
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular photoswitches in aqueous environments
Jana
Volarić
 a,
Wiktor
Szymanski
ab,
Nadja A.
Simeth
*ac and
Ben L.
Feringa
*a
a,
Wiktor
Szymanski
ab,
Nadja A.
Simeth
*ac and
Ben L.
Feringa
*a
aCentre for Systems Chemistry, Stratingh Institute for Chemistry, Faculty for Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; nadja.simeth@uni-goettingen.de
bDepartment of Radiology, Medical Imaging Center, University of Groningen, University Medical Centre Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands
cInstitute for Organic and Biomolecular Chemistry, University of Göttingen, Tammannstr. 2, 37077 Göttingen, Germany
First published on 30th September 2021
Abstract
Molecular photoswitches enable dynamic control of processes with high spatiotemporal precision, using light as external stimulus, and hence are ideal tools for different research areas spanning from chemical biology to smart materials. Photoswitches are typically organic molecules that feature extended aromatic systems to make them responsive to (visible) light. However, this renders them inherently lipophilic, while water-solubility is of crucial importance to apply photoswitchable organic molecules in biological systems, like in the rapidly emerging field of photopharmacology. Several strategies for solubilizing organic molecules in water are known, but there are not yet clear rules for applying them to photoswitchable molecules. Importantly, rendering photoswitches water-soluble has a serious impact on both their photophysical and biological properties, which must be taken into consideration when designing new systems. Altogether, these aspects pose considerable challenges for successfully applying molecular photoswitches in aqueous systems, and in particular in biologically relevant media. In this review, we focus on fully water-soluble photoswitches, such as those used in biological environments, in both in vitro and in vivo studies. We discuss the design principles and prospects for water-soluble photoswitches to inspire and enable their future applications.
 Jana Volarić | Jana Volarić was born in Zagreb, Croatia in 1992. She obtained her BSc at the University of Zagreb with Prof. Hrvoj Vančik. She did her MSc at the University of Groningen with Prof. Ben Feringa on the synthesis of dynamic bisthiourea molecular motor-based catalysts. She spent 6 months in Boston in the group of prof. Hidde Ploegh (Boston Children's Hospital, Harvard Medical School) working on site-specific labeling of influenza A virus. In 2017 Jana started her PhD under supervision of prof. Ben L. Feringa on incorporating photoswitches into biosystems in order to control biomolecular function. |
 Wiktor Szymanski | Wiktor Szymanski received his PhD degree from The Warsaw University of Technology, Poland, in 2008, working under the supervision of Prof. Ryszard Ostaszewski. He spent two years working on the use of biotransformations in organic chemistry with Prof. Ben L. Feringa and Prof. Dick B. Janssen at the University of Groningen. Since 2010 he has been working on the construction of photoactive protein–peptide– and DNA-bioconjugates and photopharmacology in the Feringa Labs. In 2014, he joined the Department of Radiology, University Medical Center Groningen, where he was appointed in 2019 as associate professor. |
 Nadja A. Simeth | Nadja A. Simeth pursued her doctorate studies with Burkhard König at the University of Regensburg and defended her thesis in summer 2018 with summa cum laude. She then joined the group of Ben L. Feringa at the University of Groningen as a postdoc supported by a Feodor-Lynen Fellowship of the Humboldt Foundation. In autumn 2021, she was appointed as assistant professor at the University of Göttingen. She is interested in the design of smart drugs, biochemical probes and labels, as well as photoresponsive supramolecular architectures and biohybrid systems. |
 Ben L. Feringa | Ben L. Feringa obtained his PhD degree in 1978 at the University of Groningen in the Netherlands under the guidance of Prof. Hans Wynberg. After working as a research scientist at Shell he was appointed full professor at the University of Groningen in 1988 and named the distinguished Jacobus H. van't Hoff Professor of Molecular Sciences in 2004. He was elected foreign honorary member of the American Academy of Arts and Sciences and member of the Royal Netherlands Academy of Sciences. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, photopharmacology, self-assembly and nanosystems. |
1 Introduction
Water is the solvent, reagent, and mediator of a wide range of chemical transformations, assemblies and metabolic processes in every living organism.1,2 In order to dynamically regulate the complex, multifaceted processes taking place in aqueous media, a powerful strategy is to apply an externally triggered control element. Specifically, light is a favourable stimulus as it is not natively present in a wide range of systems (except of course the process of vision and photosynthesis) and thus, can be applied orthogonally to their function.3–5 Moreover, it can be tuned over a wide range of wavelengths and is, especially in the visible to near-infrared (NIR) light range, harmless for most living organisms.6–8 Finally, it can be precisely delivered, allowing for high spatiotemporal control of a process of choice.3–5,9–11The photons applied externally to the chosen system need to be translated into a signal the system can respond to.4,5 This can be achieved by choosing a suitable photoactuator as a mediator. Such mediators can be small, organic molecules that are able to interact with light, i.e., organic photocleavable protecting groups (photocages) or photoswitches.3,12–15 While photocages are photolabile protecting groups that are cleaved upon irradiation and hence, can only be activated once,13,16,17 photoswitches can be reversibly interconverted between different (functional) isomers.3,18 This reversible nature makes them interesting candidates for a wide range of applications, including those that take place in water, where they act as photoresponsive amphiphiles,19–27 hydrogel constituents,28,29 probes for chemical biology,30–32 or bioactive agents in photopharmacology.6,7,30,33
Molecular photoswitches are typically organic molecules bearing extended aromatic systems to make them responsive to (visible) light.3,12 As a consequence they are inherently lipophilic. However, to apply photoswitches in aqueous media or in particular in biological systems, they need to be sufficiently water-soluble.34–38 While general strategies for solubilizing organic molecules, for instance in the context of medicinal chemistry, are well established,39–41 (Section 2 of this review), a similar broad set of approaches is yet to be applied to photoswitchable molecules. Moreover, rendering photoswitches water-soluble can have a serious impact on their photophysical and biological properties,6,7 which must be taken into consideration when designing new systems (Fig. 1). Therefore, successfully applying molecular organic photoswitches to aqueous systems is challenging.
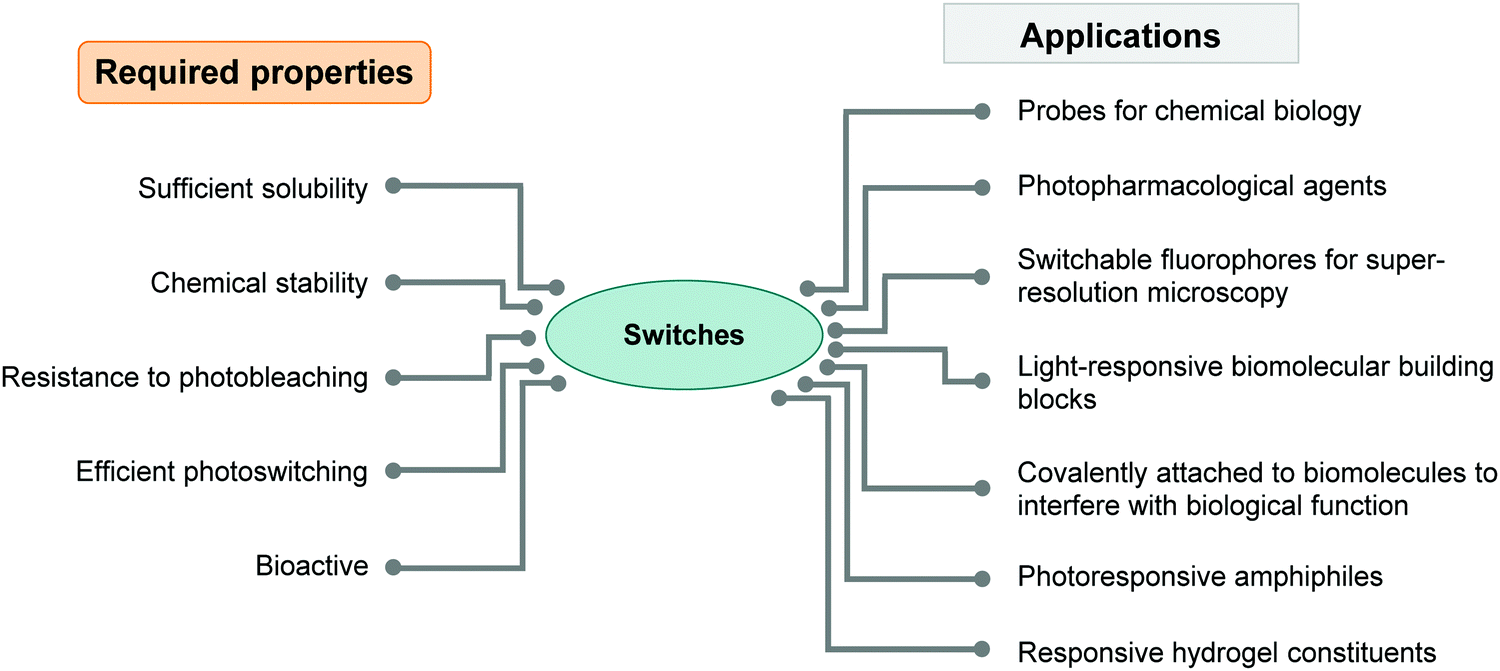 | ||
| Fig. 1 Diagram summarizing the required properties of photoswitches needed for successful applications in water. | ||
In this review, we discuss the design principles and prospects for water-soluble photoswitches as a source of inspiration and to enable their future applications. The first part of the review focuses on general principles to solubilize organic molecules in water and is followed by a section introducing different types of organic photoswitches and the impact of polar protic solvents on their properties. In the second part, we highlight selected illustrative examples of different types of photoswitches and their implementation in aqueous environment to underline the importance and impact of water-solubility on the desired application. As water is the universal solvent of life, the challenge of insufficient water-solubility of organic molecules is usually faced in a bio-inspired, biological, or medicinal context, and most strategies to overcome solubility issues are derived from this research area. Naturally, the applications and examples discussed in this review are coming from these fields. Consequently, photoswitches which are designed to be intentionally amphiphilic or lipophilic to form (supramolecular) (self-) assemblies, nano-, and microstructures in aqueous environment,21,42–45 or (hydro)gels,46–51 do not face the same obstacles as the ones intended to be fully water-soluble and hence, lie outside the scope of this review and we refer the reader to our recent overview.21
2 General aspects of the solubility of organic molecules in water and solubilizing strategies
Before discussing strategies used to solubilize organic molecules in water, we describe the definition of water-solubility, parameters that affect it and are used to predict it, as well as methods to determine the aqueous solubility of a compound of interest.2.1 Aqueous solubility
| Descriptive term | Approximate volume of solvent per mass of solute (mL g−1) |
|---|---|
| Very soluble | <1 |
| Freely soluble | 1–10 |
| Soluble | 10–30 |
| Sparingly soluble | 30–100 |
| Slightly soluble | 100–1000 |
| Very slightly soluble | 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Practically insoluble | >10000 |
For a molecule to dissolve, it first needs to be removed from the solid form by breaking the intermolecular interactions holding the solid together (see Fig. 2). This applies to both the solids being in an amorphous and in a crystalline form. However, the more stable the (crystal) structure or lattice is, the more energy is needed to detach an individual molecule from it. The molecule is simultaneously forming stabilizing bonds with the surrounding medium. The energy released upon this so-called dissolution process in water is called hydration energy and the rate at which the solvation is proceeding is called dissolution rate, with both values being specific for each compound.55
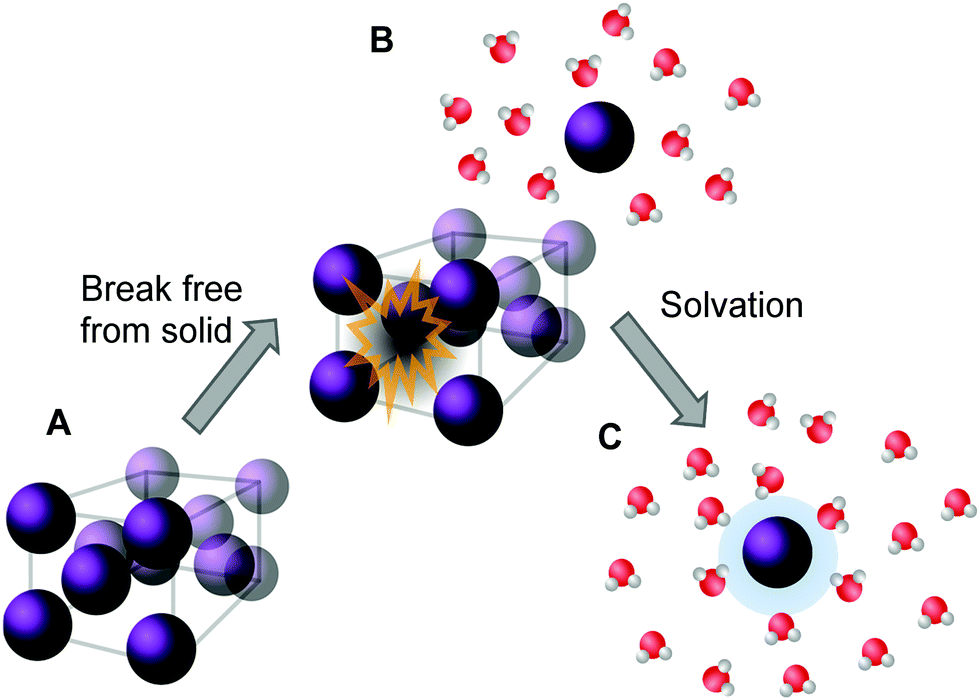 | ||
| Fig. 2 Descriptive representation of the dissolution process of a crystalline solid in water. Energy is needed to first remove the molecule from its crystal structure [from (A) to (B)] and subsequently the molecule is solvated by water. Once solvated, the molecule forms stabilizing interactions with water (C), represented by the blue halo, which results in a release of energy. (Figure adapted with permission from ref. 56. Copyright 2006, Elsevier.) | ||
In a thermodynamically stable saturated solution, the solid in its most stable crystalline form (characterized by its highest possible melting point) is in equilibrium with the dissolved species. This type of solubility is called the thermodynamic or equilibrium solubility.39,41 Since solubility depends on the solid form of the compound, it is possible to dissolve larger amounts of material by modifying the solid state of the compound into an amorphous or a less stable crystal structure via crystal engineering (taking advantage of polymorphism) (see Fig. 3A and B).57 This can be achieved, for instance, by dissolving the molecule into an organic solvent, often DMSO, and then diluting it into an aqueous solution until precipitation, or the “cloud point”, is reached.39 The type of solubility achieved in this way is called the kinetic (a.k.a. dynamic or apparent) solubility (Fig. 3D), and is higher than the thermodynamic one (Fig. 3C). However, the solid will eventually assume its most stable form and possibly crystallize from solution, which is particularly problematic in biological applications.41 Thermodynamic and kinetic solubility are general terms for both ionizable and non-ionic compounds. Additionally, the intrinsic solubility of ionizable compounds can be determined at the pH where the species is uncharged.58
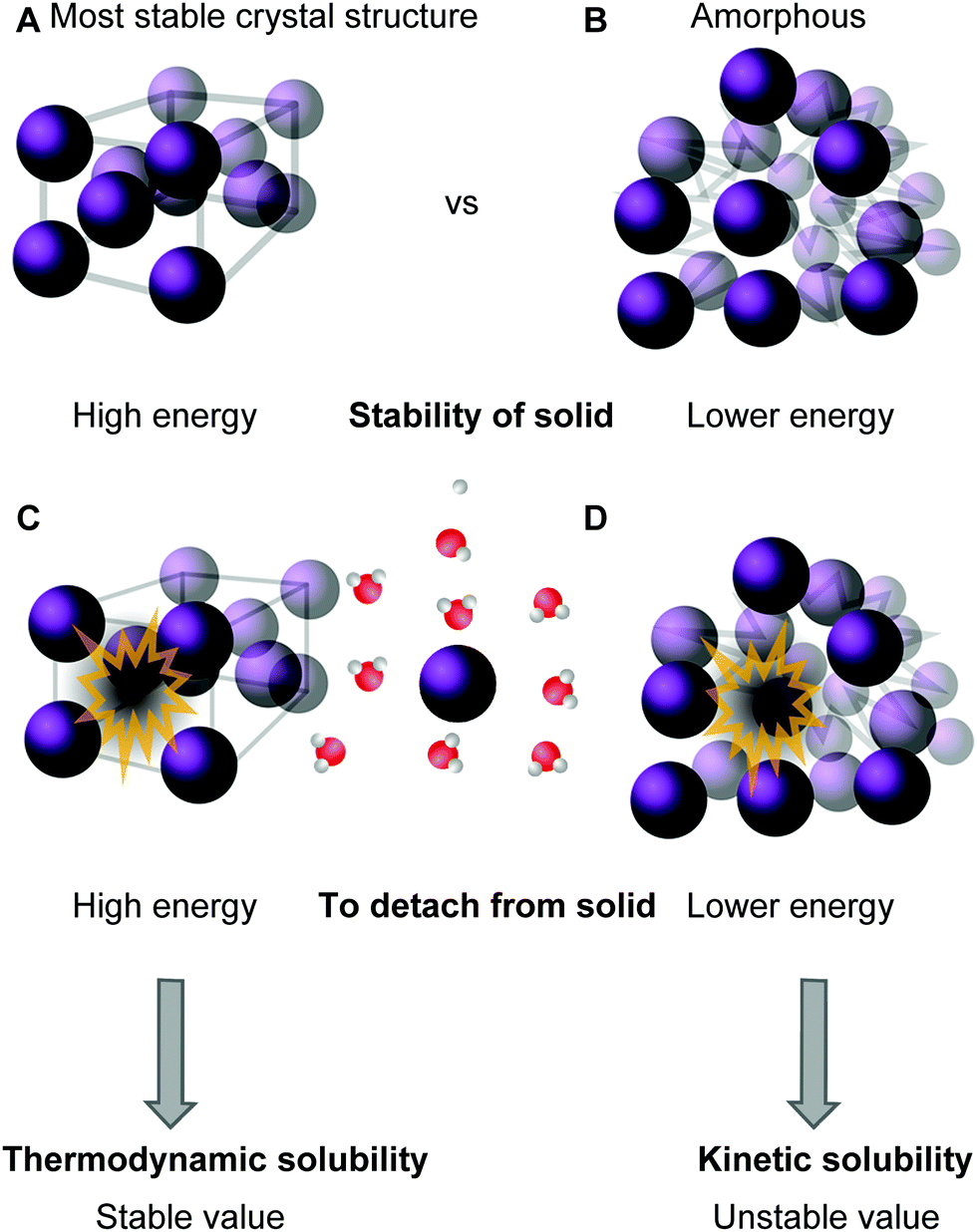 | ||
| Fig. 3 Descriptive representation of solubility types: the compound is either in its most stable crystalline solid form (A) or in a less stable, amorphous, form (B). High amount of energy is needed to remove the molecule from the stable solid (C), while a lower amount of energy is needed to remove it from the less stable amorphous form (D, see ref. 60). | ||
In the context of thermodynamics, the first step in dissolving a compound in water requires energy input to remove the molecule from the solid lattice (Fig. 2A–B). The energy released in the subsequent step, i.e., solvation (Fig. 2B–C), often compensates for the first process through the formation of favorable interactions of the dissolved molecule with water. However, depending on the nature of the compound, in some examples the solvation energy is lower than the energy required for the detachment, therefore making the overall dissolving process endothermic. In the instances when the overall process is slightly endothermic, the general entropy increase, caused by the disorder increase upon solubilizing a compound, becomes relevant and ultimately drives the process. Moreover, for general aspects regarding solubility, solvent parameters and solutions, see chapter 3 of ref. 59.
Due to the numerous parameters affecting water-solubility, there is no single model which would provide a general formula to predict the solubility of a given molecule, posing a challenge for computational studies.60–62 The most commonly applied model is the general solubility equation (GSE), reported by Yalkowski.63,64 The GSE correlates the stability of the crystal, reflected by its melting point, and the lipophilicity of the compound described by the octanol/water partition coefficient:
| logSw = −0.01(Tmp − 25 °C) − logP + 0.50 | (1) |
P describes how well the molecule interacts with water and represents the contribution of hydration to solubility. The melting point, on the other hand, describes the stabilizing intermolecular interactions within the crystalline form of the compound.39 While logP is used to describe non-ionic molecules, for ionizable ones logD is applied. Here, D is the water/octanol distribution coefficient that considers a sum of the concentration of all species of the compound, both dissociated and non-disassociated, in each phase, at a given pH: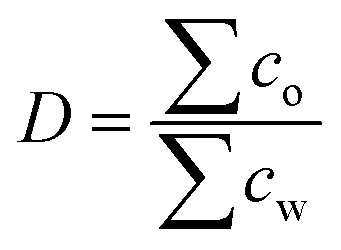 | (2) |
2.2 General strategies to dissolve organic molecules in water
Many organic molecules are lipophilic structures that require modifications to successfully dissolve in water. The most common chemical approach is to attach additional moieties, such as charged or polar units, which increase the molecule's water-solubility.39 Furthermore, if the molecule is an acid or a base, the simplest solubilization approach is to form a salt.75 In addition, non-planar molecules often exhibit increased water-solubility when compared to their rigid flat isosteres.41 Next to the chemical approaches, there is a plethora of physical methods available to improve solubility such as application of co-solvents, formulation of hydrotropes, particle size reduction and different delivery systems, such as liposomes or micelles.55,76,77 A schematic representation of the various methods is given in Fig. 4.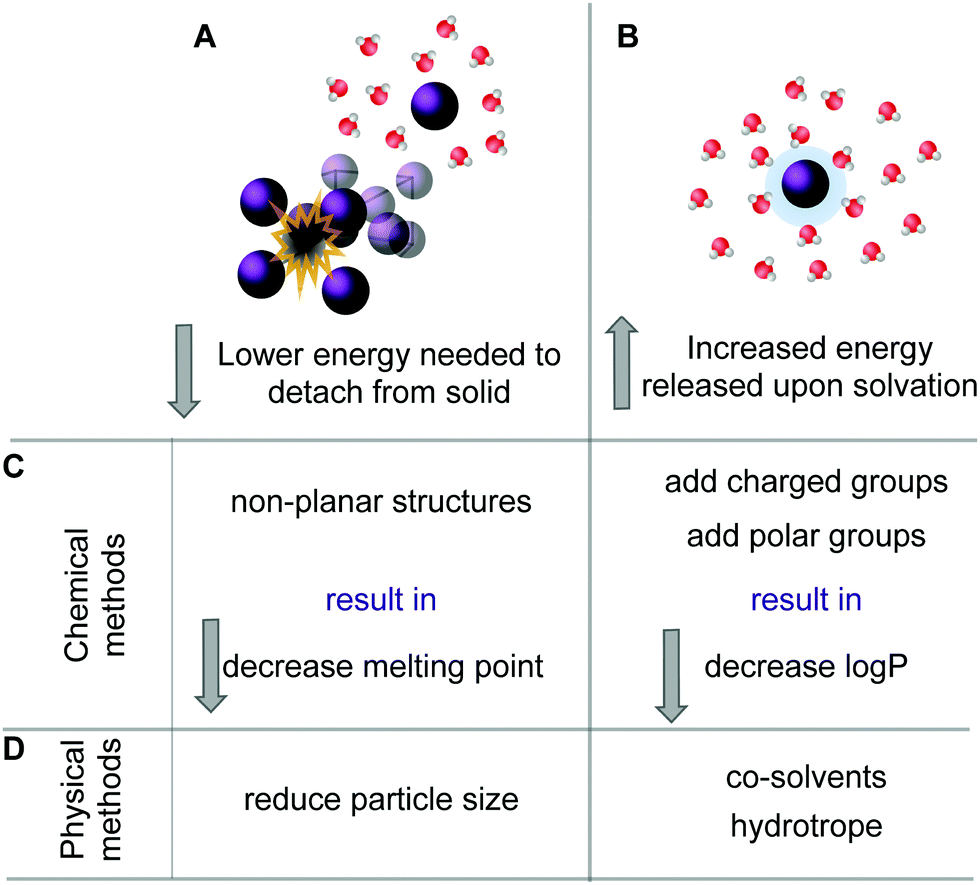 | ||
| Fig. 4 Two possible processes can be influenced to increase solubility of a specific compound. One involves lowering the energy needed to remove a molecule from its solid (A). The second approach is to increase the energy that is released upon solvation (B). The chemical methods (C) to decrease the melting point thus lowering the energy needed for (A) are, e.g., by designing non-planar structures and by addition of charged or polar solubilizing groups increases the stabilizing interactions with the solvent. The physical methods (D) to increase solubility involve reduction of particle size thus creating a larger surface or by adding co-solvents or hydrotropes (see also ref. 60). | ||
In the following sections, we provide a brief overview of different strategies which have been employed to solubilize organic molecules in water using illustrative examples. Several of those solubilization strategies have also been used for photoswitches and will be discussed in Section 4. Nevertheless, some solubilization strategies are underrepresented, or have even never been applied to photoswitchable systems.
P value.39 Such solubilizing moieties include charged and non-charged groups and are described below.
Charged groups. Some common charged groups used to achieve water-solubility are phosphates, phosphonates, sulfonates, and ammonium groups. A crucial aspect of the solubility of ionic molecules is the effect of the counter ion, which is often overlooked. This was, for example, shown for the anthraquinone sulfonate displayed in Fig. 5A and is used in redox flow batteries.78 Specifically, the disulfonate 2.1a (Fig. 5A) had the highest solubility in water followed by disulfonated 2.1c and monosulfonated 2.1b with similar values. Furthermore, for all three compounds (2.1a–c) the Mg2+ salts exhibited the highest solubility and the Ba2+ salts the lowest. This phenomenon is explained by the high lattice energies of the barium and calcium salts, compared to the magnesium salt, which lead to lower water-solubility. Next to the counter-ion, the location of the solubilizing group in the molecule can also play an important role, as seen for the disulfonate anions 2.1a and 2.1c. In particular, 2.1c showed more complex interactions with the cation in the crystal structure, which increases the lattice energy and lowers solubility.
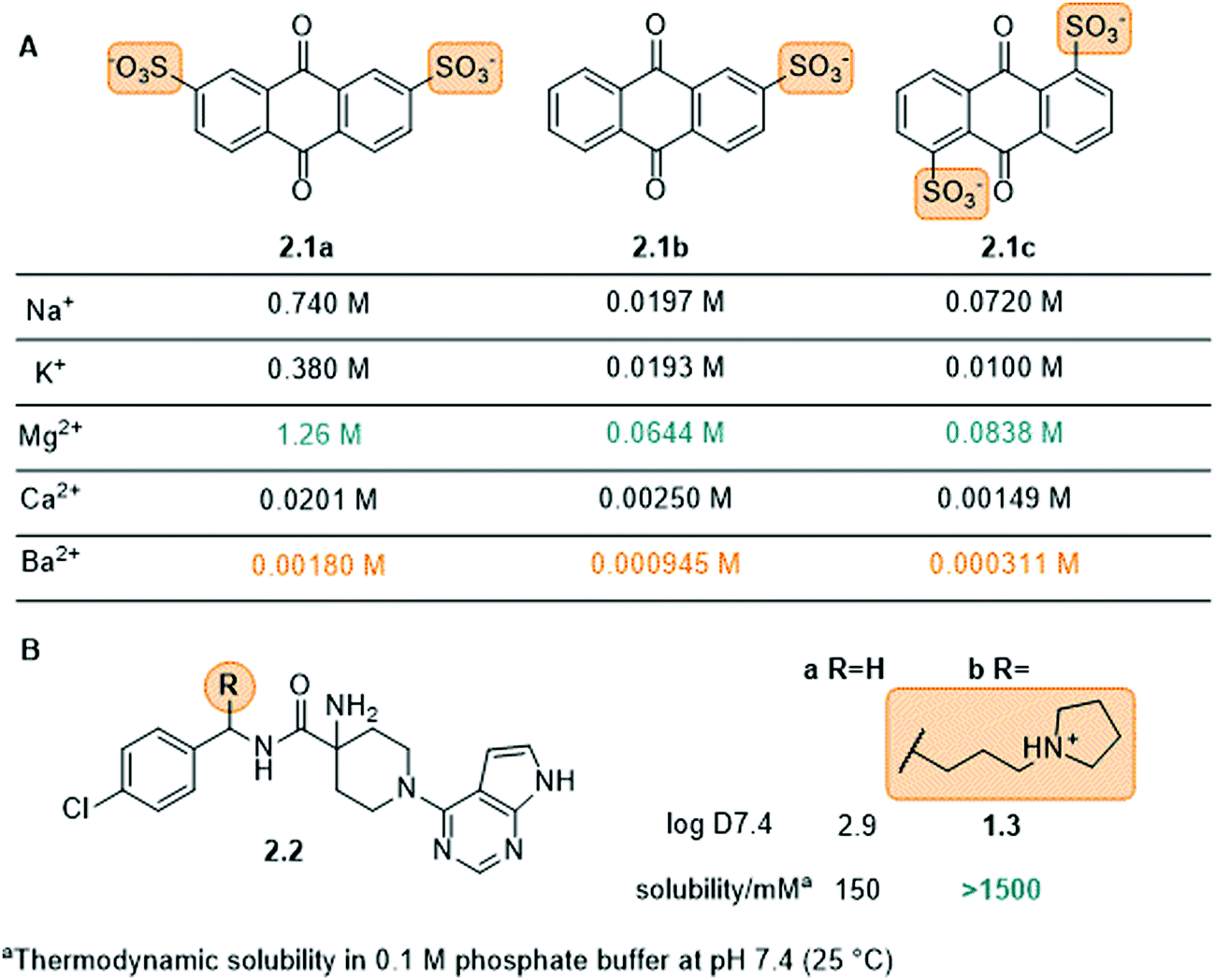 | ||
| Fig. 5 The use of charged groups to increase water solubility. (A) Sulfonated anthraquinones and the solubility of the corresponding ion pairs in water at room temperature, (B) Akt-kinase inhibitors 2.2 with amine solubilizing group, their respective logD value and solubility in 0.1 M phosphate buffer at pH 7.4 at 25 °C. | ||
Amine moieties with different substituents can be used to solubilize organic molecules in water as their pKbs cause them to be protonated under physiological conditions.39 For example, the benzylic site of the Akt-kinase inhibitor 2.2 was modified with an additional amine moiety (Fig. 5B).79 The pyrrolidine group (2.2b) increased the solubility 10-fold with the nitrogen being protonated at neutral pH (ammonium pKa ≈ 10).
Non-ionic groups. In many cases, it is not necessary to attach a charged moiety to increase the water-solubility and it is sufficient to introduce an additional electronegative atom, such as oxygen or nitrogen, into the scaffold to increase the dipole moment of the whole molecule and hence, its propensity to interact with water (Fig. 6A and B, 2.3 and 2.4). Moreover, these heteroatoms often constitute additional hydrogen bond donors/acceptors to interact with solvent and further increase solubility.39,80 This was, for example, shown in the development of cytokine-mediated STAT1 signal transduction inhibitor 2.3 (Fig. 6A). By replacing the naphthalene (2.3a) with a quinoline moiety (2.3b), the solubility increased drastically, namely by 1400-fold.80 Also in 2.4, the introduction of an electronegative atom, here a fluorine, increased the solubility of the compound by lowering the melting point and decreasing log
P (Fig. 6B).39,81
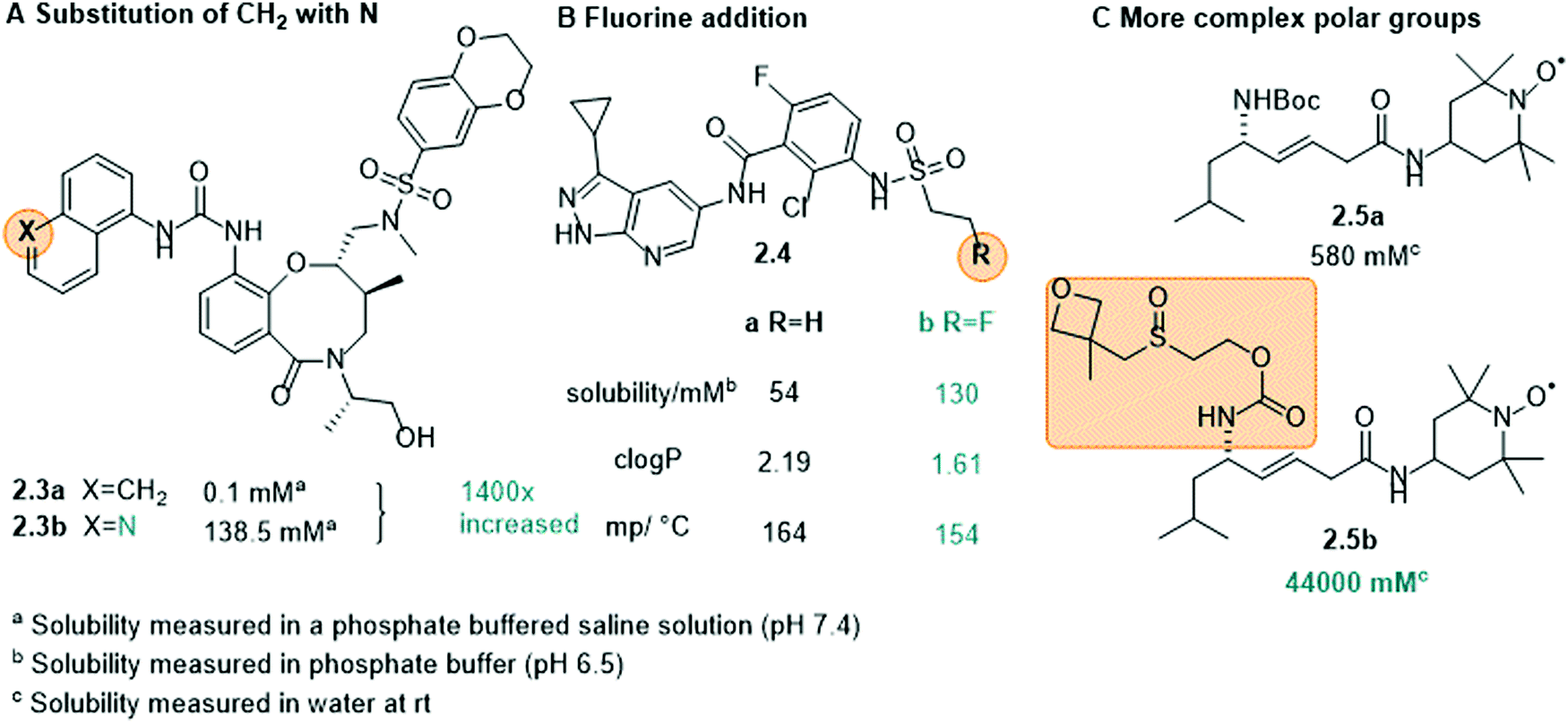 | ||
| Fig. 6 Examples of using non-ionic groups to enhance solubility (A) substitution of the naphthalene moiety (2.3a) of the STAT1 signal transduction inhibitor 2.3 with a quinoline moiety (2.3b) significantly increased the solubility, (B) the addition of a fluorine atom to 2.4 and its effect on solubility, clogP (calculated logP) and the melting point, (C) the oxetanyl sulfoxide group caused a 76-fold increase in solubility of 2.5. | ||
Other uncharged polar groups have been applied to increase aqueous solubility of organic compounds, such as polyethylene glycol (PEG) chains82,83 or the oxetanyl sulfoxide moiety (2.5, Fig. 6C).84
 | ||
| Fig. 7 The difference in water-solubility, both kinetic and intrinsic, of the carboxylic acid and sodium salt form of diclofenac 2.6. | ||
P, however it drastically lowered the melting point thus indicating a significantly less stable crystal lattice and better solubility in water (Fig. 8C)39,93,94 (see chapter 3 in ref. 60).
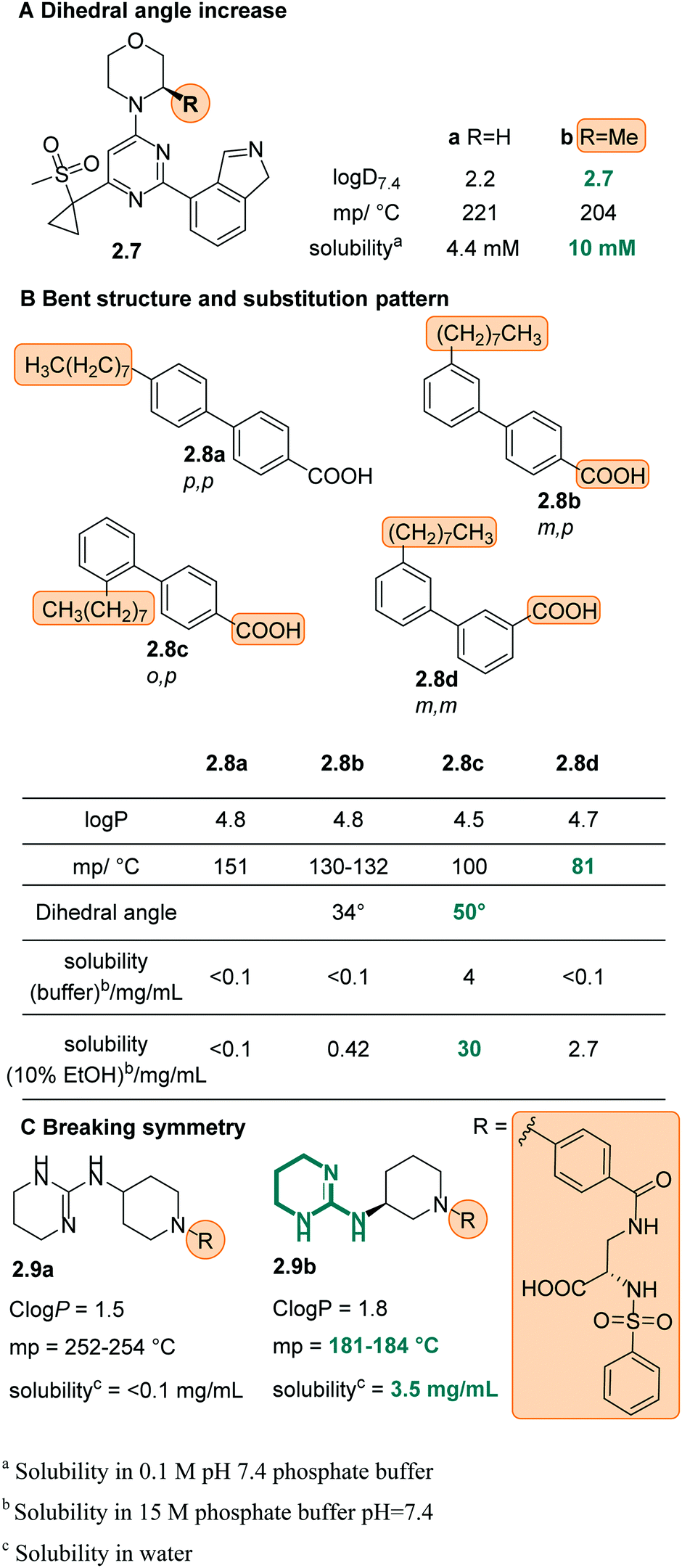 | ||
| Fig. 8 Increased dihedral angle of 2.7 (A) upon addition of a methyl group, as well as the ortho substituents in 2.8b and c (B) cause a decrease of melting point and increased solubility in water. (C) Introduction of a stereocenter in 2.9, thus breaking the symmetry of the compound, significantly lowered the melting point and caused increased solubility in water. | ||
The increase of ionic strength is commonly used in the “salting out” approach, where large concentrations of salt in aqueous solution force (organic) compounds with lower solubility to precipitate out of solution. The opposite effect, namely “salting in”, can be achieved by addition of so-called hydrotropes.95–98 Hydrotropes are ionic organic salts which are added to a poorly water-soluble compound to increase its aqueous solubility in diverse ways, for instance, by forming micellar-like structures.98 This approach is frequently used to formulate pharmaceuticals,98 however, in many cases the amount of the hydrotropic salt needed can reach high levels and cause toxicity.55
Other physical solubilization methods include, particle size reduction by mechanical micronization99 to increase the surface of the compound, nanoionisation,55 dispersion in carriers, such as micelles,55,76 or complexation in cyclodextrins.100
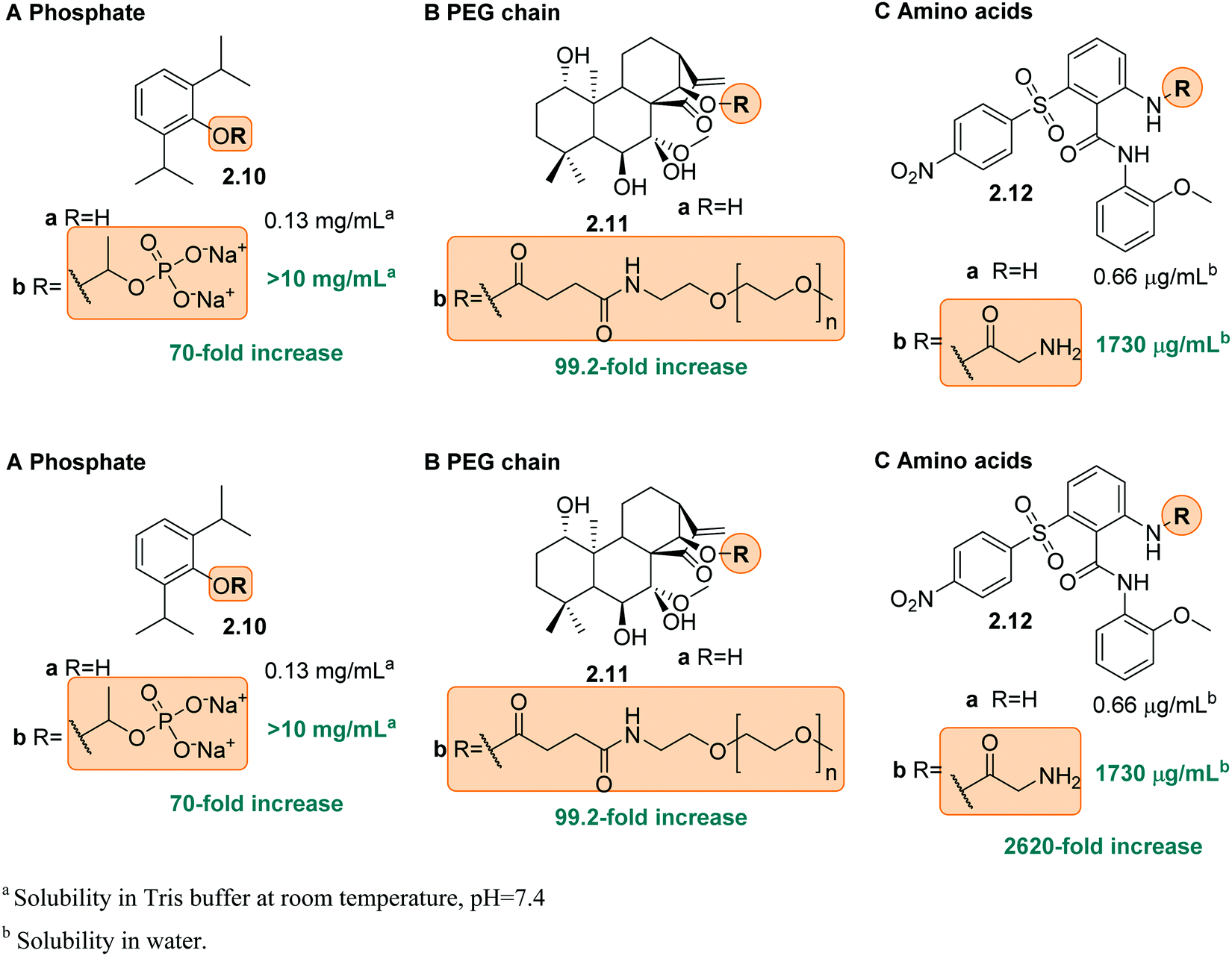 | ||
| Fig. 9 Illustrative examples of prodrugs containing solubilizing groups: phosphate (A), PEG chains (B) and amino acid moiety (C) and their respective effect on solubility in water. | ||
3 Molecular photoswitches
3.1 General definition of photoswitches and introduction of terminology describing their function and properties
A molecular photoswitch is a molecule which can be interconverted between two or more (stable) states by light.3 This photochemical isomerization is accompanied by structural and electronic changes and leads to usually well-defined differences in the UV-Vis absorption spectra of the isomeric states, rendering them photochromic. Most of these molecules show positive photochromism, with the photochemically generated species exhibiting an absorption maximum λmax(metastable) bathochromically shifted with respect to λmax(stable).106 If the initial photoisomer experiences a decolourization (bleaching) upon photoisomerization, with λmax(stable) > λmax(metastable), the compound shows negative or inverse photochromism.106 Specifically, pronounced band separation facilitates selective switching between the photoisomers with light of different colours. Generally, photoswitching is a reversible, non-destructive process. However, the performance can be lost over several switching cycles due to side-reactions, typically photo degradation, irreversible cyclization, dimerization, etc. and can be described by the cyclability Z50 (the number of cycles in which the absorbance is reduced to 50% of the initial one).106The extent of the isomerization process is characterized by a series of different parameters. The photostationary state (PSS) describes the dynamic equilibrium between two or more photoisomers under irradiation with light of a certain wavelength. At the PSS, depending on the system, the photoisomers will be present in a certain ratio, which we will refer to as photostationary state distribution (PSD; N.B., there is as of yet no consistency in the literature regarding this term). The photoisomerization quantum yield (QY) Φ quantifies the number of molecules of product formed per number of photons absorbed and hence, is an indication on how efficient the switch undergoes a photoisomerization process.107 Practically, fast photoisomerization processes are observed when both Φ and the molar attenuation coefficient, ε(λ) are high at the wavelength of excitation.107
Upon irradiation, the chromophore enters an excited state from where multiple relaxation pathways can take place, as shown in Fig. 10A, among them most importantly isomerization to the metastable isomer (Fig. 10B). The thermodynamically stable photoswitch is promoted into the first or second singlet excited state, then vibrationally relaxes, and eventually funnels through a region where a conical intersection resides, back to the ground state of another isomer, thereby undergoing an isomerization (Fig. 10B and C). The newly formed photoisomer is higher in energy than the original one and hence, it is metastable and isomerizes back to the stable state in a first order process with rate constant k. For convenience, this process is further characterized by a thermal lifetime τ (defined as 1/k) or a half-life t1/2 (describing the time in which 50% of the molecules in the metastable state isomerize back to their thermodynamically stable form) (Fig. 10D). If the thermal activation barrier between the metastable and the stable isomer is relatively low, a significant amount of thermal back isomerization is observed at ambient conditions, rendering the photoswitch a T-type one.12,106 In the case of higher thermal activation barriers, the two photoisomers are considered bi-stable and isomerization is practically triggered exclusively using photochemical processes (P-type photoswitch).12,106,108
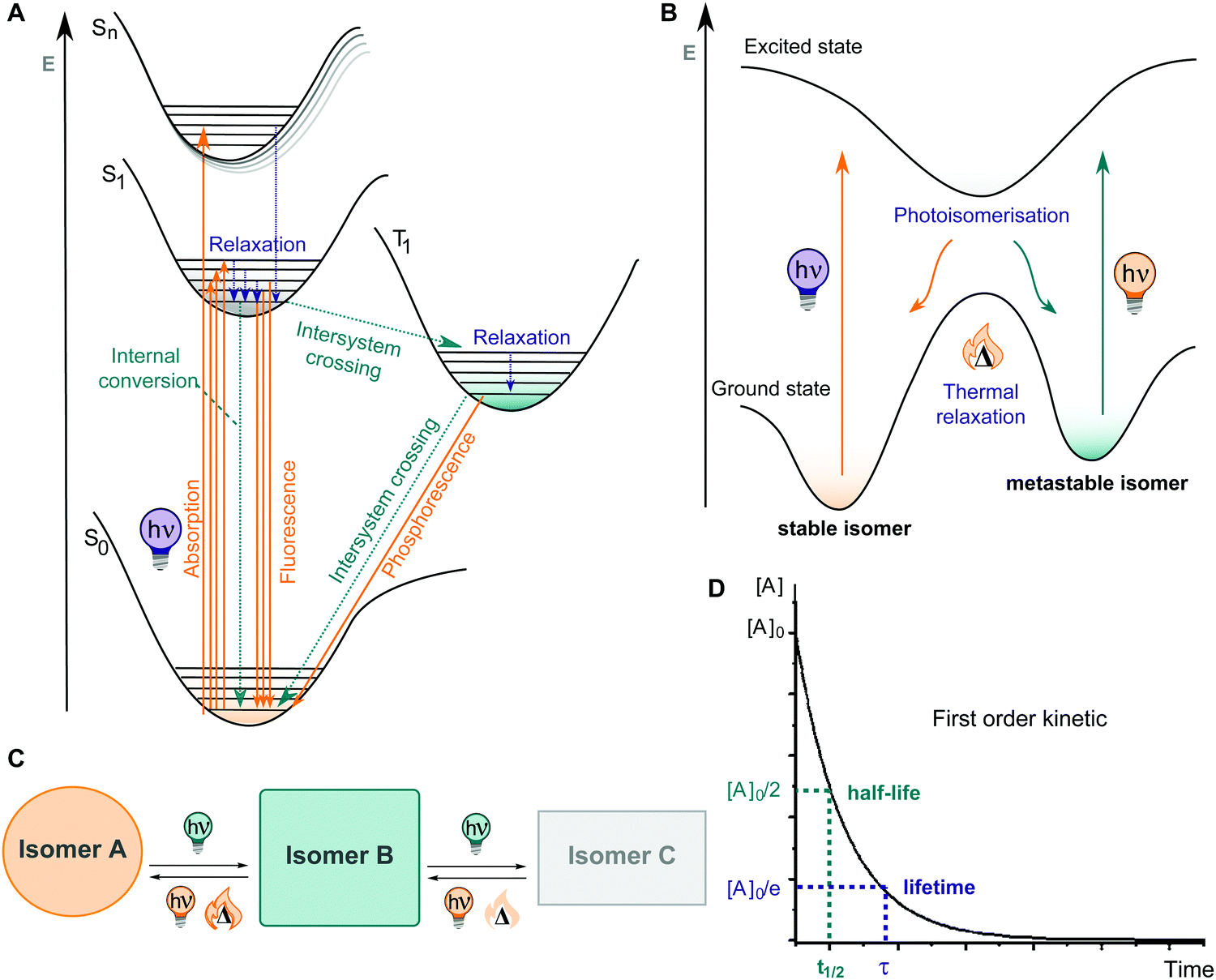 | ||
| Fig. 10 (A) The Jablonski diagram highlighting the key photophysical processes possible from excited state of a photoswitch (photochemical processes are described in C); (B) schematic representation of potential energy surfaces during isomerization of a photoswitch; (C) general scheme depicting isomerization of a photoswitch upon light irradiation. Most photoswitches reversibly isomerize from A to B with application of light and heat, while in some instances isomer B can further react to form a third isomer C; (D) graph (concentration of A vs. time) depicting the first order kinetics of photoswitching with highlighted half-life and lifetime values. | ||
In terms of application, the experimentalist ideally chooses a specific photoswitch for a certain purpose, most often based on the type of properties that are altered upon photoisomerization, and then incorporates the switch into the system of choice. However, in practice, this is not as straightforward, since the system's own characteristics will influence the properties of the switch. This delicate interplay is particularly challenging when embedding a photoswitchable molecule into a functional system in an aqueous environment. Consequently, in this section, we will introduce various kinds of photoswitches, organized according to their predominant photoisomerization mechanism, and discuss the influence of polar protic solvents, and especially that of water, on the photophysical and photochemical properties of the switch.
3.2 Photoswitches relying on the isomerization about double-bonds
Suitably substituted double bonds X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Y can be isomerized between an E and a Z form by light. They are frequently used as photoswitchable core motifs, as they are relatively small and hence, easy to incorporate into a larger molecule while introducing only small structural changes. Moreover, their effect can be easily predicted in each system due to their large geometrical changes accompanied by photoisomerization. These XY functions are often decorated with arene moieties, or in conjugation with such, to extend the π-system of the double bond and to make them responsive to light typically between 300 and 400 nm. Depending on the nature of X, Y and the substituents on the double bond, the absorption spectrum of the switch can be tuned up to the visible and near-infrared (NIR) region of the electromagnetic spectrum (vide infra). In the following, we will discuss various types of photoswitches, which isomerize about a CC, CN, or NN double bond, highlighting both frequently used and more niche structures and the effect of polar protic solvents on their characteristics. An overview of various classes of photoswitches is displayed in Fig. 11.
Y can be isomerized between an E and a Z form by light. They are frequently used as photoswitchable core motifs, as they are relatively small and hence, easy to incorporate into a larger molecule while introducing only small structural changes. Moreover, their effect can be easily predicted in each system due to their large geometrical changes accompanied by photoisomerization. These XY functions are often decorated with arene moieties, or in conjugation with such, to extend the π-system of the double bond and to make them responsive to light typically between 300 and 400 nm. Depending on the nature of X, Y and the substituents on the double bond, the absorption spectrum of the switch can be tuned up to the visible and near-infrared (NIR) region of the electromagnetic spectrum (vide infra). In the following, we will discuss various types of photoswitches, which isomerize about a CC, CN, or NN double bond, highlighting both frequently used and more niche structures and the effect of polar protic solvents on their characteristics. An overview of various classes of photoswitches is displayed in Fig. 11.
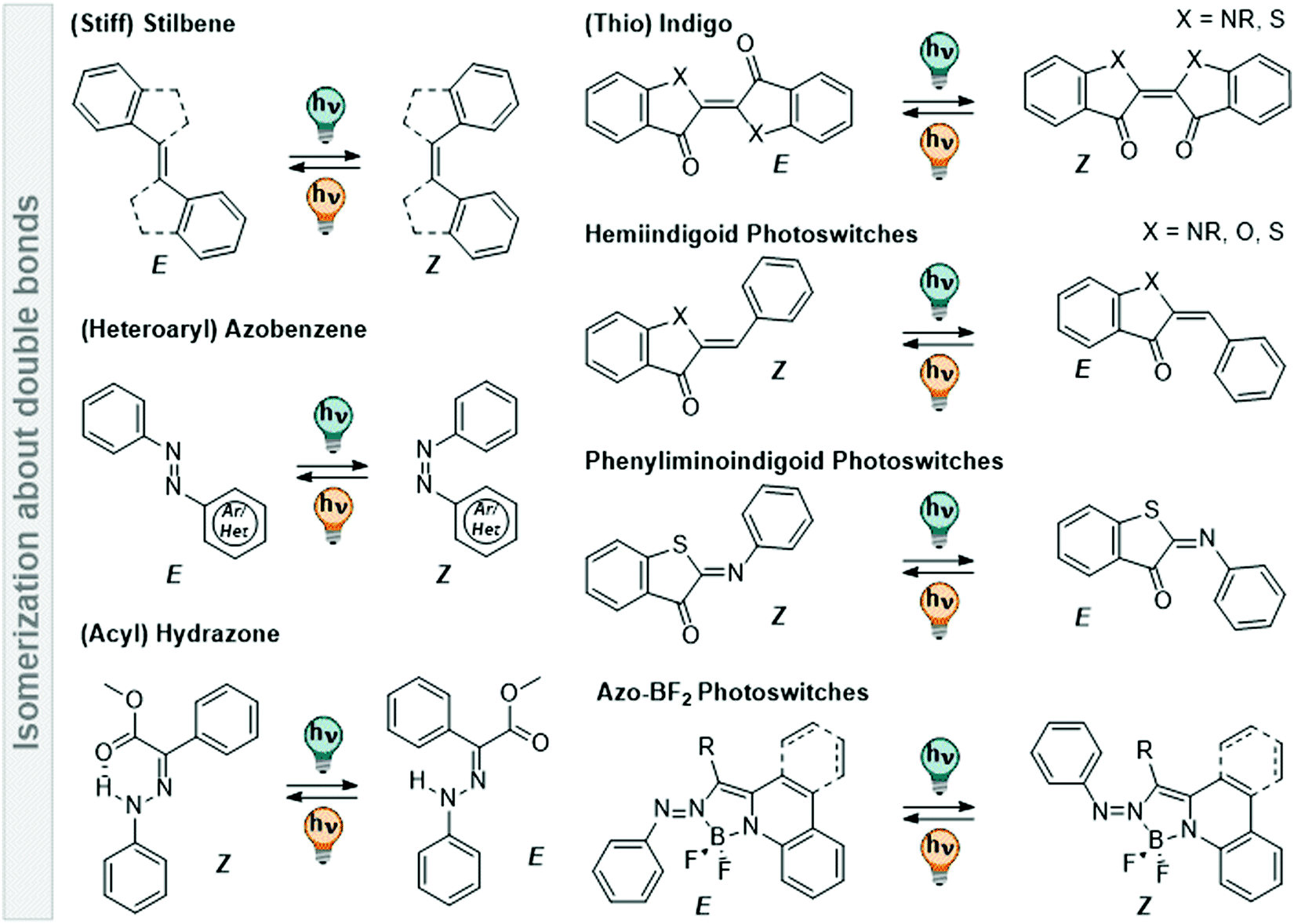 | ||
| Fig. 11 Overview of various photoswitches based on double-bond isomerization (CC, NN, and CN) that are discussed in Section 3.2. | ||
CH–Ar. They exist in a thermodynamically stable E and a photochemically generated Z form (cf.Fig. 12).109 The E isomer is, depending on its substituents, quasi-planar and has its absorption maximum at ca. 290 nm with a strong vibrionic coupling.110 Irradiation at the absorption maximum promotes the E isomer, via a π–π* transition, into its excited state, from which it relaxes to the Z form with high photoisomerization QYs (ΦE→Z is 0.40–0.60 for most (stiff) stilbene derivatives in apolar solvents).111 The Z isomer can be accumulated with PSDs around 6:4 = Z:E in cyclohexane at the PSS313nm.112 The absorption spectrum of the Z form is bathochromically shifted and smoothened with respect to the E isomer. It exhibits a twisted, helical conformation and is considered to be the metastable isomer. In contrast to other photoswitches isomerizing about double bonds, like azobenzene or hydrazones (vide infra), the metastable isomer in (stiff) stilbenes is in most cases thermodynamically stable at ambient conditions and can be isolated by simple column chromatography.109 Irradiation of the Z stilbene induces photochemical Z → E isomerization with ΦE→Z between 0.3–0.45.111
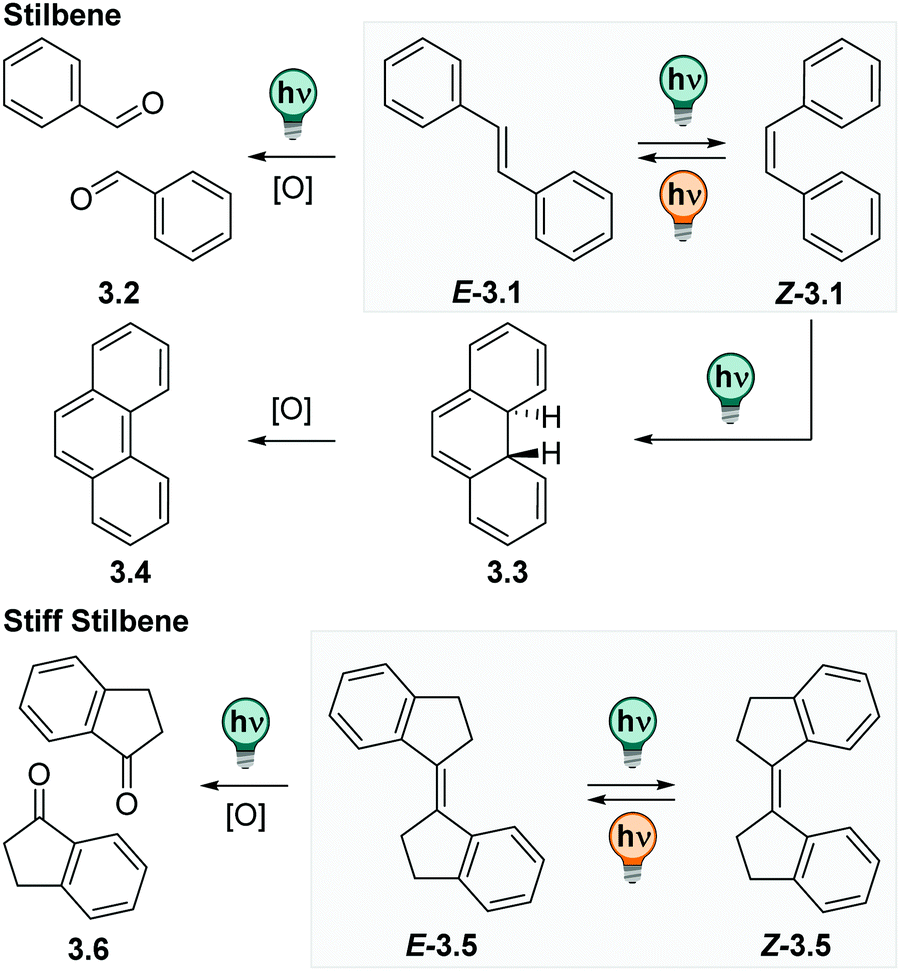 | ||
| Fig. 12 Photochemical side-reactions of stilbenes. Top: E–Z photoisomerization of stilbene and the competing photo-oxidation and electrocyclic ring-closure reaction followed by irreversible oxidation. Bottom: E–Z photoisomerization in 1,1′-bis-indanyliden (stiff stilbene) and the competing photo-oxidation. | ||
The (photo)chemistry of 1,1′-bis-indanylidens (stiff stilbenes) is analogous, yet more simplified, compared to parent stilbenes as the phenyl twisting is hindered.112,113 Originally, stiff stilbenes have been frequently used as a model system to study the parent stilbene molecule. Stiff stilbenes turned out to be photochemically more robust than stilbene itself.109,111 While the Z isomer of the parent stilbenes can undergo an electrocyclic ring-closure reaction to dihydrophenanthrene, which is then irreversibly oxidized to phenanthrene and hence lost for E–Z photoisomerization (cf.Fig. 12), this pathway is not accessible for stiff stilbenes.111,114 However, photo-induced oxidation reactions are observed for some derivatives under prolonged irradiation times (cf.Fig. 12).115,116 As a consequence, many studies are performed under inert atmosphere.110,112,117,118 This allows for E–Z photoisomerization that can be repeated over several cycles showing low fatigue independent from the solvent,117,119–122 but might be a limitation in practical applications. The absorption maximum of stiff stilbenes is between 320 and 340 nm, depending on the ring-size110 and can be shifted up to ca. 360 nm with suitable substituents.123–126 As in parent stilbenes, both photoisomers of stiff stilbenes are fluorescent with low fluorescent QYs and have been subject of a large number of theoretical and photophysical studies, however, mainly in apolar solvents.110,112,127–130 Moreover, the E–Z isomerization rate in stilbene is solvent dependent in both directions and, by trend, lower in solvents of higher viscosity or lower polarity. More importantly, it is affected by substituents, especially by those creating a dipole moment or push–pull systems.111
In last two decades, the stiff stilbene photoswitches are employed as the core motifs in molecular rotary motors (vide infra) and in a range of emerging applications,109 such as in photo-responsive supramolecular architectures,117,121,131,132 host–guest systems,119,120,122,133–135 catalysis,136 and in the context of mechanochemistry.137–140 Their properties are investigated in a wide range of solvents like chloroform,133 DCM,134 benzene,132 DMSO,120,122 MeCN,119 and MeOH.123 However, only few studies have focused on the properties of (stiff) stilbenes in water or aqueous buffers, as the 1,1′-bis-indanyliden is inherently insoluble in aqueous media.125 This is underlined by a study showing that addition of water to a solution of 5,5′,6,6′-tetrafluoro stiff stilbene in THF led to aggregation of the chromophore, which could be monitored by aggregation-induced fluorescence resulting in an increase of fluorescence QY by three orders of magnitude.125 However, recently, a few stiff stilbene derivatives were equipped with solubilizing groups and characterized in DMSO–water mixtures, buffer solutions and even in vivo.116,141–143 The compounds were reversibly switchable over several cycles,141 however, photo-induced oxidation was observed in some cases (cf.Fig. 12).116 Alternatively, heteroaromatic stilbene derivatives attracted very recently attention as alternative for classical stilbenes or azobenzenes in biological context. Specifically, styrylbenzothiazole showed promising properties in buffer and in cell studies.144
In summary, (stiff) stilbenes demonstrated their potential as water-soluble photoswitches in pioneering studies, but were to the best of our knowledge never part of a systematic study in this context. Hence, further investigations are required to reliably predict the effect of aqueous media on all photochemical properties of (stiff) stilbenes.
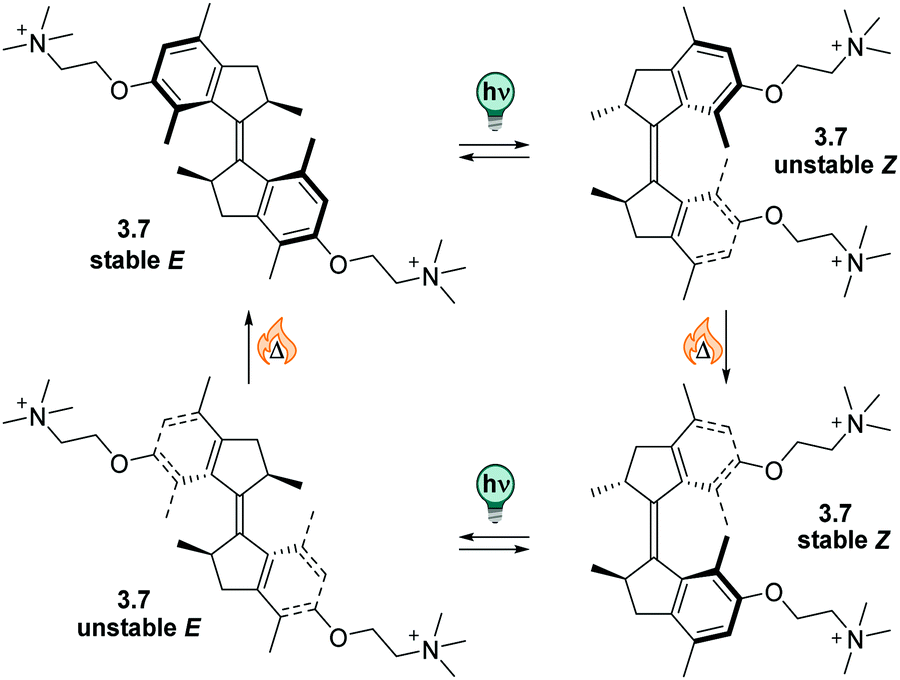 | ||
| Fig. 13 A stiff stilbene-based first generation Feringa molecular motor was equipped with alkylammonium group to promote water-solubility. The unidirectional rotation of the motor relies on photochemical E–Z isomerization (irradiation with 312 nm light) and thermal helix inversion (THI, here, five days at ambient temperature in water).152 | ||
The unfunctionalized motor molecules are very lipophilic and cannot be brought into an aqueous environment, unless dissolved in micelles.152 However, after introduction of suitable substituents, the compounds can be investigated in water and buffer (as in the example shown in Fig. 13).152–154 So far, there is only one study focusing on the photochemical and photophysical properties of fully water-soluble molecular motors.152 Upon irradiation with light of a suitable wavelength (ca. 310 nm for first generation motors and 365 nm for second generation motors), water-soluble molecular motors can be photoisomerized in aqueous media showing high PSDs and only little fatigue, similarly to their performance in organic solvents.152,153 However, in a systematic study on a second-generation molecular motor, the THI was found to be strongly solvent-dependent.155 In particular, in alkane solvents, the t1/2 of the THI increased with increasing chain length. More polar solvents of the same size also gave longer half-lives, as did alcohols with increasing chain lengths.155 A similar molecular motor dissolved in micelles in water showed a shorter half-life than in short alkanes, while the analogous motor functionalized with water-soluble groups gave a 10-times longer half-live of the metastable state when dissolved in buffer.152
In conclusion, molecular motors can be readily applied in aqueous environment and have been demonstrated to serve as fully water-soluble switches at low concentrations.152 Similar as (stiff) stilbenes, they are insufficiently studied in water and more research is needed to realize their full potential.
N–Ar, where Ar can be an aryl ring or a heteroaryl unit.156–158 They became frequently used photoswitches and can be found in a diverse range of applications due to their small size, reliable photochemical behaviour, and the versatility of methods available to synthesize and functionalize them.156–160 As a result, (heteroaryl)azobenzenes are quite well understood and were the subject of extensive fundamental studies. Like with the previously described stilbenes, the large geometrical change that azobenzenes undergo upon double bond isomerization significantly alters their chemical and photochemical properties.3,156–158 In comparison to stilbene, the absorption maximum of its diazo analogue is bathochromically shifted to around 330 nm in parent azobenzene.111,158 This allows operating the system with UV-A instead of UV-B light, which is less harmful and enables more selective addressability with respect to other chromophores present in the system.156–158 Hence, it became a popular approach to turn any stilbene-based system into the corresponding azo-analogue, a design principle termed azologization particularly in the context of photopharmacology.9
Classical azobenzenes exist in a thermodynamically stable E form, which is planar and apolar, and undergo E → Z isomerization after irradiation with light of a suitable wavelength (Fig. 14). The photochemically generated, metastable Z form has a helical conformation and a dipole moment of around 3D and can be converted back into the E form upon irradiation or in a first-order thermal process.156–158 The higher polarity of the Z isomer influences the solubility of the unsubstituted azobenzene in polar (protic) solvents, such as water. While the E isomer has more propensity towards π-interactions with other azobenzene molecules due to its flat conformation, the Z isomer is less able to interact in the same way with other extended arenes.156,161 Furthermore, its strong dipole moment enables the molecules to interact better with a polar environment and they show higher intrinsic solubility (Fig. 14). Specifically, a study measuring the solubility of the E and Z forms of several azobenzene derivatives showed that the Z isomer is 6–40 times more soluble in water than the E isomer. This behaviour is also seen for semi fluorinated azobenzenes and peptide conjugates.161 The different preferences in intermolecular interactions between the two photoisomers have been employed in the construction and the dynamic control of nanoarchitectures,162,163 amphiphiles,19,20 and photopharmacological agents.164
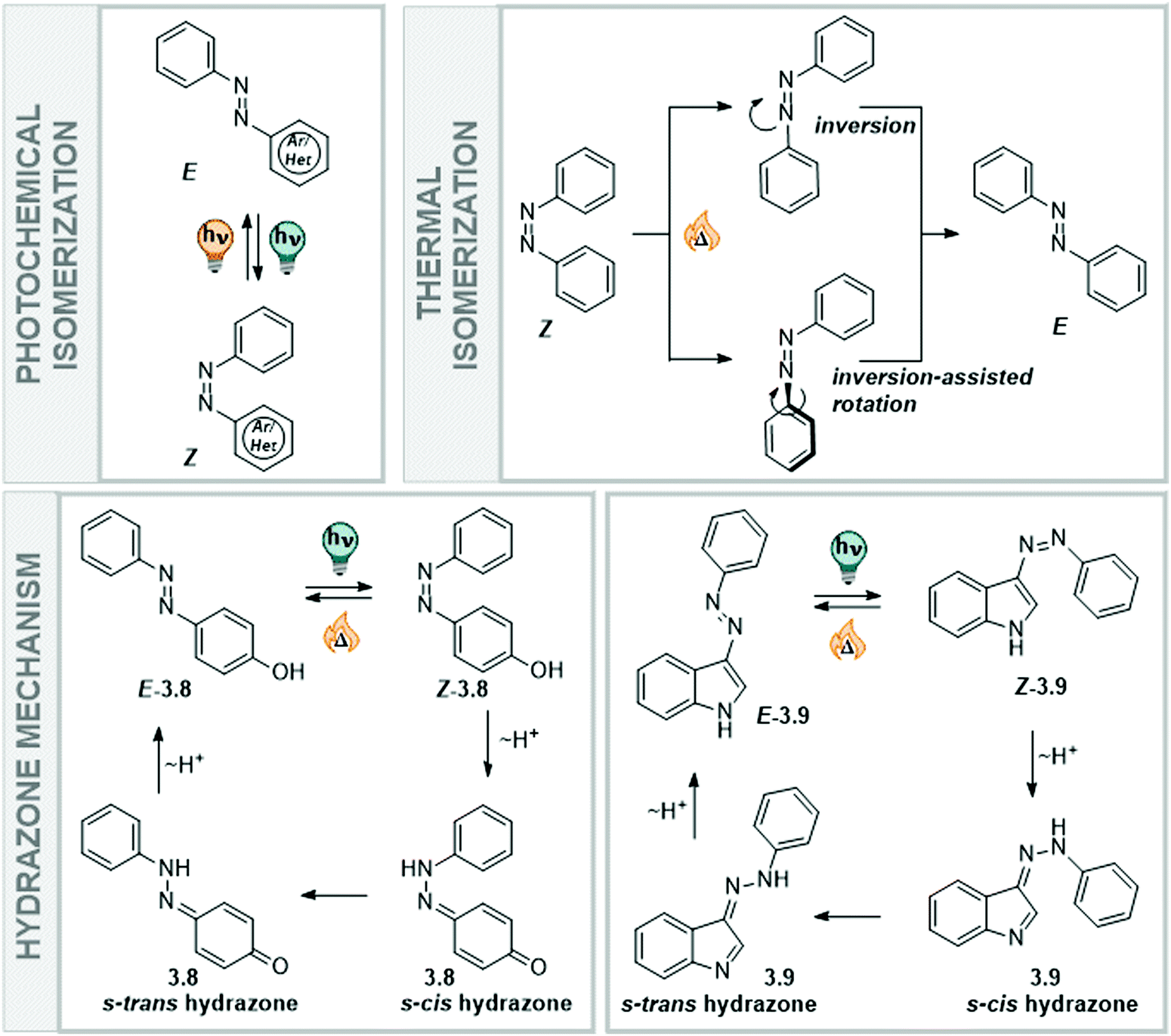 | ||
| Fig. 14 Overview over photochemical and thermal isomerization pathways in azobenzenes. | ||
Heteroaryl azoswitches do not show this behavior to the same extent, as also the E isomer is polarized even in unsubstituted derivatives due to the presence of the heteroatom(s).157,158 As in classical azobenzenes, the E form is planar and the Z is twisted, adopting either a helical conformation in the case of 6-membered rings, and a T-shaped one if a 5-membered heteroaryl ring is directly attached to the diazo bond.157,158,165 Moreover, the photophysical properties are influenced by the presence of the hetero atoms, rendering azoswitches based on pyridine,166–170 indole,171–174 indazole,175 purine,176,177 pyrimidine,178,179 pyrrole,165 imidazole,165,180–182 pyrazole,46,165,183–188 oxazole,189 thiazole,180 thiophene,190–192etc., generally responsive to light of longer wavelength. In contrast, in classical azobenzenes the UV-Vis absorption spectrum can be tuned by introducing, for instance, (push–pull) substituents in the para position156,193,194 or tetra-ortho substituents195–205 and thereby influencing the position of either the π–π* transition,199,205 the n–π* transition,195–197 or both.156,201
Also, in heteroaryl azobenzenes, the E → Z isomerization is triggered by light, while the back-isomerization can occur photochemically and thermally. Both homo and heteroaryl azoswitches show little-to-no fatigue over many switching cycles in different solvents.156–158 However, the NN double bond in parent azobenzenes carrying electron-poor substituents has been shown to be sensitive to reduction, an important parameter to be considered during synthesis and in applications.206,207 In the context of water-soluble photoswitches, this aspect is of greatest importance in biological systems, as the reducing agent glutathione is present in the cell.6,30 Similar observation have also been made for some heteroaromatic analogues, like purines177 and thiazoles,180 while arylazopyrazoles appear to be less sensitive to these conditions.157,185
Moreover, the nature of the solvent has an influence on the thermal stability of the metastable form, as it stabilizes or destabilizes the transition state geometry of the reisomerization reaction. The thermal Z → E isomerization is a ground state process, and proceeds via an inversion transition state, an inversion-assisted rotation transition state, or follows a mechanism with a hydrazone intermediate (Fig. 14). The pathway the switch eventually employs depends on the type of the azoswitch, its substitution pattern, and the solvent in which the isomerization proceeds, and has been a subject of several studies with both homo and heteroaryl azobenzenes.156–158 In general, push–pull substituted azobenzenes isomerize thermally via the faster inversion-assisted rotation transition state pathway, yielding shorter lifetimes. In polar solvents, the push–pull effect is more prominent than in apolar systems, significantly affecting the thermal lifetimes of azobenzenes isomerizing via this pathway, when moving to aqueous environment.156,194 In contrast, azoswitches isomerizing via an inversion transition state show longer lifetimes in polar solvents.208 Moreover, azobenzenes bearing a –OH, –SH, or –NHR as part of the heteroarene or as a substituent, have access to an alternative thermal relaxation pathway in polar protic solvents. The acidic proton can be tautomerized onto the NN double bond of the azo-function to result in a single bond between the nitrogens about which one of the former aryl ring can easily rotate to eventually result in the energetically more stable E isomer (Fig. 14), which leads to extremely shortened lifetimes.171–173 Proton transfer in the hydrazone mechanism is solvent-assisted and hence, favoured in protic or aqueous media, if the energy gained by accessing this pathway compensates for the loss of aromaticity.172 However, recent computational and experimental work on NH-containing hetero azoswitches shows that potentially acidic moieties only at specific positions will result in shorter lifetimes in water or other protic media. If the “breaking” of aromaticity is not compensated by the low-energy single bond rotation of the hydrazone formed, the switches will isomerize via inversion or rotation. Consequently, they show stable Z isomers in polar protic solvents, such as alcohols, water, buffer or even cell medium.172,183 The same behaviour was shown earlier for substituted parent azobenzenes, comparing the thermal half-lives of the Z isomers of 2-, 3-, and 4-hydroxyazobenzene.209 While the Z isomers of ortho- and para-OH azobenzene rapidly isomerized through tautomerism back to the thermodynamically favoured E form, the meta-substituted analogue behaved like the parent azobenzene.209
Not only the rate constant of the thermal Z → E reaction is affected when moving into aqueous environment, but also the photoisomerization QYs are solvent-dependent. For parent azobenzene, ΦE→Z is higher in alcohols and water than in apolar solvents if the isomerization is triggered via the S1(n–π*) and S2(π–π*), while ΦZ→E (n–π*) is in general lower.156 However, this trend in behaviour can change with different substituent patters and scaffolds.156–158 As studying the fundamental properties of the switch used is often out of the scope in application studies, a comprehensive overview is missing. This makes it not trivial to predict the properties of a photoactuator when moving between different solvent systems. Specifically, using an azobenzene, which was thoroughly studied in a non-polar solvent, in an aqueous environment will not necessarily result in the same behaviour. As apolar solvents are frequently used to study azoswitches, this gap of understanding hampers the rational design of water-soluble azobenzenes.
The strain that the E isomer experiences results in t1/2 of a few hours in the original CH2–CH2-bridged diazocines.210 More recently, CH2–X bridged diazocines were investigated, with X being O, S, or NR (R = Me, Et, H, Ac, cf.Fig. 15), and were found to feature bathochromically shifted UV-Vis absorption spectra, ranging up to the NIR.211,217 The thermal half-life in CH2–O derivatives in in the seconds range, while CH2–S analogous are thermally stable over a few days.211 Also, the influence of different substituents218 and solvents on the switching properties has been investigated.211,219 A recent study showed that, when moving from MeCN to pure water as a solvent, the absorption spectra of both photoisomers are affected with λmax shifting by 10–20 nm in opposite directions, thereby decreasing the band separation. While t1/2 is in some cases more than doubled in aqueous medium, the PSDs are increasingly lower with higher amounts of water added to MeCN. In the investigated series, the CH2–NAc stood out as its properties were relatively independent from the solvents and the compounds was well-soluble in pure water (up to 1.27 mM at ambient temperature).219
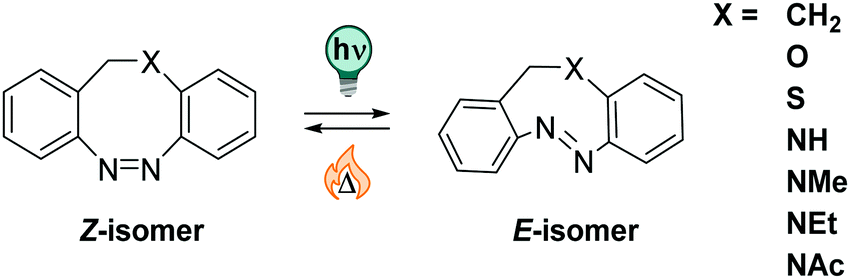 | ||
| Fig. 15 Thermally stable Z and photochemically generated E isomer in diazocines with various substituents X in the bridging unit tuning their properties. | ||
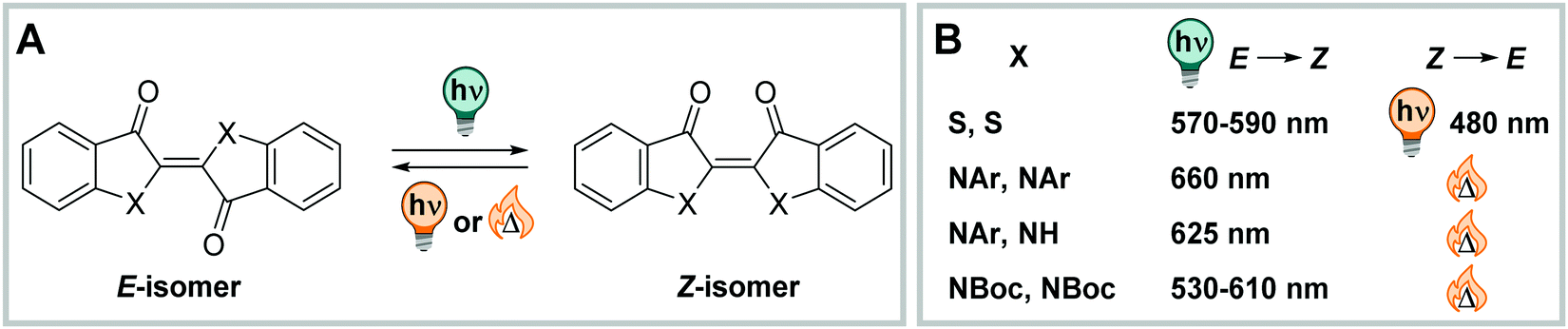 | ||
| Fig. 16 (Photo)switching in indigo photoswitches. (A) Thermally stable E and metastable Z isomer in (thio)indigo derivatives. (B) Different photochemical and thermodynamic properties of (thio)indigo switches depending on the substituent X. | ||
Both photoisomers are luminescent, the E weaker than the Z, and fluorescence QYs are significantly lower than in thioindigo, while photoisomerization QYs were comparable (vide infra).228 The Z isomers are often short-lived and could only be studied with pump–probe experiments.226,228 On the contrary, bis-Boc and (di-)arylated indigos showed prolonged thermal lifetimes of the photochemically generated Z-indigo, with t1/2 = 1–400 min (in MeCN at ambient temperature) in di-arylated and up to a few days in bis-Boc derivatives, depending on the substituents and solvent used. The half-life can be tuned by altering the substituent pattern and could be rationalized in a Hammett analysis for both compound groups.227,230 Alternatively, the thermal Z → E isomerization rate can be tuned by varying amounts of water in mono-arylated indigo. In particular, adding increasing amounts of water to a THF solution of a mono-arylated derivative deceases t1/2 from 1000 s to only 5 s, while the thermal half-life of the corresponding bis-arylated indigo derivate stays unaltered.231 In bis-Boc functionalized compounds, the half-life is significantly shortened in MeCN/water mixtures, compared to toluene or pure MeCN. Also, the QYs are lower in the presence of water for the E → Z but higher for the Z → E photo isomerization.227
Thioindigo (Fig. 16) is the sulphur-analogue of parent indigo and was studied in various contexts, for instance in semiconducting films,232 taking advantage of its redox switching233 and modulation of electro-optical properties.234 For this reason its (photophysical) characteristics and substitution effects were the subject of several experimental and theoretical studies.232,234–236 The negative photochromism237 of thioindigo was observed already around 80 years ago.221,238,239 The molecule could be switched in both direction with visible light showing good PSDs and QYs (PSDE→Z = 63%Z, PSDZ→E = 74%E for 6,6′-di-isopropoxythioindigo in epoxy resin; ΦE→Z = 0.20, ΦZ→E = 0.38 for 6,6′-di-isopropoxythioindigo in toluene).240 A limiting factor for the study of thioindigos is their “very low solubility in almost all conventional organic solvents and polymers”240 for which reason suitably substituted derivatives are usually applied.233,234,240 In this manner, it was recently possible to study the photochromic behaviour of these compounds in aqueous media.241,242 The compounds could be solubilized and investigated in water and buffer, however, addition of 20% MeCN was needed to aid solubilization and 1% TFA stabilized the Z isomer.241 Alternatively, buffer/acetone mixtures were used.242 Reversible photoswitching could be maintained and did not show any fatigue over several cycles. QYs in both directions were lower in pure water than with co-solvent (ΦZ→E = 0.05 and ΦE→Z = 0.19, and ΦZ→E = 0.21 and ΦE→Z = 0.41 with 80% MeCN in water), which was attributed to an aggregation tendency of the dye in the absence of organic (co)solvent.241 This observation agrees with the general solvent and concentration dependency of both QY and fluorescence observed in various thioindigo derivatives241,243 and is also in line with the thermal isomerization kinetics, which have a close-to-sigmoidal behaviour in pure water and a (normal) exponential one in 80% MeCN in water. The apparent half-life was around 120 min in both solvent (mixtures) at ambient temperature.241 By adding phenol substituents at suitable positions, the Z isomer could be further stabilized, and negligible thermal Z → E isomerization was observed over the course of several hours. However, very high amounts of co-solvent were needed to solubilize the molecules.242
In summary, both indigo and thioindigo derivatives can be reversibly photoisomerized in the presence of water and show robust and stable photoswitching properties with medium to good PSDs and QYs. Both scaffolds are very lipophilic, tend to aggregate in water and have to be solubilized with suitable substituents and sufficient amounts of co-solvents. The photophysical and (photo)chemical properties of both molecules are influenced by the presence of water, which results in many cases in higher thermal Z → E isomerization rates, lower QYs and lower PSDs.
C double bond as photoactive motif (Fig. 17).220 Also, from a photophysical perspective, HI and HTI show a compromise of the UV-Vis properties of their parent photoswitches, having their absorption maximum usually around 450 nm, while parent stilbene shows λmax around 290 nm and (thio)indigo between 550 nm and 650 nm (vide supra).230,241,244 The absorption maximum of the metastable Z isomer is typically bathochromically shifted with respect to the E and hence, their photochromic behaviour is closer to stilbene than to (thio)indigo, which shows negative photochromism.230,237,244 Both HI and HTI can be photoswitched in both directions using visible light.220,245
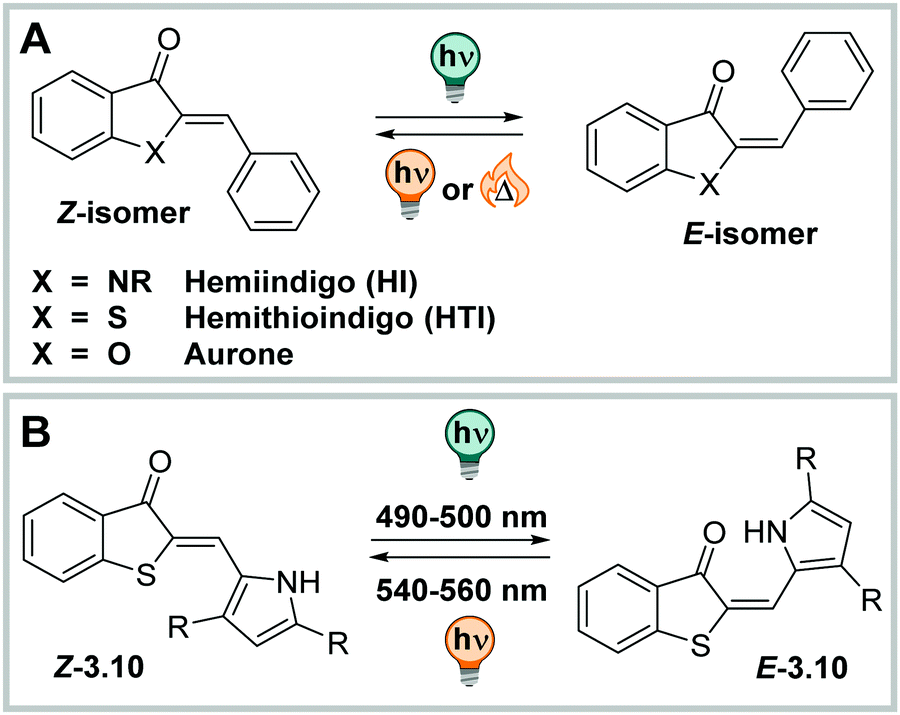 | ||
| Fig. 17 (A) Chemical structures of hemiindigo (HI), hemithioindigo (HTI), and aurone. (B) Photochemical E–Z isomerization of HTI 3.10. | ||
HI has its absorption maximum around 450 nm in the thermodynamically stable Z configuration,246 and it can be shifted up to ca. 530 nm by careful substituent choice and taking advantage of the solvent effects.220 The molecule exhibits good band separation and irradiation with blue lights results in formation of the metastable E isomer, which has λmax between 510 nm and 590 nm.245 HI shows little fatigue over many switching cycles,220 has moderate QYs (ΦZ→E = 0.16–0.23 and ΦE→Z = 0.02–0.11, depending on the derivative), high PSDs (>80%) for the Z → E in and shows close to quantitative conversion for the E → Z photoisomerization, independent from the solvent (studied in toluene, THF, and DMSO),245 in strong contrast to the HTI family which shows strong solvent dependency in photoswitching (vide infra).247–249 The thermal half-life of the metastable E isomer is in the range of several years at ambient conditions in various solvents. However, when the indolic nitrogen bears substituents other than H, the two photoisomers in HI, Z and E, become energetically very similar. Experimentally, heating the pure (photochemically generated) isomers results in a mixture of E and Z forms independent of the initial configuration.245,250 Also, this property is in strong contrast to most photoswitches isomerizing about double bonds, even comparing to the closely related HTI, aurone, or ITI (vide infra).220,251,252 Also, HI could be switched reversibly in mixtures of water and organic solvents (water/DMSO 9:1 or water/THF 8:2, with a drop of TEA to increase the switching performance) and showed good to very good photochromic properties.245 However, the authors noted that solubility at higher concentrations was an issue and more detailed analysis, e.g. by NMR spectroscopy, could not be performed. By attaching more efficiently solubilizing substituents,253–255 several HI derivatives could be investigated in pure water with no or little amounts of organic co-solvents (2% (v/v) EtOH or 10% (v/v) DMSO). The PSDZ→E was around 80%E and back-isomerization was close to quantitative. The photoisomerization QYs were lower than in organic solvents or aqueous solutions with higher contents of cosolvent (ΦZ→Eca. 0.03 and ΦE→Zca. 0.002).253 The E isomers were stable in solution and showed thermal lifetimes in the range of several days for many derivatives at ambient temperature. While low band separation and suppressed fluorescence was observed in water,253,255 isomer-specific binding to biomolecules could reinstall the emissive properties reversibly.255,256
HTI can be photoisomerized in various solvents with high PSDs in both directions (>80% photochemical generated isomer) in methanol as well as DCM. The thermal half-life t1/2 of the metastable E isomer is ca. 140 h in DCM and around 20 h in MeOH.257 HTIs show very fast E–Z photoisomerization kinetics as single compounds or when attached to macromolecules244,248 and the isomerization mechanism, solvent and substituent effects were studied in detail.258 Decoration with suitable substituents can accelerate the light-induced isomerization even more. When the thioindigo fragment is used as acceptor and the stilbene fragment, functionalized with electron-donating substituents in ortho and para-position, is used as a donor moiety, the photoisomerization occurs in the low ps-range. The photoisomerization QYs were strongly depended on the substituents with ΦZ→E = 0.14–0.23 and ΦE→Z = 0.05–0.33 in DCM.247 Moreover, HTI is fluorescent in aprotic solvents, but the fluorescence can be quenched by protic solvents as shown for some derivatives.246 Replacing the phenyl moiety with aryl-extended pyrroles results in an improved band separation and leads to quantitative photoisomerization in both directions (in DCM, see Fig. 17). H-Bonding of the pyrrole NH to the carbonyl moiety leads to a significant bathochromic shift of the compounds in both photoisomers.259 For instance, a bis-para-methoxy-phenyl substituted pyrrole moiety facilitated Z → E photoconversion with red light, yielding 80% of the E isomer (623 nm light to trigger photoisomerization in DCM).259,260 Also, a functionalized HTI, modified with multiple methoxy and hydroxy groups which contribute to a non-planar structure, could be investigated in the presence of water. Thereby, the PSDZ→E in 75% DMSO in PBS buffer contained 65% E, while at the PSSE→Z 97% Z was formed. Photoswitching was stable over several cycles and the metastable E form had a half-life of ca. 30 min at 37 °C in the DMSO-buffer mixture. The same compounds could also be photoisomerized in cellulo, however, this conclusion was made on the basis of altered biological activity and not derived from photophysical measurements.261 In a follow-up study, para-hydroxyphenyl moieties lead to very short thermal lifetimes of less than 1 min in DMSO and in the milli-second range in protic media,262 an effect often observed in azobenzenes, where it is attributed to a tautomerization mechanism that can, however, not occur in an analogous manner in unsubstituted HTI (vide supra).172,263 Several different HTI derivatives have successfully been employed as unnatural amino acids244 and were tested in cellulo.264
Moreover, HTIs were used to build more machine-like systems with several HTI units to generate complex motion,265,266 in information processing,267 or to function as photoresponsive receptors.268,269 The scaffold was further functionalized to construct unidirectional molecular motors,270–276 which behaviour is highly solvent-dependent249 and influenced by the presence of water.277
Aurones (Fig. 17) are synthetic and natural compounds belonging to the flavonoid family,278–280 mainly studied as highly tuneable fluorescent dyes251,281,282 and regarding their intrinsic biological activity.278,283,284 Alongside these studies, their photoswitching properties were investigated in chloroform and toluene. Z → E isomerization could be triggered with 400 nm light and was accompanied by formation of a bathochromically shifted band that was attributed to the metastable E isomer (PSDZ→E = 55–60%E). Consequently, they show similar photochromic behaviour as the structurally related hemi(thio)indigos.285,286 The half-life of the metastable isomer was around 40 min.286E → Z isomerization was induced through irradiation with 480 nm in CHCl3 to regenerate >95% of the Z isomer. Also, the fluorescence intensities in chloroform depended on the photoisomer present, with the E isomer (measured at the PSS, not isolated) showing weaker intensities.285 While the fluorescence, used to visualize cells, was recorded in Tris buffer in the presence of serum albumin and salmon sperm DNA,285 the photochromic properties of these compounds have to the best of our knowledge only been so far studied in organic solvents285,286 and in polymer films,251 but not in aqueous environment.
N double bond, which makes ITI more polar than the CC-containing analogue, HTI. As other indigoid photoswitches ((thio)indigo, HI, HTI and aurone),254 ITI is an all-visible photoswitch. It undergoes photochemical Z → E isomerization when irradiated with blue light (455 nm, PSDZ→Eca. 65% E).252 The process is accompanied by pronounced photochromism with the absorption maximum of the newly formed isomer being ca. 100 nm shifted compared to the thermodynamically favoured one. The metastable E isomer can be switched back photochemically by irradiation with orange light (595 nm) or relaxes thermally with lifetimes in the millisecond range via inversion about the CN nitrogen.252 Addition of acid lead to protonation of the CN bond and enforced an altered isomerization pathway simultaneously prolonging the thermal half-life of the switch to the second–minute range.287 The photochemical process in the neutral ITI can be triggered in a wide range of solvents, was also observed in buffer (1.7–6.7% DMSO for solubilization, depending on the substituents), and remained unaltered in the presence of the cellular reducing-agent glutathione.252
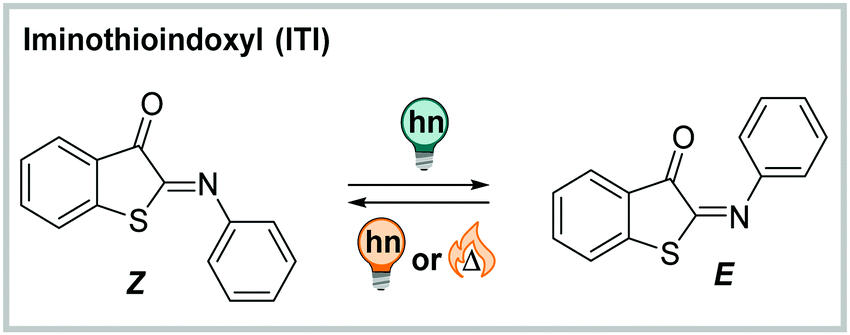 | ||
| Fig. 18 Thermally stable Z and metastable E isomer of Iminothioindoxyl (ITI). | ||
N–N motif.288–290 Isomerization about the CN double bond can be triggered by pH,291–295 metal ions296–298 or light.288,299 Their multi-responsive nature, together with their isomer-dependent emission properties, make (acyl)hydrazones interesting candidates for constructing more complex systems combing acid/base chemistry, metal-ion binding and photochemical processes as triggers.288,300–305 Thereby, the thermodynamically stable isomer in acyl (3.11 in Fig. 19) and phenylhydrazones (3.12–14 in Fig. 19) can have an E or Z absolute configuration, depending on the substituents.299 Photoirradiation induces E–Z isomerization to the metastable isomer, which in acylhydrazones quickly isomerizes back to the thermally stable form (3.11 in Fig. 19).288 However, if a suitable H-bond acceptor, such as a pyridine (3.12), or a carbonyl group (3.13), or both (3.14), are stabilizing the metastable isomer through H-bonding to the hydrazone hydrogen,306,307 bi-stable (photo)switches with very long thermal lifetimes (up to a few thousand years)308 in various solvents and the solid state can be obtained.308–311 Moreover, hydrazone-based photoswitches show good to excellent PSDs311,312 and fatigue resistance.311–313 Attachment of different substituents,311,312,314 as well as introduction of strain314,315 can tune the properties of the (acyl)hydrazones further and results in strongly bathochromically shifted311,312,314 switches, giving access to a broad variety of thermal lifetimes of the metastable isomer.315
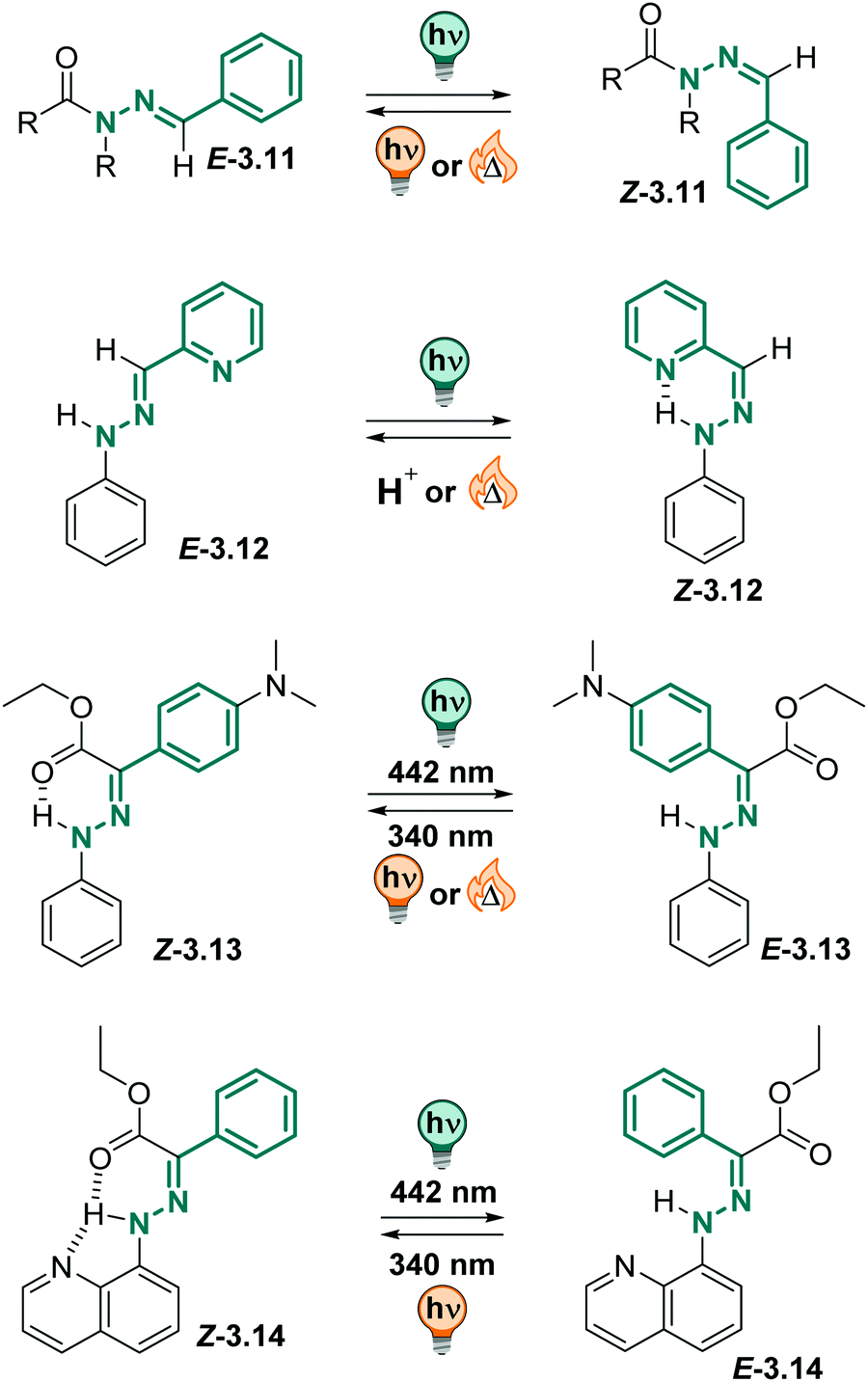 | ||
| Fig. 19 Overview of various (acyl)hydrazone derivatives and their photochemically and thermodynamic properties. | ||
Recently, compound 3.13 attracted attention, as it could be switched efficiently between both photoisomers with light, it showed strongly switchable fluorescence properties and could be excited both in 1- and 2-photon absorption processes.305,316,317 The metastable E isomer was stable over many years in toluene (t1/2 = 75 years). Importantly, the photochemical behaviour could be translated into the solid state and was also observed in different aqueous media.305,317 As in toluene and the solid state, the compound showed negative photochromism in the aqueous environment (λmax = 395 nm in the Z to 343 nm in the E) and good to excellent PSDs. The photoisomerization QYs were somewhat lower in serum than in toluene (ΦZ→E = 0.19 instead of 0.32, ΦE→Z = 0.07 instead of 0.15) while the fluorescence QY of the Z form slightly increased (ΦF = 0.035 instead of 0.007). The metastable E isomer showed good thermal stability, however, the exact thermal lifetime could not be assigned accurately in serum. The authors pointed out that they observed hydrolysis of the ester functional group at elevated temperatures which were required to study the thermal isomerization kinetics.305
Also, arylhydrazones can be used in stimuli-responsive crystals,318 are tuneable regarding their isomerization kinetics319 and can be employed in functional supramolecular systems.304,320,321 Similarly to phenylhydrazones, they could be successfully switched in water and buffer solutions, maintaining good photochemical properties.295,304,321,322 Hence, both acyl and phenylhydrazone switches can be readily employed in aqueous environment.305,322
:1 mixture) showing no fatigue after several switching cycles. However, the authors observed hydrolysis to the starting hydrazone when the compounds were kept in the solvent mixture for a few hours,324 potentially limiting the range of applications in aqueous environment.
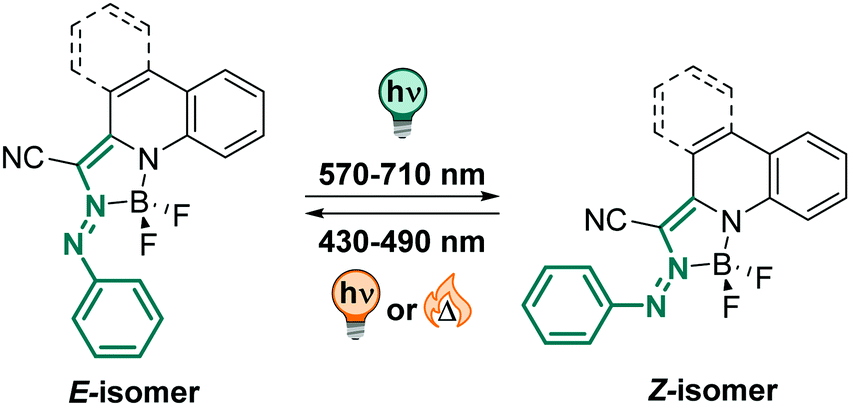 | ||
| Fig. 20 Thermodynamically stable E and metastable Z-isomer of an azo-BF2 switch. | ||
3.3 Isomerization based on photochemical cyclization reactions
A large group of photoswitches is based on light-induced electrocyclizations, which are pericyclic reactions and involve the formation or cleavage for a chemical bond (Fig. 21).3 They follow the Woodward–Hoffmann rules and can occur thermally, but usually require high temperatures (>100 °C), or proceed photochemically. Hence, the stereochemical outcome is determined by torquoselectivity, and the photochemically formed bond can typically only be opened by a light-stimulus and is thermally very stable. In the context of photoswitches, the 3,3′-electrocyclic ring-closure or opening reaction is the most frequently observed, and it constitutes the key process in diarylethenes, fulgides, fulgimides, and spiropyrans, etc.3,327–330 In this section, we only discuss molecules that relay purely on electrocyclization (see Fig. 21), while compounds isomerizing in mixed mechanisms are subject of the next section.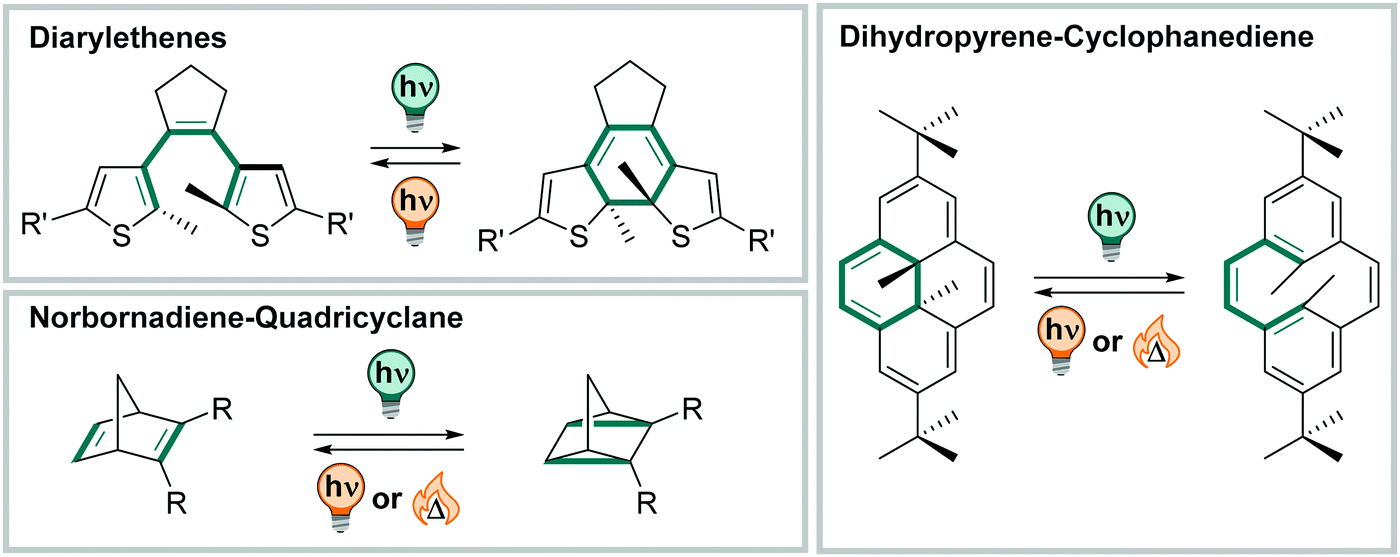 | ||
| Fig. 21 Overview of molecules discussed in Section 3.3. | ||
C bond is often part of a (perfluorinated) cyclopentadiene ring,332 but can also be fused to more complex structures, like maleimide (vide infra),333–335 imidazole,336 thiazole,332 phenanthroline,337 benzoquinone (vide infra),338,339etc. to tune the properties of the switch or enable additional function.333,340–342 The aryl rings are often (benzo)thiophenes, resulting in dithienylethenes (DTEs),3,328,331 as switching between the open and the closed form gives the best compromise between the loss of aromaticity and the gain in stabilization upon cyclization to result in bi-stable photoswitches. Furthermore, also other (hetero)aromatic systems such as pyridine,343,344 benzene,328 naphthalene,345 indole,333 (benzo)furan,328,331,344 pyrrole,328 oxazole,346 imidazole,347 and thiazole,340,348etc., have been employed.
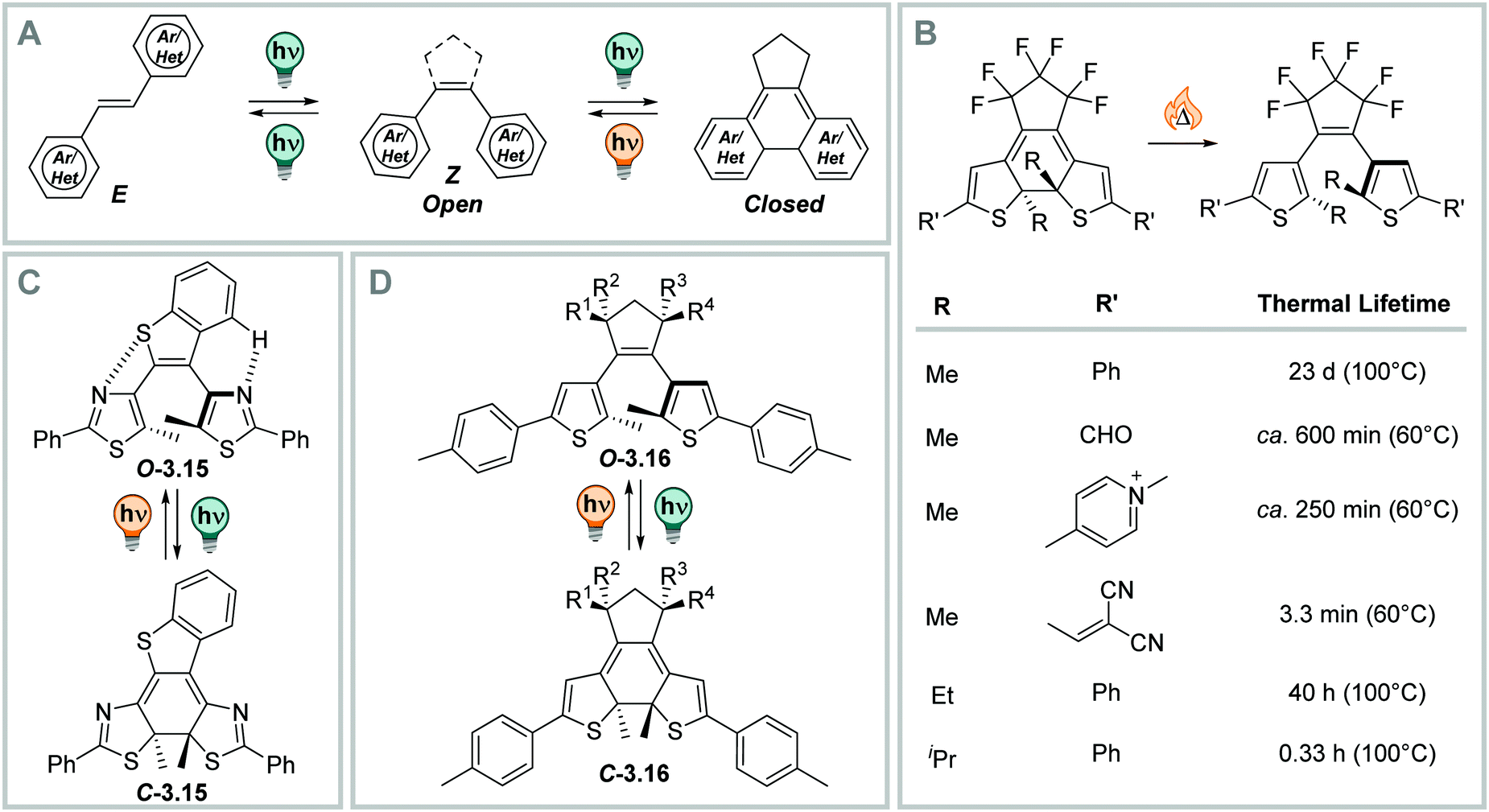 | ||
| Fig. 22 (Photo)switching in diarylethenes (DAEs). (A) Schematic representation of different photochemical mechanisms accessible to DAEs.328,331 (B) The influence of different substituents R on the thermal lifetime of the closed isomer.331,350 (C) DAE 3.15 shows close to quantitative photoisomerization quantum yields.351,352 (D) DTE 3.16 show close to quantitative PSDs in both directions and high, solvent independent ring-closure quantum yields.353 | ||
Upon irradiation of DAEs with UV light, a ring-closure reaction takes place. The flexible, less coloured open isomer is converted into the more rigid closed (C) form (Fig. 22B). The C form is strongly coloured due to the extended π-system.328,331 The absorption maximum in the visible range is between ca. 450 and 700 nm.349 The reverse ring-opening reaction can be triggered with a suitable source of visible light and usually proceeds quantitatively, as the resulting open form is not absorbing in the visible-range.328,331 The conrotatory ring-closure reaction makes a thermal ring-opening forbidden according to the Woodward–Hoffman rules, and results in thermally bi-stable photoswitches for many derivatives based on 5-membered heterocycles.350 On the other hand, benzene328 or pyridine-substituted343,344 diarylethenes feature thermally labile closed forms, as the difference in aromaticity between the O and C form in the 6-membered ring-systems is too large.328 This instability of the closed isomer overcomes the Woodward–Hoffmann rules and results in metastable C forms.350 Similarly, DTEs with electron-donating substituents show thermally induced C → O interconversion at elevated temperatures (illustrative examples are displayed in Fig. 22B).350
The properties of DTEs, such as QYs,351,353,354 PSDs,353 and wavelength of irradiation for O–C photo-interconversion355,356 were subject of extensive study and optimization. The characteristics and the efficiency of the O–C interconversion strongly depends on structural properties, such as the type of bridging moiety,333,335,338,357 the nature of the (hetero)aromatic ring or the substituents of the aryl rings358,359 or at the reactive carbon,354,360 and the influence of the used solvent.357 While the QY of the closing reaction ΦO→C is between 0.21 and 0.59 for differently substituted perfluoro cyclopentene bridged derivatives in n-hexane,331 and QYs up to 1.0 were measured for more optimized derivatives (Fig. 22C) in n-hexane351,352 and in the solid state,361 the QY for this reaction is generally lower in polar solvents (vide infra).351,357 QYs of the inverse reaction are, by trend, lower than the ones of the closing reaction of the same molecules, namely 0.03–0.47 in n-hexane,331,351 however, they can be tuned through introduction of substituents other than methyl in the 2-position of the heteroaryl ring, resulting in the C → O process becoming the more efficient one.354 On the other hand, sterically demanding substituent at this position influences the thermal lifetime of the closed form. The sterical strain makes it less stable and leads to thermal instability of the C-isomer (vide supra, Fig. 22B).362 Also, the PSDO→C strongly varies with different structural properties ranging between low344 to almost quantitative.353
Moreover, many derivatives of DAEs generally show a very high fatigue resistance over many switching cycles, i.e. large Z50 values.328,331,363 However, in thiophene-based diarylethenes, a photochemical by-product formation from the singlet excited state, which is competing with the O → C reaction, can take place, limiting its performance (Fig. 23A). This reaction is irreversible and hence, build-up of this component featuring a five-membered central ring can be observed after prolonged irradiation times with UV-light or repeated switching cycles (Fig. 23A).331,363 The fatigue resistance can be improved, for instance, by introducing steric hindrance (Fig. 23B),363–366 by attaching electron withdrawing substituents to the aryl-moieties (Fig. 23B),367–369 or by (intra- or inter-) molecular triplet sensitization (Fig. 23B).356,360
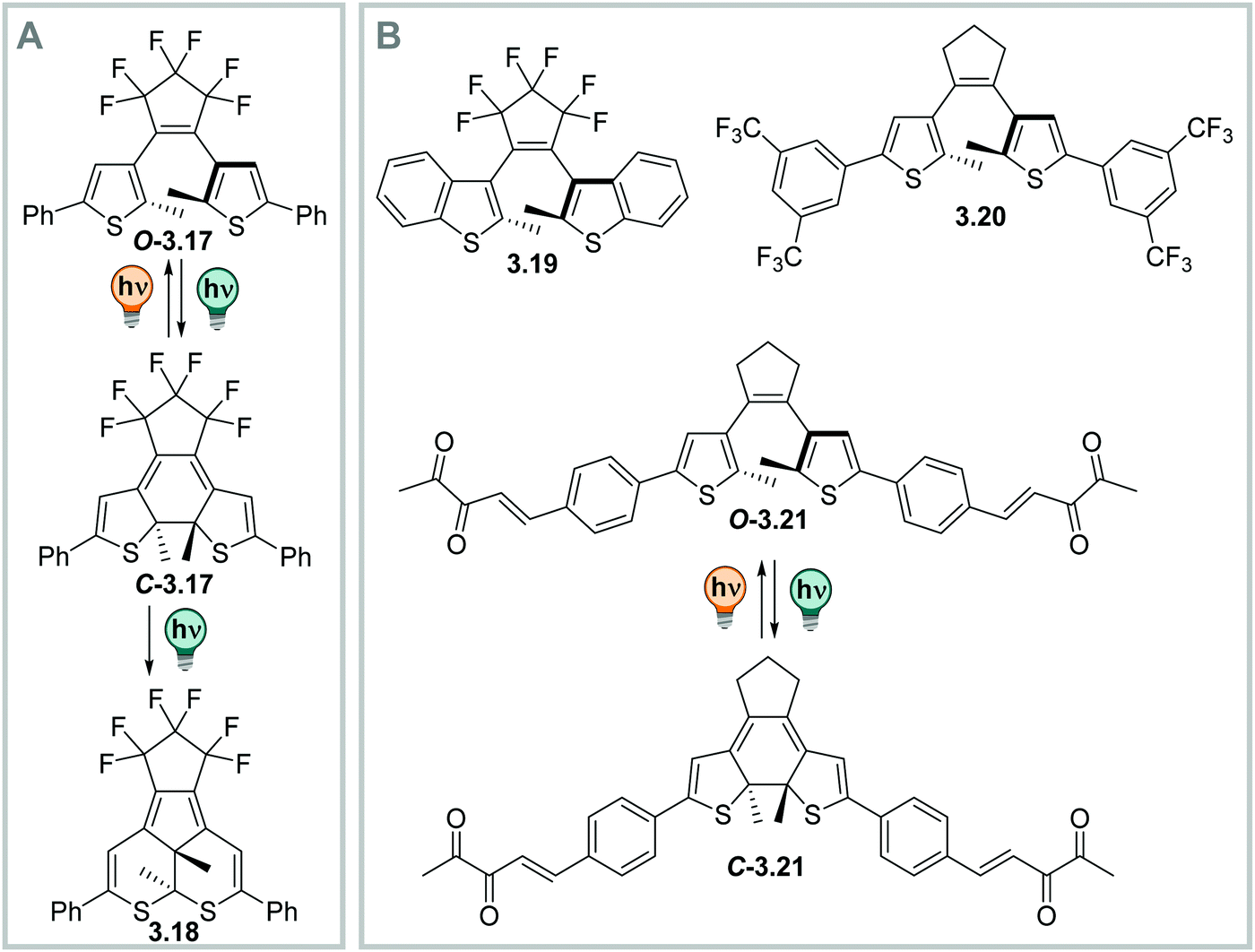 | ||
| Fig. 23 Tuning the photochemical stability of DAEs. (A) Photochemical side-product formation upon prolonged irradiation times with UV-light.331 (B) Scaffolds that prevent side-product formation: benzothiophene-based DAEs, e.g.3.19,331 DTEs with electron-poor substituents such as 3.20,367 and DTEs isomerizing upon excitation of an intramolecular triplet sensitizer like 3.21.356 | ||
DAEs with (perfluorinated) cyclopentadiene bridges can be switched reversibly over several cycles in the solid state331,361 and in a wide range of solvents, including water,358 aqueous buffer, and349,370 in living cells.371 However, by changing the bridging moiety to electron-acceptor moieties such as maleimide,333 maleic anhydride,357 or benzoquinone,338 the O → C reaction is suppressed in polar solvents and hence, cannot proceed in water and water/co-solvent mixtures (Fig. 24)333 Here, the predominantly occurring photochemical process is the population of a twisted intramolecular charge transfer (TICT) state.357,372 Thereby, the heteroaryl rings, such as (benzo)thiophene335,338,357,372 and indole,333 serve as donor and the bridging group functions as acceptor. This state is increasingly favoured with increasing polarity of the solvent and is mirrored by a decrease of ΦO→C in the same compound (0.13 in hexane and 0.003 in MeCN, respectively).357 On the other hand, the inverse C → O reaction can proceed in polar solvents quantitatively333,335,338,357 resulting in a visible-light responsive one-way switchable system with negative photochromism.333,335,338
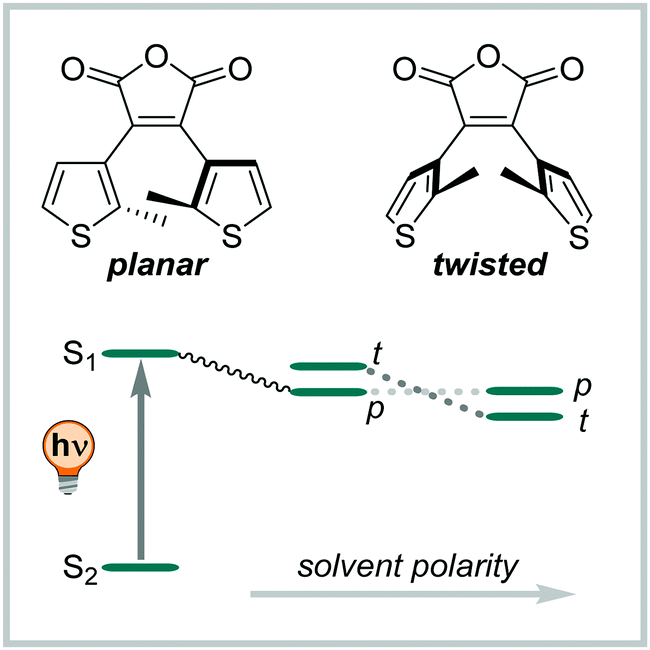 | ||
| Fig. 24 Schematic representation of the population of a twisted intramolecular charge transfer (TICT) state depending on the solvent polarity. After excitation to the S1 the compound relaxes preferably into a state where the thiophene rings have a planar (p) or twisted (t) conformation, depending on the solvent polarity.357 | ||
While photoswitches based on double-bond isomerization are often used by taking advantage of the geometrical changes between the photoisomers, switches isomerizing via an electrocyclic reaction can be used to construct systems providing both steric and electronic modulation. In the more flexible open form, the two aryl groups are orientated perpendicular to the ethene bridge. However, in the closed form, the conjugation in the π-system extends throughout the whole backbone of the switch (Fig. 25A). This altered π-conjugation is, on the one hand, responsible for the colour of the C isomer, and on the other hand allows for electronic communication between the two sides of the molecule.373,374 In this way, a photoswitchable pyridoxal 5′-phosphate mimic was constructed (Fig. 25A).375 Only in the closed form, the aldehyde function is conjugated to the pyridinium ring and hence becomes electrophilic enough for imine formation and catalysis. Both photoisomerization and catalysis were fully compatible with aqueous media (water:EtOH, 1:1). Alternatively, the DAE scaffold can be merged with an electronically active molecule, such as benzoquinone, and used as a multi-stimuli responsive switch or as photoswitchable oxidant (Fig. 25B).338,339 Here, the C isomer incorporates a part of the benzoquinone electrons in the conjugated backbone and hence, shifts its redox potential in a photo-dependent manner. The closed form is not available in redox reactions but can be activated by light and serve as oxidant with, for instance, Hantzsch ester or in isolated mitochondria demonstrating the compatibility of the concept with an aqueous environment. While the difference redox properties of the two isomers stayed intact, the O form could not be ring-closed in the same solvent due to the presence of a TICT (vide supra) and somewhat limits this system.338
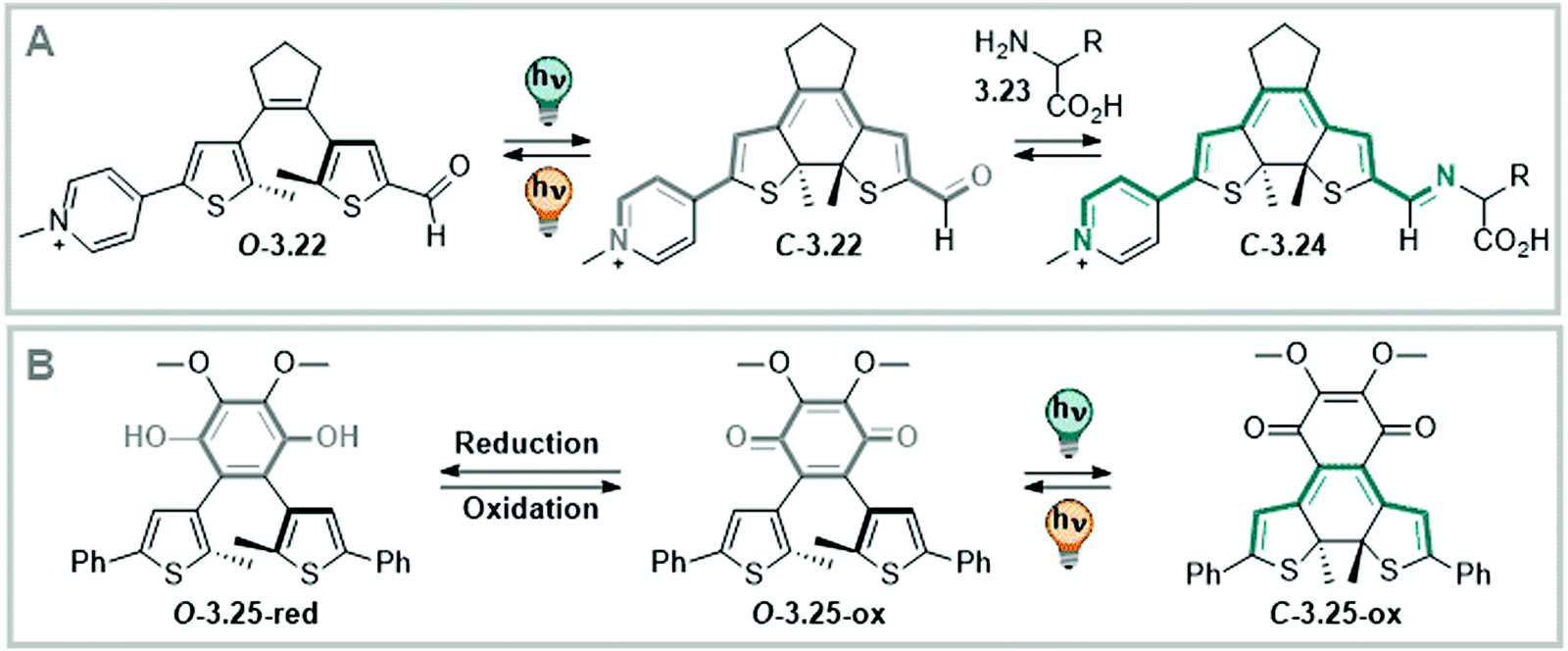 | ||
| Fig. 25 Photoswitching electronic properties in DTEs. (A) Switching between electronically insulated and connected isomers, where the closed form is able to serve as catalyst after imine formation.375 (B) A photoswitchable redox cofactor bearing a benzoquinone bridging unit that can be toggled between an oxidative (open) and non-oxidative form (closed).338 | ||
Another interesting feature in DAEs is that some derivatives exhibit switchable fluorescence, rendering them interesting candidates for molecular logic and smart materials.365,376 Moreover, switchable fluorescence can be employed for super-resolution in vivo imaging (see Section 4.3.3).358,371 Alternatively, DAEs can be used to construct systems for (internal) fluorescence quenching via FRET in one but not the other photoisomer when covalently linked to a suitable fluorophore.377
:2).379 The absorption spectrum of the DHP–CPD system can be further tuned by attaching substituents to the ethene bridge,380,381 the aryl moieties,381 by replacing the methyl groups by other functional groups,382 or by extending the conjugated π-system of the backbone.383 Specifically, attaching donor–acceptor substituents in conjugation over the whole switch structure leads to absorption bands tailing to 900 nm.381
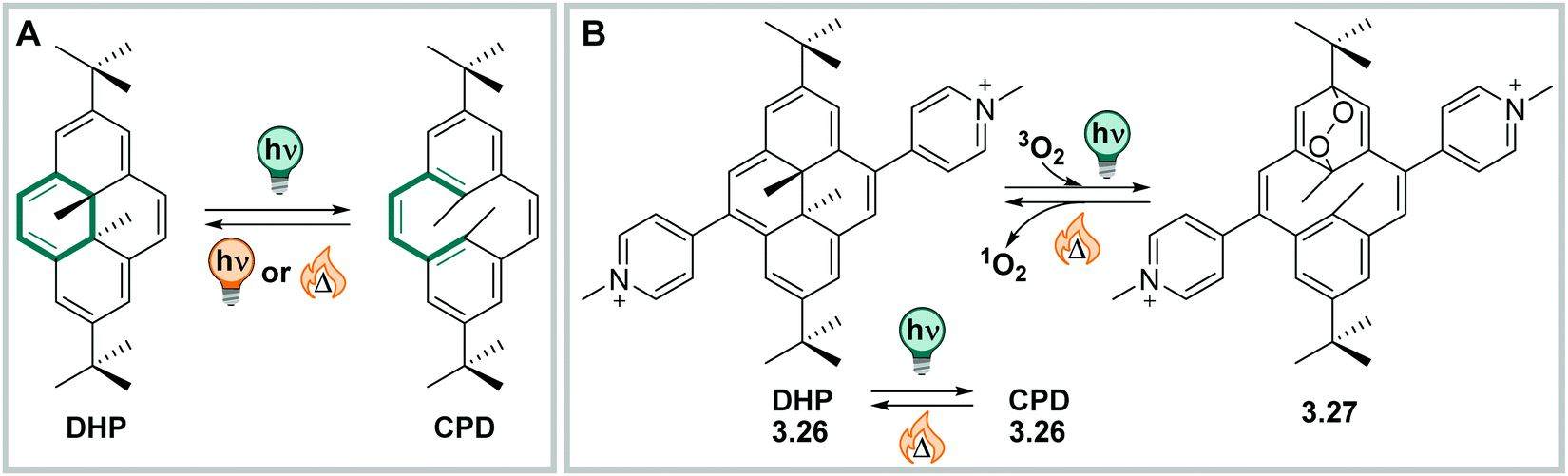 | ||
| Fig. 26 The dihydropyrene/cyclophanediene photochromic couple. (A) (Photo)switching between the two isomers. (B) Photoswitching in the presence of oxygen can lead to endoperoxide derivative 3.27, which then thermally releases 1O2. | ||
Moreover, irradiation under air generates an endoperoxide derivative, that thermally releases 1O2 while simultaneously forming the starting material without any degradation. This opens possibilities for the construction of multi-functional photoactuators and photodynamic therapy (PDT) agents.378 The switch has also been reported in the context of switchable redox properties,383 cooperative switching events in a homodimer,384 as light-responsive ligand in metalorganic complexes,385 and as ligand in biological systems (see Section 4.1).386 The last of these examples demonstrates the possibility to reversible switch between DHP and CPD under assay conditions, indicating the compatibility of the photoswitch with aqueous media. However, a study on a more diverse set of structural derivatives is required to correlate its properties with, for instance, the related DAEs and to understand the full potential and limitations of the DHP–CPD couple in various solvents.
The thermally stable norbornadiene is a T-type photoswitch that undergoes a photochemical [2+2] cyclization reaction upon irradiation with light, showing negative photochromism (Fig. 27).108 The formed quadricyclane absorption is strongly hypsochromically shifted compared to the starting material.387–389 The isomerization can be reverted photochemically,390 thermally, or (electro)chemically,391–393 for instance by adding a metal catalyst394,395 or acid,396 and (photo)switching is very robust over many cycles.388,397,398 Norbornadiene and quadricyclane are similar in size and volume and therefore do not offer many possibilities for applications where the photoswitch should induce large geometrical changes in a system.387 However, the photochemically generated quadricyclane is a small, light molecule and consists out of five highly strained rings, making the metastable isomer highly energetic and thus an excellent candidate for (solar) thermal energy storage that can be used in films or on surfaces.388,397,399–403 By attaching different substituents R (Fig. 27B), the absorption profile of norbornadiene can be tuned up to the green region of the visible spectrum.389,404–406 The thermal lifetime of quadricyclane strongly depends on the substituents and the solvent or matrix.398,402,405,406 Most studies naturally focus on characterizing the compounds’ properties in film or toluene, however, photoisomerization was also reported in MeCN, revealing lower QYs ΦNB→QC (0.03 instead of 0.28) than in toluene, but longer thermal lifetimes (t1/2 = 29 h instead of 18 h) for the same compound.407 To the best of our knowledge, the compound's photochromism was never studied in aqueous environment and the acid-sensitivity396 of the quadricyclane as well as small geometrical changes upon isomerization387 limit its potential applications.
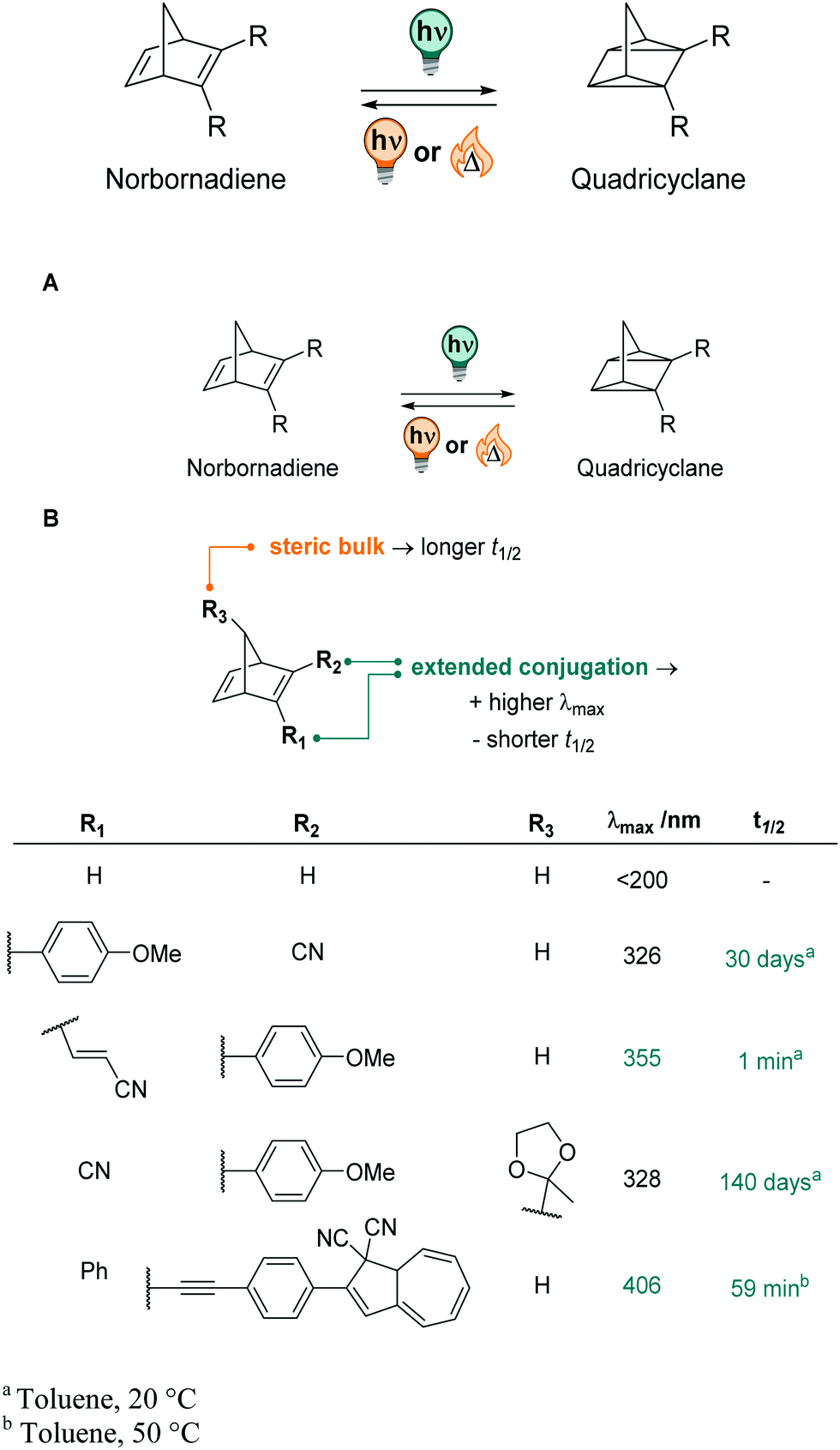 | ||
| Fig. 27 (A) The norbornadiene/quadricyclane photochromic couple, (B) the effect of substituents on the photochemical properties, including the position of the absorption band and the half-life of the metastable quadricyclane. | ||
3.4 Mixed mechanisms
In this section, we highlight photoswitches based on electrocyclic ring-closing and opening reactions, which undergo additional (photo)chemical isomerization processes. This typically results in a more complex isomerization mechanisms, involving multiple steps and several intermediates that often cannot be studied independently. An overview of the photoactuators discussed is displayed in Fig. 28.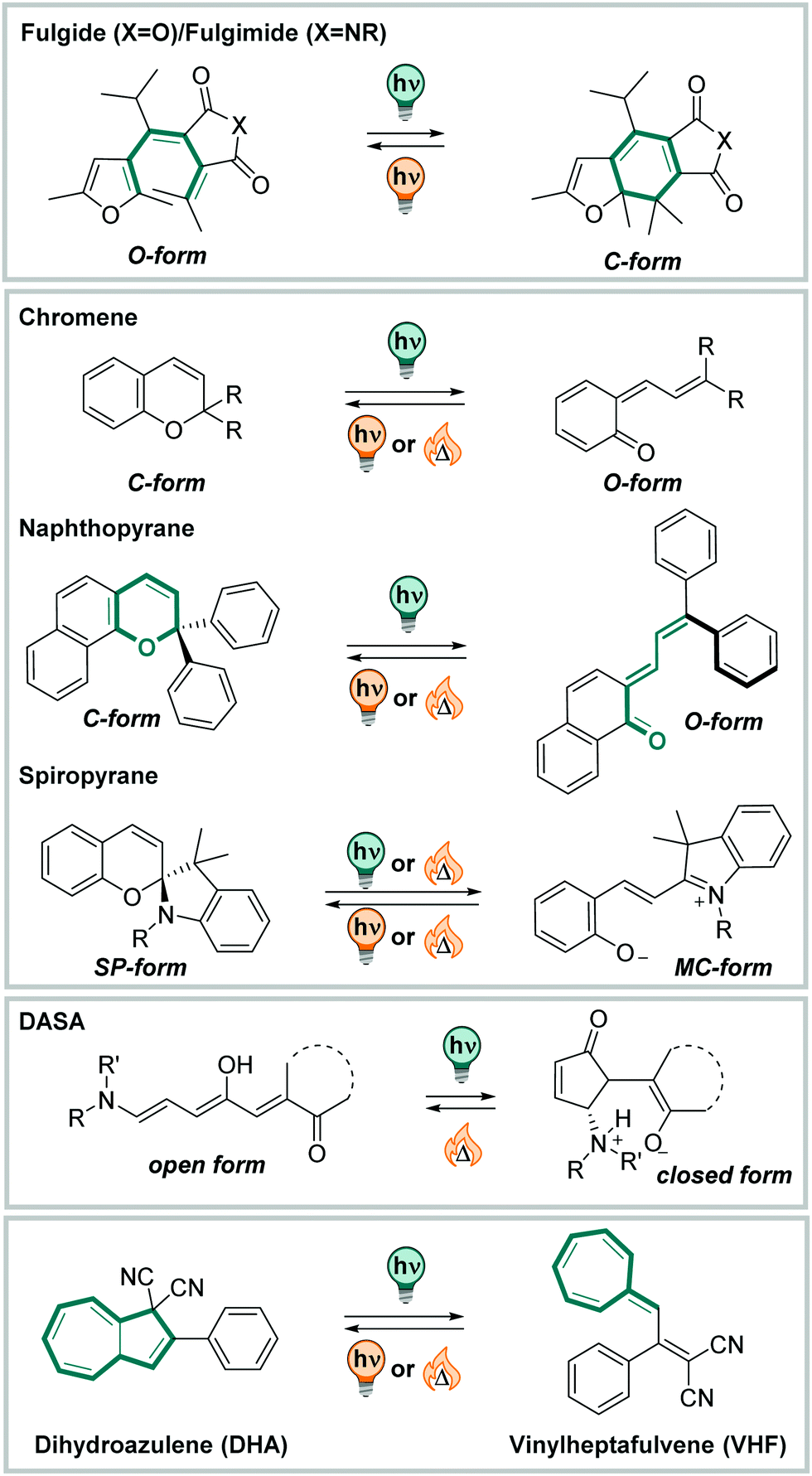 | ||
| Fig. 28 Overview of the molecules discussed in Section 3.4: fulgides, fulgimides, chromenes, naphthopyrans, spiropyrans, DASAs, and dihydroazolenes. | ||
O, Fig. 29) to build a fulgide, or a succinimide (when YNR, Fig. 29) and is then called fulgimide.410
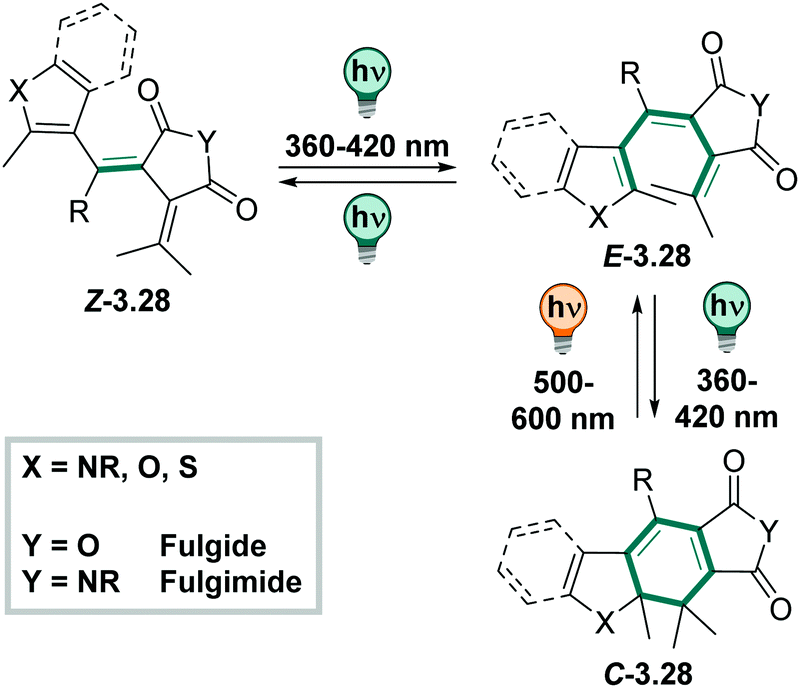 | ||
| Fig. 29 General switching of fulgides and fulgimides and the various isomers involved. | ||
Two exocyclic double-bonds assemble the full molecule, condensing it to the heteroaromatic substructure.410 Thereby, the R-substituent in the bridging part has a crucial role. Both, fulgides and fulgimides were synthesized with a large variety of R-groups in the bridge. For instance, RMe,408,412iPr,408,412 CF3,412,417tBu,412 COOH,416,417 have been employed. While smaller substituents allow for reversible E–Z isomerization of the ring-open isomers, the larger substituents, like iPr, or cyclic variations, do not.408,418 Consequently, the ring-open isomers can be separated and the isometrically pure E can be used to photoswitch exclusively via an electrocyclic mechanism.418 Fulgides with smaller substituents, such as RMe, undergo reversible E–Z isomerization under irradiation with the same wavelength of light (Fig. 29) as the one used for the ring-closure reaction, because the UV-Vis spectra of both E and Z form are significantly overlapping.412 However, the electrocyclic reaction still performs well, as it is irreversible under the same conditions and hence, functions as a photochemical sink.370 The ring-closing reaction in fulgides proceeds with varying quantum yields, determined to be ΦE→C = 0.12–0.54 for different (benzo)furans in apolar solvents, and significantly lower for indolyl fulgides (ΦE→C = 0.04–0.07).412,414 Moreover, QY and PSD values are strongly dependent on different substituents and sensitive to both steric and electronic aspects and hence, various structural modifications were explored.412,418 In contrast, moving from a fulgide to the corresponding fulgimide leaves the photochemical properties mainly unaltered.412 However, the spectra are bathochromically shifted compared to the corresponding fulgides.412,419 The closed isomer is strongly coloured due to its large π-system, conjugated throughout several ring-structures and hence, it absorbs in the visible range around 450 nm to 550 nm in fulgides and up to around 600 nm in fulgimides.408,410,412,413 Both switches are thermally very stable and show no significant decolouration reaction after several days at 50 °C in solution.410 However, the C form benzene-based fulgides is thermally labile due to the higher degree of aromaticity of the benzene scaffold.409
Irradiation of the C isomer with visible light leads to quantitative bleaching and electrocyclic ring-opening, and results in complete conversion to the E isomer.412 The reaction in general proceeds in (benzo)furan-based fulgides less efficiently than in indolyl fulgides.412 As for the closure reaction, the efficiency of the C → E interconversion is strongly substituent-dependent with quantum yields ΦC→E = 0.05–0.42 for isomerization under irradiation with visible light.412,414 Photoisomerization can be repeated several times in various solvents, in polymer films and the solid state in both directions without formation of side-products, and is very robust.409–411,414 Moreover, many fulgides and fulgimides show fluorescence,408,420 which can vary between the two photoisomers.416,421,422 Fulgides and fulgimides are used in a wide range of applications, for instance, as switchable polymer films,413,420 on surfaces,423 and in biological applications.370,411
Fulgides and fulgimides are less lipophilic than the structurally related DAEs and exhibit the rather polar succinyl moiety. Hence, they are generally better soluble in polar organic media and water than many DAE representatives, without the need for any further functionalization. To spontaneously dissolve in pure water, though, further solubilizing groups need to be attached.408,410,416,417 While in DAMs the closing reaction is suppressed by a TICT in polar (protic) media, such as water (vide supra), fulgides and fulgimides can be reversibly photoisomerized in solvent–water mixtures (aqueous ethanol (1:1))410 and aqueous buffer.370,408,411,416 Both the thermal stability remains unaltered408,410 and the photochemical properties stay intact and were subject of detailed mechanistic studies.416 Quantum yields (ΦO→C = 0.16 or 0.17 and ΦC→E = 0.10 or 0.15 in DMSO and HEPES buffer, respectively, for the same benzofuranyl fulgimide)370 as well as the PSDO→C are similar in buffer compared to polar organic solvents like DMSO (36%C in DMSO vs. 35%C in HEPES buffer, 31%C in DMSO vs. 32%C in phosphate buffer, and 66%C vs. 73%C in 30% DMSO in phosphate buffer of the respectively same fulgimide).370,408 On the other hand, the photochemical ring-opening reaction proceeds quantitatively, independent from the used solvent, as only the closed isomer is absorbing in the visible region and can hence be addressed selectively.370,408,410,411 However, fulgides undergo hydrolysis in alkaline solutions, while fulgimides are stable under the same conditions over days.410 Also, in fulgimides having a CF3 attached to the bridging moiety, it was demonstrated that this functional group can hydrolyse to the corresponding COOH under slightly basic conditions (phosphate buffer, pH 7.4, Fig. 30A).417 On the other hand, fulgimides bearing a carboxylic acid group at the same position can be switched reversibly in the same buffer (3.30 in Fig. 30B)416,417 but showed some sensitivity to acidic conditions. There, the molecules lose the COOH function to result in the non-photochromic H-bridged derivative C-3.31 (Fig. 30A).417
 | ||
| Fig. 30 Stability of differently functionalized fulgimides. (A) Hydrolysis of a CF3-substitutent in the bridging unit under slightly basic conditions and subsequent loss of CO2H upon acid conditions. (B) Photoswitching of fulgimide 3.30 with CO2H group in the bridge. | ||
 | ||
| Fig. 31 General photoswitching mechanism in chromene and naphthopyran. | ||
Naphthopyrans are benzo-extended chromenes showing similar photochromic properties as the parent compound (Fig. 31).108,426,427 The larger π-system induces a bathochromic shift of the absorption spectrum and makes the compound responsive to visible light.429 Additional substituents or scaffold hopping to heteroarenes allows further tuning of the UV-Vis absorption properties.430–433 Similarly to chromene, the open form of naphthopyran exists as several isomers, which can be converted into each another photochemically and thermally. Attachment of substituents or bridging between the pyran ring and the naphthalenic core by a fused alkyl chain can assist to direct the equilibrium between the isomers towards one species. However, this is usually synthetically laborious and compromises other properties, such as the lifetime of the open forms.430 Depending on the substituents, the decolorization reaction occurs in microseconds to a few hours.430,431,433,434 Attachment of suitable functional groups, like the positively charged N-alkylated picolinium, makes naphthopyrans soluble in buffer in the presence of small amounts of organic co-solvent (e.g. 5% DMSO).427 It was shown that photoisomerization can be triggered in the aqueous media427,429 and shows very good fatigue resistance over several cycles.429
Spiropyrans (Fig. 32), i.e., pyrans exhibiting a spiro-junction in α-position next to the oxygen, were originally reported as thermochromic compounds.329 However, only after their photochromism was reported in a derivative consisting of an indoline and a chromene moiety, which are condensed via a spiro-bond and oriented perpendicular to each other, the class of molecules attracted more attention. Since then, this structure is associated with the term spiropyran in the context of (photo)responsive molecules, a terminology which we will follow in the subsequent discussion. Spiropyran can be reversibly interconverted into its merocyanine form in a ground state or excited state process.329,435 Their isomerization can be triggered with a whole range of stimuli, including heat, pH, electrons, and light, and hence, makes them prominent candidates for a large variety of applications.329 Upon irradiation with UV-light, the chromene moiety of the ring-closed spiropyran (SP) opens in a 6π electrocyclic ring-opening reaction (vide supra) to give the436–441 ring-open merocyanine (MC) form.329,442 The central double-bond of the system undergoes further E–Z isomerization upon irradiation with the same light source, and its configuration can be separately controlled only if the phenolic oxygen is protonated or alkylated (vide infra).439,443 In this isomer, the indoline nitrogen is in conjugation to the pyran oxygen. Hence, the ring-open merocyanine form can exists in a neutral and a zwitterionic form (cf.Fig. 32). For thermochromic spiropyrans, it was reported that the electron-withdrawing (EWG, often –NO2) substituents in para-position to the pyran oxygen stabilize the zwitterionic form (cf.Fig. 32), while an electron-donating substituent (EDG) promotes the neutral merocyanine form.329 This electronic property is the key to the multi-responsive nature of the switch, making it the subject of many studies as was reviewed in detail elsewhere.329,442 The MC form is strongly coloured and has an absorption maximum in the visible range around 500–600 nm,439,440,444,445 shows solvatochromism,329 and was extensively studied also from a theoretical point of view.446,447 Heating or irradiation with a suitable light source result in the isomerization back the SP form, which is the thermally more stable one in most organic solvents (in contrast to aqueous media, vide infra).448,449 The QYs of both photochemical reactions are highly solvent- and substituent-dependent.329 For instance, QYs for the ring-opening range from 0.11 in MeCN up to 0.83 in methylcyclohexane for para-NO2 SP. The reversible (photo)isomerization can be repeated over several cycles without showing significant fatigue in a wide range of solvents.329,450
 | ||
| Fig. 32 Overview of various characteristics in the spiropyran/merocyanine photochromic couple, using a representative structure (3.32 and 3.35) bearing an NO2 substituents in para-position to the pyran oxygen. Central box (C): SP-3.32 can be photoisomerized via the Z-MC intermediate displayed its two resonance structures in (A) into the corresponding MC-3.32, which is sensitive to hydrolysis (B). Both the SP and the MC form can be protonated, with the open isomer being the weaker acid making the system a photo acid generator (PAG) upon ring-closure (D). The MC-form can undergo selective E–Z photoisomerization when the phenolic oxygen is protonated or alkylated (E). | ||
An interesting aspect in the SP-MC photochromic couple is its sensitivity towards acids: the charged MC can not only serve as complexation site (Lewis base) for metal ions,451 but also as Brønsted base.448,452,453 The phenolic oxygen can be protonated at lower pH to result in MCH+, exhibiting pKa-values between 3.7 and 4.4 depending on the substituent in the para position on the corresponding indoline moiety. Specifically, EWGs in para-position to the phenolic oxygen lower the pKa by stabilization of the zwitterionic structure (for instance, –NO2 in Fig. 32).448 Protonation leads to a significant energetic stabilization of the open form, to such an extent that the spiropyran can be interconverted quantitatively to the MC form by protonation, resulting in acidochromism.440 The characteristics depend, for instance, on substituents, solvents properties and interaction with other (macro)molecules.440,454 Protonated MCH+ shows negative photochromism upon irradiation with visible light.455 The corresponding closed form SPH+ has pKa-values between 0.4 and 1.6,448i.e. it is more acidic than the merocyanine form and hence, the SP-MC pair can serve as a photo-acid-generator (PAG) in the right environment.303,456 The anionic pyran oxygen does not only play an important role in metal complexation and as a base, but is also crucially involved in the hydrolytic stability of the photoswitch. The attack of water as a nucleophile on the ene-iminium cation has been shown to promote hydrolysis into the corresponding Fischer base and aldehyde, and is mediated by the negatively charged oxygen (Fig. 32).445,448,457 Hence, the kinetics of merocyanine hydrolysis is substituent (=stabilization of neutral and zwitterionic form) and pH dependent: at neutral pH, water can hydrolyse the MC, while in basic pH the OH− serves as nucleophile. At the same time, MCH+ is relatively stable towards hydrolysis.448 Another interesting feature of the protonated MC is that it can undergo selective, reversible E–Z isomerization upon irradiation with different light-sources, a process that can classically not be studied isolated from the SP-MC interconversion (Fig. 32).439 Similarly, the E–Z isomerization can be accessed when ring-closure is blocked by a photo-labile protecting group on the phenolic oxygen (Fig. 32).443
SP is frequently used in aqueous environment, like buffer,437,445,450,458–461 or in cells462 as a photo-acid generator (PAG) or classical photoswitch. Irradiation with UV-light results in photochemical formation of the MC form (PSDC→O = 38–85%MC and ΦC→O = 0.01–0.08 in aqueous solution or buffer).437,445,459,461 In contrast to organic solvents, where the thermal equilibrium is completely on the side of the SP form, the rates for the thermal equilibration in both directions are very similar in water due to the highly polar nature the solvent that stabilizes the zwitterionic MC form.448,449,459,463 Thermal equilibration (t1/2 = 1–6 h) results in the formation of 33–80% MC.445,459,461,462 In many examples, the rate of the competing hydrolysis reaction of the MC form is usually 2–10 times lower.445,459 The isomerization kinetics can be modulated, for instance, by introduction of suitable substituents. In this manner, an ortho-COOH derivative showed thermal SP-MC equilibration in one minute while the corresponding para-NO2 compound required ca. two hours. The hydrolysis rates stayed the same for both compounds in the applied buffer (t1/2 = 4 h).445 Irradiation with visible light allows to quantitatively access the closed isomer with ΦO→C 0.005–0.1, comparable to that in organic solvents.459,462
Spiropyrans can be functionalized at positions other than the indoline nitrogen to enhance solubility or introduce further functional groups. For instance, introduction of a pyridine, or pyridinium scaffold (Py-SP-Me, see 3.36 in Fig. 33) leads to highly polar compounds and enhances the water-solubility of both isomers.437,458,460 The scaffold can be robustly reversibly photoisomerizated (PSDC→O 26%MC, PSDO→C 100%SP, ΦC→O 0.033, ΦO→Cca. 0.2 in PBS buffer).437,458 Moreover, Py-SPs are relatively stable towards hydrolysis and can be stored in aqueous solution for several weeks and for a few days in cell extract, which can be of advantage for biological studies involving spiropyrans for longer periods of time.458,460 Moreover, they have been used as PAGs, analogous to SP.464
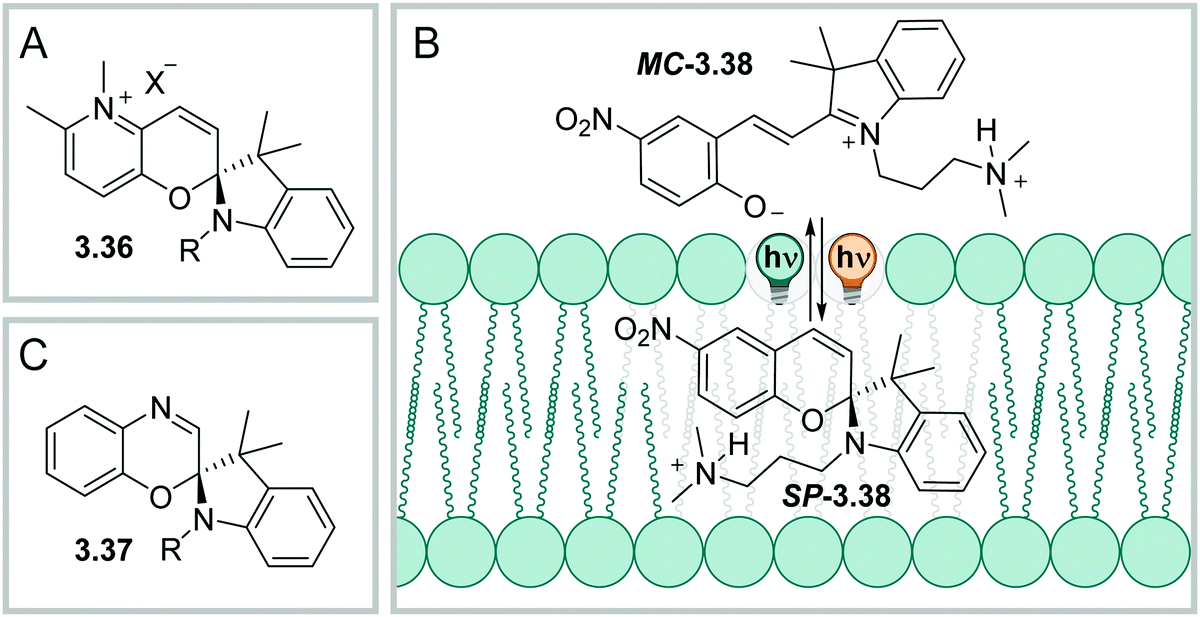 | ||
| Fig. 33 Chemical structure of a Py-SP (A) and spirooxacine (C). (B) Photoisomerization of 3.38, with SP-3.38 being more hydrophobic than MC-3.38, allowing it to diffuse through lipid bilayers. | ||
Spiropyrans found many applications, e.g., as photocontrolled binders to biomolecules, often DNA,435,459,461 in high-resolution imaging,465 and in fluorescence466,467 or conductance switching.468–470 A key feature is the interconversion between a charged and a neutral species, which has great impact on the solubility of the compound in different media. For instance, it was shown that the neutral spiropyran 3.38 is better soluble in uncharged liposomes than its zwitterionic merocyanine form (Fig. 33).444 Moreover, the neutral spiropyran can enter living cells passing the membrane and can be switched inside.462,471
While many studies focus on substituent effect on the core, also the central CC bond of the hexatriene systems can be modified. In spirooxazine 3.37, the CC is replaced by a CN bond (Fig. 33)3 which results in a molecule exhibiting an absorption profile that is bathochromically shifted with respect to parents SP.472,473 Upon irradiation with UVA-light, up to ca. 50% MC can be accumulated in DMF.472 The isomerization process is more efficient than in SP as no triplet states are involved.472 The quantum yields associated with this process are highly solvent dependent and reach from ΦC→O = 0.25 (MeOH) to 0.71 (toluene) for the same molecule. Also, the rates of the thermal back-reaction strongly depend on the solvent and are faster in polar (protic) than apolar media, spanning from a few hundred milliseconds to tens of seconds.473,474 The thermal rates are significantly slower in polymer films475 or gels.476 Alternatively, the open form can be stabilized by metal complexation.477 Most derivatives can be reversibly isomerized over several cycles without showing fatigue.473,476
 | ||
| Fig. 34 Overview of the (photo)isomerization in DASA (A) and general structures of generation 1, 2, and 3 derivatives (B). | ||
DASAs are characterized by strong inverse photochromism, with the less coloured, cyclic form hardly showing any absorbance above 300 nm. Upon irradiation of the open form, the π–π* transition of the push–pull substituted triene rapidly bleaches, and new transition bands attributed to the formation of the cyclic form arise.491 The open triene form is a strongly coloured, hydrophobic, extended structure. On the other hand, the closed form is colourless, hydrophilic, and significantly more compact.478,495 Both the band separation of the two photoisomers, as well as the wavelength of irradiation in the UV-B and C region of the electromagnetic spectrum, made photochemical back-isomerization so far not practical, rendering DASAs efficiently T-type switches for any applications.491 The photochemically induced isomerization mechanism involves multiple steps and intermediates and was thoroughly studied in recent years.483,484,486,487,496 A simplified picture is given in Fig. 35. Irradiation induces photochemical Z → E isomerization of C2C3 (A → A′, Fig. 35).483,484 Thereby, the hydroxy group facilities a pre-selection of this double bond by steric interactions and by influencing its bond length.487 The CC isomerization is followed by a rotation around the C3–C4 bond (A′ → A′′, Fig. 35), a thermal ring-closure reaction (A′′ → B′, Fig. 35), and a concluding H+ transfer onto the donor or acceptor moiety, respectively (B′ → B, Fig. 35).483,484 The hydroxy group on C2 is also crucially involved in the cyclization step, as non-hydroxy DASAs only showed unselective E–Z isomerization of several double bonds but no productive cyclization reaction.487 Different solvents influence the overall behaviour strongly (see Fig. 36A), but the actinic step is only slightly perturbated and the same key intermediate (A′) is generated, indicating that the thermal step is the overall rate-limiting one.486
 | ||
| Fig. 35 Simplified (photo)isomerization mechanism of DASA photoswitches. | ||
 | ||
| Fig. 36 Stable isomers of DASA. (A) Solvent-dependent thermal equilibrium between the open and the closed isomer in DASA. (B) Non-photochromic derivatives.495 | ||
The closed form is thermally metastable and goes back to the open triene in the range of minutes to a few hours.497 The thermal lifetimes are strongly solvent-dependent. Generation 1 DASAs exist almost exclusively in their linear form in solution.478,495,497 In contrast, substituted anilines in 2nd generation DASAs exist to a substantial amount in their cyclic form at the thermodynamic equilibrium,497 a characteristic which was further studied and improved in 3rd generation DASA photoswitches.493
Moreover, both QYs and thermal isomerization rates in DASAs are concentration depended. The ΦL→C decreases from ca. 20% to around 3% at 1000 times higher concentrations, while the thermal isomerization rate becomes ca. three times faster.498 Further studies showed that the effects of the solution-state dielectric and intermolecular interactions on the degree of charge separation could provide a rationalization for this behaviour. The stronger the zwitterionic character of the respective open and closed isomers, the more pronounced are the concentration effects.494,499
Some first generation DASA derivatives, such as 3.41 and 3.42 in Fig. 36B, could not be photoisomerized in any of the media studied so far. The origin of this behaviour is still under investigation.495 Furthermore, DASAs bearing a dialkylamine donor moiety switch reversibly mainly in aromatic apolar solvents,478,497 the secondary aniline, present in generation 2 and 3 DASAs, allows for switching in a wider range of solvents, including THF,492,493 dioxane,497 halogenated solvents489,492,493,497 and, MeCN,492,493,497 and in polymer matrices.500–502
In strong contrast, in polar protic solvents such as methanol and water, the cyclic, hydrophilic form is energetically more stable than the linear isomer and does not isomerize to the linear state.491 Also, on magnetic nanoparticles, the formation of the cyclic form is irreversible.503 Different strategies have been explored to stabilize the linear form in polar protic solvents, including the addition of molecular receptors,504 or peptides,505 which could so far only slow down the cyclization reaction in the dark. A better stabilization of the linear isomer is realized in the presence of a PdII-based molecular vessel.506 The open form can even be spontaneously generated in aqueous media, but the photochemically induced ring-formation was suppressed inside the cavity.506 However, reversibility in water and PBS buffer was observed when the DASA photoswitch was part of a micelle-forming homo-polymer.507
The pronounced changes in their geometry and polarity make DASAs interesting candidates for a wide range of applications.490,491 For instance, they have already been used in responsive polymers,501,502,508 on surfaces,509,510 to photocontrol wettability,500,511 in sensing and,512,513 for dynamic, self-regulating flows.514 Furthermore, DASAs are responsive to multiple stimuli505,512,513 which allows to further tailor their switching behavior. However, their uncontrolled behavior in water currently limits their application in this medium.
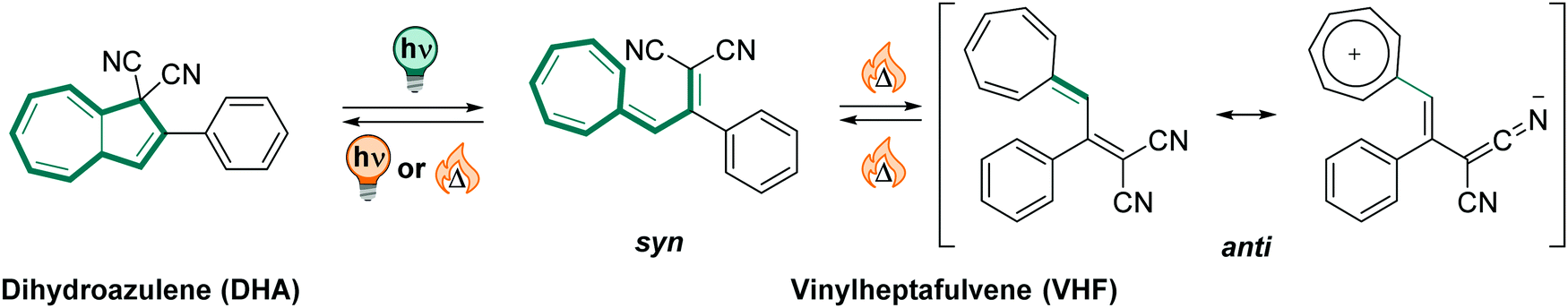 | ||
| Fig. 37 General mechanism of the (photo)isomerization in the dihydroazulene/vinylheptafulvene photochromic couple. | ||
4 Illustrative examples of photoswitches in aqueous media
As described in Section 2, various strategies have been applied to solubilize organic molecules in water. Next to the effect of aqueous media on the photochemical properties of the photoswitch and the thermal lifetime of the metastable form (see Section 3), chemical stability in water and in biological environment also plays an important role. For instance, some azobenzenes are stable in the presence of biological antioxidants, such as glutathione,529–531 while others are susceptible to reduction. They thus lose their functionality and form potentially toxic side products like diarylhydrazines and anilines.532 Furthermore, enzymes naturally present in living organisms, particularly azidoreductases that are active in hypoxic cancerous tissue with low oxygen levels, can also reduce azobenzenes.533 Especially, the Z isomer of azobenzene was found to be more prone to reduction than the less polarized E form.534 Instability towards reduction can be circumvented by attaching different substituents to the azobenzene core200 or by heteroaryl design resulting in many systems that are stable under physiological conditions in timeframes required for their application.6,11,200,535–537 However, the introduction of different solubilizing moieties often has consequences for biological activity and uptake, such as selective accumulation in different sub-cellular compartments538–540 or even hindered cellular uptake of freely dissolved molecules.541,542Keeping all the aforementioned limitations in mind, in this section we will discuss different solubilization approaches applied to photoswitches in aqueous media.
4.1 Small molecules that act as ligands in biological systems
In biomedical research and the pharmaceutical world, small molecule actuators are extensively applied to regulate biological processes.543–546 In this way, a wide range of biomolecules, such as proteins,547–549 DNA and RNA,550 as well as oligosaccharides,551–553 can be targeted. Small molecules are usually applied remotely and reach the actual target site by diffusion. Consequently, the local concentration at the target site is often low, unless the molecule exhibits specific uptake (like phosphonate into mitochondria)538 or an explicit delivery technique, for instance using liposomes, is used.554 However, increasing the overall concentration of the bioactive molecule would affect not only the target site but also others. Eventually, side effects, as often observed in cancer therapy, or accumulation in the environment, which is one of the causes for growing multi-resistance in bacteria, often outweigh the beneficial effects of drug-therapy.For these reasons, the use of an external stimulus, such as light, to enable local and temporal control of bioactivity, became a very promising approach in recent years.7 Still spatiotemporal control is limited by diffusion of the light-responsive molecule. Thus, systems which can only be turned on once, such as those employing photocleavable protecting groups (photocages), are not ideal for many applications.7 In contrast, molecular photoswitches can be reversibly activated specifically at the target site and deactivated to protect the surroundings.30 Depending on the nature of the photoswitch, the deactivation process can be achieved by light or thermally (auto-inactivation, see Section 3 for different types of photoswitches). In this section, we discuss different solubilization approaches of photoswitchable molecules used as small molecular actuators in biological systems and highlight selected examples illustrating how to control biological function.
In some cases, however, the charged unit affects the properties of the compound beyond just providing the required solubility. For instance, it can simultaneously enable binding to the target or interfere with cellular uptake. On the one hand, a positively charged group can increase the adherence of the molecule to cellular membranes, which are negatively charged on the headgroups of lipids, thus increasing the cellular uptake.555 However, charged moieties can also hinder the internalization pathway by diffusion through the lipid bilayer due to insufficient lipophilicity of the compound.556 These contradicting effects, improved adherence and decreased diffusion through the membrane, illustrate the importance of a balanced design of drug-like molecules.
Except for diffusion and passive uptake, some compounds are actively uptaken by protein transporters and receptors.557 Here, the charged moiety can either be a part of the transporter/receptor substrate or it does not interfere with the uptake process. Hence, introduction of a charged group to enhance aqueous solubility might seem straightforward, however, it often is accompanied by consequences for several other key parameters that need to be taken into consideration.541,557
In the following sections, we discuss frequently used charged groups in photoswitchable biologically active ligands.
4.1.1.1 Ammonium ion. The most explored moieties used for solubilizing photoswitches with a charged group are the protonated tri-alkyl amine or a tetra-alkyl-ammonium groups. Both are positively charged in aqueous media at a pH < 9558,559 and consequently, provide good aqueous solubility. Moreover, the (permanently) charged moieties can serve as binding groups towards different biomolecules. They can, for instance, act as an inhibitor of voltage gated channels,560,561 which are involved in communication between cells, in transport, and self-defense mechanisms.562 In this context, such compounds were used to photocontrol, for example, insulin release563 and to block channels in neuronal tissue.215,561,564,565 In another example, photoswitches carrying ammonium groups were used to restore vision in degenerated photoreceptor layers of mice. In particular, two azobenzenes, one with a diethylammonium group (4.1a, Fig. 38A) and one with a permanently positively charged triethylammonium group (4.1b, Fig. 38A), were investigated for this purpose.566 The charged photoswitches blocked ion channels in neuronal ganglions in their E form (Fig. 38C), while after E → Z photoisomerization with 480 nm light the molecules were sterically too hindered to be accommodated by the channel and hence, activate it (Fig. 38C). Both 4.1a and 4.1b are well soluble in water, however, they target different cell types in the retina566 due to distinct internalization mechanisms. The presence of receptors in certain cell types leads to active uptake of the permanently charged 4.1b566 while non-permanently charged 4.1a is mainly passively uptaken.566 This observation indicated that introduction of a permanent or non-permanent charge not only renders the molecules water-soluble, but also has consequences for their activity as well as their uptake. To further investigate this, similar photoswitches were designed to test the effect of one or multiple charges on their ability to inhibit voltage-gated potassium channels.567 Interestingly, 4.2b possessing two permanent charges (Fig. 38B) exhibited no activity on cells due to poor uptake and impaired diffusion through the cellular wall. On the other hand, 4.2a, possessing only a single positive charge, could be taken up and successfully bind to its target. These examples clearly show that designing a photoswitch with permanently charged tri-alkylammonium groups brings water-solubility, however, the molecules need to be able to reach their specific target and therefore, should be carefully designed on a per-case basis.
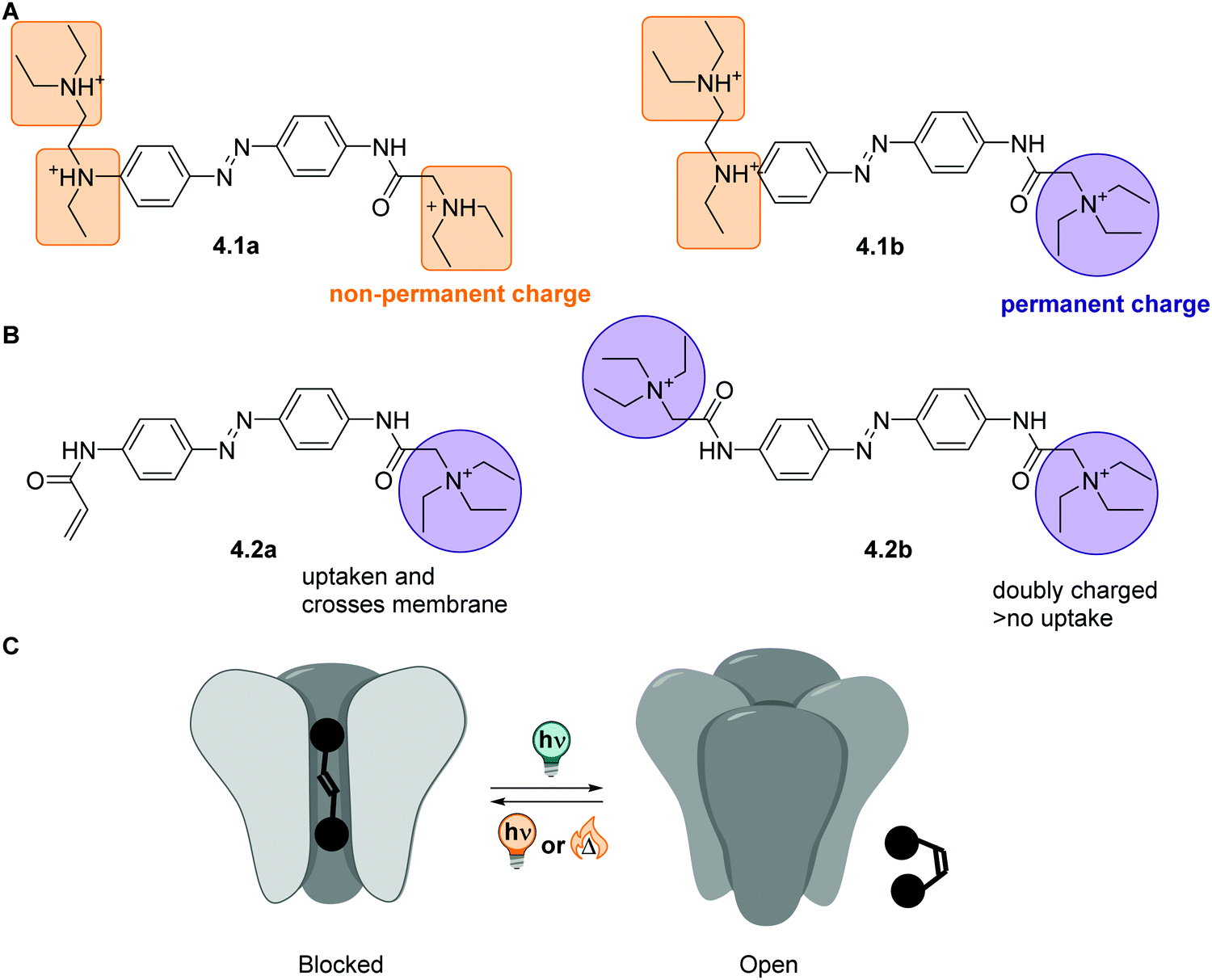 | ||
| Fig. 38 Azobenzene photoswitches furnished with permanently charged (A and B) (4.1b, 4.2a, 4.2b) ammonium groups or with non-permanently charged alkylamines (4.1a and 4.1b). (C) The charged elongated E isomer can easily fit into the channel cavity thus blocking it, while the Z isomer does not fit into the cavity leaving the channel open. | ||
Moreover, positively charged nitrogen units are frequently used to bind to RNA568,569 and DNA,461,568–573 by interacting with the negatively charged phosphate backbone. For instance, trimethylammonium and aminomethaniminium groups have been applied to both interact with DNA and solubilize spiropyrans (Fig. 39A and B).461,568–570 Spiropyrans 4.3a and 4.3b were used as photoswitchable DNA intercalators (Fig. 39). The closed spiropyran (SP) isomer, having a non-charged and non-planar core structure, is not suitable for DNA binding, despite bearing a permanent charge at the substituent. On the other hand, the merocyanine (MC) form is aromatic and planar, which is favorable for DNA intercalation. Protonation of the phenol group in slightly acidic environment, such as often found in in cancerous cells,574,575 additionally improved binding to negatively charged DNA.576
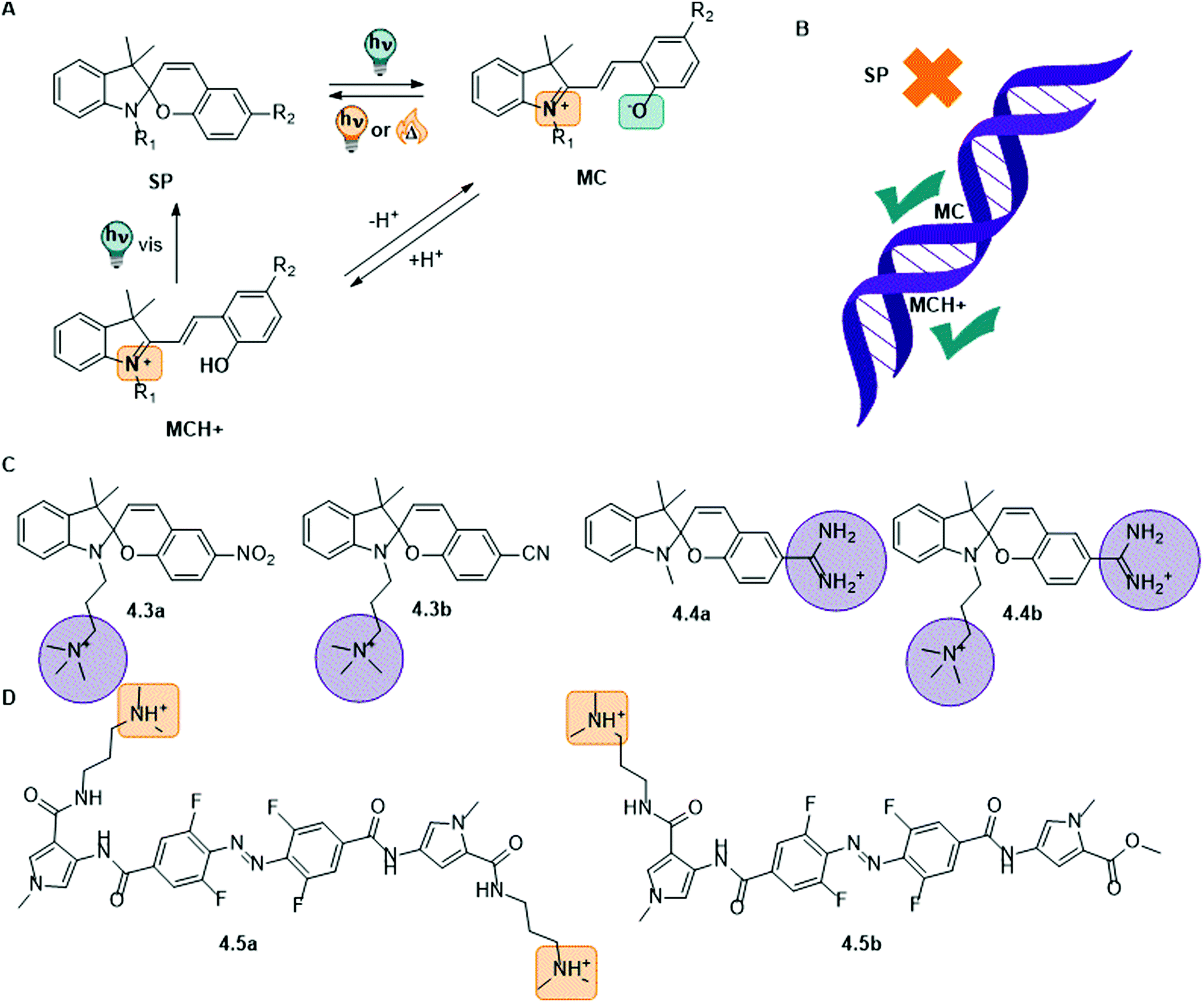 | ||
| Fig. 39 (A) The spiropyran photoswitch SP undergoes a ring opening reaction upon irradiation with light thus forming the open zwitterionic MC isomer. Under acidic conditions, MC is protonated, forming the positively charged MCH+ isomer which can be ring-closed upon irradiation with visible light. (B) Both charged isomers MC and MCH+ bind to double stranded DNA, while the SP does not interact with DNA. (C) Spiropyrans 4.3a and b, carrying a nitro and cyano groups, were further modified to carry one positive charge (4.4a) or two positive charges (4.4b) to explore the effect of additional charge on their DNA binding properties. (D) Visible light-controlled azobenzenes 4.5a, carrying two positive charges, and azobenzene 4.5b, carrying one positive charge, were designed to modulate DNA. | ||
To further explore the design, two molecules containing a positively charged amidine group, known to bind to DNA,576–578 were investigated (compounds 4.4a and 4.4b).459 Switches 4.4a and 4.4b (Fig. 39) were carefully designed to match the assay conditions suitable for DNA binding studies, where at pH 5–9 at room temperature the thermal equilibrium is reached within 5h (20:80 = SP:MC for 4.4a) or 1h (85:15 = SP:MC for 4.4b).459,579 It was found that MCH+ binds 35 times (4.4a) or 66 times (4.4b) stronger to DNA compared to their corresponding unprotonated MC forms. While 4.4a MC, 4.4b MC and 4.4b MCH+ intercalate DNA, 4.4a MC showed a less specific binding mode.459
Besides spiropyrans, also suitably substituted azobenzenes can be used as visible-light responsive DNA modulators. The visible light-responsive tetra-ortho fluoro azobenzene motif was functionalized taking inspiration of the DNA minor groove binder netropsin and was used to target nucleosomal DNA (Fig. 39D, 4.5a and 4.5b).580 Upon irradiation with 520 nm light in DMSO, relatively high PSDs were reached (70:30 = Z:E for 4.5a, 73:27 = Z:E for 4.5b) and by irradiation with 405 nm light the PSDs were 7:93 = Z:E (4.5a) and 3:97 = Z:E (4.5b), respectively, in DMSO. Most importantly, the Z-4.5a isomer showed good thermal stability (after 9 h 77:23 = Z:E) in Tris buffer with 2–5% DMSO. Furthermore, the E isomer 4.5a was successfully bound to nucleosomes while the Z isomer did not.
4.1.1.2 Other nitrogen salts. Similar to ammonium groups, pyridinium moieties often increase water-solubility, while also enhancing the affinity to DNA. In this context, both azobenzene581 and DTE-based582 switches were used to develop light-responsive DNA binders. In particular, pyridinium-functionalized photoswitches, selectively binding to G-quadruplex DNA, attracted considerable attention in the last years.116,349,583–587 G-quadruplexes are four stranded secondary structures of guanine-rich DNA that form stacked tetrads and are involved in a plethora of biological processes, ranging from transcription, replication, genome stability to cancer growth.588 In this context, azobenzenes with a permanently charged methylated piperidine, pyridine or morpholine group on each end were synthesized (4.6,7 and 10, Fig. 40A and C). The molecules induced formation of G quadruplexes in the E form, while the Z isomer caused dissociation. However, this effect was significantly diminished in the presence of metal cations, such as those present at physiological conditions (4.6, Fig. 40A.)583,584 A similar photoswitch, stiff stilbene 4.7 (Fig. 40A) behaved analogously to azobenzene 4.6. and induced reversible formation of G quadruplexes.116 The Z isomers of stiff stilbenes are thermally more stable than Z-azobenzenes, which could be beneficial form certain applications. However, irradiation with 400 nm light caused refolding triggered by irreversible photo-induced oxidation and degradation of the stilbene (Fig. 40B, structures 4.7a and 4.7b).116 This photochemically induced side-reaction has been reported previously115,589 and limits the applicability of the stiff stilbene-based modulators in the long term. The stilbenes were further modified to introduce a more flexible piperazine group (4.8, Fig. 40C), which resulted in affinity for G4 DNA over duplex DNA facilitating toxicity towards cancer cells,590,591 while remaining less toxic towards a non-malignant mammalian cell line.585 In contrast, the photophysical properties of the structurally related aryl-stilbazolium ligands (4.9, Fig. 40C) were hampered in the presence of DNA rendering them unable to switch due to too strong binding affinity.586
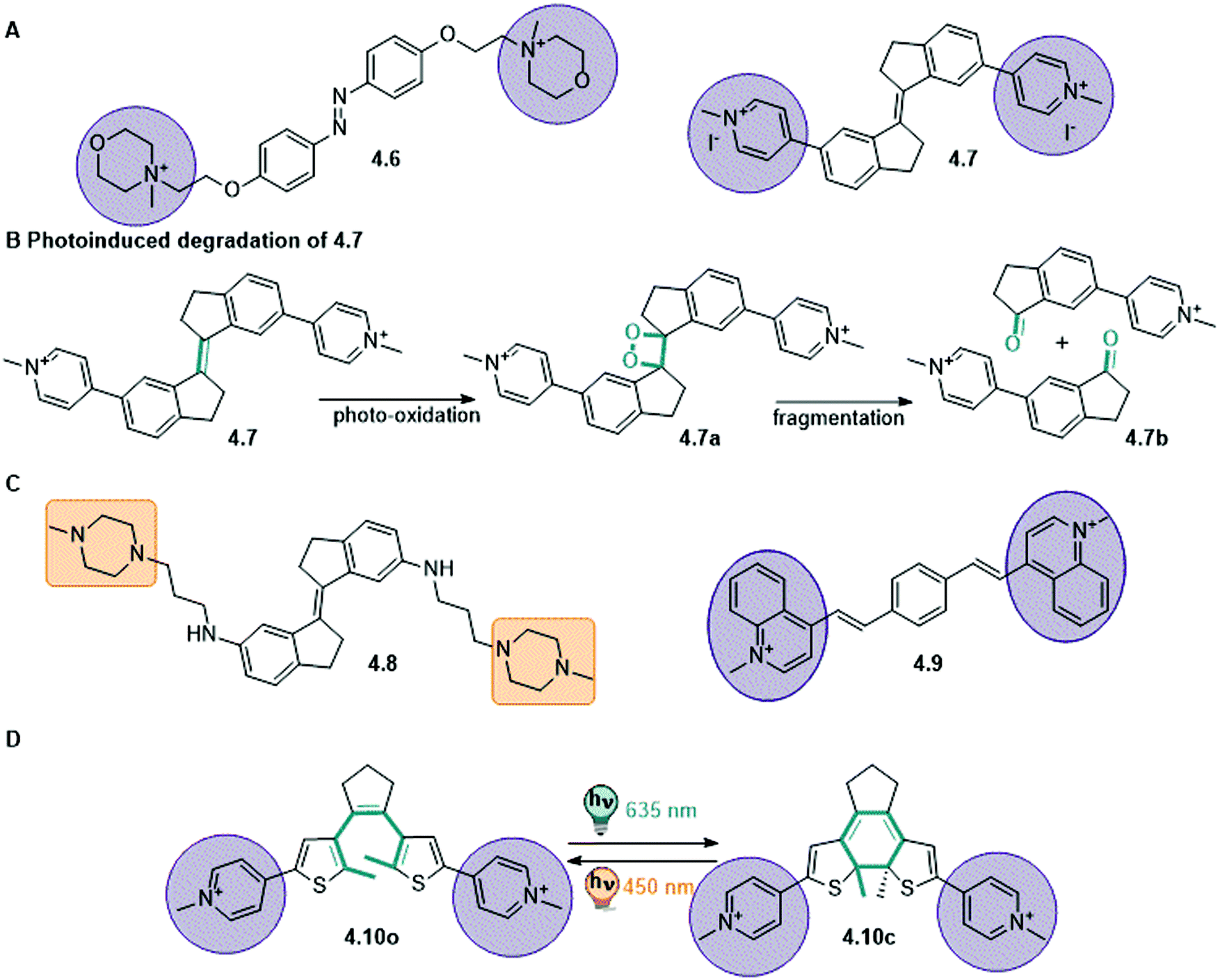 | ||
| Fig. 40 Photoswitches solubilized with positively charged cyclic amines and heteroatoms containing nitrogen atom. (A) Azobenzene 4.6, carrying two methylated morpholine moieties, and stilbene 4.7 with two pyridinium groups were used to modulate G quadruplex DNA structures. (B) Irreversible photooxidation of stilbene 4.7 into 4.7bvia4.7a. (C) Stilbene 4.8 functionalized with two piperazine groups exhibited affinity for G4 DNA over duplex DNA, facilitating toxicity towards cancer cells while the structurally related 4.9 was bound too strongly to DNA prohibiting photoswitching. (D) The photoisomerization of a DTE photoswitch carrying two pyridinium groups of which the open form 4.10o showed higher toxicity towards cells opposed to the closed form 4.10c. | ||
Very recently, a DTE furnished with pyridinium moieties (structure 4.10, Fig. 40) was reported to bind to different G4 topologies in the closed and open form. The compound was readily soluble in phosphate buffer and the switching could be reversibly performed with visible light (450 and 635 nm).349 The two photoisomers exhibited toxicity towards cancer cells, with the closed form being two-fold more potent than the open one (IC50 of 4.10c = 10 mM, 4.10o = 23 mM). However, in a study on a very similar DTE switch containing a hexafluorocyclopentane group, it was reported that the pyridinium-DTEs are potentially toxic to eukaryotic organisms.587 Switching was successfully performed in vivo in a transparent nematode organism (Caenorhabditis elegans) with sufficient oral uptake of both open and closed forms. The striking observation was that nematodes fed with the ring-closed form appeared to be paralyzed. Upon visible light irradiation, the paralysis was significantly less present. However, it seemed that both DTEs were toxic to the organism. Nevertheless, it must be noted that the tested concentrations were very high (up to 12 mM in 10% DMSO in phosphate buffer, pH = 7).
Another photoswitch that was functionalized with a N-alkylated picolinium moiety to bind to dsDNA, is chromene 4.11 (Fig. 41).427,592 Under continuous irradiation with UV light, the two interconverting open isomers, called 4.11TC and 4.11TT, are formed. They showed the ability to intercalate to the DNA double helix, which in return electrostatically stabilized the meta-stable photoisomer.427 When the light stimulus was removed, the open form undergoes ring-closure and the chromene could no longer interact with DNA. As expected, the solubility was suboptimal due to the highly aromatic nature of chromene. Upon addition of the DNA in buffer, the switch precipitated and therefore, a 5% DMSO solution had to be used for the experiments.
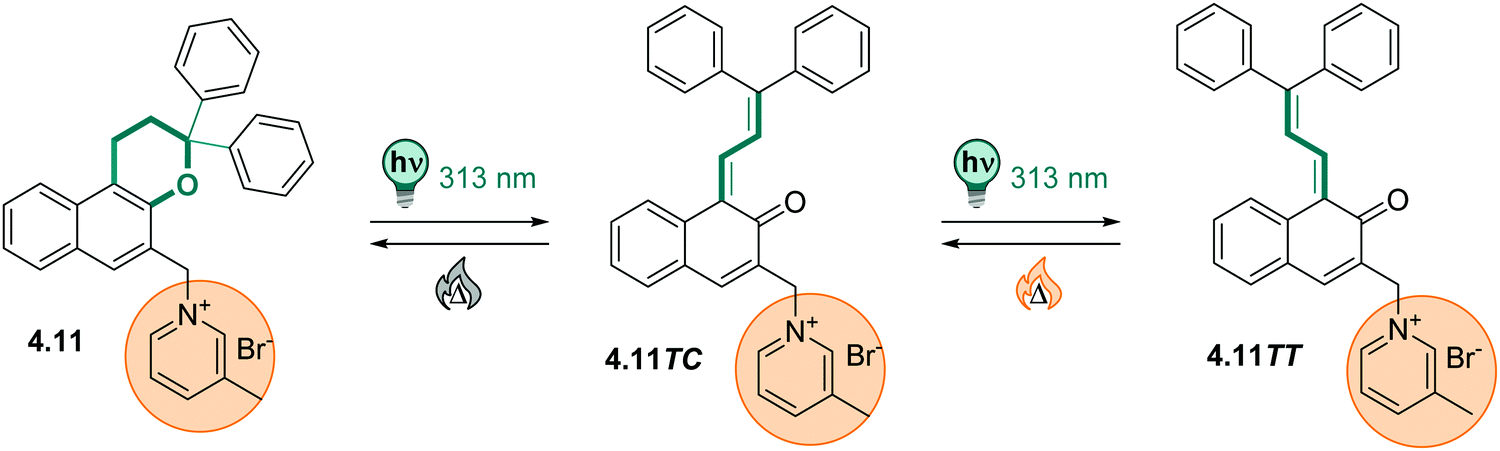 | ||
| Fig. 41 The chromene photoswitch 4.11 forms the open 4.11TC isomer upon UV irradiation, which interconverts to the TT isomer (4.11TT). | ||
4.1.1.3 Sulfonate, phosphate and phosphonate. Phosphonate, phosphates, and sulfonates, as well as related functional groups, are negatively charged when dissolved in water and can be readily employed to increase water-solubility.78 Sulfonated azobenzenes are often used as food colorants and dyes, such as carmoisine593 or Congo Red (Fig. 42).594 Besides their coloring properties, the molecules were also studied regarding their activity as DNA binders or as inhibitor of an HIV1 protease. However, the photochemical switching properties of the sulfonated dyes are to the best of our knowledge not reported.
 | ||
| Fig. 42 Sulfonated azobenzene dyes, carmoisine and Congo red. | ||
On the other hand, a phosphate-containing lipid with an incorporated azobenzene unit was used as a ligand for the G-protein coupled receptor (GPCR) S1P1-5 to photocontrol pain hypersensitivity in mice.595 Alternatively, photoswitchable ATP analogues bearing multiple phosphate groups were used to interfere with the kinesin-microtubule systems.596 Also, in designing azobenzene-based antagonists (4.13, Fig. 43) of the AMPA receptor, which is involved in neuronal firing, the introduction of a phosphonate group was key for assuring solubility under physiological conditions.597 A previously reported non-light active AMPA antagonist, NBQX, based on a quinoxalinedione moiety failed at clinical trials due to its insufficient solubility, which resulted in renal toxicity (Fig. 43).598 Attaching a methylphosphonate group (structure 4.12, Fig. 43) increased the solubility, and was adopted in the design of the photoswitchable analogue.598,599
 | ||
| Fig. 43 Introduction of a methyphosphonate group (4.12) to the AMPA antagonist NBQX ensured water-solubility. Molecule 4.12 was modified to incorporate a light-responsive azobenzene switch (4.13) as photocontrolled AMPA receptor antagonist. | ||
Moreover, phosphonate and phosphate groups have been attached to DTE photoswitches to ensure both water-solubility and inhibitory activity towards a Mycobacterium tuberculosis (MTB)-specific enzyme.600 Phosphoribosyl isomerase A (mtPriA), an enzyme unique to MTB species, possesses a symmetric active site with two phosphate-binding sites. Since a specific geometry and distance between both sites is crucial for competitive inhibition, mtPriA is an excellent target for photoswitchable inhibitors whose geometry and flexibility can be altered by light. Five bistable DTE switches containing phosphonate or phosphate groups at different positions were studied regarding their binding properties towards the active site of mtPriA (Fig. 44).600 DTE 4.14m with meta-phosphate groups featured the largest difference in binding constants between the open (Ki = 0.5 μM) and closed form (Ki = 4.4 μM). The same DTE switches (4.14m, 4.14p, 4.15m, 4.15p) were applied as inhibitors for the imidazole glycerol phosphate synthase (ImGP-S) multienzyme complex and were able to reversibly interrupt the allosteric communication between the enzymes in the multienzyme complex.601 Recently, alkyl phosphate-functionalized azobenzenes were used to control the tryptophan synthase (TS) multienzyme complex in a similar fashion. The solubility of the compounds strongly depended on the length of the alkyl-linker between the azo-function and the phosphate group. Consequently, stock-solutions for photochemistry and biochemical evaluation were prepared in pure water and the concentration was adjusted to the respective solubility. Only one derivative required the addition of 15% DMSO to be studied by UV-Vis spectroscopy and was excluded for biological evaluation.602
 | ||
| Fig. 44 DTE-base mtPriA inhibitors with phosphate and phosphonate groups on different positions of the phenyl ring. | ||
4.1.1.4 Carboxylates. Like sulfonates, phosphonates and phosphates, also carboxylates are anionic under physiological conditions, which facilitates solubility. Accordingly, the sodium salts of two carboxylic acid functionalized DAE derivatives (4.16a and b, Fig. 45) were prepared to solubilize the photoswitches in buffer. While the previous examples discussed photoswitches that alter their biological activity by a photoinduced structural change, this example showcases that photoisomerization process can be linked to drug release. Maleimide, a small molecule that can modulate DNA topoisomerase II by what is supposed to be a covalent attachment to a cysteine side chain,603 was condensed with a furyl-containing DAE in a Diels–Alder reaction.604 Photoinduced O → C isomerization locks the bioactive molecule and no maleimide is released. The reverse Diels–Alder reaction takes place at physiological conditions and body temperature and is induced by visible light. Maleimide is slowly released (50% release in 92 h at 40 °C), generating a daily dose of 46 μM (4.16a,o) and 5 μM (4.16b,o), which corresponds to the bioactive dosage window for pharmaceutical applications.603
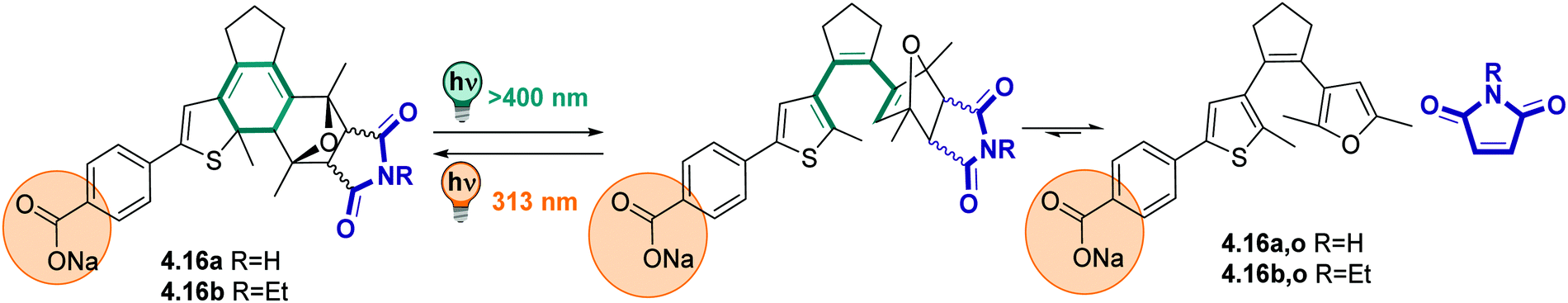 | ||
| Fig. 45 The prodrug based on a DAE forms the ring-open isomer upon irradiation with 400 nm light which can undergo reverse Diels–Alder reaction releasing maleimide and the open furyl-substituted DAE. | ||
4.1.1.5 Sulfonylurea. Sulfonylurea-based drugs are often used to treat diabetes,605 despite their relatively limited solubility in water, and are hence often solubilized using co-solvents.606 Azobenzene sulfonylurea 4.17 with a pKa of 4.76 readily dissolves in water at sufficiently high concentrations to be tested in biological assays when diluted from a 50 mM DMSO stock solution.34 The acidity of 4.17 ensures its deprotonation and solubility under physiological conditions. Moreover, its anionic nature makes it unlikely that the compound would interact with DNA, due to the negatively charged phosphate backbone of the macromolecule. Furthermore, 4.17 was tested in vivo to photo-control of glucose homeostasis using a mouse model.607 In these studies, the potential toxicity of 4.17 was investigated in detail, since a previous study reported that many amino group-bearing azobenzenes are mutagenic and carcinogenic.608 However, compound 4.17 was proven as reasonably safe, presumably due to its negatively charged character in water (Fig. 46).
 | ||
| Fig. 46 The conjugate acid of azobenzene sulfonylurea 4.17 with a pKa of 4.76 is deprotonated and thus negatively charged in aqueous solutions. | ||
4.1.2.1 Hydroxamic acids. Hydroxamic acids are highly polar moieties and assist water-solubility of a compound through ionic and hydrogen-bond interactions with the solvent. Moreover, they can be found as the active moiety in biologically relevant molecules.609 In this context, classical azobenzenes 4.18a–h (Fig. 47A) were functionalized with hydroxamic acids and studied as photoswitchable histone deacetylase (HDAC) inhibitors in cancer cell essays to develop potential therapeutics.610 The molecules were irradiated in DMSO stock solutions and subsequently diluted into the assay buffer showing good solubility. The reported switches had varying half-lives, ranging from 4 min to over 8 h in 1% DMSO in HDAC assay buffer at room temperature, thereby offering a selection for a short-lived compound that deactivates quickly after light activation or, a more stable version which remains in its light-activated isomer for a longer time. Their stability towards photobleaching and reduction in cellular environment was demonstrated by performing multiple switching cycles in the presence of glutathione. Also, arylazopyrazoles 4.19a–c (Fig. 47B) were designed as inhibitors of amidohydrolase, which is closely related to HDAC and present in many hospital-acquired bacteria.611 In particular, the hydroxamic acid group interacts with the protein by forming a complex with the Zn2+ ion, which facilitates binding of the ligand. It additionally allows the otherwise lipophilic photoswitches to readily dissolve in phosphate buffer in the case of 4.19a and 4.19b, while 4.19c could not be solubilized. The arylazopyrazole derivatives proved to exhibit excellent photophysical properties with high PSDs (from 85–90% Z to 93–97% Z). The metastable forms of switches 4.19a and 4.19b had relatively long half-lives of 17.8 h and 19.4 h, respectively (30 °C, phosphate buffer). Both types of azobenzene-based HDAC inhibitors show that a balanced design consisting of an ionic effector group, that also facilitates water-solubility, and an aromatic photoswitch can results in drug-like compounds holding promise for cellular studies and beyond.
 | ||
| Fig. 47 (A) Selected examples of photoswitchable hydroxamic acid-based HDAC inhibitors. (B) Arylazopyrazoles carrying a hydroxamic acid moiety functionalized with different substituents and their effect on solubility. | ||
4.1.2.2 PEG chain. As described in Section 2.2.1, a common approach to solubilize organic molecules in water is the attachment of hydrophilic PEG chains.102 This approach is often applied for solubilizing photoswitchable self-assembly systems.162,612
Dihydropyrenes are insoluble in water but could be solubilized in aqueous media by attaching PEG5-10 chains to their core. RNA aptamers, which are single stranded RNA molecules that bind to target molecules,386,613 were optimized by iterative in vitro selection to differently bind to isomers of dihydropyrenes. Dihydropyrenes were chosen as aptamer ligands, since both isomers are conformationally very rigid and therefore feature distinctly different shapes.383 Molecule 4.20c(Fig. 48) was identified as the most promising hit and was used to photocontrol the assembly of a ribozyme,386 a catalytically active RNA cluster.614 Binding of the pegylated 4.20c to the aptamer stabilized the ribozyme's catalytic site allosterically and showed a 900-fold difference in catalytic rates compared to 4.20o.
 | ||
| Fig. 48 The water-soluble dihydropyrene photoswitch, carrying a PEG chain, undergoes a ring opening reaction upon irradiation with visible light and the ring-closing process under UV irradiation. | ||
Also, PEGylation together with attachment of a guanidium group was successfully used in the design of a complex molecular glue, a molecular structure that glues two (or more) other molecules together through strong non-covalent interactions.615–617 Azobenzene photoswitch 4.21 was modified with multiple adherence guanidinium groups that bind to oxyanion-rich areas on the surface of the target protein via salt bridges (Fig. 49).618 Additionally, the azobenzene featured a sulfonamide group on the other side, acting as an inhibitor towards a carbonic anhydrase. Azobenzene 4.21 only functioned as inhibitor in its E configuration, since the Z geometry prohibited the molecular glue from accessing the active site. In this example, both the PEG-chains and the guanidinium moieties are crucial for water-solubility. Additionally, the presence of multiple guanidiniums ensured superb cell-membrane permeability.619 The large molecule could be reversibly photoisomerized in aqueous solution and had a half-life of 2.3 h in HEPES buffer at 37 °C.
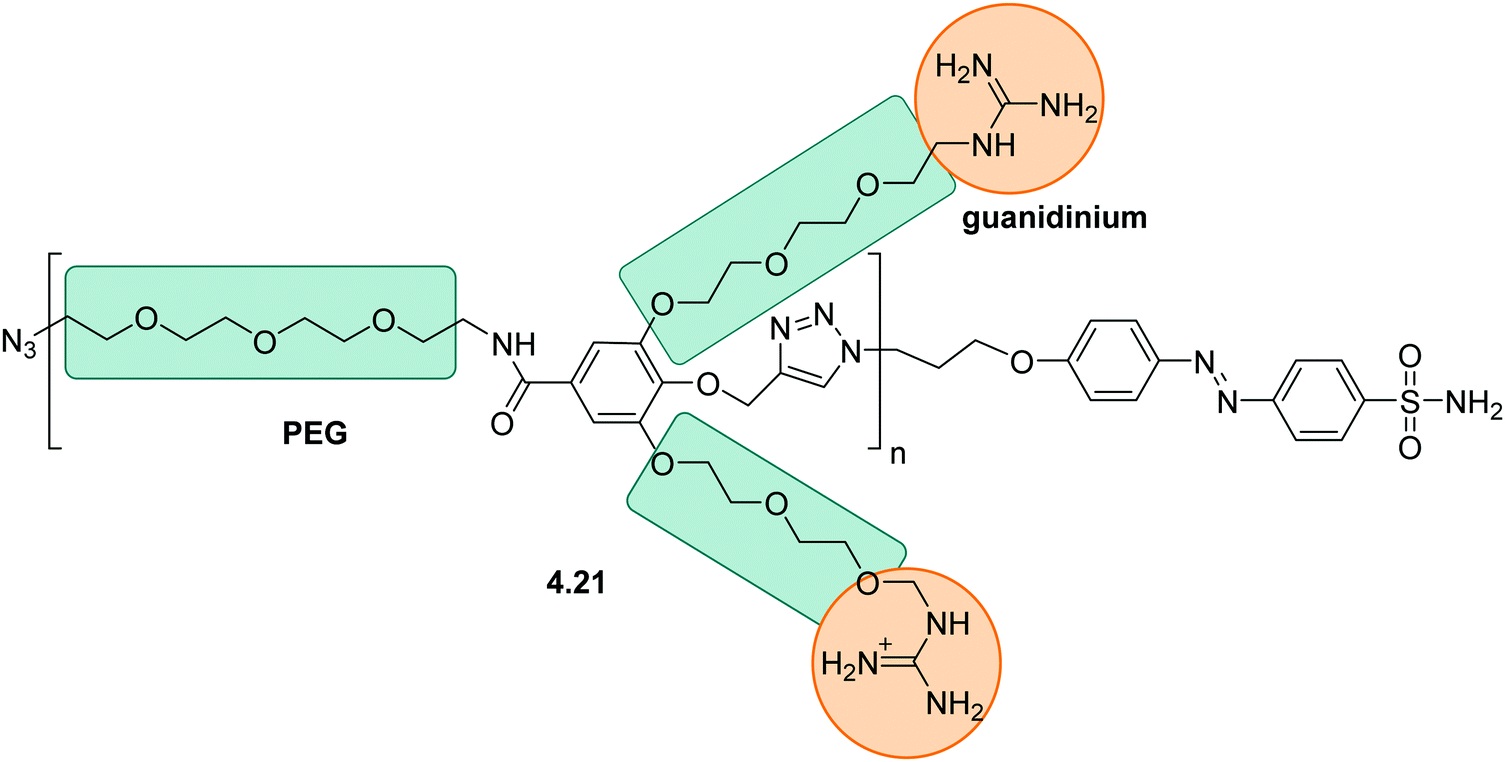 | ||
| Fig. 49 Azobenzene 4.21 carries multiple PEG linkers as well as guanidinium groups which enable water-solubility as well as enable binding to the target protein via salt bridges. | ||
4.1.3.1 Azobenzenes and heteroaryl azobenzenes. Scaffold hopping in classical azobenzenes has been applied to achieve improved photophysical properties in water and better water-solubility. In one example, azo-analogues 4.22a–b (Fig. 50A), which were designed as inhibitors of CENP-E, a protein involved in chromosome movement, were very poorly soluble under aqueous conditions. In contrast, the phenylazopyrazole photoswitch 4.22c was much more soluble and could be tested on cells at >100 μM in buffer with 0.1% DMSO.622 In a 1
:1 water:acetonitrile mixture, the half-life of 4.22c was longer than 24 h at 37 °C and the PSDs were 93%Z (PSS365nm) and 86%E (PSS510nm). E-4.1.22c (IC50 0.29 μM) had a 10-fold higher inhibitory activity than the Z form (IC50 2.4 μM). Therefore, cell proliferation could be inhibited or slowed down upon irradiation with green light (510 nm), forming the E isomer, when testing the compound on living cells (HeLa). Compounds 4.22a and 4.22b, based on classical azobenzenes, had to be tested in vitro/in the enzyme essay for inhibition of CENP-E with 10% DMSO reaching maximum concentration of only 5 μM and no difference in activity between their E and Z isomers could be observed.
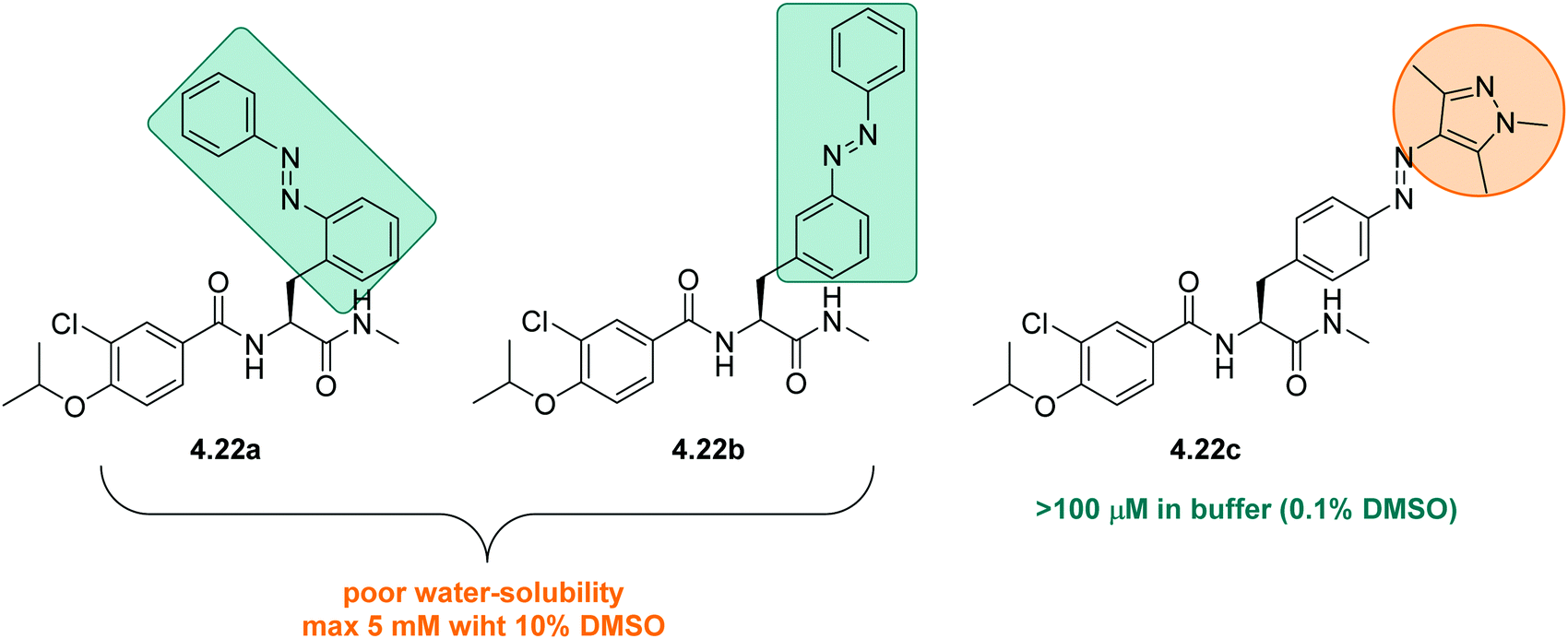 | ||
| Fig. 50 Photoswitchable CENP-E inhibitors containing an azobenzene were poorly water-soluble (4.22a and b) while the heteroaromatic arylazopyrazole photoswitch 4.22c exhibited much better aqueous solubility. | ||
Next to arylazopyrazole switches, other heteroaryl-azobenzene derivatives were also studied in biological context. One example is the azo-functionalized pyrazolopyrimidine 4.23c, which was applied in cellulo to photocontrol the RET transmembrane tyrosine kinase receptor.623 The photoswitch was investigated next to azobenzene 4.23b, which had a Z form with a lifetime of only 2 min in water, and a stilbene variant 4.23a, which irreversibly decomposed upon irradiation. Photoswitch 4.23c had good photochemical properties in water where it achieved 87% of Z at the PSS365nm with a sufficiently long lifetime applicable for biological studies (τ = 9.7 h. at 37 °C). This example shows how a minor change, such as an additional heteroatom in the ring or a stilbene scaffold instead of an azobenzene, can have a great impact on the photochemical properties, as well as water-solubility (as seen for 4.23, Fig. 51) The azoswitch 4.23c could undergo multiple photoswitching cycles without fatigue in the presence of glutathione, showing its resistance to reduction. Interestingly, the observed difference in activity of the inhibitor of the Z form was 3.8 (enzyme essay) and 3.2 (in cells) times higher than the thermally stable E isomer.
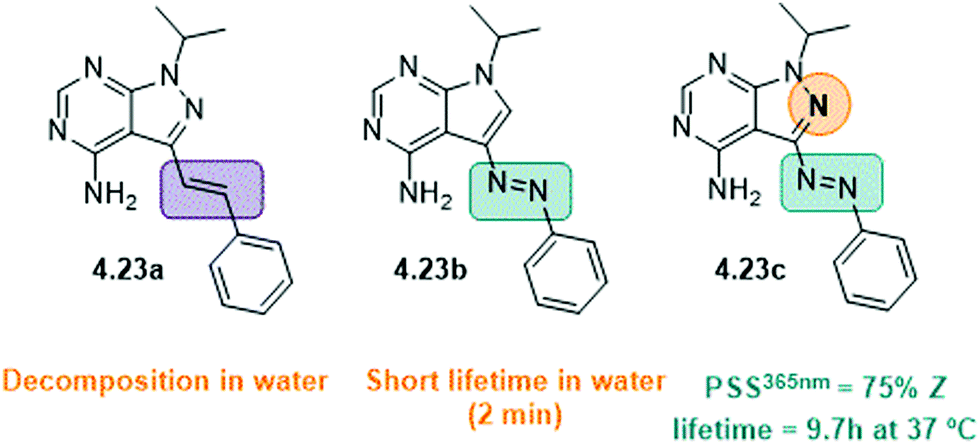 | ||
| Fig. 51 Inhibitors of RET transmembrane tyrosine kinase receptor, illustrating the effect of a different switch system (stilbene 4.23avs. azobenzene 4.23c) and an additional heteroatom (4.23b and c) on the (photo)chemical properties in aqueous solutions. | ||
Like pyrazole, also pyridine motifs can contribute to water-solubility in azobenzenes. For instance, compound 4.24 has been applied to photocontrol GPCRs624 as well as inhibitor of alpha-chymotrypsin. To study this compound sufficient aqueous solubility was crucial to achieve high-enough concentrations to determine the IC50 values.35 Moreover, a set of azobenzene- and azopyridine-based sirtuin inhibitors was designed (Fig. 52). Most of the compounds of the library had the required photophysical properties in water and long thermal half-lives of the metastable Z isomers (>300 h).625 However, phenyl azopyridines carrying a hydroxy or amino group (such as 4.25a and 4.25b) exhibited shortened half-lives due to tautomerization (see Section 3.2.3).626,627 The respective azopyridines were consequently modified by methylation of the aniline (example 4.25d) or introduction of methyl groups in the ortho positions (example 4.25c) as attempt to increase the thermal half-lives. However, this often rendered the compounds insufficiently water-soluble and caused aggregation.36,37 Compound 4.25e, bearing a C7 chain, had better water-solubility which in return affected the inhibitors activity. For longer fatty-acids (such as 4.25f), precipitation during the assay was an issue. Based on these observations, the authors concluded that in some examples the higher inhibitory activity of the Z form, which was found to be up to 21-times more potent than the E isomer, was in fact not due to its helical geometry but due to its better availability caused by higher solubility in water. This example demonstrates the complexity encountered in attempts that aim to encompass excellent water-solubility, improved bioactivity, and maintain advantageous photochemical properties.
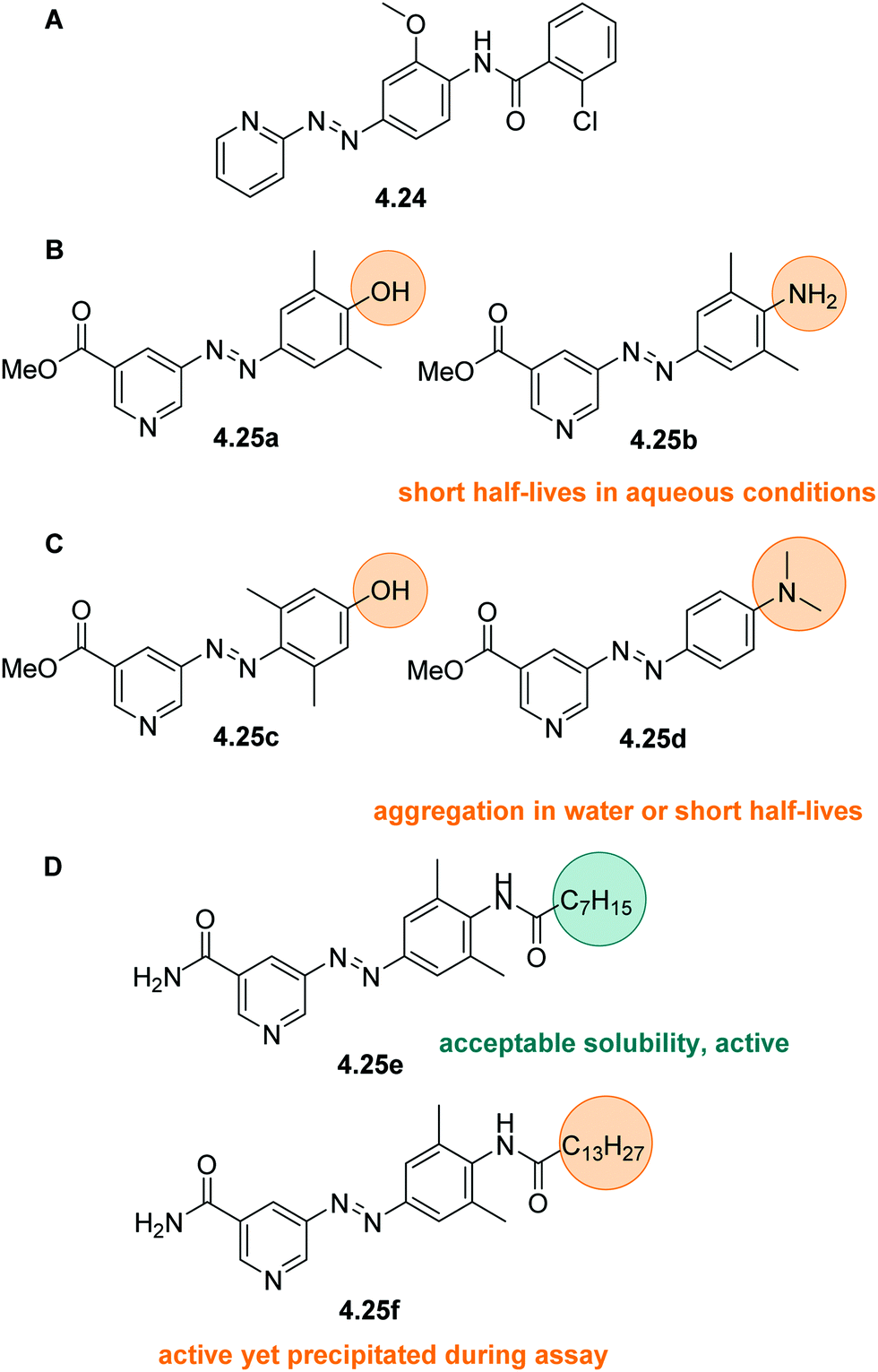 | ||
| Fig. 52 Different substituents in azopyridine-based sirtuin inhibitors and their effect on their water-solubility, (photo)chemical properties and activity. | ||
4.1.3.2 DAE and fulgimides. Classical DAEs are highly apolar structures and, thus, are often decorated with charged groups such as carboxylate and phosphate anions to enable solubilization (vide supra). Alternatively, modification in the core structure or moving to the related class of fulgides or fulgimides can be considered, similar to the heteroaryl design in azobenzenes.157 Especially in DAEs, such structural modifications are affecting the photophysical and photochemical properties significantly (for more details, see Section 3.3.1) and are discussed with illustrative examples in the following section.
In compound 4.27 (Fig. 53), the typically used central cyclopentadiene ring was replaced by a maleimide, mimicking the structure of bisindoylmaleimide 4.26 (Fig. 53), a potent sirtuin inhibitor.335 Introduction of this motif induced a bathochromic shift in the UV-profile in comparison to the cyclopentadiene-bridged analogue. The compound was tested for its inhibitory activity towards Sirt2 and exhibited a 22-fold difference in activity between the open (IC50 4.2 μM) and the closed form (IC50 92.3 μM), featuring a PSD of 98:2 = C:O under irradiation with 312 nm light in DMSO. Due to the TICT taking place, this reaction could not be induced in polar protic solvent, such as the assay buffer, and O → C photoisomerization needed to be performed in the DMSO stock solutions, highlighting another limitation that can be met when certain DTEs are used in aqueous media. However, photoinduced ring-opening was possible in situ.335 Therefore, the use of the structurally related indolyl fulgimides was explored for the same target.408 Fulgimides are more flexible and polarized and hence, in general, better soluble than DTEs in aqueous environment. Moreover, the open forms (Fig. 53, Z-4.28 and E-4.28) isomerize upon 400 nm light irradiation and only the E form further cyclizes to the closed form C-4.28. Several derivatives were studied and showed superior photophysical and photochemical properties compared to the previously developed DTEs, such as reversible photoswitching in the assay buffer. However, 4.28 exhibited only low inhibitory activity with a 1.5-fold difference between the open form and the PSS400nm sample (ca. 30% closed form). Similar observations were made in a later study comparing DTEs based on maleimide and fulgimides as light-controlled dopamine receptor ligands.628 Again, the DTEs showed a larger difference in activity between the two photoisomers (11-fold compared to 4-fold) but could not be isomerized reversibly in aqueous solution. These examples showcase that optimization of photochemically properties and solubility in water by simultaneously retaining the biological potency is not trivial.
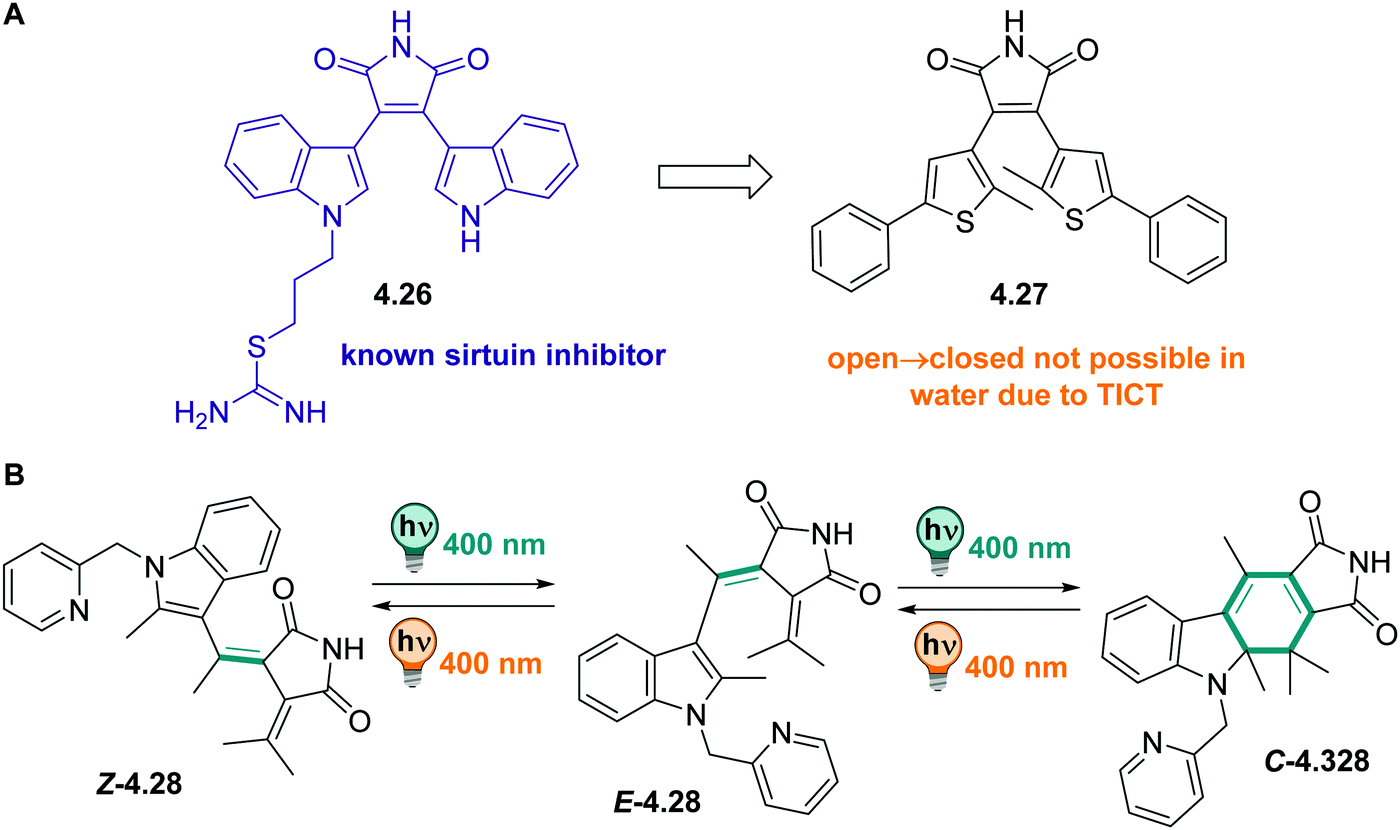 | ||
| Fig. 53 (A) The potent sirtuin inhibitor 4.26 was redesigned into the photoswitchable system 4.27. (B) Since 4.27 could not undergo ring-closing in aqueous solution, the system was further redesigned into photoswitchable fulgimides 4.28. | ||
DTEs and fulgimide photoswitches were also studied together as light-controlled inhibitors of zinc dependent HDACs.418 Avoiding donor–acceptor designs and hence, a TICT like in the examples discussed before, was the key design principle in obtaining a small library of compounds that could be addressed reversibly in polar protic solvents by light. However, some of the DTE derivatives were unstable upon irradiation, especially in aqueous solutions. DTEs 4.29a and 4.29b formed a byproduct upon isomerization, bearing a central five-member ring as displayed in Fig. 54 (see also Section 3.3.1).333,629 While 4.29a very quickly converted almost completely to the byproduct, 4.29b had a higher fatigue resistance and could form 80% closed-ring isomer after 60 s of irradiation with only small amounts of byproduct formed.418 DTE 4.29b showed inhibition of HDAC, however, with small differences in affinity between the photoisomers. As an alternative, fulgide photoswitches were synthesized. Both fulgides 4.30a and 4.30b reached over 95% of the closed isomer at the PSS365nm in DMSO. The properties were similar in buffer with 1% DMSO, however, minor fatigue was noticed. In enzyme activity assays, fulgimide 4.30b showed nanomolar affinity for HDAC, but the differences in activity of the two isomers were low also for these derivatives. Again, the photochemical behavior can be adjusted by modifying the photoswitchable core, a translation to the biological potency of the compounds is however not obvious.
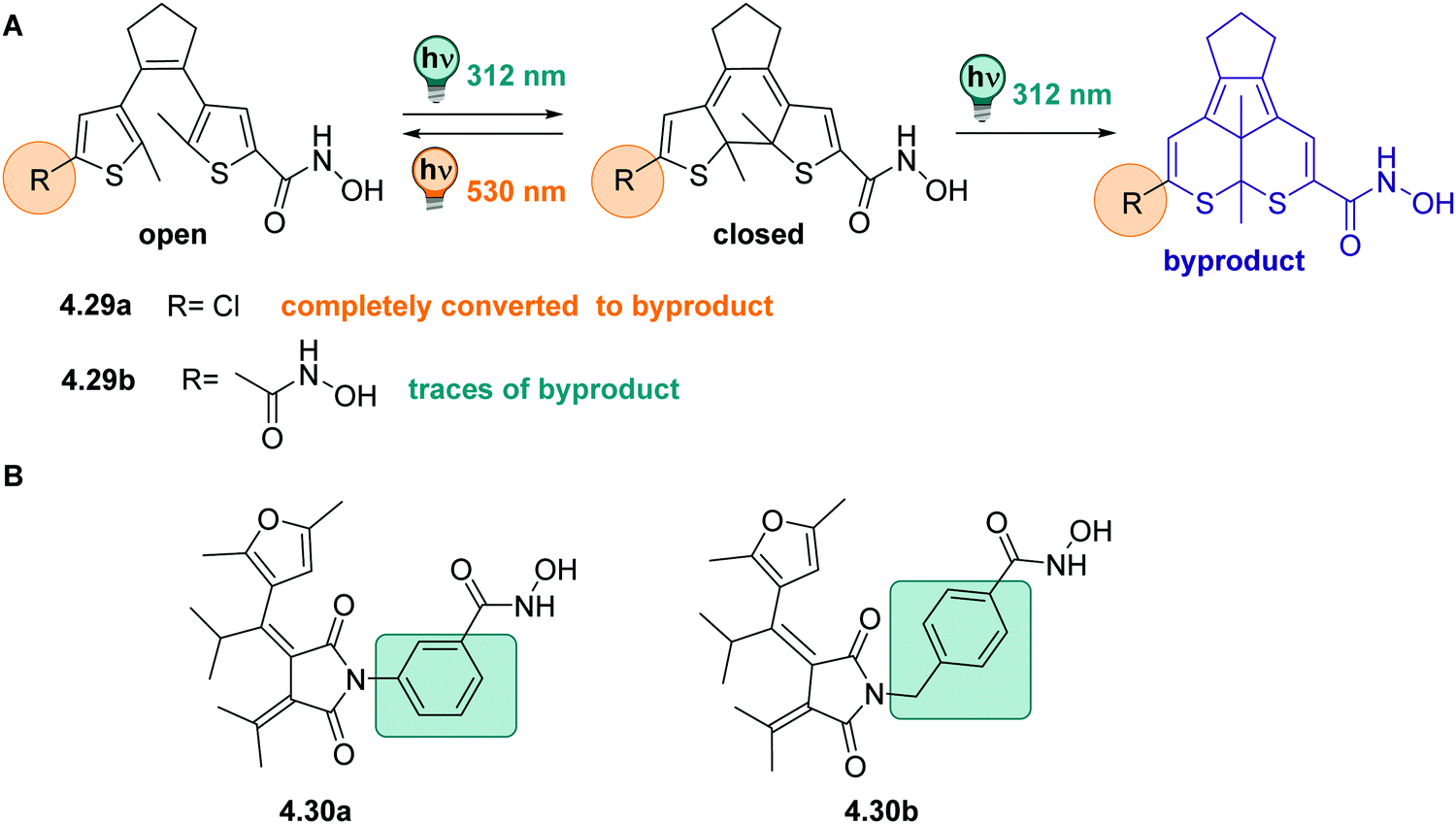 | ||
| Fig. 54 Light-controlled inhibitors of zinc dependent HDACs. (A) DTE 4.29a was unstable upon irradiation in water, quickly forming the byproduct, while DTE 4.29b formed 80% if the closed isomer with minimal byproduct formed. (B) Fulgides 4.30a and b showed excellent photochemical properties in water however the difference in activity was much smaller compared to the DTE switches. | ||
 | ||
| Fig. 55 Breaking planarity to increase water-solubility. (A) Azobenzene-based photoswitches where the Z isomer was 20-times (4.31) or 1.4-times (4.32) more soluble in phosphate buffer then the E form. (B) Introduction of three methoxy groups increases water-solubility and disables aggregate formation. | ||
The planarity in photoswitches can also be affected by attaching suitable substituents to the photoswitchable core. In this way, a library of hemithioindigo inhibitors (Fig. 55B) was designed based on a known compound colchicine.261 While some compounds were not sufficiently water-soluble and caused aggregation and toxicity (4.33a aggregation at 10 μM), the tri-methoxy-substituted compounds (4.33b and 4.33c) dissolved much better in water since the molecule's planarity was broken with a methoxy group sticking out of plane. In this manner, π-staking was prevented, and the water-solubility was increased.261
 | ||
| Fig. 56 Photoswitchable glutamate receptors carrying anionic amino acid moieties. Azobenzene 4.34, featuring an aspartate moiety, could be solubilized at concentrations of 0.1 mM in phosphate buffer (A), while the glutamate azo-derivative 4.35 was poorly soluble in water and required heating and addition of DMSO in the stock solutions despite carrying a glutamate moiety (B). | ||
Alternatively, sugar moieties were attached to photoswitches and lead to significant increase of their solubility in water.640–644 Specific sugar moieties can be recognized by a selective carbohydrate-binding protein from E. coli called lectin.645 In this manner, photoswitchable lectin binders were studied regarding the differences in binding affinities of the photoisomers bearing either a single or multiple sugar groups. (Fig. 57, 4.36a–d)640,641 Incorporation of multiple carbohydrates simultaneously increased binding to lectin and solubility in water.641 The half-lives of the Z forms of these azobenzenes were longer in aqueous environment, compared to DMSO solutions.640 As a general trend, symmetric molecules with sugar moieties on each side (4.36a and 4.36b) isomerized faster than unsymmetric derivatives (4.36c and 4.36d).640 Beyond this study, azobenzene systems with incorporated sugar moieties were used to control bacterial adhesion with light,642 inhibition of beta-galactose,643 and as glycosphingolipids for cancer immunotherapy.644
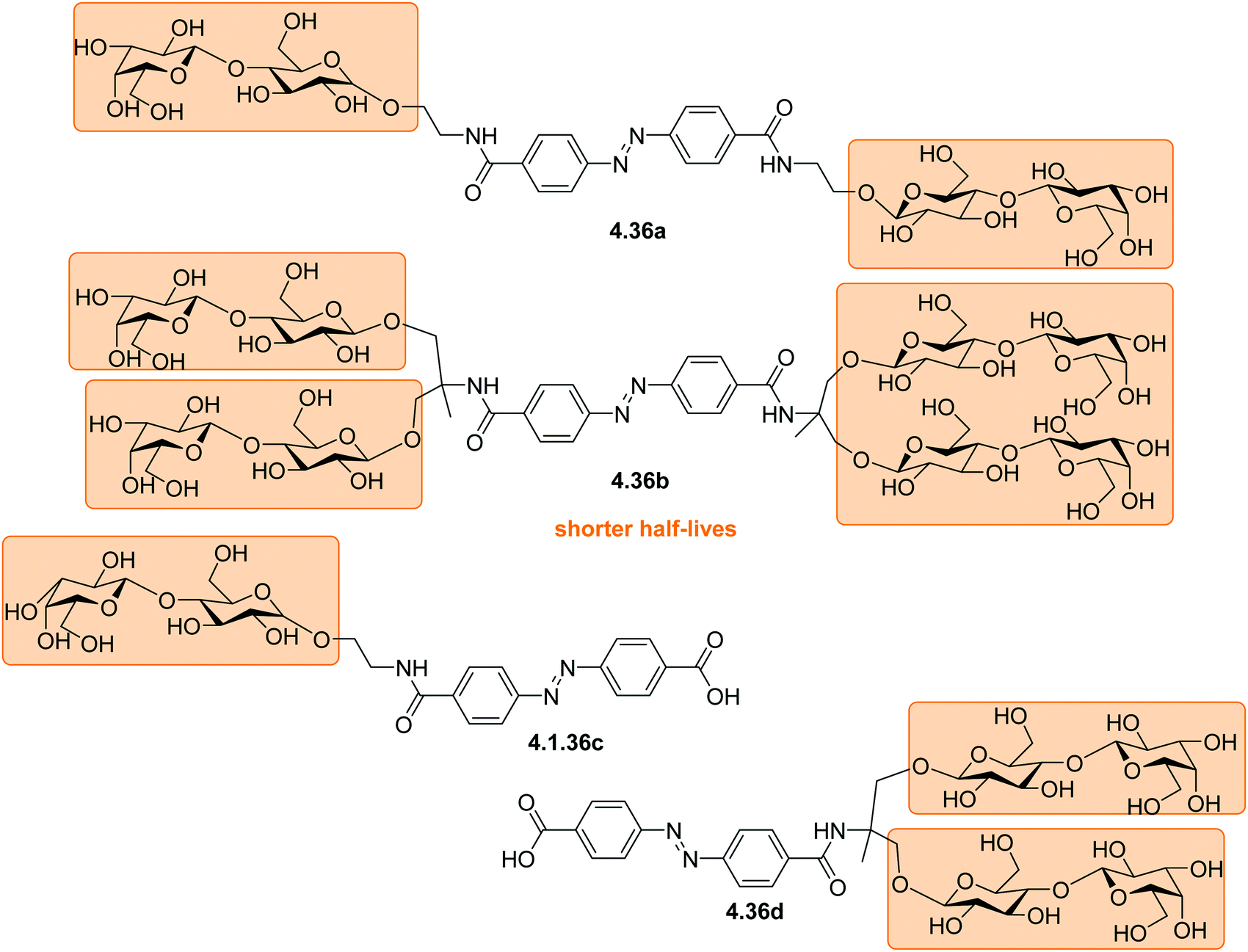 | ||
| Fig. 57 Azobenzene photoswitches decorated with multipole sugar moieties with increased water-solubility. | ||
Besides functionalizing photoswitches with amino acids and sugars, bioactive small molecules, such as antibiotics, were attached to light-responsive compounds. In this manner, ciprofloxacin was connected to an azobenzene and a spiropyran, and both compounds could be readily dissolved and studied in a microbial growth assay medium. (Fig. 58A, 4.37 and 4.38).646 The spirociprofloxacin was efficient against Gram negative Escherichia coli, while the azociprofloxacine showed activity on Gram-positive Micrococcus luteus. The azo version could be switched over multiple cycles without degradation, but the spirociprofloxacin exhibited fatigue in aqueous media, possibly caused by instability of merocyanine in water (see Section 3. Spiropyran).445
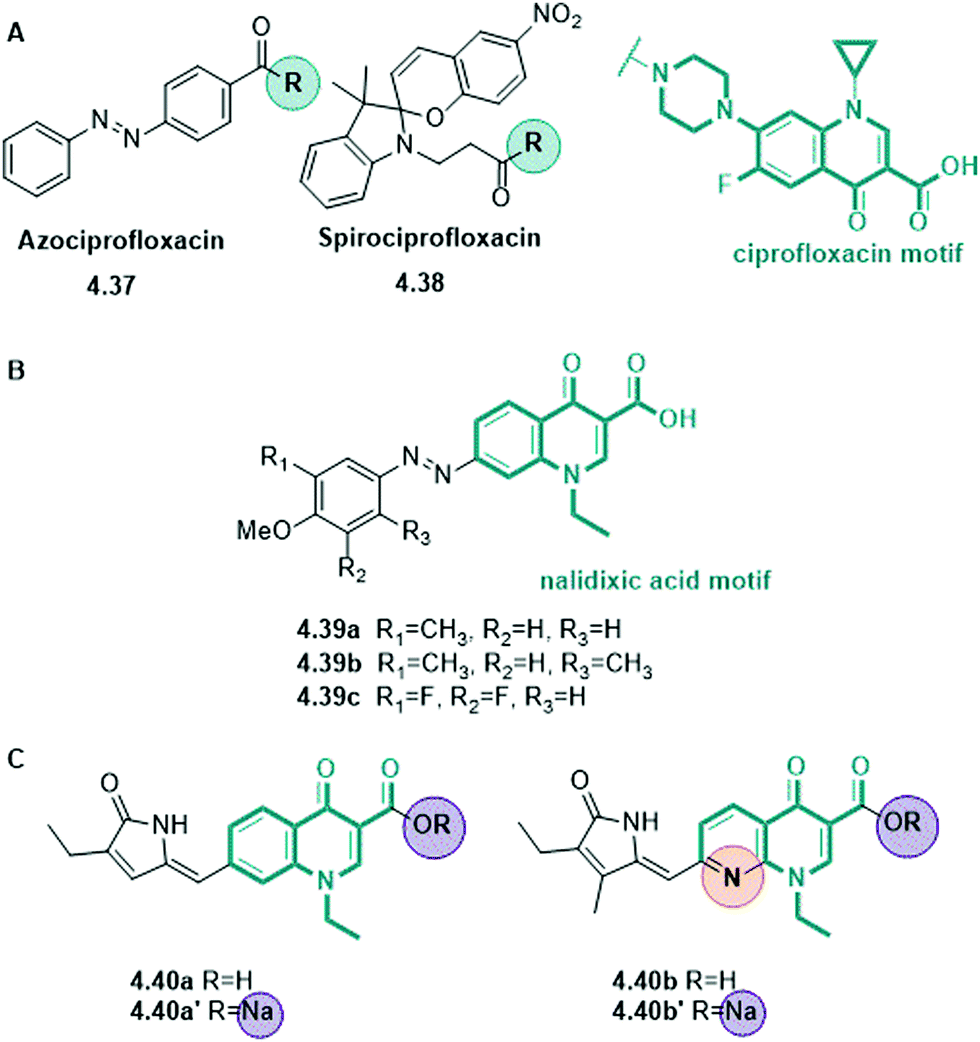 | ||
| Fig. 58 Photoswitchable antibiotics. (A) Ciprofloxacin was modified to incorporate an azobenzene (4.37) or a spiropyran photoswitch. (4.38). Azobenzenes 4.39a–c (B), hydantoin 4.40a and phytochrome 4.40b (C) contain the nalidixic acid motif ensuring antimicrobial activity. | ||
Alternatively, azobenzenes were structurally merged with known antibiotics,6,7,30 such as nalidixic acid (Fig. 58B. structures 4.39a–c)647 The photoswitchable antibiotics were designed for the spatiotemporally control of their activity with light. Specifically, the active Z isomer is temporarily formed upon irradiation and then thermally relaxes back to the inactive E isomer within a few hours. Despite its high potency as antibacterial agents, nalidixic acid has already limited solubility in water.648 Therefore, it comes as no surprise that the MIC value determination of the light-controlled derivatives had to be performed at low concentrations (64 μg mL−1) due to solubility restrictions.647 In another study, using photoswitchable hydantoin649 and phytochrome,650 it was shown that the limiting water-solubility can be improved by transforming the carboxylic acids to sodium salts (Fig. 58C, 4.40 and 4.40b).651
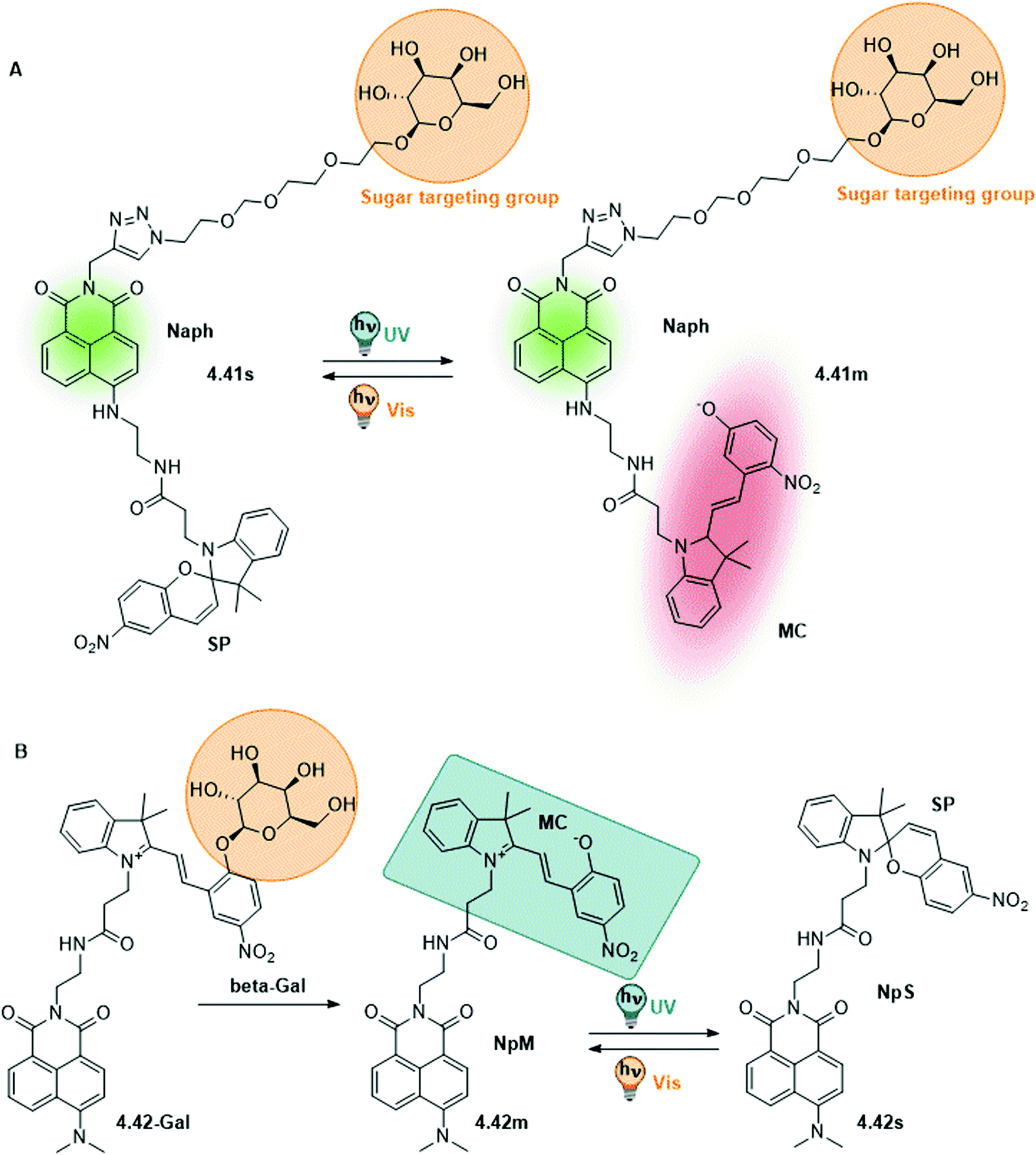 | ||
| Fig. 59 (A) Spiropyran photoswitch with a fluorescent linker and a sugar targeting group. (B) Spiropyran based photoswitch carrying a fluorescent moiety and a galactose group locking it in the merocyanine form. Upon cleavage of the sugar, the photoswitch can reversibly switch from MC to SP form in water. | ||
Very recently, a similar system was reported, where a photoresponsive probe containing Naph and a modified merocyanine were used for high-resolution fluorescence microscopy (see Section 4.3.4) to visualize the distribution of β-galactosidase in cells (Fig. 59, 4.42).661 The probe contained a galactose moiety attached directly to the phenol ring of the merocyanine group, thus preventing photoisomerization to the closed spiropyran isomer. Upon cleavage of the sugar moiety by the β-galactosidase, the red fluorescence increased due to formation of the free merocyanine. Merocyanine could then photoswitch to the fluorescence-silent spiropyran form and was used for high-resolution fluorescence microscopy. The protein–probe complex between the dye and human serum albumin (HSA) ensured solubility in water, minimal phototoxicity, and good cellular uptake.661
4.2 Water-soluble molecular photoswitches as biomolecular building blocks
To achieve light-controlled activity of biomolecules, molecular photoswitches have been designed as biomolecular building blocks, such as amino acids and nucleotides, and were incorporated into the backbone of biomacromolecules.662 Several strategies can be applied and for each there are different requirements regarding water-solubility. For short peptides synthesized by solid phase peptide synthesis (SPPS) on resin, it is feasible to use organic solvents throughout the synthesis. Therefore, there are no requirements for aqueous solubility of the photoswitchable building block, as long as the final product dissolves appropriately under the required conditions. In contrast, aqueous solubility is of great importance for photoswitchable building blocks which need to be uptaken and processed by a living organism before they are incorporated into a biomolecule. Insufficient solubility can induce aggregate formation possibly resulting in toxicity.6634.2.1.1 Genetic code expansion. The 20 canonical amino acids are building blocks of all living beings. Incorporation of unnatural amino acids in vivo offers the possibility to structurally modify proteins and to control and study their activity. Genetic code expansion is a technique that allows the integration of such unnatural building blocks by hijacking the synthetic machinery of the target organism.664–669 Also, genetic code expansion can be applied to introduce photoresponsive building blocks into a complex protein.670–672 It is of pivotal importance that the photoswitchable substrate is sufficiently water-soluble, non-toxic, and successfully uptaken by the organism for efficient incorporation.673,674
In the context of photoswitchable unnatural amino acids, to our knowledge, only azobenzene-based structures have been incorporated via genetic code expansion. Classical azobenzene-based amino acids were used to control DNA binding activity670 and to explore the mechanism of an allostery model bi-enzyme complex.671,672 Furthermore, visible light-controlled azobenzenes with tetra-ortho substituents have been successfully incorporated into proteins such as the green fluorescent protein (GFP)668 and firefly luciferase669 to reversibly control their properties.
Moreover, employing photoswitchable amino acids with an additional thiol-reactive group provides a novel approach of crosslinking peptides. After in vivo incorporation of the functionalized photoactuator into a protein of choice, the unnatural amino acid can react with a nearby moiety, such as a cysteine side chain, and crosslink two sides of the protein (Fig. 59). Both classical azobenzenes with a reactive anchoring group in the meta position (Fig. 60A, 4.43, 4.44)675 and a penta-fluoro-azobenzene amino acid 4.45 (Fig. 60B)676 were successfully incorporated into both E. coli and mammalian cells.
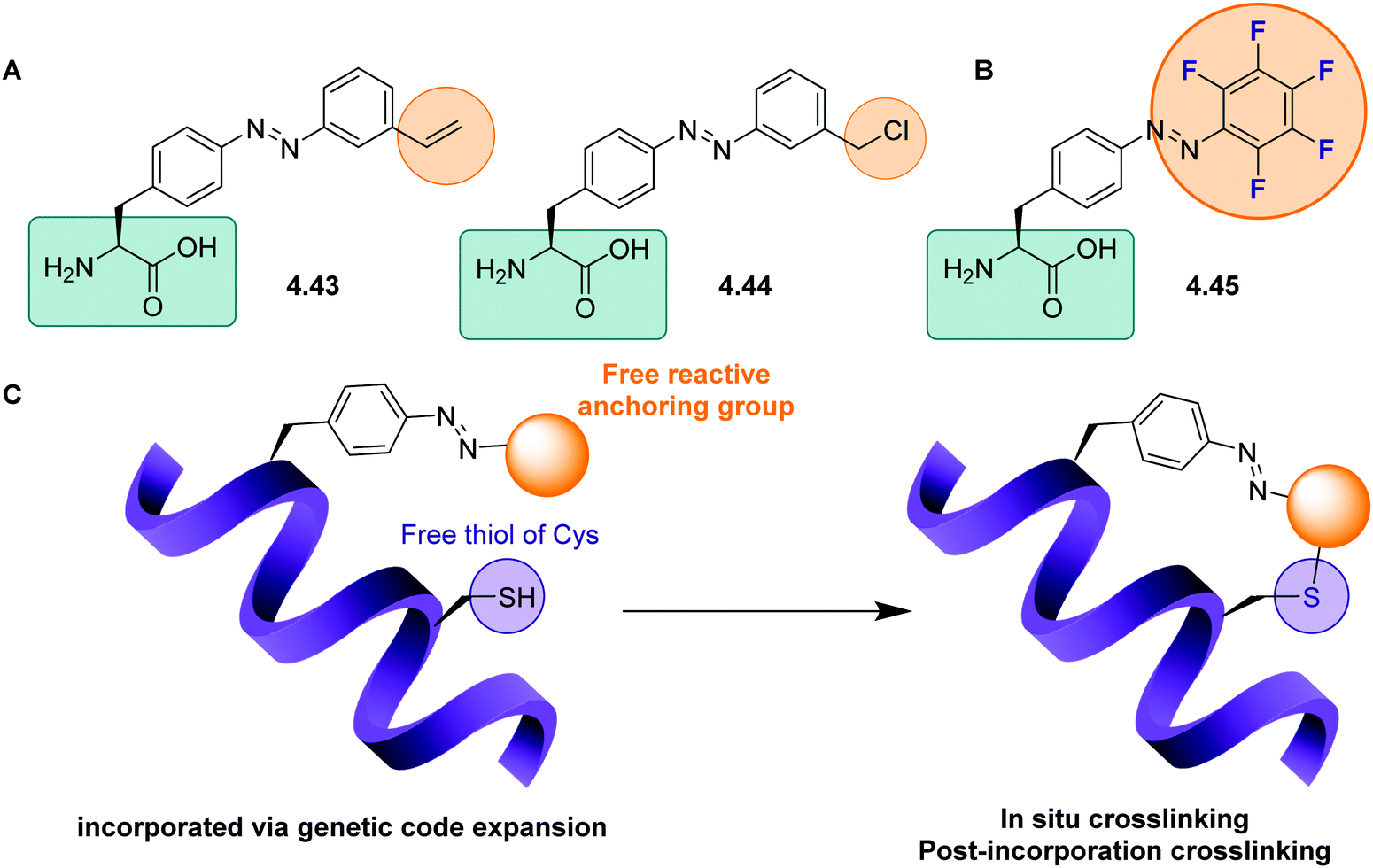 | ||
| Fig. 60 Azobenzene-based amino acids with a covalent attachment point on the opposite side of the photoswitch, both controlled by UV light (A) and by visible light (B). (C) After the light responsive amino acid is incorporated into the protein, the anchoring group reacts with a free thiol moiety nearby thus crosslinking the protein. | ||
4.2.1.2 Solid phase peptide synthesis. Solid phase peptide and oligonucleotide synthesis have been employed to synthesize known biomolecules with unnatural moieties such as photoswitches and even molecular motors,153 on a resin by stepwise coupling of each building block.677–680
Peptides with a photoswitch in the backbone. Azobenzenes with two biphenyl rings were incorporated via SPPS into a highly water-soluble short peptide chain to study the photochemical behavior of the construct in water (Fig. 61A, 4.46).681 The stability towards reduction of the incorporated azobenzene 4.46 was tested by incubation with 10 mM glutathione (GSH) as the environment in mammalian cells is highly reducing.682 No degradation was observed, however, GSH catalyzed the Z → E isomerization of the short model peptide and significantly shortened its thermal half-life. Already 50% of the Z isomer relaxed to the E at 37 °C in 60 min, while in the absence of GSH hardly any isomerization was observed. The mechanism of this phenomenon is yet unknown, though, the authors speculated that a GSH radical or thiolate anion reversibly attacks the azo double bond thereby reducing it and allowing free rotation.682
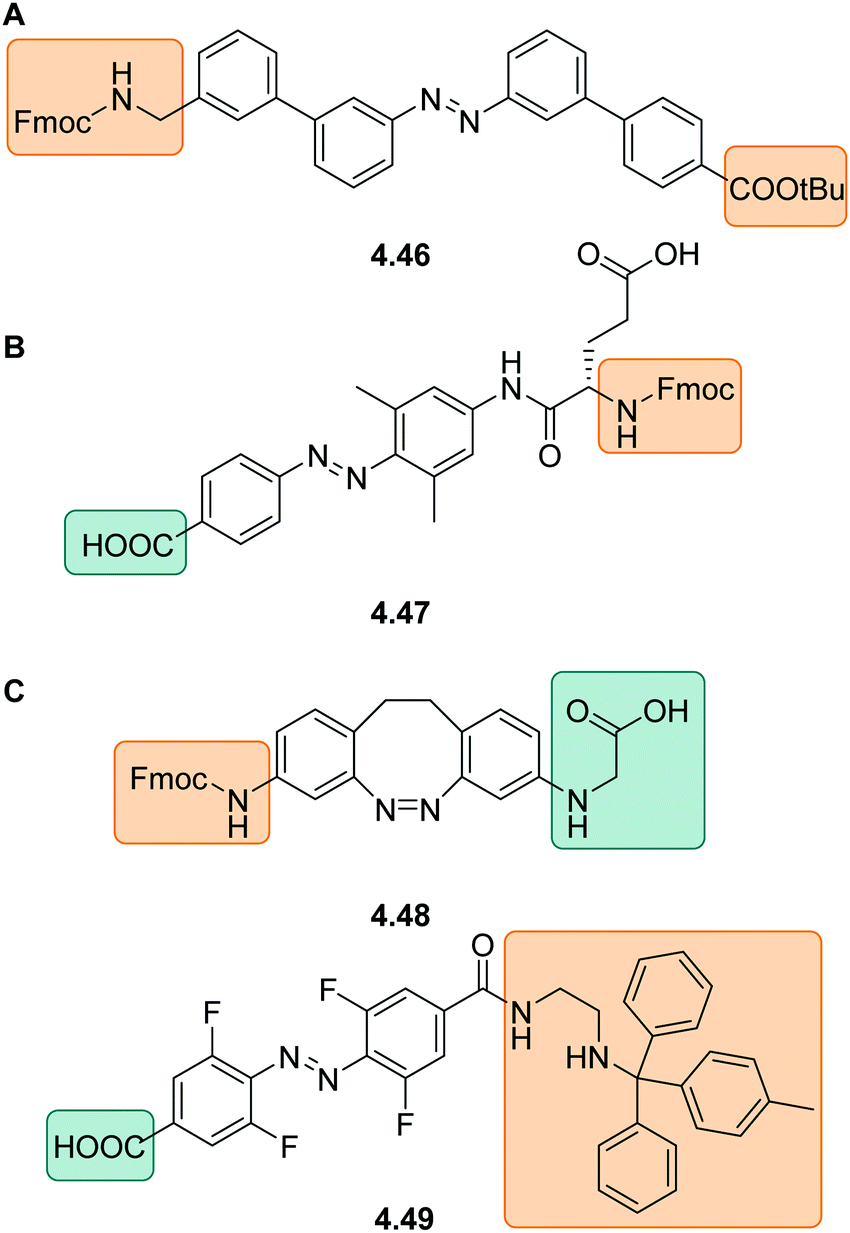 | ||
| Fig. 61 Azobenzene-based amino acids for SPPS with free (amino) acid groups highlighted in green and protected ones highlighted in orange. | ||
While for most classical azobenzenes incorporation via SPPS was successful,681,683 the visible light-responsive tetra-ortho-fluoro azobenzene was a more challenging substrate.684 Both diazocine and tetra-ortho-fluoro azobenzene amino acids (4.48 and 4.49, Fig. 61) were incorporated into a peptide backbone to study protein–protein interactions. While diazocine 4.48 was compatible with SPPS, the fluorine groups of tetra-ortho-fluorinated azobenzene core were prone to nucleophilic aromatic substitution under deprotection conditions required for Fmoc removal during SPPS procedures. Therefore, molecule 4.49 was equipped with a modified trityl group and incorporated into the peptide in solution.
Cell-penetrating peptides. For most photoswitchable short peptides synthesized by SPPS, the water-solubility enabled by the peptide chain is sufficient to use the construct in diverse applications. However, for the class of cell penetrating peptides (CPP), the delicate interplay between water-solubility, lipophilicity and polarity plays a crucial role in their activity.685–687 CPPs are often cyclic, short peptides which are readily uptaken into cells and are further able to carry various cargo.688,689 A major obstacle for clinical use of CPPs is that they can exhibit systemic toxicity and cause side effects due to poor selectivity and disruption of cell membranes.690,691 For this reason, achieving activation with spatio-temporal control using light is an appealing approach. However, introduction of an aromatic, lipophilic photoswitch into the backbone of a CPP has a great influence on its cellular uptake. Hence, the modified CPP needs to compromise between solubility and lipophilicity and needs to take the impact of all isomers of a photoswitch into account.692,693
While in most other applications azobenzenes are frequently used, for CPPs the DTEs completely dominate the field, with only one example of a hemithioindigo analogue.694 DTEs have proven to be the best choice due to the difference in rigidity of the two isomers which can be translated efficiently to photocontrol cellular uptake. Incorporation of a DTE into the backbone of the CPP enables formation of a hydrophobic, flexible open form that exhibits good membrane affinity. In particular, the O-DTE switch serves as a substitute for a β turn in the peptide sequence (Fig. 62A).695 Upon irradiation with light and formation of the more rigid C isomer, the hydrophobicity decreases and significantly deviates from the original CPP, therefore hindering cellular uptake. Additionally, the CPP was modified with fluorescein, attached via a long linker to minimize the effect of the large fluorescent organic molecule on permeability (Fig. 62B, 4.51). While it was shown that CPPs can tolerate attachment of relatively large organic molecules while retaining their properties,692 for 4.51 labeling of the peptide with the hydrophilic fluorophore decreased the cell–membrane permeability.693 Specifically, the open isomer of DTE 4.51 had a high tendency to form aggregates which inhibited internalization.693
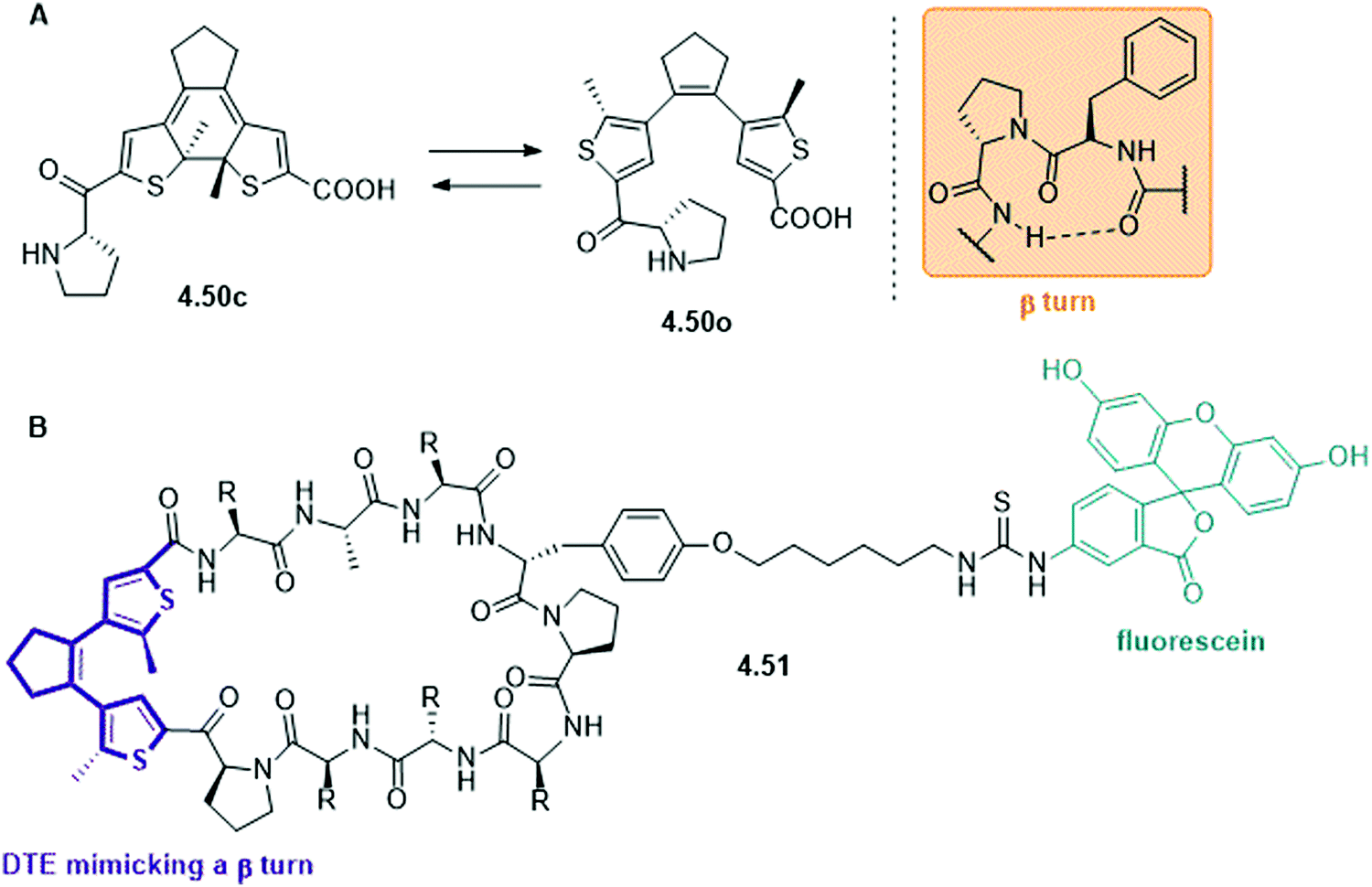 | ||
| Fig. 62 The open isomer of a DTE-based amino acid was used to mimic a β-turn (A) in a cell penetrating peptide by incorporation into the cyclic peptide backbone and modified it with a fluorescein group for visualization (B). The fluorescent molecule was attached via a long flexible linker to not compromise the cellular permeability. | ||
In another approach, gramicidin S (Fig. 63A), a CPP and a short cyclic peptide with strong antimicrobial properties, was used. Besides its use as an antibiotic, gramicidin S is also a potent cancer therapeutic agent. However, it exhibits systemic toxicity showing high mortality rates in mouse studies,696,697 probably due to blood lysis.698 To locally activate the molecule by light and potentially limit side-effects, a DTE has been incorporated into the structure of gramicidin S analogues (Fig. 63B).699 The photoswitchable peptides 4.52 and 4.53 were studied for their antimicrobial and anticancer properties both in cells698 and in living zebrafish embryos.700 As 4.52 does not show significant hemolytic activity compared to other tested peptides, it suggests that hemolysis is not the major mechanism of toxicity. However, this derivative was one of the most hydrophobic ones and was poorly water-soluble, forming aggregates in aqueous solutions.698,700 The second analogue, the gramicidin S derivative 4.53 could be further tested on an animal tumor model701 to successfully target cancer cells in its more cytotoxic open form, while the closed form was 5.5 to 8-fold less active. In contrast to the parent compound, 4.53 was stable in blood and resistant to proteolysis in vivo. Moreover, introduction of light-control by the DTE minimized the toxicity in vitro in mice with eight times lower activity of the closed isomer. Unfortunately, in living mice, the activity of the light-responsive version 4.53 was lower than the one of parent compound (30% compared to 60–80%).696
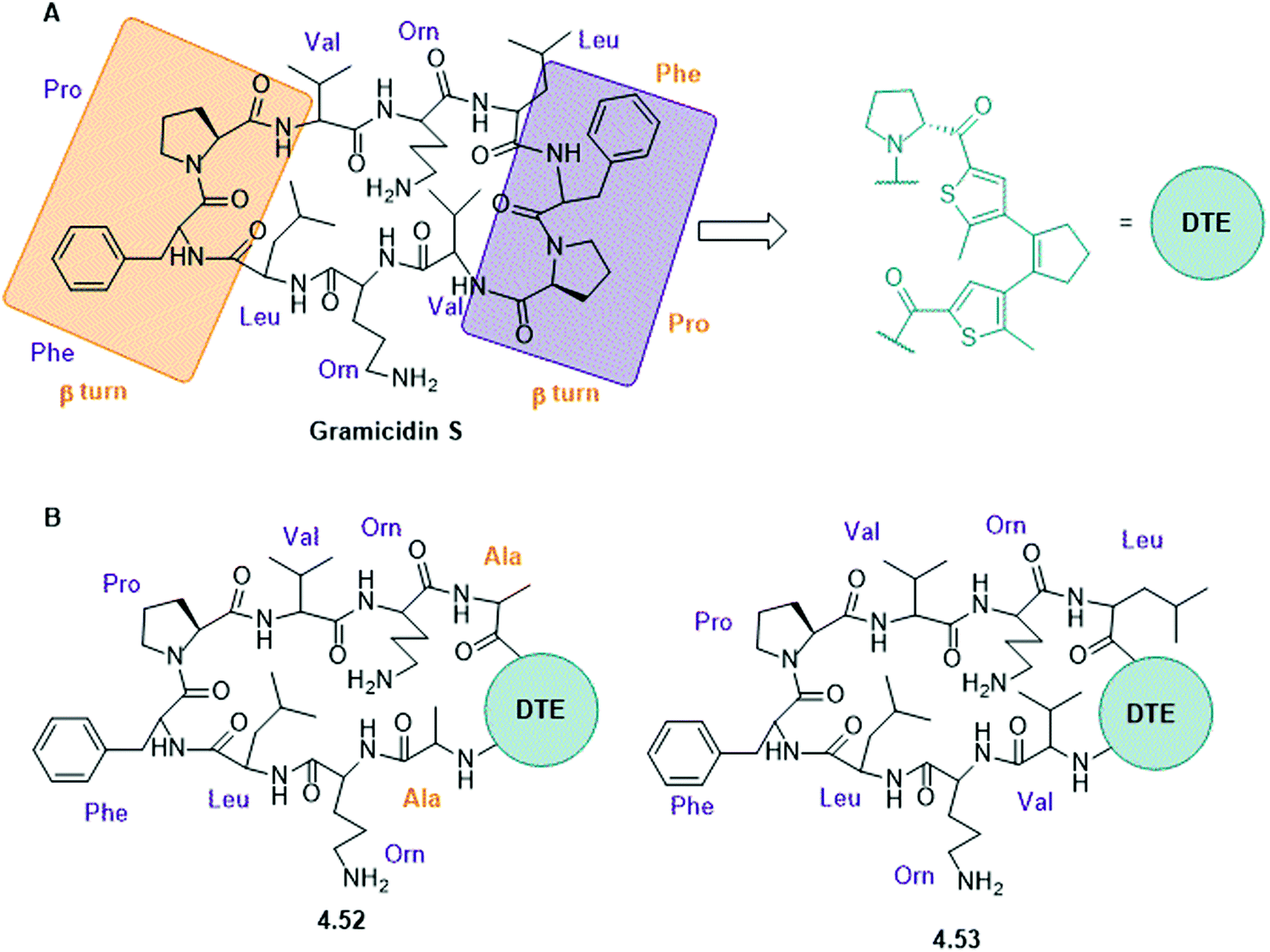 | ||
| Fig. 63 (A) Gramicidin S, a cell penetrating cyclic peptide with antibiotic and anticancer properties contains two β-turns (in orange), of which one (in blue) was replaced with a photoswitchable DTE amino acid. (B) Two modified photoswitchable Gramicidin S analogues, where the amino acids shown in orange, are substituted in the original structure CPP 4.52 was the less water-soluble design due to too high hydrophobicity while 4.53 was the most successful and subsequently was tested in animal studies. | ||
Different azobenzenes have been successfully incorporated both into DNA703,704 and RNA.704,705 Here, the E isomer is able to participate in π-stacking and allows helix formation, whereas the Z form destabilizes the helix. Azobenzene-based nucleosides 4.54p and 4.54m were used to photocontrol RNA duplex formation and they readily dissolved in PBS buffer (5–7 μM, pH = 7.4) also as nucleosides up to 0.6 (4.54p) and 0.4 mg mL−1 (4.54m). The photochemical properties were excellent, reaching a PSD365nm of 91% Z for 4.54p and 88%Z for 4.54m (Fig. 64)705 and exhibiting long thermal half-lives of the Z isomers (97 h for 4.54p and 113 h for 4.54m, PBS buffer at 37 °C). However, incorporation into the RNA strands affected the thermal stability of the Z forms. By trend, the half-lives of the RNA-azobenzene constructs were longer than for the non-bound nucleosides (138 h for 4.54p, 196 h and 155 h for 4.54m, depending on the used RNA strand), except for one example, namely 4.54p, which had a shorter thermal half-life when incorporated into RNA (86 h). This suggests that both the location and orientation of the photoswitch within the RNA strand influences the thermal relaxation rate. This finding was confirmed in a similar study, where the thermal Z → E isomerization was generally faster when the switches were incorporated into DNA compared to RNA.704 Recently, a visible-light-controlled system has been reported, where a tetra-ortho-chloro azobenzene structure 4.55 was incorporated into the backbone of RNA.706
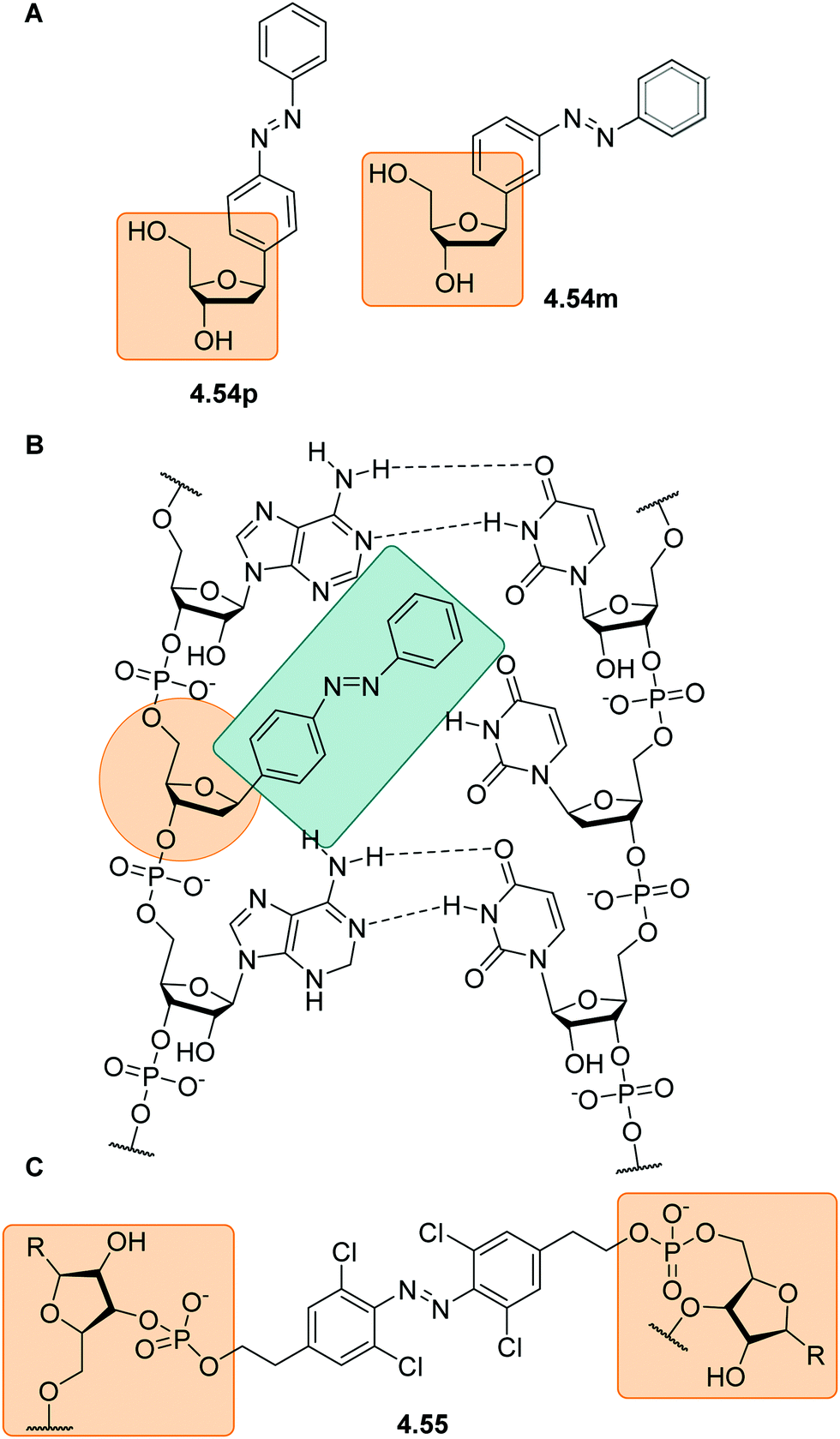 | ||
| Fig. 64 (A) Azobenzene-based nucleosides 4.54p and 4.54m used to photocontrol RNA duplex formation and their location upon incorporation into the RNA strand (B). Visible light-responsive tetra-ortho-chloro substituted azobenzene 4.55 incorporated into RNA (C). | ||
A photoswitchable nucleotide based on a DAE with an adenosine instead of the typical thiophene group (4.56, Fig. 65A) was designed to form Watson–Crick base pairs.707 In 4.56, very low amounts of the closed isomer could be obtained in aqueous solvents. Consequently, the design was improved by using an uracil or cytosine group instead of adenosine, as the pyrimidine-based systems could reversibly switch in aqueous environments. (Fig. 65B, 4.57-C and -U) The photochemical properties were tested in a water/ethanol mixture (9/1) in which the compounds could be dissolved up to 300 μM. These systems showed no fatigue over several irradiation cycles (60 μM, 366 nm and visible light) and a PSD366nm of 83%C (4.57-U) or 98%C (4.57-C), respectively.708 While the closed form of 4.57-C quickly switched to the open isomer at room temperature, 4.57-U was stable after 60 min at 90 °C. Interestingly, after incorporation into a DNA strand the photochemical properties of 4.57-U remained unaffected.
 | ||
| Fig. 65 Photoswitchable nucleotides with an incorporated DEA switch. DEA 4.56 (A) with an adenosine did not undergo efficient cyclization reaction in water, while the uracil (4.57-U) and the cytosine (4.57-C)-based DEA nucleotides had favorable properties in aqueous solutions. | ||
Spiropyrans have been widely applied as DNA intercalators (see Section 4.1), however, they are rarely being covalently attached to DNA702,709,710 due the hydrolytic instability of many derivatives in aqueous conditions.711 Spiropyran 4.58 (Fig. 66A) was attached to the 2′-position on a nucleotide, placing the switch in the minor groove of the DNA strand.712 The close proximity to the DNA bases appeared to quench the triplet state involved in the photoswitching process of the spiropyran by energy or electron transfer.712 To avoid this issue, a 6-nitrospiropyran 4.59 was designed.450 Nucleobase analogue 4.59 was incorporated into DNA on resin and in the final construct could form up to 40% of the open merocyanine form upon irradiation. It exhibits an S configured C3′ as a stereogenic element providing the adequate geometry to match the helical backbone structure of DNA. As 4.59 hydrolyzed in its MC form, its analog 4.60 was synthesized to further improve the design. Its synthesis via the phosphoramidite route was inefficient, therefore, the reversible aldol reaction was used to substitute the aldehyde part 4.60ald by post synthetic modification of the spiropyran while it was already attached to DNA (Fig. 66B). The surrounding DNA bases had a positive effect on the thermal stability of MC-4.60 increasing its lifetime from a few minutes in the non-bound spiropyran unit, to more than two hours when incorporated into the oligonucleotide.450
 | ||
| Fig. 66 (A) Spiropyran 4.58 was attached to the 2′-position on a nucleotide in the minor groove of the DNA strand, placing it too close to the DNA bases thus diminishing its photoswitching properties. The design was improved by attaching the spiropyran 4.59(B), which could form 40% of the open merocyanine isomer upon irradiation which was prone to hydrolysis. By utilizing the reversible aldol reaction, the aldehyde part of the photoswitch 4.59 was replaced with 4.60ald after it was incorporated into DNA, resulting in 4.60 with improved thermal stability. | ||
4.3 Covalent attachment to biomolecules
Photoswitches have been covalently attached to different biological targets at strategically selected locations to photocontrol bioactivity and for fluorescent labeling.634,702,713,714 For achieving photocontrol over the target biosystem, the photoresponsive molecule can: (1) act as a steric obstruction in one of is configurations; (2) have one side of the light-controlled molecule acting as a ligand, which can access the binding pocket of the biological target in only one configuration; or (3) induce a change in charge upon switching, which disturbs the activity or stability of the biological target. For some smaller systems, such as peptides obtained by solid phase peptide synthesis, the use of organic solvents to introduce modifications is often customary and it permits attachment of water-insoluble organic molecules (see Section 4.2).662 However, larger and more complex biomolecules are more sensitive to environmental conditions.715–718 Hence, aqueous solubility of the label becomes of key importance, as too high amounts of organic co-solvent can potentially unfold, inactivate or even destroy the target (biomacro) molecule.715,719–721 Moreover, covalent attachment of a photoswitch should not cause aggregation of the modified target. Finally, for labelling or application of the modified biomolecule in cellulo or in vivo, (partial) insolubility, aggregation and the use of organic co-solvent are not tolerated.Over the past decades, an extensive toolbox of methods for covalent attachment of small organic molecules to various proteins, has been developed. Labeling can be performed in both random and highly specific ways, depending on the application requirements.722,723 Typical chemical methods, which were also successfully used to attach photoswitches, are trapping of natural or artificially introduced cysteine side-chains with α-halogen-acetyl or maleimide functions,724 reactions of lysine residues or the N-terminus of peptides with different reactive groups such as indoles or activated NHS esters,725–727 (photo)click reactions on biomolecules pre-functionalized with an alkyne or azide unit or the Staudinger–Bertozzi ligation,728,729 among others. Except for chemical modifications, there are various enzymatic labeling techniques available such as sequence recognition-based sortase A ligation, subtiligase that catalyzes formation of peptide bonds, or different transferases, ligases and oxidoreductases.730–732
Having chosen the appropriate covalent attachment strategy, a suitable location where the photoswitch will be placed has to be selected. For single covalent attachment, the site the photoswitch will be placed is of great importance because the structural change of the switch must directly interfere with the target's activity. In contrast, for crosslinkers, the general strategy to employ the switch is to reversibly disrupt and/or stabilize the secondary structure of the target protein (vide infra).
 | ||
| Fig. 67 Schematic representation of the role of the photoswitch when interfering with the target biosystem itself, or its interaction with another entity (neurotransmitter molecule, small biomolecule interacting, bacterial cell, etc.). | ||
:95, DMSO). The formed Z isomer did no longer hinder the activity of FraC enabling successful lysis of blood as well as cancer cells selectively upon irradiation.
 | ||
| Fig. 68 (A) Azobenzene photoswitches with water-solubilizing groups (4.61c and d) were successfully used to photocontrol nanopore assembly of the FraC toxin. (B) Azobenzene 4.62, carrying a mannoside group, was incorporated onto the surface of human cells; the Z isomer did not allow bacterial adhesion, while the elongated E isomer enabled it. | ||
Alternatively, azobenzene-based pendants can be used to control cell–cell interactions. Also in this example, the water-solubility of the photoswitch-labels is crucial, since labeling takes place in vivo. Within a living organism, insolubility of the organic molecule can cause aggregation of the bioconjugate, exhibiting fatal toxicity for the living organism.55,736 In this context, the surface of human cells was equipped with azide-handles by metabolic oligosaccharide engineering (Fig. 68B).737 These handles served as an attachment point for glycosylated azobenzene 4.62via its alkyne moiety, in a copper-catalyzed click-reaction.737 (Fig. 68B).738 Azobenzene 4.62 readily dissolved in buffer, due to the presence of the mannoside and thus allowed for the click reaction to efficiently proceed in the cell medium and for successful labeling of the living human cells. The sugar unit also carried a functional role for cell–cell interaction with E. coli cells via lectin (compound 4.62 in Fig. 68B). If the photoswitch is in its Z form after irradiation with UV light, the mannoside is sterically not accessible for bacteria, which in turn cannot bind to the human cell (see Fig. 68B). Upon irradiation with 488 nm light, the E isomer is recovered, and its straight geometry presents the sugar moiety that now points away from the human cell membrane allowing bacterial adhesion. The aim of this fundamental study was to elucidate the mechanism of cell-to-cell interactions by a light-responsive tool and to further increase the understanding of bacterial adhesion to human cells. The photophysical properties of 4.62 were only reported in DMSO, showing high PSDs (PSDE→Z (λ = 365 nm) >90% Z) and long thermal half-lives of the metastable isomer (>30 h).737
4.3.2.1 Photoswitchable labels generating a charge upon switching. In an illustrative example a spiropyran switch, that photoisomerizes to the charged merocyanine form was used to control the mechano-selective channel of large conductance (MscL) with light.740 MscL is a channel protein in the bacterial membrane serving as a safety valve that opens upon increased pressure in the cell.741 It was reported that introduction of charged groups inside the hydrophobic pore causes the channel to be permanently open.742 Thus, attaching a spiropyran photoswitch that can be reversibly toggled between uncharged and a charged form by irradiation with light results in photo-controlled opening and closing of the channel (compound 4.63, Fig. 69A).740 Not only the charge repulsion forces the channel to open, but also the hydrophilicity of the cavity is enhanced, thus attracting water into the channel. Five photoswitchable labels were attached to the channel protein via a halo acetyl moiety connected to a cysteine side chain introduced (using genetic modification) and precisely positioned in the constriction zone of the protein channel. Sufficient solubility of the functionalized photoswitch was achieved by treating the matrix-bound MscL protein with a solution of 4.63 in water, using 20% DMSO as organic cosolvent. In this example, the presence of co-solvent was tolerated due to the stability of the protein and the labelling being performed while the MscL was bound via histidine tag to the Nickel column.740,743 The 4.63-modified MscL channel was also incorporated into liposomal vesicles for on demand light-triggered drug delivery.743 The protein channel remained closed in the dark and upon irradiation with UV light the channel opened due to the formation of the charged merocyanine 4.63m.
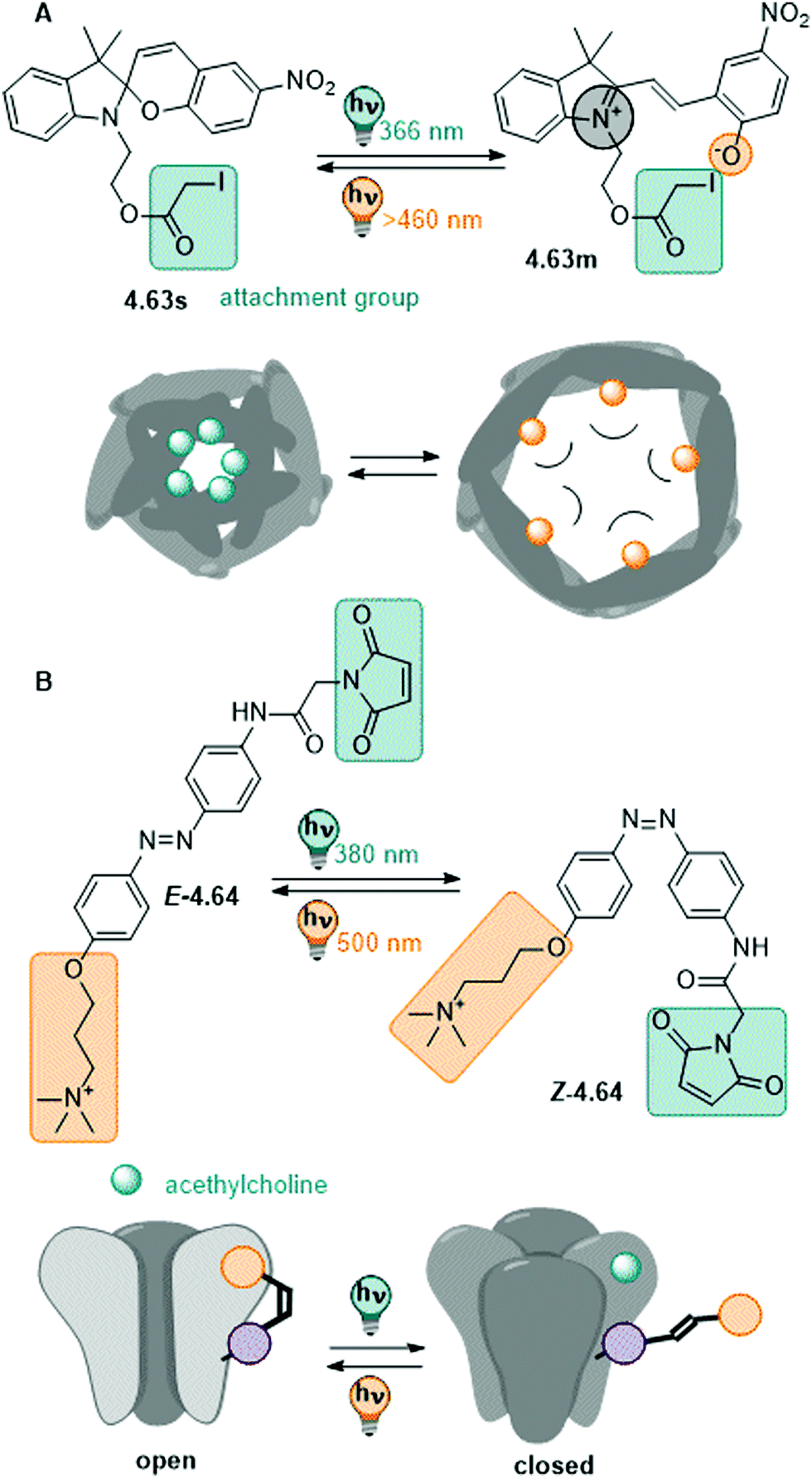 | ||
| Fig. 69 (A) Five spiropyran 4.63 units covalently attached to the MscL channel. Upon photoisomerization the charged merocyanine isomers force the channel to open, while the uncharged isomer 4.63 allows the channel to remain closed. (B) Azobenzene carrying a homocholine group was attached to the ligand-gated channel nAChR in the Z orientation preventing the binding of the natural ligand acethylcholine and leaving the channel open. Upon irradiation with 380 nm light the E isomer does not block the binding site of acethylcholine resulting in closure of the channel. | ||
Though the use of spiropyran photoswitches as a masked charge carriers to photocontrol membrane channels is a very elegant approach, it is not frequently used. This might be due to the fact that it is responsive to multiple stimuli in addition to light, such as pH and heat (see Section 3.4.2), which makes spiropyrans less controllable than other switches, like azobenzenes. However, its unique switching of polarity/charge, together with inverse photochromism in the visible range, make spiropyran a valuable photoswitchable label and control element.
4.3.2.2 Photoswitchable labels functionalized with ligands. Ligand-gated channels have been a target for covalent single-point attachment of photoswitches for decades and this methodology is now established as the most frequently used strategy when using photoswitches as covalent label.744 The first attempt was already reported in the 1980s, where a reduced disulfide bridge was alkylated with a bromomethyl-azobenzene.745 Since then, this strategy was further developed, employing various photoswitches decorated with different bioactive moieties, such as the positively charged tri-alkylammonium group. Especially, the cysteine thiol-maleimide reaction has been used to introduce various azobenzenes with specific binder moieties attached to the photoswitch. In this manner, light-responsive ligand-gated channels, such as the G protein-coupled dopamine receptor,746 the gamma-aminobutyric acid (GABAa) receptor,747 and ionotropic glutamate receptors, which feature a glutamate-modified azobenzene, have been developed.748–753
Another successful application in this context is the photocontrol of neuronal nicotinic acetylcholine receptors (nAChRs).754 The nAChRs are ligand-gated channels present in the mammalian nervous system, that are presumably involved in nicotine addiction and various psychiatric disorders.755 To regulate these channels with light, azobenzene-pendants bearing a positively charged homocholine moiety were designed (Fig. 69B, compound 4.64).756 Homocholine acts as agonist towards nAChRs and competes with the naturally present neurotransmitter acetylcholine, which contains a trimethylammonium group as binding motif.757 The E form of the antagonist 4.64 does not occupy the binding pocket, therefore allowing the naturally present neurotransmitter acetylcholine to bind and activate nAChR.754 Upon irradiation, the homocholine moiety of the formed Z isomer blocks the binding pocket, causing inhibition of nAChR. Herein, the homocholine carries a dual role, both acting as an antagonist of nAChR and as a water-solubilizing group for photoswitch 4.64. Moreover, due to its aqueous solubility, azobenzene 4.64 could be successfully applied in vivo where it was incorporated into the specific reward system region of the mouse brain. Here, viral transfection was used to introduce the cysteine mutant of nAChR in vivo, which was subsequently labeled with 4.64.758
Though compound 4.64 could be used for initial studies in mice, photoisomerization required illumination with UV light. As this is damaging for tissue and has limited penetration depth, researchers strive to find better solutions in the long-term moving to photoactuators responsive to visible and NIR.195,215,537,759,760 A great challenge in this context is to redesign photoswitches or modify them in a suitable manner while retaining the necessary water-solubility as showcased in the following example.
A strategy to circumvent UV light, is to use near-infrared light two-photon (2p) excitation to induce E → Z isomerization.761,762 The first reported maleimide-azobenzene-glutamate probes (4.65, Fig. 70A) themselves had a very limited response to two-photon excitation.763 To improve this, a 2p absorbing antennae, namely a naphthalene or pyrene moiety, respectively, was attached (Fig. 70B), compounds 4.66a and 4.66b.763 The large aromatic antennae rendered the whole assembly poorly water-soluble, impairing both the efficient attachment to the protein and the binding to the glutamate receptor.763,764 Consequently, the antenna strategy was not perused further. Instead, an electronically asymmetric azobenzene core was designed recently, which was reported to be a more efficient 2p absorbing variant of 4.64.765 This structure, bearing an electron-donating group at the para, as well as fluoro substituents in ortho positions, significantly enhanced the 2p absorption (Fig. 70C, structures 4.67avs.4.67b). Avoiding the necessity for including insoluble aromatic antennae in their structure, these probes could be covalently attached to the receptor in sufficient yields.765 They showed thermal isomerization half-lives of around 10 min in aqueous conditions (1% DMSO in PBS buffer). The new probes 4.67a and b could activate the ionotropic glutamate receptor reversibly and, in contrast to the previous designs, allowed studies both in mature neurons in vitro as well as in vivo in C. elegans due to the enhanced water-solubility.
 | ||
| Fig. 70 The maleimide-azobenzene-glutamate probe which can undergo two-photon (2p) excitation thus enabling photoswitching with near-infrared light, 4.65, had a limited response to two-photon excitation (A). Therefore, 2p absorbing antennae were introduced to the design by incorporating naphthalene (4.66a) or pyrene (4.66b) groups (B). However, the large aromatic antennae drastically reduced the water-solubility of the constructs leading to the design of 4.67 (C). Azobenzene 4.67b contains an electron-donating group at the para position and fluoro substituents in ortho position which significantly enhance the 2p absorption. | ||
 | ||
| Fig. 71 Fluorescence photoswitching for super-resolution microscopy. (A) Fluorescence switching of cyanine dyes in the presence of primary thiol-based reducing agents or TCEP; (B) fluorescence switching in functionalized rhodamine dyes which undergo a reversible transition from open to closed form (and vice versa) from the fluorescent zwitterionic form to the dark lactone. | ||
Besides cyanines, rhodamines and spyropirans, DTE switches have been successfully applied to super-resolution spectroscopy. DTEs are known for their high thermal stability of both the open and closed-ring states, as well as for their good fatigue resistance enduring numerous switching cycles (see Section 3.3.1).331 These properties make DTEs applicable for high-resolution fluorescence microscopy, such as reversible saturable optical linear fluorescence transitions (RESOLFT), where switchable fluorescent probes are required for consecutive activation/deactivation by short laser pulses772–775 However, the main obstacle for using DTEs in bioimaging is their intrinsically hydrophobic core, which renders them insoluble in water. Despite various attempts to suitably functionalize DTEs, it has been challenging to obtain fully water-soluble switches.774 Introduction of sulfonates774 or sugar moieties776–778 and incorporation of PEG chains proved insufficient779 to reach this goal. While the PEGylated DTEs were soluble in water, they were unstable in aqueous media and tended to self-associate, which led to fluorescence quenching.779,780 Furthermore, attachment of the lipophilic DTE to a protein to acquire solubility through the target does still not provide full water-solubility, and significant amounts of co-solvents (30% EtOH in water) are needed as shown for fluorescence probe 4.70 (Fig. 72A).781
 | ||
| Fig. 72 DTE fluorescent probes are lipophilic molecules that require significant amounts of co-solvents during attachment to the target protein (30% EtOH in water for 4.70). DTE derivatives containing benzothiophene dioxide groups with perfluorocyclopentene as the bridging unit were decorated with carboxylic acid moieties to enable sufficient water-solubility (B). Multiple carboxylated probes were sufficiently soluble for application in in celullo STORM fluorescence imaging. | ||
More recently, DTE derivatives containing benzothiophene dioxide groups with perfluorocyclopentene as the bridging unit were decorated with carboxylic acid moieties to enable water-solubility (Fig. 72B, 4.71 and 4.72).371 These compounds are highly fluorescent in the ring-closed and non-fluorescent in the ring-open isomer.782 DTEs 4.71 and 4.72 can be dissolved in an aqueous buffer in up to micromolar concentration, without the need for the addition of cosolvent. Both switches retained their photochromic properties in water, such as reversible switching with UV and visible light over numerous cycles. However, fluorescence QYs decreased and emission lifetimes were shortened by ca. 20–30%, compared to those in organic media.371 For covalent attachment to free lysine residues of a secondary antibody, one of the acid moieties on the DTE was converted to an NHS ester.201 The most abundant antibody type, immunoglobulin G (IgG) regularly features around 16 lysine amino acids exposed to the solvent and available for modification.783 Therefore mouse-specific secondary antibodies, which are antibodies that bind to all other antibodies produced by mice, were labeled with DTEs 4.71 and 4.72 and used in RESOLFT microscopy imaging of tubulin filaments and nucleopore complexes.784 Even though the DTE switches could be successfully applied for RESOLFT methods, single-molecule localization microscopy (SMLM) could not yet be achieved with compounds 4.71 and 4.72.785 For SMLM, the ‘blinking’ fluorophores require a very high fluorescence QYs, high brightness, and an efficient ring-closure reaction to efficiently switch off the fluorescence. To increase the electronic push–pull character between the bridging unit and the aryl rings, electron-donating groups, i.e. para-methoxy substituents in the phenyl moiety (4.73 and 4.74 in Fig. 72B), were introduced to improve the photophysical properties of the dyes. The probes were successfully attached to secondary antibodies via the NHS ester attachment strategy and applied to obtain clear STORM images of tubulin and vimentin structures in cellulo.655 This development demonstrated how optimizing the water-solubility of a given photoswitch, together with fine-tuning its photophysical properties, lead to the rapid development of a highly efficient dye for fluorescent imaging of living cells with super-resolution microscopy.
Since the photoswitch is often attached to a comparably large water-soluble protein, in most cases the solubility of the biomolecule is enough to dissolve the construct. Thus, lipophilic crosslinkers can be used, as long as the protein can tolerate the required amounts of organic co-solvent.254,789 The latter is very target-dependent and, unfortunately, not feasible for some proteins. In these cases, water-soluble photoswitches need to be applied to avoid, for instance, aggregation of the construct.
4.3.5.1 Water-insoluble switches. Hydrophobic azobenzene switches, like the frequently used 4.75 (Fig. 73A), could be attached to different model peptides and proteins, and the target itself was usually able to ensure water-solubility of the whole construct.49,790–793 However, in other examples, such as for collagen peptides, the protein's water-solubility was not sufficient to solubilize the labelled target.794,795 Consequently, organic co-solvents were added to achieve photocontrol of folding of the triple helical structure employing an azobenzene crosslinker carrying alkyne bridges (4.76, Fig. 73B).794,795
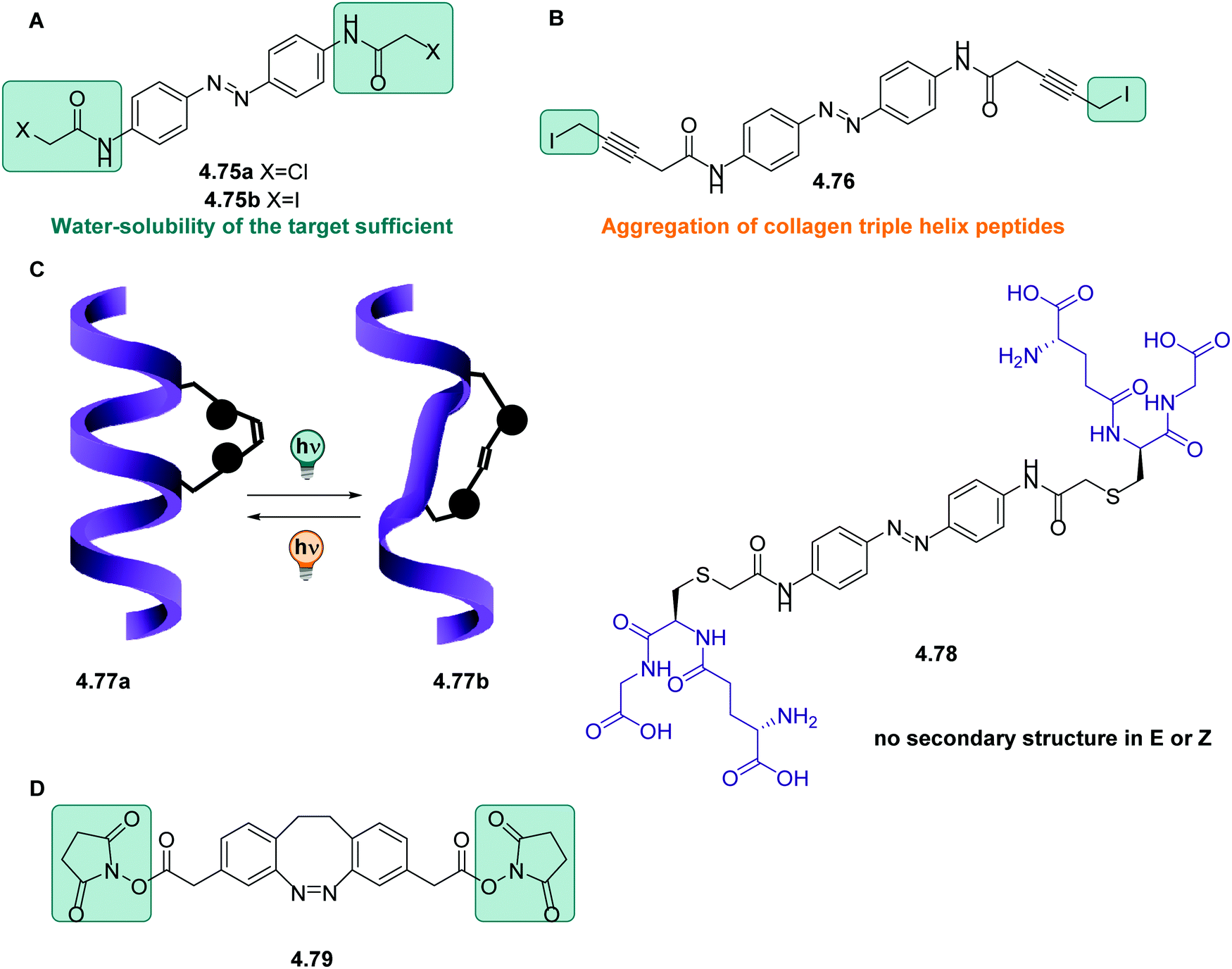 | ||
| Fig. 73 While for many applications the hydrophobic azobenzene crosslinker 4.75 could be successfully applied as the target itself ensured water-solubility of the crosslinker-protein construct (A), for other examples, such as the collagen peptides with photoswitch 4.76, the resulting crosslinked biomolecule aggregated due to insufficient aqueous solubility (B). The Z isomer of azobenzene 4.77 allowed retention of the secondary structure of the α helical cell-penetrating peptide upon crosslinking, while the E isomer disrupted the secondary structure (C). The Z isomer of the diazocine 4.79 allowed formation of the Trp fold, while the extended E isomer destabilized it. | ||
In another study, the reversible (de)stabilization of an α helical cell-penetrating peptide (CPP) was achieved by the simple azobenzene crosslinker 4.75a described above. (Fig. 73A).796 In the E form, the α helix was disrupted, and the internalization was prevented, while the Z isomer allowed the peptide to from the α helix. While in many biological applications, the aggregation of the modified protein can cause toxicity, in this work the formation of different sizes of aggregates was used to study internalization of the CPP into HeLa cells. Strikingly, the Z isomer formed much larger aggregates (300 nm size) compared to the E isomer (1.5 nm) which indicates that the internalization mechanism of the two isomers is different due to aggregate formation.796
The attachment of a photoswitchable crosslinker does not only affect the properties of the target molecules, but mutually influences the properties of the label (similar observations have also been made for non-covalent binders, see Section 4.2). To study the effect of the secondary protein structure on the photochemical properties of the photoswitch (Fig. 73C), azobenzene crosslinker 4.75b was covalently attached to both a model α helix peptide (4.77a and b) and furnished with two glutathione moieties on both ends (4.78).797 The glutathione-functionalized derivative cannot form an α helix and served as a control. The constructs were studied in aqueous conditions (20 μM in 10 mM sodium phosphate at pH 7, 25 °C) regarding both their photochemical and thermodynamic properties. The quantum yield of the Z → E isomerization of the α helix-bound azobenzene 4.77 was higher than the control 4.78 (Φ = 0.49 compared to 0.38), while the value for E → Z was similar (Φ = 0.1 compared to 0.12). Furthermore, the thermal half-lives of Z-azobenzenes bound to an α helix were longer than the Z-glutathione-azobenzene construct 4.78 (t1/2 = 26.7 compared to 11.5 min at 25 °C), presumably due to an enforced rotational mechanism caused by the constrains of the covalently linked protein.
While in classical azobenzenes the planar E isomer can lead to aggregation due to π–π stacking,798 tetra-ortho substituted derivatives do not show the same tendency, since the steric clash of the ortho substituents leads to a slightly twisted conformation in the E form. Moreover, they can be efficiently operated with biocompatible visible light in both directions. However, for tetra-ortho-methoxy substituted derivatives, the compounds were found to degrade in the presence of glutathione.199 Diazocines, on the other hand, are stable under these conditions and also have a non-planar conformation in both isomeric forms thus preventing aggregation. Hence, they are a valid alternative to build visible-light responsive crosslinkers. Consequently, diazocine crosslinker 4.79 was recently used to staple the α helix in a Trp-cage protein, where the shorter Z isomer allowed formation of the Trp fold and the extended E isomer destabilized it.799
While diazocines generally reach very high PSD in organic solvents, with more than 90% of the metastable E, it was observed in other studies that in water the PSD values drop significantly to 16–60% depending on the derivative.215,800 For diazocine 4.79 the PSD385nm was 46:54 = E:Z and the half-life of the E form was 6.2 h (0.5 mM in 10% D2O in water, pH = 5.5, at room temperature).
4.3.5.2 Water-soluble switches. Application of water-soluble photoswitches extended the substrate scope for photoresponsive crosslinkers by enabling the two-point labeling of sensitive targets which do not tolerate organic cosolvents.789,801 In particular, the water-soluble sulfonated azobenzene 4.80802,803(Fig. 74) has been successfully applied to crosslink numerous targets, such as peptides involved in protein–protein interactions,804–806 transcription factors,807 complex targets like clathrin adhesion proteins,808 and have even been applied in vivo in zebrafish studies.535 The sulfonate moieties were placed in meta position to the azo bond to allow for water-solubility without interfering with the photochemical properties of the azobenzene.809 However, they have been shown to influence or even inhibit cellular uptake of the modified target.807,810
To crosslink over larger distances, azobenzene 4.80 has been extended with a biphenyl group (4.81) or alkyne linkages (4.82).809,811 Both modifications increased the lipophilicity of the molecules significantly so that the azobenzenes were insoluble in water without the presence of the sulfonate groups, emphasizing key elements required for waterborne crosslinking (Fig. 74).809,811
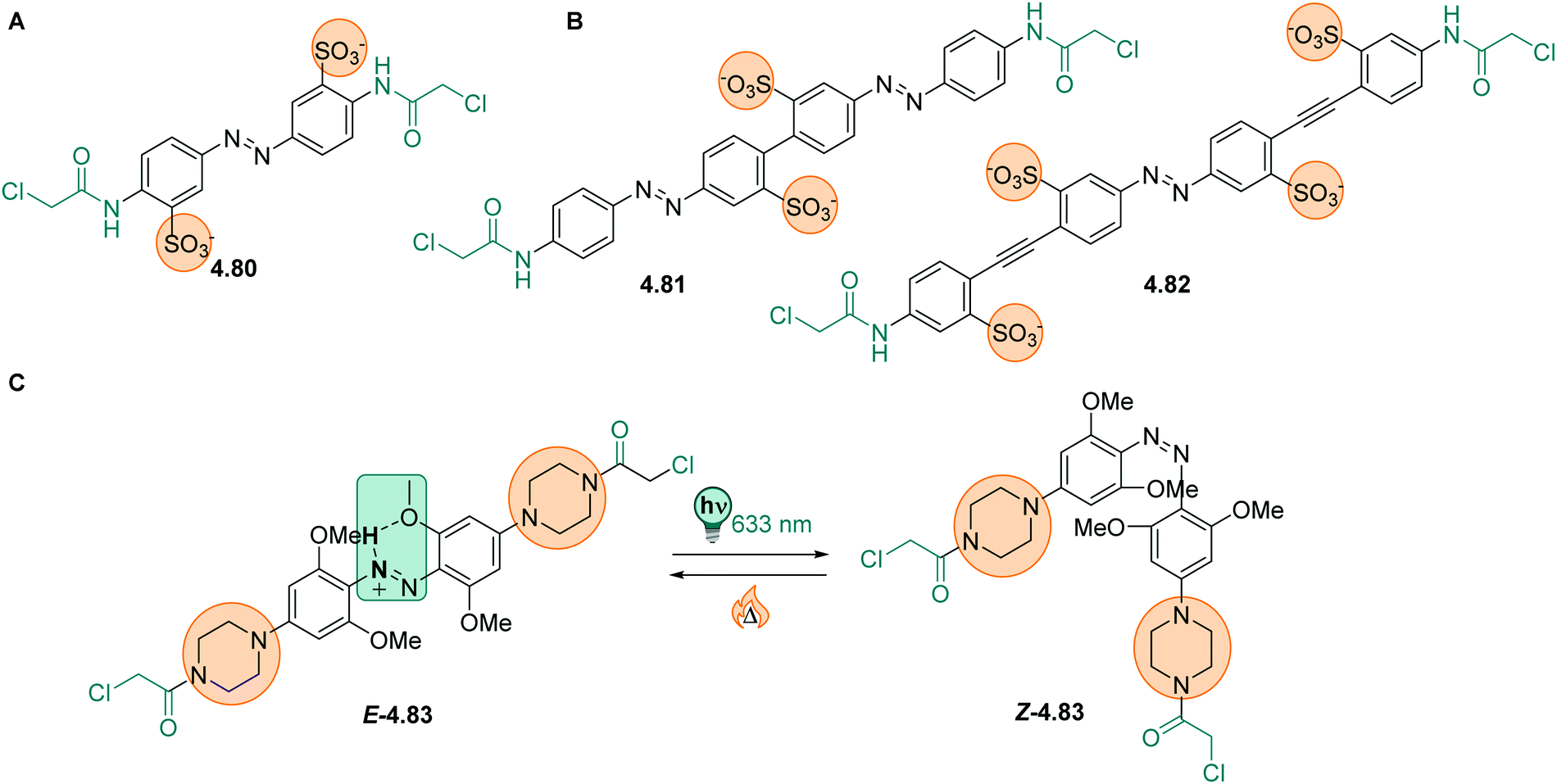 | ||
| Fig. 74 Water-soluble crosslinkers. (A) Sulfonated azobenzene 4.80 has been successfully applied to crosslink numerous targets and was subsequently multiplied and elongated in designs 4.81 and 4.82 to achieve larger end-to-end distance change upon switching (B). The azonium-based azobenzene 4.83 with tetra-ortho-methoxy substituents can be switched with 633 nm light and has a protonated nitrogen atom which forms a stabilizing hydrogen bond with the adjacent methoxy group only in the E isomer. | ||
To move from UV-light to visible-light responsive azobenzenes, tetra-ortho-methoxy azobenzene 4.83, containing an azonium bond, was developed. Protonated azobenzenes form an intramolecular H-bond between a protonated nitrogen of the azo bond and the neighboring oxygen of the methoxy group in the E form more readily than in the Z form.812,813
The switch was used to crosslink fynomers, small antibody mimetics812 and could be isomerized upon irradiation with 633 nm light while being bound to the protein. The thermal half-lives of the Z isomers were in the range of seconds (t1/2 = 2–9 s) for most targets, except for one example. Here, the protein environment at the attachment site was positively charged, forcing the azobenzene to deprotonate to the neutral state and as a consequence significantly elongating the half-life (from 7.2 s to ∼100 s). Hence, azonium-based azobenzenes are an interesting system in which the protein environment at the attachment location can be adjusted to modify the half-life of the switch.
Next to the sulfonate group, carbohydrate moieties have been used to ensure water-solubility. They were attached on each side of the photoswitch and carried a reactive group, such as chloroacetyl, allyl, propargyl or azide, for attachment to a target (Fig. 75, 4.84–88).814,815 The advantage of using glycoconjugates of azobenzenes is their biocompatibility, high water-solubility, and possibility of attaching the reactive handle at various positions onto the carbohydrate moiety.
 | ||
| Fig. 75 Azobenzene photoswitches modified with carbohydrate moieties to ensure water-solubility. The sugar groups have one or two reactive handles at various positions for attachment to a target. | ||
5 Conclusion
Over the past decades, light responsive molecules have taken the spotlight as a powerful tool to manipulate and interfere with numerous (bio) molecular systems and materials both in organic and aqueous media. However, photoswitches are inherently lipophilic organic molecules and thus naturally poorly soluble or insoluble water. Solubility of organic molecules in the “universal solvent of life” is of critical importance, especially for biological and medicinal applications most prominently seen in photopharmacology, as its absence can compromise bioavailability, resulting in aggregation or even toxicity.As with other organic molecules, photoswitches can be solubilized using different approaches, the development of which is mainly driven by the pharmacological field (Section 2.2). Common solubilization approaches, including the attachment of charged and non-charged polar moieties, or the conversion of acids and bases into their respective salts (Sections 2.2.1 and 2.2.2), are also applied in the context of photoswitches. Other methods, such as distorting the planarity of flat molecules, designing unsymmetrical structures or the functionalization with a cleavable solubilizing group (prodrug strategy, see Section 4.1.2), are less frequently used to solubilize light-responsive molecules.
However, even after the photoswitch has been successfully solubilized in water, this polar protic solvent can have a tremendous effect on the photophysical and photochemical properties of the photoresponsive molecule. As described in Section 3, water often has a strong impact on the half-life of the metastable form of the photoswitch, for instance, in push–pull azobenzenes and azobenzenes bearing –SH, –OH, –NHR substituents (Section 3.2.3). Moreover, the polar protic solvent can also hamper the reversibility of photoswitching in the case of DAEs (Section 3.3.1) and DASAs (Section 3.4.3). While azobenzenes show typically high photoswitching QYs in polar solvents, for some indigos and thioindigos the QY decrease in water (Section 3.2.5). Also, photophysical properties, such as the absorption maxima of the two photoisomers, are influenced, which leads for instance in diazocines to a reduced band separation and thus lower PSDs (Section 3.2.4). Finally, the stability of the switch itself can be decreased in water and for some compounds, like azo-BF3 switches (Section 3.2.9) and some spiropyrans (Section 3.4.1), hydrolysis has been observed.
Nevertheless, many successful and fascinating examples of applications of photoswitches in water have been described. In Section 4.1, we discuss how different solubilization strategies are applied on small photoswitches acting as ligands in biological systems. Here, the attachment of solubilizing groups is most frequently used. However, the installation of a solubilizing group, both carrying a charge or not, can often have a strong effect on the localization of the bioactive molecule in cellulo and in vivo. This was, for example, extensively reported for ion channel inhibitors solubilized with ammonium groups (see Section 4.1.1). Furthermore, the photoswitchable molecule needs to be sufficiently water-soluble for achieving bioavailability, while remaining lipophilic enough to remain bioactive. At the same time, excellent photochemical properties need to be retained. This fine balance of various parameters was illustrated for cell-penetrating peptides (Section 4.1.3.1). In this case, toxicity due to aggregation was avoided upon water-solubilization of hemithioindigo inhibitors by disrupting the planarity of the molecules upon simple introduction of an additional methoxy group.
In the case of applications where the photoswitch is designed as a biomolecular building block, such as an amino acid or a nucleotide, the photoactuator is typically incorporated via SPPS in organic solvents. Hence, the solubility of the switch itself is less important for the attachment process, however, the final construct needs to exhibit the right solubility properties matching the characteristics of the assembled biomolecule (see Section 4.2). If, for instance, the resulting peptide is relatively small, lipophilic and large aromatic structures can cause unwanted aggregation, as seen for the cell penetrating peptides. For photoswitches incorporated into the backbone of DNA and RNA, it was observed that the vicinity of the biomolecule can hamper the photochemical properties of the integrated photoswitch. In contrast, photoswitches which are attached to a target molecule after its synthesis, or to a naturally occurring target, need to fulfil different criteria regarding to their solubility, while not affecting the native activity of the biomolecule. Specifically, the required water-solubility is dependent on the tolerance of the biomolecule towards co-solvents. The MscL channel could withstand 20% DMSO in water to label it with a spiropyran, while the nanopore-forming toxin FraC required the use of fully water-soluble azobenzenes, to only name two examples. Moreover, in a biologically more complex setting, like in cellulo or in vivo, such as needed for super-resolution fluorescence microscopy in living cells, the water-solubility of the photoswitchable fluorophore need to be extremely high as any aggregate formation results in high toxicity (Section 4.3.4).
Based on this literature overview, we summarized the available water-solubilizing strategies in Fig. 76. Herein we propose a decision tree to aid the reader in the design of the water-soluble light-responsive construct. The type and location of the photoswitchable entity requires specific considerations and leads to key decisions which further dictate the solubilizing approach. While the water-solubilizing groups can be summarized in a generalized fashion, the photoswitch selection has to be performed on a case-to-case basis, considering that many of them have not been studied in water in detail. Nevertheless, it is of crucial importance to consider the photochemical properties of the chosen photoswitch in an aqueous environment.
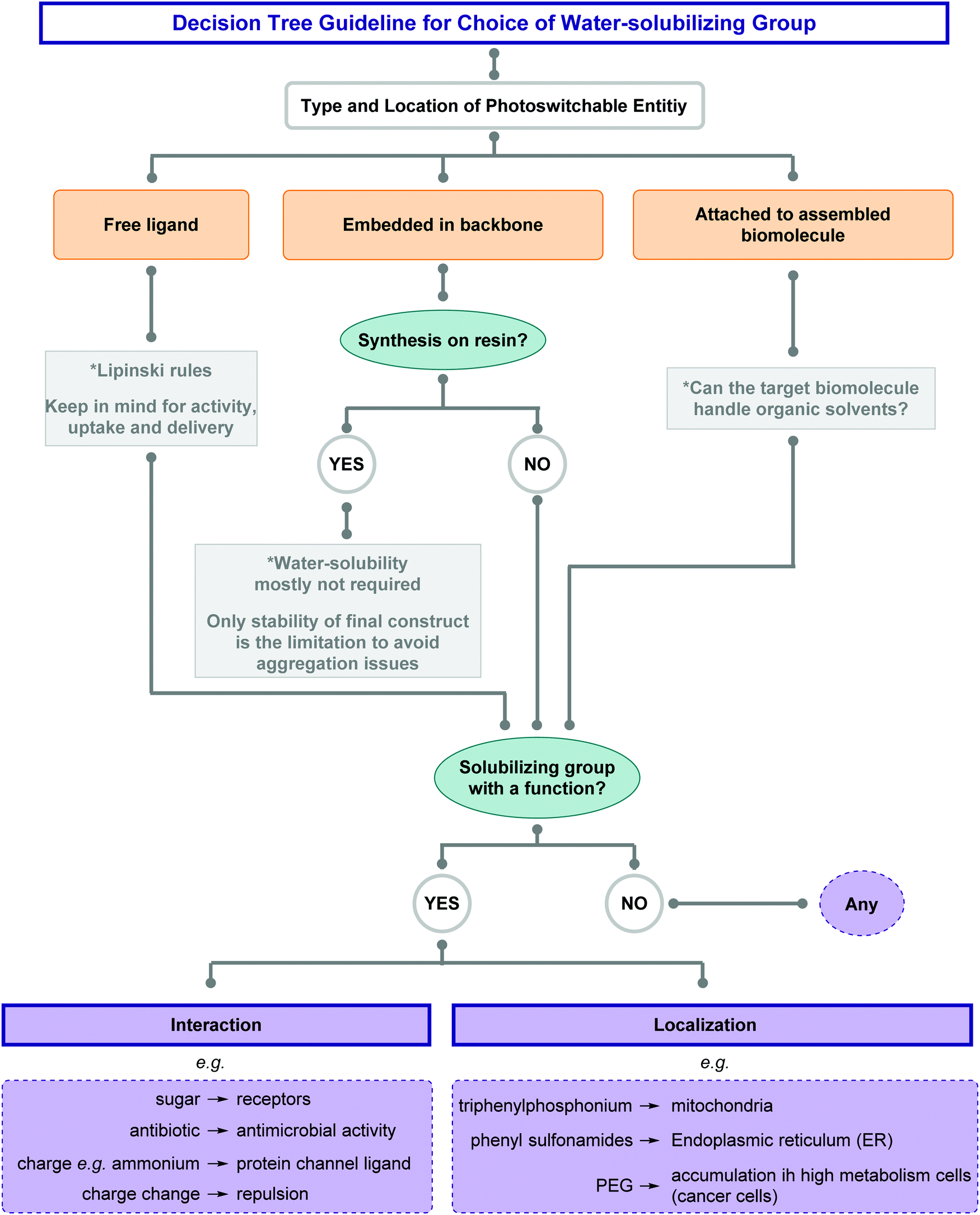 | ||
| Fig. 76 Decision tree guideline for choosing the water-solubilizing group for a specific molecule based on the type and location of the photoswitchable entity (in orange). Grey boxes describe general considerations to keep in mind while designing a construct, mint-green circles ask key questions for the design and purple areas possible roles of the solubilizing group with illustrative examples. | ||
In summary, various aspects need to be considered to successfully apply a photoswitch in aqueous media and to establish a system that operates efficiently in water, shows hydrolytic stability, is (bio)available, retains its desired activity while preserving excellent photobistability. Furthermore, the influence of the labelled biomolecule on the photoswitch properties (e.g. energy transfer) and vice versa need to be taken into account. To tackle this complexity, both a suitable photoswitch and an appropriate solubilization strategy need to be chosen. In the examples discussed here, mostly azobenzenes and DAEs are used as photoactuators. Other classes (spiropyrans, heteroaryl azobenzenes, H(T)Is, fulgides, stilbenes) are to a lesser extent applied, while some switches (hydrazones, DHPs), have been hardly used in aqueous environment. This is often because they are in general not extensively investigated and less well understood, they are not (thoroughly) studied in polar protic or aqueous media, or there are no suitable soluble analogous yet available. Consequently, there is a strong need to rationally explore the photophysical and photochemical proprieties of a wider range of photoswitches in aqueous media. Moreover, suitable solubilizing strategies need to be found for both the underexplored switches and for the more frequently used ones. Currently, solubility problems are often overcome using cosolvents, and with that the issue of kinetic solubility. However, this can lead to aggregation and precipitation of the compound and hence, hamper the development of advanced applications, such as in vivo studies. Therefore, research needs more focus on the development of photoswitches showing high thermodynamic solubility in water. This can be achieved using classical approaches such as the introduction of polar or charged groups. However, more challenging solubilizing strategies need to be systematically explored and involved in the design of novel photoswitches, for example, by favouring the incorporation of 3D over 2D moieties, a current trend that has shown some promising results in drug development.41 Also, prodrug strategies or the formation of hydrotropes could lead to major advances designing truly water-soluble photoswitches. Having these novel methods readily available together with a more detailed understanding of photoswitching in aqueous media, it will pave the way to numerous applications in the chemical biology, pharmaceutical and medicinal fields, but will also have a strong influence on other research areas, opening tremendous opportunities for surface functionalization, smart soft-materials as well as responsive and adaptive systems compatible with living systems.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Dr Stefano Crespi (University of Groningen) and Dr Jeffrey Buter (University of Groningen) for fruitful discussion during the preparation of this review. We gratefully acknowledge the generous support from the Humboldt Foundation (Feodor-Lynen scholarship to NAS), the Horizon 2020 Framework Program (ERC Advanced Investigator Grant No. 694345 to BLF), the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to BLF), and the Netherlands Organization for Scientific Research (NWO, VIDI Grant No. 723.014.001 for WS).References
- K. Reichardt and L. C. Timm, Soil, Plant and Atmosphere, Springer International Publishing, Cham, 2020, pp. 7–13 Search PubMed.
- A. Pohorille and L. R. Pratt, Orig. Life Evol. Biosph., 2012, 42, 405–409 CrossRef CAS PubMed.
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- P. Suppan, Chemistry and Light, Royal Society of Chemistry, Cambridge, 2007 Search PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski, B. L. Feringa, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- A. M. Ali, G. A. Woolley, G. A. Woolley, C. Pedone, G. Colombo, L. D. D’Andrea, R. Fattorusso, G. A. Woolley and S. J. Korsmeyer, Org. Biomol. Chem., 2013, 11, 5325–5331 RSC.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- K. Nakatani, J. Piard, P. Yu and R. Métivier, Photochromic Materials: Preparation, Properties and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 1–45 Search PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- L. Josa-Culleré and A. Llebaria, ChemPhotoChem, 2021, 5, 298–316 CrossRef.
- T. Slanina, Photochemistry: Volume 45, PI Group (UK) Ltd, Croydon, 2018, pp. 175–190 Search PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- S. Kaneko, K. Asakura and T. Banno, Chem. Commun., 2017, 53, 2237–2240 RSC.
- E. Chevallier, C. Monteux, F. Lequeux and C. Tribet, Langmuir, 2012, 28, 2308–2312 CrossRef CAS PubMed.
- S. Chen, R. Costil, F. K. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed.
- S. Chen, F. K.-C. Leung, M. C. A. Stuart, C. Wang and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 10163–10172 CrossRef CAS PubMed.
- P. M. Erne, P. Štacko, D. J. van Dijken, J. Chen, M. C. A. Stuart and B. L. Feringa, Chem. Commun., 2016, 52, 11697–11700 RSC.
- D. J. van Dijken, J. Chen, M. C. A. Stuart, L. Hou and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 660–669 CrossRef CAS PubMed.
- P. M. Erne, L. S. van Bezouwen, P. Štacko, D. J. van Dijken, J. Chen, M. C. A. Stuart, E. J. Boekema and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 15122–15127 CrossRef CAS PubMed.
- J. T. van Herpt, J. Areephong, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 1737–1742 CrossRef CAS PubMed.
- M. A. Schoondorp, A. J. Schouten, J. B. E. Hulshof and B. L. Feringa, Langmuir, 1993, 9, 1323–1329 CrossRef CAS.
- R. Kou, T. Wang, Z. Chen, R. Zhu, H. Cai, H. Pang, S. Xuan and G. Liu, Langmuir, 2019, 35, 13110–13115 CrossRef CAS PubMed.
- L. Ji, G. Ouyang and M. Liu, Langmuir, 2017, 33, 12419–12426 CrossRef CAS PubMed.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672–11691 RSC.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- H.-M. Lee, D. R. Larson and D. S. Lawrence, ACS Chem. Biol., 2009, 4, 409–427 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- J. Broichhagen, M. Schönberger, S. C. Cork, J. A. Frank, P. Marchetti, M. Bugliani, A. M. J. J. Shapiro, S. Trapp, G. A. Rutter, D. J. Hodson and D. Trauner, Nat. Commun., 2014, 5, 5116 CrossRef CAS PubMed.
- D. Pearson, N. Alexander and A. D. Abell, Chem. – Eur. J., 2008, 14, 7358–7365 CrossRef CAS PubMed.
- J. M. Kuiper and J. B. F. N. Engberts, Langmuir, 2004, 20, 1152–1160 CrossRef CAS PubMed.
- M. Shimomura, R. Ando and T. Kunitake, Ber. Buns. Phys. Chem., 1983, 87, 1134–1143 CrossRef CAS.
- S. L. Niu, C. Massif, G. Ulrich, R. Ziessel, P.-Y. Renard and A. Romieu, Org. Biomol. Chem., 2011, 9, 66–69 RSC.
- M. A. Walker, Expert Opin. Drug Discovery, 2014, 9, 1421–1433 CrossRef CAS PubMed.
- D. Jornada, G. dos Santos Fernandes, D. Chiba, T. de Melo, J. dos Santos and M. Chung, Molecules, 2015, 21, 42 CrossRef PubMed.
- M. Ishikawa and Y. Hashimoto, J. Med. Chem., 2011, 54, 1539–1554 CrossRef CAS PubMed.
- D. Frath, S. Yokoyama, T. Hirose and K. Matsuda, J. Photochem. Photobiol., C, 2018, 34, 29–40 CrossRef CAS.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520 RSC.
- Z. Ding, Y. Zhang, Y. Gao, B. Yang and S. Jiang, Nanoscale, 2020, 12, 2071–2080 RSC.
- F. Xu, L. Pfeifer, S. Crespi, F. K.-C. Leung, M. C. A. Stuart, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 5990–5997 CrossRef CAS PubMed.
- G. Davidson-Rozenfeld, L. Stricker, J. Simke, M. Fadeev, M. Vázquez-González, B. J. Ravoo and I. Willner, Polym. Chem., 2019, 10, 4106–4115 RSC.
- Y.-L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef CAS PubMed.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- A. Myrhammar, D. Rosik and A. E. Karlström, Bioconjugate Chem., 2020, 31, 622–630 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- H. Birch, A. D. Redman, D. J. Letinski, D. Y. Lyon and P. Mayer, Anal. Chim. Acta, 2019, 1086, 16–28 CrossRef CAS PubMed.
- S. Stegemann, F. Leveiller, D. Franchi, H. de Jong and H. Lindén, Eur. J. Pharm. Sci., 2007, 31, 249–261 CrossRef CAS PubMed.
- US Pharmacopeia, USP 30-NF 25, 2007.
- F. L. O. Da Silva, M. B. D. F. Marques, K. C. Kato and G. Carneiro, Expert Opin. Drug Discovery, 2020, 15, 853–864 CrossRef CAS PubMed.
- S. N. Bhattachar, L. A. Deschenes and J. A. Wesley, Drug Discovery Today, 2006, 11, 1012–1018 CrossRef CAS PubMed.
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS PubMed.
- E. Baka, J. E. A. Comer and K. Takács-Novák, J. Pharm. Biomed. Anal., 2008, 46, 335–341 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Mill Valley, 2006 Search PubMed.
- A. Lusci, G. Pollastri and P. Baldi, J. Chem. Inf. Model., 2013, 53, 1563–1575 CrossRef CAS PubMed.
- G. Klopman and H. Zhu, J. Chem. Inf. Comput. Sci., 2001, 41, 439–445 CrossRef CAS PubMed.
- J. S. Delaney, Drug Discovery Today, 2005, 10, 289–295 CrossRef CAS PubMed.
- T. Sanghvi, N. Jain, G. Yang and S. Yalkowsky, QSAR Comb. Sci., 2003, 22, 258–262 CrossRef CAS.
- A. Avdeef and M. Kansy, Mol. Pharmaceutics, 2020, 17, 3930–3940 CrossRef CAS PubMed.
- L. Mendez, G. Henriquez, S. Sirimulla and M. Narayan, Molecules, 2017, 22, 1397 CrossRef PubMed.
- K. S. Sidhu, J. M. Goodfellow and J. Z. Turner, J. Chem. Phys., 1999, 110, 7943–7950 CrossRef CAS.
- H. Lee, F. Dehez, C. Chipot, H.-K. Lim and H. Kim, J. Chem. Theory Comput., 2019, 15, 1538–1545 CrossRef CAS PubMed.
- G. Graziano, J. Phys. Chem. B, 2001, 105, 2632–2637 CrossRef CAS.
- Disordered Pharmaceutical Materials, ed. M. Descamps, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016 Search PubMed.
- J. E. Ricci and T. W. Davis, J. Am. Chem. Soc., 1940, 62, 407–413 CrossRef CAS.
- OECD, Test No. 105: Water Solubility, OECD Guidelines for the Testing of Chemicals, Section 1, OECD Publishing, Paris, 1995, DOI:10.1787/9789264069589-en.
- C. Lipinski and A. Hopkins, Nature, 2004, 432, 855–861 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS PubMed.
- A. T. M. Serajuddin, Adv. Drug Delivery Rev., 2007, 59, 603–616 CrossRef CAS PubMed.
- A. K. Nayak and P. P. Panigrahi, ISRN Phys. Chem., 2012, 2012, 1–5 CrossRef.
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403–416 CrossRef CAS PubMed.
- J. Mao, W. Ruan and Q. Chen, J. Electrochem. Soc., 2020, 167, 070522 CrossRef CAS.
- M. Addie, P. Ballard, D. Buttar, C. Crafter, G. Currie, B. R. Davies, J. Debreczeni, H. Dry, P. Dudley, R. Greenwood, P. D. Johnson, J. G. Kettle, C. Lane, G. Lamont, A. Leach, R. W. A. Luke, J. Morris, D. Ogilvie, K. Page, M. Pass, S. Pearson and L. Ruston, J. Med. Chem., 2013, 56, 2059–2073 CrossRef CAS PubMed.
- S. S. Scully, A. J. Tang, M. Lundh, C. M. Mosher, K. M. Perkins and B. K. Wagner, J. Med. Chem., 2013, 56, 4125–4129 CrossRef CAS PubMed.
- S. Wenglowsky, D. Moreno, J. Rudolph, Y. Ran, K. A. Ahrendt, A. Arrigo, B. Colson, S. L. Gloor and G. Hastings, Bioorg. Med. Chem. Lett., 2012, 22, 912–915 CrossRef CAS PubMed.
- Z. A. Parray, M. I. Hassan, F. Ahmad and A. Islam, Polym. Test., 2020, 82, 106316 CrossRef CAS.
- R. B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Adv. Drug Delivery Rev., 2003, 55, 217–250 CrossRef CAS PubMed.
- E. M. Skoda, J. R. Sacher, M. Z. Kazancioglu, J. Saha and P. Wipf, ACS Med. Chem. Lett., 2014, 5, 900–904 CrossRef CAS PubMed.
- A. Llinàs, J. C. Burley, K. J. Box, R. C. Glen and J. M. Goodman, J. Med. Chem., 2007, 50, 979–983 CrossRef PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- H. Chen, Z. Yang, C. Ding, L. Chu, Y. Zhang, K. Terry, H. Liu, Q. Shen and J. Zhou, ACS Med. Chem. Lett., 2013, 4, 180–185 CrossRef CAS PubMed.
- K. M. Foote, K. Blades, A. Cronin, S. Fillery, S. S. Guichard, L. Hassall, I. Hickson, X. Jacq, P. J. Jewsbury, T. M. McGuire, J. W. M. Nissink, R. Odedra, K. Page, P. Perkins, A. Suleman, K. Tam, P. Thommes, R. Broadhurst and C. Wood, J. Med. Chem., 2013, 56, 2125–2138 CrossRef CAS PubMed.
- M. Hiramatsu, Y. Ichikawa, S. Tomoshige, M. Makishima, A. Muranaka, M. Uchiyama, T. Yamaguchi, Y. Hashimoto and M. Ishikawa, Chem. – Asian J., 2016, 11, 2210–2217 CrossRef CAS PubMed.
- B. Laprade, T. W. Mauger and H. Petersen, Drug Dev. Ind. Pharm., 1977, 3, 73–85 CrossRef CAS.
- S. Forster, G. Buckton and A. E. Beezer, Int. J. Pharm., 1991, 72, 29–34 CrossRef CAS.
- A. Gavezzotti, J. Chem. Soc., Perkin Trans. 2, 1995, 1399 RSC.
- V. V. Levterov, O. Michurin, P. O. Borysko, S. Zozulya, I. V. Sadkova, A. A. Tolmachev and P. K. Mykhailiuk, J. Org. Chem., 2018, 83, 14350–14361 CrossRef CAS PubMed.
- G. Niu, R. Zhang, J. P. C. Kwong, J. W. Y. Lam, C. Chen, J. Wang, Y. Chen, X. Feng, R. T. K. Kwok, H. H.-Y. Sung, I. D. Williams, M. R. J. Elsegood, J. Qu, C. Ma, K. S. Wong, X. Yu and B. Z. Tang, Chem. Mater., 2018, 30, 4778–4787 CrossRef CAS.
- J. O. Bockris, J. Bowler-Reed and J. A. Kitchener, Trans. Faraday Soc., 1951, 47, 184–192 RSC.
- R. Sadeghi and F. Jahani, J. Phys. Chem. B, 2012, 116, 5234–5241 CrossRef CAS PubMed.
- L. I. N. Tomé, S. P. Pinho, M. Jorge, J. R. B. Gomes and J. A. P. Coutinho, J. Phys. Chem. B, 2013, 117, 6116–6128 CrossRef PubMed.
- V. Dhapte and P. Mehta, St. Petersbg. Polytech. Univ. J. Phys. Math., 2015, 1, 424–435 Search PubMed.
- P. Khadka, J. Ro, H. Kim, I. Kim, J. T. Kim, H. Kim, J. M. Cho, G. Yun and J. Lee, Asian J. Pharm. Sci., 2014, 9, 304–316 CrossRef.
- S. Rawat and S. K. Jain, Eur. J. Pharm. Biopharm., 2004, 57, 263–267 CrossRef CAS PubMed.
- H. Kumpulainen, T. Järvinen, A. Mannila, J. Leppänen, T. Nevalainen, A. Mäntylä, J. Vepsäläinen and J. Rautio, Eur. J. Pharm. Sci., 2008, 34, 110–117 CrossRef CAS PubMed.
- J. Shen, D. Zhang, Z. Zhao, L. Jia, D. Zheng, G. Liu, L. Hao, Q. Zhang, X. Tian, C. Li and H. Guo, Int. J. Pharm., 2013, 456, 80–86 CrossRef CAS PubMed.
- M. Zhou, R.-H. Luo, X.-Y. Hou, R.-R. Wang, G.-Y. Yan, H. Chen, R.-H. Zhang, J.-Y. Shi, Y.-T. Zheng, R. Li and Y.-Q. Wei, Eur. J. Med. Chem., 2017, 129, 310–324 CrossRef CAS PubMed.
- B. M. A. Sanches and E. I. Ferreira, Int. J. Pharm., 2019, 568, 118498 CrossRef CAS PubMed.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- H. Bouas-Laurent and H. Dürr, Pure Appl. Chem., 2001, 73, 639–665 CAS.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- H. Dürr and H. Bouas-Laurent, Photochromism: Molecules and Systems, 1st edn, Elsevier Science, Amsterdam, 2003 Search PubMed.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef PubMed.
- M. Oelgemöller, B. Brem, R. Frank, S. Schneider, D. Lenoir, N. Hertkorn, Y. Origane, P. Lemmen, J. Lex and Y. Inoue, J. Chem. Soc., Perkin Trans. 2, 2002, 1760–1771 RSC.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- M. Quick, F. Berndt, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, C. Knie, R. Mahrwald, D. Lenoir, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 1389–1402 CrossRef CAS PubMed.
- W. Fuß, C. Kosmidis, W. E. Schmid and S. A. Trushin, Angew. Chem., Int. Ed., 2004, 43, 4178–4182 CrossRef PubMed.
- D. C. Todd and G. R. Fleming, J. Chem. Phys., 1993, 98, 269–279 CrossRef CAS.
- R. Lechner, S. Kümmel and B. König, Photochem. Photobiol. Sci., 2010, 9, 1367–1377 CrossRef CAS PubMed.
- M. P. O’Hagan, S. Haldar, M. Duchi, T. A. A. Oliver, A. J. Mulholland, J. C. Morales and M. C. Galan, Angew. Chem., Int. Ed., 2019, 58, 4334–4338 CrossRef PubMed.
- Y. Wang, Y. Tian, Y.-Z. Chen, L.-Y. Niu, L.-Z. Wu, C.-H. Tung, Q.-Z. Yang and R. Boulatov, Chem. Commun., 2018, 54, 7991–7994 RSC.
- J.-X. Wang, L.-Y. Niu, P.-Z. Chen, Y.-Z. Chen, Q.-Z. Yang and R. Boulatov, Chem. Commun., 2019, 55, 7017–7020 RSC.
- T. Shimasaki, S. Kato, K. Ideta, K. Goto and T. Shinmyozu, J. Org. Chem., 2007, 72, 1073–1087 CrossRef CAS PubMed.
- S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 324–327 CrossRef CAS PubMed.
- Y. Li, E. C. M. Tse, C. J. Barile, A. A. Gewirth and S. C. Zimmerman, J. Am. Chem. Soc., 2015, 137, 14059–14062 CrossRef CAS PubMed.
- J. Jong, B. L. Feringa and S. J. Wezenberg, ChemPhysChem, 2019, 20, 3306–3310 CrossRef PubMed.
- T.-G. Zhan, H.-H. Yin, S.-T. Zheng, W.-C. Lin, N.-L. Shen, J. Cui, L.-C. Kong, L.-J. Liu and K.-D. Zhang, Chem. Commun., 2018, 54, 9356–9359 RSC.
- N. Zhu, X. Li, Y. Wang and X. Ma, Dyes Pigm., 2016, 125, 259–265 CrossRef CAS.
- Y.-H. Wu, K. Huang, S.-F. Chen, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Sci. China: Chem., 2019, 62, 1194–1197 CrossRef CAS.
- C. Cao, G. Chen and Y. Wu, Sci. China: Chem., 2011, 54, 1735–1744 CrossRef CAS.
- M. de Wergifosse, A. L. Houk, A. I. Krylov and C. G. Elles, J. Chem. Phys., 2017, 146, 144305 CrossRef PubMed.
- G. Hohlneicher, R. Wrzal, D. Lenoir and R. Frank, J. Phys. Chem. A, 1999, 103, 8969–8975 CrossRef CAS.
- S. H. Courtney and G. R. Fleming, J. Chem. Phys., 1985, 83, 215–222 CrossRef CAS.
- J. F. Letard, R. Lapouyade and W. Rettig, J. Am. Chem. Soc., 1993, 115, 2441–2447 CrossRef CAS.
- X. Yan, J.-F. Xu, T. R. Cook, F. Huang, Q.-Z. Yang, C.-H. Tung and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8717–8722 CrossRef CAS PubMed.
- R. Costil, S. Crespi, L. Pfeifer and B. L. Feringa, Chem. – Eur. J., 2020, 26, 7783–7787 CrossRef CAS PubMed.
- S. Lee and A. H. Flood, J. Phys. Org. Chem., 2013, 26, 79–86 CrossRef CAS.
- S. Olsson, C. Schäfer, M. Blom and A. Gogoll, ChemPlusChem, 2018, 83, 1169–1178 CrossRef CAS PubMed.
- S. J. Wezenberg and B. L. Feringa, Nat. Commun., 2018, 9, 1984 CrossRef PubMed.
- Z. S. Kean, S. Akbulatov, Y. Tian, R. A. Widenhoefer, R. Boulatov and S. L. Craig, Angew. Chem., Int. Ed., 2014, 53, 14508–14511 CrossRef CAS PubMed.
- T. J. Kucharski and R. Boulatov, J. Mater. Chem., 2011, 21, 8237 RSC.
- Z. Huang, Q.-Z. Yang, D. Khvostichenko, T. J. Kucharski, J. Chen and R. Boulatov, J. Am. Chem. Soc., 2009, 131, 1407–1409 CrossRef CAS PubMed.
- S. Akbulatov, Y. Tian, Z. Huang, T. J. Kucharski, Q.-Z. Yang and R. Boulatov, Science, 2017, 357, 299–303 CrossRef CAS PubMed.
- S. Akbulatov, Y. Tian and R. Boulatov, J. Am. Chem. Soc., 2012, 134, 7620–7623 CrossRef CAS PubMed.
- H. Zhu, L. Shangguan, D. Xia, J. H. Mondal and B. Shi, Nanoscale, 2017, 9, 8913–8917 RSC.
- M. P. O’Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Chem. – Eur. J., 2020, 26, 6224–6233 CrossRef PubMed.
- K. A. Rotstan, M. M. Abdelsayed, L. F. Passalacqua, F. Chizzolini, K. Sudarshan, A. R. Chamberlin, J. Míšek and A. Luptak, eLife, 2020, 9, e51737 CrossRef CAS PubMed.
- L. Gao, J. C. M. Meiring, Y. Kraus, M. Wranik, T. Weinert, S. D. Pritzl, R. Bingham, E. Ntouliou, K. I. Jansen, N. Olieric, J. Standfuss, L. C. Kapitein, T. Lohmüller, J. Ahlfeld, A. Akhmanova, M. O. Steinmetz and O. Thorn-Seshold, Cell Chem. Biol., 2021, 28, 228–241 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS PubMed.
- D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423–9431 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79–124 CrossRef PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- J. C. M. Kistemaker, A. S. Lubbe and B. L. Feringa, Mater. Chem. Front, 2021, 5, 2900–2906 RSC.
- A. S. Lubbe, C. Böhmer, F. Tosi, W. Szymanski and B. L. Feringa, J. Org. Chem., 2018, 83, 11008–11018 CrossRef CAS PubMed.
- C. Poloni, M. C. A. Stuart, P. van der Meulen, W. Szymanski and B. L. Feringa, Chem. Sci., 2015, 6, 7311–7318 RSC.
- A. S. Lubbe, Q. Liu, S. J. Smith, J. W. De Vries, J. C. M. M. Kistemaker, A. H. De Vries, I. Faustino, Z. Meng, W. Szymanski, A. Herrmann and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 5069–5076 CrossRef CAS PubMed.
- A. S. Lubbe, J. C. M. Kistemaker, E. J. Smits and B. L. Feringa, Phys. Chem. Chem. Phys., 2016, 18, 26725–26735 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- N. A. Simeth and S. Crespi, (Hetero)aryl azoswitches and their application, Photochemistry, 2020, vol. 48, pp. 344–375 Search PubMed.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- D. Larsen, P. M. Bjerre and S. R. Beeren, Chem. Commun., 2019, 55, 15037–15040 RSC.
- C. Brown, S. K. K. Rastogi, S. L. L. Barrett, H. E. E. Anderson, E. Twichell, S. Gralinski, A. McDonald and W. J. J. Brittain, J. Photochem. Photobiol., A, 2017, 336, 140–145 CrossRef CAS.
- J. W. Fredy, A. Méndez-Ardoy, S. Kwangmettatam, D. Bochicchio, B. Matt, M. C. A. Stuart, J. Huskens, N. Katsonis, G. M. Pavan and T. Kudernac, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11850–11855 CrossRef CAS PubMed.
- D. Bochicchio, S. Kwangmettatam, T. Kudernac and G. M. Pavan, ACS Nano, 2019, 13, 4322–4334 CrossRef CAS PubMed.
- S. K. Rastogi, Z. Zhao, S. L. Barrett, S. D. Shelton, M. Zafferani, H. E. Anderson, M. O. Blumenthal, L. R. Jones, L. Wang, X. Li, C. N. Streu, L. Du and W. J. Brittain, Eur. J. Med. Chem., 2018, 143, 1–7 CrossRef CAS PubMed.
- J. Calbo, C. E. Weston, A. J. P. White, H. S. Rzepa, J. Contreras-García and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS PubMed.
- J. Garcia-Amorós, S. Nonell and D. Velasco, Chem. Commun., 2011, 47, 4022 RSC.
- J. Garcia-Amorós, S. Nonell and D. Velasco, Chem. Commun., 2012, 48, 3421 RSC.
- J. Otsuki, K. Harada and K. Araki, Chem. Lett., 1999, 269–270 CrossRef CAS.
- J. Otsuki and K. Narutaki, Bull. Chem. Soc. Jpn., 2004, 77, 1537–1544 CrossRef CAS.
- S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sonnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- N. A. Simeth, S. Crespi, M. Fagnoni and B. König, J. Am. Chem. Soc., 2018, 140, 2940–2946 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth, A. Bellisario, M. Fagnoni and B. König, J. Phys. Chem. A, 2019, 123, 1814–1823 CrossRef CAS PubMed.
- N. A. Simeth, A. Bellisario, S. Crespi, M. Fagnoni and B. König, J. Org. Chem., 2019, 84, 6565–6575 CrossRef CAS PubMed.
- N. Jacob, L. Guillemard and J. Wencel-Delord, Synthesis, 2020, 574–580 CAS.
- R. Travieso-Puente, S. Budzak, J. Chen, P. Stacko, J. T. B. H. Jastrzebski, D. Jacquemin and E. Otten, J. Am. Chem. Soc., 2017, 139, 3328–3331 CrossRef CAS PubMed.
- D. Kolarski, W. Szymanski and B. L. Feringa, Org. Lett., 2017, 19, 5090–5093 CrossRef CAS PubMed.
- D. Kolarski, A. Sugiyama, T. Rodat, A. Schulte, C. Peifer, K. Itami, T. Hirota, B. L. Feringa and W. Szymanski, Org. Biomol. Chem., 2021, 19, 2312–2321 RSC.
- L. Čechová, J. Filo, M. Dračínský, C. Slavov, D. Sun, Z. Janeba, T. Slanina, J. Wachtveitl, E. Procházková and M. Cigáň, Angew. Chem., Int. Ed., 2020, 59, 15590–15594 CrossRef PubMed.
- J. Garcia-Amorós, M. Díaz-Lobo, S. Nonell and D. Velasco, Angew. Chem., Int. Ed., 2012, 51, 12820–12823 CrossRef PubMed.
- M. Schehr, C. Ianes, J. Weisner, L. Heintze, M. P. Müller, C. Pichlo, J. Charl, E. Brunstein, J. Ewert, M. Lehr, U. Baumann, D. Rauh, U. Knippschild, C. Peifer and R. Herges, Photochem. Photobiol. Sci., 2019, 18, 1398–1407 CrossRef CAS PubMed.
- T. Wendler, C. Schütt, C. Näther and R. Herges, J. Org. Chem., 2012, 77, 3284–3287 CrossRef CAS PubMed.
- J. Otsuki, K. Suwa, K. K. Sarker and C. Sinha, J. Phys. Chem. A, 2007, 111, 1403–1409 CrossRef CAS PubMed.
- K. Rustler, P. Nitschke, S. Zahnbrecher, J. Zach, S. Crespi and B. König, J. Org. Chem., 2020, 85, 4079–4088 CrossRef CAS PubMed.
- C. Chu, L. Stricker, T. M. Kirse, M. Hayduk and B. J. Ravoo, Chem. – Eur. J., 2019, 25, 6131–6140 CrossRef CAS PubMed.
- L. Stricker, M. Böckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem. – Eur. J., 2018, 24, 8639–8647 CrossRef CAS PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS PubMed.
- C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- S. Devi, M. Saraswat, S. Grewal and S. Venkataramani, J. Org. Chem., 2018, 83, 4307–4322 CrossRef CAS PubMed.
- P. Kumar, A. Srivastava, C. Sah, S. Devi and S. Venkataramani, Chem. – Eur. J., 2019, 25, 11924–11932 CrossRef CAS PubMed.
- P. R. Huddleston, V. V. Volkov and C. C. Perry, Phys. Chem. Chem. Phys., 2019, 21, 1344–1353 RSC.
- C. Slavov, C. Yang, A. H. Heindl, H. A. Wegner, A. Dreuw and J. Wachtveitl, Angew. Chem., Int. Ed., 2020, 59, 380–387 CrossRef CAS PubMed.
- A. H. Heindl and H. A. Wegner, Chem. – Eur. J., 2020, 26, 13730–13737 CrossRef CAS PubMed.
- F. Aleotti, A. Nenov, L. Salvigni, M. Bonfanti, M. M. El-Tahawy, A. Giunchi, M. Gentile, C. Spallacci, A. Ventimiglia, G. Cirillo, L. Montali, S. Scurti, M. Garavelli and I. Conti, J. Phys. Chem. A, 2020, 124, 9513–9523 CrossRef CAS PubMed.
- S. Kobayashi, H. Yokoyama and H. Kamei, Chem. Phys. Lett., 1987, 138, 333–338 CrossRef CAS.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 CrossRef CAS PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos, G. A. Woolley, F. Renth, F. Temps and D. Trauner, Chem. Commun., 2013, 49, 10314 RSC.
- O. Sadovski, A. A. Beharry, F. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS PubMed.
- D. Martínez-López, A. Babalhavaeji, D. Sampedro and G. A. Woolley, Beilstein J. Org. Chem., 2019, 15, 3000–3008 CrossRef PubMed.
- M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 13514–13518 CrossRef CAS PubMed.
- D. B. Konrad, J. A. Frank and D. Trauner, Chem. – Eur. J., 2016, 22, 4364–4368 CrossRef CAS PubMed.
- D. Martínez-López, A. Babalhavaeji, D. Sampedro and G. A. Woolley, Beilstein J. Org. Chem., 2019, 15, 3000–3008 CrossRef PubMed.
- C. Boulègue, M. Löweneck, C. Renner and L. Moroder, ChemBioChem, 2007, 8, 591–594 CrossRef PubMed.
- S. Zbaida, Drug Metab. Rev., 1995, 27, 497–516 CrossRef CAS PubMed.
- J. Dokić, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef PubMed.
- S. Steinwand, T. Halbritter, D. Rastädter, J. M. Ortiz-Sánchez, I. Burghardt, A. Heckel and J. Wachtveitl, Chem. – Eur. J., 2015, 21, 15720–15731 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- P. Lentes, E. Stadler, F. Röhricht, A. Brahms, J. Gröbner, F. D. Sönnichsen, G. Gescheidt and R. Herges, J. Am. Chem. Soc., 2019, 141, 13592–13600 CrossRef CAS PubMed.
- R. Siewertsen, J. B. Schönborn, B. Hartke, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2011, 13, 1054–1063 RSC.
- T. Tellkamp, J. Shen, Y. Okamoto and R. Herges, Eur. J. Org. Chem., 2014, 5456–5461 CrossRef CAS.
- W. Moormann, D. Langbehn and R. Herges, Synthesis, 2017, 3471–3475 CAS.
- J. B. Trads, K. Hüll, B. S. Matsuura, L. Laprell, T. Fehrentz, N. Görldt, K. A. Kozek, C. D. Weaver, N. Klöcker, D. M. Barber and D. Trauner, Angew. Chem., Int. Ed., 2019, 58, 15421–15428 CrossRef CAS PubMed.
- H. Sell, C. Näther and R. Herges, Beilstein J. Org. Chem., 2013, 9, 1–7 CrossRef CAS PubMed.
- M. Schehr, D. Hugenbusch, T. Moje, C. Näther and R. Herges, Beilstein J. Org. Chem., 2018, 14, 2799–2804 CrossRef CAS PubMed.
- W. Moormann, D. Langbehn and R. Herges, Beilstein J. Org. Chem., 2019, 15, 727–732 CrossRef CAS PubMed.
- P. Lentes, P. Frühwirt, H. Freißmuth, W. Moormann, F. Kruse, G. Gescheidt and R. Herges, J. Org. Chem., 2021, 86, 4355–4360 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- R. Brode and G. M. Wyman, J. Res. Natl. Bur. Stand., 1951, 47, 170–178 CrossRef.
- G. M. Wyman, Chem. Rev., 1955, 55, 625–657 CrossRef CAS.
- J. Pina, D. Sarmento, M. Accoto, P. L. Gentili, L. Vaccaro, A. Galvão and J. S. Seixas de Melo, J. Phys. Chem. B, 2017, 121, 2308–2318 CrossRef CAS PubMed.
- S. Yamazaki, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2011, 13, 1618–1628 RSC.
- I. Iwakura, A. Yabushita and T. Kobayashi, Chem. Phys. Lett., 2010, 484, 354–357 CrossRef CAS.
- C. Giuliano, L. Hess and J. Margerum, J. Am. Chem. Soc., 1968, 90, 587–594 CrossRef CAS.
- B. Koeppe and V. R. F. Schröder, ChemPhotoChem, 2019, 3, 613–618 CrossRef CAS.
- D. Farka, M. Scharber, E. D. Głowacki and N. S. Sariciftci, J. Phys. Chem. A, 2015, 119, 3563–3568 CrossRef CAS PubMed.
- D. Pinheiro, A. M. Galvão, M. Pineiro and J. S. S. de Melo, J. Phys. Chem. B, 2021, 125, 4108–4119 CrossRef CAS PubMed.
- C.-Y. Huang, A. Bonasera, L. Hristov, Y. Garmshausen, B. M. Schmidt, D. Jacquemin and S. Hecht, J. Am. Chem. Soc., 2017, 139, 15205–15211 CrossRef CAS PubMed.
- L. A. Huber, P. Mayer and H. Dube, ChemPhotoChem, 2018, 2, 458–464 CrossRef CAS.
- S. C. Dahlberg and C. B. Reinganum, J. Chem. Phys., 1981, 75, 2429–2431 CrossRef CAS.
- H. Kurata, S. Kim, K. Matsumoto, T. Kawase and M. Oda, Chem. Lett., 2007, 386–387 CrossRef CAS.
- J. Z. Vlahakis, K. E. Maly and R. P. Lemieux, J. Mater. Chem., 2001, 11, 2459–2464 RSC.
- G. Boice, B. O. Patrick, R. McDonald, C. Bohne and R. Hicks, J. Org. Chem., 2014, 79, 9196–9205 CrossRef CAS PubMed.
- D. Jacquemin, J. Preat, V. Wathelet, M. Fontaine and E. A. Perpète, J. Am. Chem. Soc., 2006, 128, 2072–2083 CrossRef CAS PubMed.
- J. Sühnel and K. Gustav, J. Prakt. Chem., 1978, 320, 917–921 CrossRef.
- E. I. Stearns, J. Opt. Soc. Am., 1942, 32, 282 CrossRef CAS.
- R. Brode and G. M. Wyman, J. Am. Chem. Soc., 1951, 73, 1487–1493 CrossRef.
- D. L. Ross, Appl. Opt., 1971, 10, 571–576 CrossRef CAS PubMed.
- B. Koeppe and F. Römpp, Chem. – Eur. J., 2018, 24, 14382–14386 CrossRef CAS PubMed.
- B. Koeppe, S. Rühl and F. Römpp, ChemPhotoChem, 2019, 3, 71–74 CrossRef CAS.
- R. C. Pereira, M. Pineiro, A. M. Galvão and J. S. Seixas de Melo, Dyes Pigm., 2018, 158, 259–266 CrossRef CAS.
- S. Kitzig, M. Thilemann, T. Cordes and K. Rück-Braun, ChemPhysChem, 2016, 17, 1252–1263 CrossRef CAS PubMed.
- C. Petermayer, S. Thumser, F. Kink, P. Mayer and H. Dube, J. Am. Chem. Soc., 2017, 139, 15060–15067 CrossRef CAS PubMed.
- X. Zhao and S. Yang, J. Lumin., 2020, 220, 116993 CrossRef CAS.
- B. Maerz, S. Wiedbrauk, S. Oesterling, E. Samoylova, A. Nenov, P. Mayer, R. de Vivie-Riedle, W. Zinth and H. Dube, Chem. – Eur. J., 2014, 20, 13984–13992 CrossRef CAS PubMed.
- S. Wiedbrauk and H. Dube, Tetrahedron Lett., 2015, 56, 4266–4274 CrossRef CAS.
- S. Wiedbrauk, B. Maerz, E. Samoylova, A. Reiner, F. Trommer, P. Mayer, W. Zinth and H. Dube, J. Am. Chem. Soc., 2016, 138, 12219–12227 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 13558–13561 CrossRef CAS PubMed.
- V. Smokal, O. Kharchenko, Y. Karabets, N. Iukhymenko, A. Kysil, O. Krupka and A. Kolendo, Mol. Cryst. Liq. Cryst., 2018, 672, 11–17 CrossRef CAS.
- M. W. H. Hoorens, M. Medved’, A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. Jan Buma and W. Szymanski, Nat. Commun., 2019, 10, 2390 CrossRef PubMed.
- D. V. Berdnikova, Beilstein J. Org. Chem., 2019, 15, 2822–2829 CrossRef CAS PubMed.
- E. Carrascosa, C. Petermayer, M. S. Scholz, J. N. Bull, H. Dube and E. J. Bieske, ChemPhysChem, 2020, 21, 680–685 CrossRef CAS PubMed.
- D. V. Berdnikova, Chem. Commun., 2019, 55, 8402–8405 RSC.
- M. Ikegami and T. Arai, Chem. Lett., 2005, 492–493 CrossRef CAS.
- W. Steinle and K. Rück-Braun, Org. Lett., 2003, 5, 141–144 CrossRef CAS PubMed.
- F. F. Graupner, T. T. Herzog, F. Rott, S. Oesterling, R. de Vivie-Riedle, T. Cordes and W. Zinth, Chem. Phys., 2018, 515, 614–621 CrossRef CAS.
- J. E. Zweig, T. A. Ko, J. Huang and T. R. Newhouse, Tetrahedron, 2019, 75, 130466 CrossRef CAS.
- J. E. Zweig and T. R. Newhouse, J. Am. Chem. Soc., 2017, 139, 10956–10959 CrossRef CAS PubMed.
- A. Sailer, F. Ermer, Y. Kraus, F. H. Lutter, C. Donau, M. Bremerich, J. Ahlfeld and O. Thorn-Seshold, ChemBioChem, 2019, 20, 1305–1314 CrossRef CAS PubMed.
- A. Sailer, F. Ermer, Y. Kraus, R. Bingham, F. H. Lutter, J. Ahlfeld and O. Thorn-Seshold, Beilstein J. Org. Chem., 2020, 16, 125–134 CrossRef CAS PubMed.
- M. Poutanen, Z. Ahmed, L. Rautkari, O. Ikkala and A. Priimagi, ACS Macro Lett., 2018, 7, 381–386 CrossRef CAS PubMed.
- M. Füllbeck, E. Michalsky, I. S. Jaeger, P. Henklein, H. Kuhn, K. Rück-Braun and R. Preissner, Genome Inform., 2006, 17, 141–151 Search PubMed.
- K. Hoffmann, M. Guentner, P. Mayer and H. Dube, Org. Chem. Front., 2019, 6, 1244–1252 RSC.
- S. Wiedbrauk, T. Bartelmann, S. Thumser, P. Mayer and H. Dube, Nat. Commun., 2018, 9, 1456 CrossRef PubMed.
- F. Kink, M. P. Collado, S. Wiedbrauk, P. Mayer and H. Dube, Chem. – Eur. J., 2017, 23, 6237–6243 CrossRef CAS PubMed.
- M. Guentner, E. Uhl, P. Mayer and H. Dube, Chem. – Eur. J., 2016, 22, 16433–16436 CrossRef CAS PubMed.
- G. Moncelsi, L. Escobar, H. Dube and P. Ballester, Chem. – Asian J., 2018, 13, 1632–1639 CrossRef CAS PubMed.
- L. A. Huber, K. Hoffmann, S. Thumser, N. Böcher, P. Mayer and H. Dube, Angew. Chem., Int. Ed., 2017, 56, 14536–14539 CrossRef CAS PubMed.
- E. Uhl, P. Mayer and H. Dube, Angew. Chem., Int. Ed., 2020, 59, 5730–5737 CrossRef CAS PubMed.
- K. Hoffmann, P. Mayer and H. Dube, Org. Biomol. Chem., 2019, 17, 1979–1983 RSC.
- R. Wilcken, M. Schildhauer, F. Rott, L. A. Huber, M. Guentner, S. Thumser, K. Hoffmann, S. Oesterling, R. de Vivie-Riedle, E. Riedle and H. Dube, J. Am. Chem. Soc., 2018, 140, 5311–5318 CrossRef CAS PubMed.
- A. Gerwien, P. Mayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 16442–16445 CrossRef CAS PubMed.
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Nat. Commun., 2015, 6, 8406 Search PubMed.
- M. Schildhauer, F. Rott, S. Thumser, P. Mayer, R. de Vivie-Riedle and H. Dube, ChemPhotoChem, 2019, 3, 365–371 CrossRef CAS.
- A. Gerwien, P. Mayer and H. Dube, Nat. Commun., 2019, 10, 4449 CrossRef PubMed.
- C. Zwergel, F. Gaascht, S. Valente, M. Diederich, D. Bagrel and G. Kirsch, Nat. Prod. Commun., 2012, 7, 389–394 CrossRef CAS PubMed.
- A. V. Popova, S. P. Bondarenko and M. S. Frasinyuk, Chem. Heterocycl. Compd., 2019, 55, 285–299 CrossRef CAS.
- D. Rambabu, S. Srinivas, K. Manjulatha, S. Basavoju, M. V. B. Rao and M. Pal, Mol. Cryst. Liq. Cryst., 2013, 577, 83–94 CrossRef CAS.
- Y. Xue, Y. Dou, L. An, Y. Zheng, L. Zhang and Y. Liu, RSC Adv., 2016, 6, 7002–7010 RSC.
- K. Muñoz-Becerra, N. Villegas-Escobar, C. Zúñiga-Loyola, D. Cortés-Arriagada and A. Toro-Labbé, Mol. Phys., 2019, 117, 1451–1458 CrossRef.
- J. R. Pires, C. Saito, S. L. Gomes, A. M. Giesbrecht and A. T. Amaral, J. Med. Chem., 2001, 44, 3673–3681 CrossRef CAS PubMed.
- C. Espinosa-Bustos, D. Cortés-Arriagada, M. A. Soto-Arriaza, J. Robinson-Duggon, N. Pizarro, A. R. Cabrera, D. Fuentealba and C. O. Salas, Photochem. Photobiol. Sci., 2017, 16, 1268–1276 CrossRef CAS PubMed.
- N. Shanker, O. Dilek, K. Mukherjee, D. W. McGee and S. L. Bane, J. Fluoresc., 2011, 21, 2173–2184 CrossRef CAS PubMed.
- M. Li, S. Yang, W. Liang, X. Zhang and D. Qu, Dyes Pigm., 2019, 166, 239–244 CrossRef CAS.
- M. Medved’, M. W. H. Hoorens, M. Di Donato, A. D. Laurent, J. Fan, M. Taddei, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymanski, Chem. Sci., 2021, 12, 4588–4598 RSC.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- L. A. Tatum, X. Su and I. Aprahamian, Acc. Chem. Res., 2014, 47, 2141–2149 CrossRef CAS PubMed.
- I. Aprahamian, Chem. Commun., 2017, 53, 6674–6684 RSC.
- S. M. Landge and I. Aprahamian, J. Am. Chem. Soc., 2009, 131, 18269–18271 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benitez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Org. Lett., 2011, 13, 30–33 CrossRef CAS PubMed.
- X. Su, S. Voskian, R. P. Hughes and I. Aprahamian, Angew. Chem., Int. Ed., 2013, 52, 10734–10739 CrossRef CAS PubMed.
- M. J. Moran, M. Magrini, D. M. Walba and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 13623–13627 CrossRef CAS PubMed.
- M. L. Croteau, X. Su, D. E. Wilcox and I. Aprahamian, ChemPlusChem, 2014, 79, 1214–1224 CrossRef CAS.
- X. Su and I. Aprahamian, Org. Lett., 2013, 15, 5952–5955 CrossRef CAS PubMed.
- J. T. Foy, D. Ray and I. Aprahamian, Chem. Sci., 2015, 6, 209–213 RSC.
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed.
- B. Shao and I. Aprahamian, ChemPhotoChem, 2019, 3, 361–364 CrossRef CAS.
- S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed.
- B. Shao, N. Stankewitz, J. A. Morris, M. D. Liptak and I. Aprahamian, Chem. Commun., 2019, 55, 9551–9554 RSC.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef CAS PubMed.
- I. Cvrtila, H. Fanlo-Virgós, G. Schaeffer, G. Monreal Santiago and S. Otto, J. Am. Chem. Soc., 2017, 139, 12459–12465 CrossRef CAS PubMed.
- B. Shao, M. Baroncini, H. Qian, L. Bussotti, M. Di Donato, A. Credi and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 12323–12327 Search PubMed.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- P. Courtot, R. Pichon and J. Le Saint, Tetrahedron Lett., 1976, 17, 1181–1184 CrossRef.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
- L.-Q. Zheng, S. Yang, J. Lan, L. Gyr, G. Goubert, H. Qian, I. Aprahamian and R. Zenobi, J. Am. Chem. Soc., 2019, 141, 17637–17645 CrossRef CAS PubMed.
- A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS PubMed.
- B. Shao, H. Qian, Q. Li and I. Aprahamian, J. Am. Chem. Soc., 2019, 141, 8364–8371 CrossRef CAS PubMed.
- B. Mravec, J. Filo, K. Csicsai, V. Garaj, M. Kemka, A. Marini, M. Mantero, A. Bianco and M. Cigáň, Phys. Chem. Chem. Phys., 2019, 21, 24749–24757 RSC.
- X. Guo, T. Mao, Z. Wang, P. Cheng, Y. Chen, S. Ma and Z. Zhang, ACS Cent. Sci., 2020, 6, 787–794 CrossRef CAS PubMed.
- B. Shao and I. Aprahamian, ChemistryOpen, 2020, 9, 191–194 CrossRef CAS PubMed.
- Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829–11835 CrossRef CAS PubMed.
- A. Iagatti, B. Shao, A. Credi, B. Ventura, I. Aprahamian and M. Di Donato, Beilstein J. Org. Chem., 2019, 15, 2438–2446 CrossRef CAS PubMed.
- X. Guo, B. Shao, S. Zhou, I. Aprahamian and Z. Chen, Chem. Sci., 2020, 11, 3016–3021 RSC.
- P. Gupta, T. Panda, S. Allu, S. Borah, A. Baishya, A. Gunnam, A. Nangia, P. Naumov and N. K. Nath, Cryst. Growth Des., 2019, 19, 3039–3044 CrossRef CAS.
- K. C. Hall, A. T. Franks, R. C. McAtee, M. S. Wang, V. I. Lu and K. J. Franz, Photochem. Photobiol. Sci., 2017, 16, 1604–1612 CrossRef CAS PubMed.
- G. Vantomme and J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 3940–3943 CrossRef CAS PubMed.
- S. Yang, D. Larsen, M. Pellegrini, S. Meier, D. F. Mierke, S. R. Beeren and I. Aprahamian, Chem, 2021, 7, 2190–2200 CrossRef CAS PubMed.
- Y.-X. Yuan and Y.-S. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 7303–7310 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190–13193 CrossRef CAS PubMed.
- H. Qian, Y.-Y. Wang, D.-S. Guo and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 1037–1040 CrossRef CAS PubMed.
- H. Qian, B. Shao and I. Aprahamian, Tetrahedron, 2017, 73, 4901–4904 CrossRef CAS.
- M. Irie and M. Morimoto, Photochemistry, Royal Society of Chemistry, 2020, vol. 47, pp. 457–497 Search PubMed.
- M. Irie, Y. Yokoyama, M. Irie, Y. Yokoyama and M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- M. Irie, Diarylethene Molecular Photoswitches, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2021 Search PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- T. Nakagawa, Y. Hasegawa and T. Kawai, J. Phys. Chem. A, 2008, 112, 5096–5103 CrossRef CAS PubMed.
- C. Fleming, P. Remón, S. Li, N. A. Simeth, B. König, M. Grøtli and J. Andréasson, Dyes Pigm., 2017, 137, 410–420 CrossRef CAS.
- D. Wutz, C. Falenczyk, N. Kuzmanovic and B. König, RSC Adv., 2015, 5, 18075–18086 RSC.
- C. Falenczyk, M. Schiedel, B. Karaman, T. Rumpf, N. Kuzmanovic, M. Grøtli, W. Sippl, M. Jung and B. König, Chem. Sci., 2014, 5, 4794–4799 RSC.
- P. H.-M. Lee, C.-C. Ko, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 6058–6059 CrossRef CAS PubMed.
- V. W.-W. Yam, C.-C. Ko and N. Zhu, J. Am. Chem. Soc., 2004, 126, 12734–12735 CrossRef CAS PubMed.
- N. A. Simeth, A. C. Kneuttinger, R. Sterner and B. König, Chem. Sci., 2017, 8, 6474–6483 RSC.
- E. Saito, T. Ako, Y. Kobori and A. Tsuda, RSC Adv., 2017, 7, 2403–2406 RSC.
- I. Colombier, S. Spagnoli, A. Corval, P. L. Baldeck, M. Giraud, A. Leaustic, P. Yu and M. Irie, J. Chem. Phys., 2007, 126, 011101 CrossRef CAS PubMed.
- E. C. Harvey, B. L. Feringa, J. G. Vos, W. R. Browne and M. T. Pryce, Coord. Chem. Rev., 2015, 282–283, 77–86 CrossRef CAS.
- N. M.-W. Wu, M. Ng, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 15142–15150 CrossRef CAS PubMed.
- C. Zheng, C. Fan, S. Pu, B. Ben Chen and B. Ben Chen, J. Mol. Struct., 2016, 1123, 355–359 CrossRef CAS.
- X. Li, S. Pu, H. Li and G. Liu, Dyes Pigm., 2014, 105, 47–56 CrossRef CAS.
- R. Wang, S. Pu, G. Liu and S. Cui, Beilstein J. Org. Chem., 2012, 8, 1018–1026 CrossRef CAS PubMed.
- G. Liu, M. Liu, S. Pu, C. Fan and S. Cui, Tetrahedron, 2012, 68, 2267–2275 CrossRef CAS.
- A. G. Lvov, M. Mörtel, A. V. Yadykov, F. W. Heinemann, V. Z. Shirinian and M. M. Khusniyarov, Beilstein J. Org. Chem., 2019, 15, 2428–2437 CrossRef CAS PubMed.
- L. Gundogdu, M. Kose, S. Takeuchi, Y. Yokoyama and E. Orhan, J. Lumin., 2018, 203, 568–575 CrossRef CAS.
- M. P. O’Hagan, J. Ramos-Soriano, S. Haldar, S. Sheikh, J. C. Morales, A. J. Mulholland and M. C. Galan, Chem. Commun., 2020, 56, 5186–5189 RSC.
- S. Nakamura, S. Yokojima, K. Uchida, T. Tsujioka, A. Goldberg, A. Murakami, K. Shinoda, M. Mikami, T. Kobayashi, S. Kobatake, K. Matsuda and M. Irie, J. Photochem. Photobiol., A, 2008, 200, 10–18 CrossRef CAS.
- S. Fukumoto, T. Nakashima and T. Kawai, Angew. Chem., Int. Ed., 2011, 50, 1565–1568 CrossRef CAS PubMed.
- R. Li, T. Nakashima, O. Galangau, S. Iijima, R. Kanazawa and T. Kawai, Chem. – Asian J., 2015, 10, 1725–1730 CrossRef CAS PubMed.
- R. Göstl, B. Kobin, L. Grubert, M. Pätzel and S. Hecht, Chem. – Eur. J., 2012, 18, 14282–14285 CrossRef PubMed.
- K. Morimitsu, S. Kobatake and M. Irie, Mol. Cryst. Liq. Cryst., 2005, 431, 451–454 CrossRef CAS.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- S. Fredrich, R. Göstl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- M. Irie and K. Sayo, J. Phys. Chem., 1992, 96, 7671–7674 CrossRef CAS.
- K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS PubMed.
- I. Hamdi, G. Buntinx, A. Perrier, O. Devos, N. Jaïdane, S. Delbaere, A. K. Tiwari, J. Dubois, M. Takeshita, Y. Wada and S. Aloïse, Phys. Chem. Chem. Phys., 2016, 18, 28091–28100 RSC.
- S. Fredrich, T. Morack, M. Sliwa and S. Hecht, Chem. – Eur. J., 2020, 26, 7672–7677 CrossRef CAS PubMed.
- M. Irie and M. Morimoto, Pure Appl. Chem., 2009, 81, 1655–1665 CAS.
- S. Kawai, T. Nakashima, K. Atsumi, T. Sakai, M. Harigai, Y. Imamoto, H. Kamikubo, M. Kataoka and T. Kawai, Chem. Mater., 2007, 19, 3479–3483 CrossRef CAS.
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–750 RSC.
- Y.-C. Jeong, D. G. Park, I. S. Lee, S. I. Yang and K.-H. Ahn, J. Mater. Chem., 2009, 19, 97–103 RSC.
- Y.-C. Jeong, S. I. Yang, E. Kim and K.-H. Ahn, Tetrahedron, 2006, 62, 5855–5861 CrossRef CAS.
- P. D. Patel, I. A. Mikhailov, K. D. Belfield and A. E. Masunov, Int. J. Quantum Chem., 2009, 109, 3711–3722 CrossRef CAS.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- Y.-C. Jeong, D. G. Park, E. Kim, K.-H. Ahn and S. I. Yang, Chem. Commun., 2006, 1881 RSC.
- J. J. D. de Jong, L. N. Lucas, R. Hania, A. Pugzlys, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Eur. J. Org. Chem., 2003, 1887–1893 CrossRef CAS.
- D. Lachmann, A. Konieczny, M. Keller and B. König, Org. Biomol. Chem., 2019, 17, 2467–2478 RSC.
- B. Roubinet, M. L. Bossi, P. Alt, M. Leutenegger, H. Shojaei, S. Schnorrenberg, S. Nizamov, M. Irie, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 15429–15433 CrossRef CAS PubMed.
- T. Yamaguchi, K. Uchida and M. Irie, J. Am. Chem. Soc., 1997, 119, 6066–6071 CrossRef CAS.
- N. Ziebart, F. Schröder and K. Rück-Braun, ChemPhotoChem, 2019, 3, 396–402 CrossRef CAS.
- M. Kathan, F. Eisenreich, C. Jurissek, A. Dallmann, J. Gurke and S. Hecht, Nat. Chem., 2018, 10, 1031–1036 CrossRef CAS PubMed.
- D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431–5434 CrossRef CAS PubMed.
- J.-M. L. Alvaro Fernández-Acebes, A. Fernández-Acebes and J.-M. Lehn, Chem. – Eur. J., 1999, 5, 3285–3292 CrossRef.
- F. Schweighöfer, L. Dworak, C. A. Hammer, H. Gustmann, M. Zastrow, K. Rück-Braun and J. Wachtveitl, Sci. Rep., 2016, 6, 28638 CrossRef PubMed.
- S. Cobo, F. Lafolet, E. Saint-Aman, C. Philouze, C. Bucher, S. Silvi, A. Credi and G. Royal, Chem. Commun., 2015, 51, 13886–13889 RSC.
- K. Klaue, Y. Garmshausen and S. Hecht, Angew. Chem., Int. Ed., 2018, 57, 1414–1417 CrossRef CAS PubMed.
- E. Lognon, M. Heitz, A. Bakkar, S. Cobo, F. Loiseau, E. Saint-Aman, G. Royal and M. Boggio-Pasqua, ChemPhysChem, 2020, 21, 1571–1577 CrossRef CAS PubMed.
- K. Klaue, W. Han, P. Liesfeld, F. Berger, Y. Garmshausen and S. Hecht, J. Am. Chem. Soc., 2020, 142, 11857–11864 CrossRef CAS PubMed.
- B. Saima, N. Khan, Y. S. S. Al-Faiyz, R. Ludwig, W. Rehman, M. Habib-ur-Rehman, N. S. Sheikh and K. Ayub, J. Mol. Graph. Model., 2019, 88, 261–272 CrossRef CAS PubMed.
- R. H. Mitchell, T. R. Ward, Y. Chen, Y. Wang, S. A. Weerawarna, P. W. Dibble, M. J. Marsella, A. Almutairi and Z.-Q. Wang, J. Am. Chem. Soc., 2003, 125, 2974–2988 CrossRef CAS PubMed.
- P. Liesfeld, Y. Garmshausen, S. Budzak, J. Becker, A. Dallmann, D. Jacquemin and S. Hecht, Angew. Chem., Int. Ed., 2020, 59, 19352–19358 CrossRef CAS PubMed.
- M. Jacquet, L. M. Uriarte, F. Lafolet, M. Boggio-Pasqua, M. Sliwa, F. Loiseau, E. Saint-Aman, S. Cobo and G. Royal, J. Phys. Chem. Lett., 2020, 11, 2682–2688 CrossRef CAS PubMed.
- H.-W. Lee, S. G. Robinson, S. Bandyopadhyay, R. H. Mitchell and D. Sen, J. Mol. Biol., 2007, 371, 1163–1173 CrossRef CAS PubMed.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- M. Mansø, A. U. Petersen, Z. Wang, P. Erhart, M. B. Nielsen and K. Moth-Poulsen, Nat. Commun., 2018, 9, 1945 CrossRef PubMed.
- M. Jevric, Z. Wang, A. U. Petersen, M. Mansø, C. J. Sumby, M. B. Nielsen and K. Moth-Poulsen, Eur. J. Org. Chem., 2019, 2354–2361 CrossRef CAS.
- A. Dreos, Z. Wang, B. E. Tebikachew, K. Moth-Poulsen and J. Andréasson, J. Phys. Chem. Lett., 2018, 9, 6174–6178 CrossRef CAS PubMed.
- M. Bertram, F. Waidhas, M. Jevric, L. Fromm, C. Schuschke, M. Kastenmeier, A. Görling, K. Moth-Poulsen, O. Brummel and J. Libuda, J. Chem. Phys., 2020, 152, 044708 CrossRef CAS PubMed.
- O. Brummel, D. Besold, T. Döpper, Y. Wu, S. Bochmann, F. Lazzari, F. Waidhas, U. Bauer, P. Bachmann, C. Papp, H.-P. Steinrück, A. Görling, J. Libuda and J. Bachmann, ChemSusChem, 2016, 9, 1424–1432 CrossRef CAS PubMed.
- O. Brummel, F. Waidhas, U. Bauer, Y. Wu, S. Bochmann, H.-P. Steinrück, C. Papp, J. Bachmann and J. Libuda, J. Phys. Chem. Lett., 2017, 8, 2819–2825 CrossRef CAS PubMed.
- U. Bauer, L. Fromm, C. Weiß, P. Bachmann, F. Späth, F. Düll, J. Steinhauer, W. Hieringer, A. Görling, A. Hirsch, H.-P. Steinrück and C. Papp, J. Phys. Chem. C, 2019, 123, 7654–7664 CrossRef CAS.
- H. Hogeveen and H. C. Volger, J. Am. Chem. Soc., 1967, 89, 2486–2487 CrossRef CAS.
- W. G. Dauben and R. L. Cargill, Tetrahedron, 1961, 15, 197–201 CrossRef CAS.
- B. E. Tebikachew, F. Edhborg, N. Kann, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2018, 20, 23195–23201 RSC.
- A. Dreos, Z. Wang, J. Udmark, A. Ström, P. Erhart, K. Börjesson, M. B. Nielsen and K. Moth-Poulsen, Adv. Energy Mater., 2018, 8, 1703401 CrossRef.
- C. Schuschke, C. Hohner, M. Jevric, A. Ugleholdt Petersen, Z. Wang, M. Schwarz, M. Kettner, F. Waidhas, L. Fromm, C. J. Sumby, A. Görling, O. Brummel, K. Moth-Poulsen and J. Libuda, Nat. Commun., 2019, 10, 2384 CrossRef PubMed.
- M. Jevric, A. U. Petersen, M. Mansø, S. Kumar Singh, Z. Wang, A. Dreos, C. Sumby, M. B. Nielsen, K. Börjesson, P. Erhart and K. Moth-Poulsen, Chem. – Eur. J., 2018, 24, 12767–12772 CrossRef CAS PubMed.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
- A. U. Petersen, A. I. Hofmann, M. Fillols, M. Mansø, M. Jevric, Z. Wang, C. J. Sumby, C. Müller and K. Moth-Poulsen, Adv. Sci., 2019, 6, 1900367 CrossRef PubMed.
- F. Waidhas, M. Jevric, L. Fromm, M. Bertram, A. Görling, K. Moth-Poulsen, O. Brummel and J. Libuda, Nano Energy, 2019, 63, 103872 CrossRef CAS.
- M. D. Kilde, M. Mansø, N. Ree, A. U. Petersen, K. Moth-Poulsen, K. V. Mikkelsen and M. B. Nielsen, Org. Biomol. Chem., 2019, 17, 7735–7746 RSC.
- M. Mansø, B. E. Tebikachew, K. Moth-Poulsen and M. B. Nielsen, Org. Biomol. Chem., 2018, 16, 5585–5590 RSC.
- M. Mansø, M. D. Kilde, S. K. Singh, P. Erhart, K. Moth-Poulsen and M. B. Nielsen, Phys. Chem. Chem. Phys., 2019, 21, 3092–3097 RSC.
- M. Quant, A. Hamrin, A. Lennartson, P. Erhart and K. Moth-Poulsen, J. Phys. Chem. C, 2019, 123, 7081–7087 CrossRef CAS.
- N. A. Simeth, L.-M. M. Altmann, N. Wössner, E. Bauer, M. Jung and B. König, J. Org. Chem., 2018, 83, 7919–7927 CrossRef CAS PubMed.
- R. K. Weerasekara, H. Uekusa and C. V. Hettiarachchi, Cryst. Growth Des., 2017, 17, 3040–3047 CrossRef CAS.
- R. Matsushima and H. Sakaguchi, J. Photochem. Photobiol., A, 1997, 108, 239–245 CrossRef CAS.
- K. Rustler, G. Maleeva, A. M. J. Gomila, P. Gorostiza, P. Bregestovski and B. König, Chem. – Eur. J., 2020, 26, 12722–12727 CrossRef CAS PubMed.
- F. Renth, R. Siewertsen and F. Temps, Int. Rev. Phys. Chem., 2013, 32, 1–38 Search PubMed.
- Y. Kohno, Y. Tamura and R. Matsushima, J. Photochem. Photobiol., A, 2009, 201, 98–101 CrossRef CAS.
- M. Seibold, M. Handschuh, H. Port and H. C. Wolf, J. Lumin., 1997, 72–74, 454–456 CrossRef CAS.
- J. Andréasson, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2008, 130, 11122–11128 CrossRef PubMed.
- C. Slavov, C. Boumrifak, C. A. Hammer, P. Trojanowski, X. Chen, W. J. Lees, J. Wachtveitl and M. Braun, Phys. Chem. Chem. Phys., 2016, 18, 10289–10296 RSC.
- X. Chen, N. I. Islamova, R. V. Robles and W. J. Lees, Photochem. Photobiol. Sci., 2011, 10, 1023–1029 CrossRef CAS PubMed.
- D. Wutz, D. Gluhacevic, A. Chakrabarti, K. Schmidtkunz, D. Robaa, F. Erdmann, C. Romier, W. Sippl, M. Jung and B. König, Org. Biomol. Chem., 2017, 15, 4882–4896 RSC.
- M. Kose, E. Orhan, M. Köse and E. Orhan, Turk. J. Chem., 2009, 33, 579–588 CAS.
- N. Sivasankaran and K. Palaninathan, Polym. Degrad. Stab., 2013, 98, 1852–1861 CrossRef CAS.
- E. N. Shepelenko, N. I. Makarova, V. A. Podshibyakin, K. S. Tikhomirova, A. D. Dubonosov, A. V. Metelitsa, V. A. Bren and V. I. Minkin, Russ. J. Org. Chem., 2017, 53, 366–370 CrossRef CAS.
- V. P. Rybalkin, N. I. Makarova, S. Y. Pluzhnikova, L. L. Popova, A. V. Metelitsa, V. A. Bren’ and V. I. Minkina, Russ. Chem. Bull., 2014, 63, 1780–1784 CrossRef CAS.
- M. Vlajić, W. Unger, J. Bruns and K. Rueck-Braun, Appl. Surf. Sci., 2019, 465, 686–692 CrossRef.
- A. Yassar, H. Jaafari, N. Rebière-Galy, M. Frigoli, C. Moustrou, A. Samat and R. Guglielmetti, Eur. Phys. J. Appl. Phys., 2002, 18, 3–8 CrossRef CAS.
- R. S. Becker and J. Michl, J. Am. Chem. Soc., 1966, 88, 5931–5933 CrossRef CAS.
- B. H. Strudwick, C. O’Bryen, H. J. Sanders, S. Woutersen and W. J. Buma, Phys. Chem. Chem. Phys., 2019, 21, 11689–11696 RSC.
- S. V. Paramonov, V. Lokshin, H. Ihmels and O. A. Fedorova, Photochem. Photobiol. Sci., 2011, 10, 1279–1282 CrossRef CAS PubMed.
- A. Migani, P. L. Gentili, F. Negri, M. Olivucci, A. Romani, G. Favaro and R. S. Becker, J. Phys. Chem. A, 2005, 109, 8684–8692 CrossRef CAS PubMed.
- S. Kumar, D. Hernandez, B. Hoa, Y. Lee, J. S. Yang and A. McCurdy, Org. Lett., 2008, 10, 3761–3764 CrossRef CAS PubMed.
- Y. Inagaki, Y. Kobayashi, K. Mutoh and J. Abe, J. Am. Chem. Soc., 2017, 139, 13429–13441 CrossRef CAS PubMed.
- C. M. Sousa, J. Berthet, S. Delbaere and P. J. Coelho, Dyes Pigm., 2019, 169, 118–124 CrossRef CAS.
- Y. Fang, Q. Meng, Z. Wang, G. Wang, H. Jiang, H. Zhao and Y. Wang, J. Sol–Gel Sci. Technol., 2015, 73, 293–298 CrossRef CAS.
- C. M. Sousa, J. Berthet, S. Delbaere, A. Polónia and P. J. Coelho, J. Org. Chem., 2015, 80, 12177–12181 CrossRef CAS PubMed.
- M. Zayat and D. Levy, J. Mater. Chem., 2003, 13, 727–730 RSC.
- D. Avagliano, P. A. Sánchez-Murcia and L. González, Phys. Chem. Chem. Phys., 2019, 21, 8614–8618 RSC.
- Y. Li, K. M.-C. Wong, A. Y.-Y. Tam, L. Wu and V. W.-W. Yam, Chem. – Eur. J., 2010, 16, 8690–8698 CrossRef CAS PubMed.
- C. Kaiser, T. Halbritter, A. Heckel and J. Wachtveitl, ChemistrySelect, 2017, 2, 4111–4123 CrossRef CAS.
- L. Kortekaas, O. Ivashenko, J. T. van Herpt and W. R. Browne, J. Am. Chem. Soc., 2016, 138, 1301–1312 CrossRef CAS PubMed.
- L. Kortekaas, J. Chen, D. Jacquemin and W. R. Browne, J. Phys. Chem. B, 2018, 122, 6423–6430 CrossRef CAS PubMed.
- J. T. C. Wojtyk, A. Wasey, N.-N. Xiao, P. M. Kazmaier, S. Hoz, C. Yu, R. P. Lemieux and E. Buncel, J. Phys. Chem. A, 2007, 111, 2511–2516 CrossRef CAS PubMed.
- J. D. Steen, D. R. Duijnstee, A. S. Sardjan, J. Martinelli, L. Kortekaas, D. Jacquemin and W. R. Browne, J. Phys. Chem. A, 2021, 125, 3355–3361 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- C. L. Fleming, S. Li, M. Grøtli and J. Andréasson, J. Am. Chem. Soc., 2018, 140, 14069–14072 CrossRef CAS PubMed.
- F. Jonsson, T. Beke-Somfai, J. Andréasson and B. Nordén, Langmuir, 2013, 29, 2099–2103 CrossRef CAS PubMed.
- T. Stafforst and D. Hilvert, Chem. Commun., 2009, 287–288 RSC.
- F. Liu and K. Morokuma, J. Am. Chem. Soc., 2013, 135, 10693–10702 CrossRef CAS PubMed.
- S. F. Bittmann, R. Dsouza, K. M. Siddiqui, S. A. Hayes, A. Rossos, G. Corthey, M. Kochman, V. I. Prokhorenko, R. S. Murphy, H. Schwoerer and R. J. D. Miller, Phys. Chem. Chem. Phys., 2019, 21, 18119–18127 RSC.
- M. Hammarson, J. R. Nilsson, S. Li, T. Beke-Somfai and J. Andréasson, J. Phys. Chem. B, 2013, 117, 13561–13571 CrossRef CAS PubMed.
- H. Görner, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC.
- C. Brieke and A. Heckel, Chem. – Eur. J., 2013, 19, 15726–15734 CrossRef CAS PubMed.
- S. Scarmagnani, Z. Walsh, C. Slater, N. Alhashimy, B. Paull, M. Macka and D. Diamond, J. Mater. Chem., 2008, 18, 5063 RSC.
- M. Schnurbus, M. Kabat, E. Jarek, M. Krzan, P. Warszynski and B. Braunschweig, Langmuir, 2020, 36, 6871–6879 CrossRef CAS PubMed.
- C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457–8468 RSC.
- D. Avagliano, P. A. Sánchez-Murcia and L. González, Chem. – Eur. J., 2020, 26, 13039–13045 CrossRef CAS PubMed.
- M. J. Feeney and S. W. Thomas, Macromolecules, 2018, 51, 8027–8037 CrossRef CAS.
- X. Xie, G. Mistlberger and E. Bakker, J. Am. Chem. Soc., 2012, 134, 16929–16932 CrossRef CAS PubMed.
- X. Wang, J. Hu, Y. Li, J. Jie, A. Xia, S. Zhu, Y. Wang and S. X.-A. Zhang, J. Phys. Chem. C, 2016, 120, 598–605 CrossRef CAS.
- C. Özçoban, T. Halbritter, S. Steinwand, L.-M. Herzig, J. Kohl-Landgraf, N. Askari, F. Groher, B. Fürtig, C. Richter, H. Schwalbe, B. Suess, J. Wachtveitl and A. Heckel, Org. Lett., 2015, 17, 1517–1520 CrossRef PubMed.
- M. Hammarson, J. R. Nilsson, S. Li, P. Lincoln and J. Andréasson, Chem. – Eur. J., 2014, 20, 15855–15862 CrossRef CAS PubMed.
- J. Kohl-Landgraf, M. Braun, C. Özçoban, D. P. N. Gonçalves, A. Heckel and J. Wachtveitl, J. Am. Chem. Soc., 2012, 134, 14070–14077 CrossRef CAS PubMed.
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836–11837 CrossRef CAS PubMed.
- J. R. Nilsson, S. Li, B. Önfelt and J. Andréasson, Chem. Commun., 2011, 47, 11020–11022 RSC.
- Y. Shiraishi, M. Itoh and T. Hirai, Phys. Chem. Chem. Phys., 2010, 12, 13737–13745 RSC.
- T. Halbritter, C. Kaiser, J. Wachtveitl and A. Heckel, J. Org. Chem., 2017, 82, 8040–8047 CrossRef CAS PubMed.
- D. Hu, Z. Tian, W. Wu, W. Wan and A. Li, Microsc. Microanal., 2009, 15, 840–841 CrossRef.
- I. S. Park, Y.-S. Jung, K.-J. Lee and J.-M. Kim, Chem. Commun., 2010, 46, 2859–2861 RSC.
- B. Seefeldt, R. Kasper, M. Beining, J. Mattay, J. Arden-Jacob, N. Kemnitzer, K. H. Drexhage, M. Heilemann and M. Sauer, Photochem. Photobiol. Sci., 2010, 9, 213–220 CrossRef CAS PubMed.
- D. Dulić, S. J. van der Molen, T. Kudernac, H. T. Jonkman, J. J. D. de Jong, T. N. Bowden, J. van Esch, B. L. Feringa and B. J. van Wees, Phys. Rev. Lett., 2003, 91, 207402 CrossRef PubMed.
- S. Garg, H. Schwartz, M. Kozlowska, A. B. Kanj, K. Müller, W. Wenzel, U. Ruschewitz and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 1193–1197 CrossRef CAS PubMed.
- A. B. Kanj, A. Chandresh, A. Gerwien, S. Grosjean, S. Bräse, Y. Wang, H. Dube and L. Heinke, Chem. Sci., 2020, 11, 1404–1410 RSC.
- S. K. Rastogi, Z. Zhao, M. B. Gildner, B. A. Shoulders, T. L. Velasquez, M. O. Blumenthal, L. Wang, X. Li, T. W. Hudnall, T. Betancourt, L. Du and W. J. Brittain, Tetrahedron, 2021, 80, 131854 CrossRef CAS.
- F. Krohm, J. Kind, R. Savka, M. Alcaraz Janßen, D. Herold, H. Plenio, C. M. Thiele and A. Andrieu-Brunsen, J. Mater. Chem. C, 2016, 4, 4067–4076 RSC.
- Q. Zou, X. Li, J. Zhou, K. Bai and H. Ågren, Dyes Pigm., 2014, 107, 174–181 CrossRef CAS.
- S. Coleman, R. Byrne, S. Minkovska and D. Diamond, Phys. Chem. Chem. Phys., 2009, 11, 5608–5614 RSC.
- L. Raboin, M. Matheron, J. Biteau, T. Gacoin and J.-P. Boilot, J. Mater. Chem., 2008, 18, 3242–3248 RSC.
- R. Pardo, M. Zayat and D. Levy, J. Mater. Chem., 2009, 19, 6756–6760 RSC.
- P. R. Sahoo and S. Kumar, Sens. Actuators, B, 2016, 226, 548–552 CrossRef CAS.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- A. D. Laurent, M. Medveď and D. Jacquemin, ChemPhysChem, 2016, 17, 1846–1851 CrossRef CAS PubMed.
- D. M. Sanchez, U. Raucci, K. N. Ferreras and T. J. Martínez, J. Phys. Chem. Lett., 2020, 11, 7901–7907 CrossRef CAS PubMed.
- M. Ugandi and M. Roemelt, J. Phys. Chem. A, 2020, 124, 7756–7767 CrossRef CAS PubMed.
- C. García-Iriepa, M. Marazzi and D. Sampedro, ChemPhotoChem, 2019, 3, 866–873 CrossRef.
- M. Di Donato, M. M. Lerch, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, S. P. Ihrig, M. Medved’, D. Jacquemin, W. Szymański, W. J. Buma, P. Foggi and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 15596–15599 CrossRef CAS PubMed.
- M. M. Lerch, S. J. Wezenberg, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 6344–6347 CrossRef CAS PubMed.
- J. N. Bull, E. Carrascosa, N. Mallo, M. S. Scholz, G. da Silva, J. E. Beves and E. J. Bieske, J. Phys. Chem. Lett., 2018, 9, 665–671 CrossRef CAS PubMed.
- M. M. Lerch, M. Di Donato, A. D. Laurent, M. Medved’, A. Iagatti, L. Bussotti, A. Lapini, W. J. Buma, P. Foggi, W. Szymański and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 8063–8068 CrossRef CAS PubMed.
- M. M. Lerch, M. Medved', A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, W. Szymański, W. J. Buma, P. Foggi, M. Di Donato and B. L. Feringa, J. Phys. Chem. A, 2018, 122, 955–964 CrossRef CAS PubMed.
- R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, Chem. – Eur. J., 2018, 24, 9170–9186 CrossRef CAS PubMed.
- N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242–8252 RSC.
- Y. Duan, H. Zhao, C. Xiong, L. Mao, D. Wang and Y. Zheng, Chin. J. Chem., 2021, 39, 985–998 CrossRef CAS.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- J. R. Hemmer, Z. A. Page, K. D. Clark, F. Stricker, N. D. Dolinski, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2018, 140, 10425–10429 CrossRef CAS PubMed.
- M. M. Sroda, F. Stricker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Chem. – Eur. J., 2021, 27, 4183–4190 CrossRef CAS PubMed.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- H. Zulfikri, M. A. J. Koenis, M. M. Lerch, M. Di Donato, W. Szymański, C. Filippi, B. L. Feringa and W. J. Buma, J. Am. Chem. Soc., 2019, 141, 7376–7384 CrossRef CAS PubMed.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. C. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Chem. Commun., 2016, 52, 13576–13579 RSC.
- B. F. Lui, N. T. Tierce, F. Tong, M. M. Sroda, H. Lu, J. Read de Alaniz and C. J. Bardeen, Photochem. Photobiol. Sci., 2019, 18, 1587–1595 CrossRef CAS PubMed.
- J. Alves, S. Wiedbrauk, C. Barner-Kowollik and J. P. Blinco, ChemPhotoChem, 2021, 5, 1–6 CrossRef.
- H. Zhao, D. Wang, Y. Fan, M. Ren, S. Dong and Y. Zheng, Langmuir, 2018, 34, 15537–15543 CrossRef CAS PubMed.
- J. Lee, M. M. Sroda, Y. Kwon, S. El-Arid, S. Seshadri, L. F. Gockowski, E. W. Hawkes, M. T. Valentine and J. Read de Alaniz, ACS Appl. Mater. Interfaces, 2020, 12, 54075–54082 CrossRef CAS PubMed.
- J. E. Yap, N. Mallo, D. S. Thomas, J. E. Beves and M. H. Stenzel, Polym. Chem., 2019, 10, 6515–6522 RSC.
- J. Ahrens, T. Bian, T. Vexler and R. Klajn, ChemPhotoChem, 2017, 1, 230–236 CrossRef CAS.
- N. Mallo, A. Tron, J. Andréasson, J. B. Harper, L. S. D. Jacob, N. D. McClenaghan, G. Jonusauskas and J. E. Beves, ChemPhotoChem, 2020, 4, 407–412 CrossRef CAS.
- C. Zheng, Y. Yu, K. Shi, B. Zhu, H. Zhou, S. Zhang, J. Yang, L. Shi and C. Ran, 2020, Prepr. from bioRxiv, DOI:10.1101/2020.10.04.325696.
- R. Saha, A. Devaraj, S. Bhattacharyya, S. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2019, 141, 8638–8645 CrossRef CAS PubMed.
- J. E. Yap, L. Zhang, J. T. Lovegrove, J. E. Beves and M. H. Stenzel, Macromol. Rapid Commun., 2020, 41, 2000236 CrossRef CAS PubMed.
- Q. Chen, Y. J. Diaz, M. C. Hawker, M. R. Martinez, Z. A. Page, S. Xiao-An Zhang, C. J. Hawker and J. Read de Alaniz, Macromolecules, 2019, 52, 4370–4375 CrossRef CAS.
- S. H. Mostafavi, W. Li, K. D. Clark, F. Stricker, J. R. de Alaniz and C. J. Bardeen, Macromolecules, 2019, 52, 6311–6317 CrossRef CAS.
- G. Sobczak, I. Misztalewska-Turkowicz and V. Sashuk, J. Phys. Chem. C, 2021, 125, 5306–5314 CrossRef CAS.
- S. Singh, K. Friedel, M. Himmerlich, Y. Lei, G. Schlingloff and A. Schober, ACS Macro Lett., 2015, 4, 1273–1277 CrossRef CAS.
- Y.-D. Cai, T.-Y. Chen, X. Q. Chen and X. Bao, Org. Lett., 2019, 21, 7445–7449 CrossRef CAS PubMed.
- Y. Duan, H. Zhao, G. Xue, Z. Wang, C. Xiong, L. Mao, C. He, D. Wang and Y. Zheng, Prepr. Res. Sq., 2020 DOI:10.21203/RS.3.RS-43551/V1.
- S. Seshadri, L. F. Gockowski, J. Lee, M. Sroda, M. E. Helgeson, J. Read de Alaniz and M. T. Valentine, Nat. Commun., 2020, 11, 2599 CrossRef CAS PubMed.
- H. Gotfredsen, M. D. Kilde, M. Santella, A. Kadziola and M. B. Nielsen, Mol. Syst. Des. Eng., 2019, 4, 199–205 RSC.
- S. L. Broman and M. B. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 21172–21182 RSC.
- N. Shahzad, R. U. Nisa and K. Ayub, Struct. Chem., 2013, 24, 2115–2126 CrossRef CAS.
- A. U. Petersen, M. Jevric, R. J. Mandle, E. J. Davis, S. J. Cowling, J. W. Goodby and M. B. Nielsen, RSC Adv., 2015, 5, 89731–89744 RSC.
- H. Torres-Pierna, C. Roscini, A. Vlasceanu, S. L. Broman, M. Jevric, M. Cacciarini and M. B. Nielsen, Dyes Pigm., 2017, 145, 359–364 CrossRef CAS.
- A. Vlasceanu, M. Cacciarini and M. B. Nielsen, Tetrahedron, 2018, 74, 6635–6646 CrossRef CAS.
- M. D. Kilde, R. Kristensen, G. Olsen, J. O. Jeppesen and M. B. Nielsen, Eur. J. Org. Chem., 2019, 5532–5539 CrossRef CAS.
- M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. Brøndsted Nielsen, Chem. – Eur. J., 2015, 21, 7454–7461 CrossRef CAS PubMed.
- M. H. Hansen, J. Elm, S. T. Olsen, A. N. Gejl, F. E. Storm, B. N. Frandsen, A. B. Skov, M. B. Nielsen, H. G. Kjaergaard and K. V. Mikkelsen, J. Phys. Chem. A, 2016, 120, 9782–9793 CrossRef CAS PubMed.
- A. Perrier, F. Maurel and D. Jacquemin, Phys. Chem. Chem. Phys., 2011, 13, 13791–13799 RSC.
- A. S. Gertsen, S. T. Olsen, S. L. Broman, M. B. Nielsen and K. V. Mikkelsen, J. Phys. Chem. C, 2017, 121, 195–201 CrossRef CAS.
- A. U. Petersen, S. L. Broman, S. T. Olsen, A. S. Hansen, L. Du, A. Kadziola, T. Hansen, H. G. Kjaergaard, K. V. Mikkelsen and M. Brøndsted Nielsen, Chem. – Eur. J., 2015, 21, 3968–3977 CrossRef CAS PubMed.
- A. G. Lvov and A. Bredihhin, Org. Biomol. Chem., 2021, 19, 4460–4468 RSC.
- M. Å. Petersen, A. S. Andersson, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2009, 1855–1858 CrossRef CAS.
- B. Cheng, J. Morstein, L. K. Ladefoged, J. B. Maesen, B. Schiøtt, S. Sinning and D. Trauner, ACS Chem. Neurosci., 2020, 11, 1231–1237 CrossRef CAS PubMed.
- R. M. Evans and D. J. Mangelsdorf, Cell, 2014, 157, 255–266 CrossRef CAS PubMed.
- W. A. Velema, M. van der Toorn, W. Szymanski and B. L. Feringa, J. Med. Chem., 2013, 56, 4456–4464 CrossRef CAS PubMed.
- M. A. Brown and S. C. De Vito, Crit. Rev. Environ. Sci. Technol., 1993, 23, 249–324 CrossRef CAS.
- D. Mulatihan, T. Guo and Y. Zhao, Photochem. Photobiol., 2020, 96, 1163–1168 CrossRef CAS PubMed.
- C. Renner and L. Moroder, ChemBioChem, 2006, 7, 868–878 CrossRef CAS PubMed.
- A. A. Beharry, L. Wong, V. Tropepe and G. A. Woolley, Angew. Chem., Int. Ed., 2011, 50, 1325–1327 CrossRef CAS PubMed.
- S. Samanta, A. Babalhavaeji, M. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127–14130 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- D. Kand, L. Pizarro, I. Angel, A. Avni, D. Friedmann-Morvinski and R. Weinstain, Angew. Chem., Int. Ed., 2019, 58, 4659–4663 CrossRef CAS PubMed.
- P. Mishra, B. Nayak and R. K. Dey, Asian J. Pharm. Sci., 2016, 11, 337–348 CrossRef.
- N. J. Yang and M. J. Hinner, Site-Specific Protein Labeling, Humana Press, New York, 2015, pp. 29–53 Search PubMed.
- A. Keppler, C. Arrivoli, L. Sironi and J. Ellenberg, Biotechniques, 2006, 41, 167–175 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- X. Xue, A. Lindstrom, H. Qu and Y. Li, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1607 Search PubMed.
- N. Berdigaliyev and M. Aljofan, Future Med. Chem., 2020, 12, 939–947 CrossRef CAS PubMed.
- Z. Chen, S.-H. Yoo and J. S. Takahashi, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 231–252 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- D. Bojadzic and P. Buchwald, Curr. Top. Med. Chem., 2018, 18, 674–699 CrossRef CAS PubMed.
- A. D. Bondarev, M. M. Attwood, J. Jonsson, V. N. Chubarev, V. V. Tarasov and H. B. Schiöth, Expert Opin. Drug Discovery, 2020, 15, 1291–1307 CrossRef CAS PubMed.
- A.-M. Yu, Y. H. Choi and M.-J. Tu, Pharmacol. Rev., 2020, 72, 862–898 CrossRef CAS PubMed.
- T. P. Kogan, B. Dupre, K. M. Keller, I. L. Scott, H. Bui, R. V. Market, P. J. Beck, J. A. Voytus, B. M. Revelle and D. Scott, J. Med. Chem., 1995, 38, 4976–4984 CrossRef CAS PubMed.
- A. Bernardi and P. Cheshev, Chem. – Eur. J., 2008, 14, 7434–7441 CrossRef CAS PubMed.
- N. Jain, K. Tamura, G. Déjean, F. Van Petegem and H. Brumer, ACS Chem. Biol., 2021 DOI:10.1021/acschembio.1c00063.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. del, P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- C. Rousselle, P. Clair, J. Temsamani and J.-M. Scherrmann, J. Drug Target, 2002, 10, 309–315 CrossRef CAS PubMed.
- F. Y. Aoki and K. E. Doucette, Expert Opin. Pharmacother., 2001, 2, 1671–1683 CrossRef PubMed.
- R. Zhang, X. Qin, F. Kong, P. Chen and G. Pan, Drug Delivery, 2019, 26, 328–342 CrossRef CAS PubMed.
- Y. Takeda, K. Samejima, K. Nagano, M. Watanabe, H. Sugeta and Y. Kyogoku, Eur. J. Biochem., 1983, 130, 383–389 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, 3rd edn, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006 Search PubMed.
- A. Mourot, T. Fehrentz, Y. Le Feuvre, C. M. Smith, C. Herold, D. Dalkara, F. Nagy, D. Trauner and R. H. Kramer, Nat. Methods, 2012, 9, 396–402 CrossRef CAS PubMed.
- M. Schoenberger, A. Damijonaitis, Z. Zhang, D. Nagel and D. Trauner, ACS Chem. Neurosci., 2014, 5, 514–518 CrossRef CAS PubMed.
- D. E. Clapham, Nature, 2003, 426, 517–524 CrossRef CAS PubMed.
- T. Fehrentz, F. M. E. Huber, N. Hartrampf, T. Bruegmann, J. A. Frank, N. H. F. Fine, D. Malan, J. G. Danzl, D. B. Tikhonov, M. Sumser, P. Sasse, D. J. Hodson, B. S. Zhorov, N. Klöcker and D. Trauner, Nat. Chem. Biol., 2018, 14, 764–767 CrossRef CAS PubMed.
- P. Leippe, N. Winter, M. P. Sumser and D. Trauner, ACS Chem. Neurosci., 2018, 9, 2886–2891 CrossRef CAS PubMed.
- J. Broichhagen, I. Jurastow, K. Iwan, W. Kummer and D. Trauner, Angew. Chem., Int. Ed., 2014, 53, 7657–7660 CrossRef CAS PubMed.
- K. Hüll, T. Benster, M. B. Manookin, D. Trauner, R. N. Van Gelder and L. Laprell, Sci. Rep., 2019, 9, 1–12 Search PubMed.
- M. R. Banghart, A. Mourot, D. L. Fortin, J. Z. Yao, R. H. Kramer and D. Trauner, Angew. Chem., Int. Ed., 2009, 48, 9097–9101 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, ChemBioChem, 2008, 9, 1225–1228 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, Y.-L. Ying, H. Tian and Y.-T. Long, Chem. Sci., 2014, 5, 2642 RSC.
- M. Hammarson, J. Andersson, S. Li, P. Lincoln and J. Andréasson, Chem. Commun., 2010, 46, 7130–7132 RSC.
- A. Estévez-Torres and D. Baigl, Soft Matter, 2011, 7, 6746–6756 RSC.
- A. Venancio-Marques, A. Bergen, C. Rossi-Gendron, S. Rudiuk and D. Baigl, ACS Nano, 2014, 8, 3654–3663 CrossRef CAS PubMed.
- A. Bergen, S. Rudiuk, M. Morel, T. Le Saux, H. Ihmels and D. Baigl, Nano Lett., 2016, 16, 773–780 CrossRef CAS PubMed.
- The Tumour Microenvironment: Causes and Consequences of Hypoxia and Acidity, ed. J. A. Goode and D. J. Chadwick, John Wiley & Sons, Ltd, Chichester, UK, 2001 Search PubMed.
- P. Montcourrier, P. H. Mangeat, C. Valembois, G. Salazar, A. Sahuquet, C. Duperray and H. Rochefort, J. Cell Sci., 1994, 107, 2381–2391 CrossRef PubMed.
- T. L. I. Ihmels, Materials Science of DNA Chemistry, 1st edn, Jin JI. CRC Press, Boca Raton, 2011 Search PubMed.
- S. Özden, D. Atabey, S. Yıldız and H. Göker, Bioorg. Med. Chem., 2005, 13, 1587–1597 CrossRef PubMed.
- W. D. Wilson, F. A. Tanious, D. Ding, A. Kumar, D. W. Boykin, P. Colson, C. Houssier and C. Bailly, J. Am. Chem. Soc., 1998, 120, 10310–10321 CrossRef CAS.
- K. C. Gross and P. G. Seybold, Int. J. Quantum Chem., 2001, 85, 569–579 CrossRef CAS.
- B. Heinrich, K. Bouazoune, M. Wojcik, U. Bakowsky and O. Vázquez, Org. Biomol. Chem., 2019, 17, 1827–1833 RSC.
- C. Dohno, S. N. Uno and K. Nakatani, J. Am. Chem. Soc., 2007, 129, 11898–11899 CrossRef CAS PubMed.
- A. Mammana, G. T. Carroll, J. Areephong and B. L. Feringa, J. Phys. Chem. B, 2011, 115, 11581–11587 CrossRef CAS PubMed.
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Angew. Chem., Int. Ed., 2010, 122, 5433–5437 CrossRef.
- X. Xing, X. Wang, L. Xu, Y. Tai, L. Dai, X. Zheng, W. Mao, X. Xu and X. Zhou, Org. Biomol. Chem., 2011, 9, 6639–6645 RSC.
- M. P. O’Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Chem. – Eur. J., 2020, 26, 6224–6233 CrossRef PubMed.
- I. Czerwinska and B. Juskowiak, Int. J. Biol. Macromol., 2012, 51, 576–582 CrossRef CAS PubMed.
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966–15967 CrossRef CAS PubMed.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed.
- C. A. Zoto, M. G. Ucak-Astarlioglu and R. E. Connors, J. Mol. Struct., 2016, 1105, 396–402 CrossRef CAS.
- J. R. Franco, G. Cecchi, G. Priotto, M. Paone, A. Diarra, L. Grout, R. C. Mattioli and D. Argaw, PLoS Negl. Trop. Dis, 2017, 11, e0005585 CrossRef PubMed.
- M. P. Barrett, R. J. Burchmore, A. Stich, J. O. Lazzari, A. C. Frasch, J. J. Cazzulo and S. Krishna, Lancet, 2003, 362, 1469–1480 CrossRef.
- T. Kubař, M. Hanus, F. Ryjáček and P. Hobza, Chem. – Eur. J., 2006, 12, 280–290 CrossRef PubMed.
- A. Basu and G. Suresh Kumar, J. Agric. Food Chem., 2014, 62, 317–326 CrossRef CAS PubMed.
- W. H. Ojala, C. R. Ojala and W. B. Gleason, Antivir. Chem. Chemother., 1995, 6, 25–33 CrossRef CAS.
- J. Morstein, R. Z. Hill, A. J. E. Novak, S. Feng, D. D. Norman, P. C. Donthamsetti, J. A. Frank, T. Harayama, B. M. Williams, A. L. Parrill, G. J. Tigyi, H. Riezman, E. Y. Isacoff, D. M. Bautista and D. Trauner, Nat. Chem. Biol., 2019, 15, 623–631 CrossRef CAS PubMed.
- T. Kamei, T. Fukaminato and N. Tamaoki, Chem. Commun., 2012, 48, 7625–7627 RSC.
- D. M. Barber, S. A. Liu, K. Gottschling, M. Sumser, M. Hollmann and D. Trauner, Chem. Sci., 2016, 8, 611–615 RSC.
- L. Turski, A. Huth, M. Sheardown, F. McDonald, R. Neuhaus, H. H. Schneider, U. Dirnagl, F. Wiegand, P. Jacobsen and E. Ottow, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10960–10965 CrossRef CAS PubMed.
- L. Turski, H. H. Schneider, R. Neuhaus, F. McDonald, G. H. Jones, B. Löfberg, H. Schweinfurth, A. Huth, M. Krüger and E. Ottow, Restor. Neurol. Neurosci., 2000, 17, 45–59 CAS.
- B. Reisinger, N. Kuzmanovic, P. Löffler, R. Merkl, B. König and R. Sterner, Angew. Chem., Int. Ed., 2014, 53, 595–598 CrossRef CAS PubMed.
- A. C. Kneuttinger, M. Winter, N. A. Simeth, K. Heyn, R. Merkl, B. König and R. Sterner, ChemBioChem, 2018, 19, 1750–1757 CrossRef CAS PubMed.
- N. A. Simeth, T. Kinateder, C. Rajendran, J. Nazet, R. Merkl, R. Sterner, B. König and A. C. Kneuttinger, Chem. – Eur. J., 2021, 27, 2439–2451 CrossRef CAS PubMed.
- L. H. Jensen, A. Renodon-Corniere, I. Wessel, S. W. Langer, B. Søkilde, E. V. Carstensen, M. Sehested and P. B. Jensen, Mol. Pharmacol., 2002, 61, 1235–1243 CrossRef CAS PubMed.
- R. Göstl and S. Hecht, Chem. – Eur. J., 2015, 21, 4422–4427 CrossRef PubMed.
- D. Sola, L. Rossi, G. P. C. Schianca, P. Maffioli, M. Bigliocca, R. Mella, F. Corlianò, G. P. Fra, E. Bartoli and G. Derosa, Arch. Med. Sci., 2015, 4, 840–848 CrossRef PubMed.
- N. Seedher and M. Kanojia, Pharm. Dev. Technol., 2009, 14, 185–192 CrossRef CAS PubMed.
- Z. B. Mehta, N. R. Johnston, M. S. Nguyen-Tu, J. Broichhagen, P. Schultz, D. P. Larner, I. Leclerc, D. Trauner, G. A. Rutter and D. J. Hodson, Sci. Rep., 2017, 7, 1–11 CrossRef CAS PubMed.
- H. Mori, Y. Mori, S. Sugie, N. Yoshimi, M. Takahashi, H. Ni-i, H. Yamazaki, K. Toyoshi and G. M. Williams, Cancer Res., 1986, 46, 1654–1658 CAS.
- D. Pal and S. Saha, J. Adv. Pharm. Technol. Res, 2012, 3, 92 CrossRef CAS PubMed.
- W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Chem. – Eur. J., 2015, 21, 16517–16524 CrossRef CAS PubMed.
- C. E. Weston, A. Krämer, F. Colin, Ö. Yildiz, M. G. J. Baud, F. J. Meyer-Almes and M. J. Fuchter, ACS Infect. Dis., 2017, 3, 152–161 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, F. Chen, W. Zhang and H. Zhao, Phys. Chem. Chem. Phys., 2015, 17, 12215–12221 RSC.
- Z. Zhuo, Y. Yu, M. Wang, J. Li, Z. Zhang, J. Liu, X. Wu, A. Lu, G. Zhang and B. Zhang, Int. J. Mol. Sci., 2017, 18, 2142 CrossRef PubMed.
- D. Steele, A. Kertsburg and G. A. Soukup, Am. J. PharmacoGenom., 2003, 3, 131–144 CrossRef CAS PubMed.
- R. Mogaki, K. Okuro and T. Aida, Chem. Sci., 2015, 6, 2802–2805 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- K. Okuro, K. Kinbara, K. Tsumoto, N. Ishii and T. Aida, J. Am. Chem. Soc., 2009, 131, 1626–1627 CrossRef CAS PubMed.
- R. Mogaki, K. Okuro and T. Aida, J. Am. Chem. Soc., 2017, 139, 10072–10078 CrossRef CAS PubMed.
- P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- H.-J. Böhm, A. Flohr and M. Stahl, Drug Discovery Today Technol., 2004, 1, 217–224 CrossRef PubMed.
- Y. Hu, D. Stumpfe and J. Bajorath, J. Med. Chem., 2017, 60, 1238–1246 CrossRef CAS PubMed.
- N. N. Mafy, K. Matsuo, S. Hiruma, R. Uehara and N. Tamaoki, J. Am. Chem. Soc., 2020, 142, 1763–1767 CrossRef CAS PubMed.
- R. Ferreira, J. R. Nilsson, C. Solano, J. Andréasson and M. Grøtli, Sci. Rep., 2015, 5, 9769 CrossRef CAS PubMed.
- S. Pittolo, X. Gómez-Santacana, K. Eckelt, X. Rovira, J. Dalton, C. Goudet, J.-P. P. Pin, A. Llobet, J. Giraldo, A. Llebaria and P. Gorostiza, Nat. Chem. Biol., 2014, 10, 813–815 CrossRef CAS PubMed.
- C. W. Grathwol, N. Wössner, S. Behnisch-Cornwell, L. Schulig, L. Zhang, O. Einsle, M. Jung and A. Link, ChemMedChem, 2020, 15, 1480–1489 CrossRef CAS PubMed.
- A. A. Blevins and G. J. Blanchard, J. Phys. Chem. B, 2004, 108, 4962–4968 CrossRef CAS.
- W. R. Brode, J. H. Gould and G. M. Wyman, J. Am. Chem. Soc., 1952, 74, 4641–4646 CrossRef CAS.
- D. Lachmann, C. Studte, B. Männel, H. Hübner, P. Gmeiner and B. König, Chem. – Eur. J., 2017, 23, 13423–13434 CrossRef CAS PubMed.
- T. Yamaguchi and M. Irie, Chem. Lett., 2004, 1398–1399 CrossRef CAS.
- O. Pieroni, A. Fissi, J. L. Houben and F. Ciardelli, J. Am. Chem. Soc., 1985, 107, 2990–2991 CrossRef CAS.
- H. Yamamoto, A. Nishida, T. Takimoto and A. Nagai, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 67–74 CrossRef CAS.
- A. Nojiri, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 2137–2141 CrossRef CAS PubMed.
- R. Zhang, Y.-J. Ji, L. Yang, Y. Zhang and G.-C. Kuang, Phys. Chem. Chem. Phys., 2016, 18, 9914–9917 RSC.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- E. Merlo, R. Freudenthal and A. Romano, Neuroscience, 2002, 112, 161–172 CrossRef CAS PubMed.
- M. Ishikawa, T. Ohzono, T. Yamaguchi and Y. Norikane, Sci. Rep., 2017, 7, 3–8 CrossRef PubMed.
- M. W. H. Hoorens, H. Fu, R. H. Duurkens, G. Trinco, V. Arkhipova, B. L. Feringa, G. J. Poelarends, D. J. Slotboom and W. Szymanski, Adv. Ther, 2018, 1, 1800028 CrossRef.
- P. Leippe and J. A. Frank, Curr. Opin. Struct. Biol., 2019, 57, 23–30 CrossRef CAS PubMed.
- L. Laprell, E. Repak, V. Franckevicius, F. Hartrampf, J. Terhag, M. Hollmann, M. Sumser, N. Rebola, D. A. DiGregorio and D. Trauner, Nat. Commun., 2015, 6, 8076 CrossRef CAS PubMed.
- O. Srinivas, N. Mitra, A. Surolia and N. Jayaraman, Glycobiology, 2005, 15, 861–873 CrossRef CAS PubMed.
- O. Srinivas, N. Mitra, A. Surolia and N. Jayaraman, J. Am. Chem. Soc., 2002, 124, 2124–2125 CrossRef CAS PubMed.
- V. Chandrasekaran, K. Kolbe, F. Beiroth and T. K. Lindhorst, Beilstein J. Org. Chem., 2013, 9, 223–233 CrossRef CAS PubMed.
- K. Rustler, M. J. Mickert, J. Nazet, R. Merkl, H. H. Gorris and B. König, Org. Biomol. Chem., 2018, 16, 7430–7437 RSC.
- N. Hartrampf, T. Seki, A. Baumann, P. Watson, N. A. Vepřek, B. E. Hetzler, A. Hoffmann-Röder, M. Tsuji and D. Trauner, Chem. – Eur. J., 2020, 26, 4476–4479 CrossRef CAS PubMed.
- G. P. Espino-Solis, Rev. Vitae, 2015, 22, 9–11 Search PubMed.
- W. A. Velema, M. J. Hansen, M. M. Lerch, A. J. M. Driessen, W. Szymanski and B. L. Feringa, Bioconjugate Chem., 2015, 26, 2592–2597 CrossRef CAS PubMed.
- W. A. Velema, J. P. Van Der Berg, M. J. Hansen, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924–928 CrossRef CAS PubMed.
- S. S. Tous, A. M. El Sayed, M. G. Abd El Mohsen, M. N. Agban and M. F. Boushra, J. Drug Deliv. Sci. Technol., 2012, 22, 341–346 CrossRef CAS.
- D. Martínez-López, M.-L. Yu, C. García-Iriepa, P. J. Campos, L. M. Frutos, J. A. Golen, S. Rasapalli and D. Sampedro, J. Org. Chem., 2015, 80, 3929–3939 CrossRef PubMed.
- A. T. Ulijasz and R. D. Vierstra, Curr. Opin. Plant Biol., 2011, 14, 498–506 CrossRef CAS PubMed.
- E. Contreras-García, D. Martínez-López, C. A. Alonso, C. Lozano, C. Torres, M. A. Rodríguez, P. J. Campos and D. Sampedro, Eur. J. Org. Chem., 2017, 4719–4725 CrossRef.
- E. A. Specht, E. Braselmann and A. E. Palmer, Annu. Rev. Physiol., 2017, 79, 93–117 CrossRef CAS PubMed.
- P. Debie and S. Hernot, Front. Pharmacol., 2019, 10, 510 CrossRef CAS PubMed.
- Q. Hu, Y. Shi, X. Yang, J. Zhang, B. Li, X. Liao and X. Liu, Sheng Wu Gong Cheng Xue Bao, 2020, 36, 1051–1059 Search PubMed.
- B. Roubinet, M. Weber, H. Shojaei, M. Bates, M. L. Bossi, V. N. Belov, M. Irie and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 6611–6620 CrossRef CAS PubMed.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef PubMed.
- Y. Fu, H. H. Han, J. Zhang, X. P. He, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2018, 140, 8671–8674 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- X. Chai, H.-H. Han, A. C. Sedgwick, N. Li, Y. Zang, T. D. James, J. Zhang, X.-L. Hu, Y. Yu, Y. Li, Y. Wang, J. Li, X.-P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 18005–18013 CrossRef CAS PubMed.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262–12277 RSC.
- M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed.
- C. Z. Chung, K. Amikura and D. Söll, Front. Bioeng. Biotechnol., 2020, 8, 598577 CrossRef PubMed.
- P. Arranz-Gibert, K. Vanderschuren and F. J. Isaacs, Curr. Opin. Chem. Biol., 2018, 46, 203–211 CrossRef CAS PubMed.
- J. W. Chin, Annu. Rev. Biochem., 2014, 83, 379–408 CrossRef CAS PubMed.
- A. R. Nödling, L. A. Spear, T. L. Williams, L. Y. P. Luk and Y.-H. Tsai, Essays Biochem., 2019, 63, 237–266 CrossRef PubMed.
- A. A. John, C. P. Ramil, Y. Tian, G. Cheng and Q. Lin, Org. Lett., 2015, 17, 6258–6261 CrossRef CAS PubMed.
- J. Luo, S. Samanta, M. Convertino, N. V. Dokholyan and A. Deiters, ChemBioChem, 2018, 19, 2178–2185 CrossRef CAS PubMed.
- M. Bose, D. Groff, J. Xie, E. Brustad and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 388–389 CrossRef CAS PubMed.
- A. C. Kneuttinger, K. Straub, P. Bittner, N. A. Simeth, A. Bruckmann, F. Busch, C. Rajendran, E. Hupfeld, V. H. Wysocki, D. Horinek, B. König, R. Merkl and R. Sterner, Cell Chem. Biol., 2019, 26, 1501–1514 CrossRef CAS PubMed.
- A. C. Kneuttinger, C. Rajendran, N. A. Simeth, A. Bruckmann, B. König and R. Sterner, Biochemistry, 2020, 59, 2729–2742 CrossRef CAS PubMed.
- J. Lee, K. E. Schwieter, A. M. Watkins, D. S. Kim, H. Yu, K. J. Schwarz, J. Lim, J. Coronado, M. Byrom, E. V. Anslyn, A. D. Ellington, J. S. Moore and M. C. Jewett, Nat. Commun., 2019, 10, 5097 CrossRef PubMed.
- T. Yanagisawa, M. Kuratani, E. Seki, N. Hino, K. Sakamoto and S. Yokoyama, Cell Chem. Biol., 2019, 26, 936–949 CrossRef CAS PubMed.
- C. Hoppmann, V. K. Lacey, G. V. Louie, J. Wei, J. P. Noel and L. Wang, Angew. Chem., Int. Ed., 2014, 53, 3932–3936 CrossRef CAS PubMed.
- C. Hoppmann, I. Maslennikov, S. Choe and L. Wang, J. Am. Chem. Soc., 2015, 137, 11218–11221 CrossRef CAS PubMed.
- D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 CrossRef CAS PubMed.
- S. Chandrudu, P. Simerska and I. Toth, Molecules, 2013, 18, 4373–4388 CrossRef CAS PubMed.
- A. G. Molina and Y. S. Sanghvi, Curr. Protoc. Nucleic Acid Chem., 2019, 77, e82 Search PubMed.
- R. Eritja, Int. J. Pept. Res. Ther., 2007, 13, 53–68 CrossRef CAS.
- R. F. Standaert and S. B. Park, J. Org. Chem., 2006, 71, 7952–7966 CrossRef CAS PubMed.
- L. D. DeLeve and N. Kaplowitz, Pharmacol. Ther., 1991, 52, 287–305 CrossRef CAS PubMed.
- A. Prestel and H. M. Möller, Chem. Commun., 2016, 52, 701–704 RSC.
- L. Albert, A. Peñalver, N. Djokovic, L. Werel, M. Hoffarth, D. Ruzic, J. Xu, L. O. Essen, K. Nikolic, Y. Dou and O. Vázquez, ChemBioChem, 2019, 20, 1417–1429 CrossRef CAS PubMed.
- F. Milletti, Drug Discovery Today, 2012, 17, 850–860 CrossRef CAS PubMed.
- H. de Jong, K. M. Bonger and D. W. P. M. Löwik, RSC Chem. Biol., 2020, 1, 192–203 RSC.
- I. V. Komarov and I. V. Bakanovych, Chem. Heterocycl. Compd., 2020, 56, 719–721 CrossRef CAS.
- L. Ulysse, J. Cubillos and J. Chmielewski, J. Am. Chem. Soc., 1995, 117, 8466–8467 CrossRef CAS.
- R. Behrendt, M. Schenk, H.-J. J. Musiol and L. Moroder, J. Pept. Sci., 1999, 5, 519–529 CrossRef CAS PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed.
- S. Dissanayake, W. A. Denny, S. Gamage and V. Sarojini, J. Controlled Release, 2017, 250, 62–76 CrossRef CAS PubMed.
- D. Birch, M. V. Christensen, D. Staerk, H. Franzyk and H. M. Nielsen, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 2483–2494 CrossRef CAS PubMed.
- S. F. Hedegaard, M. S. Derbas, T. K. Lind, M. R. Kasimova, M. V. Christensen, M. H. Michaelsen, R. A. Campbell, L. Jorgensen, H. Franzyk, M. Cárdenas and H. M. Nielsen, Sci. Rep., 2018, 8, 6327 CrossRef PubMed.
- T. Lougheed, V. Borisenko, T. Hennig, K. Rück-Braun and G. A. Woolley, Org. Biomol. Chem., 2004, 2, 2798–2801 RSC.
- T. Schober, I. Wehl, S. Afonin, O. Babii, A. Iampolska, U. Schepers, I. V. Komarov and A. S. Ulrich, ChemPhotoChem, 2019, 3, 384–391 CrossRef CAS.
- O. Babii, S. Afonin, T. Schober, L. V. Garmanchuk, L. I. Ostapchenko, V. Yurchenko, S. Zozulya, O. Tarasov, I. Pishel, A. S. Ulrich and I. V. Komarov, Future Drug Discovery, 2020, 2, FDD28 CrossRef.
- K. Okamoto, Y. Tomita, H. Yonezawa, T. Hirohata, R. Ogura and N. Izumiya, Oncology, 1984, 41, 43–48 CrossRef CAS PubMed.
- O. Babii, S. Afonin, A. Y. Ishchenko, T. Schober, A. O. Negelia, G. M. Tolstanova, L. V. Garmanchuk, L. I. Ostapchenko, I. V. Komarov and A. S. Ulrich, J. Med. Chem., 2018, 61, 10793–10813 CrossRef CAS PubMed.
- O. Babii, S. Afonin, M. Berditsch, S. Reier, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392–3395 CrossRef CAS PubMed.
- S. Afonin, O. Babii, A. Reuter, V. Middel, M. Takamiya, U. Strähle, I. V. Komarov and A. S. Ulrich, Beilstein J. Org. Chem., 2020, 16, 39–49 CrossRef CAS PubMed.
- O. Babii, S. Afonin, L. V. Garmanchuk, V. V. Nikulina, T. V. Nikolaienko, O. V. Storozhuk, D. V. Shelest, O. I. Dasyukevich, L. I. Ostapchenko, V. Iurchenko, S. Zozulya, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2016, 128, 5583–5586 CrossRef.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC.
- H. Nishioka, X. Liang, T. Kato and H. Asanuma, Angew. Chem., Int. Ed., 2012, 51, 1165–1168 CrossRef CAS PubMed.
- T. Goldau, K. Murayama, C. Brieke, H. Asanuma and A. Heckel, Chem. – Eur. J., 2015, 21, 17870–17876 CrossRef CAS PubMed.
- T. Goldau, K. Murayama, C. Brieke, S. Steinwand, P. Mondal, M. Biswas, I. Burghardt, J. Wachtveitl, H. Asanuma and A. Heckel, Chem. – Eur. J., 2015, 21, 2845–2854 CrossRef CAS PubMed.
- M. L. Hammill, G. Islam and J. P. Desaulniers, ChemBioChem, 2020, 21, 2367–2372 CrossRef CAS PubMed.
- M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef CAS PubMed.
- H. Cahová and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 3186–3190 CrossRef PubMed.
- H. Asanuma, K. Shirasuka, T. Yoshida, T. Takarada, X. Liang and M. Komiyama, Chem. Lett., 2001, 108–109 CrossRef CAS.
- C. Beyer and H.-A. Wagenknecht, J. Org. Chem., 2010, 75, 2752–2755 CrossRef CAS PubMed.
- S. Barrois and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2012, 8, 905–914 CrossRef CAS PubMed.
- C. Beyer and H.-A. Wagenknecht, Synlett, 2010, 1371–1376 CAS.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- Z. L. Pianowski, Chemistry, 2019, 25, 5128–5144 CrossRef CAS PubMed.
- T. Arakawa, Y. Kita and S. N. Timasheff, Biophys. Chem., 2007, 131, 62–70 CrossRef CAS PubMed.
- J. Roche and C. A. Royer, J. R. Soc., Interface, 2018, 15, 20180244 CrossRef PubMed.
- P. L. Privalov, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 281–306 CrossRef CAS PubMed.
- R. M. Daniel, M. Dines and H. H. Petach, Biochem. J., 1996, 317, 1–11 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1991, 1078, 231–235 CrossRef CAS.
- K. Griebenow and A. M. Klibanov, J. Am. Chem. Soc., 1996, 118, 11695–11700 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, 3rd edn, Academic Press, 2013 Search PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- S. T. Larda, D. Pichugin and R. S. Prosser, Bioconjugate Chem., 2015, 26, 2376–2383 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- D. G. Rawale, K. Thakur, S. R. Adusumalli and V. Rai, Eur. J. Org. Chem., 2019, 6749–6763 CrossRef CAS.
- W. Szymański, B. Wu, C. Poloni, D. B. Janssen and B. L. Feringa, Angew. Chem., Int. Ed., 2013, 52, 2068–2072 CrossRef PubMed.
- C. Poloni, W. Szymański, L. Hou, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 946–951 CrossRef CAS PubMed.
- Y. Zhang, K.-Y. Park, K. F. Suazo and M. D. Distefano, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- M. Rashidian, J. K. Dozier and M. D. Distefano, Bioconjugate Chem., 2013, 24, 1277–1294 CrossRef CAS PubMed.
- J. A. Shadish and C. A. DeForest, Matter, 2020, 2, 50–77 CrossRef.
- M. Dal Peraro and F. G. van der Goot, Nat. Rev. Microbiol., 2016, 14, 77–92 CrossRef PubMed.
- N. L. Mutter, J. Volarić, W. Szymanski, B. L. Feringa and G. Maglia, J. Am. Chem. Soc., 2019, 141, 14356–14363 CrossRef CAS PubMed.
- S. Loudwig and H. Bayley, J. Am. Chem. Soc., 2006, 128, 12404–12405 CrossRef CAS PubMed.
- G. Lukinavičius, G. Y. Mitronova, S. Schnorrenberg, A. N. Butkevich, H. Barthel, V. N. Belov and S. W. Hell, Chem. Sci., 2018, 9, 3324–3334 RSC.
- L. Möckl, A. Müller, C. Bräuchle and T. K. Lindhorst, Chem. Commun., 2016, 52, 1254–1257 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- T. Matsuzaki, Y. Tajika, N. Tserentsoodol, T. Suzuki, T. Aoki, H. Hagiwara and K. Takata, Anat. Sci. Int., 2002, 77, 85–93 CrossRef PubMed.
- A. Kocer, A. Koçer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef CAS PubMed.
- E. S. Haswell, R. Phillips and D. C. Rees, Structure, 2011, 19, 1356–1369 CrossRef CAS PubMed.
- A. Anishkin, C.-S. Chiang and S. Sukharev, J. Gen. Physiol., 2005, 125, 155–170 CrossRef CAS PubMed.
- A. Koçer, M. Walko and B. L. Feringa, Nat. Protoc., 2007, 2, 1426–1437 CrossRef PubMed.
- W. Szymanski, D. Yilmaz, A. Koçer and B. L. Feringa, Acc. Chem. Res., 2013, 46, 2910–2923 CrossRef CAS PubMed.
- H. A. Lester, M. E. Krouse, M. M. Nass, N. H. Wassermann and B. F. Erlanger, J. Gen. Physiol., 1980, 75, 207–232 CrossRef CAS PubMed.
- P. C. Donthamsetti, N. Winter, M. Schönberger, J. Levitz, C. Stanley, J. A. Javitch, E. Y. Isacoff and D. Trauner, J. Am. Chem. Soc., 2017, 139, 18522–18535 CrossRef CAS PubMed.
- L. Yue, M. Pawlowski, S. S. Dellal, A. Xie, F. Feng, T. S. Otis, K. S. Bruzik, H. Qian and D. R. Pepperberg, Nat. Commun., 2012, 3, 1095 CrossRef PubMed.
- M. Volgraf, P. Gorostiza, R. Numano, R. H. Kramer, E. Y. Isacoff and D. Trauner, Nat. Chem. Biol., 2006, 2, 47–52 CrossRef CAS PubMed.
- C. Wyart, F. Del Bene, E. Warp, E. K. Scott, D. Trauner, H. Baier and E. Y. Isacoff, Nature, 2009, 461, 407–410 CrossRef CAS PubMed.
- G. Kauwe and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9142–9147 CrossRef CAS PubMed.
- J. Levitz, C. Pantoja, B. Gaub, H. Janovjak, A. Reiner, A. Hoagland, D. Schoppik, B. Kane, P. Stawski, A. F. Schier, D. Trauner and E. Y. Isacoff, Nat. Neurosci., 2013, 16, 507–516 CrossRef CAS PubMed.
- G. Anderson, A. Noorian, G. Taylor, M. Anitha, D. Bernhard, S. Srinivasan and J. Greene, Exp. Neurol., 2007, 207, 4–12 CrossRef CAS PubMed.
- P. Gorostiza, M. Volgraf, R. Numano, S. Szobota, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10865–10870 CrossRef CAS PubMed.
- I. Tochitsky, M. R. Banghart, A. Mourot, J. Z. Yao, B. Gaub, R. H. Kramer and D. Trauner, Nat. Chem., 2012, 4, 105–111 CrossRef CAS PubMed.
- A. Taly, P.-J. Corringer, D. Guedin, P. Lestage and J.-P. Changeux, Nat. Rev. Drug Discovery, 2009, 8, 733–750 CrossRef CAS PubMed.
- C. Gotti, B. Balestra, M. Moretti, G. E. Rovati, L. Maggi, G. Rossoni, F. Berti, L. Villa, M. Pallavicini and F. Clementi, Br. J. Pharmacol., 1998, 124, 1197–1206 CrossRef CAS PubMed.
- M. R. Picciotto, M. J. Higley and Y. S. Mineur, Neuron, 2012, 76, 116–129 CrossRef CAS PubMed.
- R. D. De Cuttoli, S. Mondoloni, F. Marti, D. Lemoine, C. Nguyen, J. Naudé, T. D’izarny-Gargas, S. Pons, U. Maskos, D. Trauner, R. H. Kramer, P. Faure and A. Mourot, Elife, 2018, 7, 1–23 Search PubMed.
- M. Wegener, M. J. Hansen, A. J. M. Driessen, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 17979–17986 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- R. Prakash, O. Yizhar, B. Grewe, C. Ramakrishnan, N. Wang, I. Goshen, A. M. Packer, D. S. Peterka, R. Yuste, M. J. Schnitzer and K. Deisseroth, Nat. Methods, 2012, 9, 1171–1179 CrossRef CAS PubMed.
- G. Bort, T. Gallavardin, D. Ogden and P. I. Dalko, Angew. Chem., Int. Ed., 2013, 52, 4526–4537 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascón-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, È. Ferrer, R. Alibés, F. Busqué, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS PubMed.
- M. Gascón-Moya, A. Pejoan, M. Izquierdo-Serra, S. Pittolo, G. Cabré, J. Hernando, R. Alibés, P. Gorostiza and F. Busqué, J. Org. Chem., 2015, 80, 9915–9925 CrossRef PubMed.
- G. Cabré, A. Garrido-Charles, M. Moreno, M. Bosch, M. Porta-de-la-Riva, M. Krieg, M. Gascón-Moya, N. Camarero, R. Gelabert, J. M. Lluch, F. Busqué, J. Hernando, P. Gorostiza and R. Alibés, Nat. Commun., 2019, 10, 907 CrossRef PubMed.
- S. Pujals, N. Feiner-Gracia, P. Delcanale, I. Voets and L. Albertazzi, Nat. Rev. Chem., 2019, 3, 68–84 CrossRef.
- H. Li and J. C. Vaughan, Chem. Rev., 2018, 118, 9412–9454 CrossRef CAS PubMed.
- G. T. Dempsey, M. Bates, W. E. Kowtoniuk, D. R. Liu, R. Y. Tsien and X. Zhuang, J. Am. Chem. Soc., 2009, 131, 18192–18193 CrossRef CAS PubMed.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS PubMed.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS PubMed.
- J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS PubMed.
- T. A. Klar, S. Jakobs, M. Dyba, A. Egner and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8206–8210 CrossRef CAS PubMed.
- S. W. Hell, Phys. Lett. A, 2004, 326, 140–145 CrossRef CAS.
- S. M. Polyakova, V. N. Belov, M. L. Bossi and S. W. Hell, Eur. J. Org. Chem., 2011, 3301–3312 CrossRef CAS.
- R. Sharma, M. Singh and R. Sharma, Spectrochim. Acta, Part A, 2020, 231, 117715 CrossRef CAS PubMed.
- Y. Shoji, A. Yagi, M. Horiuchi, M. Morimoto and M. Irie, Isr. J. Chem., 2013, 53, 303–311 CrossRef CAS.
- M. Takeshita and M. Irie, Chem. Commun., 1997, 2265–2266 RSC.
- M. Takeshita, N. Kato, S. Kawauchi, T. Imase, J. Watanabe and M. Irie, J. Org. Chem., 1998, 63, 9306–9313 CrossRef CAS.
- T. Hirose, K. Matsuda and M. Irie, J. Org. Chem., 2006, 71, 7499–7508 CrossRef CAS PubMed.
- T. Hirose, M. Irie and K. Matsuda, Adv. Mater., 2008, 20, 2137–2141 CrossRef CAS.
- N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206–5208 RSC.
- K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 13558–13564 CrossRef CAS PubMed.
- J. R. McCombs and S. C. Owen, AAPS J., 2015, 17, 339–351 CrossRef CAS PubMed.
- S. W. Hell, S. Jakobs and L. Kastrup, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 859–860 CrossRef CAS.
- T. Fukaminato, T. Sasaki, T. Kawai, N. Tamai and M. Irie, J. Am. Chem. Soc., 2004, 126, 14843–14849 CrossRef CAS PubMed.
- M. Zhu and H. Zhou, Org. Biomol. Chem., 2018, 16, 8434–8445 RSC.
- K. Fujimoto, M. Kajino, I. Sakaguchi and M. Inouye, Chem. – Eur. J., 2012, 18, 9834–9840 CrossRef CAS PubMed.
- A. V. Strizhak, O. Babii, S. Afonin, I. Bakanovich, T. Pantelejevs, W. Xu, E. Fowler, R. Eapen, K. Sharma, M. O. Platonov, V. V. Hurmach, L. Itzhaki, M. Hyvönen, A. S. Ulrich, D. R. Spring and I. V. Komarov, Org. Biomol. Chem., 2020, 18, 5359–5369 RSC.
- G. M. Fahy, T. H. Lilley, H. Linsdell, M. S. J. Douglas and H. T. Meryman, Cryobiology, 1990, 27, 247–268 CrossRef CAS PubMed.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS PubMed.
- P. Glock, J. Broichhagen, S. Kretschmer, P. Blumhardt, J. Mücksch, D. Trauner and P. Schwille, Angew. Chem., Int. Ed., 2018, 57, 2362–2366 CrossRef CAS PubMed.
- K. M. Blacklock, B. J. Yachnin, G. A. Woolley and S. D. Khare, J. Am. Chem. Soc., 2018, 140, 14–17 CrossRef CAS PubMed.
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS PubMed.
- U. Kusebauch, S. A. Cadamuro, H.-J. Musiol, M. O. Lenz, J. Wachtveitl, L. Moroder and C. Renner, Angew. Chem., Int. Ed., 2006, 45, 7015–7018 CrossRef CAS PubMed.
- U. Kusebauch, S. A. Cadamuro, H.-J. Musiol, L. Moroder and C. Renner, Chem. – Eur. J., 2007, 13, 2966–2973 CrossRef CAS PubMed.
- G. C. Kim, J. H. Ahn, J. H. Oh, S. Nam, S. Hyun, J. Yu and Y. Lee, Biomacromolecules, 2018, 19, 2863–2869 CrossRef CAS PubMed.
- V. Borisenko and G. A. Woolley, J. Photochem. Photobiol., A, 2005, 173, 21–28 CrossRef CAS.
- A. M. Ali, M. W. Forbes and G. A. Woolley, ChemBioChem, 2015, 16, 1757–1763 CrossRef CAS PubMed.
- N. Preußke, W. Moormann, K. Bamberg, M. Lipfert, R. Herges and F. D. Sönnichsen, Org. Biomol. Chem., 2020, 18, 2650–2660 RSC.
- G. Cabré, A. Garrido-Charles, À. González-Lafont, W. Moormann, D. Langbehn, D. Egea, J. M. Lluch, R. Herges, R. Alibés, F. Busqué, P. Gorostiza and J. Hernando, Org. Lett., 2019, 21, 3780–3784 CrossRef PubMed.
- R. Santucci, E. Laurenti, F. Sinibaldi and R. P. Ferrari, Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol, 2002, 1596, 225–233 CrossRef CAS.
- Z. Zhang, D. C. Burns, J. R. Kumita, O. S. Smart, G. A. Woolley, Z. Zhang, D. C. Burns, J. R. Kumita, O. S. Smart and G. A. Woolley, Bioconjugate Chem., 2003, 14, 824–829 CrossRef CAS PubMed.
- D. C. Burns, F. Zhang and G. A. Woolley, Nat. Protoc., 2007, 2, 251–258 CrossRef CAS PubMed.
- L. Nevola, A. Martín-Quirõs, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt, P. Gorostiza, A. Martín-Quirós, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704–7708 CrossRef CAS PubMed.
- R. J. Mart, R. J. Errington, C. L. Watkins, S. C. Chappell, M. Wiltshire, A. T. Jones, P. J. Smith and R. K. Allemann, Mol. BioSyst., 2013, 9, 2597–2603 RSC.
- P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B. M. Whittaker, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 7644–7647 CrossRef CAS PubMed.
- F. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943–3946 CrossRef CAS PubMed.
- R. S. Ritterson, K. M. Kuchenbecker, M. Michalik and T. Kortemme, J. Am. Chem. Soc., 2013, 135, 12516–12519 CrossRef CAS PubMed.
- S. Samanta and G. A. Woolley, ChemBioChem, 2011, 12, 1712–1723 CrossRef CAS PubMed.
- J. L. Turnbull, B. R. Benlian, R. P. Golden and E. W. Miller, J. Am. Chem. Soc., 2021, 143, 6194–6201 CrossRef CAS PubMed.
- F. Zhang, O. Sadovski and G. A. Woolley, ChemBioChem, 2008, 9, 2147–2154 CrossRef CAS PubMed.
- N. Yasuike, K. M. Blacklock, H. Lu, A. S. I. Jaikaran, S. McDonald, M. Uppalapati, S. D. Khare and G. A. Woolley, ChemPhotoChem, 2019, 3, 431–440 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kálmán and G. A. Woolley, Chem. Commun., 2015, 51, 12981–12984 RSC.
- A. Müller, H. Kobarg, V. Chandrasekaran, J. Gronow, F. D. Sönnichsen and T. K. Lindhorst, Chem. – Eur. J., 2015, 21, 13723–13731 CrossRef PubMed.
- A. Müller and T. K. Lindhorst, Eur. J. Org. Chem., 2016, 1669–1672 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |