
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic routes to anti-viral agents and their synthetic intermediates
Sjoerd
Slagman
 * and
Wolf-Dieter
Fessner
*
* and
Wolf-Dieter
Fessner
*
Institut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Germany. E-mail: sjoerdslagman@gmail.com; fessner@tu-darmstadt.de
First published on 16th December 2020
Abstract
With recent outbreaks of COVID-19 and Ebola, health and healthcare have once more shown to be heavily burdened by the lack of generally effective anti-viral therapies. Initial scientific ventures towards finding anti-viral agents are soon to be followed by challenges regarding their mass production. Biocatalysis offers mild, highly selective, and environmentally benign synthetic strategies for the production of pharmaceuticals in a sustainable fashion. Here we summarise biocatalytic methods that have been applied to the production of FDA-approved anti-viral drugs and their intermediates. Exemplary are the enzymatic asymmetric synthesis of amino acid components, the fermentative production of structurally complex intermediates of anti-influenza drugs and the fully enzymatic, large-scale synthesis of a potential block-buster HIV drug. With many enzyme classes being uncharted with regards to the synthesis of anti-viral agents, there is still a large unopened toolbox waiting to be unlocked. Additionally, by discussing biocatalytic strategies towards potential anti-viral agents against SARS-CoV-2, we hope to contribute to the development of novel synthetic routes to aid in the mass production of a future treatment of COVID-19.
 Sjoerd Slagman | Sjoerd Slagman's main interest is biocatalytic method development, both in solution and at the solid/liquid interface. He currently holds a position as Post-Doc in the lab of prof. W.-D. Fessner at TU Darmstadt (DE) in which he focusses on the discovery and application of novel reductive aminases. He obtained his PhD from Wageningen University & Research (NL) where he studied laccase as surface modification tool in the lab of prof. H. Zuilhof and Dr M. Franssen. Prior, he finished his undergraduate education during which he was trained as synthetic organic chemist (University of Amsterdam, NL). |
 Wolf-Dieter Fessner | Wolf-Dieter Fessner studied chemistry and received his PhD in 1986 at Freiburg University followed by postdocs at Harvard University with George M. Whitesides and at University of Southern California with George A. Olah. Returning to Freiburg, he completed his Habilitation studying the potential of aldolases for diastereoselective organic synthesis. Following a position as Associate Professor at RWTH Aachen, he has been Professor of (Bio)Organic Chemistry at Technical University Darmstadt since 1998. His current research interest focuses on the discovery, engineering and application of enzymes from various classes for organic synthesis, with a special focus on asymmetric carboligation, chiral aminoalcohol and oligosaccharide synthesis. |
1. Introduction
Viral infections have a tremendous impact on society; recent outbreaks of COVID-19 (SARS-CoV-2) and the Ebola virus have resulted in nearly 1 million casualties worldwide, a number still rising rapidly at the time of writing (September 2020).1,2 With currently over 30 million confirmed COVID-19 cases and numerous hospitalised, the capabilities of the healthcare sector are being stretched beyond limits.3,4 Let alone the burden on those that have no adequate access to medical professionals.5,6 Apart from the impact on our physical well-being, COVID-19-related lockdowns and quarantine-measures take their toll with regards to mental fitness and welfare.7Viral outbreaks are not restricted to the present as numerous smallpox and cholera epidemics, the Spanish flu pandemic and more recently the onset of AIDS/HIV have shaped human life throughout history.8,9 With an estimated reservoir of 1.67 million unknown viruses in mammals and birds alone, viral infections are likely to be of continuous concern in the future too.10,11 In accordance to being struck by numerous pandemics, respective countermeasures also go way back. The word ‘quarantine’, for example, originates from the Venetian ‘quarantena’, which designated the 40 days required for ships to be in isolation prior to allowing its passengers to go ashore during the plague epidemic.12 The history of anti-viral drugs, however, spans only about 60 years. In 1963 the first anti-viral drug idoxuridine was FDA-approved for the treatment of herpes. A better understanding of viruses, and especially the emergence of retroviruses such as HIV, spurred the development of the field in the 80's. Up to date, approximately 100 anti-viral drugs have been approved by the FDA.
So far, no anti-viral drug has been proven to be effective for treating patients infected with COVID-19. With the proverbial clock ticking, research towards novel anti-viral agents and repurposing of known pharmaceuticals has sky-rocketed.13,14 A similarly big challenge awaits when a treatment is found, namely the large-scale production of the pharmaceutical agent. For this matter, access to a large synthetic toolbox, full of efficient and robust stereoselective technologies, is essential.15 These synthetic tools have to meet increasingly higher demands due to a rise in complexity of marketed drugs.16,17 While many of the initial anti-viral drugs were nucleoside analogues based on (deoxy)sugar building blocks from the chiral pool or simply non-chiral, novel anti-viral drugs are often more complex, multicyclic or macrocyclic, and possess multiple chiral centres that are difficult to install.18–20 To access such chiral centres, organic chemists commonly revert to transition metal catalysis which is, however, often burdened with highly sensitive chemical species, accumulation of toxic waste and non-optimal stereoselectivity. Synthesising molecules through fermentation or by employing (purified) enzymes on the other hand, usually proceeds with high (stereo)selectivity and allows for benign reaction conditions to be employed. The use of water as solvent, mild reaction conditions with regards to temperature and pH, and generation of mostly biodegradable waste make biotransformations particularly attractive.21–23 As such, biocatalysis thus not only offers an environmentally benign alternative to chemical synthesis, but also often proves to be the most economically viable choice.
In line with an increasing awareness of its potential, biocatalytic production of pharmaceuticals has become increasingly popular over the last decades.24–33 Additionally, in times of crisis and scarcity (of medicine) one requires tools to rapidly access the chemicals in need, a recent 90 day pressure test revealed biocatalysis to be excellently suited for achieving this.34
To aid in the continuous development of biocatalytic tools for anti-viral agent synthesis and to hopefully also contribute to the rapid production of a treatment for COVID-19, we hereby present a comprehensive compilation of research on the biocatalytic synthesis of anti-viral agents. After a brief introduction to the pharmacological targets of anti-viral drugs, the full breath of biocatalytic methods applied for accessing those drugs will be covered extensively. This includes an evaluation of the pertinent patent literature as well as industrial microbial production of starting materials. Both small scale academic ventures and industrial processes will be thoroughly discussed. This review largely covers FDA-approved drugs as to ensure that the highlighted biocatalytic pathways result in the effective production of active pharmaceutical agents. All drugs will be jointly discussed according to their structural similarity, which to a large degree is in agreement with a similarity in the addressed pharmacological target. Where possible, retrosynthetic analysis is applied to be able to pick-and-mix the mentioned biocatalytic syntheses of an intermediate with (chemical) routes to other intermediates of the drug of interest. The analysis of published literature on the topic is appended by an overview of biocatalytic strategies amenable towards the synthesis of promising agents against COVID-19. We will finalise with an outlook on the potential of novel biocatalytic methods that have not, or only scarcely, been employed to the synthesis of anti-viral agents. For the use of biocatalysis as drug discovery tool, the reader is referred to other outstanding works as this is considered to be outside the scope of this review.30,31
2. Mechanism of action of anti-viral agents
A virus is an infectious particle that can only replicate inside the cells of a host organism. After infection, the host cell is forced to rapidly produce numerous copies of the virus to be released from the exhausted cell for subsequent infection of other cells. Anti-viral agents are designed to interrupt, or at least disturb, the viral life cycle. Several approaches to tackling viral infections have been adapted throughout the short history of anti-viral drug development. Most drugs have been targeted at the replication machinery of the viral genome. Exemplary are drugs that are incorporated in the viral DNA upon phosphorylation in vivo, preventing further DNA replication by DNA polymerase (Fig. 1A).19 Several (acyclic) nucleoside analogues such as brivudine, act in this fashion. Other (acyclic) nucleoside analogues, like didanosine and lamivudine, are phosphorylated in vivo to subsequently compete with natural deoxynucleotides for incorporation into the (viral) DNA. Chain elongation through reverse transcription can hereby be prevented. These drugs are referred to as nucleoside reverse transcriptase inhibitors (NRTIs, Fig. 1B). The hepatitis C virus (HCV) non-structural protein NS5B acts as RNA polymerase that can be inhibited by incorporation of the triphosphorylated form of a pro-drug such as sofosbuvir to terminate RNA chain elongation (Fig. 1C). Similarly, ribavirin inhibits RNA polymerisation of influenza viruses. NS5A, although not shown to be inherently enzymatically active, among others, modulates NS5B activity and is inhibited by drugs such as ledipasvir. Peptidomimetic protease inhibitors inhibit proteolytic cleavage of proteins that are essential to viral reproduction (Fig. 1D). HIV-1 protease inhibitors, for example, prevent cleavage of the precursor protein gag p55.35 Integrase inhibitors prevent insertion of viral DNA into chromosomal DNA of the host cell (Fig. 1E). Entry inhibitors such as maraviroc block the virus from entering the host cell (Fig. 1F). The anti-influenza agents, oseltamivir, peramivir, zanamivir and laninamivir inhibit viral neuraminidase to prevent budding (release) of the virus from the host cell (Fig. 1G). Drugs interfering with viral uncoating or viral maturation have to this date not been FDA-approved or produced biocatalytically.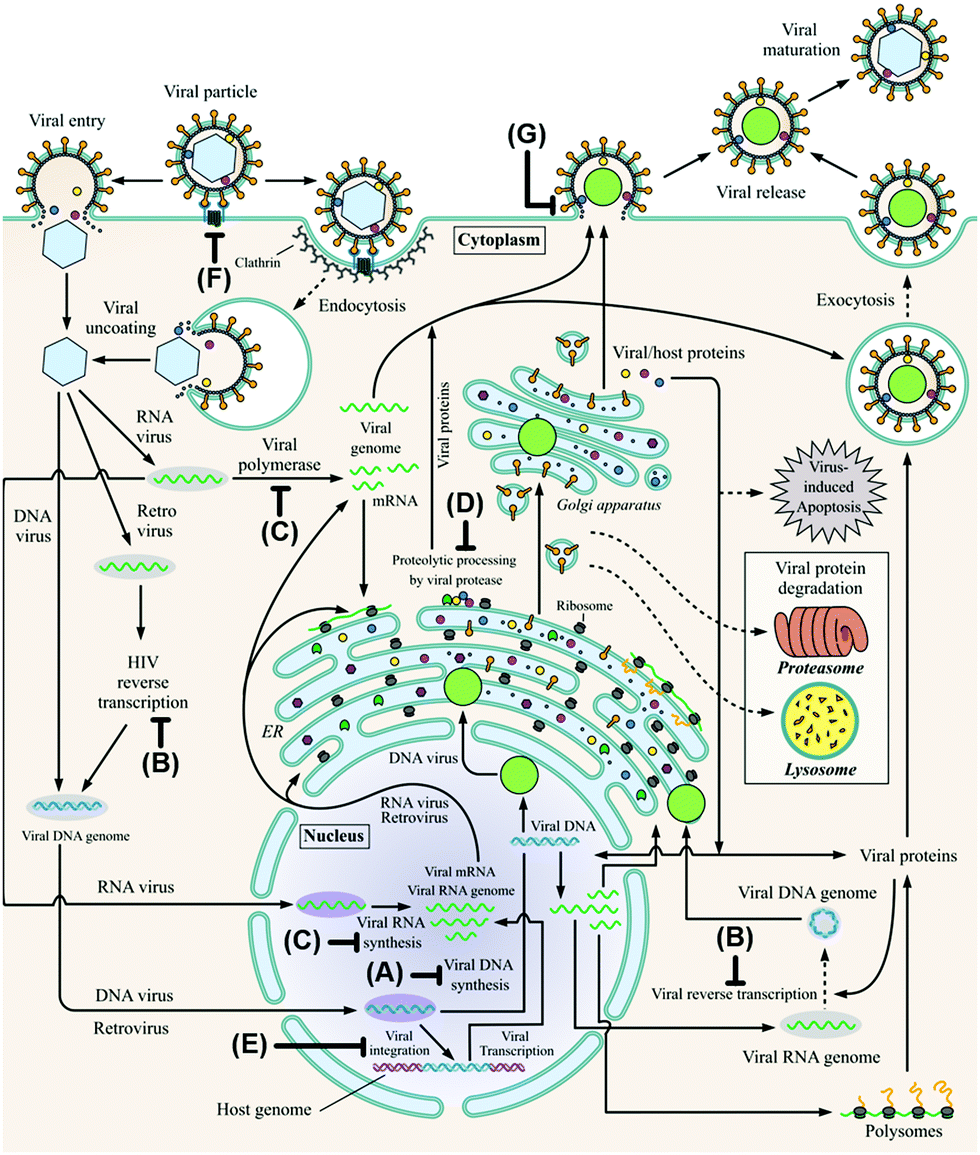 | ||
| Fig. 1 The viral life cycle upon host cell infestation including depiction of drug interaction sites: polymerase inhibitors (A), (nucleoside) reverse transcriptase inhibitors ((N)RTIs, B), RNA polymerase inhibitors including NS5A and NS5B inhibitors (C), protease inhibitors (D), integrase inhibitors (E), entry inhibitors (F) and neuraminidase inhibitors (G). ER: endoplasmic reticulum and HIV: human immunodeficiency virus. Adapted from De Clercq and Li (reproduced with permission).19 | ||
Anti-viral drug therapies are designed to reduce the virus replication rate and thus the total virus load in order to ameliorate symptoms caused by infection. The drug itself cannot destroy the virus to cure the disease, which requires involvement of the humoral immune system.36 While there is an urgent need for new effective anti-viral drugs against COVID-19, the SARS-CoV-2 virus is likely to persist as a constant threat to mankind, even once vaccination will become possible.
3. FDA-approved anti-viral agents
3.1 Nucleoside analogues
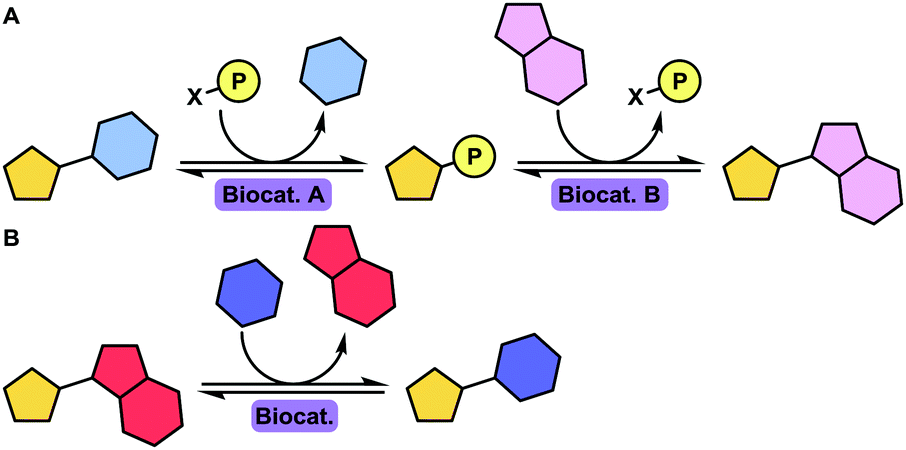 | ||
| Scheme 1 Schematic depiction of dual (A) or single (B) enzyme-catalysed transglycosylation towards the synthesis of nucleoside analogues. Enzymes have been coloured according to their enzyme commission number, e.g. transferases are represented by purple colouration. Shading is varied for subclasses; for example, phosphotransferases (EC 2.7) are shaded darker than transketolases (EC 2.2). | ||
Pivotal in the development of anti-viral drugs was the approval of idoxuridine, an iodinated derivative of 2′-deoxyuridine (1), in 1963.19 Idoxuridine is used for treating retinal herpes simplex virus (HSV) infections through its incorporation into viral DNA. Its structural analogues 2′-trifluorothymidine (trifluridine) and brivudine (Scheme 2) were approved for treating HSV infections in 1980 and 2000, respectively. While idoxuridine was initially prepared through iodination of 1,43 it can nowadays be obtained efficiently by biocatalytic means. By depriving either pyrimidine nucleoside phosphorylase (PyNP) or thymidine phosphorylase (TP) from an additional phosphate source, only transient phosphorylation of the nucleoside sugar unit is possible, allowing for transglycosylation with a suitable nucleobase.44 By doing so, 1 could be converted to idoxuridine, trifluridine and brivudine using either of the immobilised enzymes. Immobilisation on a macro-sized matrix ensures facile isolation of the catalyst which enables its straightforward recycling. Alternatively, trifluridine could be obtained through the use of a 2′-deoxyribosyltransferase.45
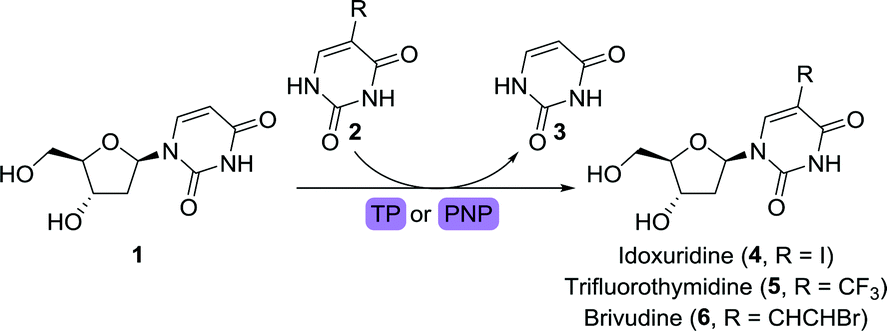 | ||
| Scheme 2 Synthesis of anti-viral 2′-deoxynucleosides with thymidine phosphorylase (TP) or pyrimidine nucleoside phosphorylase (PyNP). For brevity and clarity, equilibrium arrows and cleaved/replaced components will usually only be displayed in reaction schemes when of importance for the mechanistic understanding. | ||
 | ||
| Scheme 3 Biocatalytic synthesis of vidarabine. | ||
Fernández-Lucas and co-workers instead utilised immobilised 2′-deoxyribosyltransferases for the synthesis of vidarabine.51 This work was recently extended through the use of a highly stable and remarkably versatile purine nucleoside 2′-deoxyribosyltransferase (PDT) from Trypanosoma brucei for the synthesis of this drug.52 Mutagenesis through random or site-directed sequence variation allows for small modifications of an enzyme, resulting in alterations in its properties. Properties such as activity, selectivity and/or thermostability can hereby be tuned. In the case of this PDT enzyme, the valine in position 11 was replaced by alanine or serine (V11A or V11S), resulting in variants that showed an almost threefold increase in catalytic activity.
The second wave of FDA-approved anti-viral agents largely consisted of nucleoside analogues active against the human immunodeficiency virus (HIV). Didanosine (2′,3′-dideoxyinosine), for example, acts on viral reverse transcriptase and competes with dATP to inhibit DNA synthesis.53 The versatility of the PDT from Trypanosoma brucei was further highlighted by the fact that it could also convert 2′,3′-dideoxyadenine to didanosine.52 Alternatively, this anti-viral nucleoside could be obtained by means of phosphorylase catalysis or the use of whole cells expressing such enzymes.48,54,55 Highly interesting with regards to didanosine synthesis is the full bioretrosynthetic pathway construction published by Birmingham and co-workers (Scheme 4).56 Drawing inspiration from sugar and nucleoside metabolism, the group engineered three enzymes involved in converting 2′,3′-dideoxyribose (10) to didanosine, namely ribokinase (RK), 1,5-phosphopentomutase (PPM) and purine nucleoside phosphorylase (PNP). Inhibition by the co-factor-related by-product ADP, however, limited the efficiency of the overall cascade. Inhibition could be suppressed by allowing for ATP recycling through addition of adenylate kinase (AK) and pyruvate kinase (PK, Scheme 4A). An unexpected bypass was found upon engineering RK, eliminating the need for PPM (Scheme 4B). Use of the shortened cascade resulted in a fiftyfold increase in didanosine production (milligram scale).
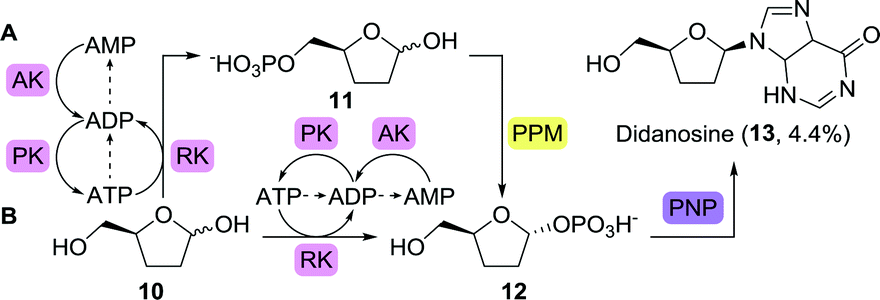 | ||
| Scheme 4 Multi-enzymatic bioretrosynthetic pathway construction towards the synthesis of didanosine. Two routes have been engineered, either including 1,5-phosphopentomutase (PPM, A) or bypassing this enzyme (B). Arrows between ATP, ADP and AMP show likely routes of degradation.56 RK: ribokinase, PNP: purine nucleoside phosphorylase, AK: adenylate kinase and PK: pyruvate kinase. | ||
Two other HIV drugs, zidovudine and stavudine, were synthesised from the common nucleoside intermediate 5-methyluridine (17).57,58 Like the previously mentioned nucleoside analogues, 17 has been obtained through the use of isolated phosphorylases or whole cells overexpressing such enzymes (Scheme 5).59–62
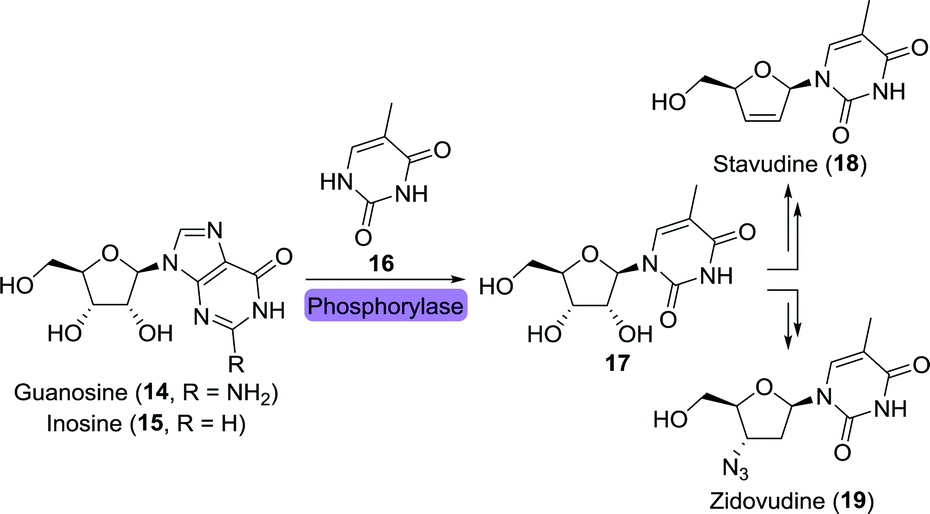 | ||
| Scheme 5 Biocatalytic synthesis of the nucleoside intermediate 5-methyluridine (17) as common intermediate towards the synthesis of stavudine and zidovudine. | ||
Arguably, hydrolases are the most commonly applied enzymes in the industrial synthesis of small-molecules.63 This class comprises enzymes capable of cleaving bonds through attack of a water molecule. Dependent on the enzyme and the reaction conditions, the reverse reaction is also feasible. In this way, ester, organophosphate or amide bonds can either be broken or made. Many of these hydrolases, such as lipases for example, are highly regio-, chemo- and stereoselective. The latter allows for kinetic resolution of a racemic mixture resulting in the production of a single stereoisomer. As this feature comes with a very broad substrate scope, application of hydrolase enzymes will be thoroughly discussed throughout this work.
Regioselective de-acetylation with Candida rugosa lipase (CRL) allowed for the synthesis of 3-hydroxy-5-acetyl-thymidine (21) from the diacetate 20 in 90% yield (<1 g, Scheme 6).64 This intermediate could be chemically converted to zidovudine via a Mitsunobu double inversion protocol followed by deprotection.
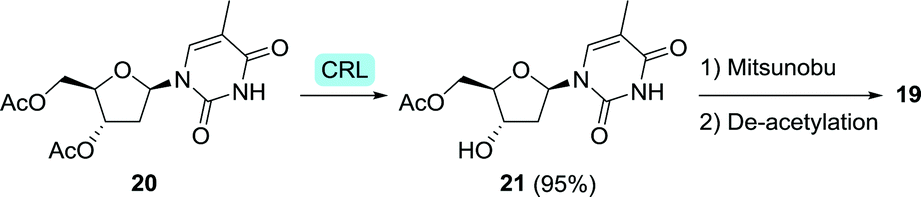 | ||
| Scheme 6 Regioselective de-acetylation of an intermediate (20) of zidovudine (19) with Candida rugosa lipase (CRL).64 | ||
Ribavirin is a nucleoside analogue composed of a ribose sugar and 1,2,4-triazolecarboxamide (23) as alternative nucleobase (Scheme 7). It is FDA-approved for a number of viral infections including those caused by the hepatitis C virus and the respiratory syncytial virus (RSV).65 Its biocatalytic synthesis has been pursued extensively, allowing for a large toolbox of enzymes capable of converting a wide variety of nucleosides to ribavirin through transglycosylation.66–72 Recent examples include the use of immobilised bacteria for the synthesis of this drug.73,74
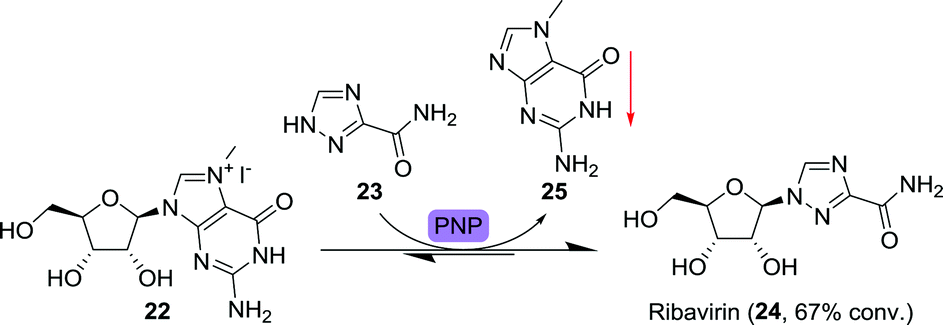 | ||
| Scheme 7 Precipitation-driven purine nucleoside phosphorylase (PNP)-catalysed synthesis of ribavirin.75 | ||
An important feature of most enzymatic reactions is that they are reversible, which often limits formation of the desired product. Strategies for pushing the reaction equilibrium to the product side are highly important and will thus be discussed extensively using pertinent examples.
Ubiali and co-workers applied a two-pronged equilibrium shifting strategy towards the synthesis of ribavirin by using 7-methylguanosine iodide (22) as starting material (Scheme 7).75 PNP from Aeromonas hydrophila did catalyse the transfer of 23 to 22, but not the reverse reaction as the formed nucleobase, 7-methylguanine (25), was not accepted by the enzyme. Due to its poor solubility, 7-methylguanine precipitated, further limiting the backwards reaction. This allowed for good (67%) conversion to the product ribavirin. Taribavirin, a pro-drug of ribavirin currently undergoing phase III clinical trials,76 was likewise prepared by biocatalytic glycosyltransfer between inosine and 3-cyano-1,2,4-triazole using Brevibacterium acetylicum ATCC 39311 followed by aminolysis at 20 g scale.77
En route to sofosbuvir's nucleoside moiety, Cleary and co-workers employed highly versatile Candida antarctica lipase B (CAL-B) to resolve a mixture of aldol products. By doing so, enantiomerically pure intermediate 2880 was obtained in the desired configuration by starting from 33 g of aldol product (Scheme 8).81 This intermediate was initially synthesised in enantiomerically enriched form through lithium diisopropylamide (LDA)-catalysed aldol addition of ethyl 2-fluoropropanoate (27) to (R)-glyceraldehyde acetonide (26). The latter can be accessed through oxidation of (S)-glycerol acetonide (32), which in turn, can be obtained by resolution of glycerol acetonide esters (31) using various free esterases or whole cells overexpressing such enzymes (Scheme 9).82–85
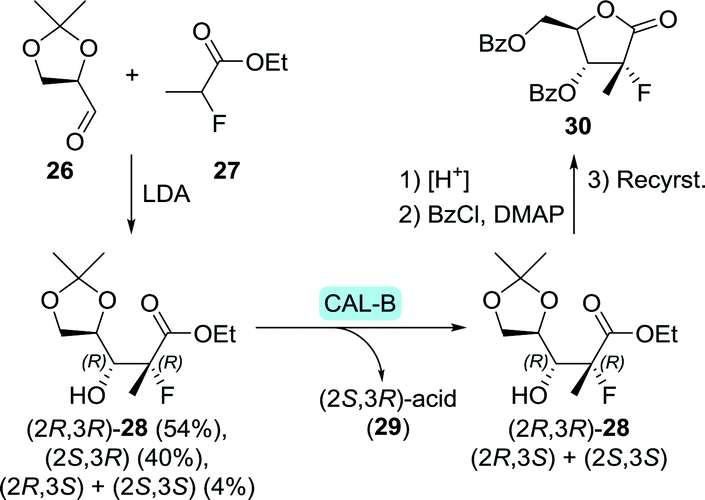 | ||
| Scheme 8 Candida antarctica lipase B (CAL-B)-catalysed resolution of the sofosbuvir aldol intermediate 28.80 LDA: lithium diisopropylamide and DMAP: N,N-dimethyl-4-aminopyridine. | ||
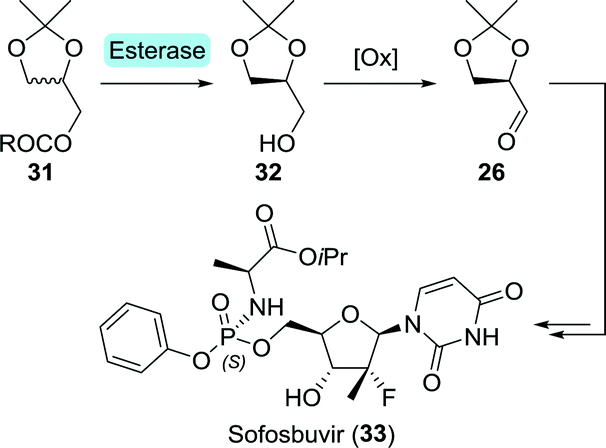 | ||
| Scheme 9 Esterase-catalysed resolution of glycerol acetonide esters. | ||
Swiss-based HC-Pharma used CAL-B for a mild regioselective deacetylation of the C6-hydroxy group of the bis-acetylated nucleoside precursor of sofosbuvir. With the C4-acetate left untouched, this mono-acetate intermediate could be coupled to the phosphoramidate without competing reactivity.86
The phosphoramidate moiety of sofosbuvir is chiral and only the (S)-configurated enantiomer of the phosphoramidate drug is FDA-approved. Raushel and co-workers used an engineered Pseudomonas diminuta phosphotriesterase87 for the kinetic resolution of 34 (Scheme 10). A G60A mutation allowed for almost 200-fold selectivity towards hydrolysis of (R)-34.88 Coupling of (S)-34 and the sugar derivative through nucleophilic substitution at the phosphorus centre led to the desired stereoinversion.89
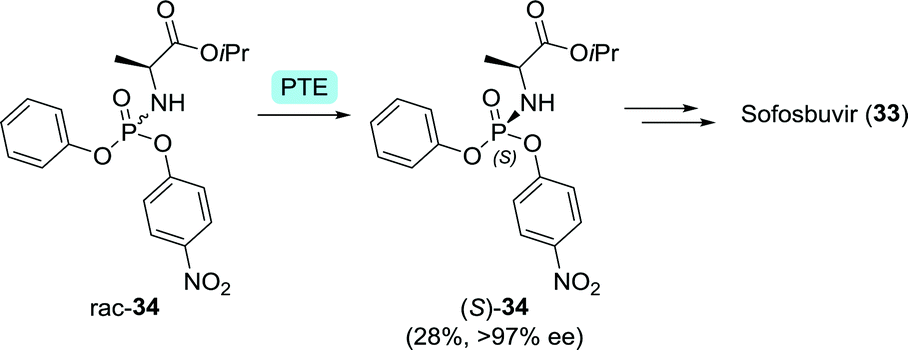 | ||
| Scheme 10 Resolution of a Pro-Tide phosphoramidate intermediate of sofosbuvir with Pseudomonas diminuta phosphotriesterase (PTE).88 | ||
An illustrative example of the value of industrial biocatalytic anti-viral production is the recently published synthesis of islatravir by Merck (Scheme 11).90 Islatravir is a reverse transcriptase translocation inhibitor currently in phase III clinical trials for the treatment of HIV. The team developed a telescoped two-step, five-enzyme approach for the synthesis 38 from 2-ethynylpropane-1,2,3-triol (35), which is followed by a one-pot cascade containing four enzymes to achieve the synthesis of islatravir from 38.
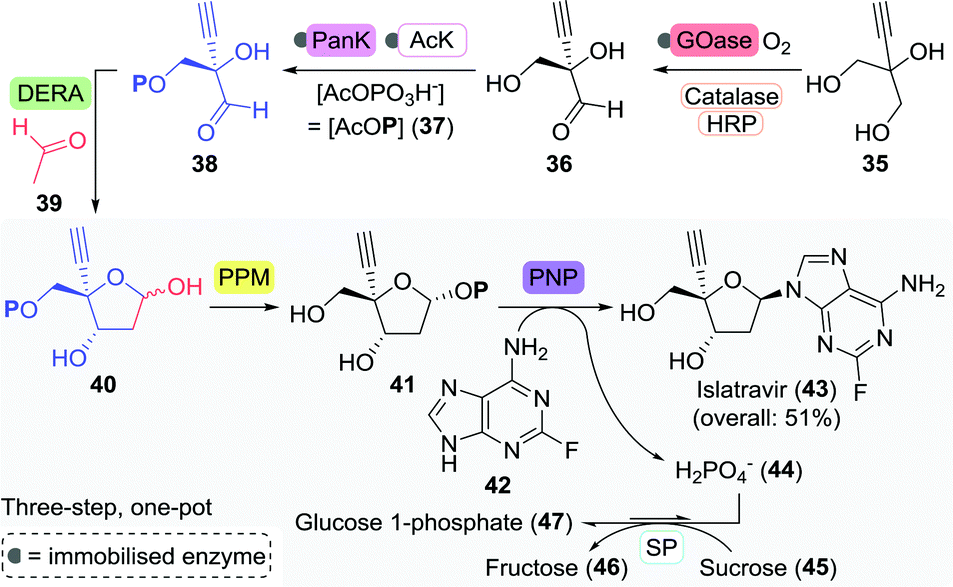 | ||
| Scheme 11 Fully enzymatic synthesis of islatravir as developed by Merck.90 GOase: galactose oxidase, HRP: horseradish peroxidase, PanK: pantothenate kinase, AcK: acetate kinase, DERA: deoxyribose 5-phosphate aldolase, PPM: 1,5-phosphopentomutase, PNP: purine nucleoside phosphorylase and SP: sucrose phosphorylase. | ||
Enzymes offer a clear advantage over ‘traditional’ catalysts here as they all operate under similar conditions (solvent, pH, and temperature) and show distinct non-competitive reactivities which allows their use in a one-pot cascade. The first step of the reaction sequence posed the biggest challenge as the selected galactose oxidase (GOase) from Fusarium graminearum (engineered F2 variant) had to undergo complete reversal of stereoselectivity in order to oxidise 35 to the (R)-enantiomer of 36. This feat was eventually achieved through 12 rounds of evolution and a staggering 34 amino acid mutations, concomitantly providing an 11-fold increase in enzyme activity. Directed evolution of ATP-dependent pantothenate kinase (PanK) subsequently allowed for the phosphorylation of 36. Employment of an acetate kinase (AcK) ensured regeneration of the co-factor ATP.
The subsequent four-enzyme cascade started with deoxyribose 5-phosphate aldolase (DERA)-catalysed formation of the aldol product 40 from 38 and acetaldehyde (39). Two rounds of directed evolution were sufficient for increasing the tolerance of DERA from Shewanella halifaxensis to 400 mM acetaldehyde. Aldol product 40 was converted to islatravir through the use of E. coli PPM and PNP. Directed evolution of these latter two enzymes resulted in a 70- and 350-fold increase in activity towards 40 and the fluorinated nucleobase 42, respectively. The equilibrium of the four-enzyme cascade was pushed to the product side by implementation of sucrose phosphorylase (SP) from Alloscardovia omnicolens, which consumed phosphate through the formation of glucose 1-phosphate (47).
Employment of this series of heavily engineered enzymes in just a two-staged process led to the isolation of islatravir in a high overall yield of 51%. This approach eliminates purification steps, recycles expensive co-factors, and couples favourable and unfavourable reactions. Stereochemical purity was amplified at every enzymatic step along the non-natural, nine-enzyme reaction sequence, and the final synthesis was both atom economical and cost effective.91
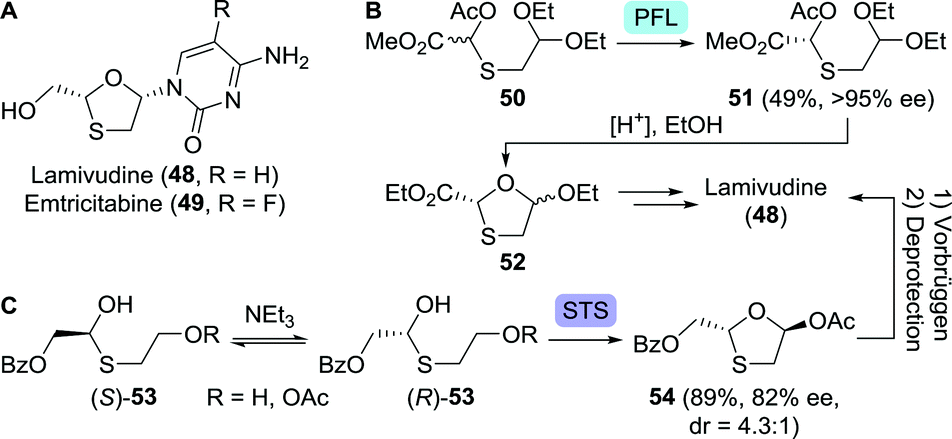 | ||
| Scheme 12 Lamivudine and emtricitabine (A). Lipase-catalysed (Pseudomonas fluorescens; PFL) resolution of an acyclic lamivudine/emtricitabine intermediate (B).93 Dynamic kinetic resolution to get to the protected lamivudine/emtricitabine core (C).99 Base-catalysed interconversion of the acyclic thioether enantiomers as depicted above serves as a tentative mechanistic proposal by the authors of this work. STS: surfactant-treated subtilisin Carlsberg. | ||
In an early approach towards lamivudine, Rayner and co-workers performed lipase-catalysed resolution (Pseudomonas fluorescens) on the acylated O,S-acetal 50, allowing for spontaneous cyclisation upon acid-mediated deprotection (Scheme 12B).93 Because the heterocyclic product 52 was synthesised as a mixture of diastereomers, their separation, upon coupling to the nucleobase, resulted in a low yield of diastereomerically pure lamivudine. In an alternative route, 5′-nucleotidase from snake venom was used to resolve lamivudine-5′-phosphate, requiring additional (enzymatic) dephosphorylation.94 Final stage kinetic resolution of a racemic mixture of lamivudine using cytidine deaminase eliminated the need for its 5′-phosphate analogue.95 The structurally related anti-viral agent, emtricitabine could be obtained in enantiopure form through pig liver esterase-catalysed resolution of its 5′-butanoate ester as shown by Schinazi and co-workers.96
Various other resolution approaches to the enantiomerically pure protected 1,3-oxathiolane core have been reported since, however, all intrinsically restricted to 50% yield.97,98 These limitations could be lifted by employing the principle of dynamic kinetic resolution (DKR). DKR relies on in situ racemisation of the starting material and selective (enzymatic) conversion of one of the isomers to the desired product. Ramström and co-workers employed dynamic interconversion of a hemi-thioacetal (53) using triethylamine and subsequent stereoselective ring-closing using surfactant-treated subtilisin Carlsberg (STS, a protease from Bacillus licheniformis) to afford the protected 1,3-oxathiolane 54 in 89% yield (Scheme 12C).99 Chemical Vorbrüggen coupling to the nucleobase and subsequent deprotection furnished lamivudine. By coupling STS-catalysed ring-closing to CAL-B-catalysed resolution, enantiopurity of the protected 1,3-oxathiolane intermediate could be further enhanced.100
Abacavir is an anti-viral nucleoside analogue in which a cyclopentene unit constitutes the furanose mimic (Scheme 13).101 The drug is commonly being prepared from Vince-lactam (55) which acts as a versatile building block for a variety of pharmaceuticals.102 Stereoselective ring-opening of the racemic Vince-lactam can be achieved using either a (+)-lactamase followed by chemical hydrolysis of the remaining material or a (−)-lactamase leading directly to the desired (1S,4R)-γ-amino acid (1S,4R)-56. Over the past 30 years, numerous lactamases have been isolated and applied in the ring-opening of Vince-lactam.103–118 Resolution could, alternatively, be achieved using a variety of other enzymes including CAL-B and a non-haem chloroperoxidase from Streptomyces viridochromogenes.119–122 CAL-B resolution efficiency could be increased by making use of the activated N-hydroxymethyl Vince-lactam. In this way, Fülöp and co-workers realised enhanced reactivity and higher enantiomeric excess (ee) of the remaining (+)-lactam in comparison to CAL-B-catalysed resolution of the non-activated Vince-lactam.123 Spontaneous cleavage of the activating group eliminated the need for chemical deprotection. The use of Savinase, a protease commonly employed in laundry detergents, allowed for stereoselective ring opening of N-protected Vince-lactams.124 Upon (enzymatic) synthesis of protected (1S,4R)-56, Abacavir could be obtained through several chemical steps.125
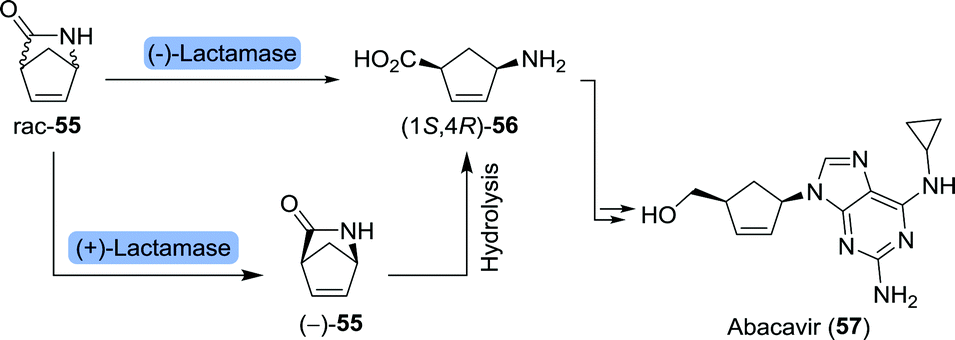 | ||
| Scheme 13 Lactamase-catalysed resolution of Vince-lactam to obtain the corresponding (1S,4R)-γ-amino acid. This amino acid served as an intermediate towards abacavir and peramivir (vide infra). | ||
The versatility of CAL-B was further exemplified through the resolution of a structural isomer of Vince-lactam, 6-azabicyclo[3.2.0]hept-3-en-7-one (58, Scheme 14). This allowed for palladium-catalysed nucleobase (60) coupling after tosylation, leading up to the synthesis of abacavir.126 Although much of the research described above has been conducted at laboratory scale, protease-catalysed Vince-lactam ring-opening is nowadays performed on industrial scale, too.
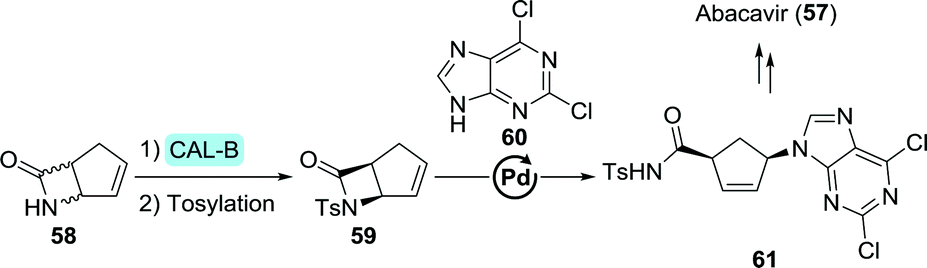 | ||
| Scheme 14 Resolution of a β-lactam abacavir intermediate with Candida antarctica lipase B (CAL-B).126 | ||
Entecavir is an anti-HBV drug characterised by an unusual 4-methylenecyclopent-1-ene core, thus bearing a exocyclic terminal double bond.127 Bristol Myers Squibb furnished entecavir through desymmetrisation of either di-acetate 62 or its parent diol 64 at multigram scale using a Pseudomonas sp. lipase (Scheme 15).128 The formed complementary mono-acetates (1R,4S,5R)-63 and (1S,4R,5S)-63 could both be converted to Intermediate 65.129 Chemical synthesis subsequently provided entecavir. Desymmetrisation of 62 could also be achieved using electric eel acetylcholinesterase.130 Although the esterase was slightly more selective towards the mono-acetate, use of bacterial enzymes is preferred due to a lack of efficient expression systems for animal-derived enzymes.
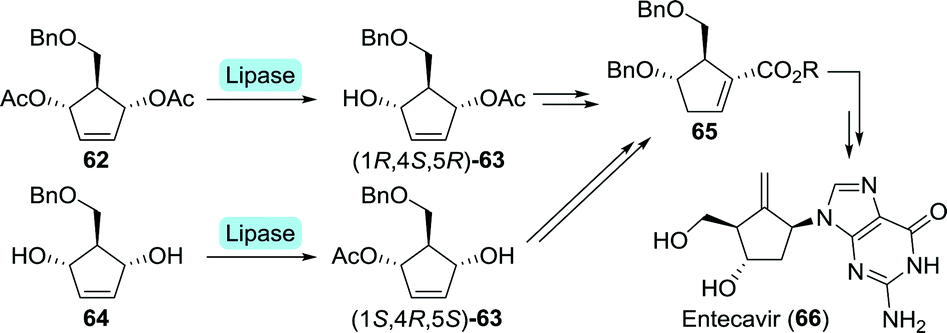 | ||
| Scheme 15 Lipase-catalysed resolution of entecavir intermediates. | ||
Alternative routes to entecavir as also explored by Bristol Myers Squibb proceeded through the Corey lactone 70.129 The latter has proven to be accessible through resolution of racemic bicyclo[3.2.0]hept-2-en-6-one (67) which could be selectively reduced to (R)-alcohol 68 using baker's yeast (Scheme 16). Re-oxidation to the ketone and subsequent chemical steps furnished 70.131
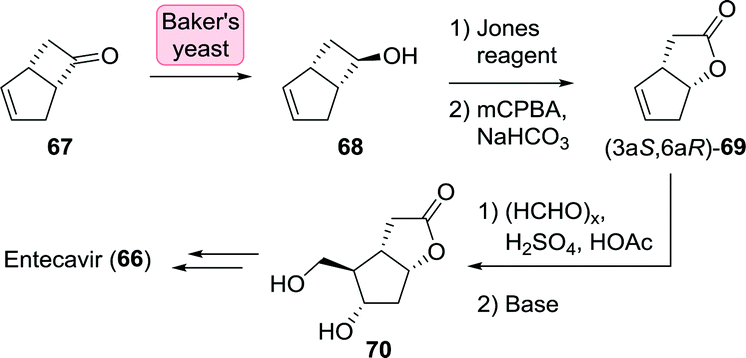 | ||
| Scheme 16 Synthesis of the Corey lactone 70 through stereoselective reduction of bicyclo[3.2.0]hept-2-en-6-one with baker's yeast.131 | ||
Intermediate (3aS,6aR)-69 could also be accessed directly from (−)-67 through enzymatic Baeyer–Villiger oxidation using a double mutant (F255A/F443V) of (2,2,3-trimethyl-5-oxocyclopent-3-enyl)acetyl-CoA 1,5-monooxygenase (OTEMO) from Pseudomonas putida.132 Although the non-desired (3aR,6aS)-69 has previously been generated from racemic 67 using Baeyer–Villiger oxidases, the OTEMO variant could not convert the racemic substrate with sufficient stereoselectivity.
3.2 Acyclic nucleoside analogues
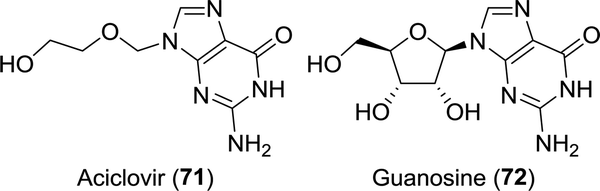 | ||
| Chart 1 Guanosine-mimicking anti-viral agent aciclovir. | ||
Valaciclovir is a pro-drug of aciclovir in which its hydroxy moiety is esterified with L-valinate (Chart 2).134 This could be achieved mildly using a Bacillus protease.135 Analogously, valganciclovir (Chart 2) is the L-valinate ester pro-drug of ganciclovir. Selective protease-catalysed mono-de-esterification of the di-N-Cbz-valine ester of ganciclovir afforded the mono-ester valganciclovir.136
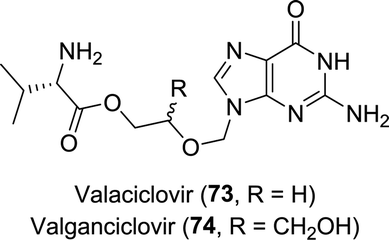 | ||
| Chart 2 Aciclovir pro-drugs valaciclovir and valganciclovir. | ||
Although the active drugs themselves are non-chiral, enantiopure, isotopically labelled anti-herpetics famciclovir and penciclovir have been prepared enzymatically.137 Lipase from Candida cylindracea was used to resolve racemic 13C-labelled methyl 4-(benzyloxy)-2-(hydroxymethyl)butanoate (rac-75) on small scale (Scheme 17). The remaining (R)-75 and the formed (S)-76 could subsequently be converted to either labelled (R)-famciclovir and (R)-penciclovir, or (S)-famciclovir and (S)-penciclovir, respectively. These isotopically labelled drugs could be of use to determine whether stereorecognition plays a role during in vitro phosphorylation.
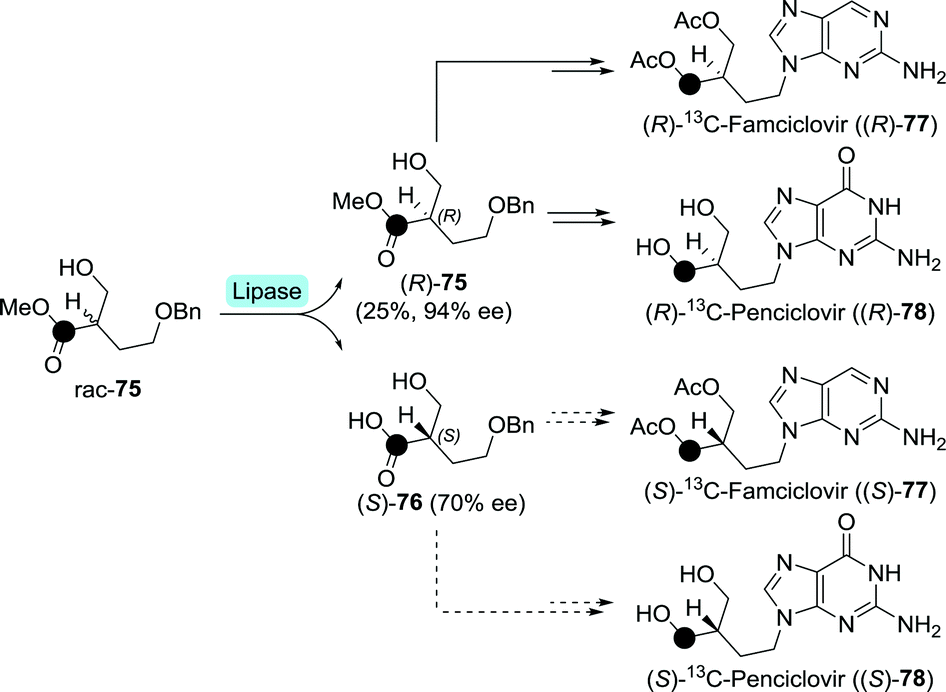 | ||
| Scheme 17 Lipase-catalysed resolution of 13C-labelled (•) methyl 4-(benzyloxy)-2-(hydroxymethyl)butanoate towards the synthesis of radiolabelled penciclovir and famciclovir.137 | ||
Tenofovir (85) is a hydrolytically stable phosphonate that mimics mono-phosphorylated nucleosides, but suffers from low bioavailability upon oral administration due to its ionic charge.138 In order for tenofovir to be employed as a drug, one requires masking of this ionic charge in the form of pro-drugs. Tenofovir disoproxil and tenofovir alafenamide are such pro-drugs which are used for treating HIV (Scheme 18).139,140 Tenofovir disoproxil is a diester, whereas tenofovir alafenamide makes use of the Pro-Tide technology discussed under sofosbuvir (Section 3.1.3).79
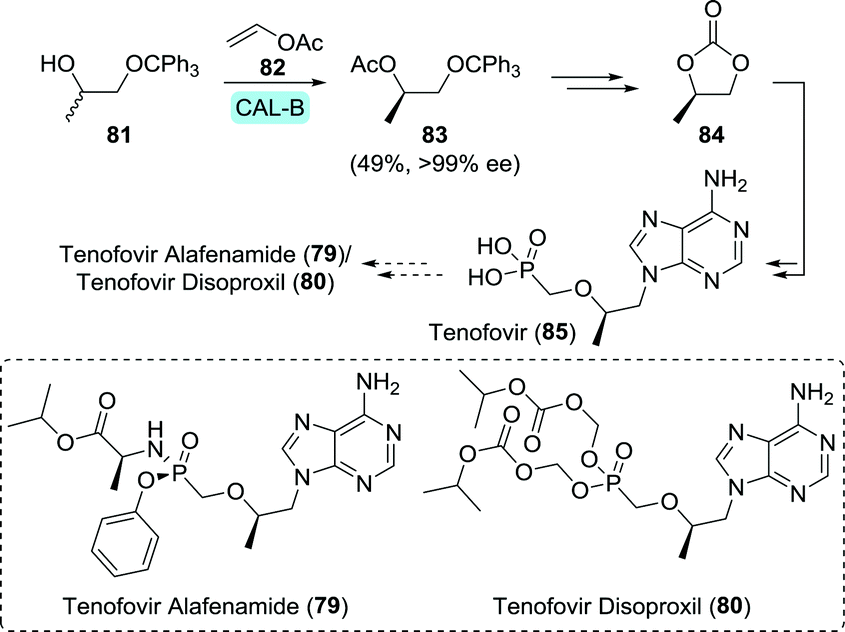 | ||
| Scheme 18 Candida antarctica lipase B (CAL-B)-catalysed resolution of 1-(trityloxy)propan-2-ol towards the synthesis of (R)-propylene carbonate, which is an intermediate of tenofovir.143 | ||
Both tenofovir derivatives are commonly obtained by first synthesising tenofovir itself.141,142 Edlin and co-workers did so by using the pivotal building block (R)-propylene carbonate (84) to convey chirality.141 Using immobilised CAL-B (Novozym 435) to resolve 1-(trityloxy)propan-2-ol (81), 84 could be produced in gram scale in flow (Scheme 18).143
As illustrated for famciclovir and penciclovir, radio-labelled pharmaceuticals are indispensable tools for studying pharmacological properties.144 Using a ketoreductase (KRED), Rivera et al. synthesised perdeuterated and mono-tritium-labelled (R)-9-(2-hydroxypropyl)adenine (87) as tenofovir intermediates (85, Scheme 19A).145 Deuterium-labelled [D6]-87 was produced through H/D exchange of its acidic protons. Additional labelling was subsequently achieved by enzymatic ketone reduction using the (R)-selective Codexis KRED P1B02 (>99% ee for non-labelled 87) via substrate-coupled co-factor recycling with 2-deutero-isopropanol (2-D-89) as the deuteride source (Scheme 19A). Loss of volatile acetone during the course of the reaction additionally pushed the reaction equilibrium to the product side.
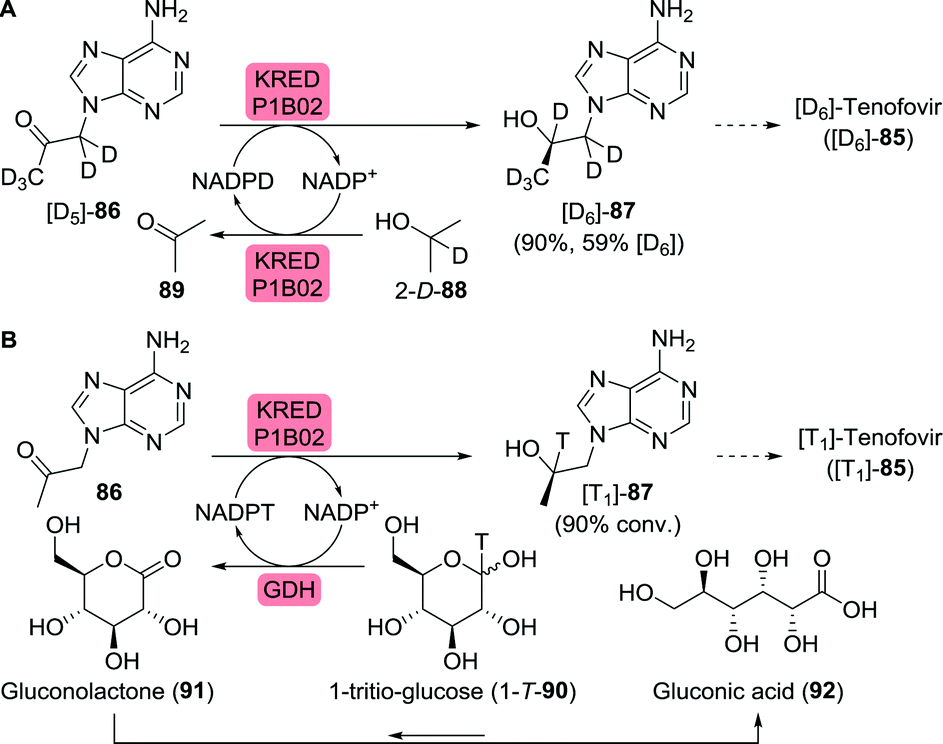 | ||
| Scheme 19 Biocatalytic radiolabelling of tenofovir intermediates with deuterium (A) or tritium (B).145 KRED: ketoreductase, GDH: glucose dehydrogenase. | ||
Initially, the authors envisioned a similar approach for enzymatic tritium labelling but substantial amounts of acetone in the tritium-labelled isopropanol rendered this approach unsuccessful. Instead, glucose dehydrogenase and 1-tritio-glucose (1-T-90) were employed to generate the required NADPT (Scheme 19B). The released gluconolactone (91) spontaneously hydrolysed to gluconic acid (92), further driving the reaction.
This is just one example of a (multi-)enzymatic co-factor recycling cascade. Unlike co-factors such as PLP or FAD, nicotinamide co-factors such as NAD(P)H are not tightly bound to the enzyme. Stoichiometric use of co-factors such as NAD(P)H beyond the multimilligram scale is not economically viable, thus requiring co-factor recycling in situ. Commonly, when using isolated enzymes, an artificial recycling cascade such as the one described above is employed to regenerate NAD(P)H back from NAD(P)+ using either formate/formate dehydrogenase or glucose/glucose dehydrogenase. When using whole cell biocatalysis, the living organism ensures recycling of the co-factor. Several recycling strategies will be discussed throughout this review.
Minamiura and co-workers resolved 3-chloro-1,2-propanediol (rac-93) using Pseudomonas sp., which selectively consumed the (S)-enantiomer (Scheme 20A).148,149 Treatment with base afforded (R)-glycidol with excellent ee. Resolution of 3-chloro-1,2-propanediol or acetylated analogues thereof could also be achieved with a variety of other biocatalysts including glycerol kinases and lipases, although the latter required sequential resolutions to achieve satisfactory enantiopurity.150,151
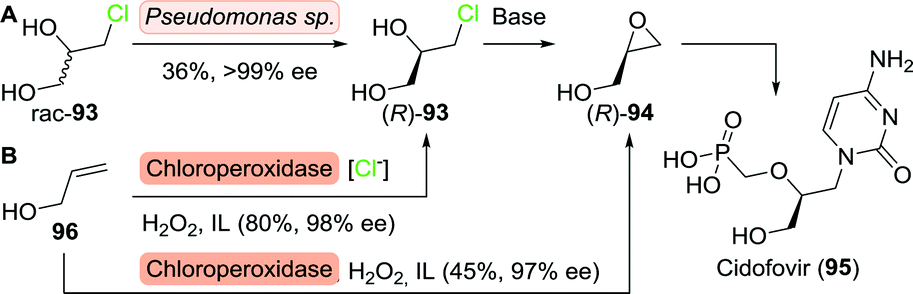 | ||
| Scheme 20 Resolution of 3-chloro-1,2-propanediol upon consumption of the (S)-enantiomer by Pseudomonas sp. followed by conversion to (R)-glycidol as a precursor to cidofovir (A).148 Chloroperoxidase-catalysed synthesis of either (R)-3-chloro-1,2-propanediol or (R)-glycidol, dependent on the presence or absence of chloride ions (B).157 IL: ionic liquid. | ||
Duine et al. employed quinohaemoproteins from a variety of sources for direct resolution of glycidol.152,153 These enzymes selectively oxidised (S)-glycidol to glycidic acid, however, only with poor enantioselectivity. Resolution of the butyrate ester of glycidol by lipase from Rhizopus oryzae proved to be more effective, resulting in >99% ee for (R)-glycidol, although highly dependent on the solvent and method of enzyme immobilisation.154 It is worth mentioning that some commercial enzyme formulations consist of a mixture of enzymes. Fernandez-Lafuente and co-workers used such a mixture, porcine pancreas lipase (PPL), to resolve the butyrate ester of glycidol and managed to prepare (R)-glycidol with 96% ee under optimal conditions.155 Interestingly, the use of a lipase-like enzyme named 25L, a specific isolate from this mixture, resulted in a significant stereoselectivity improvement (>99% ee).156
In a complementary approach, Zhai and co-workers employed the chloroperoxidase from Caldariomyces fumago for functionalisation of allyl alcohol (96) in ionic liquids.157 While the enzyme selectively produced (R)-3-chloro-1,2-propanediol ((R)-93) in the presence of a chlorine source, omission of the latter led to the formation of (R)-glycidol ((R)-94), (Scheme 20B). Both products could be obtained with excellent ee. This modular approach thus allowed for the production of two highly functionalised enantiopure C3-building blocks from simple achiral starting material.
3.3 Peptide mimics
Since the FDA-approval of saquinavir (Chart 3) in 1995, around a dozen peptidomimetic protease inhibitors have been authorised as anti-viral agents.19,158 These drugs structurally resemble small peptides and thus require amino acids as key building blocks. Because biocatalytic production of (non-)proteinogenic amino acids is generally well established and has been thoroughly reviewed, the subject will not be addressed here.159 Where of interest, biocatalytic production of synthetically challenging unnatural amino acids will be discussed. | ||
| Chart 3 Protease inhibitor saquinavir; the first FDA-approved anti-viral peptide mimic. | ||
After various marginally successful attempts at resolution of (protected) L-tert-leucine, Turner and co-workers developed an interesting DKR approach in which racemic tert-leucine was resolved from its rapidly interconverting (R)- and (S)-enantiomeric oxazolone derivatives (98, Scheme 21).164Mucor miehei lipase (MML) selectively catalysed ring-opening of the (S)-enantiomer in the presence of butanol and triethylamine (NEt3). Subsequent two-step chemoenzymatic deprotection afforded L-tert-leucine (100) with >99% ee in 94% yield. More recently, kinetic resolution of N-phenylacetyl- and N-acetyl-protected L-tert-leucine was achieved using Kluyvera citrophila penicillin G acylase and Mycobacterium sp. JX009, respectively.165,166
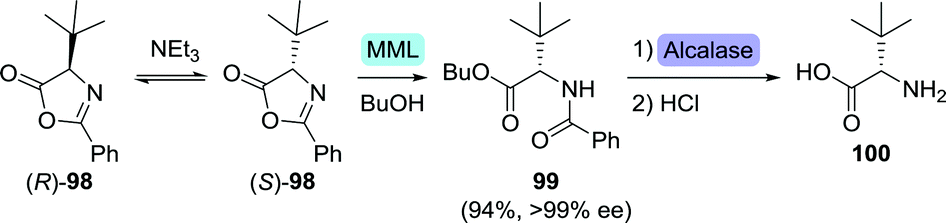 | ||
| Scheme 21 Dynamic kinetic resolution through base-catalysed isomerisation of L-tert-leucine-based oxazolone enantiomers and stereoselective ring-opening with Mucor miehei lipase (MML).164 | ||
Although dynamic kinetic resolution is elegant, single step asymmetric synthesis is a more attractive strategy. Wandrey and co-workers at Degussa were successful in employing a Bacillus cereus leucine dehydrogenase (LeuDH) in the synthesis of L-tert-leucine from trimethylpyruvate (101, Scheme 22).167 Recycling of the polymer-bound co-factor was enabled through the use of formate dehydrogenase. The use of a membrane reactor allowed for retention of both high-molecular mass co-factor and enzyme but permitted formed product to pass. By doing so, space-time yields of 638 g (L d)−1 could be achieved from a continuously operated process for several months. Later, the use of whole cells overexpressing both enzymes eliminated the need for addition of external co-factor and proved to be equally efficient in producing L-tert-leucine with >99% ee.168 Recent developments include the use of LeuDHs from other organisms and replacement of formate dehydrogenase-catalysed co-factor recycling by the more efficient glucose dehydrogenase system.169–173 Directed evolution of the LeuDH from L. aphaericus resulted in a two-fold increase in specific activity as compared to the wild-type enzyme, which was met with a concomitant increase in space-time yield from 666 to 1170 g (L d)−1.174
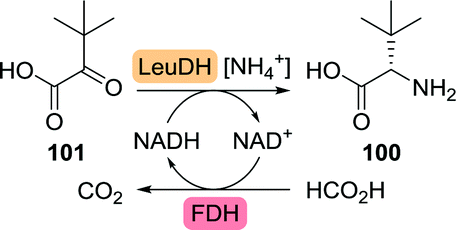 | ||
| Scheme 22 Enzymatic synthesis of L-tert-leucine with Leucine dehydrogenase (LeuDH) and co-factor recycling with formate dehydrogenase (FDH). | ||
Complementary to the use of LeuDH, several transaminases were also found capable of converting trimethylpyruvate (101) to L-tert-leucine (100). Transaminases (also called aminotransferases) catalyse the transfer of an amine group from a donor to an amine acceptor, which is a ketone or aldehyde. Most transaminases will accept α-amino acids as substrate (α-transaminases), but several can additionally convert primary amines not bearing an α-carboxylic acid moiety (ω-transaminases, sometimes also called aminotransaminases).
Similar to most (enzymatic) reactions, transaminase-catalysed reductive amination is reversible. This intrinsic equilibrium situation requires the use of an external driving force in order to favour product formation. Several approaches to shift the equilibrium for the synthesis of L-tert-leucine have been proposed. Fotheringham et al. employed an E. coli branched-chain amino acid aminotransferase (BCAT) to convert trimethylpyruvate to L-tert-leucine using glutamic acid (102) as the amine donor (Scheme 23).175 The formed side product, α-ketoglutarate (103), could be converted back to glutamic acid using a Bacillus subtilis ornithine aminotransferase. Ornithine aminotransferase requires ornithine (104) as amine donor and converts it to glutamate-γ-semialdehyde (105). The latter is susceptible to spontaneous, virtually irreversible, cyclisation, thereby resulting in 73% yield as compared to 31% in the absence of the recycling cascade.
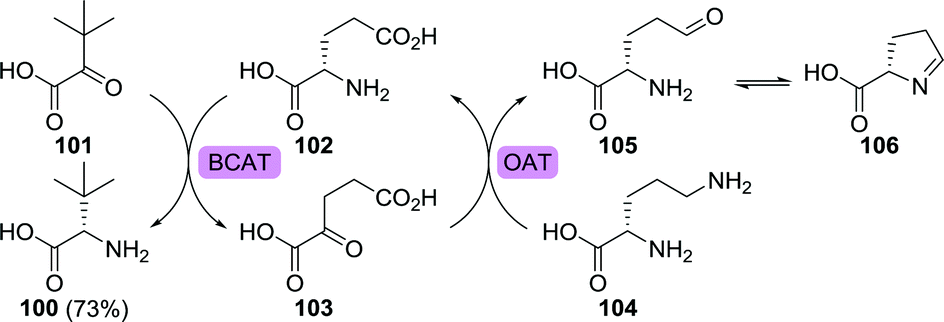 | ||
| Scheme 23 Synthesis of L-tert-leucine using a branched-chain amino acid aminotransferase (BCAT) and amine donor recycling/equilibrium shifting with ornithine aminotransferase (OAT).175 | ||
In a similar approach, Gefflaut and co-workers replaced ornithine aminotransferase with Thermosinus carboxydivorans Nor1 α-transaminase which was capable of employing glutamine as amine donor.176 The resulting α-ketoacid spontaneously cyclised, again shifting the reaction equilibrium to the product side. In an alternative approach, the authors employed Megasphaera elsdenii DSM20460 α-transaminase as this enzyme was shown to accept both trimethylpyruvate and glutamine as substrates. By doing so, the use of a single enzyme thus catalysed the formation of product and it ensured a favourable shift of the reaction equilibrium towards product formation. Alternative equilibrium shifting approaches involved transaminase-catalysed amine donor regeneration coupled to pyruvate decarboxylase or the use of an amino acid dehydrogenase for amine donor regeneration coupled to alcohol dehydrogenase-catalysed co-factor recycling at the expense of isopropanol.177,178
Using an enzymatic fuel cell, chemical energy can be converted to electrical energy. In an intriguing proof-of-concept study involving integration of enzymatic electrocatalysis in the enzymatic fuel cell, Minteer and co-workers were able to generate high added-value chemicals such as L-tert-leucine (100) from largely elementary starting materials (Scheme 24).179 In the anodic chamber, Pyrococcus furiosus soluble [NiFe] hydrogenase I (SHI) reduced methyl viologen (MV2+, 1072+) to MV˙+ (107˙+) via oxidation of hydrogen gas to protons. The protons migrated over the semi-permeable membrane to the cathodic chamber. Re-oxidation of MV˙+ at the anode resulted in the production of electrons, enabling reduction of MV2+ at the cathode. Nitrogen gas was subsequently converted to ammonia at the expense of MV˙+ using a nitrogenase. The presence of ammonia finally allowed LeuDH to convert trimethylpyruvate (101) to L-tert-leucine (100). In situ NADH co-factor recycling was achieved by oxidation of MV˙+ using a diaphorase from Geobacillus stearothermophilus.
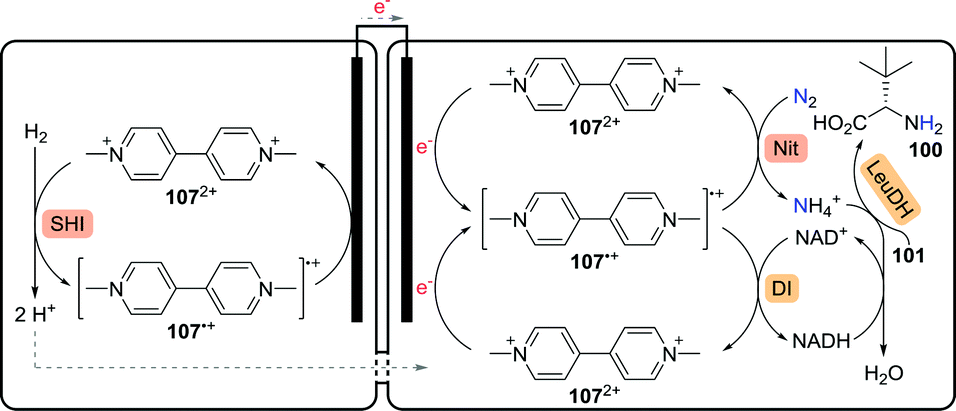 | ||
| Scheme 24 Enzymatic fuel cell-based synthesis of L-tert-leucine.179 SHI: soluble [NiFe] hydrogenase I, Nit: nitrogenase, DI: diaphorase and LeuDH: leucine dehydrogenase. | ||
Whereas most anti-viral peptidomimetic drugs are designed to combat HIV, telaprevir and boceprevir are used against the hepatitis C virus. From a biocatalytic perspective, telaprevir provides for a particularly interesting synthetic target. When retrosynthetically disassembled, one can envision its synthesis from six building blocks, which include three unnatural amino acids (Chart 4).23,180,181 One is the amino acid L-tert-leucine (100) discussed above and the other two are L-cyclohexylglycine (110) and the bicyclic proline analogue 111.
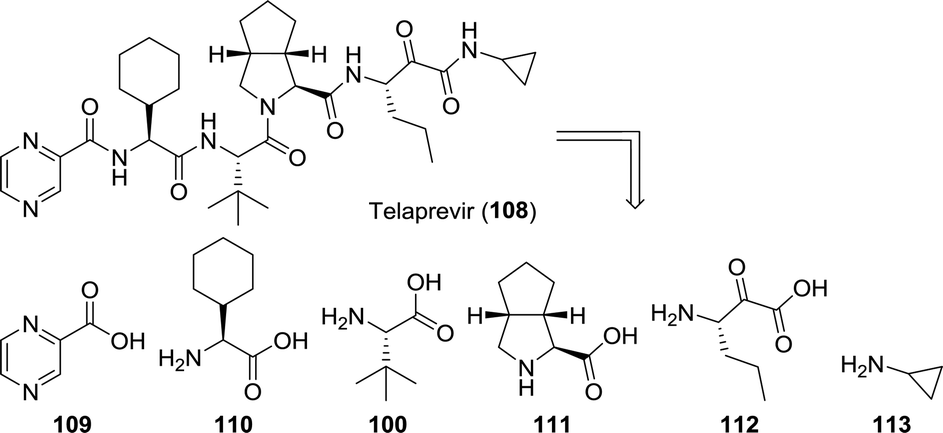 | ||
| Chart 4 Retrosynthetic analysis of telaprevir. | ||
Remarkably, Sorm and co-workers reported the resolution of N-acetyl-L-cyclohexylglycine using hog renal acylase I as early as 1966.182 More recently, Holla and co-workers described the resolution of racemic N-acetyl-L-cyclohexylglycine using a microbial acylase (Aspergillus sp.).183 Various other N-protected L-cyclohexylglycine derivatives could be resolved using, for example, penicillin G acylase.183 Although enzymatic resolution is most commonly achieved using acylases or lipases, other biocatalysts have been shown to be effective, too. Lin and Wang applied Rhodococcus sp. AJ270 cells to resolve numerous α-amino nitriles resulting in the formation of a wide variety of (unnatural) amino acids including L-cyclohexylglycine.184 An (S)-selective amidase and a non-stereoselective nitrile hydratase were shown to catalyse this microbial transformation. Again, kinetic resolution being limited to 50% yield makes asymmetric synthesis of the amino acid a more attractive approach. Such a synthesis was achieved by a team from Asymchem through enzymatic reductive amination of pro-chiral 2-cyclohexyl-2-oxoacetate using a LeuDH from Bacillus sphaericus.185
The third unnatural amino acid building block of telaprevir is the bicyclic proline analogue 111. Turner and co-workers employed desymmetrisation of commercially available 114 using a monoamine oxidase (MAO) variant to yield the corresponding 1Δ-pyrroline (115, Scheme 25A).186 MAO catalyses the oxidative deamination of amines to ketones and aldehydes. The employed fungal MAO-N D5 from Aspergillus niger, however, is a more versatile engineered variant that also catalyses reductive amination and oxidation of cyclic amines, for example.187 Hydrocyanation of 115, followed by nitrile hydrolysis subsequently furnished 111. In collaboration with the group of Orru, an Ugi-type 3-component reaction was developed for the synthesis of various prolyl peptides by starting from a carboxylic acid component, a nitrile component and a cyclic imine.188 This approach was ultimately employed for the synthesis of telaprevir through the use of an elegant late stage 3-component coupling involving 116, 117 and 115, thereby eliminating the need to access amino acid 111 separately (Scheme 25B).189
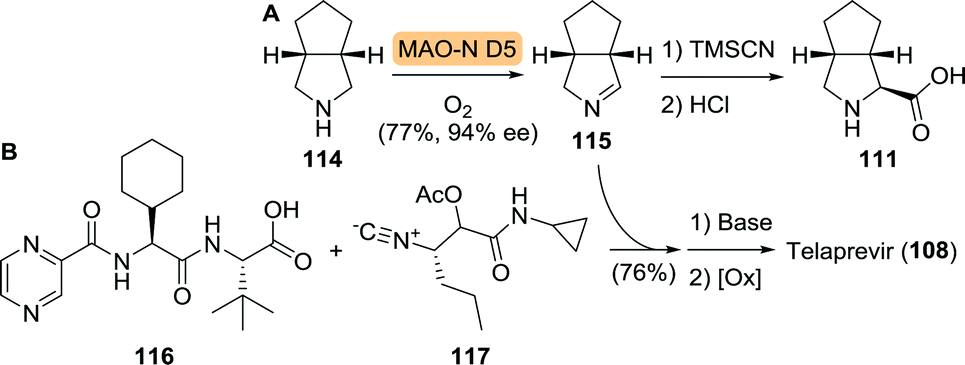 | ||
| Scheme 25 Oxidation of cis-3-aza-bicyclo[3.3.0]octane (114) by monoamine oxidase variant D5 (MAO-N D5) and subsequent chemical synthesis to the corresponding amino acid 111 (A).186 Late stage Ugi-type 3-component reaction towards the synthesis of telaprevir (B).189 | ||
The MAO-N D5 variant used in the synthesis of 115 had been employed in the millimolar range, which would not be viable on industrial scale. A collaboration between Merck and Codexis resulted in the engineering of a highly active MAO-N variant capable of resisting temperatures up to 50 °C and allowing substrate concentrations above 1 M.190
Similar to telaprevir, boceprevir could be built up from an unnatural bicyclic proline analogue (120, Scheme 26). Use of the Merck/Codexis MAO-N variant resulted in oxidation of 118 with high yield and ee. In situ capture of the corresponding 1Δ-pyrroline 119 using sodium bisulfite and subsequent chemical transformations led to an efficient synthesis of the methyl ester of 120 (120-Me) in 56% overall yield and >99% ee.
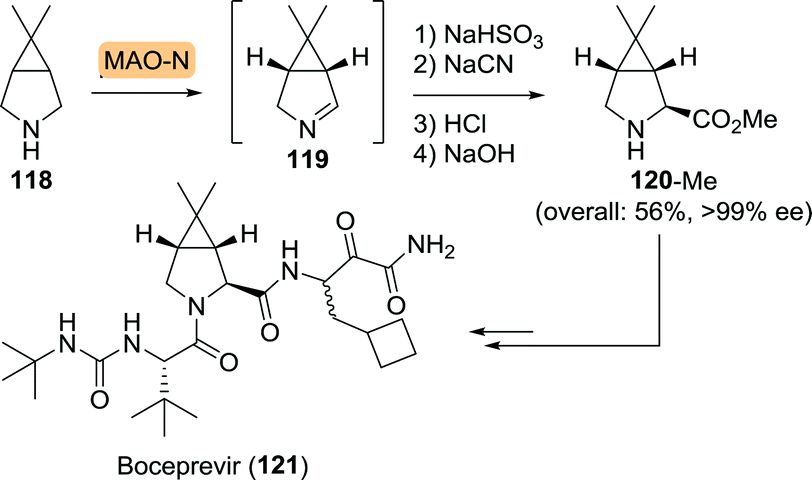 | ||
| Scheme 26 Monoamine oxidase (MAO-N) variant-catalysed synthesis of boceprevir intermediate.190 | ||
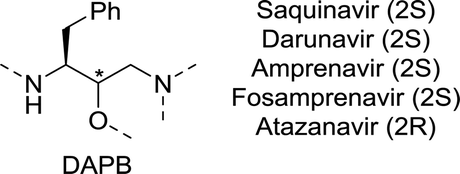 | ||
| Chart 5 (3S)-1,3-diamino-4-phenylbutan-2-ol (DAPB) core of saquinavir, darunavir, (Fos)amprenavir and atazanavir. | ||
A team at Roche approached the synthesis of saquinavir from several directions.196 Retrosynthetically, saquinavir was disconnected as such that its building blocks could be assembled largely through simple amide coupling (Scheme 27A). In one of these approaches, the final disconnection involves nucleophilic attack of the decahydroisoquinoline building block 125 on the protected aminodiol 124 resulting in the formation of the DAPB core.
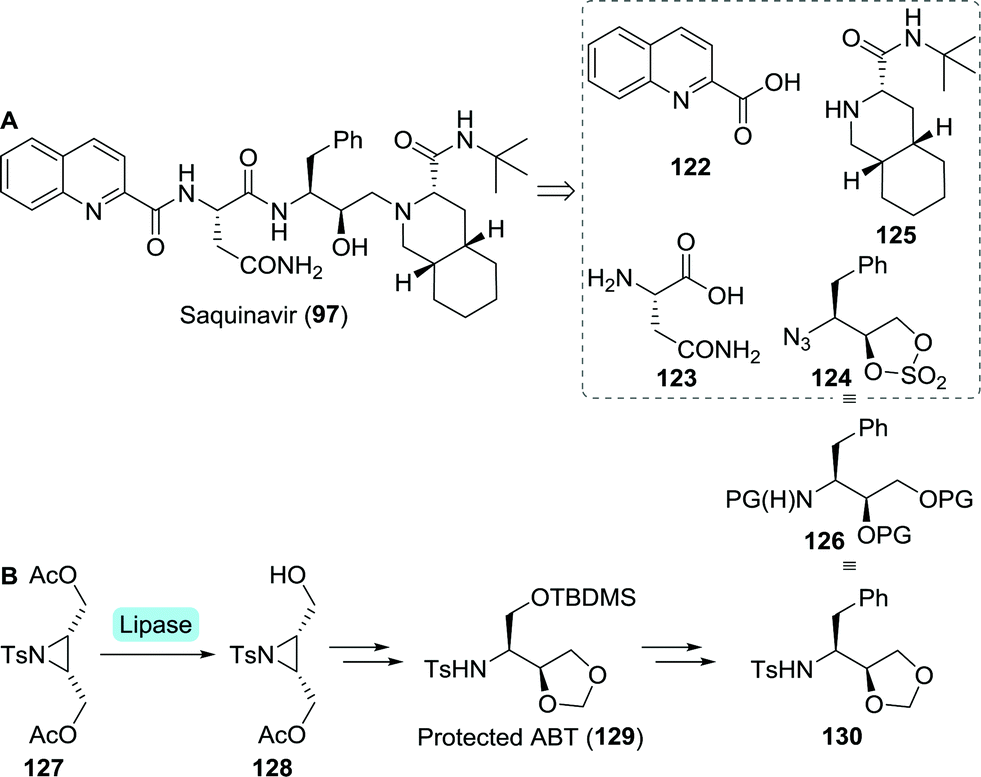 | ||
| Scheme 27 Retrosynthetic analysis of saquinavir (A). Lipase-catalysed desymmetrisation of a tosylated aziridine intermediate towards protected 3-aminobutane-1,2,4-triol (ABT) and protected (2S,3S)-3-amino-4-phenylbutane-1,2-diol (B).198 | ||
Sugiura and co-workers employed desymmetrisation of a bis-acetylated tosyl aziridine (127) using Pseudomonas sp. lipase for the synthesis of its mono-acetylated counterpart 128 (Scheme 27B).197 Several subsequent chemical steps led to the formation of protected 3-aminobutane-1,2,4-triol (ABT, 129), a key building block of nelfinavir (vide infra).198129 could subsequently be converted to the desired protected aminodiol 130. As protection of 130 is analogous to that of 124, this could serve as building block for saquinavir too.
Due to concerns regarding the stability of the azido-protected (2S,3S)-3-amino-4-phenylbutane-1,2-diol derivative 124, Roche abandoned this route.196 Instead, replacing the sulphate protected diol by an epoxide was considered favourable to facilitate the subsequent coupling step.
Chemoenzymatic synthesis of chiral epoxide 133 (PG = Boc) was initially explored at multigram scale by Bristol Myers Squibb (Scheme 28).199 Microbial reduction of 131 by Streptomyces nodosus SC 13149 resulted in the formation of chlorohydrin (2R)-132 with excellent enantio- and diastereomeric purity, which, upon treatment with base, would afford (2R)-133. As a follow-up of this work, Bristol Myers Squibb investigated several Rhodococcus strains for the production of (2R)-132 and employed the building block in the synthesis of atazanavir.200 Subjecting 131 to reduction by a ketoreductase from Ralstonia sp. led to the interesting observation that the enzyme's stereoselectivity could be reversed by switching the N-protecting group.201 Use of Boc-protected 131 resulted in the formation of (2R)-132, whereas the use of its Cbz-protected counterpart resulted in the formation of (2S)-132. Recently, Shao et al. subjected a short-chain dehydrogenase from Novosphingobium aromaticivorans to multiple rounds of mutagenesis to obtain a variant (G141V/I195L) that was approximately four times more active against 131 than the wild-type enzyme.202
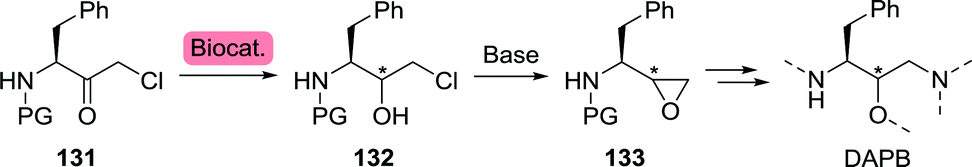 | ||
| Scheme 28 Stereoselective biocatalytic reduction towards the construction of the (3S)-1,3-diamino-4-phenylbutan-2-ol (DAPB) core of saquinavir, darunavir, (Fos)amprenavir and atazanavir. For readability and continuity, atoms have been numbered as to match numbering for 1,3-diamino-4-phenylbutan-2-ol (DAPB). | ||
The central scaffold of nelfinavir differs from DAPB in that the phenyl moiety is replaced by a thiophenol.203 As with DAPB, the nelfinavir core could be accessed through the thiophenol equivalent of 131,204 which in turn allowed for KRED catalysis to furnish the corresponding (2R)-chlorohydrin as reported by Pace and co-workers.205 It was shown that, as for the synthesis of 131 from L-phenylalanine, S-phenyl-L-cysteine could serve as starting material for the corresponding α-chloroketone.204,205 Although several patented procedures are available for the biocatalytic synthesis of the non-proteinogenic amino acid S-phenyl-L-cysteine, at the time productivity did not exceed laboratory scale. Mitsui Toatsu Chemicals, however, developed a scalable procedure using tryptophan synthase-producing E. coli to convert L-serine and thiophenol to S-phenyl-L-cysteine.206
By re-engineering the cysteine biosynthetic pathway in Escherichia coli, Maier developed a strain capable of producing a large variety of unnatural amino acids including S-phenyl-L-cysteine (137, Scheme 29).207 The wild-type microbial strain converted glucose (90) to serine (134) and thereafter to O-acetylserine (135) using serine acetyltransferase (SAT). With sulfide as the nucleophile, the enzyme O-acetylserine sulfhydrylase subsequently converted O-acetylserine to cysteine (136). O-Acetylserine sulfhydrylase was shown to also accept other nucleophiles instead of sulfide to produce unnatural amino acids in low yield. The low yield was attributed to inhibition of SAT by cysteine, which thereby limited the overall concentration of O-acetylserine. Screening of several SAT variants led to the discovery of an enzyme that was less susceptible to inhibition by cysteine while retaining 50% activity (SATfbr). Implementation of the gene encoding for SATfbr allowed for effective microbial production of S-phenyl-L-cysteine (137) using thiophenol as nucleophile (72% yield from O-acetylserine).
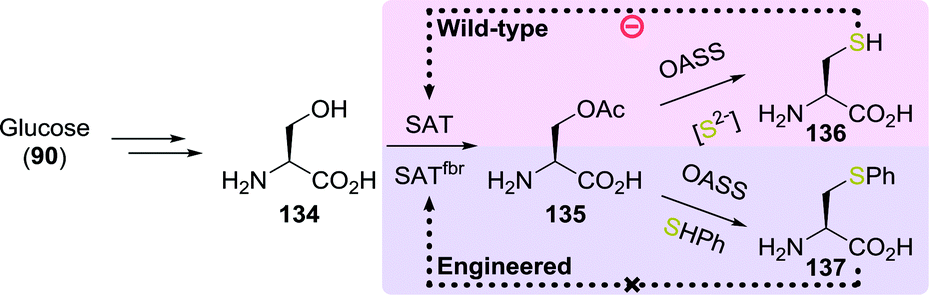 | ||
| Scheme 29 Fermentation with wild-type and engineered E. coli for the production of L-cysteine and S-phenyl-L-cysteine.207 SAT: serine acetyltransferase and OASS: O-acetylserine sulfhydrylase. | ||
However, both the high costs of S-phenyl-L-cysteine and the possibility to access the arylthioether through late-stage substitution make other synthetic strategies more attractive. Most routes instead involve the use of protected 3-aminobutane-1,2,4-triol (ABT, 138, Scheme 30).208–210 Suitably protected equivalents of ABT could be obtained efficiently through biocatalytic resolution. Faure and co-workers employed either single-step or two-step lipase-catalysed resolution of N-protected ABT to obtain (2S,3R)-144 from 143 (Scheme 31A). Improved enantiomeric purity was observed for the two-step-resolution approach (>99% vs. <98%).211 Noto and co-workers analogously used immobilised Amano PS lipase (a Burkholderia cepacia (BCL) preparation) to resolve dioxepanes 145 and 146 at laboratory scale (Scheme 31B).212
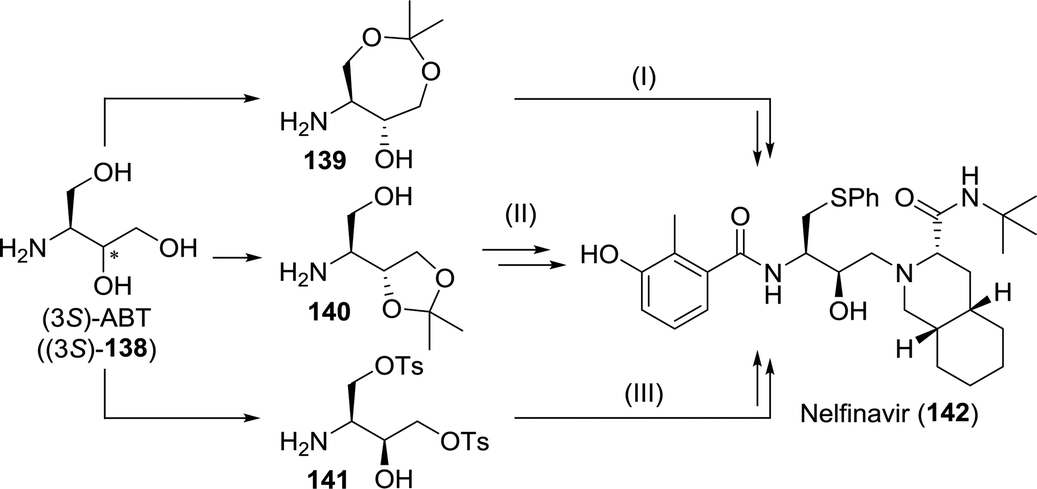 | ||
| Scheme 30 Synthesis of nelfinavir from suitably protected 3-aminobutane-1,2,4-triol (ABT) as described by Uchida et al. (I),208 Cho and co-workers (II)209 and Borer et al.210 | ||
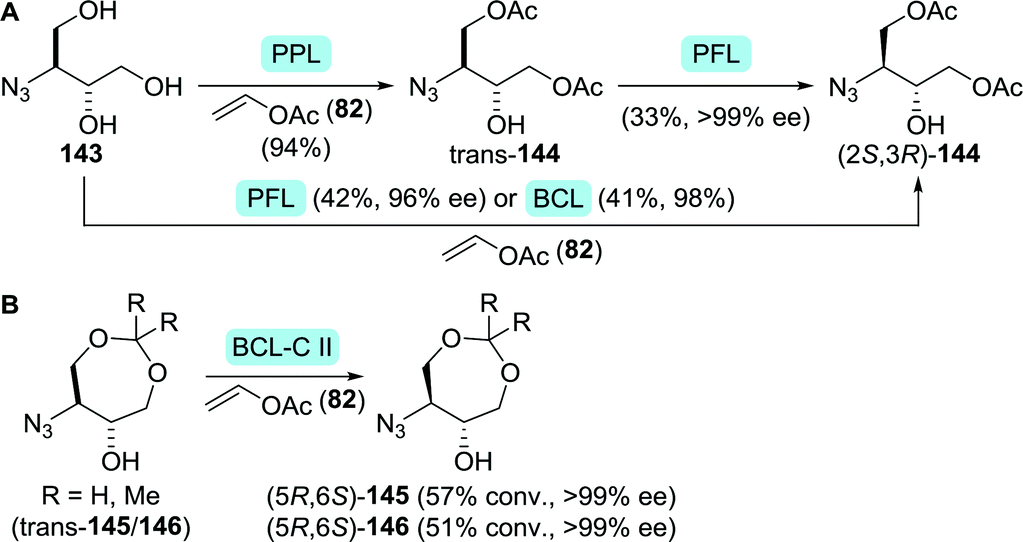 | ||
| Scheme 31 Resolution of protected ABT derivatives 143 (A)211 and 145/146 (B).212 PPL: porcine pancreas lipase, PFL: Amano AK lipase (P. fluorescens), BCL: Amano PS lipase (Burkholderia cepacia) and BCL-C II: BCL immobilised on ceramic instead of diatomite. | ||
Bare ABT 138 itself also provides for an attractive target for biocatalytic production due to its high solubility and the presence of two adjacent chiral centres in a small molecule. Lye and co-workers developed a two-step enzymatic cascade for the synthesis of ABT.213 Transketolase catalyses the interconversion of two phosphorylated C2–C5 ketoses/aldoses. Several transketolase types, however, can also accept non-phosphorylated sugar derivatives. E. coli transketolase was used to synthesise L-erythrulose (148) from glycolaldehyde (147) and hydroxypyruvate (149) by carboligation (Scheme 32A). Decarboxylative coupling of an α-ketoacid such as hydroxypyruvate results in the (irreversible) formation of CO2, which provides a strong driving force for product formation. An ω-transaminase from Pseudomonas aeruginosa was used to transfer the amine moiety from (S)-α-methylbenzylamine ((S)-150) to the intermediate L-erythrulose. A laboratory scale two-step, one-pot cascade reaction was enabled by overexpressing the endogenous transketolase and the transaminase in E. coli.
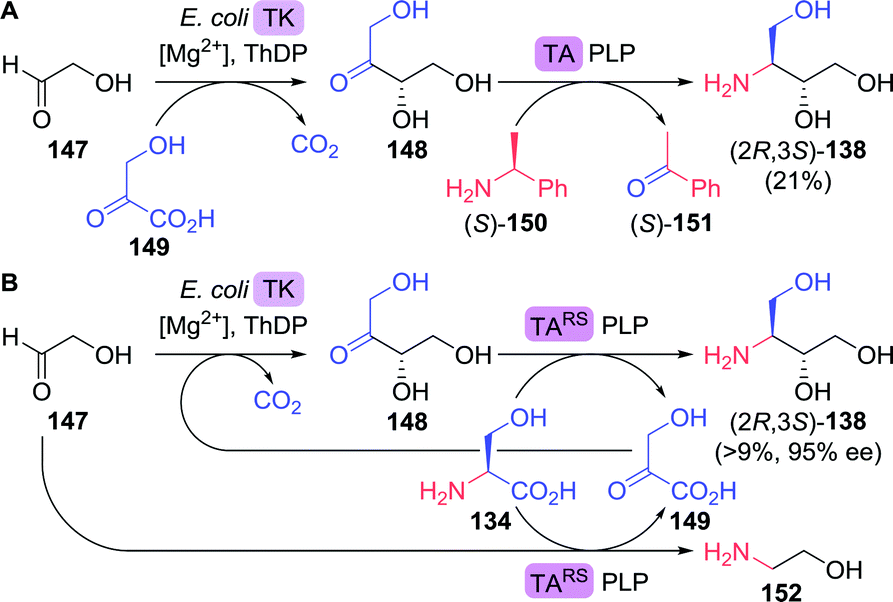 | ||
| Scheme 32 Two-step enzymatic synthesis of 3-aminobutane-1,2,4-triol (A)213 and inclusion of in situ hydroxypyruvate formation/amine donor recycling (B).216 TK: transketolase, ThDP: thiamine diphosphate, TA: transaminase and PLP = pyridoxal phosphate. | ||
Although constituting a very attractive cascade, no enantiomeric and/or diastereomeric purity was reported, the transaminase reaction was in equilibrium, (S)-α-methylbenzylamine proved an inconvenient amine donor and hydroxypyruvate is chemically unstable.214 By switching to a highly versatile ω-transaminase from Chromobacterium violaceum capable of employing isopropylamine as simple donor, formation of volatile acetone pushed the reaction equilibrium in favour of product formation and side reactions could be suppressed. This optimised cascade resulted in the formation of (2R,3S)-138 as a single stereoisomer (>95% ee).215 Addition of a second α-transaminase enzyme from Deinococcus geothermalis DSM11300 allowed for the in situ generation of hydroxypyruvate from L-serine (134), which enabled continuous dosing of this labile intermediate.216
By using a single transaminase from Rhodobacter sphaeroides ATCC17025 (TARS) with specificity for both L-erythrulose (148) and L-serine, simultaneous generation of hydroxypyruvate (149) and ABT led to a simplified semi closed-loop cascade (Scheme 32B). Although the required initial formation of hydroxypyruvate was ensured as TARS also utilised glycolaldehyde as an amine acceptor, significant amination of this substrate led to a limitation of the overall ABT yield to a maximum of 9%. As the complex reaction system requires careful adjustments, further process optimisation is being pursued extensively.217–219
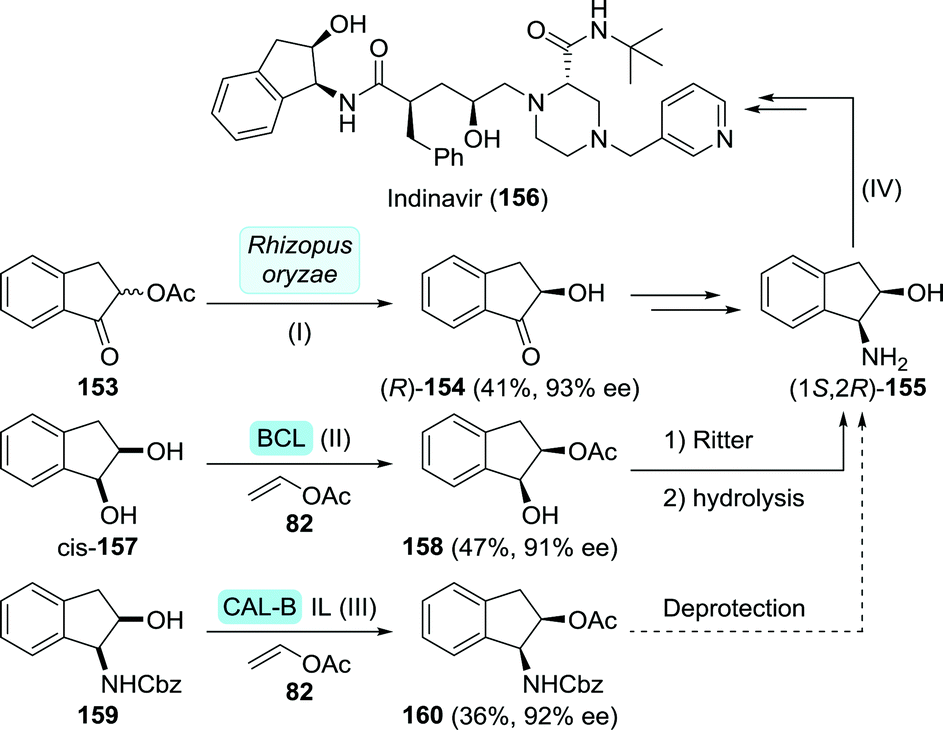 | ||
| Scheme 33 Synthesis of (1S,2R)-1-amino-2-indanol via biocatalytic resolution as described by Ozgul et al. (I),221 Nohira et al. (II)222 and Afonso and co-workers (III),223 and the synthesis of indinavir from this intermediate as described by Zhong and co-workers (IV).225 BCL: Amano PS lipase (Burkholderia cepacia), IL: ionic liquid and CAL-B: Candida antarctica lipase B. | ||
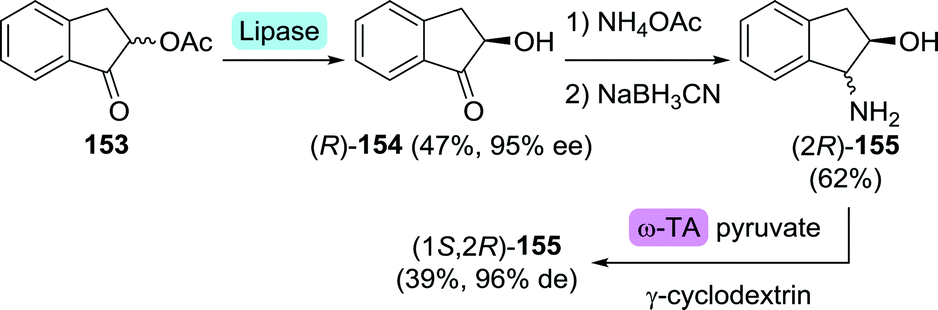 | ||
| Scheme 34 Four-step route to (1S,2R)-amino-2-indanol including lipase and transaminase (TA)-catalysed resolution.224 | ||
Earlier attempts at resolving intermediates of (1S,2R)-1-amino-2-indanol involved the use of Pseudomonas putida to achieve dihydroxylation of indene (161, Scheme 35).226,227 As the reaction initially yielded both enantiomers of cis-indane-1,2-diol (157), dihydroxylation was suggested to be catalysed by an insufficiently stereoselective toluene dioxygenase. With extended reaction duration, enantiomeric excess increased to >90% in favour of (1S,2R)-157, however. Based on the concomitant formation of α-hydroxyindanone (154), the authors suggested the presence of a cis-glycol dehydrogenase that exclusively oxidised (1R,2S)-157. Alternatively, (1S,2R)-Indane-1,2-diol could be accessed by reduction of indane-1,2-dione using Trichosporon cutanaeum MY1506.228 Synthesis of the target (1S,2R)-1-amino-2-indanol from (1S,2R)-indane-1,2-diol could be achieved using a Ritter reaction.229
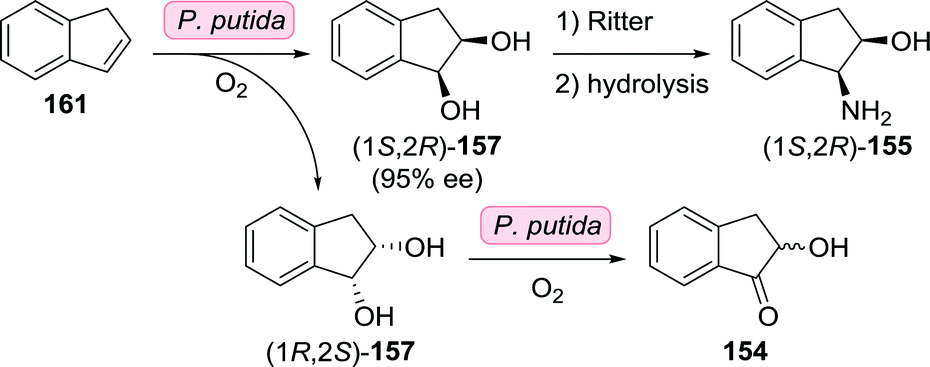 | ||
| Scheme 35 Oxidation of indene by P. putida towards the synthesis of (1S,2R)-1-amino-2-indanol.226,227 | ||
Indinavir's piperazine fragment could also be accessed biocatalytically. Hydrolysis of racemic piperazine-2-carboxamide using an amidase from Klebsiella terrigena DSM 9174 afforded (S)-piperazine-2-carboxylic acid in 99% ee, albeit in low yield (22%).230
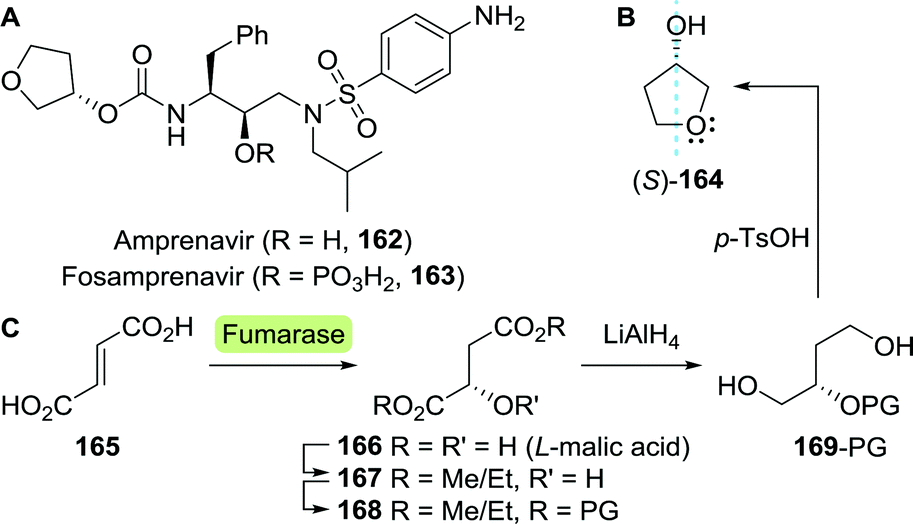 | ||
| Scheme 36 Structures of amprenavir and fosamprenavir (A). 3-Hydroxytetrahydrofuran including plain of symmetry indicating the near absent difference in size of the left- and right-hand ring fragments (B). Fumarase-catalysed hydration of fumarate, subsequent protection, reduction to the 2-protected (S)-1,2,4-butanetriol and acid-mediated deprotection/cyclisation to yield (S)-3-hydroxytetrahydrofuran (C). PG: protecting group. | ||
Whereas screening of over 100 hydrolytic enzymes by Bornscheuer et al. resulted in the discovery of candidates capable of resolving three other challenging substrates, resolution of acetylated (S)-3-hydroxytetrahydrofuran remained elusive.231 Engineering of a Bacillus stearothermophilus esterase, however, effected a more than two-fold increase in stereoselectivity (E-factor 4.3 to 10.4).232 Although a significant improvement, this remains insufficient for practical applications.
Reetz and co-workers instead focused on biocatalytic reduction of pro-chiral oxolan-3-one. Initially, reduction with Thermoethanolicus brockii alcohol dehydrogenase afforded (R)-3-hydroxytetrahydrofuran with only 23% ee. Directed evolution of this highly stable enzyme was pursued in order to reverse and improve its stereoselectivity. The triple mutant I86V/W110L/L294Q proved most effective as it was able to fully convert oxolan-3-one with 94% ee in favour of the (S)-enantiomer.233
The groups of Wynberg and Izawa showed that (S)-3-hydroxytetrahydrofuran could be prepared from the hydroxy-protected acyclic precursor L-malate dialkyl ester (168).234,235 The latter was synthesised from L-malic acid (166), which is commercially produced by fermentation on industrial scale through fumarase-catalysed hydration of fumarate (165, Scheme 36C).236L-Malic acid can also be prepared from the racemate through kinetic resolution.237
The chemical synthesis of (S)-3-hydroxytetrahydrofuran proceeded through protected (S)-1,2,4-butanetriol (169-PG). As shown by Chung and co-workers, production of 1,2,4-butanetriol could also be achieved from renewable resources through the use of an engineered E. coli strain capable of fermenting xylose (Scheme 37).238 Introduction of heterologous genes encoding for Caulobacter crescentus xylose dehydrogenase (XDH) allowed for the oxidation of xylose (170) to xylonic acid (171). C3-dehydration by E. coli's native D-xylonic acid dehydratase (DHT) followed by decarboxylation with benzoylformate decarboxylase (mdlC) from Pseudomonas putida afforded 3,4-dihydroxybutanal (173). The latter could be reduced to 1,2,4-butanetriol (169) using native KREDs. Disruption of the genes encoding for 2-keto-3-deoxy-D-xylonate aldolase (kdxA) and the enzymes responsible for xylose isomerisation (xylA and xylB) resulted in an additional improvement of the cascade to yield 1,2,4-butanetriol in 0.88 g L−1 (13% overall yield). In comparison, a two-step, two-microbe system for the production of (S)-169 as reported by Frost and co-workers (initial production of 171 with XDH from P. fragi), proved to be more efficient (1.6 g L−1, 18% overall yield and >99% ee).239 Through re-engineering of the malate pathway, glucose could also be used as carbon-source for the production of 1,2,4-butanetriol, albeit in titres of only 55 mg L−1.240 Alternatively, resolution of racemic 1,2,4-butanetriol provided access to both enantiomers.150
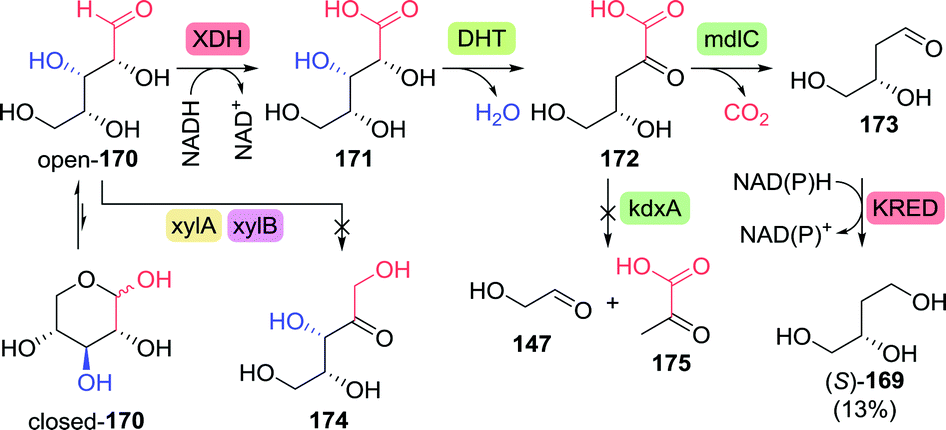 | ||
| Scheme 37 Production of (S)-1,2,4-butanetriol through fermentation of xylose with engineered E. coli.238 XDH: xylose dehydrogenase, DHT: D-xylonic acid dehydratase, mdlC: benzoylformate decarboxylase, xylA/xylB: xylose isomerisation enzymes, kdxA: 2-keto-3-deoxy-D-xylonate aldolase and KRED: ketoreductase. | ||
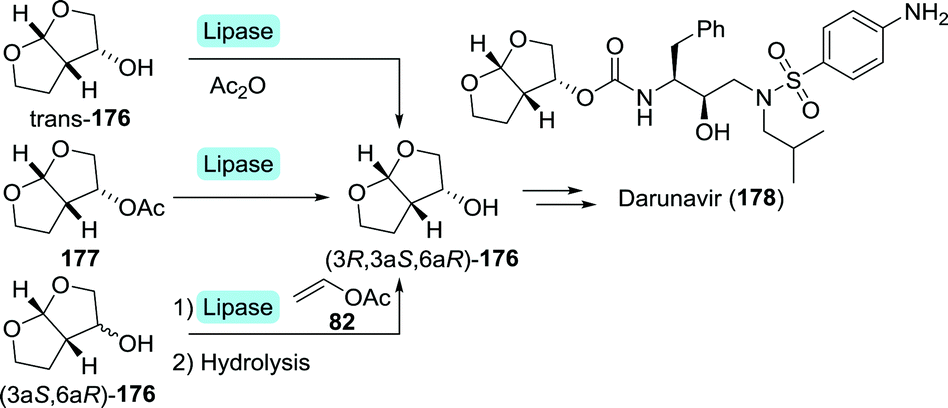 | ||
| Scheme 38 Enzymatic resolution of (acetylated) 3-OH-bis-THF with lipase. | ||
Several multi-step, one-pot procedures that include lipase resolution have been published for the synthesis of 3-OH-bis-THF. Itoh and co-workers devised a strategy involving organocatalytic cycloaddition of glycolaldehyde (147) and 1,2-dihydrofuran (179) to obtain 3-OH-bis-THF as a mixture of diastereomers.247 The latter was subsequently resolved using BCL (Scheme 39A). Around the same time, Opatz et al. reported a three-step, one-pot telescoping approach to (3R,3aS,6aR)-176,248 making use of a stereoselective [2+2]-photocycloaddition between furan (181) and Cbz-protected glycolaldehyde (182) to afford the bicyclic oxetane 183 (Scheme 39B). Hydrogenative deprotection resulted in the spontaneous rearrangement of ring-strained 184 to yield trans-176. The latter was subsequently resolved using porcine pancreas lipase (PPL) and propionic anhydride as acylating agent. PPL showed enhanced enantioselectivity as compared to Pseudomonas fluorescens lipase and CAL-B, although preferentially for the undesired acylated product. Interestingly, switching from acetic anhydride to propionic anhydride resulted in an increase of ee from 93% to >99%. The use of propionic anhydride also shortened reaction times by 24 h as compared to other donors.
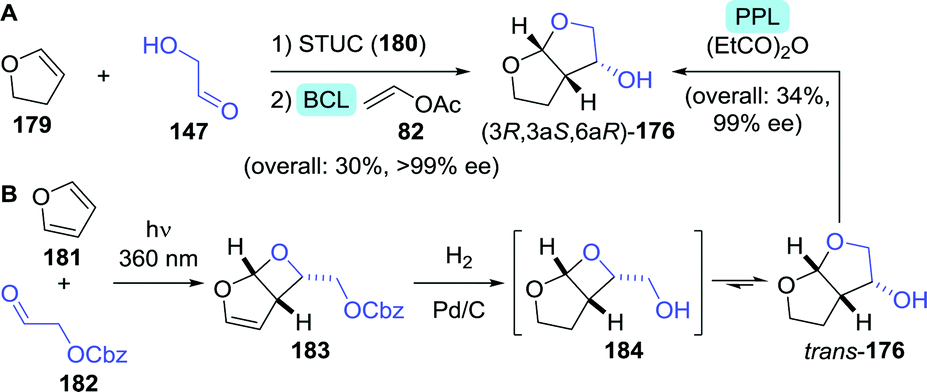 | ||
| Scheme 39 Two-step, one-pot organocatalytic cyclisation followed by lipase-catalysed resolution (A)247 and three-step, one-pot photocatalytic cyclisation, followed by deprotection and lipase-catalysed resolution (B)248 for the synthesis of 3-OH-bis-THF. BCL: Amano PS lipase (Burkholderia cepacia), STUC: Schreiner's thiourea catalyst and PPL: porcine pancreas lipase. | ||
3.4 Bis-cyclopropyl-based protease inhibitors
Among the anti-viral agents classified as peptide mimics, the bis-cyclopropyl-based drugs form a defined sub-class of protease inhibitors for combating the hepatitis C virus. Again, a large degree of structural similarity is observed among these agents. All share a cyclopropylsulfonamide linked to a (1R,2S)-1-aminocyclopropanecarboxylic acid (ACCA) moiety flanked by a hydroxyproline derivative or analogue thereof. An additional common feature is that most are macrocyclic, except for asunaprevir and vaniprevir (Chart 6).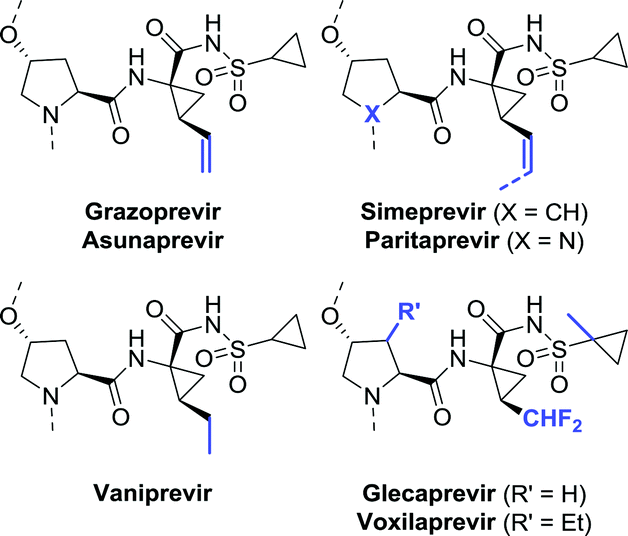 | ||
| Chart 6 Common structure of biscyclopropyl-based protease inhibitors. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 7.1 g L−1 of L-hydroxyproline could be produced from glucose.
:1), 7.1 g L−1 of L-hydroxyproline could be produced from glucose.
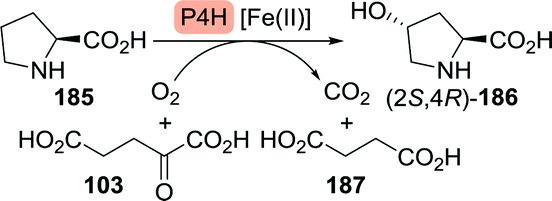 | ||
| Scheme 40 Hydroxylation of L-proline with proline 4-hydroxylase (P4H). | ||
Clapés and co-workers recently developed a de novo chemoenzymatic approach towards hydroxyproline by starting from simple building blocks.254 Aldol addition of pyruvate (175) to Cbz-protected 2-aminoacetaldehyde (188) catalysed by 2-keto-3-deoxy-L-rhamnonate aldolase (YfaU) was followed by treatment of the condensation product with palladium on carbon. The latter resulted in amine deprotection, spontaneous cyclisation and subsequent reductive amination to yield hydroxyproline as a mixture of stereoisomers (cis-186, 40% de, Scheme 41). YfaU-catalysed carboligation did not occur stereoselectively, likely due to the achiral nature of the protected 2-aminoacetaldehyde and the equilibrium nature of the conversion favouring thermodynamic over kinetic control.
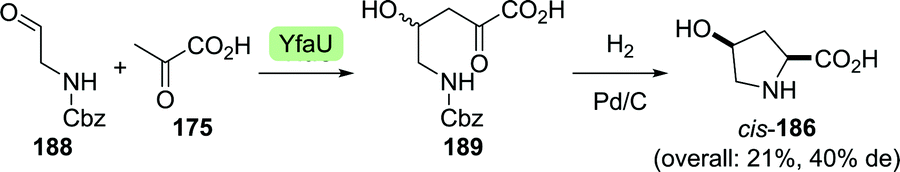 | ||
| Scheme 41 Condensation of N-Cbz-protected 2-aminoacetyaldehyde and pyruvate catalysed by 2-keto-3-deoxy-L-rhamnonate aldolase (YfaU) followed by deprotection/cyclisation to obtain racemic hydroxyproline.254 | ||
In a similar approach, Franssen et al. used 2-keto-3-deoxygluconate aldolase from Sulfolobus acidocaldarius to condense 2-azidoacetaldehyde and pyruvate in a stereoselective fashion.255 Zinc-catalysed deprotection and reduction resulted in the generation of hydroxyproline as a mixture of diastereomers in which the undesired (4S)-isomer prevailed. Hydroxylation of L-proline is thus the preferred route to the desired (2S,4R)-hydroxyproline isomer.
The synthesis of vinyl-ACCA as outlined by Boehringer Ingelheim in 2005 is regarded as the gold standard for the production of this compound (Scheme 42A).264 Their synthetic strategy involved resolution of the N-Boc-protected vinyl-ACCA methyl ester (cis-190) by Alcalase 2.4L (a Bacillus licheniformis protease formulation). Although applicable on large scale, two consecutive resolution steps were required in order to reach 99% ee for the desired remaining isomer (1R,2S)-190, resulting in a complex, time-consuming approach. In an independent study, resolution of the N-Boc protected vinyl-ACCA ethyl ester with Sphingomonas aquatilis whole cells afforded the remaining (1R,2S)-configured ester with 88% ee.265 Strikingly, resolution of unprotected racemic vinyl-ACCA was claimed to result in a reversal of selectivity; the formed (1R,2S)-configured ester was isolated in 99.6% ee.
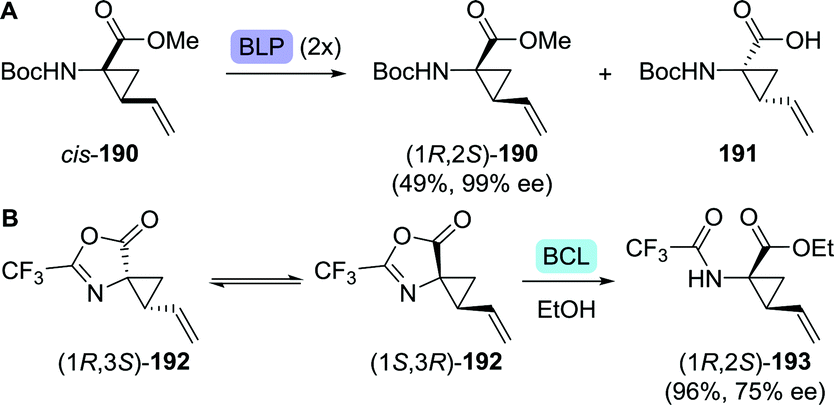 | ||
| Scheme 42 Resolution of the N-Boc-protected amino-2-vinylcyclopropanecarboxylic acid (vinyl-ACCA) methyl ester with Alcalase 2.4L (a Bacillus licheniformis protease (BLP) formulation) (A).264 Dynamic kinetic resolution with Amano PS lipase (Burkholderia cepacia, BCL) to obtain protected vinyl-ACCA (B).266 | ||
Kroll and co-workers protected racemic vinyl-ACCA as its trifluoromethyl-substituted azlactone (192, Scheme 42B).266 Interconversion of both enantiomers of 192, which was proposed to be enabled through an unusual [3,3]-sigmatropic oxadivinylcyclopropane rearrangement, allowed for dynamic kinetic resolution through lipase-catalysed ethanolysis. Unfortunately, using BCL for hydrolysis of the desired stereoisomer resulted in only 75% ee and 96% yield after 18 days. Ethyl-ACCA, a vaniprevir fragment, could be obtained through reduction of vinyl-ACCA.267,268
Glecaprevir and voxilaprevir both contain a difluoromethyl-ACCA fragment. A team at Abbvie developed a synthetic route to glecaprevir in which difluoromethyl-ACCA was derived from vinyl-ACCA via multi-step chemical conversion.269 As the yield was moderate and the synthesis required a complicated work-up procedure, this process proved not to be viable on large scale. The authors disclosed that alternative synthetic procedures had been developed.270 One of these included the preparation of enantiomerically enriched difluoromethyl-ACCA through two consecutive enzymatic resolution steps (Scheme 43).271 Dipropyl 2-(difluoromethyl)cyclopropane-1,1-dicarboxylate (rac-194) would first be subjected to resolution with Thermomyces lanuginosus lipase (AH45) to isolate the desired (R)-enantiomer (R)-194 (Scheme 43). Subsequent desymmetrisation with an esterase from either Bacillus subtilis (yvaK) or Bacillus stearothermophilus (BsteE), followed by Curtius rearrangement and hydrolysis afforded N-Boc-protected difluoromethyl-ACCA (196).
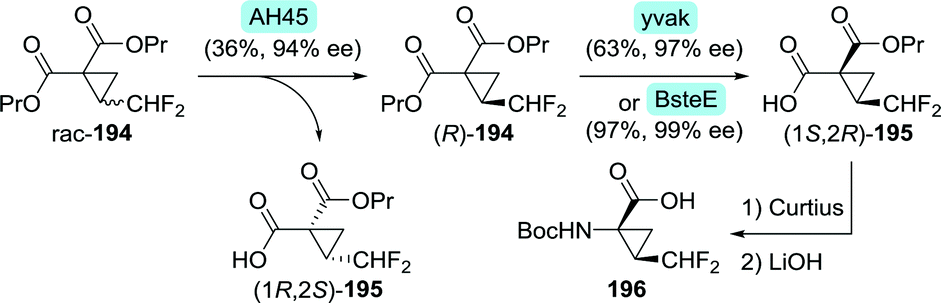 | ||
| Scheme 43 Two-step enzymatic resolution of a difluoromethyl-ACCA intermediate.271 AH45: Thermomyces lanuginosus lipase, yvaK: Bacillus subtilis esterase and BsteE: Bacillus stearothermophilus esterase. | ||
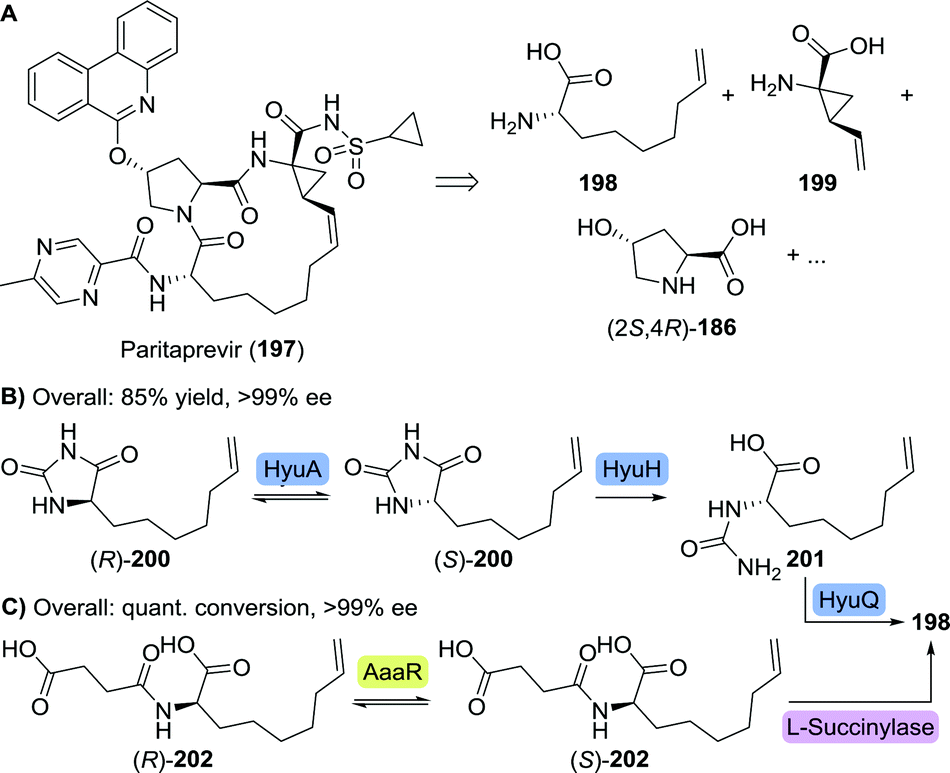 | ||
| Scheme 44 Retrosynthetic analysis of paritaprevir (A). Enzymatic dynamic kinetic resolution for the synthesis of L-6-heptenylglycine through its hydantoin (B)274 or its N-succinate (C).275 HyuA: hydantoin racemase from Agrobacterium radiobacter, HyuH: L-hydantoinase from Arthrobacter aurescens, HyuQ: L-carbamoylase from Bacillus stearothermophilus, aaaR: N-acylamino acid racemase and quant. conv.: quantitative conversion. | ||
Dutch life-sciences company DSM developed a dynamic kinetic resolution process comprising of three enzymes, which required L-6-heptenylglycine to be prepared as its hydantoin derivative (200).274 An L-selective hydantoinase from Arthrobacter aurescens (HyuH) ensured enantioselective ring-opening and the L-carbamoylase from Bacillus stearothermophilus (HyuQ) subsequently cleaved the resulting urea moiety (Scheme 44B). Rapid interconversion of both hydantoin enantiomers was enabled by a hydantoin racemase from Agrobacterium radiobacter (HyuA). In a complementary approach, Japanese Kaneka applied a N-acylamino acid racemase (AaaR)-catalysed interconversion of racemic N-succinyl 6-heptenylglycine (202), followed by kinetically controlled enantioselective amide cleavage by L-succinylase to furnish the desired enantiopure amino acid (198, Scheme 44C).275
Finally, a team at Merck developed an efficient four-step asymmetric synthesis of L-6-heptenylglycine via 2-oxonon-8-enoic acid (204) which in turn was obtained through Grignard chemistry (Scheme 45).276 Use of a leucine dehydrogenase from Codexis converted the ketoacid 204 to L-6-heptenylglycine (198) with >99% ee. Subsequent Boc protection and isolation of 205 as its dicyclohexylamine salt allowed for an overall yield of 60% starting from 7-bromoheptene (203), thereby clearly outcompeting non-dynamic resolution approaches as such strategies would result in a maximum theoretical yield of 50%.
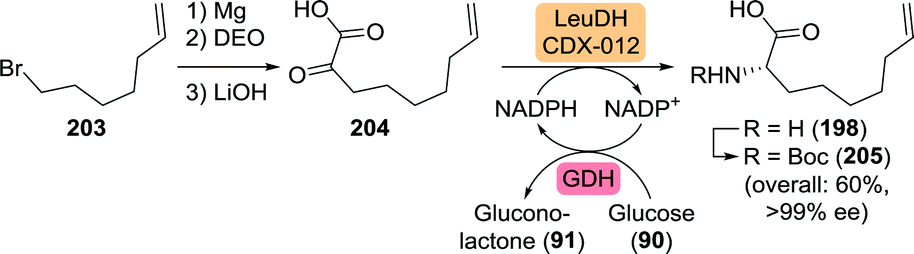 | ||
| Scheme 45 Concise synthesis of 2-oxonon-8-enoic acid and subsequent reductive amination catalysed by a leucine dehydrogenase (LeuDH).276 GDH: glucose dehydrogenase and DEO: diethyl oxalate. | ||
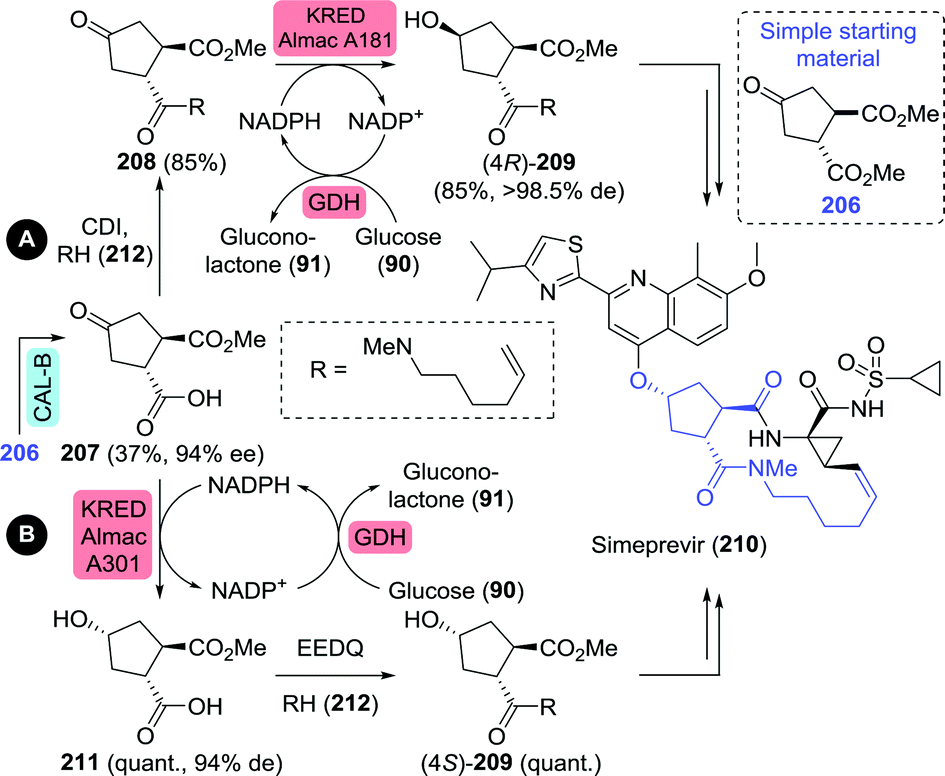 | ||
| Scheme 46 Chemoenzymatic synthesis of simeprevir precursors as proposed by Janssen Pharmaceuticals.277 CAL-B: Candida antarctica lipase B, CDI: carbonyldiimidazole, KRED: ketoreductase, quant.: quantitative yield, GDH: glucose dehydrogenase and EEDQ: N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline. | ||
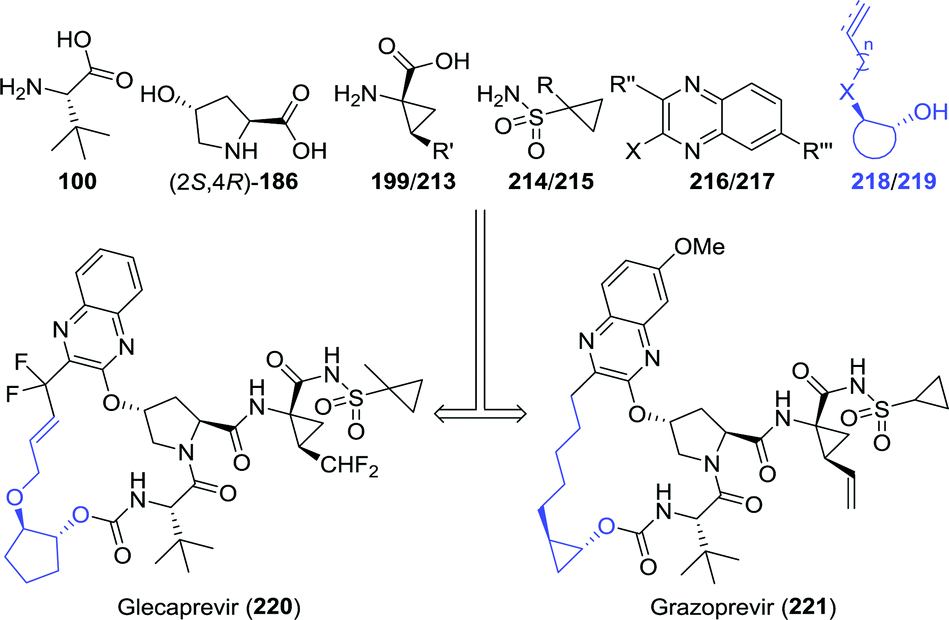 | ||
| Chart 7 Retrosynthetic analysis of glecaprevir and grazoprevir. | ||
Controlled hydrolytic resolution of cyclopentane-1,2-diyl diacetate (222) with BCL, O-alkylation with allyl bromide and subsequent deprotection afforded 218 in 14% overall yield and 96% ee (Scheme 47A).279 However, running regioselective lipase-catalysed resolution up to complete mono-deacetylation is challenging, thus often requiring termination of the reaction at a the stage of sub-optimal conversion. The same Abbvie team that optimised the synthesis of difluoromethyl-ACCA also managed to improve the synthesis of (1R,2R)-2-(allyloxy)cyclopentanol.269 Ring-opening of cyclopentene oxide (224) with allyl alcohol, acetylation and subsequent resolution with CAL-B allowed for isolation of the target molecule in 32% overall yield and >99% ee at pilot scale (Scheme 47B).
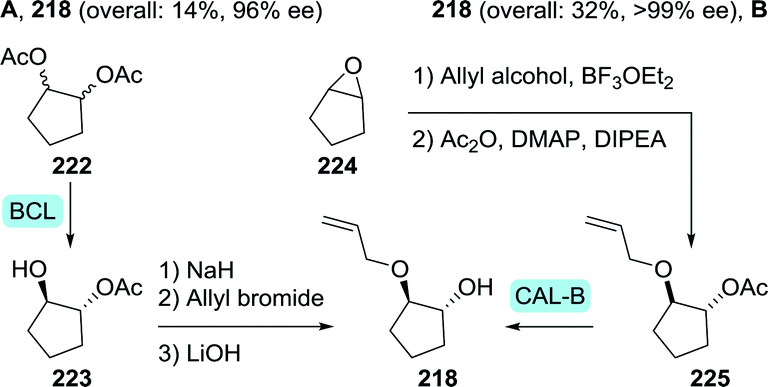 | ||
| Scheme 47 Two complementary chemoenzymatic routes for the synthesis of (1R,2R)-2-(allyloxy)cyclopentanol by enzymatic resolution, effected either initially (A) or as the final step (B).269 BCL: Amano PS lipase (Burkholderia cepacia), CAL-B: Candida antarctica lipase B and DIPEA: N,N-diisopropyl-N-ethylamine. | ||
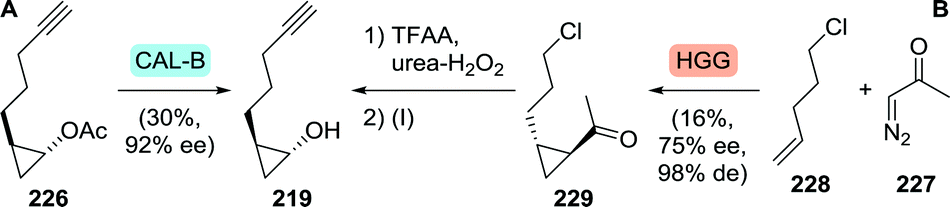
Multikilogram scale resolution of 2-(pent-4-ynyl)cyclopropyl acetate (226) with CAL-B as performed by Merck afforded the grazoprevir intermediate (1R,2R)-2-(pent-4-ynyl)cyclopropanol (219) with 92% ee (Scheme 48A).281 As asymmetric synthesis would, however, be the preferred synthetic strategy, Merck also investigated biocatalytic cyclopropanation towards this fragment.282 The team envisioned recently discovered new-to-nature carbene transfer onto activated alkenes catalysed by haemoproteins to achieve cyclopropanation. Use of the non-activated 5-chloropentene (228), however, proved to be challenging, but a mutant of Hells’ Gate globin I (HGG) from Methylacidophilium infernorum was found to catalyse the desired reaction. Four rounds of directed evolution were required to obtain the Q50V/L54A/F43V/N45S mutant, which was sufficiently active and showed reasonable stereoselectivity (Scheme 48B). Preparative scale proof-of-concept cyclopropanation performed on 227 and 228 led to only 16% yield and 75% ee (99:1 trans-selectivity). Several chemical steps are thereafter needed to complete the synthesis of (1R,2R)-2-(pent-4-ynyl)cyclopropanol (219).281 The promising nature of biocatalytic carbene transfer will be further discussed in chapter 5.
3.5 Fused multicyclic integrase inhibitors
In 2007, raltegravir was the first of several structurally related integrase inhibitors to be approved for combating HIV.283 Raltegravir is built up from three (hetero)cyclic fragments connected by short flexible linkers. The latest additions to this class of anti-viral agents are elvitegravir, dolutegravir and bictegravir, which instead are rigid fused multicycles. Additionally, all three of these integrase inhibitors contain an amino alcohol fragment that could be synthesised biocatalytically (Chart 8).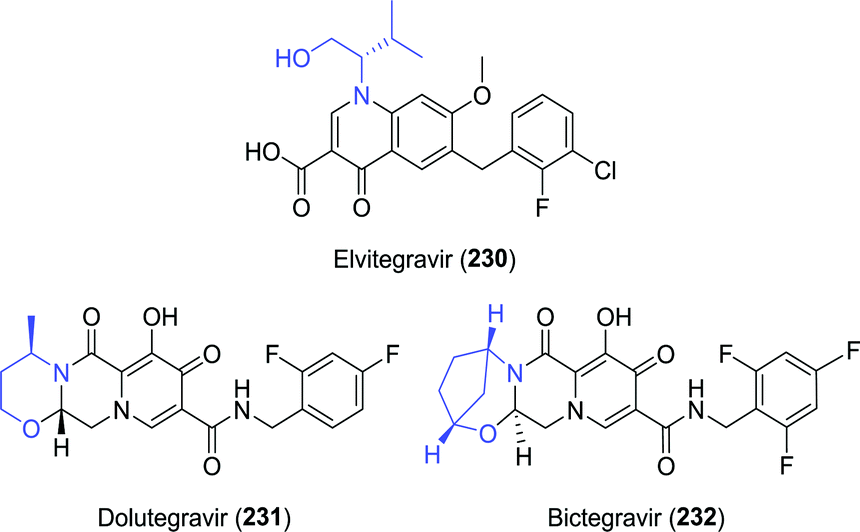 | ||
| Chart 8 Structures of the fused multicyclic integrase inhibitors elvitegravir, dolutegravir and bictegravir. | ||
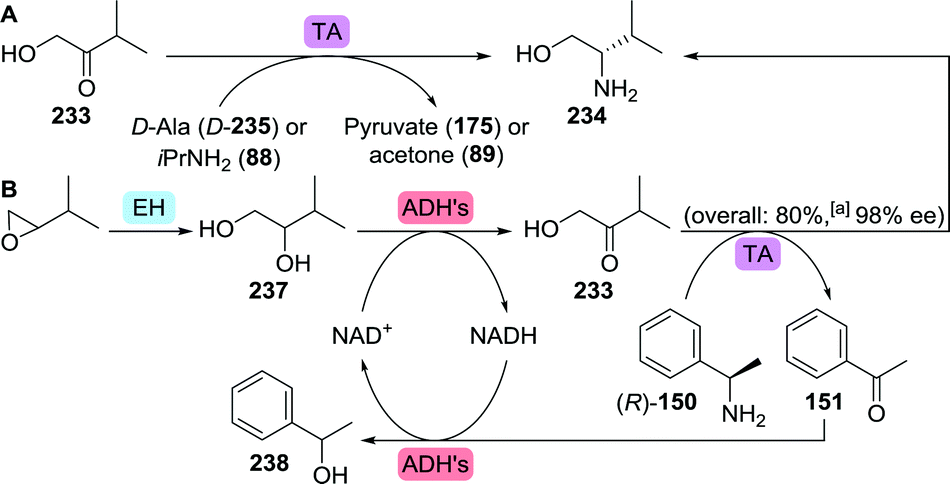
The amino alcohol moiety of elvitegravir, L-valinol, was produced on ton scale through reduction of L-valine.284 Several exemplary biocatalytic strategies have been developed that allowed for its synthesis from achiral starting material, however. Kroutil and co-workers discovered four transaminases capable of converting hydroxy-3-methyl-2-butanone (233) to L-valinol (234) using either isopropylamine or D-alanine as amine donor (Scheme 49A).285 More recently, a transaminase from Mycobacterium vanbaalenii was shown to catalyse this transformation, however, requiring the less sustainable aromatic (R)-α-methylbenzylamine ((R)-150) as amine donor.286 An interesting small scale cascading approach was taken by co-expressing the latter enzyme with an epoxide hydrolase and two alcohol dehydrogenases (ADHs) to allow for the conversion of epoxides to amino alcohols (Scheme 49B).287 Complete hydrolysis of racemic 2-isopropyloxirane (236) was achieved with a Sphingomonas sp. HXN-200 epoxide hydrolase. Subsequent oxidation of the resulting (R)- and (S)-237 required the use of two ADHs with complementary (R)- and (S)-selectivity, namely 2,3-butanediol dehydrogenase from Bacillus subtilis and polyol dehydrogenase from Gluconobacter oxydans, respectively. The hydroxy-3-methyl-2-butanone product (233) was finally converted to L-valinol (234) by enantioselective transamination, which liberated acetophenone (151) as side-product. Its reduction by the ADHs allowed for closed-loop recycling of co-factor NAD+. In the search for complementary reductive amination biocatalysts, several Lysinibacillus fusiformis LeuDH variants288 were shown capable of also converting α-hydroxyketones to produce, among others, L-valinol with up to 96% ee.289
 | ||
| Scheme 49 Transaminase (TA)-catalysed synthesis of L-valinol (A)285 and incorporation of a transaminase in a multi-enzymatic cascade for the synthesis of L-valinol from 2-isopropyloxirane (B).287 EH: epoxide hydrolase and ADH: alcohol dehydrogenase.a Conversion to product. | ||
Also dolutegravir's (R)-3-aminobutanol building block can be accessed through reduction of the corresponding acid or its ester. The latter has been prepared through hydrolytic resolution with lipase or aminopeptidase, for example.290–293
More interesting, however, is its asymmetric synthesis from readily available ethyl 3-oxobutanoate (239). Kroutil et al. have used an (R)-selective transaminase from Arthrobacter sp. (ATA-117) to synthesise ethyl (R)-3-aminobutanoate ((R)-240-Et) with 98% yield and 98% ee from 239 (Scheme 50).294 The use of D-alanine (D-235) as amine donor allowed coupling to a lactate dehydrogenase to convert the resulting pyruvate (175) to lactate (242) to shift the equilibrium to the product side. The same team also reported on several other transaminases capable of catalysing this transformation, including ArRmut11.295 The latter is a mutant of ATA-117 jointly developed by Merck and Codexis to perform late stage biocatalytic reductive amination towards the synthesis of the anti-diabetic sitagliptin.296 The vast majority of transaminases are (S)-selective, tolerate only a single large substituent on a ketone substrate and are specific for amino acid-based amine donors. In contrast, ArRmut11 is highly (R)-selective, accepts bulky substrates and can employ isopropylamine as amine donor. Its extensive engineering by directed evolution is considered to be one of the hallmarks of biocatalytic drug production.
 | ||
| Scheme 50 Transaminase (ATA-117)-catalysed synthesis of (R)-3-aminobutanoate.294 GDH: glucose dehydrogenase and LDH: lactate dehydrogenase. | ||
By making use of a transaminase from Actinobacteria sp., 4-hydroxy-2-butanone could be converted to (R)-3-aminobutanol directly, eliminating the need for additional carboxyl reduction.297 In order to further optimise (R)-3-aminobutanol production, Wei and co-workers mutated Aspergillus terreus transaminase to obtain the H55A/G126F/S215P triple mutant, which showed a 10-fold increase in kcat·KM−1 and improved thermostability as compared to the wild-type.298
In an alternative approach to (R)-3-aminobutanoic acid (240), Wu, Janssen and co-workers performed biocatalytic hydroamination of crotonic acid (244).299Bacillus sp. YM55-1 aspartase was shown to be solely capable of catalysing the deamination of aspartate (243) to fumarate (165, Scheme 51). Screening of thousands of its mutants for expanded substrate scope proved to only be marginally successful.300,301 Therefore, the team engaged in a in silico modelling study to pin-point possible mutations that would likely support conversion of the target substrates. For the production of (R)-3-aminobutanoic acid, a small “smart library” of 34 enzyme variants was constructed, which, upon expression, resulted in 14 active enzymes. Use of the quadruple mutant B19 (T187C/M321I/K324L/N326A) allowed for the synthesis of (R)-3-aminobutanoic acid from crotonic acid with 92% yield and 99% ee on kilogram scale.
 | ||
| Scheme 51 Mutation of a highly specific Aspartase to a Lyase capable of converting crotonic acid to (R)-3-aminobutanoic acid.299 | ||
The third fused multicyclic integrase inhibitor to be discussed is bictegravir, which contains a (1R,3S)-3-aminocyclopentanol fragment that could be obtained through resolution. Resolution by transesterification with pancreatin or Pseudomonas lipase afforded the N-protected 4-aminocyclopent-2-en-1-yl acetate in the desired configuration (248/249, Scheme 52).302,303 In a complementary fashion, 4-acetamidocyclopent-2-en-1-yl acetate (247) could be resolved through chemoselective hydrolysis of its ester functionality with electric eel acetylcholinesterase.302 Olefin reduction and deprotection would subsequently afford (1R,3S)-3-aminocyclopentanol (250).
 | ||
| Scheme 52 Enzymatic kinetic resolution catalysed by electric eel acetylcholinesterase, Pseudomonas sp. lipase (PSL)302 or pancreatin (PC)303 in the production of (1R,3S)-3-aminocyclopentanol, which serves as an intermediate in the synthesis of bictegravir. | ||
3.6 Anti-influenza amino sugar derivatives
The first drug to be approved for treating influenza was achiral amantadine (adamantylamine).304 However, because most influenza viruses became resistant against this agent, it is no longer recommended for treatment of the common flu. The same holds for the analogous rimantadine (1-(adamantanyl)ethanamine). With the FDA-approval of the structurally more complex neuraminidase inhibitors oseltamivir and zanamivir in 1999, the number of anti-viral agents available for treating influenza has been doubled.305 Most influenza A and B viruses are currently still susceptible to these agents. Viral neuraminidase is involved in budding of the virus from its host cell upon hydrolysis of N-acetylneuraminic acid (Neu5Ac, 252), a process that proceeds through a boat-shaped sialosyl cation intermediate (253, Chart 9).306 Modern-day neuraminidase inhibitors such as oseltamivir (254), zanamivir (255) and peramivir (256), act as mimics of this sialosyl cation.307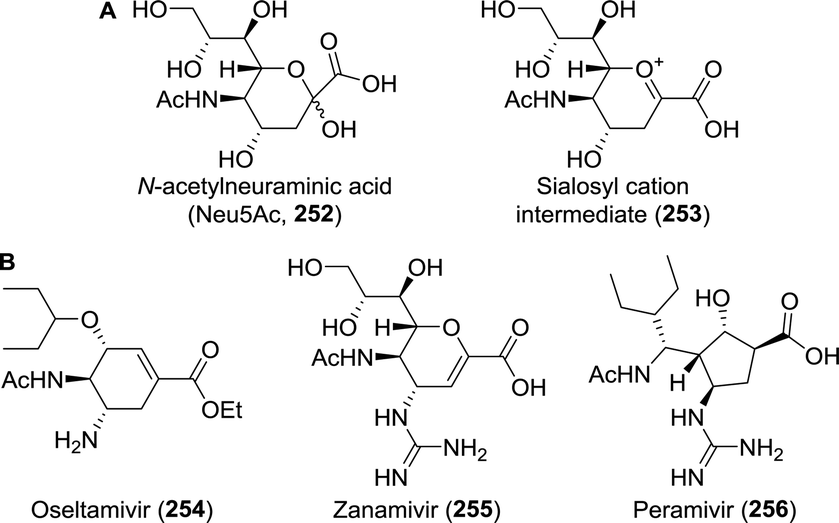 | ||
| Chart 9 Neu5Ac and the intermediate structure of the sialosyl cation upon dehydration by (viral) neuraminidase (A). Anti-influenza neuraminidase inhibitors indicating their functioning as transition state mimics (B).307 | ||
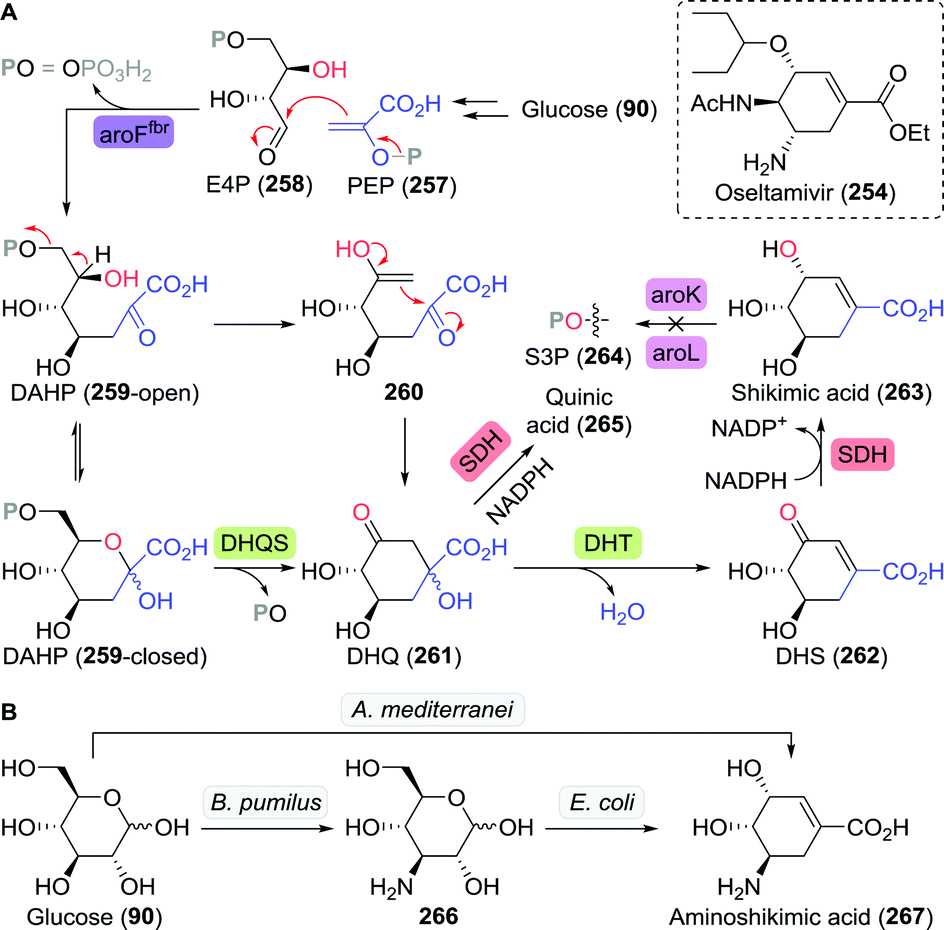 | ||
| Scheme 53 Production of the oseltamivir (see insert) intermediate Shikimic acid through fermentation with an engineered E. coli strain (A).312 Production of aminoshikimic acid through fermentation with either multiple strains or a single bacterium (B).321 PEP: phosphor-enol pyruvate, E4P: erythrose-4-phosphate, aroFfbr: 3-deoxy-D-arabino-heptulosonic acid 7-phosphate synthase mutant, DAHP: 3-deoxy-D-arabino-heptulosonic acid 7-phosphate, DHQS: 3-dehydroquinate synthase, DHQ: 3-dehydroquinic acid, SDH: shikimate dehydrogenase, DHT: 3-dehydroquinate dehydratase, DHS: 3-dehydroshikimic acid, aroK/aroL: shikimate phosphorylation enzymes and S3P: shikimic acid 3-phopshate. | ||
Active equilibration of the formed shikimic acid to quinic acid limited purity of the non-isolated product. The yield of shikimic acid could be optimised by, among others, carefully controlling the amount of glucose in the medium.313 Through optimisation of the glycolysis pathway, PEP production could be maximised, which thereby allowed for further optimisation of shikimic acid production up to 87 g L−1 and 36% yield from glucose.314 The by-product quinic acid could also serve as a source of shikimic acid through strategic application of several Gluconobacter oxydans enzymes.315 Metabolic engineering aspects for further optimisation of microbial shikimic acid production continue to arouse attention.316–319
Oseltamivir (254) could, alternatively, be synthesised from aminoshikimic acid (267, Scheme 53B),320 which in turn can be produced through fermentation employing either a single engineered microbe or two microbes in tandem.321
Oxidation of bromobenzene (268) or ethyl benzoate (269) catalysed by organisms expressing toluene dioxygenases (TDOs) led to the formation of the corresponding cis-diols (Scheme 54).322–325 Either of both diols has been employed as starting material for the synthesis of oseltamivir.324–327
 | ||
| Scheme 54 Oxidative fermentation of bromobenzene and ethyl benzoate catalysed by microbes expressing toluene dioxygenases (TDOs). | ||
As for many of the molecules discussed throughout this work, enzymatic resolution/desymmetrisation has proven to be an effective tool for generating optically active oseltamivir intermediates too.328–330 The most intriguing example of which has been reported by Wirz and co-workers as it involves stereoselective hydrolysis of a densely functionalised intermediate (Scheme 55).328 The synthesis started from 2,6-dimethoxyphenol (272) which was chemically converted to 273. Ruthenium-catalysed all-cis-hydrogenation of 273 followed by demethylation afforded 275. Subsequent pig liver esterase (PLE)-catalysed desymmetrisation resulted in the mono-acid 276 in 98% yield and excellent enantiopurity (>96% ee). Subsequent chemical steps furnished oseltamivir as its phosphate salt.
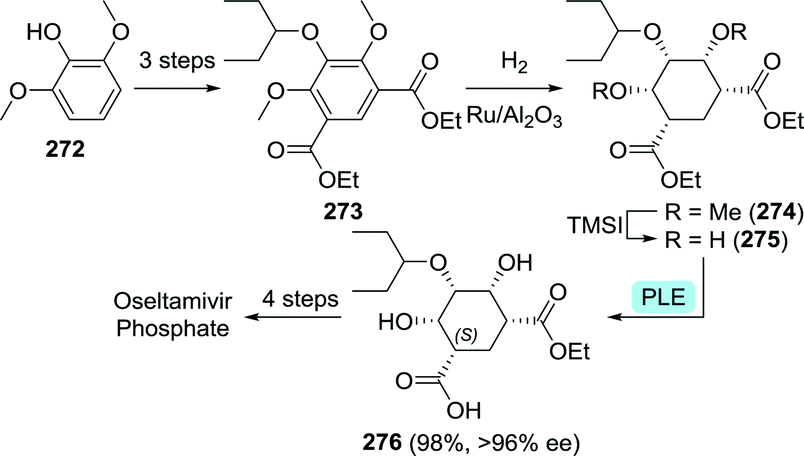 | ||
| Scheme 55 Chemoenzymatic route towards oseltamivir as developed by Wirz and co-workers. TMSI: iodo(trimethyl)silane and PLE: pig liver esterase.328 | ||
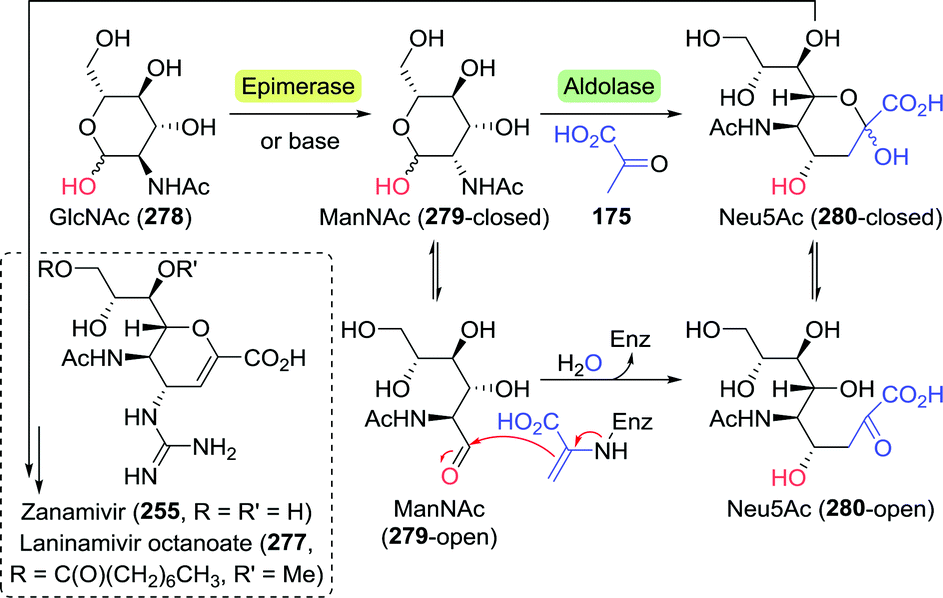 | ||
| Scheme 56 (Chemo)enzymatic synthesis of N-acetylneuraminic acid (Neu5Ac). GlcNAc: N-acetylglucosamine and ManNAc: N-acetylmannosamine. | ||
The pro-drug laninamivir octanoate (277) is an analogue of zanamivir that has been approved for the treatment of influenza in Japan.350 It differs from zanamivir in that the hydroxy group in the 7-position is methylated and its 9-hydroxy group is esterified to the corresponding octanoate ester. Analogous to the synthesis of zanamivir, a Neu5Ac aldolase-catalysed addition of pyruvate to 4-OMe-ManNAc afforded the 7-OMe-Neu5Ac intermediate required for the synthesis of laninamivir.351
3.7 Miscellaneous compounds
 | ||
| Scheme 57 Structure of ledipasvir (A). Enzymatic resolution of N-Boc-protected esters of the ledipasvir proline derivatives (B).354,355 CAL-B: Candida antarctica lipase B and PPL: porcine pancreas lipase. | ||
Resolution of the corresponding ester or amide has been achieved using various enzymes.358–362 Many lipases are known to show enhanced activity on the interface of water and an immiscible co-solvent. Gröger et al. exploited this feature by employing BCL to resolve the β-phenylalanine butyl ester in “mini-emulsions” generated through ultrasonication.363 This technique allowed for substrate concentrations to exceed 3 M, while still maintaining excellent ee (>99%) and high conversion. Concomitant in situ racemisation, as developed by Bäckvall and co-workers, allowed for DKR of the β-phenylalanine ethyl ester (286) with CAL-A with 89% or 96% ee using a ruthenium or palladium catalyst, respectively (Scheme 58A).364,365 Although amides are usually not substrates for lipases, Fülöp and co-workers achieved highly stereoselective resolution of strained 4-phenylazetidin-2-one (289) at elevated temperatures to furnish the pure (S)-β-lactam (S)-289 as a precursor to the desired L-β-phenylalanine (L-290, Scheme 58B).366
 | ||
| Scheme 58 Enzymatic resolution of β-phenylalanine (derivatives). Dynamic kinetic resolution of the β-phenylalanine ethyl ester with Candida antarctica lipase A (CAL-A, A),364,365 resolution of 4-phenylazetidin-2-one with Candida antarctica lipase B (CAL-B, B),366 and bienzymatic resolution of β-phenylalanine with a phenylalanine aminomutase (PAM) mutant and phenylalanine ammonia lyase (PAL, C).367aConversion to product. | ||
Janssen and co-workers developed a mutated phenylalanine aminomutase (PAM) capable of enantioselectively converting D-β-phenylalanine to L-phenylalanine (292), which could thus be employed to resolve racemic β-phenylalanine.367 Quasi-irreversible conversion of L-phenylalanine to cinnamic acid (293) by phenylalanine ammonia lyase (PAL) provided for the driving force and allowed for facile isolation of the L-β-phenylalanine product (L-290, Scheme 58C). The large structural similarity between PAM and PAL drove the research team to engineer another PAM mutant (R92S) that showed additional PAL activity,368 which allowed for the resolution of racemic β-phenylalanine by direct deamination of D-β-phenylalanine (48% yield, >97% ee).
The aspartase that had been mutated to a lyase capable of producing (R)-3-aminobutanoic acid (Section 3.5, Scheme 51) could also be mutated as such that it would catalyse the hydroamination of cinnamic acid to form L-β-phenylalanine (>99% ee).299 However, conversion did not exceed 50% due to the unfavourable reaction equilibrium.
L-β-Phenylalanine could also be produced directly by transamination of 3-oxo-3-phenylpropanoic acid (295), which did prove to be a rather chemically unstable substrate. Such limitations could, however, be partially offset through lipase-catalysed in situ saponification of ethyl 3-oxo-phenylpropanoate (294, Scheme 59).369 In the presence of an (S)-transaminase from E. aerosaccus the β-oxoacid was subsequently converted to L-290 at the expense of 3-aminobutyric acid (240) as the amine donor (82% conversion to product, >99% ee). (S)-transaminases from Sphaerobacter thermophilus,370,371Polaromonas sp.372 and a β-amino acid dehydrogenase from Candidatus cloacamonas acidaminovorans have also been proven capable of catalysing said transformation.373 Replacing lipases with nitrilases in the cascade conversion allowed for the use of 3-oxo-3-phenylpropanenitrile as alternative starting material.370,372,373 In those cases where isolated yields have been reported for the lipase- or nitrilase-transaminase cascade, however, the amount of isolated material from these small scale experiments remains low.
 | ||
| Scheme 59 In situ de-esterification with lipase and subsequent transaminase (TA)-catalysed synthesis of L-β-phenylalanine.369aConversion to product. | ||
In order to circumvent the use of the unstable β-oxoacid, Rudat and co-workers employed a mutated transaminase to convert the corresponding ethyl ester, but the enantiopure L-β-phenylalanine ethyl ester could only be isolated in 9% yield.374
(S)-3-Amino-3-phenylpropanol can also be converted to maraviroc upon conversion of the alcohol to a good leaving group.357 Resolution of various protected 3-amino-3-phenylpropanol derivatives could be achieved using either lipase or penicillin G acylase.375,376
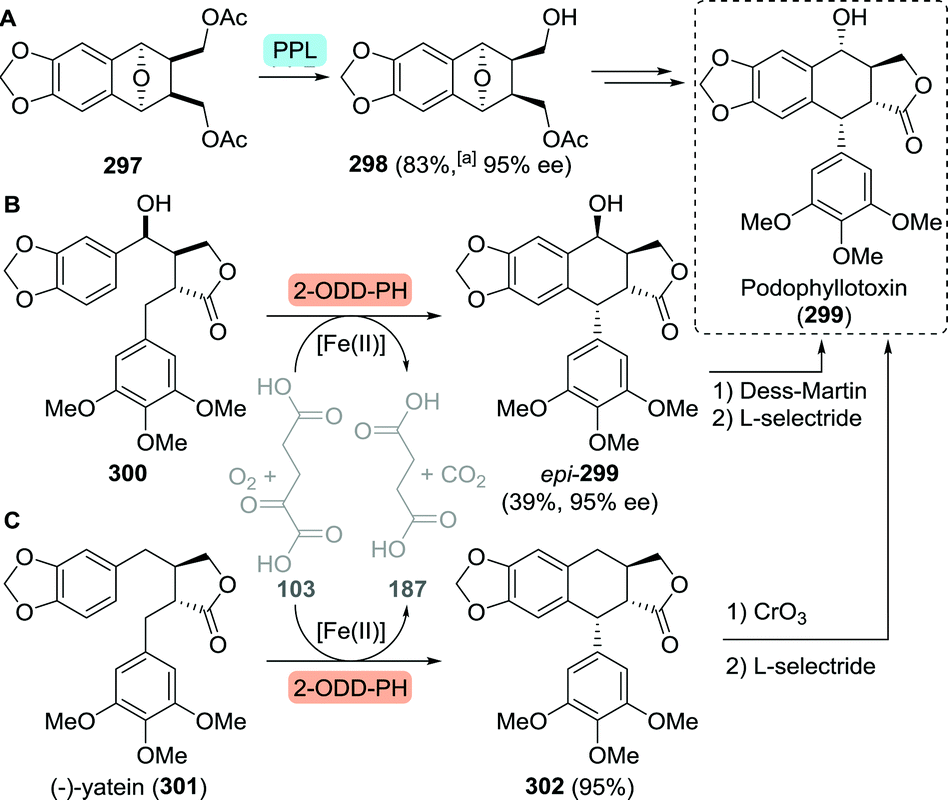 | ||
| Scheme 60 Desymmetrisation of a podophyllotoxin intermediate with porcine pancreas lipase (PPL, A).378 Two concurrent routes to podophyllotoxin based on (hydroxy)-yatein cyclisation with the 2-oxoglutarate-dependent dioxygenase from Podofyllum hexandrum (2-ODD-PH, B and C).380,381aBased on recovered starting material. | ||
Elucidation of the biosynthetic route to podophyllotoxin in 2015 revealed a 2-oxoglutarate-dependent dioxygenase (2-ODD) that catalysed the crucial cyclisation of yatein to deoxy-podophyllotoxin.379 Recently, both Fuchs et al. and Renata et al. have independently employed the dioxygenase from Podofyllum hexandrum (2-ODD-PH) to achieve the synthesis of podophyllotoxin itself.380,381 Multigram scale resolution of racemic hydroxy-yatein (300) through selective conversion of (−)-hydroxy-yatein to epi-podophyllotoxin (epi-299) using 2-ODD-PH had been shown by Fuchs and co-workers (Scheme 60B). Subsequent chemical steps furnished podophyllotoxin (299) in 32% overall yield. The synthesis by Renata and co-workers involved chemical synthesis of (−)-yatein (301), its 2-ODD-PH-catalysed conversion to deoxy-podophyllotoxin (302), subsequent chemical oxidation and stereoselective reduction to podophyllotoxin (28% overall yield, Scheme 60C). These chemoenzymatic routes could thereby compete with the most efficient chemical syntheses.382
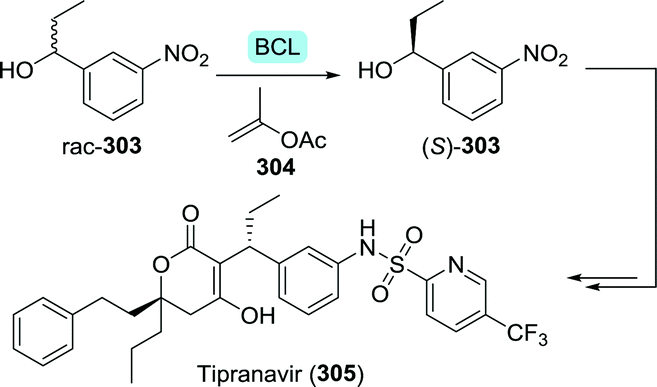 | ||
| Scheme 61 Kinetic resolution of the tipranavir intermediate 1-(3-nitrophenyl)propanol with Burkholderia cepacia Amano PS lipase (BCL).384 | ||
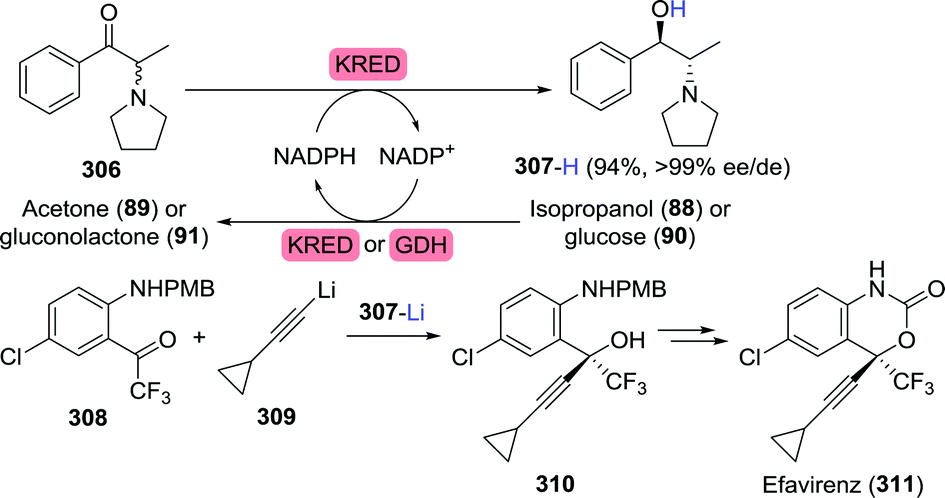 | ||
| Scheme 62 Synthesis of (1R,2S)-phenyl-2-(pyrrolidinyl)propanol through reduction with a ketoreductase (KRED) and its implementation as chirality inducer in the synthesis of an efavirenz intermediate.386,387 GDH: glucose dehydrogenase and PMB: p-methoxybenzyl. | ||
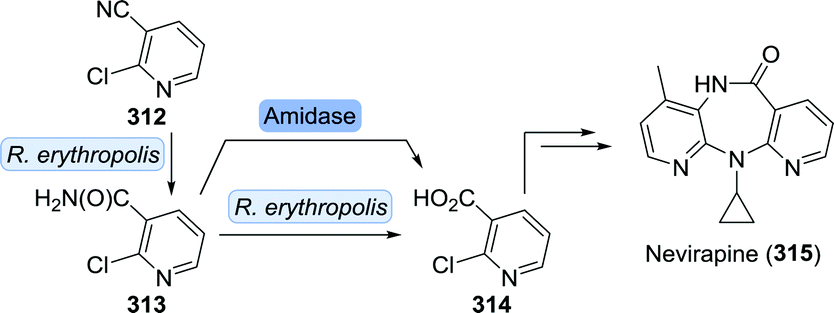 | ||
| Scheme 63 Biocatalytic synthesis of the nevirapine intermediate 2-chloronicotinic acid.389,390 | ||
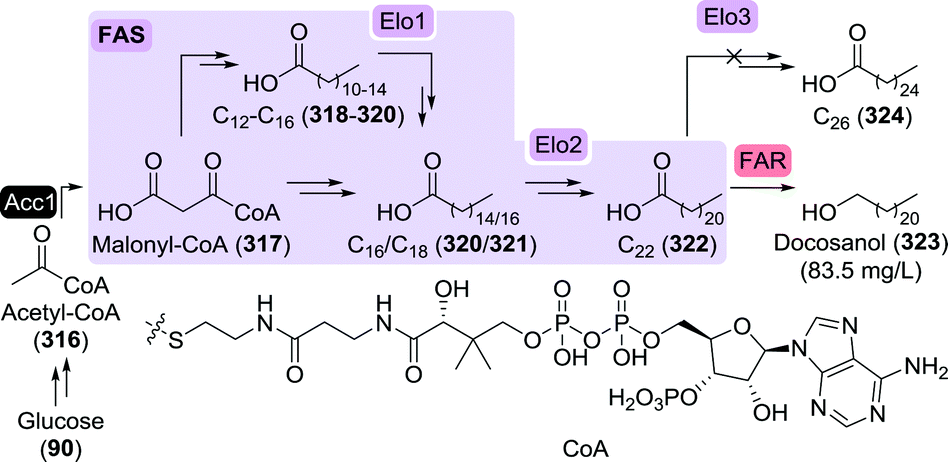 | ||
| Scheme 64 Docosanol production through fermentation with heavily engineered microbes.392 Acc1: acetyl-CoA carboxylase, FAS: fatty acid synthase, Elo1-3: elongase 1-3 and FAR: fatty acid reductase. | ||
4. Biocatalytic production of potential SARS-CoV-2 anti-viral agents
The immense impact and current lack of an effective cure for COVID-19 has led to numerous global ventures aimed at repurposing (anti-viral) small-molecule drugs for the treatment of this SARS-CoV-2-induced disease.394,395 The sheer size of the COVID-19 pandemic with the need for treatment of millions of patients requires significant scale-up of the potential anti-viral agents of interest. Economical scale-up of such drug candidates has only been the topic of a limited number of studies.394Emergence of new forms of influenza such as the highly pathogenic and life-threatening avian influenza virus H5N1 has already identified critical supply issues for the only orally effective drug oseltamivir.396 Bottlenecks in its synthesis were caused by the limited availability of the required natural feedstock chemicals (quinic acid or shikimic acid; see Section 3.6.1, Scheme 53), potentially hazardous intermediates, and demanding purification steps. Scarcity of the drug thereby spurred the development of several novel synthetic routes with improved scalability.397,398 Notwithstanding the need for bulk supply of starting materials, biocatalysis might offer a sustainable solution to the scalable mass production of pharmaceuticals, as emphasised throughout this manuscript. Crucial factors regarding the effectiveness of biocatalysis include the high selectivity of enzymes and the self-replicating nature of biological systems used for their production.
Here we would like to highlight some biocatalytic opportunities and strategies towards the synthesis of anti-viral agents against SARS-CoV-2 that underwent, or are currently undergoing, phase III clinical trials. As no definitive conclusion can yet be drawn regarding the effect of any such agents, we have chosen a diversity-oriented approach, focussing on various targets that would highlight the potential of biocatalysis. Inclusion in this panel should not be regarded as proof of treatment effectiveness.
4.1 Remdesivir
Gilead's remdesivir had been shown active against the RNA virus Ebola and is currently considered to be one of the most promising drugs for treating patients infected with SARS-CoV-2, which too is an RNA virus.399 Remdesivir is an investigational drug that has not been FDA-approved but it is the first medicine against COVID-19 to be recommended for authorisation in the EU as of late June 2020.400 It is also authorised in the United States under an Emergency Use Authorization (EUA) for the treatment of patients with severe COVID-19.401 Resembling AMP, it is a nucleotide pro-drug that has the major benefit of being resistant to proofreading exonucleases occurring in coronaviruses, thus evading the repair mechanism for preservation of its anti-viral activity.402Apart from the heteroaromatic moiety mimicking the nucleobase, remdesivir (327) is made up from a sugar and chiral phosphoramidate building block. Remdesivir has been synthesised by starting from perbenzylated ribonolactone (D-ribono-1,4-lactone (326), Scheme 65).403 Ribonolactone can be obtained through enzymatic oxidation of ribose with Agaricus meleagris pyranose dehydrogenase.404Yarrowia lipolytica short-chain dehydrogenase also proved capable of oxidising 5-O-trityl-D-ribose.405
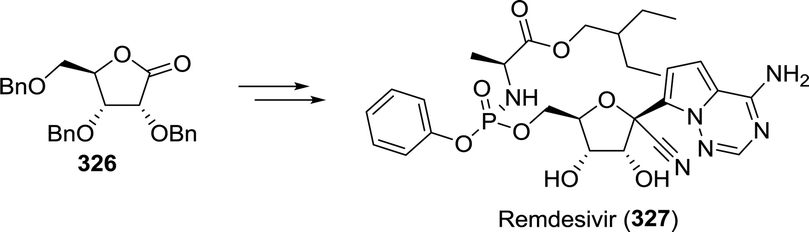 | ||
| Scheme 65 Gilead developed the synthesis of remdesivir from Ribonolactone.399,403 | ||
Remdesivir bears a Pro-Tide ligand allowing for improved bioavailability. It was shown that the related Pro-Tide ligand of sofosbuvir could be enzymatically resolved to obtain the desired (S)-enantiomer.89 Appreciating that the remdesivir phosphoramidate only differs from its sofosbuvir counterpart through the presence of a slightly elongated ester fragment (2-ethylbutyl vs. isopropyl alanine ester), makes it highly likely that this fragment could be obtained through enzymatic resolution as well.
A critical step in the synthesis of remdesivir is the installation of the cyanide group at the pseudo-anomeric centre. Currently, adequate biocatalytic methods for installing the chiral quaternary centre are lacking. Novel C–C bond forming enzymes are required to catalyse the desired transformation, since neither the enzymes involved in the biosynthesis of C-nucleosides,406 nor known oxynitrilases match the requirements.407
4.2 (Hydroxy)chloroquine
The anti-malarials chloroquine (328) and hydroxychloroquine (329) (Scheme 66A) had been speculated to be potential anti-viral agents against SARS-CoV-2 soon after the outbreak of the virus.408 However, their efficiency and potential have not remained undisputed.409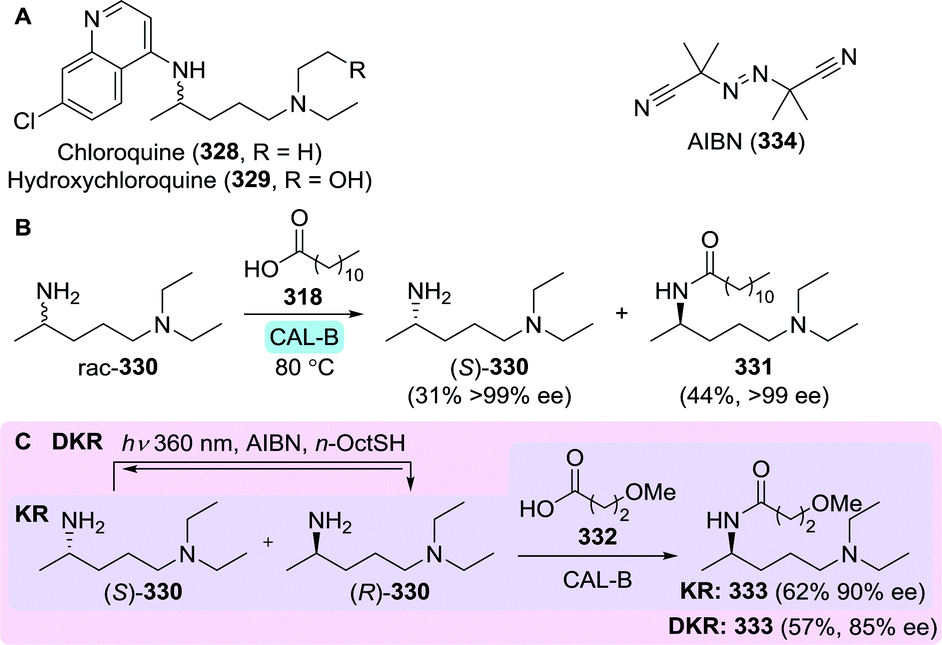 | ||
| Scheme 66 Structures of chloroquine and hydroxychloroquine (A). Resolution of N1,N1-diethylpentane-1,4-diamine catalysed by Candida antarctica lipase B (CAL-B) and dodecanoic acid as acyl donor (B).412 Kinetic resolution (KR) and dynamic kinetic resolution (DKR) of N1,N1-diethylpentane-1,4-diamine to produce the corresponding (R)-amide (C).414 | ||
Because both anti-malarial agents are used as racemates, they have as such also been tested in racemic form during COVID-19 trials. However, it is unknown whether both enantiomers can act as active pharmaceutical ingredient. It has been proposed to repurpose these drugs via a chiral switch strategy, thus either as enantiopure (R)- or (S)- (hydroxy)chloroquine.410,411
Gil and co-workers have shown that the diamine fragment of chloroquine, N1,N1-diethylpentane-1,4-diamine (330), could rapidly be resolved with immobilised CAL-B (Novozym 435, Scheme 66B) and dodecanoic acid (318) as acyl donor.412 Both the remaining (S)-amine (S)-330 and the formed (R)-amide 331 were isolated with >99% ee at small scale. The unique stability features of CAL-B were highlighted once more as resolution was best performed at 80 °C. The same group also managed to perform dynamic kinetic resolution through thiyl radical-mediated racemisation to produce the (R)-amide 333 (resolution with CAL-B, Scheme 66C).413,414 As thiyl radical formation was performed photocatalytically, DKR reaction times could easily be tuned by turning irradiation on and off. By employing a three-step, one-pot procedure, starting with initial (non-dynamic) kinetic resolution, the (R)-amide could be isolated in 62% yield and 90% ee. Although this DKR procedure was shown to be efficient for the formation of other (R)-amides, DKR of N1,N1-diethylpentane-1,4-diamine (330) proved to be less efficient as compared to conventional CAL-B-catalysed resolution. Biocatalytic reductive amination catalysed by transaminases or recently discovered reductive aminases (vide infra) would allow for the use of a non-chiral ketone as starting material and might result in enhanced stereoselectivity, even at room temperature.
4.3 Azithromycin
Azithromycin is an FDA-approved anti-bacterial macrolide that, commonly in combination with hydroxychloroquine, has gone through various clinical trials in order to establish its effectiveness in combating COVID-19.408,409,415 This semisynthetic drug (335) is obtained from erythromycin A (336) through chemical oxime formation, Beckmann rearrangement and N-methylation (Chart 10).416 Erythromycin A, also used as antibiotic, is in turn commonly produced through fermentation.417,418 Strains that have proven capable of naturally accumulating erythromycin A, such as Streptomyces coelicolor and Streptomyces lividans do so in small amounts only. Transfer of gene clusters from these strains to optimised overexpressing strains (S. erythraea) led to an approximate 50-fold increase in titres for erythromycin analogues.419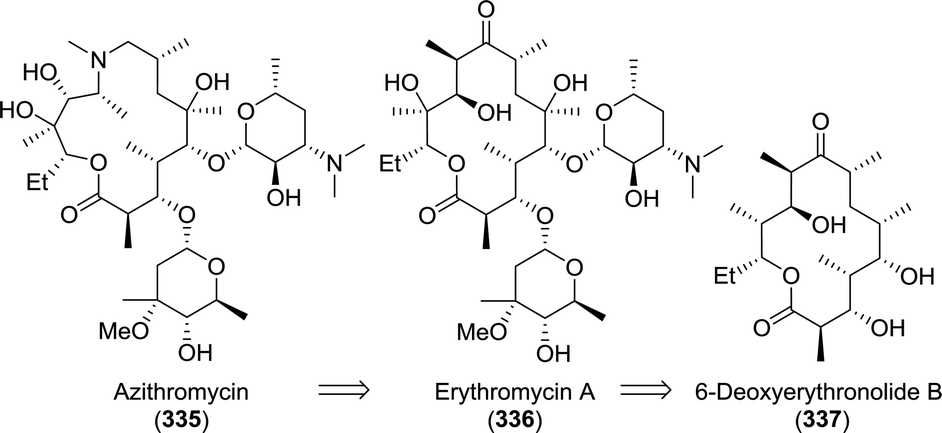 | ||
| Chart 10 Bioretrosynthetic analysis of azithromycin. | ||
The azithromycin precursor erythromycin is commonly isolated as a mixture of the congeners A–C. Because erythromycin A purity is essential for effective azithromycin production, minimising the production of the congeners B and C is crucial. Accumulation of erythromycin B and C could be almost completely abolished through regulation of the EryK and EryG genes, coding for a P450 hydroxylase and an (S)-adenosylmethionine-dependent O-methyltransferase, respectively.420
A crucial intermediate towards the biosynthesis of erythromycin A is the polyketide 6-deoxyerythronolide B (337, Chart 10). This non-glycosylated precursor could either be obtained through the use of metabolically engineered microorganisms or isolated enzymes.421,422 While 6-deoxyerythronolide B could be produced efficiently (>1 g L−1) in an E. coli host,423,424 erythromycin production using this bacterium has proven to be much less effective (<0.1 g L−1).425,426
4.4 Dexamethasone
Very recently (mid 2020), the corticosteroid dexamethasone received considerable attention. In preliminary studies the use of the drug resulted in a reduction of the COVID-19 mortality rate for patients on mechanical ventilation by about one third.427 This result is not based on anti-viral activity but on the potent anti-inflammatory effects of the steroid. Patients with severe COVID-19 can develop a systemic inflammatory response that can lead to lung injury and multisystem organ dysfunction, and it has been proposed that dexamethasone might prevent or mitigate these deleterious effects.Production of this drug starts from other steroid precursors and commonly involves biocatalysis, as is typical for the steroid field, which often requires regio- and stereospecific functionalisation at unreactive aliphatic positions.428,429 Although detailed procedures are scarce, early stage microbial dehydrogenation of 341 and subsequent chemical steps have allowed for the production of Dexamethasone (340, Scheme 67).430 Late stage microbial dehydrogenation at the Δ1-position and deacetylation of 338 afforded 339,431 which, upon HF-mediated epoxide ring-opening432 also led to dexamethasone. Deacetylation of dexamethasone acetate has been achieved with Penicillium decumbens ATCC 10436, albeit in only 5% yield.433
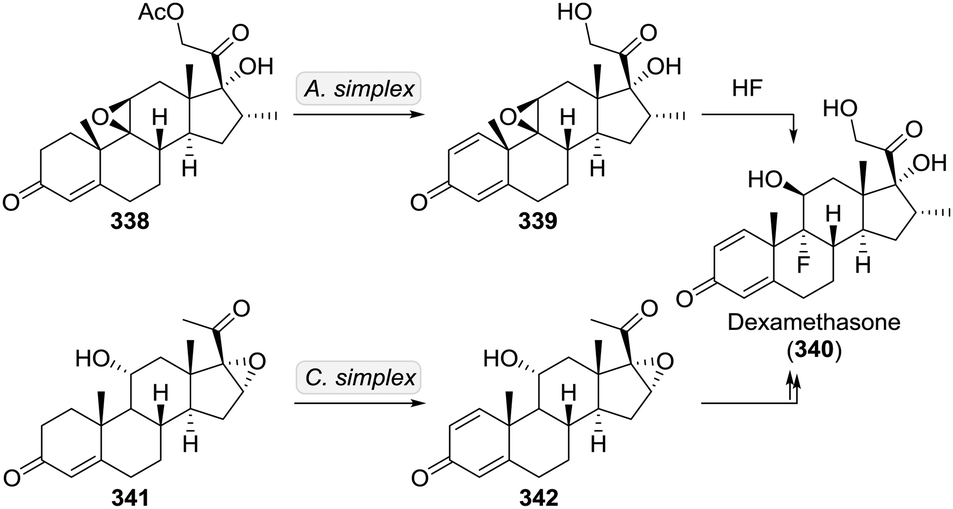 | ||
| Scheme 67 Chemoenzymatic synthesis of dexamethasone from steroid precursors. | ||
4.5 Darunavir and TMC-310911
The peptidomimetic anti-viral agent darunavir has already been discussed above related to its FDA-approval for the treatment of HIV infections (Sections 3.3.2 and 3.3.5). TMC-310911 (also called ASC-09) is an analogue of darunavir developed by Johnson & Johnson, which was intended for the treatment of HIV. The latter is now also being trialled for treating SARS-CoV-2-infected patients (Chart 11).434 Thus, all biocatalytic strategies towards the chiral building blocks of darunavir also apply to TMC-310911. Both the biocatalytic synthesis of its (2S,3S)-1,3-diamino-4-phenylbutan-2-ol (DAPB) core as well as resolution approaches towards 3-OH-bis-THF have been extensively researched. | ||
| Chart 11 Darunavir and TMC-310911 with the (2S,3S)-1,3-diamino-4-phenylbutan-2-ol (DAPB) core and the 3-OH-bis-THF capping ligand highlighted in blue. | ||
4.6 Danoprevir
Danoprevir is a bis-cyclopropyl-based protease inhibitor that is currently being considered for treating SARS-CoV-2 infections (Chart 12).435 As discussed previously for its structural analogue paritaprevir, all amino acid components from which danoprevir's macrocyclic fragment is built-up, could be obtained through biocatalysis (see Section 3.4). This includes, for example, proline 4-hydroxylase-catalysed hydroxylation of to obtain L-hydroxyproline and the synthesis of L-6-heptenylglycine with leucine dehydrogenase. Several approaches towards the enzymatic resolution of (1R,2S)-amino-2-vinylcyclopropanecarboxylic acid (vinyl-ACCA) have additionally been developed. | ||
| Chart 12 Danoprevir with its unnatural amino acid fragments highlighted; L-6-heptenylglycine and (1R,2S)-amino-2-vinylcyclopropanecarboxylic acid (vinyl-ACCA) in blue and L-hydroxyproline in red. | ||
4.7 Ruxolitinib
Faster clinical improvement was observed in COVID-19 patients treated with ruxolitinib (349), although quantitative evidence for the effect of this drug is yet lacking.436,437 The (S)-3-cyclopentyl-3-hydroxypropanenitrile precursor (346) could be obtained biocatalytically. Enzymatic reduction of 3-cyclopentyl-3-oxopropanenitrile (345) with a yeast short-chain dehydrogenase (YMR226C) resulted in the formation of the hydroxylated fragment in 89% yield and >99% ee (Scheme 68). Subsequent coupling with 4-bromo-1H-pyrazole (347) under Mitsunobu conditions allowed for the desired reversal of stereochemistry.438 | ||
| Scheme 68 Chemoenzymatic synthesis of a ruxolitinib intermediate with a yeast short-chain dehydrogenase.438 | ||
5. Emerging enzyme classes – challenges and opportunities in the synthesis of anti-viral agents
As shown through the examples discussed above, biocatalysis has evidently matured into an essential tool for modern, cost effective manufacturing of (chiral) anti-viral agents and their intermediates. There is, however, still a huge untapped potential as numerous enzymes, and their corresponding activities, have yet to be explored to match the available reaction scope of chemical conversions. Additionally, the synthesis of only a limited number of industrially relevant molecules have been pursued by means of enzyme catalysis, leaving the vast majority of demanding molecular architectures still to be investigated.The renewable nature of enzyme catalysts not only brings significant benefits from an environmental perspective but also stabilises catalyst costs.439 Although biocatalysis thus provides considerable merits as a complementary technology to ‘traditional’ synthesis/catalysis, it is subject to certain limitations and challenges, too. While numerous vendors nowadays provide various off-the-shelf chemical catalysts, the number of catalogued enzymes of a certain type can be rather narrow or absent. A few enzyme classes such as lipases or KREDs are well established. There are, however, still gaps in the biocatalytic toolbox, although many recently discovered enzyme classes are being rapidly developed in both academia and industry.33
Of particular significance are nucleoside analogues and peptidomimetics as these types of anti-viral agents directly interfere with unique stages of the viral replication cycle (Fig. 1). By discussing specific requirements of these drug structures as well as the technologies for making and coupling suitable synthetic intermediates, this chapter serves as an outlook into the future needs of biocatalytic anti-viral drug development, highlighting the potential of novel or engineered enzymes.
The importance of nucleoside analogues in treating viral infections lies in their versatility. These drugs typically target viral replication at stages A to C (Fig. 1), where they mimic the natural substrates by variation of both the nucleobase and the sugar core components.
While enzymatic approaches towards several nucleoside sugar moieties have been developed throughout recent years, biocatalytic nucleobase synthesis has only been marginally explored. Currently, synthesis of nucleobase analogues mostly relies on conventional cyclocondensation reactions using various simple building blocks.440–442 Recent studies showed that biocatalytic synthesis of simple unsaturated/aromatic N-heterocycles can be achieved using transaminases and imine reductases.443,444 In the future, such enzymatic conversions might also become applicable for the synthesis of analogues of purines and pyrimidines.
Functionalisation of the nucleobase is often required in order for the nucleoside analogue to show improved affinity/selectivity (i.e. islatravir and abacavir). Biocatalytic alternatives to metal-catalysed functionalisation strategies might be of value here, too. Currently, several enzymes have already been shown to be capable of functionalising N-heterocycles such as tryptophan and indole, for example.445–447 The importance of other enzymatic (late-stage) C–H functionalisation will be elaborated on in more detail towards the end of this chapter.
Similarly, decoration of the nucleoside's sugar core is often required in order for the drug to selectively inhibit viral protein targets. Examples of nucleoside analogues bearing such additional functional groups are sofosbuvir, remdesivir and islatravir, which contain fluoro, cyano or alkynyl branching, respectively. Biocatalytic access to such non-natural structural elements requires novel enzymes with innovative reactivity and tolerance for unusual substrates.
The synthesis of Sofosbuvir's fluorinated sugar core through chemical aldol addition, as discussed previously, provides an excellent example for highlighting the potential of enzyme catalysis upon prospective protein engineering. Sofosbuvir's sugar fragment has been prepared through a lithium diisopropylamide (LDA)-promoted aldol addition of ethyl 2-fluoropropanoate (27) to (R)-glyceraldehyde acetonide (26).80 However, this reaction resulted in a mixture of stereoisomers that required additional resolution and down-stream purification in order to isolate (2R,3R)-28 in pure form (Scheme 69A). This type of transformation would thus likely profit from the use of a biocatalyst as these often provide excellent stereoselectivity. Chang and co-workers have recently reported on the aldolase-catalysed addition of fluoropyruvic acid (352) to unprotected (R)-glyceraldehyde (351). Treatment of the resulting aldol product with hydrogen peroxide resulted in the formation of carboxylic acid 353, which is structurally similar to 28 (Scheme 69B).448 Use of either an aldolase from Sphingomonas wittichii (SwHpcH1) or E. coli (EcHpcH) resulted in quantitative conversion of starting material with a strong preference towards the anti-product. By mutating the leucine in position 212 to phenylalanine, EcHpcH was made to favour formation of the desired (3R)-product, albeit at the expense of enzyme activity.
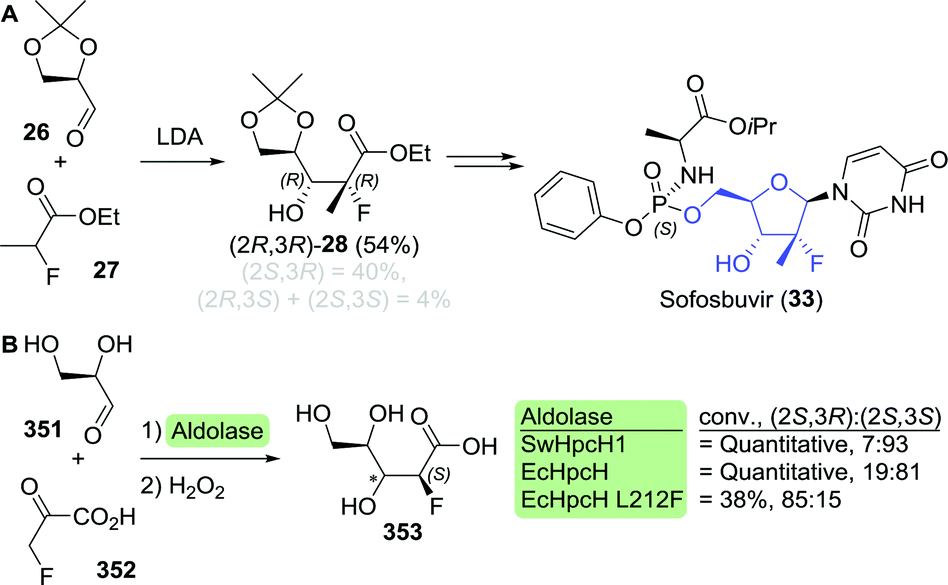 | ||
| Scheme 69 Lithium diisopropylamide (LDA)-catalysed aldol addition towards the sofosbuvir intermediate (2R,3R)-28 as developed by HC-Pharma (A).80 Enzymatic aldol reaction of (R)-glyceraldehyde and fluoropyruvic acid as developed by Chang et al.448 SwHpcH1: Sphingomonas wittichii aldolase and EcHpcH: E. coli aldolase. | ||
Clearly, significant further development is needed to arrive at the sugar core of sofosbuvir, including enzyme engineering to adapt for fluorinated oxobutanoate as nucleophile. Nonetheless, the use of an enzyme-catalysed reaction to furnish sofosbuvir's sugar core would be a more sustainable alternative to harsh wasteful chemical aldol addition. Indeed, aldolases are of particular synthetic value as they allow for the stereoselective construction of complex molecular architectures through C–C bond formation as exemplified by the stereospecific construction of islatravir's and zanamivir's central frameworks from simple building blocks.
In order to attain the correct oxidation state of the aldose moiety in sofosbuvir, the resultant fluorocarboxylic acid intermediate (analogous to 353) would need to be reduced to a fluoroaldehyde. Aldehyde functionalities are sensitive and readily oxidise to carboxylic acids, which is why selective oxidation or reduction to an aldehyde species can be challenging to control. The ability of enzymes to perform orthogonal reactions in water allows for in situ generation of (labile) intermediates to enable cascade reactions.449 Carboxylic acid reductases are a class of enzymes that only recently emerged as biocatalytic tools that can catalyse the selective reduction of a wide variety of carboxylic acids to aldehydes.450
Not only the biocatalytic synthesis of the heavily decorated sugar fragments themselves has been proven to be challenging, their enzymatic coupling to the nucleobase is often highly demanding, too. The ethynyl group on islatravir's core, for example, hampered biocatalytic coupling efficiency considerably. The latter only could be effected upon significant engineering of the involved enzymes.90 As discussed above, potential biocatalytic synthesis of remdesivir would likely pose even bigger challenges due to the presence of an additional cyano substituent that renders the nucleobase linkage non-glycosidic. Natural glycosyltransferases and phosphorylases are incapable of coupling such non-natural components, which would require significant engineering of existing biocatalysts or the discovery of novel enzymes for this purpose.
Peptidomimetic drugs are the second largest class of anti-viral agents. These drugs act as protease and integrase inhibitors and thereby suppress viral replication (Fig. 1D/E). Their main building blocks are amino acids of which several are non-canonical. Notable examples are bulky amino acids such as L-tert-leucine, L-6-heptenylglycine and L-cyclohexylglycine. The latter are accessible in enantiomerically pure form through various well established biocatalytic resolution or deracemisation approaches. In addition, the biocatalytic asymmetric synthesis of amino acids from pro-chiral ketones is becoming more and more routine (vide supra) using, e.g., leucine dehydrogenases or transaminases.
Over the past five years reductive aminases have been developed as another class of enzymes capable of performing cofactor-driven reductive aminations between an amine and a carbonyl component.451 Reductive aminases are a sub-class of imine reductases that catalyse both sequential steps of imine formation and reduction. Even though these enzymes are only a very recent addition to the biocatalysis toolbox, first processes are already employed on industrial scale.452,453
Despite the multitude of available methods applied to the biocatalytic synthesis of non-canonical amino acids the preparation of (S)-2-amino-4-morpholinobutanoic acid, which is a cobicistat building block, is lacking. Cobicistat (354, Chart 13A) is part of several multi-drug formulations, but not an anti-viral agent itself. It acts as an inhibitor of liver enzymes that are responsible for eliminating the active drug by metabolising it for clearance. The net result of cobicistat combinations is an increase in the effective drug concentration, which thus allows for lower dosaging.454 The morpholine containing unit is commonly prepared through oxidation of L-homoserine (or a peptidomimetic fragment containing this amino acid) to the corresponding aldehyde and subsequent reductive amination with morpholine.455 The use of any of the described biocatalytic transamination/reductive amination strategies for the synthesis of, for example, (S)-2-amino-4-morpholinobutanoic acid and ruxolitinib (Scheme 68) would be of considerable interest.
 | ||
| Chart 13 Structures of cobicistat with its morpholine-based amino acid highlighted in blue (A) and (1R,2S)-1-aminocyclopropanecarboxylic acid (ACCA) derivatives (B). | ||
Other amino acids, for example those bearing an α-quaternary centre, cannot be accessed through reductive amination or transamination. Their direct biocatalytic synthesis would thus require different enzymes. Exemplary is the bis-cyclopropyl-based protease inhibitors’ distinctive ACCA-moiety (Chart 13B) which is currently obtained through wasteful enzymatic kinetic resolution. Enzymatic cyclopropanation has briefly been touched upon regarding a marginally successful synthesis of the grazoprevir intermediate (1R,2R)-2-(pent-4-ynyl)cyclopropanol (Section 3.4.6). The scope of cytochrome P411-mediated (carbene transfer) reactions, however, is far bigger than mere cyclopropanation of such scarcely functionalised substrates.456–458 Synthesis of ACCA by cytochrome-catalysed cyclopropanation would potentially allow for its single step synthesis from achiral starting material. The promising nature of these enzymes will be highlighted once more towards the end of this chapter.
The (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol building block (361) is the central core of ritonavir and lopinavir (Kaletra), an anti-HIV combination drug that is currently also trialled against SARS-CoV-2 (Scheme 70).459,460 Rudolph and co-workers have developed one of the shortest chemical routes towards 361 by combining asymmetric aldol addition to α-amino aldehyde 359 and reductive amination.461 Whereas the degree of chiral induction had not been reported, reductive amination of similar test substrates resulted in 86% de, at best. With the possible integration of in situ aldehyde generation, stereoselective aldol addition and transamination, the potential enzymatic synthesis of 361 might provide for an excellent showcase of biocatalysis. Although yet a pipe dream, recent protein engineering initiatives have already shown that enzymatic transamination of similarly bulky substrates is possible.296,462
 | ||
| Scheme 70 Ritonavir and lopinavir, the active pharmaceutical ingredients of Kaletra (A). Chemical synthesis of (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol as developed by Rudolph and co-workers (B).461 PMP: para-methoxyphenyl and CAN: cerium ammonium nitrate. | ||
Coupling of biocatalytically generated amino acid (analogues) through enzymatic amide bond formation would mean yet another advancement in the sustainable nature of biocatalytic anti-viral drug production. Currently, coupling is generally achieved using (harsh) chemical methods.463,464 This not only holds for amide bonds between amino acid fragments but also for those present in integrase inhibitors such as dolutegravir and bictegravir. Hydrolases are a well-established class of enzymes capable of catalysing kinetically controlled amide forming reactions.465–467 More recently, the use of ATP-dependent enzymes for the formation of amide bonds has harnessed considerable attention, too. Exemplary is the use of amide bond synthetase McbA for the synthesis of the monoamine oxidase A inhibitor moclobemide.468 The amidation could even be performed at near stoichiometric amounts of amine. The potential, but also the challenges, of biocatalytic amide bond formation have recently been reviewed by Petchey and Grogan.469 For examples of biocatalytic amidation reactions on industrial scale the reader is referred to a recent review by Dorr and Fuerst.470
Although not discussed previously, a common chemical route to the synthesis of darunavir's capping fragment 3-OH-bis-THF ((3R,3aS,6aR)-176) involves the (in situ) formation of 3,4-dihydroxy-2-(2-hydroxyethyl)butanal (364) or suitable derivatives thereof (Scheme 71). Application of acidic conditions trigger spontaneous double cyclisation to furnish 3-OH-bis-THF. Non-stereoselective synthesis of protected analogues of 364 required additional (enzymatic) resolution or down-stream purification of 3-OH-bis-THF.471 Complementary to a complex stereoselective chemical synthesis of the desired (2S,3R)-364,472–474 an enzymatic aldol addition would pose an interesting short synthetic strategy. With various examples discussed throughout this manuscript, biocatalytic carboligation has clearly been shown to be a powerful tool capable of rapidly accessing the complex architectures present in both anti-viral nucleoside analogues and peptidomimetics.
 | ||
| Scheme 71 Formal synthesis of (2S,3R)-3,4-dihydroxy-2-(2-hydroxyethyl)butanal, followed by cyclisation towards the darunavir intermediate 3-OH-bis-THF. | ||
The value of biocatalytic C–H functionalisation at a late stage in the synthesis route deserves specific attention as it allows the use of simple (non-activated) non-functionalised starting materials. Exemplary are flavin-dependent halogenases which catalyse site-selective C–H halogenation of aromatic compounds. They offer potential synthetic utility through preparative-scale halogenation of building blocks, sequential halogenation/cross-coupling or late-stage product functionalisation.475 Throughout the last years numerous enzymes have been discovered/developed to achieve oxyfunctionalisation, such as novel peroxygenases.476,477 Cytochrome P450 enzymes, which possess the unique ability of activating inert sp3-hybridised C–H bonds, offer an attractive synthetic tool for both regioselective oxyfunctionalisation as well as late-stage scaffold diversification.478,479 Although P450-catalysed processes have often been deemed unsuited for industrial scale-up, recent developments by Innosyn/DSM have led to the production of 4-hydroxy-α-isophorone on kilogram scale.480 Catalytic anti-Markovnikov oxidation of alkene feedstocks by engineered cytochrome P450 enzymes could simplify synthetic routes to many important molecules and solve a long-standing challenge in chemistry.481 Beyond the use of α-ketoglutarate-dependent non-haem iron oxygenases/hydroxylases such as those employed in the synthesis of L-hydroxyproline and podophyllotoxin, the use of related halogenases for C–H functionalisation has been rapidly developed over the last years.482
Apart from challenges regarding the specific chemistries discussed above, other limitations towards the wide-spread implementation of biocatalysis involve process-related parameters. One of the key advantages of biocatalysts is their activity in water. The use of aqueous solvent systems does, however, impede the use of hydrophobic substrates, a limitation that can partially be relieved by the use of co-solvents or emulsions.363 However, in conjunction with extreme pH and elevated temperatures, the use of co-solvents is likely to result in a loss of enzyme activity. A viable remedy is to employ naturally (thermo)stable enzymes from extremophilic organisms that live in harsh environments. Such enzymes are commonly also stable against organic co-solvents.483–486
Despite the challenges, the adoption of biocatalysis in the pharmaceutical industry continues to expand as a result of an increased ability of engineering such enzymes to meet the demands of ideal industrial process conditions. In order to become compatible with the time pressure related to pharmaceutical process development, dramatic increases in the speed of protein engineering are needed to reduce the lead times for biocatalytic process optimisation.487 Conventional experiments for generating proteins with improved properties by directed evolution are iterative, lengthy and costly. Novel ultrahigh-throughput microfluidic screening technologies488 can dramatically accelerate the discovery of superior biocatalysts against the desired criteria from a single round of genetic randomisation.489
Further acceleration is likely to be gained by applying machine learning technologies and artificial intelligence.488,490 Indeed, the augmented ability of tailoring enzyme function by in vitro evolution (enhancing specific activity, substrate scope, stereoselectivity, tolerance to high co-solvent/substrate/reagent/product loading, long-term kinetic and thermal stability) or even inventing entirely new-to-nature activities that match chemical precedence has enormous potential for future pharmaceutical process development.491
6. Conclusions
The outbreak of COVID-19 has once more emphasised the impact of viral infections on human health and healthcare. Research towards the development and repurposing of anti-viral agents has sky-rocketed concomitantly. Upon finding an effective drug, mass production of this agent will prove to be the next challenge. This comprehensive overview reveals that biocatalytic synthesis of anti-viral agents is much more widespread than apparent from earlier surveys on the topic; altogether over 60% of all FDA-approved anti-viral agents (or intermediates thereof) are accessible through biocatalysis. As such, this key enabling technology has a bright prospect of being a crucial sustainability factor in the synthesis of future COVID-19 drugs.The structural diversity among anti-viral drug molecules is evidently matched well by a broad range of biocatalytic strategies that have been applied to make them. While enzymatic kinetic resolution is still used frequently, ingenious strategies for direct asymmetric synthesis clearly dominate recent developments to obtain enantiomerically pure anti-viral agents. Microbial engineering furthermore enabled numerous effective fermentative procedures. More and more of these synthetic strategies have been scaled up from academic ventures to industrial processes.33 Generally, the field made a transition from single step hydrolytic transformations to the development of efficient complex reaction cascades in vitro.492 The recent development of an entirely non-natural enzymatic reaction sequence for the synthesis of islatravir is, in our opinion, an absolute masterwork that heralds a paradigm shift in the synthesis of complex chiral small molecule drugs by exploiting the potential of synthetic biology.90,91 This seems to suggest that there is (almost) no limit to what can be achieved through dedicated biocatalysis.
Although the number of biocatalytically accessible molecules is rising due a continuous enlargement of the enzymatic toolbox, the process of achieving this remains challenging. There are still a large number of enzyme classes and their corresponding transformations that are underrepresented in the class of anti-viral pharmaceuticals, or that have not yet been applied at all. Reasons might stem from a limited early development stage of novel enzyme types or a lack of enzyme activity matching the desired substrate structures. Protein engineering, and especially rapid screening for improved protein variants, is currently one of the largest bottlenecks towards the routine application of novel enzymes. Lifting such limitations is a crucial step in the further development of the field of biocatalysis. Because of our limited understanding of the protein folding problem and related structure–activity–function relationships, research at the forefront of the science is involving artificial intelligence by machine-learning approaches jointly with ultra-high-throughput screening platforms to accelerate enzyme discovery and engineering.491
Perhaps the largest limitation towards a more profound implementation of biotechnological processes is not a practical one, but rather the result of a lack of incorporation of biocatalytic methods into the synthetic chemistry curriculum. To be able to realise the benefits of biocatalysis, students need to be familiarised with the synthetic potential of biocatalytic methods, and they should be taught early on how these can be interfaced with chemical routes. Biocatalysis and ‘traditional’ synthetic chemistry are not two separate philosophies,23 and more and more scientists from both fields are actively involved in research efforts to integrate the two in view of the enormous opportunities for novel creative solutions and IP generation.
Given the demand from a continuously rising number of chiral (anti-viral) drugs, the impact of biocatalysis on industrial pharmaceutical synthesis will likely experience a similar growth. As the market is currently going through a transition imposed by the COVID-19 pandemic, scientists around the globe are urged to seize the momentum by implementing these novel biocatalytic technologies in order to sustainably revolutionise pharmaceutical production. We strongly believe that a crisis such as the one caused by COVID-19 will also open up golden opportunities for those considering biocatalytic synthesis in the search for the proverbial silver bullet against this, and future, viral diseases.
Conflicts of interest
Within the framework of the Tralaminol project the authors co-operate with BASF SE, which is involved in the production of anti-viral intermediates discussed in this work. However, the authors did not consult with BASF staff on this topic.Acknowledgements
This work was supported by The German Federal Ministry of Education and Research (BMBF) under Grant Agreement No 031B0595 and the initiative ERA CoBioTech. ERA CoBioTech has received funding from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No 722361.References
- WHO Coronavirus Disease (COVID-19) Dashboard, https://covid19.who.int/, accessed 22/09, 2020.
- Ebola situation reports: Democratic Republic of the Congo, World Health Organization, 2019.
- S. M. Moghadas, A. Shoukat, M. C. Fitzpatrick, C. R. Wells, P. Sah, A. Pandey, J. D. Sachs, Z. Wang, L. A. Meyers, B. H. Singer and A. P. Galvani, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9122–9126 CrossRef CAS PubMed.
- E. J. Emanuel, G. Persad, R. Upshur, B. Thome, M. Parker, A. Glickman, C. Zhang, C. Boyle, M. Smith and J. P. Phillips, N. Engl. J. Med., 2020, 382, 2049–2055 CrossRef.
- J. Hopman, B. Allegranzi and S. Mehtar, JAMA, J. Am. Med. Assoc., 2020, 323, 1549–1550 CrossRef CAS PubMed.
- M. Gilbert, G. Pullano, F. Pinotti, E. Valdano, C. Poletto, P.-Y. Boëlle, E. D'Ortenzio, Y. Yazdanpanah, S. P. Eholie, M. Altmann, B. Gutierrez, M. U. G. Kraemer and V. Colizza, Lancet, 2020, 395, 871–877 CrossRef CAS.
- B. Pfefferbaum and C. S. North, N. Engl. J. Med., 2020, 383, 510–512 CrossRef CAS PubMed.
- B. A. Cunha, Infect. Dis. Clin., 2004, 18, 141–155 CrossRef.
- M. H. Merson, J. O'Malley, D. Serwadda and C. Apisuk, Lancet, 2008, 372, 475–488 CrossRef.
- D. Carroll, P. Daszak, N. D. Wolfe, G. F. Gao, C. M. Morel, S. Morzaria, A. Pablos-Méndez, O. Tomori and J. A. K. Mazet, Science, 2018, 359, 872–874 CrossRef CAS PubMed.
- S. J. Anthony, J. H. Epstein, K. A. Murray, I. Navarrete-Macias, C. M. Zambrana-Torrelio, A. Solovyov, R. Ojeda-Flores, N. C. Arrigo, A. Islam, S. Ali Khan, P. Hosseini, T. L. Bogich, K. J. Olival, M. D. Sanchez-Leon, W. B. Karesh, T. Goldstein, S. P. Luby, S. S. Morse, J. A. K. Mazet, P. Daszak and W. I. Lipkin, mBio, 2013, 4, e00598 CrossRef.
- R. Henry, Emerging Infect. Dis., 2013, 19, 263 CrossRef.
- J. M. Sanders, M. L. Monogue, T. Z. Jodlowski and J. B. Cutrell, JAMA, J. Am. Med. Assoc., 2020, 323, 1824–1836 CrossRef CAS.
- L. Dong, S. Hu and J. Gao, Drug Discoveries Ther., 2020, 14, 58–60 CrossRef CAS.
- M. A. Hardy, B. A. Wright, J. L. Bachman, T. B. Boit, H. M. S. Haley, R. R. Knapp, R. F. Lusi, T. Okada, V. Tona, N. K. Garg and R. Sarpong, ACS Cent. Sci., 2020, 6, 1017–1030 CrossRef CAS PubMed.
- O. Méndez-Lucio and J. L. Medina-Franco, Drug Discovery Today, 2017, 22, 120–126 CrossRef PubMed.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS PubMed.
- R. M. Gulick, Top. Antivir. Med., 2018, 25, 127–132 Search PubMed.
- E. De Clercq and G. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 CrossRef PubMed.
- S. Chaudhuri, J. A. Symons and J. Deval, Antiviral Res., 2018, 155, 76–88 CrossRef CAS PubMed.
- J. M. Woodley, Appl. Microbiol. Biotechnol., 2019, 103, 4733–4739 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- N. J. Turner and L. Humphreys, Biocatalysis in organic synthesis: The retrosynthesis approach, Royal Society of Chemistry,London, 2018 Search PubMed.
- J. M. Woodley, Trends Biotechnol., 2008, 26, 321–327 CrossRef CAS PubMed.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- R. N. Patel, Bioorg. Med. Chem., 2018, 26, 1252–1274 CrossRef CAS PubMed.
- P. Grunwald, Pharmaceutical Biocatalysis: Chemoenzymatic Synthesis of Active Pharmaceutical Ingredients, CRC Press,Boca Raton, 2019 Search PubMed.
- K. Rosenthal and S. Lütz, Curr. Opin. Green Sustainable Chem., 2018, 11, 58–64 CrossRef.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 1063–1080 CrossRef CAS.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- Y. Wang, Y. Cao, Y. Li, J. Jin, J. Li and H. Song, Tetrahedron: Asymmetry, 2017, 28, 745–757 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2020, 59, 2–34 CrossRef.
- A. Casini, F.-Y. Chang, R. Eluere, A. M. King, E. M. Young, Q. M. Dudley, A. Karim, K. Pratt, C. Bristol, A. Forget, A. Ghodasara, R. Warden-Rothman, R. Gan, A. Cristofaro, A. E. Borujeni, M.-H. Ryu, J. Li, Y.-C. Kwon, H. Wang, E. Tatsis, C. Rodriguez-Lopez, S. O’Connor, M. H. Medema, M. A. Fischbach, M. C. Jewett, C. Voigt and D. B. Gordon, J. Am. Chem. Soc., 2018, 140, 4302–4316 CrossRef CAS PubMed.
- E. O. Freed, Virology, 1998, 251, 1–15 CrossRef CAS.
- A. Kapoor and S. Raju, Illustrated medical pharmacology, JP Medical Ltd, New Delhi, 1st edn, 2013 Search PubMed.
- J. Holguin, R. Cardinaud and C. A. Salemink, Eur. J. Biochem., 1975, 54, 515–520 CrossRef CAS.
- T. A. Krenitsky, G. W. Koszalka and J. V. Tuttle, Biochemistry, 1981, 20, 3615–3621 CrossRef CAS PubMed.
- J. F. Lucas and M.-J. C. Rius, Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives, John Wiley & Sons,Hoboken, 2019 Search PubMed.
- E. S. Lewkowicz and A. M. Iribarren, Curr. Pharm. Des., 2017, 23, 6851–6878 CrossRef CAS PubMed.
- J. Fernández-Lucas, Appl. Microbiol. Biotechnol., 2015, 99, 4615–4627 CrossRef PubMed.
- M. Ferrero and V. Gotor, Chem. Rev., 2000, 100, 4319–4348 CrossRef CAS PubMed.
- W. H. Prusoff, Biochim. Biophys. Acta, 1959, 32, 295–296 CrossRef CAS.
- I. Serra, T. Bavaro, D. A. Cecchini, S. Daly, A. M. Albertini, M. Terreni and D. Ubiali, J. Mol. Catal. B: Enzym., 2013, 95, 16–22 CrossRef CAS.
- A. Fresco-Taboada, I. Serra, J. Fernández-Lucas, C. Acebal, M. Arroyo, M. Terreni and I. De la Mata, Molecules, 2014, 19, 11231–11249 CrossRef.
- R. Whitley, C. Alford, F. Hess and R. Buchanan, Drugs, 1980, 20, 267–282 CrossRef CAS.
- T. Utagawa, H. Morisawa, F. Yoshinaga, A. Yamazaki, K. Mitsugi and Y. Hirose, Agric. Biol. Chem., 1985, 49, 1053–1058 CAS.
- I. Serra, D. Ubiali, J. Piškur, S. Christoffersen, E. S. Lewkowicz, A. M. Iribarren, A. M. Albertini and M. Terreni, ChemPlusChem, 2013, 78, 157–165 CrossRef CAS.
- I. Serra, S. Daly, A. R. Alcantara, D. Bianchi, M. Terreni and D. Ubiali, RSC Adv., 2015, 5, 23569–23577 RSC.
- L. Tamborini, C. Previtali, F. Annunziata, T. Bavaro, M. Terreni, E. Calleri, F. Rinaldi, A. Pinto, G. Speranza, D. Ubiali and P. Conti, Molecules, 2020, 25, 1223 CrossRef CAS PubMed.
- J. Fernández-Lucas, R. Harris, I. Mata-Casar, A. Heras, I. de la Mata and M. Arroyo, J. Ind. Microbiol. Biotechnol., 2013, 40, 955–966 CrossRef PubMed.
- E. Pérez, P. A. Sánchez-Murcia, J. Jordaan, M. D. Blanco, J. M. Mancheño, F. Gago and J. Fernández-Lucas, ChemCatChem, 2018, 10, 4406–4416 CrossRef.
- C. M. Perry and S. Noble, Drugs, 1999, 58, 1099–1135 CrossRef CAS.
- S. Christoffersen, I. Serra, M. Terreni and J. Piškur, Nucleosides, Nucleotides Nucleic Acids, 2010, 29, 445–448 CrossRef CAS PubMed.
- M. Nóbile, M. Terreni, E. Lewkowicz and A. M. Iribarren, Biocatal. Biotransform., 2010, 28, 395–402 CrossRef.
- W. R. Birmingham, C. A. Starbird, T. D. Panosian, D. P. Nannemann, T. M. Iverson and B. O. Bachmann, Nat. Chem. Biol., 2014, 10, 392–399 CrossRef CAS.
- B.-C. Chen, S. L. Quinlan and J. Gregory Reid, Tetrahedron Lett., 1995, 36, 7961–7964 CrossRef CAS.
- R. H. Spector, B.-C. Chen and S. L. Quinlan, US Pat., US5608049A, 1997 Search PubMed.
- N. Hori, M. Watanabe, Y. Yamazaki and Y. Mikami, Agric. Biol. Chem., 1989, 53, 197–202 CAS.
- M. Ishii, H. Shirae and K. Yokozeki, Agric. Biol. Chem., 1989, 53, 3209–3218 CAS.
- N. Hori, M. Watanabe, K. Sunagawa, K. Uehara and Y. Mikami, J. Biotechnol., 1991, 17, 121–131 CrossRef CAS.
- D. F. Visser, K. J. Rashamuse, F. Hennessy, G. E. R. Gordon, P. J. Van Zyl, K. Mathiba, M. L. Bode and D. Brady, Biocatal. Biotransform., 2010, 28, 245–253 CrossRef CAS.
- M.-S. Daniel, L.-I. María and G.-F. Vicente, Curr. Org. Chem., 2016, 20, 1186–1203 CrossRef.
- C. Callaghan, M. Redmond, R. C. Alnoch, C. Mateo, M. Filice and J. M. Palomo, ChemCatChem, 2017, 9, 2536–2543 CrossRef CAS.
- E. Thomas, M. G. Ghany and T. J. Liang, Antiviral Chem. Chemother., 2012, 23, 1–12 CrossRef CAS.
- H. Shirae, K. Yokozeki and K. Kubota, Agric. Biol. Chem., 1988, 52, 1499–1504 CAS.
- H. Shirae, K. Yokozeki and K. Kubota, Agric. Biol. Chem., 1988, 52, 1233–1237 CAS.
- H. Shirae, K. Yokozeki, M. Uchiyama and K. Kubota, Agric. Biol. Chem., 1988, 52, 1777–1783 CAS.
- H. Shirae and K. Yokozeki, Agric. Biol. Chem., 1991, 55, 1849–1857 CAS.
- H. Shirae, K. Yokozeki and K. Kubota, Agric. Biol. Chem., 1991, 55, 605–607 CAS.
- V. N. Barai, A. I. Zinchenko, L. A. Eroshevskaya, E. N. Kalinichenko, T. I. Kulak and I. A. Mikhailopulo, Helv. Chim. Acta, 2002, 85, 1901–1908 CrossRef CAS.
- I. D. Konstantinova, N. A. Leont'eva, G. A. Galegov, O. I. Ryzhova, D. V. Chuvikovskii, K. V. Antonov, R. S. Esipov, S. A. Taran, K. N. Verevkina, S. A. Feofanov and A. I. Miroshnikov, Russ. J. Bioorg. Chem., 2004, 30, 553–560 CrossRef CAS PubMed.
- E. C. De Benedetti, C. W. Rivero and J. A. Trelles, J. Mol. Catal. B: Enzym., 2015, 121, 90–95 CrossRef CAS.
- C. W. Rivero, E. C. De Benedetti, M. E. Lozano and J. A. Trelles, Process Biochem., 2015, 50, 935–940 CrossRef CAS PubMed.
- M. Rabuffetti, T. Bavaro, R. Semproli, G. Cattaneo, M. Massone, C. F. Morelli, G. Speranza and D. Ubiali, Catalysts, 2019, 9, 355 CrossRef CAS.
- F. Poordad, E. Lawitz, M. L. Shiffman, T. Hassanein, A. J. Muir, B. R. Bacon, J. Heise, D. Halliman, E. Chun and J. Hammond, Hepatology, 2010, 52, 1208–1215 CrossRef CAS.
- H. Ren, L. Lu, M. Zheng, R. Ou, X. Lin, G. Huang and Y. Ning, PRC Pat., CN102978264A, 2013 Search PubMed.
- I. Gentile, A. E. Maraolo, A. R. Buonomo, E. Zappulo and G. Borgia, Exp. Opin. Drug Discovery, 2015, 10, 1363–1377 CrossRef CAS PubMed.
- D. Cahard, C. McGuigan and J. Balzarini, Mini-Rev. Med. Chem., 2004, 4, 371–381 CrossRef CAS PubMed.
- P. Zhang, H. Iding, M. Cedilote, S. Brunner, T. Williamson and T. P. Cleary, Tetrahedron: Asymmetry, 2009, 20, 305–312 CrossRef CAS.
- P. Wang, B.-K. Chun, S. Rachakonda, J. Du, N. Khan, J. Shi, W. Stec, D. Cleary, B. S. Ross and M. J. Sofia, J. Org. Chem., 2009, 74, 6819–6824 CrossRef CAS.
- F. Molinari, O. Brenna, M. Valenti and F. Aragozzini, Enzyme Microb. Technol., 1996, 19, 551–556 CrossRef CAS.
- M. J. Dröge, R. Bos and W. J. Quax, Eur. J. Biochem., 2001, 268, 3332–3338 CrossRef PubMed.
- D. Romano, F. Falcioni, D. Mora, F. Molinari, A. Buthe and M. Ansorge-Schumacher, Tetrahedron: Asymmetry, 2005, 16, 841–845 CrossRef CAS.
- L. F. Godinho, C. R. Reis, P. G. Tepper, G. J. Poelarends and W. J. Quax, Appl. Environ. Microbiol., 2011, 77, 6094–6099 CrossRef CAS.
- M. Gaboardi, G. Pallanza, M. Baratella, G. Castaldi and M. Castaldi, World Pat., WO2017144423A1, 2017 Search PubMed.
- C. Roodveldt and D. S. Tawfik, Protein Eng., Des. Sel., 2005, 18, 51–58 CrossRef CAS.
- D. F. Xiang, A. N. Bigley, E. Desormeaux, T. Narindoshvili and F. M. Raushel, Biochemistry, 2019, 58, 3204–3211 CrossRef CAS PubMed.
- B. S. Ross, P. Ganapati Reddy, H.-R. Zhang, S. Rachakonda and M. J. Sofia, J. Org. Chem., 2011, 76, 8311–8319 CrossRef CAS PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- E. O'Reilly and J. Ryan, Science, 2019, 366, 1199–1200 CrossRef PubMed.
- C. Orkin, J. M. Llibre, S. Gallien, A. Antinori, G. M. N. Behrens and A. Carr, HIV Med., 2018, 19, 18–32 CrossRef CAS.
- J. Milton, S. Brand, M. F. Jones and C. M. Rayner, Tetrahedron Lett., 1995, 36, 6961–6964 CAS.
- R. Storer, I. R. Clemens, B. Lamont, S. A. Noble, C. Williamson and B. Belleau, Nucleosides Nucleotides, 1993, 12, 225–236 CrossRef CAS.
- M. Mahmoudian, B. S. Baines, C. S. Drake, R. S. Hale, P. Jones, J. E. Piercey, D. S. Montgomery, I. J. Purvis, R. Storer, M. J. Dawson and G. C. Lawrence, Enzyme Microb. Technol., 1993, 15, 749–755 CrossRef CAS.
- L. K. Hoong, L. E. Strange, D. C. Liotta, G. W. Koszalka, C. L. Burns and R. F. Schinazi, J. Org. Chem., 1992, 57, 5563–5565 CrossRef CAS.
- R. P. C. Cousins, M. Mahmoudian and P. M. Youds, Tetrahedron: Asymmetry, 1995, 6, 393–396 CrossRef CAS.
- Y. Chen, X. Zhang, G. Zheng and S. Gao, Process Biochem., 2019, 81, 77–84 CrossRef CAS.
- L. Hu, F. Schaufelberger, Y. Zhang and O. Ramström, Chem. Commun., 2013, 49, 10376–10378 RSC.
- Y. Ren, L. Hu and O. Ramström, Mol. Catal., 2019, 468, 52–56 CrossRef CAS.
- P. S. Hervey and C. M. Perry, Drugs, 2000, 60, 447–479 CrossRef CAS PubMed.
- R. Singh and R. Vince, Chem. Rev., 2012, 112, 4642–4686 CrossRef CAS PubMed.
- S. J. C. Taylor, R. McCague, R. Wisdom, C. Lee, K. Dickson, G. Ruecroft, F. O'Brien, J. Littlechild, J. Bevan, S. M. Roberts and C. T. Evans, Tetrahedron: Asymmetry, 1993, 4, 1117–1128 CrossRef CAS.
- X. Qin, J. Wang and G. Zheng, Appl. Biochem. Biotechnol., 2010, 162, 2345–2354 CrossRef CAS PubMed.
- S. Zhu, C. Gong, D. Song, S. Gao and G. Zheng, Appl. Environ. Microbiol., 2012, 78, 7492–7495 CrossRef CAS PubMed.
- Z. Assaf, E. Eger, Z. Vitnik, W. M. F. Fabian, D. Ribitsch, G. M. Guebitz, K. Faber and M. Hall, ChemCatChem, 2014, 6, 2517–2521 CrossRef CAS.
- J. Wang, G. Zhao, Z. Zhang, Q. Liang, C. Min and S. Wu, Appl. Microbiol. Biotechnol., 2014, 98, 6991–7001 CrossRef CAS PubMed.
- J. Wang, J. Zhu, C. Min and S. Wu, BMC Biotechnol., 2014, 14, 40 CrossRef PubMed.
- S. Gao, S. Zhu, R. Huang, Y. Lu and G. Zheng, Bioorg. Med. Chem. Lett., 2015, 25, 3878–3881 CrossRef CAS PubMed.
- L. Ren, S. Zhu, Y. Shi, S. Gao and G. Zheng, Appl. Biochem. Biotechnol., 2015, 176, 170–184 CrossRef CAS PubMed.
- J. Wang, J. Zhu and S. Wu, Appl. Microbiol. Biotechnol., 2015, 99, 4691–4700 CrossRef CAS PubMed.
- T.-Y. Xue, G.-C. Xu, R.-Z. Han and Y. Ni, Appl. Biochem. Biotechnol., 2015, 176, 1687–1699 CrossRef CAS PubMed.
- S. Gao, R. Huang, S. Zhu, H. Li and G. Zheng, Appl. Microbiol. Biotechnol., 2016, 100, 9543–9553 CrossRef CAS PubMed.
- S. Zhu, R. Huang, S. Gao, X. Li and G. Zheng, J. Biosci. Bioeng., 2016, 121, 484–490 CrossRef CAS PubMed.
- Y. Su, S. Gao, H. Li and G. Zheng, Process Biochem., 2018, 72, 96–104 CrossRef CAS.
- M. Yang, Q. Gao, S. Wu, J. Wang and G. Zheng, Biotechnol. Lett., 2012, 34, 275–279 CrossRef CAS.
- S. Zhu, L. Ren, S. Yu, C. Gong, D. Song and G. Zheng, Bioorg. Med. Chem. Lett., 2014, 24, 4899–4902 CrossRef CAS PubMed.
- Y. Sun, H. Zhao, J. Wang, J. Zhu and S. Wu, Appl. Microbiol. Biotechnol., 2015, 99, 7559–7568 CrossRef CAS PubMed.
- H. S. Toogood, R. C. Brown, K. Line, P. A. Keene, S. J. C. Taylor, R. McCague and J. A. Littlechild, Tetrahedron, 2004, 60, 711–716 CrossRef CAS.
- J.-G. Yin, Y. Gong, X.-Y. Zhang, G.-W. Zheng and J.-H. Xu, Catal. Sci. Technol., 2016, 6, 6305–6310 RSC.
- E. Forro and F. Fueloep, Eur. J. Org. Chem., 2008, 5263–5268 CrossRef CAS.
- L. L. Torres, A. Schliessmann, M. Schmidt, N. Silva-Martin, J. A. Hermoso, J. Berenguer, U. T. Bornscheuer and A. Hidalgo, Org. Biomol. Chem., 2012, 10, 3388–3392 RSC.
- Z. Galla, E. Forro and F. Fueloep, Tetrahedron: Asymmetry, 2016, 27, 729–731 CrossRef CAS.
- M. Mahmoudian, A. Lowdon, M. Jones, M. Dawson and C. Wallis, Tetrahedron: Asymmetry, 1999, 10, 1201–1206 CrossRef CAS.
- S. M. Daluge, M. T. Martin, B. R. Sickles and D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 297–327 CrossRef CAS PubMed.
- G. A. Boyle, C. D. Edlin, Y. Li, D. C. Liotta, G. L. Morgans and C. C. Musonda, Org. Biomol. Chem., 2012, 10, 1870–1876 RSC.
- T. Shaw and S. Locarnini, Exp. Rev. Anti-Infect. Ther., 2004, 2, 853–871 CrossRef CAS PubMed.
- R. N. Patel, A. Banerjee, Y. R. Pendri, J. Liang, C.-P. Chen and R. Mueller, Tetrahedron: Asymmetry, 2006, 17, 175–178 CrossRef CAS.
- Y. R. Pendri, C.-P. H. Chen, S. S. Patel, J. M. Evans, J. Liang, D. R. Kronenthal, G. L. Powers, S. J. Prasad, J. T. Bien and Z. Shi, World Pat., WO2004052310, 2006 Search PubMed.
- D. A. Griffith and S. J. Danishefsky, J. Am. Chem. Soc., 1996, 118, 9526–9538 CrossRef CAS.
- I. J. S. Fairlamb, J. M. Dickinson, R. O'Connor, S. Higson, L. Grieveson and V. Marin, Bioorg. Med. Chem., 2002, 10, 2641–2656 CrossRef CAS.
- K. Balke, M. Bäumgen and U. T. Bornscheuer, ChemBioChem, 2017, 18, 1627–1638 CrossRef CAS.
- A. J. Wagstaff, D. Faulds and K. L. Goa, Drugs, 1994, 47, 153–205 CrossRef CAS PubMed.
- D. Ormrod, L. J. Scott and C. M. Perry, Drugs, 2000, 59, 839–863 CrossRef CAS PubMed.
- K. McClean, C. Preston, D. Spence, P. W. Sutton and J. Whittall, Tetrahedron Lett., 2011, 52, 215–218 CrossRef CAS.
- M. K. Sethi, S. R. Bhandya, R. Shukla, A. Kumar, N. Maddur, V. S. N. J. Mittapalli, V. S. Rawat and R. K. Yerramalla, J. Mol. Catal. B: Enzym., 2014, 108, 77–81 CrossRef CAS.
- J. T. Sime, R. D. Barnes, S. W. Elson, R. L. Jarvest and K. J. O'Toole, J. Chem. Soc., Perkin Trans. 1, 1992, 1653–1658, 10.1039/P19920001653.
- C. Callebaut, G. Stepan, Y. Tian and M. D. Miller, Antimicrob. Agents Chemother., 2015, 59, 5909–5916 CrossRef CAS.
- B. P. Kearney, J. F. Flaherty and J. Shah, Clin. Pharmacokinet., 2004, 43, 595–612 CrossRef CAS.
- A. S. Ray, M. W. Fordyce and M. J. Hitchcock, Antiviral Res., 2016, 125, 63–70 CrossRef CAS PubMed.
- D. L. Riley, D. R. Walwyn and C. D. Edlin, Org. Process Res. Dev., 2016, 20, 742–750 CrossRef CAS.
- S. Thatipally, R. A. O. Dammalapati Venkata Lakshmi Narasimha, R.Ramakrishna Kallam Venkata Siva, A. Gorantla Seeta Rama and S. Chava, World Pat., WO2015040640A2, 2015 Search PubMed.
- N. S. Suveges, A. A. Rodriguez, C. C. Diederichs, S. P. de Souza, R. A. C. Leão, L. S. M. Miranda, B. A. C. Horta, S. F. Pedraza, O. V. de Carvalho, K. C. Pais, J. H. C. Terra and R. O. M. A. de Souza, Eur. J. Org. Chem., 2018, 2931–2938 CrossRef CAS.
- E. M. Isin, C. S. Elmore, G. N. Nilsson, R. A. Thompson and L. Weidolf, Chem. Res. Toxicol., 2012, 25, 532–542 Search PubMed.
- N. R. Rivera, J. Moore, D. J. Schenk, H. Wang, D. Hesk and I. Mergelsberg, Tetrahedron Lett., 2016, 57, 1090–1092 CrossRef CAS.
- G. L. Plosker and S. Noble, Drugs, 1999, 58, 325–345 CrossRef CAS.
- P. R. Brodfuehrer, H. G. Howell, C. Sapino and P. Vemishetti, Tetrahedron Lett., 1994, 35, 3243–3246 CrossRef CAS.
- T. Suzuki, N. Kasai, R. Yamamoto and N. Minamiura, Appl. Microbiol. Biotechnol., 1993, 40, 273–278 CrossRef CAS.
- N. Kasai, T. Suzuki and Y. Furukawa, Chirality, 1998, 10, 682–692 CrossRef CAS.
- H. K. Chenault, L. F. Chafin and S. Liehr, J. Org. Chem., 1998, 63, 4039–4045 CrossRef CAS.
- L. Poppe, L. Novák, M. Kajtár-Peredy and C. Szántay, Tetrahedron: Asymmetry, 1993, 4, 2211–2217 CrossRef CAS.
- A. Geerlof, J. A. Jongejan, T. J. G. M. van Dooren, P. C. Raemakers-Franken, W. J. J. van den Tweel and J. A. Duine, Enzyme Microb. Technol., 1994, 16, 1059–1063 CrossRef CAS.
- A. Geerlof, J. B. A. Van Tol, J. A. Jongejan and J. A. Duine, Biosci., Biotechnol., Biochem., 1994, 58, 1028–1036 CrossRef CAS.
- J. M. Palomo, R. L. Segura, G. Fernandez-Lorente, J. M. Guisán and R. Fernández-Lafuente, Tetrahedron: Asymmetry, 2004, 15, 1157–1161 CrossRef CAS.
- M. Pregnolato, M. Terreni, I. E. de Fuentes, A. R. Alcantara Leon, P. Sabuquillo, R. Fernández-Lafuente and J. M. Guisán, J. Mol. Catal. B: Enzym., 2001, 11, 757–763 CrossRef CAS.
- J. M. Palomo, R. L. Segura, C. Mateo, M. Terreni, J. M. Guisán and R. Fernández-Lafuente, Tetrahedron: Asymmetry, 2005, 16, 869–874 CrossRef CAS.
- Y. Liu, Y. Wang, Y. Jiang, M. Hu, S. Li and Q. Zhai, Biotechnol. Prog., 2015, 31, 724–729 CrossRef CAS PubMed.
- A. A. Agbowuro, W. M. Huston, A. B. Gamble and J. D. A. Tyndall, Med. Res. Rev., 2018, 38, 1295–1331 CrossRef CAS PubMed.
- Y.-P. Xue, C.-H. Cao and Y.-G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561 RSC.
- K. F. Croom, S. Dhillon and S. J. Keam, Drugs, 2009, 69, 1107–1140 CrossRef CAS PubMed.
- S. Venkatraman, Trends Pharmacol. Sci., 2012, 33, 289–294 CrossRef CAS PubMed.
- A. D. Kwong, R. S. Kauffman, P. Hurter and P. Mueller, Nat. Biotechnol., 2011, 29, 993–1003 CrossRef CAS PubMed.
- A. S. Bommarius, M. Schwarm, K. Stingl, M. Kottenhahn, K. Huthmacher and K. Drauz, Tetrahedron: Asymmetry, 1995, 6, 2851–2888 CrossRef CAS.
- N. J. Turner, J. R. Winterman, R. McCague, J. S. Parratt and S. J. C. Taylor, Tetrahedron Lett., 1995, 36, 1113–1116 CrossRef CAS.
- S. L. Liu, Q. X. Song, D. Z. Wei, Y. W. Zhang and X. D. Wang, Prep. Biochem. Biotechnol., 2006, 36, 235–241 CrossRef CAS.
- J.-Z. Jin, D.-L. Chang and J. Zhang, Appl. Biochem. Biotechnol., 2011, 164, 376–385 CrossRef CAS.
- C. Wandrey and B. Bossow, Biotechnol. Bioind., 1986, 1, 8–13 CrossRef.
- A. Menzel, H. Werner, J. Altenbuchner and H. Groger, Eng. Life Sci., 2004, 4, 573–576 CrossRef CAS.
- J. Li, J. Pan, J. Zhang and J.-H. Xu, J. Mol. Catal. B: Enzym., 2014, 105, 11–17 CrossRef CAS.
- W. Liu, H. Ma, J. Luo, W. Shen, X. Xu, S. Li, Y. Hu and H. Huang, Biochem. Eng. J., 2014, 91, 204–209 CrossRef CAS.
- W. Jiang and B. Fang, Sci. Rep., 2016, 6, 30462 CrossRef CAS.
- W. Jiang, D. Sun, J. Lu, Y. Wang, S. Wang, Y. Zhang and B. Fang, Eng. Life Sci., 2016, 16, 283–289 CrossRef CAS.
- J. Lu, Y. Zhang, D. Sun, W. Jiang, S. Wang and B. Fang, Appl. Biochem. Biotechnol., 2016, 180, 1180–1195 CrossRef CAS PubMed.
- L. Zhu, Z. Wu, J.-M. Jin and S.-Y. Tang, Appl. Microbiol. Biotechnol., 2016, 100, 5805–5813 CrossRef CAS PubMed.
- T. Li, A. B. Kootstra and I. G. Fotheringham, Org. Process Res. Dev., 2002, 6, 533–538 CrossRef CAS.
- E. Heuson, F. Charmantray, J.-L. Petit, V. de Berardinis and T. Gefflaut, Adv. Synth. Catal., 2019, 361, 778–785 CrossRef CAS.
- Y.-M. Seo and H. Yun, J. Microbiol. Biotechnol., 2011, 21, 1049–1052 CrossRef CAS PubMed.
- H. Zhou, L. Meng, X. Yin, Y. Liu, G. Xu, J. Wu, M. Wu and L. Yang, Eur. J. Org. Chem., 2019, 6470–6477 CrossRef CAS.
- H. Chen, M. B. Prater, R. Cai, F. Dong, H. Chen and S. D. Minteer, J. Am. Chem. Soc., 2020, 142, 4028–4036 CrossRef CAS PubMed.
- A. P. Green and N. J. Turner, Perspect. Sci., 2016, 9, 42–48 CrossRef.
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem. – Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed.
- K. Eisler, J. Rudinger and F. Sorm, Collect. Czech. Chem. Commun., 1966, 31, 4563–4580 CrossRef CAS.
- M. Beller, M. Eckert, H. Geissler, B. Napierski, H.-P. Rebenstock and E. W. Holla, Chem. – Eur. J., 1998, 4, 935–941 CrossRef CAS.
- M.-X. Wang and S.-J. Lin, J. Org. Chem., 2002, 67, 6542–6545 CrossRef CAS.
- H. Hong, C. Zheng and G. Lina, US Pat., US20160319312A1, 2016 Search PubMed.
- V. Köhler, K. R. Bailey, A. Znabet, J. Raftery, M. Helliwell and N. J. Turner, Angew. Chem., Int. Ed., 2010, 49, 2182–2184 CrossRef.
- C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224–2225 CrossRef CAS PubMed.
- A. Znabet, E. Ruijter, F. J. J. de Kanter, V. Köhler, M. Helliwell, N. J. Turner and R. V. A. Orru, Angew. Chem., Int. Ed., 2010, 49, 5289–5292 CrossRef CAS PubMed.
- A. Znabet, M. M. Polak, E. Janssen, F. J. J. de Kanter, N. J. Turner, R. V. A. Orru and E. Ruijter, Chem. Commun., 2010, 46, 7918–7920 RSC.
- T. Li, J. Liang, A. Ambrogelly, T. Brennan, G. Gloor, G. Huisman, J. Lalonde, A. Lekhal, B. Mijts, S. Muley, L. Newman, M. Tobin, G. Wong, A. Zaks and X. Zhang, J. Am. Chem. Soc., 2012, 134, 6467–6472 CrossRef CAS PubMed.
- S. Noble and D. Faulds, Drugs, 1996, 52, 93–112 CrossRef CAS PubMed.
- K. McKeage, C. M. Perry and S. J. Keam, Drugs, 2009, 69, 477–503 CrossRef CAS.
- S. Noble and K. L. Goa, Drugs, 2000, 60, 1383–1410 CrossRef CAS.
- T. M. Chapman, G. L. Plosker and C. M. Perry, Drugs, 2004, 64, 2101–2124 CrossRef CAS PubMed.
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172–5208 CrossRef CAS PubMed.
- K. E. B. Parkes, D. J. Bushnell, P. H. Crackett, S. J. Dunsdon, A. C. Freeman, M. P. Gunn, R. A. Hopkins, R. W. Lambert and J. A. Martin, J. Org. Chem., 1994, 59, 3656–3664 CrossRef CAS.
- K. Fuji, T. Kawabata, Y. Kiryu, Y. Sugiura, T. Taga and Y. Miwa, Tetrahedron Lett., 1990, 31, 6663–6666 CrossRef CAS.
- K. Fuji, T. Kawabata, Y. Kiryu and Y. Sugiura, Heterocycles, 1996, 2, 701–722 CrossRef.
- R. N. Patel, A. Banerjee, C. G. McNamee, D. B. Brzozowski and L. J. Szarka, Tetrahedron: Asymmetry, 1997, 8, 2547–2552 CrossRef CAS.
- R. N. Patel, L. Chu and R. Mueller, Tetrahedron: Asymmetry, 2003, 14, 3105–3109 CrossRef CAS.
- A. S. de Miranda, R. C. Simon, B. Grischek, G. C. de Paula, B. A. C. Horta, L. S. M. de Miranda, W. Kroutil, C. O. Kappe and R. O. M. A. de Souza, ChemCatChem, 2015, 7, 984–992 CrossRef CAS.
- K. Wu, K. Zheng, L. Xiong, Z. Yang, Z. Jiang, X. Meng and L. Shao, Appl. Microbiol. Biotechnol., 2019, 103, 4417–4427 CrossRef CAS.
- A. Bardsley-Elliot and G. L. Plosker, Drugs, 2000, 59, 581–620 CrossRef CAS PubMed.
- S. W. Kaldor, V. J. Kalish, J. F. Davies, B. V. Shetty, J. E. Fritz, K. Appelt, J. A. Burgess, K. M. Campanale, N. Y. Chirgadze, D. K. Clawson, B. A. Dressman, S. D. Hatch, D. A. Khalil, M. B. Kosa, P. P. Lubbehusen, M. A. Muesing, A. K. Patick, S. H. Reich, K. S. Su and J. H. Tatlock, J. Med. Chem., 1997, 40, 3979–3985 CrossRef CAS PubMed.
- L. Castoldi, L. Ielo, P. Hoyos, M. J. Hernáiz, L. De Luca, A. R. Alcántara, W. Holzer and V. Pace, Tetrahedron, 2018, 74, 2211–2217 CrossRef CAS.
- K. Fukuta and N. Fukuhara, US Pat., US005756319A, 1998 Search PubMed.
- T. H. P. Maier, Nat. Biotechnol., 2003, 21, 422–427 CrossRef CAS.
- T. Inaba, A. G. Birchler, Y. Yamada, S. Sagawa, K. Yokota, K. Ando and I. Uchida, J. Org. Chem., 1998, 63, 7582–7583 CrossRef CAS.
- T. Inaba, Y. Yamada, H. Abe, S. Sagawa and H. Cho, J. Org. Chem., 2000, 65, 1623–1628 CrossRef CAS PubMed.
- K. F. Albizati, S. Babu, A. Birchler, J. K. Busse, M. Fugett, A. Grubbs, A. Haddach, M. Pagan, B. Potts, T. Remarchuk, D. Rieger, R. Rodriguez, J. Shanley, R. Szendroi, T. Tibbetts, K. Whitten and B. C. Borer, Tetrahedron Lett., 2001, 42, 6481–6485 CrossRef CAS.
- G. Iacazio, D. Martini, S. Sanchez and B. Faure, Tetrahedron: Asymmetry, 2000, 11, 1313–1321 CrossRef CAS.
- M. Gruttadauria, P. L. Meo, S. Riela, F. Giacalone and R. Noto, Tetrahedron: Asymmetry, 2006, 17, 3128–3134 CrossRef CAS.
- C. U. Ingram, M. Bommer, M. E. B. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. J. Lye, Biotechnol. Bioeng., 2007, 96, 559–569 CrossRef CAS PubMed.
- M. Lorillière, M. De Sousa, F. Bruna, E. Heuson, T. Gefflaut, V. de Berardinis, T. Saravanan, D. Yi, W.-D. Fessner, F. Charmantray and L. Hecquet, Green Chem., 2017, 19, 425–435 RSC.
- L. Rios-Solis, M. Halim, A. Cázares, P. Morris, J. M. Ward, H. C. Hailes, P. A. Dalby, F. Baganz and G. J. Lye, Biocatal. Biotransform., 2011, 29, 192–203 CrossRef CAS.
- M. F. Villegas-Torres, R. J. Martinez-Torres, A. Cázares-Körner, H. Hailes, F. Baganz and J. Ward, Enzyme Microb. Technol., 2015, 81, 23–30 CrossRef CAS PubMed.
- S. Matosevic, G. J. Lye and F. Baganz, J. Biotechnol., 2011, 155, 320–329 CrossRef CAS PubMed.
- A. Abdul Halim, N. Szita and F. Baganz, J. Biotechnol., 2013, 168, 567–575 CrossRef CAS PubMed.
- P. Gruber, F. Carvalho, M. P. C. Marques, B. O'Sullivan, F. Subrizi, D. Dobrijevic, J. Ward, H. C. Hailes, P. Fernandes, R. Wohlgemuth, F. Baganz and N. Szita, Biotechnol. Bioeng., 2018, 115, 586–596 CrossRef CAS PubMed.
- G. L. Plosker and S. Noble, Drugs, 1999, 58, 1165–1203 CrossRef CAS PubMed.
- A. S. Demir, H. Hamamci, F. Doganel and E. Ozgul, J. Mol. Catal. B: Enzym., 2000, 9, 157–161 CrossRef CAS.
- S. Nakano, Y. Igarashi and H. Nohira, Tetrahedron: Asymmetry, 2001, 12, 59–62 CrossRef CAS.
- N. M. T. Lourenço, S. Barreiros and C. A. M. Afonso, Green Chem., 2007, 9, 734–736 RSC.
- H. Yun, J. Kim, K. Kinnera and B.-G. Kim, Biotechnol. Bioeng., 2006, 93, 391–395 CrossRef CAS PubMed.
- A. G. Myers, J. K. Barbay and B. Zhong, J. Am. Chem. Soc., 2001, 123, 7207–7219 CrossRef CAS PubMed.
- N. Connors, R. Prevoznak, M. Chartrain, J. Reddy, R. Singhvi, Z. Patel, R. Olewinski, P. Salmon, J. Wilson and R. Greasham, J. Ind. Microbiol. Biotechnol., 1997, 18, 353–359 CrossRef CAS.
- B. C. Buckland, S. W. Drew, N. C. Connors, M. M. Chartrain, C. Lee, P. M. Salmon, K. Gbewonyo, W. Zhou, P. Gailliot, R. Singhvi, R. C. Olewinski, W.-J. Sun, J. Reddy, J. Zhang, B. A. Jackey, C. Taylor, K. E. Goklen, B. Junker and R. L. Greasham, Metab. Eng., 1999, 1, 63–74 CrossRef CAS PubMed.
- S. Stahl, N. Ikemoto, A. King, R. Greasham and M. Chartrain, J. Biosci. Bioeng., 1999, 88, 495–499 CrossRef CAS PubMed.
- C. H. Senanayake, L. M. DiMichele, J. Liu, L. E. Fredenburgh, K. M. Ryan, F. Edward Roberts, R. D. Larsen, T. R. Verhoeven and P. J. Reider, Tetrahedron Lett., 1995, 36, 7615–7618 CrossRef CAS.
- E. Eichhorn, J.-P. Roduit, N. Shaw, K. Heinzmann and A. Kiener, Tetrahedron: Asymmetry, 1997, 8, 2533–2536 CrossRef CAS.
- M. Baumann, B. H. Hauer and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2000, 11, 4781–4790 CrossRef CAS.
- A. Nobili, M. G. Gall, I. V. Pavlidis, M. L. Thompson, M. Schmidt and U. T. Bornscheuer, FEBS J., 2013, 280, 3084–3093 CrossRef CAS PubMed.
- Z. Sun, R. Lonsdale, A. Ilie, G. Li, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1598–1605 CrossRef CAS.
- V. K. Tandon, A. M. Van Leusen and H. Wynberg, J. Org. Chem., 1983, 48, 2767–2769 CrossRef CAS.
- Y. Honda, S. Katayama, M. Kojima, T. Suzuki, N. Kishibata and K. Izawa, Org. Biomol. Chem., 2004, 2, 2061–2070 RSC.
- A. Liese, K. Seelbach and C. Wandrey, Industrial biotransformations, John Wiley & Sons,Hoboken, 2006 Search PubMed.
- A. Liljeblad and L. T. Kanerva, Tetrahedron: Asymmetry, 1999, 10, 4405–4415 CrossRef CAS.
- K. N. G. Valdehuesa, H. Liu, K. R. M. Ramos, S. J. Park, G. M. Nisola, W.-K. Lee and W.-J. Chung, Process Biochem., 2014, 49, 25–32 CrossRef CAS.
- W. Niu, M. N. Molefe and J. W. Frost, J. Am. Chem. Soc., 2003, 125, 12998–12999 CrossRef CAS PubMed.
- X. Li, Z. Cai, Y. Li and Y. Zhang, Sci. Rep., 2014, 4, 5541 CrossRef CAS PubMed.
- A. K. Ghosh and Y. Chen, Tetrahedron Lett., 1995, 36, 505–508 CrossRef CAS.
- R. H. Yu, R. P. Polniaszek, M. W. Becker, C. M. Cook and L. H. L. Yu, Org. Process Res. Dev., 2007, 11, 972–980 CrossRef CAS.
- W. L. Canoy, B. E. Cooley, J. A. Corona, T. C. Lovelace, A. Millar, A. M. Weber, S. Xie and Y. Zhang, Org. Lett., 2008, 10, 1103–1106 CrossRef CAS PubMed.
- A. K. Ghosh, J. F. Kincaid, D. E. Walters, Y. Chen, N. C. Chaudhuri, W. J. Thompson, C. Culberson, P. M. D. Fitzgerald, H. Y. Lee, S. P. McKee, P. M. Munson, T. T. Duong, P. L. Darke, J. A. Zugay, W. A. Schleif, M. G. Axel, J. Lin and J. R. Huff, J. Med. Chem., 1996, 39, 3278–3290 CrossRef CAS PubMed.
- R. A. C. Leão, R. O. de Lopes, M. A. D. M. Bezerra, M. N. Muniz, B. B. Casanova, S. C. B. Gnoatto, G. Gosmann, L. Kocsis, R. O. M. A. de Souza and L. S. M. de Miranda, J. Flow Chem., 2015, 5, 216–219 CrossRef.
- Y. L. Khmelnitsky, P. C. Michels, I. C. Cotterill, M. Eissenstat, V. Sunku, V. R. Veeramaneni, H. Cittineni, G. R. Kotha, S. R. Talasani, K. K. Ramanathan, V. K. Chitineni and B. R. Venepalli, Org. Process Res. Dev., 2011, 15, 279–283 CrossRef CAS.
- T. Kanemitsu, M. Inoue, N. Yoshimura, K. Yoneyama, R. Watarai, M. Miyazaki, Y. Odanaka, K. Nagata and T. Itoh, Eur. J. Org. Chem., 2016, 1874–1880 CrossRef CAS.
- A. Sevenich, G.-Q. Liu, A. J. Arduengo, B. F. Gupton and T. Opatz, J. Org. Chem., 2017, 82, 1218–1223 CrossRef CAS PubMed.
- T. Shibasaki, H. Mori, S. Chiba and A. Ozaki, Appl. Environ. Microbiol., 1999, 65, 4028–4031 CrossRef CAS.
- T. Shibasaki, W. Sakurai, A. Hasegawa, Y. Uosaki, H. Mori, M. Yoshida and A. Ozaki, Tetrahedron Lett., 1999, 40, 5227–5230 CrossRef CAS.
- T. Shibasaki, H. Mori and A. Ozaki, Biosci., Biotechnol., Biochem., 2000, 64, 746–750 CrossRef CAS PubMed.
- C. Klein and W. Hüttel, Adv. Synth. Catal., 2011, 353, 1375–1383 CrossRef CAS.
- F. Falcioni, B. Bühler and A. Schmid, Biotechnol. Bioeng., 2015, 112, 322–330 CrossRef CAS PubMed.
- K. Hernández, A. Gómez, J. Joglar, J. Bujons, T. Parella and P. Clapés, Adv. Synth. Catal., 2017, 359, 2090–2100 CrossRef.
- M. Schurink, S. Wolterink-van Loo, J. van der Oost, T. Sonke and M. C. R. Franssen, ChemCatChem, 2014, 6, 1073–1081 CrossRef CAS.
- G. M. Keating, Drugs, 2016, 76, 617–624 CrossRef CAS PubMed.
- Y. N. Lamb, Drugs, 2017, 77, 1797–1804 CrossRef CAS PubMed.
- J. G. Taylor, S. Zipfel, K. Ramey, R. Vivian, A. Schrier, K. K. Karki, A. Katana, D. Kato, T. Kobayashi and R. Martinez, Bioorg. Med. Chem. Lett., 2019, 29, 2428–2436 CrossRef CAS PubMed.
- P. M. Scola, L.-Q. Sun, A. X. Wang, J. Chen, N. Sin, B. L. Venables, S.-Y. Sit, Y. Chen, A. Cocuzza and D. M. Bilder, J. Med. Chem., 2014, 57, 1730–1752 CrossRef CAS PubMed.
- J. A. McCauley, C. J. McIntyre, M. T. Rudd, K. T. Nguyen, J. J. Romano, J. W. Butcher, K. F. Gilbert, K. J. Bush, M. K. Holloway and J. Swestock, J. Med. Chem., 2010, 53, 2443–2463 CrossRef CAS PubMed.
- Å. Rosenquist, B. Samuelsson, P.-O. Johansson, M. D. Cummings, O. Lenz, P. Raboisson, K. Simmen, S. Vendeville, H. de Kock and M. Nilsson, J. Med. Chem., 2014, 57, 1673–1693 CrossRef PubMed.
- K. F. McDaniel, Y.-Y. Ku, Y. Sun, H.-J. Chen, J. Shanley, T. Middleton, Y. S. Or and D. Kempf, in HCV: The Journey from Discovery to a Cure, ed. M. J. Sofia, Springer, Cham, 2019, vol. I, pp. 389–413 Search PubMed.
- D. D. Caspi, R. D. Cink, D. Clyne, M. Diwan, K. M. Engstrom, T. Grieme, J. Mei, R. W. Miller, C. Mitchell, J. G. Napolitano, N. Nere, M. M. Ravn, A. Sheikh, S. Wagaw and H. Zhang, Tetrahedron, 2019, 75, 4271–4286 CrossRef CAS.
- P. L. Beaulieu, J. Gillard, M. D. Bailey, C. Boucher, J.-S. Duceppe, B. Simoneau, X.-J. Wang, L. Zhang, K. Grozinger, I. Houpis, V. Farina, H. Heimroth, T. Krueger and J. Schnaubelt, J. Org. Chem., 2005, 70, 5869–5879 CrossRef CAS PubMed.
- S. Zhu, Y. Shi, X. Zhang and G. Zheng, Appl. Biochem. Biotechnol., 2018, 184, 500–512 CrossRef CAS PubMed.
- D. A. Chaplin, M. E. Fox and S. H. B. Kroll, Chem. Commun., 2014, 50, 5858–5860 RSC.
- C. Fliche, J. Braun and F. C. Le Goffic, Synth. Commun., 1994, 24, 2873–2876 CrossRef CAS.
- Z. J. Song, D. M. Tellers, M. Journet, J. T. Kuethe, D. Lieberman, G. Humphrey, F. Zhang, Z. Peng, M. S. Waters, D. Zewge, A. Nolting, D. Zhao, R. A. Reamer, P. G. Dormer, K. M. Belyk, I. W. Davies, P. N. Devine and D. M. Tschaen, J. Org. Chem., 2011, 76, 7804–7815 CrossRef CAS PubMed.
- R. D. Cink, K. A. Lukin, R. D. Bishop, G. Zhao, M. J. Pelc, T. B. Towne, B. D. Gates, M. M. Ravn, D. R. Hill, C. Ding, S. C. Cullen, J. Mei, M. R. Leanna, J. Henle, J. G. Napolitano, N. K. Nere, S. Chen, A. Sheikh and J. M. Kallemeyn, Org. Process Res. Dev., 2020, 24, 183–200 CrossRef CAS.
- R. D. Cink, K. A. Lukin, M. R. Leanna, M. J. Pelc, T. B. Towne, D. S. Welch, K. M. Engstrom, M. M. Ravn, R. D. Bishop and G. Zhao, US Pat., US9809576B1, 2017 Search PubMed.
- M. J. Abrahamson, A. B. Kielbus, W. T. Riordan, D. R. Hill, S. R. Chemburkar, R. E. Reddy, T. B. Towne, J. Mei and G. J. Brown, US Pat., US010316338B1, 2019 Search PubMed.
- X.-J. Wang, L. Zhang, L. L. Smith-Keenan, I. N. Houpis and V. Farina, Org. Process Res. Dev., 2007, 11, 60–63 CrossRef CAS.
- N. Nishino, G. M. Shivashimpi, P. B. Soni, M. P. I. Bhuiyan, T. Kato, S. Maeda, T. G. Nishino and M. Yoshida, Bioorg. Med. Chem., 2008, 16, 437–445 CrossRef CAS.
- M. Rusnak-Müller, O. May, P. J. Hermsen, H. M. M. G. Straatman, W. Skranc, W. H. J. Boesten, D. Heemskerk, B. De Lange and G. Sarakinos, World Pat., WO2008067981A2, 2010 Search PubMed.
- M. Nojiri, T. Nishiyama and N. Taoka, US Pat., US009464306B2, 2016 Search PubMed.
- J. Park, J. C. Moore and F. Xu, Org. Process Res. Dev., 2016, 20, 76–80 CrossRef CAS.
- D. P. M. Depré, D. J. Ormerod, A. Horvath, T. S. Moody, M. Brossat, O. Riant, N. Vriamont, S. F. E. Lemaire and S. N. J. Hermant, US Pat., US20180093943A1, 2018 Search PubMed.
- D. Banner, M. Ceccarelli Simona, U. W. E. Grether, W. Haap, H. Hilpert, H. Kuehne, H. Mauser, J.-M. Plancher and A. Sanchez Ruben, World Pat., WO2010142650A1, 2010 Search PubMed.
- S. U. N. Or Yat, J. U. N. Ma, G. Wang, J. Long and B. I. N. Wang, World Pat., WO2012040167A1, 2012 Search PubMed.
- J. Kuethe, Y.-L. Zhong, N. Yasuda, G. Beutner, K. Linn, M. Kim, B. Marcune, S. D. Dreher, G. Humphrey and T. Pei, Org. Lett., 2013, 15, 4174–4177 CrossRef CAS PubMed.
- E. M. Bassan, C. A. Baxter, G. L. Beutner, K. M. Emerson, F. J. Fleitz, S. Johnson, S. Keen, M. M. Kim, J. T. Kuethe, W. R. Leonard, P. R. Mullens, D. J. Muzzio, C. Roberge and N. Yasuda, Org. Process Res. Dev., 2012, 16, 87–95 CrossRef CAS.
- T. Kim, A. M. Kassim, A. Botejue, C. Zhang, J. Forte, D. Rozzell, M. A. Huffman, P. N. Devine and J. A. McIntosh, ChemBioChem, 2019, 20, 1129–1132 CrossRef CAS PubMed.
- C. Hicks and R. M. Gulick, Clin. Infect. Dis., 2009, 48, 931–939 CrossRef CAS PubMed.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- C. S. Fuchs, R. C. Simon, W. Riethorst, F. Zepeck and W. Kroutil, Bioorg. Med. Chem., 2014, 22, 5558–5562 CrossRef CAS PubMed.
- J.-D. Zhang, J.-W. Zhao, L.-L. Gao, H.-H. Chang, W.-L. Wei and J.-H. Xu, J. Biotechnol., 2019, 290, 24–32 CrossRef CAS PubMed.
- J.-D. Zhang, X.-X. Yang, Q. Jia, J.-W. Zhao, L.-L. Gao, W.-C. Gao, H.-H. Chang, W.-L. Wei and J.-H. Xu, Catal. Sci. Technol., 2019, 9, 70–74 RSC.
- F.-F. Chen, G.-W. Zheng, L. Liu, H. Li, Q. Chen, F.-L. Li, C.-X. Li and J.-H. Xu, ACS Catal., 2018, 8, 2622–2628 CrossRef CAS.
- F.-F. Chen, S. C. Cosgrove, W. R. Birmingham, J. Mangas-Sanchez, J. Citoler, M. P. Thompson, G.-W. Zheng, J.-H. Xu and N. J. Turner, ACS Catal., 2019, 9, 11813–11818 CrossRef CAS.
- P. Flores-Sánchez, J. Escalante and E. Castillo, Tetrahedron: Asymmetry, 2005, 16, 629–634 CrossRef.
- T. Heck, D. Seebach, S. Osswald, M. K. J. ter Wiel, H.-P. E. Kohler and B. Geueke, ChemBioChem, 2009, 10, 1558–1561 CrossRef CAS PubMed.
- H. Rangel, M. Carrillo-Morales, J. M. Galindo, E. Castillo, A. Obregón-Zúñiga, E. Juaristi and J. Escalante, Tetrahedron: Asymmetry, 2015, 26, 325–332 CrossRef CAS.
- H. Mäenpää, L. T. Kanerva and A. Liljeblad, ChemCatChem, 2016, 8, 1226–1232 CrossRef.
- D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell and W. Kroutil, Adv. Synth. Catal., 2008, 350, 2761–2766 CrossRef CAS.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- X.-L. Tang, N.-N. Zhang, G.-Y. Ye and Y.-G. Zheng, J. Biotechnol., 2019, 295, 49–54 CrossRef CAS PubMed.
- X. Gao, X. Zhang, N. Zhu, Y. Mou, H. Zhang, X. Liu and P. Wei, Appl. Microbiol. Biotechnol., 2020, 104, 3959–3969 CrossRef CAS PubMed.
- R. Li, H. J. Wijma, L. Song, Y. Cui, M. Otzen, Y. E. Tian, J. Du, T. Li, D. Niu, Y. Chen, J. Feng, J. Han, H. Chen, Y. Tao, D. B. Janssen and B. Wu, Nat. Chem. Biol., 2018, 14, 664–670 CrossRef CAS PubMed.
- Y. Asano, I. Kira and K. Yokozeki, Biomol. Eng., 2005, 22, 95–101 CrossRef CAS PubMed.
- A. Vogel, R. Schmiedel, U. Hofmann, K. Gruber and K. Zangger, ChemCatChem, 2014, 6, 965–968 CrossRef CAS.
- M. J. Mulvihill, J. L. Gage and M. J. Miller, J. Org. Chem., 1998, 63, 3357–3363 CrossRef CAS.
- J. C. Barrow, K. L. Glass, H. G. Selnick, R. M. Freidinger, R. S. L. Chang, S. S. O'Malley and C. Woyden, Bioorg. Med. Chem. Lett., 2000, 10, 1917–1920 CrossRef CAS PubMed.
- J. A. Englund, R. E. Champlin, P. R. Wyde, H. Kantarjian, R. L. Atmar, J. Tarrand, H. Yousuf, H. Regnery, A. I. Klimov and N. J. Cox, Clin. Infect. Dis., 1998, 26, 1418–1424 CrossRef CAS PubMed.
- R. J. Jackson, K. L. Cooper, P. Tappenden, A. Rees, E. L. Simpson, R. C. Read and K. G. Nicholson, J. Infect., 2011, 62, 14–25 CrossRef.
- M. Von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS PubMed.
- M. R. Richards, M. G. Brant, M. J. Boulanger, C. W. Cairo and J. E. Wulff, MedChemComm, 2014, 5, 1483–1488 RSC.
- J. C. Rohloff, K. M. Kent, M. J. Postich, M. W. Becker, H. H. Chapman, D. E. Kelly, W. Lew, M. S. Louie, L. R. McGee, E. J. Prisbe, L. M. Schultze, R. H. Yu and L. Zhang, J. Org. Chem., 1998, 63, 4545–4550 CrossRef CAS.
- S. Abrecht, P. Harrington, H. Iding, M. Karpf, R. Trussardi, B. Wirz and U. Zutter, Chimia, 2004, 58, 621–629 CrossRef CAS.
- M. Krämer, J. Bongaerts, R. Bovenberg, S. Kremer, U. Müller, S. Orf, M. Wubbolts and L. Raeven, Metab. Eng., 2003, 5, 277–283 CrossRef.
- S. Ghosh, Y. Chisti and U. C. Banerjee, Biotechnol. Adv., 2012, 30, 1425–1431 CrossRef CAS PubMed.
- K. M. Draths, D. R. Knop and J. W. Frost, J. Am. Chem. Soc., 1999, 121, 1603–1604 CrossRef CAS.
- D. R. Knop, K. M. Draths, S. S. Chandran, J. L. Barker, R. von Daeniken, W. Weber and J. W. Frost, J. Am. Chem. Soc., 2001, 123, 10173–10182 CrossRef CAS PubMed.
- S. S. Chandran, J. Yi, K. M. Draths, R. V. Daeniken, W. Weber and J. W. Frost, Biotechnol. Prog., 2003, 19, 808–814 CrossRef CAS PubMed.
- O. Adachi, Y. Ano, H. Toyama and K. Matsushita, Biosci., Biotechnol., Biochem., 2006, 70, 2579–2582 CrossRef CAS PubMed.
- J. O. Ahn, H.-W. Lee, R. Saha, M. S. Park, J.-K. Jung and D.-Y. Lee, J. Microbiol. Biotechnol., 2008, 18, 1773–1784 CAS.
- A. Escalante, R. Calderón, A. Valdivia, R. de Anda, G. Hernández, O. T. Ramírez, G. Gosset and F. Bolívar, Microb. Cell Fact., 2010, 9, 21 CrossRef PubMed.
- A. Rodriguez, J. A. Martínez, J. L. Báez-Viveros, N. Flores, G. Hernández-Chávez, O. T. Ramírez, G. Gosset and F. Bolivar, Microb. Cell Fact., 2013, 12, 86 CrossRef PubMed.
- J. A. Martínez, F. Bolívar and A. Escalante, Front. Bioeng. Biotechnol., 2015, 3 Search PubMed.
- M. Karpf and R. Trussardi, US Pat., US20060047002A1, 2006 Search PubMed.
- J. Guo and J. W. Frost, Org. Lett., 2004, 6, 1585–1588 CrossRef CAS PubMed.
- D. T. Gibson, J. R. Koch, C. L. Schuld and R. E. Kallio, Biochemistry, 1968, 7, 3795–3802 CrossRef CAS PubMed.
- D. R. Boyd, M. V. Hand, N. D. Sharma, J. Chima, H. Dalton and G. N. Sheldrake, J. Chem. Soc., Chem. Commun., 1991, 1630–1632, 10.1039/C39910001630.
- B. Sullivan, I. Carrera, M. Drouin and T. Hudlicky, Angew. Chem., Int. Ed., 2009, 48, 4229–4231 CrossRef CAS PubMed.
- L. Werner, A. Machara and T. Hudlicky, Adv. Synth. Catal., 2010, 352, 195–200 CrossRef CAS.
- M. Matveenko, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2008, 49, 7018–7020 CrossRef CAS.
- J.-J. Shie, J.-M. Fang and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 5788–5791 CrossRef CAS PubMed.
- U. Zutter, H. Iding, P. Spurr and B. Wirz, J. Org. Chem., 2008, 73, 4895–4902 CrossRef CAS PubMed.
- S. Silva and C. D. Maycock, Org. Chem. Front., 2017, 4, 236–240 RSC.
- H. Li, S.-J. Shen, C.-L. Zhu and H. Xu, J. Am. Chem. Soc., 2018, 140, 10619–10626 CrossRef CAS PubMed.
- M. von Itzstein, W.-Y. Wu and B. Jin, Carbohydr. Res., 1994, 259, 301–305 CrossRef CAS.
- M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright and C. Williamson, J. Chem. Soc., Perkin Trans. 1, 1995, 1173–1180, 10.1039/P19950001173.
- B. R. Lundgren and C. N. Boddy, Org. Biomol. Chem., 2007, 5, 1903–1909 RSC.
- J. Kang, P. Gu, Y. Wang, Y. Li, F. Yang, Q. Wang and Q. Qi, Metab. Eng., 2012, 14, 623–629 CrossRef CAS PubMed.
- M. E. Horsman, B. R. Lundgren and C. N. Boddy, Chem. Eng. Commun., 2016, 203, 1326–1335 CrossRef CAS.
- D. G. Comb and S. Roseman, J. Am. Chem. Soc., 1958, 80, 497–499 CrossRef CAS.
- C. Augé, S. David and C. Gautheron, Tetrahedron Lett., 1984, 25, 4663–4664 CrossRef.
- M. J. Kim, W. J. Hennen, H. M. Sweers and C. H. Wong, J. Am. Chem. Soc., 1988, 110, 6481–6486 CrossRef CAS.
- C. Augé, C. Gautheron, S. David, A. Malleron, B. Cavayé and B. Bouxom, Tetrahedron, 1990, 46, 201–214 CrossRef.
- M. D. Bednarski, H. K. Chenault, E. S. Simon and G. M. Whitesides, J. Am. Chem. Soc., 1987, 109, 1283–1285 CrossRef CAS.
- U. Kragl, D. Gygax, O. Ghisalba and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1991, 30, 827–828 CrossRef.
- S. Blayer, J. M. Woodley, M. D. Lilly and M. J. Dawson, Biotechnol. Prog., 1996, 12, 758–763 CrossRef CAS.
- M. Mahmoudian, D. Noble, C. S. Drake, R. F. Middleton, D. S. Montgomery, J. E. Piercey, D. Ramlakhan, M. Todd and M. J. Dawson, Enzyme Microb. Technol., 1997, 20, 393–400 CrossRef CAS.
- J.-O. Lee, J.-K. Yi, S.-G. Lee, S. Takahashi and B.-G. Kim, Enzyme Microb. Technol., 2004, 35, 121–125 CrossRef CAS.
- Y. Zhang, F. Tao, M. Du, C. Ma, J. Qiu, L. Gu, X. He and P. Xu, Appl. Microbiol. Biotechnol., 2010, 86, 481–489 CrossRef CAS PubMed.
- F. Tao, Y. Zhang, C. Ma and P. Xu, Sci. Rep., 2011, 1, 142 CrossRef.
- I. Maru, J. Ohnishi, Y. Ohta and Y. Tsukada, Carbohydr. Res., 1998, 306, 575–578 CrossRef CAS.
- X. Chen, J. Zhou, L. Zhang, Z. Pu, L. Liu, W. Shen and Y. Fan, Metab. Eng., 2018, 47, 374–382 CrossRef CAS PubMed.
- P. Xu, J. H. Qiu, Y. N. Zhang, J. Chen, P. G. Wang, B. Yan, J. Song, R. M. Xi, Z. X. Deng and C. Q. Ma, Adv. Synth. Catal., 2007, 349, 1614–1618 CrossRef CAS.
- H. Ikematsu and N. Kawai, Expert Rev. Anti-Infect. Ther., 2011, 9, 851–857 CrossRef CAS PubMed.
- T. Honda, T. Masuda, S. Yoshida, M. Arai, Y. Kobayashi and M. Yamashita, Bioorg. Med. Chem. Lett., 2002, 12, 1921–1924 CrossRef CAS PubMed.
- Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482–3486 CrossRef CAS PubMed.
- J. O. Link, J. G. Taylor, L. Xu, M. Mitchell, H. Guo, H. Liu, D. Kato, T. Kirschberg, J. Sun, N. Squires, J. Parrish, T. Keller, Z.-Y. Yang, C. Yang, M. Matles, Y. Wang, K. Wang, G. Cheng, Y. Tian, E. Mogalian, E. Mondou, M. Cornpropst, J. Perry and M. C. Desai, J. Med. Chem., 2014, 57, 2033–2046 CrossRef CAS PubMed.
- R. W. Scott, J. P. Vitale, K. S. Matthews, M. G. Teresk, A. Formella and J. W. Evans, World Pat., WO2013184702A1, 2015 Search PubMed.
- S. Dazhao, S. Qiu, H. Shuaijie, W. Canhui, Z. Mingchen, Y. Youzhe, X. Kailong and G. Peng, PRC Pat., CN105461606B, 2018 Search PubMed.
- C. M. Perry, Drugs, 2010, 70, 1189–1213 CrossRef CAS PubMed.
- S. J. Haycock-Lewandowski, A. Wilder and J. Åhman, Org. Process Res. Dev., 2008, 12, 1094–1103 CrossRef CAS.
- V. A. Soloshonok, N. A. Fokina, A. V. Rybakova, I. P. Shishkina, S. V. Galushko, A. E. Sorochinsky, V. P. Kukhar, M. V. Savchenko and V. K. Svedas, Tetrahedron: Asymmetry, 1995, 6, 1601–1610 CrossRef CAS.
- S. J. Faulconbridge, K. E. Holt, L. Garcia Sevillano, C. J. Lock, P. D. Tiffin, N. Tremayne and S. Winter, Tetrahedron Lett., 2000, 41, 2679–2681 CrossRef CAS.
- S. Gedey, A. Liljeblad, L. Lázár, F. Fülöp and L. T. Kanerva, Tetrahedron: Asymmetry, 2001, 12, 105–110 CrossRef CAS.
- H. Gröger, H. Trauthwein, S. Buchholz, K. Drauz, C. Sacherer, S. Godfrin and H. Werner, Org. Biomol. Chem., 2004, 2, 1977–1978 RSC.
- M. O’Neill, B. Hauer, N. Schneider and N. J. Turner, ACS Catal., 2011, 1, 1014–1016 CrossRef.
- H. Gröger, O. May, H. Hüsken, S. Georgeon, K. Drauz and K. Landfester, Angew. Chem., Int. Ed., 2006, 45, 1645–1648 CrossRef PubMed.
- M. Shakeri, K. Engström, A. G. Sandström and J.-E. Bäckvall, ChemCatChem, 2010, 2, 534–538 CrossRef CAS.
- K. Engström, M. Shakeri and J.-E. Bäckvall, Eur. J. Org. Chem., 2011, 1827–1830 CrossRef.
- E. Forró, T. Paál, G. Tasnádi and F. Fülöp, Adv. Synth. Catal., 2006, 348, 917–923 CrossRef.
- B. Wu, W. Szymanski, S. De Wildeman, G. J. Poelarends, B. L. Feringa and D. B. Janssen, Adv. Synth. Catal., 2010, 352, 1409–1412 CrossRef CAS.
- S. Bartsch, G. G. Wybenga, M. Jansen, M. M. Heberling, B. Wu, B. W. Dijkstra and D. B. Janssen, ChemCatChem, 2013, 5, 1797–1802 CrossRef CAS.
- X. Feng, J. Guo, R. Zhang, W. Liu, Y. Cao, M. Xian and H. Liu, ACS Omega, 2020, 5, 7745–7750 CrossRef CAS PubMed.
- S. Mathew, S. P. Nadarajan, T. Chung, H. H. Park and H. Yun, Enzyme Microb. Technol., 2016, 87–88, 52–60 CrossRef CAS PubMed.
- G.-H. Kim, H. Jeon, T. P. Khobragade, M. D. Patil, S. Sung, S. Yoon, Y. Won, S. Sarak and H. Yun, ChemCatChem, 2019, 11, 1437–1440 CrossRef CAS.
- S. Mathew, S. P. Nadarajan, U. Sundaramoorthy, H. Jeon, T. Chung and H. Yun, Biotechnol. Lett., 2017, 39, 535–543 CrossRef CAS PubMed.
- D. Zhang, X. Chen, R. Zhang, P. Yao, Q. Wu and D. Zhu, ACS Catal., 2015, 5, 2220–2224 CrossRef CAS.
- O. Buß, M. Voss, A. Delavault, P. Gorenflo, C. Syldatk, U. Bornscheuer and J. Rudat, Molecules, 2018, 23, 1211 CrossRef PubMed.
- N. W. Fadnavis, K. R. Radhika and A. Vedamayee Devi, Tetrahedron: Asymmetry, 2006, 17, 240–244 CrossRef CAS.
- O. Torre, V. Gotor-Fernández and V. Gotor, Tetrahedron: Asymmetry, 2006, 17, 860–866 CrossRef CAS.
- K. Beutner, A. Friedman-Kien, N. Artman, M. Conant, M. Illeman, R. Thisted and D. King, Lancet, 1989, 333, 831–834 CrossRef.
- D. B. Berkowitz, S. Choi and J.-H. Maeng, J. Org. Chem., 2000, 65, 847–860 CrossRef CAS PubMed.
- W. Lau and E. S. Sattely, Science, 2015, 349, 1224–1228 CrossRef CAS PubMed.
- M. Lazzarotto, L. Hammerer, M. Hetmann, A. Borg, L. Schmermund, L. Steiner, P. Hartmann, F. Belaj, W. Kroutil, K. Gruber and M. Fuchs, Angew. Chem., Int. Ed., 2019, 58, 8226–8230 CrossRef CAS PubMed.
- J. Li, X. Zhang and H. Renata, Angew. Chem., Int. Ed., 2019, 58, 11657–11660 CrossRef CAS PubMed.
- D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557–7559 CrossRef CAS PubMed.
- J. S. Orman and C. M. Perry, Drugs, 2008, 68, 1435–1463 CrossRef CAS PubMed.
- K. S. Fors, J. R. Gage, R. F. Heier, R. C. Kelly, W. R. Perrault and N. Wicnienski, J. Org. Chem., 1998, 63, 7348–7356 CrossRef CAS PubMed.
- S. M. Vrouenraets, F. W. Wit, J. V. Tongeren and J. M. Lange, Expert Opin. Pharmacother., 2007, 8, 851–871 CrossRef CAS PubMed.
- A. S. Thompson, E. G. Corley, M. F. Huntington and E. J. J. Grabowski, Tetrahedron Lett., 1995, 36, 8937–8940 CrossRef.
- T. Junhua and L. Yongtang, PRC Pat., CN104805148B, 2019 Search PubMed.
- S. Diab, D. T. McQuade, B. F. Gupton and D. I. Gerogiorgis, Org. Process Res. Dev., 2019, 23, 320–333 CrossRef CAS.
- L.-Q. Jin, Y.-F. Li, Z.-Q. Liu, Y.-G. Zheng and Y.-C. Shen, New Biotechnol., 2011, 28, 610–615 CrossRef CAS PubMed.
- L.-Q. Jin, Z.-Q. Liu, J.-M. Xu, R.-C. Zheng, Y.-G. Zheng and Y.-C. Shen, Catal. Commun., 2013, 38, 6–9 CrossRef CAS.
- D. T. Leung and S. L. Sacks, Expert Opin. Pharmacother., 2004, 5, 2567–2571 CrossRef CAS PubMed.
- T. Yu, Y. J. Zhou, L. Wenning, Q. Liu, A. Krivoruchko, V. Siewers, J. Nielsen and F. David, Nat. Commun., 2017, 8, 15587 CrossRef CAS PubMed.
- S. Shi, Y. Chen, V. Siewers and J. Nielsen, mBio, 2014, 5, e01130–01114 Search PubMed.
- C. De Savi, D. L. Hughes and L. Kvaerno, Org. Process Res. Dev., 2020, 24, 940–976 CrossRef CAS.
- B. A. Ahidjo, M. W. C. Loe, Y. L. Ng, C. K. Mok and J. J. H. Chu, ACS Infect. Dis., 2020, 6, 1624–1634 CrossRef CAS PubMed.
- V. Farina and J. D. Brown, Angew. Chem., Int. Ed., 2006, 45, 7330–7334 CrossRef CAS PubMed.
- Y. S. Hong and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 6310–6311 CrossRef PubMed.
- H. Ishikawa, T. Suzuki, H. Orita, T. Uchimaru and Y. Hayashi, Chem. – Eur. J., 2010, 16, 12616–12626 CrossRef CAS PubMed.
- T. K. Warren, R. Jordan, M. K. Lo, A. S. Ray, R. L. Mackman, V. Soloveva, D. Siegel, M. Perron, R. Bannister, H. C. Hui, N. Larson, R. Strickley, J. Wells, K. S. Stuthman, S. A. Van Tongeren, N. L. Garza, G. Donnelly, A. C. Shurtleff, C. J. Retterer, D. Gharaibeh, R. Zamani, T. Kenny, B. P. Eaton, E. Grimes, L. S. Welch, L. Gomba, C. L. Wilhelmsen, D. K. Nichols, J. E. Nuss, E. R. Nagle, J. R. Kugelman, G. Palacios, E. Doerffler, S. Neville, E. Carra, M. O. Clarke, L. Zhang, W. Lew, B. Ross, Q. Wang, K. Chun, L. Wolfe, D. Babusis, Y. Park, K. M. Stray, I. Trancheva, J. Y. Feng, O. Barauskas, Y. Xu, P. Wong, M. R. Braun, M. Flint, L. K. McMullan, S.-S. Chen, R. Fearns, S. Swaminathan, D. L. Mayers, C. F. Spiropoulou, W. A. Lee, S. T. Nichol, T. Cihlar and S. Bavari, Nature, 2016, 531, 381–385 CrossRef CAS PubMed.
- J. Wise, BMJ, 2020, 369, m2610 CrossRef PubMed.
- Food and Drug Administration, Coronavirus (COVID-19) Update: FDA issues emergency use authorization for potential COVID-19 treatment, https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment, accessed July 2020.
- M. L. Agostini, E. L. Andres, A. C. Sims, R. L. Graham, T. P. Sheahan, X. Lu, E. C. Smith, J. B. Case, J. Y. Feng, R. Jordan, A. S. Ray, T. Cihlar, D. Siegel, R. L. Mackman, M. O. Clarke, R. S. Baric and M. R. Denison, mBio, 2018, 9, e00221 CrossRef CAS PubMed.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648–1661 CrossRef CAS PubMed.
- P. Sedmera, P. Halada, E. Kubátová, D. Haltrich, V. Přikrylová and J. Volc, J. Mol. Catal. B: Enzym., 2006, 41, 32–42 CrossRef CAS.
- K. Napora, T. M. Wrodnigg, P. Kosmus, M. Thonhofer, K. Robins and M. Winkler, Bioorg. Med. Chem. Lett., 2013, 23, 3393–3395 CrossRef CAS PubMed.
- S. Gao, A. Radadiya, W. Li, H. Liu, W. Zhu, V. de Crécy-Lagard, N. G. J. Richards and J. H. Naismith, Chem. Commun., 2020, 56, 7617–7620 RSC.
- H. Griengl, H. Schwab and M. Fechter, Trends Biotechnol., 2000, 18, 252–256 CrossRef CAS PubMed.
- P. Gautret, J.-C. Lagier, P. Parola, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira and H. T. Dupont, Int. J. Antimicrob. Agents, 2020, 105949 CrossRef CAS PubMed.
- J. M. Molina, C. Delaugerre, J. Le Goff, B. Mela-Lima, D. Ponscarme, L. Goldwirt and N. de Castro, Med. Mal. Infect., 2020, 50, 30085–30088 Search PubMed.
- I. D’Acquarica and I. Agranat, Drug Discovery Today, 2020, 25, 1121–1123 CrossRef PubMed.
- G. Lentini, M. M. Cavalluzzi and S. Habtemariam, Molecules, 2020, 25, 1834 CrossRef CAS PubMed.
- M. Nechab, N. Azzi, N. Vanthuyne, M. Bertrand, S. Gastaldi and G. Gil, J. Org. Chem., 2007, 72, 6918–6923 CrossRef CAS PubMed.
- S. Escoubet, S. Gastaldi, N. Vanthuyne, G. Gil, D. Siri and M. P. Bertrand, J. Org. Chem., 2006, 71, 7288–7292 CrossRef CAS PubMed.
- F. Poulhès, N. Vanthuyne, M. P. Bertrand, S. Gastaldi and G. Gil, J. Org. Chem., 2011, 76, 7281–7286 CrossRef PubMed.
- P. Gautret, J.-C. Lagier, P. Parola, L. Meddeb, J. Sevestre, M. Mailhe, B. Doudier, C. Aubry, S. Amrane and P. Seng, Travel Med. Infect. Di., 2020, 101663 CrossRef PubMed.
- R. R. Wilkening, R. W. Ratcliffe, G. A. Doss, K. F. Bartizal, A. C. Graham and C. M. Herbert, Bioorg. Med. Chem. Lett., 1993, 3, 1287–1292 CrossRef CAS.
- A. L. Demain and J. L. Adrio, in Natural Compounds as Drugs, ed. F. Petersen and R. Amstutz, Birkhäuser Basel, Basel, 1st edn, 2008, vol. I, pp. 251–289, DOI:10.1007/978-3-7643-8117-2_7.
- C. Olano, F. Lombó, C. Méndez and J. A. Salas, Metab. Eng., 2008, 10, 281–292 CrossRef CAS PubMed.
- E. Rodriguez, Z. Hu, S. Ou, Y. Volchegursky, C. R. Hutchinson and R. McDaniel, J. Ind. Microbiol. Biotechnol., 2003, 30, 480–488 CrossRef CAS PubMed.
- Y. Chen, W. Deng, J. Wu, J. Qian, J. Chu, Y. Zhuang, S. Zhang and W. Liu, Appl. Environ. Microbiol., 2008, 74, 1820–1828 CrossRef CAS PubMed.
- R. Pieper, G. Luo, D. E. Cane and C. Khosla, Nature, 1995, 378, 263–266 CrossRef CAS PubMed.
- A. R. Reeves, I. A. Brikun, W. H. Cernota, B. I. Leach, M. C. Gonzalez and J. Mark Weber, Metab. Eng., 2007, 9, 293–303 CrossRef CAS PubMed.
- J. Lau, C. Tran, P. Licari and J. Galazzo, J. Biotechnol., 2004, 110, 95–103 CrossRef CAS PubMed.
- X. Gao, P. Wang and Y. Tang, Appl. Microbiol. Biotechnol., 2010, 88, 1233–1242 CrossRef CAS PubMed.
- H. Zhang, Y. Wang, J. Wu, K. Skalina and B. A. Pfeifer, Chem. Biol., 2010, 17, 1232–1240 CrossRef CAS PubMed.
- M. Jiang and B. A. Pfeifer, Metab. Eng., 2013, 19, 42–49 CrossRef CAS PubMed.
- P. Horby, W. S. Lim, J. Emberson, M. Mafham, J. Bell, L. Linsell, N. Staplin, C. Brightling, A. Ustianowski, E. Elmahi, B. Prudon, C. Green, T. Felton, D. Chadwick, K. Rege, C. Fegan, L. C. Chappell, S. N. Faust, T. Jaki, K. Jeffery, A. Montgomery, K. Rowan, E. Juszczak, J. K. Baillie, R. Haynes and M. J. Landray, N. Engl. J. Med., 2020 DOI:10.1056/NEJMoa2021436.
- M. V. Donova and O. V. Egorova, Appl. Microbiol. Biotechnol., 2012, 94, 1423–1447 CrossRef CAS PubMed.
- S. B. Mahato and S. Garai, Steroids, 1997, 62, 332–345 CrossRef CAS.
- J.-L. Barredo and I. Herráiz, Microbial steroids: Methods and protocols, Springer, Cham, 2017 Search PubMed.
- Yan, F. Y. Hua, P. Y. Liang, H. Xiong, L. M. Hua, L. Hui and Z. H. Ming, PRC Pat., CN106520896A, 2016 Search PubMed.
- H. A. Gerber, US Pat., US3086032A, 1963 Search PubMed.
- H. L. Holland, D. H. Nguyen and N. M. Pearson, Steroids, 1995, 60, 646–649 CrossRef CAS PubMed.
- A Randomized, Open, Controlled Clinical Study to Evaluate the Efficacy of ASC09F and Ritonavir for 2019-nCoV Pneumonia, https://ClinicalTrials.gov/show/NCT04261270, accessed June, 2020.
- Evaluation of Ganovo (Danoprevir) Combined With Ritonavir in the Treatment of SARS-CoV-2 Infection, https://ClinicalTrials.gov/show/NCT04291729, accessed June, 2020.
- Y. Cao, J. Wei, L. Zou, T. Jiang, G. Wang, L. Chen, L. Huang, F. Meng, L. Huang, N. Wang, X. Zhou, H. Luo, Z. Mao, X. Chen, J. Xie, J. Liu, H. Cheng, J. Zhao, G. Huang, W. Wang and J. Zhou, J. Allergy Clin. Immunol., 2020, 146, 137–146 CrossRef CAS PubMed.
- J. Stebbing, A. Phelan, I. Griffin, C. Tucker, O. Oechsle, D. Smith and P. Richardson, Lancet Infect. Dis., 2020, 20, 400–402 CrossRef CAS.
- W. Yong, L. Ligang, X. Jie, Y. Shiqiong, L. Qian and K. Litao, PRC Pat., CN104496904B, 2017 Search PubMed.
- J. M. Woodley, Curr. Opin. Green Sustainable Chem., 2020, 21, 22–26 CrossRef.
- M. Legraverend, Tetrahedron, 2008, 64, 8585–8603 CrossRef CAS.
- M. Radi, S. Schenone and M. Botta, Org. Biomol. Chem., 2009, 7, 2841–2847 RSC.
- M. Mahfoudh, R. Abderrahim, E. Leclerc and J.-M. Campagne, Eur. J. Org. Chem., 2017, 2856–2865 CrossRef CAS.
- J. L. Galman, I. Slabu, F. Parmeggiani and N. J. Turner, Chem. Commun., 2018, 54, 11316–11319 RSC.
- A. Al-Shameri, N. Borlinghaus, L. Weinmann, P. N. Scheller, B. M. Nestl and L. Lauterbach, Green Chem., 2019, 21, 1396–1400 RSC.
- B. R. K. Menon, J. Latham, M. S. Dunstan, E. Brandenburger, U. Klemstein, D. Leys, C. Karthikeyan, M. F. Greaney, S. A. Shepherd and J. Micklefield, Org. Biomol. Chem., 2016, 14, 9354–9361 RSC.
- C. Schnepel and N. Sewald, Chem. – Eur. J., 2017, 23, 12064–12086 CrossRef CAS PubMed.
- R. Médici, J. I. Garaycoechea, L. A. Dettorre, A. M. Iribarren and E. S. Lewkowicz, Biotechnol. Lett., 2011, 33, 1999–2003 CrossRef PubMed.
- J. Fang, D. Hait, M. Head-Gordon and M. C. Y. Chang, Angew. Chem., Int. Ed., 2019, 58, 11841–11845 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961 CrossRef CAS PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- A. Bornadel, S. Bisagni, A. Pushpanath, S. L. Montgomery, N. J. Turner and B. Dominguez, Org. Process Res. Dev., 2019, 23, 1262–1268 CrossRef CAS.
- L. Xu, H. Liu, B. P. Murray, C. Callebaut, M. S. Lee, A. Hong, R. G. Strickley, L. K. Tsai, K. M. Stray, Y. Wang, G. R. Rhodes and M. C. Desai, ACS Med. Chem. Lett., 2010, 1, 209–213 CrossRef CAS PubMed.
- M. C. Desai, H. Liu and L. Xu, US Pat., US9891239B2, 2018 Search PubMed.
- L. Xie, K. Chen, H. Cui, N. Wan, B. Cui, W. Han and Y. Chen, ChemBioChem, 2020, 21, 1820–1825 CrossRef CAS PubMed.
- A. Li, C. G. Acevedo-Rocha, L. D'Amore, J. Chen, Y. Peng, M. Garcia-Borràs, C. Gao, J. Zhu, H. Rickerby, S. Osuna, J. Zhou and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 2–9 CrossRef.
- Z.-J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- S. Walmsley, B. Bernstein, M. King, J. Arribas, G. Beall, P. Ruane, M. Johnson, D. Johnson, R. Lalonde, A. Japour, S. Brun and E. Sun, N. Engl. J. Med., 2002, 346, 2039–2046 CrossRef CAS PubMed.
- B. Cao, Y. Wang, D. Wen, W. Liu, J. Wang, G. Fan, L. Ruan, B. Song, Y. Cai, M. Wei, X. Li, J. Xia, N. Chen, J. Xiang, T. Yu, T. Bai, X. Xie, L. Zhang, C. Li, Y. Yuan, H. Chen, H. Li, H. Huang, S. Tu, F. Gong, Y. Liu, Y. Wei, C. Dong, F. Zhou, X. Gu, J. Xu, Z. Liu, Y. Zhang, H. Li, L. Shang, K. Wang, K. Li, X. Zhou, X. Dong, Z. Qu, S. Lu, X. Hu, S. Ruan, S. Luo, J. Wu, L. Peng, F. Cheng, L. Pan, J. Zou, C. Jia, J. Wang, X. Liu, S. Wang, X. Wu, Q. Ge, J. He, H. Zhan, F. Qiu, L. Guo, C. Huang, T. Jaki, F. G. Hayden, P. W. Horby, D. Zhang and C. Wang, N. Engl. J. Med., 2020, 382, 1787–1799 CrossRef PubMed.
- D. Menche, F. Arikan, J. Li and S. Rudolph, Org. Lett., 2007, 9, 267–270 CrossRef CAS PubMed.
- H. Land, F. Ruggieri, A. Szekrenyi, W.-D. Fessner and P. Berglund, Adv. Synth. Catal., 2020, 362, 812–821 CrossRef CAS.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- M. J. García, F. Rebolledo and V. Gotor, Tetrahedron, 1994, 50, 6935–6940 CrossRef.
- R. N. Lima, C. S. dos Anjos, E. V. M. Orozco and A. L. M. Porto, Mol. Catal., 2019, 466, 75–105 CrossRef CAS.
- M. Koreishi, K. Tani, Y. Ise, H. Imanaka, K. Imamura and K. Nakanishi, Biosci., Biotechnol., Biochem., 2007, 71, 1582–1586 CrossRef CAS PubMed.
- M. R. Petchey, B. Rowlinson, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, ACS Catal., 2020, 10, 4659–4663 CrossRef CAS PubMed.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- B. M. Dorr and D. E. Fuerst, Curr. Opin. Chem. Biol., 2018, 43, 127–133 CrossRef CAS PubMed.
- T. Ikemoto and Y. Watanabe, US Pat., US7951977B2, 2011 Search PubMed.
- G. L. Moore, R. W. Stringham, D. S. Teager and T.-Y. Yue, Org. Process Res. Dev., 2017, 21, 98–106 CrossRef CAS PubMed.
- Y. Hayashi, T. Aikawa, Y. Shimasaki, H. Okamoto, Y. Tomioka, T. Miki, M. Takeda and T. Ikemoto, Org. Process Res. Dev., 2016, 20, 1615–1620 CrossRef CAS.
- A. K. Ghosh, J. Li and R. S. Perali, Synthesis, 2006, 3015–3018 CrossRef CAS.
- B. F. Fisher, H. M. Snodgrass, K. A. Jones, M. C. Andorfer and J. C. Lewis, ACS Cent. Sci., 2019, 5, 1844–1856 CrossRef CAS PubMed.
- C. K. Prier and B. Kosjek, Curr. Opin. Chem. Biol., 2019, 49, 105–112 CrossRef CAS PubMed.
- M.-C. Sigmund and G. J. Poelarends, Nat. Catal., 2020, 3, 690–702 CrossRef CAS.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- N. D. Fessner, ChemCatChem, 2019, 11, 2226–2242 CrossRef PubMed.
- I. Kaluzna, T. Schmitges, H. Straatman, D. van Tegelen, M. Müller, M. Schürmann and D. Mink, Org. Process Res. Dev., 2016, 20, 814–819 CrossRef CAS.
- S. C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges and F. H. Arnold, Science, 2017, 358, 215–218 CrossRef CAS PubMed.
- T. Hayashi, M. Ligibel, E. Sager, M. Voss, J. Hunziker, K. Schroer, R. Snajdrova and R. Buller, Angew. Chem., Int. Ed., 2019, 58, 18535–18539 CrossRef CAS PubMed.
- F. Rigoldi, S. Donini, A. Redaelli, E. Parisini and A. Gautieri, APL Bioeng., 2018, 2, 011501 CrossRef PubMed.
- B. V. Adalbjörnsson, H. S. Toogood, A. Fryszkowska, C. R. Pudney, T. A. Jowitt, D. Leys and N. S. Scrutton, ChemBioChem, 2010, 11, 197–207 CrossRef PubMed.
- A. O. Magnusson, A. Szekrenyi, H.-J. Joosten, J. Finnigan, S. Charnock and W.-D. Fessner, FEBS J., 2019, 286, 184–204 CrossRef CAS PubMed.
- F. S. Aalbers, M. J. Fürst, S. Rovida, M. Trajkovic, J. R. G. Castellanos, S. Bartsch, A. Vogel, A. Mattevi and M. W. Fraaije, eLife, 2020, 9, e54639 CrossRef CAS PubMed.
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS PubMed.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun and R. T. Kennedy, Angew. Chem., Int. Ed., 2020, 59, 4470–4477 CrossRef CAS PubMed.
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS.
- S. Mazurenko, Z. Prokop and J. Damborsky, ACS Catal., 2020, 10, 1210–1223 CrossRef CAS.
- Y. V. Sheludko and W.-D. Fessner, Curr. Opin. Struct. Biol., 2020, 63, 123–133 CrossRef CAS PubMed.
- W.-D. Fessner, New Biotechnol., 2015, 32, 658–664 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |