater. Interfaces, 2017, 9, 33446–33454 CrossRef CAS PubMed.
- R. N. Sampaio, B. N. DiMarco and G. J. Meyer, ACS Energy Lett., 2017, 2, 2402–2407 CrossRef CAS.
- D. Moia, M. Abe, P. Wagner, H. Saguchi, N. Koumura, J. Nelson, P. R. F. Barnes and S. Mori, J. Phys. Chem. C, 2020, 124, 6979–6992 CrossRef CAS.
- E. C. Brigham and G. J. Meyer, J. Phys. Chem. C, 2014, 118, 7886–7893 CrossRef CAS.
- P. Xu, C. L. Gray, L. Xiao and T. E. Mallouk, J. Am. Chem. Soc., 2018, 140, 11647–11654 CrossRef CAS PubMed.
- R. N. Sampaio, L. Troian-Gautier and G. J. Meyer, Angew. Chem., Int. Ed., 2018, 57, 15390–15394 CrossRef CAS PubMed.
- R. E. Bangle and G. J. Meyer, J. Phys. Chem. C, 2019, 123, 25967–25976 CrossRef CAS.
- R. E. Bangle, J. Schneider, E. J. Piechota, L. Troian-Gautier and G. J. Meyer, J. Am. Chem. Soc., 2020, 142, 674–679 CrossRef CAS PubMed.
- B. N. DiMarco, L. Troian-Gautier, R. N. Sampaio and G. J. Meyer, Chem. Sci., 2018, 9, 940–949 RSC.
- L. Troian-Gautier, B. N. DiMarco, R. N. Sampaio, S. L. Marquard and G. J. Meyer, J. Am. Chem. Soc., 2018, 140, 3019–3029 CrossRef CAS PubMed.
- R. N. Sampaio, R. M. O'Donnell, T. J. Barr and G. J. Meyer, J. Phys. Chem. Lett., 2014, 5, 3265–3268 CrossRef CAS PubMed.
- R. M. O'Donnell, R. N. Sampaio, T. J. Barr and G. J. Meyer, J. Phys. Chem. C, 2014, 118, 16976–16986 CrossRef.
- B. N. DiMarco, R. M. O'Donnell and G. J. Meyer, J. Phys. Chem. C, 2015, 119, 21599–21604 CrossRef CAS.
- K. Hu, A. D. Blair, E. J. Piechota, P. A. Schauer, R. N. Sampaio, F. G. L. Parlane, G. J. Meyer and C. P. Berlinguette, Nat. Chem., 2016, 8, 853–859 CrossRef CAS PubMed.
- R. N. Sampaio, E. J. Piechota, L. Troian-Gautier, A. B. Maurer, K. Hu, P. A. Schauer, A. D. Blair, C. P. Berlinguette and G. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7248–7253 CrossRef CAS PubMed.
- E. J. Piechota, L. Troian-Gautier, R. N. Sampaio, M. K. Brennaman, K. Hu, C. P. Berlinguette and G. J. Meyer, J. Am. Chem. Soc., 2018, 140, 7176–7186 CrossRef CAS PubMed.
- E. J. Piechota, R. N. Sampaio, L. Troian-Gautier, A. B. Maurer, C. P. Berlinguette and G. J. Meyer, J. Phys. Chem. C, 2019, 123, 3416–3425 CrossRef CAS.
- F. Labat, T. Le Bahers, I. Ciofini and C. Adamo, Acc. Chem. Res., 2012, 45, 1268–1277 CrossRef CAS PubMed.
- M. Pastore, Computation, 2017, 5, 5 CrossRef.
- F. De Angelis, Acc. Chem. Res., 2014, 47, 3349–3360 CrossRef CAS PubMed.
- N. Martsinovich and A. Troisi, Energy Environ. Sci., 2011, 4, 4473–4495 RSC.
- J. P. Menzel, A. Papadopoulos, J. Belić, H. J. M. de Groot, L. Visscher and F. Buda, J. Phys. Chem. C, 2020, 124, 27965–27976 CrossRef CAS.
- J. Massin, M. Bräutigam, S. Bold, M. Wächtler, M. Pavone, A. B. Muñoz-García, B. Dietzek, V. Artero and M. Chavarot-Kerlidou, J. Phys. Chem. C, 2019, 123, 17176–17184 CrossRef CAS.
- S. Piccinin, D. Rocca and M. Pastore, J. Phys. Chem. C, 2017, 121, 22286–22294 CrossRef CAS.
- H. S. Yu, S. L. Li and D. G. Truhlar, J. Chem. Phys., 2016, 145, 130901 CrossRef PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- K. Burke, J. Chem. Phys., 2012, 136, 150901 CrossRef PubMed.
- P. Huang and E. A. Carter, Annu. Rev. Phys. Chem., 2008, 59, 261–290 CrossRef CAS PubMed.
- M. S. Hybertsen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5390–5413 CrossRef CAS PubMed.
- F. Bruneval and X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085125 CrossRef.
- J. Harl, L. Schimka and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 115126 CrossRef.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- F. De Angelis, S. Fantacci, A. Selloni and M. K. Nazeeruddin, Chem. Phys. Lett., 2005, 415, 115–120 CrossRef CAS.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS PubMed.
- S. A. Mewes, F. Plasser, A. Krylov and A. Dreuw, J. Chem. Theory Comput., 2018, 14, 710–725 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS PubMed.
- A. Ottochian, C. Morgillo, I. Ciofini, M. J. Frisch, G. Scalmani and C. Adamo, J. Comput. Chem., 2020, 41, 1242–1251 CrossRef CAS PubMed.
- N. Santhanamoorthi, C.-M. Lo and J.-C. Jiang, J. Phys. Chem. Lett., 2013, 4, 524–530 CrossRef CAS PubMed.
- A. Amat, C. Miliani, A. Romani and S. Fantacci, Phys. Chem. Chem. Phys., 2015, 17, 6374–6382 RSC.
- J. Massin, S. Lyu, M. Pavone, A. B. Muñoz-García, B. Kauffmann, T. Toupance, M. Chavarot-Kerlidou, V. Artero and C. Olivier, Dalton Trans., 2016, 45, 12539–12547 RSC.
- D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed.
- J. Wen, B. Han, Z. Havlas and J. Michl, J. Chem. Theory Comput., 2018, 14, 4291–4297 CrossRef CAS PubMed.
- M. Pastore, F. De Angelis and C. Angeli, Theor. Chem. Acc., 2016, 135, 108 Search PubMed.
- C. Suellen, R. G. Freitas, P.-F. Loos and D. Jacquemin, J. Chem. Theory Comput., 2019, 15, 4581–4590 CrossRef CAS PubMed.
- F. Rizzo, M. Cavazzini, S. Righetto, F. D. Angelis, S. Fantacci and S. Quici, Eur. J. Org. Chem., 2010, 4004–4016 CrossRef CAS.
- S. Lyu, J. Massin, M. Pavone, A. B. Muñoz-García, C. Labrugère, T. Toupance, M. Chavarot-Kerlidou, V. Artero and C. Olivier, ACS Appl. Energy Mater., 2019, 2, 4971–4980 CrossRef CAS.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- G. García, C. Adamo and I. Ciofini, Phys. Chem. Chem. Phys., 2013, 15, 20210–20219 RSC.
- S. D. Sousa, S. Lyu, L. Ducasse, T. Toupance and C. Olivier, J. Mater. Chem. A, 2015, 3, 18256–18264 RSC.
- M. Olaru, E. Rychagova, S. Ketkov, Y. Shynkarenko, S. Yakunin, M. V. Kovalenko, A. Yablonskiy, B. Andreev, F. Kleemiss, J. Beckmann and M. Vogt, J. Am. Chem. Soc., 2020, 142, 373–381 CrossRef CAS PubMed.
- L. Huet, A. Perfetto, F. Muniz-Miranda, M. Campetella, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2020, 16, 4543–4553 CrossRef CAS PubMed.
- E. Mosconi, J.-H. Yum, F. Kessler, C. J. Gómez García, C. Zuccaccia, A. Cinti, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, J. Am. Chem. Soc., 2012, 134, 19438–19453 CrossRef CAS PubMed.
- S. M. Feldt, P. W. Lohse, F. Kessler, M. K. Nazeeruddin, M. Grätzel, G. Boschloo and A. Hagfeldt, Phys. Chem. Chem. Phys., 2013, 15, 7087–7097 RSC.
- K. B. Ørnsø, E. Ö. Jónsson, K. W. Jacobsen and K. S. Thygesen, J. Phys. Chem. C, 2015, 119, 12792–12800 CrossRef.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- J. A. Lemkul, J. Huang, B. Roux and A. D. MacKerell, Chem. Rev., 2016, 116, 4983–5013 CrossRef CAS PubMed.
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700–733 CrossRef CAS.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- J. Preat, A. Hagfeldt and E. A. Perpète, Energy Environ. Sci., 2011, 4, 4537–4549 RSC.
- S. Yun, A. Hagfeldt and T. Ma, Adv. Mater., 2014, 26, 6210–6237 CrossRef CAS PubMed.
- C.-T. Li, H.-Y. Chang, Y.-Y. Li, Y.-J. Huang, Y.-L. Tsai, R. Vittal, Y.-J. Sheng and K.-C. Ho, ACS Appl. Mater. Interfaces, 2015, 7, 28254–28263 CrossRef CAS PubMed.
- S. Tontapha, W. Sang-aroon, T. Promgool, S. Kanokmedhakul, W. Maiaugree, E. Swatsitang, V. Homrahad and V. Amornkitbumrung, Mater. Today Commun., 2020, 22, 100742 CrossRef CAS.
- X. Cui, J. Xiao, Y. Wu, P. Du, R. Si, H. Yang, H. Tian, J. Li, W.-H. Zhang, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2016, 55, 6708–6712 CrossRef CAS PubMed.
- E. German, R. Faccio and A. W. Mombrú, Appl. Surf. Sci., 2018, 428, 118–123 CrossRef CAS.
- I. de, P. R. Moreira, F. Illas and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155102 CrossRef.
- Z. M. Gibbs, F. Ricci, G. Li, H. Zhu, K. Persson, G. Ceder, G. Hautier, A. Jain and G. J. Snyder, npj Comput. Mater., 2017, 3, 1–7 CrossRef CAS.
- E. Schiavo, C. Latouche, V. Barone, O. Crescenzi, A. B. Muñoz-García and M. Pavone, Phys. Chem. Chem. Phys., 2018, 20, 14082–14089 RSC.
- B. A. Odeke, G. D. Chung, J. A. Fajemisin, K. S. Suraj, D. K. Tonui, A. R. Tobi, T. C. Bewaale, J. A. Ajibola and N. Y. Dzade, Materials, 2020, 13, 5765 CrossRef CAS PubMed.
- T. Olsen, C. E. Patrick, J. E. Bates, A. Ruzsinszky and K. S. Thygesen, npj Comput. Mater., 2019, 5, 1–23 CrossRef.
- M. C. Toroker, D. K. Kanan, N. Alidoust, L. Y. Isseroff, P. Liao and E. A. Carter, Phys. Chem. Chem. Phys., 2011, 13, 16644–16654 RSC.
- H. Chen, Y. Gong, Á. Vázquez-Mayagoitia, J. Zhang and J. M. Cole, ACS Appl. Energy Mater., 2020, 3, 423–430 CrossRef CAS.
- P. Umari, L. Giacomazzi, F. De Angelis, M. Pastore and S. Baroni, J. Chem. Phys., 2013, 139, 014709 CrossRef CAS PubMed.
- O. V. Kontkanen, M. Niskanen, T. I. Hukka and T. T. Rantala, Phys. Chem. Chem. Phys., 2016, 18, 14382–14389 RSC.
- M. Planells, L. Pellejà, J. N. Clifford, M. Pastore, F. D. Angelis, N. López, S. R. Marder and E. Palomares, Energy Environ. Sci., 2011, 4, 1820–1829 RSC.
- M. Wykes, F. Odobel, C. Adamo, I. Ciofini and F. Labat, J. Mol. Model., 2016, 22, 289 CrossRef PubMed.
- A. B. Muñoz-García, L. Caputo, E. Schiavo, C. Baiano, P. Maddalena and M. Pavone, Front. Chem., 2019, 7, 158 CrossRef PubMed.
- K. J. Chen, A. D. Laurent, F. Boucher, F. Odobel and D. Jacquemin, J. Mater. Chem. A, 2016, 4, 2217–2227 RSC.
- A. B. Muñoz-García and M. Pavone, Phys. Chem. Chem. Phys., 2015, 17, 12238–12246 RSC.
- A. Carella, R. Centore, F. Borbone, M. Toscanesi, M. Trifuoggi, F. Bella, C. Gerbaldi, S. Galliano, E. Schiavo, A. Massaro, A. B. Muñoz-García and M. Pavone, Electrochim. Acta, 2018, 292, 805–816 CrossRef CAS.
- C. Dong, X. Li, W. Zhao, P. Jin and J. Qi, J. Phys. Chem. C, 2013, 117, 9092–9103 CrossRef CAS.
- W. Ma, Y. Jiao, H. Li, H. Guo, E. Kaxiras and S. Meng, ACS Appl. Mater. Interfaces, 2020, 12, 49174–49181 CrossRef CAS PubMed.
- M. Pastore and F. De Angelis, J. Phys. Chem. Lett., 2013, 4, 956–974 CrossRef CAS PubMed.
- S. I. Allec, A. Kumar and B. M. Wong, Dye-Sensitized Solar Cells, Academic Press, 2019, pp. 171–201 Search PubMed.
- R. Sánchez-de-Armas, J. Oviedo López, M. A. San-Miguel, J. F. Sanz, P. Ordejón and M. Pruneda, J. Chem. Theory Comput., 2010, 6, 2856–2865 CrossRef PubMed.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 113, 4496–4565 CrossRef CAS PubMed.
- Q. Huaulmé, V. M. Mwalukuku, D. Joly, J. Liotier, Y. Kervella, P. Maldivi, S. Narbey, F. Oswald, A. J. Riquelme, J. A. Anta and R. Demadrille, Nat. Energy, 2020, 5, 468–477 CrossRef.
- Z. Yang, K. Li, C. Lin, L. R. Devereux, W. Zhang, C. Shao, J. M. Cole and D. Cao, ACS Appl. Energy Mater., 2020, 3, 4367–4376 CrossRef CAS.
- F. Li, X. Peng, Z. Wang, Y. Zhou, Y. Wu, M. Jiang and M. Xu, Energy Environ. Mater., 2019, 2, 280–291 CrossRef.
- J. Westermayr and P. Marquetand, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00749.
- L. Ju, M. Li, L. Tian, P. Xu and W. Lu, Mater. Today Commun., 2020, 25, 101604 CrossRef CAS.
- Q. Arooj and F. Wang, Sol. Energy, 2019, 188, 1189–1200 CrossRef CAS.
- V. Venkatraman, R. Raju, S. P. Oikonomopoulos and B. K. Alsberg, J. Cheminf., 2018, 10, 18 Search PubMed.
- H. N. Tsao and M. Grätzel, ACS Appl. Mater. Interfaces, 2018, 10, 36602–36607 CrossRef CAS PubMed.
- Y. Wen, L. Fu, G. Li, J. Ma and H. Ma, Sol. RRL, 2020, 4, 2000110 CrossRef CAS.
- C. B. Cooper, E. J. Beard, Á. Vázquez-Mayagoitia, L. Stan, G. B. G. Stenning, D. W. Nye, J. A. Vigil, T. Tomar, J. Jia, G. B. Bodedla, S. Chen, L. Gallego, S. Franco, A. Carella, K. R. J. Thomas, S. Xue, X. Zhu and J. M. Cole, Adv. Energy Mater., 2019, 9, 1802820 CrossRef.
- R. Pollice, G. dos Passos Gomes, M. Aldeghi, R. J. Hickman, M. Krenn, C. Lavigne, M. Lindner-D'Addario, A. Nigam, C. T. Ser, Z. Yao and A. Aspuru-Guzik, Acc. Chem. Res., 2021, 54, 849–860 CrossRef CAS PubMed.
- Q. Zhang, D. Myers, J. Lan, S. A. Jenekhe and G. Cao, Phys. Chem. Chem. Phys., 2012, 14, 14982–14998 RSC.
- P. M. Sommeling, B. C. O'Regan, R. R. Haswell, H. J. P. Smit, N. J. Bakker, J. J. T. Smits, J. M. Kroon and J. A. M. van Roosmalen, J. Phys. Chem. B, 2006, 110, 19191–19197 CrossRef CAS PubMed.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- H.-S. Kim, S.-B. Ko, I.-H. Jang and N.-G. Park, Chem. Commun., 2011, 47, 12637–12639 RSC.
- A. Yella, S. Mathew, S. Aghazada, P. Comte, M. Grätzel and M. K. Nazeeruddin, J. Mater. Chem. C, 2017, 5, 2833–2843 RSC.
- B. Roose, S. Pathak and U. Steiner, Chem. Soc. Rev., 2015, 44, 8326–8349 RSC.
- K. Kakiage, H. Osada, Y. Aoyama, T. Yano, K. Oya, S. Iwamoto, J.-I. Fujisawa and M. Hanaya, Sci. Rep., 2016, 6, 35888 CrossRef CAS PubMed.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- F. Sauvage, D. Chen, P. Comte, F. Huang, L.-P. Heiniger, Y.-B. Cheng, R. A. Caruso and M. Graetzel, ACS Nano, 2010, 4, 4420–4425 CrossRef CAS PubMed.
- E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215–219 CrossRef CAS PubMed.
- W. Li, Z. Wu, J. Wang, A. A. Elzatahry and D. Zhao, Chem. Mater., 2014, 26, 287–298 CrossRef CAS.
- M. Pazoki, N. Taghavinia, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2014, 118, 16472–16478 CrossRef CAS.
- M. Pazoki, J. Oscarsson, L. Yang, B. W. Park, E. M. J. Johansson, H. Rensmo, A. Hagfeldt and G. Boschloo, RSC Adv., 2014, 4, 50295–50300 RSC.
- D. Chen, L. Cao, F. Huang, P. Imperia, Y.-B. Cheng and R. A. Caruso, J. Am. Chem. Soc., 2010, 132, 4438–4444 CrossRef CAS PubMed.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- Q. Zhang, C. S. Dandeneau, X. Zhou and G. Cao, Adv. Mater., 2009, 21, 4087–4108 CrossRef CAS.
- R. Vittal and K.-C. Ho, Renewable Sustainable Energy Rev., 2017, 70, 920–935 CrossRef CAS.
- Q. Wali, A. Fakharuddin and R. Jose, J. Power Sources, 2015, 293, 1039–1052 CrossRef CAS.
- J. Wan, L. Tao, B. Wang, J. Zhang, H. Wang and P. D. Lund, J. Power Sources, 2019, 438, 227012 CrossRef CAS.
- T. Peng, J. Xu and R. Chen, Chem. Phys. Lett., 2020, 738, 136902 CrossRef CAS.
- N. Memarian, I. Concina, A. Braga, S. M. Rozati, A. Vomiero and G. Sberveglieri, Angew. Chem., Int. Ed., 2011, 50, 12321–12325 CrossRef CAS PubMed.
- Y.-F. Wang, K.-N. Li, W.-Q. Wu, Y.-F. Xu, H.-Y. Chen, C.-Y. Su and D.-B. Kuang, RSC Adv., 2013, 3, 13804–13810 RSC.
- L. Tao, Z. Sun, L. Chen, M. Liang and S. Xue, Chem. Commun., 2020, 56, 5042–5045 RSC.
- S. S. Shin, J. H. Suk, B. J. Kang, W. Yin, S. J. Lee, J. H. Noh, T. K. Ahn, F. Rotermund, I. S. Cho and S. I. Seok, Energy Environ. Sci., 2019, 12, 958–964 RSC.
- V. Ganapathy, B. Karunagaran and S.-W. Rhee, J. Power Sources, 2010, 195, 5138–5143 CrossRef CAS.
- C.-Y. Cho, S. Baek, K. Kim and J. H. Moon, RSC Adv., 2016, 6, 74003–74008 RSC.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C.-h. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- H. Ozawa, Y. Okuyama and H. Arakawa, Dalton Trans., 2012, 41, 5137–5139 RSC.
- H. Ozawa, T. Sugiura, T. Kuroda, K. Nozawa and H. Arakawa, J. Mater. Chem. A, 2016, 4, 1762–1770 RSC.
- T. Kono, N. Masaki, M. Nishikawa, R. Tamura, H. Matsuzaki, M. Kimura and S. Mori, ACS Appl. Mater. Interfaces, 2016, 8, 16677–16683 CrossRef CAS PubMed.
- L. E. Polander, A. Yella, B. F. E. Curchod, N. Ashari Astani, J. Teuscher, R. Scopelliti, P. Gao, S. Mathew, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Angew. Chem., Int. Ed., 2013, 52, 8731–8735 CrossRef CAS PubMed.
- S. Aghazada, P. Gao, A. Yella, G. Marotta, T. Moehl, J. Teuscher, J.-E. Moser, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2016, 55, 6653–6659 CrossRef CAS PubMed.
- S. Aghazada, Y. Ren, P. Wang and M. K. Nazeeruddin, Inorg. Chem., 2017, 56, 13437–13445 CrossRef CAS PubMed.
- K.-L. Wu, A. J. Huckaba, J. N. Clifford, Y.-W. Yang, A. Yella, E. Palomares, M. Grätzel, Y. Chi and M. K. Nazeeruddin, Inorg. Chem., 2016, 55, 7388–7395 CrossRef CAS PubMed.
- K.-L. Wu, Y. Hu, C.-T. Chao, Y.-W. Yang, T.-Y. Hsiao, N. Robertson and Y. Chi, J. Mater. Chem. A, 2014, 2, 19556–19565 RSC.
- M. V. Bobo, A. Paul, A. J. Robb, A. M. Arcidiacono, M. D. Smith, K. Hanson and A. K. Vannucci, Inorg. Chem., 2020, 59, 6351–6358 CrossRef CAS PubMed.
- T. Yamaguchi, T. Miyabe, T. Ono and H. Arakawa, Chem. Commun., 2010, 46, 5802–5804 RSC.
- K.-L. Wu, S.-T. Ho, C.-C. Chou, Y.-C. Chang, H.-A. Pan, Y. Chi and P.-T. Chou, Angew. Chem., Int. Ed., 2012, 51, 5642–5646 CrossRef CAS PubMed.
- T. Kinoshita, K. Nonomura, N. Joong Jeon, F. Giordano, A. Abate, S. Uchida, T. Kubo, S. I. Seok, M. K. Nazeeruddin, A. Hagfeldt, M. Grätzel and H. Segawa, Nat. Commun., 2015, 6, 8834 CrossRef CAS PubMed.
- S. Fuse, S. Sugiyama, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, R. Katoh, T. Kaiho and T. Takahashi, Chem. – Eur. J., 2014, 20, 10685–10694 CrossRef CAS PubMed.
- K. Matsumura, S. Yoshizaki, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, S. Fuse, H. Tanaka and T. Takahashi, Chem. – Eur. J., 2015, 21, 9742–9747 CrossRef CAS PubMed.
- D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS.
- S. Tamba, R. Fujii, A. Mori, K. Hara and N. Koumura, Chem. Lett., 2011, 40, 922–924 CrossRef CAS.
- W. Wang, X. Li, J. Lan, D. Wu, R. Wang and J. You, J. Org. Chem., 2018, 83, 8114–8126 CrossRef CAS PubMed.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376–16379 CrossRef CAS PubMed.
- X. Kang, J. Zhang, D. O'Neil, A. J. Rojas, W. Chen, P. Szymanski, S. R. Marder and M. A. El-Sayed, Chem. Mater., 2014, 26, 4486–4493 CrossRef CAS.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059–8078 CrossRef CAS.
- P.-H. Lin, T.-J. Lu, D.-J. Cai, K.-M. Lee and C.-Y. Liu, ChemSusChem, 2015, 8, 3222–3227 CrossRef CAS PubMed.
- N. P. Liyanage, A. Yella, M. Nazeeruddin, M. Grätzel and J. H. Delcamp, ACS Appl. Mater. Interfaces, 2016, 8, 5376–5384 CrossRef CAS PubMed.
- A. Baumann, J. Watson and J. H. Delcamp, ChemSusChem, 2020, 13, 283–286 CrossRef CAS PubMed.
- Y.-S. Ciou, P.-H. Lin, W.-M. Li, K.-M. Lee and C.-Y. Liu, J. Org. Chem., 2017, 82, 3538–3551 CrossRef CAS PubMed.
- S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Péchy and M. Grätzel, Chem. Commun., 2008, 5194–5196 RSC.
- W. Zhang, Y. Wu, X. Li, E. Li, X. Song, H. Jiang, C. Shen, H. Zhang, H. Tian and W.-H. Zhu, Chem. Sci., 2017, 8, 2115–2124 RSC.
- J. Yang, P. Ganesan, J. Teuscher, T. Moehl, Y. J. Kim, C. Yi, P. Comte, K. Pei, T. W. Holcombe, M. K. Nazeeruddin, J. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, J. Am. Chem. Soc., 2014, 136, 5722–5730 CrossRef CAS PubMed.
- J.-H. Yum, T. W. Holcombe, Y. Kim, K. Rakstys, T. Moehl, J. Teuscher, J. H. Delcamp, M. K. Nazeeruddin and M. Grätzel, Sci. Rep., 2013, 3, 2446 CrossRef PubMed.
- R. Li, J. Liu, N. Cai, M. Zhang and P. Wang, J. Phys. Chem. B, 2010, 114, 4461–4464 CrossRef CAS PubMed.
- H. Cheema, J. Watson, A. Peddapuram and J. H. Delcamp, Chem. Commun., 2020, 56, 1741–1744 RSC.
- P. Brogdon, F. Giordano, G. A. Puneky, A. Dass, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel, G. S. Tschumper and J. H. Delcamp, Chem. – Eur. J., 2016, 22, 694–703 CrossRef CAS PubMed.
- P. Wang, L. Yang, H. Wu, Y. Cao, J. Zhang, N. Xu, S. Chen, J.-D. Decoppet, S. M. Zakeeruddin and M. Grätzel, Joule, 2018, 2, 2145–2153 CrossRef CAS.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed.
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS PubMed.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef PubMed.
- W. Zhang, Y. Wu, H. W. Bahng, Y. Cao, C. Yi, Y. Saygili, J. Luo, Y. Liu, L. Kavan, J.-E. Moser, A. Hagfeldt, H. Tian, S. M. Zakeeruddin, W.-H. Zhu and M. Grätzel, Energy Environ. Sci., 2018, 11, 1779–1787 RSC.
- Y. Liu, Y. Cao, W. Zhang, M. Stojanovic, M. I. Dar, P. Péchy, Y. Saygili, A. Hagfeldt, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2018, 57, 14125–14128 CrossRef CAS PubMed.
- L. Yang, S. Chen, J. Zhang, J. Wang, M. Zhang, X. Dong and P. Wang, J. Mater. Chem. A, 2017, 5, 3514–3522 RSC.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799–3802 CrossRef CAS PubMed.
- Y. Ren, D. Sun, Y. Cao, H. N. Tsao, Y. Yuan, S. M. Zakeeruddin, P. Wang and M. Grätzel, J. Am. Chem. Soc., 2018, 140, 2405–2408 CrossRef CAS PubMed.
- H. Wu, X. Xie, Y. Mei, Y. Ren, Z. Shen, S. Li and P. Wang, ACS Photonics, 2019, 6, 1216–1225 CrossRef CAS.
- L. Zhang, X. Yang, W. Wang, G. G. Gurzadyan, J. Li, X. Li, J. An, Z. Yu, H. Wang, B. Cai, A. Hagfeldt and L. Sun, ACS Energy Lett., 2019, 4, 943–951 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379–6381 RSC.
- R. R. Rodrigues, A. Peddapuram, A. L. Dorris, N. I. Hammer and J. H. Delcamp, ACS Appl. Energy Mater., 2019, 2, 5547–5556 CrossRef CAS.
- R. R. Rodrigues, H. Cheema and J. H. Delcamp, Angew. Chem., Int. Ed., 2018, 57, 5472–5476 CrossRef CAS PubMed.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- S. H. Kang, M. J. Jeong, Y. K. Eom, I. T. Choi, S. M. Kwon, Y. Yoo, J. Kim, J. Kwon, J. H. Park and H. K. Kim, Adv. Energy Mater., 2017, 7, 1602117 CrossRef.
- Y. K. Eom, S. H. Kang, I. T. Choi, Y. Yoo, J. Kim and H. K. Kim, J. Mater. Chem. A, 2017, 5, 2297–2308 RSC.
- S. Mathew, N. A. Astani, B. F. E. Curchod, J. H. Delcamp, M. Marszalek, J. Frey, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem. A, 2016, 4, 2332–2339 RSC.
- J. Luo, M. Xu, R. Li, K.-W. Huang, C. Jiang, Q. Qi, W. Zeng, J. Zhang, C. Chi, P. Wang and J. Wu, J. Am. Chem. Soc., 2014, 136, 265–272 CrossRef CAS PubMed.
- J. Luo, J. Zhang, K.-W. Huang, Q. Qi, S. Dong, J. Zhang, P. Wang and J. Wu, J. Mater. Chem. A, 2016, 4, 8428–8434 RSC.
- H.-H. Chou, K. S. K. Reddy, H.-P. Wu, B.-C. Guo, H.-W. Lee, E. W.-G. Diau, C.-P. Hsu and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2016, 8, 3418–3427 CrossRef CAS PubMed.
- Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., Int. Ed., 2014, 53, 10779–10783 CrossRef CAS PubMed.
- Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055–14058 CrossRef CAS PubMed.
- Y. Tang, Y. Wang, X. Li, H. Ågren, W.-H. Zhu and Y. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 27976–27985 CrossRef CAS PubMed.
- H. Zhou, J.-M. Ji, S. H. Kang, M. S. Kim, H. S. Lee, C. H. Kim and H. K. Kim, J. Mater. Chem. C, 2019, 7, 2843–2852 RSC.
- G. Yang, Y. Tang, X. Li, H. Ågren and Y. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 36875–36885 CrossRef CAS PubMed.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, L. Han, I. Bedja, R. K. Gupta and A. Islam, J. Phys. Chem. C, 2017, 121, 6464–6477 CrossRef CAS.
- W. Li, Z. Liu, H. Wu, Y.-B. Cheng, Z. Zhao and H. He, J. Phys. Chem. C, 2015, 119, 5265–5273 CrossRef CAS.
- T. Higashino, Y. Fujimori, K. Sugiura, Y. Tsuji, S. Ito and H. Imahori, Angew. Chem., Int. Ed., 2015, 54, 9052–9056 CrossRef CAS PubMed.
- L. Si, H. He and K. Zhu, New J. Chem., 2014, 38, 1565–1572 RSC.
- C.-C. Chen, J.-S. Chen, V. S. Nguyen, T.-C. Wei and C.-Y. Yeh, Angew. Chem., Int. Ed., 2021, 60, 4886–4893 CrossRef CAS PubMed.
- C.-L. Wang, M. Zhang, Y.-H. Hsiao, C.-K. Tseng, C.-L. Liu, M. Xu, P. Wang and C.-Y. Lin, Energy Environ. Sci., 2016, 9, 200–206 RSC.
- H. Cheema, A. Baumann, E. K. Loya, P. Brogdon, L. E. McNamara, C. A. Carpenter, N. I. Hammer, S. Mathew, C. Risko and J. H. Delcamp, ACS Appl. Mater. Interfaces, 2019, 11, 16474–16489 CrossRef CAS PubMed.
- S. Chakraborty, H.-C. You, C.-K. Huang, B.-Z. Lin, C.-L. Wang, M.-C. Tsai, C.-L. Liu and C.-Y. Lin, J. Phys. Chem. C, 2017, 121, 7081–7087 CrossRef CAS.
- K. Zeng, W. Tang, C. Li, Y. Chen, S. Zhao, Q. Liu and Y. Xie, J. Mater. Chem. A, 2019, 7, 20854–20860 RSC.
- K. Zeng, Y. Lu, W. Tang, S. Zhao, Q. Liu, W. Zhu, H. Tian and Y. Xie, Chem. Sci., 2019, 10, 2186–2192 RSC.
- K. Zeng, Y. Chen, W.-H. Zhu, H. Tian and Y. Xie, J. Am. Chem. Soc., 2020, 142, 5154–5161 CrossRef CAS PubMed.
- R. Bisht, M. F. Mele Kavungathodi and J. Nithyanandhan, Chem. – Eur. J., 2018, 24, 16368–16378 CrossRef CAS PubMed.
- A. Alagumalai, M. K. Munavvar Fairoos, M. C. Sil and J. Nithyanandhan, ACS Appl. Mater. Interfaces, 2016, 8, 35353–35367 CrossRef CAS PubMed.
- J. H. Delcamp, Y. Shi, J.-H. Yum, T. Sajoto, E. Dell'Orto, S. Barlow, M. K. Nazeeruddin, S. R. Marder and M. Grätzel, Chem. – Eur. J., 2013, 19, 1819–1827 CrossRef CAS PubMed.
- F. M. Jradi, X. Kang, D. O'Neil, G. Pajares, Y. A. Getmanenko, P. Szymanski, T. C. Parker, M. A. El-Sayed and S. R. Marder, Chem. Mater., 2015, 27, 2480–2487 CrossRef CAS.
- F. M. Jradi, D. O'Neil, X. Kang, J. Wong, P. Szymanski, T. C. Parker, H. L. Anderson, M. A. El-Sayed and S. R. Marder, Chem. Mater., 2015, 27, 6305–6313 CrossRef CAS.
- T. Maeda, S. Arikawa, H. Nakao, S. Yagi and H. Nakazumi, New J. Chem., 2013, 37, 701–708 RSC.
- S. Paek, H. Choi, C. Kim, N. Cho, S. So, K. Song, M. K. Nazeeruddin and J. Ko, Chem. Commun., 2011, 47, 2874–2876 RSC.
- J.-Y. Li, C.-Y. Chen, W.-C. Ho, S.-H. Chen and C.-G. Wu, Org. Lett., 2012, 14, 5420–5423 CrossRef CAS PubMed.
- V. Punitharasu, M. F. Mele Kavungathodi, A. K. Singh and J. Nithyanandhan, ACS Appl. Energy Mater., 2019, 2, 8464–8472 CrossRef CAS.
- C. Qin, Y. Numata, S. Zhang, X. Yang, A. Islam, K. Zhang, H. Chen and L. Han, Adv. Funct. Mater., 2014, 24, 3059–3066 CrossRef CAS.
- G. H. Rao, A. Venkateswararao, L. Giribabu, L. Han, I. Bedja, R. K. Gupta, A. Islam and S. P. Singh, Phys. Chem. Chem. Phys., 2016, 18, 14279–14285 RSC.
- Y. Cao, Y. Liu, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Joule, 2018, 2, 1108–1117 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, T. Kyomen and M. Hanaya, Chem. Commun., 2015, 51, 6315–6317 RSC.
- Y. Hao, W. Yang, L. Zhang, R. Jiang, E. Mijangos, Y. Saygili, L. Hammarström, A. Hagfeldt and G. Boschloo, Nat. Commun., 2016, 7, 13934 CrossRef CAS PubMed.
- J.-M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 RSC.
- H. Cheema, J. Watson and J. H. Delcamp, Sol. Energy, 2020, 208, 747–752 CrossRef CAS.
- H. Cheema, J. Watson, P. S. Shinde, R. R. Rodrigues, S. Pan and J. H. Delcamp, Chem. Commun., 2020, 56, 1569–1572 RSC.
- H. Cheema and J. H. Delcamp, Adv. Energy Mater., 2019, 9, 1900162 CrossRef.
- A. Baumann, C. Curiac and J. H. Delcamp, ChemSusChem, 2020, 13, 2503–2512 CrossRef CAS PubMed.
- Y. Wu, W.-H. Zhu, S. M. Zakeeruddin and M. Grätzel, ACS Appl. Mater. Interfaces, 2015, 7, 9307–9318 CrossRef CAS PubMed.
- J. Sobuś, B. Gierczyk, G. Burdziński, M. Jancelewicz, E. Polanski, A. Hagfeldt and M. Ziółek, Chem. – Eur. J., 2016, 22, 15807–15818 CrossRef PubMed.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem. – Eur. J., 2013, 19, 17075–17081 CrossRef CAS PubMed.
- Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910–9919 CrossRef CAS PubMed.
- N. Asim, K. Sopian, S. Ahmadi, K. Saeedfar, M. A. Alghoul, O. Saadatian and S. H. Zaidi, Renewable Sustainable Energy Rev., 2012, 16, 5834–5847 CrossRef CAS.
- M. S. Su'ait, M. Y. A. Rahman and A. Ahmad, Sol. Energy, 2015, 115, 452–470 CrossRef.
- S. Ahmad, E. Guillén, L. Kavan, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2013, 6, 3439–3466 RSC.
- S. Mastroianni, I. Asghar, K. Miettunen, J. Halme, A. Lanuti, T. M. Brown and P. Lund, Phys. Chem. Chem. Phys., 2014, 16, 6092–6100 RSC.
- M. I. Asghar, K. Miettunen, J. Halme, P. Vahermaa, M. Toivola, K. Aitola and P. Lund, Energy Environ. Sci., 2010, 3, 418–426 RSC.
- M. K. Kashif, J. C. Axelson, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, J. Am. Chem. Soc., 2012, 134, 16646–16653 CrossRef CAS PubMed.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- S. Mozaffari, M. R. Nateghi and M. B. Zarandi, Renewable Sustainable Energy Rev., 2017, 71, 675–686 CrossRef CAS.
- G. P. Salvador, D. Pugliese, F. Bella, A. Chiappone, A. Sacco, S. Bianco and M. Quaglio, Electrochim. Acta, 2014, 146, 44–51 CrossRef CAS.
- C.-Y. Hsu, Y.-C. Chen, R. Y.-Y. Lin, K.-C. Ho and J. T. Lin, Phys. Chem. Chem. Phys., 2012, 14, 14099–14109 RSC.
- S. M. Feldt, G. Wang, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115, 21500–21507 CrossRef CAS.
- U. B. Cappel, S. M. Feldt, J. Schöneboom, A. Hagfeldt and G. Boschloo, J. Am. Chem. Soc., 2010, 132, 9096–9101 CrossRef CAS PubMed.
- L. Kavan, Curr. Opin. Electrochem., 2017, 2, 88–96 CrossRef CAS.
- J.-W. Shiu, Y.-C. Chang, C.-Y. Chan, H.-P. Wu, H.-Y. Hsu, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, J. Mater. Chem. A, 2014, 3, 1417–1420 RSC.
- Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC.
- S. Tatay, S. A. Haque, B. O'Regan, J. R. Durrant, W. J. H. Verhees, J. M. Kroon, A. Vidal-Ferran, P. Gaviña and E. Palomares, J. Mater. Chem., 2007, 17, 3037–3044 RSC.
- M. Freitag, J. Teuscher, Y. Saygili, X. Zhang, F. Giordano, P. Liska, J. Hua, S. M. Zakeeruddin, J.-E. Moser, M. Grätzel and A. Hagfeldt, Nat. Photonics, 2017, 11, 372–378 CrossRef CAS.
- T. J. Zerk, C. T. Saouma, J. M. Mayer and W. B. Tolman, Inorg. Chem., 2019, 58, 14151–14158 CrossRef CAS PubMed.
- S. C. Pradhan, A. Hagfeldt and S. Soman, J. Mater. Chem. A, 2018, 6, 22204–22214 RSC.
- M. Magni, R. Giannuzzi, A. Colombo, M. P. Cipolla, C. Dragonetti, S. Caramori, S. Carli, R. Grisorio, G. P. Suranna, C. A. Bignozzi, D. Roberto and M. Manca, Inorg. Chem., 2016, 55, 5245–5253 CrossRef CAS PubMed.
- R. R. Rodrigues, J. M. Lee, N. S. Taylor, H. Cheema, L. Chen, R. C. Fortenberry, J. H. Delcamp and J. W. Jurss, Dalton Trans., 2020, 49, 343–355 RSC.
- H. Michaels, I. Benesperi, T. Edvinsson, A. B. Muñoz-Garcia, M. Pavone, G. Boschloo and M. Freitag, Inorganics, 2018, 6, 53 CrossRef.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- M. Freitag, W. Yang, L. A. Fredin, L. D'Amario, K. M. Karlsson, A. Hagfeldt and G. Boschloo, ChemPhysChem, 2016, 17, 3845–3852 CrossRef CAS PubMed.
- W. Yang, N. Vlachopoulos, Y. Hao, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2015, 17, 15868–15875 RSC.
- J. Burschka, V. Brault, S. Ahmad, L. Breau, M. K. Nazeeruddin, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2012, 5, 6089–6097 RSC.
- C.-H. Tsai, C.-Y. Lu, M.-C. Chen, T.-W. Huang, C.-C. Wu and Y.-W. Chung, Org. Electron., 2013, 14, 3131–3137 CrossRef CAS.
- F. Bella and R. Bongiovanni, J. Photochem. Photobiol., C, 2013, 16, 1–21 CrossRef CAS.
- A. Gagliardi, D. Gentilini and A. D. Carlo, J. Phys. Chem. C, 2012, 116, 23882–23889 CrossRef CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2008, 38, 115–164 RSC.
- Z. Yu, N. Vlachopoulos, M. Gorlov and L. Kloo, Dalton Trans., 2011, 40, 10289–10303 RSC.
- Z. Yu, M. Gorlov, J. Nissfolk, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2010, 114, 10612–10620 CrossRef CAS.
- M. Gorlov, H. Pettersson, A. Hagfeldt and L. Kloo, Inorg. Chem., 2007, 46, 3566–3575 CrossRef CAS PubMed.
- Y. Liu, J. R. Jennings, Y. Huang, Q. Wang, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 18847–18855 CrossRef CAS.
- M. K. Kashif, M. Nippe, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 5527–5531 CrossRef CAS PubMed.
- H. Ellis, S. K. Eriksson, S. M. Feldt, E. Gabrielsson, P. W. Lohse, R. Lindblad, L. Sun, H. Rensmo, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2013, 117, 21029–21036 CrossRef CAS.
- S. Koussi-Daoud, D. Schaming, L. Fillaud, G. Trippé-Allard, F. Lafolet, E. Polanski, K. Nonomura, N. Vlachopoulos, A. Hagfeldt and J.-C. Lacroix, Electrochim. Acta, 2015, 179, 237–240 CrossRef CAS.
- M. Nasr-Esfahani, M. Zendehdel, N. Y. Nia, B. Jafari and M. K. Babadi, RSC Adv., 2014, 4, 15961–15967 RSC.
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CrossRef CAS PubMed.
- C. Dragonetti, M. Magni, A. Colombo, F. Melchiorre, P. Biagini and D. Roberto, ACS Appl. Energy Mater., 2018, 1, 751–756 CrossRef CAS.
- J. Cong, D. Kinschel, Q. Daniel, M. Safdari, E. Gabrielsson, H. Chen, P. H. Svensson, L. Sun and L. Kloo, J. Mater. Chem. A, 2016, 4, 14550–14554 RSC.
- E. Benazzi, M. Magni, A. Colombo, C. Dragonetti, S. Caramori, C. A. Bignozzi, R. Grisorio, G. P. Suranna, M. P. Cipolla, M. Manca and D. Roberto, Electrochim. Acta, 2018, 271, 180–189 CrossRef CAS.
- T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS PubMed.
- I. A. Rutkowska, A. Andrearczyk, S. Zoladek, M. Goral, K. Darowicki and P. J. Kulesza, J. Solid State Electrochem., 2011, 15, 2545–2552 CrossRef CAS.
- T. Daeneke, A. J. Mozer, T.-H. Kwon, N. W. Duffy, A. B. Holmes, U. Bach and L. Spiccia, Energy Environ. Sci., 2012, 5, 7090–7099 RSC.
- I. R. Perera, A. Gupta, W. Xiang, T. Daeneke, U. Bach, R. A. Evans, C. A. Ohlin and L. Spiccia, Phys. Chem. Chem. Phys., 2014, 16, 12021–12028 RSC.
- S. Carli, E. Benazzi, L. Casarin, T. Bernardi, V. Bertolasi, R. Argazzi, S. Caramori and C. A. Bignozzi, Phys. Chem. Chem. Phys., 2016, 18, 5949–5956 RSC.
- K. Oyaizu, N. Hayo, Y. Sasada, F. Kato and H. Nishide, Dalton Trans., 2013, 42, 16090–16095 RSC.
- A. Apostolopoulou, M. Vlasiou, P. A. Tziouris, C. Tsiafoulis, A. C. Tsipis, D. Rehder, T. A. Kabanos, A. D. Keramidas and E. Stathatos, Inorg. Chem., 2015, 54, 3979–3988 CrossRef CAS PubMed.
- X. Li, Z. Ku, Y. Rong, G. Liu, L. Liu, T. Liu, M. Hu, Y. Yang, H. Wang, M. Xu, P. Xiang and H. Han, Phys. Chem. Chem. Phys., 2012, 14, 14383–14390 RSC.
- L. Liu, X. Li, J. Chen, Y. Rong, Z. Ku and H. Han, Sci. Rep., 2013, 3, 2413 CrossRef PubMed.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS.
- F. Kato, A. Kikuchi, T. Okuyama, K. Oyaizu and H. Nishide, Angew. Chem., Int. Ed., 2012, 51, 10177–10180 CrossRef CAS PubMed.
- Y. Liu, J. R. Jennings and Q. Wang, ChemSusChem, 2013, 6, 2124–2131 CrossRef CAS PubMed.
- M. Cheng, X. Yang, C. Chen, J. Zhao, F. Zhang and L. Sun, Phys. Chem. Chem. Phys., 2013, 15, 15146–15152 RSC.
- N. Flores-Díaz, A. Soto-Navarro, M. Freitag, G. Lamoureux and L. W. Pineda, Sol. Energy, 2018, 167, 76–83 CrossRef.
- H. Iftikhar, G. G. Sonai, S. G. Hashmi, A. F. Nogueira and P. D. Lund, Materials, 2019, 12, 1998 CrossRef CAS PubMed.
- Q. Yu, D. Zhou, Y. Shi, X. Si, Y. Wang and P. Wang, Energy Environ. Sci., 2010, 3, 1722–1725 RSC.
- M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and X. Hao, Prog. Photovoltaics, 2020, 28, 629–638 Search PubMed.
- G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS.
- Y. Saygili, M. Stojanovic, N. Flores-Díaz, S. M. Zakeeruddin, N. Vlachopoulos, M. Grätzel and A. Hagfeldt, Inorganics, 2019, 7, 30 CrossRef CAS.
- H. Ellis, I. Schmidt, A. Hagfeldt, G. Wittstock and G. Boschloo, J. Phys. Chem. C, 2015, 119, 21775–21783 CrossRef CAS.
- M. K. Kashif, R. A. Milhuisen, M. Nippe, J. Hellerstedt, D. Z. Zee, N. W. Duffy, B. Halstead, F. D. Angelis, S. Fantacci, M. S. Fuhrer, C. J. Chang, Y.-B. Cheng, J. R. Long, L. Spiccia and U. Bach, Adv. Energy Mater., 2016, 6, 1600874 CrossRef.
- W. Xiang, W. Huang, U. Bach and L. Spiccia, Chem. Commun., 2013, 49, 8997–8999 RSC.
- C. Dong, W. Xiang, F. Huang, D. Fu, W. Huang, U. Bach, Y.-B. Cheng, X. Li and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 6933–6937 CrossRef CAS PubMed.
- H. Ellis, R. Jiang, S. Ye, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2016, 18, 8419–8427 RSC.
- S. Venkatesan, W.-H. Lin, H. Teng and Y.-L. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 42780–42789 CrossRef CAS PubMed.
- K. D. Karlin and J. K. Yandell, Inorg. Chem., 1984, 23, 1184–1188 CrossRef CAS.
- M. Magni, A. Colombo, C. Dragonetti and P. Mussini, Electrochim. Acta, 2014, 141, 324–330 CrossRef CAS.
- A. Colombo, C. Dragonetti, M. Magni, D. Roberto, F. Demartin, S. Caramori and C. A. Bignozzi, ACS Appl. Mater. Interfaces, 2014, 6, 13945–13955 CrossRef CAS PubMed.
- A. Colombo, G. Di Carlo, C. Dragonetti, M. Magni, A. Orbelli Biroli, M. Pizzotti, D. Roberto, F. Tessore, E. Benazzi, C. A. Bignozzi, L. Casarin and S. Caramori, Inorg. Chem., 2017, 56, 14189–14197 CrossRef CAS PubMed.
- W. L. Hoffeditz, M. J. Katz, P. Deria, G. E. Cutsail III, M. J. Pellin, O. K. Farha and J. T. Hupp, J. Phys. Chem. C, 2016, 120, 3731–3740 CrossRef CAS.
- Y. Wang and T. W. Hamann, Chem. Commun., 2018, 54, 12361–12364 RSC.
- M. Karpacheva, F. J. Malzner, C. Wobill, A. Büttner, E. C. Constable and C. E. Housecroft, Dyes Pigm., 2018, 156, 410–416 CrossRef CAS.
- A. M. Spokoyny, T. C. Li, O. K. Farha, C. W. Machan, C. She, C. L. Stern, T. J. Marks, J. T. Hupp and C. A. Mirkin, Angew. Chem., 2010, 122, 5467–5471 CrossRef.
- M. F. Hawthorne, J. I. Zink, J. M. Skelton, M. J. Bayer, C. Liu, E. Livshits, R. Baer and D. Neuhauser, Science, 2004, 303, 1849–1851 CrossRef CAS PubMed.
- T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580–4582 CrossRef CAS PubMed.
- J. Cong, Y. Hao, G. Boschloo and L. Kloo, ChemSusChem, 2015, 8, 264–268 CrossRef CAS PubMed.
- J. N. de Freitas, A. F. Nogueira and M.-A. D. Paoli, J. Mater. Chem., 2009, 19, 5279–5294 RSC.
- Z. Chen, F. Li, H. Yang, T. Yi and C. Huang, ChemPhysChem, 2007, 8, 1293–1297 CrossRef CAS PubMed.
- Y. Rong, Z. Ku, M. Xu, L. Liu, M. Hu, Y. Yang, J. Chen, A. Mei, T. Liu and H. Han, RSC Adv., 2014, 4, 9271–9274 RSC.
- C.-P. Lee and K.-C. Ho, Eur. Polym. J., 2018, 108, 420–428 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, P. Comte, I. Exnar and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 1166–1167 CrossRef CAS PubMed.
- L. Fan, S. Kang, J. Wu, S. Hao, Z. Lan and J. Lin, Energy Sources Part Recovery Util. Environ. Eff., 2010, 32, 1559–1568 CAS.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS PubMed.
- F. Bella, N. Vlachopoulos, K. Nonomura, S. M. Zakeeruddin, M. Grätzel, C. Gerbaldi and A. Hagfeldt, Chem. Commun., 2015, 51, 16308–16311 RSC.
- C.-L. Chen, T.-W. Chang, H. Teng, C.-G. Wu, C.-Y. Chen, Y.-M. Yang and Y.-L. Lee, Phys. Chem. Chem. Phys., 2013, 15, 3640–3645 RSC.
- Masud, K. M. Kim and H. K. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 42067–42080 CrossRef CAS PubMed.
- K. M. Kim, Masud, J.-M. Ji and H. K. Kim, ACS Appl. Energy Mater., 2021, 4, 1302–1312 CrossRef CAS.
- J. N. de Freitas, A. d. S. Gonçalves, M.-A. De Paoli, J. R. Durrant and A. F. Nogueira, Electrochim. Acta, 2008, 53, 7166–7172 CrossRef.
- W. Kubo, K. Murakoshi, T. Kitamura, S. Yoshida, M. Haruki, K. Hanabusa, H. Shirai, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2001, 105, 12809–12815 CrossRef CAS.
- F. Bella, J. R. Nair and C. Gerbaldi, RSC Adv., 2013, 3, 15993–16001 RSC.
- F. Bella, D. Pugliese, J. R. Nair, A. Sacco, S. Bianco, C. Gerbaldi, C. Barolo and R. Bongiovanni, Phys. Chem. Chem. Phys., 2013, 15, 3706–3711 RSC.
- F. Bella, A. Lamberti, A. Sacco, S. Bianco, A. Chiodoni and R. Bongiovanni, J. Membr. Sci., 2014, 470, 125–131 CrossRef CAS.
- F. Bella, E. D. Ozzello, A. Sacco, S. Bianco and R. Bongiovanni, Int. J. Hydrogen Energy, 2014, 39, 3036–3045 CrossRef CAS.
- F. Bella, R. Bongiovanni, R. S. Kumar, M. A. Kulandainathan and A. M. Stephan, J. Mater. Chem. A, 2013, 1, 9033–9036 RSC.
- A. Chiappone, F. Bella, J. R. Nair, G. Meligrana, R. Bongiovanni and C. Gerbaldi, ChemElectroChem, 2014, 1, 1350–1358 CrossRef CAS.
- K. S. Lee, Y. Jun and J. H. Park, Nano Lett., 2012, 12, 2233–2237 CrossRef CAS PubMed.
- H.-S. Lee, C.-H. Han, Y.-M. Sung, S. S. Sekhon and K.-J. Kim, Curr. Appl. Phys., 2011, 11, S158–S162 CrossRef.
- T. Stergiopoulos, I. M. Arabatzis, G. Katsaros and P. Falaras, Nano Lett., 2002, 2, 1259–1261 CrossRef CAS.
- J. Li, H. Wang, G. Zhou and Z.-S. Wang, Chem. Commun., 2013, 49, 9446–9448 RSC.
- F. Bella, N. N. Mobarak, F. N. Jumaah and A. Ahmad, Electrochim. Acta, 2015, 151, 306–311 CrossRef CAS.
- S. Venkatesan and Y.-L. Lee, Coord. Chem. Rev., 2017, 353, 58–112 CrossRef CAS.
- S.-J. Seo, H.-J. Cha, Y. S. Kang and M.-S. Kang, Electrochim. Acta, 2014, 145, 217–223 CrossRef CAS.
- I.-P. Liu, W.-N. Hung, H. Teng, S. Venkatesan, J.-C. Lin and Y.-L. Lee, J. Mater. Chem. A, 2017, 5, 9190–9197 RSC.
- S. Venkatesan, I.-P. Liu, C.-W. Li, C.-M. Tseng-Shan and Y.-L. Lee, ACS Sustainable Chem. Eng., 2019, 7, 7403–7411 CrossRef CAS.
- I.-P. Liu, Y.-S. Cho, H. Teng and Y.-L. Lee, J. Mater. Chem. A, 2020, 8, 22423–22433 RSC.
- S. Venkatesan, I.-P. Liu, W.-N. Hung, H. Teng and Y.-L. Lee, Chem. Eng. J., 2019, 367, 17–24 CrossRef CAS.
- S. Venkatesan, I.-P. Liu, C.-M. Tseng Shan, H. Teng and Y.-L. Lee, Chem. Eng. J., 2020, 394, 124954 CrossRef CAS.
- S. Venkatesan, E. Surya Darlim, M.-H. Tsai, H. Teng and Y.-L. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 10955–10964 CrossRef CAS PubMed.
- N. Zebardastan, M. H. Khanmirzaei, S. Ramesh and K. Ramesh, Electrochim. Acta, 2016, 220, 573–580 CrossRef CAS.
- S. M. Seo, C. K. Kim and H. K. Kim, J. Mater. Chem. A, 2019, 7, 14743–14752 RSC.
- A. Hagfeldt and N. Vlachopoulos, The Future of Semiconductor Oxides in Next-Generation Solar Cells, Elsevier, 2018, pp. 183–239 Search PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- M. S. Michael, M. M. E. Jacob, S. R. S. Prabaharan and S. Radhakrishna, Solid State Ionics, 1997, 98, 167–174 CrossRef CAS.
- K. M. Abraham, Z. Jiang and B. Carroll, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS.
- Y. Li, H. Li, C. Zhong, G. Sini and J.-L. Brédas, npj Flex. Electron., 2017, 1, 1–8 CrossRef CAS.
- I. Yavuz and K. N. Houk, J. Phys. Chem. C, 2017, 121, 993–999 CrossRef CAS.
- J. Melas-Kyriazi, I.-K. Ding, A. Marchioro, A. Punzi, B. E. Hardin, G. F. Burkhard, N. Tétreault, M. Grätzel, J.-E. Moser and M. D. McGehee, Adv. Energy Mater., 2011, 1, 407–414 CrossRef CAS.
- Y. Zhang, K. Cao, X. Zhu, X. Li, X. Qiao, G. Tu, B. Zhang, D. Huang, Y. Shen and M. Wang, RSC Adv., 2013, 3, 14037–14043 RSC.
- M. Rawolle, K. Sarkar, M. A. Niedermeier, M. Schindler, P. Lellig, J. S. Gutmann, J.-F. Moulin, M. Haese-Seiller, A. S. Wochnik, C. Scheu and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2013, 5, 719–729 CrossRef CAS PubMed.
- M. Juozapavicius, B. C. O'Regan, A. Y. Anderson, J. V. Grazulevicius and V. Mimaite, Org. Electron., 2012, 13, 23–30 CrossRef CAS.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N.-L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133, 18042–18045 CrossRef CAS PubMed.
- S. Benhattab, R. Nakar, J. W. Rodriguez Acosta, N. Berton, J. Faure-Vincent, J. Bouclé, F. Tran Van and B. Schmaltz, Dyes Pigm., 2018, 151, 238–244 CrossRef CAS.
- P. Liu, B. Xu, K. M. Karlsson, J. Zhang, N. Vlachopoulos, G. Boschloo, L. Sun and L. Kloo, J. Mater. Chem. A, 2015, 3, 4420–4427 RSC.
- B. Xu, H. Tian, D. Bi, E. Gabrielsson, E. M. J. Johansson, G. Boschloo, A. Hagfeldt and L. Sun, J. Mater. Chem. A, 2013, 1, 14467–14470 RSC.
- T. Malinauskas, D. Tomkute-Luksiene, R. Sens, M. Daskeviciene, R. Send, H. Wonneberger, V. Jankauskas, I. Bruder and V. Getautis, ACS Appl. Mater. Interfaces, 2015, 7, 11107–11116 CrossRef CAS PubMed.
- Y. Hua, J. Zhang, B. Xu, P. Liu, M. Cheng, L. Kloo, E. M. J. Johansson, K. Sveinbjörnsson, K. Aitola, G. Boschloo and L. Sun, Nano Energy, 2016, 26, 108–113 CrossRef CAS.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo, I. McPherson and H. J. Snaith, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- D.-G. Ha, J.-J. Kim and M. A. Baldo, AIP Adv., 2016, 6, 045221 CrossRef.
- H. Uratani, S. Kubo, K. Shizu, F. Suzuki, T. Fukushima and H. Kaji, Sci. Rep., 2016, 6, 39128 CrossRef CAS PubMed.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- M. Degbia, B. Schmaltz, J. Bouclé, J. V. Grazulevicius and F. Tran-Van, Polym. Int., 2014, 63, 1387–1393 CrossRef CAS.
- B. Xu, E. Sheibani, P. Liu, J. Zhang, H. Tian, N. Vlachopoulos, G. Boschloo, L. Kloo, A. Hagfeldt and L. Sun, Adv. Mater., 2014, 26, 6629–6634 CrossRef CAS PubMed.
- M. Planells, A. Abate, D. J. Hollman, S. D. Stranks, V. Bharti, J. Gaur, D. Mohanty, S. Chand, H. J. Snaith and N. Robertson, J. Mater. Chem. A, 2013, 1, 6949–6960 RSC.
- L. Yang, B. Xu, D. Bi, H. Tian, G. Boschloo, L. Sun, A. Hagfeldt and E. M. J. Johansson, J. Am. Chem. Soc., 2013, 135, 7378–7385 CrossRef CAS PubMed.
- W. Yuan, H. Zhao and G. L. Baker, Org. Electron., 2014, 15, 3362–3369 CrossRef CAS.
- P. Liu, B. Xu, Y. Hua, M. Cheng, K. Aitola, K. Sveinbjörnsson, J. Zhang, G. Boschloo, L. Sun and L. Kloo, J. Power Sources, 2017, 344, 11–14 CrossRef CAS.
- B. Xu, H. Tian, L. Lin, D. Qian, H. Chen, J. Zhang, N. Vlachopoulos, G. Boschloo, Y. Luo, F. Zhang, A. Hagfeldt and L. Sun, Adv. Energy Mater., 2015, 5, 1401185 CrossRef.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- B. Kim, J. K. Koh, J. Kim, W. S. Chi, J. H. Kim and E. Kim, ChemSusChem, 2012, 5, 2173–2180 CrossRef CAS PubMed.
- J. Zhang, L. Häggman, M. Jouini, A. Jarboui, G. Boschloo, N. Vlachopoulos and A. Hagfeldt, ChemPhysChem, 2014, 15, 1043–1047 CrossRef CAS PubMed.
- J. Zhang, N. Vlachopoulos, Y. Hao, T. W. Holcombe, G. Boschloo, E. M. J. Johansson, M. Grätzel and A. Hagfeldt, ChemPhysChem, 2016, 17, 1441–1445 CrossRef CAS PubMed.
- W.-C. Chen, Y.-H. Lee, C.-Y. Chen, K.-C. Kau, L.-Y. Lin, C.-A. Dai, C.-G. Wu, K.-C. Ho, J.-K. Wang and L. Wang, ACS Nano, 2014, 8, 1254–1262 CrossRef CAS PubMed.
- Q. Liu, C. Li, K. Jiang, Y. Song and J. Pei, Particuology, 2014, 15, 71–76 CrossRef CAS.
- M. Chevrier, H. Hawashin, S. Richeter, A. Mehdi, M. Surin, R. Lazzaroni, P. Dubois, B. Ratier, J. Bouclé and S. Clément, Synth. Met., 2017, 226, 157–163 CrossRef CAS.
- N. Alonso-Vante, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1994, 1649–1654 RSC.
- H. Yan, X. Wang, M. Yao and X. Yao, Prog. Nat. Sci.: Mater. Int., 2013, 23, 402–407 CrossRef.
- K. Rajeshwar, Encyclopedia of Electrochemistry, American Cancer Society, 2007 Search PubMed.
- B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C.-Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. H. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed.
- H. Sakamoto, S. Igarashi, M. Uchida, K. Niume and M. Nagai, Org. Electron., 2012, 13, 514–518 CrossRef CAS.
- E. V. A. Premalal, N. Dematage, G. R. R. A. Kumara, R. M. G. Rajapakse, M. Shimomura, K. Murakami and A. Konno, J. Power Sources, 2012, 203, 288–296 CrossRef CAS.
- M. Freitag, Q. Daniel, M. Pazoki, K. Sveinbjörnsson, J. Zhang, L. Sun, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2015, 8, 2634–2637 RSC.
- Y. Cao, Y. Saygili, A. Ummadisingu, J. Teuscher, J. Luo, N. Pellet, F. Giordano, S. M. Zakeeruddin, J.-E. Moser, M. Freitag, A. Hagfeldt and M. Grätzel, Nat. Commun., 2017, 8, 15390 CrossRef PubMed.
- K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed.
- M. Wałesa-Chorab, R. Banasz, D. Marcinkowski, M. Kubicki and V. Patroniak, RSC Adv., 2017, 7, 50858–50867 RSC.
- Z. Wei, J. Fan, C. Dai, Z. Pang and S. Han, ACS Omega, 2018, 3, 6874–6879 CrossRef CAS PubMed.
- T. M. Koh, K. Nonomura, N. Mathews, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, J. Phys. Chem. C, 2013, 117, 15515–15522 CrossRef CAS.
- T. Stergiopoulos, E. Rozi, C.-S. Karagianni and P. Falaras, Nanoscale Res. Lett., 2011, 6, 307 CrossRef PubMed.
- P. Ferdowsi, Y. Saygili, S. M. Zakeeruddin, J. Mokhtari, M. Grätzel, A. Hagfeldt and L. Kavan, Electrochim. Acta, 2018, 265, 194–201 CrossRef CAS.
- Y. Hao, W. Yang, M. Karlsson, J. Cong, S. Wang, X. Li, B. Xu, J. Hua, L. Kloo and G. Boschloo, ACS Energy Lett., 2018, 3, 1929–1937 CrossRef CAS.
- E. M. J. Johansson, L. Yang, E. Gabrielsson, P. W. Lohse, G. Boschloo, L. Sun and A. Hagfeldt, J. Phys. Chem. C, 2012, 116, 18070–18078 CrossRef CAS.
- J.-Y. Kim, J. Y. Kim, D.-K. Lee, B. Kim, H. Kim and M. J. Ko, J. Phys. Chem. C, 2012, 116, 22759–22766 CrossRef CAS.
- L. Yang, R. Lindblad, E. Gabrielsson, G. Boschloo, H. Rensmo, L. Sun, A. Hagfeldt, T. Edvinsson and E. M. J. Johansson, ACS Appl. Mater. Interfaces, 2018, 10, 11572–11579 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- J. Krüger, R. Plass, M. Grätzel, P. J. Cameron and L. M. Peter, J. Phys. Chem. B, 2003, 107, 7536–7539 CrossRef.
- J. Krüger, R. Plass, L. Cevey, M. Piccirelli, M. Grätzel and U. Bach, Appl. Phys. Lett., 2001, 79, 2085–2087 CrossRef.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS PubMed.
- J. Luo, J. Xia, H. Yang, L. Chen, Z. Wan, F. Han, H. A. Malik, X. Zhu and C. Jia, Energy Environ. Sci., 2018, 11, 2035–2045 RSC.
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H.-P. Steinrück, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358, 1192–1197 CrossRef CAS PubMed.
- U. B. Cappel, T. Daeneke and U. Bach, Nano Lett., 2012, 12, 4925–4931 CrossRef CAS PubMed.
- J. Burschka, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2013, 25, 2986–2990 CrossRef CAS.
- D.-Y. Chen, W.-H. Tseng, S.-P. Liang, C.-I. Wu, C.-W. Hsu, Y. Chi, W.-Y. Hung and P.-T. Chou, Phys. Chem. Chem. Phys., 2012, 14, 11689–11694 RSC.
- M. Xu, Y. Rong, Z. Ku, A. Mei, X. Li and H. Han, J. Phys. Chem. C, 2013, 117, 22492–22496 CrossRef CAS.
- B. Xu, E. Gabrielsson, M. Safdari, M. Cheng, Y. Hua, H. Tian, J. M. Gardner, L. Kloo and L. Sun, Adv. Energy Mater., 2015, 5, 1402340 CrossRef.
- X. Yang, W. Wang, Y. Zhang and L. Sun, Sol. Energy, 2018, 170, 1001–1008 CrossRef CAS.
- W. Wang, X. Yang, J. Li, H. Wang, J. An, L. Zhang, X. Jiang, Z. Yu and L. Sun, Energy Technol., 2018, 6, 752–758 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan, G. Luo, Y. Lin, Y. Xie and Y. Wei, Chem. Soc. Rev., 2017, 46, 5975–6023 RSC.
- L. Wang, M. Al-Mamun, P. Liu, Y. Wang, H. G. Yang, H. F. Wang and H. Zhao, NPG Asia Mater., 2015, 7, e226 CrossRef CAS.
- Z. Tang, J. Wu, M. Zheng, J. Huo and Z. Lan, Nano Energy, 2013, 2, 622–627 CrossRef CAS.
- G. Calogero, P. Calandra, A. Irrera, A. Sinopoli, I. Citro and G. D. Marco, Energy Environ. Sci., 2011, 4, 1838–1844 RSC.
- L. Kavan, H. Krysova, P. Janda, H. Tarabkova, Y. Saygili, M. Freitag, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Electrochim. Acta, 2017, 251, 167–175 CrossRef CAS.
- I. K. Popoola, M. A. Gondal, J. M. AlGhamdi and T. F. Qahtan, Sci. Rep., 2018, 8, 12864 CrossRef PubMed.
- H. Ellis, N. Vlachopoulos, L. Häggman, C. Perruchot, M. Jouini, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2013, 107, 45–51 CrossRef CAS.
- C. H. Yoon, R. Vittal, J. Lee, W.-S. Chae and K.-J. Kim, Electrochim. Acta, 2008, 53, 2890–2896 CrossRef CAS.
- G. Yue, J. Wu, Y. Xiao, M. Huang, J. Lin, L. Fan and Z. Lan, Electrochim. Acta, 2013, 92, 64–70 CrossRef CAS.
- J. G. Nam, Y. J. Park, B. S. Kim and J. S. Lee, Scr. Mater., 2010, 62, 148–150 CrossRef CAS.
- S. Suresh, G. E. Unni, M. Satyanarayana, A. Sreekumaran Nair and V. P. Mahadevan Pillai, J. Colloid Interface Sci., 2018, 524, 236–244 CrossRef CAS PubMed.
- R. S. Moraes, E. Saito, D. M. G. Leite, M. Massi and A. S. da Silva Sobrinho, Appl. Surf. Sci., 2016, 364, 229–234 CrossRef CAS.
- X. Li, D. Zhang, S. Chen, H. Zhang, Z. Sun, S. Huang and X. Yin, Nano-Micro Lett., 2011, 3, 195–199 CrossRef CAS.
- J. Briscoe and S. Dunn, Adv. Mater., 2016, 28, 3802–3813 CrossRef CAS PubMed.
- W. Wei, H. Wang and Y. H. Hu, Int. J. Energy Res., 2014, 38, 1099–1111 CrossRef CAS.
- M. Wu and T. Ma, ChemSusChem, 2012, 5, 1343–1357 CrossRef CAS PubMed.
- A. Hauch and A. Georg, Electrochim. Acta, 2001, 46, 3457–3466 CrossRef CAS.
- G. Wang, Y. Lin, X. Xiao, X. Li and W. Wang, Surf. Interface Anal., 2004, 36, 1437–1440 CrossRef CAS.
- E. Olsen, G. Hagen and S. Eric Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 63, 267–273 CrossRef CAS.
- A. R. Oganov, R. J. Hemley, R. M. Hazen and A. P. Jones, Rev. Mineral. Geochem., 2013, 75, 47–77 CrossRef CAS.
- L. Kavan, J.-H. Yum and M. Grätzel, Nano Lett., 2011, 11, 5501–5506 CrossRef CAS PubMed.
- Y. Gao, L. Chu, M. Wu, L. Wang, W. Guo and T. Ma, J. Photochem. Photobiol., A, 2012, 245, 66–71 CrossRef CAS.
- M. J. Ju, I.-Y. Jeon, H. M. Kim, J. I. Choi, S.-M. Jung, J.-M. Seo, I. T. Choi, S. H. Kang, H. S. Kim, M. J. Noh, J.-J. Lee, H. Y. Jeong, H. K. Kim, Y.-H. Kim and J.-B. Baek, Sci. Adv., 2016, 2, e1501459 CrossRef PubMed.
- I.-Y. Jeon, H. M. Kim, D. H. Kweon, S.-M. Jung, J.-M. Seo, S.-H. Shin, I. T. Choi, Y. K. Eom, S. H. Kang, H. K. Kim, M. J. Ju and J.-B. Baek, Nano Energy, 2016, 30, 867–876 CrossRef CAS.
- C. K. Kim, H. M. Kim, M. Aftabuzzaman, I.-Y. Jeon, S. H. Kang, Y. K. Eom, J. B. Baek and H. K. Kim, Mater. Today Energy, 2018, 9, 67–73 CrossRef.
- C. K. Kim, H. Zhou, T. Kowalewski, K. Matyjaszewski and H. K. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 2093–2102 CrossRef CAS PubMed.
- K. Saranya, M. Rameez and A. Subramania, Eur. Polym. J., 2015, 66, 207–227 CrossRef CAS.
- J. Xia, L. Chen and S. Yanagida, J. Mater. Chem., 2011, 21, 4644–4649 RSC.
- X. Fang, T. Ma, M. Akiyama, G. Guan, S. Tsunematsu and E. Abe, Thin Solid Films, 2005, 472, 242–245 CrossRef CAS.
- Y. Saito, T. Kitamura, Y. Wada and S. Yanagida, Chem. Lett., 2002, 31, 1060–1061 CrossRef.
- H. N. Tsao, J. Burschka, C. Yi, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2011, 4, 4921–4924 RSC.
- M. Dinari, M. M. Momeni and M. Goudarzirad, J. Mater. Sci., 2016, 51, 2964–2971 CrossRef CAS.
- T. Xu, W. Cao, D. Kong, X. Qin, J. Song, K. Kou, L. Chen, Q. Qiao and W. Huang, Mater. Today Commun., 2020, 25, 101313 CrossRef CAS.
- E. Benazzi, J. Mallows, G. H. Summers, F. A. Black and E. A. Gibson, J. Mater. Chem. C, 2019, 7, 10409–10445 RSC.
- A. Nattestad, I. Perera and L. Spiccia, J. Photochem. Photobiol., C, 2016, 28, 44–71 CrossRef CAS.
- M. Green, Third Generation Photovoltaics: Advanced Solar Energy Conversion, Springer-Verlag, Berlin Heidelberg, 2003 Search PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- Y. Farré, M. Raissi, A. Fihey, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, ChemSusChem, 2017, 10, 2618–2625 CrossRef PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y.-B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- H. Sato, T. Minami, S. Takata and T. Yamada, Thin Solid Films, 1993, 236, 27–31 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and N. D. Nicholson, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 643–661 RSC.
- A. Nattestad, M. Ferguson, R. Kerr, Y.-B. Cheng and U. Bach, Nanotechnology, 2008, 19, 295304 CrossRef PubMed.
- Q. Wu, Y. Shen, L. Li, M. Cao, F. Gu and L. Wang, Appl. Surf. Sci., 2013, 276, 411–416 CrossRef CAS.
- M. Awais, E. Gibson, J. G. Vos, D. P. Dowling, A. Hagfeldt and D. Dini, ChemElectroChem, 2014, 1, 384–391 CrossRef.
- S. Uehara, S. Sumikura, E. Suzuki and S. Mori, Energy Environ. Sci., 2010, 3, 641–644 RSC.
- M. Bonomo, G. Naponiello, I. Venditti, V. Zardetto, A. D. Carlo and D. Dini, J. Electrochem. Soc., 2017, 164, H137 CrossRef CAS.
- C. J. Flynn, E. E. Oh, S. M. McCullough, R. W. Call, C. L. Donley, R. Lopez and J. F. Cahoon, J. Phys. Chem. C, 2014, 118, 14177–14184 CrossRef CAS.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. L. Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS.
- R. Brisse, R. Faddoul, T. Bourgeteau, D. Tondelier, J. Leroy, S. Campidelli, T. Berthelot, B. Geffroy and B. Jousselme, ACS Appl. Mater. Interfaces, 2017, 9, 2369–2377 CrossRef CAS PubMed.
- P. Qin, H. Zhu, T. Edvinsson, G. Boschloo, A. Hagfeldt and L. Sun, J. Am. Chem. Soc., 2008, 130, 8570–8571 CrossRef CAS PubMed.
- L. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef CAS PubMed.
- S. Mori, S. Fukuda, S. Sumikura, Y. Takeda, Y. Tamaki, E. Suzuki and T. Abe, J. Phys. Chem. C, 2008, 112, 16134–16139 CrossRef CAS.
- L. D'Amario, J. Föhlinger, G. Boschloo and L. Hammarström, Chem. Sci., 2017, 9, 223–230 RSC.
- P. Ho, L. Q. Bao, K.-S. Ahn, R. Cheruku and J. H. Kim, Synth. Met., 2016, 217, 314–321 CrossRef CAS.
- L. D'Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 33470–33477 CrossRef PubMed.
- C. Xin, Y. Wang, S. Zhang, L. Xu, Y. Yu, H. Xiang, W. Wu and J. Hua, Phys. Status Solidi RRL, 2017, 11, 1700258 CrossRef.
- G. Natu, Z. Huang, Z. Ji and Y. Wu, Langmuir, 2012, 28, 950–956 CrossRef CAS PubMed.
- Y. Yu, X. Li, Z. Shen, X. Zhang, P. Liu, Y. Gao, T. Jiang and J. Hua, J. Colloid Interface Sci., 2017, 490, 380–390 CrossRef CAS PubMed.
- C. J. Flynn, S. M. McCullough, L. Li, C. L. Donley, Y. Kanai and J. F. Cahoon, J. Phys. Chem. C, 2016, 120, 16568–16576 CrossRef CAS.
- L. Favereau, Y. Pellegrin, L. Hirsch, A. Renaud, A. Planchat, E. Blart, G. Louarn, L. Cario, S. Jobic, M. Boujtita and F. Odobel, Adv. Energy Mater., 2017, 7, 1601776 CrossRef.
- Z. Huang, X. Zeng, H. Wang, W. Zhang, Y. Li, M. Wang, Y.-B. Cheng and W. Chen, RSC Adv., 2014, 4, 60670–60674 RSC.
- M. Zannotti, C. J. Wood, G. H. Summers, L. A. Stevens, M. R. Hall, C. E. Snape, R. Giovanetti and E. A. Gibson, ACS Appl. Mater. Interfaces, 2015, 7, 24556–24565 CrossRef CAS PubMed.
- G. Natu, P. Hasin, Z. Huang, Z. Ji, M. He and Y. Wu, ACS Appl. Mater. Interfaces, 2012, 4, 5922–5929 CrossRef CAS PubMed.
- L. D'Amario, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. C, 2014, 118, 19556–19564 CrossRef.
- H. Yang, G. H. Guai, C. Guo, Q. Song, S. P. Jiang, Y. Wang, W. Zhang and C. M. Li, J. Phys. Chem. C, 2011, 115, 12209–12215 CrossRef CAS.
- M. Zannotti, E. Benazzi, L. A. Stevens, M. Minicucci, L. Bruce, C. E. Snape, E. A. Gibson and R. Giovannetti, ACS Appl. Energy Mater., 2019, 2, 7345–7353 CrossRef CAS.
- J. Bai, X. Xu, L. Xu, J. Cui, D. Huang, W. Chen, Y. Cheng, Y. Shen and M. Wang, ChemSusChem, 2013, 6, 622–629 CrossRef CAS PubMed.
- Z. Huang, M. He, M. Yu, K. Click, D. Beauchamp and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 6857–6861 CrossRef CAS PubMed.
- Z. Yu, I. R. Perera, T. Daeneke, S. Makuta, Y. Tachibana, J. J. Jasieniak, A. Mishra, P. Bäuerle, L. Spiccia and U. Bach, NPG Asia Mater., 2016, 8, e305 CrossRef.
- O. Langmar, C. R. Ganivet, P. Schol, T. Scharl, G. de la Torre, T. Torres, R. D. Costa and D. M. Guldi, J. Mater. Chem. C, 2018, 6, 5176–5180 RSC.
- T. Jiang, M. Bujoli-Doeuff, Y. Farré, Y. Pellegrin, E. Gautron, M. Boujtita, L. Cario, S. Jobic and F. Odobel, RSC Adv., 2016, 6, 112765–112770 RSC.
- O. Langmar, C. R. Ganivet, G. de la Torre, T. Torres, R. D. Costa and D. M. Guldi, Nanoscale, 2016, 8, 17963–17975 RSC.
- O. Langmar, C. R. Ganivet, T. Scharl, G. de la Torre, T. Torres, R. D. Costa and D. M. Guldi, ACS Appl. Energy Mater., 2018, 1, 6388–6400 CrossRef.
- O. Langmar, C. R. Ganivet, A. Lennert, R. D. Costa, G. de la Torre, T. Torres and D. M. Guldi, Angew. Chem., Int. Ed., 2015, 54, 7688–7692 CrossRef CAS PubMed.
- S. Du, P. Cheng, P. Sun, B. Wang, Y. Cai, F. Liu, J. Zheng and G. Lu, Chem. Res. Chin. Univ., 2014, 30, 661–665 CrossRef CAS.
- T. Jiang, M. Bujoli-Doeuff, E. Gautron, Y. Farré, L. Cario, Y. Pellegrin, M. Boujtita, F. Odobel and S. Jobic, J. Alloys Compd., 2018, 769, 605–610 CrossRef CAS.
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939–942 CrossRef CAS.
- J. Bandara and J. P. Yasomanee, Semicond. Sci. Technol., 2006, 22, 20–24 CrossRef.
- J. Ahmed, C. K. Blakely, J. Prakash, S. R. Bruno, M. Yu, Y. Wu and V. V. Poltavets, J. Alloys Compd., 2014, 591, 275–279 CrossRef CAS.
- D. Ursu, M. Miclau, R. Banica and N. Vaszilcsin, Mater. Lett., 2015, 143, 91–93 CrossRef CAS.
- D. Xiong, Z. Xu, X. Zeng, W. Zhang, W. Chen, X. Xu, M. Wang and Y.-B. Cheng, J. Mater. Chem., 2012, 22, 24760–24768 RSC.
- D. Xiong, H. Wang, W. Zhang, X. Zeng, H. Chang, X. Zhao, W. Chen and Y.-B. Cheng, J. Alloys Compd., 2015, 642, 104–110 CrossRef CAS.
- M. Yu, T. I. Draskovic and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 5026–5033 RSC.
- T. Zhu, Z. Deng, X. Fang, Z. Huo, S. Wang, W. Dong, J. Shao, R. Tao, C. Song and L. Wang, J. Alloys Compd., 2016, 685, 836–840 CrossRef CAS.
- M. Yu, G. Natu, Z. Ji and Y. Wu, J. Phys. Chem. Lett., 2012, 3, 1074–1078 CrossRef CAS PubMed.
- Z. Xu, D. Xiong, H. Wang, W. Zhang, X. Zeng, L. Ming, W. Chen, X. Xu, J. Cui, M. Wang, S. Powar, U. Bach and Y.-B. Cheng, J. Mater. Chem. A, 2014, 2, 2968–2976 RSC.
- A. Renaud, L. Cario, P. Deniard, E. Gautron, X. Rocquefelte, Y. Pellegrin, E. Blart, F. Odobel and S. Jobic, J. Phys. Chem. C, 2014, 118, 54–59 CrossRef CAS.
- D. Ursu, N. Vaszilcsin, R. Bănica and M. Miclau, J. Mater. Eng. Perform., 2016, 25, 59–63 CrossRef CAS.
- S. Powar, et al., J. Phys. Chem. C, 2014, 118(30), 16375–16379 CrossRef CAS.
- X. Xu, J. Cui, J. Han, J. Zhang, Y. Zhang, L. Luan, G. Alemu, Z. Wang, Y. Shen, D. Xiong, W. Chen, Z. Wei, S. Yang, B. Hu, Y. Cheng and M. Wang, Sci. Rep., 2014, 4, 3961 CrossRef PubMed.
- D. Xiong, W. Zhang, X. Zeng, Z. Xu, W. Chen, J. Cui, M. Wang, L. Sun and Y.-B. Cheng, ChemSusChem, 2013, 6, 1432–1437 CrossRef CAS PubMed.
- U. Daniel, D. Anamaria, I. Sebarchievicia and M. Miclau, Energy Procedia, 2017, 112, 497–503 CrossRef CAS.
- D. Xiong, Q. Zhang, S. K. Verma, H. Li, W. Chen and X. Zhao, J. Alloys Compd., 2016, 662, 374–380 CrossRef CAS.
- A. Renaud, L. Cario, Y. Pellegrin, E. Blart, M. Boujtita, F. Odobel and S. Jobic, RSC Adv., 2015, 5, 60148–60151 RSC.
- Z. Shi, H. Lu, Q. Liu, K. Deng, L. Xu, R. Zou, J. Hu, Y. Bando, D. Golberg and L. Li, Energy Technol., 2014, 2, 517–521 CrossRef CAS.
- Z. Shi, H. Lu, Q. Liu, F. Cao, J. Guo, K. Deng and L. Li, Nanoscale Res. Lett., 2014, 9, 608 CrossRef PubMed.
- F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4, 2551–2564 CrossRef CAS.
- K. Aiempanakit, P. Rakkwamsuk and S. Dumrongrattana, Agric. Nat. Resour., 2008, 42, 351–356 Search PubMed.
- S. Das, S. Saha, D. Sen, U. K. Ghorai, D. Banerjee and K. K. Chattopadhyay, J. Mater. Chem. C, 2014, 2, 1321–1330 RSC.
- J. Ghijsen, L. H. Tjeng, J. van Elp, H. Eskes, J. Westerink, G. A. Sawatzky and M. T. Czyzyk, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 11322–11330 CrossRef CAS PubMed.
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- J. Piippo, T. Saario, T. Laitinen, M. Bojinov and J. Hinttala, Mater. Sci. Forum, 1998, 289–292, 429–438 CAS.
- B. J. Ingram, T. O. Mason, R. Asahi, K. T. Park and A. J. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 155114 CrossRef.
- M. Lee, D. Kim, Y. T. Yoon and Y. I. Kim, Bull. Korean Chem. Soc., 2014, 35, 3261–3266 CrossRef CAS.
- F. A. Benko and F. P. Koffyberg, J. Phys. Chem. Solids, 1987, 48, 431–434 CrossRef CAS.
- K. Ueda and H. Hosono, J. Appl. Phys., 2002, 91, 4768–4770 CrossRef CAS.
- K. Ueda, S. Inoue, H. Hosono, N. Sarukura and M. Hirano, Appl. Phys. Lett., 2001, 78, 2333–2335 CrossRef CAS.
- B. Cui, H. Lin, Y.-z. Liu, J.-b. Li, P. Sun, X.-c. Zhao and C.-j. Liu, J. Phys. Chem. C, 2009, 113, 14083–14087 CrossRef CAS.
- Y. Pellegrin, L. Le Pleux, E. Blart, A. Renaud, B. Chavillon, N. Szuwarski, M. Boujtita, L. Cario, S. Jobic, D. Jacquemin and F. Odobel, J. Photochem. Photobiol., A, 2011, 219, 235–242 CrossRef CAS.
- F. Brunner, N. Marinakis, C. Wobill, M. Willgert, C. D. Ertl, T. Kosmalski, M. Neuburger, B. Bozic-Weber, T. Glatzel, E. C. Constable and C. E. Housecroft, J. Mater. Chem. C, 2016, 4, 9823–9833 RSC.
- Y. Dong, L. Wei, R. Fan, Y. Yang and P. Wang, RSC Adv., 2016, 6, 39972–39981 RSC.
- G. Alemu, J. Cui, K. Cao, J. Li, Y. Shen and M. Wang, RSC Adv., 2014, 4, 51374–51380 RSC.
- J. Cui, J. Lu, X. Xu, K. Cao, Z. Wang, G. Alemu, H. Yuang, Y. Shen, J. Xu, Y. Cheng and M. Wang, J. Phys. Chem. C, 2014, 118, 16433–16440 CrossRef CAS.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, J. Fortage, G. Boschloo, E. Blart, Y. Pellegrin, F. Odobel, A. Hagfeldt and L. Hammarström, Angew. Chem., Int. Ed., 2009, 48, 4402–4405 CrossRef CAS PubMed.
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS.
- S. Wrede and H. Tian, Phys. Chem. Chem. Phys., 2020, 22, 13850–13861 RSC.
- M. Buchalska, J. Kuncewicz, E. Świętek, P. Łaabuz, T. Baran, G. Stochel and W. Macyk, Coord. Chem. Rev., 2013, 257, 767–775 CrossRef CAS.
- C. J. Wood, K. C. D. Robson, P. I. P. Elliott, C. P. Berlinguette and E. A. Gibson, RSC Adv., 2014, 4, 5782–5791 RSC.
- Z. Ji, G. Natu and Y. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 8641–8648 CrossRef CAS PubMed.
- Z. Ji, G. Natu, Z. Huang, O. Kokhan, X. Zhang and Y. Wu, J. Phys. Chem. C, 2012, 116, 16854–16863 CrossRef CAS.
- S. Lyu, Y. Farré, L. Ducasse, Y. Pellegrin, T. Toupance, C. Olivier and F. Odobel, RSC Adv., 2016, 6, 19928–19936 RSC.
- N. Marinakis, M. Willgert, E. C. Constable and C. E. Housecroft, Sustainable Energy Fuels, 2017, 1, 626–635 RSC.
- M. He, Z. Ji, Z. Huang and Y. Wu, J. Phys. Chem. C, 2014, 118, 16518–16525 CrossRef CAS.
- M. Gennari, F. Légalité, L. Zhang, Y. Pellegrin, E. Blart, J. Fortage, A. M. Brown, A. Deronzier, M.-N. Collomb, M. Boujtita, D. Jacquemin, L. Hammarström and F. Odobel, J. Phys. Chem. Lett., 2014, 5, 2254–2258 CrossRef CAS PubMed.
- A. Sinopoli, F. A. Black, C. J. Wood, E. A. Gibson and P. I. P. Elliott, Dalton Trans., 2017, 46, 1520–1530 RSC.
- A. Sinopoli, C. J. Wood, E. A. Gibson and P. I. P. Elliott, Eur. J. Inorg. Chem., 2016, 2887–2890 CrossRef CAS.
- A. Sinopoli, C. J. Wood, E. A. Gibson and P. I. P. Elliott, Dyes Pigm., 2017, 140, 269–277 CrossRef CAS.
- P. Qin, M. Linder, T. Brinck, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Mater., 2009, 21, 2993–2996 CrossRef CAS.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CrossRef CAS.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915–3918 RSC.
- Q.-Q. Zhang, K.-J. Jiang, J.-H. Huang, C.-W. Zhao, L.-P. Zhang, X.-P. Cui, M.-J. Su, L.-M. Yang, Y.-L. Song and X.-Q. Zhou, J. Mater. Chem. A, 2015, 3, 7695–7698 RSC.
- Z. Liu, W. Li, S. Topa, X. Xu, X. Zeng, Z. Zhao, M. Wang, W. Chen, F. Wang, Y.-B. Cheng and H. He, ACS Appl. Mater. Interfaces, 2014, 6, 10614–10622 CrossRef CAS PubMed.
- Z. Liu, D. Xiong, X. Xu, Q. Arooj, H. Wang, L. Yin, W. Li, H. Wu, Z. Zhao, W. Chen, M. Wang, F. Wang, Y.-B. Cheng and H. He, ACS Appl. Mater. Interfaces, 2014, 6, 3448–3454 CrossRef CAS PubMed.
- P. Naik, A. Planchat, Y. Pellegrin, F. Odobel and A. Vasudeva Adhikari, Sol. Energy, 2017, 157, 1064–1073 CrossRef CAS.
- S. Karamshuk, S. Caramori, N. Manfredi, M. Salamone, R. Ruffo, S. Carli, C. A. Bignozzi and A. Abbotto, Energies, 2016, 9, 33 CrossRef.
- R. Brisse, C. Praveen, V. Maffeis, T. Bourgeteau, D. Tondelier, T. Berthelot, B. Geffroy, T. Gustavsson, J. M. Raimundo and B. Jousselme, Sustainable Energy Fuels, 2018, 2, 648–654 RSC.
- A. R. Marri, F. A. Black, J. Mallows, E. A. Gibson and J. Fielden, Dyes Pigm., 2019, 165, 508–517 CrossRef CAS.
- C. J. Wood, M. Cheng, C. A. Clark, R. Horvath, I. P. Clark, M. L. Hamilton, M. Towrie, M. W. George, L. Sun, X. Yang and E. A. Gibson, J. Phys. Chem. C, 2014, 118, 16536–16546 CrossRef CAS.
- E. Sheibani, L. Zhang, P. Liu, B. Xu, E. Mijangos, G. Boschloo, A. Hagfeldt, L. Hammarström, L. Kloo and H. Tian, RSC Adv., 2016, 6, 18165–18177 RSC.
- N. Grzegorzek, A. Zieleniewska, A. Schür, C. Maichle-Mössmer, M. S. Killian, D. M. Guldi and E. T. Chernick, ChemPlusChem, 2019, 84, 766–771 CrossRef CAS PubMed.
- F. Wu, S. Zhao, C. Zhong, Q. Song and L. Zhu, RSC Adv., 2015, 5, 93652–93658 RSC.
- F. Wu, L. Zhu, S. Zhao, Q. Song and C. Yang, Dyes Pigm., 2016, 124, 93–100 CrossRef CAS.
- L. Zhu, H. B. Yang, C. Zhong and C. M. Li, Dyes Pigm., 2014, 105, 97–104 CrossRef CAS.
- K. A. Click, D. R. Beauchamp, B. R. Garrett, Z. Huang, C. M. Hadad and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 26103–26111 RSC.
- Z. Ji, G. Natu, Z. Huang and Y. Wu, Energy Environ. Sci., 2011, 4, 2818–2821 RSC.
- H. D. Weldekirstos, M.-C. Kuo, S.-R. Li, W.-L. Su, M. A. Desta, W.-T. Wu, C.-H. Kuo and S.-S. Sun, Dyes Pigm., 2019, 163, 761–774 CrossRef CAS.
- N. T. Z. Potts, T. Sloboda, M. Wächtler, R. A. Wahyuono, V. D'Annibale, B. Dietzek, U. B. Cappel and E. A. Gibson, J. Chem. Phys., 2020, 153, 184704 CrossRef CAS PubMed.
- J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50, 5258–5260 RSC.
- G. H. Summers, J.-F. Lefebvre, F. A. Black, E. S. Davies, E. A. Gibson, T. Pullerits, C. J. Wood and K. Zidek, Phys. Chem. Chem. Phys., 2015, 18, 1059–1070 RSC.
- Y. Higashino, S. Erten-Ela and Y. Kubo, Dyes Pigm., 2019, 170, 107613 CrossRef CAS.
- F. Wu, J. Liu, X. Li, Q. Song, M. Wang, C. Zhong and L. Zhu, Eur. J. Org. Chem., 2015, 2015, 6850–6857 CrossRef CAS.
- Y.-S. Yen, W.-T. Chen, C.-Y. Hsu, H.-H. Chou, J. T. Lin and M.-C. P. Yeh, Org. Lett., 2011, 13, 4930–4933 CrossRef CAS PubMed.
- C.-H. Chang, Y.-C. Chen, C.-Y. Hsu, H.-H. Chou and J. T. Lin, Org. Lett., 2012, 14, 4726–4729 CrossRef CAS PubMed.
- B. Jin, W. Wu, X. Zhang, F. Guo, Q. Zhang and J. Hua, Chem. Lett., 2013, 42, 1271–1272 CrossRef CAS.
- Y. Farré, F. Maschietto, J. Föhlinger, M. Wykes, A. Planchat, Y. Pellegrin, E. Blart, I. Ciofini, L. Hammarström and F. Odobel, ChemSusChem, 2020, 13, 1844–1855 CrossRef PubMed.
- F. A. Black, C. J. Wood, S. Ngwerume, G. H. Summers, I. P. Clark, M. Towrie, J. E. Camp and E. A. Gibson, Faraday Discuss., 2017, 198, 449–461 RSC.
- G. H. Summers, G. Lowe, J.-F. Lefebvre, S. Ngwerume, M. Bräutigam, B. Dietzek, J. E. Camp and E. A. Gibson, ChemPhysChem, 2017, 18, 406–414 CrossRef CAS PubMed.
- K. Yun, S. Zhang, F. Yu, H. Ye and J. Hua, J. Energy Chem., 2018, 27, 728–735 CrossRef.
- J. Warnan, Y. Pellegrin, E. Blart, L. Zhang, A. Brown, L. Hammarström, D. Jacquemin and F. Odobel, Dyes Pigm., 2014, 105, 174–179 CrossRef CAS.
- E. Benazzi, G. H. Summers, F. A. Black, I. V. Sazanovich, I. P. Clark and E. A. Gibson, Philos. Trans. R. Soc., A, 2019, 377, 20180338 CrossRef CAS PubMed.
- Y. M. Klein, N. Marinakis, E. C. Constable and C. E. Housecroft, Crystals, 2018, 8, 389 CrossRef.
- J. Warnan, J. Gardner, L. Le Pleux, J. Petersson, Y. Pellegrin, E. Blart, L. Hammarström and F. Odobel, J. Phys. Chem. C, 2014, 118, 103–113 CrossRef CAS.
- Y. Farré, L. Zhang, Y. Pellegrin, A. Planchat, E. Blart, M. Boujtita, L. Hammarström, D. Jacquemin and F. Odobel, J. Phys. Chem. C, 2016, 120, 7923–7940 CrossRef.
- D. Ameline, S. Diring, Y. Farre, Y. Pellegrin, G. Naponiello, E. Blart, B. Charrier, D. Dini, D. Jacquemin and F. Odobel, RSC Adv., 2015, 5, 85530–85539 RSC.
- L. Zhang, L. Favereau, Y. Farre, A. Maufroy, Y. Pellegrin, E. Blart, M. Hissler, D. Jacquemin, F. Odobel and L. Hammarström, RSC Adv., 2016, 6, 77184–77194 RSC.
- H. Tian, J. Oscarsson, E. Gabrielsson, S. K. Eriksson, R. Lindblad, B. Xu, Y. Hao, G. Boschloo, E. M. J. Johansson, J. M. Gardner, A. Hagfeldt, H. Rensmo and L. Sun, Sci. Rep., 2014, 4, 4282 CrossRef PubMed.
- A. Maufroy, L. Favereau, F. B. Anne, Y. Pellegrin, E. Blart, M. Hissler, D. Jacquemin and F. Odobel, J. Mater. Chem. A, 2015, 3, 3908–3917 RSC.
- V. Nikolaou, F. Plass, A. Planchat, A. Charisiadis, G. Charalambidis, P. A. Angaridis, A. Kahnt, F. Odobel and A. G. Coutsolelos, Phys. Chem. Chem. Phys., 2018, 20, 24477–24489 RSC.
- J. Lu, Z. Liu, N. Pai, L. Jiang, U. Bach, A. N. Simonov, Y.-B. Cheng and L. Spiccia, ChemPlusChem, 2018, 83, 711–720 CrossRef CAS PubMed.
- M. Bonomo, N. Barbero, F. Matteocci, A. D. Carlo, C. Barolo and D. Dini, J. Phys. Chem. C, 2016, 120, 16340–16353 CrossRef CAS.
- G. H. Summers and E. A. Gibson, ChemPhotoChem, 2018, 2, 498–506 CrossRef CAS.
- M. Bonomo, A. Di Carlo, R. Centore, D. Dini and A. Carella, Sol. Energy, 2018, 169, 237–241 CrossRef CAS.
- S. Powar, Q. Wu, M. Weidelener, A. Nattestad, Z. Hu, A. Mishra, P. Bäuerle, L. Spiccia, Y.-B. Cheng and U. Bach, Energy Environ. Sci., 2012, 5, 8896–8900 RSC.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed.
- A. Morandeira, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. B, 2005, 109, 19403–19410 CrossRef CAS PubMed.
- A. Morandeira, J. Fortage, T. Edvinsson, L. Le Pleux, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. C, 2008, 112, 1721–1728 CrossRef CAS.
- M. Borgström, E. Blart, G. Boschloo, E. Mukhtar, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. B, 2005, 109, 22928–22934 CrossRef PubMed.
- C. J. Wood, C. A. McGregor and E. A. Gibson, ChemElectroChem, 2016, 3, 1827–1836 CrossRef CAS.
- H. Liu, W. Xiang and H. Tao, J. Photochem. Photobiol., A, 2017, 344, 199–205 CrossRef CAS.
- E. A. Gibson, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS PubMed.
- X. Xu, B. Zhang, J. Cui, D. Xiong, Y. Shen, W. Chen, L. Sun, Y. Cheng and M. Wang, Nanoscale, 2013, 5, 7963–7969 RSC.
- S. Powar, R. Bhargava, T. Daeneke, G. Götz, P. Bäuerle, T. Geiger, S. Kuster, F. A. Nüesch, L. Spiccia and U. Bach, Electrochim. Acta, 2015, 182, 458–463 CrossRef CAS.
- Y. Farré, M. Raissi, A. Fihey, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, Dyes Pigm., 2018, 148, 154–166 CrossRef.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, L. Hammarström, F. Odobel, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115, 9772–9779 CrossRef CAS.
- S. Powar, T. Daeneke, M. T. Ma, D. Fu, N. W. Duffy, G. Götz, M. Weidelener, A. Mishra, P. Bäuerle, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 602–605 CrossRef CAS PubMed.
- L. L. Pleux, A. L. Smeigh, E. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, Energy Environ. Sci., 2011, 4, 2075–2084 RSC.
- W. Xiang, J. Marlow, P. Bäuerle, U. Bach and L. Spiccia, Green Chem., 2016, 18, 6659–6665 RSC.
- T. T. T. Pham, S. K. Saha, D. Provost, Y. Farré, M. Raissi, Y. Pellegrin, E. Blart, S. Vedraine, B. Ratier, D. Aldakov, F. Odobel and J. Bouclé, J. Phys. Chem. C, 2017, 121, 129–139 CrossRef CAS.
- B. Xu, S. Wrede, A. Curtze, L. Tian, P. B. Pati, L. Kloo, Y. Wu and H. Tian, ChemSusChem, 2019, 12, 3243–3248 CrossRef CAS PubMed.
- B. Xu, L. Tian, A. S. Etman, J. Sun and H. Tian, Nano Energy, 2019, 55, 59–64 CrossRef CAS.
- L. Tian, J. Föhlinger, Z. Zhang, P. B. Pati, J. Lin, T. Kubart, Y. Hua, J. Sun, L. Kloo, G. Boschloo, L. Hammarström and H. Tian, Chem. Commun., 2018, 54, 3739–3742 RSC.
- S. Mastroianni, A. Lanuti, S. Penna, A. Reale, T. M. Brown, A. D. Carlo and F. Decker, ChemPhysChem, 2012, 13, 2925–2936 CrossRef CAS PubMed.
- J. Park, P. Lee and M. J. Ko, Int. J. Precis. Eng. Manuf.-Green Tech., 2019, 6, 125–131 CrossRef.
- R. Jiang, A. Anderson, P. R. F. Barnes, L. Xiaoe, C. Law and B. C. O'Regan, J. Mater. Chem. A, 2014, 2, 4751–4757 RSC.
- H. Rui, J. Shen, Z. Yu, L. Li, H. Han and L. Sun, Angew. Chem., Int. Ed., 2021, 60, 16156–16163 CrossRef CAS PubMed.
- T. Ueda, ChemElectroChem, 2018, 5, 823–838 CrossRef CAS.
- H. E. Moll, F. A. Black, C. J. Wood, A. Al-Yasari, A. R. Marri, I. V. Sazanovich, E. A. Gibson and J. Fielden, Phys. Chem. Chem. Phys., 2017, 19, 18831–18835 RSC.
- T. M. A. Bakker, S. Mathew and J. N. H. Reek, Sustainable Energy Fuels, 2018, 3, 96–100 RSC.
- L. Zhang, G. Boschloo, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2016, 18, 5080–5085 RSC.
- L. Tian, J. Föhlinger, P. B. Pati, Z. Zhang, J. Lin, W. Yang, M. Johansson, T. Kubart, J. Sun, G. Boschloo, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2017, 20, 36–40 RSC.
- J. Bandara and H. Weerasinghe, Sol. Energy Mater. Sol. Cells, 2005, 85, 385–390 CrossRef CAS.
- A. L. Smeigh, L. L. Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2011, 48, 678–680 RSC.
- F. A. Black, C. A. Clark, G. H. Summers, I. P. Clark, M. Towrie, T. Penfold, M. W. George and E. A. Gibson, Phys. Chem. Chem. Phys., 2017, 19, 7877–7885 RSC.
- Z. Huang, G. Natu, Z. Ji, M. He, M. Yu and Y. Wu, J. Phys. Chem. C, 2012, 116, 26239–26246 CrossRef CAS.
- T. Daeneke, Z. Yu, G. P. Lee, D. Fu, N. W. Duffy, S. Makuta, Y. Tachibana, L. Spiccia, A. Mishra, P. Bäuerle and U. Bach, Adv. Energy Mater., 2015, 5, 1401387 CrossRef.
- R. J. Dillon, L. Alibabaei, T. J. Meyer and J. M. Papanikolas, ACS Appl. Mater. Interfaces, 2017, 9, 26786–26796 CrossRef CAS PubMed.
- M. Bonomo, D. Gatti, C. Barolo and D. Dini, Coatings, 2018, 8, 232 CrossRef.
- X. L. Zhang, Z. Zhang, F. Huang, P. Bäuerle, U. Bach and Y.-B. Cheng, J. Mater. Chem., 2012, 22, 7005–7009 RSC.
- H. Zhu, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2007, 111, 17455–17458 CrossRef CAS.
- Z. Huang, G. Natu, Z. Ji, P. Hasin and Y. Wu, J. Phys. Chem. C, 2011, 115, 25109–25114 CrossRef CAS.
- Z. Bian, T. Tachikawa, S.-C. Cui, M. Fujitsuka and T. Majima, Chem. Sci., 2012, 3, 370–379 RSC.
- O. Langmar, D. Saccone, A. Amat, S. Fantacci, G. Viscardi, C. Barolo, R. D. Costa and D. M. Guldi, ChemSusChem, 2017, 10, 2385–2393 CrossRef CAS PubMed.
- Q. Liu, L. Wei, S. Yuan, X. Ren, Y. Zhao, Z. Wang, M. Zhang, L. Shi and D. Li, J. Mater. Sci., 2015, 50, 6668–6676 CrossRef CAS.
- M. Bonomo, N. Barbero, G. Naponiello, M. Giordano, D. Dini and C. Barolo, Front. Chem., 2019, 7, 99 CrossRef CAS PubMed.
- M. Bonomo, C. Magistris, R. Buscaino, A. Fin, C. Barolo and D. Dini, ChemistrySelect, 2018, 3, 1066–1075 CrossRef CAS.
- L. Zhang, L. Favereau, Y. Farré, E. Mijangos, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Phys. Chem. Chem. Phys., 2016, 18, 18515–18527 RSC.
- L. D'Amario, L. J. Antila, B. Pettersson Rimgard, G. Boschloo and L. Hammarström, J. Phys. Chem. Lett., 2015, 6, 779–783 CrossRef PubMed.
- J. C. Freys, J. M. Gardner, L. D'Amario, A. M. Brown and L. Hammarström, Dalton Trans., 2012, 41, 13105–13111 RSC.
- O. Langmar, E. Fazio, P. Schol, G. de la Torre, R. D. Costa, T. Torres and D. M. Guldi, Angew. Chem., Int. Ed., 2019, 58, 4056–4060 CrossRef CAS PubMed.
- A. Kolay, P. Ghosal and M. Deepa, ACS Sustainable Chem. Eng., 2020, 8, 8593–8603 CrossRef CAS.
- I. C. Kaya, S. Akin, H. Akyildiz and S. Sonmezoglu, Sol. Energy, 2018, 169, 196–205 CrossRef CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- J. A. Treadway, J. A. Moss and T. J. Meyer, Inorg. Chem., 1999, 38, 4386–4387 CrossRef CAS PubMed.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307–7309 RSC.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- H. Luo, W. Song, P. G. Hoertz, K. Hanson, R. Ghosh, S. Rangan, M. K. Brennaman, J. J. Concepcion, R. A. Binstead, R. A. Bartynski, R. Lopez and T. J. Meyer, Chem. Mater., 2013, 25, 122–131 CrossRef CAS.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347–2350 CrossRef CAS.
- W. Song, A. K. Vannucci, B. H. Farnum, A. M. Lapides, M. K. Brennaman, B. Kalanyan, L. Alibabaei, J. J. Concepcion, M. D. Losego, G. N. Parsons and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 9773–9779 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 10974–10982 CrossRef CAS PubMed.
- L. Zhang, Y. Gao, X. Ding, Z. Yu and L. Sun, ChemSusChem, 2014, 7, 2801–2804 CrossRef CAS PubMed.
- L. Alibabaei, B. D. Sherman, M. R. Norris, M. K. Brennaman and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5899–5902 CrossRef CAS PubMed.
- D. L. Ashford, B. D. Sherman, R. A. Binstead, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2015, 54, 4778–4781 CrossRef CAS PubMed.
- R. L. House, N. Y. M. Iha, R. L. Coppo, L. Alibabaei, B. D. Sherman, P. Kang, M. K. Brennaman, P. G. Hoertz and T. J. Meyer, J. Photochem. Photobiol., C, 2015, 25, 32–45 CrossRef CAS.
- F. Li, K. Fan, L. Wang, Q. Daniel, L. Duan and L. Sun, ACS Catal., 2015, 5, 3786–3790 CrossRef CAS.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- B. D. Sherman, D. L. Ashford, A. M. Lapides, M. V. Sheridan, K.-R. Wee and T. J. Meyer, J. Phys. Chem. Lett., 2015, 6, 3213–3217 CrossRef CAS.
- J. R. Swierk, N. S. McCool and T. E. Mallouk, J. Phys. Chem. C, 2015, 119, 13858–13867 CrossRef CAS.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- N. Armaroli and V. Balzani, Chem. – Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- R. W. Call, L. Alibabaei, R. J. Dillon, R. R. Knauf, A. Nayak, J. L. Dempsey, J. M. Papanikolas and R. Lopez, ACS Appl. Mater. Interfaces, 2016, 8, 12282–12290 CrossRef CAS PubMed.
- M. V. Sheridan, B. D. Sherman, R. L. Coppo, D. Wang, S. L. Marquard, K.-R. Wee, N. Y. Murakami Iha and T. J. Meyer, ACS Energy Lett., 2016, 1, 231–236 CrossRef CAS.
- B. D. Sherman, J. J. Bergkamp, C. L. Brown, A. L. Moore, D. Gust and T. A. Moore, Energy Environ. Sci., 2016, 9, 1812–1817 RSC.
- B. D. Sherman, M. V. Sheridan, K.-R. Wee, S. L. Marquard, D. Wang, L. Alibabaei, D. L. Ashford and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 16745–16753 CrossRef CAS PubMed.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- L. Alibabaei, R. J. Dillon, C. E. Reilly, M. K. Brennaman, K.-R. Wee, S. L. Marquard, J. M. Papanikolas and T. J. Meyer, ACS Appl. Mater. Interfaces, 2017, 9, 39018–39026 CrossRef CAS PubMed.
- M. V. Sheridan, D. J. Hill, B. D. Sherman, D. Wang, S. L. Marquard, K.-R. Wee, J. F. Cahoon and T. J. Meyer, Nano Lett., 2017, 17, 2440–2446 CrossRef CAS PubMed.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- O. Suryani, Y. Higashino, J. Y. Mulyana, M. Kaneko, T. Hoshi, K. Shigaki and Y. Kubo, Chem. Commun., 2017, 53, 6784–6787 RSC.
- D. Wang, B. H. Farnum, M. V. Sheridan, S. L. Marquard, B. D. Sherman and T. J. Meyer, ACS Appl. Mater. Interfaces, 2017, 9, 33533–33538 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 3, 944–951 CrossRef CAS.
- D. Wang, S. L. Marquard, L. Troian-Gautier, M. V. Sheridan, B. D. Sherman, Y. Wang, M. S. Eberhart, B. H. Farnum, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 719–726 CrossRef CAS PubMed.
- P. Xu, T. Huang, J. Huang, Y. Yan and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6946–6951 CrossRef CAS PubMed.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- D. Wang, L. Wang, M. D. Brady, C. J. Dares, G. J. Meyer, T. J. Meyer and J. J. Concepcion, J. Phys. Chem. C, 2019, 123, 30039–30045 CrossRef CAS.
- D. Wang, J. Hu, B. D. Sherman, M. V. Sheridan, L. Yan, C. J. Dares, Y. Zhu, F. Li, Q. Huang, W. You and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13256–13260 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, P. G. Hoertz and T. J. Meyer, Angew. Chem., Int. Ed., 2009, 48, 9473–9476 CrossRef CAS PubMed.
- D. L. Ashford, W. Song, J. J. Concepcion, C. R. K. Glasson, M. K. Brennaman, M. R. Norris, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 19189–19198 CrossRef CAS PubMed.
- D. M. Ryan, M. K. Coggins, J. J. Concepcion, D. L. Ashford, Z. Fang, L. Alibabaei, D. Ma, T. J. Meyer and M. L. Waters, Inorg. Chem., 2014, 53, 8120–8128 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- K. Hanson, D. A. Torelli, A. K. Vannucci, M. K. Brennaman, H. Luo, L. Alibabaei, W. Song, D. L. Ashford, M. R. Norris, C. R. K. Glasson, J. J. Concepcion and T. J. Meyer, Angew. Chem., Int. Ed., 2012, 51, 12782–12785 CrossRef CAS PubMed.
- H. Lee, L. J. Kepley, H. G. Hong and T. E. Mallouk, J. Am. Chem. Soc., 1988, 110, 618–620 CrossRef CAS.
- H. Lee, L. J. Kepley, H. G. Hong, S. Akhter and T. E. Mallouk, J. Phys. Chem., 1988, 92, 2597–2601 CrossRef CAS.
- B. Shan, M. K. Brennaman, L. Troian-Gautier, Y. Liu, A. Nayak, C. M. Klug, T.-T. Li, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 10390–10398 CrossRef CAS PubMed.
- R. J. Kamire, K. L. Materna, W. L. Hoffeditz, B. T. Phelan, J. M. Thomsen, O. K. Farha, J. T. Hupp, G. W. Brudvig and M. R. Wasielewski, J. Phys. Chem. C, 2017, 121, 3752–3764 CrossRef CAS.
- L. Wang, D. E. Polyansky and J. J. Concepcion, J. Am. Chem. Soc., 2019, 141, 8020–8024 CrossRef CAS PubMed.
- C. J. Flynn, S. M. McCullough, E. Oh, L. Li, C. C. Mercado, B. H. Farnum, W. Li, C. L. Donley, W. You, A. J. Nozik, J. R. McBride, T. J. Meyer, Y. Kanai and J. F. Cahoon, ACS Appl. Mater. Interfaces, 2016, 8, 4754–4761 CrossRef CAS PubMed.
- S. M. McCullough, J. M. Evans, T. Moot, A. D. Taggart, L. Troian-Gautier and J. F. Cahoon, ACS Appl. Energy Mater., 2020, 3, 1496–1505 CrossRef CAS.
- A. D. Taggart, J. M. Evans, L. Li, K. J. Lee, J. L. Dempsey, Y. Kanai and J. F. Cahoon, ACS Appl. Energy Mater., 2020, 3, 10702–10713 CrossRef CAS.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2016, 8, 541–549 RSC.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2018, 9, 1439–1447 RSC.
- C. E. Creissen, J. Warnan, D. Antón-García, Y. Farré, F. Odobel and E. Reisner, ACS Catal., 2019, 9, 9530–9538 CrossRef CAS PubMed.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- B. Shan, S. Vanka, T.-T. Li, L. Troian-Gautier, M. K. Brennaman, Z. Mi and T. J. Meyer, Nat. Energy, 2019, 4, 290–299 CrossRef CAS.
- D. F. Watson, A. Marton, A. M. Stux and G. J. Meyer, J. Phys. Chem. B, 2003, 107, 10971–10973 CrossRef CAS.
- E. M. James, T. J. Barr and G. J. Meyer, ACS Appl. Energy Mater., 2018, 1, 859–867 CrossRef CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- T.-T. Li, B. Shan and T. J. Meyer, ACS Energy Lett., 2019, 4, 629–636 CrossRef CAS.
- L. Wu, M. K. Brennaman, A. Nayak, M. Eberhart, A. J. M. Miller and T. J. Meyer, ACS Cent. Sci., 2019, 5, 506–514 CrossRef CAS PubMed.
- L. Wu, M. Eberhart, B. Shan, A. Nayak, M. K. Brennaman, A. J. M. Miller, J. Shao and T. J. Meyer, ACS Appl. Mater. Interfaces, 2019, 11, 4560–4567 CrossRef CAS PubMed.
- D. L. Ashford, C. R. K. Glasson, M. R. Norris, J. J. Concepcion, S. Keinan, M. K. Brennaman, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2014, 53, 5637–5646 CrossRef CAS PubMed.
- D. L. Ashford, M. K. Brennaman, R. J. Brown, S. Keinan, J. J. Concepcion, J. M. Papanikolas, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2015, 54, 460–469 CrossRef CAS PubMed.
- L. Wang, D. W. Shaffer, G. F. Manbeck, D. E. Polyansky and J. J. Concepcion, ACS Catal., 2020, 10, 580–585 CrossRef CAS.
- C. Decavoli, C. L. Boldrini, N. Manfredi and A. Abbotto, Eur. J. Inorg. Chem., 2020, 2020, 978–999 CrossRef CAS.
- V. Nikolaou, A. Charisiadis, G. Charalambidis, A. G. Coutsolelos and F. Odobel, J. Mater. Chem. A, 2017, 5, 21077–21113 RSC.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion and T. J. Meyer, Dalton Trans., 2011, 40, 3789–3792 RSC.
- M. R. Norris, J. J. Concepcion, D. P. Harrison, R. A. Binstead, D. L. Ashford, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2080–2083 CrossRef CAS PubMed.
- M. R. Norris, J. J. Concepcion, Z. Fang, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 13580–13583 CrossRef CAS PubMed.
- J. J. Concepcion, D. K. Zhong, D. J. Szalda, J. T. Muckerman and E. Fujita, Chem. Commun., 2015, 51, 4105–4108 RSC.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170–6193 RSC.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Suriñach, M. Z. Ertem and A. Llobet, J. Am. Chem. Soc., 2020, 142, 5068–5077 CrossRef CAS PubMed.
- Y. Xie, D. W. Shaffer, A. Lewandowska-Andralojc, D. J. Szalda and J. J. Concepcion, Angew. Chem., Int. Ed., 2016, 55, 8067–8071 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- T. E. Rosser, M. A. Gross, Y.-H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- B. Shan, B. D. Sherman, C. M. Klug, A. Nayak, S. L. Marquard, Q. Liu, R. M. Bullock and T. J. Meyer, J. Phys. Chem. Lett., 2017, 8, 4374–4379 CrossRef CAS PubMed.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
- M. D. Brady, R. N. Sampaio, D. Wang, T. J. Meyer and G. J. Meyer, J. Am. Chem. Soc., 2017, 139, 15612–15615 CrossRef CAS PubMed.
- M. K. Gish, A. M. Lapides, M. K. Brennaman, J. L. Templeton, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. Lett., 2016, 7, 5297–5301 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- D. R. Ort, S. S. Merchant, J. Alric, A. Barkan, R. E. Blankenship, R. Bock, R. Croce, M. R. Hanson, J. M. Hibberd, S. P. Long, T. A. Moore, J. Moroney, K. K. Niyogi, M. A. J. Parry, P. P. Peralta-Yahya, R. C. Prince, K. E. Redding, M. H. Spalding, K. J. van Wijk, W. F. J. Vermaas, S. von Caemmerer, A. P. M. Weber, T. O. Yeates, J. S. Yuan and X. G. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8529–8536 CrossRef CAS PubMed.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- H. Pettersson, K. Nonomura, L. Kloo and A. Hagfeldt, Energy Environ. Sci., 2012, 5, 7376–7380 RSC.
- J. Takashima, T. Fujii and K. Furusaki, Meet. Abstr., 2008, MA2008-02, 478 Search PubMed.
- S. Yun, P. D. Lund and A. Hinsch, Energy Environ. Sci., 2015, 8, 3495–3514 RSC.
- N. Jiang, T. Sumitomo, T. Lee, A. Pellaroque, O. Bellon, D. Milliken and H. Desilvestro, Sol. Energy Mater. Sol. Cells, 2013, 119, 36–50 CrossRef CAS.
- M. Flasque, A. N. V. Nhien, J. Swiatowska, A. Seyeux, C. Davoisne and F. Sauvage, ChemPhysChem, 2014, 15, 1126–1137 CrossRef CAS PubMed.
- F. Bella, S. Galliano, C. Gerbaldi and G. Viscardi, Energies, 2016, 9, 384 CrossRef.
- N. Kato, Y. Takeda, K. Higuchi, A. Takeichi, E. Sudo, H. Tanaka, T. Motohiro, T. Sano and T. Toyoda, Sol. Energy Mater. Sol. Cells, 2009, 93, 893–897 CrossRef CAS.
- S. Dai, J. Weng, Y. Sui, S. Chen, S. Xiao, Y. Huang, F. Kong, X. Pan, L. Hu, C. Zhang and K. Wang, Inorg. Chim. Acta, 2008, 361, 786–791 CrossRef CAS.
- H. Matsui, K. Okada, T. Kitamura and N. Tanabe, Sol. Energy Mater. Sol. Cells, 2009, 93, 1110–1115 CrossRef CAS.
- Y. Rong, X. Li, Z. Ku, G. Liu, H. Wang, M. Xu, L. Liu, M. Hu, P. Xiang, Z. Zhou, T. Shu and H. Han, Sol. Energy Mater. Sol. Cells, 2012, 105, 148–152 CrossRef CAS.
- N. Kato, K. Higuchi, H. Tanaka, J. Nakajima, T. Sano and T. Toyoda, Sol. Energy Mater. Sol. Cells, 2011, 95, 301–305 CrossRef CAS.
- A. Hinsch, W. Veurman, H. Brandt, R. L. Aguirre, K. Bialecka and K. F. Jensen, Prog. Photovoltaics, 2012, 20, 698–710 CAS.
- H. Pettersson and T. Gruszecki, Sol. Energy Mater. Sol. Cells, 2001, 70, 203–212 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- N. Mariotti, M. Bonomo, L. Fagiolari, N. Barbero, C. Gerbaldi, F. Bella and C. Barolo, Green Chem., 2020, 22, 7168–7218 RSC.
- J. M. Kroon, N. J. Bakker, H. J. P. Smit, P. Liska, K. R. Thampi, P. Wang, S. M. Zakeeruddin, M. Grätzel, A. Hinsch, S. Hore, U. Würfel, R. Sastrawan, J. R. Durrant, E. Palomares, H. Pettersson, T. Gruszecki, J. Walter, K. Skupien and G. E. Tulloch, Prog. Photovoltaics, 2007, 15, 1–18 CAS.
- R. Sastrawan, J. Beier, U. Belledin, S. Hemming, A. Hinsch, R. Kern, C. Vetter, F. M. Petrat, A. Prodi-Schwab, P. Lechner and W. Hoffmann, Prog. Photovoltaics, 2006, 14, 697–709 CAS.
- D. Magistri, A. Lembo, V. Mirruzzo, G. Viscardi, P. Mariani, R. Manfredi, V. Liparoti, A. Lanuti, C. Bignozzi, T. M. Brown, L. Bonandini, E. Busatto, D. Colonna, A. Di Carlo, A. Giglielmotti, A. Guidobaldi, R. Boaretto, D. Prencipe, C. Barolo, R. Tagliaferro, S. Caramori, L. Vesce, G. Soscia, A. Smarra, R. Riccitelli and A. Reale, HOPV15, 2015 Search PubMed.
- M. L. Parisi, S. Maranghi, L. Vesce, A. Sinicropi, A. Di Carlo and R. Basosi, Renewable Sustainable Energy Rev., 2020, 121, 109703 CrossRef.
- N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhôte, H. Pettersson, A. Azam and M. Grätzel, J. Electrochem. Soc., 1996, 143, 3099 CrossRef CAS.
- H. Pettersson, T. Gruszecki, L.-H. Johansson and P. Johander, Sol. Energy Mater. Sol. Cells, 2003, 77, 405–413 CrossRef CAS.
- S. Burnside, S. Winkel, K. Brooks, V. Shklover, M. Gratzel, A. Hinsch, R. Kinderman, C. Bradbury, A. Hagfeldt and H. Pettersson, J. Mater. Sci.: Mater. Electron., 2000, 11, 355–362 CrossRef CAS.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC.
- N. Yan, C. Zhao, S. You, Y. Zhang and W. Li, Chin. Chem. Lett., 2020, 31, 643–653 CrossRef CAS.
- M. Li, C. Zhao, Z.-K. Wang, C.-C. Zhang, H. K. H. Lee, A. Pockett, J. Barbé, W. C. Tsoi, Y.-G. Yang, M. J. Carnie, X.-Y. Gao, W.-X. Yang, J. R. Durrant, L.-S. Liao and S. M. Jain, Adv. Energy Mater., 2018, 8, 1801509 CrossRef.
- H. K. H. Lee, J. Wu, J. Barbé, S. M. Jain, S. Wood, E. M. Speller, Z. Li, F. A. Castro, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC.
- H. Matsui, K. Yamamoto and K. Okada, OPTICS & PHOTONICS International Congress 2019, 2019.
- O. Pekkola, C. Lungenschmied, P. Fejes, A. Handreck, W. Hermes, S. Irle, C. Lennartz, C. Schildknecht, P. Schillen, P. Schindler, R. Send, S. Valouch, E. Thiel and I. Bruder, Sci. Rep., 2018, 8, 9208 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |