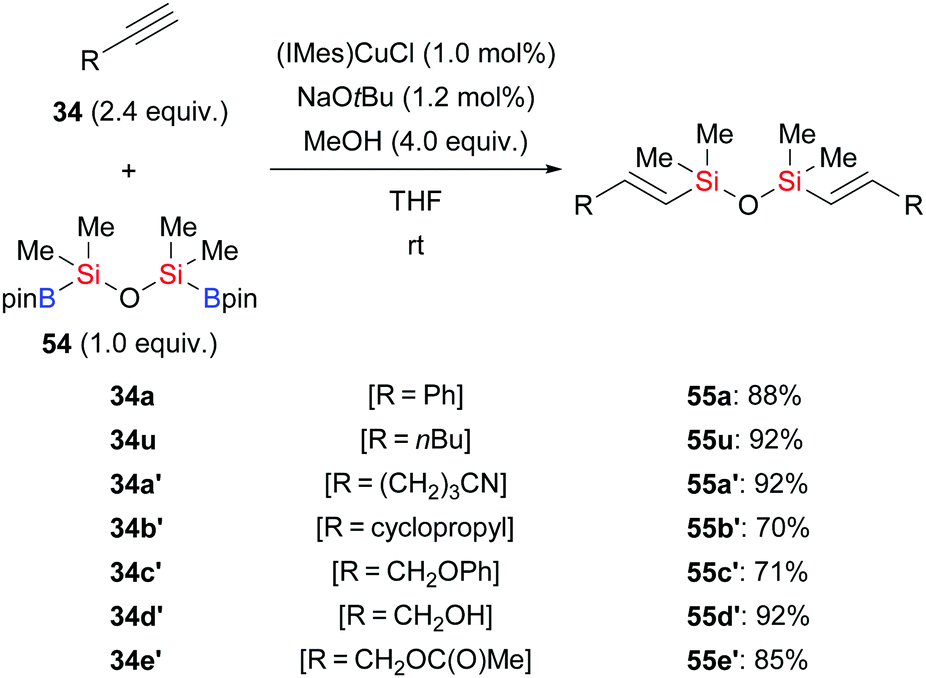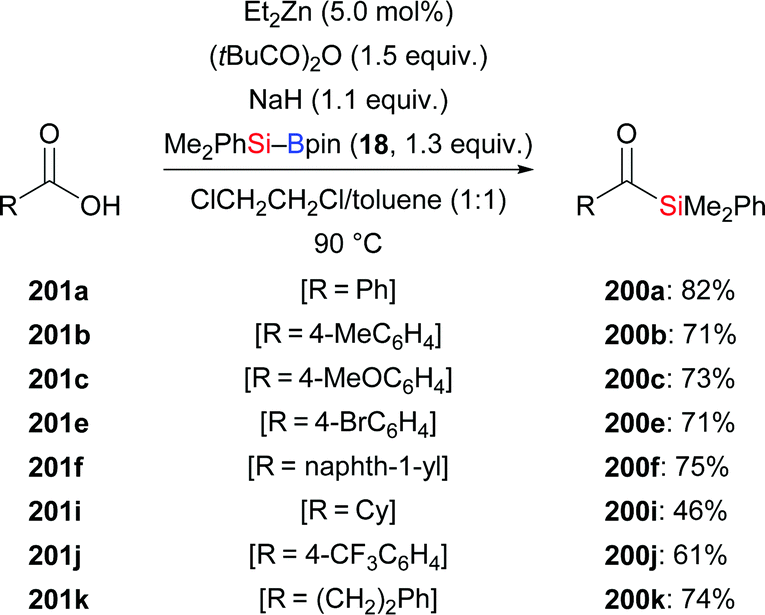DOI:
10.1039/D0CS00965B
(Review Article)
Chem. Soc. Rev., 2021,
50, 2010-2073
Activation of the Si–B interelement bond related to catalysis
Received
29th July 2020
First published on 16th December 2020
Abstract
Si–B reagents, namely silylboronic esters and silylboranes, have become increasingly attractive as versatile reagents to introduce silicon and boron atoms into organic frameworks. Diverse transformations through transition-metal-catalysed or transition-metal-free Si–B bond activation have become available. This Review summarises the recent developments in the now broad field of Si–B chemistry and covers the literature from the last seven years as an update of our review on the same topic published in early 2013 (M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441). It mainly focuses on new applications of Si–B reagents but new methods of their preparation and, where relevant, reaction mechanisms are also discussed.

Jian-Jun Feng
| Jian-Jun Feng (born in 1985 in Jingdezhen/China) received his PhD degree in chemistry from the East China Normal University in 2013 under the supervision of Professor Junliang Zhang. He spent 2013–2014 as a senior synthetic chemist at the WuXi AppTec and 2014–2017 as a Lecturer at East China Normal University. He then moved to the Technische Universität Berlin in 2017 to pursue postdoctoral training in silicon chemistry in Professor Oestreich's group funded by the Alexander von Humboldt-Stiftung. He joined Hunan University as a full professor in 2020. His research interests include the development of catalytic asymmetric reactions and sustainable catalysis. |

Wenbin Mao
| Wenbin Mao (born in 1991 Danyang/China) studied chemistry at Soochow University (2009–2013 and 2014–2017). He obtained his bachelor's degree (2013) with Professor Baolong Li and master's degree (2017) with Professor Chen Zhu. He is currently pursuing graduate research in the group of Martin Oestreich at the Technische Universität Berlin funded by China Scholarship Council. |

Liangliang Zhang
| Liangliang Zhang (born in 1991 in Shandong/China) studied chemistry at the Shandong Normal University (2010–2014) and Xiamen University (2014–2017). He obtained his bachelor's (2014) and master's degrees (2017) with Professor Guo Tang. He is currently pursuing graduate research in the group of Martin Oestreich at the Technische Universität Berlin funded by China Scholarship Council. |

Martin Oestreich
| Martin Oestreich (born in 1971 in Pforzheim/Germany) is Professor of Organic Chemistry at the Technische Universität Berlin. He received his diploma degree with Paul Knochel (Marburg, 1996) and his doctoral degree with Dieter Hoppe (Münster, 1999). After a two-year postdoctoral stint with Larry E. Overman (Irvine, 1999–2001), he completed his habilitation with Reinhard Brückner (Freiburg, 2001–2005) and was appointed as Professor of Organic Chemistry at the Westfälische Wilhelms-Universität Münster (2006–2011). He also held visiting positions at Cardiff University in Wales (2005), at The Australian National University in Canberra (2010), and at Kyoto University in Japan (2018). Martin recently edited a monograph entitled Organosilicon Chemistry: Novel Approaches and Reactions together with Tamejiro Hiyama. |
1. Introduction
Silylboronic esters and silylboranes are a class of interelement compounds that contain an Si–B bond. The electronegativity difference between silicon (EN = 1.8) and boron (EN = 2.0) allows for chemoselective activation of that bond. Activation can be achieved by nucleophiles/bases as well as transition-metal catalysts. Pioneering contributions by Nöth and Y. Ito with Suginome to the synthesis of reasonably stable Si–B derivatives paved the way for their routine use in organic main-group chemistry. These reagents do not only serve as silicon pronucleophiles but also as boron sources. The rapid development of this field is documented by a steadly increasing number of publications. In early 2013, we published a comprehensive review on Si–B chemistry in Chemical Reviews.1,2 Since then, important progress has been made as reflected by numerous reviews published in recent years, often covering certain aspects of this chemistry.3–9 However, a full treatise of this timely topic such as our initial review is missing. The present Review is an update that completely summarises the literature published from January 2013 to April 2020 following our earlier organization. Older references have been included when needed for clarification or explanation.
2. Preparation of Si–B compounds
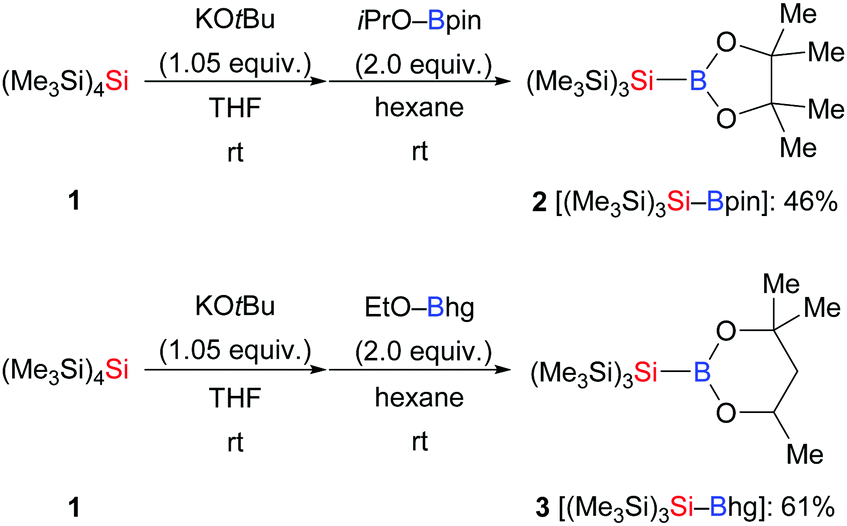
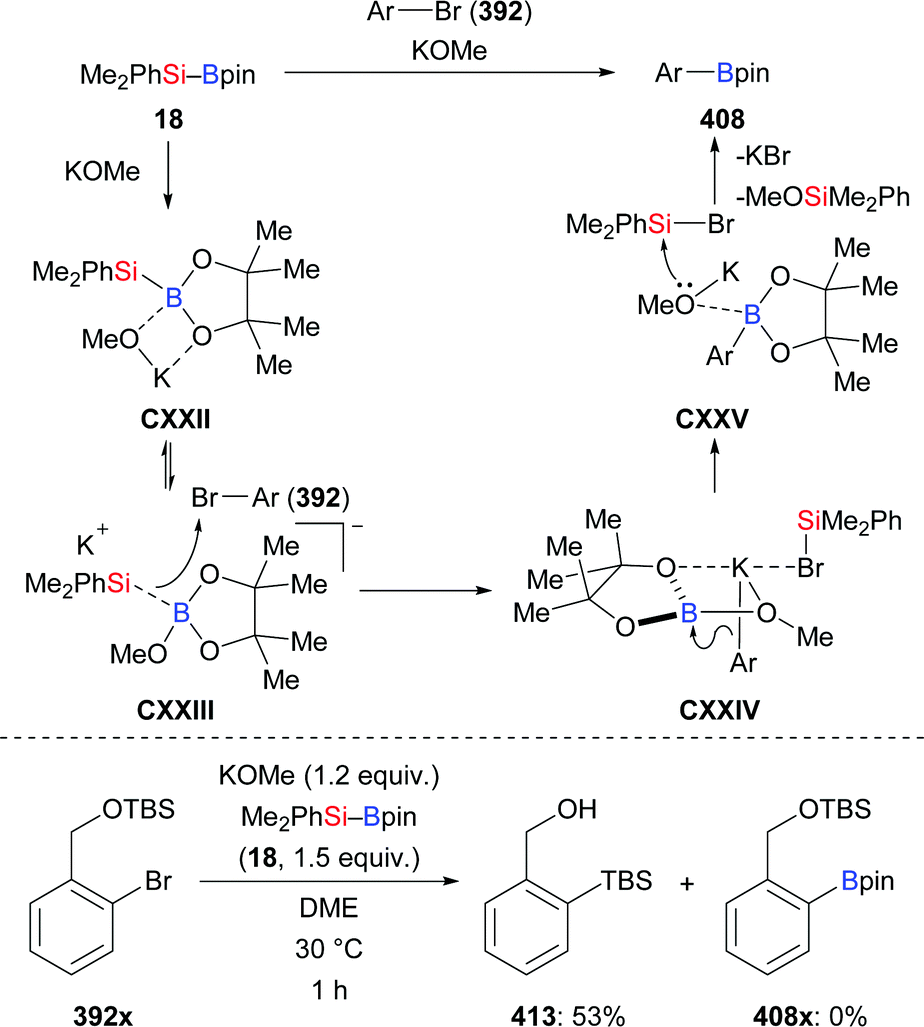
In general, the preparation of Si–B compounds can be achieved by nucleophilic substitution at boron with silyllithium reagents, intramolecular reductive coupling of chlorosilanes and chloroboranes, and transition-metal-catalysed borylation of tertiary hydrosilanes.1,10 Especially the synthetic contributions made by Nöth,10a,c Tanaka,10b and Y. Ito and Suginome10d,e as well as Hartwig10f have enabled the widespread use of several Si–B compounds such as Me2PhSi–Bpin, Me2PhSi–B(NR2)2 and Et3Si–Bpin. However, as these well-established Si–B compounds are unstable to air and moisture, the laboratory of H. Ito developed two bulky tris(trimethylsilyl)silylboronic esters (Me3Si)3Si–Bpin (2) and (Me3Si)3Si–Bhg (3) by nucleophilic displacement at the corresponding boron electrophiles with tris(trimethylsilyl)silylpotassium generated from 1 (Scheme 1).11 Notably, these Si–B compounds were readily purified by column chromatography over silica gel and exhibit high stability to air.
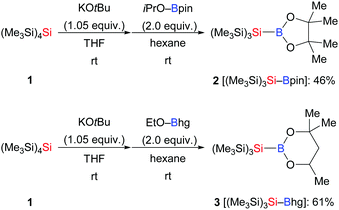 |
| | Scheme 1 Synthesis of air-stable (Me3Si)3Si–B(OR)2. | |
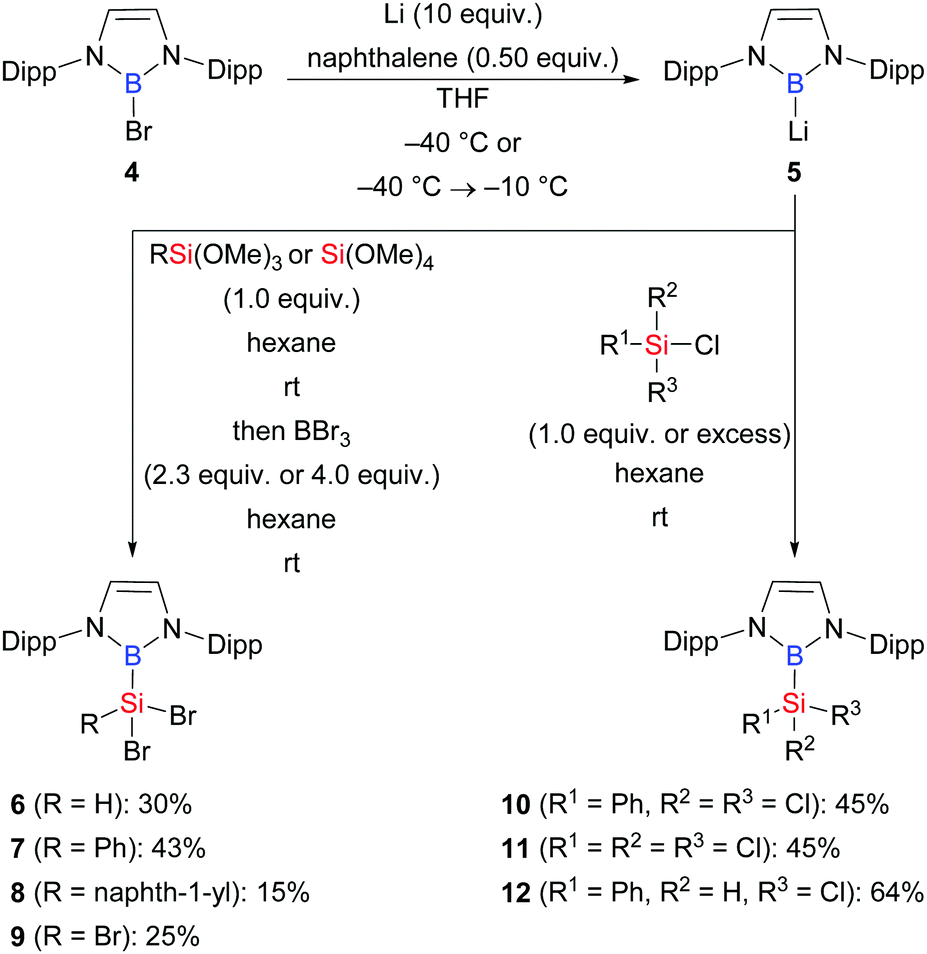
In 2020, a reverse route employing a boryl anion and silicon electrophiles was systematically investigated by the laboratory of Cui (Scheme 2).12 These authors showed that Si–B compounds 6–12 are accessible in moderate to good yields from reactions of 5 with a series of chloro- and alkoxysilanes.
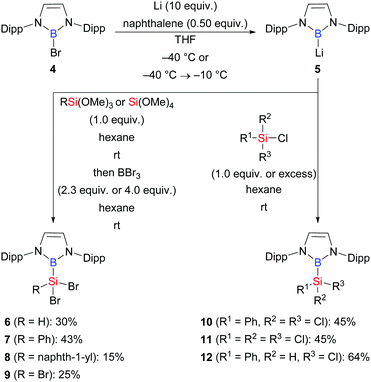 |
| | Scheme 2 Synthesis of Si–B compounds from the reaction of a boryl anion with chloro- and alkoxysilanes. | |
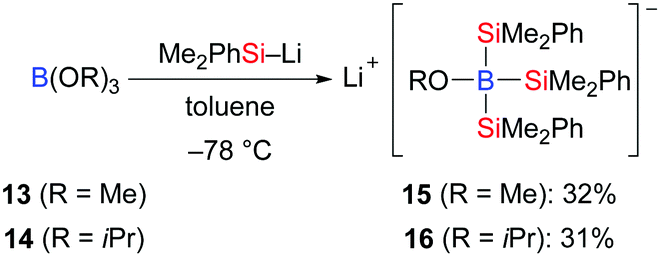
In contrast to the silylboronic esters, studies on structurally characterised silylborates are still rare. Tsurusaki and Kyushin successfully accomplished the preparation of lithium alkoxytris(dimethylphenylsilyl)borates by the reaction of trialkyl borates B(OR)313 (R = Me) and 14 (R = iPr) with dimethylphenylsilyllithium (Scheme 3).13 These authors also solved the molecular structures of the contact ion pair 15 and the solvent-separated ion pair 16 by X-ray diffraction (not shown).
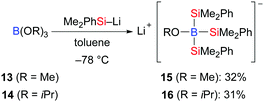 |
| | Scheme 3 Synthesis of lithium alkoxytris- (dimethylphenylsilyl)borates. | |
3. Mechanisms of Si–B bond activation
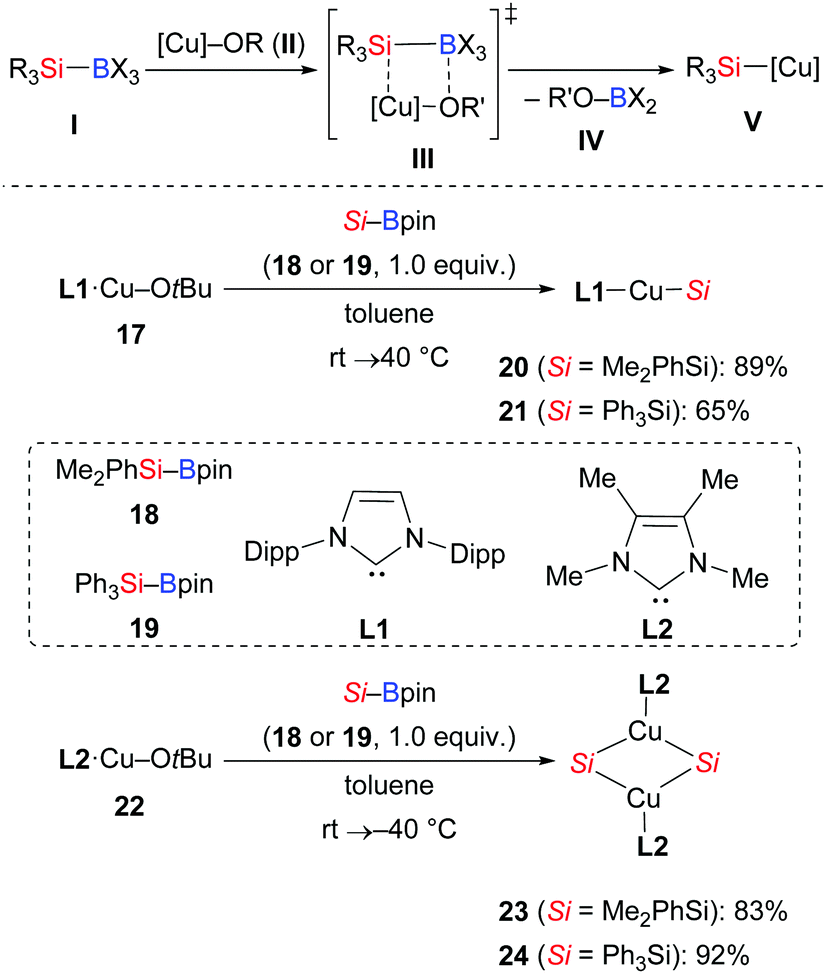
The mechanisms for the chemoselective cleavage of Si–B bonds are diverse, ranging from oxidative addition, transmetalation, Lewis base activation, and carbenoid insertion to photochemical radical processes.1,14 Among these, transmetalation of the Si–B linkage at a Cu–O bond has been most frequently used to release silylcopper nucleophiles V in recent years (Scheme 4, top). To gain insight into this activation, Kleeberg and co-workers studied an [(NHC)Cu–OtBu] system, and their results showed great influence of the steric properties of the NHC (Scheme 4, bottom).15 As a consequence, sterically demanding NHC ligands such as IDipp L1 led to monomeric, linear complexes [(NHC)Cu–SiR3] 20 and 21, while with a less demanding ITMe ligand L2, the dinuclear, μ-SiR3-bridged complexes 23 and 24 with ultrashort Cu⋯Cu distances were observed.
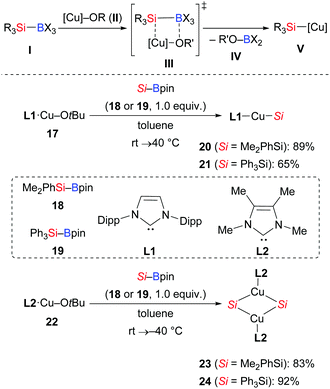 |
| | Scheme 4 Si–B bond activation by [(NHC)CuOtBu] complexes. | |
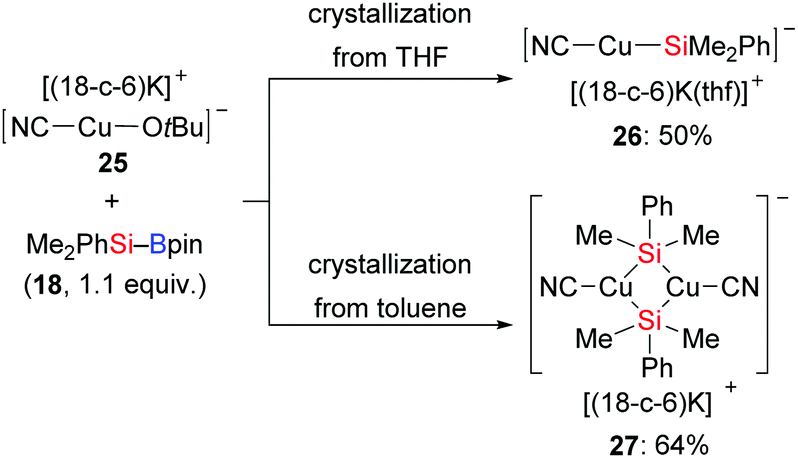
Shortly thereafter, the same group synthesised the alkoxycyanocuprate [(18-c-6)K][NC–Cu–OtBu] 25 as a well-defined catalyst model to mimick another established catalytic system, CuCN/NaOMe (Scheme 5).16 Interestingly, a linear, two-coordinated copper complex 26 was obtained from THF but a solvent free dimeric μ-silyl-bridged complex 27 with a very short Cu⋯Cu distance formed in toluene.
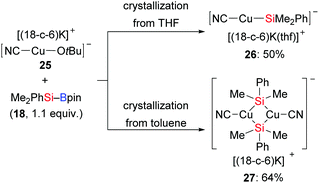 |
| | Scheme 5 Si–B bond activation by [(18-c-6)K][NC-Cu(I)-OtBu] (with 18-c-6 = 18-crown-6). | |
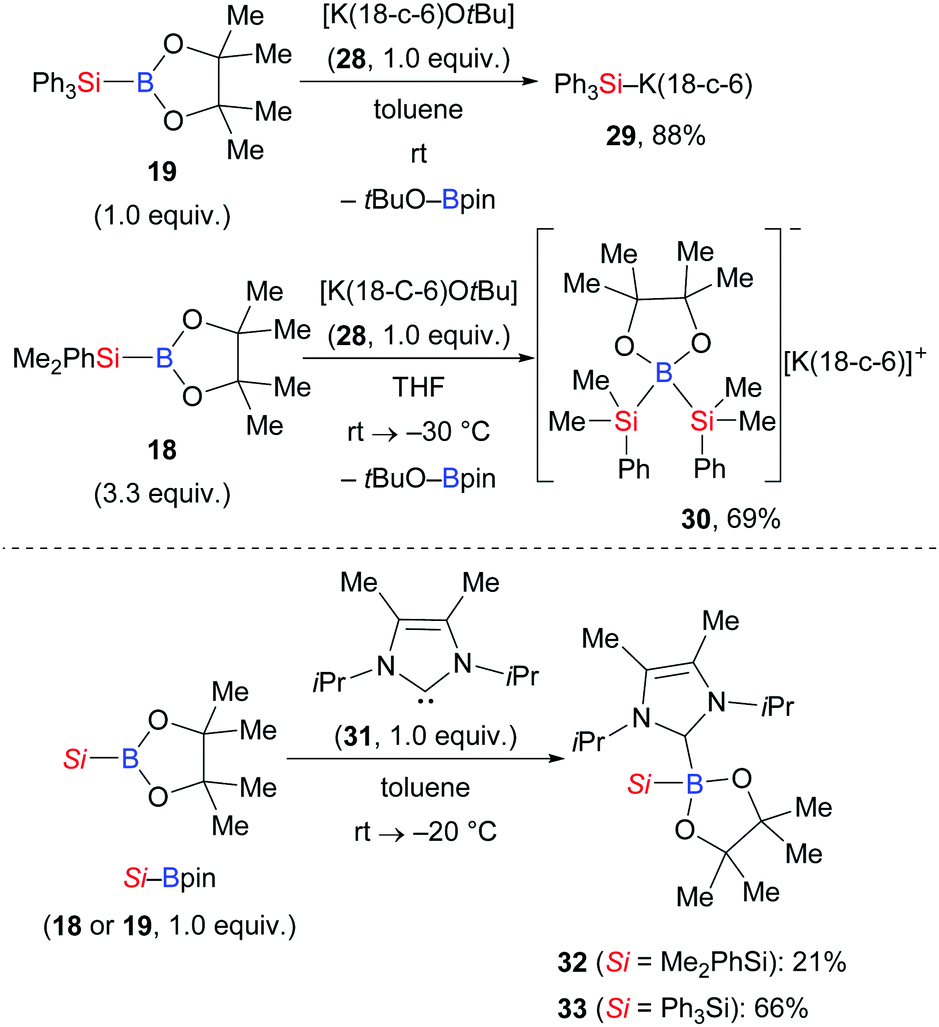
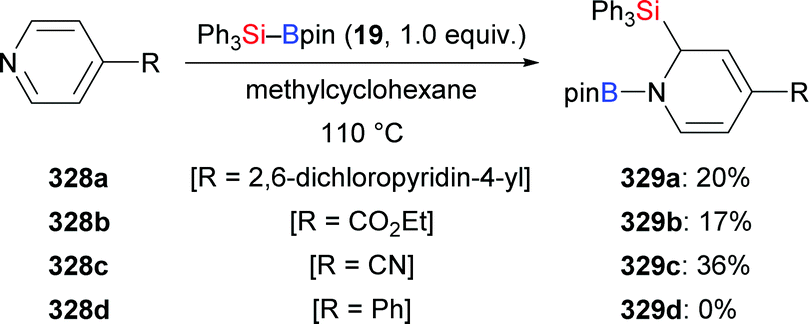
Apart from this, Kleeberg and co-workers also looked into the activation of Si–B bonds with Lewis bases.17 These authors performed a comparative study on the treatment of Si–B compounds, e.g. Ph3Si–Bpin (19) and Me2PhSi–Bpin (18), with potassium(18-crown-6) tert-butoxide (28) and 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (31), respectively (Scheme 6). The reaction with K(18-crown-6) tert-butoxide led to the activation of the Si–B bond, providing either the silylpotassium complex [K(18-c-6)SiPh3] (29)18 or [K(18-c-6)(thf)2][pinB(SiMe2Ph)2] (30), the formal Lewis pair of [K(18-c-6)SiMe2Ph] and Me2PhSi–Bpin (18). Both complexes basically reacted as sources of nucleophilic silyl moieties in reactions with selected electrophiles (not shown). Conversely, the use of Lewis base 31 resulted in the formation of the isolable Lewis acid/base adducts 32 and 33, which do not react as sources of nucleophilic silyl moieties.
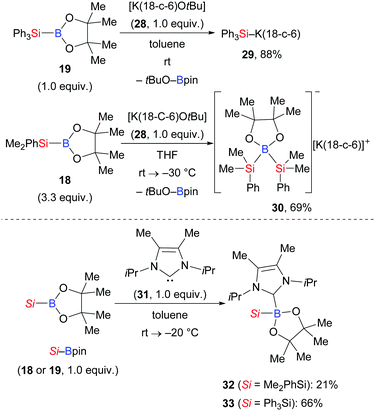 |
| | Scheme 6 Si–B bond activation promoted by Lewis bases. | |
4. Functionalization of unsaturated compounds
4.1. 1,2-Addition to isolated C–C multiple bonds
4.1.1. Alkynes.
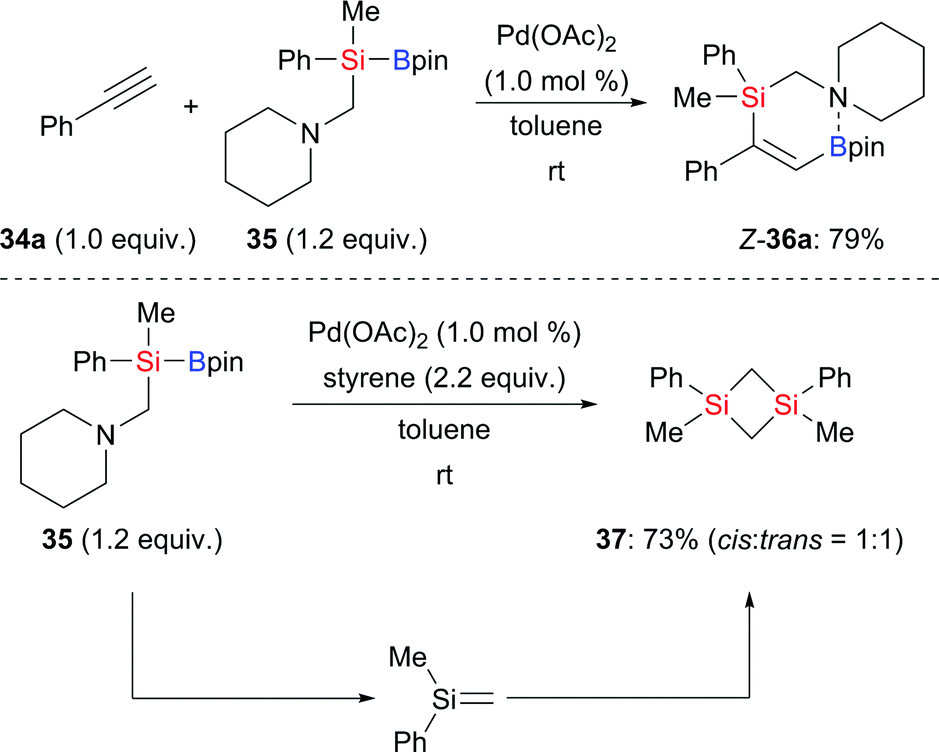
Alkyne silaboration by 1,2-addition of a Si–B bond is a powerful tool to construct multisubstituted alkenes. Palladium catalysis is one of the most important among a variety of alkyne silaborations.19 In 2017, Ohmura, Suginome and co-worker reported a palladium-catalysed silaboration of phenylacetylene, employing silylboronic ester 35 with an aminomethyl group attached to the silicon atom (34a → Z-36a, Scheme 7, top).20 This reaction took place under mild conditions without the need for an added ligand. The 11B NMR chemical shift of Z-36a revealed Lewis pair formation between the boron atom and the nitrogen atom. There was no reaction with Me2PhSi–Bpin (18) under the same setup. This difference suggests that the nitrogen atom in 35 may coordinate to palladium, thereby facilitating the activation of the Si–B bond. Interestingly, these authors also found that Pd(OAc)2 promotes β-elimination of 35 in the presence of styrene; the thus-formed silene led to the formation of 1,3-disilacyclobutanes (35 → 37, Scheme 7, bottom).
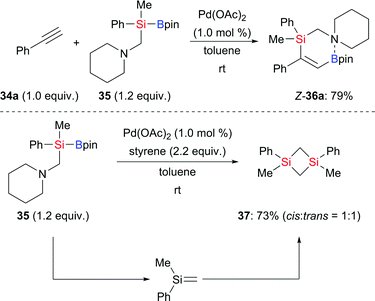 |
| | Scheme 7 Ligand-free palladium-catalysed silaboration of phenyl-acetylene. | |
In 2010, Suginome and co-workers reported the first abnormal regioselective silaboration of terminal alkynes, wherein the silyl moiety is transferred to the terminus of the alkyne. A very bulky (η3-C3H5)PdCl[o-biphenyl(tBu)2P] complex was used as catalyst (not shown).21 After that, gold-, platinum-, zinc-, and copper-based catalysts have been shown to enable the same transformation. In 2014, Stratakis and co-workers disclosed a gold-catalysed addition of silylboranes to terminal alkynes (34b–e → E-39b–e, Scheme 8, top).22 The abnormal regioselectivity has been attributed to steric factors exerted by the gold nanoparticle. This alkyne silaboration proceeds under mild conditions without any external ligands or additives. Interestingly, disproportionation of Me2PhSi–Bpin was observed, furnishing the corresponding disilane and diborane by σ-bond metathesis (not shown). Later, a platinum-catalysed method in the presence of supported platinum nanoparticles was reported by Grirrane and co-workers (34a → E-39a, Scheme 8, bottom).23 As in Stratakis's work, no additives or ligands were needed.
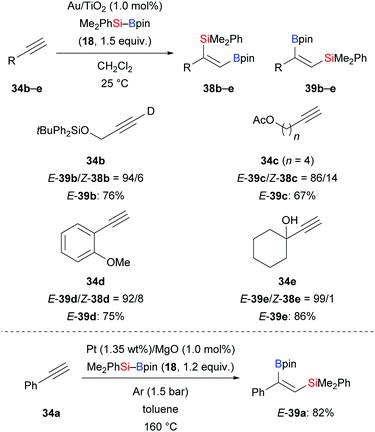 |
| | Scheme 8 Silaboration of terminal alkynes in the presence of nano-particles. | |
Activation of silylboronic esters by palladium, platinum, and gold catalysts involves oxidative addition of the Si–B bond to the metal center. The thus-generated silyl–M–boryl intermediate then undergoes migratory insertion with alkynes.22 An important contribution to these transition-metal-catalysed silaborations that does not involve oxidative addition was reported by Uchiyama and co-workers. This reaction proceeds by in situ formation of highly reactive silylzinc species in the presence of a dialkylzinc reagent, a phosphine, and a silylborane. This combination of reagents reacts with terminal alkynes to afford various trisubstituted alkenes with high regio- and stereocontrol (Scheme 9, top).24 The phosphine seems to greatly effect the regioselectivity. Without, normal regioselectivity where the boryl group is connected to the alkyne terminus was obtained (34f → Z-38f). When Ph3P was used, opposite regioselectivity was obtained (34f → E-39f). A tentative mechanism was proposed for this transformation (Scheme 9, bottom). Reaction of Me2PhSi–Bpin (18), Me2Zn, and Ph3P yields intermediate VI, which further converts into the borate complex VII by transfer of a methyl group. Subsequent transfer of the silyl group from the boron to the zinc atom releases the silylzinc species VIII and MeBpin (IX). Addition of the Zn–Si bond across the C–C triple bond gives complex X with a newly formed C–Zn bond. This reacts with the previously formed MeBpin (IX), thereby delivering the desired product and regenerating Me2Zn. Furthermore, intermediate X was trapped by electrophiles such as allyl bromides, methyl iodide, and N-iodosuccinimide (not shown).
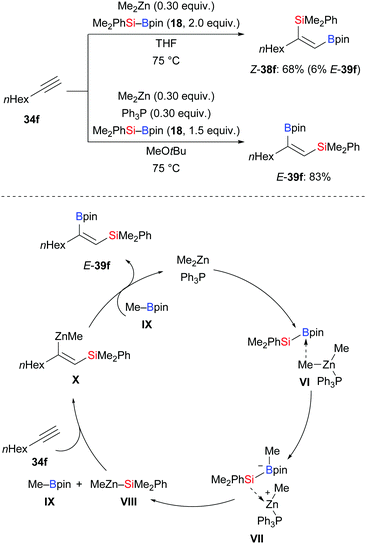 |
| | Scheme 9 Silaboration of terminal alkynes involving an in situ-generated silylzinc reagent. | |
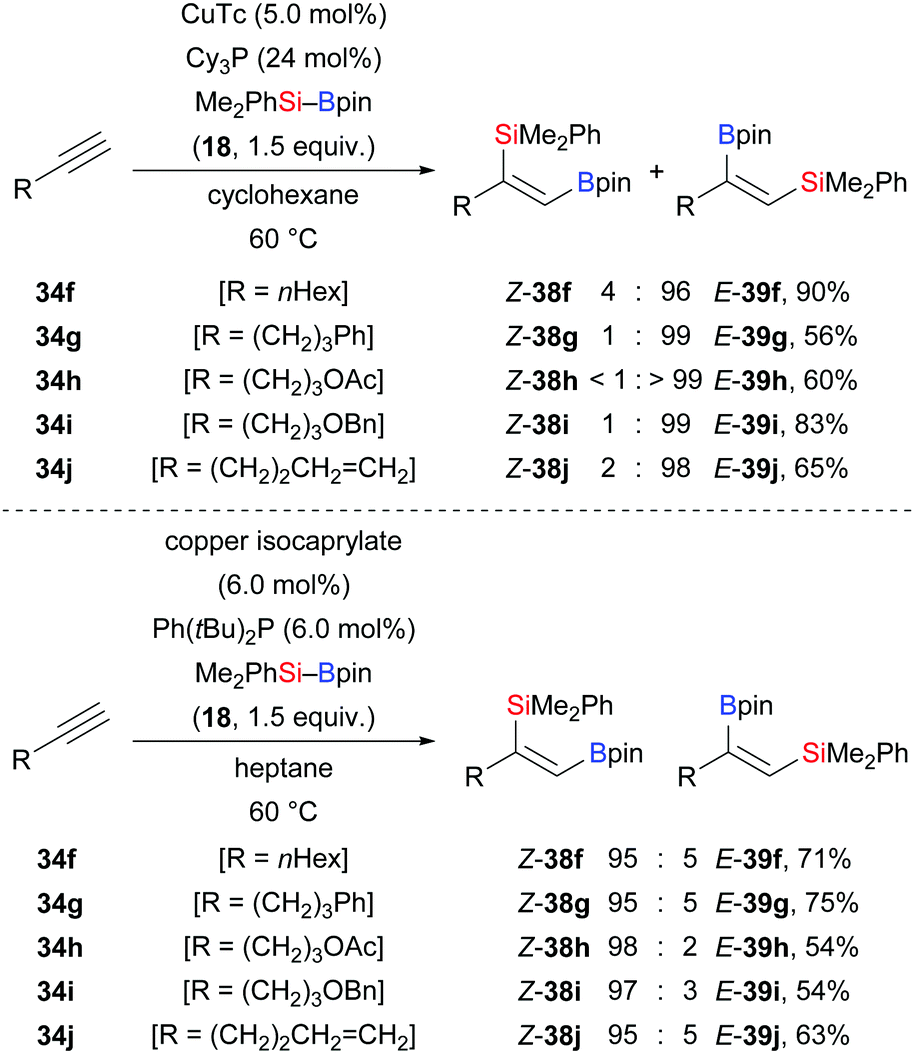
Similar regiodivergent alkyne silaborations were also found to be controllable by tuning of copper catalysts and phosphine ligands as reported by Xu and co-workers (Scheme 10).25 When CuTc and Cy3P were employed in the reaction, abnormal regioselectivity with the silyl group at the terminus was seen (34f–j → E-39f–j). In turn, the use of bulky Ph(tBu)2P as ligand and copper isocaprylate led to the opposite regioselectivity (34f–j → Z-38f–j). Bulky ligands seem to favour normal regioselectivity which stands in contrast to the aforementioned palladium-catalysed silaboration.21 When phenylacetylene was used in the reaction, only a poor yield of the desired product was detected along with hydrosilylation and gem-diborylated vinylsilane products (53% and 32%, respectively). A deuterium-labelling experiment showed that the C(sp)–H bond may serve as a proton resource during the hydrosilylation (not shown).
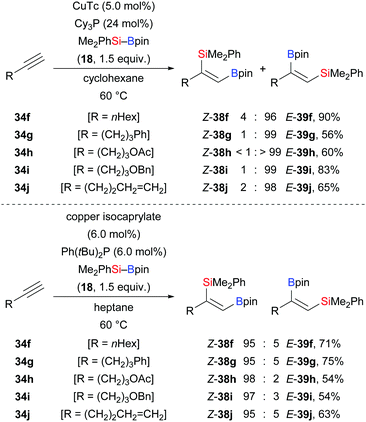 |
| | Scheme 10 Ligand-dependent regiodivergent copper-catalysed silaboration of terminal alkynes. Yields are for the major product. | |
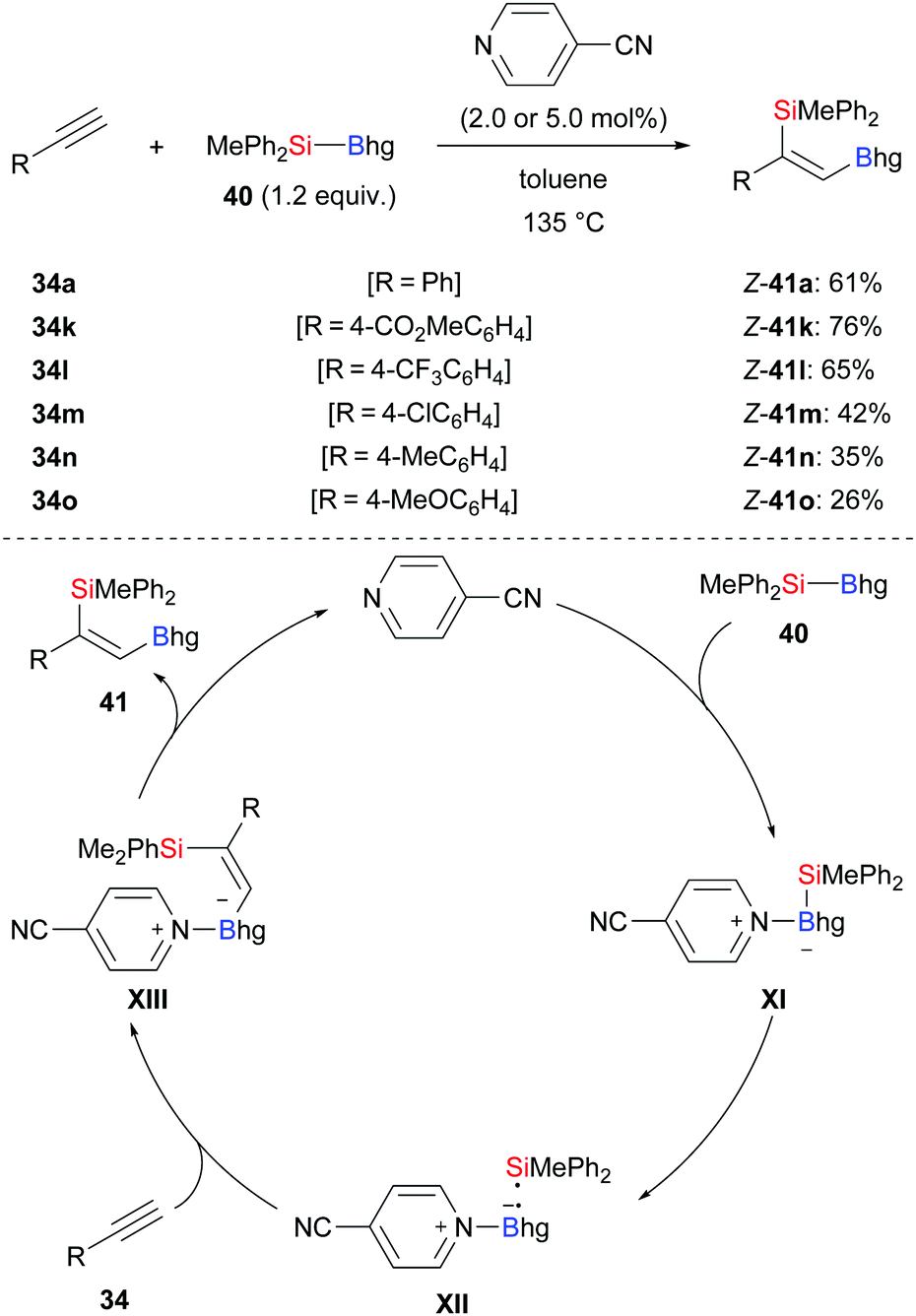
Transition-metal-free, that is organocatalytic, Si–B bond activation is a promising alternative to the previously discussed approaches. Suginome and co-workers applied pyridine-based organocatalysts to the silaboration of phenylacetylenes (34a, k–o → Z-41a, k–o, Scheme 11).26 With 4-cyanopyridine as catalyst, the reaction required 135 °C to add MePh2Si–Bhg (40) across the C–C triple bond with consistently high regio- and stereoselectivity. The proposed mechanism commences with coordination of 4-cyanopyridine to the silylborane to form adduct XI, which undergoes homolytic cleavage to afford radical pair XII. That radical pair instantaneously adds to the C–C triple bond, affording new intermediate XIII. Its dissociation gives the desired product 41 and regenerates the catalyst.
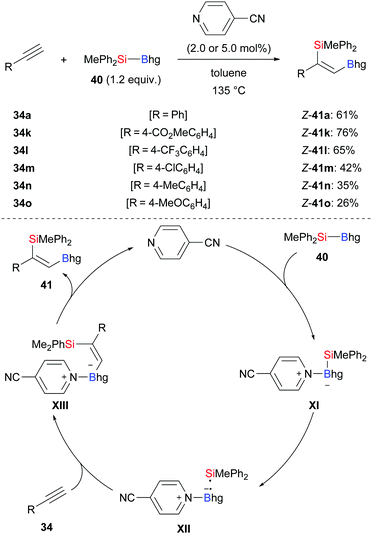 |
| | Scheme 11 Transition-metal-free silaboration of terminal alkynes catalysed by a pyridine derivative. | |
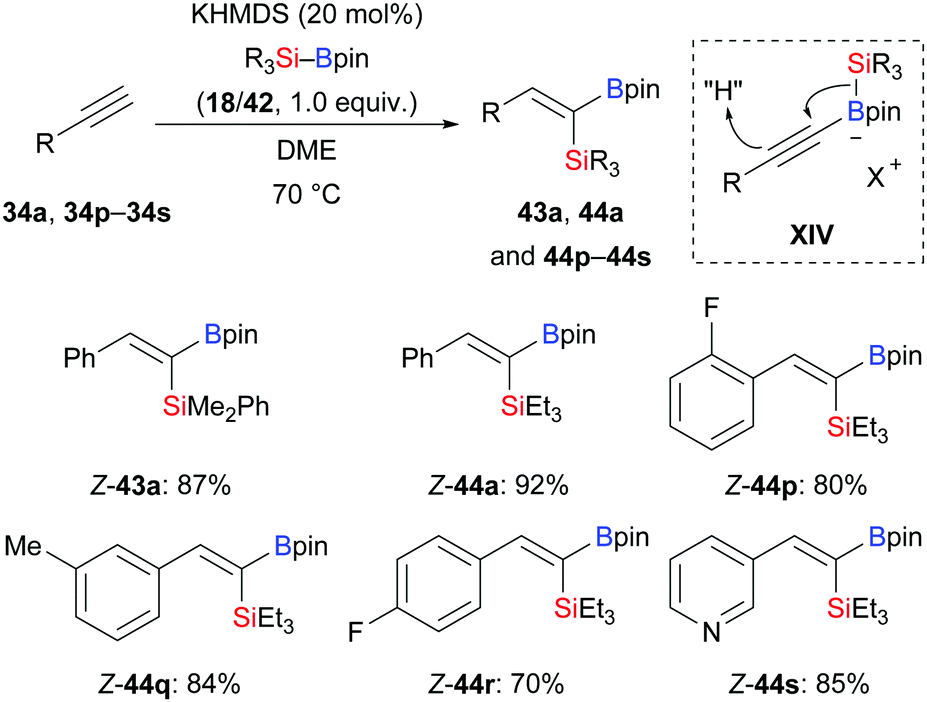
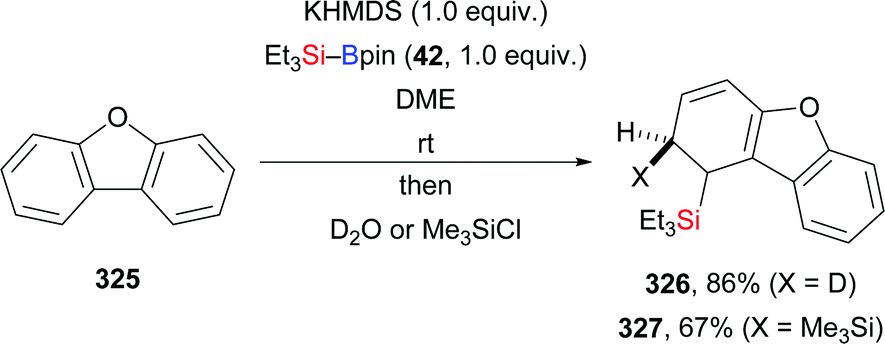
In 2020, Martin and co-workers presented a base-catalysed stereoselective 1,1-silaboration of terminal alkynes. This process proceeds with catalytic amounts of KHMDS to yield gem-silylborylated alkenes (34a, p–s → Z-43a and Z-44a, p–s, Scheme 12).27 Deuterium-labelling experiments revealed that this alkyne silaboration passes through initial deprotonaton of the C(sp)–H bond (pKa = 23) by KHMDS (pKa = 27) to then add to Et3Si–Bpin (42; not shown). The resulting ate complex XIV is believed to convert into the product in concerted fashion. The synthetic potential of this atom-economical protocol was illustrated by selective functionalization of the distinguishable C–Si and C–B bonds (not shown). It ought to be mentioned that 1,1-silaboration of ethyl propiolate mediated by a catalytic amount of an organocatalyst such as nBu3P, KOtBu or ICy was reported by Suginome and co-workers one year before (see Section 4.5).26
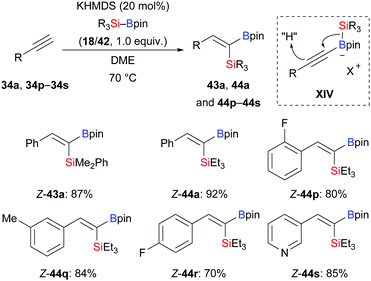 |
| | Scheme 12 Stereoselective base-catalysed 1,1-silaboration of terminal alkynes. | |
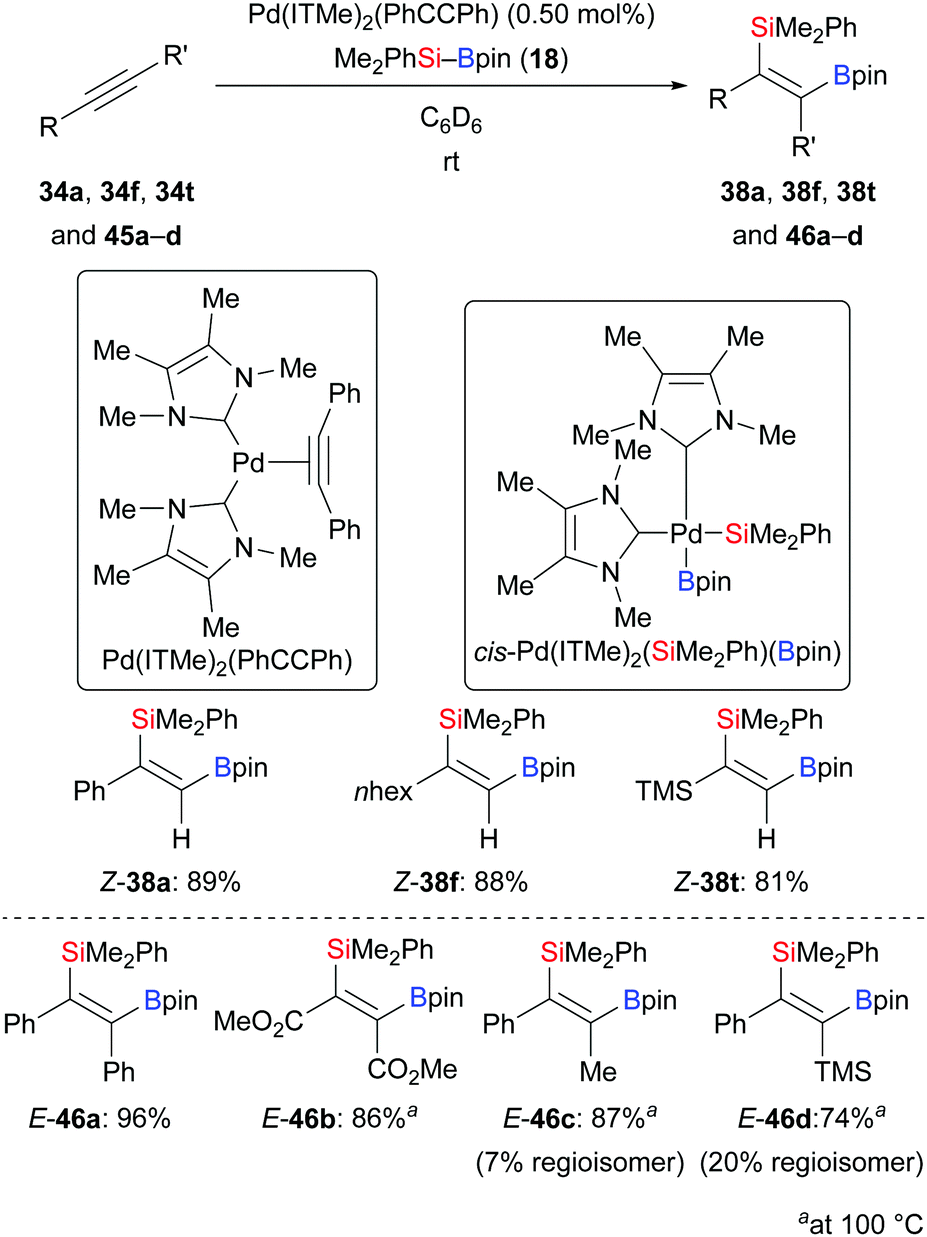
Elevated reaction temperature seems to be typical for palladium-catalysed alkyne silaborations. By applying the palladium complex Pd(ITMe)2(PhCCPh) as precatalyst, Navarro and co-workers accomplished the title reaction at room temperature (34a, f, t → 38a, f, t, Scheme 13, top).28 Low catalyst loadings and short reaction time showcased the high reactivity of this palladium complex. This method was also applicable to internal alkynes yet requiring 100 °C; exclusive cis-stereoselectivity was seen (45a–d → 46a–d, Scheme 13, bottom). Regioisomers were found when unsymmetrically substituted internal alkynes were used (45c–d → 46c–d). Notably, oxidative addition complex cis-Pd(ITMe)2(SiMe2Ph)(Bpin) was isolated in 69% yield from the reaction of Pd(ITMe)2(PhCCPh) with two equivalents of Me2PhSi–Bpin (18) in toluene, and single crystals were obtained and characterised by X-ray diffraction.
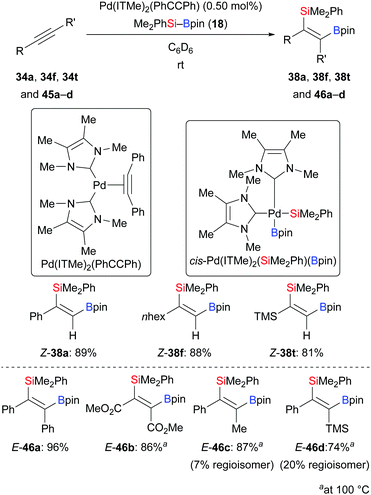 |
| | Scheme 13 Palladium-catalysed silaboration of alkynes at room temperature (terminal) and 100 °C (internal). | |
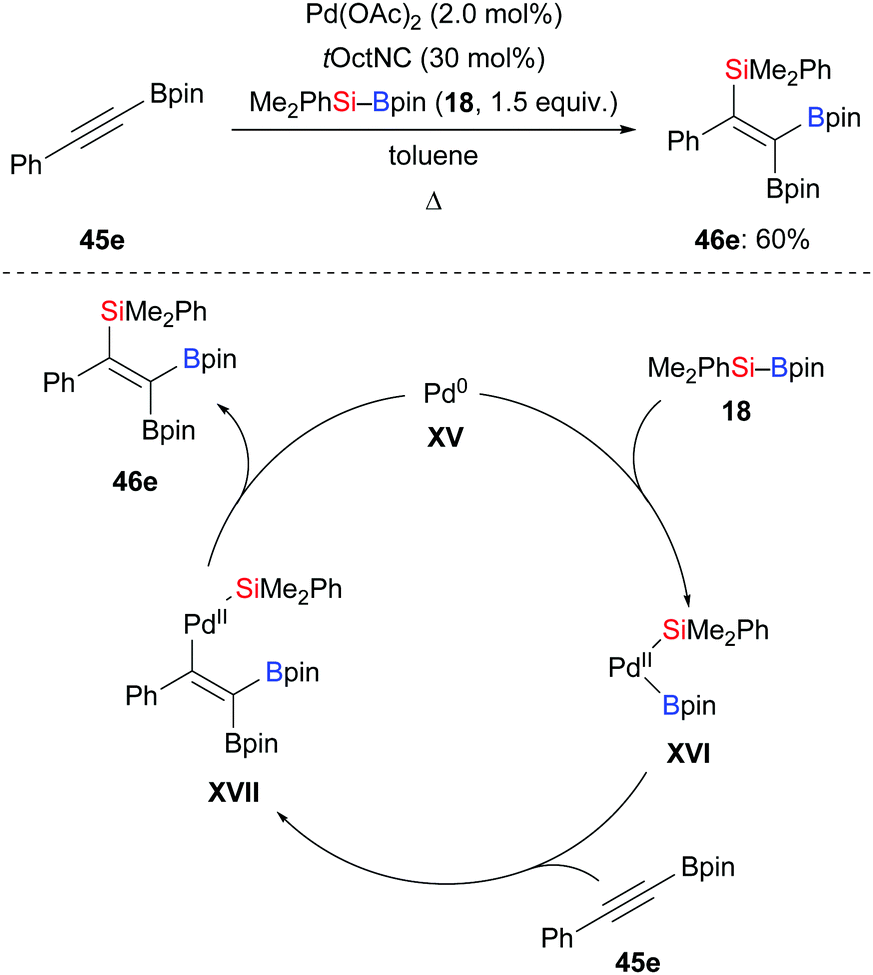
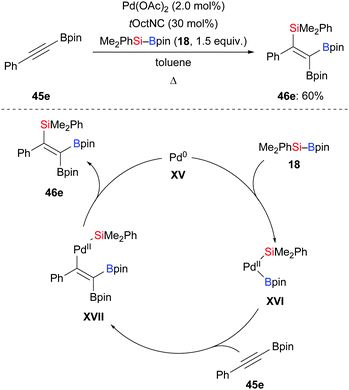
Based on the seminal report by Y. Ito and Suginome in 1996,29 Nishihara and co-workers disclosed a palladium-catalysed highly regio- and stereoselective silaboration of an alkynylboronate in 2013 (45e → 46e, Scheme 14, top).30 This palladium-catalysed addition of Me2PhSi–Bpin (18) across the C–C triple bond of the alkynylboronate afforded a tetrasubstituted alkene. This was converted into a variety of tetraarylated alkenes by chemoselective Suzuki–Miyaura cross-coupling reactions (not shown). A plausible mechanism was proposed (Scheme 14, bottom). Oxidative addition of 18 to a palladium(0) complex XV generates palladium(II) complex XVI, which undergoes regioselective migratory insertion with alkyne 45e to form XVII. Finally, the product 46e is obtained after reductive elimination, along with the regeneration of the palladium(0) catalyst. One year later, Murakami and co-workers used a similar catalytic system to synthesise 2-silyl-1-alkenyl-boronates, which were reacted with aldehydes to construct homoallylic alcohols (not shown).31
 |
| | Scheme 14 Palladium-catalysed regio- and stereoselective silaboration of an alkynylboronate. | |
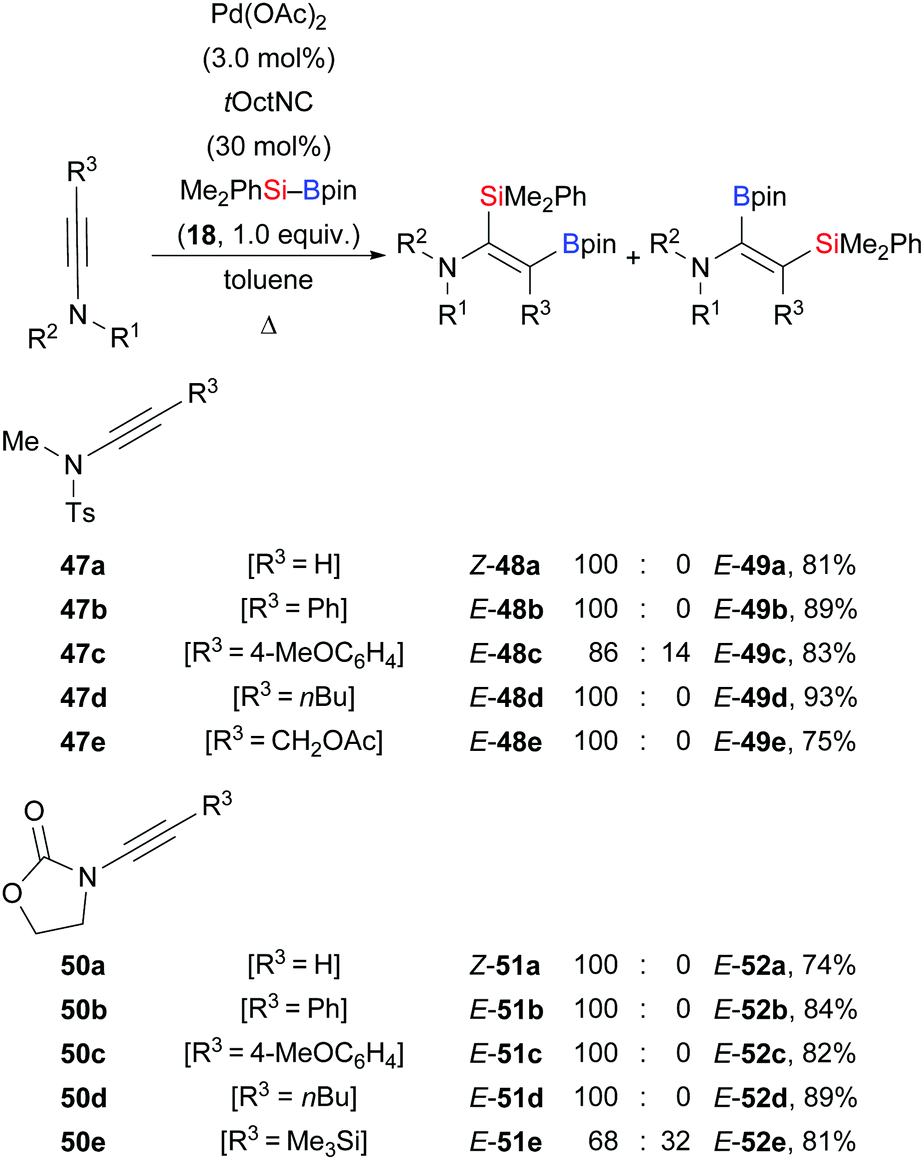
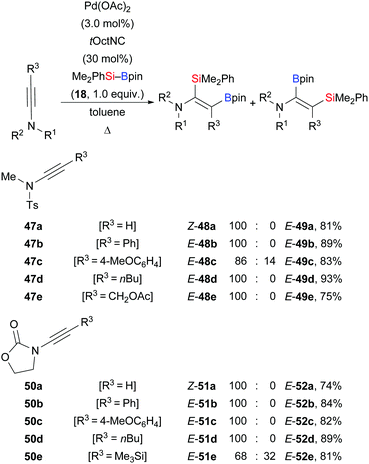
In 2013, Sato and co-workers reported a palladium-catalysed highly regio- and stereoselective silaboration of ynamides as an entry to multi-substituted enamide derivatives (Scheme 15).32 This procedure is amenable to a variety of tosylamide- (47a–e → 48a–e) and oxazolidinone-derived ynamides (50a–e → 51a–e). In the majority of cases, the silyl group was transferred to the C(sp) position α to the nitrogen atom, providing the enamides as single isomers.
 |
| | Scheme 15 Palladium-catalysed regio- and stereoselective silaboration of ynamides. Yields are for the mixture of regioisomers. | |
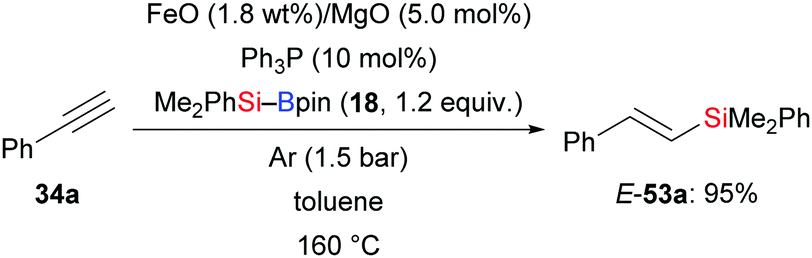
Hydrosilylation is one of the most important and powerful methods for the preparation of silicon-containing compounds in laboratory and industry settings. In 2015, Grirrane and co-workers introduced a highly regio- and stereoselective silylation of phenylacetylene using Me2PhSi–Bpin (18) as the silicon source and Fe/MgO nanoparticles as catalyst (34a → E-53a, Scheme 16).23 A blank experiment showed that FeO is crucial to the high yield.
 |
| | Scheme 16 Silylation of phenylacetylene in the presence of nano-particles. | |
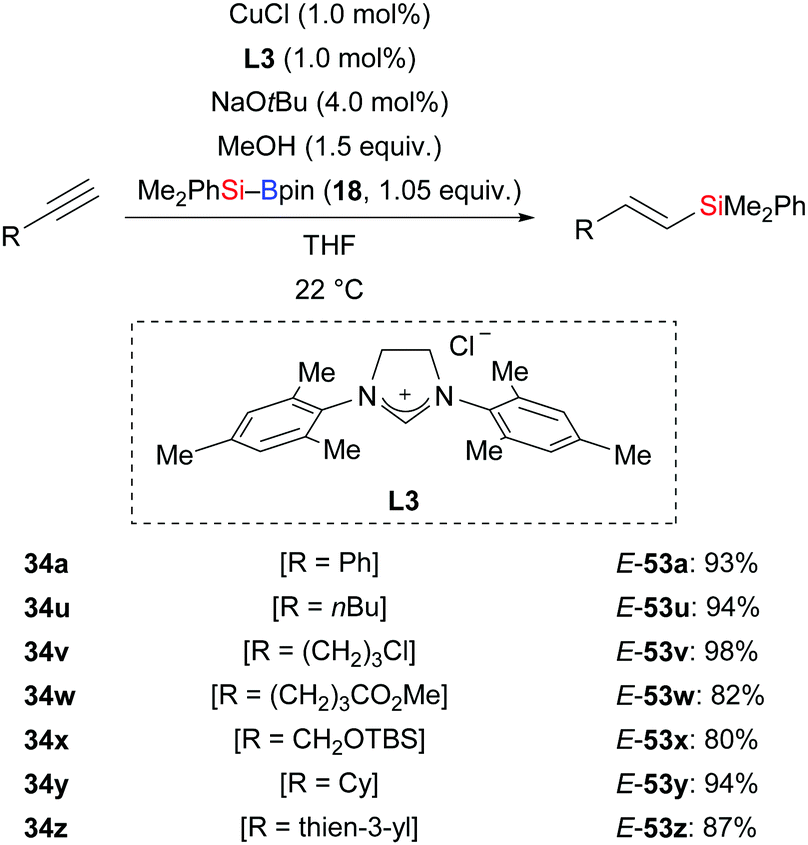
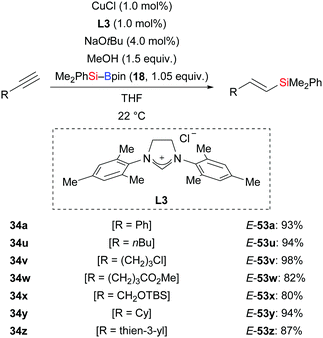
In 2013, Hoveyda and co-workers developed a protosilylation of terminal alkynes catalysed by an NHC–copper complex; the site- and stereoselectivity was high (34a, u–z → E-53a, u–z, Scheme 17).33 Me2PhSi–Bpin (18) was used as the silicon pronucleophile. Under the standard protocol, both aryl- and alkyl-substituted alkynes were converted into the corresponding vinylsilanes with the silyl group attached to the terminus. These results hint that the regioselectivity is governed by steric factors. MeOH serves as proton source in this reaction.
 |
| | Scheme 17 Copper-catalysed regio- and stereoselective silylation of terminal alkynes. | |
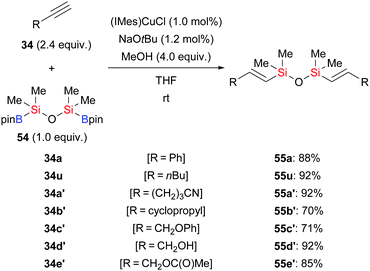
By using 1,1,3,3-tetramethyl-1,3-(pinacolboryl)disiloxane (55) as the silicon source, Zhou and co-workers developed a general and practical procedure to provide access to a wide variety of vinyldisiloxanes in highly regio- and stereoselective fashion (34a, u, a′–e′ → E-55a, u, a′–e′, Scheme 18).34a The potential of this method has been highlighted by subsequent palladium-catalysed Hiyama cross-coupling to provide 1,2-disubstituted (E)-alkenes. A similar procedure was reported by these authors two years later, using a conjugated microporous polymer functionalised with an NHC–copper complex (not shown).34b This novel catalyst could be recycled at least four times without any loss of activity.
 |
| | Scheme 18 Copper-catalysed silylation of terminal alkynes with a disiloxane-based Si–B reagent. | |
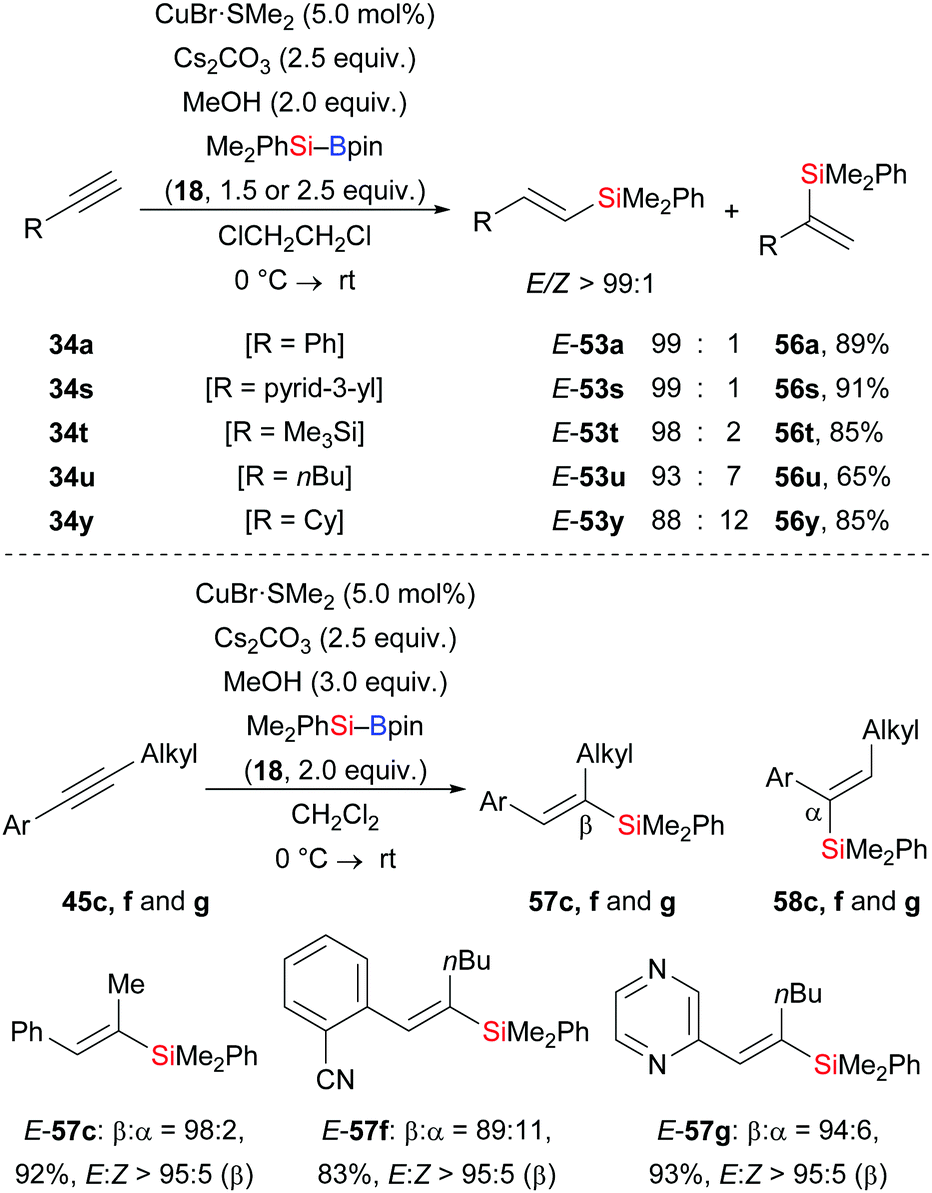
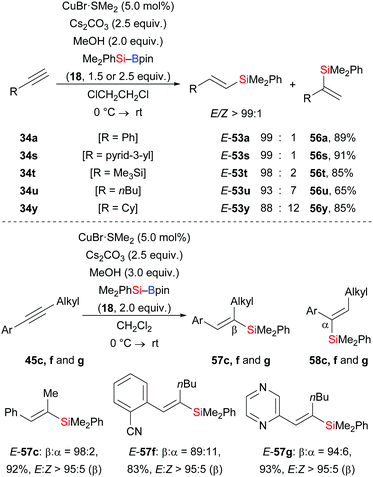
At about the same time, a related regioselective protosilylation of terminal alkynes was presented by Oestreich and co-workers (34a, s–u, y → E-53a, s–u, y, Scheme 19).35 Unlike the above reports about ligand-controlled regioselectivity, commercially available CuBr·SMe2 was used as a catalyst to reach high regioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) without any need for external ligands. Moreover, the solvent showed great influence on the regioisomeric ratio. The reaction of 34a in different solvents led to β:α = 85:15 in CH2Cl2, β:α = 96:4 in THF, and β:α = 99:1 in ClCH2CH2Cl. Furthermore, that catalytic system could be applied in the silaboration of unsymmetrically substituted internal alkynes with synthetically useful regiocontrol (45c, f, g → E-57c, f, g, Scheme 19).
:1) without any need for external ligands. Moreover, the solvent showed great influence on the regioisomeric ratio. The reaction of 34a in different solvents led to β:α = 85:15 in CH2Cl2, β:α = 96:4 in THF, and β:α = 99:1 in ClCH2CH2Cl. Furthermore, that catalytic system could be applied in the silaboration of unsymmetrically substituted internal alkynes with synthetically useful regiocontrol (45c, f, g → E-57c, f, g, Scheme 19).
 |
| | Scheme 19 Copper-catalysed regio- and stereoselective silylation of terminal and internal alkynes. Yields are for the mixture of regioisomers (top). | |
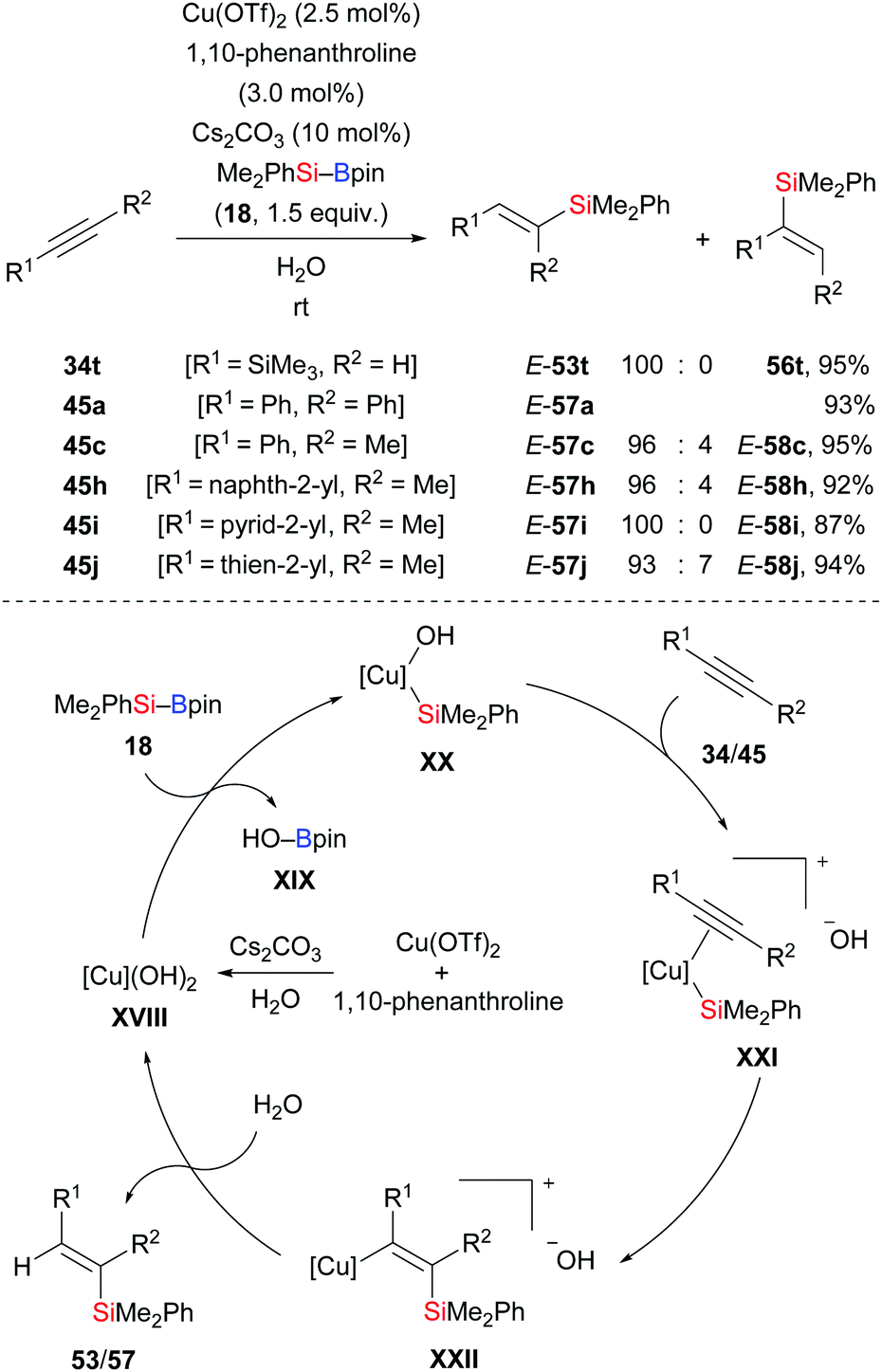
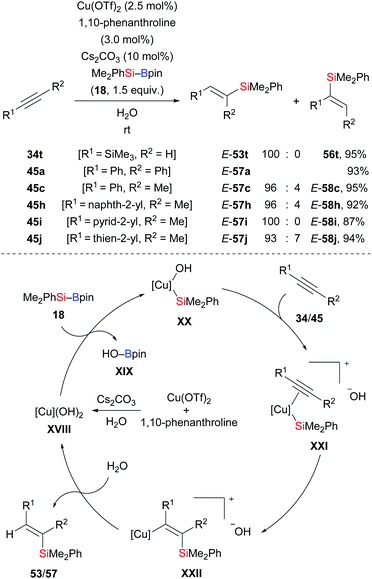
In 2015, Li and co-workers disclosed an efficient method to prepare trisubstituted vinylsilanes by copper-catalysed addition of Me2PhSi–Bpin (18) across internal alkynes (34t → E-53t; 45a, c, h–j → E-57a, c, h–j, Scheme 20).36 Water used in the reaction serves as solvent and proton source as supported by a deuterium-labelling experiment. The reaction begins with formation of LCu(OH)2 catalyst XVIII from LCu(OTf)2, cesium carbonate, and water. Activation of the Si–B bond in 18 by the copper hydroxide through a σ-bond metathesis generates the nucleophilic Cu–Si species XX. After alkyne coordination, the C–C triple bond inserts into the Cu–Si bond in XX to form the vinylcopper intermediate XXII. Hydrolysis affords the vinylsilane and regenerates the LCu(OH)2 catalyst XVIII.
 |
| | Scheme 20 Copper-catalysed regio- and stereoselective silylation of internal alkynes. Yields are for the mixture of regioisomers. | |
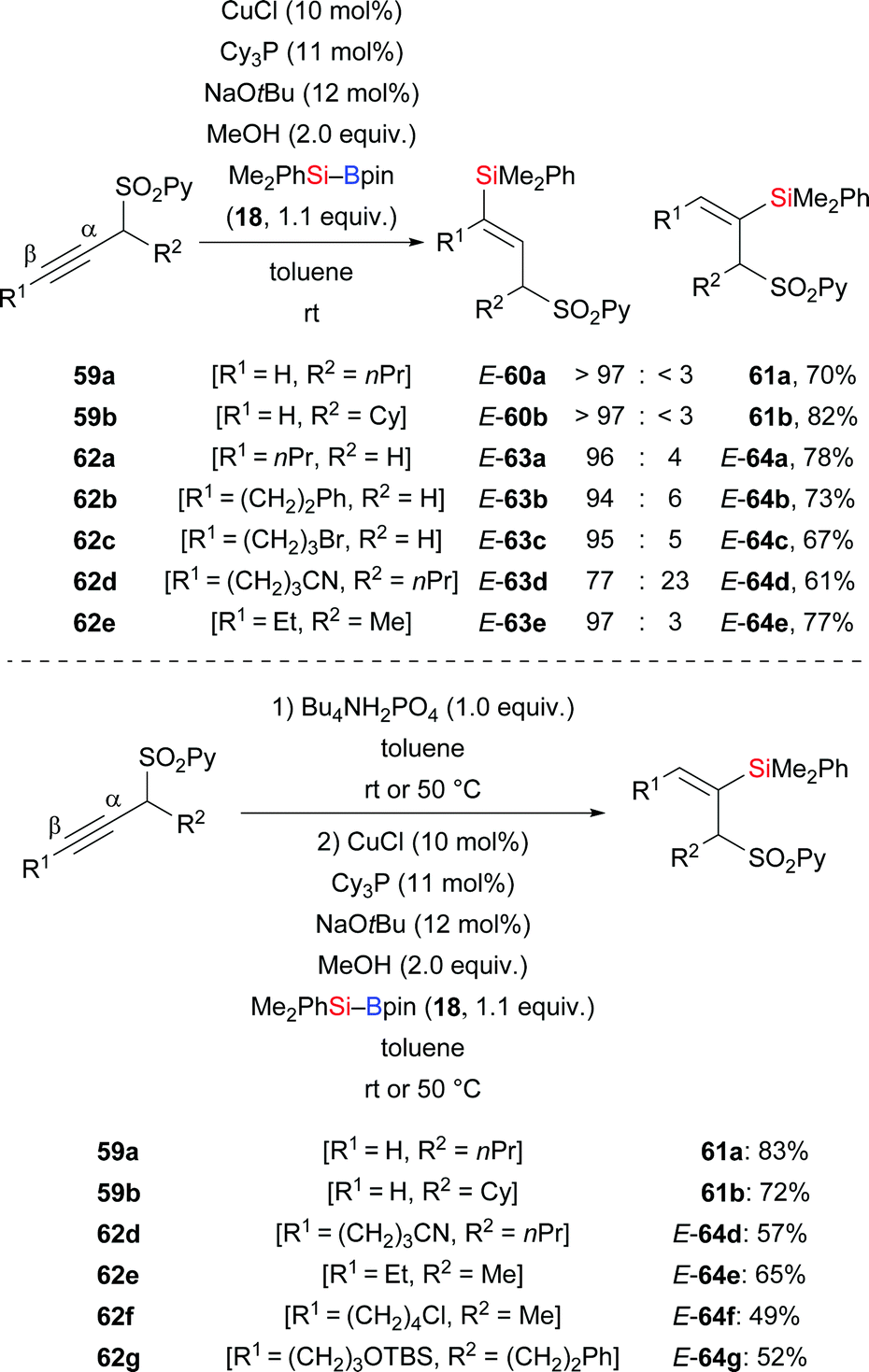
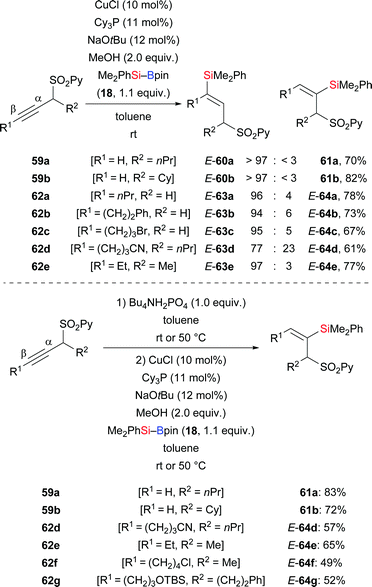
A copper-catalysed silylation of alkynes bearing a pyrid-2-yl sulfonyl group (SO2Py) in the propargylic position was disclosed by Carretero and co-workers in 2015.37 Their mild method led to a large library of di- and trisubstituted alkenes with excellent regio- and stereoselectivity (Scheme 21). The SO2Py group is a removable directing group that allowed for accessing either regioisomer from the same substrate under different reaction conditions. A combination of CuCl and Cy3P resulted in β-regioselectivity (59a–b → E-60a–b; 62a–e → E-63a–e, Scheme 21, top). Conversely, a one-pot, two-step procedure was elaborated to reverse the regioselectivity in this formal hydrosilylation. To achieve α-regioselectivity, the propargylic sulfones are isomerized into the corresponding allenylic sulfones prior to the addition of the silicon nucleophile to the central allene carbon atom (59a–b → 61a–b; 62d–g → E-64d–g, Scheme 21, bottom). Chemoselective transformations of the SO2Py group and the silyl group were performed for further elaboration of silylation products (not shown).
 |
| | Scheme 21 Copper-catalysed silylation of alkynes bearing a 2-pyridyl sulfonyl group (SO2Py) in the propargylic position. Yields are for the mixture of regioisomers. | |
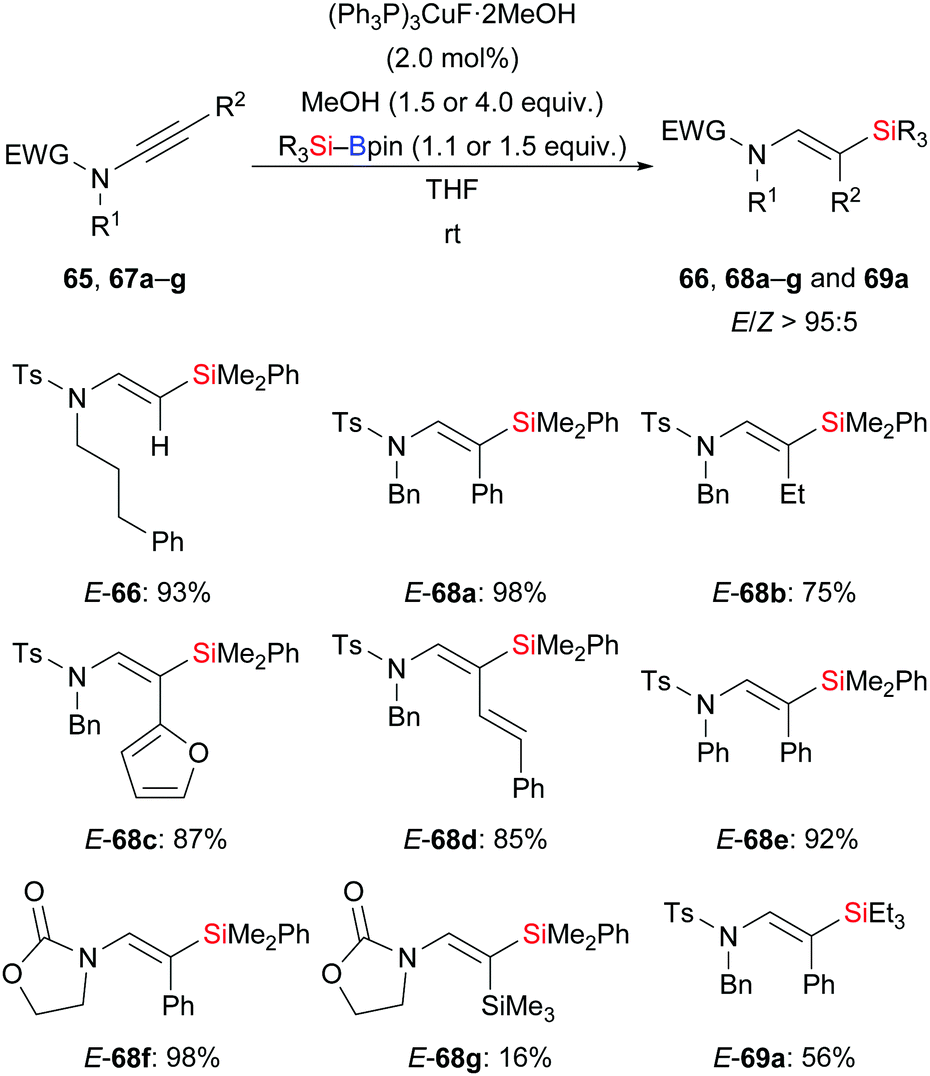
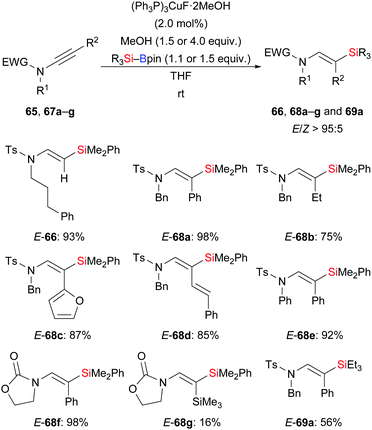
A general and efficient procedure for the copper-catalysed addition of silylboronic esters to ynamides was demonstrated by Evano and co-workers in 2016 (65 → E-66; 67a–g → E-68a–g or E-69a, Scheme 22).38 This mild protosilylation of ynamides gives access to various enamides with high regio- and stereocontrol. It is noteworthy that a single isomer was formed exclusively which has been attributed to a directing effect of the amide group. Alternatively, the polarization of the C–C triple bond by the amide group could explain this outcome.
 |
| | Scheme 22 Copper-catalysed silylation of ynamides. | |
Reactions based on silylmetalation of alkynes and subsequent coupling with an electrophile have emerged as an attractive and useful method for the assembly of polysubstituted vinylsilanes. In 2015, Takaki and co-workers disclosed a copper-catalysed three-component coupling between an alkynes, a silylborane, and a stannyl ether to afford a range of trisubstituted silastannylated alkenes in highly regio- and stereoselective manner (34a, u, a′, f′–i′ → Z-71a, Z-70u, a′, f′–h′, Z-71i′, Scheme 23).39 Compared to those of palladium-catalysed silastannylation with silylstannanes,40 the opposite regioselectivity was obtained in most of cases. When the substrate was phenylacetylene (34a) or a proparylic ether such as 34i′, the silastannylation proceeded with opposite regioselectivity.
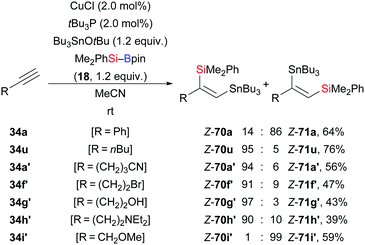 |
| | Scheme 23 Copper-catalysed formal silastannylation of terminal alkynes. Yields are for the mixture of regioisomers. | |
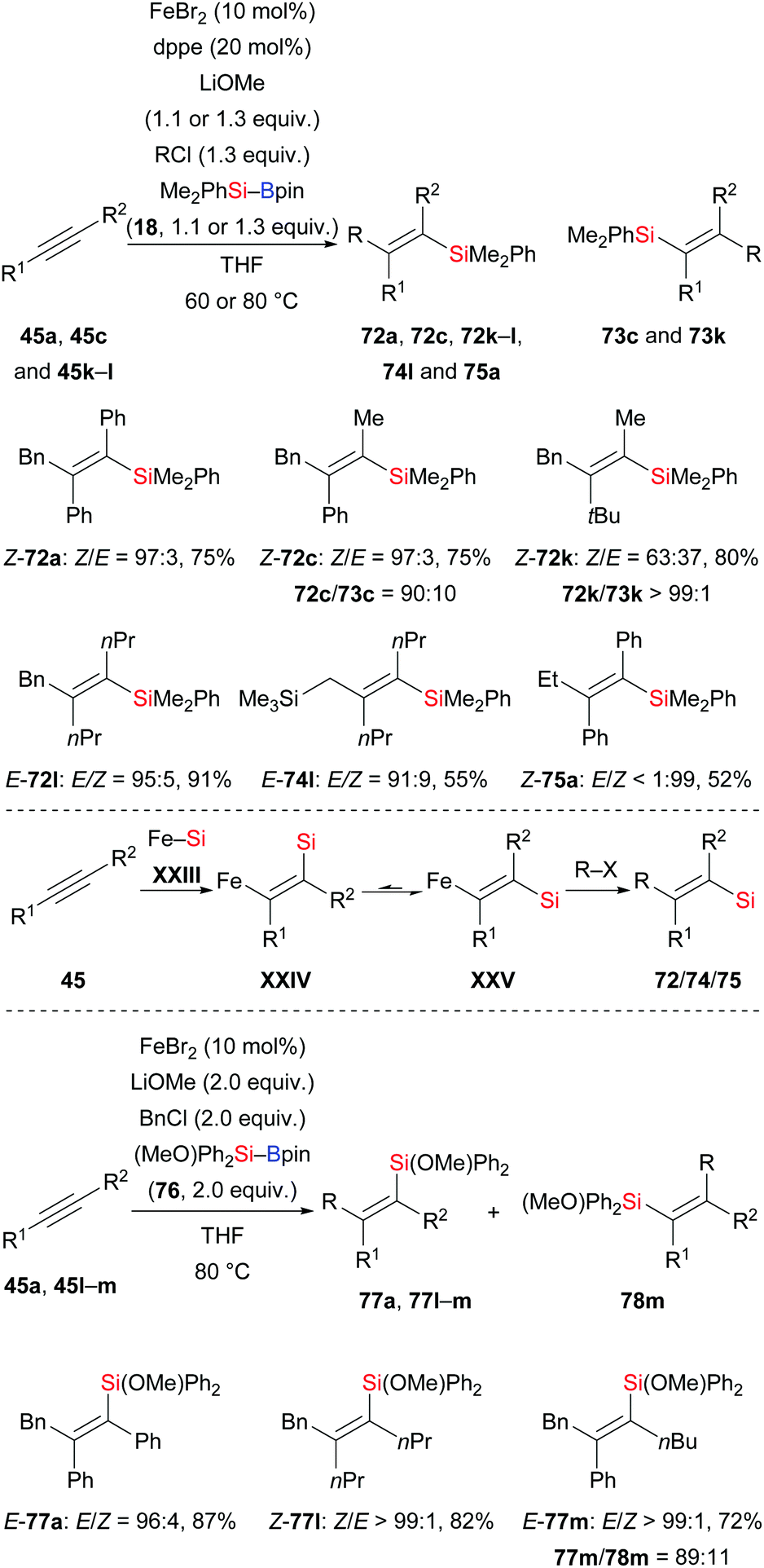
In 2017, an iron-catalysed anti-selective carbosilylation of internal alkynes with silylboranes and alkyl halides was reported by Nakamura and co-workers (45a, c, k–l → 72a, c, k–l, 74l, 75a, Scheme 24, top).41 This new strategy is a straightforward way to add a silicon nucleophile and a carbon electrophile across an alkyne in one pot, thereby forming a number of tetrasubstituted alkenes. When two equivalents of MeOH were used instead of the alkyl halide, moderate syn-selectivity was found for the hydrosilylation product under the reaction setup of the anti-carbosilylation. Moreover, exclusive syn-selectivity was obtained in the absence of the dppe ligand. These findings suggest E/Z isomerization of the ferrasilylation intermediate with the E-isomer XXV more likely to undergo the alkylation (Scheme 24, middle). Based on these results, these authors further developed a syn-selective carbosilylation of internal alkynes by employing the heteroatom-substituted silylborane (MeO)Ph2Si–Bpin (76) (45a, l–m → 77a, l–m, Scheme 24, bottom). The oxygen atom of 76 coordinates to the iron center to form a chelated and thus more stable Z-isomer which participates in the alkylation step to afford carbosilylation products syn-selectively.
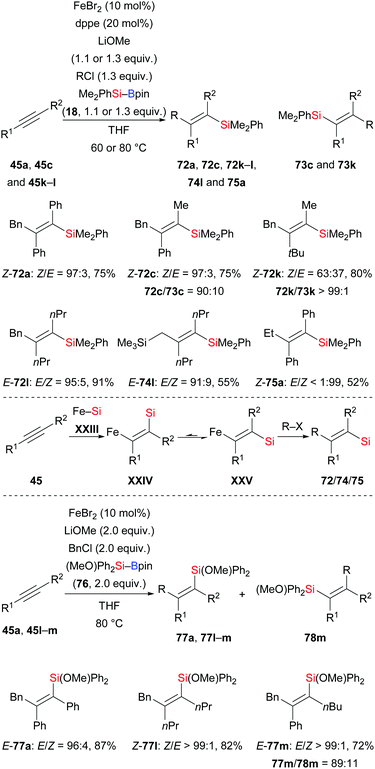 |
| | Scheme 24 Iron-catalysed anti- or syn-selective carbosilylation of internal alkynes. | |
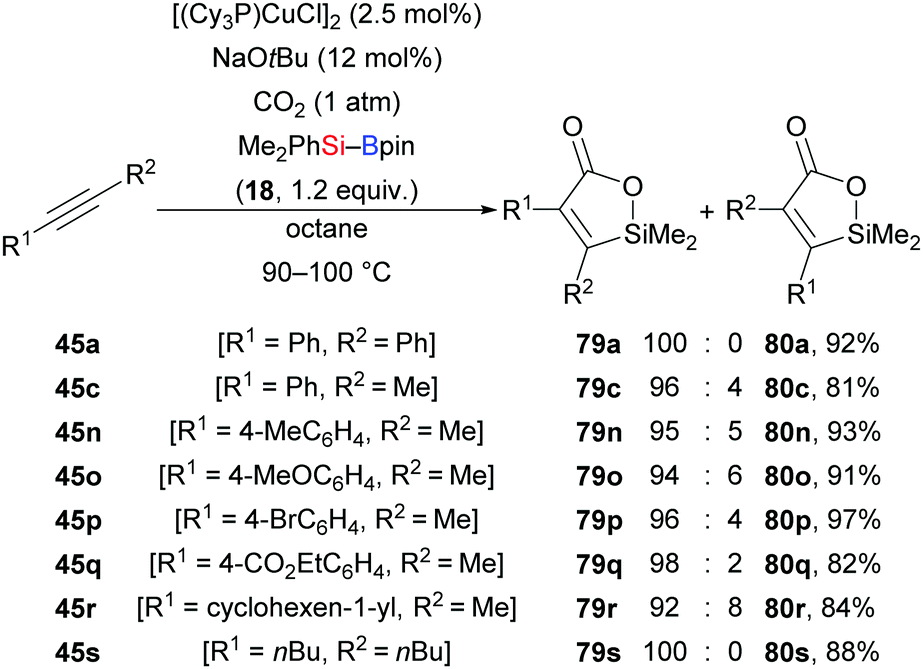
A copper-catalysed highly regio- and stereoselective silacarboxylation of internal alkynes employing carbon dioxide and silylboranes was first reported by the Tsuji group in 2012 (45a, c, n–s → 79a, c, n–s, Scheme 25).42 This method was carried out under atmospheric pressure of CO2 to yield silalactones with high regiocontrol. Further elaboration of these silalactones was done by Hiyama cross-coupling (not shown).43 Reactions with Et3Si–Bpin (42) instead of Me2PhSi–Bpin (18) were less efficient, only furnishing trace amounts of the product along with the formation of a mixture of β-silyl-α,β-unsaturated carboxylic acids (not shown). Control experiments excluded a stepwise pathway, proceeding through silaboration of the alkynes followed by carboxylation of vinylboronic esters.
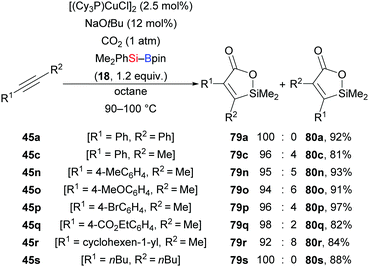 |
| | Scheme 25 Copper-catalysed silacarboxylation of internal alkynes by employing carbon dioxide as an electrophile. Yields are for the mixture of regioisomers. | |
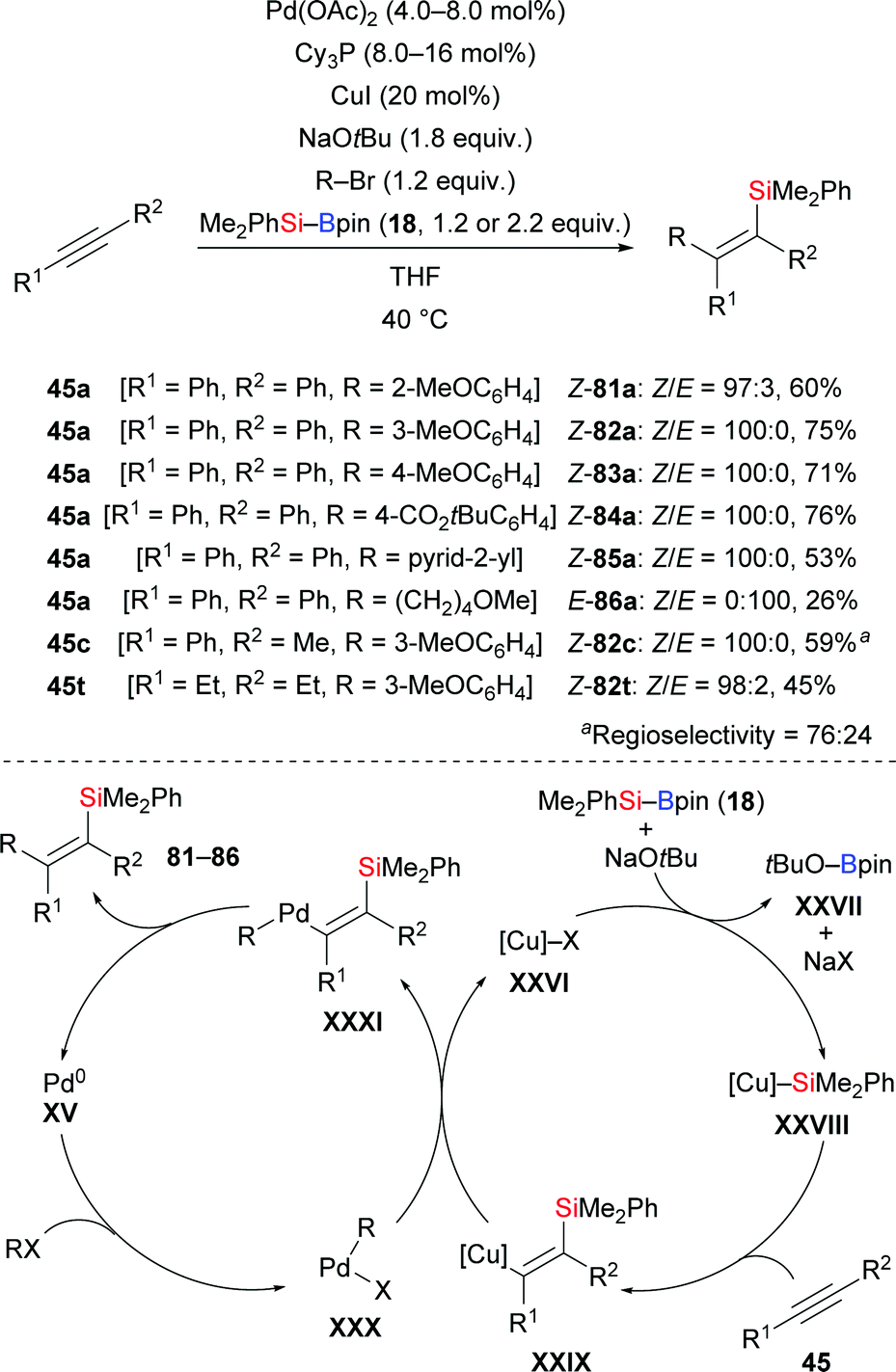
Another cross-coupling of alkynes, silylboronic esters, and aryl halides co-catalysed by copper and palladium was presented by Nozaki and co-workers in 2016 (45a, c, t → 81–86a, 82c, 82t, Scheme 26).44 A silyl group and an aryl group were added to the C–C triple bond in syn-fashion. The products were then further processed by desilylative bromination (not shown). A catalytic cycle was proposed. After the usual formation of intermediate XXVIII migratory insertion to form a Cu–C bond in XXIX occurs. Transmetalation between this Cu–C complex and ArPdIIX (XXX), generated by oxidative addition of an aryl halide to Pd0, affords the PdII–C complex XXXI and regenerates catalyst XXVI. The carbosilylated product is released after reductive elimination of XXXI concomitant with the regeneration of the Pd0 catalyst XV.
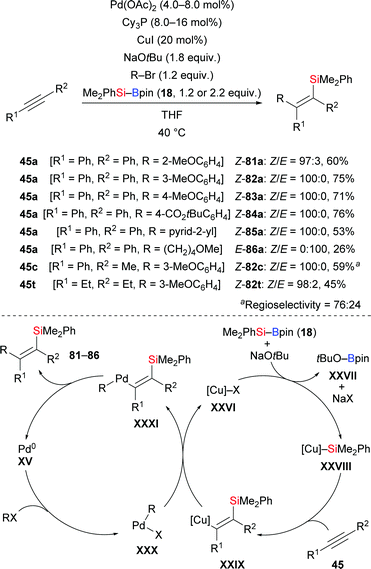 |
| | Scheme 26 Copper/palladium co-catalysed carbosilylation of internal alkynes. | |
4.1.2. Alkenes.
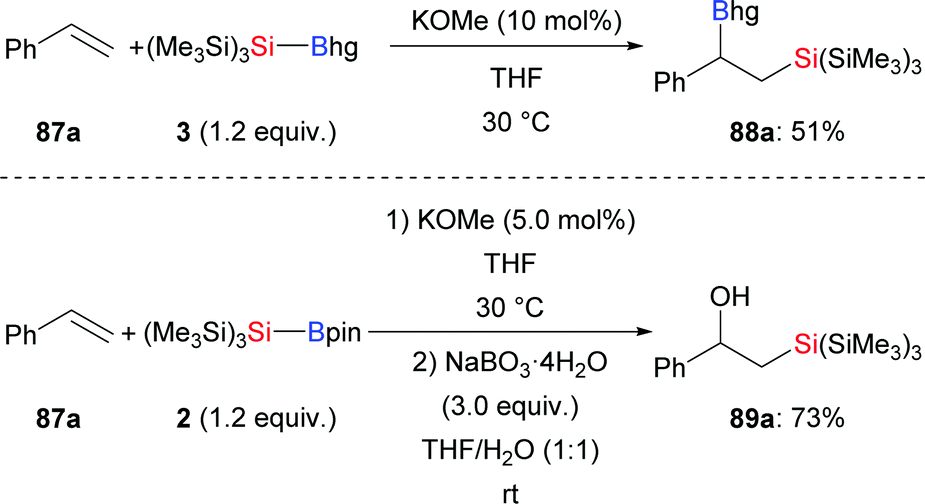
Since Y. Ito and co-workers first reported the silaboration of alkenes, significant progress has been made.45 In 2017, H. Ito and co-workers realised the preparation of two new bulky, air- and moisture-stable supersilylboronic esters through the coupling of tris(trimethylsilyl)silylpotassium and boron electrophiles (cf.Scheme 1). Both are reactive in the reaction with styrene by using a catalytic amount of KOMe for activation. The corresponding products were formed in moderate to good yields (87a → 88a and 89a, Scheme 27).11 It is noteworthy that Et3Si–Bpin does not react under the same setup.
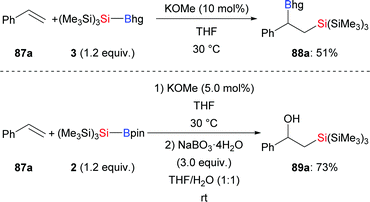 |
| | Scheme 27 Silaboration of styrene with Si–B reagents catalysed by KOMe. | |
As part of their work on nickel-catalysed hydroboration of alkenes with B2pin2, Kamei and co-workers reported three examples of a formal hydrosilylation of alkenes using a silylborane as the silicon source and water as the proton source (87b, 87c, 91a → 90b, 90c, 92a, Scheme 28).46 No hydroboration product was detected.
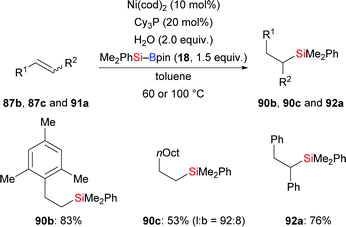 |
| | Scheme 28 Nickel-catalysed hydrosilylation of alkenes. | |
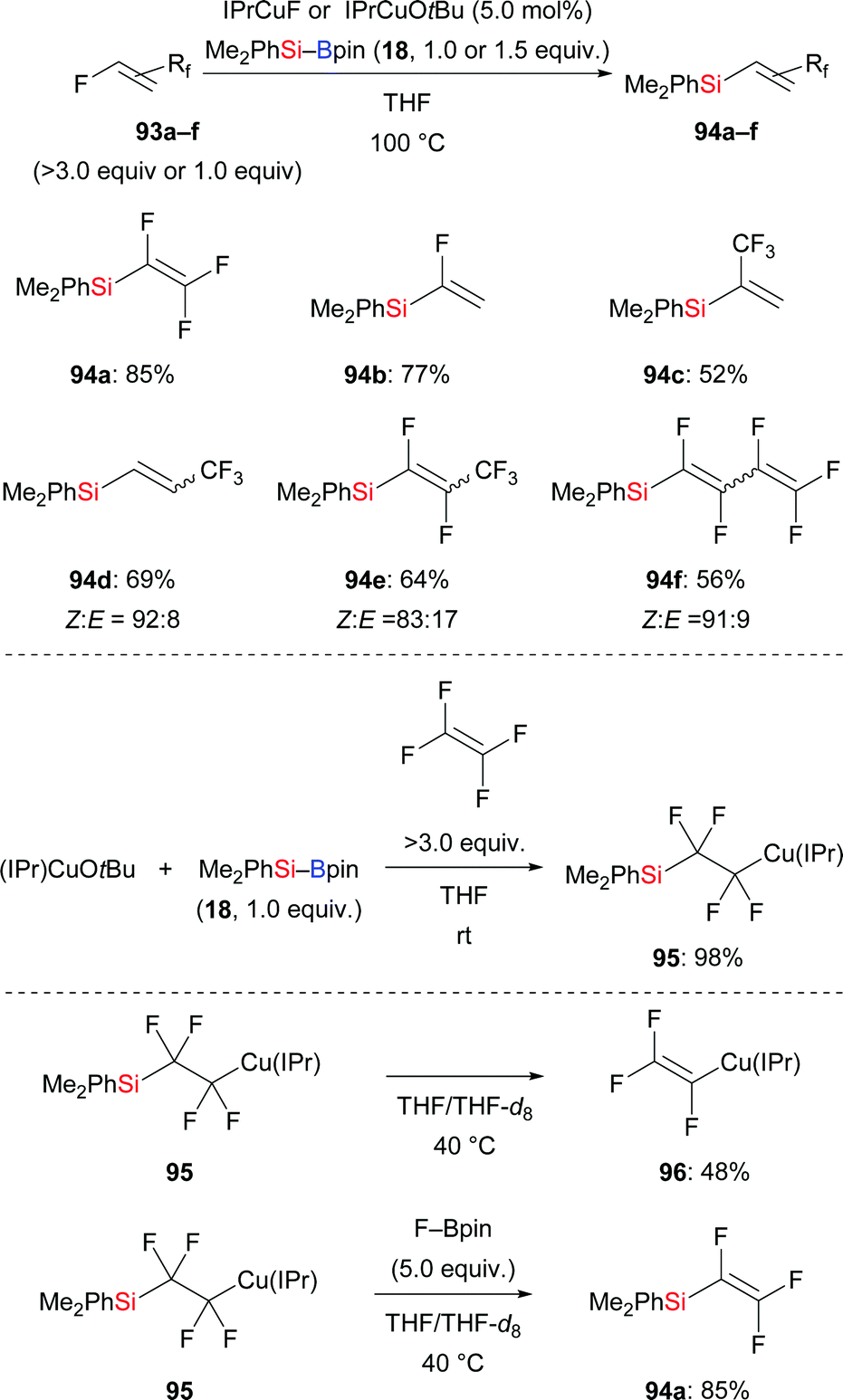
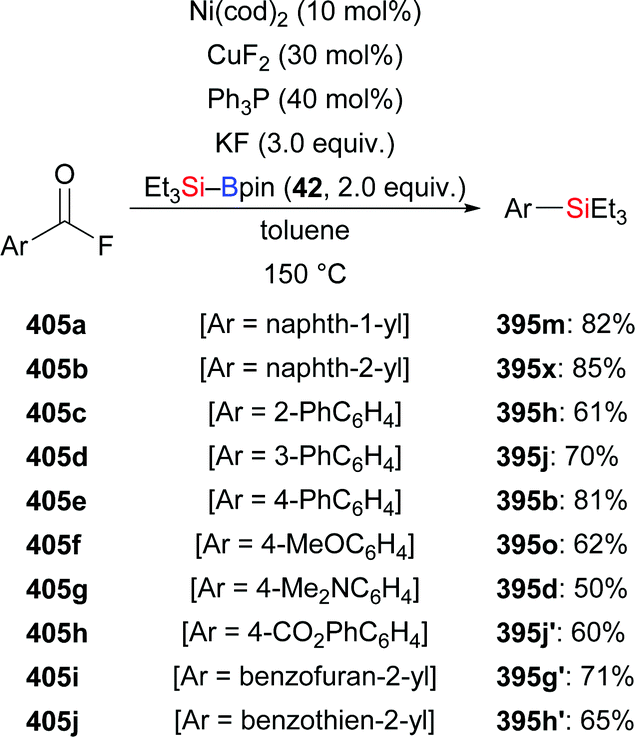
Defluorosilylation is currently attracting considerable attention. In 2018, the Ogoshi group developed a general method for the preparation of fluorinated vinylsilanes by a copper-catalysed defluorosilylation of fluoroalkenes with Me2PhSi–Bpin (18) (93a–f → 94a–f, Scheme 29, top).47 The resulting fluorinated vinylsilanes are synthetically useful building blocks for further elaboration. For example, a copper-catalysed cross-coupling of 94a and iodobenzene was performed, furnishing α,β,β-trifluorostyrene in 52% yield (not shown). To clarify the mechanism of this transformation, a possible intermediate 2-silyl-1,1,2,2-tetrafluoroalkylcopper(I) complex 95 was prepared in 98% yield (Scheme 29, middle). A series of stoichiometric reactions were conducted, suggesting that in situ-generated F–Bpin plays a key role in the β-elimination of fluoride (Scheme 29, bottom). F–Bpin serves as a Lewis acid in this E2 reaction. Later, Wang and co-workers reported a similar defluorosilylation of gem-difluoroalkenes with Et3Si–Bpin (42) to prepare monofluorinated vinylsilanes, thereby expanding the utility of this strategy further (not shown).48
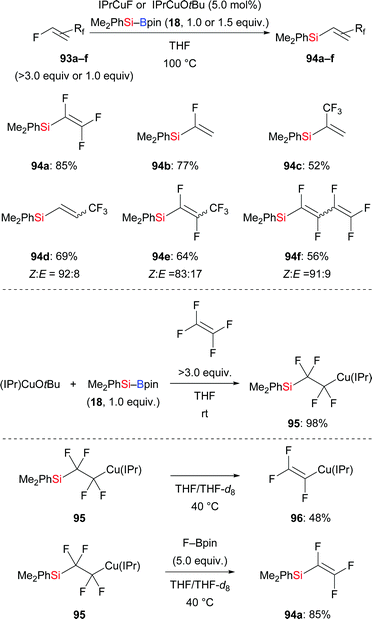 |
| | Scheme 29 Copper-catalysed defluorosilylation of fluoroalkenes. | |
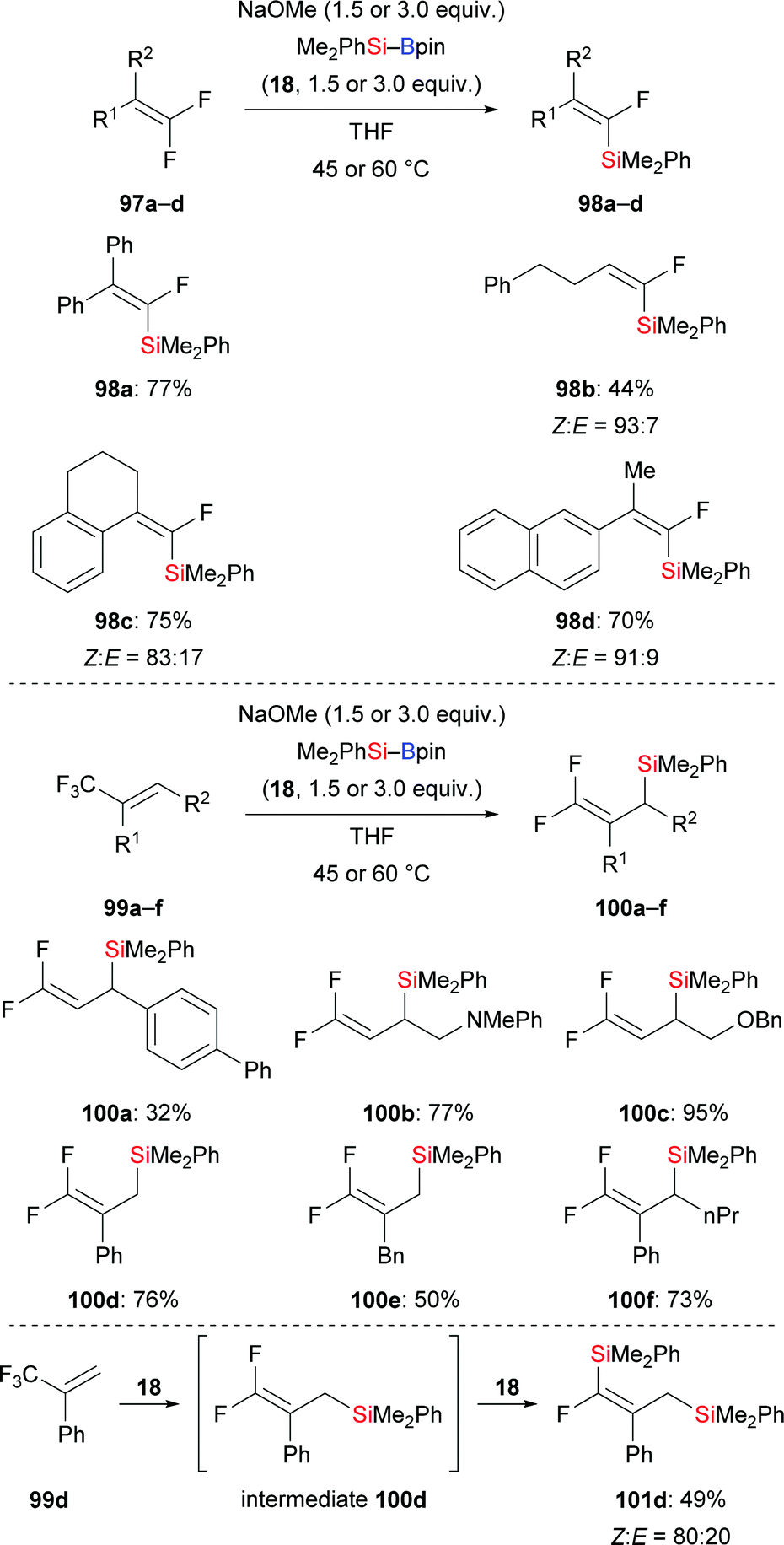
Very recently, a transition-metal-free defluorosilylation of a variety of fluoroalkenes with silylboronic esters in the presence of NaOMe was published by Shi and co-workers, opening a door to the formation of various silylated fluoroalkenes with C(sp2)–Si and C(sp3)–Si bonds (Scheme 30).49 Both gem-difluoroalkenes and trifluoromethylalkenes are suitable substrates under the standard setup. Vinylsilanes (97a–d → 98a–d, Scheme 30, top) and allylsilanes (99a–f → 100a–f, Scheme 30, middle) were formed in synthetically useful yields. It is worthy of note that gem-difluoroalkenes with an allylic silyl group stemming from the defluorosilylation of trifluoromethyl-substituted alkenes can be engaged in another defluorosilylation when excess silylborane is used (99d → 101d, Scheme 30, bottom). Et3Si–Bpin (42) is also reactive in the defluorosilylation of 99c, affording the corresponding allylsilane in 95% yield (not shown). The authors’ mechanistic analysis and DFT calculations suggest an SN2′ substitution and SNV substitution, respectively.
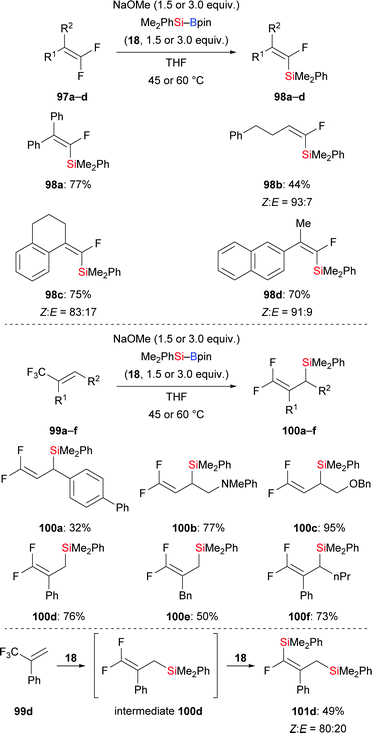 |
| | Scheme 30 Transition-metal-free defluorosilylation of fluoroalkenes and trifluoromethyl-substituted alkenes. | |
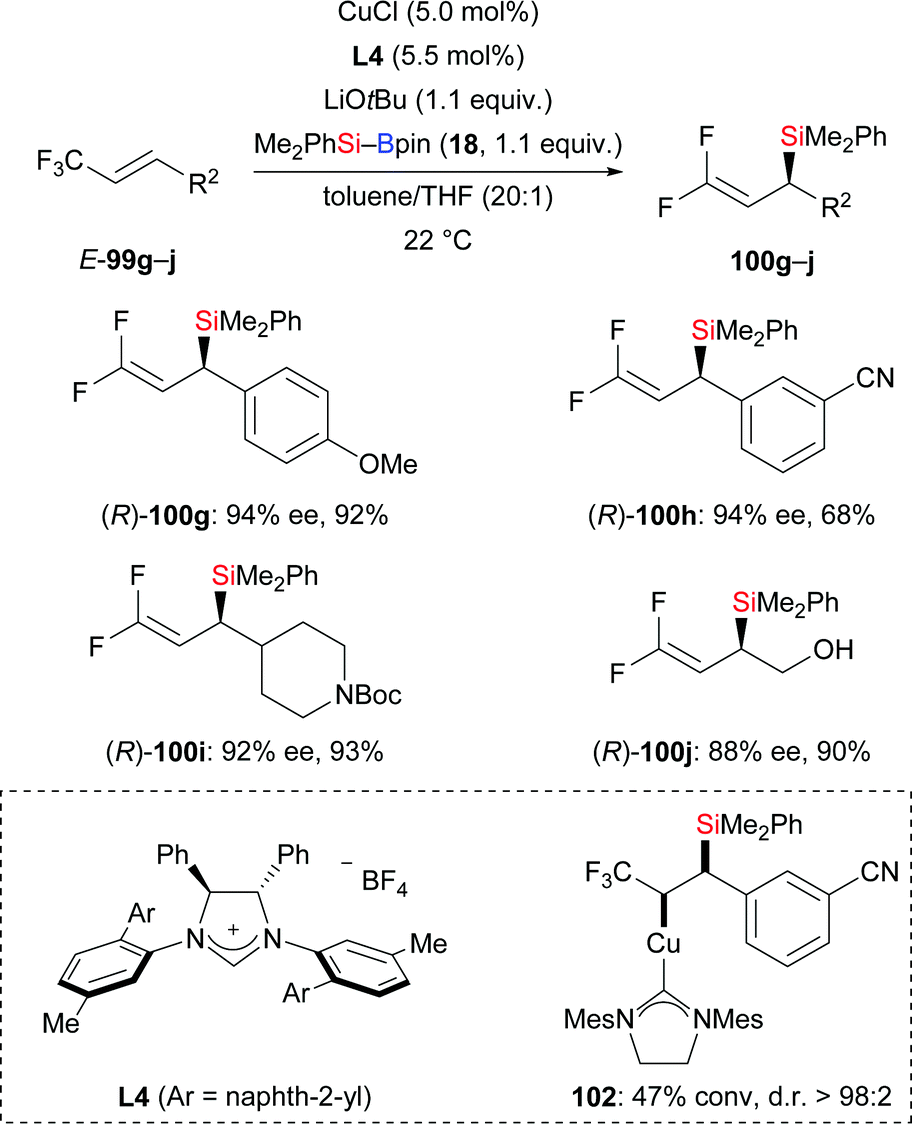
Later, the Hoveyda group presented an enantioselective silyl substitution by using NHC–copper catalysis, allowing for a practical and general entry into the synthesis of chiral allylic silanes with ≤94% ee (99g–j → (R)-100g–j, Scheme 31).50 The mechanism was probed experimentally. For this, the NHC–Cu–Si complex 102 was prepared and characterised under N2 atmosphere. The authors found that the Cu–Si addition step is much faster than the following β-elimination of fluoride which is facilitated by either a mild Lewis acid or a nucleophilic promoter. DFT studies were conducted to rationalize the stereoinduction as well as anti-selectivity observed in the aforementioned β-elimination.
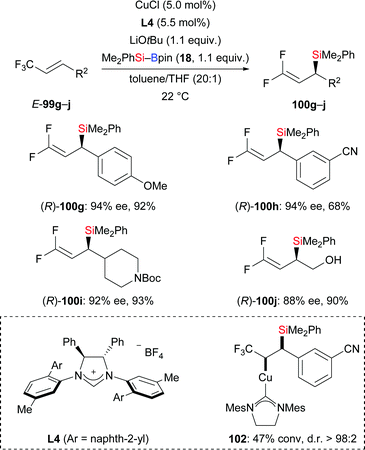 |
| | Scheme 31 Copper-catalysed enantioselective silyl substitution. | |
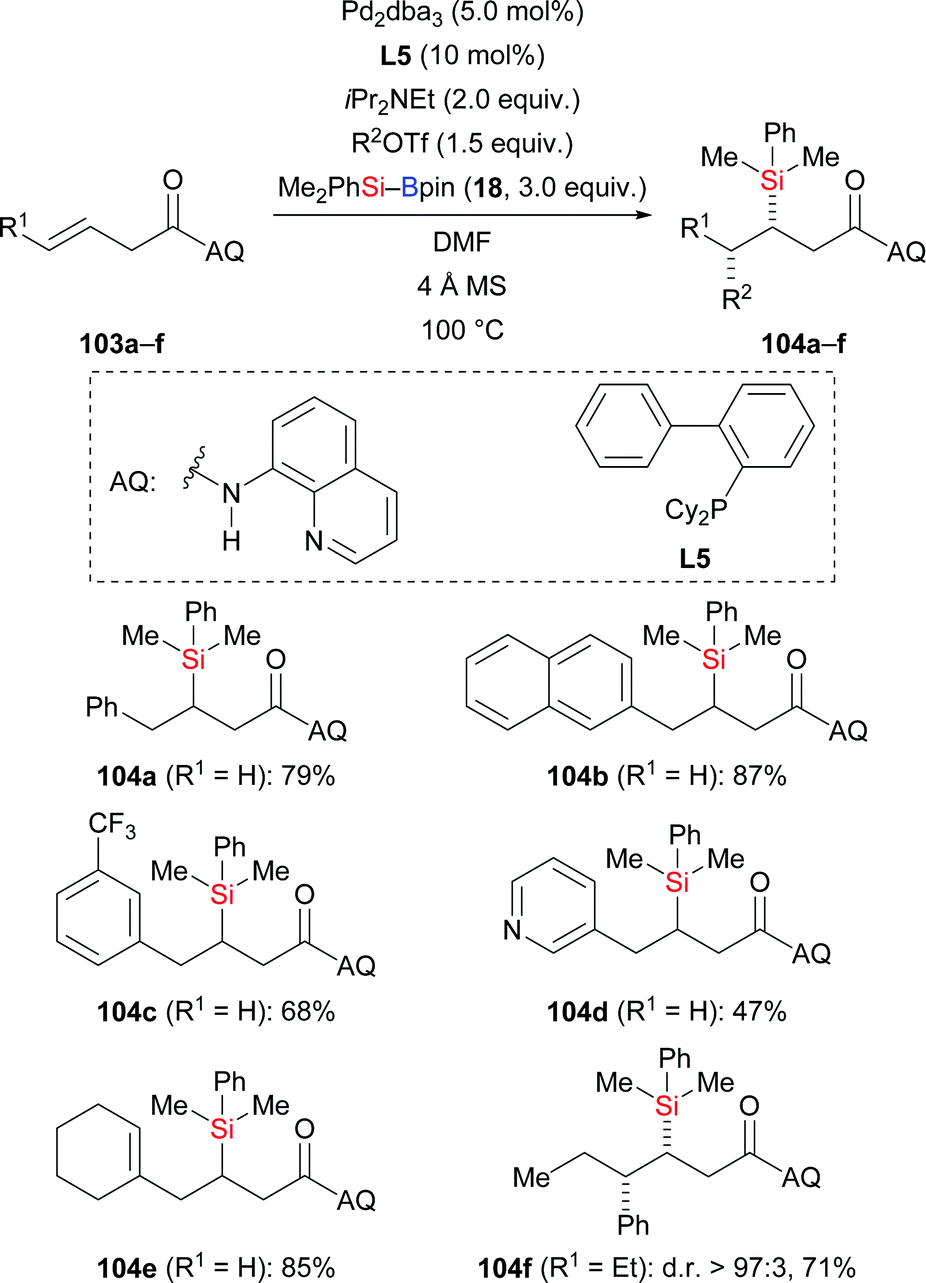
A palladium-catalysed highly regio- and stereoselective carbosilylation of β,γ-unsaturated carbonyl compounds using Me2PhSi–Bpin (18) as the silicon pronucleophile and aryl/alkenyl triflates as electrophiles was presented by Engle and co-workers in 2019 (103a–f → 104a–f, Scheme 32).51 The reaction involves a highly regioselective Heck-type aryl- or alkenylpalladation followed by C–Si bond formation at the palladium-bearing carbon atom. A removable directing group (AQ) was installed to steer the incorporation of the silyl group to the proximal position through the formation of palladacycle intermediates. In this transformation, a small amount of carboboration products was detected as byproduct; other side reactions were arylsilylation and hydroarylation.
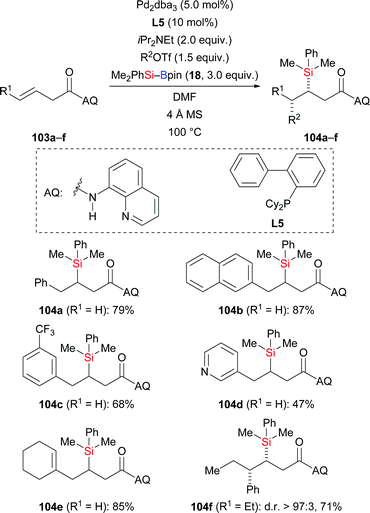 |
| | Scheme 32 Palladium-catalysed directed syn-1,2-carbosilylation. | |
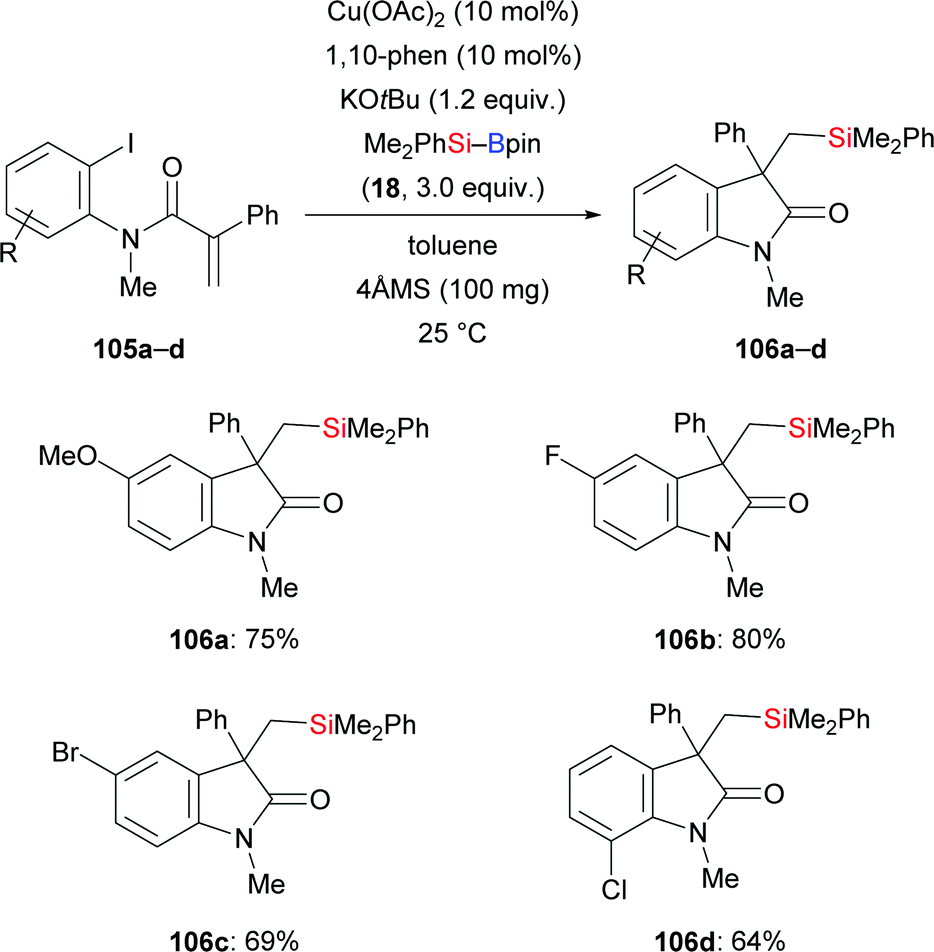
Jia and co-workers disclosed a copper-catalysed carbosilylation of α,β-unsaturated ortho-iodoanilides to construct oxindoles containing a quaternary carbon center (105a–d → 106a–d, Scheme 33).52 The authors speculated that this involves an intermolecular silylcupration of the C–C double bond followed by an intramolecular coupling of the thus-formed alkylcopper intermediate and the aryl iodide.
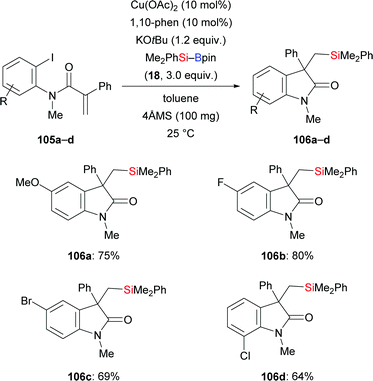 |
| | Scheme 33 Copper-catalysed carbosilylation of α,β-unsaturated ortho-iodoanilides. | |
4.2. 1,2-, 1,4- and 1,n-addition to C–C multiple bond systems
4.2.1. 1,3-Dienes.
A copper-catalysed highly regioselective three-component coupling reaction employing 1,3-dienes, silylboronic esters, and nitriles as reactants was presented by the Fujihara group in 2020 (107a–b → 108a–b, 109–113a, Scheme 34, top).53 This mild and efficient reaction is a new procedure for the synthesis of β,γ-unsaturated ketones with a silyl group attached to the α-substituent. When 2-tolunitrile was applied in the reaction, a stable iminium salt precipitated after acidic hydrolysis (not shown). Aliphatic nitriles were not suitable substrates in this process, giving the desired products in low yields. Unsymmetric dienes such as isoprene are compatible when using a different phosphine ligand, and the corresponding products are formed with high regioselectivity (107c → 108c, Scheme 34, bottom). Et3Si–Bpin (42) is reactive in the reaction with isoprene and benzonitrile, affording the product in moderate yield with high regiocontrol (not shown).
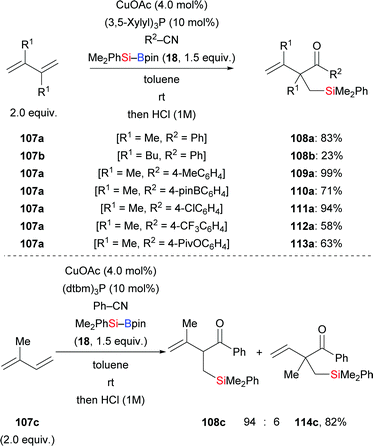 |
| | Scheme 34 Copper-catalysed three-component coupling reactions of 1,3-dienes. Yield is for the mixture of regioisomers (bottom). | |
4.2.2. 1,6- and 1,7-diynes as well as 1,3- and 1,7-enynes54.
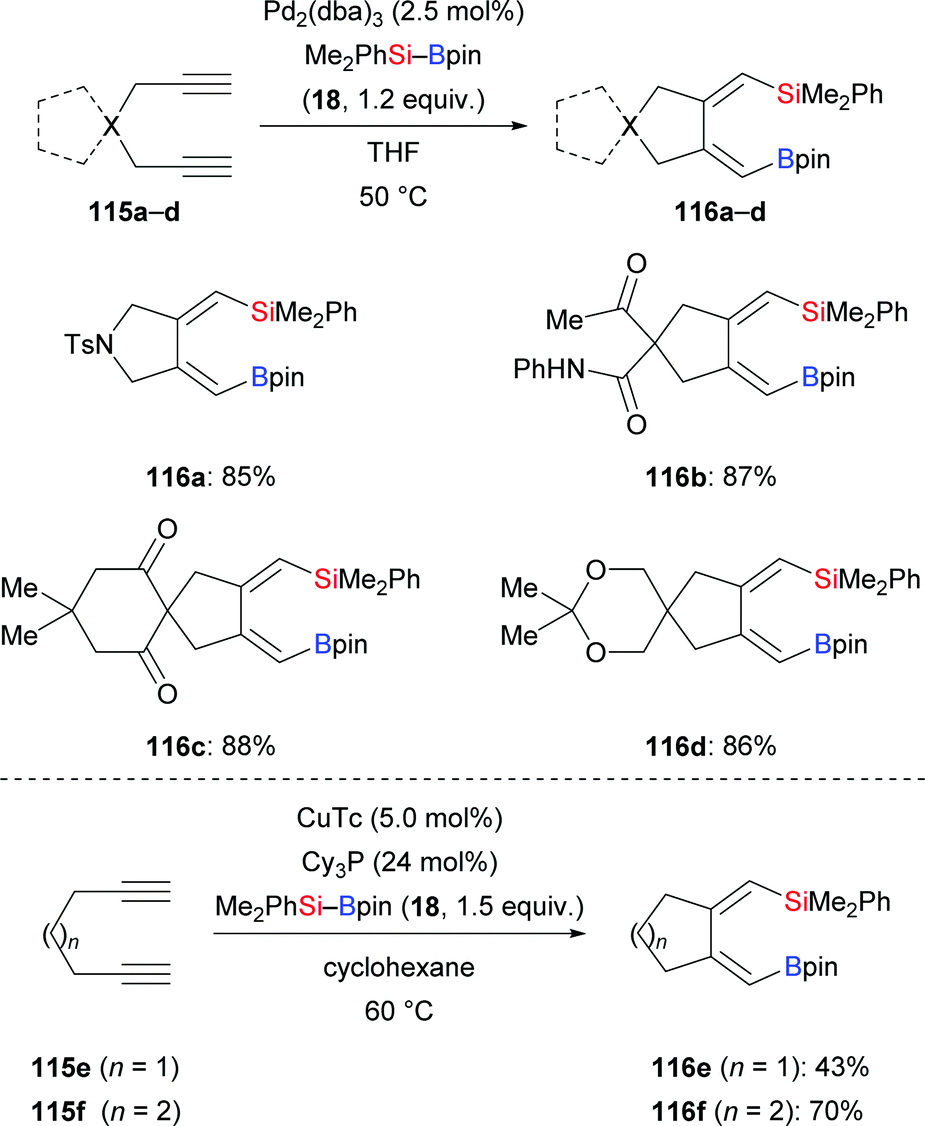
In 2018, Loh and co-workers introduced a general protocol for the palladium-catalysed silaborative carbocyclization of 1,6-diynes to access 1,2-dialkylidenecyclopentanes (115a–d → 116a–d, Scheme 35, top).55 This method showed broad substrate scope and compatibility with functional groups. The resulting products are synthetically useful building blocks as illustrated by several follow-up reactions (not shown). Octa-1,7-diyne did not react under these reaction conditions. In the following year, the Xu group reported a copper-catalysed silaborative carbocyclization of diynes, again with high regioselectivity (115e–f → 116e–f, Scheme 35, bottom).25 In this work, both hepta-1,6-diyne and octa-1,7-diyne were suitable substrates, furnishing the corresponding products in 43% and 70% yield, respectively.
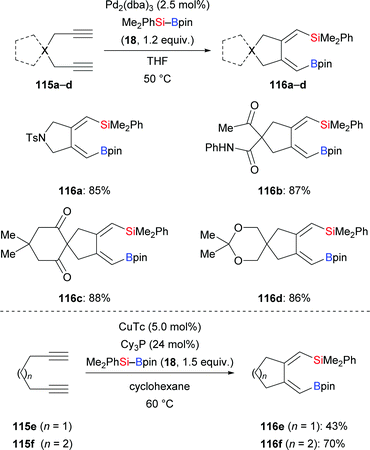 |
| | Scheme 35 Silaborative carbocyclization of diynes under palladium and copper catalysis. | |
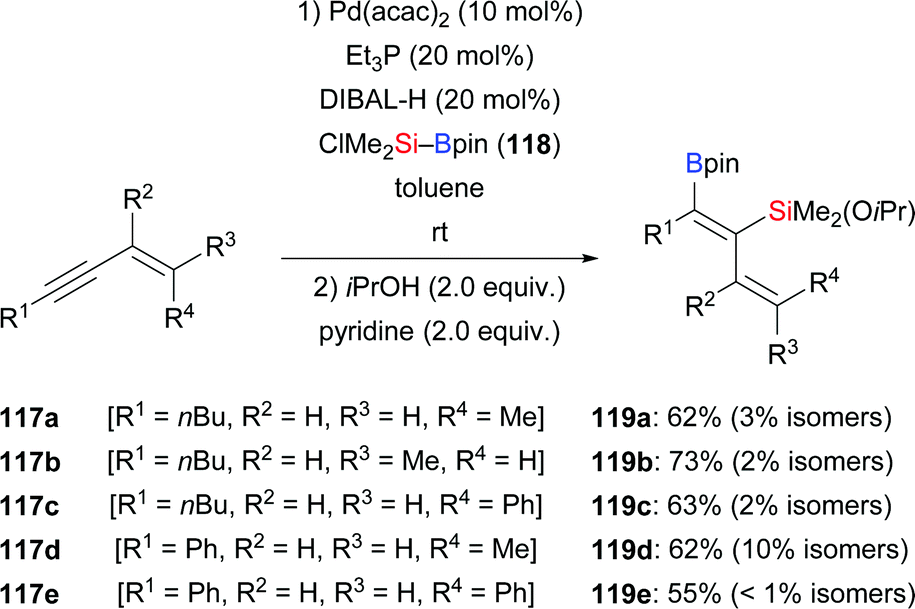
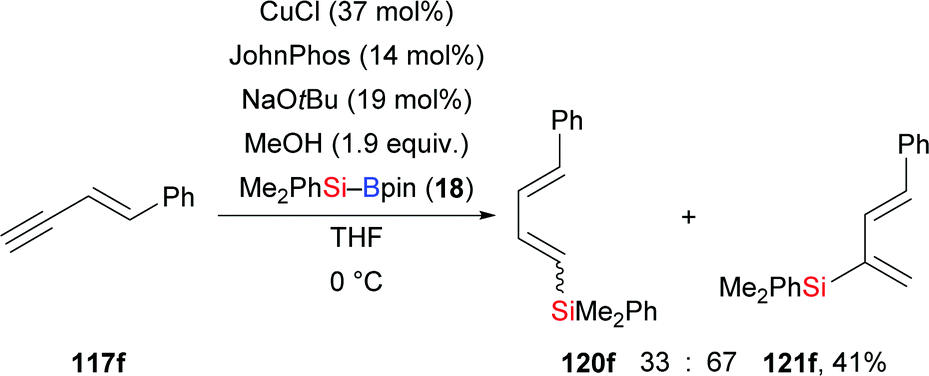
A palladium-catalysed highly regio- and stereoselective silaboration of 1,3-enynes with more reactive (chlorodimethylsilyl)pinacolborane (118) was developed by Moberg and co-workers in 2012. A variety of functionalised 1,3-dienes 119a–e was derived from the chemoselective addition of Si–B reagent 118 across the C–C triple bond, leaving the C–C double bond unreacted (117a–e → 119a–e, Scheme 36).56a The ClMe2Si group was subsequently converted into the corresponding silyl ether by alcoholysis with iPrOH and pyridine. These products are versatile platforms for further elaboration by Suzuki–Miyaura and Hiyama–Denmark cross-coupling reactions to afford multisubstituted 1,3-dienes, allenes, and conjugated trienes.56b In 2014, the Welker group reported a hydrosilylation of E-4-phenyl-3-buten-1-yne with silylborane by using CuCl/JohnPhos in THF (117f → 120f and 121f, Scheme 37).57 The silyl group was again transferred to the C–C triple bond, yet with little regiocontrol.
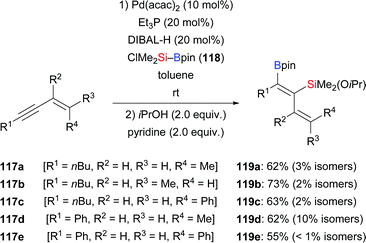 |
| | Scheme 36 Palladium-catalysed silaboration of 1,3-enynes. | |
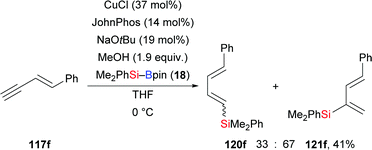 |
| | Scheme 37 Copper-catalysed silaboration of a 1,3-enyne. Yield is for the mixture of regioisomers. | |
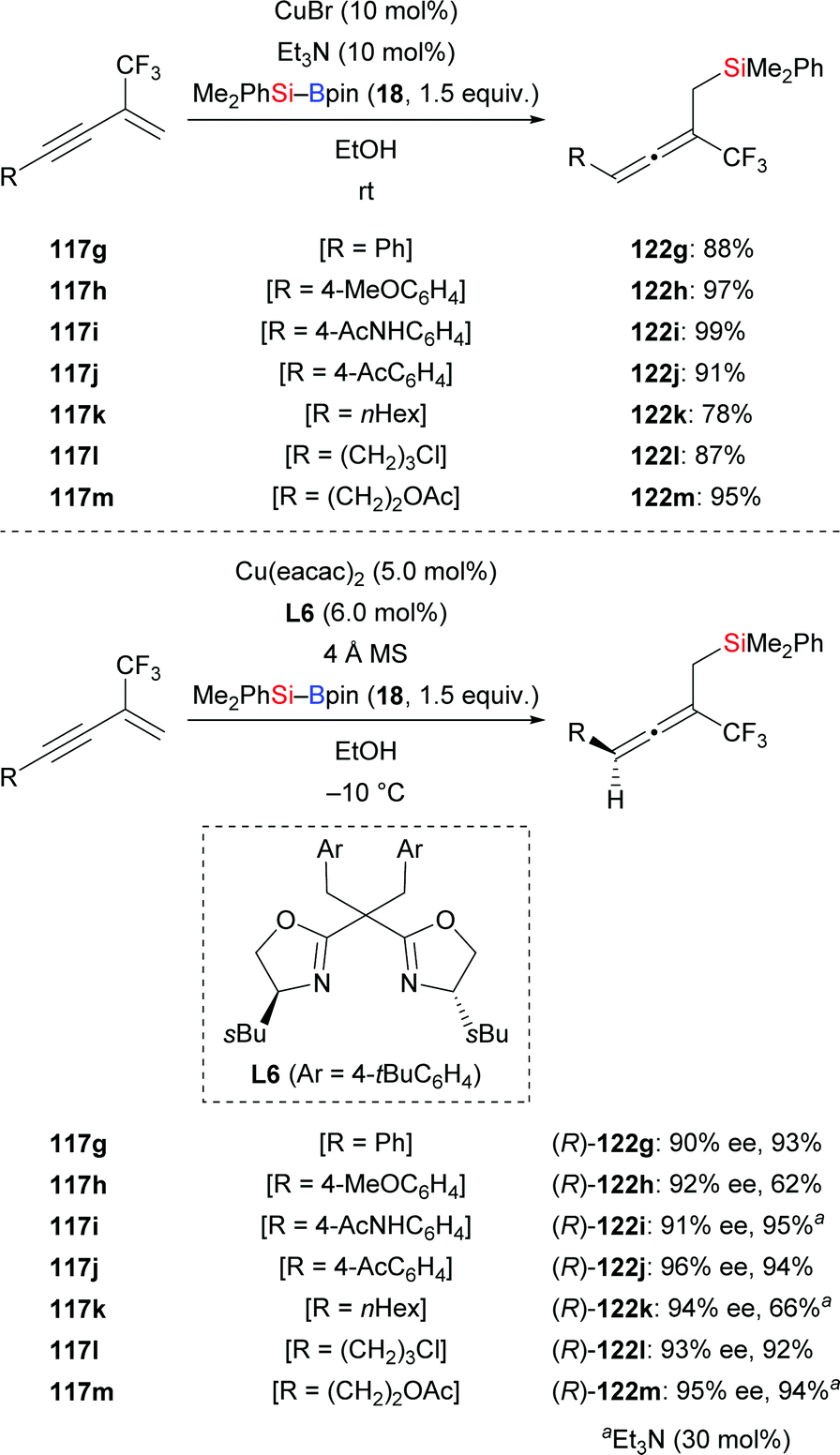
Just recently, Xu and co-workers developed a copper-catalysed regio- and enantioselective protosilylation of trifluoromethyl-substituted 1,3-enynes to access a variety of trisubstituted allenes (117g–m → 122g–m, Scheme 38).58 The racemic version was carried out at room temperature using CuCl as precatalyst; the corresponding products were formed in good to excellent yields. By applying chiral Box ligand L6 as ligand at −10 °C, these authors realised this transformation in asymmetric fashion, affording chiral allenes in good yields and with excellent enantioinduction. A gram-scale synthesis of an enantioenriched allenylsilane was carried out without any loss of enantioselectivity, highlighting the synthetic utility of this method (not shown).
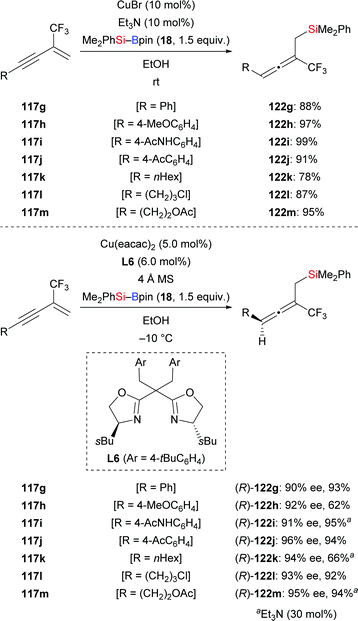 |
| | Scheme 38 Copper-catalysed (enantioselective) silylation of CF3-substituted 1,3-enynes. | |
A palladium-catalysed silaborative carbocyclization of selected 1,7-enynes was presented by Moberg. This reaction allows for the synthesis of chromane and tetrahydroquinoline derivatives (123a–b → 124a–b; 125 → 126, Scheme 39, top).59 Cyclization precursors with a stereocenter in the propargylic position undergo the ring closure with moderate diastereoselectivity (127a–b → 128a–b, Scheme 39, middle). The level of diastereocontrol is governed by the length of the tether between the alkyne and alkene moieties (83:17 for 1,7-enyne and 63:37 for 1,6-enyne). Rigid systems with trans relative configuration reacted with high diastereoselectivity (129a–b → 130a–b, Scheme 39, bottom) while related cis-substituted 1,7-enynes were less reactive (not shown).
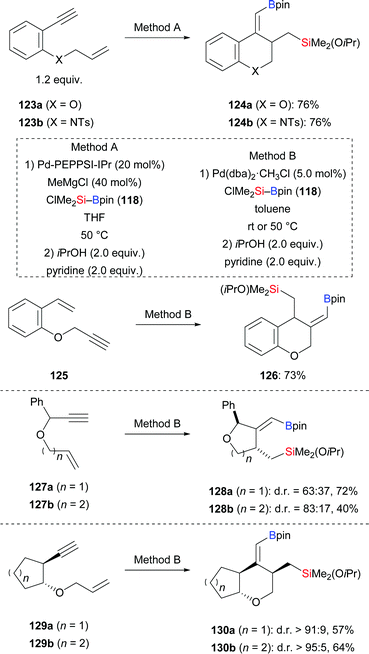 |
| | Scheme 39 Copper-catalysed silaborative carbocyclization of 1,7-enynes. | |
4.2.3. Allenes.
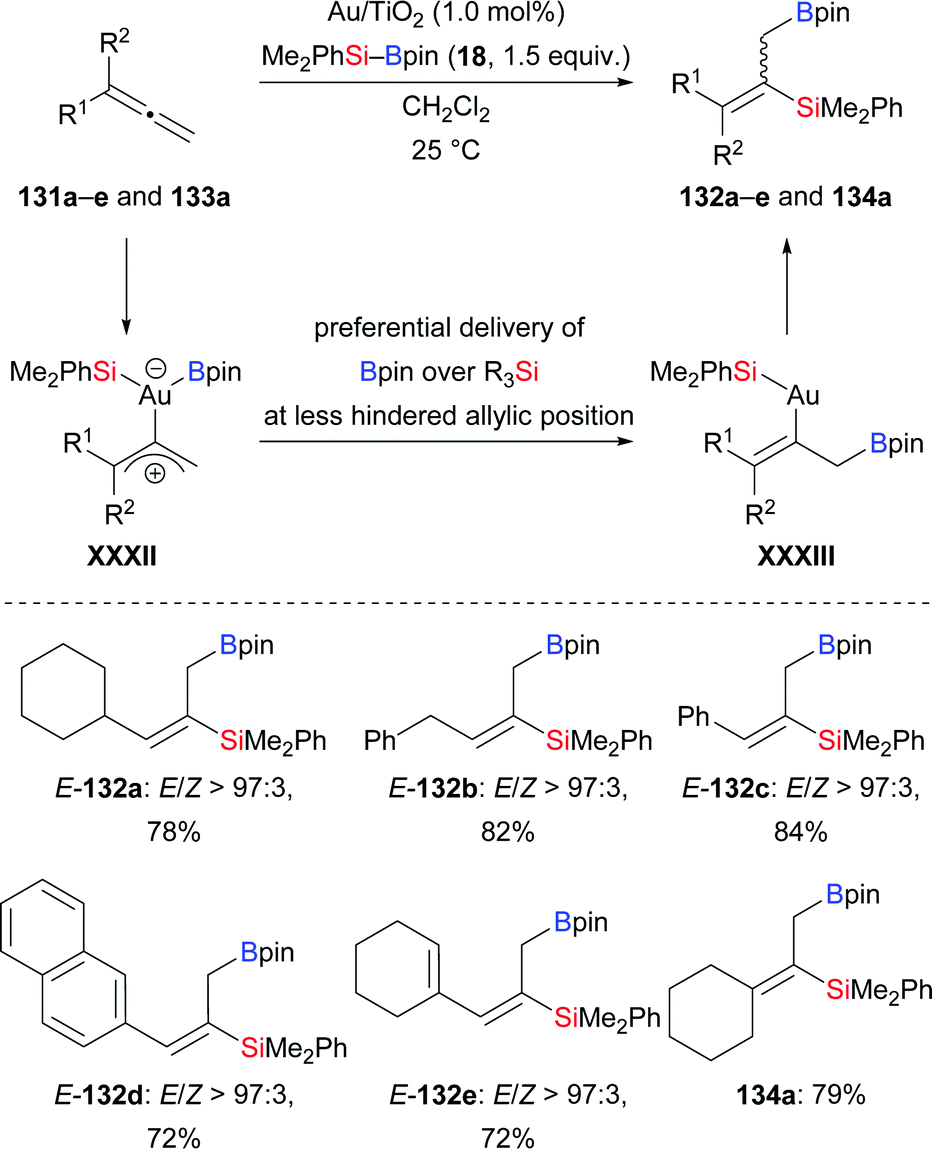
A gold-catalysed regio- and stereoselective silaboration of allenes in the presence of Au/Ti2O nanoparticles was developed by the Stratakis group in 2018 (131a–e → 132a–e; 133a → 134a, Scheme 40).60 The commercially available Au/Ti2O nanoparticles used in the reaction are recyclable. Of note, no ligands or additives are necessary. This silaboration occurs exclusively at the terminal C–C double bond for steric reasons. The silaboration of a cyclopropyl-substituted allene afforded a mixture of cyclopropyl-substituted product and ring-opening product in a 1:1 ratio (not shown), suggesting the existence of intermediate XXXII. C–B bond formation is thermodynamically favoured over the formation of the C–Si bond (XXXII → XXXIII).
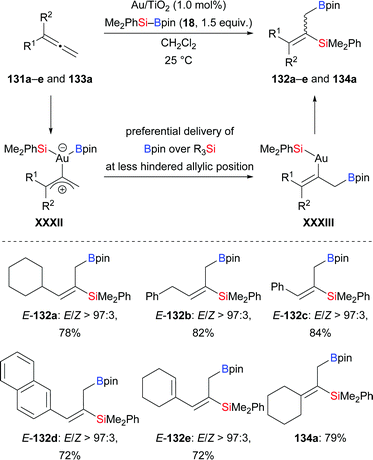 |
| | Scheme 40 Gold-catalysed regioselective silaboration of terminal allenes. | |
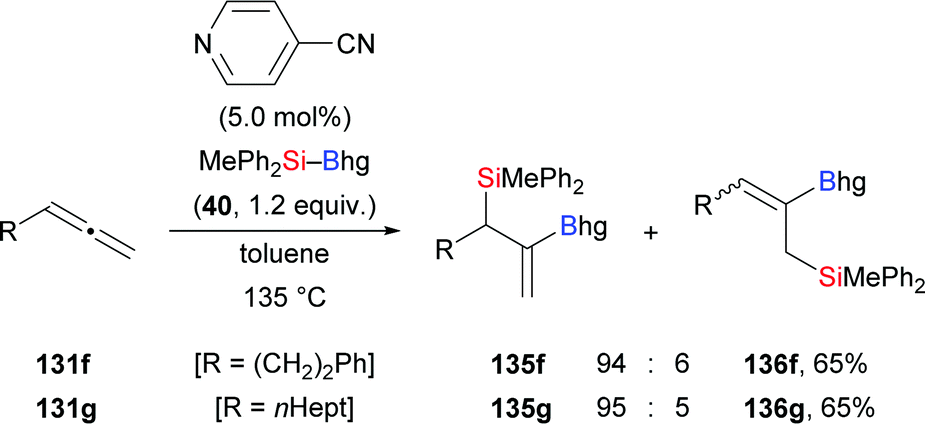
As noted earlier, pyridine-based organocatalysts have been used in the regio- and syn-selective silaboration of terminal alkynes (cf.Scheme 11).26 In that work, terminal allenes were subjected to the same reaction conditions (131f–g → 135f–g, Scheme 41). The silyl and boryl groups are delivered to the internal C–C double bond with the boryl moiety attached on the former C(sp) atom. Small amounts of regioisomers were only seen in two cases.
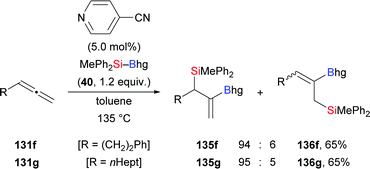 |
| | Scheme 41 Transition-metal-free silaboration of terminal allenes. | |
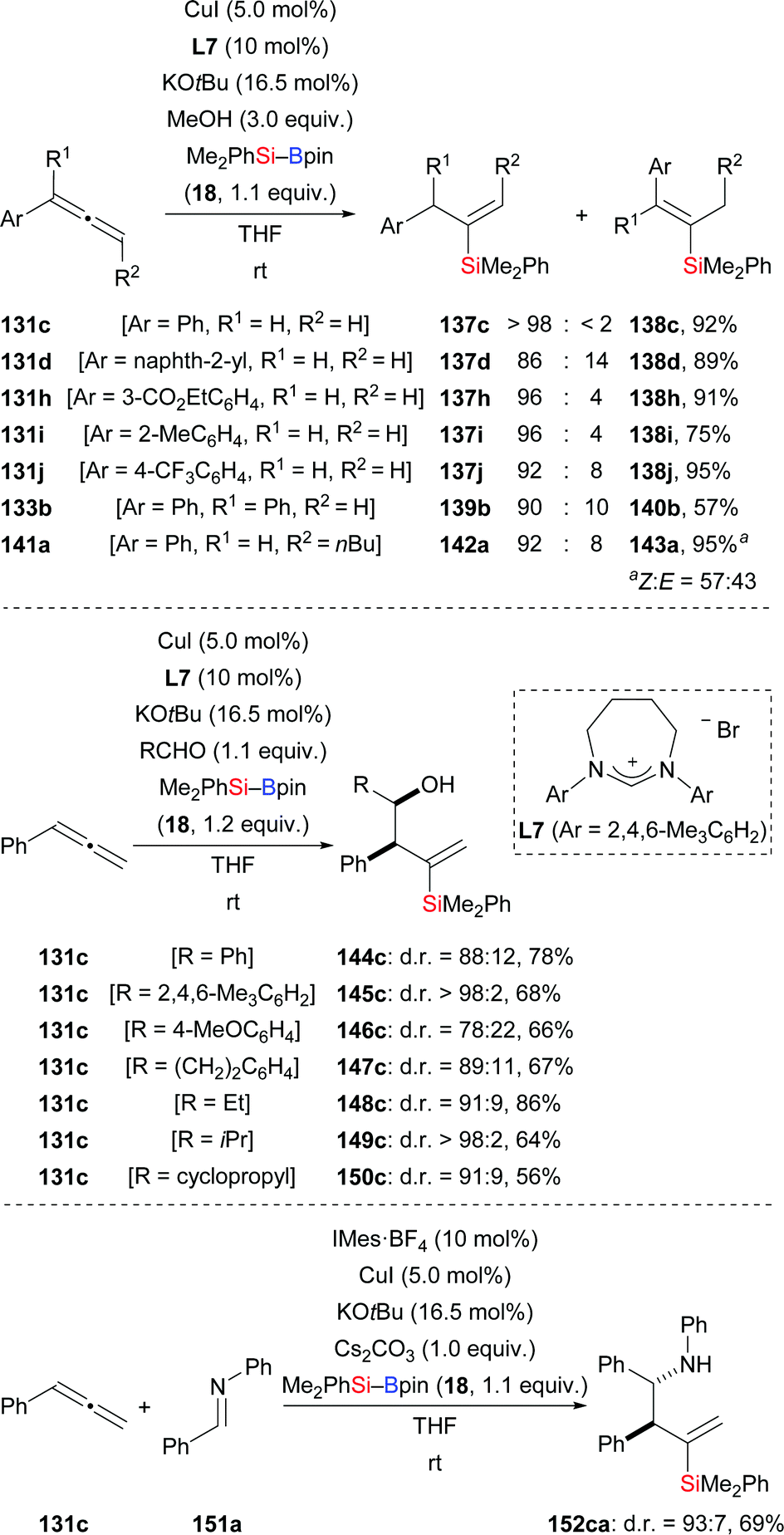
A highly regio- and stereoselective protosilylation of allenes catalysed by an NHC–copper complex was reported by Procter and co-workers (131c, d, h–j → 137c, d, h–j; 133b → 139b; 141a → 142a, Scheme 42, top).61a The seven-membered NHC ligand derived from L7 was shown to be crucial to achieve high regioselectivity. When CD3OD is used instead of MeOH in this hydrosilylation, significant H/D scrambling is evidence for MeOH acting as the proton source. By intercepting the assumed allylic copper intermediate with aldehydes, a three-component coupling reaction was achieved (131c → 144c–150c, Scheme 42, middle). This three-component reaction proceeded smoothly with excellent regiocontrol and diastereoselectivity. Two years later, this strategy was extended to the assembly of homoallylic amines by a three-component coupling of terminal allenes, silylboranes, and imines (131c → 152ca, Scheme 42, bottom).61b
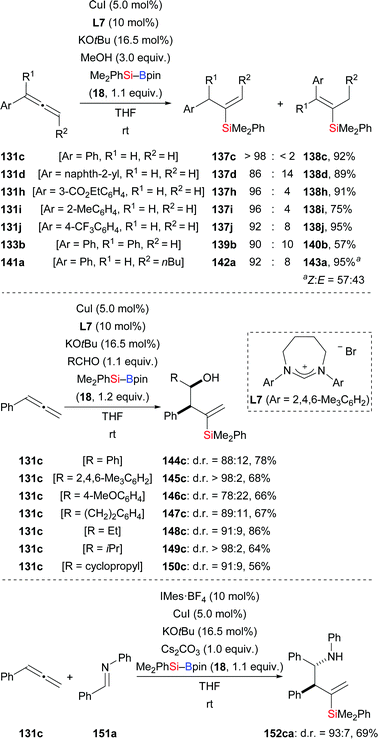 |
| | Scheme 42 Silylation of allenes catalysed by an NHC–copper complex. Yields are for the mixture of regioisomers (top). | |
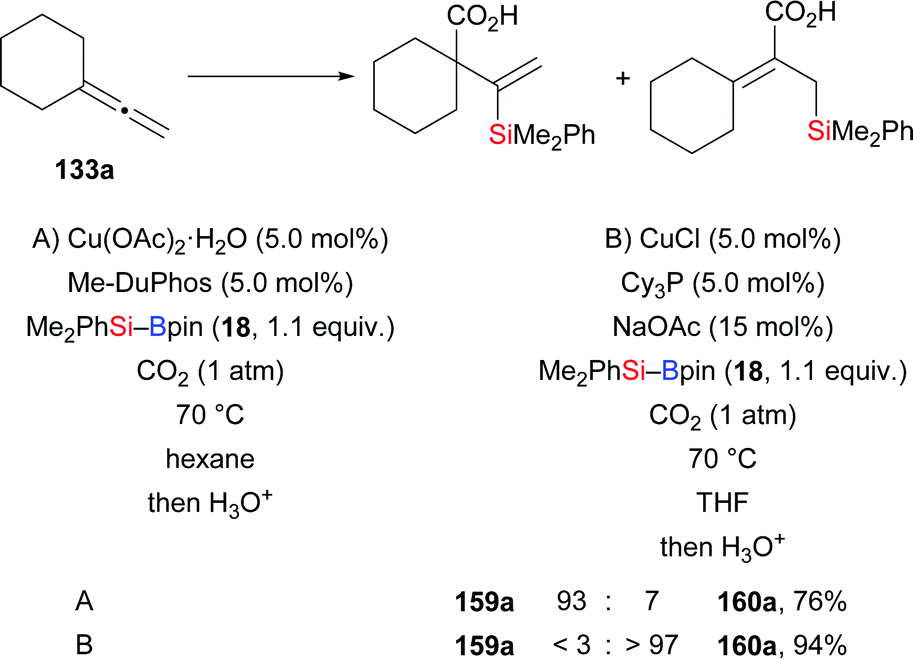
A switch of regiochemistry was observed by the Tsuji group in the three-component coupling reaction of terminal allenes, Et3Si–Bpin (42), and ketones (131a, k → 154aa, 156aa–af, 156kg, Scheme 43, top).62 Various E-configured homoallylic alcohols were obtained with excellent regioselectivity. For example, allene 131a yielded the corresponding product in 62% under the optimized setup (131a → 154aa). Aside from Et3Si–Bpin (42), commercially available Me2PhSi–Bpin (18) underwent the coupling with 131a and acetophenone in 72% yield (dppp was used as ligand; not shown). Mechanistic investigations led the authors to propose a reasonable pathway consisting of silylcupration of the allene followed by addition to the carbonyl compound (not shown). The same group of authors later developed a copper-catalysed silaformylation of allenes by using formate esters as the formyl source yet with opposite regioselectivity (131l → 157l; 133a, c–e → 158a, c–e, Scheme 43, bottom).63 The avoidance of toxic CO gas makes this reaction operationally simple. A wide range of functional groups, including C–C double bonds as well as acetal and ester moieties, were compatible in this transformation. To further illustrate the synthetic utility of this method, a gram-scale silaformylation of 133a was carried out, affording the corresponding product 158a in 95% yield. Mechanistic studies showed that (free) CO is not involved in this transformation.
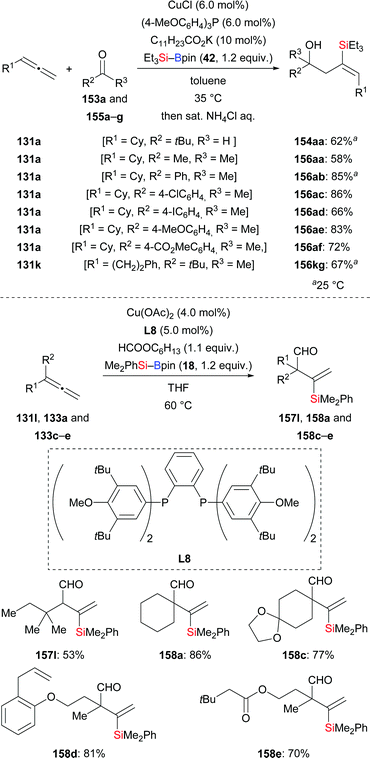 |
| | Scheme 43 Copper-catalysed three-component coupling of terminal allenes, silylboranes, and carbonyl compounds. | |
Another contribution of the Tsuji laboratory from 2014 is about a copper-catalysed regiodivergent silacarboxylation of allenes with silylboranes under an atmosphere of CO2 (133a → 159a and 160a, Scheme 44).64a When Cu(OAc)2·2H2O/Me-DuPhos was used as precatalyst (A), various vinylsilanes were obtained with high regioselectivity. Conversely, CuCl/Cy3P as precatalyst (B) reversed the regioselectivity completely to yield allylsilanes. An enantioselective silacarboxylation was attempted by using (R,R)-Me-DuPhos but enantioinduction was low (18% ee; not shown). Control experiments in the absence of CO2 helped excluding a stepwise process involving silaboration of the allene and carboxylation of the silaboration product (not shown). As before,62 silylcupration of the allene and downstream carboxylation of the vinyl- or allylcopper intermediate are believed to lead to either of the products. The regioselectivity is set in that silylcupration step as a result of the different steric demands of the phosphine ligands Me-DuPhos and Cy3P, respectively. Detailed insight was later gained by DFT calculations.64b
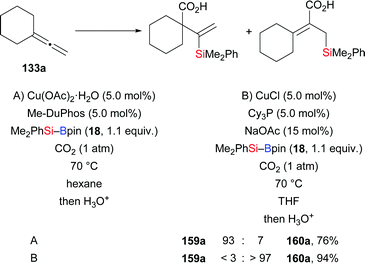 |
| | Scheme 44 Copper-catalysed regiodivergent silacarboxylation of allenes. Yields are for the mixture of regioisomers. | |
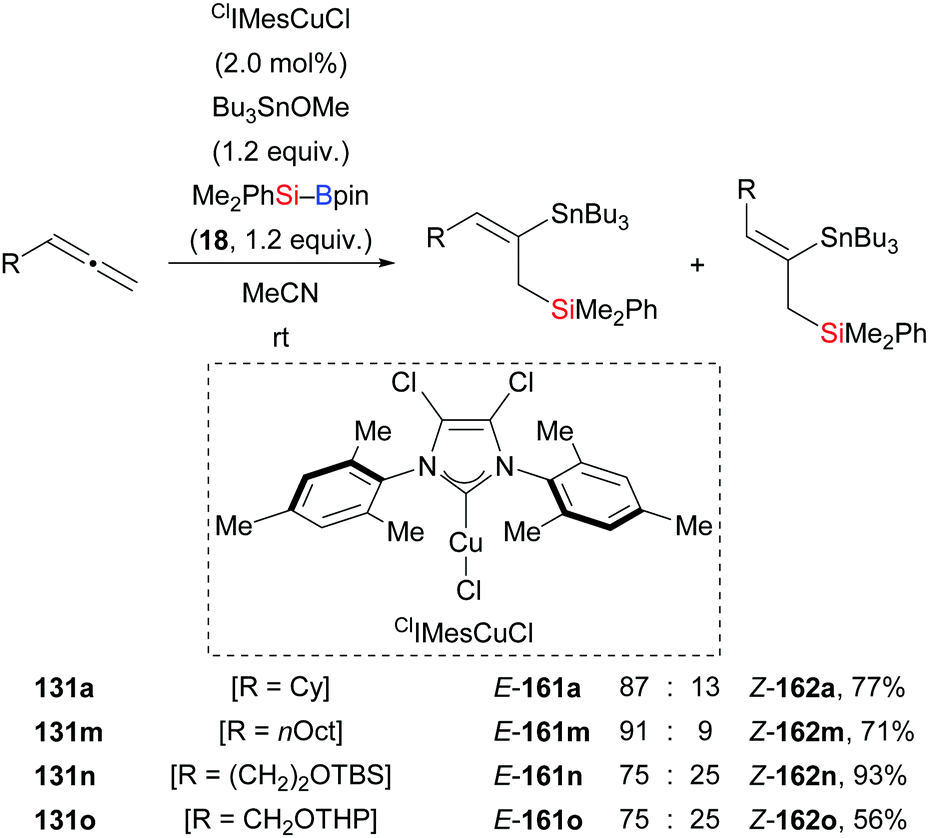
A formal silastannylation of terminal allenes catalysed by an NHC–copper complex was disclosed by Takaki and co-workers in 2015 (131a, m–o → 161a, m–o, Scheme 45).39 A silylboronic ester was used as the silicon source and a stannyl ether served as the tin electrophile. This reaction proceeded with reverse regioselectivity, compared to that of an earlier palladium-catalysed silastannylation of allenes with silylstannanes.65
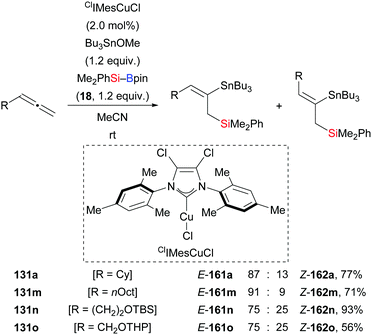 |
| | Scheme 45 Copper-catalysed formal silastannylation of terminal allenes. Yields are for the mixture of isomers. | |
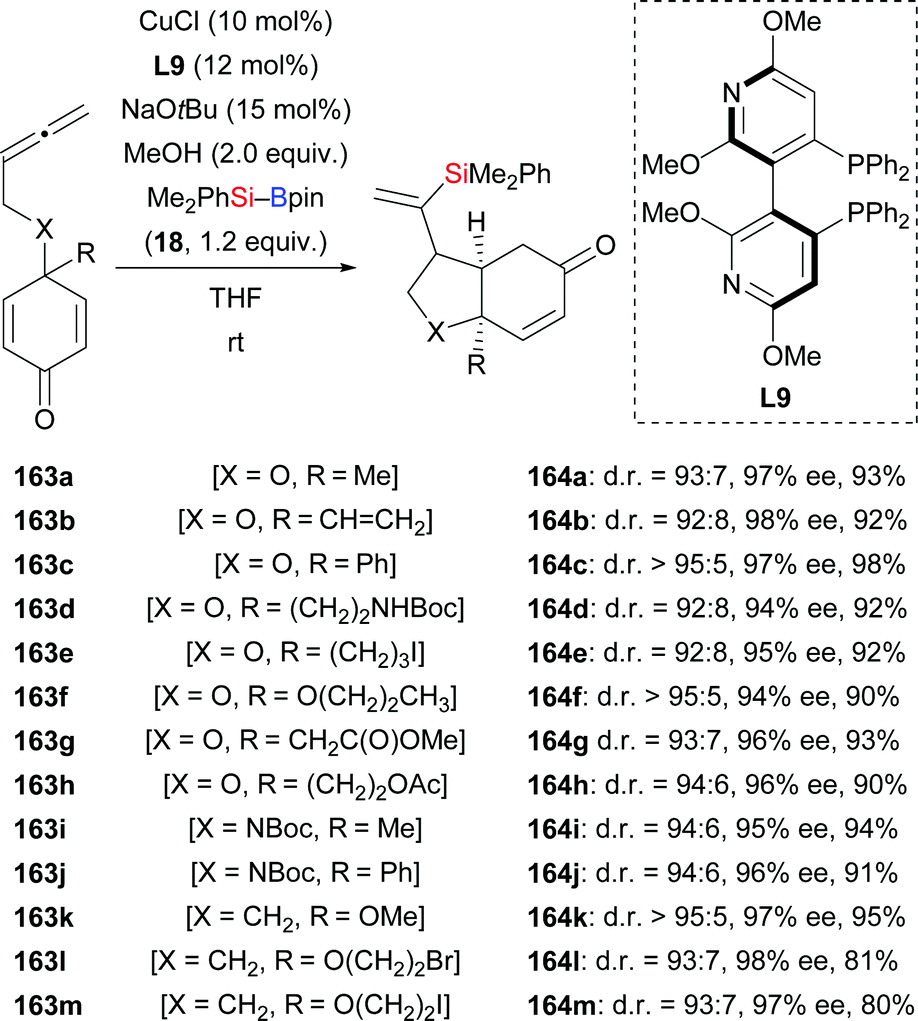
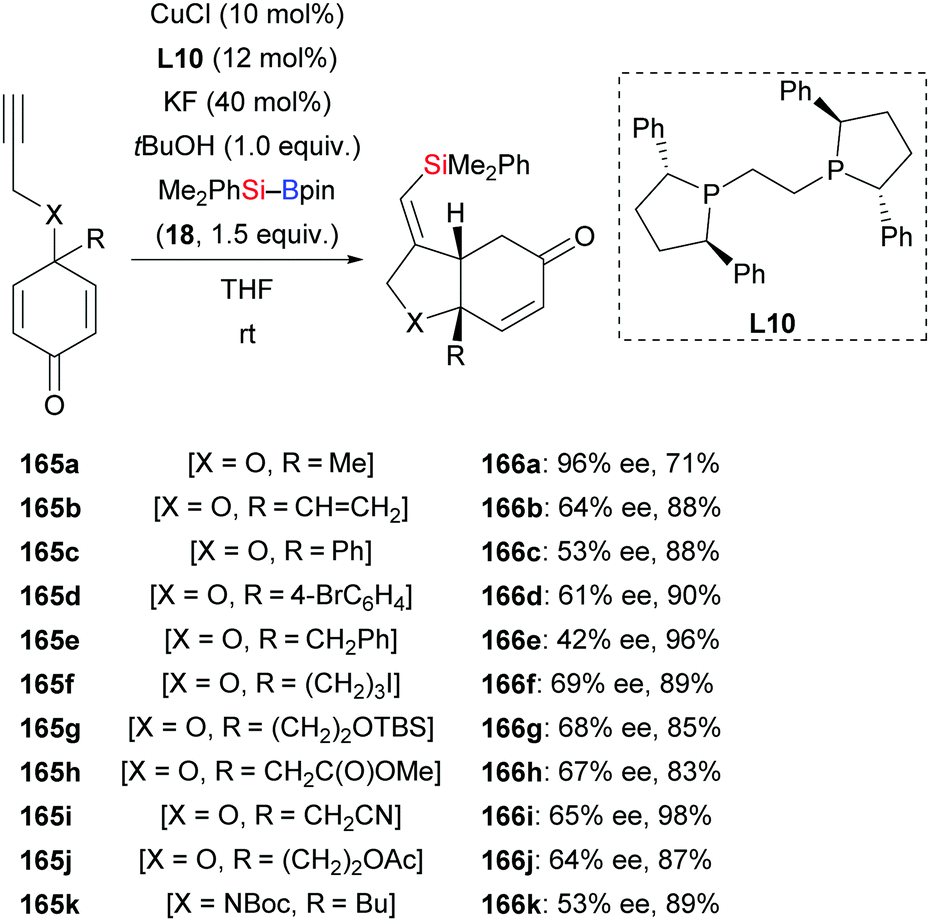
In 2015, Lin and co-workers demonstrated a copper-catalysed enantio- and diastereoselective silylative cyclization of allenes tethered to a cyclohexadienone unit.66 The resulting chiral bicyclo[4.3.0]nonanes were formed with high regiocontrol (163a–h → 164a–h, Scheme 46). This domino reaction proceeds by regioselective intermolecular silylcupration of the allene and subsequent enantioselective intramolecular 1,4-addition to the α,β-unsaturated acceptor. Next to the oxygen tether, nitrogen- and methylene linkers work equally well (163i–m → 164i–m). These authors also reported a copper-catalysed enantioselective silylative cyclization of 1,6-enynes in 2019.67 Yields were excellent throughout but enantioinduction moderate (165a–k → 168a–k, Scheme 47). Here, the authors speculated that coordination of the copper catalyst to the propargylic ether oxygen atom (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) steers the regioselectivity of the silylcupration of the alkyne.
O) steers the regioselectivity of the silylcupration of the alkyne.
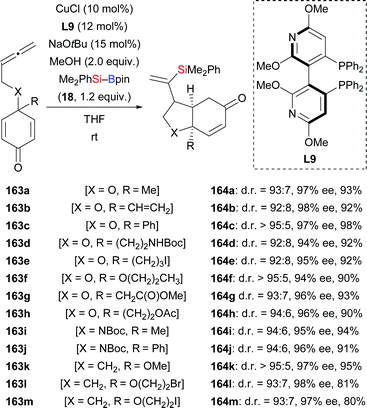 |
| | Scheme 46 Copper-catalysed enantioselective silylative cyclization of allenes to access bicyclo[4.3.0]nonanes. | |
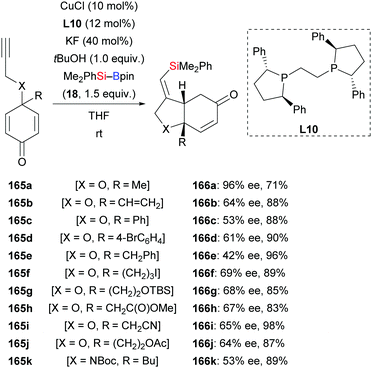 |
| | Scheme 47 Copper-catalysed enantioselective silylative cyclization of alkynes to access bicyclo[4.3.0]nonanes. | |
4.3. 1,2-Addition to C–Het double bonds
4.3.1. Aldehydes and ketones.
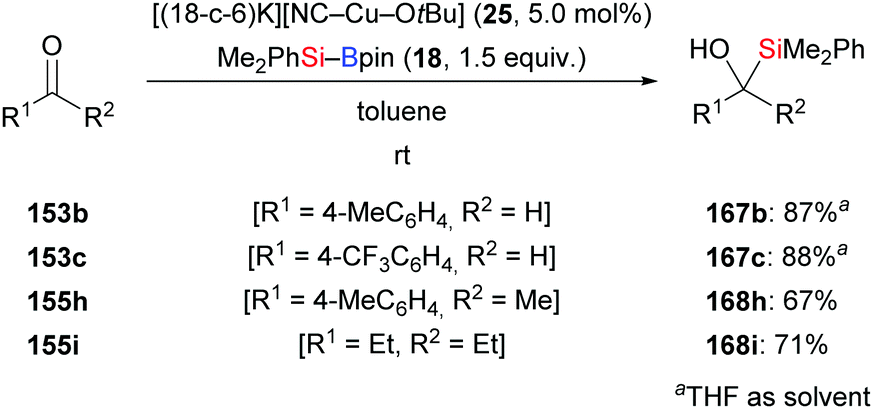
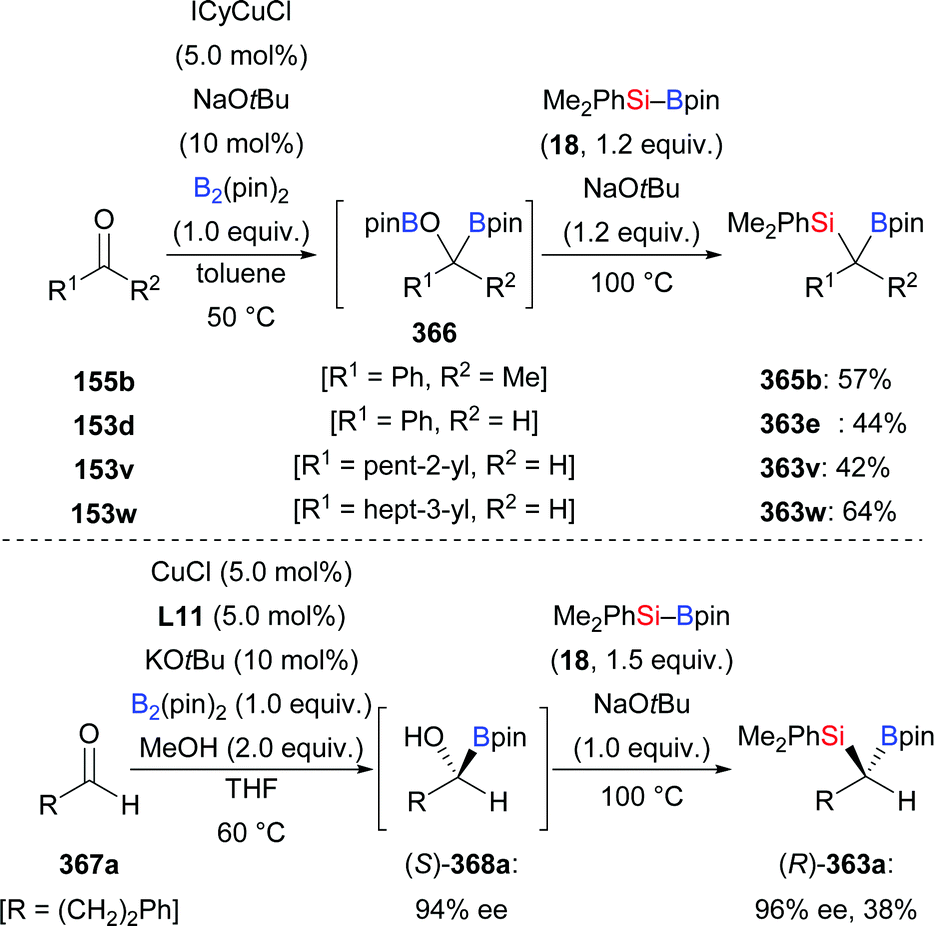
The new [(18-c-6)K][NC–Cu–OtBu] salt which can be used for the activation of silylboronic esters was prepared and characterised by Kleeberg and co-workers. It was found to be an efficient precatalyst in the 1,2-addition of silicon nucleophiles to selected aldehydes and ketones (153b–c → 167b–c; 155h–i → 168h–i, Scheme 48).16 An alcohol additive was usually not necessary. Imines are also suitable substrates but require the presence of iPrOH (not shown).
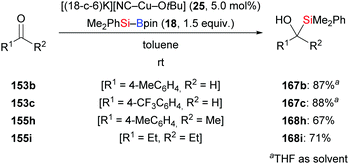 |
| | Scheme 48 Copper-catalysed addition of silylboranes to aldehydes and ketones. | |
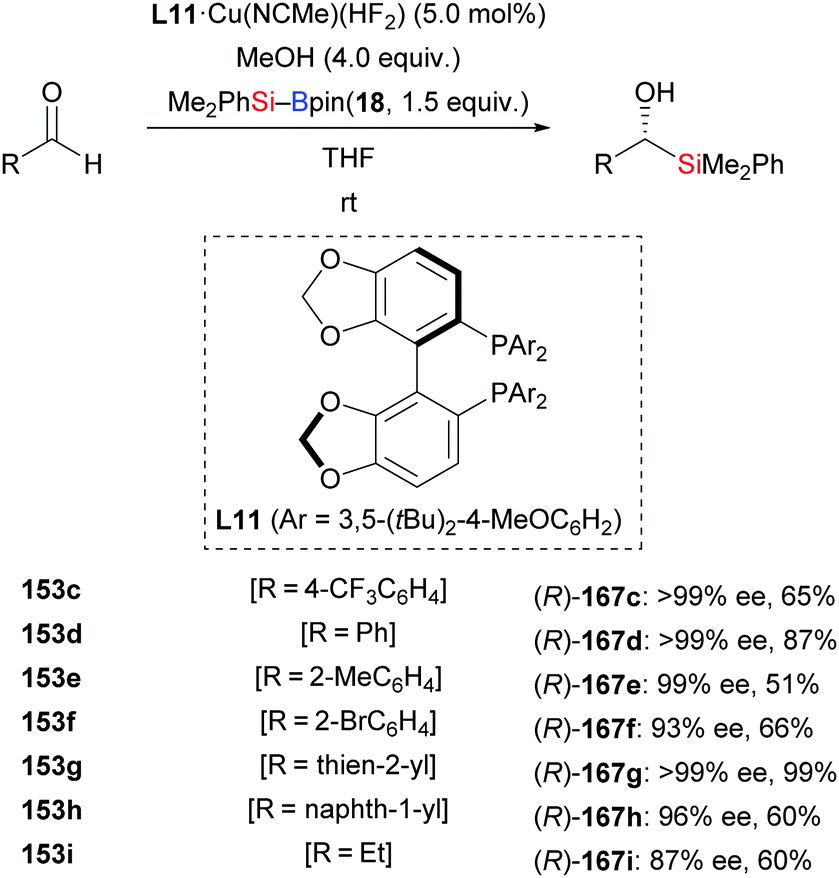
In 2013, the Riant group accomplished an enantioselective addition of silylboranes to aldehydes by employing the newly developed complex L11·Cu(NCMe)(HF2) (153c–i → (R)-167c–i, Scheme 49).68 The HF2− anion is believed to assist the activation of the Si–B bond, thereby avoiding the need of alkoxide additives. Various aromatic (except those bearing an ortho substituent) and aliphatic aldehydes proved to be suitable substrates, giving the corresponding α-hydroxysilanes in good to high yields with excellent enantiocontrol. As probed with α,β-unsaturated citral, 1,2- is favoured over 1,4-addition, affording the 1,2-adduct in 30% yield with 66% ee (not shown).
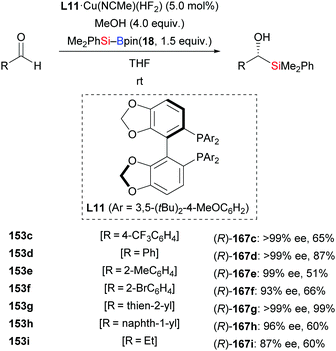 |
| | Scheme 49 Copper-catalysed enantioselective 1,2-addition of an Si–B reagent to aldehydes. | |
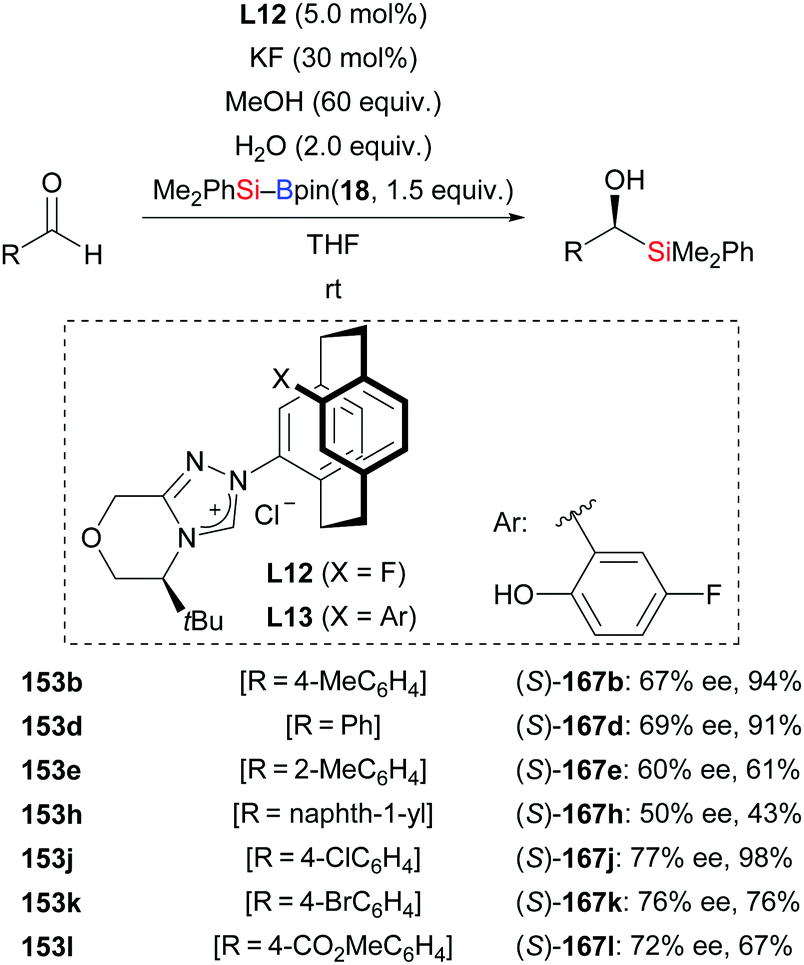
An operationally simple and efficient method for the transition-metal-free enantioselective addition of silylboronic esters to aromatic aldehydes was disclosed by Ma and co-workers (153b, d, e, h, j–l → (S)-167b, d, e, h, j–l, Scheme 50).69a These authors used a [2.2]paracyclophane-based NHC derived from L12 as catalyst. The reaction proceeded with the aid of water to afford a number of chiral α-hydroxysilanes in good to high yields with moderate enantioselectivity. With no water, there was no product formation. The ligand design continued in the Ma group, and led to the development of the chiral triazolium ligand precursor L13 in 2018.69b It was used in the same transformation (not shown).
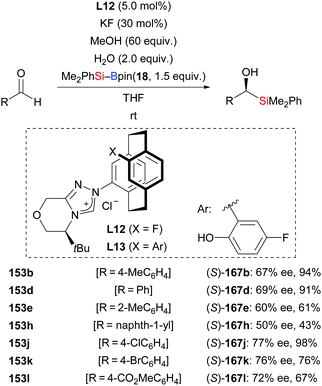 |
| | Scheme 50 NHC-catalysed enantioselective addition of silylboranes to aromatic aldehydes. | |
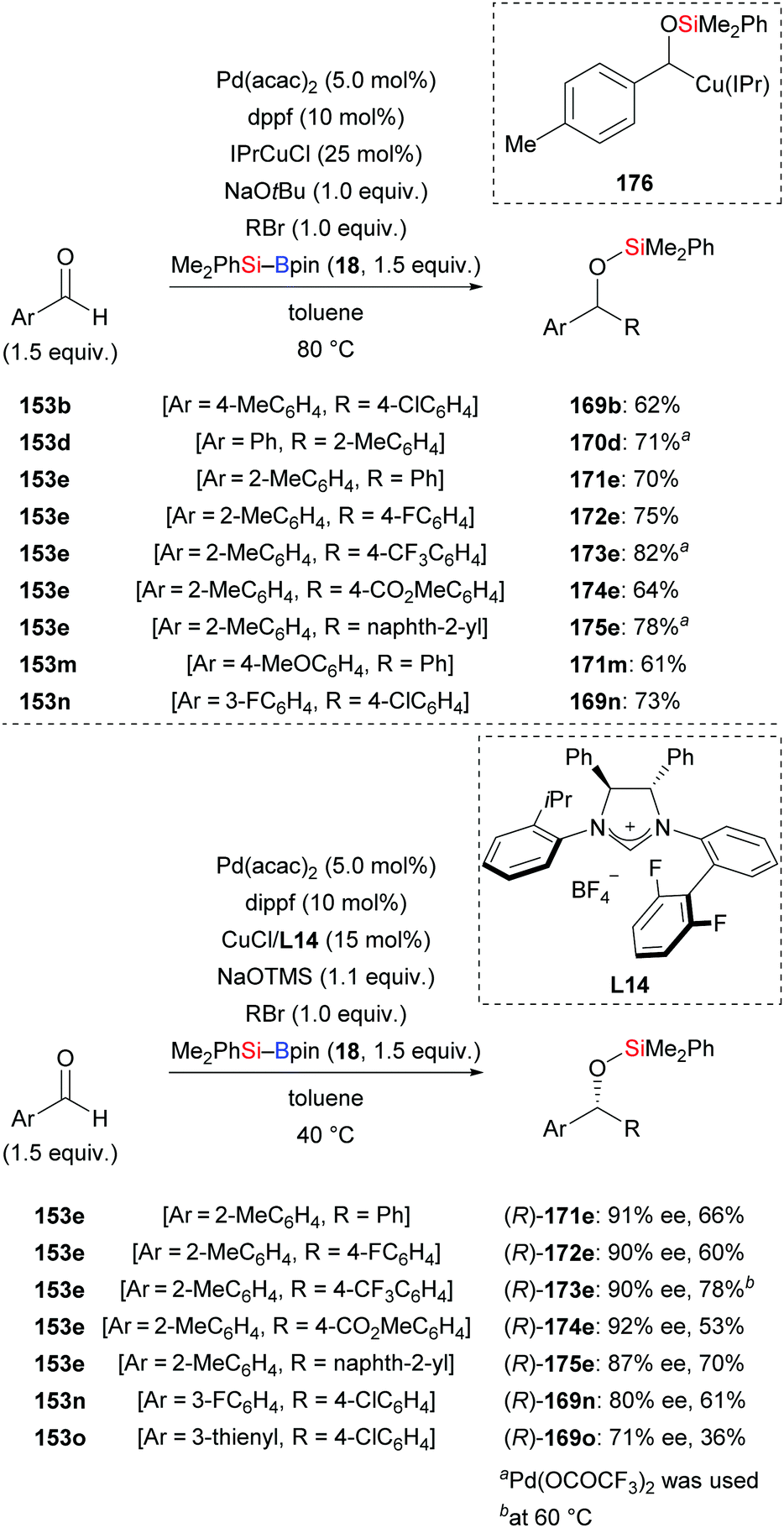
In 2013, Oestreich, Kleeberg, and co-workers clarified the occurrence of a 1,2-Brook rearrangement in the addition of silylboronic esters to aldehydes.70 Ohmiya and co-workers then made great progress in the application of this finding in three-component reactions. In 2018, these authors accomplished a synergistic palladium/copper-catalysed three-component cross-coupling reaction of aromatic aldehydes, silylboranes, and aryl halides (153b → 169b; 153d → 170d; 153e → 171–175e; 153m → 171m; 152n → 169n, Scheme 51, top).71a An asymmetric version was also accomplished by using a chiral NHC–copper complex derived from L14 and a bisphosphine–palladium catalyst. The products were formed in moderate to good yields with high enantioselectivity (153e → (R)-171–175e; 153n–o → (R)-169n–o, Scheme 51, bottom).71b A wide range of functional groups were tolerated in both the racemic and enantioselective version. As a latent α-alkyloxyalkyl anion equivalent, the aldehyde engages in the aforementioned 1,2-addition followed by 1,2-Brook rearrangement, eventually arriving at the α-silyloxybenzylcopper complex 176. To gain further insight, key intermediate 176 was synthesised and reacted with 1-bromo-4-chlorobenzene to deliver the desired product in 68% yield (not shown). Based on this finding, a plausible mechanism was proposed by Ohmiya and co-workers as depicted in Scheme 52. Activation of the Si–B bond mediated by CuX XXVI and base affords the silylcopper XXVIII, which then undergoes addition to the aldehyde. The (enantioenriched) 1,2-adduct XXXIV subsequently rearranges to XXXV with retention of the configuration. Parallel to this, oxidative addition of an aryl halides to Pd0 takes place to generate PdII intermediate XXX, which then engages in a stereospecific transmetalation with the α-chiral α-silyloxybenzylcopper complex XXXV. Palladium complex XXXVI and copper catalyst XXVI are (re)formed in this step. Reductive elimination of XXXVI liberates the coupling and regenerates the palladium catalyst XV.
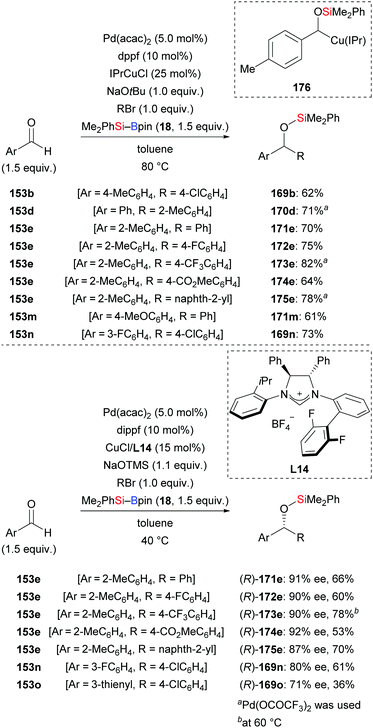 |
| | Scheme 51 Synergistic palladium/copper-catalysed three-component cross-coupling reaction with aryl bromides. | |
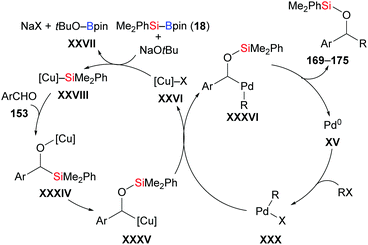 |
| | Scheme 52 Mechanism for synergistic Pd/Cu-catalysed three component cross-coupling reaction. | |
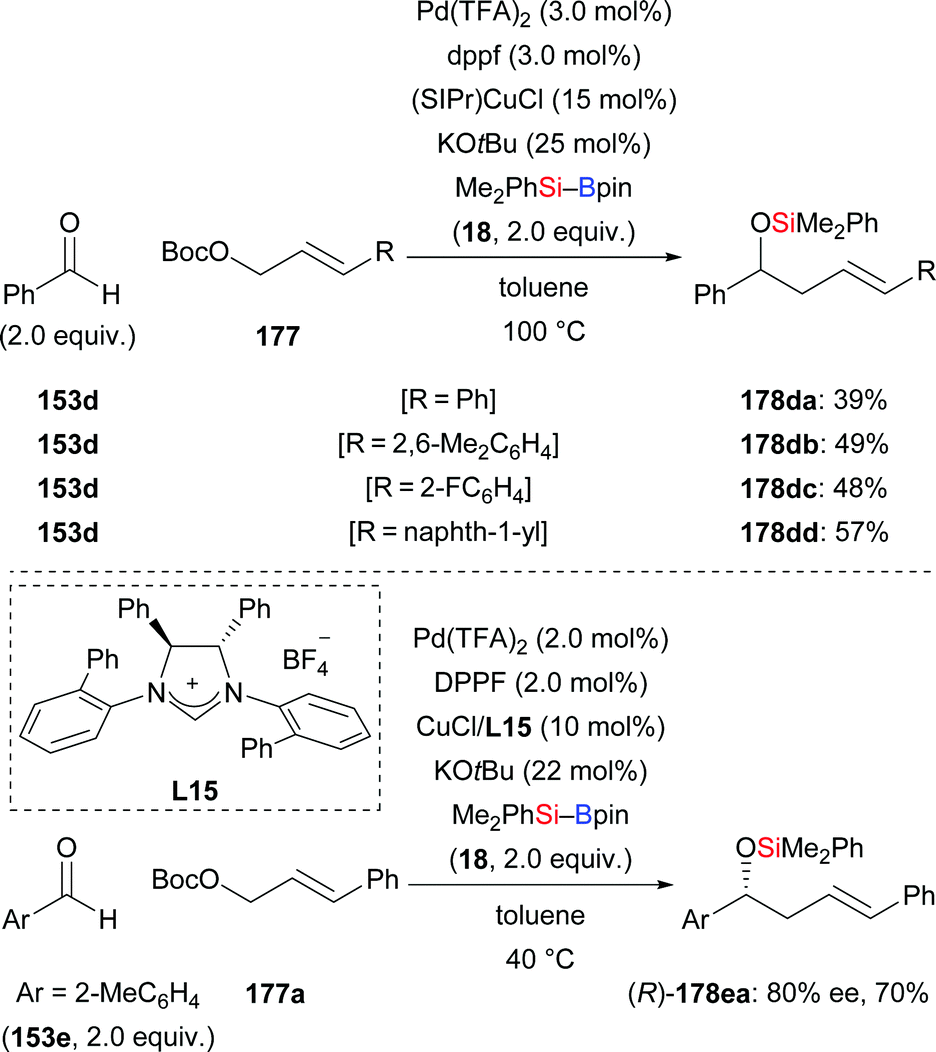
This synergistic palladium/copper-catalysed umpolung strategy is also applicable to allylic electrophiles as reaction partners. Employing allylic carbonates, the same group recently published a method for the synthesis of homoallylic alcohols in moderate yields (153d → 178da–dd, Scheme 53, top).72 With the chiral NHC derived from L15 as ligand, the reaction of ortho-tolualdehyde (153e) with Me2PhSi–Bpin (18) and Boc-protected (E)-3-phenylprop-2-en-1-ol 177a afforded the desired product with good enantioselectivity (153e → 178ea, Scheme 53, bottom).71b
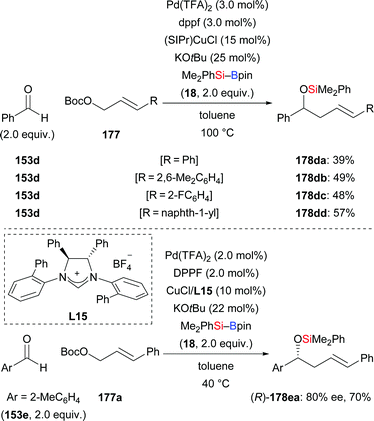 |
| | Scheme 53 Synergistic palladium/copper-catalysed three-component cross-coupling reaction with allylic carbonates. | |
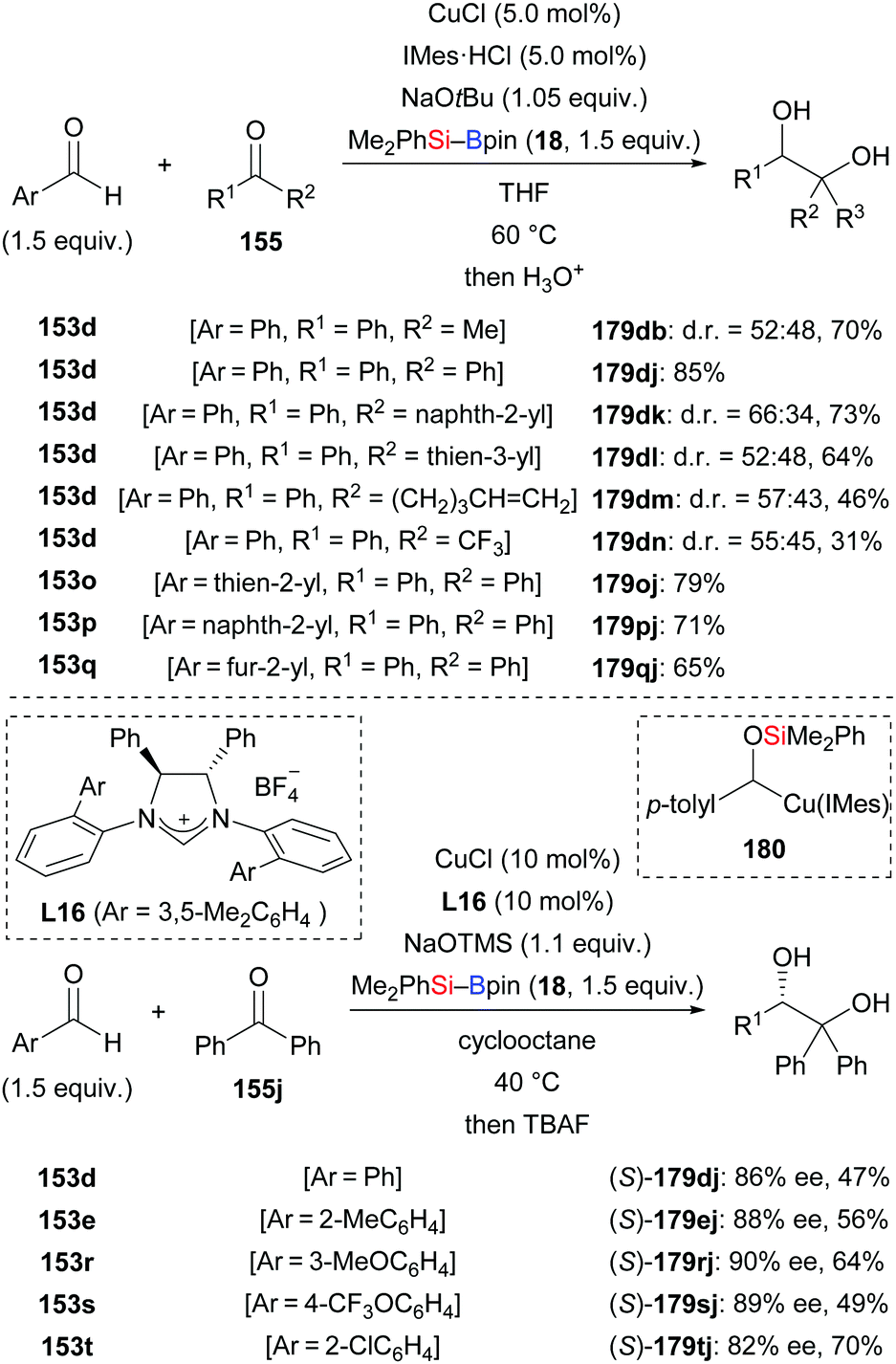
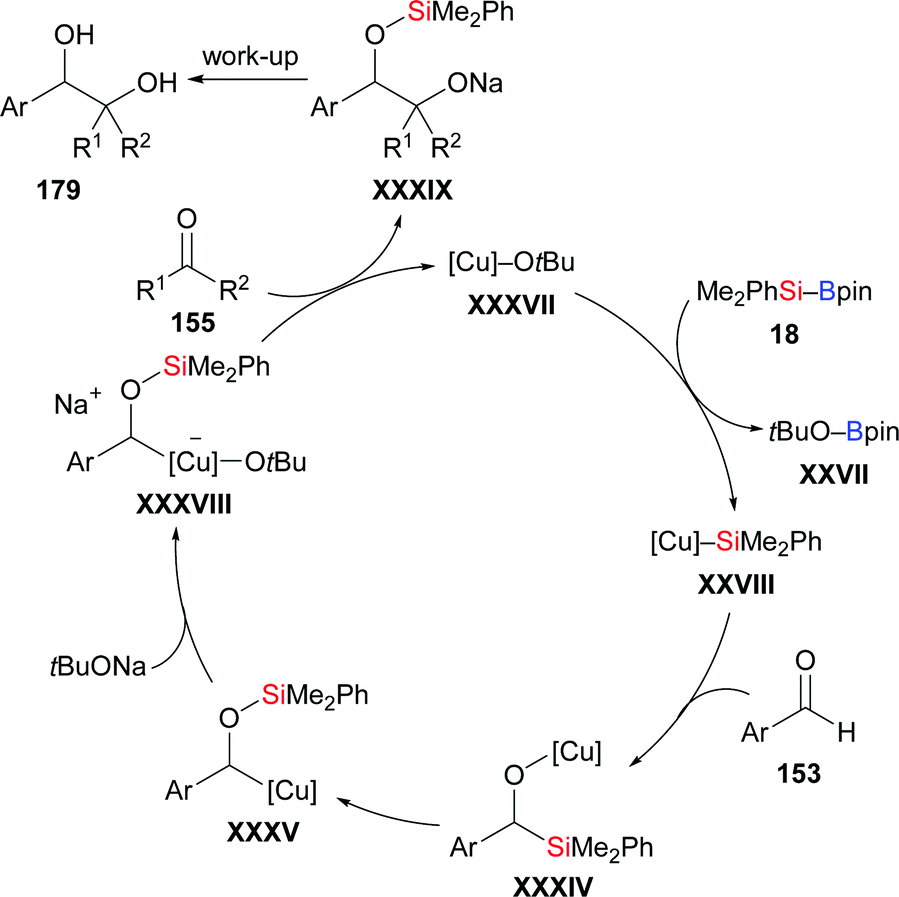
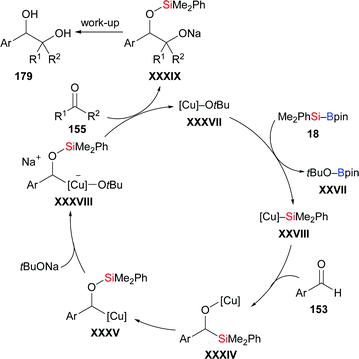
In 2019, a reductive cross-coupling between aromatic aldehydes and ketones to produce 1,2-diols was also accomplished by Ohmiya and co-workers (153d → 179db; 179dj–dn; 153o–q → 179oj–qj, Scheme 54, top).73 An asymmetric variant was achieved by employing a chiral copper catalyst generated from CuCl and NHC precursor L16. The desired chiral diol derivatives formed in moderate to good yields with high enantioselectivity (153d, e, r–t → 179dj, ej, rj–tj, Scheme 54, bottom). As discussed above, the α-silyloxybenzylcopper complex 180 is believed to be the key intermediate. When prepared independently and reacted with diphenyl ketone, the desired product did form in 33% yield in the presence of 10 equivalents of NaOtBu. Of note, no product was obtained in the absence of NaOtBu, suggesting the alkoxide is intimately involved in the coupling step, that is by coordination to the copper center in 180. A proposed catalytic cycle is depicted in Scheme 55. First, [Cu]–OtBu (XXXVII), formed by the reaction of CuCl, IMes·HCl, and NaOtBu, activates the Si–B bond to generate the silicon nucleophile XXVIII. Addition of XXVIII to the aldehyde gives the 1,2-adduct XXXIV, which undergoes a 1,2-Brook rearrangement to form key intermediate XXXV (cf.180). In the presence of NaOtBu, this complex XXXV converts into the ‘activated’ sodium cuprate species XXXVIII. Its addition to ketones and subsequent hydrolysis affords the desired product along with regenerated catalyst XXXVII.
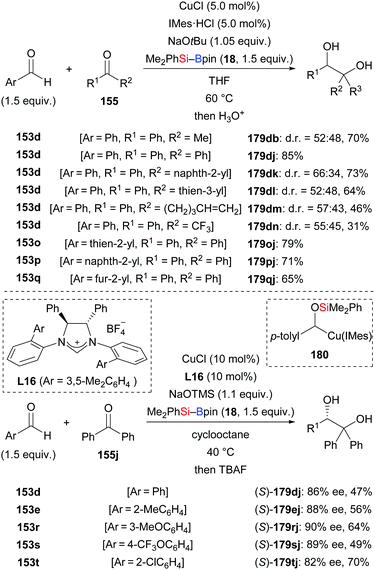 |
| | Scheme 54 Copper-catalysed reductive coupling of aromatic aldehydes and ketones. | |
 |
| | Scheme 55 Mechanism of copper-catalysed reductive coupling of aromatic aldehydes and ketones. | |
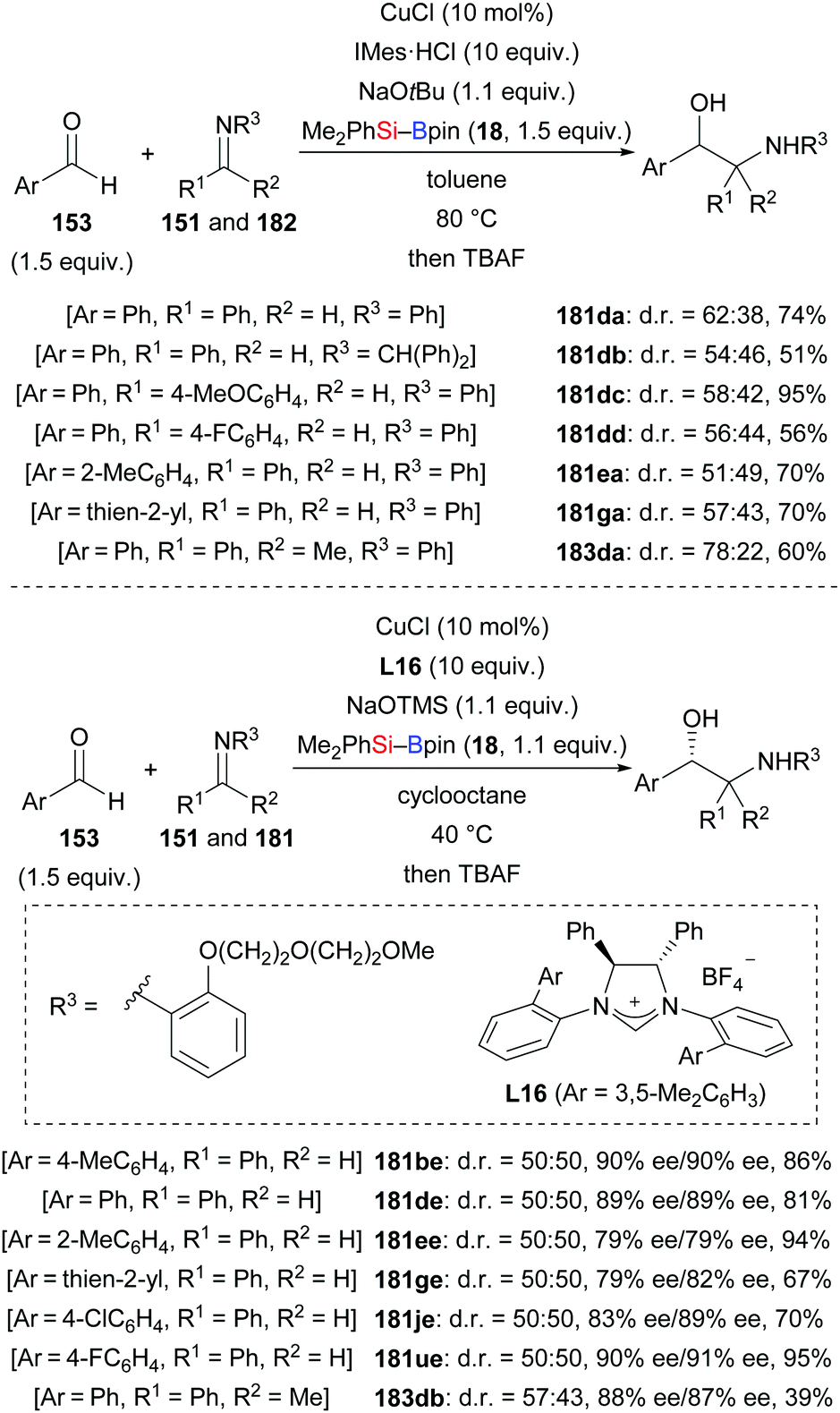
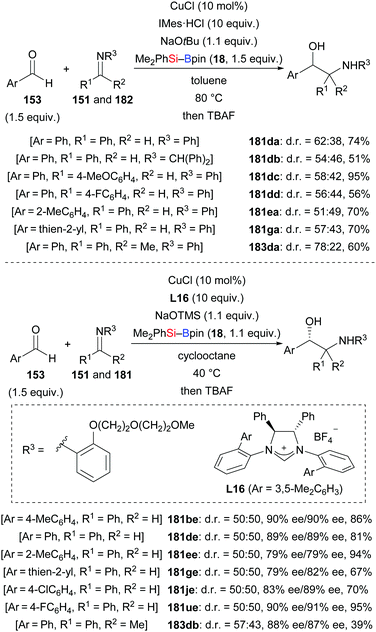
A related three-component reductive coupling of aromatic aldehydes, silylboranes, and imines to access β-amino alcohols was also developed by these authors in both racemic and asymmetric versions (153d → 181da–dd; 153e → 181ea; 153g → 181ga; 153d → 183da, Scheme 56).74 To obtain good enantioselectivity, an aryl group decorated with an ethylene-glycol tether had to be used as an imine protecting group (153b, d, e, g, j, and u → 181be, de, ee, ge, je, ue, and 183db). Its ability to coordinate to the catalyst rather than its steric demand is believed to account for the improved enantioselectivity.
 |
| | Scheme 56 Copper-catalysed reductive coupling of aromatic aldehydes and imines. | |
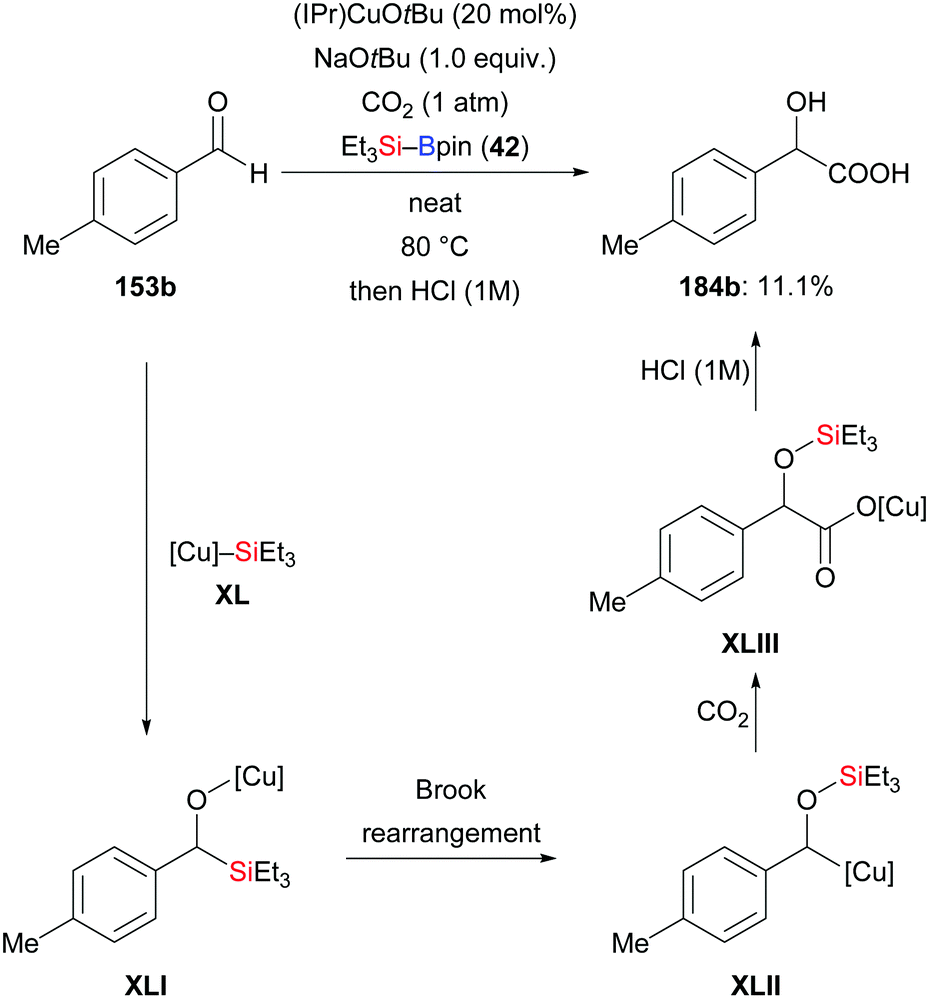
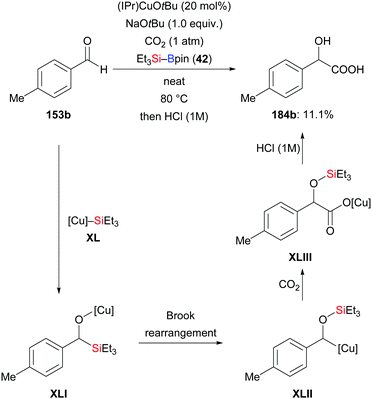
Using the same strategy, the Nozaki group realised a copper-catalysed reductive coupling reaction of p-tolualdehyde and CO2 with Et3Si–Bpin in 2020; the yield is low though (153b → 184b, Scheme 57).75
 |
| | Scheme 57 Copper-catalysed reductive coupling of aldehydes and CO2. | |
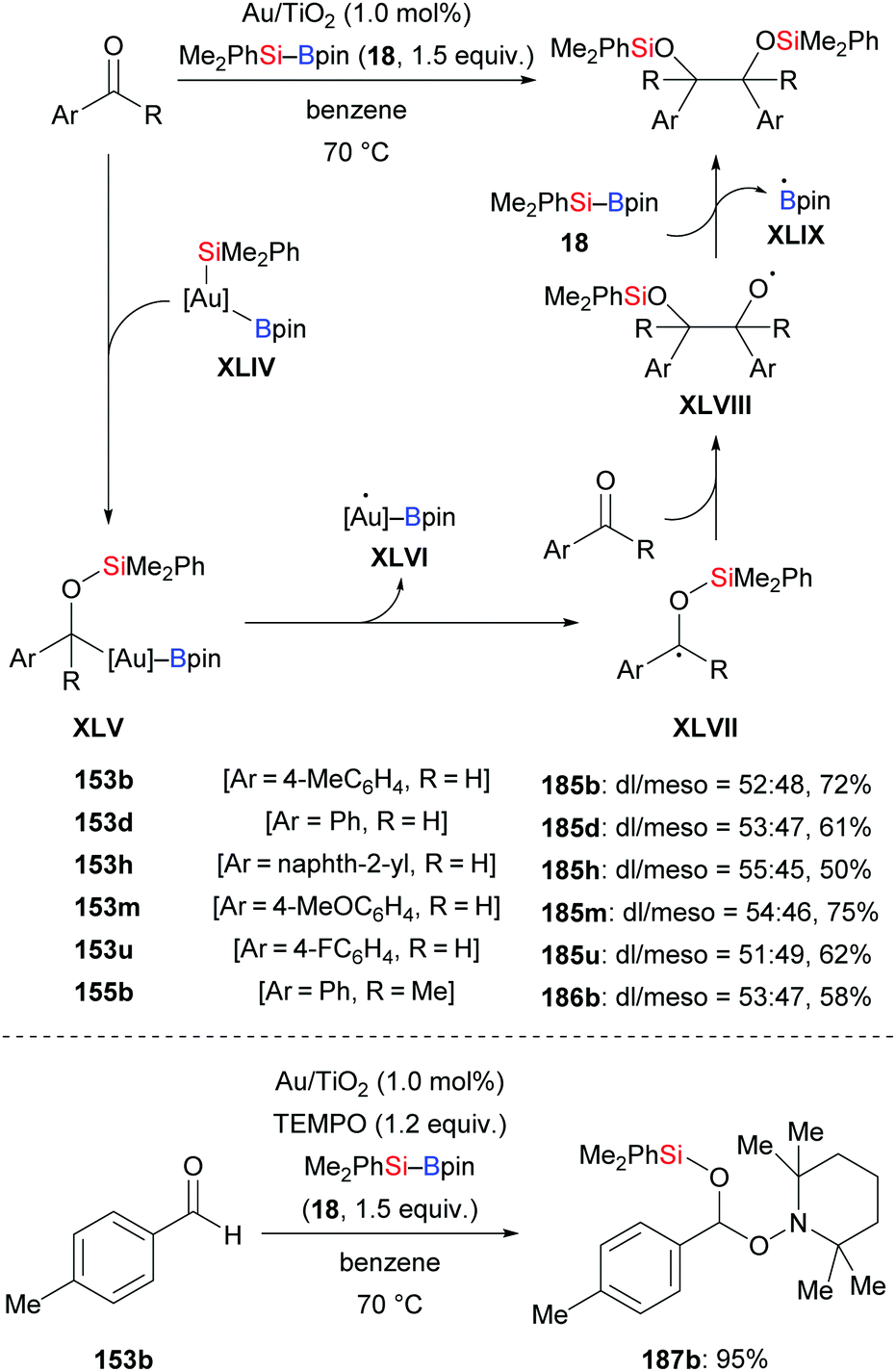
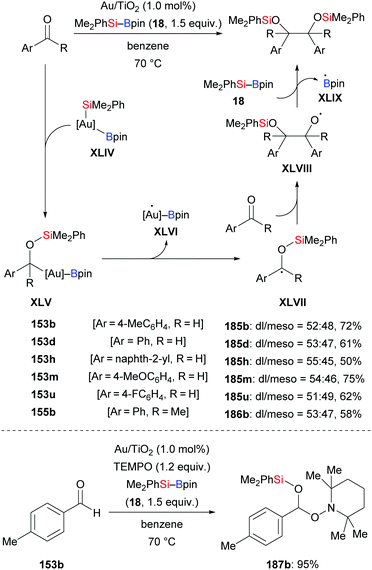
In 2018, a silylative pinacol-type reductive dimerization of aromatic aldehydes and acetophenones mediated by Au/TiO2 nanoparticles was accomplished by Stratakis and co-workers (153b, d, h, m, u → 185b, d, h, m, u; 155b → 186b, Scheme 58, top).76 Aromatic aldehydes bearing substituents in the ortho-position and aliphatic aldehydes did not participate in this transformation. The same outcome was obtained with bulky acetophenones and aliphatic ketones. When TEMPO as a radical scavenger was added to the reaction, only 5% of the product formed, and the TEMPO-trapped adduct was isolated in 95% yield (153b → 187b, Scheme 58, bottom). This suggests that this reaction proceeds by a radical mechanism (cf.Scheme 59). B2pin2 was detected by GC-MS analysis as the byproduct of this reaction.
 |
| | Scheme 58 Gold-catalysed silylatvie pinacol-type reductive dimerization via a radical pathway. Yields are for the mixture of isomers (top). | |
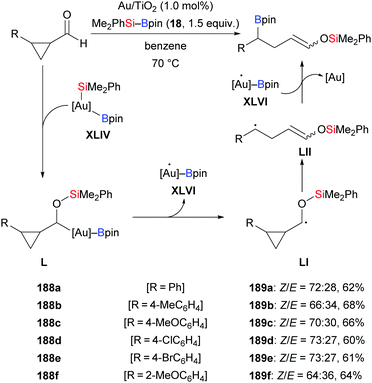 |
| | Scheme 59 Gold-catalysed silaboration of aryl-substituted cyclopropyl aldehydes involving ring opening. Yields are for the mixture of isomers. | |
A year after that, this research group took advantage of this radical pathway in a gold-catalysed silaboration of aryl-substituted cyclopropyl aldehydes, which involves a radical-clock-type rearrangement (188a–f → 189a–f, Scheme 59).77 This gold-catalysed procedure proceeded smoothly in the presence of Au/Ti2O nanoparticles, providing a library of β-boronate-tethered silyl enol ethers in synthetically useful yields. This silaboration products were found to be labile during the purification, delivering the corresponding aldehydes by hydrolytic deprotection. No pinacol-type products were detected in this transformation. Aliphatic substituted cyclopropyl aldehydes and cyclopropanecarboxaldehyde shows no reactivity, probably because aryl groups assume the role of stabilizing the carbon-centered radical during the ring-opening progress.
4.3.2. Imines.
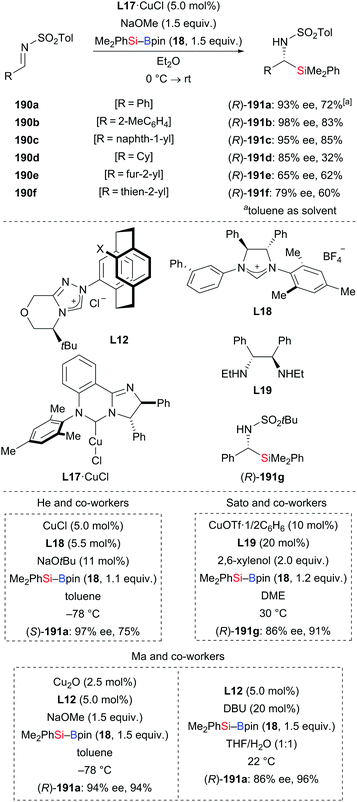
In 2014, Oestreich and co-workers reported a copper-catalysed enantioselective addition of silylboranes to aldimines, establishing a new access to α-silylated amines (190a–f → 191a–f, Scheme 60, top).78 Employing the preformed NHC–copper complex L17·CuCl developed by McQuade and co-workers,79 this method proceeded with a high level of enantiocontrol. A catalytic amount of NaOMe with 4 equivalents of MeOH instead of 1.5 equivalents of NaOMe in THF afforded 191a in 81% yield with 77% ee. These authors also found that the solvent had significant influence on the asymmetric induction. MePh2Si–Bpin (192) proved to be less effective, affording the corresponding α-silylated amine in 57% yield with 60% ee (not shown). Shortly thereafter, He and co-workers,80 Sato and co-workers,81 and Ma and co-workers82 developed different catalytic systems to also promote the enantioselective transfer of the silicon nucleophile to imines (Scheme 60, bottom). Ketimines were tested under He's setup but yields were low and enantiomeric excesses moderate. In Sato's work, a gram-scale silylation was conducted to deliver (R)-191g in 52% yield with >99% ee after two recrystallizations; these authors illustrated the synthetic value of these products by stereospecific carboxylation to yield α-amino acids (not shown). Two kinds of chiral ligands, NHCs and diamines, were successfully employed; a [2,2]-paracyclophane-based NHC used in Ma's work did either serve as ligand or acted directly as a catalyst in the α-silylation of N-tosylaldimines.
 |
| | Scheme 60 Enantioselective 1,2-addition of an Si–B reagent to imines. | |
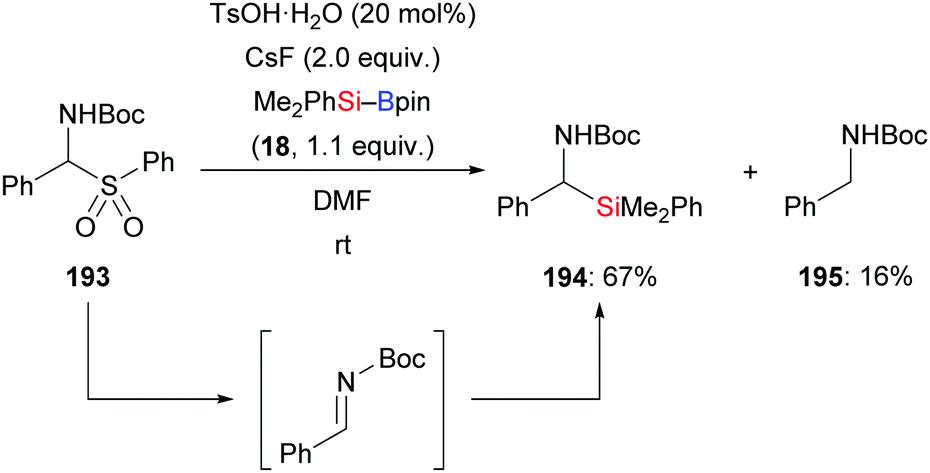
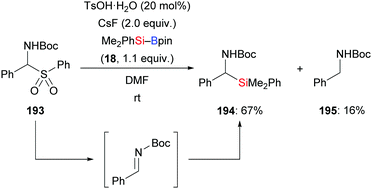
An investigation described by the Sato group in 2012, put forth a new one-pot synthesis of α-amino acids starting from α-aryl α-amido sulfones such as 193 and CO2 as precursors (not shown). The reaction sequence passes through the in situ formation of the corresponding Boc-protected imines followed by α-silylation and the aforementioned carboxylation. By running the reaction at room temperature in the absence of CO2, the silylation product did form in 67% yield (193 → 194 and 195, Scheme 61).83 A protic additive was found to be necessary. The formation of the α-aminosilane as an intermediate confirmed the α-silylation step.
 |
| | Scheme 61 Metal-free synthesis of α-aminosilanes from imine precursors. | |
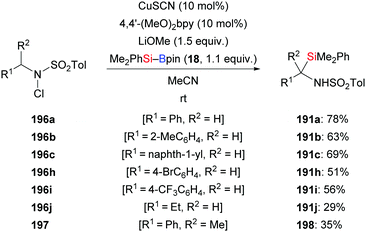
In 2018, Feng and Oestreich introduced a copper-catalysed silylation of C–H bonds adjacent to an amide, making yet another way available for the synthesis of α-aminosilanes (196a–c, h–j → 191a–c, h–j; 197 → 198, Scheme 62).84 Silylboranes such as Me2PhSi–Bpin (18) and MePh2Si–Bpin (192) reacted well whereas Et3Si–Bpin (42) was unreactive. Control experiments showed that this reaction proceeds through imines as intermediates (not shown).
 |
| | Scheme 62 Copper-catalysed synthesis of α-aminosilanes from imine precursors. | |
4.3.3. Acids and anhydrides.
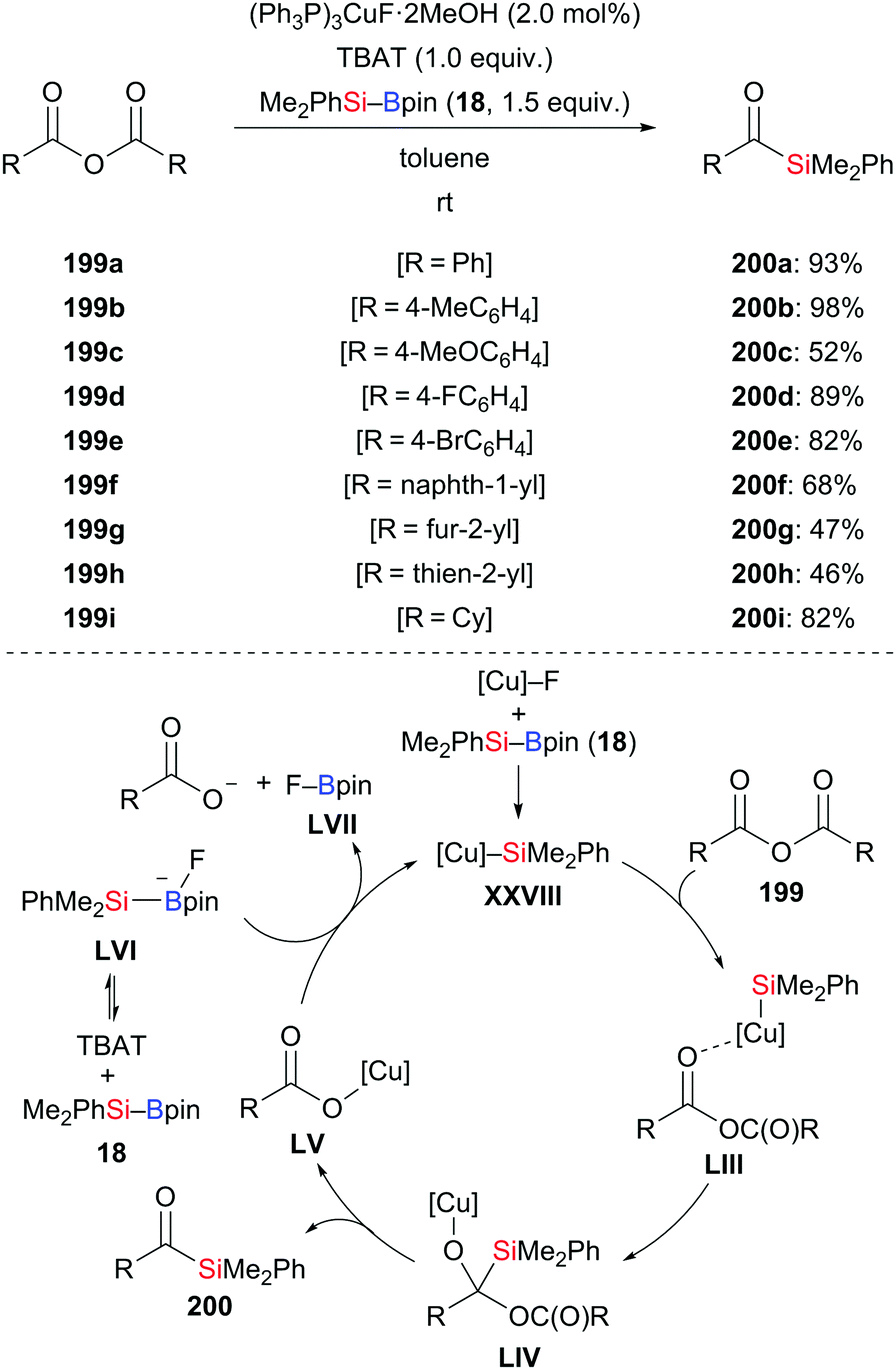
A mild and practical procedure for the synthesis of acylsilanes by the addition of silylboronic esters to acid derivatives was documented by Riant and co-workers in 2013 (199a–i → 200a–i, Scheme 63, top).85 A screening of different acid derivatives identified symmetrical anhydrides to be the best choice as substrates. A large number of functional groups such as halo (199d and 199e), naphthyl (199f), furyl (199g), and thienyl (199h), were compatible in this reaction, furnishing the corresponding acylsilanes in good to excellent yields. No strongly basic precursors are needed. A tentative catalytic cycle is illustrated in Scheme 63 (bottom). As usual, the copper catalyst reacts with silyboranes to afford the copper-based silicon nucleophile XXVIII. Its coordination to the carbonyl oxygen atom of the anhydride facilitates the 1,2-addition to give the tetrahedral intermediate LIV. The product is obtained after collapse of LIV along with the formation of copper carboxylate LV. The catalytic cycle closes with reformation of the silicon nucleophile XXVIII.
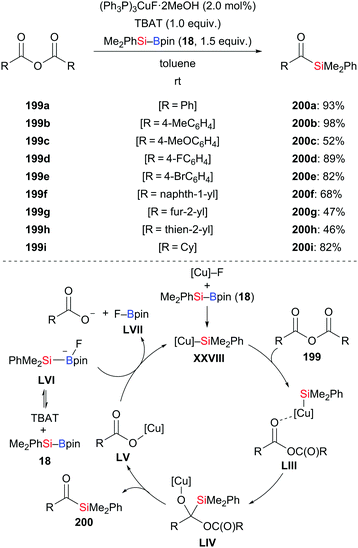 |
| | Scheme 63 Copper-catalysed addition of silylboranes to symmetrical anhydrides. | |
In 2019, the Fujihara group disclosed a zinc-mediated addition of silylboronic esters to carboxylic acids (201a–f, i–k → 200a–f, i–k, Scheme 64).86 The substrate scope of this acylsilanes synthesis with respect to the electrophile proved to be quite general and a number of functional groups were tolerated. In turn, Me2PhSi–Bpin (18) could not be replaced by Et3Si–Bpin (42). NaH serves as base to deprotonate the free acid to the sodium carboxylate. Control experiments showed that the added pivalic anhydride converts that carboxylate into a mixed anhydride which is regioselectively silylated.
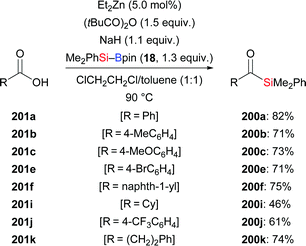 |
| | Scheme 64 Copper-catalysed formal addition of Si–B reagents to carboxylic acids. | |
4.4. 1,2-Addition to Het–Het double bonds
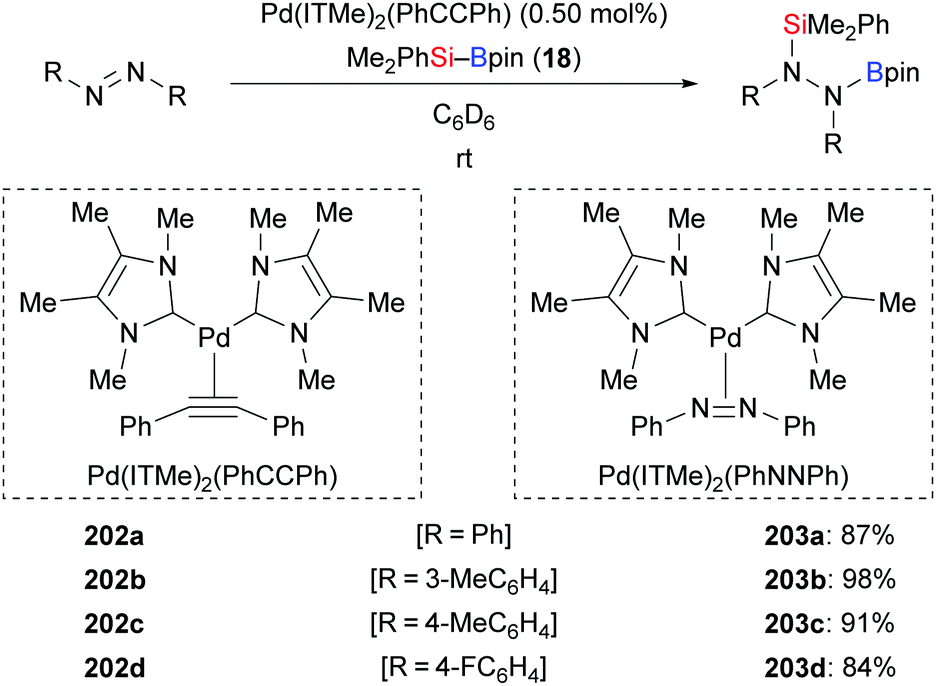
A palladium-catalysed silaboration of azobenzenes was described by Spencer and co-workers in 2016 (202a–d → 203a–d, Scheme 65).87 The resulting functionalised hydrazines are stable towards air and moisture and are easily purified by filtration after precipitation from water. Unsymmetric azobenzenes were compatible in this transformation but led to mixtures of regioisomers (not shown). Low catalyst loading, short reaction time, room temperature, and straightforward purification of the silaborylated products emphasise the utility of this procedure. In 2018, Navarro and co-workers reported the new palladium complex Pd(ITMe)2(PhNNPh), which also promotes this reaction.28b
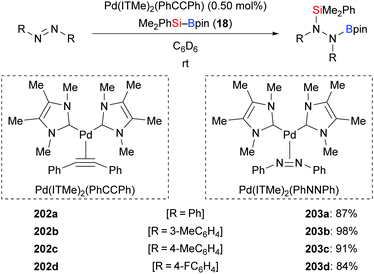 |
| | Scheme 65 Palladium-catalysed addition of silylboranes across NN bonds. | |
4.5. 1,4- and 1,6-addition to α,β-unsaturated carbonyl and carboxyl compounds
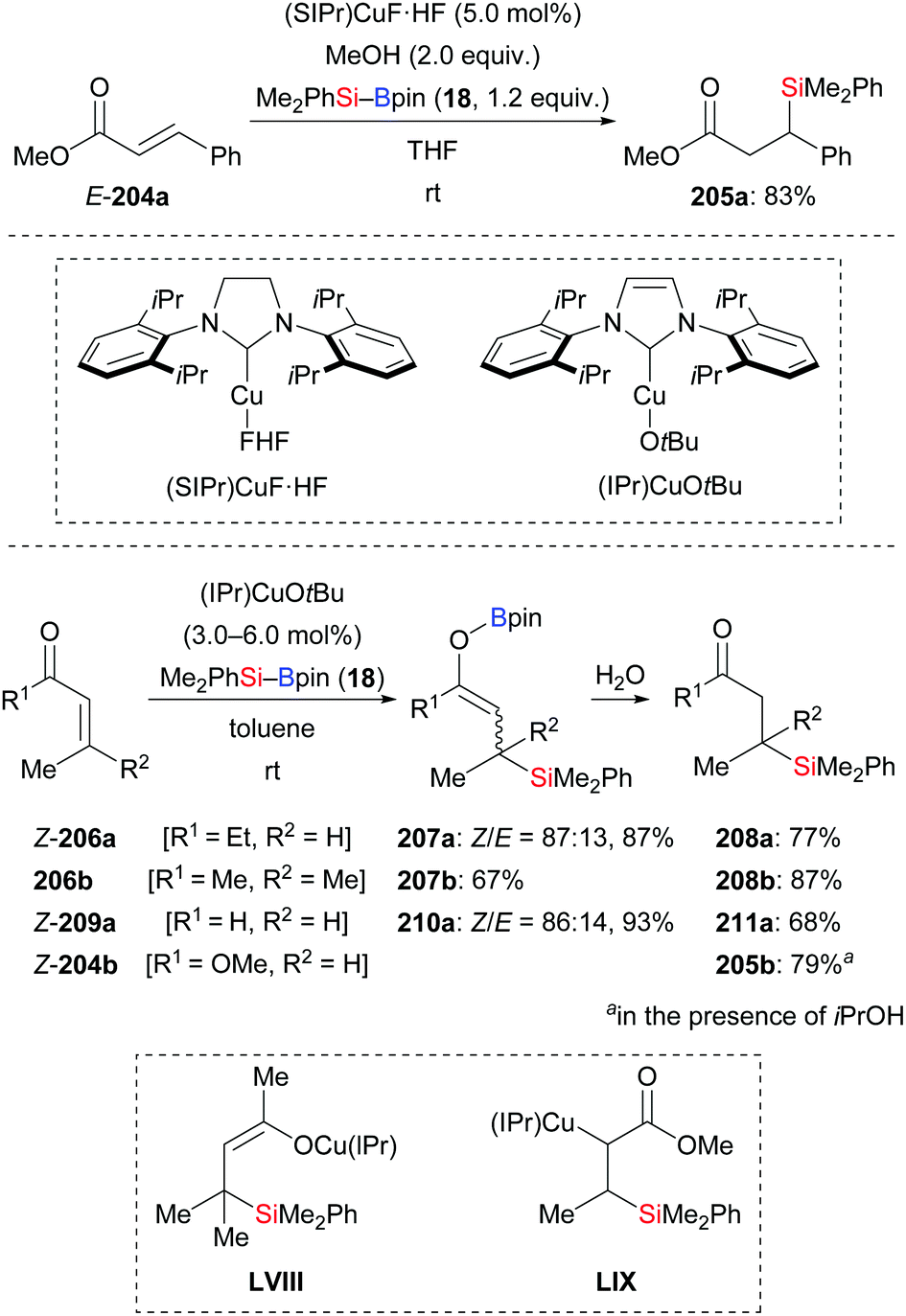
By using the newly developed NHC–copper complex (SIPr)CuF·HF, Riant, Leyssens, and co-workers realised the conjugate addition of Me2PhSi–Bpin (18) to E-204a to yield product 205a in 83% (Scheme 66, top).88a Two years later, the Kleeberg group realised a similar transformation by using (IPr)CuOtBu (Scheme 66, bottom).88b β-Silylated boron enolates were found in the reaction of α,β-unsaturated ketones (206a–b → 208a–b) and an aldehyde (209a → 211a), but not in the reaction of ester 204b. These authors also performed stoichiometric experiments where (IPr)Cu–SiMe2Ph was reacted with α,β-unsaturated ketone 206b to yield β-silyl copper enolate LVIII as the product; LVIII was confirmed by NMR spectroscopy and its molecular structure was determined by X-ray diffraction. Conversely, the reaction of α,β-unsaturated ester Z-204b and (IPr)Cu–SiMe2Ph did not give the O-enolate but instead the C-enolate LIX. This explains why β-silyl boron enolates were not detected in the reaction with esters.
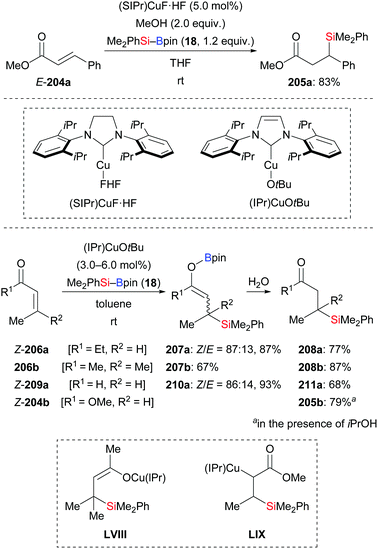 |
| | Scheme 66 Copper-catalysed conjugate addition of silylboronic esters with isolation of enolate intermediates. | |
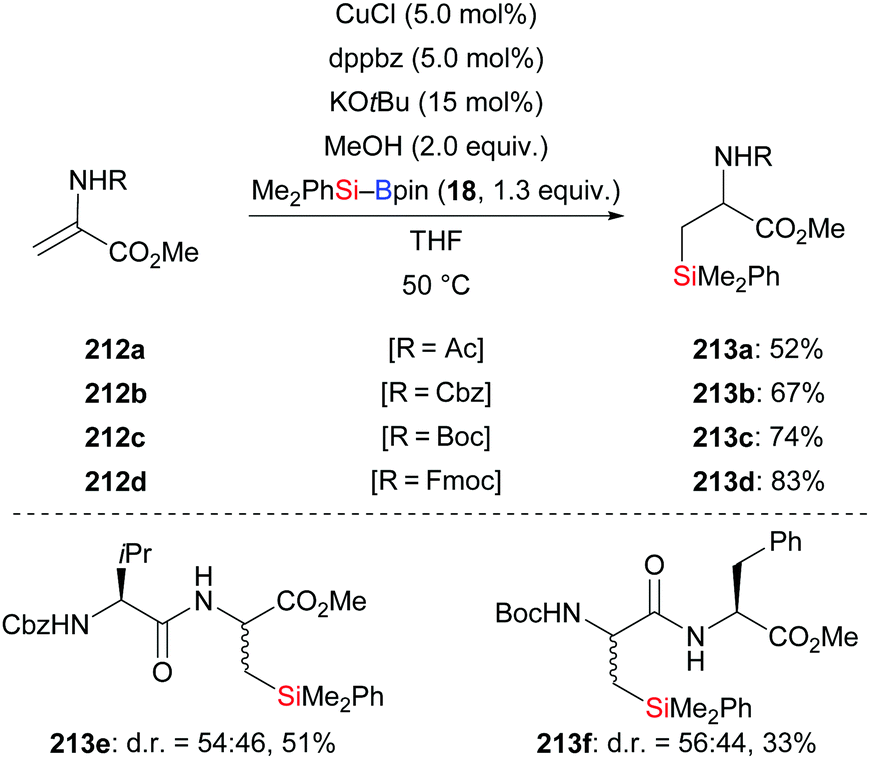
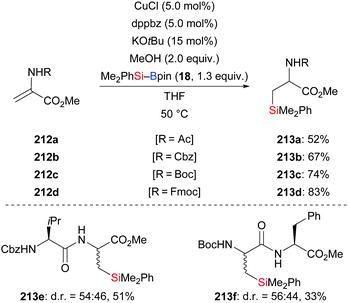
A copper-based catalytic system for the conjugate addition of silylboronic esters to unactivated protected dehydroalanine derivatives was developed by Piersanti and co-workers in 2015 (212a–d → 213a–d, Scheme 67).89 Several dehydropeptides which can be easily prepared without any purification, were compatible under the standard setup, e.g.213e and 213f. However, poor diastereoselectivity was found in these transformation because of the lack of stereocontrol in the enolate protonation step.
 |
| | Scheme 67 Copper-catalysed conjugate addition to dehydroalanine. | |
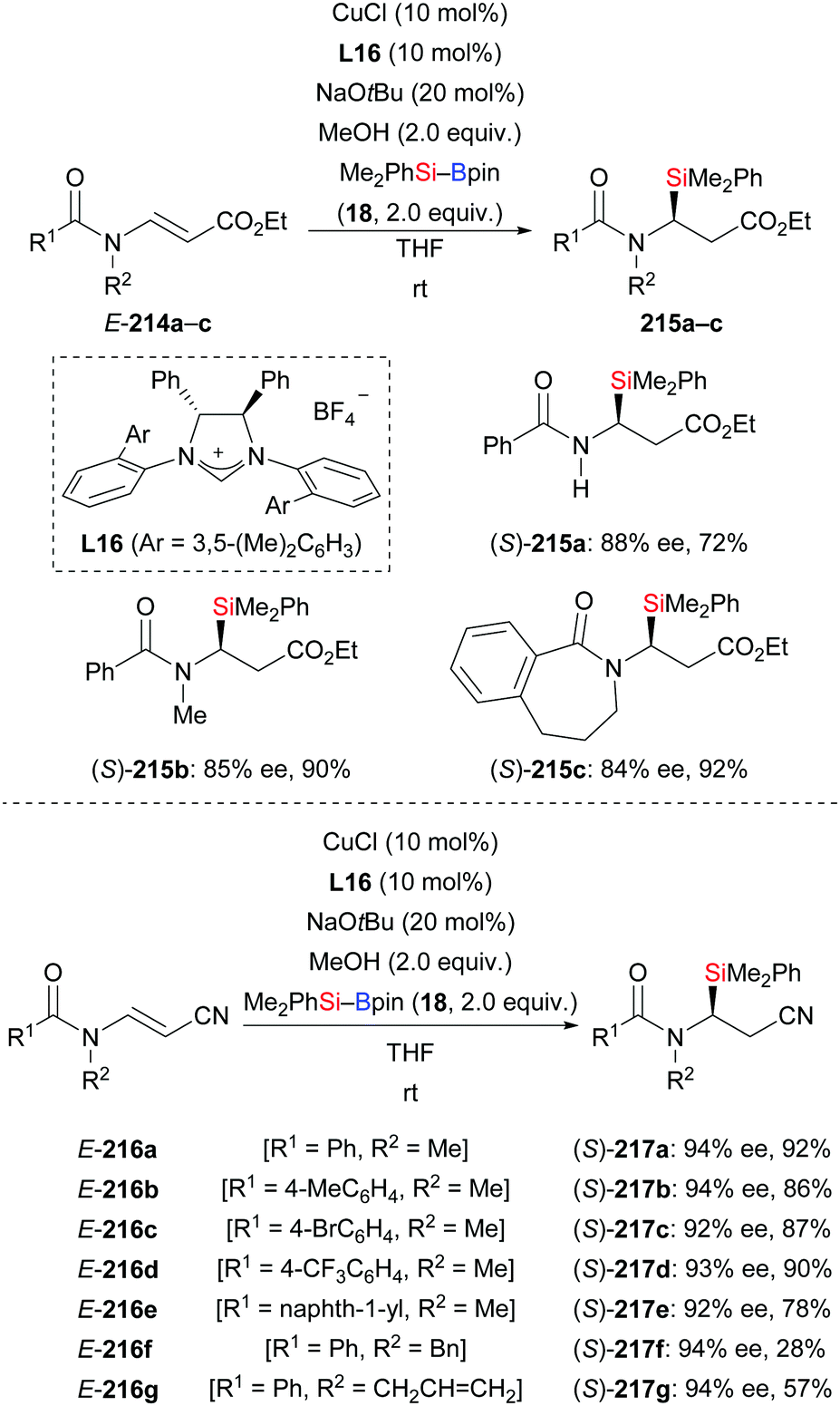
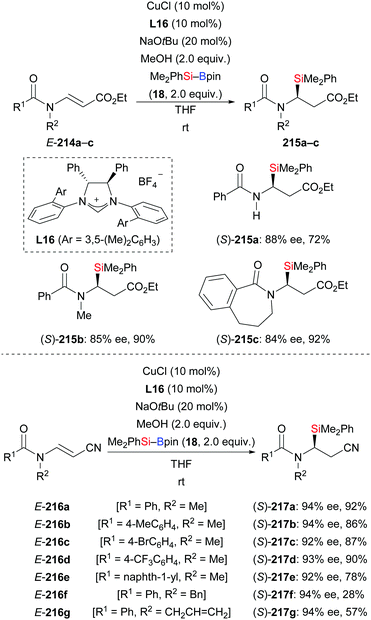
An NHC–copper complex also promoted the conjugate silylation of β-aminoacrylates and β-aminoacrylonitriles (214a–c → 215a–c; 216a–g → 217a–g, Scheme 68).90 This method was established by Xu and co-workers in 2019. At room temperature, it provided a variety of chiral α-aminosilanes in good yields with high enantioinduction (cf. Section 4.3.2). It is noteworthy that substrate E-214a with a free N–H bond reacted cleanly to the desired product 215a in 72% yield with 88% ee.
 |
| | Scheme 68 NHC–copper-catalysed enantioselective silylation of β-aminoacrylates and β-aminoacrylonitriles. | |
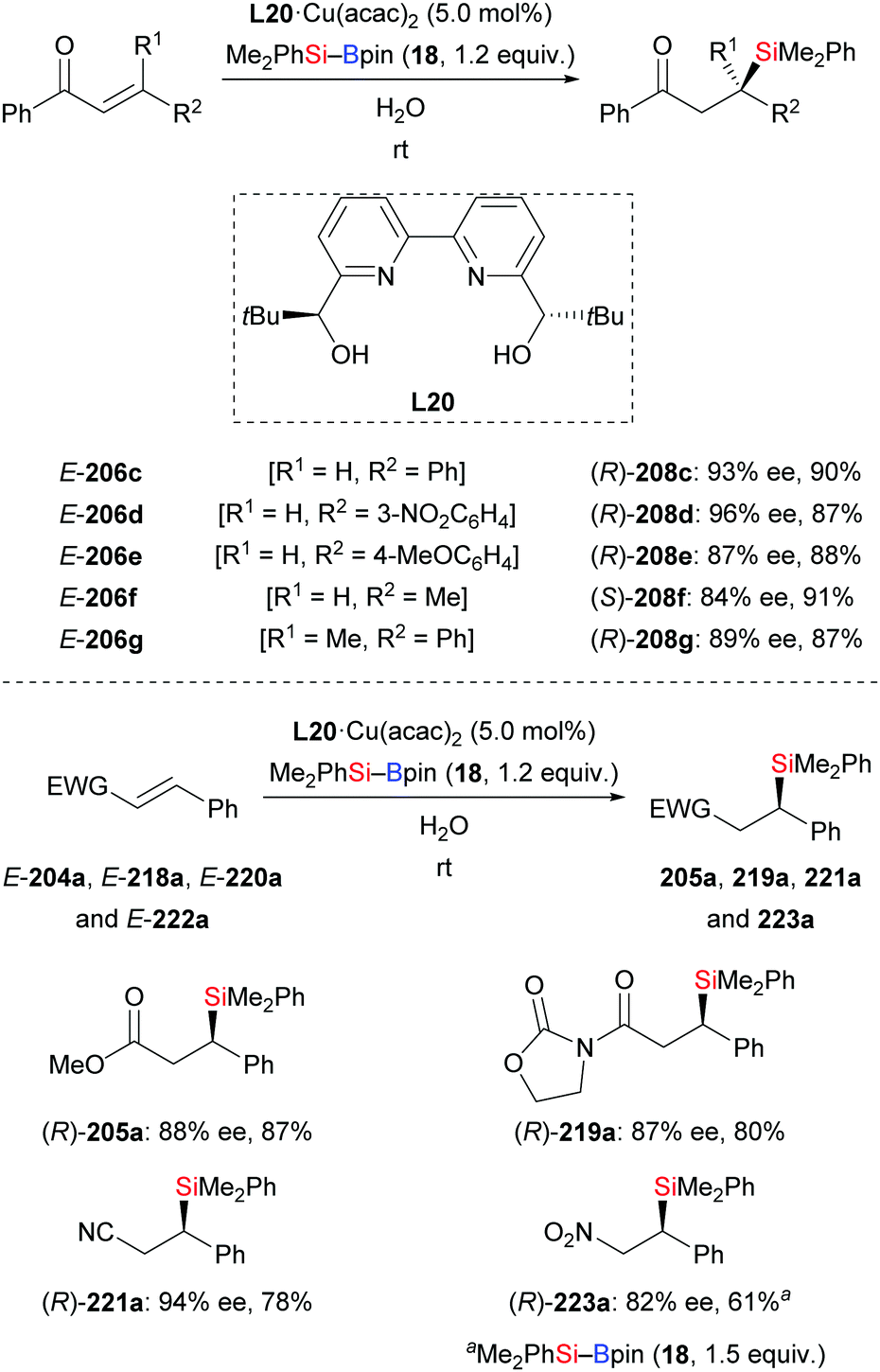
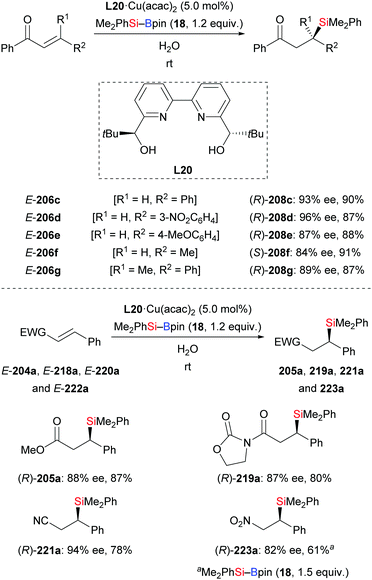
An asymmetric conjugate addition of silylboronic esters to α,β-unsaturated carbonyl compounds in water using L20·Cu(acac)2 as precatalyst was documented by Kobayashi and co-workers (206c–h → 208c–h, Scheme 69, top).91 Despite the insolubility of the catalyst, water has been the optimal solvent for this reaction. The catalyst could be easily separated after the reaction and then used for a second run, leading to the desired products with hardly any loss in yield and enantioselectivity (not shown). Substrates with an electron-withdrawing group (206d) or an electron-donating group (206e) were tolerated. Of note, this method could be used to construct a quaternary carbon center as in (R)-208g. Other α,β-unsaturated compounds such as ester E-204a, amide E-218a, nitrile E-220a, and β-nitrostyrene E-222a were compatible, furnishing the corresponding products with good enantioselectivity (Scheme 69, bottom). The influence of catalyst solubility in this transformation was investigated by using mixtures of THF and water in various ratios. The authors detected an increase in enantioselectivity when the amount of water was raised.
 |
| | Scheme 69 Copper-catalysed enantioselective conjugate silylation in water. | |
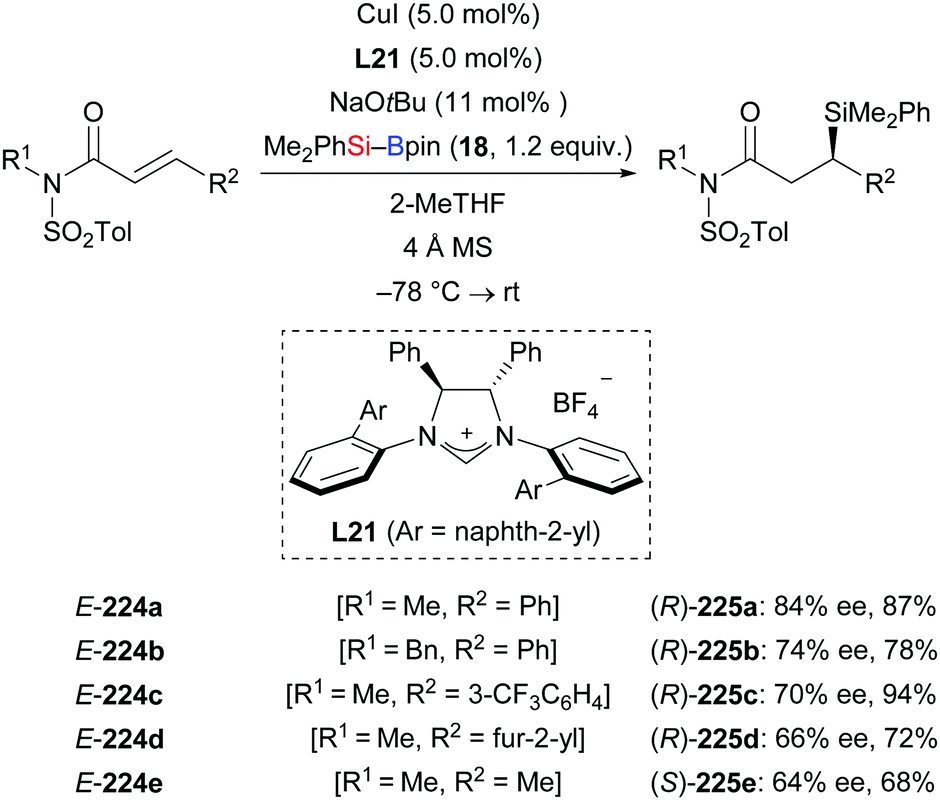
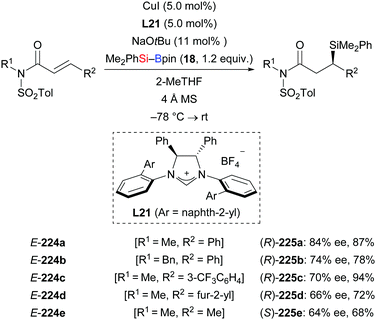
In 2013, Procter and co-workers reported an NHC–copper catalysis to achieve the enantioselective silylation of N-tosylated α,β-unsaturated amides (Scheme 70).92 Aryl (E-224a–c), furyl (E-224d), and alkyl (E-224e) substituents in the β-position were tolerated; ee values were moderate throughout. This NHC–copper catalysis was also applicable to the kinetic resolution of α,β-unsaturated lactams (not shown).
 |
| | Scheme 70 NHC–copper-catalysed asymmetric silyl transfer to N-tosylated α,β-unsaturated amides. | |
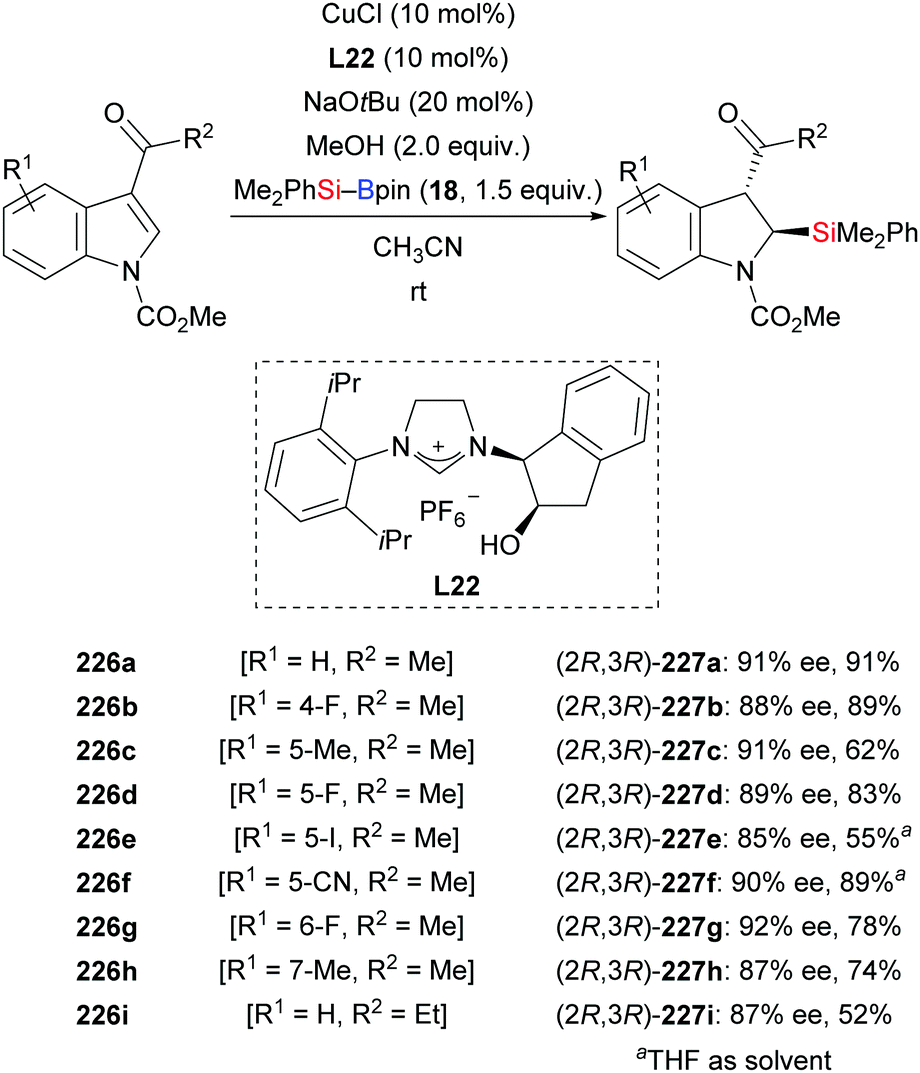
An NHC–copper catalysis also enabled an enantioselective dearomative silylation of indoles bearing an acyl group at C3 (226a–i → 227a–i, Scheme 71).93 Developed by Xu and co-workers in 2018, this procedure is yet another approach to the synthesis of α-aminosilanes with high levels of regio- and enantioselectivity (cf. Section 4.3.2). While the kinetically favoured cis-configured products were observed when monitoring the reaction by NMR spectroscopy, these had completely epimerized to the thermodynamically more stable trans-configured products upon isolation. A mechanistic model has been proposed which is in agreement with the stereochemical outcome of the reaction.
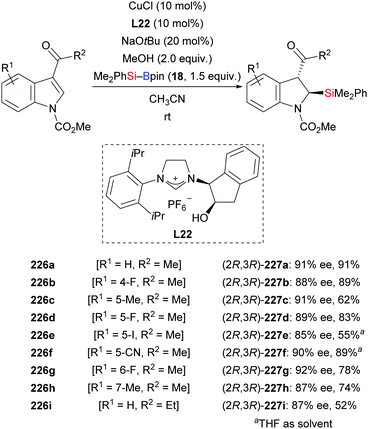 |
| | Scheme 71 Copper-catalysed enantioselective dearomative silylation of indoles. | |
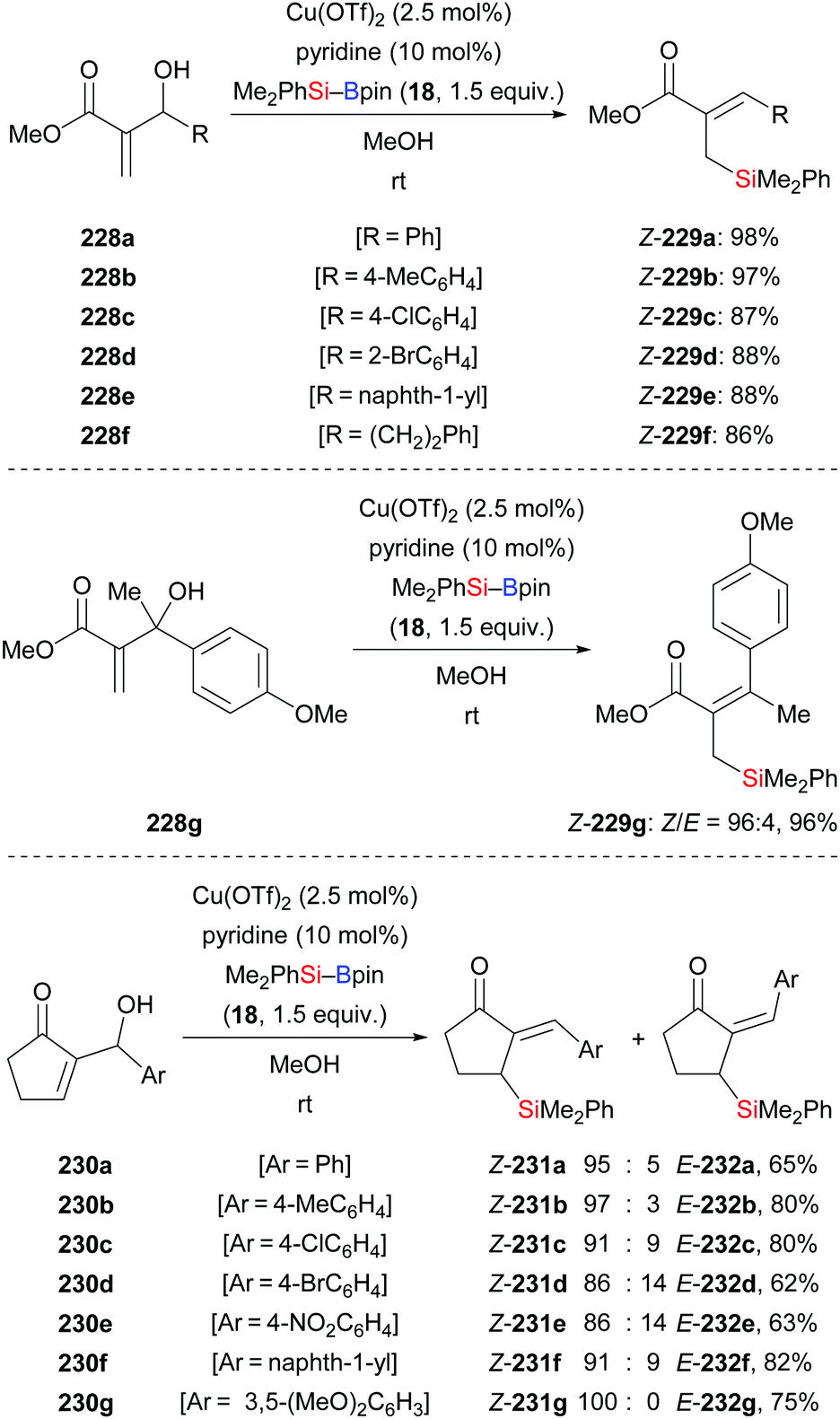
A general and efficient method for the synthesis of functionalised allylsilanes by copper-catalysed silylation of acyclic Morita–Baylis–Hillman alcohols with silylboranes was accomplished by Li and co-workers (228a–f → 229a–f, Scheme 72, top).94a The substrate scope of this method is broad. Substrates bearing electron-withdrawing or electron-donating groups on the aryl ring reacted smoothly, exclusively furnishing the Z-configured silylation products in high yields. 2-Methyl-1-phenylprop-2-en-1-ol devoid of the alkoxycarbonyl group did not react. The Riant group later expanded this protocol to tertiary alcohols (228g → 229g, Scheme 72, middle).94b Li's catalysis is also amenable to cyclic Morita–Baylis–Hillman alcohols but the diastereoselectivity was low in a few cases (230a–g → 231a–g, Scheme 72, bottom).
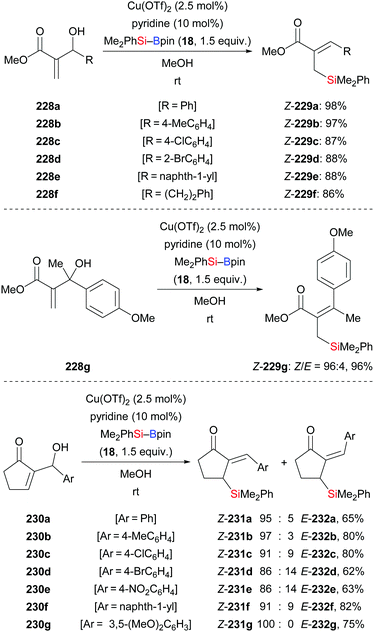 |
| | Scheme 72 Copper-catalysed silylation of Morita–Baylis–Hillman alcohols. Yields are for the mixture of isomers. | |
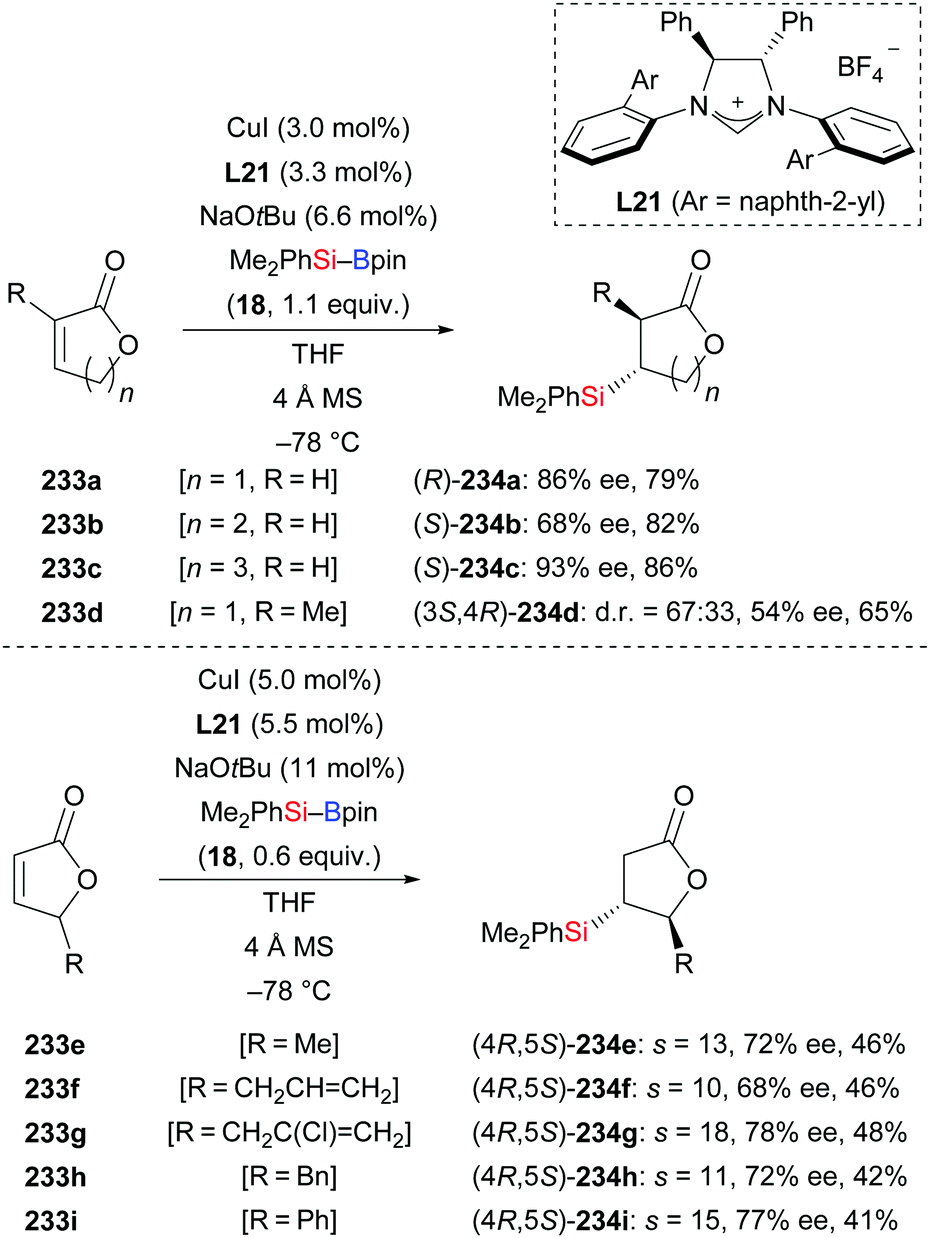
In 2013, Procter and co-workers applied their methodology92 to α,β-unsaturated lactones (233a–d → 234a–d, Scheme 73, top).95 Various γ-butyro, δ-valero, and ε-caprolactones were suitable substrates, furnishing the desired products with good enantioselectivity. The 8-membered derivative did not react, likely due to conformational effects. This NHC–copper catalysis was again92 utilized in the kinetic resolution of racemic mixtures of 5-substituted γ-butyrolactones (233e–i → 234e–i, Scheme 73, bottom). Because of the increased steric hindrance around the β-carbon atom, higher catalyst and silylborane loadings and longer reaction times were necessary. To demonstrate the value of this method, a three-step synthesis of (+)-blastmycinone starting from (4R,5S)-234e was elaborated (not shown).
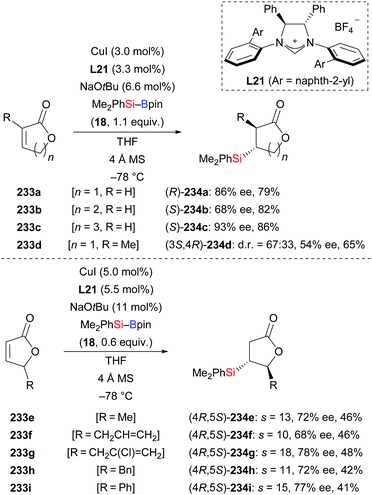 |
| | Scheme 73 NHC–Cu-catalysed asymmetric silyl transfer to unsaturated lactones and application in kinetic resolution. s = selectivity factor. | |
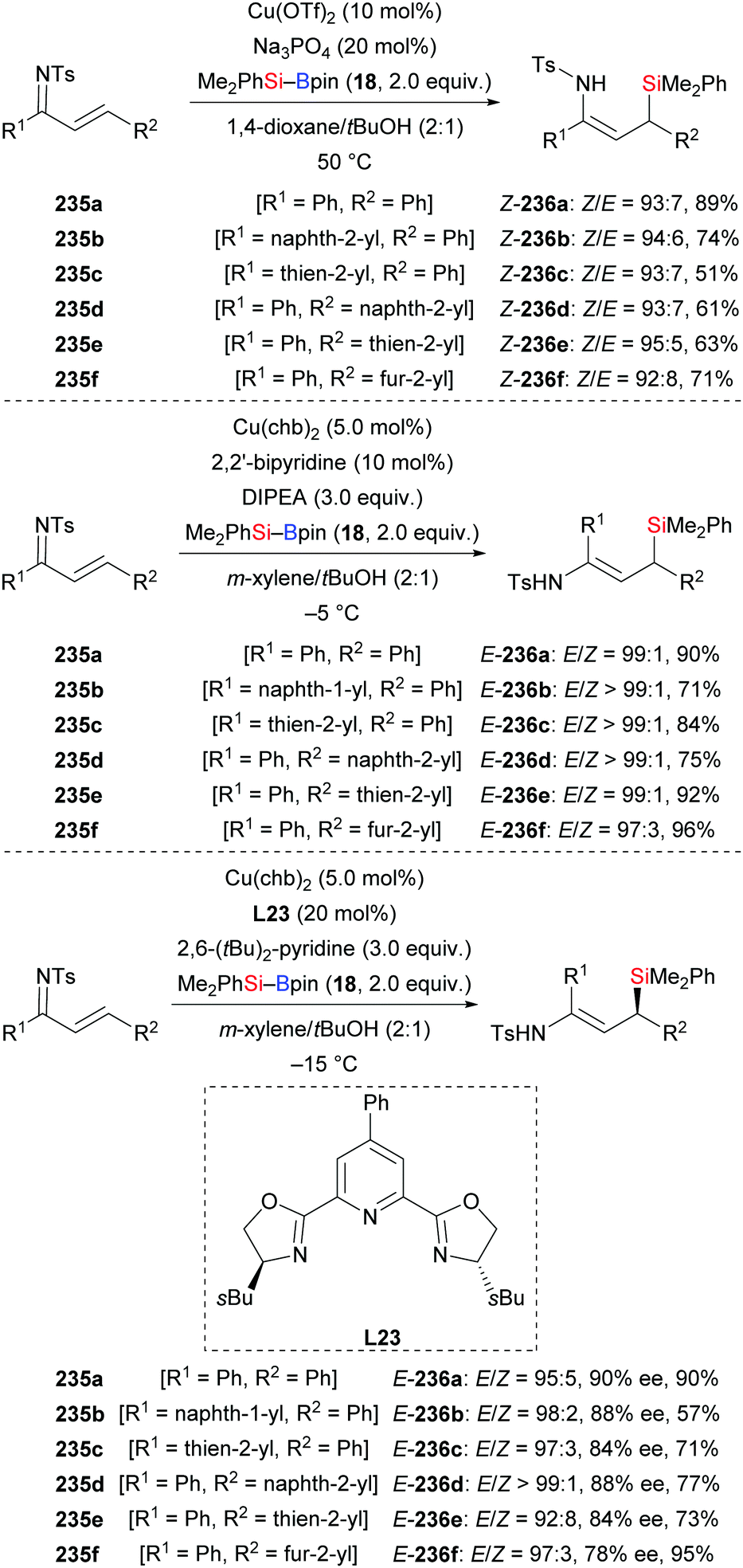
A concise method for the synthesis of enamide-containing allylsilanes by conjugate addition of silylboronic esters to α,β-unsaturated ketimines was developed by Loh and co-workers in 2018 (235a–f → 236a–f, Scheme 74).96 By subtle modification of reaction conditions, E- and Z-configured formal hydrosilylation products could be accessed from the same starting material with high levels of stereocontrol (top and middle). With the chiral Pybox ligand L23, an enantioselective conjugate silyl transfer was achieved; ee values were good (Scheme 74, bottom). Two transition states were established to explain the enantioinduction in this reaction (not shown). To demonstrate the utility of this method, the 1,4-addition to 235a was scaled up, yielding the enantioenriched product E-236a without any loss of yield and enantioselectivity.
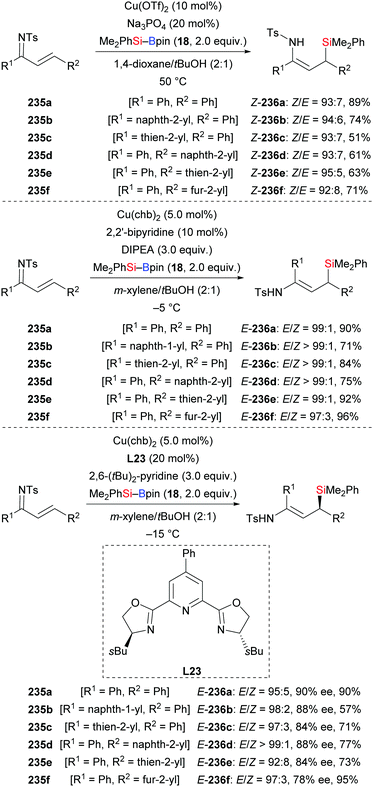 |
| | Scheme 74 Copper-catalysed stereo- and enantioselective conjugate addition to α,β-unsaturated ketimines. Cu(chb)2 = copper bis(4-cyclohexylbutyrate). | |
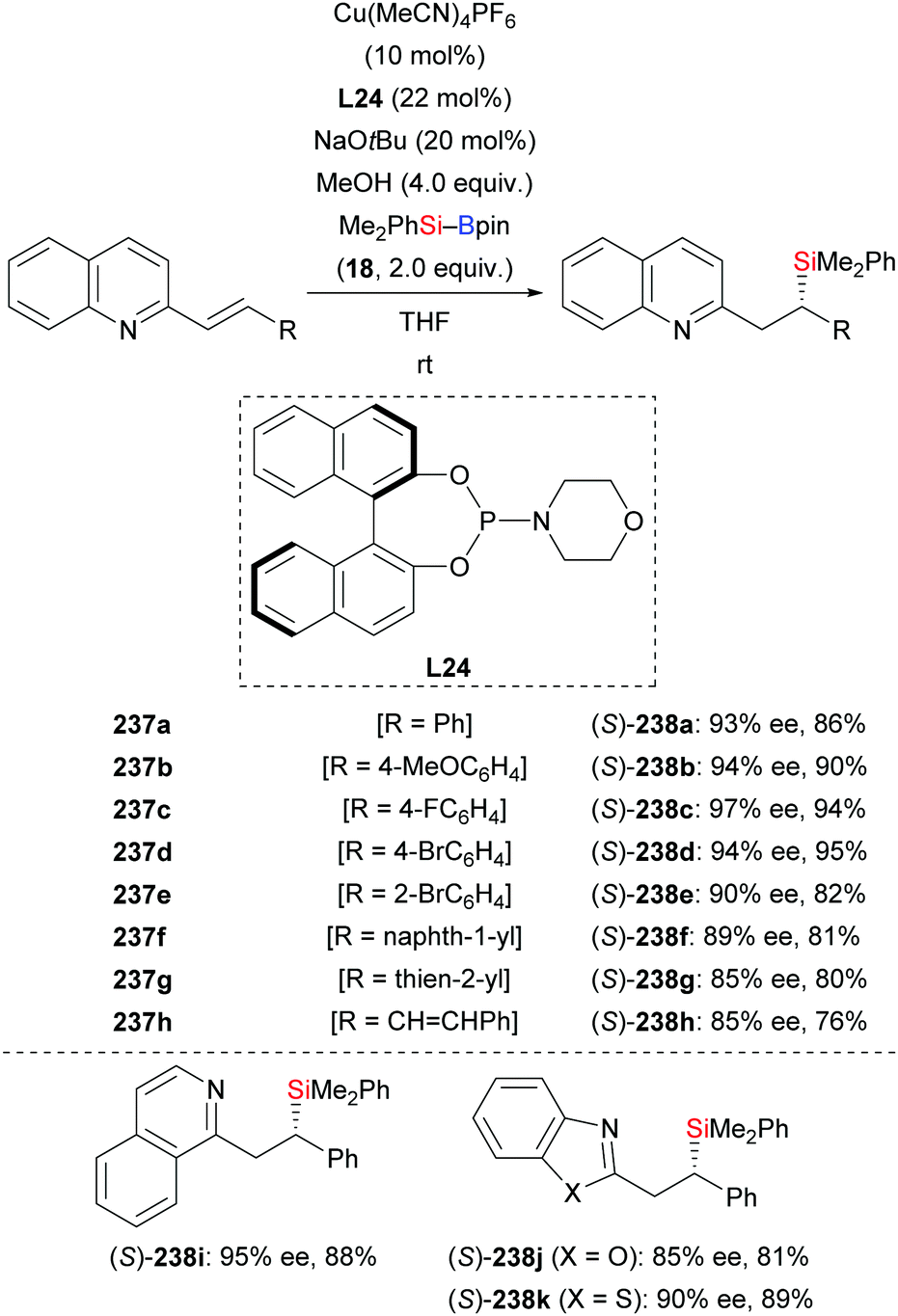
In 2019, Oestreich and co-workers reported a copper-catalysed enantioselective conjugate addition of silicon Grignard reagents to alkenyl-substituted nitrogen-containing heteroarenes with a chiral Josiphos ligand (not shown).97 This transformation was also found to be feasible by using silylboronic esters as the silicon pronucleophile as described by Liu and co-workers in 2020 (237a–k → 238a–k, Scheme 75).98 Alkenyl-substituted quinolines decorated with various aromatic groups in the β-position reacted smoothly to afford the corresponding products in high yields and with excellent enantiocontrol (Scheme 75, top). Other heteroarenes such as isoquinolines, benzoxazoles, and benzothiazoles were also possible motifs (Scheme 75, bottom). A gram-scale reaction of 237d proceeded with no loss in yield and enantioselectivity. To illustrate the synthetic utility of this method, the silyl group in (S)-238d was oxidatively degraded to a hydroxy group by a stereospecific Tamao–Fleming oxidation (not shown).
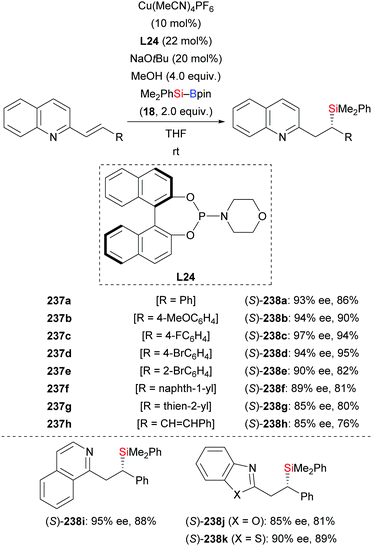 |
| | Scheme 75 Copper-catalysed asymmetric silyl transfer to alkenes activated by azaaryl groups. | |
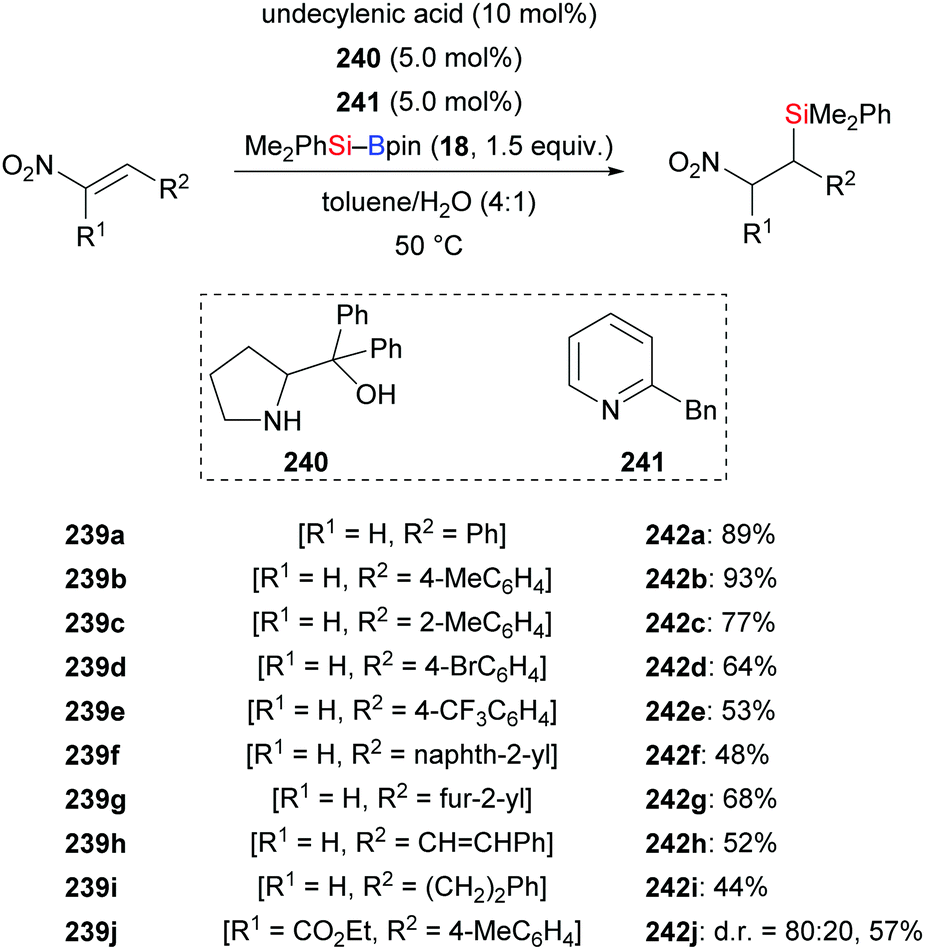
In 2016, the He group reported a convenient procedure for the preparation of β-nitro-substituted silanes (239a–j → 242a–j, Scheme 76), which can be converted into β-silylamines by reduction with Zn/HCl (not shown).99 This transition-metal-free reaction proceeded in a toluene/water solvent mixture. The method was compatible with several functional groups; substrates bearing an alkyl group in the β-position as in 239i were less reactive. The trisubstituted nitroalkene 239j reacted with Me2PhSi–Bpin to afford the silylation product with a diastereomeric ratio of 80:20.
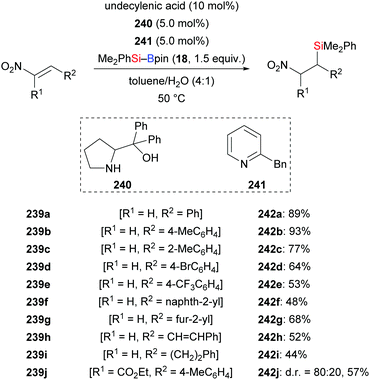 |
| | Scheme 76 Transition-metal-free silylation of nitroalkenes with silyl-boranes. | |
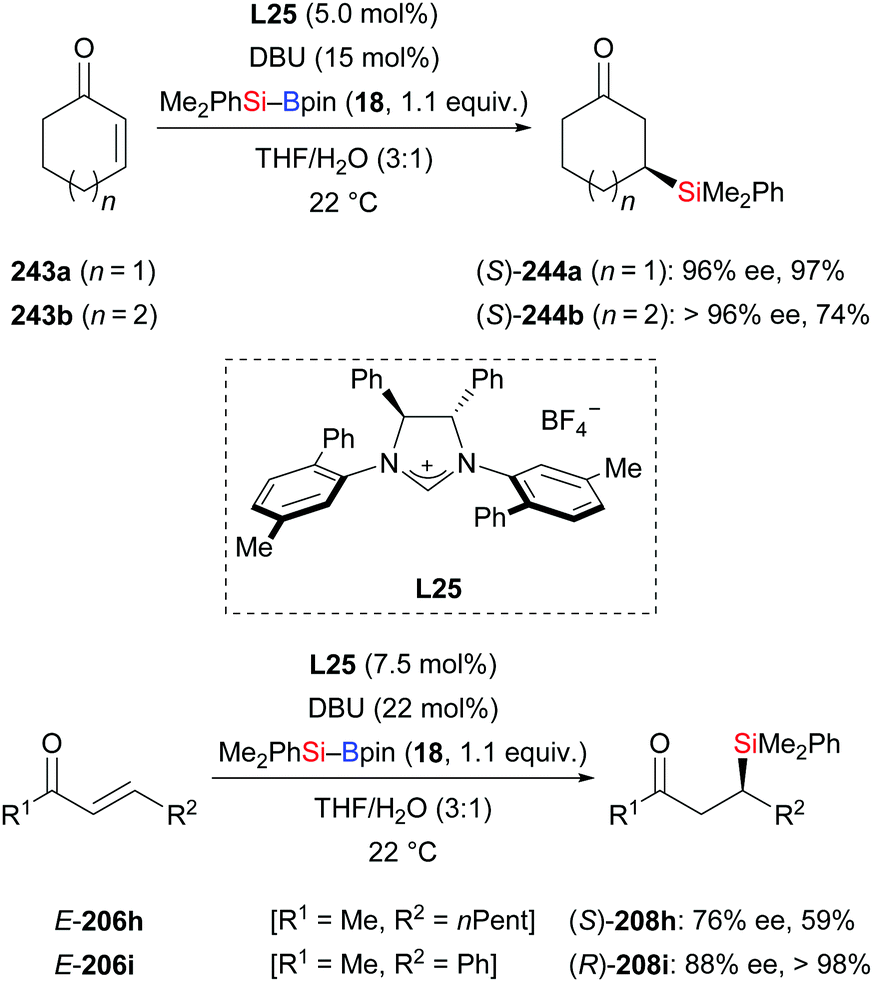
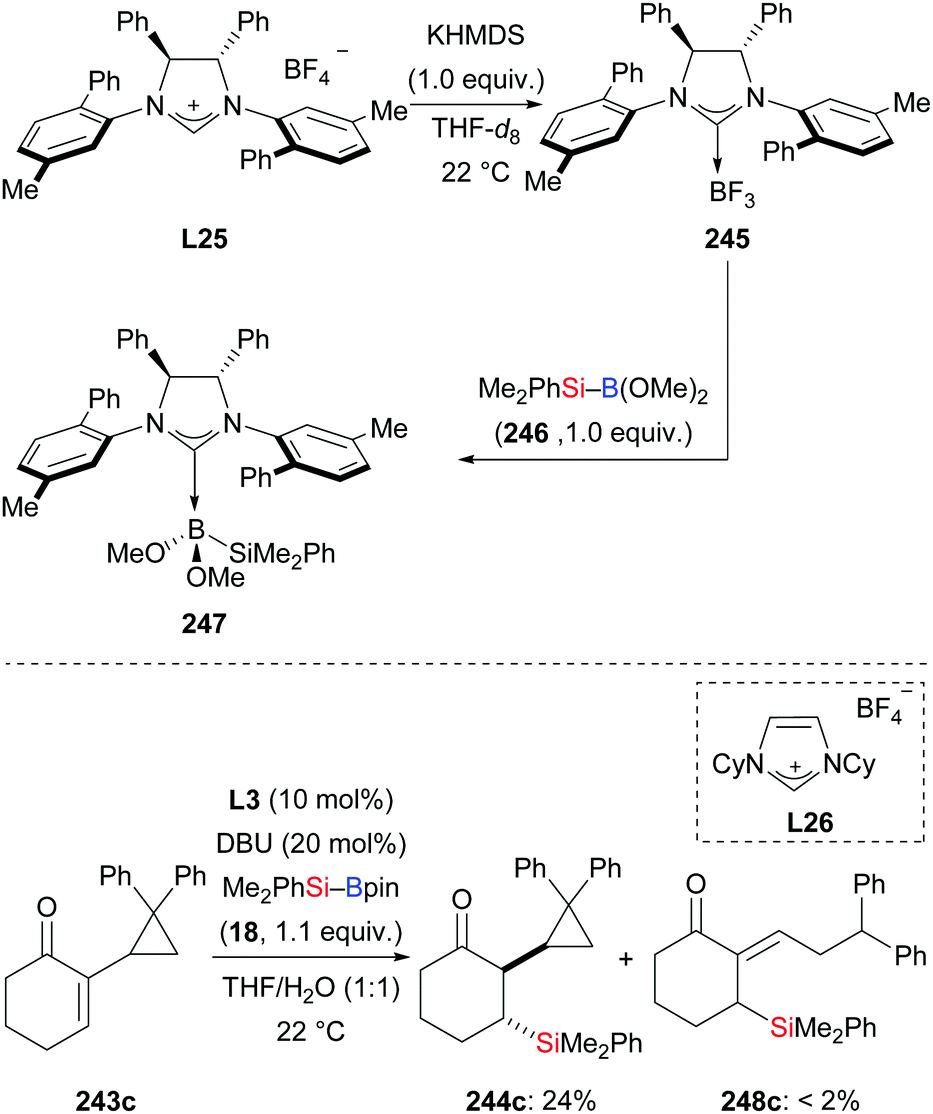
In 2011, the Hoveyda group introduced NHC-catalysed C–Si bond formation (Scheme 77).100 These authors found the same sense of enantioinduction in conjugate silylation of both cyclic and linear substrates, which stands in contrast to the results of the NHC-catalysed borylation with B–B reagents. To clarify the origin of this dichotomy, four transition-state models were computed (not shown). Based on this, the difference was explained by the bigger silyl group (compared to Bpin) and the longer Si–B bond (compared to B–B bonds). With easy-to-make silylboronic ester 246, the NHC adduct 247 was detected in the 11B NMR spectrum at −0.4 ppm and the 13C NMR spectrum at 181.2 ppm (Scheme 78, top). Using the NHC precursor L26, a radical-clock experiment was conducted without the formation of the ring-opened product 248c. This excluded a radical mechanism in this NHC-catalysed conjugate silylation (243c → 244c, Scheme 78, bottom). Lower efficiency was found in the absence of water. Both water and excess DBU accelerate the hydrolysis of the sterically hindered Bpin into the B(OH)2 unit. DBU plays an important role in the process, transferring the hydroxide ions into the organic phase in the form of HDBU+OH−.
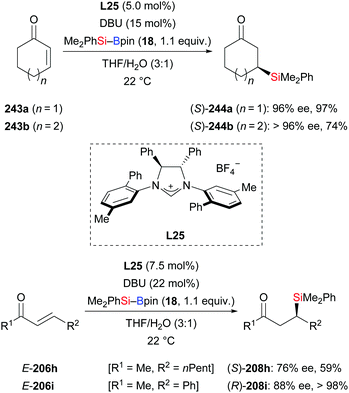 |
| | Scheme 77 NHC-catalysed conjugate addition of Me2PhSi–Bpin to enones. | |
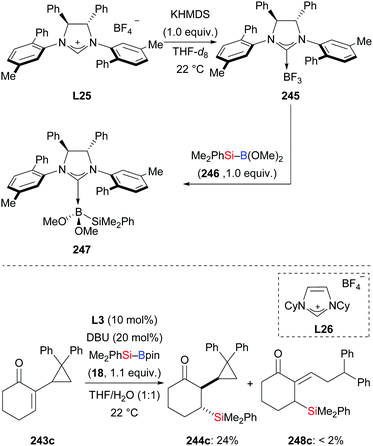 |
| | Scheme 78 Mechanism analysis of the NHC-catalysed addition of silylboranes to α,β-unsaturated carbonyl compounds. | |
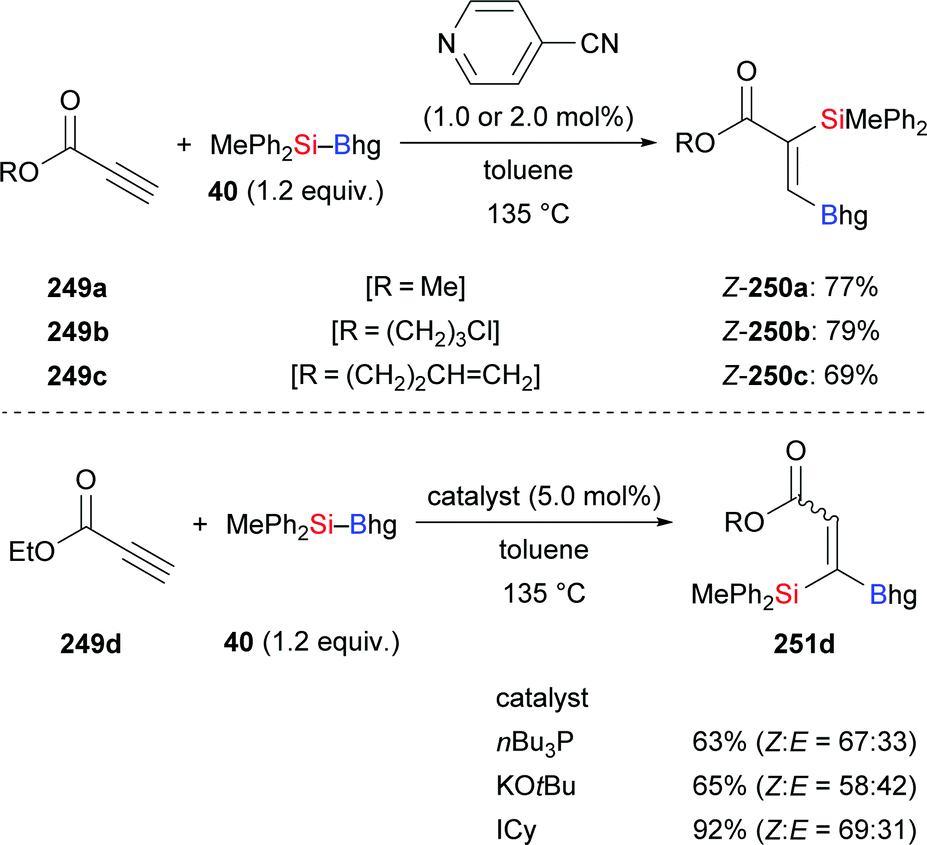
As previously discussed (cf.Schemes 11 and 41), 4-cyanopyridine can be used as a catalyst for the regioselective silaboration of terminal alkynes and allenes.26 This protocol is also applicable to the addition of silylboronic esters to propiolates (249a–c → 250a–c, Scheme 79, top).26 Propiolate esters of primary alcohols reacted in good yields whereas the tert-butyl ester did not convert at all. Me2PhSi–Bpin (18), MePh2Si–Bpin (192), and Ph3Si–Bpin (19) did also form the desired products albeit in low yields (not shown). When nBu3P, KOtBu or ICy were used as organocatalysts 1,1-silaboration proceeded26 (249d → 251d, Scheme 79, bottom; cf.Scheme 12).
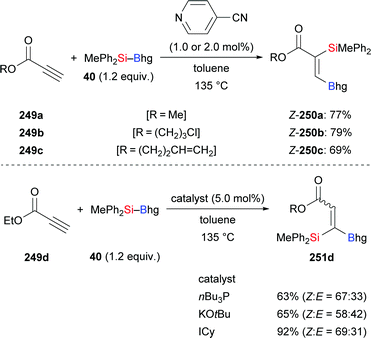 |
| | Scheme 79 Transition-metal-free silaboration of propiolates. | |
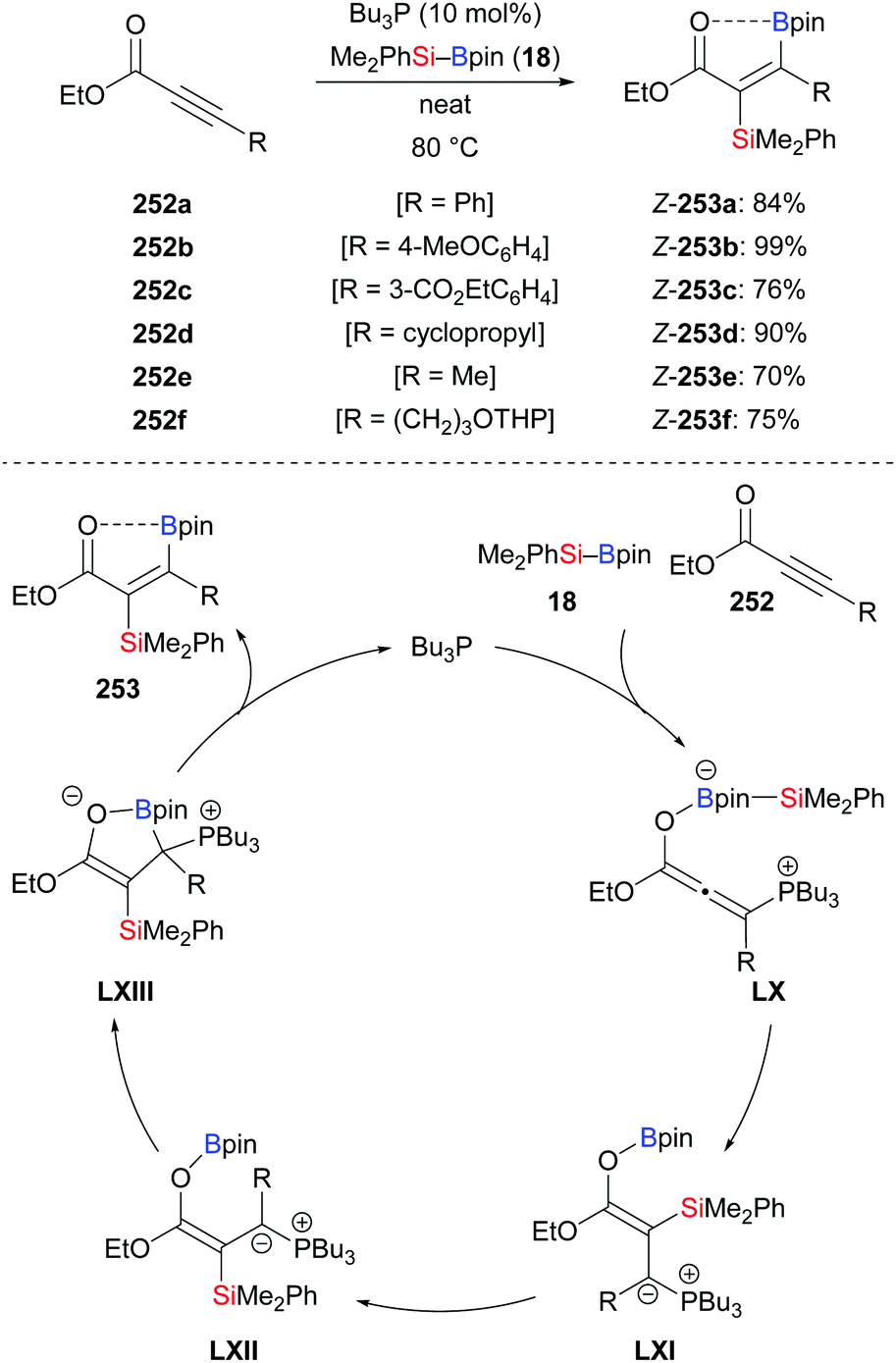
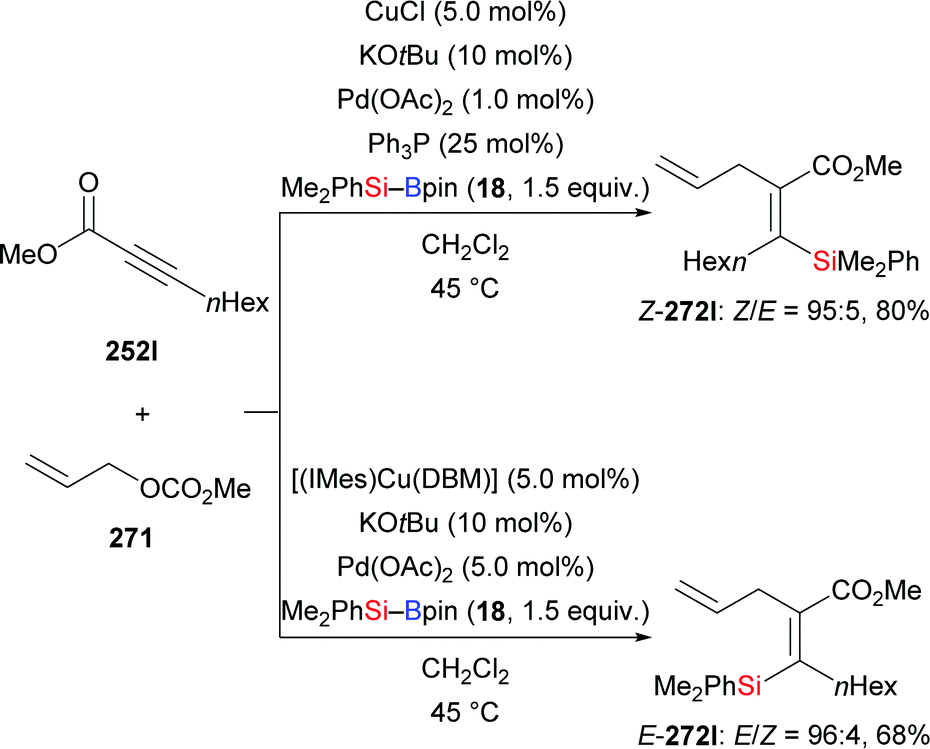
An anti-selective silaboration across polar C–C triple bonds was established by Sawamura and co-workers in 2015, offering a straightforward method for the preparation of β-boryl-α-silyl acrylates in good to excellent yields (252a–f → Z-253a–f, Scheme 80, top).101 The 11B NMR spectra of the products indicated that the carbonyl oxygen atom is coordinated to the boron atom. The observed regioselectivity requires transfer of the electrophilic boryl moiety to the positively polarized β-position. Several functional groups were tolerated. The silyl and boryl groups in the silaboration products can be differentiated and further derivatised to access tetrasubstituted alkenes (not shown). A plausible mechanism is depicted in Scheme 80 (bottom). A zwitterionic allenolate intermediate LX is formed by the reaction of nBu3P, the silylborane and the alkynoate. The silyl group is then transferred to the C(sp)-hybridised β-carbon atom of LX to form ylides LXI/LXII. The carbanion in ylide LXII attacks the boron atom to form the cyclic intermediate LXIII from which the phosphine catalyst is released.
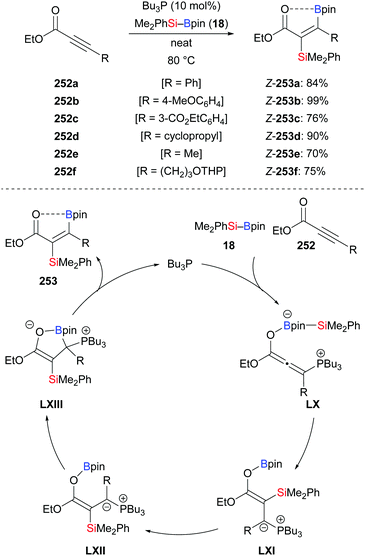 |
| | Scheme 80 Phosphine-catalysed trans-selective silaboration of alkynoates. | |
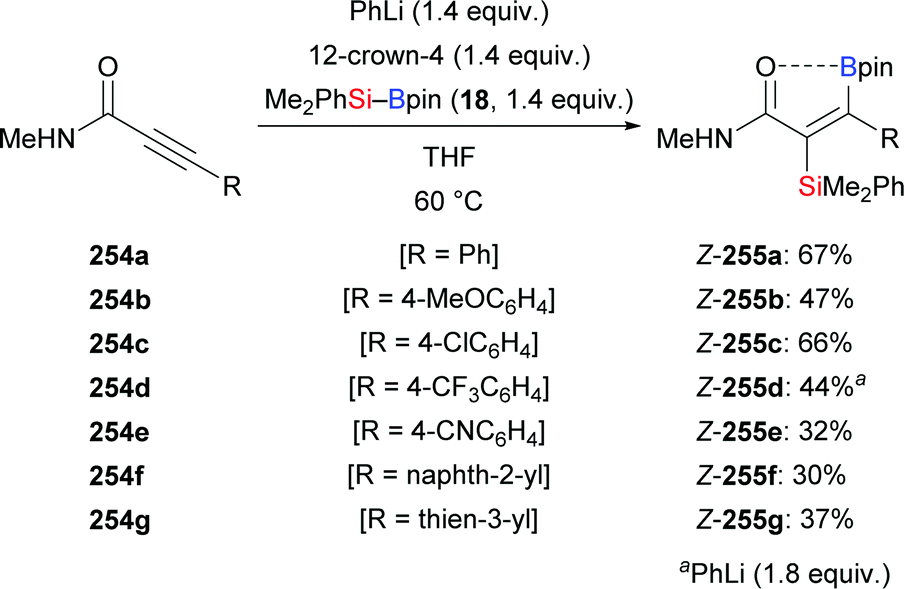
A transition-metal-free silaboration of acetylenic amides mediated by a Brønsted base was reported by Santos and co-workers (254a–g → 255a–g, Scheme 81).102 Phenyllithium and 12-crown-4 were used to activate the silylboronic ester, and the yields dropped dramatically without the chelating crown ether. This reaction was compatible with several N- and aryl-substituted secondary propargylamides, affording the trans-silaboration products in moderate yields. However, tertiary amides proved to be unreactive because a ‘naked’ Lewis basic amide is necessary to activate the Si–B reagent. Again, the trans relationship between the silyl and boryl groups has been rationalised by the coordination of the carbonyl oxygen atom to the boron center (as verified by 11B NMR spectroscopy). The synthetic usefulness of those β-boryl-α-silyl acrylamides with two linchpins for chemoselective derivatisation was also demonstrated (not shown).
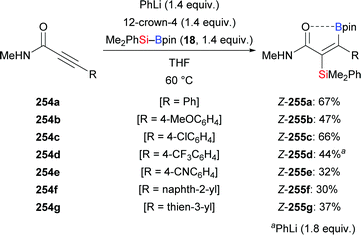 |
| | Scheme 81 Brønsted base-mediated regio- and stereoselective trans-silaboration of propargylamides. | |
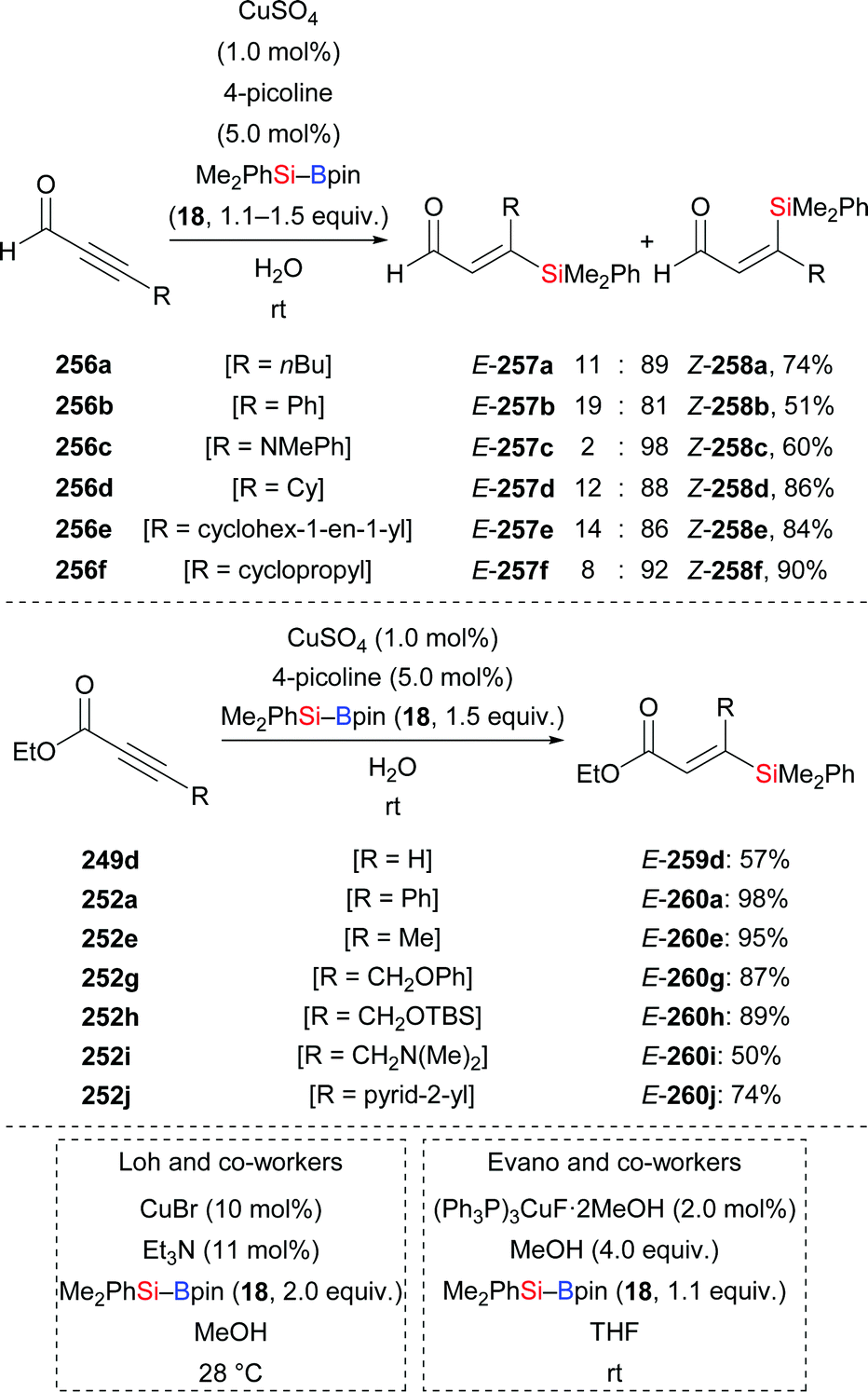
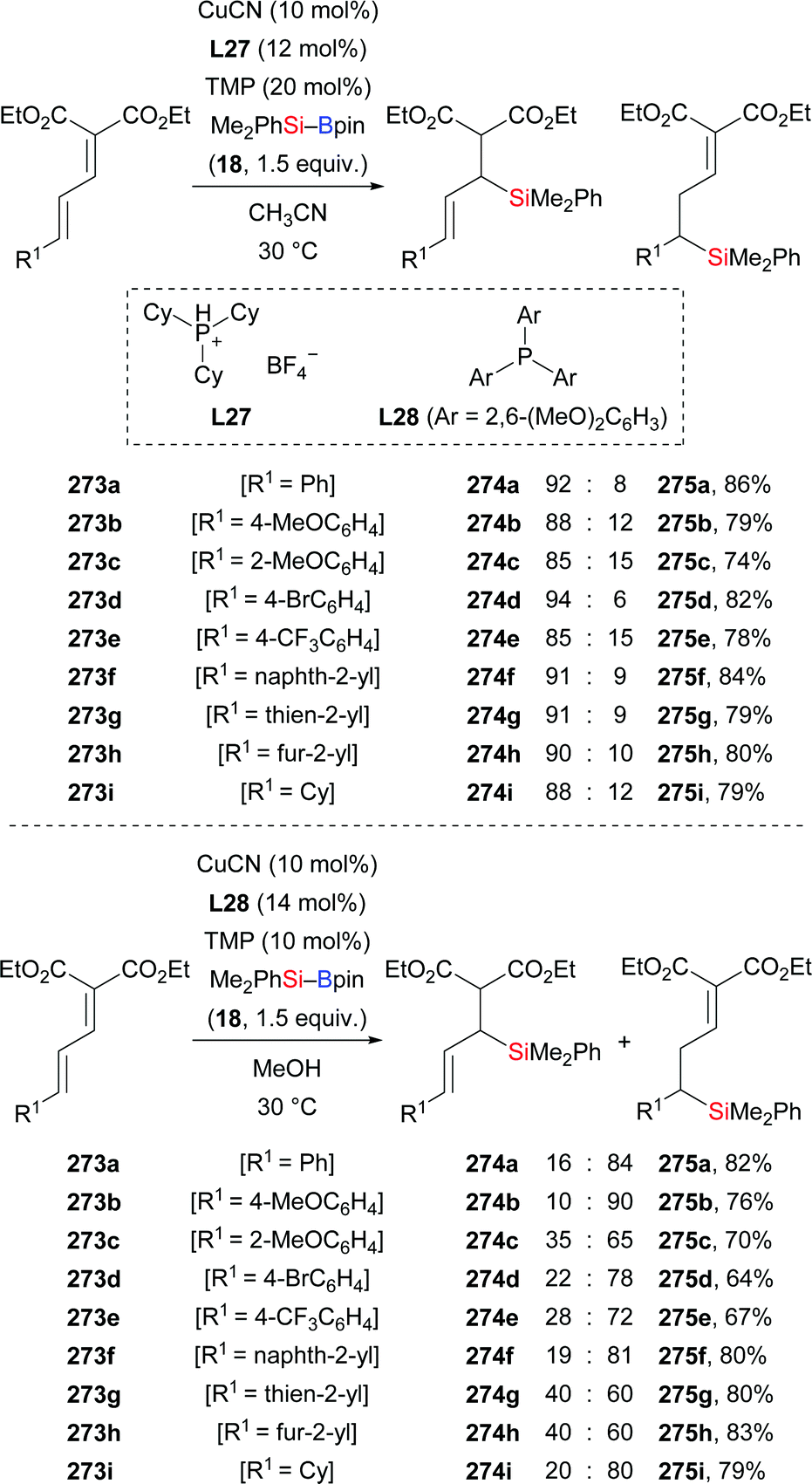
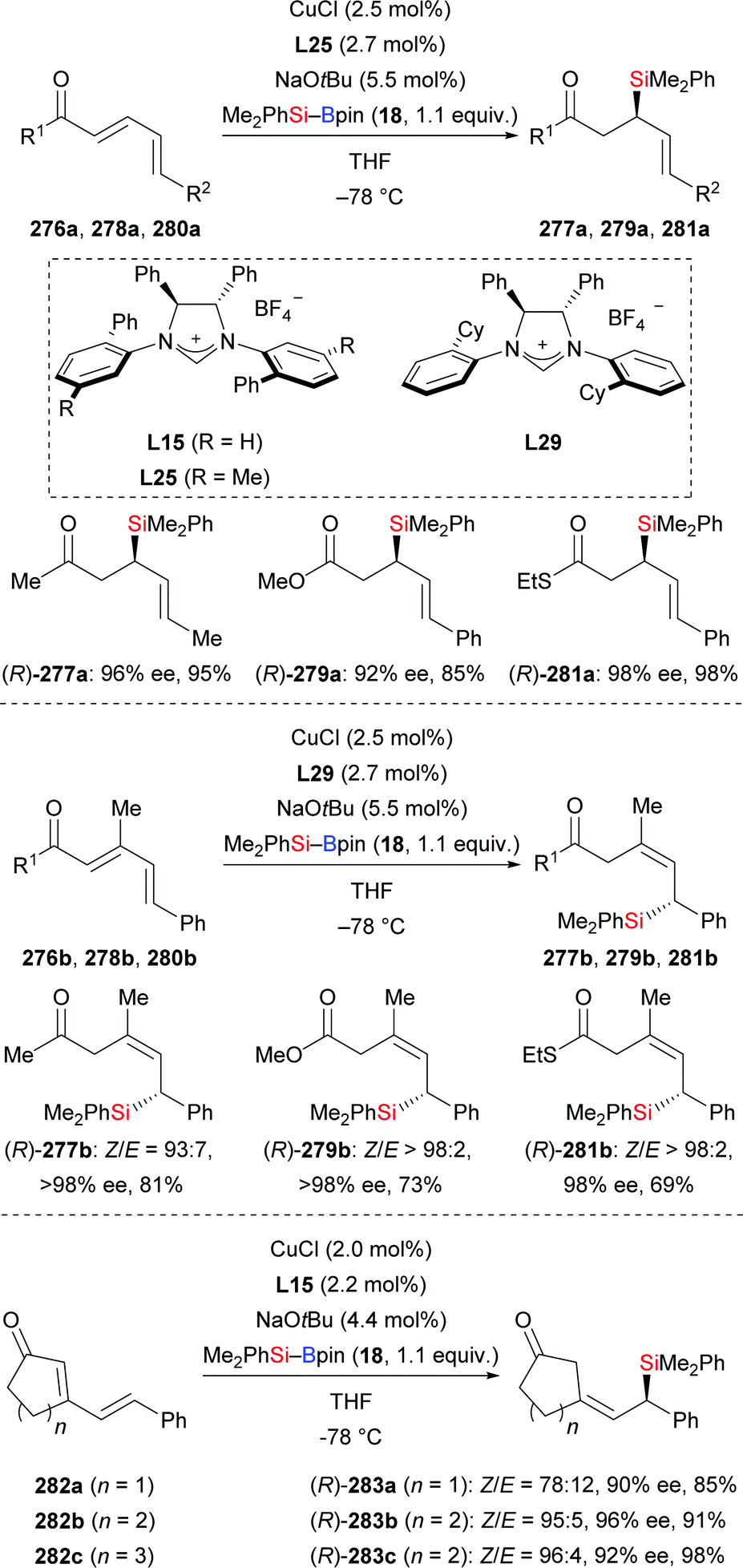
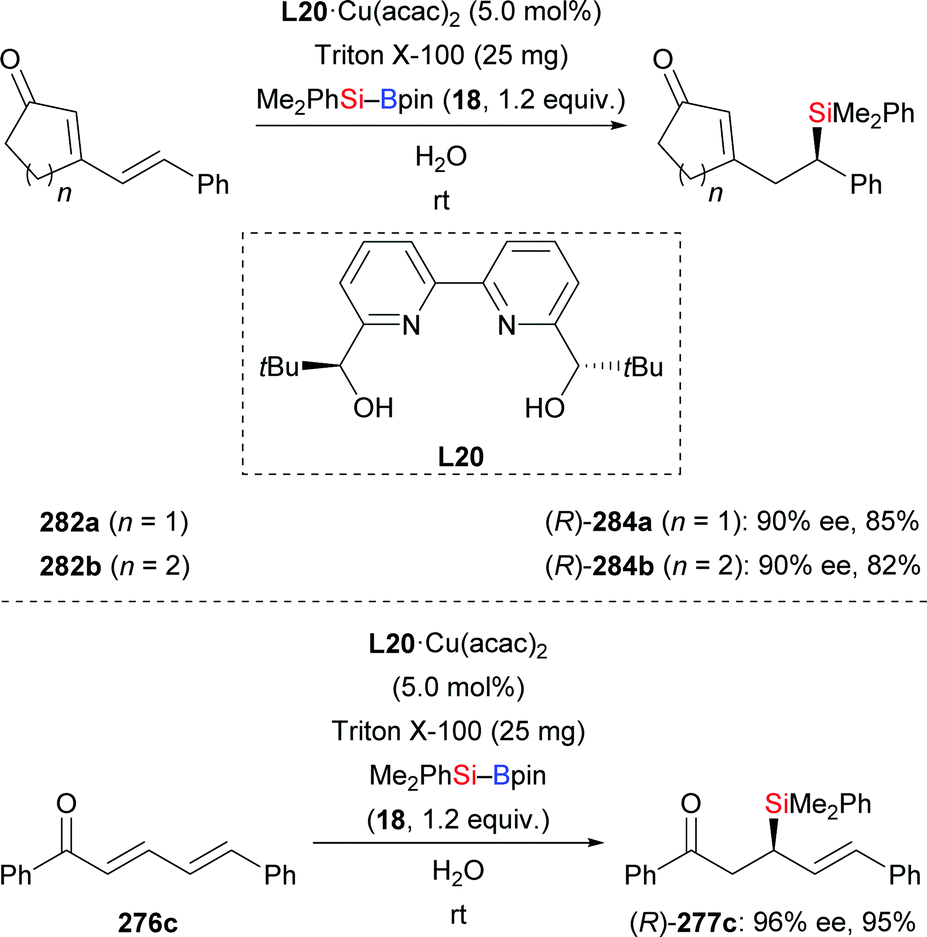
A copper-catalysed diastereodivergent formal hydrosilylation of ynones and ynoates was reported by Santos and co-workers in 2013 (Scheme 82, top and middle).103a This high-yielding conjugate addition takes place in water at room temperature. When aldehydes (256a–f → 258a–f, Scheme 82, top) and ketones (not shown) were used as reactants, Z-configured carbonyl compounds were obtained as main products. In turn, esters (249d → 259d; 252a, e, g–j → 260a, e, g–j, Scheme 82, middle) and amides (not shown) as substrates gave E-configured products exclusively. An explanation of this stereochemical outcome has been provided by the authors: (1) syn-addition of the copper-based silicon nucleophile occurs with esters and amides, generating the E-configured products whereas (2) aldehyde and ketone derivatives form allenolate intermediates which are protonated opposite to the sterically demanding silyl group, eventually leading to Z-configured products. Similar work was later reported by Loh103b and Evano38 (Scheme 82, bottom).
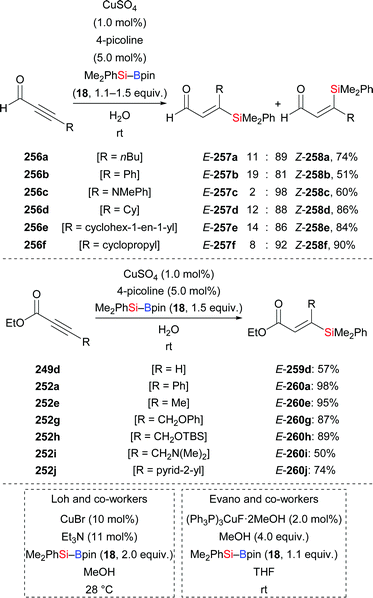 |
| | Scheme 82 Copper-catalysed diastereodivergent silylation of various ynones and ynoates. Yields are for the mixture of isomers (top). | |
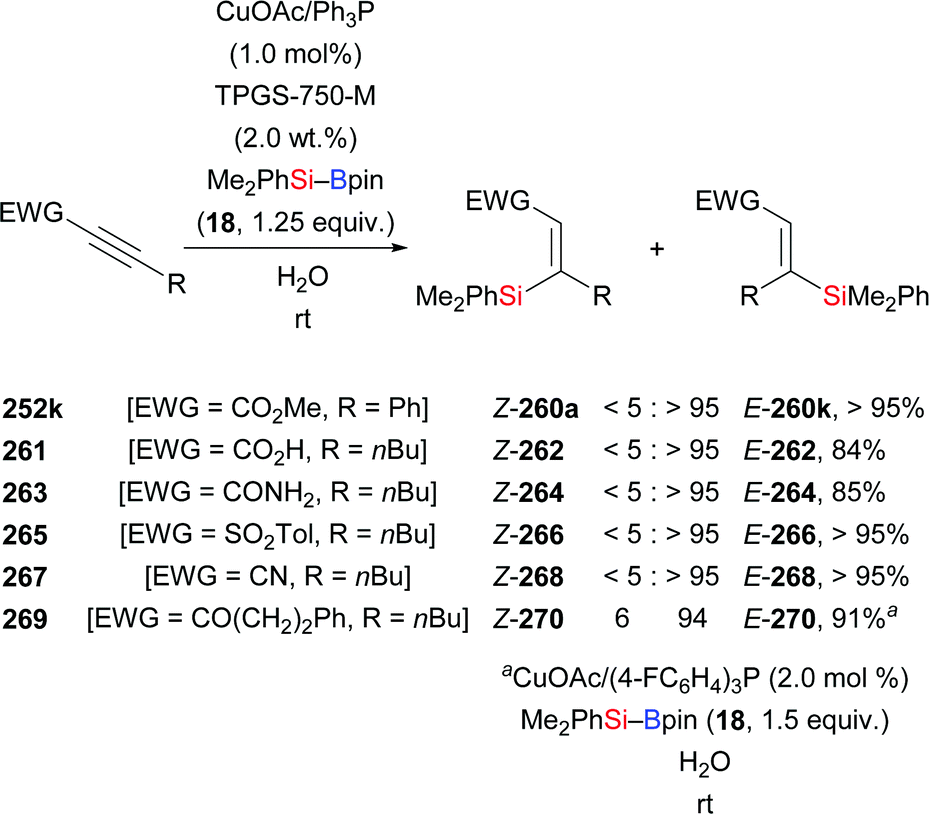
In 2013, Lipshutz and co-workers demonstrated a copper-catalysed 1,4-addition of Me2PhSi–Bpin (18) to electron-deficient alkynes in water (Scheme 83).104 All reactions were finished within minutes. Electron-withdrawing groups such as ester (252k), acid (261), amide (263), sulfone (265), cyano (267), and ketone (269) are evidence of the broad scope. The catalyst loading can be reduced to 0.01 mol%. To highlight the utility of this method, a gram-scale reaction with 252k was done, affording the desired product in slightly diminished yield of 86%. Of note, this catalytic system can be recycled for at least six times without any loss of efficiency.
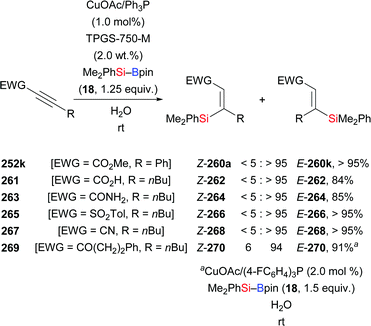 |
| | Scheme 83 Copper-catalysed silylation of electron-deficient alkynes. Yields are for the mixture of isomers. | |
A copper/palladium dual catalytic system was used by Riant and co-workers for the preparation of tetrasubstituted vinylsilanes by three-component coupling of acetylenic esters, allylic carbonates, and silylboranes in 2013 (252l → 272l, Scheme 84).105 By minor variation of the reaction conditions, E- and Z-configured coupling products were accessed in good yields with high stereoselectivity. There was no E/Z isomerization of coupling products when resubjected to the reaction (not shown). Mechanistic studies showed that E-configuration is the result of syn-addition of the silicon nucleophile across the C–C triple bond, and Z-configuration traces back to the intermediacy of an allenoate (not shown).
 |
| | Scheme 84 Copper/palladium co-catalysed three-component coupling of acetylenic esters, allylic carbonates, and silylboranes. | |
An efficient method for the synthesis of various functionalised allylsilanes by copper-catalysed conjugate silyl transfer onto diendioates was introduced by Loh and co-workers in 2019 (273a–i → 274a–i and 275a–i, Scheme 85).106 1,4- and 1,6-hydrosilylation products were obtained by changing the ligand used in the reaction. The base and ligand were found to be essential for regiocontrol, and the formation of 1,6-hydrosilylation products was more favourable with bulky ligands. The 1,4-selective reaction of 273a was carried out on a gram scale, delivering the desired product in 79% yield. Several follow-up transformations of 274a were conducted to illustrate the synthetic utility of this method (not shown).
 |
| | Scheme 85 Copper-catalysed regiodivergent 1,4- and 1,6-conjugate addition to diendioates. Yields are for the mixture of regioisomers. | |
In 2012, Hoveyda and co-workers developed a diastereo- and enantioselective silyl transfer to cyclic and acylic dienones and dienoates (276a–b → 277a–b; 278a–b → 279a–b; 280a–b → 281a–b; 282a–c → 283a–c, Scheme 86).107 The 1,6-addition took place with dienones or dienoates bearing methyl group in the β-position, giving the Z-configured silylated products with high enantioselectivity (Scheme 86, middle). Cyclic dienones also afforded the 1,6-addition products with Z-configuration (Scheme 86, bottom). Four kinds of NHC–Cu·dienone models were constructed with the aid of DFT calculations to explain the stereochemical outcome (not shown). The reason for the Z-selectivity in this transformation is still unclear.
 |
| | Scheme 86 NHC–Cu-catalysed asymmetric silyl transfer to dienones and dienoates. | |
A copper catalysed 1,6-addition of Me2PhSi–Bpin (18) to cyclic dienones was also reported by Kobayashi and co-workers in 2015 (282a–b → 284a–b, Scheme 87).91 The 1,6-addition products obtained by this method are different from 283. For acceptor 282b as an example, these authors found that initially formed (R)-283b isomerizes to (R)-284b. The L20·Cu(acac)2 complex also catalyses the silylation of an acyclic dienone but this time forming 1,4-adduct as the main product (276c → 277c, Scheme 87).
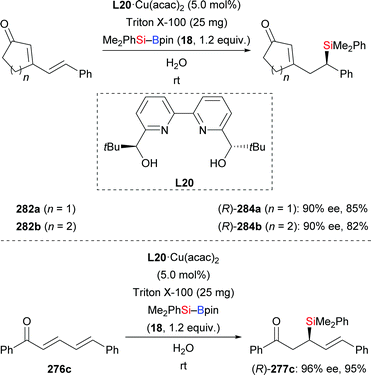 |
| | Scheme 87 Copper-catalysed conjugate silyl transfer to dienones. | |
A silylation–aromatization sequence involving copper-catalysed conjugate 1,6-addition to para-quinone methides was demonstrated by Tortosa and co-workers in 2015 (285a–i → 286a–i, Scheme 88).108 A variety of benzylsilanes, which could serve as bench-stable benzylic carbanion precursors, were obtained in good yields under this mild transformation.
 |
| | Scheme 88 Copper-catalysed silyl transfer to para-quinone methides. | |
In 2015, Xu, Loh and co-workers disclosed a copper-catalysed silyl transfer to (Z)-2-alken-4-ynoates, establishing a straightforward method for the synthesis of various di-, tri-, and tetrasubstituted allenes in good yields (287a–i → 288a–i, Scheme 89).109a This enantioselective 1,6-addition proceeded at room temperature employing the chiral Box ligand L30; the resulting silylated allenes are formed in good yields and with high enantioinduction (Scheme 89, bottom). Enynoates with aliphatic substituents attached to the C–C triple bond or to the β-position reacted smoothly, albeit with moderate enantioselectivity (e.g.287h and 287i). (E)-2-alken-4-ynoates proved to be less suitable for both the racemic and asymmetric versions (not shown).109b
 |
| | Scheme 89 Copper-catalysed silyl transfer to (Z)-2-alken-4-ynoates. | |
A copper-catalysed double protosilylation procotol for the synthesis of 1,3-disilylpropenes was acccomplished by the same laboratory in 2018 (289a–f → 290a–f; 291 → 292; 293 → 294, Scheme 90).110 This reaction proceeds through a stepwise process, involving protosilylation of the C–C triple bond followed by 1,6-protosilylation. Exclusively E-configured silylated products were obtained with this transformation. Various alkyl-substituted enynoates bearing a variety of functional groups were tolerated in this reaction (not shown). Sequential incorporation of two different silyl groups into enynoate 289a in one-pot succeeded with tBuPh2Si–Bpin (295) and Me2PhSi–Bpin (18) to furnish the desired product in 66% yield (not shown). An asymmetric version was achieved by using Phox ligand L32 (Scheme 90, bottom). Z-289a did also react under these reaction conditions, albeit in a very low yield (12%). A gram-scale synthesis of (S)-290a was also successful with no loss in efficiency.
 |
| | Scheme 90 Copper-catalysed double hydrosilylation of electron-deficient enynes. | |
One year later, these authors reported a copper-catalysed regioselective silyl transfer to the β-position of enynoates (289 → 296, Scheme 91).111 The resulting β-silyldienoates were obtained exlusively in E-configuration. This procedure was also applicable to enynamides (not shown).
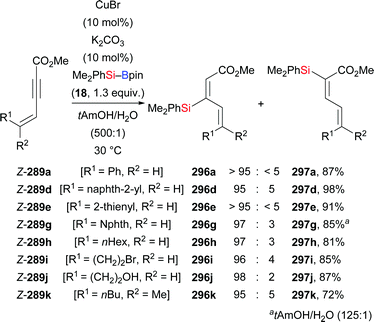 |
| | Scheme 91 Copper-catalysed silyl transfer to the C–C triple bond of enynes. Yields are for the mixture of regioisomers. | |
As shown in Scheme 92, Santos and co-workers (top) as well as and Loh and co-workers (bottom) independently reported the copper-catalysed 1,4-addition of Me2PhSiBpin to activated allenes (298a–j → 299a–j; 300 → 301; 302 → 303).103b,112 The silyl group was selectively transferred the central allene carbon atom of the allenoate in both cases. The reactions in Santos’ work were conducted in water and open to air. It is worth mentioning that other electron-withdrawing group such as tosyl (as in 300) and phosphine oxide (as in 302) were compatible under Loh's setup.
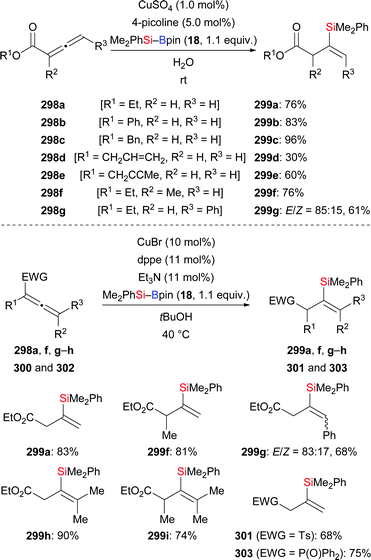 |
| | Scheme 92 Copper-catalysed regioselective formal hydrosilylation of activated allenes. | |
4.6. Allylic and propargylic substitution
4.6.1. Allylic precursors.
After Vyas and Oestreich realised the copper-catalysed branched-selective allylic substitution of linear allylic chlorides with Si–B reagents,113 yielding α-chiral allylic silanes in racemic form, Oestreich and co-workers continued to explore the asymmetric allylic silylation.114 These authors tested both alkene isomers with chloride and phosphate as leaving groups and found that both alkene geometries with phosphate as leaving group led to high enantiocontrol (E-305a → γ-306a; Z-305a → γ-306a, Scheme 93, top). It was achieved by using preformed NHC–copper(I) complex L17·CuCl as catalyst. Various E-configured allylic phosphates with aryl and alkyl groups, including a sterically more demanding isopropyl group as R1, yielded α-chiral allylic silanes with high selectivity (E-305b–f → γ-306b–f, Scheme 93, bottom). However, a further increase of steric hindrance with a tert-butyl group as R1 resulted in the formation of the undesired linear allylic silane α-307g. Also, an allylic phosphate with an R2 group led to poor regioselectivity and moderate enantioselectivity (E-305h → γ-306h, Scheme 93, bottom). However, the use of a bulkier version of catalyst, L33·CuCl instead of L17·CuCl, together with a solvent change and a different reaction temperature led to a significant improvement (E-305h → γ-306h, Scheme 94).78b Moreover, a substrate with two substituents in the γ-position converted regioselectively into an allylic silane with a ‘quaternary’ carbon atom (E-305i → γ-306i, Scheme 94).
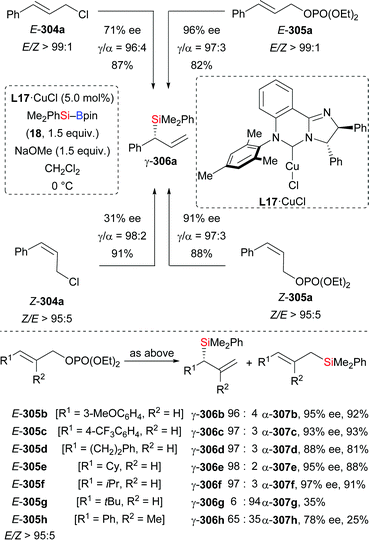 |
| | Scheme 93 Copper(I)-catalysed branched-selective allylic substitution (part I). Yields are for the mixture of regioisomers. | |
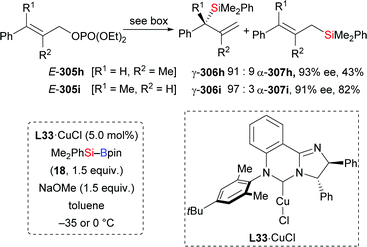 |
| | Scheme 94 Copper(I)-catalysed branched-selective allylic substitution (part II). Yields are for the mixture of regioisomers. | |
With a switch to chloride as the leaving group, the allylic silylation of substrates E-308 bearing a protected hydroxy group in the δ-position occurred diastereoselectively to generate δ-hydroxy α-chiral allylic silanes 309 as single regioisomers with anti-relative stereochemistry (Scheme 95).115 Various protecting groups such as TBDMS (as in 308a–c), TES (as in 308d–f), and a simple methyl group (as in 308g–i) led to diastereomeric ratios ranging from 85:15 to nearly perfect >99:1 for the transfer of the silyl group to aryl- or alkyl-substituted precursors (E-308a–i → 309a–i, Scheme 95).
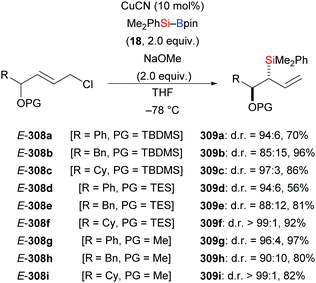 |
| | Scheme 95 Copper(I)-catalysed allylic substitution of δ-hydroxy allylic chlorides. | |
Later, the Oestreich group conducted a full experimental analysis of the silylation of racemic cyclic allylic phosphate rac-310 as this transformation was achieved in near-quantitative yield and with high enantiomeric excess employing precatalyst L17·CuCl (Scheme 96).116 The detailed mechanistic analysis revealed that the catalytic system delivered the silicon nucleophile in either syn-SN2′ or anti-SN2′ fashion to (R)-310 and (S)-310, respectively. As a consequence, both enantiomers of rac-310 led to the same absolute configuration in the final product (rac-310 → (S)-311, Scheme 96), qualifying this reaction as a direct enantioconvergent transformation.
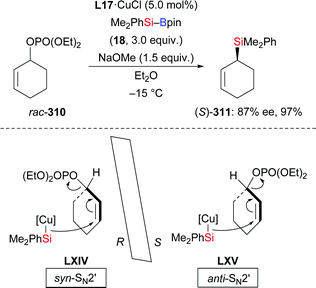 |
| | Scheme 96 Enantioconvergent allylic silylation of a racemic cyclic allylic phosphate. | |
A method for the regio- and enantioselective allylic silylation of linear allylic phosphates was also developed by Hayashi and Shintani with an in situ-generated NHC–Cu(I)–OH catalyst formed by deprotonation of chiral imidazolinium salt L34 and CuCl (Scheme 97).117 A screening of bases showed that NaOH outperformed all of the tested sodium and potassium alkoxides, resulting in higher enantioinduction and site selectivity. The reaction is general for various aryl and alkyl substituents in the γ-position (E-312a, e, g → γ-306a, e, g, Scheme 97) and even allylic phosphate E-312g with a terminal tert-butyl group reacts well with excellent enantioselectivity and a bit lower γ/α ratio. Moreover, a γ,γ-disubstituted substrate can also be converted into the corresponding allylic silanes with a ‘quaternary’ stereocenter with slightly lower γ/α ratio (E-305i–j → γ-306i–j, Scheme 97).
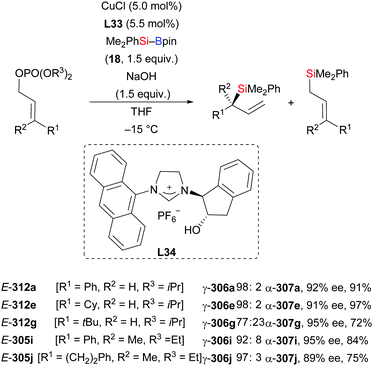 |
| | Scheme 97 Copper(I)-catalysed branched-selective allylic substitution (part III). Yields are for the mixture of isomers. | |
In 2019, Liu and co-workers developed a nickel/copper-catalysed regiodivergent synthesis of allylsilanes directly from allylic alcohols controlled by the steric and electronic properties of the ligands (Scheme 98).118 Interestingly, the branched products 306 were formed predominantly in the presence of less hindered ligands such as Et3P (L35) without an additional base. In turn, the use of bulky 8-(diphenylphosphanyl)quinoline (L36) resulted in a selectivity switch to give the linear products 307 in good yield and high stereoselectivity regardless of primary or secondary allylic alcohols.
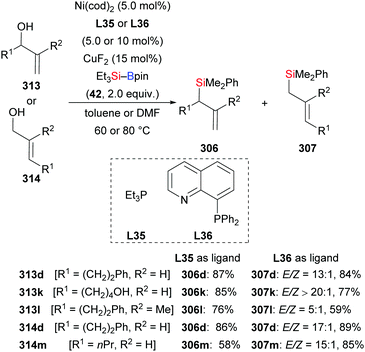 |
| | Scheme 98 Ligand-controlled regiodivergent silylation of allylic alcohols by nickel/copper catalysis. Yields are for the major product. | |
Unlike the reported transition-metal-catalysed allylic substitution reactions above, Nozaki and Shintani found that the cyclopropanation products are obtained from allylic phosphates and Si–B reagents in the presence of KHMDS (Scheme 99).119 The method is quite general and applicable to substrates with alkyl and aryl substitution (E-305a, b, n–q, 305r → 315a, b, n–q, 315r); the cyclization of γ,γ-disubstituted substrate 305r to a cyclopropane bearing a quaternary carbon center is particularly remarkable. With regard to the mechanism, a silylpotassium is thought to be the active silicon nucleophile that selectively attacks at the β-position of the allylic substrate.
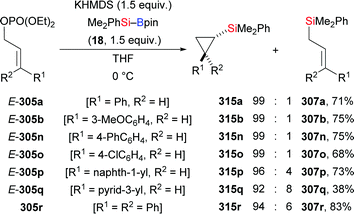 |
| | Scheme 99 Transition-metal-free silylative cyclopropanation of allylic phosphates with Me2PhSi–Bpin. Yields are for the mixture of isomers. | |
4.6.2. Propargylic precursors.
Propargylic silylation is another SN2′-type displacement of propargylic electrophiles with transition-metal-based silicon nucleophiles generated from Si–B bonds by transmetalation. In 2017, the laboratory of Xu and Loh introduced propargylic epoxides as substrates (Scheme 100).120 By variation of the reaction conditions, the authors could selectively obtain tri- and tetrasubstituted functionalised allenols and alkenes in moderate to high yields (316 → 317; 316 → 318; 316 → 319, Scheme 100, top). The tri- and tetrasubstituted alkenes were generated from the 2,3-allenol intermediate by another silylcupration of either of the allenic double bonds followed β-elimination of the hydroxy group or protonation (Scheme 100, bottom).
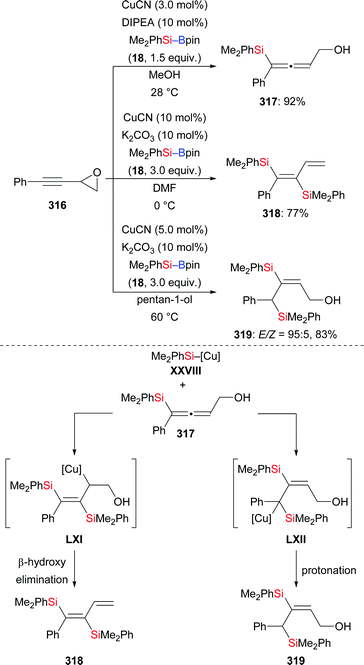 |
| | Scheme 100 Copper-catalysed silylation of propargylic epoxides with Me2PhSi–Bpin. | |
Very recently, an extension of that work was reported by Kleij and co-workers.121 These authors employed alkyne-substituted cyclic carbonates as propargylic acceptors, which undergo decarboxylation after the substitution event (320a–g → 321a–g, Scheme 101). The practical protocol is rather general, allowing for the introduction of various substituents at fully substituted 2,3-allenols.
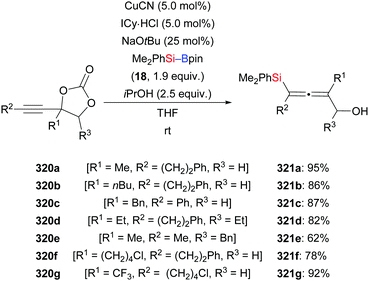 |
| | Scheme 101 Copper-catalysed decarboxylative silylation of alkyne-substituted cyclic carbonates with Me2PhSi–Bpin. | |
In 2019, Xu and Loh accomplished an enantioselective propargylic silylation of propargylic dichlorides by copper catalysis with Box ligand L37 (Scheme 102).122 Propargylic substitution occurred in the presence of CuF2/L37/TMP/MeOH to yield a series of enantioenriched chloro-substituted allenylsilanes. The reaction is general for various aryl-substituted propargyl dichlorides, providing chiral allenylsilanes in moderate to high yields and with good ee values (322a–e → 323a–e, Scheme 102, top). However, alkyl-substituted propargyl dichlorides were transformed in good yields but with somewhat lower enantioinduction (322f, g → 323f, g, Scheme 102, top). Notably, the asymmetric disilylation of a propargylic dichloride was also achieved in one pot, providing the bissilylated allene in good yield and with high enantiomeric purity (322c → 324c, Scheme 102, bottom). As to the mechanism, the allenylic chloride 323c is assumed to be an intermediate to undergo syn-addition and subsequent anti-elimination to form 324c.123
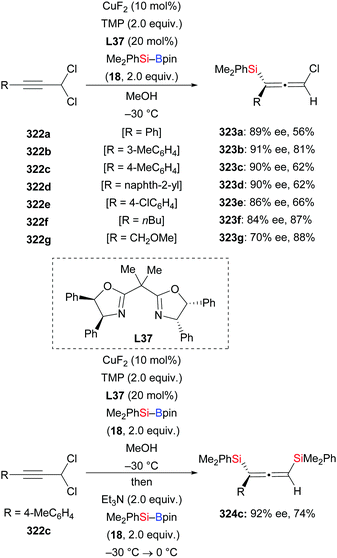 |
| | Scheme 102 Copper-catalysed asymmetric silylation of propargyl dichlorides with Me2PhSi–Bpin. | |
4.7. Dearomatization of dibenzo[b,d]furan and pyridines
A dearomatization process can convert simple 2D compounds into more complex 3D molecules and is therefore an intrinsically attractive goal.124 However, there are just few reported examples on silylative dearomatization to date.
When studying the C–H bond silylation of pyridines, Martin and co-workers found that the dearomatization products 326 or 327 could be formed after quenching a mixture of dibenzo[b,d]furan (325), Et3Si–Bpin (42), and KHMDS with D2O or Me3SiCl (Scheme 103).125 Unexpected dearomatization products were also observed when the laboratory of Ohmura and Suginome studied the mechanism of the pyridine-catalysed silaboration of terminal alkynes and allenes (cf.Schemes 11 and 41).26 These authors found pyridines undergo 1,2-addition of Ph3Si–Bpin (19) at 110 °C to afford dearomatization products in low yields (328a–c → 329a–c, Scheme 104).
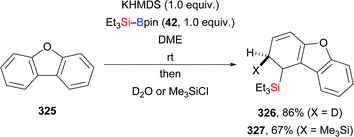 |
| | Scheme 103 Dearomatization of dibenzo[b,d]furan with Et3Si–Bpin. | |
 |
| | Scheme 104 Dearomatization of 4-substituted pyridines with Ph3Si–Bpin. | |
5. Functionalization of strained-ring compounds
5.1. Methylenecyclopropanes (MCPs)
MCPs are highly strained and reactive molecules. Their multiform reactivities based on the three σ-bonds in the cyclopropane ring and the C–C double bond may result in formation of a variety of products. Especially Suginome and co-workers have done pioneering work in this field.126 Among those alternative reaction channels, the silaboration of the unsymmetrical 1-substituted 2-methylenecyclopropanes is particularly difficult to control. In 2015, Suginome and co-workers developed a platinum-catalysed phosphine-free silaborative C–C bond cleavage of 1-substituted-2-methylenecyclopropanes with Si–B compounds under mild conditions, giving 3-substituted 2-boryl-4-silylbut-1-enes through selective cleavage of the less hindered proximal C–C bond (bond a) of the cyclopropane ring (331a–e → 332a–e, Scheme 105).127 Concerning the substrate scope, 1-alkyl-substituted substrates could be transformed in high yield but the reaction of the 1-phenyl-substituted derivative resulted in a small amount of desired product (331e → 332e, Scheme 105). The steric hindrance on the silicon atom is also critical for efficient formation of the silaboration products, and less bulky Si–B compounds lead to the formation of the product with cleavage of bond b of the cyclopropane ring in different proportions.
 |
| | Scheme 105 Platinum-catalysed regioselective silaborative C–C cleavage of MCPs. | |
In 2018, Suginome and co-workers synthesised chiral-guest-responsive helical polymer ligands, e.g. the ternary PQXphos (360/20*/![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif)
![[0 with combining macron]](https://www.rsc.org/images/entities/char_0030_0304.gif) ) L38, bearing both boronyl and 2-[bis(3,5-dimethylphenyl)phosphino]phenyl pendants as coordinating groups (Scheme 106).128 An unprotected 1,2-aminoalcohol was added as a chiral guest. This induced helically chiral macromolecular scaffold provided an efficient asymmetric reaction environment in the palladium-catalysed asymmetric silaborative C–C bond cleavage of the meso-configured MCP 333, leading to the ring-opened product 334 in good yield and with high enantiomeric excess. Later, these authors realised a highly efficient chirality transfer from the solvent limonene to the macromolecular main chain of PQXs (Scheme 107).129 A good level of enantioselectivity was again achieved in the above C–C bond cleavage of that MCP.
) L38, bearing both boronyl and 2-[bis(3,5-dimethylphenyl)phosphino]phenyl pendants as coordinating groups (Scheme 106).128 An unprotected 1,2-aminoalcohol was added as a chiral guest. This induced helically chiral macromolecular scaffold provided an efficient asymmetric reaction environment in the palladium-catalysed asymmetric silaborative C–C bond cleavage of the meso-configured MCP 333, leading to the ring-opened product 334 in good yield and with high enantiomeric excess. Later, these authors realised a highly efficient chirality transfer from the solvent limonene to the macromolecular main chain of PQXs (Scheme 107).129 A good level of enantioselectivity was again achieved in the above C–C bond cleavage of that MCP.
 |
| | Scheme 106 Palladium-catalysed asymmetric silaborative C–C bond cleavage of a meso-configured MCP using a macromolecular chiral ligand (part I). | |
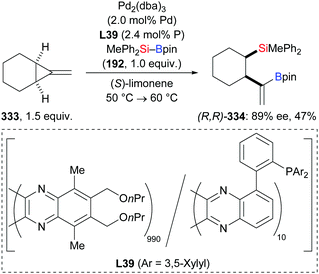 |
| | Scheme 107 Palladium-catalysed asymmetric silaborative C–C bond cleavage of a meso-configured MCP using a macromolecular chiral ligand (part II). | |
5.2. Vinylidenecyclopropanes
Allenes attached to small strained rings can also engage in ring-opening processes rather than a simple 1,2- or 2,3-addition of an Si–B bond under transition-metal catalysis. In 2019, Chen and co-workers reported a copper-catalysed ring-opening silylation of vinylidenecyclopropanes and obtained propargylic silanes as a result of C–Si bond formation at the allene terminus followed by ring opening (335a–f → 336a–f, Scheme 108).130 Hence, the proposed mechanism is believed to pass through initial addition of copper-based silicon nucleophile XXVIII to the vinylidenecyclopropane 335 to form the vinylcopper intermediate LXVIII (Scheme 109, top). The copper C- and O-enolates LXIX and LXX are formed by β-carbon elimination. Enolate protonation to yield 336 or addition of another electrophile to yield 337 terminates the reaction (Scheme 109, top). When using ethyl bromoacetate as the trapping reagent, product 337a was obtained in 67% yield (Scheme 109, bottom).
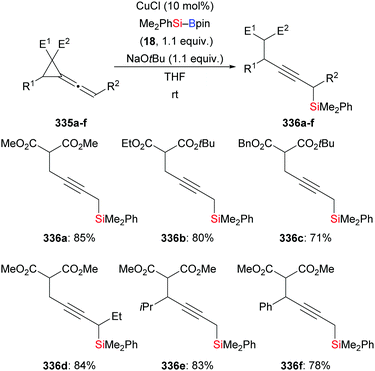 |
| | Scheme 108 Copper-catalysed silylation of vinylidenecyclopropanes. | |
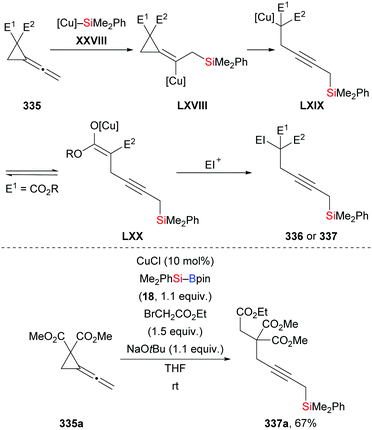 |
| | Scheme 109 Assumed mechanism of the copper-catalysed silylation of vinylidenecyclopropanes. | |
5.3. Cyclopropenes and 7-oxa-/7-azabenzonorbornadienes
Examples of the addition of Si–B compounds to cycloalkenes are still scarce. In 2019, Cao and co-workers achieved a copper-catalysed formal hydrosilylation of 1-substituted 3,3-difluorocyclopropenes (Scheme 110).131 The reaction proceeded smoothly, and the difluorocyclopropyl-substituted silanes were obtained in good to excellent yields with the diastereoselectivity in favour of the trans-isomer (338a–e → 339a–e, Scheme 110).
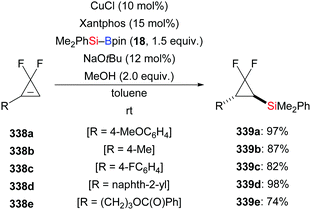 |
| | Scheme 110 Copper-catalysed diastereoselective formal hydrosilylation of difluorocyclopropenes with Me2PhSi–Bpin. | |
On the way to an asymmetric version of this transformation, Zhang and Oestreich tested dozens of chiral ligands in combination with Cu(CH3CN)4PF6, tBuONa, and MeOH.132 Excellent stereoselectivity and high conversion were achieved with Segphos ligand L40 for a series of 3,3-disubstituted cyclopropenes and various Si–B compounds (340a–i → 341a–i, Scheme 111). A bulkier alkyl group instead of the methyl group at C3 of the cyclopropene had no influence on the enantioselectivity but was slightly detrimental to diastereoselectivity and yield (340b–e → 341b–e, Scheme 111). These results imply that the reaction proceeds without relying on a coordinating/directing group and that the diastereoselectivity is affected by the steric discrimination of the geminal substituents. Deuterium-labelling experiments and nOe measurements confirmed a syn-addition across the C–C double bond and showed that the proton originates from the alcohol additive.
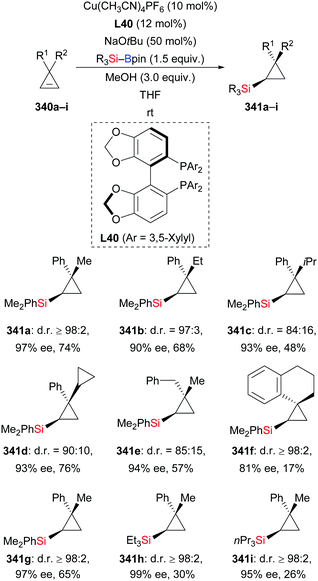 |
| | Scheme 111 Copper-catalysed enantio- and diastereoselective addition of R3Si–Bpin to 3,3-disubstituted cyclopropenes. | |
Very recently, Cui and Oestreich successfully extended this asymmetric catalysis to the formal hydrosilylation of 7-oxa- and 7-azabenzonorbornadiene derivatives with a change of the ligand from Segphos derivative L40 to R-QuinoxP* (L41) (Scheme 112).133exo-Selective addition of the silicon moiety across these strained alkenes was achieved, and no ring opening was observed in these reactions. The reaction was general for various 7-oxa- and 7-azabenzonorbornadiene derivatives and Si–B compounds (342a–e → 344a–e; 345a, b → 346a, b, Scheme 112). However, other substrates such as benzonorbornadiene, norbornadiene, or norbornene did not participate under these reaction conditions.
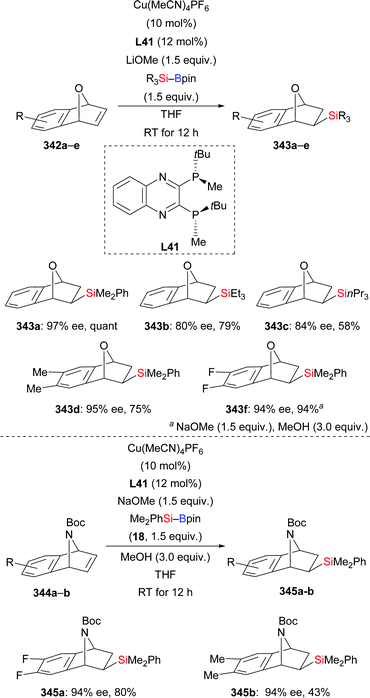 |
| | Scheme 112 Copper-catalysed enantio- and exo-selective addition of R3Si–Bpin to 7-oxa- and 7-azabenzonorbornadiene derivatives. | |
5.4. Bicyclo[1,1,1]pentanes
Recently, Uchiyama and co-workers accomplished the silaboration of [1.1.1]propellane introducing both a boryl and a silyl group onto the bicyclo[1.1.1]pentane scaffold (346 → 347, Scheme 113, top).134 Silaborated 347 was obtained in high yield under mild, additive-free condition; a gram-scale reaction even eliminated the need for purification by column chromatography. To reach a better understanding of the mechanism, the authors added 2.0 equivalents of 9,10-dihydroanthracene to the reaction mixture as a radical inhibitor, and only a trace amount of the desired product was detected (Scheme 113, middle). Also, the presence or absence of air in the system leads to huge yield differences (Scheme 113, bottom). These experimental results and DFT calculations indicated that the silaboration of [1.1.1]propellane follows a radical-chain mechanism, probably initiated by oxygen.
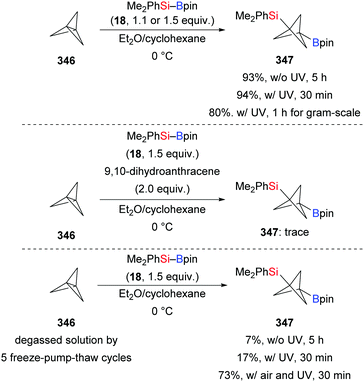 |
| | Scheme 113 Additive-free silaboration of [1.1.1]propellane with Me2PhSi–Bpin. | |
5.5. Diazabicycles, aziridines, oxetanes, and epoxides
Reports on the reactions of heterocycles with Si–B compounds are rare. A single example of a copper-catalysed silylative ring opening of diazabicycles was reported by Yun and co-workers (348 → 349, Scheme 114).135 The transformation occurred by C–N bond cleavage enabled by a copper/base/MeOH catalyst system, where MeOH fulfils the role of the proton source. By analogy, recently our group reported one example of a silylative ring-opening reaction of an aziridine (350 → 351, Scheme 115).136 Interestingly, when 2.0 equiv. LiCl was added to the reaction, the yield of this reaction increased from 38% to 82%. This indicated that LiCl serves as a Lewis acid, aiding the C–N bond cleavage.
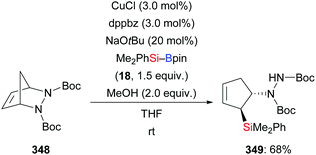 |
| | Scheme 114 Copper-catalysed silylative ring opening of diazabicycles. | |
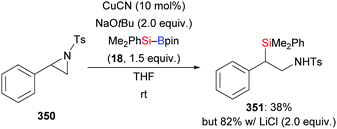 |
| | Scheme 115 Copper-catalysed ring opening of aziridine. | |
A contribution to the silylative ring opening by C–O bond cleavage was made by Stratakis and co-workers in 2016 (Scheme 116).137 These authors accomplished the silaboration of strained cyclic ethers (oxetanes and epoxides) to form γ- and β-silyloxy boronates using supported gold nanoparticles as recyclable catalysts. During the silaboration process, the boron moiety acts as a nucleophile and the silyl group is introduced as an electrophile. A series of substituted oxetanes including the parent compound and epoxides participated in this addition reaction in good yields (352a–f → 353a–f; 354a–c → 355a–c). It is worth mentioning that 2-aryl-substituted oxetanes underwent the silaboration highly regioselectively with the boryl group attached to the benzylic position (352e–f → 353e–f).
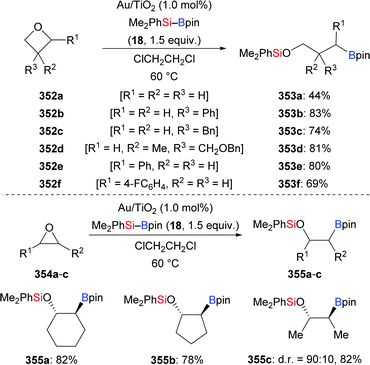 |
| | Scheme 116 Silaboration of oxetanes (top) and unactivated epoxides (bottom) catalysed by gold nanoparticles. | |
6. (2+2+1)- and (4+2+1)-cycloadditions of multiple-bond systems
Encouraged by the performance of the palladium-based catalytic system in alkyne–alkyne–silylene (2+2+1) cycloaddition,138 Ohmura and Suginome tried to explore a way to achieve an alkene–alkyne–silylene (2+2+1) cycloaddition. After many attempts, the authors finally realised this with a new rhodium catalysis, where a silylboronic ester bearing an alkoxy group participated in the reaction as synthetic equivalents of silylene. This was applicable to the conversion of a range of 1,6- and 1,7-enynes to bicyclic 1-silacyclopent-2-enes (357a–f → 358a–f, Scheme 117).139 The authors proposed a mechanism (Scheme 118) in which the transmetalation between [Rh]–X LXXI and Si–B compound takes place to produce a rhodium-based silylenoid LXXIII. Then, the 1,6-enyne inserts into the Rh–Si bond to form the alkenylrhodium complex LXXIV regioselectively. Subsequent insertion of the C–C double bond into the Rh–C bond occurs and an alkylrhodium complex LXXV is formed. Finally, the silicon-containing five-membered ring is formed by either nucleophilic substitution on the silicon center or σ-bond metathesis between Rh–C and Si–X bonds with LXXI returning to the catalytic cycle.
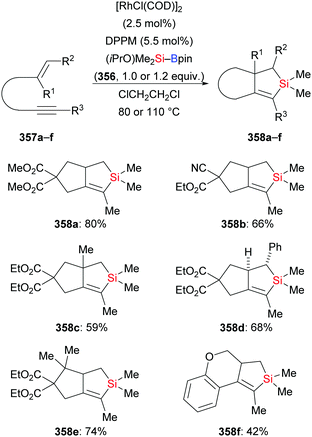 |
| | Scheme 117 Rhodium-catalysed (2+2+1) cycloaddition. | |
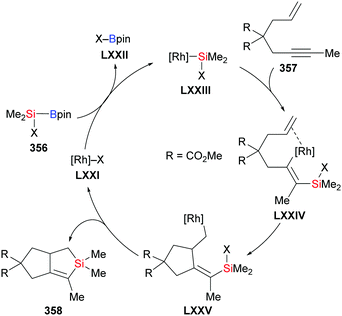 |
| | Scheme 118 Proposed mechanism of rhodium-catalysed (2+2+1) cycloaddition. | |
Just recently, Ohmura and Suginome extended this rhodium catalysis to a (4+2+1) cycloaddition.140 Based on the key intermediate, that is the “rhodium silylenoid”, in the reaction, the authors successfully developed a (4+2+1) cycloaddition of nona-1,3-dien-8-yne derivatives 359 and boryl(isopropoxy)silane (356) in the presence of a rhodium catalyst and dppm as ligand (Scheme 119, top). A series of silicon-containing seven-membered rings was obtained (359a–g → 360a–g). It should be noted that the (2+2+1) cycloaddition still takes place under this rhodium catalysis for the substrate 359h bearing 4-phenylbuta-1,3-dien-1-yl moiety (359h → 361h, Scheme 119, bottom). Unlike the mechanism of (2 +2 + 1) cycloaddition, the (4+2+1) cycloaddition involves the π-allylrhodium intermediate LXXVII, which is formed through intramolecular insertion of the diene moiety of LXXVI into the Rh–C bond (Scheme 120). As a result, a seven-membered ring as in 360 is produced through σ-bond metathesis or a five-membered ring as in 361 is obtained avoiding steric repulsion with the terminal substituent.
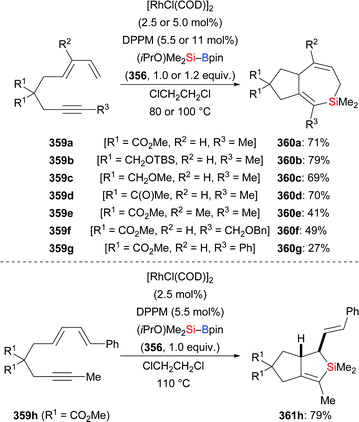 |
| | Scheme 119 Rhodium-catalysed (4+2+1) cycloaddition. | |
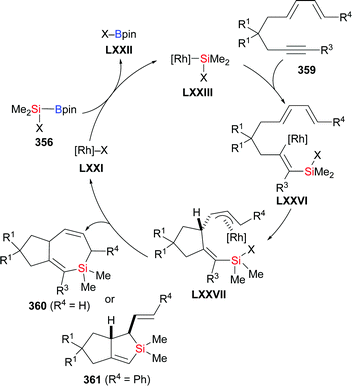 |
| | Scheme 120 Proposed mechanism of rhodium-catalysed (4+2+1) cycloaddition. | |
7. Functionalization of carbenoids and related compounds
7.1. Carbenoids
The carbenoid insertion into Si–B bonds to result in geminal functionalization of carbenoids has been well investigated, yet simple and mild methods are still worth exploring. In 2014, Wang and co-workers reported a practical protocol for the synthesis of 1-boryl-1-silylalkanes through the reaction of easy-to-make N-tosylhydrazones with Me2PhSi–Bpin under transition metal-free conditions (362a–d → 363a–d, Scheme 121, top).141 A possible reaction mechanism is depicted in Scheme 121 (bottom). First, the diazo compound LXXVII is generated in situ from N-tosylhydrazone 362 after treatment with NaH. Second, the nucleophilic diazo carbon atom will attack the electron-deficient boron atom of the Si–B reagent to form the boron ate complex LXXIX. Third, N2 is released with a simultaneous 1,2-shift of the silicon group from boron to carbon, providing formal Si–B insertion products.
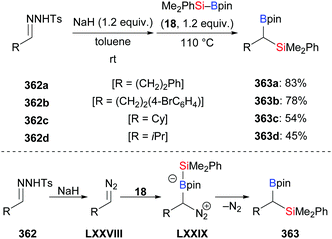 |
| | Scheme 121 Formal carbon insertion of N-tosylhydrazone into an Si–B bond for geminal silaboration. | |
There is also a single example by Xu and Wang of the synthesis of a chiral 1,1-silylboronate esters with a fully substituted benzylic center in one step (Scheme 122).142 These authors treated the enantioenriched Hoppe-type carbamate (S)-364b with sec-BuLi in Et2O followed by the addition of Me2PhSi–Bpin (18) at −78 °C. This afforded the 1,1-silylboronate ester (R)-365b in 95% isolated yield and with 92% ee.
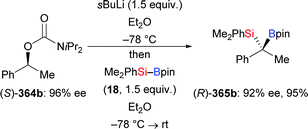 |
| | Scheme 122 Synthesis of enantioenriched 1,1-silylboronate ester. | |
Almost at the same time, Liu and Lan reported a geminal silaboration of aldehydes and ketones by a stepwise operation, where B2pin2 is initially reacted with those carbonyls followed by silylation with Me2PhSi–Bpin (Scheme 123, top).143 An ate complex formed from 366 by the addition of the silicon nucleophile undergoes 1,2-migration with OBpin as the leaving group. The stepwise strategy was applicable to several aldehydes and ketones in good yields (155b, 153d, v, w → 365b, 363e, v, w, Scheme 123, top). When chiral ligand L11 was present in the first step, the products were obtained with high ee values (367a → (R)-363a, Scheme 123, bottom). The second step is stereospecific and proceeded with inversion at the asymmetrically substituted carbon center.
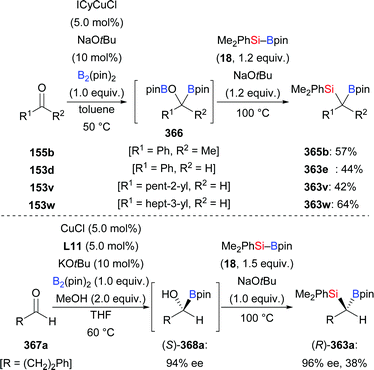 |
| | Scheme 123
gem-Silaboration of aldehydes and ketones. | |
Shortly thereafter, the laboratory of Aggarwal applied the strategy of the carbenoid insertion into Si–B bonds to the stereocontrolled synthesis of polypropionate fragments.144 The authors subjected carbamate 369 and Me2PhSi–Bpin (18) to a Hoppe-type reaction sequence using sBuLi together with (+)- or (−)-sparteine (Scheme 124). The 1,1-silylboronates 370 formed with high diastereocontrol, and all four stereoisomers were efficiently prepared (369 or ent-369 → 370 or ent-370).
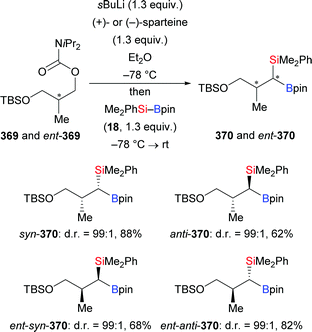 |
| | Scheme 124 Silaboration of a chiral Hoppe carbamate and preparation of all four stereoisomers. | |
8. C–Si and C–B cross-coupling
Over the past eight years, Si–B reagents have been employed as versatile pronucleophiles in the cross-coupling arena. As illustrated in the following sections, the combination of Si–B reagents and electrophilic reaction partners can be a robust and reliable approach toward the selective formation of C–Si and C–B bonds.
8.1. C(sp3)–Si cross-coupling
Hydrosilylation is useful for the synthesis of linear alkylsilanes. However, problems with isomerization and regioselectivity are often encountered in the hydrosilylation of structurally unbiased internal alkenes for the synthesis of α-branched alkylsilanes.145 Recently, the cross-coupling of alkyl electrophiles and Si–B reagents has become an alternative route for C(sp3)–Si bond formation. The issue of regioselectivity is overcome by prefunctionalization.146
In 2013, as part of their work on an enantioselective synthesis of allylsilanes by allylic substitution, Hayashi and co-workers reported one example of a copper-catalysed cross-coupling of an activated alkyl electrophile with a silylboronic ester.117 The reaction of primary benzylic phosphate 371a with Me2PhSi–Bpin (18) in the presence of CuCl, chiral NHC precursor L34, and a base, e.g. KOtBu, NaOtBu or NaOMe, afforded the benzylic silane 372a in moderate yield. It is noteworthy that a base-promoted uncatalysed reaction is observed in the absence of a copper(I) source and the NHC ligand (Scheme 125).
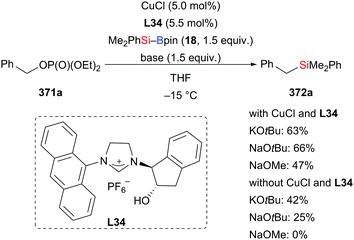 |
| | Scheme 125 Copper-catalysed cross-coupling reaction of Me2PhSi–Bpin with a benzylic phosphate. | |
Later, an efficient nickel/copper-catalysed silylation of benzylic pivalates 373a–c with silicon pronucleophile 42 was described by Martin and co-workers in 2014. Both primary and secondary benzylic pivalates were coupled efficiently (Scheme 126, top).147a In an extension of this work, Martin successfully applied this cross-coupling of silicon pronucleophile 42 to benzylic ether 375 by using a ligand-free nickel-catalysed system (Scheme 126, bottom).147b
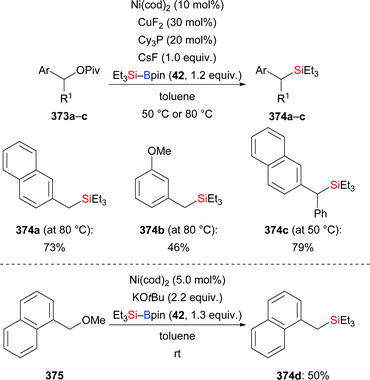 |
| | Scheme 126 Cross-coupling reactions of Et3Si–Bpin with benzylic electrophiles involving C–O bond cleavage. | |
In 2016, Loh and co-workers reported an efficient palladium-catalysed silylation reaction of benzylic halides with silylboronic esters (Scheme 127).148 The substrate scope was generally good with various electron-donating and -withdrawing groups attached to the aryl ring, including a few heterocycles. Aside from primary benzylic bromides 376a–j, secondary benzylic chlorides 376k–l afforded the corresponding benzylsilanes 372a–l. Moreover, the authors synthesised enantioenriched substrate 376l (75% ee) which racemized in the reaction (not shown). On the basis of this observations, the plausible catalytic cycle was proposed by the authors. After oxidative addition of the C(sp3)–X bond to Pd(0) (XV), the intermediate LXXX is believed to form LXXXI with a Pd(II)–[O] bond by the reaction with silver oxide. LXXXI then engages in a σ-bond metathesis with the Si–B reagent, forming a Pd(II)–Si bond (LXXXI → LXXXII → LXXXIV). Reductive elimination from LXXXIV liberates the desired product and regenerates the Pd(0) catalyst (Scheme 128).
 |
| | Scheme 127 Palladium-catalysed cross-coupling reaction of Me2PhSi–Bpin with benzylic halides. | |
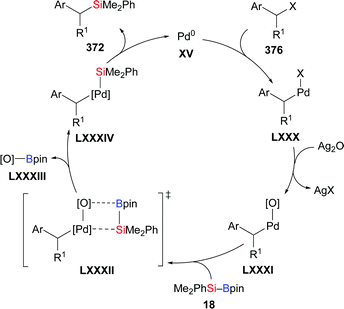 |
| | Scheme 128 Assumed mechanism of the palladium-catalysed cross-coupling reaction of silylboronic esters with benzylic halides. | |
A deaminative C(sp3)–Si cross-coupling with silylboronic esters had remained unknown until the Oestreich group used benzylic ammonium triflates as electrophiles. In the presence of CuBr and NaOtBu, several primary (such as 377a) and secondary (such as 377b–n) electrophiles converted into the corresponding benzylic silanes in moderate to good yields (Scheme 129, top).149 A tertiary benzylic ammonium salt decomposed under these reaction conditions (not shown). Of note, cyclopropylsubstituted 377n yielded 372y in 42% with no ring opening. This supports an ionic mechanism and likely excludes the intermediacy of a benzyl radical. While Et3Si–Bpin (42) was suitable as a pronucleophile, the yield eroded with MePh2Si–Bpin (192) (not shown). In the light of the limited examples of the synthesis of α-chiral silanes by cross-coupling, these authors extended the racemic variant to readily available enantioenriched benzylic ammonium salts. A number of highly enantioenriched benzylammonium triflates (such as 377b–d, g, i, k) were transformed into the desired benzylsilanes with inversion of the configuration and essentially no loss of enantiomeric purity (Scheme 129, bottom). These results further support an SN2-type mechanism.
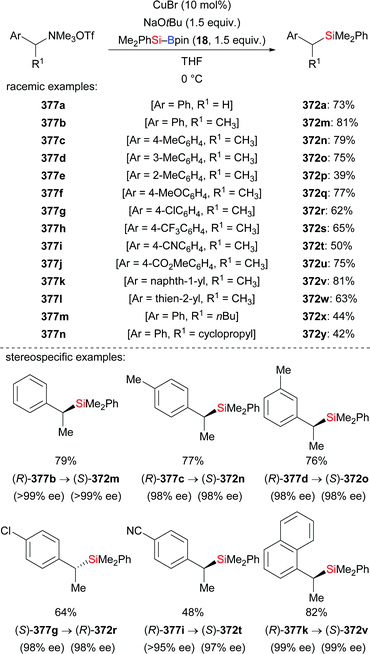 |
| | Scheme 129 Copper-catalysed cross-coupling reaction of silylboronic esters with (chiral) benzylic ammonium triflates. | |
Apart from benzylic electrophiles, the same laboratory succeeded in developing the C(sp3)–Si coupling of unactivated alkyl electrophiles with silylboronic esters. Using a combination of CuCN and NaOtBu, various functionalised primary alkyl triflates 378a–h were converted into the corresponding tetraorganosilanes in good to high isolated yields while substrates with halide leaving groups gave lower yields; alkyl phosphates and tosylates did not react (Scheme 130).150
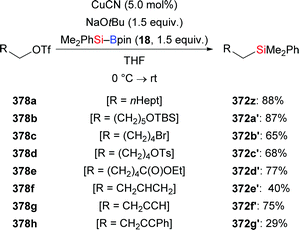 |
| | Scheme 130 Copper-catalysed cross-coupling reaction of Me2PhSi–Bpin with primary alkyl triflates. | |
That reaction was limited to primary electrophiles as secondary substrates are prone to facile β-elimination. The authors assumed an ionic mechanism. To expand the substrate scope and gain insight into the mechanism, Oestreich and co-workers reported an enantiospecific silylation of α-triflyloxy nitriles (such as 378i) and esters (such as 378j). These reactions proceed with inversion of the configuration, thus supporting the postulated SN2 mechanism (Scheme 131).151
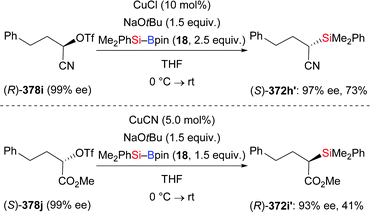 |
| | Scheme 131 Enantiospecific cross-coupling reaction of Me2PhSi–Bpin with activated secondary alkyl electrophiles. | |
Oestreich and co-workers then identified a catalytic setup that allows for the cross-coupling of unactivated alkyl iodides (Scheme 132).152 The catalyst system is a combination of CuSCN, a bipyridine ligand, and LiOtBu in a solvent system consisting of THF and DMF in a 9:1 ratio. The reaction produced primary and secondary alkylsilanes with various functional groups in the alkyl chain in good to high yields. Among tertiary substrates only adamantyl iodide 379c reacted in acceptable yield. More bulky MePh2Si–Bpin (192) and less reactive Et3Si–Bpin (42) also furnished the corresponding products (not shown), albeit with yields lower than that for Me2PhSi–Bpin (18). It is noteworthy that, in this case, the C–Si bond-forming process proceeded through a radical mechanism. This was supported by the following facts: (1) the diastereomerically pure pregnenolone-derived iodide 379f underwent the C(sp3)–Si coupling with complete epimerization (d.r. > 98:2 for 379f to d.r. = 50:50 for 380f); (2) TEMPO inhibited the coupling of cyclohexyl iodide, and the cyclohexyl/TEMPO adduct was detected. These results together with DFT calculations performed by Qu and Grimme were merged into a detailed mechanistic picture. It is initiated by heterolytic Si–B bond cleavage to form Me2PhSi−, and transfer of Li(THF)3+ to Me2PhSi− then leads to Me2PhSi–Li(THF)3 (LXXXVI) which can enter the catalytic cycle as a nucleophile (Scheme 133, top). The cationic copper(I) catalyst LXXXVII is formed through dissociation of the counteranion and coordination of two bipyridine ligands. Transmetalation produces the nucleophilic Cu(I)–SiMe2Ph complex (LXXXVIII); an SET process between LXXXVIII and alkyl iodide 379 to generate an alkyl radical intermediate is followed by a radical coupling to form the silylation product with regeneration of the cationic copper(I) complex LXXXVII (Scheme 133, bottom). The radical nature of this copper catalysis prompted these authors to intercept the C(sp3)–Si bond formation by a radical cyclization onto a tethered alkene. The silylative radical cyclization of precursors 381a–c gave the corresponding mono- or bicyclic silylation compounds 382a–c in modest to good yields with high stereoselectivity (Scheme 132, bottom).
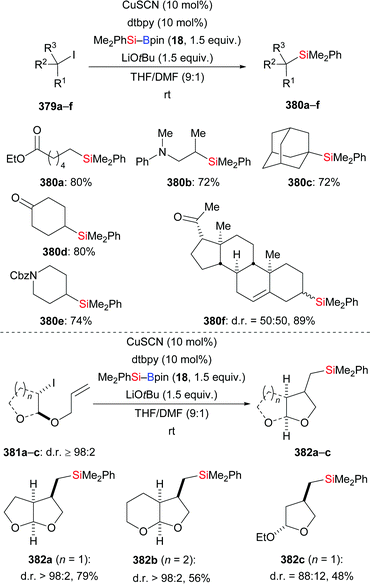 |
| | Scheme 132 Copper-catalysed cross-coupling of unactivated alkyl iodides with Me2PhSi–Bpin, including silylative radical cyclizations. | |
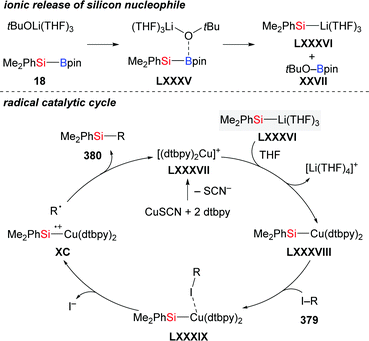 |
| | Scheme 133 Copper-catalysed silyl substitution of alkyl iodides through a radical mechanism. | |
In an extension of this work, Xue and Oestreich disclosed a copper-catalysed decarboxylative radical silylation of redox-active esters 383 derived from N-hydroxyphthalimide (Scheme 134).153 Various copper catalysts, ligands, bases, and solvents were suitable; however, the best results were achieved in THF/NMP (9:1) at room temperature with copper(I) thiophene-2-carboxylate (CuTc) as the precatalyst together with dtbpy and Cy3P as ligands and NaOEt or LiOtBu as bases. Changing to other redox-active esters as leaving groups was not successful. Again, both primary and secondary alkyl electrophiles were successfully coupled with high selectivity and comparable functional-group tolerance. Adamantyl-substituted 380c was the only product with a tertiary alkyl residue that was accessible. It is noteworthy that the reactions enabled the synthesis of α-aminosilanes starting from α-amino acid derivatives 380l–o (cf. Section 4.3.2). While changing from Me2PhSi–Bpin (18) to MePh2Si–Bpin (192) still afforded 384, less reactive Et3Si–Bpin (42) did not react. Racemization and radical-trapping experiments were consistent with a radical mechanism in analogy to the previous dehalogenative silylation (cf.Scheme 133).
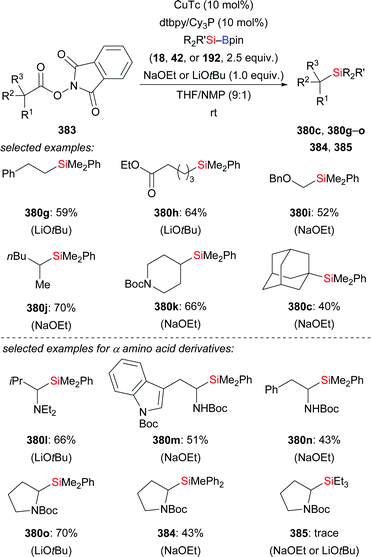 |
| | Scheme 134 Copper-catalysed decarboxylative silylation of N-hydroxyphthalimide esters with Me2PhSi–Bpin. | |
Just recently, Sakamoto, Maruoka and co-workers transferred this newly developed radical silylation concept to the coupling of alkylsilyl peroxides 386a–g and Me2PhSi–Bpin (18). After extensive screening of reaction conditions, the authors discovered that the reaction of various cyclic alkylsilyl peroxides in the presence of CuI and 1,10-phenanthroline at 130 °C furnishes ketone-containing alkylsilanes in moderate to good yields (Scheme 135).154 In contrast, acyclic alkylsilyl peroxides produced corresponding alkylsilane products without that ketone moiety through cleavage of the oxygen–oxygen and C(sp3)–C(sp3) bonds (not shown).
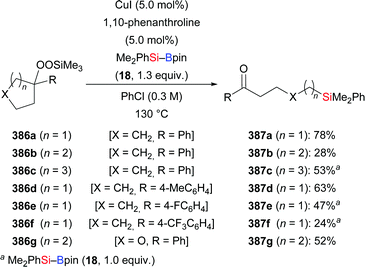 |
| | Scheme 135 Copper-catalysed silylation of cyclic alkylsilyl peroxides with Me2PhSi–Bpin. | |
When using geminal dibromides as electrophilic reaction partners, a combination of those two mechanistically different approaches (nucleophilic substitution and radical cross-coupling) was accomplished by Hazrati and Oestreich (Scheme 136).155 This copper-catalysed process worked well with several functionalised terminal dibromides with no branching in the proximity (388a–c → 389a–c). While branching closer to the reaction site as in 388d with a cyclohexyl group did not thwart the silylation, the yield dropped substantially with tert-butyl or phenyl as the R group (388e → 389e; 388f → 389f). To probe the mechanism, the authors designed the radical-clock experiment outlined in Scheme 136 (bottom). Upon treatment of 388g under the standard setup, ring-opened bissilylated product 390 was isolated in 54% yield. These findings strongly support the involvement of an ionic SN2 process in first C(sp3)–Si bond formation and a radical pathway in the construction of second C(sp3)–Si bond.
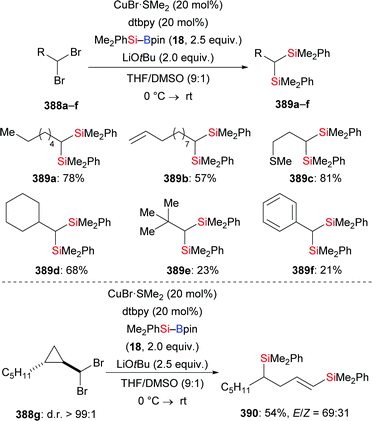 |
| | Scheme 136 Copper-catalysed double C(sp3)–Si cross-coupling of geminal dibromides with Me2PhSi–Bpin. | |
Beyond these metal-catalysed protocols, Shibata and co-workers introduced metal-free defluorosilylation reactions of alkyl fluorides 391a–i that relies on the chemoselective activation of the Si–B bond using KOtBu (Scheme 137, top).156 Concerning the substrate scope, primary (such as 391a–e, i) and secondary (such as 391f–h) benzylic fluorides were cleanly converted into corresponding benzylsilanes. While the linear alkyl fluoride 391i was smoothly transformed into the corresponding product using only KOtBu as the activation agent, defluorosilylation of tertiary (1-fluorocyclobutyl)benzene was achieved using Ni(cod)2 as the catalyst (not shown). Later, Martin and co-workers further extended the scope of this reaction for linear alkyl fluorides by applying LiHMDS as the activation agent (Scheme 137, bottom).157 Both authers proposed a mechanism involving SN2 attack of the in situ generated silyl anion species on alkyl fluoride.
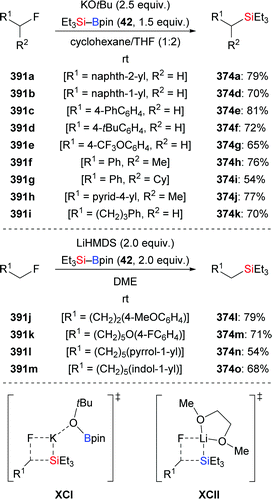 |
| | Scheme 137 Base-mediated defluorosilylation of C(sp3)–F bonds. | |
8.2. C(sp2)–Si and C(sp2)–B cross-coupling
In recent years, Si–B bond activation applied to C(sp2)–Si cross-coupling reactions with various electrophiles has been developed as an efficient method for the synthesis of functionalised arylsilanes. In 2015, a seminal example of a palladium-catalysed cross-coupling between aryl or heteroaryl halides and silylboronic esters was reported by He and co-workers (Scheme 138).158 A catalyst and base screening revealed that (Ph3P)4Pd and K2CO3 as base are an effective combination for this transformation. As shown in Scheme 138, electron-deficient (such as 392d–g, i) and electron-rich (such as 392b–c, h, j) aryl bromides were tolerated under this reaction setup. A wide range of functional groups including groups such as hydroxy (392c), cyano (392f), ester (392g), and amino (392j) was compatible, and generally good yields were obtained. Even though the authors mainly focused on aryl bromides, their work contains an example for the formation of corresponding arylsilane from an aryl chloride (392l) and an aryl iodide (392m), respectively. The transformation is restricted to Me2PhSi–Bpin (18); Et3Si–Bpin (42) did not react, possibly due to difficulties associated with the transmetalation step. A preliminary mechanistic study showed that the preformed Ph–Pd(II)–Br complex XCIII undergoes smooth cross-coupling with Me2PhSi–Bpin (18) to give the desired aryl silane 393a in a high yield. This observation suggested that the C–Si bond formation likely follows a pathway similar to that of the classic Suzuki–Miyaura cross-coupling.
 |
| | Scheme 138 Palladium-catalysed cross-coupling reactions of aryl halides and Me2PhSi–Bpin. | |
Very recently, Feng and co-workers extended the substrate scope to functionalised (hetero)aryl chlorides by applying a more sustainable iron catalytic system (Scheme 139).159 Again, a broad substrate scope and good functional-group tolerance were realised. Moreover, the iron-catalysed silylation reaction was further demonstrated by its applicability in the late-stage functionalization of some pharmaceuticals (not shown). Notably, the iron-catalysed silylation of aryl chlorides with Et3Si–Bpin (42) led to clean formation of desired product while Me2PhSi–Bpin (18) is not a suitable silicon pronucleophile, which is complementary to He's method.
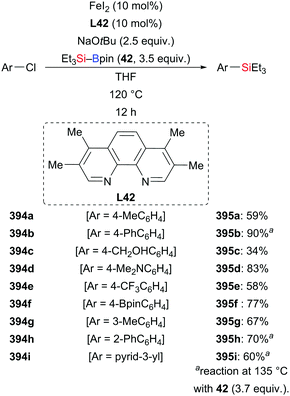 |
| | Scheme 139 Iron-catalysed cross-coupling reactions of aryl chlorides and Et3Si–Bpin. | |
Apart from aryl bromides and chlorides, C(sp2)–Si cross-coupling of fluoroarenes with silylboronates was accomplished by Shibata and co-workers (Scheme 140).156 The method relies on cleavage of unactivated C(sp2)–F bonds by a nickel catalyst. After extensive optimization, a ligand-free protocol based on a combination of Ni(cod)2 and KOtBu in a binary solvent system (cyclohexane/THF = 1:2) at room temperature provided the best result. No reaction was observed in the absence of the nickel catalyst or the base. The reaction conditions were applied to a variety of π-extended (such as 396a–f) and non-π-extended (such as 396g–l) fluoroarenes with broad functional-group tolerance, e.g., allowing for the formation of amine 395d or boronic ester 395p in moderate to good yields. Fluorinated heteroaromatic substrates did also convert under these reaction conditions. Substrates bearing more than one fluorine atom such as 396k–l were successfully monosilylated by carefully adjusting the stoichiometry of the reaction. The reaction is not limited to Et3Si–Bpin (42); using Me2PhSi–Bpin (18) as the silicon pronucleophile gave the desired product, albeit in decreased yield. Interestingly, fluoroarenes led to clean formation of silylated product while chlorine-, bromine-, and iodine-substituted arenes afforded a mixture of silylated and borylated compounds, whereby the borylated products were the major products (not shown). Several mechanistic experiments, including the utilization of a radical clock and silicon radical scavengers were conducted to gain insight into the mechanism of this transformation. These results indicated no involvement of radical intermediates in this system. Based on control experiments and 11B and 19F NMR measurements, two plausible pathways have been proposed by the authors involving nucleophilic aromatic substitution (SNAr) for π-extended fluoroarenes and nonclassical oxidative addition for non-π-extended substrates, respectively (not shown). These mechanisms are in accord with that of Martin's silylation of aryl methyl ethers.147b
 |
| | Scheme 140 Nickel-catalysed cross-coupling reactions of aryl fluorides and Si–B reagents. | |
Later, Martin and co-workers reported the defluorosilylation of a wide selection of fluoroarenes employing Si–B compounds (Scheme 141, top).157 This LiHMDS-mediated process works without the aid of a transition-metal catalyst. The reaction is heavily influenced by the choice of the base and the solvent. The replacement of LiHMDS by common (in)organic bases such as KOtBu, KOMe, Cs2CO3, and LDA or changing DME to other ethereal solvents failed to provide the target products; countercations other than Li+ resulted in significant erosion in yield. The substrate scope mainly focused on π-extended fluoroarenes such as 396a–e, m; nonconjugated fluoroarenes such as 396g, n afforded the corresponding products by using KOtBu as the base. It is noteworthy that no regioisomers of these silylated products were detected, indicating that aryne intermediates are not involved in these reactions. A key intermediate in the proposed mechanism is the solvent-separated ion pair XCIV, formed upon exposure of Et3Si–Bpin (42) to LiHMDS and DME. Such species might subsequently act as silyl anion surrogates in a concerted nucleophilic substitution at the ipso-C(sp2)–F site to access arylsilanes XCV. Similarly, NaOtBu-mediated defluorosilylation of fluoroarene with Me2PhSi–Bpin was reported by Wang, Uchiyama, and co-workers (Scheme 141, bottom).160 The concerted nucleophilic aromatic substitution pathway was supported by DFT calculations.
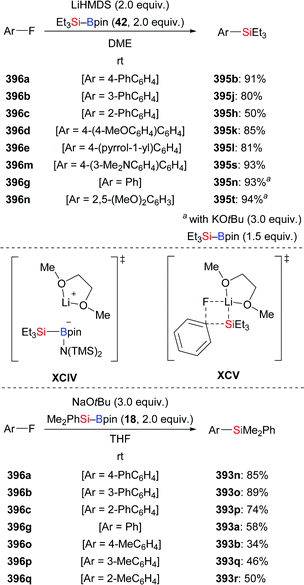 |
| | Scheme 141 Base-mediated cross-coupling reactions of aryl fluorides and Si–B reagents. | |
Recently, the use of C–O electrophiles has gained momentum as alternatives to aryl halides in cross-coupling reactions.161 Apart from the inertness of the C(aryl)–O bond, the selectivity between the cleavage of C(aryl)–O and C(acyl)–O bonds needs to be addressed when developing cross-coupling reactions for aryl ester substrates. C(acyl)–O bonds are more reactive than C(aryl)–O bonds in terms of bond dissociation energies. However, the use of bulky acyl groups such as a pivaloyl group is beneficial for achieving the selective transformation of C(aryl)–O bonds of aryl esters. In 2014, Martin's group developed an elegant C(sp2)–Si cross-coupling reaction of aryl esters involving C(aryl)–O bond activation (Scheme 142).147a An efficient catalytic system based on Ni(cod)2 and Cy3P with CuF2/CsF additives allowed for the silylation of aromatic pivalates 397a–g with Et3Si–Bpin (42). A variety of naphthyl and phenyl pivalates, regardless of electronic effects on the aryl ring, could be coupled in moderate to excellent yields; ortho-substituents (397d) did not hamper the reaction if conducted at 80 °C.
 |
| | Scheme 142 Nickel-catalysed cross-coupling reactions aryl pivalates and Et3Si–Bpin. | |
To elucidate the mechanism and to clarify the role of the CuF2/CsF additives, Martin in collaboration with Gómez-Bengoa, Burés, and co-workers undertook a detailed mechanistic study four years later.162 Results from spectroscopic measurements, stoichiometric experiments, VTNA kinetic studies, and DFT calculations were merged into a detailed mechanistic picture (Scheme 143). After oxidative addition of aryl esters to the nickel(0) complex by C(sp2)–O cleavage (XCVI → XCVII), an unusual dinickel(μ-η2-arene) complex XCVIII is formed that is equilibrium with the mononickel Ni(II) complex XCVII and Ni(0). Subsequent transmetalation of the mononickel Ni(II) complex with silylboronic esters gives rise to C and PivOBpin (XCIX). Finally, reductive elimination delivers the aryl silanes and regenerates the Ni(0) catalyst. The PivOBpin (XCIX) byproduct is captured by fluoride ions to form strong B–F bonds and insoluble fluoroborates in the presence of CuF2/CsF additives, thus ensuring catalyst turnover.
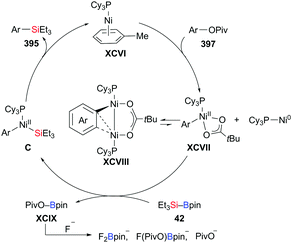 |
| | Scheme 143 Mechanistic picture for the silylation of aryl pivalates. | |
C(aryl)–O bonds in aryl ethers are much less reactive than aryl esters, which makes the development of such reactions even more challenging. Again, ipso-silylation of aryl methyl ethers by selective cleavage of the C(aryl)–OMe bonds was achieved by Martin and co-workers (Scheme 144, top).147b Different from previous C–C and carbon–heteroatom cross-coupling reactions of aryl methyl ethers at high temperature and excessive amounts of added ligands, it was nicely shown by the authors that the combination of Ni(cod)2 and KOtBu affords the desired products at room temperature (or even at 0 °C under certain circumstances) and without recourse to any external ligands. Under the standard protocol, both anisole derivatives without π-extended backbones (such as 398a–c) and π-extended aryl ethers (such as 398d–h) were converted into the corresponding aryl silanes in moderate to good yields. However, functional groups such as bromo, alkynyl, ketone, acid, and pyrazole residues were not tolerated (not shown). Apart from Et3Si–Bpin (42), dimethylphenyl-, tert-butyldimethyl-, and tripropyl-substituted silicon pronucleophiles reacted smoothly to afford the corresponding products, albeit higher catalyst loadings and elevated temperatures were required in these cases (not shown). Of note, the optimised conditions were applied to other substituted naphth-2-yl ethers including ethyl-, benzyl-, isopropyl-, and tert-butyl naphthyl ethers yet in decreased yields; the benzyl ether in axially chiral naphthamide 399 was directly transformed into arylsilane 395y without loss of enantiopurity (Scheme 144, bottom).163 The substrate scope could be extended to acyclic vinyl ethers such as 400a–b, thus giving access to E-configured vinylsilanes regardless of the substrate's double bond geometry. Moreover, no particularly significant Z/E isomerization was found for (Z)-400a in the absence of 42. These results indicated that the involvement of a “classical” oxidative addition of the C(sp2)–OMe bond to Ni(0) might be unlikely in this system (Scheme 145).
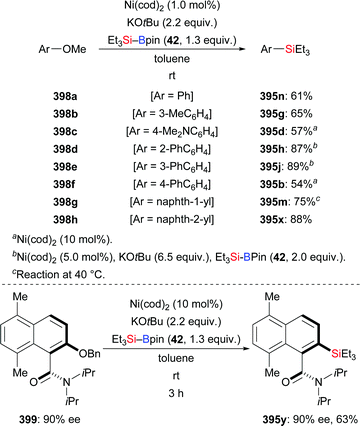 |
| | Scheme 144 Nickel-catalysed cross-coupling reactions of aryl ethers and Si–B reagents. | |
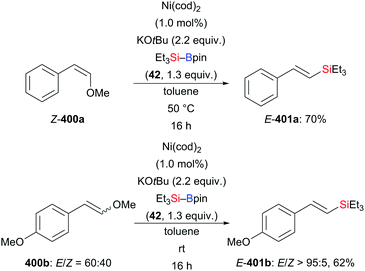 |
| | Scheme 145 Nickel-catalysed cross-coupling reactions of vinyl ethers and Si–B reagents. | |
Several experiments were performed to investigate the possibility of a radical-mediated mechanism. The lack of inhibition in the presence of stoichiometric amounts of silyl radical scavengers and failure to verify radicals with radical clock molecules made a radical-type pathway highly unlikely. Therefore, two plausible mechanistic pathways were proposed by Martin (Scheme 146). First, Et3SiK or the ate complex [Et3Si–Bpin(OtBu)]K (CI) are generated from the reaction between KOtBu and Et3Si–Bpin (42). Transmetalation with Ni(cod)2 generates the active silylnickel(0) ate complex CII, which may react further in two different ways. In path a, an internal metal-catalysed nucleophilic aromatic substitution assisted by complexation of the K+ counterion with the lone pair of the ether oxygen atom would give the arylsilanes and regenerate the Ni(0) catalyst (CII → CIII). Alternatively, the C–O cleavage can occur by a “nonclassical” oxidative pathway as shown in path b (CII → CIV). Very recently, Fu, Yu and co-workers investigated this mechanism using DFT methods on the basis of oxidative addition for non-π-extended aromatic systems.164a These authors found that the activation of the C–O bond proceeds by oxidative addition through a three-centered transition state. Interestingly, Avasare calculated two different mechanistic pathways involving internal nucleophilic substitution and nonclassical oxidative addition with the π-extended aryl ethers.164b In this case, internal nucleophilic substitution pathway is more facile and feasible than a normal or a nonclassical oxidative addition. The formation of silylnickel(0) ate complexes is supported by both cases.
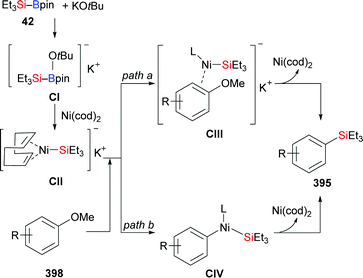 |
| | Scheme 146 Proposed reaction pathways for the nickel-catalysed silylation of aryl methyl ethers. | |
In contrast to Martin's silylation of aryl pivalates by C(aryl)–O bond activation, Rueping165 and Shi166 independently succeeded in nickel/copper-co-catalysed decarbonylative silylation of phenolic esters by C(acyl)–O bond cleavage. Rueping's group found that a combination of Ni(cod)2, CuF2, KF, and monodentate nBu3P in toluene at 160 °C gave the best result (Scheme 147, conditions A). A wide range of phenolic esters derived from naphthols (such as 395x, z, m), phenols (such as 395b, a′, d), and heteroarenes (such as 395b′–d′) were efficiently converted into the corresponding arylsilanes by employing silylboronic ester 42. It is notable that substrate 402a′ with a methyl ester group was compatible with this method, giving 395a’ in 85% yield. Apart from Et3Si–Bpin (42), the author applied MeEt2Si–Bpin, tBuMe2Si–Bpin and Me2PhSi–Bpin to the coupling of phenyl 2-naphthoate (402x) with similar success, except for nPr3Si–Bpin (not shown). In their mechanistic proposal, the authors assumed that oxidative addition of the nickel(0) catalyst into an ester C(acyl)–O bond is the first step (CV → CVI). Subsequent transmetalation involving the in situ-generated copper silane complex XL may deliver the key intermediate CX that can undergo decarbonylation to generate complex CXI. Finally, reductive elimination of CXI yields the desired arylsilanes 395, regenerating nickel(0) catalyst CV (Scheme 148).
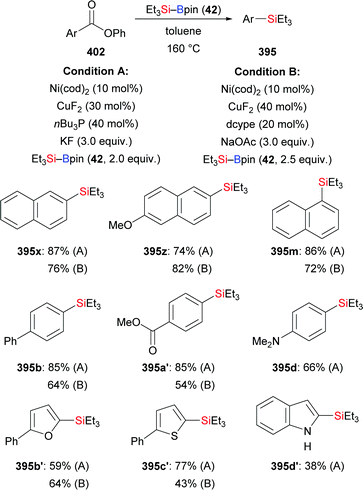 |
| | Scheme 147 Nickel/copper-catalysed decarbonylative silylation of phenolic esters. | |
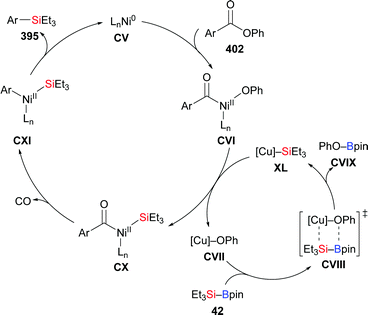 |
| | Scheme 148 Proposed mechanism of Rueping's decarbonylative silylation of phenolic esters. | |
Different from Rueping's work, Shi described an efficient nickel/copper-catalysed decarbonylative silylation of phenolic esters with Et3Si–Bpin (42) using NaOAc as base and bidentate dcype as the supporting ligand. The substrate scope mainly focused on π-extended phenolic esters and heteroaryl esters (Scheme 147, conditions B). Based on a catalytic cycle for decarbonylative borylation of esters and stoichiometric decarbonylative reactions of esters with Ni(cod)2 (not shown), Shi proposed a mechanism different from the one proposed by Rueping (Scheme 148). As shown in Scheme 149, it is believed that the decarbonylation occurs prior to transmetalation.166
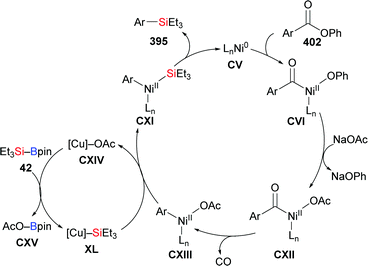 |
| | Scheme 149 Proposed mechanism of Shi's decarbonylative silylation of phenolic esters. | |
Guided by the above mechanistic hypothesis, Rueping as well as Nishihara were able to extend the scope of electrophiles. Reuping expanded the decarbonylative silylation strategy to arylamides by C(acyl)–N bond cleavage (Scheme 150).167 Again, using the same reactions conditions as for the decarbonylative silylation of phenolic esters, various amides 403 underwent the transformation in good yields. A change to other amides as leaving groups was not successful.
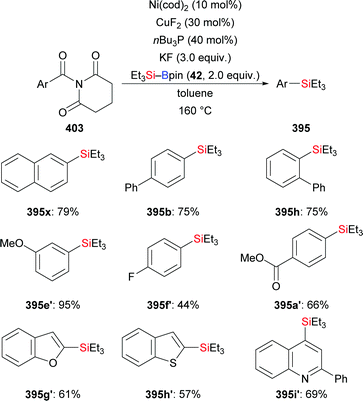 |
| | Scheme 150 Nickel/copper-catalysed decarbonylative silylation of amides. | |
Later, as part of their work on the decarbonylative cross-coupling reaction of aldehydes, Rueping and co-workers reported three examples of a nickel-catalysed decarbonylative silylation of aldehydes 153 with Me2PhSi–Bpin (18) (Scheme 151).168 The key to success for this reaction was the use of ketone 404 as a hydride acceptor that intercepts the nickel hydride to form a nickel alkoxide which, in turn, engages in the transmetalation.
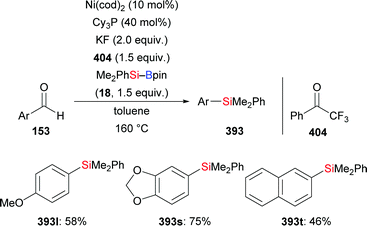 |
| | Scheme 151 Nickel/copper-catalysed decarbonylative silylation of alde-hydes. | |
Recently, reactions of acyl fluorides serving as aryl sources in a decarbonylative process for C(aryl)–Si bond formation were investigated by Nishihara and co-workers (Scheme 152).169 Optimization studies led to a catalytic setup composed of Ni(cod)2, CuF2, KF, and Ph3P. Again, a wide range of aryl- and heteroarylsilanes were synthesised by this strategy, also demonstrating high functional-group tolerance. Of note, the phenolic ester group in acyl fluoride 405h was also compatible, which was reported as a reactive electrophile under a similar nickel/copper catalytic system (cf.Scheme 147).165,166 However, alkenyl and aliphatic acyl fluorides failed to participate in this transformation.
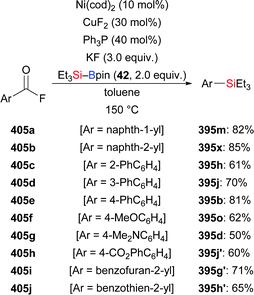 |
| | Scheme 152 Nickel/copper-catalysed decarbonylative silylation of acyl fluorides. | |
Aside from the aforementioned construction of C(aryl)–Si and C(alkenyl)–Si bonds, attempts towards the formation of C(acyl)–Si bonds employing a copper-catalysed carbonylative silylation of unactivated alkyl halides with silylboronic esters have been made by Mankad and co-workers (Scheme 153).170 The major challenge to overcome with this approach was the competitive direct silylation of alkyl halides reported by the Oestreich group.152 The salient feature of the new protocol is the use of IPrCuCl together with 6 atm CO at 60 °C in 1,4-dioxane; IPr as a ligand and NaOPh as the base were crucial for this successful carbonylative silylation, whereas the less sterically hindered IMes ligand and other bases such as NaOtBu or NaOMe gave no product, and the alkylsilane was generated as the major product. This protocol was applicable to primary (such as 407a–c), secondary alkyl iodides (such as 407d–f), and tertiary alkyl bromides (such as 407g–i); tertiary alkyl iodides were prone to facile β-elimination under the basic conditions. A variety of functional groups such as chloroalkyl (as in 407a), terminal alkenes (as in 407b), and cyano (not shown) can be tolerated. Furthermore, apart from Me2PhSi–Bpin (18) as a silicon pronucleophile, the less reactive Et3Si–Bpin (42) also proved to be a good coupling partner at elevated temperature, although the yield eroded with more bulky MePh2Si–Bpin (192) under the standard reaction conditions (not shown).
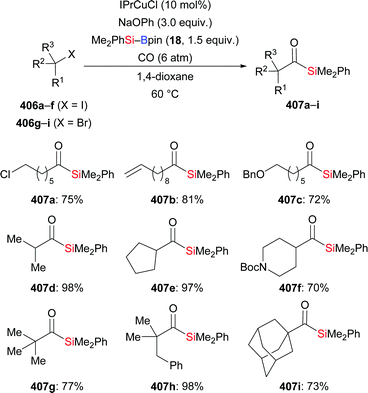 |
| | Scheme 153 Copper-catalysed carbonylative silylation of alkyl bromides and iodides. | |
Radical-trapping experiments utilizing TEMPO and radical-clock experiments supported that a radical mechanism is likely to be operative (not shown). First, transmetalation of IPrCu–OPh, which is formed from IPrCuCl and NaOPh, with Me2PhSi–Bpin (18) may deliver the silylcopper(I) complex CXVII. Subsequent SET between complex CXVII and the alkyl halide affords the alkyl radical R˙ and the silylcopper(II) intermediate CXIX, which is consistent with the radical silylation of alkyl halides developed by Oestreich.152 The resulting alkyl radical R˙ can undergo a carbonylation to arrive at the acyl radical species CXX. The copper(III) intermediate CXXI is then generated by the reaction of silylcopper(II) intermediate CXIX with species CXX. Finally, reductive elimination gives the alkyl-substituted acylsilanes and regenerates the copper(I) catalyst CXVI (Scheme 154, top). This mechanism was further supported by the stoichiometric coupling of IPrCu–SiMe2Ph with alkyl halides under CO (Scheme 154, bottom).
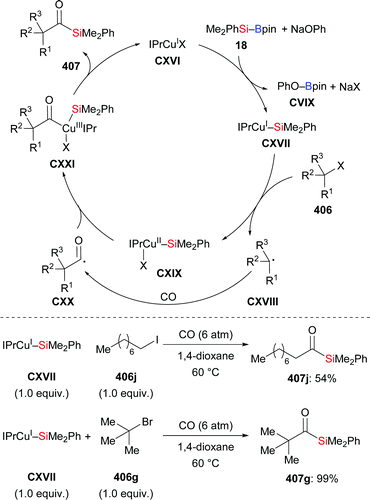 |
| | Scheme 154 Copper-catalysed carbonylative silylation of alkyl bromides and iodides through a radical mechanism. | |
Because the sp2-hybridised boron atom in the Si–B compounds has a higher Lewis acidity than the sp3-hybridised silicon atom with a triorganosilyl moiety, nucleophiles or bases can attack at the boron center to form borate complexes. From this complex, activation of the Si–B bond may deliver silicon nucleophiles, which undergo the aforementioned cross-couplings with a variety of electrophiles to generate tetraorganosilicon compounds. A fascinating transition-metal-free borylation of organic halides with a silyborane has been realised by H. Ito and co-workers.171 Counterintuitive chemoselective borylation (rather than silylation) of aryl bromides occurred in the presence of KOMe and DME to yield the borylated product in high yield with an excellent borylation/silylation ratio (typical B/Si ratios range from 90:10 to 96:4; Scheme 155, top). The use of the less bulky alkoxide base and ethereal solvent was important to ensure this good result. With the present system, the utilization of aryl iodides afforded the arylboronates in yields and B/Si ratios comparable to those for aryl bromides. Conversely, less reactive aryl chlorides gave the desired product in lower yield. The KOMe-mediated system showed good functional group compatibility and facilitated the borylation of electron-rich (e.g.392n), electron-poor (e.g.392p), and even sterically hindered (het)aryl bromides (e.g.392s) at slightly elevated temperature and in a short reaction time. However, aryl bromides containing nitro, ketone or terminal alkyne functional groups resulted in very low yields or complex mixtures as these substrates are prone to react with the in situ-generated silicon nucleophile (not shown). Alkyl boronates are accessible in good NMR yields and B/Si ratios from the corresponding alkyl bromides, although the reaction required higher temperatures than those for the synthesis of aryl boronates (Scheme 155, bottom). In some cases, contamination by the silyl-substituted product and decomposition of the borylated product during the purification process were observed. Thus, a sequential boryl substitution/Suzuki–Miyaura coupling was developed to use these unpurified borylated products, demonstrating the practical utility of this reaction (not shown).
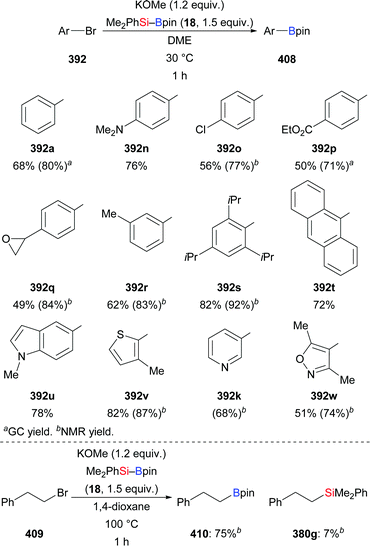 |
| | Scheme 155 KOMe-mediated borylation of aryl and alkyl bromides and iodides. | |
Vinyl halides were less suitable in the presence of KOMe, giving vinylboronates in moderate yields. After extensive screening of bases, high yields and B/Si ratios were restored with NaOEt as the base and by increasing the amount of Me2PhSi–Bpin to 2.0 equivalents relative to the substrate (Scheme 156).171c Again, good functional-group compatibility was observed. Both acyclic (such as 412a–f) and cyclic (such as 412g, h) vinyl bromides and iodides were tolerated. Sterically hindered cyclic 411h containing an n-butyl ester also provided the desired borylated product 412h in good yield. It is noteworthy that the reaction proceeded in a perfectly stereoretentive manner under the optimised setup, thereby making a mechanism involving radicals unlikely and suggesting a pathway through carbanions for this transformation. This was further corroborated by no significant retardation of the reaction rate in the presence of a radical scavenger (not shown) and by an intramolecular retro-Brook rearrangement of 392x (Scheme 157, bottom). After systematic experimental and theoretical studies, the authors proposed a mechanism in which the silylboronic ester/KOMe ate complex CXXII is formed, followed by reversible Si–B bond cleavage (CXXII → CXXIII). Attack of the silicon nucleophile at the halogen atom of the aryl bromide generates the potassium complex CXXIV. Subsequently, this aryl anion would attack the boron electrophile rather than the Me2PhSiBr generated in situ to afford the corresponding boron ate complex CXXV. Finally, the resulting organoborate species CXXV reacts with the Me2PhSiBr, providing the target aryl boronates together with MeOSiMe2Ph and KBr as by-products (Scheme 157, top).171d
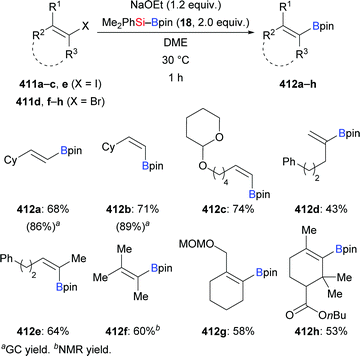 |
| | Scheme 156 NaOEt-mediated borylation of vinyl bromides and iodides. | |
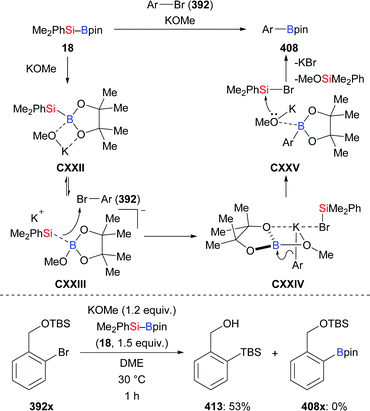 |
| | Scheme 157 Reaction mechanism of boryl substitution of aryl bromides. | |
The base-mediated borylation with silylboronic ester (“BBS strategy”) provides a facile access to various aryl-, heteroaryl-, alkenyl-, and alkylboronates at mild reaction temperature and in a short reaction time. However, the application of this strategy has been limited by the formation of unwanted silyl-substituted products, which are assumed to arise from the nucleophilic attack of the aryl anion at the silicon electrophile in intermediate CXXIV. This disadvantage was recently overcome by the utilization of (Me3Si)3Si–Bhg (3) bearing a bulky silyl group, which improves the B/Si selectivity (up to 99:1) by suppressing the silylation pathway (Scheme 158).11
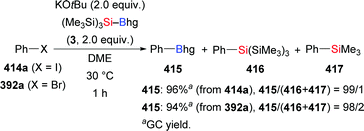 |
| | Scheme 158 KOtBu-mediated borylation of aryl halides using an Si–B reagent with a sterically shielded silyl group. | |
Recently, H. Ito and co-workers were able to further extend the borylation to a silylborane bearing a dimesitylboryl group (Scheme 159).171e Screening of the reaction conditions led to a procedure with silylborane MePh2Si–BMes2 (418, 1.5 equiv.) and NaOtBu (1.2 equiv.) in a solvent system consisting of a 1:1 mixture of 1,4-dioxane and hexane at 50 °C. Again, the reactions of aryl bromides or iodides with 418 afforded the desired aryl dimesitylboranes in moderate to high yields and with B/Si ratios from 67:33 to 96:4. Moreover, a site-selective dimesitylborylation of dibromoarenes was achieved. For example, using 1,4-dibromo-2-methylbenzene (392a′) as the substrate, C4-selective dimesitylborylation was achieved under the standard borylation conditions. For comparison, conventional lithiation–borylation procedures provided the inseparable borylated products with low site selectivity (C1/C4 = 66:34). The utility of this method was demonstrated by the synthesis of a D–π–A aryl dimesitylborane with a non-symmetrical bi(hetero)aryl spacer (not shown).
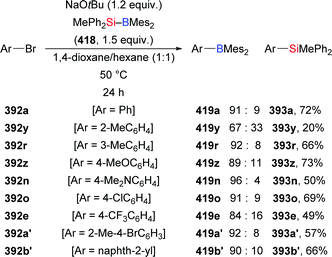 |
| | Scheme 159 NaOtBu-mediated borylation of aryl bromides using MePh2Si–BMes2. Yields are for the major product. | |
9. C–H bond silylation and borylation
9.1. C(sp2)–H bond silylation and borylation
In terms of step and atom economy, catalytic silylation of C–H bonds is an attractive strategy for accessing highly valuable organosilanes.172 Compared to using hydrosilanes and disilanes as silylation agents, C–H silylation using Si–B reagents as a reaction partner is relatively rare. In 2014, Kanai, Kuninobu and co-workers reported the C–H silylation of 2-phenylpyridines with (Et2N)Ph2Si–Bpin (421). Key to success was the treatment of 421 with KF before addition of the palladium catalyst [Pd(MeCN)4](BF4)2 and the strong oxidant K2S2O8. A broad variety of F(Ph)2Si-substituted products containing electron-withdrawing and -donating groups was obtained by this method. Good yield was also achieved with a quinoline directing group, although other directing groups, such as pyrazolyl and N-methylimidazolyl groups, were not effective. Interestingly, X-ray diffraction revealed a Lewis acid–base interaction between the silicon and nitrogen atoms in the F(Ph)2Si-substituted 2-phenylpyridine products which are fluorescent due to the expansion of the π-conjugated system through this interaction (Scheme 160).173
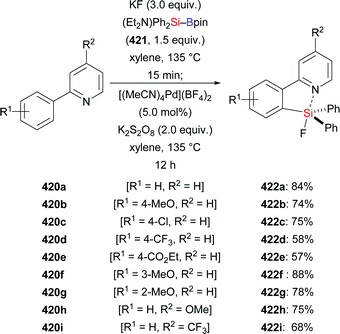 |
| | Scheme 160 Palladium-catalysed C–H silylation of 2-phenylpyridines. | |
Beyond metal-catalysed protocols, Martin and co-workers introduced an elegant C(sp2)–H silylation of azines that relies on the chemoselective activation of the Si–B bond using KHMDS.125 The reaction of azines such as pyridines 424a–g, quinolines 424h, imidazo[1,2-b]pyridazines 424i, imidazo[1,2-a]pyrimidines 424j, pyrazines 424k, and pyridimines 424l with Et3Si–Bpin (42) in the presence of KHMDS in DME yielded the corresponding C–H silylated products in good yields and with synthetically useful site selectivities (Scheme 161, top). Of note, the site selectivity can be modulated by a judicious choice of the solvent employed. For example, C4-silylated pyridine 424m was predominantly observed by using DME as the solvent, while C2-silylated pyridine 424m′ was obtained as major product in 1,4-dioxane. The high site selectivity was rationalised by a solvent-separated ion pair CXXVI and a contact ion pair CXVII, respectively (Scheme 161, bottom). This mild protocol features high site selectivity and broad substrate scope, enabling the late-stage silylation of azine drugs and thus providing a good method to access valuable motifs for medicinal chemistry (not shown).
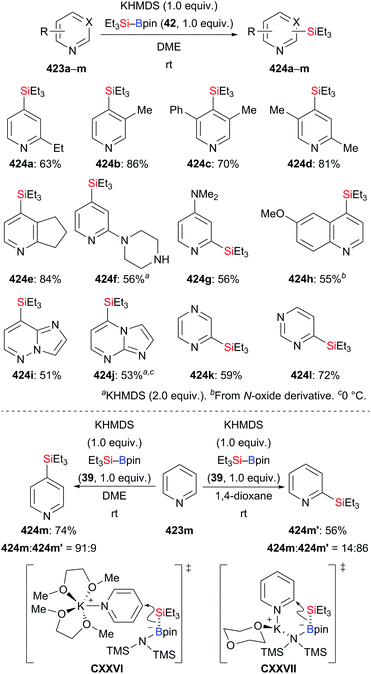 |
| | Scheme 161 Site-selective in the KHMDS-mediated C–H silylation of azines. | |
Recently, as part of their work on dehydrogenative borylation of styrenes, Mankad and co-workers reported a copper-catalysed dehydrogenative silylation of styrenes in moderate to high yields, using ketone additives as sacrificial oxidants. Apart from 1,2-disubstituted vinylsilanes, trisubstituted vinylsilanes that would be unavailable from alkyne hydrosilylation became accessible using a combination of (SIMes)CuOtBu and 6-undecanone in toluene at 110 °C (Scheme 162).174
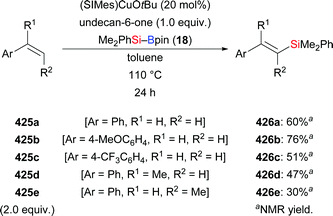 |
| | Scheme 162 Copper-catalysed dehydrogenative silylation of styrenes. | |
In contrast to the C–H bond silylation, pioneering work by the Hartwig group showed that catalytic borylation of arenes with Et3Si–Bpin as borylating reagent allows for the preparation of arylboronates with yields and regioselectivities comparable to those for the iridium-catalysed borylation of arenes with B2pin2.10f Based on these results, H. Ito and co-workers recently realised an iridium-catalysed C(sp2)–H functionalization of substituted benzofuran derivatives with MePh2Si–BMes2 (418), furnishing products with the Mes2B unit selectively attached to 2-position of benzofurans (427a–g → 428a–g, Scheme 163).175 The authors tested numerous ligands in combination with different iridium catalyst precursors. Moderate to good yields were achieved in the presence of [(coe)2IrCl]2, IMes·HCl, and KOtBu in 1,4-dioxane at 120 °C. One drawback of this method is that the dimesitylborylation is limited to substituted benzofurans; furan and benzothiophene substrates gave the corresponding products in poor yields. Competing silylation was seen as the main side reaction in selected cases.
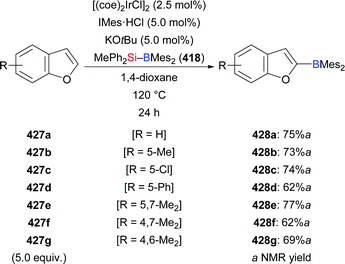 |
| | Scheme 163 Iridium-catalysed borylation of benzofurans with MePh2Si–BMes2. | |
9.2. C(sp3)–H bond borylation
Compared with arenes, borylation of unreactive aliphatic C–H bonds suffers from reactivity and selectivity problems.176 To realise the site-selective functionalization of C(sp3)–H bonds, C–H borylation occurs at activated C(alkyl)–H positions or a directing group is utilised to steer the C–H activation.177
As part of their seminal work, Hartwig and co-workers reported a benzylic C(sp3)–H borylation reaction of toluene derivatives with silylboranes without the need for a directing group in 2008 (Scheme 164, top).10f It is noteworthy that significant differences in the reactivity of methylarenes with Et3Si–Bpin and B2pin2 were observed. For example, the reaction of m-chlorotoluene 394g with Et3Si–Bpin catalysed by iridium ligated by dtbpy gave a 40:60 ratio of products from borylation at the benzylic position and the 5-position of the arene, while no benzylic borylation product was detected with B2pin2 as the borylating reagent. To further improve the chemoselectivity of this benzylic C(sp3)–H borylation, a new iridium precatalyst ligated by the electron-deficient phenanthroline L43 and a silyl ligand was developed by the same laboratory in 2015 (Scheme 164, bottom).178 With the optimised system for benzylic boronate ester products, the substrate scope of the reaction was explored. A wide range of functional groups including dialkylamino group (as in 431d), halogens (as in 431b, j, k, m), ester (as in 431e), or amide (as in 431f) was compatible with the reaction conditions, and generally good yields as well as synthetically useful benzylic versus aryl C–H functionalization ratios (Bn:Ar) were found. Electron-deficient toluene derivatives reacted generally faster than electron-rich derivatives. However, the Bn:Ar selectivity was often lower for reactions of electron-deficient methylarenes than for reactions of electron-rich congeners, suggesting that the rate of C(aryl)–H borylation is more sensitive to the electronic properties of the methylarene than the rate of C(benzyl)–H borylation. This phenomenon prompted Hartwig and co-workers to systematically investigate the mechanism by experimental and quantum-chemical calculations. The resting state of the active catalyst was demonstrated to be a diboryl monosilyl Ir(III) complex ligated by the phenanthroline-type ligand. From the resting state CXXVIII, reversible oxidative addition of a benzylic C–H bond of the methylarene occurs to form the seven-coordinate intermediate CXXIX containing a partial B–H bond. Subsequently, intermediate CXXIX undergoes an irreversible isomerization through transition state CXXX, in which the hydride is exchanged between the boryl ligands, to form intermediate CXXXI. Reductive elimination then forms the benzylic boronate ester products 429 and intermediate CXXXII, Finally, the catalyst is regenerated by transmetalation with Et3Si–Bpin (42) to liberate the Et3SiH byproduct (Scheme 165). Kinetic and computational studies suggest that the isomerization could be the rate-limiting step of the benzylic borylation, while the turnover-limiting step in the borylation of C(aryl)–H bonds is known to be C–H oxidative addition. Complex CXXVIII is more electron-deficient than the commonly used iridium trisboryl complex ligated by dtbpy. Reduction of electron density at the iridium center could significantly reduce the borylation rate of C(aryl)–H bonds while not having any significant effect on the borylation rate of C(benzyl)–H bonds. This difference resulted in the preferential reactivity of C(benzyl)–H bonds over C(aryl)–H bonds.
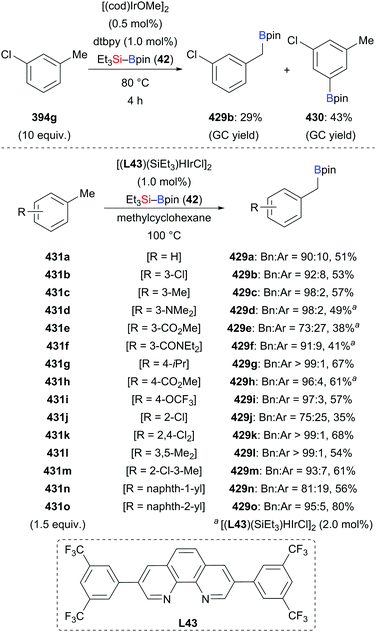 |
| | Scheme 164 Iridium-catalysed chemoselective C(sp3)–H borylation in the benzylic position of toluene derivatives. | |
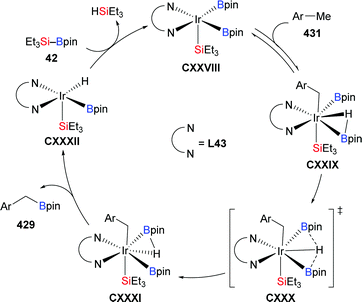 |
| | Scheme 165 Reaction mechanism of the C(sp3)–H borylation in the benzylic position of toluene derivatives. | |
In an extension of the relay-directed iridium-catalysed C–H borylation reaction with hydrosilanes as directing groups, Hartwig and co-authors applied this strategy to unactivated C(alkyl)–H bonds (Scheme 166).179 Again, optimization studies led to an effective catalytic setup composed of iridium precatalyst ligated by the π-extended phenanthroline L44 and a silyl ligand together with isooctane. Notably, the utilization of Et3Si–Bpin (42) rather than B2(pin)2 as the borylating reagent was pivotal to obtain good yields and selectivities. Under the standard protocol with two equivalents of 42, the reactions occurred with high selectivity at primary C(sp3)–H bonds γ to the hydrosilyl group to form primary alkyl bisboronate esters (432a–c → 433a–c, top). In cases of borylations in the absence of any γ primary C(sp3)–H bonds, secondary C(sp3)–H bonds were borylated with both high regioselectivity and diastereoselectivity, affording monoborylated products in good yields (434a–f → 435a–f, bottom). It is noteworthy that a vinyl boronate ester was selectively formed in the borylation reaction of a substrate containing both secondary C(alkyl)–H bonds and a C(vinyl)–H bond γ to the silicon atom (not shown).
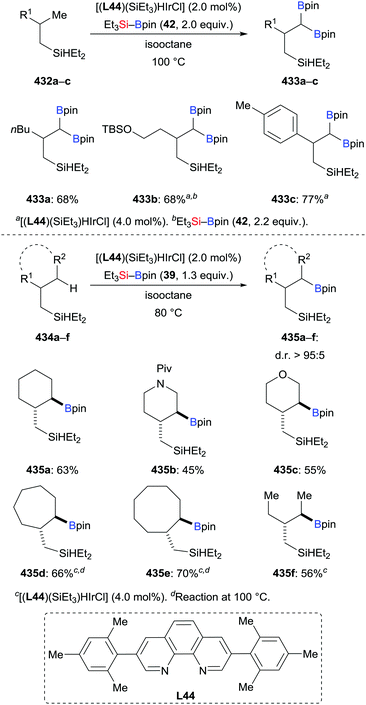 |
| | Scheme 166 Iridium-catalysed borylation of unactivated C(alkyl)–H bonds. | |
Suginome, Ohmura and co-workers developed iridium-catalysed a C(sp3)–H borylation reaction of methylchlorosilanes in 2012.180 With B2(pin)2 as the borylating reagent, methylchlorosilanes underwent C–H borylation at the methyl groups attached to the silicon atom. This method was applicable to Et3Si–Bpin, albeit in decreased yields (not shown).
10. Summary
Since our last comprehensive summary of the field published in 2013,1 synthetic Si–B chemistry has undergone tremendous growth. Established areas, especially copper-catalysed transformations involving transmetalation of the Si–B reagent,181 have fluorished. Numerous electrophile/nucleophile combinations led to the discovery of new silylation reactions with high regio- and stereocontrol. Moreover, completely new applications surfaced and have rapidly developed into highly useful tools. This is particularly true for C–Si bond formation by cross-coupling reactions and C–H silylation and borylation employing Si–B reagents as sources of silicon and boron, respectively. All of this was accomplished in just seven years but where does Si–B chemistry go?
One possible answer was given by H. Ito and co-workers when this Review was in preparation. These authors reported new methods for preparation of (1) trialkylsilylboronic esters that bear bulky alkyl groups and, most significantly, are decorated with functional groups as well as (2) dialkyl(aryl)silylboronic esters which have been difficult to synthesize by conventional methods.182 This was achieved by rhodium- or platinum-catalysed direct borylation of hydrosilanes with B2pin2 (436a–i → 437a–i, Scheme 167, top). Notably, these new Si–B reagents can be used as silicon nucleophiles in a diverse set of representative silylation reactions of alkyl and aryl electrophiles such as Oestreich's copper-catalysed radical cross-coupling,152 Martin's nickel-catalysed cross-coupling,147b and He's palladium-catalysed cross-coupling;158 Hoveyda's NHC-catalysed conjugate silylation was also successful100,183 (Scheme 167, bottom). This promising protocol significantly expands the boundaries of Si–B chemistry and is as such a major breakthrough. It opens the door to the synthesis of new silicon-containing bioactive molecules and organic materials with distinct properties, the future of silicon chemistry!
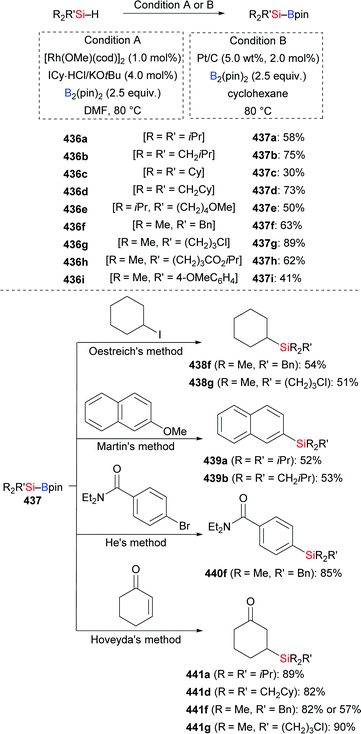 |
| | Scheme 167 Synthesis of trialkyl- and dialkyl(aryl)silylboronic esters and their utility in C–Si bond-forming reactions. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J.-J. F. is grateful to the Humboldt Foundation for a postdoctoral fellowship (2017–2020), and W. M. and L. Z. thank the China Scholarship Council for a predoctoral fellowships (both 2017–2021). M. O. is indebted to the Einstein Foundation Berlin for an endowed professorship.
Notes and references
- M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed.
- For reviews published before 2013, see:
(a)
M. Suginome and T. Ohmura, Transition Metal-Catalyzed Element-Boryl Additions to Unsaturated Organic Compounds, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011, vol. 1, pp. 171–212 Search PubMed;
(b) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29–49 CrossRef CAS;
(c) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320–2354 CrossRef CAS PubMed;
(d) M. Suginome and Y. Ito, J. Organomet. Chem., 2003, 680, 43–50 CrossRef CAS;
(e) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed.
-
K. Kubota and H. Ito, Catalytic Generation of Silicon Nucleophiles, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 1–31 Search PubMed.
- J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter, S. R. Harruff and R. Van Hoveln, ACS Catal., 2019, 9, 8961–8979 CrossRef CAS.
-
L. B. Delvos and M. Oestreich, Silylboron Reagents, in Science of Synthesis Knowledge Update 2017/1, ed. M. Oestreich, Thieme, Stuttgart, 2017, pp. 65–176 Search PubMed.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- Y. Tsuji and T. Fujihara, Chem. Rec., 2016, 16, 2294–2313 CrossRef CAS PubMed.
- A. Hensel and M. Oestreich, Top. Organomet. Chem., 2016, 58, 135–167 CrossRef CAS.
-
M. Sawamura and H. Ito, Carbon–Boron and Carbon–Silicon Bond Formation, in Copper-Catalysed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 2014, pp. 157–177 Search PubMed.
-
(a) H. Nöth and G. Höllerer, Chem. Ber., 1966, 99, 2197–2205 CrossRef;
(b) S.-Y. Onozawa, Y. Hatanaka and M. Tanaka, Chem. Commun., 1997, 1229–1230 RSC;
(c) T. Habereder and H. Nöth, Appl. Organomet. Chem., 2003, 17, 525–538 CrossRef CAS;
(d) M. Suginome, T. Matsuda and Y. Ito, Organometallics, 2000, 19, 4647–4649 CrossRef CAS;
(e) T. Ohmura, K. Masuda, H. Furukawa and M. Suginome, Organometallics, 2007, 26, 1291–1294 CrossRef CAS;
(f) T. A. Boebel and J. F. Hartwig, Organometallics, 2008, 27, 6013–6019 CrossRef CAS.
- E. Yamamoto, R. Shishido, T. Seki and H. Ito, Organometallics, 2017, 36, 3019–3022 CrossRef CAS.
- Z. Liu and C. Cui, J. Organomet. Chem., 2020, 906, 121041 CrossRef CAS.
- A. Tsurusaki, K. Yoshida and S. Kyushin, Dalton Trans., 2017, 46, 8705–8708 RSC.
- J. Moon, H. Baek and J. Kim, J. Phys. Chem. A, 2017, 121, 6531–6537 CrossRef CAS PubMed.
- J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2016, 55, 4813–4823 CrossRef CAS PubMed.
- J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2017, 56, 6671–6680 CrossRef CAS PubMed.
- C. Kleeberg and C. Borner, Eur. J. Inorg. Chem., 2013, 2799–2806 CrossRef CAS.
- C. Kleeberg, Dalton Trans., 2013, 42, 8276–8287 RSC.
- For reviews, see:
(a) M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 336, 54–77 CrossRef CAS;
(b) M. Iwasaki and Y. Nishihara, Chem. Rec., 2016, 16, 2031–2045 CrossRef CAS PubMed.
- T. Ohmura, H. Nishiura and M. Suginome, Organometallics, 2017, 36, 4298–4304 CrossRef CAS.
- T. Ohmura, K. Oshima, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2010, 132, 12194–12196 CrossRef CAS PubMed.
- C. Gryparis and M. Stratakis, Org. Lett., 2014, 16, 1430–1433 CrossRef CAS PubMed.
- A. Khan, A. M. Asiri, S. A. Kosa, H. Garcia and A. Grirrane, J. Catal., 2015, 329, 401–412 CrossRef CAS.
- Y. Nagashima, D. Yukimori, C. Wang and M. Uchiyama, Angew. Chem., Int. Ed., 2018, 57, 8053–8057 CrossRef CAS PubMed.
- M. Zhao, C.-C. Shan, Z.-L. Wang, C. Yang, Y. Fu and Y.-H. Xu, Org. Lett., 2019, 21, 6016–6020 CrossRef CAS PubMed.
- Y. Morimasa, K. Kabasawa, T. Ohmura and M. Suginome, Asian J. Org. Chem., 2019, 8, 1092–1096 CrossRef CAS.
- Y. Gu, Y. Duan, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 2061–2065 CrossRef CAS PubMed.
-
(a) M. B. Ansell, J. Spencer and O. Navarro, ACS Catal., 2016, 6, 2192–2196 CrossRef CAS;
(b) M. B. Ansell, S. K. Furfari, F. G. N. Cloke, S. M. Roe, J. Spencer and O. Navarro, Organometallics, 2018, 37, 1214–1218 CrossRef CAS.
- M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777–2778 RSC.
-
(a) J. Jiao, K. Nakajima and Y. Nishihara, Org. Lett., 2013, 15, 3294–3297 CrossRef CAS;
(b) J. Jiao, K. Hyodo, H. Hu, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 285–295 CrossRef CAS PubMed.
- T. Miura, Y. Nishida and M. Murakami, J. Am. Chem. Soc., 2014, 136, 6223–6226 CrossRef CAS PubMed.
- N. Saito, K. Saito, H. Sato and Y. Sato, Adv. Synth. Catal., 2013, 355, 853–856 CrossRef CAS.
- F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204–3214 CrossRef CAS PubMed.
-
(a) H. Zhou and Y.-B. Wang, ChemCatChem, 2014, 6, 2512–2516 CrossRef CAS;
(b) H. Zhou, Q.-Y. Zhang and X.-B. Lu, RSC Adv., 2016, 6, 44995–45000 RSC.
- C. K. Hazra, C. Fopp and M. Oestreich, Chem. – Asian J., 2014, 9, 3005–3010 CrossRef CAS PubMed.
- Q.-Q. Xuan, C.-L. Ren, L. Liu, D. Wang and C. J. Li, Org. Biomol. Chem., 2015, 13, 5871–5874 RSC.
- A. García-Rubia, J. A. Romero-Revilla, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2015, 137, 6857–6865 CrossRef PubMed.
- S. Vercruysse, K. Jouvin, O. Riant and G. Evano, Synthesis, 2016, 48, 3373–3381 CrossRef CAS.
- H. Yoshida, Y. Hayashi, Y. Ito and K. Takaki, Chem. Commun., 2015, 51, 9440–9442 RSC.
-
(a) B. L. Chenard, E. D. Laganis, F. Davidson and T. V. RajanBabu, J. Org. Chem., 1985, 50, 3666–3667 CrossRef CAS;
(b) T. N. Mitchell, H. Killing, R. Dicke and R. Wickenkamp, J. Chem. Soc., Chem. Commun., 1985, 354–355 RSC;
(c) T. N. Mitchell, R. Wickenkamp, A. Amamria, R. Dicke and U. Schneider, J. Org. Chem., 1987, 52, 4868–4874 CrossRef CAS;
(d) I. Hemeon and R. D. Singer, Chem. Commun., 2002, 1884–1885 RSC;
(e) M. Murakami, T. Matsuda, K. Itami, S. Ashida and M. Terayama, Synthesis, 2004, 1522–1526 CrossRef CAS;
(f) T. E. Nielsen, S. Le Quement and D. Tanner, Synthesis, 2004, 1381–1390 CAS.
- T. Iwamoto, T. Nishikori, N. Nakagawa, H. Takaya and M. Nakamura, Angew. Chem., Int. Ed., 2017, 56, 13298–13301 CrossRef CAS PubMed.
- T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef CAS PubMed.
-
T. Hiyama, Y. Minami and A. Mori, Transition-Metal-Catalyzed Cross-coupling of Organosilicon Compounds, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 271–332 Search PubMed.
- R. Shintani, H. Kurata and K. Nozaki, J. Org. Chem., 2016, 81, 3065–3069 CrossRef CAS PubMed.
- M. Suginome, H. Nakamura and Y. Ito, Angew. Chem., Int. Ed. Engl., 1997, 36, 2516–2518 CrossRef CAS.
- T. Kamei, S. Nishino and T. Shimada, Tetrahedron Lett., 2018, 59, 2896–2899 CrossRef CAS.
- H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef CAS PubMed.
- D.-H. Tan, E. Lin, W.-W. Ji, Y.-F. Zeng, W.-X. Fan, Q. Li, H. Gao and H. Wang, Adv. Synth. Catal., 2018, 360, 1032 CrossRef CAS.
- P. Gao, G. Wang, L. Xi, M. Wang, S. Li and Z. Shi, Chin. J. Chem., 2019, 37, 1009–1014 CrossRef CAS.
- P. H. S. Paioti, J. del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917–19934 CrossRef CAS PubMed.
- Z. Liu, J. Chen, H.-X. Lu, X. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068–17073 CrossRef CAS PubMed.
- R. Liang, R. Chen, C. Zhong, J. Zhu, Z. Cao and Y. Jia, Org. Lett., 2020, 22, 3215–3218 CrossRef CAS PubMed.
- Y. Matsuda, Y. Tsuji and T. Fujihara, Chem. Commun., 2020, 56, 4648–4651 RSC.
- For a recent summary, see: C. Moberg, Synthesis, 2020, 52, 3129–3139 CrossRef CAS.
- Q. Zhang, Q.-J. Liang, J.-L. Xu, Y.-H. Xu and T. P. Loh, Chem. Commun., 2018, 54, 2357–2360 RSC.
-
(a) H. Zhou and C. Moberg, J. Am. Chem. Soc., 2012, 134, 15992–15999 CrossRef CAS PubMed;
(b) E. Li, H. Zhou, V. Östlund, R. Hertzberg and C. Moberg, New J. Chem., 2016, 40, 6340–6346 RSC.
- P. P. Choudhury, C. S. Junker, R. R. Pidaparthi and M. E. Welker, J. Organomet. Chem., 2014, 754, 88–93 CrossRef CAS.
- C. Yang, Z.-L. Liu, D.-T. Dai, Q. Li, W.-W. Ma, M. Zhao and Y.-H. Xu, Org. Lett., 2020, 22, 1360–1367 CrossRef CAS PubMed.
- Y.-C. Xiao and C. Moberg, Org. Lett., 2016, 18, 308–311 CrossRef CAS PubMed.
- M. Kidonakis and M. Stratakis, ACS Catal., 2018, 8, 1227–1230 CrossRef CAS.
-
(a) J. Rae, Y. C. Hu and D. J. Procter, Chem. – Eur. J., 2014, 20, 13143–13145 CrossRef CAS PubMed;
(b) J. Rae, K. Yeung, J. J. W. McDouall and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 1102–1107 CrossRef CAS PubMed.
- Y. Tani, T. Yamaguchi, T. Fujihara, J. Terao and Y. Tsuji, Chem. Lett., 2015, 44, 271–273 CrossRef CAS.
- T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS PubMed.
-
(a) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef CAS PubMed;
(b) R. Yuan, R. Hu and G. Fu, Chem. – Asian J., 2016, 11, 2201–2209 CrossRef CAS PubMed.
-
(a) T. N. Mitchell and U. Schneider, J. Organomet. Chem., 1991, 407, 319–327 CrossRef CAS;
(b) A. G. M. Barrett and P. W. H. Wan, J. Org. Chem., 1996, 61, 8667–8670 CrossRef CAS;
(c) S. Shin and T. V. RajanBabu, J. Am. Chem. Soc., 2001, 123, 8416–8417 CrossRef CAS PubMed;
(d) M. Jeganmohan, M. Shanmugasundaram, K.-J. Chang and C.-H. Cheng, Chem. Commun., 2002, 2552–2553 RSC.
- Z.-T. He, X.-Q. Tang, L.-B. Xie, M. Cheng, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815–14818 CrossRef CAS PubMed.
- C.-Y. He, L.-B. Xie, R. Ding, P. Tian and G.-Q. Lin, Tetrahedron, 2019, 75, 1682–1688 CrossRef CAS.
- V. Cirriez, C. Rasson, T. Hermant, J. Petrignet, J. Díaz Álvarez, K. Robeyns and O. Riant, Angew. Chem., Int. Ed., 2013, 52, 1785–1788 CrossRef CAS PubMed.
-
(a) P. An, Y. Huo, Z. Chen, C. Song and Y. Ma, Org. Biomol. Chem., 2017, 15, 3202–3206 RSC;
(b) Y. Huo, P. Shen, W. Duan, Z. Chen, C. Songa and Y. Ma, Chin. Chem. Lett., 2018, 29, 1359–1362 CrossRef CAS.
- C. Kleeberg, E. Feldmann, E. Hartmann, D. J. Vyas and M. Oestreich, Chem. – Eur. J., 2011, 17, 13538–13543 CrossRef CAS PubMed.
-
(a) M. Takeda, K. Yabushita, S. Yasuda and H. Ohmiya, Chem. Commun., 2018, 54, 6776–6779 RSC;
(b) K. Yabushita, A. Yuasa, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 113–117 CrossRef CAS PubMed.
- A. Yuasa, K. Nagao and H. Ohmiya, Beilstein J. Org. Chem., 2020, 16, 185–189 CrossRef CAS PubMed.
- M. Takeda, A. Mitsui, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3664–3669 CrossRef CAS PubMed.
- A. Mitsui, K. Nagao and H. Ohmiya, Org. Lett., 2020, 22, 800–803 CrossRef CAS PubMed.
- K. Masada, S. Kusumoto and K. Nozaki, Org. Lett., 2020, 22, 4922–4926 CrossRef CAS PubMed.
- M. Kidonakis, A. Mullaj and M. Stratakis, J. Org. Chem., 2018, 83, 15553–15557 CrossRef CAS PubMed.
- V. Kotzabasaki, M. Kidonakis, E. Vassilikogiannaki and M. Stratakis, Eur. J. Org. Chem., 2019, 7233–7236 CrossRef CAS.
-
(a) A. Hensel, K. Nagura, L. B. Delvos and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 4964–4967 CrossRef CAS PubMed;
(b) L. B. Delvos, A. Hensel and M. Oestreich, Synthesis, 2014, 46, 2957–2964 CrossRef.
-
(a) J. K. Park, H. H. Lackey, M. D. Rexford, K. Kovnir, M. Shatruk and D. T. McQuade, Org. Lett., 2010, 12, 5008–5011 CrossRef CAS PubMed;
(b) J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410–2413 CrossRef CAS PubMed;
(c) J. K. Park and D. T. McQuade, Angew. Chem., Int. Ed., 2012, 51, 2717–2721 CrossRef CAS PubMed;
(d) J. K. Park and D. T. McQuade, Synthesis, 2012, 44, 1485–1490 CrossRef CAS.
- C. Zhao, C. Jiang, J. Wang, C. Wu, Q.-W. Zhang and W. He, Asian J. Org. Chem., 2014, 3, 851–855 CrossRef CAS.
- T. Mita, M. Sugawara, K. Saito and Y. Sato, Org. Lett., 2014, 16, 3028–3031 CrossRef CAS PubMed.
-
(a) Z. Chen, Y. Huo, P. An, X. Wang, C. Song and Y. Ma, Org. Chem. Front., 2016, 3, 1725–1737 RSC;
(b) X. Wang, Z. Chen, W. Duan, C. Song and Y. Ma, Tetrahedron: Asymmetry, 2017, 28, 783–790 CrossRef CAS.
-
(a) T. Mita, J. Chen, M. Sugawara and Y. Sato, Org. Lett., 2012, 14, 6202–6205 CrossRef CAS PubMed ; for a review, see: ;
(b) T. Mita and Y. Sato, J. Synth. Org. Chem., Jpn., 2013, 71, 1163–1171 CrossRef CAS.
- J.-J. Feng and M. Oestreich, Org. Lett., 2018, 20, 4273–4276 CrossRef CAS PubMed.
- V. Cirriez, C. Rasson and O. Riant, Adv. Synth. Catal., 2013, 355, 3137–3140 CrossRef CAS.
- K. Tatsumi, S. Tanabe, Y. Tsuji and T. Fujihara, Org. Lett., 2019, 21, 10130–10133 CrossRef CAS PubMed.
- M. B. Ansell, G. E. Kostakis, H. Braunschweig, O. Navarro and J. Spencer, Adv. Synth. Catal., 2016, 358, 3765–3769 CrossRef CAS PubMed.
-
(a) T. Vergote, F. Nahra, A. Welle, M. Luhmer, J. Wouters, N. Riant, O. Mager and T. Leyssens, Chem. – Eur. J., 2012, 18, 793–798 CrossRef CAS PubMed;
(b) J. Plotzitzka and C. Kleeberg, Organometallics, 2014, 33, 6915–6926 CrossRef CAS.
- F. Bartoccini, S. Bartolucci, S. Lucarini and G. Piersanti, Eur. J. Org. Chem., 2015, 3352–3360 CrossRef CAS.
- Y. Zhang, M. Tong, Q. Gao, P. Zhang and S. Xu, Tetrahedron Lett., 2019, 60, 1210–1212 CrossRef CAS.
- T. Kitanosono, L. Zhu, C. Liu, P. Xu and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 15422–15425 CrossRef CAS PubMed.
- V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2014, 16, 476–479 CrossRef CAS PubMed.
- Y. Shi, Q. Gao and S. Xu, J. Org. Chem., 2018, 83, 14758–14767 CrossRef CAS PubMed.
-
(a) Q.-Q. Xuan, N.-J. Zhong, C.-L. Ren, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Org. Chem., 2013, 78, 11076–11081 CrossRef CAS PubMed;
(b) C. Rasson, A. Stouse, A. Boreux, V. Cirriez and O. Riant, Chem. – Eur. J., 2018, 24, 9234–9237 CrossRef CAS PubMed.
- V. Pace, J. P. Rae, H. Y. Harb and D. J. Procter, Chem. Commun., 2013, 49, 5150–5152 RSC.
- B.-C. Da, Q.-J. Liang, Y.-C. Luo, T. Ahmad, Y.-H. Xu and T.-P. Loh, ACS Catal., 2018, 8, 6239–6245 CrossRef CAS.
- W. Mao, W. Xue, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 10723–10726 CrossRef CAS PubMed.
- Y.-L. Zeng, B. Chen, Y.-T. Wang, C.-Y. He, Z.-Y. Mu, J.-Y. Du, L. He, W.-D. Chu and Q.-Z. Liu, Chem. Commun., 2020, 56, 1693–1696 RSC.
- C. R. Jiang, C.-L. Zhao, H.-F. Guo and W. He, Chem. Commun., 2016, 52, 7862–7865 RSC.
-
(a) J. M. O’Brien and A. H. Hoveda, J. Am. Chem. Soc., 2011, 133, 7712–7715 CrossRef PubMed;
(b) H. Wu, J. M. Garcia, F. Haeffner, S. Radomkit, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 10585–10602 CrossRef CAS PubMed.
- K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 1304–1307 CrossRef CAS PubMed.
- R. Fritzemeier and W. L. Santos, Chem. – Eur. J., 2017, 23, 15534–15537 CrossRef CAS PubMed.
-
(a) J. A. Calderone and W. L. Santos, Angew. Chem., Int. Ed., 2014, 53, 4154–4158 CrossRef CAS PubMed;
(b) Y.-H. Xu, L.-H. Wu, J. Wang and T.-P. Loh, Chem. Commun., 2014, 50, 7195–7197 RSC.
- R. T. H. Linstadt, C. A. Peterson, D. J. Lippincott, C. I. Jette and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 4159–4163 CrossRef CAS PubMed.
- S. Vercruysse, L. Cornelissen, F. Nahra, L. Collard and O. Riant, Chem. – Eur. J., 2014, 20, 1834–1838 CrossRef CAS PubMed.
- T. Ahmad, Q. Li, S.-Q. Qiu, J.-L. Xu, Y.-H. Xu and T.-P. Loh, Org. Biomol. Chem., 2019, 17, 6122–6126 RSC.
- K.-s. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823–7826 CrossRef CAS PubMed.
- A. López, A. Parra, C. Jarava-Barrera and M. Tortosa, Chem. Commun., 2015, 51, 17684–17687 RSC.
-
(a) M. Wang, Z.-L. Liu, X. Zhang, P.-P. Tian, Y.-H. Xu and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 14830–14833 CrossRef CAS PubMed;
(b) W. Mao and M. Oestreich, Org. Lett., 2020, 22, 8096–8100 CrossRef CAS PubMed.
- F.-F. Meng, J.-H. Xie, Y.-H. Xu and T.-P. Loh, ACS Catal., 2018, 8, 5306–5312 CrossRef CAS.
- F.-F. Meng, Q.-Y. Xue, B. Jiang, M. Zhao, J.-H. Xie, Y.-H. Xu and T.-P. Loh, Org. Lett., 2019, 21, 2932–2936 CrossRef CAS PubMed.
- S. Pashikanti, J. A. Calderone, M. K. Nguyen, C. D. Sibley and W. L. Santos, Org. Lett., 2016, 18, 2443–2446 CrossRef CAS PubMed.
- D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513–8515 CrossRef CAS PubMed.
- L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 4650–4653 CrossRef CAS PubMed.
- C. K. Hazra, E. Irran and M. Oestreich, Eur. J. Org. Chem., 2013, 4903–4908 CrossRef CAS.
- L. B. Delvos and M. Oestreich, Synthesis, 2015, 47, 924–933 CrossRef CAS.
- M. Takeda, R. Shintani and T. Hayashi, J. Org. Chem., 2013, 78, 5007–5017 CrossRef CAS PubMed.
- Y. Gan, W. Xu and Y. Liu, Org. Lett., 2019, 21, 9652–9657 CrossRef CAS PubMed.
- R. Shintani, R. Fujie, M. Takeda and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 6546–6549 CrossRef CAS PubMed.
- X.-H. Chang, Z.-L. Liu, Y.-C. Luo, C. Yang, X.-W. Liu, B.-C. Da, J.-J. Li, T. Ahmad, T.-P. Loh and Y.-H. Xu, Chem. Commun., 2017, 53, 9344–9347 RSC.
- K. Guo and A. W. Kleij, Org. Lett., 2020, 22, 3942–3945 CrossRef CAS PubMed.
- Z.-L. Liu, C. Yang, Q.-Y. Xue, M. Zhao, C.-C. Shan, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2019, 58, 16538–16542 CrossRef CAS.
- A. M. Caporusso, A. Zampieri, L. A. Aronica and D. Banti, J. Org. Chem., 2006, 71, 1902–1910 CrossRef CAS.
- S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 7720–7738 CrossRef CAS PubMed.
- Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef CAS PubMed.
-
(a) M. Suginome, T. Matsuda and Y. Ito, J. Am. Chem. Soc., 2000, 122, 11015–11016 CrossRef CAS;
(b) T. Ohmura, H. Taniguchi, Y. Kondo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 3518–3519 CrossRef CAS PubMed;
(c) Y. Akai, T. Yamamoto, Y. Nagata, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 11092–11095 CrossRef CAS PubMed.
- T. Ohmura, H. Taniguchi and M. Suginome, ACS Catal., 2015, 5, 3074–3077 CrossRef CAS.
- T. Yamamoto, R. Murakami, S. Komatsu and M. Suginome, J. Am. Chem. Soc., 2018, 140, 3867–3870 CrossRef CAS PubMed.
- Y. Nagata, R. Takeda and M. Suginome, ACS Cent. Sci., 2019, 5, 1235–1240 CrossRef CAS PubMed.
- J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 8800–8804 CrossRef CAS PubMed.
- X. Zhao, S. Xu, J. He, Y. Zhou and S. Cao, Org. Chem. Front., 2019, 6, 2539–2543 RSC.
- L. Zhang and M. Oestreich, Chem. – Eur. J., 2019, 25, 14304–14307 CrossRef CAS PubMed.
- M. Cui and M. Oestreich, Org. Lett., 2020, 22, 3684–3687 CrossRef CAS PubMed.
- M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed.
- H. Lee, J. T. Han and J. Yun, ACS Catal., 2016, 6, 6487–6490 CrossRef CAS.
- H. Yi and M. Oestreich, Chem. – Eur. J., 2019, 25, 6505–6507 CrossRef CAS PubMed.
- E. Vasilikogiannaki, A. Louka and M. Stratakis, Organometallics, 2016, 35, 3895–3902 CrossRef CAS.
- T. Ohmura, K. Masuda and M. Suginome, J. Am. Chem. Soc., 2008, 130, 1526–1527 CrossRef CAS PubMed.
- T. Ohmura, I. Sasaki and M. Suginome, Org. Lett., 2019, 21, 1649–1653 CrossRef CAS PubMed.
- I. Sasaki, T. Ohmura and M. Suginome, Org. Lett., 2020, 22, 2961–2966 CrossRef PubMed.
- H. Li, X. Shangguan, Z. Zhang, S. Huang, Y. Zhang and J. Wang, Org. Lett., 2014, 16, 448–451 CrossRef CAS PubMed.
- H. Zhao, M. Tong, H. Wang and S. Xu, Org. Biomol. Chem., 2017, 15, 3418–3422 RSC.
- L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef CAS PubMed.
- A. Millán, P. D. Grigol Martinez and V. K. Aggarwal, Chem. – Eur. J., 2018, 24, 730–735 CrossRef PubMed.
- Recent reviews of hydrosilylation reactions:
(a) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC;
(b) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16–24 CrossRef.
-
(a) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed;
(b) C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed.
- Z.-D. Huang, R. Ding, P. Wang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2016, 52, 5609–5612 RSC.
- J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577–1580 CrossRef CAS PubMed.
- J. Scharfbier and M. Oestreich, Synlett, 2016, 27, 1274–1276 CrossRef CAS.
- J. Scharfbier, H. Hazrati and M. Oestreich, Org. Lett., 2017, 19, 6562–6565 CrossRef CAS PubMed.
- W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222–14225 CrossRef CAS PubMed.
- W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649–11652 CrossRef CAS PubMed.
- T. Seihara, S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Org. Lett., 2019, 21, 2477–2481 CrossRef CAS PubMed.
- H. Hazrati and M. Oestreich, Org. Lett., 2018, 20, 5367–5369 CrossRef CAS PubMed.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- X.-W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064–2068 CrossRef CAS PubMed.
- H. Guo, X. Chen, C. Zhao and W. He, Chem. Commun., 2015, 51, 17410–17412 RSC.
- J. Jia, X. Zeng, Z. Liu, L. Zhao, C.-Y. He, X.-F. Li and Z. Feng, Org. Lett., 2020, 22, 2816–2821 CrossRef CAS PubMed.
- K. Kojima, Y. Nagashima, C. Wang and M. Uchiyama, ChemPlusChem, 2019, 84, 277–280 CrossRef CAS PubMed.
- For selected reviews:
(a) H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen and C.-J. Li, ACS Catal., 2017, 7, 510–519 CrossRef CAS;
(b) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed;
(c) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed;
(d) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed;
(e) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC;
(f) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed;
(g) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed.
- R. J. Somerville, L. V. A. Hale, E. Gómez-Bengoa, J. Burés and R. Martin, J. Am. Chem. Soc., 2018, 140, 8771–8780 CrossRef CAS PubMed; R. J. Somerville, L. V. A. Hale, E. Gómez-Bengoa, J. Burés and R. Martin, Correction to “Intermediacy of Ni−Ni Species in sp2 C−O Bond Cleavage of Aryl Esters: Relevance in Catalytic C−Si Bond Formation”, J. Am. Chem. Soc., 2019, 141, 20565 CrossRef PubMed.
- A. J. Fugard, A. S. K. Lahdenperä, J. S. J. Tan, A. Mekareeya, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2019, 58, 2795–2798 CrossRef CAS PubMed.
-
(a) B. Wang, Q. Zhang, J. Jiang, H. Yu and Y. Fu, Chem. – Eur. J., 2017, 23, 17249–17256 CrossRef CAS PubMed;
(b) P. Jain, S. Pal and V. Avasare, Organometallics, 2018, 37, 1141–1149 CrossRef CAS.
- L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS PubMed.
- X. Pu, J. Hu, Y. Zhao and Z. Shi, ACS Catal., 2016, 6, 6692–6698 CrossRef CAS.
- S.-C. Lee, L. Guo, H. Yue, H.-H. Li and M. Rueping, Synlett, 2017, 28, 2594–2598 CrossRef CAS.
- W. Srimontree, L. Guo and M. Rueping, Chem. – Eur. J., 2020, 26, 423–427 CrossRef CAS PubMed.
- X. Wang, Z. Wang and Y. Nishihara, Chem. Commun., 2019, 55, 10507–10510 RSC.
- L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2020, 142, 80–84 CrossRef CAS PubMed.
-
(a) E. Yamamoto, K. Izumi, Y. Horita and H. Ito, J. Am. Chem. Soc., 2012, 134, 19997–20000 CrossRef CAS PubMed;
(b) E. Yamamoto, K. Izumi, Y. Horita, S. Ukigai and H. Ito, Top. Catal., 2014, 57, 940–945 CrossRef CAS;
(c) E. Yamamoto, S. Ukigai and H. Ito, Chem. Sci., 2015, 6, 2943–2951 RSC;
(d) R. Uematsu, E. Yamamoto, S. Maeda, H. Ito and T. Taketsugu, J. Am. Chem. Soc., 2015, 137, 4090–4099 CrossRef CAS PubMed;
(e) E. Yamamoto, K. Izumi, R. Shishido, T. Seki, N. Tokodai and H. Ito, Chem. – Eur. J., 2016, 22, 17547–17551 CrossRef CAS PubMed ; for a review, see: ;
(f) E. Yamamoto, S. Maeda, T. Taketsugu and H. Ito, Synlett, 2017, 28, 1258–1267 CrossRef CAS.
- Recent reviews of C–H Bond silylation:
(a) S. C. Richter and M. Oestreich, Trends Chem., 2020, 2, 13–27 CrossRef CAS;
(b) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed;
(c) Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 47, 3645–3668 CrossRef CAS.
- Q. Xiao, X. Meng, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2014, 53, 3168–3172 CrossRef CAS PubMed.
- T. J. Mazzacano and N. P. Mankad, ACS Catal., 2017, 7, 146–149 CrossRef CAS.
- R. Shishido, I. Sasaki, T. Seki, T. Ishiyama and H. Ito, Chem. – Eur. J., 2019, 25, 12924–12928 CrossRef CAS PubMed.
-
(a) G. Wang, L. Liu, H. Wang, Y.-S. Ding, J. Zhou, S. Mao and P. Li, J. Am. Chem. Soc., 2017, 139, 91–94 CrossRef CAS PubMed;
(b) X. Zou, H. Zhao, Y. Li, Q. Gao, Z. Ke and S. Xu, J. Am. Chem. Soc., 2019, 141, 5334–5342 CrossRef CAS PubMed;
(c) Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599–10604 CrossRef CAS PubMed.
- Recent reviews of C–H bond borylation:
(a) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS;
(b) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC;
(c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC;
(d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633–8643 CrossRef CAS PubMed.
- M. A. Larsen, S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 762–765 CrossRef CAS PubMed.
- T. Ohmura, T. Torigoe and M. Suginome, J. Am. Chem. Soc., 2012, 134, 17416–17419 CrossRef CAS PubMed.
- W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS PubMed.
- R. Shishido, M. Uesugi, R. Takahashi, T. Mita, T. Ishiyama, K. Kubota and H. Ito, J. Am. Chem. Soc., 2020, 142, 14125–14133 CrossRef CAS PubMed.
- K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898–2900 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Wenbin
Mao†
*ab,
Wenbin
Mao†