
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in chemical probing of protein O-GlcNAc glycosylation: structural role and molecular mechanisms
Abhijit
Saha
 a,
Davide
Bello
a and
Alberto
Fernández-Tejada
*ab
a,
Davide
Bello
a and
Alberto
Fernández-Tejada
*ab
aChemical Immunology Lab, Centre for Cooperative Research in Biosciences, CIC-bioGUNE, Basque Research and Technology Alliance (BRTA), Derio 48160, Biscay, Spain. E-mail: afernandeztejada@cicbiogune.es
bIkerbasque, Basque Foundation for Science, Bilbao 48013, Spain
First published on 2nd August 2021
Abstract
The addition of O-linked-β-D-N-acetylglucosamine (O-GlcNAc) onto serine and threonine residues of nuclear and cytoplasmic proteins is an abundant, unique post-translational modification governing important biological processes. O-GlcNAc dysregulation underlies several metabolic disorders leading to human diseases, including cancer, neurodegeneration and diabetes. This review provides an extensive summary of the recent progress in probing O-GlcNAcylation using mainly chemical methods, with a special focus on discussing mechanistic insights and the structural role of O-GlcNAc at the molecular level. We highlight key aspects of the O-GlcNAc enzymes, including development of OGT and OGA small-molecule inhibitors, and describe a variety of chemoenzymatic and chemical biology approaches for the study of O-GlcNAcylation. Special emphasis is placed on the power of chemistry in the form of synthetic glycopeptide and glycoprotein tools for investigating the site-specific functional consequences of the modification. Finally, we discuss in detail the conformational effects of O-GlcNAc glycosylation on protein structure and stability, relevant O-GlcNAc-mediated protein interactions and its molecular recognition features by biological receptors. Future research in this field will provide novel, more effective chemical strategies and probes for the molecular interrogation of O-GlcNAcylation, elucidating new mechanisms and functional roles of O-GlcNAc with potential therapeutic applications in human health.
 Abhijit Saha | Abhijit Saha received his MSc degree in Chemistry from Gauhati University in 2008 and then joined IIT Guwahati as a project assistant to work on peptide synthesis with Prof. Mandal. In 2015, he obtained his PhD in Chemical Biology from Kyoto University (Japan) working on DNA molecular recognition under the supervision of Prof. Sugiyama. He then pursued post-doctoral studies on G-Quadruplex DNA at Institut Curie (Paris, France) in the laboratory of Prof. Teulade-Fichou. In March 2020, he joined CIC bioGUNE (Biscay, Spain) as a Marie S. Curie Individual Fellow to develop his expertise in chemical immunology under the supervision of Prof. Fernández-Tejada. |
 Davide Bello | Davide Bello obtained his PhD in Chemistry from the University of St Andrews (Scotland, UK) in 2006. He then undertook a number of postdoctoral stays within several European Institutions, including the Max Planck Institute with Prof. Becker (Dortmund, Germany, 2007), IRB Barcelona with Prof. Lavilla (Spain, 2008), and the University of the Basque Country with Prof. Cossío (Spain, 2010). In 2011 he joined Prof. O’Hagan's group in St Andrews and in 2019 he moved to the University of Dundee Drug Discovery Unit (Scotland, UK) to work with Dr. Wyllie and Prof. Gilbert on molecular target identification. In September 2020, he joined the Chemical Immunology Lab at CIC bioGUNE (Biscay, Spain) led by Prof. Fernández-Tejada. |
 Alberto Fernández-Tejada | Alberto Fernández-Tejada is Ikerbasque Research Professor and Group Leader of the Chemical Immunology Lab at CIC bioGUNE (Biscay, Spain). He obtained his PhD in Chemistry with Prof. Corzana from the University of La Rioja (Spain) in 2009, and then carried out postdoctoral research with Prof. Gin and Prof. Danishefsky at Memorial Sloan Kettering Cancer Center (USA) until 2014. After further postdoctoral studies with Prof. Jiménez-Barbero (CIB-CSIC, CIC bioGUNE, Spain) and Prof. Davis at the University of Oxford (UK), in 2017 he started his independent career in CIC bioGUNE funded through an ERC Starting Grant. His research program at the chemistry–biology frontier is focused on the development of novel chemical tools to advance molecular adjuvants and vaccines and to probe protein O-GlcNAc glycosylation. |
1. Introduction
O-GlcNAc glycosylation (also known as O-GlcNAcylation) is a key posttranslational modification (PTM) based on addition of a single monosaccharide, β-O-D-N-acetylglucosamine (β-O-GlcNAc), onto serine or threonine residues of nuclear, cytoplasmic and mitochondrial proteins.1,2 It is a widespread and unique form of glycosylation that has been found in prokaryotic and eukaryotic organisms and differs from other types of protein O- and N-glycosylation involving the attachment of several glycan units to cell surface and extracellular proteins. The O-GlcNAc modification is a highly dynamic process that occurs in an analogous fashion to protein phosphorylation, and there is an extensive crosstalk between both PTMs.3 The addition and removal of O-GlcNAc in proteins takes place by the action of two tightly regulated enzymes highly conserved in eukaryotes, which make this modification reversible and serve to maintain cellular O-GlcNAc levels well-balanced (Fig. 1). O-GlcNAc transferase (OGT) catalyses the incorporation of β-O-GlcNAc using the donor substrate uridine-5′-diphospho-N-acetylglucosamine (UDP-GlcNAc),4,5 biosynthesised from glucose via the hexosamine biosynthetic pathway (HBP), while the enzyme O-GlcNAcase (OGA) catalyses the removal of O-GlcNAc from the glycosylated substrates.6,7O-GlcNAc-modified proteins are involved in multiple cellular processes, including transcription and translation, nutrient and stress sensing, neuronal function and cell cycle.8 As a nutrient sensor, human O-GlcNAc glycosylation underlies the fundamental mechanisms of chronic diseases related to metabolism and aging, such as diabetes, cancer and neurodegeneration, which has been extensively reviewed elsewhere.9–12O-GlcNAc plays fundamental roles in diabetes, where increased O-GlcNAcylation associated to hyperglycemia has been shown to be the molecular reason for glucose toxicity, insulin resistance, and impaired β-cell function.13–16 Moreover, key transcription factors controlling proinsulin expression are dynamically regulated by O-GlcNAc modification. In cancer, there is increasing evidence that O-GlcNAcylation contributes to the oncogenic features and progression of tumour cells.17–19 In general, O-GlcNAc cycling is increased in most cancer cells, which have been found to show elevated OGT expression and depleted OGA levels20 leading to increased O-GlcNAcylation. Indeed, blocking O-GlcNAc modification resulted in reduced tumour growth, invasion and angiogenesis, although the mechanistic details of these events are not fully understood.21 Recent work points to a direct role for O-GlcNAcylation in the “Warburg effect”,22 whereby accelerated glucose uptake and glycolysis in cancer cells confers them a selective growth advantage. In addition, many critical signalling pathways (e.g., PI3K/mTOR), oncoproteins (e.g., c-Myc, SV40 large T antigen), tumour suppressor proteins (e.g., p53) and transcription factors (e.g., p65/NF-κB) are O-GlcNAc-modified,23 with site-specific O-GlcNAcylation playing distinct molecular roles in their protein stability and function. Notably, O-GlcNAc is highly abundant in the brain, where decreased O-GlcNAcylation of proteins involved in neurodegeneration has been observed to be associated with Alzheimer's and Parkinson's diseases.14,24 Indeed, O-GlcNAc modification has been shown to block proteotoxic aggregation of such proteins (e.g., tau, α-synuclein) suggesting a protective role for O-GlcNAcylation in preventing neurodegeneration.25,26 Notwithstanding these important links, more detailed investigations are required to completely elucidate the precise function and molecular mechanisms of O-GlcNAc in disease, further revealing its promise as a potential clinical target.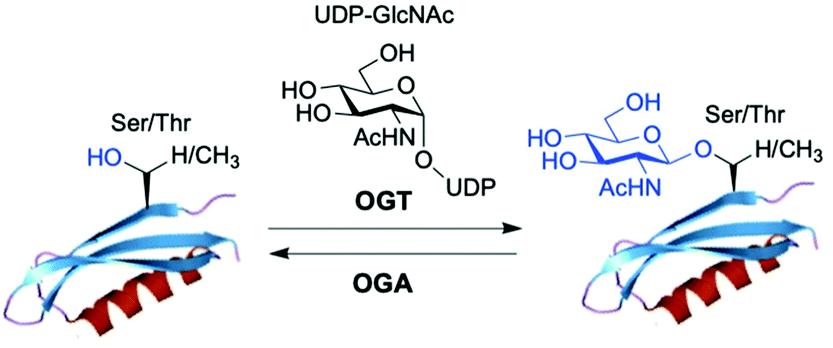 | ||
| Fig. 1 The enzyme O-GlcNAc transferase (OGT) catalyses the addition of O-GlcNAc to nuclear and cytoplasmic proteins, whereas the enzyme O-GlcNAcase (OGA) catalyses the removal of the sugar. | ||
Despite its ubiquitous nature and crucial role in biology and medicine, the molecular bases of O-GlcNAcylation have not yet been fully understood. This has been partly due to the scarcity of powerful chemical tools to identify and study this modification, which has hampered the elucidation of its functional roles and structural recognition mechanisms at the molecular level. The challenge in detecting O-GlcNAcylated proteins has been partially addressed over the years with the development of metabolic and chemoenzymatic labelling approaches, which in combination with modern mass spectrometry (MS)-based proteomics techniques have enabled the identification of many of these proteins, albeit site-mapping remains difficult.27 Nonetheless, new proteins not detected using the available strategies are likely yet to be discovered and functionally investigated as novel, improved approaches are being developed. Herein, we provide a comprehensive, critical overview of current chemical biology tools for the study of O-GlcNAc modification, including development of inhibitors of O-GlcNAc processing enzymes. Moreover, we highlight existing gaps and future perspectives in chemical probing of protein O-GlcNAc glycosylation, with particular emphasis on shedding light on the molecular mechanisms and recognition processes of O-GlcNAcylated proteins and their interacting partners. With this review, we aim to provide critical insights into the chemical prospects for molecular interrogation of O-GlcNAcylation, leading to new directions in the field and a better understanding of the key functional and structural roles of this protein modification, with implications for human health and disease.
2. Enzymes regulating O-GlcNAc cycling and chemical inhibitors
2.1. O-GlcNAc transferase (OGT)
OGT is a multidomain protein belonging to the CAZy GT-B glycosyltransferase superfamily that is encoded by a single gene (OGT) on the X chromosome. There are three different types of OGT isoforms expressed in human cells, all containing a C-terminal catalytic domain but differing in the number of N-terminal tetratricopeptide repeats (TPR) and subcellular localisation: nucleocytoplasmic OGT (ncOGT, 116 kDa), mitochondrial OGT (mOGT; 103 kDa), and short OGT (sOGT; 78 kDa).28 While the TPRs have been suggested to be involved in direct substrate recognition and glycosite selection,29,30 the precise functional roles are still being elucidated and further binding modes have also been investigated.31 Therefore, the molecular mechanisms by which OGT recognises its substrate proteins are not entirely understood as this modification occurs only in certain classes of proteins and in specific protein regions. In fact, no strict consensus sequence has been identified for OGT, albeit some structural motifs associated to sequence preferences dictate substrate selectivity.31 Crystallographic and kinetic studies with truncated human OGT (hOGT) and hOGT–substrate complexes30–34 have provided important structural insights into substrate preferences and reaction mechanisms, leading to the proposal of an ordered bi–bi kinetic mechanism for OGT glycosyl-transfer. According to this model, UDP-GlcNAc binds the active site first followed by the acceptor peptide, making a polar contact with the nucleotide-sugar α-phosphate through the amide of the amino acid that becomes glycosylated and with its hydroxyl group projecting directly into the binding site (Fig. 2a). The peptide acceptor binds OGT mainly in an extended conformation via amide backbone hydrogen bonds with adjacent TPR residues, with limited interactions through their side chains, which may explain the broad substrate tolerance of OGT.35,36 Recent biochemical and chemical strategies have revealed the importance of an asparagine ladder (conserved asparagine residues within the TPR lumen that anchor the peptide backbone of the substrates)37,38 and of luminal aspartate residues (Asp386, Asp420, Asp454) that define glycosite positioning,39 overall contributing to OGT interactions with different acceptor proteins and driving substrate selection. Moreover, interactions with adaptor/modulator proteins have also been implicated in regulating OGT function by altering its activity toward specific substrates.40 While these chemical biology and structural studies have identified important clues into substrate recognition and the existence of an electrophile migration mechanism of glycosylation,34 other binding modes of OGT may exist. Further structural and biochemical investigations are therefore required to define these interactions and to gain a better understanding of the molecular mechanisms regulating OGT activity and function.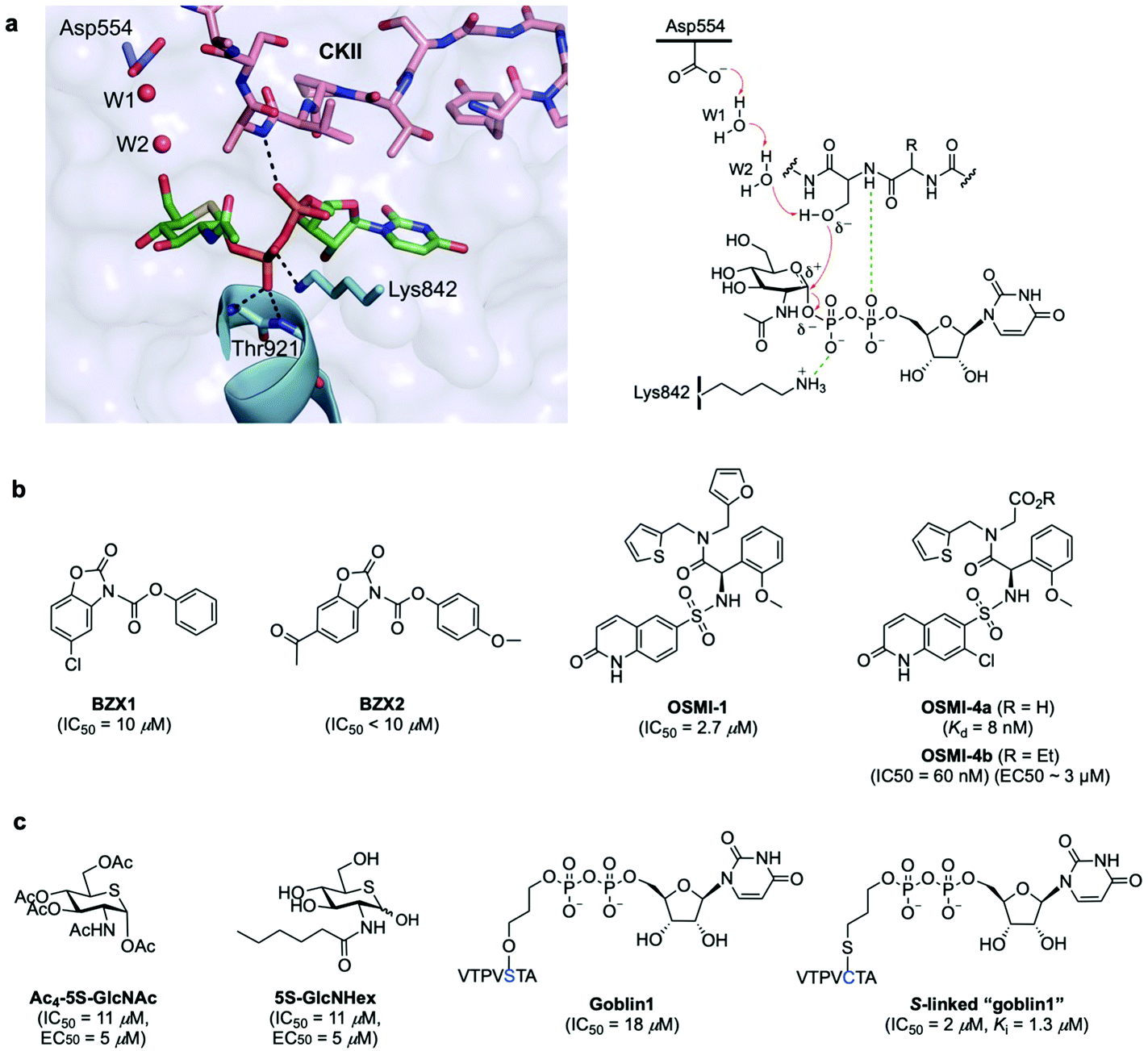 | ||
| Fig. 2 (a) Active site view of truncated OGT in complex with UDP-5S-GlcNAc and CKII peptide (PDB: 4GYY) showing key interacting residues and polar contacts (black dashed lines) (left panel);35 mechanism of O-GlcNAcylation by OGT highlighting important active site enzyme residues and interactions (right panel). (b and c) Chemical structures of some OGT inhibitors and their reported potencies: (b) BZX1, BZX2, OSMI-1, OSMI-4 identified via high-throughput screening, (c) Ac4-5S-GlcNAc, 5S-GlcNHex, goblin1 and its S-linked analogue, designed based on the structures of the UDP-GlcNAc donor substrate and the UDP reaction product. | ||
The identification of these OGT inhibitors validates HTS strategies for the discovery of other glycosyltransferase inhibitors45 and provides opportunities for the development of more potent and specific inhibitors better suited to interrogate OGT in vivo. Alternatively, analogues of UDP-GlcNAc have also been rationally designed for metabolic OGT inhibition (Fig. 2c). A cell-permeable per-O-acetylated-5S-GlcNAc (IC50 = 11 μM, EC50 = 5 μM) derivative is converted in cells to UDP-5S-GlcNAc, which acts as a potent OGT inhibitor in vitro (Ki = 8 μM) and in cell lines.46 With a view to inhibiting OGT in vivo, more water soluble, cell-permeable analogues incorporating hydrophobic N-acyl substituents were developed, such as 5S-GlcNHex (IC50 = 11 μM, EC50 = 5 μM) which is metabolically converted to the same UDP-5S-GlcNAc active compound and exhibited OGT inhibitory activity in mice.47 Despite their high potency, these analogues hijack the HBP and reduce the native UDP-GlcNAc pool within the cell, potentially causing undesired inhibition of other necessary glycosyltransferases, which might affect other cellular processes. Another example of rational development of OGT inhibitors is UDP–peptide bisubstrate conjugates such as goblin 1 (IC50 = 18 μM in vitro)48 and more potent thio-linked derivatives (IC50 = 2 μM),49 which are however not cell-permeable due to the negative charge of the diphosphate moiety, precluding their use in vivo.
Overall, these studies have provided useful OGT inhibitors for in vitro and even animal models.50 Nonetheless, novel chemical strategies are needed for the development of improved, substrate-specific inhibitors that can be used to probe O-GlcNAcylation on selected protein(s) and that show sufficiently high potency to be applied in vivo.
2.2. O-GlcNAcase (OGA)
In humans, OGA is encoded by the OGA gene in the form of two major splice variants differing by the presence or absence of a C-terminal region: a long OGA (lOGA) and a short OGA (sOGA), respectively. Human long OGA (hOGA) is made up of an N-terminal catalytic domain similar to the CAZy glycoside hydrolase family 84 (GH84) and a C-terminal pseudo-histone acetyltransferase (HAT) domain, connected by an ordered stalk domain in-between. After cloning and biochemical characterisation of hOGA in 2001,7 the matter of OGA substrate recognition beyond the GlcNAc unit has been a long-standing question. OGA follows a double-displacement, substrate-assisted catalytic mechanism, involving the formation and breakdown of a transient oxazoline intermediate facilitated by two catalytic acid–base residues (Asp174 and Asp175) (Fig. 3a).51,52 Thus, the N-acetamido group of the substrate acts as a nucleophile displacing the protein hydroxyl group to generate the transient enzyme-bound oxazoline, which then undergoes hydrolytic ring-opening to provide the corresponding GlcNAc hemiacetal. The structures of truncated hOGA in apo form and in complex with small molecule inhibitors (thiamEt-G, PUGNAc-imidazole, and VV347) as well as different glycopeptide substrates were solved in 2017 by three research groups independently.53–55 These studies revealed an unusual dimeric structure and general principles for OGA substrate recognition, whereby the glycopeptide is bound in a substrate-binding cleft created by the dimerisation of hOGA.55,56 The interactions of the GlcNAc sugar with the OGA catalytic pocket are significant and highly conserved, whereas the peptides adopt a similar V-shaped conformation binding in a bidirectional orientation, with some side chain-specific interactions with OGA residues in the cleft. Future structural studies are expected to illuminate how OGA interacts with specific protein substrates beyond the catalytic domain, yielding a more in-depth understanding of the molecular details of OGA substrate recognition. The three structures of hOGA complexed with its inhibitor thiamEt-G (thiazoline amino ethyl gluco-configured)53–55 revealed strong interactions with active site residues (e.g., Lys98, Asp174, Asp175 Asp285, Asn313) (Fig. 3b), and yielded direct structural evidence of the proposed substrate-assisted mechanism for OGA.52 Moreover, the structures of hOGA in complex with the three previous inhibitors provide valuable insights to assist future efforts in inhibitor design.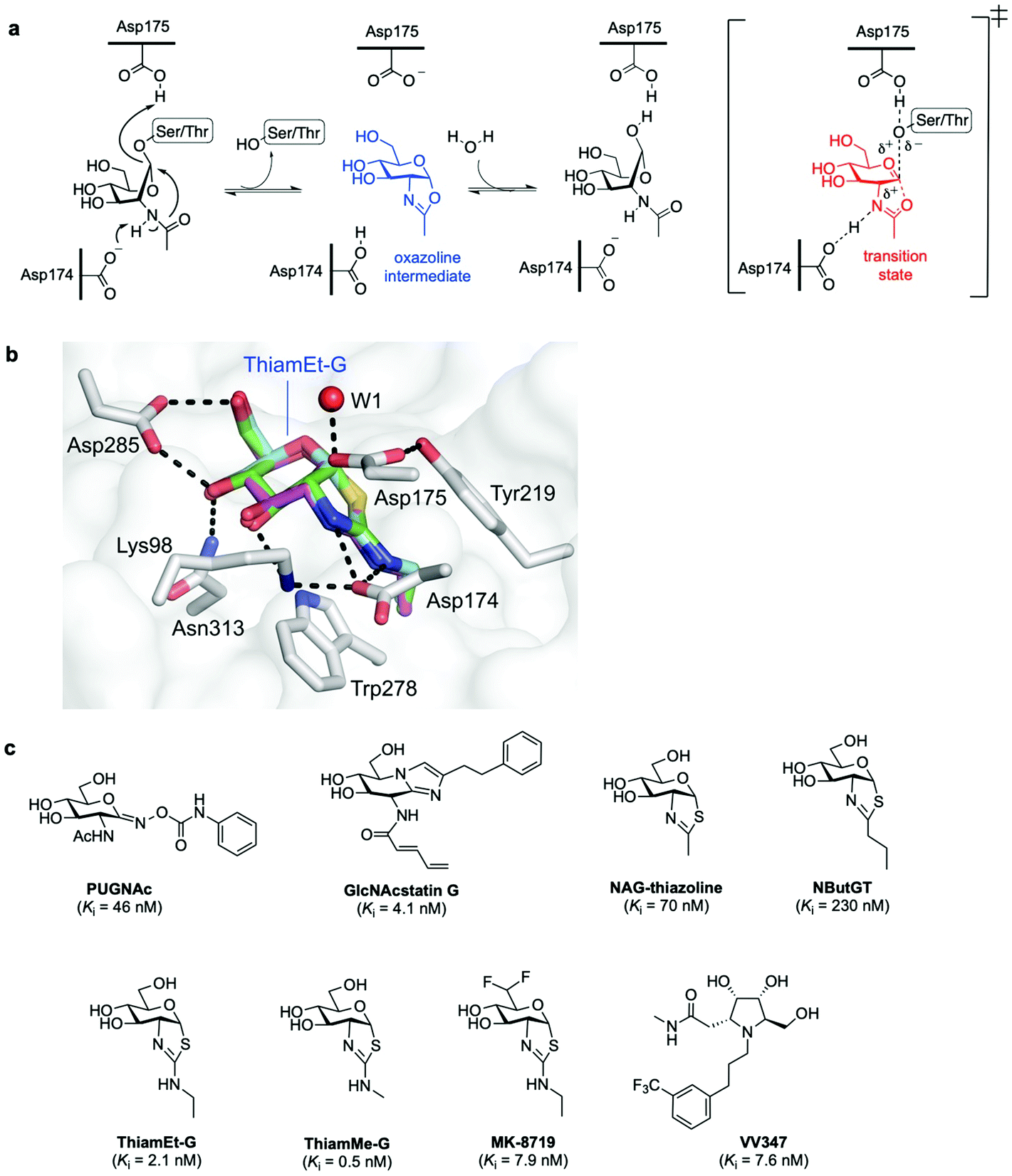 | ||
| Fig. 3 (a) Catalytic mechanism of human O-GlcNAcase (hOGA) deglycosylation (hydrolysis) proceeds through an oxazoline intermediate with the assistance of Asp174 and Asp175 as catalytic acid–base residues. (b) Active site view of OGA in complex with thiamEt-G (depicted as an overlay of three different PDB structures (5UN9, 5M7S, 5UHL)) showing key interacting residues and contacts (black dashed lines).35 (c) Chemical structure of some OGA inhibitors with their reported potencies: PUGNAc, GlcNAcstatin G, NAG-thiazoline, NButGT, thiamEt-G, its methyl analogue thiamMe-G, its difluoro congener MK-8719, and iminocyclitol derivative VV347. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold) for hOGA over β-hexosaminidases,60 inducing hyper O-GlcNAcylation in cells and emerging as a useful tool to study the role of O-GlcNAc in cellular biology. NAG-thiazoline (1,2-dideoxy-2′-methyl-α-D-glucopyranoso-[2,1-d]-Δ2′-thiazoline), designed as a mimic of the catalytic oxazoline intermediate,63 is another potent (Ki = 70 nM) but not selective OGA inhibitor. This led to the rational development and study of several derivatives bearing longer alkyl substituents in the thiazoline ring that exhibited enhanced selectivity but decreased potency, including the partially unstable inhibitor NButGT (Ki = 230 nM).51,59 A subtle modification of NButGT, in which the proximal methylene group of the alkyl chain was replaced with an amine with a view to increasing electrostatic interactions, provided the very potent inhibitor thiamEt-G (Ki = 21 nM later redetermined as 2.1 nM)64 with excellent selectivity (35000-fold) over β-hexosaminidases.65 Moreover, its further beneficial properties, namely streamlined chemical synthesis, improved stability and cell-permeability, as well as its ability to cross the blood–brain barrier, have made thiamEt-G a leading OGA inhibitor in a number of in vitro and in vivo studies.
000-fold) for hOGA over β-hexosaminidases,60 inducing hyper O-GlcNAcylation in cells and emerging as a useful tool to study the role of O-GlcNAc in cellular biology. NAG-thiazoline (1,2-dideoxy-2′-methyl-α-D-glucopyranoso-[2,1-d]-Δ2′-thiazoline), designed as a mimic of the catalytic oxazoline intermediate,63 is another potent (Ki = 70 nM) but not selective OGA inhibitor. This led to the rational development and study of several derivatives bearing longer alkyl substituents in the thiazoline ring that exhibited enhanced selectivity but decreased potency, including the partially unstable inhibitor NButGT (Ki = 230 nM).51,59 A subtle modification of NButGT, in which the proximal methylene group of the alkyl chain was replaced with an amine with a view to increasing electrostatic interactions, provided the very potent inhibitor thiamEt-G (Ki = 21 nM later redetermined as 2.1 nM)64 with excellent selectivity (35000-fold) over β-hexosaminidases.65 Moreover, its further beneficial properties, namely streamlined chemical synthesis, improved stability and cell-permeability, as well as its ability to cross the blood–brain barrier, have made thiamEt-G a leading OGA inhibitor in a number of in vitro and in vivo studies.
For instance, thiamEt-G has been used to increase O-GlcNAc levels in mouse models of Alzheimer's disease, hindering protein aggregation and reducing neurodegeneration.66 Based on the thiamEt-G structure, 2′-aminothiazoline derivatives with different alkyl groups at the 2′ position were developed as genuine transition state analogues, the most potent of which (thiamMe-G) showed subnanomolar inhibition (Ki = 0.5 nM) and even greater selectivity (1800000-fold) over β-hexosaminidases.64 Recently, medicinal chemistry optimisation of the (thiamEt-G) parent compound by reducing its polar surface area led to the identification of a highly potent (Ki = 7.9 nM) and selective difluoro-substituted congener (MK-8719), which showed improved drug-like properties and has been advanced for clinical trials.67 Another approach to potent OGA inhibitors has taken the form of stereoisomeric pyrrolidine-based iminocyclitols.68 Among them, VV347 stood out, which exhibited single digit nanomolar potency (Ki = 7.6 nM), good bioavailability and biodistribution in mice, being able to cross the blood–brain barrier.53
Taken together, the detailed understanding of the OGA catalytic mechanism and structure has made it possible to successfully develop a range of rationally designed, highly potent and selective inhibitors as useful chemical tools for further functional studies and therapeutic applications of O-GlcNAcylation.
3. Approaches for investigating O-GlcNAc modified proteins
While O-GlcNAcylation is widespread among cellular regulatory proteins, progress in understanding its molecular roles has been slow, partly due to the difficulties in identifying and studying this modification (which remained undetected until the 80s), owing to several intrinsic features. First, O-GlcNAcylation is dynamic, prevalent on low-abundance regulatory proteins, and substoichiometric. Second, the GlcNAc O-glycosidic linkage is inherently labile, both enzymatically and chemically, falling off upon fragmentation during standard mass spectrometry (MS) methods; moreover, O-GlcNAc–peptide ion signals are suppressed in favour of ions from unmodified peptides.69 Lastly, while some insights into particular motifs for O-GlcNAcylation have been provided,31,70 the lack of a definite consensus sequence for OGT has hampered the specific determination of in vivo modification sites based on the primary sequence alone. Despite these challenges, a variety of approaches have been developed over the years for the detection and site mapping of O-GlcNAc, enabling the identification of over 5000 human O-GlcNAcylated proteins and 7000 glycosylation sites.71 In the following sections, we discuss some of these strategies as well as their applications, advantages and limitations in detecting O-GlcNAc in vitro and in vivo and provide some insights into the functional significance of this modification. We also introduce future perspectives in the development of new chemical tools for identifying unknown O-GlcNAc proteins.3.1. Antibodies and lectins for O-GlcNAc research
Several antibodies able to detect O-GlcNAc in cytosolic and nuclear extracts have been developed. A number of O-GlcNAcylated proteins have been identified by Western blotting using the pan-specific O-GlcNAc monoclonal antibodies RL2 (an IgG type antibody raised against O-GlcNAc modified fragments of glycosylated nucleoporins)72 and CTD110.6 (an IgM class antibody developed using a synthetic O-GlcNAcylated repeat of the RNA polymerase II C-terminus as the immunogen).73 However, these antibodies suffer from limited specificity, cross-reactivity issues74 and relatively low binding affinities, which preclude the efficient detection of single-modified, low-abundance O-GlcNAc proteins.69 Several other anti-O-GlcNAc mouse mAbs are also known, such as HGAC39 and 85,75 10D8,76 and three IgG mAbs (18B10.C7(#3), 9D1.E4(#10), and 1F5.D6(#14)), which were developed using synthetic O-GlcNAc glycopeptide antigens.77 The specificity of these mAbs towards terminal O-GlcNAc versus other forms of exposed GlcNAc has been investigated. In particular, IgM CTD110.6 and IgG mAbs 18B10.C7(#3) and 9D1.E4(#10) were reported to detect O-GlcNAc specifically on cell surface glycoproteins,78 and together with 1F5.D6(#14), these antibodies were also able to bind terminal β-GlcNAc on N-glycans.78,79 Most recently, a new antibody mixture for O-GlcNAc has been made available for Western blotting applications (O-GlcNAc MultiMab Rabbit mAb mix #82332S by Cell Signaling Technology) that specifically recognises endogenous levels of O-GlcNAcylated proteins. Moreover, a few site-specific O-GlcNAc antibodies have been developed targeting defined O-GlcNAc epitopes within particular proteins, including c-Myc (Thr58),80 tau (Ser400),81 histone H2A (Ser40, Thr101),82 and TAB1 (Ser395),83 which have enabled more precise investigations into O-GlcNAc function. Thus, while all these antibodies have been useful in probing protein O-GlcNAcylation in a number of applications, there is a need for additional O-GlcNAc-specific IgG antibodies as powerful probes to glean mechanistic insights into site-specific O-GlcNAc modification to continue bringing fastest, significant developments in the field.In addition to antibodies, some plant lectins, and in particular wheat germ agglutinin (WGA), have been long used for research in the field as relatively specific carbohydrate-binding proteins recognising O-GlcNAc. WGA binds intracellular O-GlcNAc-modified proteins, but it also recognises N-acetyl-D-neuraminic acid (Neu5Ac) and terminal GlcNAc residues on glycoconjugates and oligosaccharides.84 Although succinylation of WGA (sWGA) increases its specificity for O-GlcNAc, it also reduces the already low affinity of unmodified WGA (Kd ∼ 2.5 mM for free GlcNAc), making this lectin useful mainly as an enrichment tool for purification and for immunoblotting.85 Thus, the low affinity and selectivity of WGA for single GlcNAc limits its further utility, unless these residues are closely clustered in a peptide, as demonstrated with the development of the WGA-based lectin weak affinity chromatography technique.86 More recently, additional lectins originally derived from fungi have been identified with stronger affinity for O-GlcNAc (Kd ∼ 200 μM).87,88 Moreover, although remarkable synthetic lectins have been developed by Davis and coworkers that show much higher affinity and specificity for O-GlcNAc89,90 (see Section 4.4.2), these promising biomimetic analogues have not yet been applied for the detection of O-GlcNAcylated proteins.
As another tool for O-GlcNAc detection, van Aalten and coworkers identified a bacterium Clostridium perfringens OGA (CpOGA), in which Asp-to-Asn mutation led to an engineered CpOGAD298N variant that lost hydrolytic activity but retained substrate binding.91 This impaired OGA mutant bound O-GlcNAc peptides with notable nanomolar affinity, although with preferential binding to Ser- over Thr-O-GlcNAc substrates, and has been successfully applied to identify a number of O-GlcNAcylated proteins in embryonic Drosophila.92
3.2. Metabolic chemical reporters (MCRs) for labelling intracellular O-GlcNAcylation
To overcome the above limitations in specificity and efficiency when probing O-GlcNAcylation, various methods have been developed through metabolic oligosaccharide engineering (MOE) (also known as metabolic glycan labelling, MGL) by using chemically synthesised metabolic chemical reporters (MCRs) incorporating unnatural chemoselective handles. The substrate UDP-GlcNAc is produced de novo by the HBP, but can also be generated by the GlcNAc salvage pathway from the exogenous GlcNAc (Fig. 4a). Notably, OGT has been found to be able to tolerate subtle structural changes in the donor substrate, recognising modified UDP-GlcNAc derivatives for further sugar transfer. This feature has allowed the development of a variety of acetylated GlcNAc-derived chemical reporters that upon internalisation into cells and metabolic processing into unnatural UDP sugar donors can be incorporated into the target proteins by OGT. Subsequent bioorthogonal reaction between the chemically tagged glycoprotein and fluorescent and/or biotinylated probes enables visualisation and/or isolation of the O-GlcNAcylated proteins.93 The first reported example of metabolic labelling of intracellular O-GlcNAcylation was demonstrated by the Bertozzi group and made use of 2-azidoacetamido-2-deoxy-glucopyranose (Ac4GlcNAz).94 The biologically inert azide group chemically incorporated into the GlcNAc acetamido moiety was shown to be well tolerated by the biosynthetic enzymes of the salvage pathway, leading to metabolic formation of UDP-GlcNAz. This unnatural sugar donor was then utilised by OGT to incorporate O-GlcNAz into Ser/Thr residues, thus generating chemically tagged proteins bearing an azide functionality for further detection by bioorthogonal reactions (e.g., Staudinger ligation or copper-catalysed alkyne−azide cycloaddition (CuAAC)). The good degree of tolerance of the GlcNAc salvage pathway and the OGT promiscuity towards the azide-modified GlcNAz moiety have been applied in chemical proteomics approaches95 to identify O-GlcNAcylated proteins in different cell lines (e.g., glucose-6-phosphatase, elF-5, galectin 3 identified in HeLa cells96), albeit weak metabolic labelling was observed.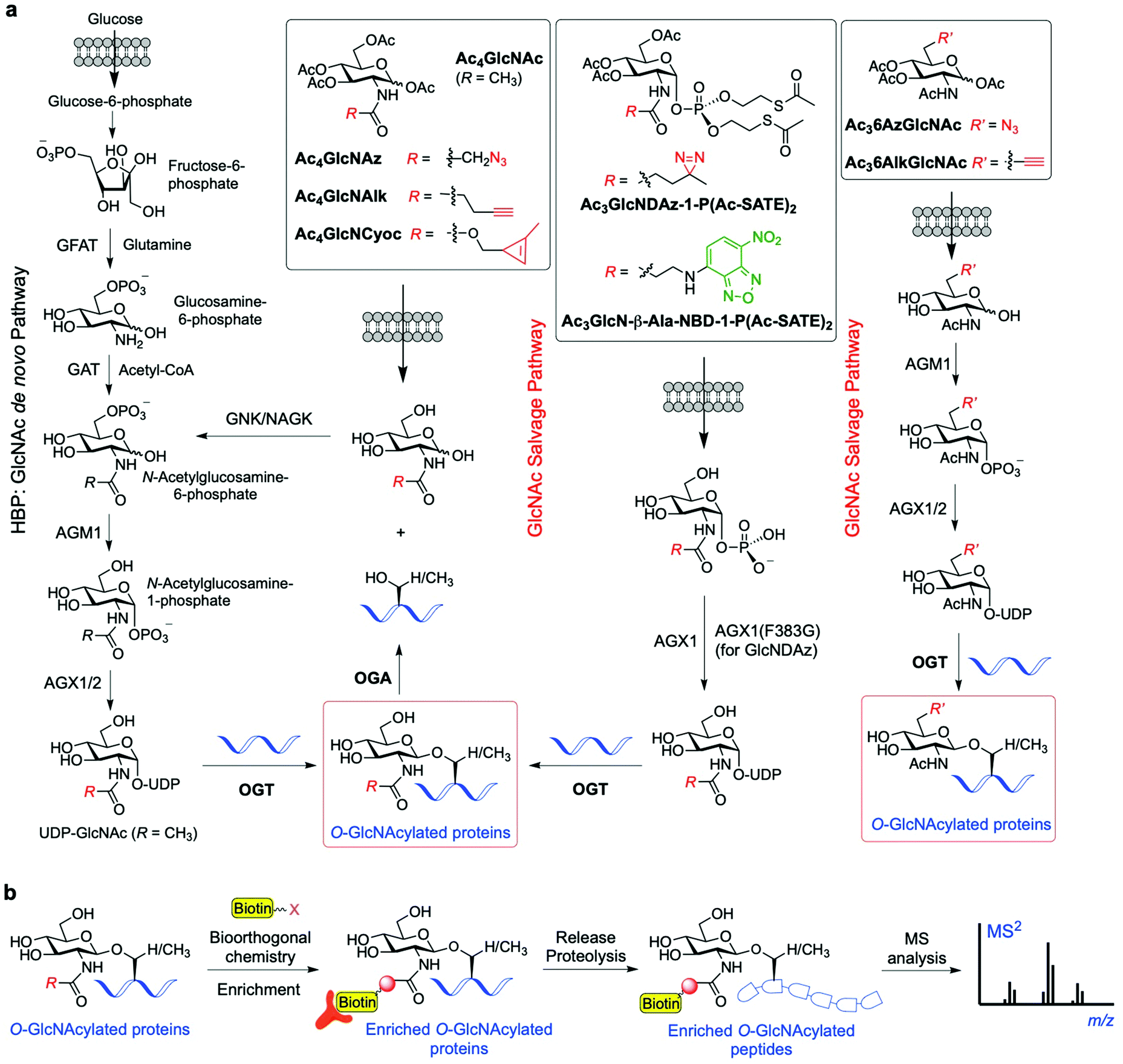 | ||
| Fig. 4 (a) Schemes of the GlcNAc de novo hexosamine biosynthetic pathway (HBP) and salvage pathway from exogenous GlcNAc, showing a range of synthetic metabolic chemical reporters (MCRs). The engineered GlcNAc analogues enter the salvage pathway and are converted to the corresponding UDP-GlcNAc derivatives in the cell; subsequent OGT-catalysed O-GlcNAcylation enables labelling of the relevant O-GlcNAc modified proteins. (b) General workflow showing how the probes are applied for the identification of the O-GlcNAcylated proteins.95,104,105 | ||
A limitation of peracetylated Ac4GlcNAz as a metabolic reporter is the relatively high degree of non-specific glycosylation. Previous studies showed that GlcNAz is incorporated not only into intracellular O-GlcNAcylated proteins, but also directly into the core pentasaccharide of N-linked glycans.97 Moreover, as UDP-GlcNAc can be interconverted to UDP-GalNAc (and vice versa) by the UDP-galactose-4′-epimerase (GALE) enzyme, the corresponding GlcNAz can also be alternatively incorporated into mucin-type O-linked glycans,98 further compromising the specificity and efficiency of the labelling. In this context, a study investigating the metabolic cross-talk between O-GlcNAcylation and the GalNAc salvage pathway found that treating cells with peracetylated GalNAz (Ac4GalNAz) resulted in UDP-GlcNAz biosynthesis via GALE and more robust labelling of O-GlcNAcylated proteins than that with Ac4GlcNAz.99 These MCR approaches have been successfully applied for the identification of many O-GlcNAcylated proteins. For instance, an elegant proteomics approach using Ac4GlcNAz and click chemistry-based protein enrichment followed by on-resin proteolysis and selective β-elimination of the O-GlcNAc-peptide moieties enabled the profiling and site-mapping of hundreds of O-GlcNAcylated proteins (e.g., the E3 ubiquitin-protein ligase CBL and ubiquilin-2 identified in HEK293 cells).100 Most recently, a tandem metabolic glycan engineering strategy has been developed by Vocadlo and coworkers to assess whether the de novo, ribosome-synthesised nascent peptide chains are substrates of OGT and cotranslationally glycosylated with O-GlcNAc.101 Ac4GalNAz was employed to incorporate O-GlcNAz into the nascent peptides (via metabolic conversion of UDP-GalNAz to UDP-GlcNAz by GALE as described above), and the cells were then treated with O-propargyl-puromycin (OPP), leading to incorporation of an alkyne moiety into the nascent peptide C-terminus and termination of translation. This strategy allowed for subsequent labelling via two sequential rounds of bioorthogonal reactions (Staudinger ligation onto the GlcNAz fragment and CuAAC on the terminal alkyne), enabling both visualisation and enrichment of the doubly tagged proteins for proteomics analysis. This technique identified around 500 O-GlcNAc candidate proteins, and 75 of them were considered to be good candidates for further validation as OGT substrates for cotranslational glycosylation. Subsequent gene ontology studies showed significant enrichment of proteins in functional categories that suggest physiological roles for cotranslational O-GlcNAcylation. Further application of this approach on three candidate proteins (Ataxin-2L, Nup153 and HCF-1) confirmed the occurrence of cotranslational O-GlcNAcylation, thus validating this strategy as a powerful tool for the identification of O-GlcNAc modified nascent proteins.
Another synthetic chemical reporter for detecting O-GlcNAcylated proteins is Ac4GlcNAlk, which was developed by Pratt and coworkers, featuring a built-in alkyne tail at the 2-acetamido position.102 GlcNAlk was found to be incorporated and removed from target proteins at rates similar to those of GlcNAz, and was not readily interconverted to GalNAlk via GALE, leading to a low level of labelled O-linked mucin glycoproteins. This makes GlcNAlk a more specific reporter of O-GlcNAcylation compared to GlcNAz and GalNAz, and was exploited in combination with bioorthogonal labelling using a cleavable biotin affinity tag for the identification of 374 O-GlcNAc proteins, most of them not previously reported (e.g., the ubiquitin ligase NEDD4-1). However, GlcNAlk was still incorporated into N-linked glycans, preventing its utility as a completely selective O-GlcNAcylation reporter. Therefore, a new acetylated MCR was synthesised bearing an azide substituent at the 6-position, Ac36AzGlcNAc,103 which enabled selective O-GlcNAc probing. Due to the absence of the C6 hydroxyl group, this compound cannot be metabolised to the corresponding UDP-sugar donor by the canonical GlcNAc salvage pathway, which begins with phosphorylation of the monosaccharide at the 6-position. Instead, 6AzGlcNAc can bypass this initial biosynthetic step through direct phosphorylation at its C1-hydroxyl by the enzyme phosphoacetylglucosamine mutase (AGM1), and then enter the salvage pathway to give the corresponding UDP-6AzGlcNAc, which can be finally used by OGT for O-6AzGlcNAcylation. Comparative proteomics experiments using peracetylated 6AzGlcNAc, GlcNAz, and GalNAz revealed that 6AzGlcNAc is highly specific towards O-GlcNAcylated proteins, while GlcNAz and GalNAz were less specific, labelling other types of glycosylation.103
Subsequently, an additional 6-modified MCR was developed incorporating an alkyne group at that position, named Ac36AlkGlcNAc.106 This alkyne GlcNAc analogue was found to label proteins with faster kinetics and improved signal-to-noise ratio than Ac36AzGlcNAc, because the reverse orientation of the CuAAC chemistry employed reduced the background labelling. The chemical proteomics experiments with Ac36AlkGlcNAc enabled the identification of more potentially O-GlcNAcylated proteins (including caspases-3 and -8) and generally with higher efficiency and selectivity than previously developed MCRs. Strikingly, other non-N-acetyl-glucosamine monosaccharides, e.g., 2- and 6-azido modified glucose analogues (2AzGlc/GlcAz and 6AzGlc), have also been reported to act as MCRs of O-GlcNAc glycosylation.107–109 This unexpected substrate tolerance of OGT, being also able to transfer glucose analogues to protein substrates, enabled the discovery of intracellular O-glucose modification of proteins using 6AzGlc.109
The approaches described above rely on widely used bioorthogonal labelling reactions (e.g., copper-catalysed (CuAAC) or strain-promoted azide–alkyne cycloaddition (SPAAC) (see Section 3.5)) that, however, suffer from several limitations. These are mainly associated to the toxicity of copper in living cells, which precludes the use of CuAAC in vivo, and the high background noise of SPAAC due to unwanted reactions of strained alkynes with intracellular thiols. A potential fully orthogonal and well tolerated method relies on the implementation of the inverse electron demand Diels–Alder (iEDDA, see Section 3.5) reaction, using chemically inert, dienophile-bearing MCRs that react rapidly with suitable electron-deficient diene derivatives. Notably, applications of metabolic labelling of glycans incorporating strained110 or terminal alkenes111 with 1,2,4,5-tetrazine tags have been demonstrated in living cells. In this context, carbamate-linked methylcyclopropene derivatives appeared to be especially well suited for bioorthogonal in vivo chemistry.112,113 A number of examples using the methylcyclopropene–tetrazine pair have been reported, culminating in the development of a strategy for direct visualisation of the glycosylation state of selected intracellular proteins within living cells by the Wittmann lab.114 In this case, a peracetylated GlcNAc incorporating a methylcyclopropene tag (Ac4GlcNCyoc) was incubated with cells expressing several proteins of interest (e.g., OGT, FOXO1, p53, and Akt1) that had been genetically fused with enhanced green fluorescent protein (EGFP). The resulting proteins incorporating GlcNCyoc were then specifically labelled with suitable tetrazine–fluorophore conjugates via an iEDDA reaction, enabling subsequent direct monitoring of protein O-GlcNAcylation by measuring Förster resonance energy transfer (FRET) between EGFP and the sugar-conjugated fluorophore by fluorescence lifetime imaging.
Another application of metabolic glycan labelling involved the development of a GlcNAc photoaffinity probe (GlcNDAz) that was used in photocrosslinking studies for the identification of binding partners of O-GlcNAc modified proteins,115 which will be discussed in detail in Section 3.3. More recently, a one-step metabolic feeding strategy was developed that enabled direct labelling of O-GlcNAc modified proteins in vitro and in live cells using a fluorescent glucosamine derivative.116 The small fluorophore 4-nitro-2,1,3-benzoxadiazole (NBD) was incorporated through the 2-acetamido position (via a short β-Ala spacer) into a suitably protected precursor for metabolic feeding, in which besides the peracetylated hydroxyl functions, the phosphate was masked with two S-acetyl-2-thioethyl (Ac-SATE) groups (Ac3GlcN-β-Ala-NBD-α-1-P(Ac-SATE)2, Fig. 4)). This advanced metabolic intermediate was deacetylated within cells to the corresponding α-phosphate sugar, which was found to be tolerated by AGX1 (the last enzyme in the GlcNAc salvage pathway) to be converted into the UDP-GlcN-β-Ala-NBD donor. Remarkably, OGT was able to transfer the GlcN-NBD conjugate, leading to fluorescent labelling of O-GlcNAcylated proteins within living cells. This powerful strategy opens up new opportunities to further explore the roles of O-GlcNAcylation in a cellular context.
While the development of MCRs has expanded the chemical toolbox and facilitated the investigation of O-GlcNAcylation, these reporters are not devoid of limitations. Some metabolic GlcNAc probes suffer from selectivity issues, which results in nonspecific labelling of other glycosylation forms, including cell-surface, O- and N-linked glycoproteins as well as intracellular O-glucosylation. Moreover, metabolic oligosaccharide engineering can perturb metabolic pathways in living cells, and thus, the captured glycosylation state of proteins may not be relevant. Most recently, reported data have alerted about unwanted side reactions occurring with peracetylated MCRs, which in addition to OGT-catalysed S-GlcNAcylation117,118 can result in non-enzymatic labelling of cysteine residues, potentially interfering and causing false positives in the proteomic identification of O-GlcNAcylated proteins.119 This artificial side reaction, termed S-glyco-modification, occurs through a mechanism that involves base-promoted β-elimination of the 3-acetoxy group to provide α,β-unsaturated aldehydes, followed by thio-Michael addition to give the S-glyco modified, 3-thiolated sugar.120 Based on this knowledge, a new unnatural monosaccharide unprotected at C3-OH [1,6-di-O-propionyl-GalNAz (1,6-Pr2GalNAz)] was rationally developed, which exhibited high labelling efficiency for protein O-GlcNAcylation without undesired S-glyco-modification.120 These partially protected metabolic glycan probes emerge therefore as a next-generation of improved MCRs of choice for a more efficient probing of O-GlcNAc in vivo.
3.3. Chemical biology strategies to identify O-GlcNAc interacting partners and reader proteins
Despite the significant pathophysiological implications arising from protein O-GlcNAcylation, the functional roles and mechanisms of this ubiquitous modification in cellular signalling remain unclear. It has been proposed that the O-GlcNAc modification of proteins may impact their downstream function by altering their interaction with specific partner proteins. To identify such potential binding partners, a MGL based, photocrosslinking approach was developed by Kohler and coworkers leading to the identification of O-GlcNAc binding proteins in live cells.115 This strategy started with the synthesis of a photoreactive GlcNAc analogue (GlcNDAz) modified at the 2-acetamido position with a diazirine moiety (Fig. 5), which under UV light generates a reactive carbene that crosslinks with amino acids of interacting proteins. Before the actual crosslinking experiments with the corresponding O-GlcNDAz modified proteins, OGT was first confirmed to be able to accept UDP-GlcNDAz and transfer the unnatural sugar into acceptor peptides. Initial attempts to make use of the GlcNAc salvage pathway to produce the photocrosslinking nucleotide sugar (UDP-GlcNDAz) in cells from peracetylated Ac4GlcNDAz were unsuccessful. To overcome this issue, a suitably protected, membrane-permeable metabolic intermediate (Ac3GlcNDAz-1P-(Ac-SATE)2) was prepared (Fig. 5) and then added to cells, which upon hydrolysis by intracellular esterases resulted in accumulation of GlcNDAz-1-P but no conversion to UDP-GlcNDAz, suggesting that GlcNDAz-1-phosphate is a poor substrate for AGX1. Structure-guided engineering of the enzyme yielded an AGX1 mutant having an expanded binding pocket for the diazirine substituent that enabled production of UDP-GlcNDAz in transfected cells. Thus, suitably engineered cells treated with Ac3GlcNDAz-1P-(Ac-SATE)2 led to O-GlcNDAz modified proteins, subsequently identified by immunoblot as FG-repeat nucleoporins, which upon UV irradiation underwent crosslinking with nuclear transport factors (e.g., transportin-1) as assessed by tandem MS analysis of the purified covalent complexes. These results suggested that O-GlcNAc modification is associated with recognition events that occur during nuclear transport, although they did not unambiguously confirm the direct involvement of the O-GlcNAc moiety in these interactions. This study validated the utility of O-GlcNDAz-based crosslinking as a tool for probing the interactions between O-GlcNAcylated proteins and their putative partners and could be applicable for investigating the functional consequences of O-GlcNAc modification. Building upon this work, mutagenesis of the UDP-GlcNAc binding pocket of OGT provided an OGT mutant (C917A) with preference for the unnatural substrate that increased enzymatic transfer and incorporation of GlcNDAz into proteins in vitro and in cells.121 This technical improvement enabled enhanced crosslinking of O-GlcNDAz modified molecules and, hence, more efficient identification of O-GlcNAc mediated protein–protein interactions, which, in turn, could be exploited to provide insights into the molecular mechanisms of O-GlcNAc function.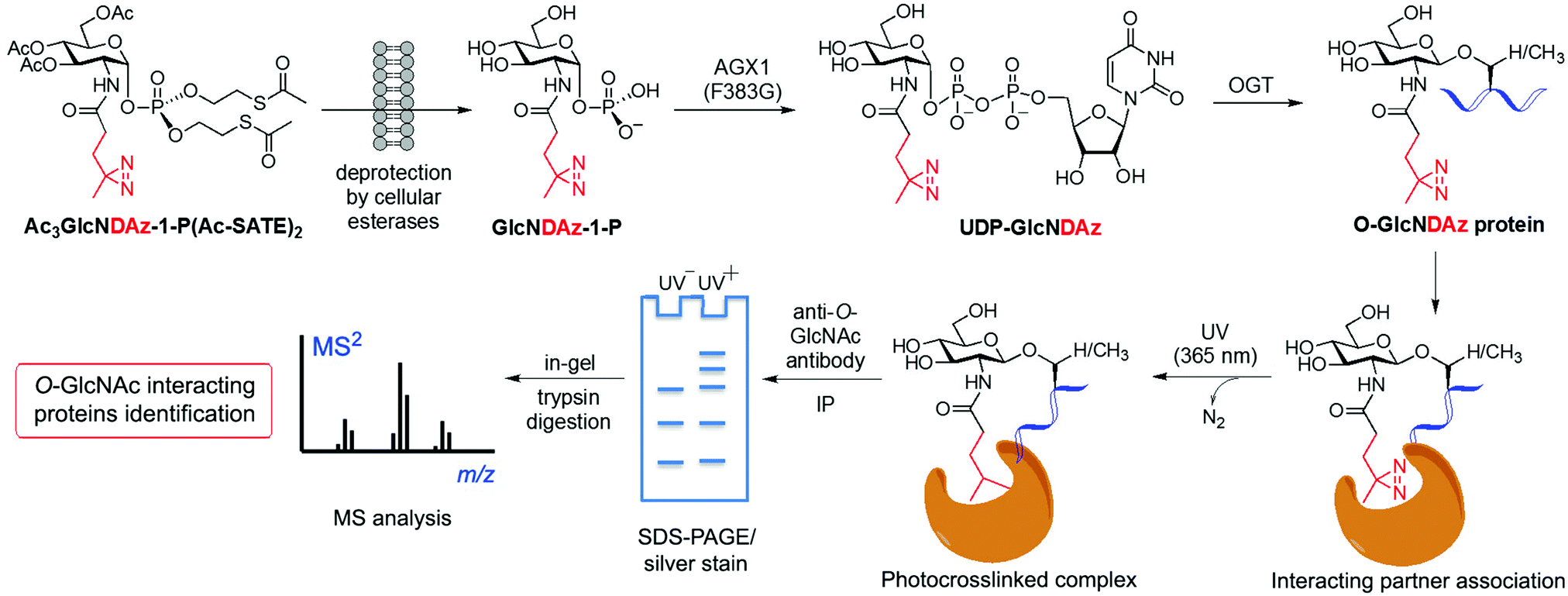 | ||
| Fig. 5 Identification of O-GlcNAc-mediated protein interactions and binding partners using a metabolic labelling photocrosslinking approach. Cells are cultured with a synthetic diazirine-functionalised, cell-permeable precursor [Ac3GlcNDAz-1-P(Ac-SATE)2], which is deprotected inside the cell by esterases and converted to UDP-GlcNDAz by an AGX1 mutant. OGT (or an engineered OGT with enhanced substrate preference) transfers GlcNDAz to its native substrates, labelling O-GlcNAcylated proteins with the unnatural modification. Short UV irradiation (365 nm) activates the diazirine for carbene-mediated crosslinking with the neighbouring binding partners. After cell lysis, immunoprecipitation of the covalent protein complex with an anti-O-GlcNAc antibody, followed by SDS-PAGE separation, in-gel tryptic digestion and subsequent proteomics analysis by LC-MS/MS, enables the identification of O-GlcNAc interacting proteins. | ||
To elucidate the structural basis of O-GlcNAc recognition, a biochemical probing strategy was subsequently developed by the Boyce lab to identify putative O-GlcNAc “reader” proteins that specifically recognise O-GlcNAc.122 By overlapping 802 mapped Ser-O-GlcNAc sites, a consensus O-GlcNAcylated peptide sequence was first deduced that encompassed a previously observed Pro-Val-Ser tripeptide, suggesting its importance as a potential motif for O-GlcNAc modification and recognition. Initial experiments demonstrated that OGT was able to O-GlcNAcylate a “bait” peptide that contained this consensus sequence and a polyethylene glycol–biotin tag, transferring one GlcNAc moiety onto the expected serine residue. This “bait” glycopeptide was then used in pulldown experiments to selectively affinity-enrich putative O-GlcNAc binding partners from cell lysates. Subsequent MS proteomics analysis enabled the identification of several endogenous nuclear and cytoplasmic proteins, including importin-β1, a mediator of nuclear cargo trafficking that had been previously found to interact with O-GlcNAcylated nucleoporins.115
Further analysis of other hit proteins within the glycopeptide-enriched pool of O-GlcNAc binders, namely human α-enolase, EBP1, and 14-3-3β/α and γ, revealed that they bind the O-GlcNAc moiety directly and specifically. Additional confirmation that these “reader” proteins interact with native OGT substrates and bind to O-GlcNAc in living cells was obtained using photocrosslinking experiments by metabolic labelling with GlcNDAz (see above). Moreover, X-ray crystal structures of the bait glycopeptide in complex with the 14-3-3β/α isoforms revealed peptide/protein backbone interactions as well as hydrogen bonds with the O-GlcNAc residue, which bound in a similar orientation as the O-phosphate group of previously solved phosphopeptide/14-3-3 complexes. These structural data provided biophysical insights into the O-GlcNAc-selective recognition by 14-3-3 to advance the investigation of their role as reader proteins in O-GlcNAc signaling, albeit the natural O-GlcNAc binding partners of 14-3-3 proteins are still unknown. This elegant biochemical approach together with complementary structural studies is expected to contribute to the identification of other candidate O-GlcNAc readers and to provide the structural basis for the recognition of O-GlcNAc by human proteins, setting the stage for further functional studies into the molecular mechanisms of O-GlcNAc signaling.
3.4. Chemoenzymatic approaches for the detection and study of O-GlcNAcylated proteins
The classical chemoenzymatic strategy developed by Hart and coworkers123 to detect O-GlcNAc modification in cells relied on the enzyme β-1,4-galactosyltransferase (GalT), which transferred a tritium labelled [3H]-galactose to the C4-hydroxyl of terminal O-GlcNAc residues in glycoproteins using UDP-[3H]-galactose as a donor. Because this method requires handling of radioactive materials and long exposure times not suitable for routine application, an improved approach was implemented by the Hsieh-Wilson lab leveraging the altered substrate tolerance of a GalT mutant (GalTY289L)124 that features a larger active site able to accommodate C2-modified galactose analogues. This engineered mutant was exploited to catalyse the transfer of 2-keto-Gal residues from the corresponding unnatural sugar-nucleotide to terminal O-GlcNAc residues without compromising enzyme specificity and catalytic efficiency125 (Fig. 6a). The unnatural ketone group was used as a chemical handle for labelling the tagged O-GlcNAc-modified proteins with aminooxy-functionalised biotin probes via oxime ligation, permitting subsequent detection of the target proteins by chemiluminescence. This biotin tagging strategy was applied to enrich O-GlcNAcylated proteins from cell lysates for further proteomics analysis, leading to the high-throughput identification of a number of functionally diverse O-GlcNAc proteins from the mammalian brain, which suggests a key role for this modification in modulating neuronal function.126 The chemoenzymatic approach was subsequently expanded to ketone-biotin labelling of glycoproteins from two cell states (e.g., stimulated versus unstimulated) followed by an additional isotope tagging step after tryptic digestion involving incorporation of light/heavy methyl groups into the amines of the peptide via reductive amination, enabling relative quantitative MS-based analysis.127 This integrated method, named quantitative isotopic and chemoenzymatic tagging (QUIC-Tag), was applied to identify O-GlcNAcylated proteins undergoing changes in glycosylation in response to cellular stimuli, and in combination with electron-transfer dissociation (ETD) fragmentation permitted site mapping of those dynamic O-GlcNAc modifications in the brain.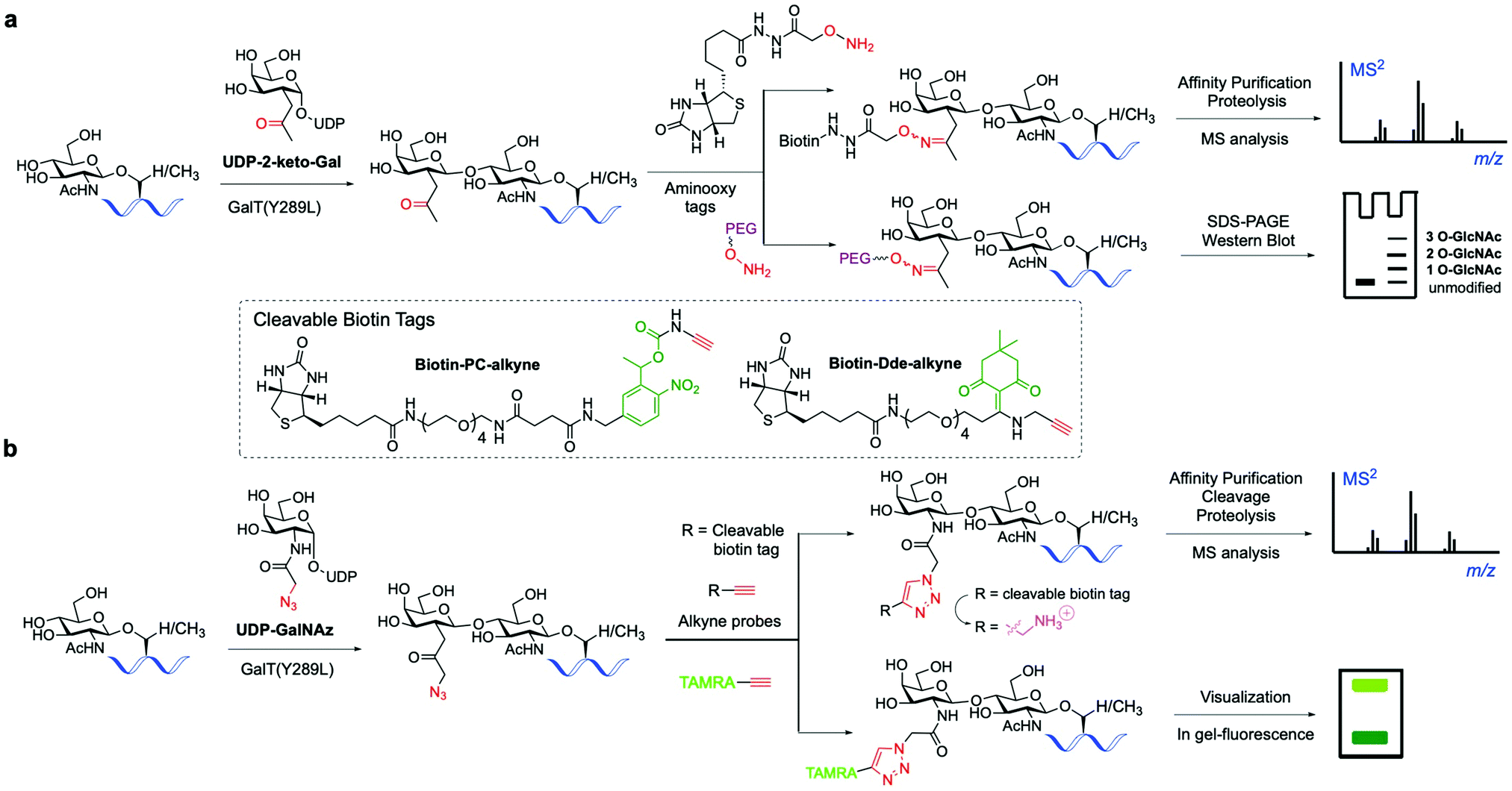 | ||
| Fig. 6 Chemo-enzymatic labelling of endogenous O-GlcNAcylated proteins by enzymatic modification with unnatural UDP-Gal analogues ((a) keto or (b) azide-functionalised) using a mutant GalT(Y289L). Subsequent chemical probing with (cleavable) biotin, fluorescent or PEG mass tags using (a) oxime or (b) click-chemistry for identification of the O-GlcNAcylated proteins.136 | ||
This study revealed the reversible and differential modulation of O-GlcNAcylation in neuronal cells and in vivo as a key regulatory PTM analogous to phosphorylation.127 Another addition to this approach is the selective installation of molecularly defined, aminooxy functionalised PEG tags (“mass tags”), which cause shifts in protein migration on SDS-PAGE that can be detected by immunoblotting (Fig. 6a). This technique enables monitoring of the glycosylation stoichiometry and state (i.e., mono-, di-, etc., O-GlcNAcylated) as well as dynamics of endogenous O-GlcNAc proteins.128 This chemoenzymatic tagging strategy in combination with biochemical and neurobiological approaches was then applied to characterise the roles of O-GlcNAc in the CREB transcription factor as a regulator of important neuronal functions and long-term memory.129
Hsieh-Wilson and co-workers further extended the scope of this probing tool by using an alternative, azide-containing galactosamine donor (UDP-GalNAz) that was well tolerated by GalT(Y289L) for transfer of the unnatural GalNAz residue to O-GlcNAcylated proteins (Fig. 6b). This modified strategy enabled sensitive, faster and more specific CuAAC labelling with alkyne-functionalised biotin and fluorescent probes in comparison with the corresponding aminooxy tags and was utilised for direct in-gel fluorescence detection and proteomic identification of O-GlcNAc-glycosylated proteins within cells.130
This advanced chemoenzymatic strategy was then applied to detect and validate O-GlcNAcylation in a number of important regulatory proteins in the nervous system and in metabolism, including phosphofructokinase 1 (PFK1), which was determined to be dynamically glycosylated at Ser529 in response to hypoxia, acting as a key metabolic regulator of glucose metabolism associated with cancer cell growth in vivo.22 UDP-GalNAz has also been used in combination with SPAAC for optimising the chemoenzymatic mass tagging protocol mentioned above for improved quantification of O-GlcNAc stoichiometry and state by Western blotting.131 To further enhance the detection capability of the method, a UV-cleavable biotin affinity probe (Biotin-PC-alkyne) was developed to improve the release of tagged peptides from streptavidin, increasing the analytical sensitivity for more efficient identification of O-GlcNAcylated proteins (Fig. 6b).132 Moreover, the moiety retained upon cleavage yielded a positively charged amino group tag, providing higher overall peptide charge and improved ionisation by ETD for precise O-GlcNAc site mapping.133,134 Despite its use in various applications, cleavage of the photolinker upon UV irradiation was not complete, hampering the quantitative release of O-GlcNAcylated proteins for proteomics analysis. Thus, a chemically cleavable biotin tag was developed containing the well-known 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (Dde) moiety, extensively used as a protecting group for lysine in peptide synthesis.135 The corresponding Dde-biotin probe showed stability to harsh denaturing conditions and could be completely cleaved under mild chemical conditions, providing a relatively small, cationic fragment that facilitated peptide sequencing by ETD-MS. This combined chemoenzymatic tagging and chemical cleavage approach enabled an improved labelling efficiency and recovery of O-GlcNAcylated proteins as well as extensive identification of O-GlcNAc sites on α-crystallin and OGT.135
Another chemoenzymatic histological method was developed by Wu and coworkers in which GalT(Y289L) accepted an alkyne-bearing GalNAc donor, UDP-N-pentynylgalactosamine (UDP-GalNAl), and transferred the modified GalNAl residue to O-GlcNAcylated proteins, albeit less efficiently than the corresponding azide analogue.137 Subsequent CuAAC with an azide-functionalised biotin probe accelerated by the Cu(I)-ligand BTTP enabled a faster and more efficient O-GlcNAc labelling in histological samples following neutravidin-HRP imaging. Because GalT(Y289L) has been reported to accept terminal GlcNAc residues in N-glycans,138 pretreatment of the tissues with PNGaseF before the chemoenzymatic tagging was necessary to increase specificity. Application of this approach led to high-resolution visual identification of distinct O-GlcNAcylation patterns and levels within various murine organs as well as normal versus diseased human histological samples, highlighting the power of this method to study O-GlcNAc biology in physiopathological processes. An additional chemoenzymatic method was also developed that combined recombinant glycosyltransferases (B3GALNT2 and OGT) and bioorthogonal chemical probing for the detection and visualisation of O-GlcNAcylation as well as of potential sites for O-GlcNAc modification in biological samples.139 B3GALNT2 is a β-1,3-N-acetylgalactosaminyltransferase that recognises terminal β-GlcNAc residues and was found to accept UDP-GalNAz and transfer the clickable GalNAz moiety with good selectivity to O-GlcNAcylated proteins (i.e. closed sites), which were detected via SPAAC with an alkyne-biotin probe and subsequently visualised with streptavidin-conjugated fluorophores. In this study, open sites (defined as potential sites for O-GlcNAcylation) were also detected by using OGT in the presence of UDP-GlcNAz followed by click-chemistry probing of the resulting O-GlcNAzylated protein. This approach for assessing open and closed O-GlcNAc sites was applied in vitro and in cells, revealing 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3) as a previously unidentified target protein for O-GlcNAcylation, which could be a novel mechanism to regulate cellular metabolism in connection with cancer. Most recently, another chemoenzymatic strategy has been reported that uses two enzymatic transformations with easily accessible enzymes to tag O-GlcNAcylated proteins, followed by hydrazide chemistry for their enrichment.140 First, wild-type GalT tags the O-GlcNAcylated proteins with natural β-1-4-galactose, whose C6-hydroxyl group is then selectively oxidised by galactose oxidase (GAO) to give the corresponding C6-aldehyde. This aldehyde serves as a chemical handle for capturing the galactosylated O-GlcNAcylated proteins using hydrazide resins, and elution with methoxyamine via oxime-bond formation then provides the corresponding released proteins for site-specific analysis of O-GlcNAcylation by LC-MS/MS. Despite the accessibility of the method and the comparable results to the original chemoenzymatic strategy, the relative promiscuity of GalT and GAO led to the identification of O-glucosylated and TF [Gal-(β-1-3)GalNAc-(α-O-Ser/Thr)]-bearing proteins, indicating the non-specificity of the technique.
These chemoenzymatic approaches and their extensions have been extensively used in probing O-GlcNAcylation at the cellular level with a good degree of success, especially in denatured protein samples, although less in native, fully folded proteins in vivo. However, the use of engineered enzymes and unnatural substrates may lead to specificity and reactivity issues inherent to the enzymes’ substrate preferences and recognition, while can also alter cellular metabolic pathways, potentially providing incomplete, inaccurate and/or non-physiologically relevant information. Therefore, there is still room for novel and improved chemoenzymatic tools for a more efficient identification and site-mapping of O-GlcNAcylated proteins to decipher their functional roles and molecular mechanisms.
3.5. Bioorthogonal ligation strategies for probing O-GlcNAcylation
Amongst the various two-step labelling strategies for detecting the O-GlcNAc modification, the first step involves the incorporation of some kind of chemical reporter group into the glucosamine residue. This unnatural chemical functionality should be stable but sufficiently reactive and display a good degree of bioorthogonality under physiological conditions to enable fast chemoselective ligation with a suitably designed molecular probe for subsequent detection of the tagged GlcNAc derivative. In studies probing O-GlcNAcylation, several reporter groups such as azide,94 ketone125,126 alkyne,102 isonitrile,141 or alkene114,142 have been used as chemical handles to label O-GlcNAc modified proteins by subsequent reaction with different fluorescent and affinity probes using a variety of bioorthogonal ligation chemistries (Fig. 7).143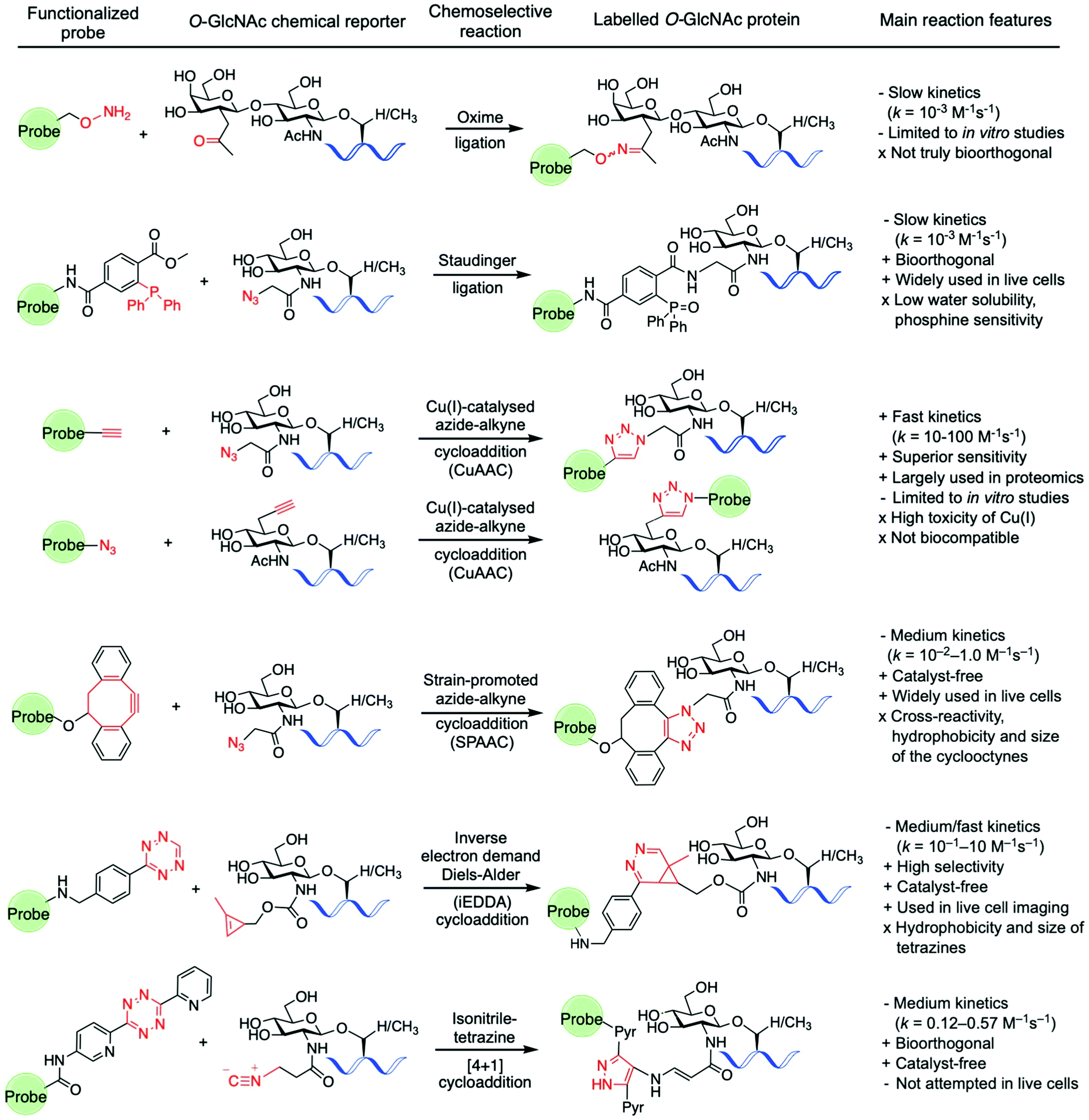 | ||
| Fig. 7 Bioorthogonal ligation reactions employed for probing O-GlcNAcylation: oxime ligation, Staudinger ligation, “click” chemistry (CuAAC and SPAAC), iEDDA reaction, and isonitrile–tetrazine ligation. | ||
For instance, as discussed above, Hsieh-Wilson and coworkers introduced a ketone functionality into the N-acetyl group of the galactosamine residue that gets enzymatically transferred to O-GlcNAcylated proteins, and then used an aminooxy-biotin affinity probe to detect the O-GlcNAc modification via oxime ligation under mild conditions.125 However, oxime ligation suffers from two major issues: firstly, the reaction is not truly bioorthogonal, as many biomolecules present ketone groups that can react non-specifically with the aminooxy affinity probe; secondly, the reaction generally proceeds with relatively slow kinetics (k = 10−3 M−1 s−1 at neutral pH),144 and slightly acidic pH values are required for efficient ligation. Despite several examples demonstrating applications of this oxime ligation based methodology in studying O-GlcNAcylation,126,128,129 these drawbacks may limit the further use of this reaction in the development of new strategies for detecting O-GlcNAcylation, especially in vivo. The Staudinger ligation developed by Bertozzi145,146 enables the formation of an amide bond by coupling an azide incorporated into the glycan structure with a specifically engineered phosphine probe. This ligation has been extensively used in chemical biology applications,147 including identification of O-GlcNAcylated proteins from cell extracts using phosphine-containing probes.94,96,99,148 While the Staudinger ligation has sufficient biocompatibility for in vivo applications, it suffers from several shortcomings, namely slow reaction kinetics (k = 10−3 M−1 s−1)149 as well as low water solubility and oxygen sensitivity of the phosphine reagents. Moreover, its optimisation for increased reaction rates and solubility is synthetically challenging, which led to the development of alternative bioorthogonal ligation methods.
As mentioned above, the CuAAC, commonly known as “click” reaction,150 has been widely used for tagging metabolically incorporated azide or alkyne-modified O-GlcNAc moieties with the corresponding alkyne or azide-functionalised probes, respectively, enabling identification of a number of O-GlcNAcylated proteins.100–103,109,151 Despite its superior kinetics (k ≈ 10–100 M−1 s−1) and sensitivity under physiological conditions,152 the toxicity of the Cu(I) ions in living cells compromises the biocompatibility of the reaction, precluding its application in vivo for live cell labelling. Thus, the copper-free non-toxic SPAAC was subsequently developed by the Bertozzi lab. This reaction is driven by the favourable ring-strain release of the cyclic alkyne and occurs readily in living cells without the need for copper catalysis.152,153 A number of structurally varied strained cyclooctyne scaffolds with different properties have been developed and further applied in bioconjugation.154,155 Among them, the aliphatic derivative DIFO and the dibenzoannulated cyclooctyne DBCO, also known as DIBAC (dibenzoazacyclooctyne), have been explored as affinity tags for labelling azide-modified glycan moieties in the detection of O-GlcNAcylated proteins.103,131,156–158 These SPAAC reagents exhibit good reactivity, with reaction rates generally lying in between those of the Staudinger ligation and the CuAAC reaction (k = 10−2–1.0 M−1 s−1).143
However, the relatively large size and hydrophobicity of the cyclooctynes together with their cross-reactivity with cellular free-thiol nucleophiles159,160 limit the use of SPAAC for detecting intracellular O-GlcNAcylation events. Comparative studies of these “click-chemistry” methods have been performed using ManNAz and GlcNAz for live cell labelling of cell surface glycoproteins152 and for O-GlcNAc proteomics analysis.157 While both ligations can efficiently label azido-modified glycoproteins, CuAAC emerged as the method of choice for proteomics due to its superior sensitivity, enabling more accurate and efficient protein identification compared to SPAAC. However, the toxicity of the copper catalyst makes it unsuitable for in vivo applications, highlighting the need for labelling strategies that are both highly sensitive and biocompatible.
For these reasons, the recent focus has shifted to the development of alternative, faster and non-toxic bioorthogonal reactions involving other chemical functionalities. The iEDDA reaction is a [4+2] cycloaddition between an electron-rich alkene and an electron-poor 1,2,4,5-tetrazine161 that has emerged as a powerful bioorthogonal tool in chemical biology due to its excellent features, fulfilling most of the criteria for bioorthogonality (e.g., biocompatible, catalyst-free, rapid, selective).162 Thus, the high selectivity and extremely fast kinetics of the reaction (with constant rates of up to 106 M−1 s−1) without the need for catalyst, together with the inherent stability of both alkene and tetrazine counterparts even in the presence of thiols, make the iEDDA a robust bioorthogonal ligation method for in vivo applications. Exploiting this chemistry, visualisation of protein specific O-GlcNAcylation could be achieved for the first time in living cells by using a metabolic labelling approach, as described above.114 Following this strategy, a methylcyclopropene-tagged GlcNAc derivative (Ac4GlcNCyoc) was metabolically incorporated into cells (and particularly into EGFP-fused, intracellular target proteins), and then labelled using fluorescent tetrazine probes in a specific iEDDA reaction in live cells. Notably, the kinetics of the iEDDA (which can range from 1 up to 106 M−1 s−1) is greatly influenced by steric and stereoelectronic effects, including the ring strain of the alkene component and the nature of the substituents on the tetrazine ring. As such, structural optimisation of both coupling partners is possible and can lead to higher reactivity and bioorthogonality for live cell applications (see ref. 162 for an excellent, extensive review on iEDDA).
Another bioorthogonal ligation involving tetrazines is the isonitrile–tetrazine “click” reaction, in which an isonitrile group undergoes a highly specific [4+1] cycloaddition with a tetrazine with a moderate reaction rate (dipyridyltetrazines reacted with primary and tertiary isonitriles with rate constants of 0.12 and 0.57 M−1 s−1, respectively).141 In one application of this ligation, unnatural ManNAc, GalNAc, and GlcNAc analogues modified at the N-acetyl substituent with the isonitrile functionality were metabolically incorporated into cell-surface glycans and specifically labelled with a tetrazin-biotin tag using this chemistry.141 Despite the promising features of this method (small size, stability, negligible toxicity and reactivity of the isonitrile moiety with tetrazine), the resulting cycloaddition conjugate is prone to degradation in aqueous media, with half-lives of 16 hours for primary isonitriles, which could compromise the overall efficiency and biological applications of this ligation. Another particular type of cycloaddition reaction that has been used in chemical biology to label alkene-modified biomolecules is the photo-induced 1,3-dipolar cycloaddition between tetrazoles and alkenes,163 also known as “photoclick chemistry”. In this method, short UV photoirradiation of 2,5-diaryltetrazoles results in a fast cycloreversion reaction with the release of N2 and in situ generation of a nitrile imine dipole, which then reacts with alkene dipolarophiles in a slower, concerted manner to provide pyrazoline cycloadducts that are themselves fluorescent.164 This reaction offers a rapid chemical ligation strategy with constant rates ranging from 10−3 to 103 M−1 s−1 depending on the substituents and the nature of the counterparts, with strained alkenes providing higher kinetics.
Using metabolic and chemoenzymatic approaches, alkene-bearing residues were incorporated into proteins165,166 and mRNA167 and then photoreacted with tetrazole-functionalised probes for biomolecular labelling. Despite the attractive features (e.g., speed, fluorogenic) and promising biological applications of this ligation, including selective dual imaging in combination with iEDDA, the generated nitrile imine has been shown to also react with a range of biological nucleophiles such as thiols, amines and acids.168 These side reactions precluded its further application as a (non) true bioorthogonal reaction and highlight the continued search for new and/or improved truly bioorthogonal ligations169 that can be mutually compatible for labelling and probing complex cellular processes, such as O-GlcNAcylation.
3.6. Methods to study discrete O-GlcNAcylation on proteins with site-specificity
Despite the useful approaches developed to investigate global O-GlcNAc glycosylation,136,170 deciphering site-specific O-GlcNAcylation remains a challenging task. In the case of PFK1, the corresponding flag-tagged protein was expressed in combination with OGT to dissect O-GlcNAc sites.22 After immunoprecipitation and digestion, O-GlcNAcylated peptides were enriched by WGA lectin affinity chromatography and then analysed by ETD-MS/MS, identifying the Ser529 glycosylation site. Naturally, site-specific mutagenesis of Ser/Thr residues to alanine represents the most commonly employed method to probe O-GlcNAcylation and its location. For instance, S529A mutation in PFK1 was shown to block O-GlcNAc glycosylation, whereas mutation of Thr527 had no impact, confirming Ser529 as the specific, primary site for O-GlcNAc. In another case, modification of Ser149 in the tumour suppressor protein p53 abolished O-GlcNAcylation but enabled Thr155 phosphorylation, highlighting the crosstalk between both PTMs at proximal sites.171 Despite its wide use, this type of loss-of-function mutation requires knowledge of the precise glycosite for each target protein and prevents analysis of competing PTMs on the same site. Thus, in recent years, a number of strategies for selective GlcNAc installation on proteins of interest have been developed, enabling controlled access to stoichiometric, protein-targeted GlcNAcylation for precise functional studies.The van Aalten lab has described a practical approach for site-specific, genetic incorporation of a hydrolytically stable thio-GlcNAc analogue into target proteins in vitro and in live cells, enabling precise studies of the effects of this modification within a living system.118 This method employs Ser/Thr to Cys mutagenesis and exploits the ability of OGT to transfer GlcNAc to cysteine residues,117 enabling OGA-resistant GlcNAcylation mimicry with high stoichiometry at selected S-glycosites. Combined with CRISPR-Cas9 technology, a genetically encoded OGA (S405C) was engineered in mouse embryonic stem cells and subsequent quantitative OGT-catalysed S-GlcNAcylation provided a hyper-S-GlcNAcylated OGA mutant with retained hydrolase activity but reduced half-life,118 which highlights the influence of O-GlcNAcylation on protein stability. Withers and coworkers developed another method for the introduction of S-GlcNAc into target proteins (e.g., tau and synuclein) in vitro by using an engineered GH20 hexoaminidase as a thioglycoligase able to install GlcNAc in cysteine residues.172 More recently, Ramirez et al. have reported a strategy for controlled, proximity-induced O-GlcNAcylation of single proteins in living cells.173 This approach leverages an OGT-fused nanobody recognising tagged or endogenous target proteins and capable of directing OGT to transfer O-GlcNAc selectively to the desired substrate, as demonstrated for α-synuclein. Despite its potential to increase O-GlcNAc levels in specific proteins of interest, overexpression of the nanobody-OGT fusion construct may impact physiological protein interactions as well as global O-GlcNAcylation, which requires appropriate control experiments to address these concerns.
Alternatively, chemical synthetic strategies have been developed to generate GlcNAc modified glycoproteins in vitro in which the GlcNAc residue can be introduced stoichiometrically at the desired position, providing practical access to homogeneously glycosylated proteins for probing site-specific GlcNAcylation (Fig. 8). One methodology established by Davis and coworkers relies on the chemical modification of endogenous or genetically introduced cysteine residues on native proteins to install GlcNAc moieties directly174 or, especially, via previous cysteine conversion to dehydroalanine (Dha)175 by an alkylating agent under denaturing conditions. Conjugate addition of thio-GlcNAc to the Dha-tagged unfolded protein followed by refolding yields the site-specifically modified target protein (Fig. 8a). This method has been employed to probe the functional consequences of histone GlcNAc modification by creating synthetic, homogeneously GlcNAcylated histones and nucleosomes bearing S-GlcNAc at defined positions. While H2A-Thr101 GlcNAcylation led to destabilisation of the H2A/H2B dimers and reduced nucleosome stability, promoting an open chromatin structure,176 H2B-Ser112 GlcNAc modification did not affect nucleosome assembly but impacted the interacting nucleosome–protein partners, modulating binding of the FACT chromatin remodelling complex as assessed by proteomics analysis.177 Expanding this Dha-based protein chemistry using carbon–carbon bond forming chemical mutagenesis, a variety of synthetic O-GlcNAcylated glycoproteins were generated as unnatural O-GlcNAc-homohomo-Ser mimics that could be recognised and cleaved by human OGA.178 This approach, although powerful, presents some limitations. These include the requirement for proteins with few native cysteines to achieve selectivity, and the resulting racemisation of the amino acid α-carbon that leads to a mixture of D-/L-stereochemistry at the site of modification, which might confound biological interpretation of the relevant functional studies.
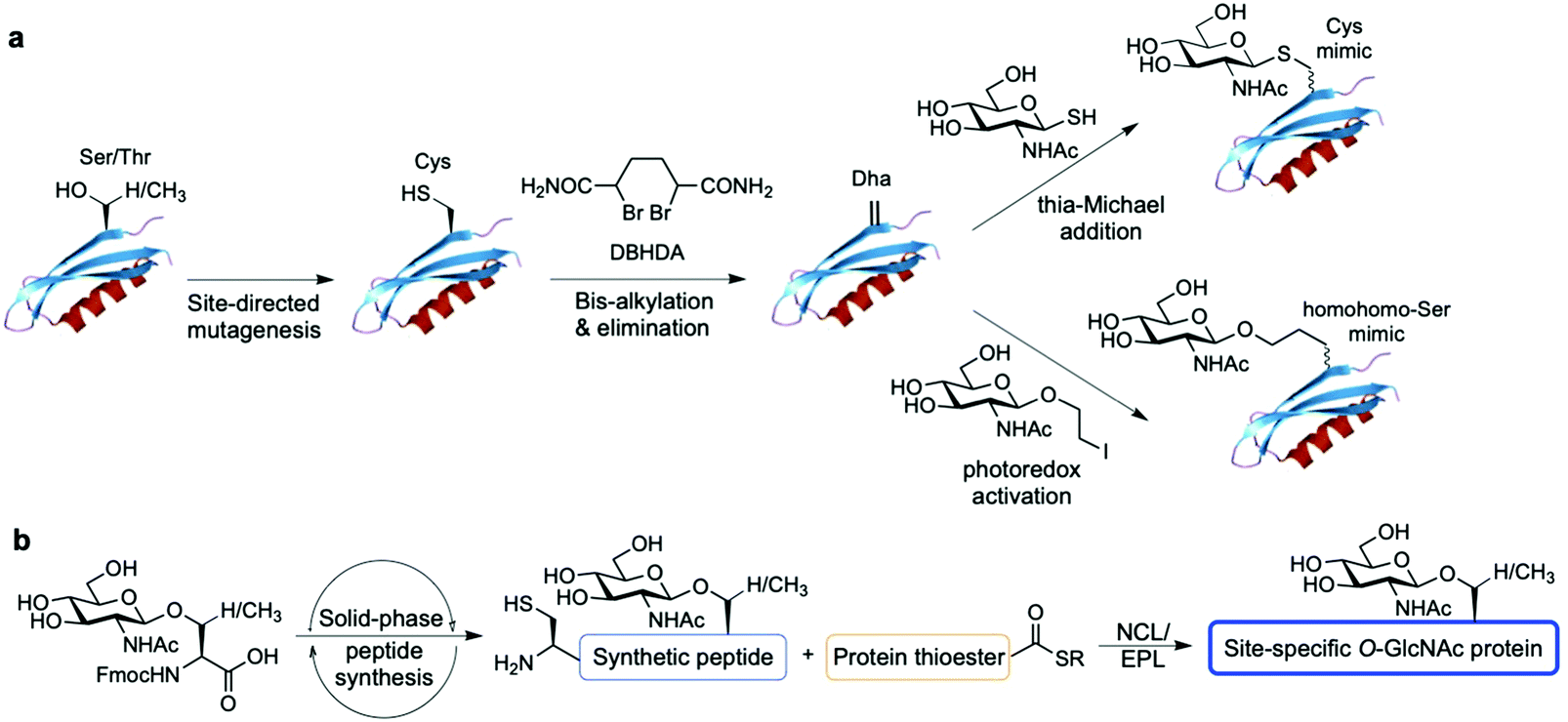 | ||
| Fig. 8 Chemical methods for site-specific installation of O-GlcNAcylation on target proteins: (a) chemical protein modification (post-translational mutagenesis) via the tag-and-modify approach using dehydroalanine (Dha) and GlcNAc mimics. (b) Synthetic glycopeptide and protein chemistry for NCL/EPL-enabled O-GlcNAc glycoprotein (semi)synthesis. | ||
Synthetic protein chemistry using native chemical ligation (NCL)-based strategies179 including total chemical synthesis and semisynthesis via expressed protein ligation (EPL)180 is another promising approach to produce well-defined O-GlcNAcylated proteins with full chemical control, enabling pure (site) probing of O-GlcNAc glycosylation (Fig. 8b). These NCL methods rely on an N-terminal Cys residue of a peptide fragment and a thioester functionality at the C-terminus of another fragment, which undergo transthioesterification and subsequent S–N acyl transfer to provide the amide-linked protein. These polypeptides can be chemically synthesised by solid phase peptide synthesis (SPPS) bearing the O-GlcNAc sugar at the desired position as the corresponding O-GlcNAcylated Ser/Thr motif, and can also be obtained by recombinant expression, most commonly as an intein-fused protein thioester.181 A number of targets incorporating site-specific PTMs have been accessed using NCL/EPL,182 although only a few include O-GlcNAc modified proteins, e.g., histone H2B,183 tau,184 α-synuclein,185–187 and three small heat shock proteins (sHSPs) (i.e., HSP27, αA-crystallin and αB-crystallin).188 Combining protein semisynthesis and biochemical experiments, Pratt and coworkers investigated the functional effects of site-specific α-synuclein O-GlcNAcylation, showing that the modification reduced protein aggregation and toxicity in vitro.185–187 Most recently, following a similar multidisciplinary approach the same group probed the impact of O-GlcNAc on the semisynthetic sHSPs mentioned above and found that glycosylation improves the chaperone activity of these proteins against amyloid formation.188 Furthermore, the non-hydrolysable S-GlcNAcylated casein kinase II (CKII) (at native Ser347)189 and α-synuclein (at native Ser87)190 have also been prepared by chemical ligation, highlighting S-GlcNAc as an enzymatically stable, suitable structural mimic of O-GlcNAc, as shown by biological studies and two-dimensional nuclear magnetic resonance (NMR) combined with computational modelling.190
Understandably, these synthetic targets can be challenging, especially those glycosylated at a position more than 50 residues from the N- or C-terminus, and require the presence of cysteine sites that otherwise need to be engineered for the ligation step and subsequently converted to the native residue via desulphurisation. Nonetheless, continuous methodological advances in chemical protein synthesis191,192 are enabling one to take full advantage of the potential of these ligation methods, which are uniquely suited to provide access to site-specifically and stoichiometrically modified O-GlcNAcylated proteins. These chemically pure glycoproteins can then be utilised for in vitro biochemical and structural biology studies to probe structure–function correlations and mechanisms of O-GlcNAcylation at the molecular level.
4. Molecular mechanisms of the role of O-GlcNAc in protein structure, function and interactions.
As explained above, the O-GlcNAc modification of nuclear and cytoplasmic proteins by OGT is strikingly different from other types of N- and O-linked glycosylation, including the O-GlcNAcylation of the extracellular domain of Notch receptors193 by another enzyme (EGF-domain O-GlcNAc transferase, EOGT),194 which involves further elongation with additional sugars to provide complex glycan structures on cell-surface proteins. Given its fast cycling and highly dynamic nature, the intracellular O-GlcNAc glycosylation shares close resemblance to protein phosphorylation, and there is an extensive crosstalk between them that is reflected in the O-GlcNAc signalling network.195 Despite their well-known regulatory interplay, the molecular mechanisms whereby O-GlcNAcylation and phosphorylation modulate the secondary structure of the underlying protein remain to be elucidated.8 Thus, in this section, we will highlight the molecular/structural role of O-GlcNAc in the conformation and stabilisation of O-GlcNAcylated peptides as well as its influence on protein structure and function. Notably, recent progress in synthetic chemistry and NMR spectroscopy in combination with computational methods has enabled access to site-specifically modified O-GlcNAc glycopeptides to gain molecular understanding of the local structure, dynamic features, and physical properties induced by O-GlcNAcylation. Finally, we will discuss representative examples of O-GlcNAc-mediated protein interactions, and provide insights into the molecular recognition by their binding partners.4.1. Conformation and structure of synthetic O-GlcNAc glycopeptides and glycoproteins
O-GlcNAcylation and O-phosphorylation are often present on loops and intrinsically disordered (ID) regions,196 with more than 70% of the nuclear O-GlcNAc being estimated to appear in ID domains. O-GlcNAc is known to crosstalk with phosphorylation to modulate gene transcription by dynamic modification of the carboxy terminal domain (CTD) of RNA polymerase II (Pol II).197,198 The CTD consists of variable tandem heptad repeats (YSPTSPS) that are reciprocally O-GlcNAcylated and phosphorylated at Ser2 and Ser5, which affects the transcriptional activity of the enzyme. Thr4 has been mapped as an O-GlcNAc site that may inhibit Ser2 and Ser5 phosphorylation of Pol II. To study the molecular basis of the role of O-GlcNAcylation in protein structure, Wong and coworkers performed pioneering studies investigating the effect of O-GlcNAc on the conformational preferences in aqueous solution of synthetic 10-mer model peptides containing the CTD heptad repeat unit.199 By combining NMR spectroscopy and computational calculations using NMR-derived constraints, they showed that O-GlcNAc glycosylation on the Thr4 of the native, randomly coiled peptide induced the formation of a β-turn structure with the carbohydrate lying over the plane of the turn. Notably, Monte Carlo and molecular dynamics (MD) simulations provided a low-energy structure in which Ser2 and Ser5 approached each other, leading to a “conformational switch” model whereby phosphate-mediated charge repulsion and glycosylation-promoted turn formation would act in a reciprocally exclusive manner. This glycan-induced conformational change was also observed in further structural studies by the same group with a synthetic octapeptide fragment of a mucin domain, in which the attachment of O-GlcNAc stabilised a turn-like structure near the glycosylation site, highlighting the conformational interplay between the carbohydrate and the peptide backbone.200 Moreover, to provide a molecular understanding of the function of glycosylated Pol II in transcription, Lu et al. investigated the process of dynamic O-GlcNAcylation in the context of full-length CTD, discovering a distributive mechanism relying on multiple binding events as an efficient mode of transcriptional regulation in response to fluctuations in the metabolic cellular status.201Another example of the complex interplay between phosphorylation and O-GlcNAcylation in ID regions is the N-terminal ID domain of murine estrogen receptor β (mER-β), where the Ser16 residue can be mutually modified by either O-phosphate or O-GlcNAc, modulating the stabilisation and activity of the protein.202 To elucidate the molecular mechanisms of the roles of both PTMs in regulating the bioactivity of mER-β, Li and coworkers studied the impact of O-GlcNAcylation and phosphorylation on the structure in aqueous solution of synthetic, N-terminal 17-mer model peptides.203 Using NMR, circular dichroism (CD) and MD simulations, the authors observed that O-GlcNAc glycosylation induced β-turn formation around the modification site, confirming that O-GlcNAcylation promotes stable, turn-like secondary structure motifs. These results are consistent with the observed functional behaviour whereby O-GlcNAcylation stabilises mER-β while O-phosphorylation promotes its degradation. Therefore, the divergent local conformational changes of both alternative PTMs at Ser16 may directly influence the disturbance of the dynamic features of the global ID region, which might be related to their reciprocal roles in modulating mER-β function.
To investigate the molecular mechanisms by which O-GlcNAc promotes turn-like structures in the underlying peptide backbone and examine the conformational differences between serine and threonine O-GlcNAcylation, Fernández-Tejada et al. carried out the structural analysis of the simplest β-O-GlcNAc-Ser/Thr model glycopeptides in water by combining NMR and MD simulations (Fig. 9).204 In both glycoamino acid diamides, the ϕp/ψp torsion angles for the peptide backbone corresponded mainly to a PPII-like conformation (around 45%), while 20% of the conformers showed values associated to helix-like structures, with the GlcNAc N-acetyl group adopting a fixed orientation relative to the sugar moiety. Notably, compared to the more flexible β-O-GlcNAc-Ser, β-O-GlcNAc-Thr is rather rigid in solution, with its side chain restricted at a χ1 torsional angle value around 60° and its O-glycosidic linkage adopting an eclipsed conformation (ψs ≈ 120°) (Fig. 9a). This conformer avoids the steric repulsion between the Thr methyl group and the anomeric H1 proton that would be present when this ψs angle is around 180°, which is the value observed for the serine glycoamino acid. The distinct glycosidic linkage conformation of the threonine derivative enables hydrogen bond formation between the sugar hydroxymethyl group and the Thr carbonyl group and offers space to accommodate a bridging water molecule between the GlcNAc N-acetyl substituent and the Thr-nitrogen (Fig. 9b). Thus, in this simple model, specific hydrogen bonds as well as water pockets contribute to explain the different relative orientation of the β-O-GlcNAc-Ser/Thr derivatives and modulate the sugar–peptide interactions, which could provide the required presentation mode of the GlcNAc residue to interact with their biological receptors.
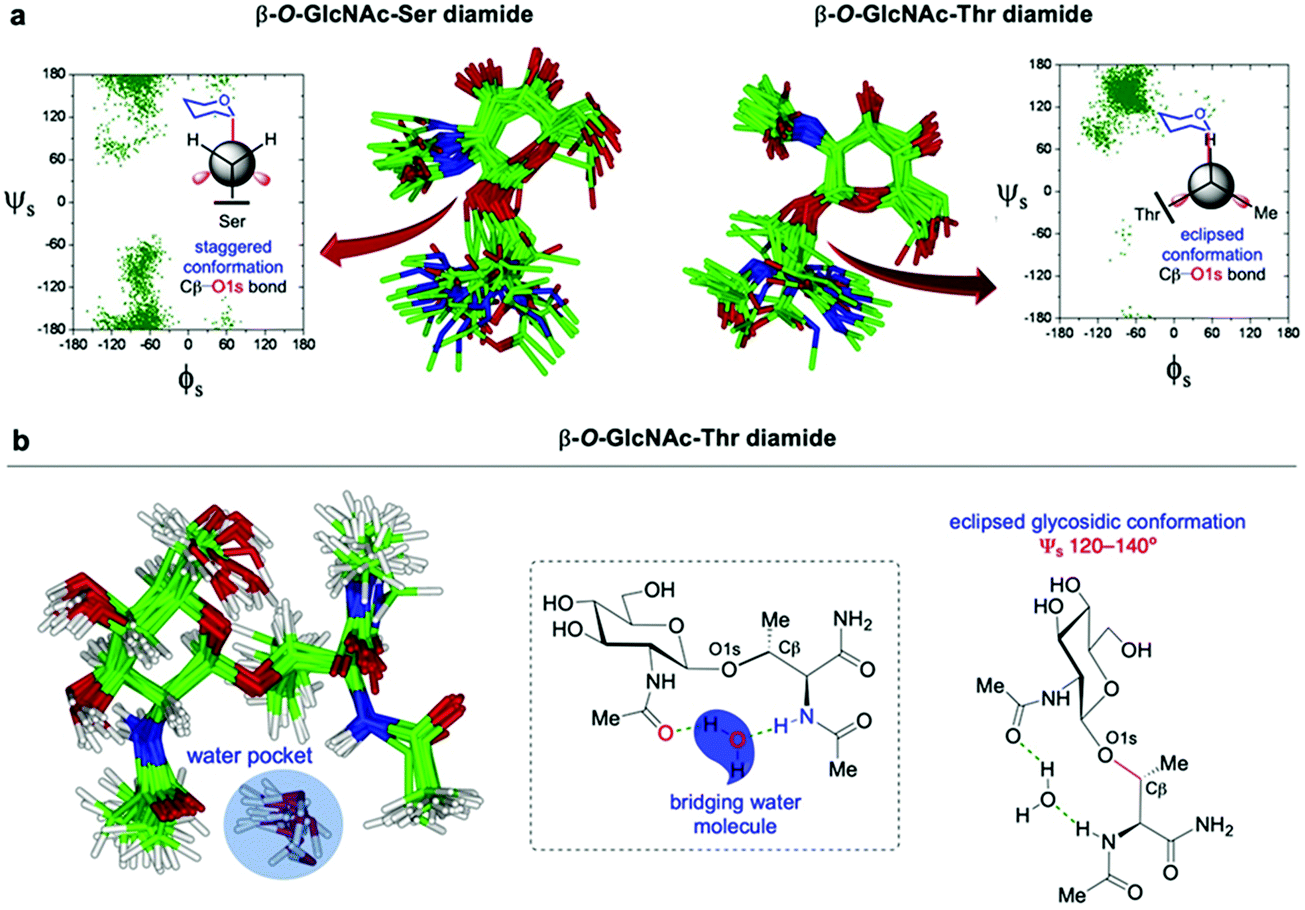 | ||
| Fig. 9 (a) Distribution of the ϕs/ψs torsion angles (glycosidic linkage) and major conformations (calculated ensembles) in solution for β-O-GlcNAc-Ser (left) and β-O-GlcNAc-Thr (right) derived from NMR-guided MD simulations. The Newman projections for the Cβ-O1s bond are also included, showing the staggered conformation (Ser derivative) and eclipsed conformation (Thr derivative). (b) Key water pocket (bridging water molecule) between the GlcNAc (N-acetyl group) and the peptide backbone (Thr-nitrogen) derived from NMR-guided MD simulations for β-O-GlcNAc-Thr due to its distinct conformational behaviour in solution. | ||
In additional studies investigating the effect of O-GlcNAcylation and phosphorylation on the conformational properties of a polypeptide, Liang et al. found that insertion of either modification in the turn region of an α-helical hairpin peptide (α-helix/turn/α-helix) slowed down the rates of β-sheet rich amyloid fibril formation by altering the backbone orientation, even though the native structure and the conformational stability of the soluble peptide were not substantially affected.205 Using another α-helical model peptide, Zondlo and coworkers showed that phosphorylation and O-GlcNAcylation at serine and threonine residues stabilised the α-helix at the N-terminus of the peptide, whereas in the internal and C-terminal regions both modifications had a destabilising effect, resulting in random coil conformations.206 Notably, the structural impact of modifications on threonine was more pronounced than that on serine residues, suggesting the potential of threonine sites as hot spots in structural modulation via protein PTMs. The same group also studied by CD and NMR the structural basis of the influence of phosphorylation and O-GlcNAcylation on the conformation of synthetic peptides derived from the proline-rich domain of tau,207 the aggregation of which leads to Alzheimer's disease. Both modifications were found to have divergent structural effects, with phosphorylation promoting conformational order and PPII formation, particularly on threonine residues, and O-GlcNAcylation inducing more subtle conformational preferences against PPII and favouring more disordered and extended conformations. These results are consistent with the observed effects of O-GlcNAc modification in opposing tau aggregation and the association between hyperphosphorylation of tau and induced conformational order leading to tau misfolding and neurofibrillary tangle formation.66,208 More recently, two systematic computational studies using mainly MD calculations have yielded critical molecular-level insights into the conformational preferences and dynamics upon phosphorylation and O-GlcNAcylation of model peptides209 and longer tau fragments.210 These simulations were generally in agreement with the related NMR data and elucidated key water-mediated and phosphate–lysine interactions underlying the differential structural effects observed for both PTMs.
NMR spectroscopy has also been used to probe the in vitro activity of OGT on recombinant tau and synthetic peptides and to assess the reciprocal relationship between phosphorylation and O-GlcNAcylation, confirming the identification of S400 as the O-GlcNAc site through a combination of bidimensional experiments and chemical shift perturbation analysis.211 Remarkably, variation of amide signals upon O-GlcNAc incorporation was only observed for adjacent residues, suggesting a limited impact of the carbohydrate moiety on the overall peptide structure of tau[393–411], which was shown by HN/Hα NOE contacts to adopt an extended conformation that was not affected by S400 glycosylation. Along the same lines, NMR studies by Vocadlo and coworkers using a C-terminal tau[353–408] fragment demonstrated that O-GlcNAc glycosylation only induced minimal local conformational and dynamic changes in the disordered peptide, without affecting its global structure.212 In the absence of major effects on tau conformation and dynamics, O-GlcNAc modification may decrease tau aggregation by enhancing its solubility or destabilising formation of fibrils or soluble aggregates. Notably, the Hackenberg group used an EPL-based semi-synthetic strategy to access a site-specifically and stoichiometrically modified S400-O-GlcNAc tau, which can serve as a useful tool to probe the molecular role of O-GlcNAcylation and its effect on tau aggregation using a chemically pure full-length variant.184 Most recently, the in vitro O-GlcNAc pattern and the molecular mechanisms of the interplay between O-GlcNAcylation and phosphorylation of recombinant tau have been investigated by NMR spectroscopy.213 In contrast to previous findings, tau was not found to be extensively O-GlcNAcylated; additionally, while phosphorylation was shown to increase direct tau glycosylation by OGT, O-GlcNAcylation did not seem to alter tau phosphorylation by kinases. Collectively, these results point to a more complex, less straightforward interplay between both PTMs, probably through indirect mechanisms that modulate the action of the enzymes regulating phosphorylation and O-GlcNAcylation in vivo. In another example, synthetic O-GlcNAc and phosphate variants of the PHF6 hexapeptide required for tau oligomerisation were used to investigate the effects of O-GlcNAcylation and phosphorylation on the aggregation properties of a native amyloid scaffold.214 Unlike the naked control peptides, both forms of modified peptides retained a random coil conformation (as assessed by CD) and aggregated less than the parent amyloid scaffold. However, in co-incubation experiments, only the glycosylated variants showed an inhibitory effect on PHF6 aggregation, probably due to interactions with the glycan residue through potential hydrogen bond formation.
Importantly, as mentioned above, the effect of O-GlcNAcylation on preventing the aggregation of proteins associated with neurodegenerative disorders is not limited to tau and Alzheimer's disease. The Pratt lab has explored the impact of site-specific O-GlcNAc modifications on the biophysical properties of α-synuclein, the Parkinson's disease counterpart of tau, using several semisynthetic O-GlcNAcylated α-synuclein variants (modified at Thr72, Thr75, Thr81, Ser87, and a triply glycosylated Thr72, Thr75 and Thr81 analogue).185–187 None of the O-GlcNAc modifications led to any significant secondary structure as shown by CD, causing no changes in the native monomeric and unfolded state of the protein in solution. Notably, O-GlcNAcylation reduced α-synuclein aggregation and toxicity in a site-specific manner, with the triply modified α-synuclein showing the strongest inhibitory effect. Furthermore, these sugar modifications, particularly at Ser87, could alter the architecture of the aggregates that formed, with site-specific differences that were consistent with the NMR structure of the α-synuclein fiber.215 These structural effects may originate from the disruption of the hydrophobic interactions required for aggregation owing to the hydrophilicity of the GlcNAc residue. A recent MD simulation study of O-GlcNAcylated synuclein proposed that O-GlcNAc modification suppresses oligomer formation by preventing intermolecular hydrogen-bonding interactions between monomers via steric effects, providing some insights into the molecular mechanism of O-GlcNAc-induced inhibition of α-synuclein oligomerisation.216 In the case of the semi-synthetic, differentially O-GlcNAcylated sHSPs prepared by Pratt and coworkers,188 the enhanced chaperone activity and aggregation-protective effect caused by the glycan were found to stem from its ability to disrupt the auto-regulatory interaction of the proteins’ IXI motif with their chaperone cleft. Using biophysical and computational modelling techniques, they showed that O-GlcNAcylation decreased the binding of the IXI sequence of HSP27 to its chaperone groove, leading to the formation of larger HSP27 oligomers and a conformational rearrangement in a more active state. This potentially results in a dynamic structure able to more easily bind to hydrophobic fragments thus preventing amyloid aggregation. Overall, these multidisciplinary studies point to a molecular mechanism for O-GlcNAc that supports an important preventive and protective role of this modification in neurodegeneration.
4.2. Synthetically engineered site-specific GlcNAcylation
The above studies highlight the utility of glycopeptide and protein chemistry to access synthetic GlcNAc chemical tools for probing the conformational effects and molecular mechanisms of site-specific O-GlcNAcylation, revealing mechanistic insights into O-GlcNAc as a potential therapeutic modification that modulates protein activity and stability and regulates the development of neurodegenerative diseases. Considering the important role of O-GlcNAc in effectively stabilising peptide and protein substrates, O-GlcNAcylated peptides have been engineered to improve the properties of therapeutic peptides and to gain a deeper understanding of their mechanisms of stabilisation. With this double purpose in mind, synthetic O-GlcNAc modified analogues of pharmacologically active peptides have been investigated via functional and structural studies. Arsequell et al. synthesised three glycosylated variants of the 17-mer N/OFQ peptide, which is the native ligand of the druggable, pain-related nociceptin opioid receptor, and examined their biological activities and conformational features by NMR and CD.217 While α-O-GalNAc glycosylation at Ser10 of nociceptin led to a slight increase in binding affinity, the corresponding β-O-GlcNAc glycopeptide showed similar affinity to the unglycosylated compound, showing a more flexible behaviour with more than one structural motif and less α-helix proportion.Based on the ability of O-GlcNAcylation to remotely prevent protein cleavage, presumably due to glycan masking of the cleavage sites,95,106,218 Pratt and coworkers have exploited this stabilisation effect by installing artificial O-GlcNAc modifications on two clinically relevant peptides, glucagon-like peptide 1 (GLP-1) and a parathyroid hormone (PTH) fragment (Fig. 10).219 The O-GlcNAcylated variants showed improved in vivo activity and stability, which were further increased by incorporation of unnatural amino acids in the GLP-1 analogues, suggesting the potential of such combined chemical modification approach to synergistically enhance the properties and clinical efficacy of therapeutic peptides. By using CD, O-GlcNAc glycosylation was found to have little influence on the peptide α-helical secondary structure, except two PTH analogues that were more unstructured, especially the most potent one. Furthermore, conformational modelling of the corresponding ligand–receptor complexes provided molecular insights into how O-GlcNAcylation affected GLP-1 and PTH binding as well as the potential interactions contributing to peptide activity, leading to the proposed mechanistic explanation of the biased agonism observed for a triply functionalised backbone- and O-GlcNAc-modified GLP-1 analogue.
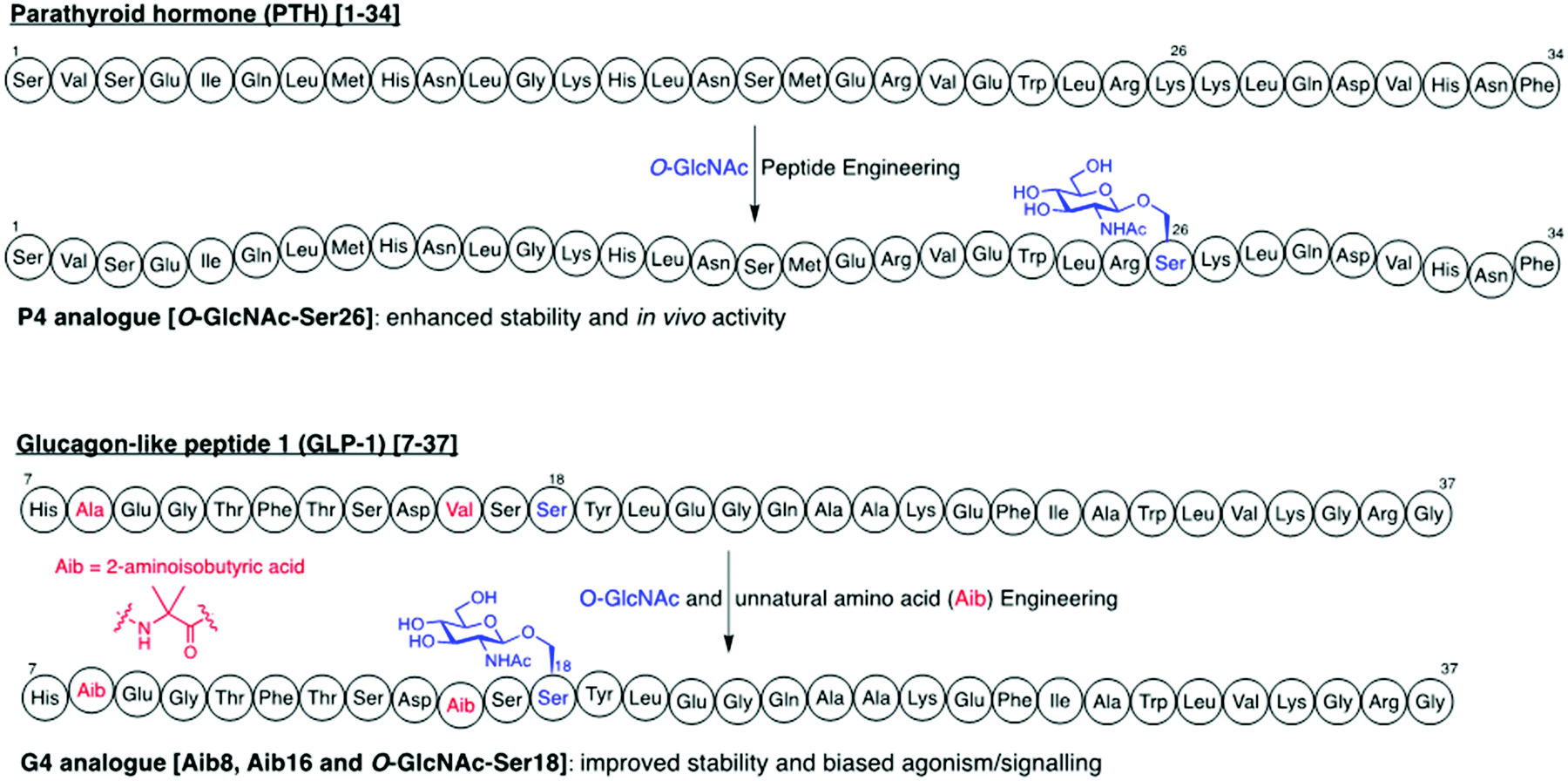 | ||
| Fig. 10 Synthetic engineering of therapeutic peptides PTH and GLP-1 via artificial O-GlcNAc installation. | ||
Overall, these results suggest that in addition to potential polar contacts with the peptide itself, O-GlcNAc may be involved in direct receptor interactions, leading to changes in downstream signalling pathways. Despite these comprehensive data, the exact molecular mechanisms of O-GlcNAcylation-promoted stabilisation are not yet fully understood and could be associated to favoured GlcNAc-driven protein interactions (see Section 4.3) instead of direct blocking of protease recognition.
While the use of synthetic, homogeneously O-GlcNAcylated peptides and proteins has enabled significant advances in probing functional and molecular roles of specific O-GlcNAc sites in vitro, extension of this chemical strategy to an intracellular setting or in vivo models is limited by the hydrolytic instability of the O-GlcNAc linkage against OGA, which may compromise site-specific modification efforts. To overcome this issue, the non-hydrolysable thioglycoside analogue of O-GlcNAc, S-linked GlcNAc, was developed220 and incorporated in a number of synthetic S-GlcNAcylated peptides and proteins produced by EPL, including CKIIα189 and α-synuclein.190 In the first example, Cole and coworkers used protein semisynthesis and prepared a range of site-specifically phospho- and S-GlcNAc-modified CKIIα variants for cellular studies of the interplay between both PTMs in CKII regulation. While phosphorylation of Thr344 was shown to improve CKIIα cellular stability by enhancing its interaction with the adaptor protein Pin1, S-GlcNAcylation at Ser347 was found to reduce Thr344 endogenous phosphorylation with concomitant CKIIα destabilisation, and also to influence CKII kinase function, presumably by altering protein substrate selectivity via potential interactions with CKII substrates through the glycan moiety. Subsequently, the Pratt lab demonstrated the in vitro stability of this modification against hOGA removal by using an S-GlcNAcylated peptide analogue from the N-terminus of mER-β and an α-synuclein variant with S-GlcNAcylation at Ser87.190 Moreover, the authors applied a combination of NMR experiments and computational modelling to show that S-GlcNAc induced similar conformational effects on the mER-β peptide secondary structure in comparison to the native modification,203 albeit CD analysis suggested greater β-turn formation. Finally, site-specific S-GlcNAcylation was shown to cause no impact on the solution secondary structure of α-synuclein and had identical effects on protein aggregation and membrane binding compared to the corresponding O-GlcNAcylated protein,190 highlighting S-GlcNAc as an enzymatically stable, good structural and functional mimic of O-GlcNAc for in vivo applications.
The above studies have demonstrated the utility of synthetic glycopeptides and glycoproteins for interrogating the molecular mechanisms of O-GlcNAcylation and its effects on the biophysical and biochemical properties of the site-specifically modified proteins themselves. Notably, O-GlcNAc can also exert its structural and functional role by regulating the ability of its protein substrates to interact with their binding partners.
4.3. O-GlcNAc-mediated protein interactions
Chemical biology approaches have been applied for probing the indirect implications of dynamic O-GlcNAcylation in terms of its impact on modulating functionally relevant protein interactions responsible for downstream signalling effects. Synthetic chemistry and biology tools have also contributed to the study of O-GlcNAc readers (lectins), writers (OGT) and erasers (OGA), as well as the relevant molecular recognition processes and binding events in combination with biophysical techniques.Given the importance of histone O-GlcNAcylation in the regulation of transcription, Davis and coworkers investigated the physical and mechanistic effect of O-GlcNAc modification on histone stability and function at the molecular level.221 As mentioned above, using post-translational chemical mutagenesis, they site-specifically installed S-GlcNAc onto cysteine-mutated recombinant histones (H2A and H2B) via previous conversion to dehydroalanine (Dha) to generate semisynthetic GlcNAcylated histones and assembled nucleosomes. Modification of H2A at Thr101 destabilised its interaction with H2B and decreased the stability of the H2A/H2B dimer in the nucleosome, providing a plausible structural basis to facilitate transcriptional elongation.176 Meanwhile, glycosylation at H2B-Ser112 and reconstitution of the GlcNAcylated nucleosome led to recruitment of and direct interaction with the FACT complex, pointing to a molecular mechanism for GlcNAc-triggered, FACS-driven increased transcription.177 The Pratt group investigated potential interactions of semisynthetic α-synuclein variants (O-GlcNAc at Thr72 and Ser87) with the protease calpain by exploring the influence of site-specific O-GlcNAc modification on protein cleavage.218O-GlcNAcylation was shown to inhibit α-synuclein proteolysis at sites far in the primary sequence from the glycosylation points, suggesting an O-GlcNAc impact on calpain binding not just related to steric hindrance. Notably, the observed divergent effects in the cleavage sites due to O-GlcNAc glycosylation, including deletion of known positions as well as appearance of new sites, point to a dual mode of regulation by this modification via favouring or disrupting protein–protein interactions.
O-GlcNAcylation modulates protein–protein interactions in response to internal and external stimuli, thereby influencing protein function and cell signalling by the regulated assembly of multiprotein complexes. Several examples of O-GlcNAc-mediated protein–protein interactions in a variety of cell biological contexts are known, in which O-GlcNAc modification on a range of substrates such as chromatin proteins or transcription factors governs a multitude of biological processes (e.g., gene expression, transcriptional signalling and control) by inhibiting or promoting such interactions.222 For instance, O-GlcNAcylation at Thr352 of the NFκB transcription factor p65 subunit interrupts its interaction with the inhibitory protein IκB, inducing the nuclear translocation of O-GlcNAcylated p65 and increasing NFκB transcriptional activation, particularly under hyperglycemic conditions, which links glucose availability to NFκB signalling via direct p65 O-GlcNAcylation.223 Analogously, the same group reported that O-GlcNAc modification of the tumour suppressor protein p53 at Ser149 disrupts the p53–MDM2 interaction required to degrade p53 through ubiquitin-mediated proteolysis, thus reducing its ubiquitination.171 This stabilisation of p53 by Ser149 O-GlcNAcylation was associated with decreased phosphorylation at Thr155, which is known to promote p53 destruction by the ubiquitin–proteasome pathway, highlighting the reciprocal crosstalk between both PTMs in modulating p53 stability and activity. In the context of neurodegeneration, as detailed above, O-GlcNAcylation of α-synuclein blocks the hydrogen-bond interaction between protein monomers due to steric effects, preventing oligomerisation and α-synuclein aggregation.216 Moreover, the Pratt group recently showed how sHSP O-GlcNAc glycosylation at the IXI motif perturbs its interaction with the chaperone cleft, a glycan-promoted disruption that prevents the ability of this motif to intramolecularly compete with substrate binding, improving sHSP chaperone activity.188
On the other hand, protein–protein interactions induced by O-GlcNAc have also been functionally characterised through directed biochemical experiments focused on specific glycoproteins of interest. In an early example, only the O-GlcNAcylated form (Thr92) of the transcription factor STAT5 was shown, upon cytokine stimulation, to bind to the coactivator of transcription CREB-binding protein (CBP), an interaction that potentiates the transactivation of STAT5 target genes and is required for STAT5-mediated transcriptional induction.224 In another study, Ruan et al. showed that OGT is recruited by the adaptor protein host cell factor 1 (HCF1) forming a complex that O-GlcNAcylates PGC1α,225 a key transcriptional regulator of gluconeogenesis. This glycosylation, in turn, promotes interaction with the deubiquitinating enzyme BAP1, resulting in stabilisation of PGC1α from proteolytic degradation and increased gluconeogenic gene expression.225 Modification by O-GlcNAc is known to prevent ubiquitination of various natural substrates, providing another mechanism for enhanced protein stability, and this example constitutes the first evidence for a direct interplay between O-GlcNAcylation and ubiquitination on a single protein. Being most abundant in the nucleus, O-GlcNAc also plays a fundamental role in chromatin regulation through two interconnected mechanisms. In addition to modulating nucleosome structure by altering its assembly upon histone glycosylation, O-GlcNAcylation also acts indirectly as a molecular mark for specific recruitment of and interaction with reader proteins to mediate various downstream functions. For instance, Fujiki et al. reported that O-GlcNAc modification of H2B at Ser112 promotes its monoubiquitination by recruiting the ubiquitin ligase BRE1A, with the glycan residue serving as an anchor, suggesting a potential mechanism for GlcNAcylation-driven transcriptional activation that awaits further structural characterisation.226 Subsequent investigations have provided additional mechanistic and functional insights into histone H2B O-GlcNAcylation, whereby the enzyme TET2 (methylcytosine dioxygenase 2) interacts with and recruits OGT to specific chromatin locations, enhancing H2B-Ser112 O-GlcNAc modification locally and upregulating gene transcription.227 Another important upstream regulator of OGT and potentially of O-GlcNAc mediated histone–protein interactions is the polycomb repressive complex 2 (PCR2) containing the enhancer of zeste homologue 2 (EZH2), which catalyses histone H3 Lys27 trimethylation to form H3K27me3, a critical histone PTM to mark the transcriptionally silenced chromatin.228 Wong and co-workers found that OGT recruits and associates with EZH2 in the PCR2 complex and modifies the H3Lys27 methyltransferase EZH2 with O-GlcNAc at Ser76, which led to increased protein stability and facilitated the formation of H3Lys27me3 to inhibit tumour suppression.229 Additional studies further revealed that O-GlcNAcylation of EZH2 at other N-terminal positions (particularly Ser73 and Ser84) also stabilised free EZH2 from ubiquitin-proteasome degradation, without affecting the formation or stability of the EZH2/PCR2 complex.230 Moreover, a glycosite-dependent regulation of EZH2 function by O-GlcNAcylation was identified, with only a C-terminal O-GlcNAc modification (at Ser729) having an impact on the methyltransferase activity to form H3Lys27me2/3, presumably by altering the subconformation of the EZH2 C-terminal region. These combined results provide a regulatory molecular mechanism by which OGT facilitates histone H3Lys27 methylation and suggest that selective inhibition of EZH2 O-GlcNAcylation may be exploited for anticancer drug discovery to block tumour progression.
These targeted biochemical studies on known glycoproteins highlight how the dynamic, O-GlcNAc mediated regulation of such multiprotein complexes governs a variety of important biological processes. However, they use standard techniques that cannot determine whether the glycan plays an essential role in binding or simply brings about indirect effects. Importantly, protein–protein interactions can also be responsible for the substoichiometric nature of O-GlcNAcylation by potentially masking modification sites on specific substrates, thus precluding subsequent glycosylation by OGT. This low O-GlcNAc stoichiometry together with the weak and transient binding events associated to these protein interactions complicates their detection using conventional strategies, requiring the development of innovative methods to characterise and identify such and new interactions involving O-GlcNAc. As described above, Kohler and coworkers developed a chemical biology approach based on metabolic labelling with diazirine-modified O-GlcNAc (O-GlcNDAz) and photocrosslinking (see Section 3.3 and Fig. 5), enabling the covalent capture of protein–protein interactions in living cells.115 Because crosslinking of the corresponding O-GlcNDAz modified proteins occurs with binding partners within a short radius from the sugar (approximately 2–4 Å), only those specific interactions directly mediated by glycan at the interface (at or near the interaction site) are identified with this system. This approach in combination with additional biological techniques has been subsequently applied by the Boyce lab to study O-GlcNAc-mediated protein–protein interactions in different experimental settings,231,232 as well as to investigate the potential interaction of candidate “reader” proteins for O-GlcNAc in living cells.
Despite early studies by Lefebvre and co-workers reporting HSP70 chaperones with lectin activity towards O-GlcNAc,233,234 and the examples above involving different signalling proteins as potential O-GlcNAc-binding lectins, precise knowledge about O-GlcNAc readers and the structure and/or function of the intracellular glycoprotein–protein complexes is scarce. Applying a newly developed biochemical approach that used synthetic, OGT-glycosylated, biotinylated “bait” peptides followed by MS proteomics (see Section 3.3), Boyce and coworkers extracted O-GlcNAc-mediated binding events from cell lysates and identified several mammalian proteins, including the 14-3-3 family or α-enolase, as O-GlcNAc-interacting partners.122 The discovered candidate readers were found to bind directly and specifically to O-GlcNAcylated (but not unmodified) peptides and proteins in vitro and in living cells, which was confirmed by metabolic labelling using GlcNDAz and UV-specific crosslinking. Further structural studies by co-crystallisation of the model glycopeptide with 14-3-3 isoforms showed a glycan-dependent interaction via extensive hydrogen bonds between the sugar and the protein binding pockets, revealing the initial molecular basis of selective O-GlcNAc recognition. Nonetheless, the biophysical features of these interactions as well as of O-GlcNAc binding are not fully elucidated. Indeed, relatively few structural studies have been performed to gain atomic resolution information on the key aspects underlying the molecular recognition of O-GlcNAc by the corresponding interacting partners, including lectins, immune receptors, and enzymes.
4.4. Molecular recognition features of O-GlcNAc with relevant biological receptors
As mentioned in Section 3.1, the plant lectin WGA and also its succinylated form (sWGA) have been long used in O-GlcNAc research as a traditional method for the enrichment and purification of endogenous O-GlcNAcylated proteins,85 and they have also shown potential in pharmaceutical applications.235 In addition to Neu5Ac residues, WGA recognises selectively GlcNAc units, both in α- and/or β-configuration with similar affinity. It exists in three variant isoforms comprising two identical 171-residue subunits that associate to form 36-kDa dimers, with each polypeptide consisting of four repetitive hevein-type domains of 43 amino acids folded similarly and stabilised by four disulfide bridges in equivalent positions (Fig. 11a). In early years, the WGA interaction with O-GlcNAc and a number of derivatives was explored by diverse experimental techniques,236–239 providing initial biophysical data and binding information. Subsequent computational studies by flexible molecular docking methods predicted the corresponding binding mode and free energies,240 reproducing well the experimental data. This good correlation is arguably based on entropy–enthalpy compensation as reported previously by Jiménez-Barbero and coworkers, who employed NMR titration and bidimensional experiments combined with computational calculations to characterise the binding and three-dimensional solution structure of the complex between the WGA-B domain and N,N′,N′′-triacetylchitobiose [(GlcNAc)3].241 Using NOE-based data and MD simulations, the trisaccharide was found to exhibit two different orientations (binding modes) within the WGA-B binding site, with slight carbohydrate-induced conformational changes observed for the complexed protein. According to their model, the association process is enthalpically stabilised by hydrogen bonding and van der Waals forces, whereas rigidification of the glycan and/or lectin side chains together with solvation effects opposes binding. This entropy loss caused a decrease in affinity for the WGA-B/(GlcNAc)3 interaction with a dissociation constant in the low millimolar range, as assessed by 1H-NMR titration experiments and isothermal titration calorimetry (ITC). An especially powerful and widely utilised NMR method to investigate glycan–protein interactions and characterise ligand binding, in particular under slow exchange conditions on the 1H-NMR timescale, is saturation transfer difference (STD) NMR. Using the WGA–GlcNAc system as a model, Angulo et al. applied a novel STD-based protocol that enabled a direct and accurate measurement of the dissociation constant (KD) for the WGA interaction with O-GlcNAc and N,N′-diacetylchitobiose [GlcNAc-β(1-4)-GlcNAc] (2.4 mM and 200 μM, respectively), with great approximation to the thermodynamic values.242
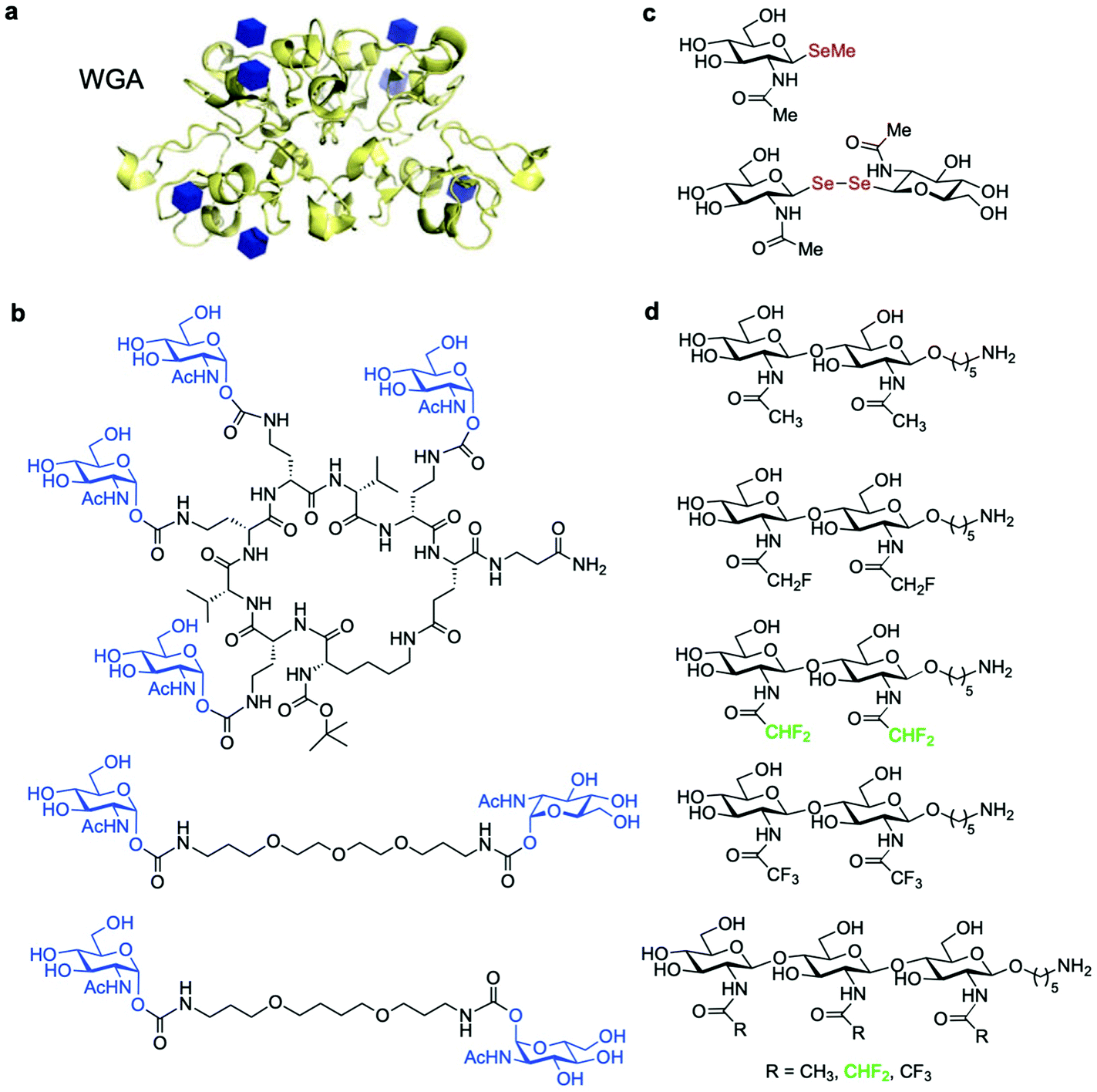 | ||
| Fig. 11 (a) Model of WGA lectin derived from the X-ray crystal structure in complex with GlcNAc (PDB: 2UVO). (b) High affinity tetravalent neoglycopeptide (IC50 = 0.9 μM) and divalent GlcNAc ligands display potent multivalent binding to WGA. (c) Selenium-based GlcNAc affinity ligands applied for detection of GlcNAc–WGA interactions by 77Se NMR. (d) Fluoroacetamide-containing chitobiose and chitotriose mimics used as NMR probes for molecular recognition studies between WGA and GlcNAc glycans. | ||
Considering the particularly low affinity of WGA for O-GlcNAc, multivalent presentation of O-GlcNAc derivatives has been a strategy to enhance binding affinity, providing glycan systems that could inhibit functionally relevant carbohydrate–protein interactions with potential implications for human diseases. The WGA dimer comprises eight total binding sites for O-GlcNAc and related oligomers thereof,243 and thus shows promise for exploiting polyvalent interaction effects. The Wittman lab first synthesised a diverse neoglycopeptide library of conformationally restricted O-GlcNAc glycoclusters where the glycan residues were attached onto an immobilised cyclopeptide scaffold in the last synthetic step.244 Screening of the binding properties using an on-bead, enzyme-immune lectin-binding assay with biotinylated WGA identified some tetra/penta/hexavalent candidates that were further analysed for WGA binding in solution by an enzyme-linked lectin assay (ELLA). The obtained IC values showed strongly increased potencies (up to ≈1000-fold) compared to monovalent O-GlcNAc, suggesting that not only the number of glycan residues, but also their spatial presentation is responsible for their high affinity to WGA. Based on this assumption, the same group then synthesised a range of additional mono-to tetravalent O-GlcNAc ligands containing α-linked glycosyl carbamate units with varying spacer length and flexibility, and identified two divalent derivatives and a tetravalent cyclic neoglycopeptide with high binding potencies (IC50 ≈ 10–50 μM and 1 μM, respectively) (Fig. 11b).245
Crystallographic studies with three WGA–ligand complexes, including the potent divalent and tetravalent ligands, the latter also complemented by NMR and MD simulations, revealed key structural insights into the molecular underpinnings of multivalent binding to WGA. The X-ray structure of the divalent compound displayed a chelating binding mode with four ligand copies occupying all the binding site pairs, and the α-linkage was also identified as a key feature for optimal binding, explaining altogether the high potency of the divalent variant. Moreover, the tetravalent glycocyclopeptide adopted an already preorganised conformation in solution that was suitable for interacting with the protein through two GlcNAc residues and was therefore maintained in the formed WGA complex.245 Overall, this comprehensive understanding of the molecular mechanisms underlying the recognition and enhanced binding affinity of such polyvalent ligands can be leveraged for the rational development of more potent glycan constructs or even lectin mimetics (see Section 4.4.2) for therapeutic or detection purposes.
To further advance the molecular characterisation of the recognition event by ligand-based NMR methods, exploiting specific, NMR-active heteronuclei (e.g., 77Se or 19F) as high-resolution detection probes in the glycan has been shown to be a powerful approach to investigate molecular binding (Fig. 11c and d). Some elegant examples of the application of these chemical tools for probing the association between several GlcNAc derivatives and WGA have been demonstrated, elucidating key structural details of the interaction process. For instance, the Widmalm lab studied the binding properties of methyl-1-seleno-GlcNAc (GlcNAcSeMe) with WGA using 77Se NMR spectroscopy and STD experiments, showing noticeable changes in the ligand parameters such as greatly decreased resonance intensities, considerable line broadening, and a marked downfield chemical shift upon lectin addition (Fig. 11c-top).246 These sharp variations can be explained by the fact that the substituted anomeric oxygen lies close to an amino acid residue in the binding pocket of the lectin crystal structure and indicated a modified chemical environment between the free and bound state.
A molecular docking simulation showed that the introduced selenomethyl group was also in proximity to a protein aromatic residue presumably interacting with the lectin, being the reason of the profound experimental changes with a potentially increased binding affinity due to the selenium atom. In a similar study, the Davis group probed the interaction of GlcNAc diselenide (GlcNAcSe)2 to WGA using NMR and computational calculations (Fig. 11c-bottom).247 A clear STD effect together with increased linewidths as well as transferred NOESY experiments demonstrated glycan binding, with a similar KD (1.6 mM) to the native GlcNAc, as determined by classical titration experiments. Combining molecular docking, STD-NMR data and CORCEMA-ST calculations the binding mode of (GlcNAcSe)2 in the primary binding site of WGA was deduced, with the GlcNAc residue occupying the same position as the corresponding monosaccharide in the crystal structure and without direct interaction between either selenium atom and the lectin. Understandably, fluorine has also been widely utilised in 19F NMR-based experiments as another privileged spectroscopic handle to report key structural information on glycan–protein interactions,248 and in particular to study molecular recognition events involving O-GlcNAc moieties. For example, the labs of Reichardt and Jiménez-Barbero have employed a set of fluoroacetamide groups as novel dual chemical tags for probing the binding of GlcNAc derivatives to WGA by 19F NMR and STD experiments.249,250 Using synthetic GlcNAc dimers (di-N-acetyl chitobiose) and trimers (tri-N-acetyl chitotriose) (Fig. 11d), one of the key contacts identified in the molecular recognition was the CH–π stacking of the (fluoro)acetamide methyl group with the tyrosine ring of the lectin, as also detected for the non-fluorinated substituent in the native GlcNAc derivative. Indeed, as shown by computational calculations, the incorporation of one (and especially two) electron-withdrawing fluorine atoms polarises the remaining C–H bond(s), enhancing the corresponding sugar–aromatic interactions (particularly of the non-reducing sugar moiety) and therefore the binding affinity. STD competition experiments with natural (GlcNAc)2 (KD ≈ 190 μM) enabled quantitative estimation of the KD of the corresponding WGA complexes, with a trifluoroacetamide compound showing the weakest binding (≈650 μM) due to unattainable CH–π stacking and the difluoroacetamide variant (Fig. 11d, –CHF2 group highlighted in green) being the best binder (≈50 μM), which makes this moiety the dual NMR tag of choice for monitoring O-GlcNAc molecular interaction processes.
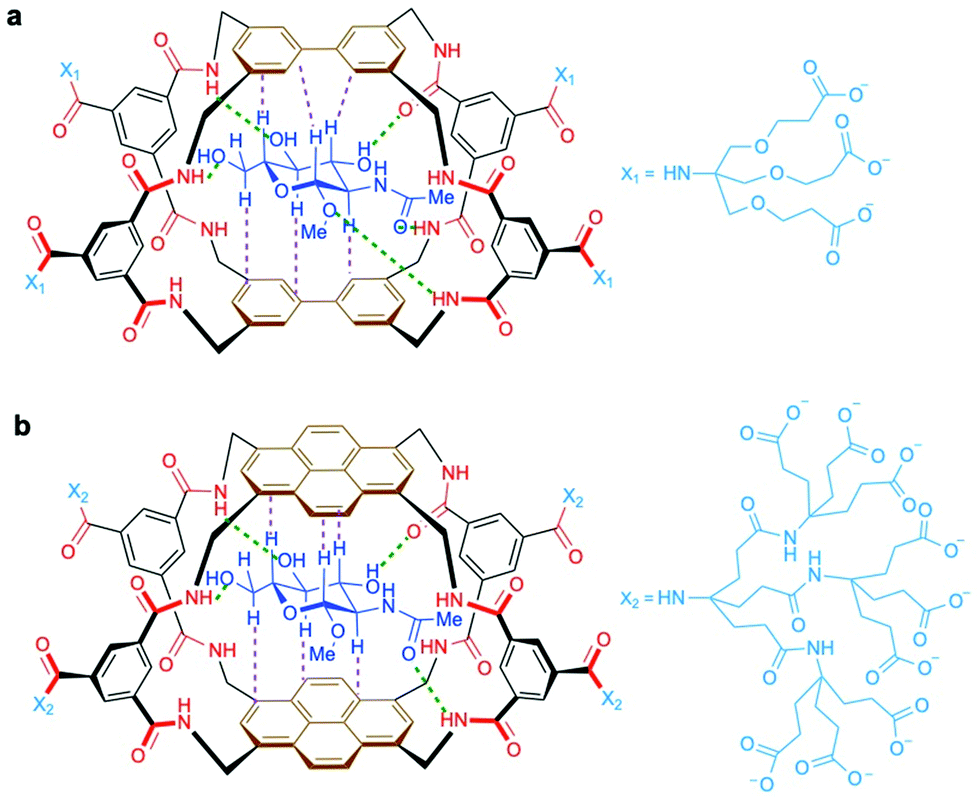 | ||
| Fig. 12 Chemical structure of synthetic lectins ((a) first-generation design, (b) second-generation “eclipsed” receptor) for β-O-GlcNAc, showing intermolecular CH-aromatic contacts (purple dashed lines) and polar hydrogen-bond interactions (green dashed lines). | ||
In their subsequent study, Rios et al. improved upon the previous receptor design by rigidifying the aromatic surfaces with planar pyrene moieties in place of the twistable biphenyl rings as well as expanding the water-solubilising side chains to ensure aqueous solubility (Fig. 12b). This new receptor was synthetically assembled in two different forms providing two isomers (“staggered” and “eclipsed”), both of which showed excellent affinities for β-O-GlcNAc derivatives in solution as measured by 1H NMR titration and/or ITC (Ka ≈ 20,000 M−1 for β-GlcNAc-OMe and Ka ≈ 70000 M−1 for the model CKII glycopeptide, respectively), considerably exceeding those of natural GlcNAc lectins (25-fold improvement).90 The three-dimensional structures of the complexes with β-GlcNAc-OMe, elucidated using a combination of NMR and computational methods, showed the carbohydrates sandwiched between the aromatic pyrene surfaces with the acetamido and methoxy substituents protruding into the portals (Fig. 12b). The structure of the glycopeptide associated to the eclipsed receptor revealed a similar orientation for the O-GlcNAc residue whereas the peptide backbone conformation could not be resolved, albeit it would presumably be involved in diverse additional contacts that could explain the increased Ka. Notably, the outstanding affinity exhibited by this synthetic host for the O-GlcNAcylated CKII peptide highlights the importance of the peptide context for the enhanced interactions leading to these high-affinity levels, which make these biomimetic receptors a promising tool for further development to be applied in different biological settings to explore the O-GlcNAc modification.
Overall, these studies highlight the utility of combining various chemical tools with NMR and computational methods as powerful probes to investigate the molecular binding properties of O-GlcNAc with lectin receptors, advancing our understanding of sugar–protein interactions for potential exploitation in biomedical applications and providing practical chemical solutions to O-GlcNAc recognition and detection in glycobiology.
O-GlcNAc-modified peptides have been identified as MHC-I binding antigens. In an early study, synthetic O-GlcNAc glycopeptide analogues of the cytotoxic T lymphocyte (CTL) epitope of Sendai Virus nucleoprotein bound to MHC-I molecules and induced glycan-specific CTL responses.254 The glycopeptide recognition by the CTL was dependent on the chemical structure of the glycan (an α-GalNAc glycopeptide analogue was not bound) as well as its position in the peptide, with key site-specific changes either abrogating or restoring peptide binding. Additional related studies suggested that the carbohydrate not only mediated binding with MHC-I but also interacted directly with the TCR, serving as an anchor point by hydrogen bond formation through the acetamido group.255 Subsequent crystal structures confirmed that the O-GlcNAc residue was solvent exposed, pointing out of the peptide binding grove, mobile and accessible to interact with the TCR (Fig. 13), suggesting the molecular basis of the recognition between the MHC-I/O-GlcNAc-glycopeptide complex and the TCR.256 Overall, these combined studies by Elliot and coworkers highlight the potential effects of peptide O-GlcNAc modification by abrogating recognition of an immunogenic peptide or converting a silent unglycosylated epitope into an O-GlcNAcylated T cell neoepitope, which could lead to the emergence of new tumour antigens during malignant transformation. Indeed, O-GlcNAc modified peptides have been recently identified as MHC-I restricted leukemia neoantigens. This finding spurred the development of the corresponding synthetic glycopeptide epitopes, which were able to induce strong cellular immune responses in humans,257 highlighting their potential as promising new targets for cancer immunotherapy. Positional analysis of these O-GlcNAc glycopeptide neoantigens from leukemia samples showed that the GlcNAc residues are positioned in the middle of the peptide. This location may be optimal for T cell recognition, exposing the glycan for recognition by glycopeptide-specific CTLs as demonstrated in the previous example. Notably, an analogous presentation mode was also observed in interaction studies with phosphopeptides, in which the T cell receptor (TCR) CDR3α lies proximal to the central phosphate group, suggesting direct TCR binding.258 Thus, altered PTMs can markedly regulate immune system recognition, creating new epitopes with novel structures that can modulate MHC as well as TCR interaction.
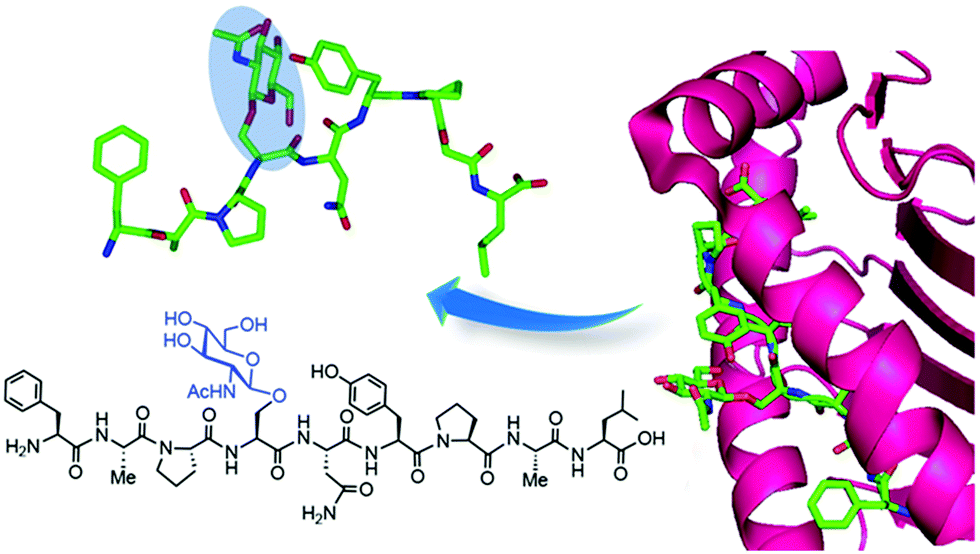 | ||
| Fig. 13 Crystal structure of the FAPS(β-O-GlcNAc)NYPAL glycopeptide (termed K3G) in complex with the MHC class I H-2Db molecule, showing one of the two major conformers for the O-GlcNAc residue. | ||
In another study, Heck and coworkers detected O-GlcNAc modified peptides bound to human leukocyte antigen (HLA) class I molecules that were surprisingly further extended with additional monosaccharides, as identified using advanced MS, enzymatic assays, oxonium ion fragmentation, and molecular modelling.259 The observed glycopeptides were proposed to possess an O-GlcNAc “stem” modified by Gal, and potentially expanded by N-acetyl lactosamine and Neu5Ac, and were mostly computationally assigned to O-GlcNAcylated source proteins. This elongation would occur through the action of glycosyltransferases along the trafficking of cytosolic HLA-I-bound O-GlcNAc-peptides to the cell membrane following the classical class I antigen presentation pathway, representing the first evidence of O-GlcNAc extension in vivo. The O-GlcNAc moieties were found only at central peptide residues with nearby proline residues particularly at the -2 and -3 sites, in line with a potential sequence motif for O-GlcNAcylation.31,70 Using docking calculations, the computed complex of an O-GlcNAc modified peptide bound to the HLA-B*07 molecule showed a solvent oriented glycosylated threonine that is not directly involved in HLA binding, leaving the carbohydrate exposed to the surface and potentially accessible to glycosyltransferases. These expanded O-GlcNAc HLA class I peptides are likely to influence immune recognition, potentially representing a functional type of T cell neoepitope with roles in O-GlcNAc mediated processes that may be dysregulated in diseases.
Taken together, these combined structural immunology studies suggest a common molecular mechanistic basis to understand how altered post-translationally modified peptides are presented to and impact the interaction with relevant immune receptors, yielding key molecular insights that could facilitate cancer therapies targeting these PTMs.
As discussed above, a wide variety of proteins have been shown to bind and direct OGT to particular substrates in response to cellular stimuli. Most of these proteins are themselves O-GlcNAcylated, with the carbohydrate either promoting or interfering with the interaction and targeting. OGA may also be directed to O-GlcNAcylated proteins by interacting partners, although more studies are required to confirm this possible mechanism. Overall, these binding proteins play an essential role in modulating the activity and localisation of both enzymes, and through their involvement in various protein complexes, OGT and OGA control the functions of O-GlcNAcylation, coordinating key cellular networks and fundamental biological processes. However, the structural and biochemical bases regulating these protein–protein interactions have not yet been fully elucidated, which limits our knowledge of the modulation of O-GlcNAc enzymes by their binding partners.
As detailed in Section 2.1, several crystallographic studies of truncated OGT complexed with sugar-donor and acceptor peptides30–34 have characterised critical structural details, revealing that the active site-bound UDP-GlcNAc interacts extensively with the peptide acceptor near the catalytic pocket, although the sugar moiety of the donor does not contribute to OGT binding. Despite this information, the exact molecular mechanisms of OGT–protein substrate interactions are not entirely defined, albeit key insights have emerged from the structural and complementary biochemical studies. While there is no strict consensus sequence, a short structural motif may serve as a molecular recognition point for OGT, with preference for amino acid residues near the glycosylation site that enforce an extended peptide conformation (such as prolines and β-branched residues).31 Moreover, the extended TPR domain mediates protein–protein interactions and contributes to substrate recognition and specificity through contacts with solvent-exposed regions of the protein backbone via a series of asparagine and aspartate residues,38,39 potentially inducing conformational changes that facilitate substrate binding at the active site. As illustrated above (see Section 4.3) through various examples, OGT substrate recognition can also be mediated by conserved adaptor proteins (e.g., HCF1) recruiting and targeting acceptor proteins to OGT depending on environmental and nutrient conditions, leading to regulation of downstream cellular pathways. Additionally, a non-specific O-GlcNAcylation mechanism has been hypothesised to confer substrate specificity to OGT,8 based on its preferential modification of substrates containing disordered, flexible regions (e.g., loops and termini) that can bind to the active site in an extended conformation exposing the amide backbone.30 These features raise the possibility that OGT can non-specifically glycosylate substrates in unstructured domains of unfolded proteins without recognising any particular sequence or structure. Because such flexible elements are not abundant in most mature proteins, this substrate selection mechanism is not expected to be operative in a normal physiological state but rather would potentially occur in response to various stress stimuli that result in increased accumulation of such unstructured substrates. As O-GlcNAc levels are elevated during cellular stress,260 this hypothesis points to a role for O-GlcNAcylation in the handling of unfolded proteins under stress conditions, inhibiting their aggregation and proteasomal degradation as well as facilitating their refolding by chaperones with O-GlcNAc-directed lectin activity (for example, HSP70). The study by Vocadlo and coworkers showing that this glycosylation also occurs co-translationally to stabilise nascent (likely unstructured) polypeptides against ubiquitination261 further supports this idea and suggests that O-GlcNAcylation maintains protein homeostasis and may regulate protein quality control through this proposed mechanism.101
On the other hand, the precise molecular details of OGA substrate recognition are much less well understood. As described in Section 2.2, recent structural studies with truncated hOGA in complex with inhibitors53,54 and several glycopeptide substrates55,56 have revealed key features that led to the proposal of a general mechanism for how OGA recognises and interacts with various substrate proteins. According to this model, OGA binds substrates tightly through the O-GlcNAc residue while also engaging in additional interactions beyond the catalytic domain. Regardless of the underlying Ser/Thr residue or the flanking amino acid sequence, the GlcNAc moiety is anchored in a conserved conformation by strong contacts with multiple active site residues, a binding mode that explains the complete OGA selectivity for hydrolysis of β-O-GlcNAc proteins versus α-O-GlcNAc or β-O-GalNAc substrates. The glycosylated peptides can be bound in different orientations within a hydrophobic cleft, with the backbones retaining a common V-shaped conformation and the side chain residues being stabilised in several ways, rationalising the ability of OGA to deglycosylate a variety of sequences. The conserved contacts between the glycan and the active site residues are essential for recognising and anchoring the glycopeptide in the substrate binding cleft, whereas prevailing interactions with the backbone amides might explain the substrate promiscuity of OGA. Moreover, peptide side-chain specific interactions with further OGA residues contribute to increased binding affinity, potentially altering hydrolysis rates. However, precise knowledge about the extent of these interactions further than the O-GlcNAc moiety is rather limited and future studies are needed to fully understand the molecular underpinnings of OGA substrate recognition, especially beyond the catalytic site.
To gain a better understanding of the structural bases and mechanisms of the molecular recognition events by which OGT and OGA select and interact with many diverse protein substrates, further structural investigations and novel biochemical approaches are needed. This future research will be critical to identify new binding modes to protein substrates and interacting partners, discover what and how recruiter proteins direct both enzymes to their substrates, as well as elucidate the impact of the protein primary sequence and/or conformational preference on the overall targeting and interaction process.
5. Conclusions and outlook
Over the last few years, the O-GlcNAc field is witnessing continuous, excellent progress contributing to advance our knowledge of this fundamental but arguably underappreciated covalent protein modification. Chemistry is definitely playing a key role in this front by providing a wide variety of chemical tools and approaches to probe the mostly unknown biochemical, structural and functional consequences of this unique form of protein glycosylation. These tools include rationally designed, potent and selective, small-molecule OGT/OGA inhibitors for O-GlcNAc elucidation and potential translational applications, as well as further chemical reporters and chemoenzymatic methods for the identification of O-GlcNAcylated proteins and discovery of new roles. Nonetheless, novel, more effective, and critically improved approaches are still required to achieve a more direct, minimally perturbing, and unbiased probing of endogenous protein O-GlcNAc modification in vivo. Such innovative technologies in combination with advanced MS techniques will facilitate dynamic tracking and interrogation of O-GlcNAcylation with site-specific precision. Chemistry is further contributing to move forward the field through the development of additional bioorthogonal ligation strategies for fastest O-GlcNAc labelling and detection, as well as enhanced protein synthesis technologies towards homogeneously modified, stoichiometric O-GlcNAcylated molecules for functional studies and therapeutic applications. Notably, despite some representative examples, few chemically pure O-GlcNAc glycoproteins have been synthesised, hindering rapid progress in elucidating the molecular consequences of this modification. Synthetic glycopeptides have also been crucial to advance O-GlcNAc research and uncover O-GlcNAcylation mechanisms through the development of site-specific antibodies, which not only enable detection and precise functional investigations, but could potentially be applied for diagnostic and clinical purposes. Chemically synthesised O-GlcNAc peptides have also been leveraged in immunological and structural studies, leading for instance to cancer neoantigen discovery and providing key insights into the mechanisms of substrate recognition by OGT and OGA. Despite their potential, few studies have been performed utilising synthetic glycopeptides and/or glycoproteins to probe the conformational effects of Ser/Thr O-GlcNAcylation on protein structure and interactions, as well as on its complex crosstalk with phosphorylation. Remarkably, even less work has been done on the implications of such O-GlcNAc impact on the molecular recognition by relevant biological receptors, which may be behind their important functions in vivo. This is clearly an underexplored area that can benefit from the power of chemistry to provide access to homogeneous synthetic substrates, which combined with the necessary NMR and computational methods should enable future key investigations at atomic-level resolution. This further research might contribute to elucidate the structural role of O-GlcNAc in protein conformation and how this influences the interaction with its corresponding binding partners, yielding critical information on the molecular mechanisms of recognition that may be at the basis of fundamental O-GlcNAc mediated cellular processes. Notably, given the emerging, important links between O-GlcNAc and the immune system,262–264 immunology is another discipline where current progress in the O-GlcNAc field is expected to provide key insights into the functional consequences of O-GlcNAc modification. Thus, chemical immunology emerges as a fertile area in which novel chemical strategies can be developed and exploited to uncover new roles of O-GlcNAc in immunity.265In conclusion, as shown in this review, chemistry will certainly continue to be a key enabling tool in the O-GlcNAc field, providing new probes, strategies and synthetic solutions to address outstanding challenges in the study of this significant protein modification. Nonetheless, it will only be through a multi- and interdisciplinary approach combining chemical, biological and structural techniques that we, as a community, will succeed in the complex, multi-faceted mission of elucidating the mechanisms and functional consequences of O-GlcNAcylation. This knowledge, in turn, will allow us to improve our understanding of the molecular roles of O-GlcNAc underlying diverse aspects of cellular physiology with implications for health and disease.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Research Council (ERC-2016-STG-716878 to A. F.-T.), the Spanish Ministry of Science and Innovation (CTQ2017-87530-R, RYC-2015-17888 to A. F.-T.; Severo Ochoa accreditation SEV-2016-0644 to CIC bioGUNE), and the European Commission (Marie S. Curie postdoctoral fellowship MSCA-2019-IF-898128 to A. S.) is gratefully acknowledged. A. F.-T. thanks Raquel Fernández for inspiration.Notes and references
- C. R. Torres and G. W. Hart, J. Biol. Chem., 1984, 259, 3308–3317 CrossRef CAS.
- G. D. Holt and G. W. Hart, J. Biol. Chem., 1986, 261, 8049–8057 CrossRef CAS.
- S. A. M. van der Laarse, A. C. Leney and A. J. R. Heck, FEBS J., 2018, 285, 3152–3167 CrossRef CAS PubMed.
- W. A. Lubas, D. W. Frank, M. Krause and J. A. Hanover, J. Biol. Chem., 1997, 272, 9316–9324 CrossRef CAS PubMed.
- L. K. Kreppel, M. A. Blomberg and G. W. Hart, J. Biol. Chem., 1997, 272, 9308–9315 CrossRef CAS PubMed.
- D. L. Y. Dong and G. W. Hart, J. Biol. Chem., 1994, 269, 19321–19330 CrossRef CAS.
- Y. Gao, L. Wells, F. I. Comer, G. J. Parker and G. W. Hart, J. Biol. Chem., 2001, 276, 9838–9845 CrossRef CAS PubMed.
- X. Yang and K. Qian, Nat. Rev. Mol. Cell Biol., 2017, 18, 452–465 CrossRef CAS PubMed.
- P. S. Banerjee, O. Lagerlöf and G. W. Hart, Mol. Aspects Med., 2016, 51, 1–15 CrossRef CAS PubMed.
- G. W. Hart, J. Biol. Chem., 2019, 294, 2211–2231 CrossRef CAS PubMed.
- H. Nie and W. Yi, J. Zhejiang Univ., Sci., B, 2019, 20, 437–448 CrossRef PubMed.
- Y. Zhu and G. W. Hart, Mol. Aspects Med., 2020, 100885 Search PubMed.
- L. Wells, K. Vosseller and G. W. Hart, Cell. Mol. Life Sci., 2003, 60, 222–228 CrossRef CAS PubMed.
- M. R. Bond and J. A. Hanover, Annu. Rev. Nutr., 2013, 33, 205–229 CrossRef CAS PubMed.
- J. Ma and G. W. Hart, Expert Rev. Proteomics, 2013, 10, 365–380 CrossRef CAS PubMed.
- S. B. Peterson and G. W. Hart, Crit. Rev. Biochem. Mol. Biol., 2016, 51, 150–161 CrossRef CAS PubMed.
- C. Slawson and G. W. Hart, Nat. Rev. Cancer, 2011, 11, 678–684 CrossRef CAS PubMed.
- Y. Fardini, V. Dehennaut, T. Lefebvre and T. Issad, Front. Endocrinol., 2013, 4, 99 Search PubMed.
- Z. Ma and K. Vosseller, Amino Acids, 2013, 45, 719–733 CrossRef CAS PubMed.
- E. Forma, P. Jóźwiak, M. Bryś and A. Krześlak, Cell. Mol. Biol. Lett., 2014, 19, 438–460 CAS.
- C. M. Ferrer, V. L. Sodi and M. J. Reginato, J. Mol. Biol., 2016, 428, 3282–3294 CrossRef CAS PubMed.
- W. Yi, P. M. Clark, D. E. Mason, M. C. Keenan, C. Hill, W. A. Goddard, E. C. Peters, E. M. Driggers and L. C. Hsieh-Wilson, Science, 2012, 337, 8146–8980 CrossRef PubMed.
- Y. Fardini, V. Dehennaut, T. Lefebvre and T. Issad, Front. Endocrinol., 2013, 4, 99 Search PubMed.
- Y. Zhu, X. Shan, S. A. Yuzwa and D. J. Vocadlo, J. Biol. Chem., 2014, 289, 34472–34481 CrossRef PubMed.
- S. A. Yuzwa and D. J. Vocadlo, Chem. Soc. Rev., 2014, 43, 6839–6858 RSC.
- P. Ryan, M. Xu, A. K. Davey, J. J. Danon, G. D. Mellick, M. Kassiou and S. Rudrawar, ACS Chem. Neurosci., 2019, 10, 2209–2221 CrossRef CAS PubMed.
- M. Worth, H. Li and J. Jiang, ACS Chem. Biol., 2017, 12, 326–335 CrossRef CAS PubMed.
- Z. G. Levine and S. Walker, Annu. Rev. Biochem., 2016, 85, 631–657 CrossRef CAS PubMed.
- S. P. N. Iyer and G. W. Hart, J. Biol. Chem., 2003, 278, 24608–24616 CrossRef CAS PubMed.
- M. B. Lazarus, Y. Nam, J. Jiang, P. Sliz and S. Walker, Nature, 2011, 469, 564–569 CrossRef CAS PubMed.
- S. Pathak, J. Alonso, M. Schimpl, K. Rafie, D. E. Blair, V. S. Borodkin, A. W. Schüttelkopf, O. Albarbarawi and D. M. F. Van Aalten, Nat. Struct. Mol. Biol., 2015, 22, 744–749 CrossRef CAS PubMed.
- M. Schimpl, X. Zheng, V. S. Borodkin, D. E. Blair, A. T. Ferenbach, A. W. Schüttelkopf, I. Navratilova, T. Aristotelous, O. Albarbarawi, D. A. Robinson, M. A. MacNaughtan and D. M. F. Van Aalten, Nat. Chem. Biol., 2012, 8, 969–974 CrossRef CAS PubMed.
- M. B. Lazarus, J. Jiang, V. Kapuria, T. Bhuiyan, J. Janetzko, W. F. Zandberg, D. J. Vocadlo, W. Herr and S. Walker, Science, 2013, 342, 1235–1239 CrossRef CAS PubMed.
- M. B. Lazarus, J. Jiang, T. M. Gloster, W. F. Zandberg, G. E. Whitworth, D. J. Vocadlo and S. Walker, Nat. Chem. Biol., 2012, 8, 966–968 CrossRef CAS PubMed.
- C. M. Joiner, H. Li, J. Jiang and S. Walker, Curr. Opin. Struct. Biol., 2019, 56, 97–106 CrossRef CAS PubMed.
- D. T. King, A. Males, G. J. Davies and D. J. Vocadlo, Curr. Opin. Chem. Biol., 2019, 53, 131–144 CrossRef CAS PubMed.
- C.-W. Hu, M. Worth, D. Fan, B. Li, H. Li, L. Lu, X. Zhong, Z. Lin, L. Wei, Y. Ge, L. Li and J. Jiang, Nat. Chem. Biol., 2017, 13, 1267–1273 CrossRef CAS PubMed.
- Z. G. Levine, C. Fan, M. S. Melicher, M. Orman, T. Benjamin and S. Walker, J. Am. Chem. Soc., 2018, 140, 3510–3513 CrossRef CAS PubMed.
- C. M. Joiner, Z. G. Levine, C. Aonbangkhen, C. M. Woo and S. Walker, J. Am. Chem. Soc., 2019, 141, 12974–12978 CrossRef CAS PubMed.
- W. D. Cheung, K. Sakabe, M. P. Housley, W. B. Dias and G. W. Hart, J. Biol. Chem., 2008, 283, 33935–33941 CrossRef CAS PubMed.
- B. J. Gross, B. C. Kraybill and S. Walker, J. Am. Chem. Soc., 2005, 127, 14588–14589 CrossRef CAS PubMed.
- J. Jiang, M. B. Lazarus, L. Pasquina, P. Sliz and S. Walker, Nat. Chem. Biol., 2012, 8, 72–77 CrossRef CAS PubMed.
- R. F. Ortiz-Meoz, J. Jiang, M. B. Lazarus, M. Orman, J. Janetzko, C. Fan, D. Y. Duveau, Z. W. Tan, C. J. Thomas and S. Walker, ACS Chem. Biol., 2015, 10, 1392–1397 CrossRef CAS PubMed.
- S. E. S. Martin, Z.-W. Tan, H. M. Itkonen, D. Y. Duveau, J. A. Paulo, J. Janetzko, P. L. Boutz, L. Törk, F. A. Moss, C. J. Thomas, S. P. Gygi, M. B. Lazarus and S. Walker, J. Am. Chem. Soc., 2018, 140, 13542–13545 CrossRef CAS PubMed.
- Z. Gao, O. G. Ovchinnikova, B.-S. Huang, F. Liu, D. E. Williams, R. J. Andersen, T. L. Lowary, C. Whitfield and S. G. Withers, J. Am. Chem. Soc., 2019, 141, 2201–2204 CrossRef CAS PubMed.
- T. M. Gloster, W. F. Zandberg, J. E. Heinonen, D. L. Shen, L. Deng and D. J. Vocadlo, Nat. Chem. Biol., 2011, 7, 174–181 CrossRef CAS PubMed.
- T. W. Liu, W. F. Zandberg, T. M. Gloster, L. Deng, K. D. Murray, X. Shan and D. J. Vocadlo, Angew. Chem., Int. Ed., 2018, 57, 7644–7648 CrossRef CAS PubMed.
- V. S. Borodkin, M. Schimpl, M. Gundogdu, K. Rafie, H. C. Dorfmueller, D. A. Robinson and D. M. F. Van Aalten, Biochem. J., 2014, 457, 497–502 CrossRef CAS PubMed.
- K. Rafie, A. Gorelik, R. Trapannone, V. S. Borodkin and D. M. F. van Aalten, Bioconjugate Chem., 2018, 29, 1834–1840 CrossRef CAS PubMed.
- R. Trapannone, K. Rafie and D. M. F. Van Aalten, Biochem. Soc. Trans., 2016, 44, 88–93 CrossRef CAS PubMed.
- M. S. Macauley, G. E. Whitworth, A. W. Debowski, D. Chin and D. J. Vocadlo, J. Biol. Chem., 2005, 280, 25313–25322 CrossRef CAS PubMed.
- N. Çetinbaş, M. S. Macauley, K. A. Stubbs, R. Drapala and D. J. Vocadlo, Biochemistry, 2006, 45, 3835–3844 CrossRef PubMed.
- C. Roth, S. Chan, W. A. Offen, G. R. Hemsworth, L. I. Willems, D. T. King, V. Varghese, R. Britton, D. J. Vocadlo and G. J. Davies, Nat. Chem. Biol., 2017, 13, 610–612 CrossRef CAS PubMed.
- N. L. Elsen, S. B. Patel, R. E. Ford, D. L. Hall, F. Hess, H. Kandula, M. Kornienko, J. Reid, H. Selnick, J. M. Shipman, S. Sharma, K. J. Lumb, S. M. Soisson and D. J. Klein, Nat. Chem. Biol., 2017, 13, 613–615 CrossRef CAS PubMed.
- B. Li, H. Li, L. Lu and J. Jiang, Nat. Struct. Mol. Biol., 2017, 24, 362–369 CrossRef CAS PubMed.
- B. Li, H. Li, C.-W. Hu and J. Jiang, Nat. Commun., 2017, 8, 666 CrossRef PubMed.
- M. S. Macauley and D. J. Vocadlo, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 107–121 CrossRef CAS PubMed.
- M. Horsch, L. Hoesch, A. Vasella and D. M. Rast, Eur. J. Biochem., 1991, 197, 815–818 CrossRef CAS PubMed.
- G. E. Whitworth, M. S. Macauley, K. A. Stubbs, R. J. Dennis, E. J. Taylor, G. J. Davies, I. R. Greig and D. J. Vocadlo, J. Am. Chem. Soc., 2007, 129, 635–644 CrossRef CAS PubMed.
- H. C. Dorfmueller, V. S. Borodkin, M. Schimpl, X. Zheng, R. Kime, K. D. Read and D. M. F. van Aalten, Chem. Biol., 2010, 17, 1250–1255 CrossRef CAS PubMed.
- H. C. Dorfmueller, V. S. Borodkin, M. Schimpl, S. M. Shepherd, N. A. Shpiro and D. M. F. van Aalten, J. Am. Chem. Soc., 2006, 128, 16484–16485 CrossRef CAS PubMed.
- H. C. Dorfmueller, V. S. Borodkin, M. Schimpl and D. M. F. van Aalten, Biochem. J., 2009, 420, 221–227 CrossRef CAS PubMed.
- S. Knapp, D. Vocadlo, Z. Gao, B. Kirk, J. Lou and S. G. Withers, J. Am. Chem. Soc., 1996, 118, 6804–6805 CrossRef CAS.
- N. Cekic, J. E. Heinonen, K. A. Stubbs, C. Roth, Y. He, A. J. Bennet, E. J. McEachern, G. J. Davies and D. J. Vocadlo, Chem. Sci., 2016, 7, 3742–3750 RSC.
- S. A. Yuzwa, M. S. Macauley, J. E. Heinonen, X. Shan, R. J. Dennis, Y. He, G. E. Whitworth, K. A. Stubbs, E. J. McEachern, G. J. Davies and D. J. Vocadlo, Nat. Chem. Biol., 2008, 4, 483–490 CrossRef CAS PubMed.
- S. A. Yuzwa, X. Shan, M. S. MacAuley, T. Clark, Y. Skorobogatko, K. Vosseller and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 393–399 CrossRef CAS PubMed.
- H. G. Selnick, J. F. Hess, C. Tang, K. Liu, J. B. Schachter, J. E. Ballard, J. Marcus, D. J. Klein, X. Wang, M. Pearson, M. J. Savage, R. Kaul, T. S. Li, D. J. Vocadlo, Y. Zhou, Y. Zhu, C. Mu, Y. Wang, Z. Wei, C. Bai, J. L. Duffy and E. J. McEachern, J. Med. Chem., 2019, 62, 10062–10097 CrossRef CAS PubMed.
- M. Bergeron-Brlek, J. Goodwin-Tindall, N. Cekic, C. Roth, W. F. Zandberg, X. Shan, V. Varghese, S. Chan, G. J. Davies, D. J. Vocadlo and R. Britton, Angew. Chem., Int. Ed., 2015, 54, 15429–15433 CrossRef CAS PubMed.
- Z. Wang and G. W. Hart, Clin. Proteomics, 2008, 4, 5–13 CrossRef CAS.
- A. C. Leney, D. El Atmioui, W. Wu, H. Ovaa and A. J. R. Heck, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7255–E7261 CrossRef CAS PubMed.
- E. Wulff-Fuentes, R. R. Berendt, L. Massman, L. Danner, F. Malard, J. Vora, R. Kahsay and S. Olivier-Van Stichelen, Sci. Data, 2021, 8, 25 CrossRef CAS PubMed.
- C. M. Snow, A. Senior and L. Gerace, J. Cell Biol., 1987, 104, 1143–1156 CrossRef CAS PubMed.
- F. I. Comer, K. Vosseller, L. Wells, M. A. Accavitti and G. W. Hart, Anal. Biochem., 2001, 293, 169–177 CrossRef CAS PubMed.
- T. Isono, PLoS One, 2011, 6, e18959 CrossRef CAS PubMed.
- J. R. Turner, A. M. Tartakoff and N. S. Greenspan, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5608–5612 CrossRef CAS PubMed.
- N. Yoshida, R. A. Mortara, M. F. Araguth, J. C. Gonzalez and M. Russo, Infect. Immun., 1989, 57, 1663–1667 CrossRef CAS PubMed.
- C. F. Teo, S. Ingale, M. A. Wolfert, G. A. Elsayed, L. G. Nöt, J. C. Chatham, L. Wells and G.-J. Boons, Nat. Chem. Biol., 2010, 6, 338–343 CrossRef CAS PubMed.
- Y. Tashima and P. Stanley, J. Biol. Chem., 2014, 289, 11132–11142 CrossRef CAS PubMed.
- R. A. Reeves, A. Lee, R. Henry and N. E. Zachara, Anal. Biochem., 2014, 457, 8–18 CrossRef CAS PubMed.
- K. Kamemura, B. K. Hayes, F. I. Comer and G. W. Hart, J. Biol. Chem., 2002, 277, 19229–19235 CrossRef CAS PubMed.
- S. A. Yuzwa, A. K. Yadav, Y. Skorobogatko, T. Clark, K. Vosseller and D. J. Vocadlo, Amino Acids, 2011, 40, 857–868 CrossRef CAS PubMed.
- M. Hirosawa, K. Hayakawa, C. Yoneda, D. Arai, H. Shiota, T. Suzuki, S. Tanaka, N. Dohmae and K. Shiota, Sci. Rep., 2016, 6, 31785 CrossRef CAS PubMed.
- S. Pathak, V. S. Borodkin, O. Albarbarawi, D. G. Campbell, A. Ibrahim and D. M. F. van Aalten, EMBO J., 2012, 31, 1394–1404 CrossRef CAS PubMed.
- M. Monsigny, A. C. Roche, C. Sene, R. Maget-Dana and F. Delmotte, Eur. J. Biochem., 1980, 104, 147–153 CrossRef CAS PubMed.
- N. E. Zachara, K. Vosseller and G. W. Hart, Curr. Protoc. Protein Sci., 2011, 66, 12.8.1–12.8.33 Search PubMed.
- K. Vosseller, J. C. Trinidad, R. J. Chalkley, C. G. Specht, A. Thalhammer, A. J. Lynn, J. O. Snedecor, S. Guan, K. F. Medzihradszky, D. A. Maltby, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2006, 5, 923–934 CrossRef CAS PubMed.
- O. Machon, S. F. Baldini, J. P. Ribeiro, A. Steenackers, A. Varrot, T. Lefebvre and A. Imberty, Glycobiology, 2017, 27, 123–128 CrossRef CAS PubMed.
- W. Liu, G. Han, Y. Yin, S. Jiang, G. Yu, Q. Yang, W. Yu, X. Ye, Y. Su, Y. Yang, G. W. Hart and H. Sun, Glycobiology, 2018, 28, 363–373 CrossRef CAS PubMed.
- Y. Ferrand, E. Klein, N. P. Barwell, M. P. Crump, J. Jiménez-Barbero, C. Vicent, G.-J. Boons, S. Ingale and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 1775–1779 CrossRef CAS PubMed.
- P. Rios, T. S. Carter, T. J. Mooibroek, M. P. Crump, M. Lisbjerg, M. Pittelkow, N. T. Supekar, G.-J. Boons and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 3387–3392 CrossRef CAS PubMed.
- D. Mariappa, N. Selvan, V. S. Borodkin, J. Alonso, A. T. Ferenbach, C. Shepherd, I. H. Navratilova and D. M. F. van Aalten, Biochem. J., 2015, 470, 255–262 CrossRef CAS PubMed.
- N. Selvan, R. Williamson, D. Mariappa, D. G. Campbell, R. Gourlay, A. T. Ferenbach, T. Aristotelous, I. Hopkins-Navratilova, M. Trost and D. M. F. van Aalten, Nat. Chem. Biol., 2017, 13, 882–887 CrossRef CAS PubMed.
- S. Cecioni and D. J. Vocadlo, Curr. Opin. Chem. Biol., 2013, 17, 719–728 CrossRef CAS PubMed.
- D. J. Vocadlo, H. C. Hang, E. J. Kim, J. A. Hanover and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9116–9121 CrossRef CAS PubMed.
- K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS PubMed.
- A. Nandi, R. Sprung, D. K. Barma, Y. Zhao, S. C. Kim, J. R. Falck and Y. Zhao, Anal. Chem., 2006, 78, 452–458 CrossRef CAS PubMed.
- Y. Zhu, J. Wu and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 9301–9305 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- M. Boyce, I. S. Carrico, A. S. Ganguli, S. H. Yu, M. J. Hangauer, S. C. Hubbard, J. J. Kohler and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3141–3146 CrossRef CAS PubMed.
- H. Hahne, N. Sobotzki, T. Nyberg, D. Helm, V. S. Borodkin, D. M. F. Van Aalten, B. Agnew and B. Kuster, J. Proteome Res., 2013, 12, 927–936 CrossRef CAS PubMed.
- Y. Zhu, L. I. Willems, D. Salas, S. Cecioni, W. B. Wu, L. J. Foster and D. J. Vocadlo, J. Am. Chem. Soc., 2020, 142, 15729–15739 CrossRef CAS PubMed.
- B. W. Zaro, Y. Y. Yang, H. C. Hang and M. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8146–8151 CrossRef CAS PubMed.
- K. N. Chuh, B. W. Zaro, F. Piller, V. Piller and M. R. Pratt, J. Am. Chem. Soc., 2014, 136, 12283–12295 CrossRef CAS PubMed.
- N. J. Pedowitz, B. W. Zaro and M. R. Pratt, Curr. Protoc. Chem. Biol., 2020, 12, e81 CAS.
- N. Darabedian and M. R. Pratt, Methods Enzymol., 2019, 622, 293–307 CAS.
- K. N. Chuh, A. R. Batt, B. W. Zaro, N. Darabedian, N. P. Marotta, C. K. Brennan, A. Amirhekmat and M. R. Pratt, J. Am. Chem. Soc., 2017, 139, 7872–7885 CrossRef CAS PubMed.
- D. L. Shen, T. W. Liu, W. Zandberg, T. Clark, R. Eskandari, M. G. Alteen, H. Y. Tan, Y. Zhu, S. Cecioni and D. Vocadlo, ACS Chem. Biol., 2017, 12, 206–213 CrossRef CAS PubMed.
- B. W. Zaro, A. R. Batt, K. N. Chuh, M. X. Navarro and M. R. Pratt, ACS Chem. Biol., 2017, 12, 787–794 CrossRef CAS PubMed.
- N. Darabedian, J. Gao, K. N. Chuh, C. M. Woo and M. R. Pratt, J. Am. Chem. Soc., 2018, 140, 7092–7100 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- A. Niederwieser, A.-K. Späte, L. D. Nguyen, C. Jüngst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- A. K. Späte, V. F. Schart, J. Häfner, A. Niederwieser, T. U. Mayer and V. Wittmann, Beilstein J. Org. Chem., 2014, 10, 2235–2242 CrossRef PubMed.
- D. M. Patterson, K. A. Jones and J. A. Prescher, Mol. BioSyst., 2014, 10, 1693–1697 RSC.
- F. Doll, A. Buntz, A. K. Späte, V. F. Schart, A. Timper, W. Schrimpf, C. R. Hauck, A. Zumbusch and V. Wittmann, Angew. Chem., Int. Ed., 2016, 55, 2262–2266 CrossRef CAS PubMed.
- S. Yu, M. Boyce, A. M. Wands, M. R. Bond, C. R. Bertozzi and J. J. Kohler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4834–4839 CrossRef CAS PubMed.
- H. Y. Tan, R. Eskandari, D. Shen, Y. Zhu, T. W. Liu, L. I. Willems, M. G. Alteen, Z. Madden and D. J. Vocadlo, J. Am. Chem. Soc., 2018, 140, 15300–15308 CrossRef CAS PubMed.
- J. C. Maynard, A. L. Burlingame and K. F. Medzihradszky, Mol. Cell. Proteomics, 2016, 15, 3405–3411 CrossRef CAS PubMed.
- A. Gorelik, S. G. Bartual, V. S. Borodkin, J. Varghese, A. T. Ferenbach and D. M. F. van Aalten, Nat. Struct. Mol. Biol., 2019, 26, 1071–1077 CrossRef CAS PubMed.
- W. Qin, K. Qin, X. Fan, L. Peng, W. Hong, Y. Zhu, P. Lv, Y. Du, R. Huang, M. Han, B. Cheng, Y. Liu, W. Zhou, C. Wang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 1817–1820 CrossRef CAS PubMed.
- K. Qin, H. Zhang, Z. Zhao and X. Chen, J. Am. Chem. Soc., 2020, 142, 9382–9388 CrossRef CAS PubMed.
- A. C. Rodriguez, S. H. Yu, B. Li, H. Zegzouti and J. J. Kohler, J. Biol. Chem., 2015, 290, 22638–22648 CrossRef CAS PubMed.
- C. A. Toleman, M. A. Schumacher, S.-H. Yu, W. Zeng, N. J. Cox, T. J. Smith, E. J. Soderblom, A. M. Wands, J. J. Kohler and M. Boyce, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5956–5961 CrossRef CAS PubMed.
- E. P. Roquemore, T. Y. Chou and G. W. Hart, Methods Enzymol., 1994, 230, 443–460 CAS.
- B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2002, 277, 20833–20839 CrossRef CAS PubMed.
- N. Khidekel, S. Arndt, N. Lamarre-Vincent, A. Lippert, K. G. Poulin-Kerstien, B. Ramakrishnan, P. K. Qasba and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2003, 125, 16162–16163 CrossRef CAS PubMed.
- N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13132–13137 CrossRef CAS PubMed.
- N. Khidekel, S. B. Ficarro, P. M. Clark, M. C. Bryan, D. L. Swaney, J. E. Rexach, Y. E. Sun, J. J. Coon, E. C. Peters and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2007, 3, 339–348 CrossRef CAS PubMed.
- J. E. Rexach, C. J. Rogers, S. H. Yu, J. Tao, Y. E. Sun and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2010, 6, 645–651 CrossRef CAS PubMed.
- J. E. Rexach, P. M. Clark, D. E. Mason, R. L. Neve, E. C. Peters and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2012, 8, 253–261 CrossRef CAS PubMed.
- P. M. Clark, J. F. Dweck, D. E. Mason, C. R. Hart, S. B. Buck, E. C. Peters, B. J. Agnew and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 11576–11577 CrossRef CAS PubMed.
- N. Darabedian, J. W. Thompson, K. N. Chuh, L. C. Hsieh-Wilson and M. R. Pratt, Biochemistry, 2018, 57, 5769–5774 CrossRef CAS PubMed.
- Z. Wang, N. D. Udeshi, M. O’Malley, J. Shabanowitz, D. F. Hunt and G. W. Hart, Mol. Cell. Proteomics, 2010, 9, 153–160 CrossRef CAS PubMed.
- Z. Wang, N. D. Udeshi, C. Slawson, P. D. Compton, K. Sakabe, W. D. Cheung, J. Shabanowitz, D. F. Hunt and G. W. Hart, Sci. Signaling, 2010, 3, ra2 Search PubMed.
- J. F. Alfaro, C. X. Gong, M. E. Monroe, J. T. Aldrich, T. R. W. Clauss, S. O. Purvine, Z. Wang, D. G. Camp, J. Shabanowitz, P. Stanley, G. W. Hart, D. F. Hunt, F. Yang and R. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7280–7285 CrossRef CAS PubMed.
- M. E. Griffin, E. H. Jensen, D. E. Mason, C. L. Jenkins, S. E. Stone, E. C. Peters and L. C. Hsieh-Wilson, Mol. BioSyst., 2016, 12, 1756–1759 RSC.
- L. C. H. Wilson, J. W. Thompson and M. E. Griffin, Methods Enzymol., 2018, 598, 101–135 Search PubMed.
- A. L. Aguilar, X. Hou, L. Wen, P. G. Wang and P. Wu, ChemBioChem, 2017, 18, 2416–2421 CrossRef CAS PubMed.
- E. Boeggeman, B. Ramakrishnan, C. Kilgore, N. Khidekel, L. C. Hsieh-Wilson, J. T. Simpson and P. K. Qasba, Bioconjugate Chem., 2007, 18, 806–814 CrossRef CAS PubMed.
- Z. L. Wu, T. J. Tatge, A. E. Grill and Y. Zou, Cell Chem. Biol., 2018, 25, 1428 CrossRef CAS PubMed.
- S. Xu, F. Sun and R. Wu, Anal. Chem., 2020, 92, 9807–9814 CrossRef CAS PubMed.
- S. Stairs, A. A. Neves, H. Stöckmann, Y. A. Wainman, H. Ireland-Zecchini, K. M. Brindle and F. J. Leeper, ChemBioChem, 2013, 14, 1063–1067 CrossRef CAS PubMed.
- A. K. Späte, V. F. Schart, J. Häfner, A. Niederwieser, T. U. Mayer and V. Wittmann, Beilstein J. Org. Chem., 2014, 10, 2235–2242 CrossRef PubMed.
- E. J. Kim, Molecules, 2018, 23, 2411 CrossRef PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef PubMed.
- R. Sprung, A. Nandi, Y. Chen, S. C. Kim, D. Barma, J. R. Falck and Y. Zhao, J. Proteome Res., 2005, 4, 950–957 CrossRef CAS PubMed.
- F. L. Lin, H. M. Hoyt, H. Van Halbeek, R. G. Bergman and C. R. Bertozzi, J. Am. Chem. Soc., 2005, 127, 2686–2695 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- W. Qin, P. Lv, X. Fan, B. Quan, Y. Zhu, K. Qin, Y. Chen, C. Wang and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6749–E6758 CrossRef CAS PubMed.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- E. J. Kim, D. W. Kang, H. F. Leucke, M. R. Bond, S. Ghosh, D. C Love, J.-S. Ahn, D.-O. Kang and J. A. Hanover, Carbohydr. Res., 2013, 377, 18–27 CrossRef CAS PubMed.
- S. Li, H. Zhu, J. Wang, X. Wang, X. Li, C. Ma, L. Wen, B. Yu, Y. Wang, J. Li and P. G. Wang, Electrophoresis, 2016, 37, 1431–1436 CrossRef CAS PubMed.
- J. Ma, W.-H. Wang, Z. Li, J. Shabanowitz, D. F. Hunt and G. W. Hart, Anal. Chem., 2019, 91, 2620–2625 CrossRef CAS PubMed.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- A. Herner and Q. Lin, Top. Curr. Chem., 2016, 374, 1 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Curr. Opin. Chem. Biol., 2014, 21, 89–95 CrossRef CAS PubMed.
- W. Song, Y. Wang, Z. Yu, C. I. R. Vera, J. Qu and Q. Lin, ACS Chem. Biol., 2010, 5, 875–885 CrossRef CAS PubMed.
- D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680–13683 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J.-S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- P. S. Banerjee, G. W. Hart and J. W. Cho, Chem. Soc. Rev., 2013, 42, 4345–4357 RSC.
- W. H. Yang, J. E. Kim, H. W. Nam, J. W. Ju, H. S. Kim, Y. S. Kim and J. W. Cho, Nat. Cell Biol., 2006, 8, 1074–1083 CrossRef CAS PubMed.
- G. Tegl, J. Hanson, H.-M. Chen, D. H. Kwan, A. G. Santana and S. G. Withers, Angew. Chem., Int. Ed., 2019, 58, 1632–1637 CrossRef CAS PubMed.
- D. H. Ramirez, C. Aonbangkhen, H. Y. Wu, J. A. Naftaly, S. Tang, T. R. O’Meara and C. M. Woo, ACS Chem. Biol., 2020, 15, 1059–1066 CrossRef CAS PubMed.
- M. Fernández-González, O. Boutureira, G. J. L. Bernardes, J. M. Chalker, M. A. Young, J. C. Errey and B. G. Davis, Chem. Sci., 2010, 1, 709–715 RSC.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- L. Lercher, R. Raj, N. A. Patel, J. Price, S. Mohammed, C. V. Robinson, C. J. Schofield and B. G. Davis, Nat. Commun., 2015, 6, 7978 CrossRef CAS PubMed.
- R. Raj, L. Lercher, S. Mohammed and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 8918–8922 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W. L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M. L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, aag1465 CrossRef PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS PubMed.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS PubMed.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS PubMed.
- S. Schwagerus, O. Reimann, C. Despres, C. Smet-Nocca and C. P. R. Hackenberger, J. Pept. Sci., 2016, 22, 327–333 CrossRef CAS PubMed.
- N. P. Marotta, Y. H. Lin, Y. E. Lewis, M. R. Ambroso, B. W. Zaro, M. T. Roth, D. B. Arnold, R. Langen and M. R. Pratt, Nat. Chem., 2015, 7, 913–920 CrossRef CAS PubMed.
- Y. E. Lewis, A. Galesic, P. M. Levine, C. A. De Leon, N. Lamiri, C. K. Brennan and M. R. Pratt, ACS Chem. Biol., 2017, 12, 1020–1027 CrossRef CAS PubMed.
- P. M. Levine, A. Galesic, A. T. Balana, A. L. Mahul-Mellier, M. X. Navarro, C. A. De Leon, H. A. Lashuel and M. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1511–1519 CrossRef CAS PubMed.
- A. T. Balana, P. M. Levine, T. W. Craven, S. Mukherjee, N. J. Pedowitz, S. P. Moon, T. T. Takahashi, C. F. W. Becker, D. Baker and M. R. Pratt, Nat. Chem., 2021, 13, 441–450 CrossRef CAS PubMed.
- M. K. Tarrant, H. S. Rho, Z. Xie, Y. L. Jiang, C. Gross, J. C. Culhane, G. Yan, J. Qian, Y. Ichikawa, T. Matsuoka, N. Zachara, F. A. Etzkorn, G. W. Hart, J. S. Jeong, S. Blackshaw, H. Zhu and P. A. Cole, Nat. Chem. Biol., 2012, 8, 262–269 CrossRef CAS PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, Biochemistry, 2017, 56, 3507–3517 CrossRef CAS PubMed.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem., 2018, 2, 122 CrossRef CAS.
- Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed.
- A. Matsuura, M. Ito, Y. Sakaidani, T. Kondo, K. Murakami, K. Furukawa, D. Nadano, T. Matsuda and T. Okajima, J. Biol. Chem., 2008, 283, 35486–35495 CrossRef CAS PubMed.
- Y. Sakaidani, T. Nomura, A. Matsuura, M. Ito, E. Suzuki, K. Murakami, D. Nadano, T. Matsuda, K. Furukawa and T. Okajima, Nat. Commun., 2011, 2, 583 CrossRef PubMed.
- G. W. Hart, C. Slawson, G. Ramirez-Correa and O. Lagerlof, Annu. Rev. Biochem., 2011, 80, 825–858 CrossRef CAS PubMed.
- I. Nishikawa, Y. Nakajima, M. Ito, S. Fukuchi, K. Homma and K. Nishikawa, Int. J. Mol. Sci., 2010, 11, 4991–5008 CrossRef CAS PubMed.
- F. I. Comer and G. W. Hart, Biochemistry, 2001, 40, 7845–7852 CrossRef CAS PubMed.
- S. M. Ranuncolo, S. Ghosh, J. A. Hanover, G. W. Hart and B. A. Lewis, J. Biol. Chem., 2012, 287, 23549–23561 CrossRef CAS PubMed.
- E. E. Simanek, D.-H. Huang, L. Pasternack, T. D. Machajewski, O. Seitz, D. S. Millar, H. J. Dyson and C.-H. Wong, J. Am. Chem. Soc., 1998, 120, 11567–11575 CrossRef CAS.
- W. Wu, L. Pasternack, D.-H. Huang, K. M. Koeller, C.-C. Lin, O. Seitz and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 2409–2417 CrossRef CAS.
- L. Lu, D. Fan, C.-W. Hu, M. Worth, Z.-X. Ma and J. Jiang, Biochemistry, 2016, 55, 1149–1158 CrossRef CAS PubMed.
- X. Cheng and G. W. Hart, J. Biol. Chem., 2001, 276, 10570–10575 CrossRef CAS PubMed.
- Y.-X. Chen, J.-T. Du, L.-X. Zhou, X.-H. Liu, Y.-F. Zhao, H. Nakanishi and Y.-M. Li, Chem. Biol., 2006, 13, 937–944 CrossRef CAS PubMed.
- A. Fernández-Tejada, F. Corzana, J. H. Busto, G. Jiménez-Osés, J. Jiménez-Barbero, A. Avenoza and J. M. Peregrina, Chem. – Eur. J., 2009, 15, 7297–7301 CrossRef PubMed.
- F.-C. Liang, R. P.-Y. Chen, C.-C. Lin, K.-T. Huang and S. I. Chan, Biochem. Biophys. Res. Commun., 2006, 342, 482–488 CrossRef CAS PubMed.
- M. B. Elbaum and N. J. Zondlo, Biochemistry, 2014, 53, 2242–2260 CrossRef CAS PubMed.
- M. A. Brister, A. K. Pandey, A. A. Bielska and N. J. Zondlo, J. Am. Chem. Soc., 2014, 136, 3803–3816 CrossRef CAS PubMed.
- F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart and C.-X. Gong, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10804–10809 CrossRef CAS PubMed.
- L. Rani and S. S. Mallajosyula, J. Phys. Chem. B, 2017, 121, 10618–10638 CrossRef CAS PubMed.
- L. Rani, J. Mittal and S. S. Mallajosyula, J. Phys. Chem. B, 2020, 124, 1909–1918 CrossRef CAS PubMed.
- C. Smet-Nocca, M. Broncel, J.-M. Wieruszeski, C. Tokarski, X. Hanoulle, A. Leroy, I. Landrieu, C. Rolando, G. Lippens and C. P. R. Hackenberger, Mol. BioSyst., 2011, 7, 1420–1429 RSC.
- S. A. Yuzwa, A. H. Cheung, M. Okon, L. P. McIntosh and D. J. Vocadlo, J. Mol. Biol., 2014, 426, 1736–1752 CrossRef CAS PubMed.
- G. Bourré, F.-X. Cantrelle, A. Kamah, B. Chambraud, I. Landrieu and C. Smet-Nocca, Front. Endocrinol., 2018, 9, 595 CrossRef PubMed.
- M. Frenkel-Pinter, M. Richman, A. Belostozky, A. Abu-Mokh, E. Gazit, S. Rahimipour and D. Segal, Chem. – Eur. J., 2018, 24, 14039–14043 CrossRef CAS PubMed.
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Y. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed.
- K. Wu, D. Li, P. Xiu, B. Ji and J. Diao, Phys. Biol., 2020, 18, 16002 CrossRef PubMed.
- G. Arsequell, M. Rosa, C. Mayato, R. L. Dorta, V. Gonzalez-Nunez, K. Barreto-Valer, F. Marcelo, L. P. Calle, J. T. Vázquez, R. E. Rodríguez, J. Jiménez-Barbero and G. Valencia, Org. Biomol. Chem., 2011, 9, 6133–6142 RSC.
- P. M. Levine, C. A. De Leon, A. Galesic, A. Balana, N. P. Marotta, Y. E. Lewis and M. R. Pratt, Bioorg. Med. Chem., 2017, 25, 4977–4982 CrossRef CAS PubMed.
- P. M. Levine, A. T. Balana, E. Sturchler, C. Koole, H. Noda, B. Zarzycka, E. J. Daley, T. T. Truong, V. Katritch, T. J. Gardella, D. Wootten, P. M. Sexton, P. McDonald and M. R. Pratt, J. Am. Chem. Soc., 2019, 141, 14210–14219 CrossRef CAS PubMed.
- Y. Ohnishi, M. Ichikawa and Y. Ichikawa, Bioorg. Med. Chem. Lett., 2000, 10, 1289–1291 CrossRef CAS PubMed.
- S. Nadal, R. Raj, S. Mohammed and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 45, 35–47 CrossRef CAS PubMed.
- H. J. Tarbet, C. A. Toleman and M. Boyce, Biochemistry, 2018, 57, 13–21 CrossRef CAS PubMed.
- W. H. Yang, S. Y. Park, H. W. Nam, D. H. Kim, J. G. Kang, E. S. Kang, Y. S. Kim, H. C. Lee, K. S. Kim and J. W. Cho, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17345–17350 CrossRef CAS PubMed.
- C. Gewinner, G. Hart, N. Zachara, R. Cole, C. Beisenherz-Huss and B. Groner, J. Biol. Chem., 2004, 279, 3563–3572 CrossRef CAS PubMed.
- H.-B. Ruan, X. Han, M.-D. Li, J. P. Singh, K. Qian, S. Azarhoush, L. Zhao, A. M. Bennett, V. T. Samuel, J. Wu, J. R. Yates and X. Yang, Cell Metab., 2012, 16, 226–237 CrossRef CAS PubMed.
- R. Fujiki, W. Hashiba, H. Sekine, A. Yokoyama, T. Chikanishi, S. Ito, Y. Imai, J. Kim, H. H. He, K. Igarashi, J. Kanno, F. Ohtake, H. Kitagawa, R. G. Roeder, M. Brown and S. Kato, Nature, 2011, 480, 557–560 CrossRef CAS PubMed.
- Q. Chen, Y. Chen, C. Bian, R. Fujiki and X. Yu, Nature, 2013, 493, 561–564 CrossRef CAS PubMed.
- R. Cao, L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage, P. Tempst, R. S. Jones and Y. Zhang, Science, 2002, 298, 1039–1043 CrossRef CAS PubMed.
- C.-S. Chu, P.-W. Lo, Y.-H. Yeh, P.-H. Hsu, S.-H. Peng, Y.-C. Teng, M.-L. Kang, C.-H. Wong and L.-J. Juan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1355–1360 CrossRef CAS PubMed.
- P.-W. Lo, J.-J. Shie, C.-H. Chen, C.-Y. Wu, T.-L. Hsu and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7302–7307 CrossRef CAS PubMed.
- N. J. Cox, G. Unlu, B. J. Bisnett, T. R. Meister, B. M. Condon, P. M. Luo, T. J. Smith, M. Hanna, A. Chhetri, E. J. Soderblom, A. Audhya, E. W. Knapik and M. Boyce, Biochemistry, 2018, 57, 91–107 CrossRef CAS PubMed.
- H. J. Tarbet, L. Dolat, T. J. Smith, B. M. Condon, E. T. O’Brien III, R. H. Valdivia and M. Boyce, eLife, 2018, 7, e31807 CrossRef PubMed.
- T. Lefebvre, C. Cieniewski, J. Lemoine, Y. Guerardel, Y. Leroy, J.-P. Zanetta and J.-C. Michalski, Biochem. J., 2001, 360, 179–188 CrossRef CAS.
- C. Guinez, J. Lemoine, J.-C. Michalski and T. Lefebvre, Biochem. Biophys. Res. Commun., 2004, 319, 21–26 CrossRef CAS PubMed.
- B. Ryva, K. Zhang, A. Asthana, D. Wong, Y. Vicioso and R. Parameswaran, Front. Oncol., 2019, 9, 100 CrossRef PubMed.
- Y. Nagata and M. M. Burger, J. Biol. Chem., 1974, 249, 3116–3122 CrossRef CAS.
- J.-P. Privat, F. Delmotte and M. Monsigny, FEBS Lett., 1974, 46, 224–228 CrossRef CAS PubMed.
- P. Midoux, J. P. Grivet, F. Delmotte and M. Monsigny, Biochem. Biophys. Res. Commun., 1984, 119, 603–611 CrossRef CAS PubMed.
- G. Bains, R. T. Lee, Y. C. Lee and E. Freire, Biochemistry, 1992, 31, 12624–12628 CrossRef CAS PubMed.
- D. Neumann, O. Kohlbacher, H.-P. Lenhof and C.-M. Lehr, Eur. J. Biochem., 2002, 269, 1518–1524 CrossRef CAS PubMed.
- J. F. Espinosa, J. L. Asensio, J. L. García, J. Laynez, M. Bruix, C. Wright, H.-C. Siebert, H.-J. Gabius, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Biochem., 2000, 267, 3965–3978 CrossRef CAS PubMed.
- J. Angulo, P. M. Enríquez-Navas and P. M. Nieto, Chem. – Eur. J., 2010, 16, 7803–7812 CrossRef CAS PubMed.
- C. S. Wright and G. E. Kellogg, Protein Sci., 1996, 5, 1466–1476 CrossRef CAS PubMed.
- V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 900–903 CrossRef CAS PubMed.
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704–8719 CrossRef CAS PubMed.
- C. Hamark, J. Landström and G. Widmalm, Chem. – Eur. J., 2014, 20, 13905–13908 CrossRef CAS PubMed.
- I. Pérez-Victoria, O. Boutureira, T. D. W. Claridge and B. G. Davis, Chem. Commun., 2015, 51, 12208–12211 RSC.
- B. Linclau, A. Ardá, N.-C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jiménez-Barbero, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- L. P. Calle, B. Echeverria, A. Franconetti, S. Serna, M. C. Fernández-Alonso, T. Diercks, F. J. Cañada, A. Ardá, N.-C. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2015, 21, 11408–11416 CrossRef CAS PubMed.
- L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2017, 23, 3957–3965 CrossRef CAS PubMed.
- F. Corzana, A. Fernández-Tejada, J. H. Busto, G. Joshi, A. P. Davis, J. Jiménez-Barbero, A. Avenoza and J. M. Peregrina, ChemBioChem, 2011, 12, 110–117 CrossRef CAS PubMed.
- A. Fernández-Tejada, F. Corzana, J. H. Busto, G. Jiménez-Osés, J. M. Peregrina and A. Avenoza, Chem. – Eur. J., 2008, 14, 7042–7058 CrossRef PubMed.
- G. D. Holt, C. M. Snow, A. Senior, R. S. Haltiwanger, L. Gerace and G. W. Hart, J. Cell Biol., 1987, 104, 1157–1164 CrossRef CAS PubMed.
- J. S. Haurum, G. Arsequell, A. C. Lellouch, S. Y. Wong, R. A. Dwek, A. J. McMichael and T. Elliott, J. Exp. Med., 1994, 180, 739–744 CrossRef CAS PubMed.
- J. S. Haurum, L. Tan, G. Arsequell, P. Frodsham, A. C. Lellouch, P. A. H. Moss, R. A. Dwek, A. J. McMichael and T. Elliott, Eur. J. Immunol., 1995, 25, 3270–3276 CrossRef CAS PubMed.
- A. Glithero, J. Tormo, J. S. Haurum, G. Arsequell, G. Valencia, J. Edwards, S. Springer, A. Townsend, Y. L. Pao, M. Wormald, R. A. Dwek, E. Y. Jones and T. Elliott, Immunity, 1999, 10, 63–74 CrossRef CAS PubMed.
- S. A. Malaker, S. A. Penny, L. G. Steadman, P. T. Myers, J. C. Loke, M. Raghavan, D. L. Bai, J. Shabanowitz, D. F. Hunt and M. Cobbold, Cancer Immunol. Res., 2017, 5, 376–384 CrossRef CAS PubMed.
- F. Mohammed, M. Cobbold, A. L. Zarling, M. Salim, G. A. Barrett-Wilt, J. Shabanowitz, D. F. Hunt, V. H. Engelhard and B. E. Willcox, Nat. Immunol., 2008, 9, 1236–1243 CrossRef CAS PubMed.
- F. Marino, M. Bern, G. P. M. Mommen, A. C. Leney, J. A. M. van Gaans-van den Brink, A. M. J. J. Bonvin, C. Becker, C. A. C. M. van Els and A. J. R. Heck, J. Am. Chem. Soc., 2015, 137, 10922–10925 CrossRef CAS PubMed.
- N. E. Zachara, N. O’Donnell, W. D. Cheung, J. J. Mercer, J. D. Marth and G. W. Hart, J. Biol. Chem., 2004, 279, 30133–30142 CrossRef CAS PubMed.
- Y. Zhu, T.-W. Liu, S. Cecioni, R. Eskandari, W. F. Zandberg and D. J. Vocadlo, Nat. Chem. Biol., 2015, 11, 319–325 CrossRef CAS PubMed.
- Y. Li, M. Xie, L. Men and J. Du, Int. J. Mol. Med., 2019, 44, 363–374 CAS.
- Y.-H. Chang, C.-L. Weng and K.-I. Lin, J. Biomed. Sci., 2020, 27, 57 CrossRef CAS PubMed.
- M. Quik, C. H. Hokke and B. Everts, Immunology, 2020, 161, 175–185 CrossRef CAS PubMed.
- A. Estevez, D. Zhu, C. Blankenship and J. Jiang, Chem. – Eur. J., 2020, 26, 12086–12100 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |