
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical tools for polymer materials
Yinjun
Chen
 a,
Gaëlle
Mellot
b,
Diederik
van Luijk
a,
Costantino
Creton
*b and
Rint P.
Sijbesma
*a
a,
Gaëlle
Mellot
b,
Diederik
van Luijk
a,
Costantino
Creton
*b and
Rint P.
Sijbesma
*a
aDepartment of Chemical Engineering & Chemistry and Institute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: r.p.sijbesma@tue.nl
bLaboratoire Sciences et Ingénierie de la Matière Molle, ESPCI Paris, PSL University, Sorbonne Université, CNRS, F-75005 Paris, France. E-mail: Costantino.Creton@espci.psl.eu
First published on 5th February 2021
Abstract
Mechanochemistry provides a unique approach to investigate macroscopic deformation, failure and healing of polymer materials. The development of mechanophores – molecular units that respond to mechanical force – has been instrumental in the success of this endeavor. This review aims to provide a critical evaluation of the large variety of mechanophores reported in literature, and to assess the molecular and macroscopic factors that determine their activation. Applications in materials science are highlighted, and challenges in polymer mechanochemistry are discussed.
 Yinjun Chen | Dr Yinjun Chen received his MSc in Chemistry Engineering from Xiamen University, China (2015). He joined the research group of Prof. Costantino Creton in 2015 and received his doctoral degree in 2018. His thesis work is focused on quantitative mapping of stress in soft materials by mechanochemistry. He is currently pursuing his post-doctoral studies in Eindhoven University of Technology under the guidance of Prof. Rint P. Sijbesma. |
 Gaëlle Mellot | Gaëlle Mellot is postdoctoral research scientist in the Soft Matter Science and Engineering Laboratory (ESPCI Paris – PSL). She is a chemical engineer graduated from ENSCR in 2016 and obtained her PhD degree at Sorbonne University (IPCM) in 2019. During her PhD she worked under supervision of Dr François Stoffelbach and Dr Jutta Rieger on the synthesis of nanofibers in water using the RAFT-mediated PISA technique and supramolecular chemistry. In 2020, she joined the group of Costantino Creton where she is carrying out research on the synthesis and incorporation of mechanophores in elastomers to investigate and understand damage in materials. |
 Diederik van Luijk | Diederik van Luijk received his BSc degree in Chemical Engineering at Eindhoven University of Technology in the group of Prof. Emiel Hensen in 2015 and his MSc degree at the same university in the group of Prof. Bert Meijer in 2018. Currently, he is a PhD candidate in the group of Prof. Rint Sijbesma pursuing the development and application of new mechanophores and mechanochemical reaction systems. |
 Costantino Creton | Costantino Creton is CNRS Directeur de Recherche in the laboratory of Soft matter Science and Engineering of ESPCI Paris – PSL. He graduated in Materials Science from the EPFL in 1985 and obtained his PhD in 1991 at Cornell University. After two post-docs he joined the ESPCI Paris in 1994 as a CNRS permanent researcher. He has developed a multidisciplinary approach (chemistry, physics, mechanics) to investigate the relationship between fracture of soft materials in the bulk and at interfaces. More recently he has been one of the pioneers in the quantitative use of mechanochemistry to locate bond scission during macroscopic fracture. |
 Rint P. Sijbesma | Rint Sijbesma is full professor in supramolecular polymer chemistry at the Eindhoven University of Technology. He received his PhD degree in 1993 with prof. Nolte on synthetic receptor molecules. After working as a postdoctoral student with Prof. Wudl (UCSB) on C60 chemistry, he moved to Eindhoven to explore supramolecular polymers with prof. Bert Meijer, and was appointed full professor in 2006. Over the years, Sijbesma has developed broad research activities in dynamic polymer systems, with topics that include self-assembled membranes, biomimetic hydrogels, and dynamic covalent polymers. He shares a fascination for polymer mechanochemistry with the other authors of this review. |
1. Introduction
Energy supplied by mechanical force may be used to drive a chemical reaction across an activation barrier – similar to how light, electricity and, most commonly, heat are used for this purpose.1–3 Many mechanochemical reactions are known in both organic4,5 and inorganic chemistry;6,7 and sometimes, these reactions proceed at room temperature via pathways that are improbable or inaccessible for conventional thermal reactions. The discovery of these reactions has enabled new efficient synthetic transformations, improved insight into the properties of materials under stress, and inspired the creation of stress-responsive materials that are programmable on the molecular level. The force required for such a mechanochemical reaction to proceed can be provided by macroscopic deformation of a material through grinding, ball-milling, compression, extension, or shearing.8 Alternatively, forces can be transduced to chemical bonds by sonication,9 atomic force microscopy (AFM),10–12 optical tweezers, or by the introduction of ring-strain.13,14Specifically within polymer materials, the use of mechanochemistry has evolved rapidly as a multi-purpose tool for characterization across length scales, and for creating materials with a novel response to force.15,16 The need to investigate the complex relationship between the molecular structure of a polymer and its mechanical properties as a material has stimulated the development of mechanophores:17 molecular units that can quantify and locate force on the molecular scale, making them unique tools for understanding and predicting macroscopic behavior.18–20 Meanwhile, smart materials can use mechanochemical reactions as triggers to change their own structure (force-responsive materials) or to produce a chemical function useful for catalysis, drug delivery, or soft robotics.21–23 Besides the use that mechanochemistry can have in functional polymer materials, polymers themselves are an excellent environment for studying mechanochemical reactions.24 Strong and flexible linear chains are commonly used to transfer force to a mechanophore, and do so both efficiently and controllably. Thus, the use of polymers as a matrix for mechanochemistry helps to advance the physical chemistry behind this useful group of reactions.
As the toolbox of mechanochemistry is expected to be opened more frequently by researchers in other disciplines, accessibility to all polymer scientists is critical for its successful application. Despite a wealth of application-oriented reviews,25–30 the selection of the correct tool is often challenging, indicating a need for guidelines to choose a suitable force-responsive group and a suitable method to incorporate the mechanophore inside a material to implement its function. This review aims to provide a ‘field guide’ for the implementation of mechanochemistry in synthetic polymers by summarizing the molecules, materials, and methods that have been investigated and applied. It is limited to the use of molecular mechanoresponsive units in polymer materials. Mechanical characterization of biomolecules, inorganic materials, and responsive materials based on microphase separation have been reviewed elsewhere.
Section 2 of this review provides an overview of mechanophores, categorizing them by output and clarifying their activation parameters. In Section 3, the synthesis and activation of mechanophores in polymer materials is discussed, starting with the different ways mechanophores can be implemented in a material, and continuing with their activation in different materials. In Section 4, applications of mechanochemistry in polymers are summarized, paying special attention to the polymer architectures and mechanophores used for each application. Together, these three sections should serve to simplify the daunting task of choosing the right mechanophore implemented in the right manner in the right material, as well as provide a comprehensive overview of the available tools for those looking to expand the available set of mechanophores. The final section concludes the review by summarizing remaining challenges and future applications.
2. Mechanophores and their responses
Mechanophores are molecular units that produce a physical or chemical response when an applied mechanical force brings about a structural rearrangement (such as a conformational change or bond scission). The rearrangement results in a response that varies from a physical signal (such as a change in absorbance or emission of light) to a chemical signal (such as enhanced catalytic activity, formation of a reactive radical or the release of a small molecule).For any application, a suitable mechanophore must meet several criteria. For each type of response, desired parameters should be identified; such as the excitation and emission wavelength of a mechanofluorescent response. A key parameter for any mechanophore is the threshold force at which it is activated. The threshold force is defined here as the offset force at which a mechanical response is observed in a single mechanophore molecule. Experimentally, this can be measured using a Single-Molecule Force Spectroscopy (SMFS) experiment in which a mechanophore is subjected to a tensile test using an AFM probe or with an optical tweezer setup. With both methods, the mechanophore must be covalently attached to two different surfaces which are then pulled apart until a mechanochemical event occurs at a given force. This way of measuring typically requires extensive synthesis to allow robust (notably, force-insensitive) surface functionalization as well as specialized and labor-intensive measurement procedures. For mechanophores of which the threshold force has not been experimentally characterized, computationally-determined force thresholds are provided instead. Most often, threshold forces have been calculated using the Constrained Geometry simulating External Force (CoGEF) method.31 In a CoGEF calculation, the mechanophore is first modelled in its unstrained state, typically using Density Functional Theory (DFT) methods. Then, two anchoring points on opposite sides of the mechanophore are selected and the distance between these points is increased in small steps, selecting for each step the geometry that minimizes the energy. The force profile is extracted from the distance increment and the computed minimal energy, and the activation force is taken as the maximum computed force before a mechanochemical event is found in the simulations. This simulation models events that occur during an SMFS measurement32,33 and generally agrees very well with experimental SMFS data across a wide range of mechanophores. Other types of force-dependent calculations – while generally less accessible – do provide additional insight into details of the potential energy surface and the transition state.15,34
For the purpose of this review, we use a hybrid classification of mechanophores based on whether activation results in a physical spectral response (mechanochromic, mechanofluorescent, mechanoluminescent) or elicits chemical reactivity (mechanocatalytic, mechanoradical, or release and rearrangement). These different classes of mechanophores are discussed with specific emphasis on a comparison of their force sensitivity. The force of activation for a large set of mechanophores has recently been evaluated with COGEF calculations.33
2.1 Mechanochromic moieties
Mechanochromic moieties change their UV-vis absorption spectrum upon mechanical activation. Generally, the change in absorption is caused by expansion of conjugation in the molecular structure, which leads to a bathochromic shift of the absorption maximum. A visible color change provides an easily observed signal for stress, strain or damage in polymeric materials. A wide range of mechanochromic moieties has been used as mechanophores in polymeric materials. For each of these mechanophores, chemical stability, synthetic accessibility, mechanical activation parameters, and changes in absorption spectrum define their suitability as a reporter of force in various polymer materials.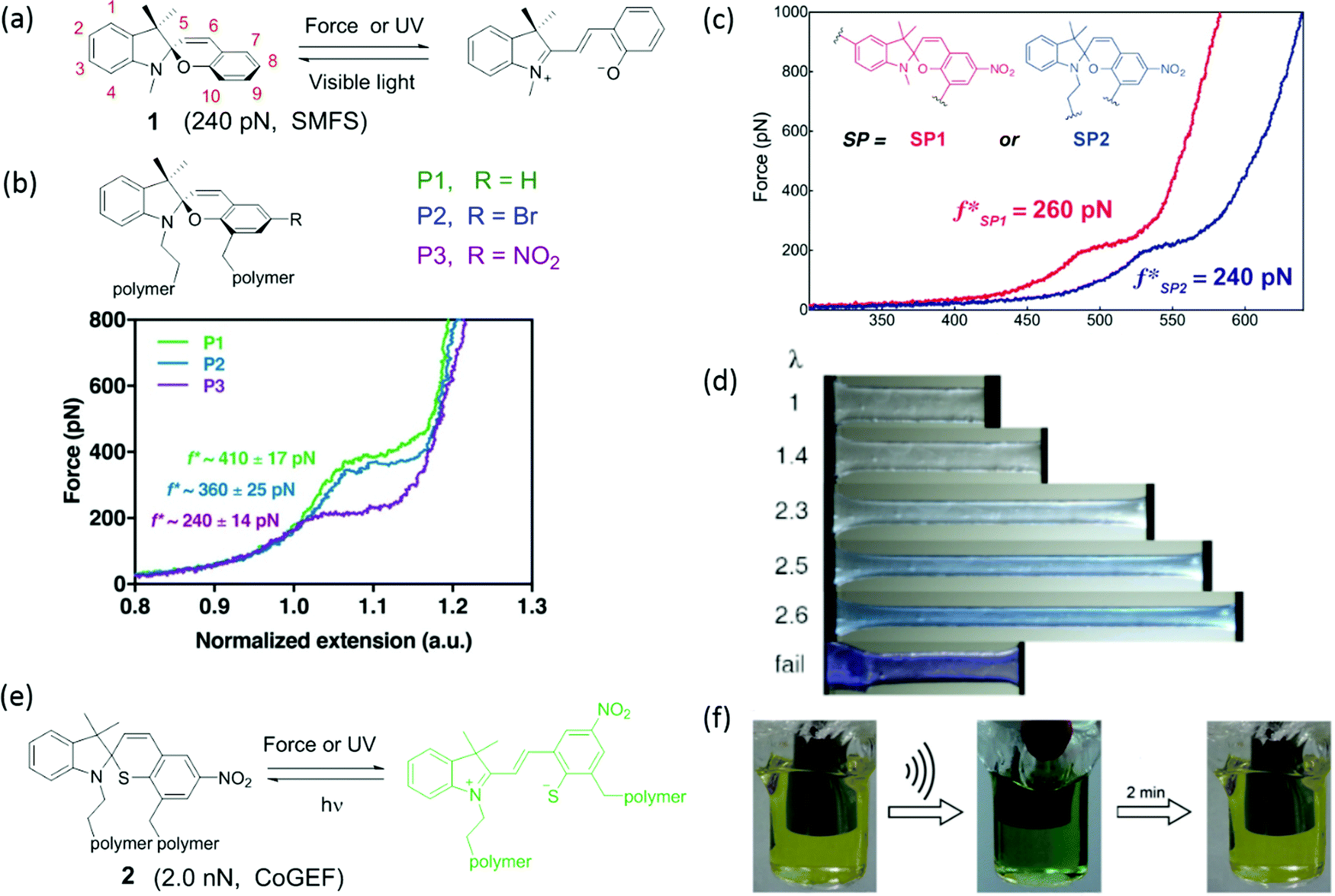 | ||
| Fig. 1 (a) Activation of spiropyran by mechanical force or UV light; the reverse reaction is accelerated by visible light. (b) Three of spiropyran derivatives and their single-molecular force spectrum.41 (c) Single-molecular force spectrum of two spiropyran derivatives with varying attachment points.36 (d) Color change of spiropyran in a multiple-network elastomer during uniaxial extension.42 (e) Activation of spirothiopyran by mechanical force or UV light and the reverse reaction by visible light. (f) Activation of spirothiopyran in the backbone of a polyester by sonication in solution.43 (b) is reprinted with permission from ref. 41, Copyright 2018 American Chemical Society. (c) is reprinted with permission from ref. 36, Copyright 2015 American Chemical Society. (d) is reprinted with permission from ref. 42, (published under a Creative Commons license, CC BY-NC), Copyright 2020 AAAS. (f) is reprinted with permission from ref. 43, Copyright 2016 Wiley-VCH. | ||
The first use of spiropyran as a mechanochromic mechanophore was reported by Moore's group in 2009.35 Colorless spiropyran was incorporated in the centre of a poly(methyl acrylate) (PMA) polymer backbone. Under uniaxial extension, spiropyran units were converted into highly colored merocyanine, resulting in a color change of the PMA material from yellow to purple. After failure, material turned red. While spiropyrans are generally colorless or yellow, the color of merocyanines in a polymer is influenced by the chemical environment,37 and can be blue, purple and red depending on the polarity of the polymer and its water content.42 Moreover, merocyanine containing polymers often show different colors in loading and unloading due to isomerization around the bonds connecting the cyclic subunits (Fig. 1d).44 The striking optical response of spiropyran containing polymers is very easily observed by eye; and as a consequence, this mechanophore has been incorporated in a variety of polymer materials. These studies will be discussed in detail in Sections 3 and 4.
Various pyran analogs such as spirothiopyran (STP),43 naphthopyran (NP),45 and bis-naphthopyran (BNP),46 have been designed with the aim of tuning reactivity, color and critical activation force. Spirothiopyran is a versatile mechanophore, as it features both mechanochromism and force-activated addition reactions of the sulfur atom (Fig. 1e). Ring opening of the thiopyran ring of STP via a 6π electrocyclic ring-opening reaction to thiomerocyanine (TMC) is accompanied by a color change from yellow to green. The nucleophilic thiolate formed after activation is a reactive partner in the thiol–ene click addition reaction with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. Weng's group reported the first example of STP-containing mechanochromic polymer materials, where STP was embedded into the backbones of a polyester and a polyurethane. A yellow solution of the polyester turned green after sonication as shown in Fig. 1f, indicating the formation of the thiomerocyanine form of the dye. In the presence of N-ethyl maleimide, the green color quickly disappeared due to reaction with the thiomerocyanine. When 1,6-bismaleimidohexane crosslinker was present, sonication of a thiospiropyran-containing polymer led to crosslinking of the linear polymers into insoluble networks.43 Calculations with CoGEF33 show that the threshold force to activate STP is 2.0 nN – distinctly lower than for SP mechanophores, which have a calculated Fmax of 2.6 nN.33,47
C double bonds. Weng's group reported the first example of STP-containing mechanochromic polymer materials, where STP was embedded into the backbones of a polyester and a polyurethane. A yellow solution of the polyester turned green after sonication as shown in Fig. 1f, indicating the formation of the thiomerocyanine form of the dye. In the presence of N-ethyl maleimide, the green color quickly disappeared due to reaction with the thiomerocyanine. When 1,6-bismaleimidohexane crosslinker was present, sonication of a thiospiropyran-containing polymer led to crosslinking of the linear polymers into insoluble networks.43 Calculations with CoGEF33 show that the threshold force to activate STP is 2.0 nN – distinctly lower than for SP mechanophores, which have a calculated Fmax of 2.6 nN.33,47
Naphthopyran (NP) is also mechanochromic, with a color change from colorless to yellow when the ring opens (Fig. 2a). The calculated threshold force to activate NP (Fmax = 3.7–4.4 nN, CoGEF) is higher than for SP. The threshold force depends on the attachment points as well as on the nature of the substituents. Fig. 2b shows three types of attachment of polymer chains; only NP5 was activated in tensile tests when covalently crosslinked into PDMS, while NP8 and NP9 were inactive (Fig. 2c).48 Varying the substituents at positions 1 and 3 (Fig. 2a and e) not only gave different colors of the activated merocyanine form, but the critical forces of activation were also different. For instance, in compound 3 the critical force was 4.1–4.4 nN to give a yellow merocyanine; while it was 3.7–3.9 nN for compound 4a–c, with a color change to purple. Each of the six NP's in PDMS studied by Robb et al. gave different mechanochromic behavior. All polymers showed color change; but differences in color intensity due to different concentrations of merocyanine illustrate the variation in threshold forces among these mechanophores (Fig. 2e).49 The compounds also show differences in fading time in the relaxed state because the merocyanines have different stabilities.
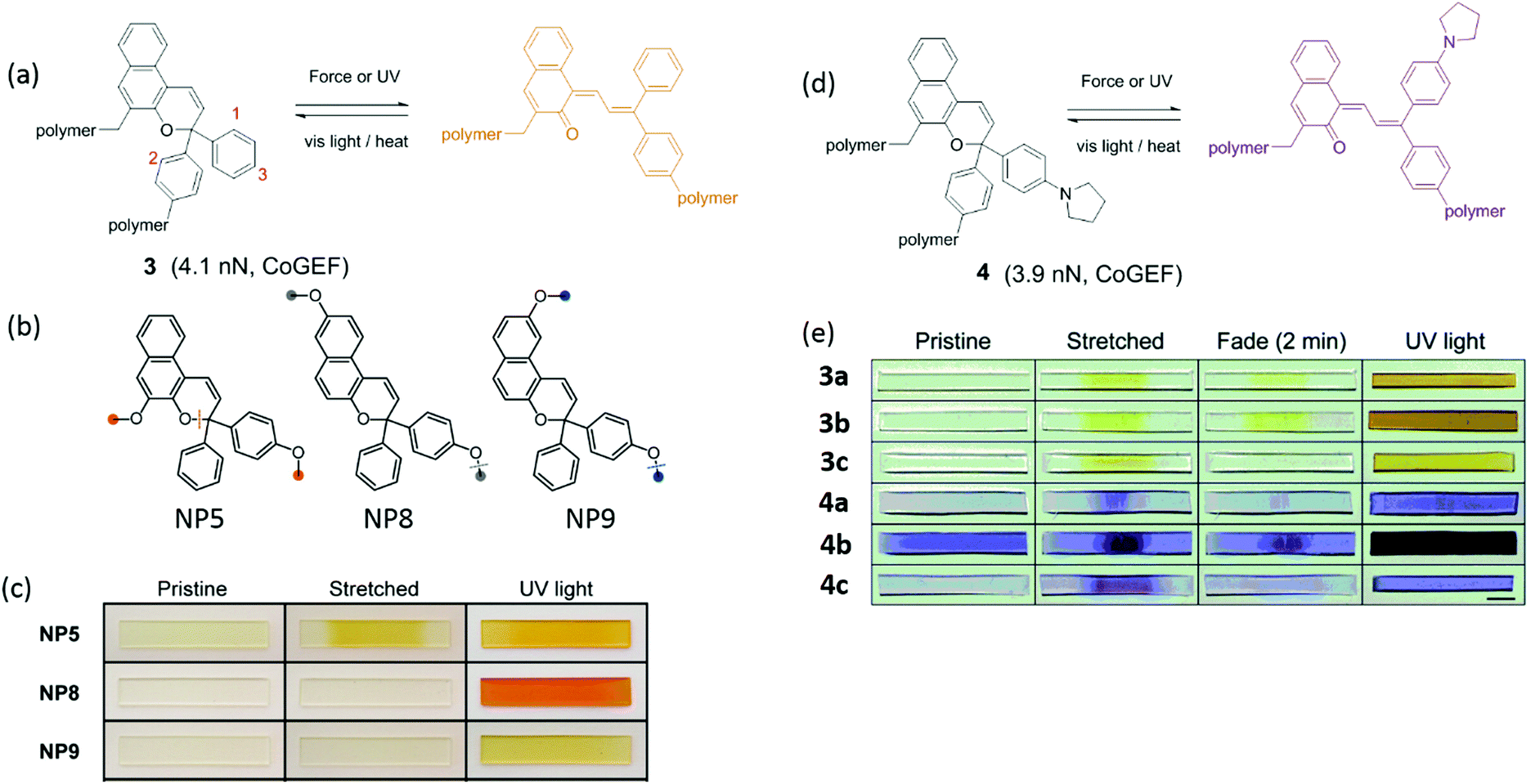 | ||
| Fig. 2 (a) Activation of naphthopyran (NP) by force and UV light; the reverse reaction is accelerated by heat or visible light. (b) Naphthopyran derivatives with varying polymer attachment points. (c) Images of polymer materials in (b) showed different mechanochromic responsiveness before and after elongation and UV irradiation.48 (d) Naphthopyran mechanophore with pyrrrolidine substituent (e) naphtopyran derivatives and their color changes in naphtopyran-containing PDMS after extension or UV irradiation.49 (b) is adapted with permission from ref. 48, Copyright 2016 American Chemical Society. (c) is adapted with permission from ref. 48, Copyright 2016 American Chemical Society. (e) is adapted with permission from ref. 49, Published by The Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). | ||
Bisnaphthopyran (BNP) combines two pyran rings and is unique among SP derivatives because it isomerizes via two 6π electrocyclic ring-opening reactions. The two pyran rings can be activated consecutively by UV light, while mechanical force directly activates both pyran rings in a single step, as has been demonstrated by Robb et al. in ultrasonication experiments.46 When one of the pyran rings is activated by UV light, the second ring can be activated by mechanical force as shown in Fig. 3. Each of the states of BNP exhibits a different color. State 1 – with one opened pyran ring – is yellow, while open–open state 2 is purple. In CoGEF simulations, the threshold force to open the first ring is 4.1 nN, while opening the second ring requires a force of at least 4.6 nN.33 This difference is not enough to selectively create a high amount of state 1 in the ultrasonication experiments. In the mechanostationary state, it was assumed that most, if not all, of the open–closed form was the product of electrocyclic ring closure of the open–open state.46
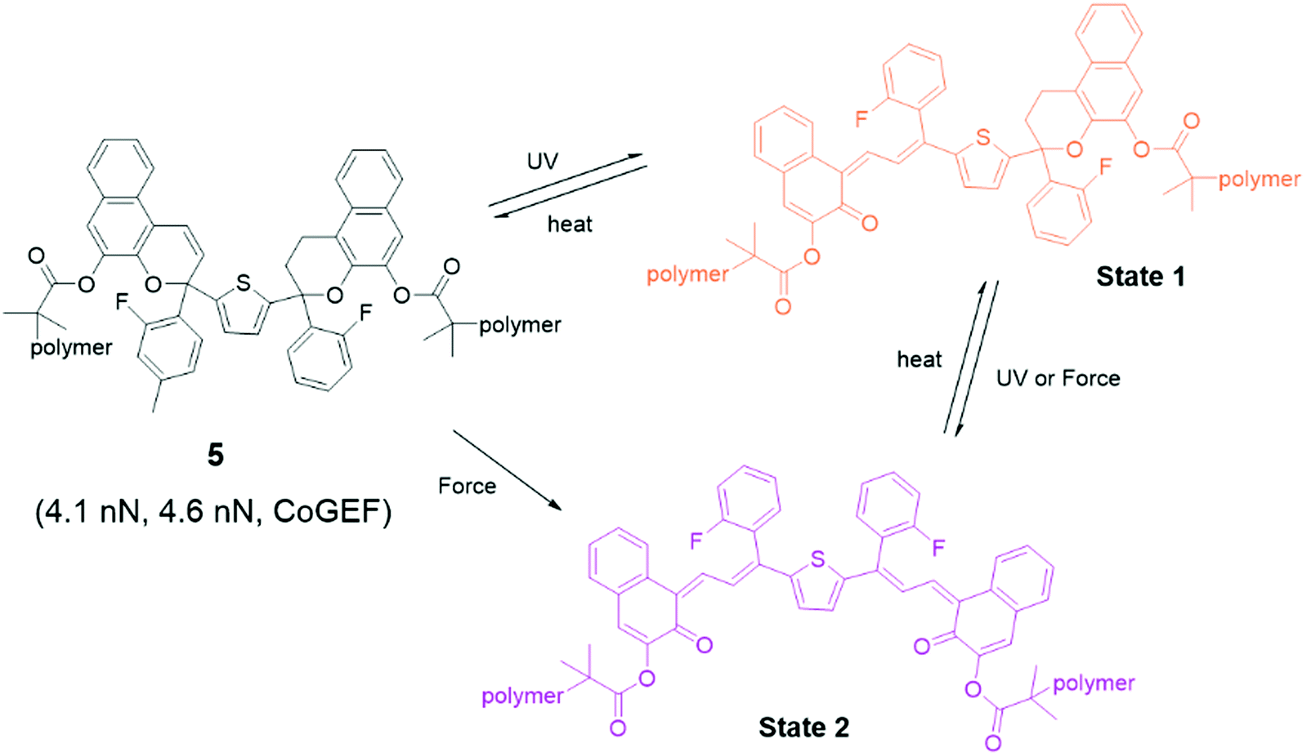 | ||
| Fig. 3 Activation of bisnaphthopyran 5. UV light leads to stepwise ring opening via state 1, while mechanical force directly converts 5 to state 2. | ||
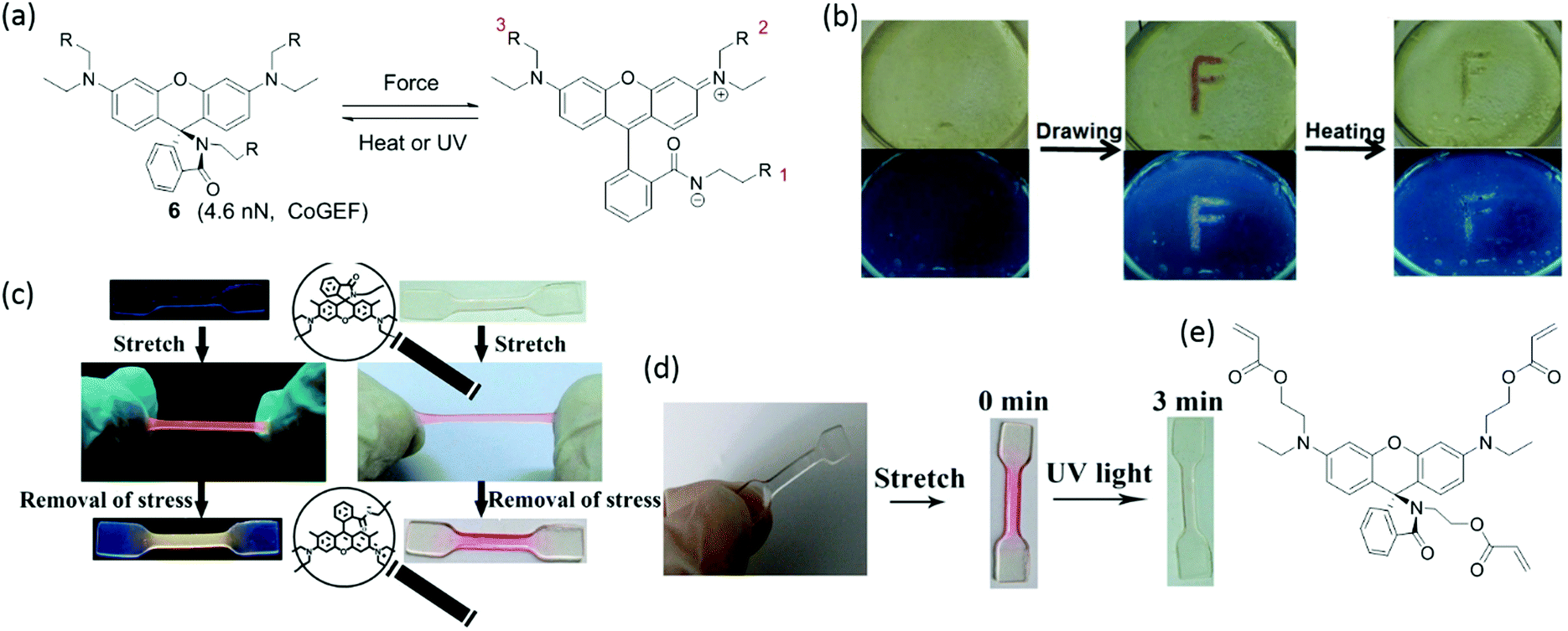 | ||
| Fig. 4 (a) Activation of rhodamine and the reverse reaction by stimulation with force and heat or UV, respectively. (b) Rhodamine was embedded into polyurethane film. The film was drawn and showed a color change that fades upon heating.50 (c) Rhodamine integrated into the filler network of a multiple-network elastomer changes color in extension; different fluorescent colors are observed in loading and unloading.52 (d) Bleaching of color in activated elastomer by UV light.52 (e) Three-arm rhodamine modified with double bonds. (b) is reprinted with permission from ref. 50, Copyright 2015 Wiley-VCH. (c) is reprinted with permission from ref. 52, Copyright 2017 American Chemical Society. (d) is adapted with permission from ref. 52, Copyright 2017 American Chemical Society. | ||
To effectively activate spirorhodamine, the mechanical force transmitted along the polymer chain should induce the scission of the C–N bond in the spirolactam. Activation is influenced by the position of the attachment points and by the electronic properties of the substituents on spirorhodamine. For example, a spirorhodamine-diol was attached to polyurethane at positions 1 and 3 in Fig. 4a.52 The resulting polymer materials show reversible mechanochromism with fluorescent emission in compression (between colorless and reddish). However, when the attachment points were changed from 1+3 to 2+3, mechanical force was not transferred across the C–N bond in spirolacatam, and the spirorhodamine was not activated by force. Furthermore, spirorhodamines with varying substituents on the xanthene part of the molecule show different photochromic responses. Rhodamine with two amino groups52 shows a strong absorption in the blue-green range of the visible spectrum, and UV light induces the transformation of the open form to the spiro form – in contrast to the spirorhodamine with a single amino group on the chromophore (Fig. 4d).50 Trifunctional spirorhodamine (Fig. 4e) was incorporated into the filler network of poly(ethyl acrylate) multiple-network elastomers. The elastomers display a UV-sensitivity that is opposite to that of the polyurethane labelled by spirorhodamine-diols. Furthermore, the elastomers have a red-shifted fluorescence, and the fluorescent color changes from red to yellow when unloading. Interestingly, the fluorescent color can be tailored by incorporating pyrene in the polymer material.53 Upon stretching, the combination of green fluorescence from the pyrene excimer, blue fluorescence from monomeric pyrene and red fluorescence from mechanochemically-activated rhodamine gives rise to white emission.
2.2 Photoluminescent mechanophores
While the mechanochromic mechanophores discussed in the previous section show some fluorescence in addition to a color change, mechanophores that are exclusively used for their emissive properties deserve separate treatment. Photoluminescent mechanophores display a change in fluorescent or phosphorescent behavior when subjected to a mechanical force. In most cases, a ‘turn-on response’ is generated, meaning that the mechanically active groups display no photoluminescent response at the measured wavelength before a dye is unveiled by a mechanical stimulus. Compared to the colorimetric response of a mechanochromic system, a photoluminescent response is far stronger if a fluorophore with a sufficiently high quantum yield is released. This allows for strong and accurate detection at mechanophore concentrations of the order of 10−6 M, and even allows for detection of single bond-breaking events.54 In addition to the parameters outlined in Section 2.1, quantum yield is therefore an important parameter for determining the suitability of a photoluminescent mechanophore.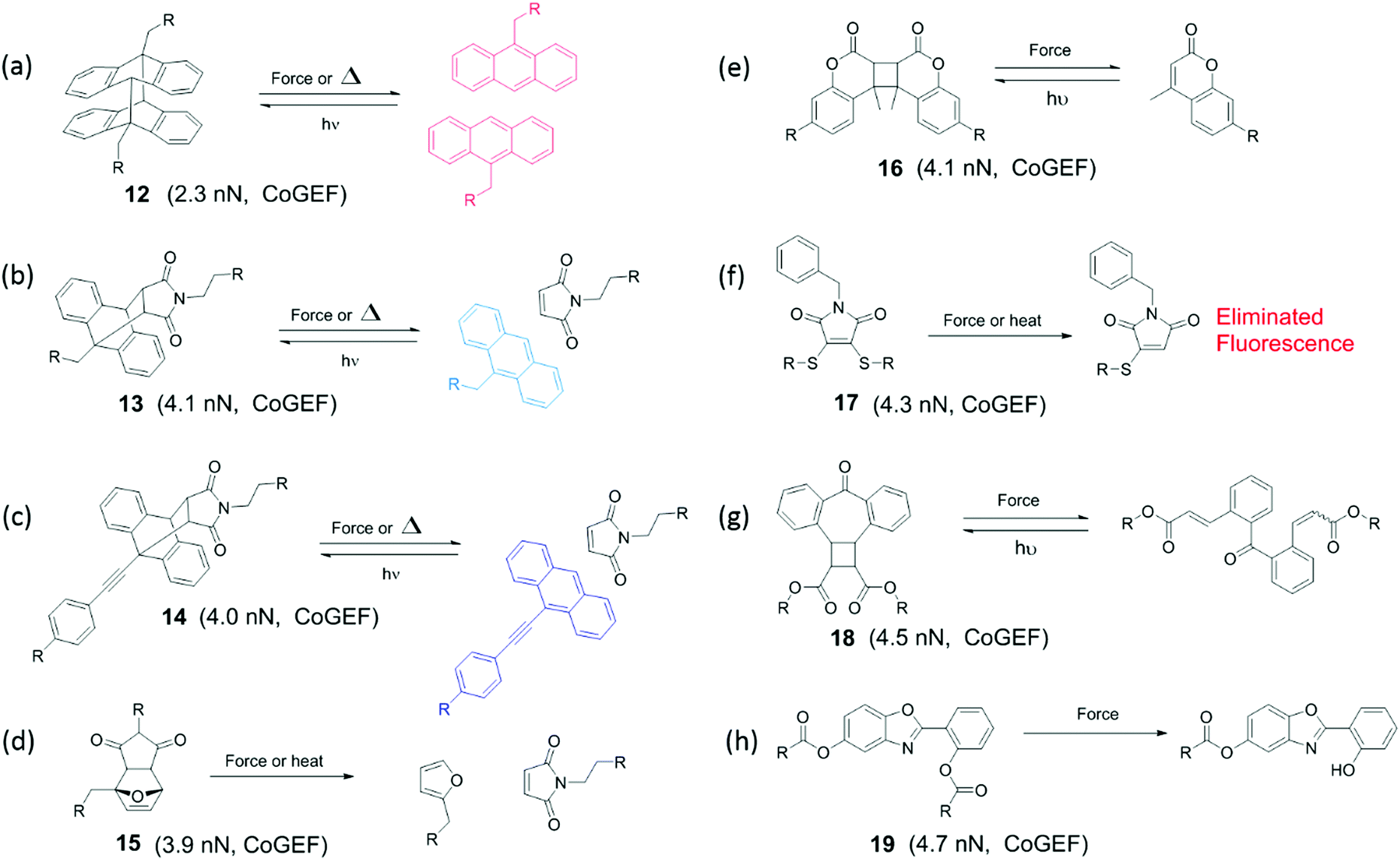 | ||
| Fig. 5 Structure of various fluorescent mechanophores that are activated by force and display covalent bond scission. (a) Anthracene dimer; (b) Anthracene–maleimide adduct; (c) π-Extended anthracene–maleimide adduct; (d) Furan–maleimide adduct;65,66 (e) Coumarin dimer; (f) Dithiomaleimide; (g) Methanone-tethered cinnamate dimer. (h) 2-(2′-Hydroxyphenyl)benzoxazole. | ||
Other categories of covalent fluorescent mechanophores reported by pioneers include the coumarin dimer (compound 16),67,68 dithiomaleimide moiety (compound 17),69 methanone-tethered cinnamate dimer (compound 18),70 and 2-(2′-hydroxyphenyl)benzoxazole (compound 19).71 Like anthracene adducts, these mechanophores lead to scission of the polymer chain, concomitant with fluorescence. Coumarin dimers have been investigated by Craig et al., who integrated coumarin dimers in the middle of poly(methyl acrylate) chains and characterized the relationship between the activation efficiency of mechanophore and molecular weight of the polymer.67 Dithiomaleimide 17 is notable for being fluorescent before cleavage, thereby being the only example of a mechanophore that loses fluorescence after mechanical activation.69 CoGEF calculations provided a force threshold of 4.3 nN and a cleavage mechanism that started with homogeneous bond scission of the C–S bond,33 although the final reaction products (structure) are not validated in experiment after activation. With the exception of anthracene adducts, many of these fluorescent mechanophores have not yet been taken full advantage of in mechanochemistry for damage research in polymer materials.
Taking this advantage to an extreme, a conjugated polymer of a fluorescent donor doped with an acceptor was shown to be highly sensitive to chain extension.54 Poly(dioctylfluorene-alt-benzothiaziazole) (F8BT) was copolymerized with a small amount of dithienyl benzothiadiazole (DTBT). Förster resonant energy transfer (FRET) between the excited donor and initially ground-state acceptor occurs in all cases; but the extent of transfer depends on the distance between donor and acceptor, which in turn is influenced by stretching the polymer materials. A threshold force of approximately 300 fN was determined experimentally – roughly four orders of magnitude lower than a typical covalent mechanofluorophore. No bonds are broken upon activation, as not a chemical bond but the conformational entropy of the coiled polymer chain is destabilized by the applied force.
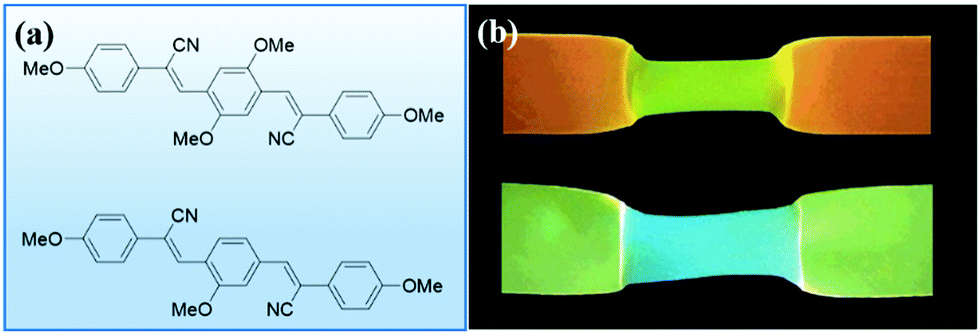
Moving towards slightly higher interaction energies, we find supramolecular complexes held together by electrostatic interaction,73 π–π stacking,72 or hydrophobic interactions.74 This category of mechanophores includes dyes of conjugated oligo(p-phenylenevinylene) derivatives (OPV) aggregated by π–π stacking interactions (Fig. 6). The aggregated excimers dissociated in response to tensile deformation and resulted in a luminescent color change from either yellow to green, or from green to blue, depending on the OPV derivatives that were used. Recently, another supramolecular mechanophore was accessed by incorporating a fluorophore–quencher pair into a mechanically interlocked rotaxane (Fig. 7a). Sagara's and Weder's groups74–76 embedded the rotaxane mechanophore into polyurethane elastomers and the elastomers displayed rapid and reversible fluorescence switching upon extension as shown in Fig. 7b. The fluorescent response correlated with the macroscopic deformation and the optical properties could be tailored by varying the chromophores in rotaxane.
 | ||
| Fig. 6 Conjugated oligo(p-phenylenevinylene) derivatives display different fluorescent colors in aggregated or dissociated states. (a) Two conjugated oligo(p-phenylenevinylene) derivatives. (b) The two dyes in (a) were physically blended into polymer matrix and the materials revealed fluorescent color change in extension.72 (b) is reprinted with permission from ref. 72, Copyright 2020 Springer Nature. | ||
 | ||
| Fig. 7 Supramolecular mechanophores prepared by locking fluorophore and quencher in a rotaxane. (a) Three rotaxane mechanophores with different fluorophores. (b) Fluorescent responses of the three rotaxane mechanophores in tensile tests.75 (a) and (b) are adapted with permission from ref. 75, Copyright 2019 American Chemical Society. | ||
Mechanically active charge-transfer complexes have also been introduced in polymer materials without the stabilization of rotaxane formation. A fluorescent pyrene group was connected to two naphthalene diimide (NDI) groups using a short covalent linker and incorporated in the backbone of a polycaprolactone chain.77 Pyrene forms a charge-transfer complex with the neighboring NDI, locally folding the chain and quenching the fluorescence of pyrene. When the polycaprolactone films were stretched, the interaction between pyrene and NDI was broken and a fluorescent response was observed. Sufficiently soft materials allowed enough mobility for the pyrene and NDI to recombine, quenching the fluorescence and displaying reversible behavior. In amorphous polycaprolactone however, an increasing response was observed even after stretching, presumably due to strain-induced ordering and crystallization of the polymer matrix. A different fluorescent mechanophore based on π–π interactions of a pyrene derivative is the sulfonated derivative (hydroxyethyl)pyrene trisulfonate (HEPTS).78 HEPTS forms aggregates in apolar solvents and in polyurethane materials, thereby shifting to a yellow-emitting fluorescence from the blue emission of non-aggregated HEPTS. Stretching materials made from HEPTS-telechelic polyurethane chains mixed in a non-functionalized material showed a shift in the emission spectrum due to dissociation of the aggregates.
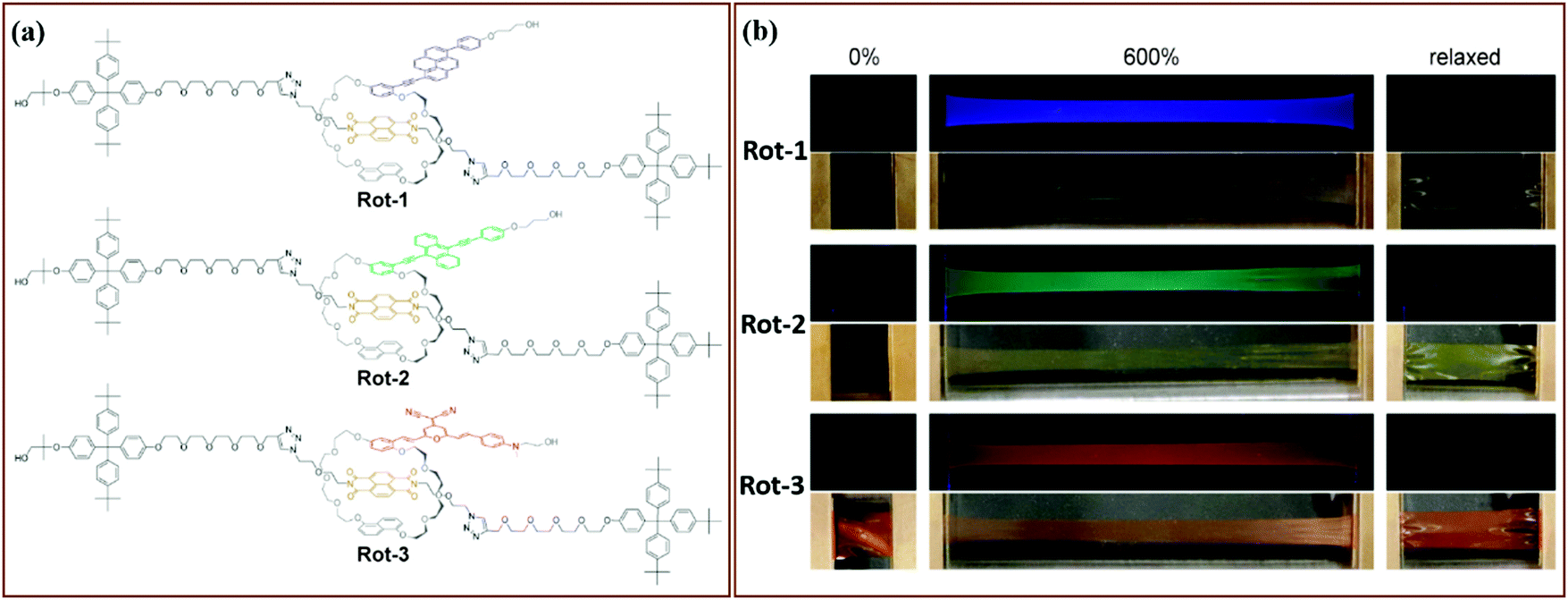
A fluorescent mechanophore stabilized by electrostatic interactions was reported by Jen et al.73 This thermodynamically unstable mechanophore is formed by a Michael addition reaction and is electrostatically-stabilized by a protonated amine (Fig. 8). Mechanical force initiated the reversible elimination, leading to the release of a conjugated dye. When the mechanophores were covalently crosslinked in an epoxy network, a color change was observed at a low onset of deformation of 0.14 accompanied with a fluorescent color change.
 | ||
| Fig. 8 Breaking an electrostatic interaction by force resulted in a molecular rearrangement. This rearrangement leads to the release of 3-cyano-4,5,5-trimethyl-5H-furan-2-ylidene malononitrile dye; and materials showed a color change, including fluorescent color switching with varying degrees of compressive strain.73 Adapted with permission from ref. 73, Copyright 2016 Wiley-VCH. | ||
Mechanophores that display photoluminescence due to reduced molecular mobility are a recent addition to the mechanochemical toolbox. Films made from hyperbranched poly(amido amine)s were reported to displayed an increased fluorescent intensity at near-zero strain (Fig. 9).79 These materials contain tertiary amines in close proximity to amide groups. The fluorescent properties of these materials, classified as unconventional macromolecular chromophores,80 are believed to derive from intramolecular electron overlap of closely clustered amines and amides. Stretching this material resulted in a linear increase of fluorescence intensity at the same wavelength, which could be activated and deactivated reversibly. The applicability of this class of mechanophores to different systems may be limited by the close proximity, and therefore high concentration, of these groups that is required for fluorescence. Additionally, a fluorescent background is present prior to activation and no significant shift in excitation wavelength is observed; so the fluorescent signal is only meaningful when it can be compared to the initial state of the material. Nevertheless, this unique approach may lead to the development of more generally applicable, highly sensitive mechanofluorophores as well as a better understanding of this unusual photophysical response.
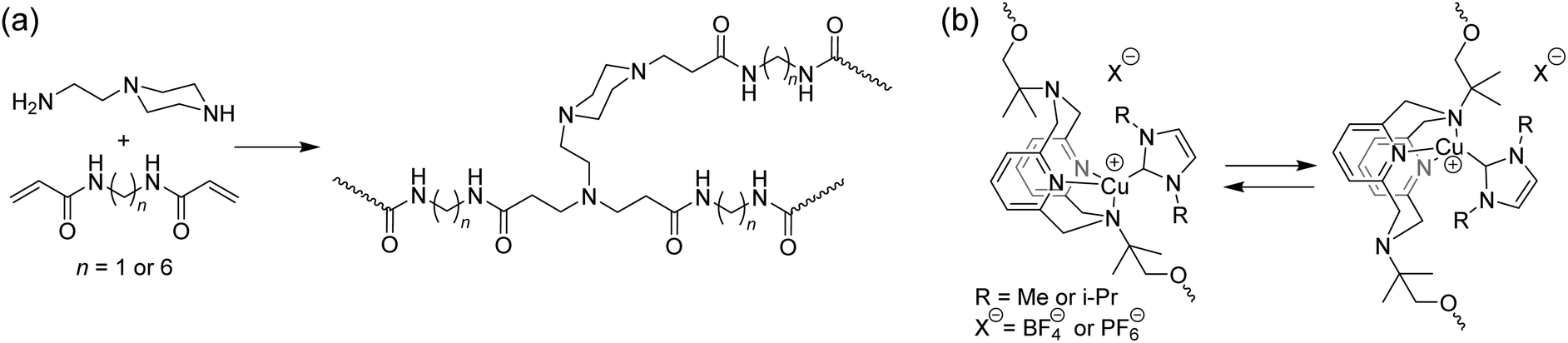 | ||
| Fig. 9 Structures of mobility-based photoluminescent mechanophores. (a) A hyperbranched network containing amide groups and tertiary amines in close proximity show enhanced fluorescence emission upon increasing strain.79 (b) Phosphorescent copper(I)-pyridinophanes ligated to an N-heterocyclic carbenes undergo a rapid ligand exchange, which is hindered by mechanical tension, thereby increasing the photoluminescent response.81 For R = i-Pr, a shift in emission spectrum is also observed.83 | ||
Filonenko and co-workers have described a chemically different, yet mechanistically similar approach for force detection in polymers with a Cu(I)–pyridinophane complex (Fig. 9).81,82 This complex contains a tetradentate ligand that binds to the metal in a tridentate fashion, leaving one tertiary amine available that can exchange rapidly with an identical copper-bound ligand. The complex displays a phosphorescent photoluminescent response; however, the dynamic ligand exchange provides a pathway for non-radiative decay, resulting in a low phosphorescence intensity. When the exchangeable ligands are mechanically activated by tensile strain in polyurethane materials, the reduced dynamicity of the system leads to a higher probability of phosphorescent emission occurring before non-radiative decay, thereby enhancing the phosphorescent signal. This normally does not produce a shift in emission wavelength, limiting its use in materials to intensity-based detection. A derivative with a more sterically-hindered co-ligand was reported that did show a hypochromic emission shift upon mechanical activation in a comparable material.83 The origin of this shift was explained by the interaction between the Cu+ center and the non-coordinating PF6− or BF4− counterions. This shift also occurred due to different stimuli that affect the ion-pair distance, such as solvent polarity or temperature changes. The reason for a change in ion-pair distance was thought to be related to the change in free volume of the polymer material. Because of the shift in emission wavelength, a ratiometric response could be measured, in contrast to other mobility-based mechanophores. However, the color change is highly dependent on the environment, and it is difficult to predict its behavior in other materials.
2.3 Mechanoluminescent mechanophores
Mechanoluminescence is the emission of light due to a mechanical stimulus. Use of bis(adamantyl)-1,2-dioxetane, shown in Fig. 10a as a mechanophore was introduced by Chen et al.84 Luminescence of this dioxetane is activated by force which cleaves the mechanophore in two adamantanone units, one of which is in an excited state. Relaxation of the excited adamantanone to its ground state is accompanied by bright-blue luminescence. Adamantyl substituents increase the thermal stability compared to other 1,2-dioxetane derivatives.85 For this reason bis(adamantyl)-1,2-dioxetane derivatives have been applied as heat or acid activated chemiluminescence probes for bio-labeling.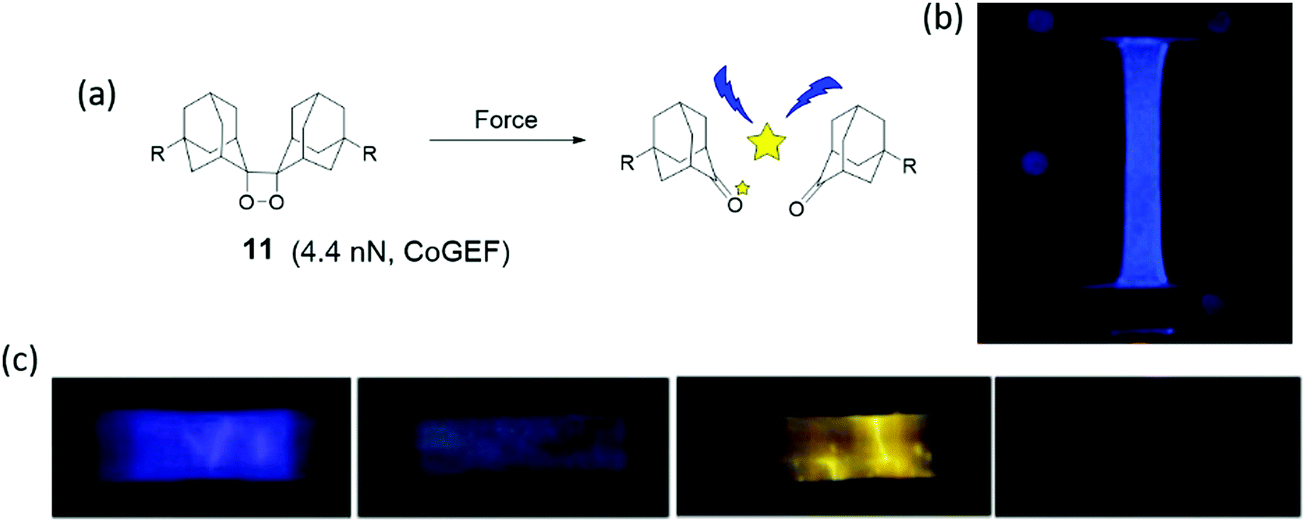 | ||
| Fig. 10 (a) Dioxetane mechanophore is activated by force and emits blue light. (b) Dioxetane mechanophore was incorporated into filler network of multiple-network elastomers, and the elastomers presented blue light in uniaxial extension.86 (c) Different dye acceptors were physically blended into poly(methacrylate) materials, and materials showed different light during elongation.84 (b) is reprinted with permission from ref. 86, (published under a Creative Commons license, CC BY-NC), Copyright 2014 AAAS. (c) is reprinted with permission from ref. 84, Copyright 2012 Springer Nature. | ||
When the dioxetane mechanophore was integrated into the center of a PMA polymer backbone or used as a crosslinker in an acrylate polymer network, emission of blue light with a maximum at 420 nm85,87 was observed during extension or sonication in polymer solution (Fig. 10b). The sensitivity and color of light emission was tuned by energy transfer to suitable acceptor dyes84 (Fig. 10c). Dioxetane luminescence has been used as a highly sensitive molecular probe to evaluate failure mechanisms,86,88 stress distribution, and stress evolution86 in polymer materials, which is discussed in Sections 3 and 4.
In an attempt to reduce the activation threshold, a cascade strategy was developed in which a Pd–NHC complex49–90 was incorporated into a (PTHF) backbone, and release of a strongly basic NHC ligand upon mechanochemical activation was used to deprotonate a precursor 1,2-dioxetane chemiluminescent probe.89 Subsequently, the 1,2-dioxetane was activated and emitted light. The cascade strategy decreases the force threshold of the dioxetane mechanophore, and elevates the sensitivity and effectivity of mechanical activation of mechanophores.
2.4 Mechanoradicals
Groups that can cleave heterolytically to form two radical species are known as mechanoradicals. Often these groups display a change of absorbance spectrum when they are activated or display photoluminescent properties, which is why they can also be considered mechanochromophores or mechanofluorophores. However, the generation of radicals has additional utility in electron paramagnetic resonance (EPR) detection and especially in chemical reactivity, as these reactive species may undergo or initiate various reactions (see Section 4). Broadly, they may be sorted into two groups: carbon-centered and heteroatomic mechanoradicals.Diarylbibenzofuranone (DABBF) is a homodimer of arylbenzofuranone connected by a central C–C bond as shown in Fig. 11a. The C–C bond with a length of 1.586 Å and a bond dissociation energy of 95.5 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 91 is longer and weaker than a normal C–C bond. Using heat or light, these motifs can dissociate into two highly stable carbon-centered radicals – a process which is accompanied by a large shift in absorbance from 346 nm to 650 nm (Fig. 11b).91,92 Interestingly, arylbenzofuranones are not oxygen-sensitive, and DABBF can easily dissociate and recombine reversibly under ambient conditions.91,93 Mechanically, the threshold force has been calculated using CoGEF to be 3.5 nN.33
91 is longer and weaker than a normal C–C bond. Using heat or light, these motifs can dissociate into two highly stable carbon-centered radicals – a process which is accompanied by a large shift in absorbance from 346 nm to 650 nm (Fig. 11b).91,92 Interestingly, arylbenzofuranones are not oxygen-sensitive, and DABBF can easily dissociate and recombine reversibly under ambient conditions.91,93 Mechanically, the threshold force has been calculated using CoGEF to be 3.5 nN.33
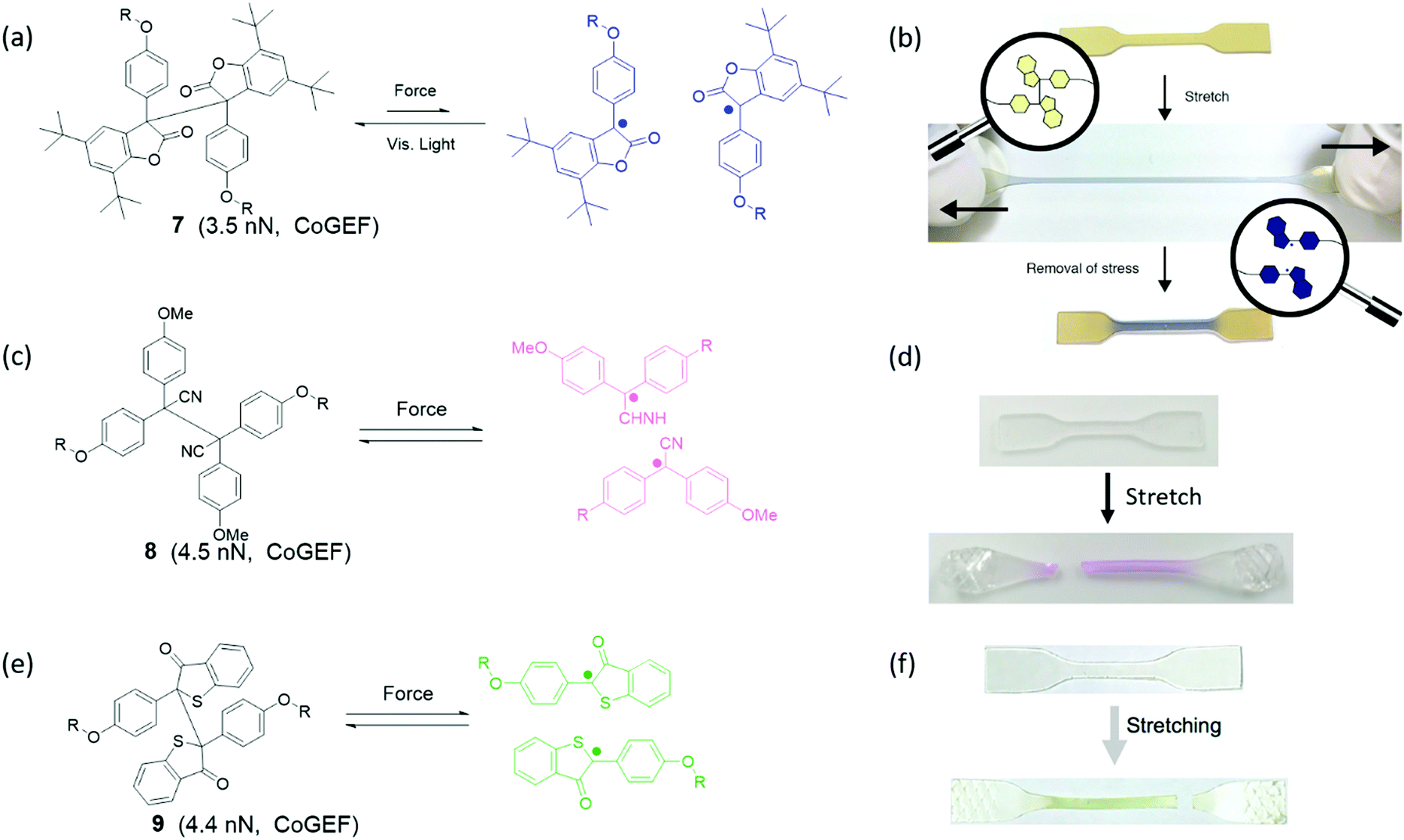 | ||
| Fig. 11 (a) Activation of DABBF by force. (b) Strain-induced color change of polyurethane with DABBF incorporated in the main chain.92 (c) Activation of TASN by force. (d) Color change from colorless to pink upon uniaxial extension of a poly(hexyl methacrylate) network containing TASN.100 (e) Activation of DBBT by force (f) strain induced color change of block polymer of poly(styrene)–poly(methyl acrylate)–poly(styrene) with DBBT incorporated into the soft domain.97 (b) is reprinted with permission from ref. 92, Copyright 2015 American Chemical Society. (d) is adapted with permission from ref. 100, Copyright 2020 American Chemical Society. (f) is reprinted with permission from ref. 97, Copyright 2018 American Chemical Society. | ||
Tetraarylsuccinonintrile (TASN) is a similar dimer in which the lactone group is replaced with a nitrile substituent (Fig. 11c). In this mechanophore the central C–C bond is slightly longer at 1.608 Å, but the bond dissociation energy increases to 26.2 kcal mol−191 resulting in a larger overall threshold force at 4.5 nN.33 Cleaved TASN exhibits an absorption maximum at 550 nm (Fig. 11d) as well as strong yellow light emission upon excitation with UV light (λ = 356 nm).94 It retains the remarkable reversibility of DABBF both in solution and in polymer networks even in the presence of oxygen.91,95
Diarylbibenzothiophenonyl (DABBT) is another dimer that undergoes homolytic cleavage of a C–C bond by light or mechanical activation into two arylbibenzothiophenonyl radicals as shown in Fig. 11e. It has an equilibrium bond length of 1.574 Å, a dissociation energy of 96 kJ mol−1,96 and a threshold force of 4.4 nN.33 These radicals display a broad absorbance peak around λ = 450 nm (Fig. 11f). These homodimers can be modified to adjust their dissociation energy and presumably also the threshold force, although the latter has not been demonstrated. For instance, when the phenyl rings are functionalized with a p-bromo substituent, the dissociation energy is lowered to 86 kJ mol−1.96
The DABBT mechanophore was first applied in a polymer material by Otsuka's group in 2018.97,98 A block copolymer was synthesized to contain soft domains with inbuilt DABBT as a damage sensor, and hard domains with another TASN mechanophore integrated within the polymer backbone. Due to the different colors of the two dissociated mechanophores, the color of polymers in response to mechanical stimulus enables discrimination between force concentration in the hard and in the soft phase, which could be actuated by stretching and grinding, respectively. However, recent research focusing on DABBT as a mechanophore is rare. Application of radicals generated from DABBT has been limited to mechanochromic force detection rather than other possibilities unlocked using these stable yet active radicals. Possible further application could lie in the initiation of a local chemical reaction in a network; for instance, a polymerization to strengthen the material.99 For now, the unique color change from light yellow to green in mechanochemistry possesses high potential for mechanochromic applications when, for instance, multiple independent color changes are required.
When two mechanoradical dimers were separately integrated into the backbone of polymer chains,92,98,101,102 the materials showed both thermal and stress sensitivity. They exhibited color change before and after extension or grinding due to the dissociation of dimers (Fig. 11b and d). The color change is different due to the different absorption of the two radicals after dissociation. Materials labelled with DABBF show a color switch from colorless to blue (Fig. 11b),92 and the color of materials with TASN changes from colorless to pink (Fig. 11d).98 The threshold force of DABBF is lower than that of TASN; and thus, DABBF is activated by force before TASN. The dissociation of DABBF in various polymer architectures was investigated by Otsuka's group102 who found more effective activation of mechanophores in longer polymer chains. Additionally, it was found that these mechanophores are more sensitive to mechanical stress in star polymers than linear polymers in bulk materials.102 Further details are presented in Section 3.
Carbon-centered radicals have also been formed by scission of two carbon–heteroatom bonds in diazo-functionalized polymers. A well-known thermal initiator for radical reactions, 4,4′-azobis(4-cyanovaleric acid), was coupled to polymers and found to be mechanically-active.103 Nitrogen gas was liberated upon sonication and two stabilized radicals were formed. A threshold force of 3.7 nN was calculated using CoGEF.33
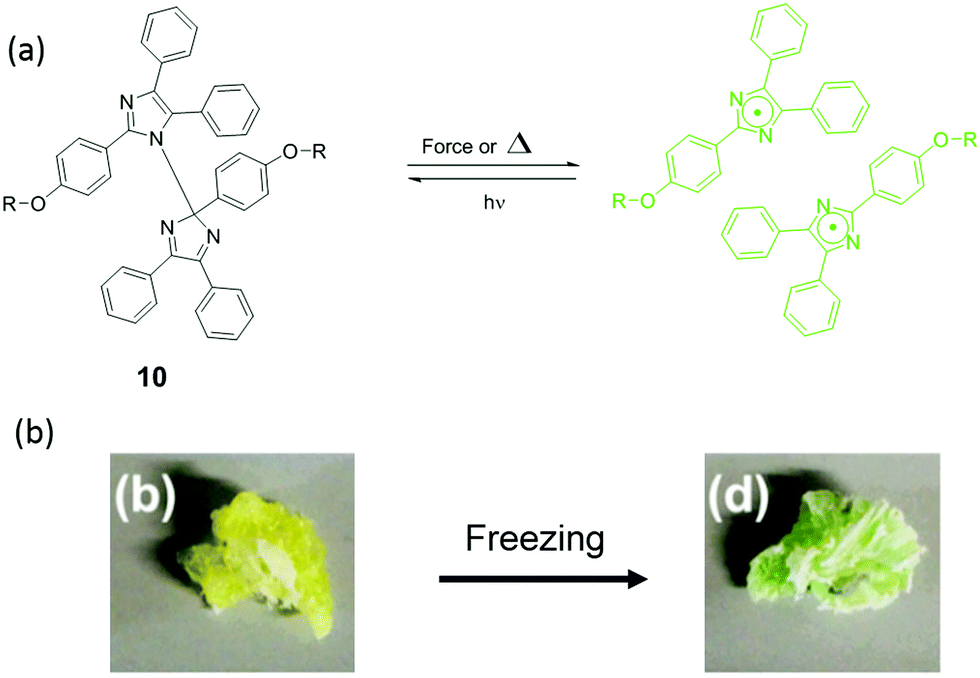 | ||
| Fig. 12 (a) Mechanism of HABI activation by force or heat (b) HABI was incorporated into poly(urethane), and the materials showed color change after freezing.104 (b) is adapted with permission from the authors of ref. 104. | ||
Weakly polarized bonds involving heteroatoms often break homolytically without requiring extensive conjugation to stabilize the formed radical. In the group of chalcogenides, S–S and Se–Se bonds are mechanically-active and have been incorporated into polymer materials. Diselenide bonds in polystyrene chains were activated by sonication and allowed for selective cleavage of the polymers.106 In addition, the radicals that were formed reacted with a diselenide bond of an added small molecule in a metathesis reaction. Mechanically-induced diselenide metathesis reactions were also used in force-responsive amphiphilic vesicles, that responded to an increase in osmotic pressure.107 The formation of selenide radicals has so far been primarily followed by its characteristic reactivity, and because of this diselenides may become important mechanophores for use in responsive materials.
Disulfide bonds are more established mechanophores and have been more thoroughly characterized and applied in polymers. In the presence of nucleophiles disulfides may react in a force-dependent SN2 reaction, in which no radicals are formed.108 At higher forces the reaction proceeds with a more radical-like character – partially due to the higher energy, and partially due to conformational changes resulting in steric hindrance.109,110 For homolytic cleavage, a threshold force of 3.6 nN was calculated for alkyl-substituted disulfides.33 The formation of radicals can be established using spin traps, allowing for a colorimetric response.111 Different responses to mechanical liberation of thiol radicals or anions allow for applications other than sensing. For instance, recently, Göstl and Herrmann et al. visualized the mechanical cleavage of disulfides with a secondary reaction that could also be potentially useful for controlled drug release applications.112 In this work, mechanically generated thiols reacted with a Michael acceptor that underwent a retro-Diels–Alder reaction to liberate a furan derivative. Upon re-aromatization, this furan derivative spontaneously dissociated to release a small molecule, in this case a fluorescent dye or a prodrug.
2.5 Mechanocatalysts
There is enormous potential for application of latent catalysts that become active under mechanical force, particularly in autonomous self-healing materials. However, only a small number of mechanically-activated catalysts have been reported, most of which are metal–ligand coordination complexes. When a coordination complex with latent catalytic activity is incorporated in a polymer backbone, extensional forces preferentially break the weak metal–ligand bond, and the newly formed free ligand or the coordinatively unsaturated metal may catalyze a reaction. The dual possibilities for catalyst activation are illustrated with two examples of mechanocatalysts based on transition metal–N-heterocyclic carbene (NHC) complexes. After mechanical activation of silver(I)–N-heterocyclic carbenes (Ag–NHCs) by ultrasound,113,114 the nucleophilic free carbene is an active catalyst for the transesterification of vinyl acetate as shown in Fig. 13a. In contrast to this, in the Ru(II)–NHC complex, (Fig. 13b) the metal is the active center of an olefin metathesis catalyst that is formed after removal of a sterically-demanding carbene ligand from the metal.114 Another type of mechanocatalyst is based on Cu(I)–NHCs.115,116 These complexes were integrated into a variety of polymer chains (polytetrahydrofuran, polystyrene, polyisobutylene) by Binder et al.116 After activation by ultrasound, the Cu(I) metal center becomes an active catalyst in the CuAAC “click” reaction of benzyl azide and phenylacetylene (Fig. 13c).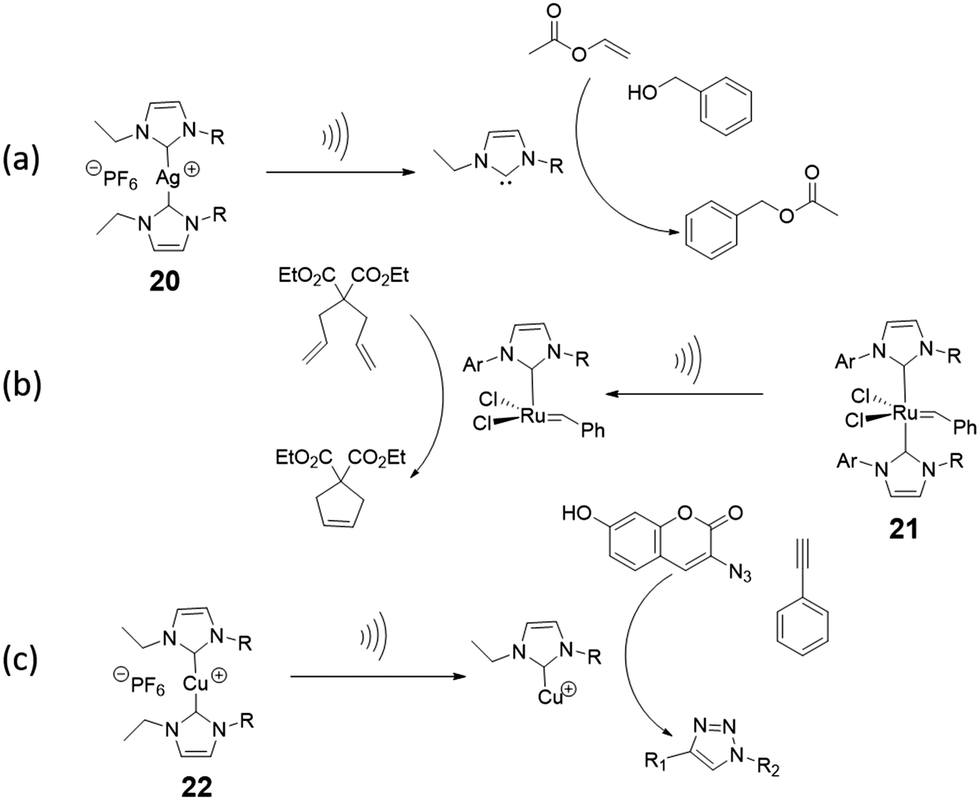 | ||
| Fig. 13 Mechanocatalysts (a) Ag(I)–NHC incorporated in poly(tetrahydrofuran) releases a N-heterocyclic carbene upon activation by ultrasound in solution, which catalyzes transesterification.114 (b) Activation of Ru(II)-NHCs incorporated in poly(tetrahydrofuran) produces a Ru(II) metal centre that catalyzes alkene metathesis reactions.114 (c) Mechanical activation of Cu(I)–NHC in a polymer leads to the formation of a Cu(I) metal centre that catalyzes an azide–acetylene cycloaddition (“click” reaction).116 | ||
2.6 Release of small molecules and structural rearrangement
Some mechanochemically responsive polymer materials release small molecules upon mechanical activation. After activation, the mechanophores in these materials undergo structural rearrangement or retro-Diels–Alder reaction and release small molecules. Exploration of such mechanophores is still in its early stages. Currently, literature on the release of small molecules includes HCl,117,118 THF,119,120 phenyltriazolinedione,44 iron ion,121 and coumarin.122 These small molecules were released from polymer materials by various modes of activation. For instance, sonication of the mechanophore shown in Fig. 14a results in release of HCl while phenyltriazolinedione was released via the initiation of a retro-Diels–Alder reaction in a polymer material under compression. The earliest example of a small molecule release involved a oxanorbornadiene mechanophore incorporated in a polyurethane (Fig. 14b).119 This polymer released small amounts of THF upon repeated compression. Similarly, Robb et al.122 integrated a furan-maleimide Diels–Alder adduct in PMA with a cargo molecule (X, in compound 27) that was covalently bound to the furan. When the mechanophore was mechanically-activated, the cargo molecule was released (Fig. 14e) This modular approach to the release of cargo molecules of choice provides a platform for drug delivery, self-healing, depolymerization and other applications.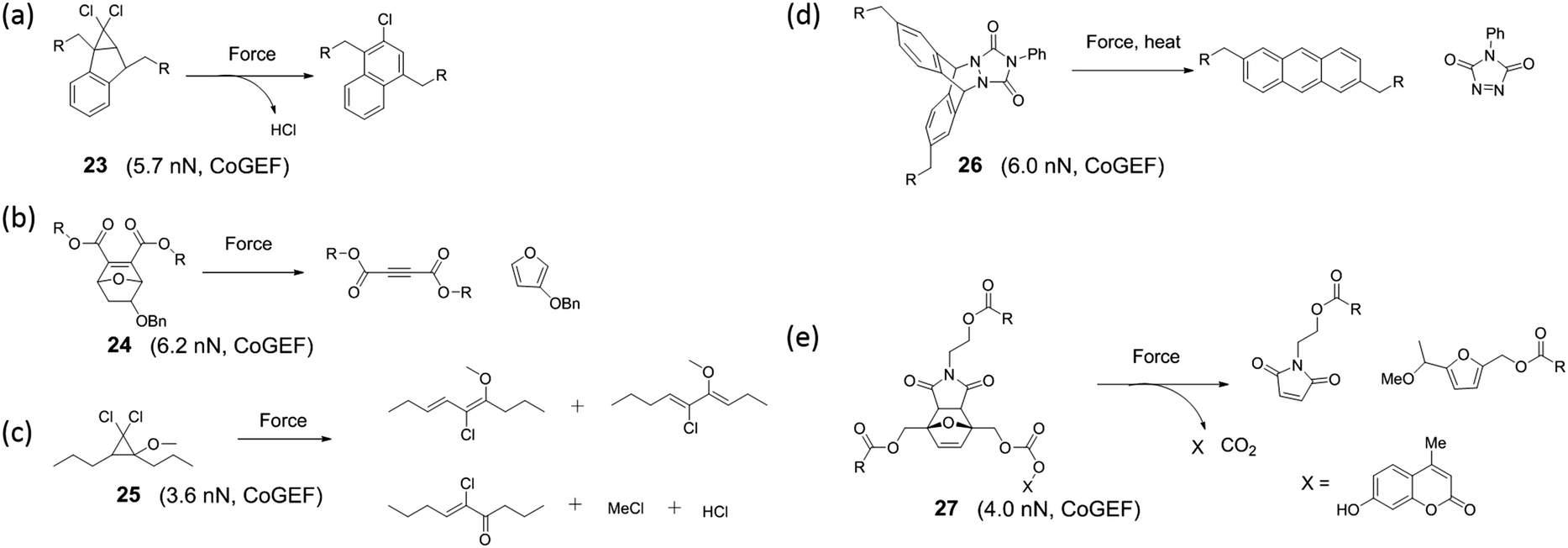 | ||
| Fig. 14 Mechanophores releasing small molecules upon activation. | ||
Mechanophores derived from cyclopropane, cyclobutene, or epoxides that undergo bond rearrangement in response to a mechanical stimulus have been shown to dramatically elevate mechanical properties or conductivity of polymer materials. Mechanical activation leads to electrocyclic ring opening and rearrangement of the structure. Upon rearrangement, cyclopropane and cyclobutene mechanophores produce double bonds in the polymer backbone (Fig. 15). For instance, gem-dihalocyclopropane mechanophores, reported by Craig's group,123–127 generate a 2,3-dihaloalkene sequence after activation as shown in Fig. 15a. Threshold forces for activation of gem-dihalocyclopropanes have been calculated by CoGEF,33 and the results are consistent with measurements by single-molecular force spectroscopy techniques. While the mechanochemical reaction does not give an optical signal or catalysis, it generates a double bond. The double bond generated upon mechanochemical ring opening of this type of mechanophore is a potential crosslinker and this feature has been used to strengthen a polymer with a mechanical stimulus.128 Epoxide mechanophores generate carbonyl ylides upon ring opening, which facilitates force-induced cross-linking by reaction with an alcohol (Fig. 15a).128 A different strategy offering high potential to strengthen polymer materials was reported by Weng's group.129 Multiple macrocyclic cinnamate dimers were integrated into a polyester. Stretching a polymer of the dimers above a critical force of 1–2 nN more than doubled its contour length and increased the strain energy that the chain absorbed before fragmenting by at least 2500kJ per mole of monomer (Fig. 15b).
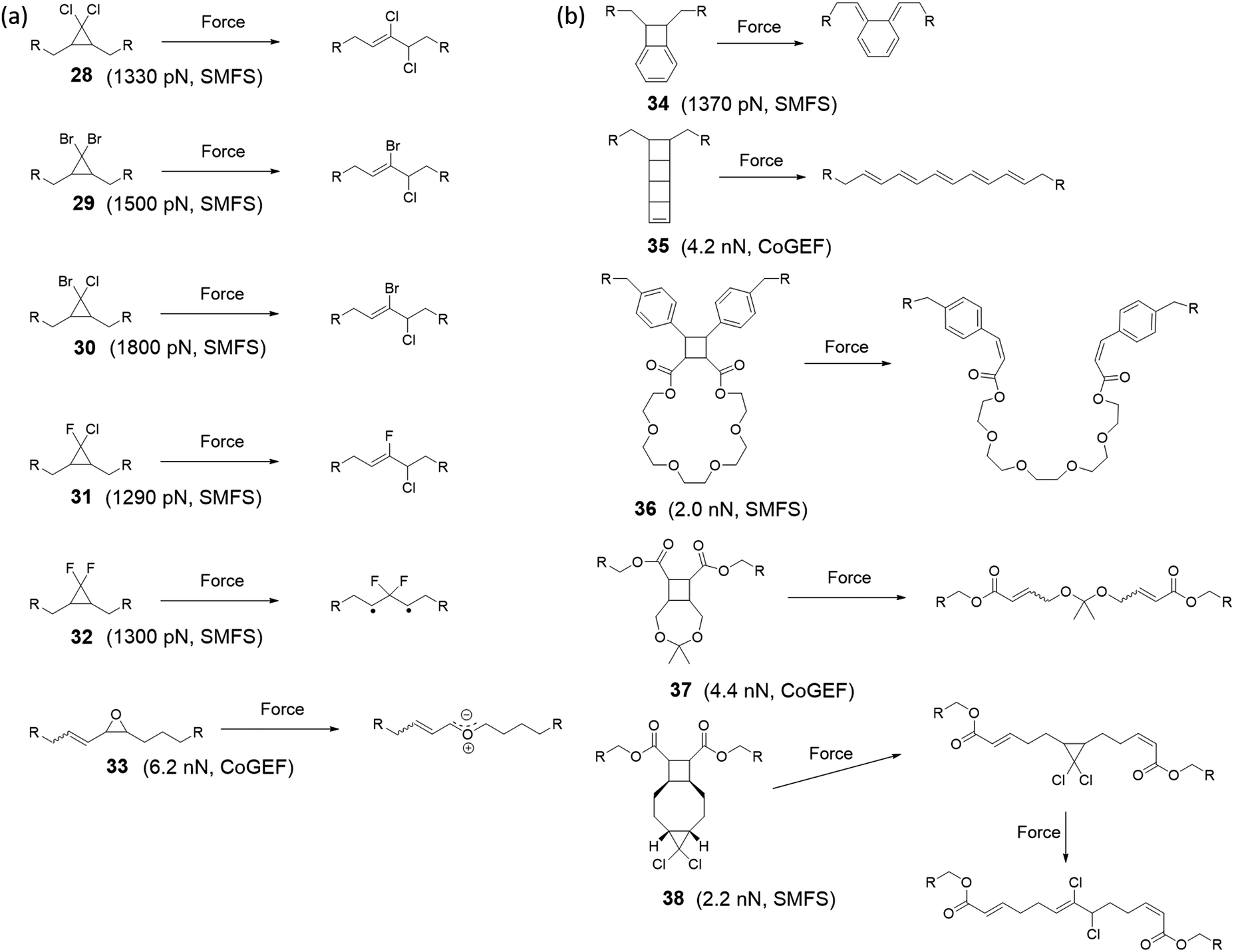 | ||
| Fig. 15 Mechanophores that rearrange upon mechanical activation. (a) Dihalocyclopropane and epoxide mechanophores rearranging via a 2π electrocyclic ring-opening reaction and corresponding threshold forces calculated with SMFS or CoGEF. (b) Cyclobutane mechanophores with their corresponding threshold force. | ||
A completely different type of functionality is unveiled in polyladderene mechanophores: soluble, non-conjugated polymers that convert to conjugated polyacetylenes upon mechanical activation. In seminal work from the group of Xia, a ladderene mechanophore consisting of four fused cyclobutene rings was used as monomer in a ring-opening metathesis polymerization to give a poly-ladderene. Upon sonication in solution, a conjugated polymer was formed (Fig. 15b), which self-assembled into semiconducting nanowires.130,131
3. Polymer materials employing mechanochemistry
Although a force-responsive molecule may be studied per se, its real usefulness arises when it is incorporated into a polymer chain, into a network, or more generally, into a material. Hence, many synthetic approaches to incorporate a mechanosensitive moiety in polymer materials have been developed.132–134 The following sections aim to exhaustively present several synthesis methods that can be used to incorporate various mechanoresponsive molecules into polymeric architectures.3.1 How to incorporate mechanophores in polymers?
In order to be useful as mechanoresponsive molecules, the mechanophores presented in Section 2 need to be coupled at both ends. They can be incorporated either in the backbone of the polymer itself as an initiator or a monomer (or oligomer) unit or in a network as a crosslinker or coupling agent. We will first review reported methods to incorporate mechanophores in the polymer chain. A summary of the different methods to incorporate mechanophores in polymers is presented in Table 1.3.1.1.1 Chain-growth polymerization process. For polymers synthesized by chain-growth polymerization, the insertion of a mechanosensitive moiety in the backbone requires the use of controlled/living polymerization techniques in solution, employing the mechanophore as an initiator.136 With this strategy, well-defined polymers with controlled molar masses and low dispersities can be synthesized and the mechanophore can be incorporated either at the chain-end (for subsequent coupling), near the middle of the chain, or even at the junction between two polymer blocks.133,136
The most described system is based on PMA.35,56,62,63,67,84,137–142 Following a suitable functionalization, the mechanophore is covalently incorporated in the polymer as an initiator using controlled radical polymerization techniques (Fig. 16). The group of Moore described the incorporation of SP and benzocyclobutene (BCB) in PMA for the first time.35,138 In this study, an α-bromo-α-methylpropionyloxy bifunctionalized SP or BCB was used to initiate the single electron transfer living radical polymerization (SET-LRP) of the methyl acrylate (MA) monomer to produce linear PMA polymers containing a SP or BCB moiety near its chain midpoint, respectively (Fig. 16). The study showed that the molar mass could be easily tuned (from 18 kDa to 287 kDa) while maintaining a low dispersity (PDI ≤ 1.3). Similarly, other mechanosensitive molecules were incorporated in the center of PMA polymer chains; namely, azobenzene,139 maleimide-anthracene Diels–Alder adduct,56,62,63,142 coumarin,67 1,2-dioxetane,84 and platinum–acetylide complex.141 This technique (Fig. 16) is not limited to the preparation of mechanophore-containing PMA. Several studies reported the use of similar procedures to incorporate mechanosensitive molecules in other linear polymers chains: poly(methyl methacrylate) (PMMA),143 and other polyacrylates,144 – including poly(ethyl acrylate) (PEA), poly(n-butyl acrylate) (PnBA), poly(iso-butyl acrylate) (PiBA) and poly(tert-butyl acrylate) (PtBA) and polystyrenes (PS).145
 | ||
| Fig. 16 General procedure for the preparation of mechanophore-linked PMA based on SET-LRP route using α-bromo-α-methylpropionyloxy bifunctionalized mechanophore as an initiator. | ||
Similarly, using the mechanophore as an initiator in a ring opening polymerization (ROP) process resulted in a mechanoresponsive polyester.146,147 As an example, O’Bryan et al.146 reported the use of indolinospiropyran diol as an initiator in ROP of ε-caprolactone leading to photo- and mechanochromic polymers. Later, Peterson et al.147 extended the preparation methods of SP-containing poly(ε-caprolactone) (PCL) to 3D printing techniques. This technology allowed, in particular, the preparation of multicomponent materials with spatially-varying mechanoresponsive properties (Fig. 17).
 | ||
| Fig. 17 Preparation of a 3D-printed multicomponent mechanoresponsive sample: (a) CAD representation, (b) Pre-elongation sample, (c) Post-elongation sample, (d) Post-elongation sample after 365 nm UV irradiation. Scale bars = 20 nm.147Fig. 17 is reprinted with permission from ref. 52, Copyright 2015 American Chemical Society. | ||
3.1.1.2 Step-growth polymerization process. Another class of polymers that are well studied in polymer mechanochemistry are poly(urethanes) (PU), because of their highly-tunable properties.43,50,74,92,104,148–160 Their synthesis is based on a step-growth polymerization process. Since the first report by Rubner, who described the preparation and the investigation of the thermo- and mechanochromic properties of polydiacetylene-containing PU,155,156 numerous systems based on mechanoresponsive thermoplastic PU have been reported. As an example, Kim and Reneker prepared segmented PU made of a copolyamide block with an azobenzene moiety in the soft segment.152 The p,p′-diaminoazobenzene mechanosensitive moiety was first reacted with 1,6-hexanediamine, succinyl chloride and glutaryl dichloride to obtain a copolyamide, which was then incorporated in small quantities (about 0.1 wt% of total weight) in the PU during the polymerization. More generally, the incorporation of mechanophores in a PU-based system is achieved by step growth polymerization, and results in multiple mechanosensitive moieties that are distributed throughout the backbone. Prior to the step-growth polymerization process, the mechanophore needs to be functionalized with one of the reactive functions involved in the polymerization.43,50,74,92,148–160 Using this strategy, Lee et al.154 described the first incorporation of SP in a PU by functionalizing the mechanophore with isocyanate moieties through the reaction of dihydroxyspiropyran with an excess of methylene diphenyldiisocyanate. The subsequent stoichiometric reaction with an hydroxyl-terminated poly(tetramethylene glycol) and a chain-extender (Fig. 18) resulted in a PU of 50–70 kDa. The versatile nature of the preparation method of mechanoresponsive PU gives access to tunable material structures and properties: preparation of segmented PU and variation of the mechanophore location – either in the soft or the hard segment without modifying the mechanical properties of the material,153 addition of supramolecular units,74,149,150 incorporation of the mechanophore in a dual (i.e. physically and chemically) cross-linked network,159 or into waterborne PU,160 design of self-healing materials,151 or stress-responsive polymers based on mechanical-force-triggered cross-linking.43 Regardless of the targeted materials properties The synthesis strategy is generally the same: the hydroxyl functions of the mechanophore are completely reacted with an excess of the suitable isocyanate, and then the functional mechanophore is incorporated into the PU chain during the step-growth polymerization.
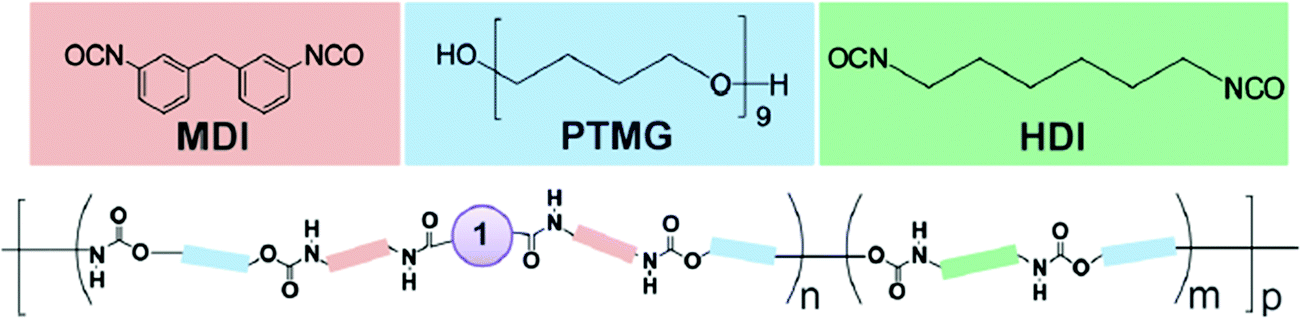 | ||
| Fig. 18 Chemical structure unsegmented PU containing multiple SP (1) units in its chain.154Fig. 18 is reprinted with permission from ref. 154, Copyright 2010 American Chemical Society. | ||
Many mechanosensitive molecules – including azobenzene,152 SP,149–151,153,154,159,160 1,2-dioxetane,148 DABBF,92 rhodamine,50 STP,43 and HABI104 – have been incorporated in PU chains by using the same chemistry (i.e. functionalization of a diol-terminated mechanophore with functions followed by step-growth polymerization), independently of the mechanophore responsiveness.
Interestingly, using a similar synthetic approach, i.e. step growth polymerization, mechanophores were incorporated into other thermoplastic polymers such as polyamides (PA) and polyesters (PES) by polyamidation or polyesterification, respectively, starting from the diol-functional mechanophore.43,70,148 Chen and Sijbesma reported the straightforward synthesis of PU, PES and PA with a tunable amount of the mechanoluminescent bis(adamantyl)dioxetane, starting from the same dihydroxyl-functionalized mechanophore precursor. The subsequent reaction of the latter with an excess of suberic acid and hydroxyl-terminated poly(tetramethylene glycol) or bis(3-aminopropyl)-terminated poly(tetrahydrofuran) lead to a mechanoresponsive PES or PA, respectively (Fig. 19).148
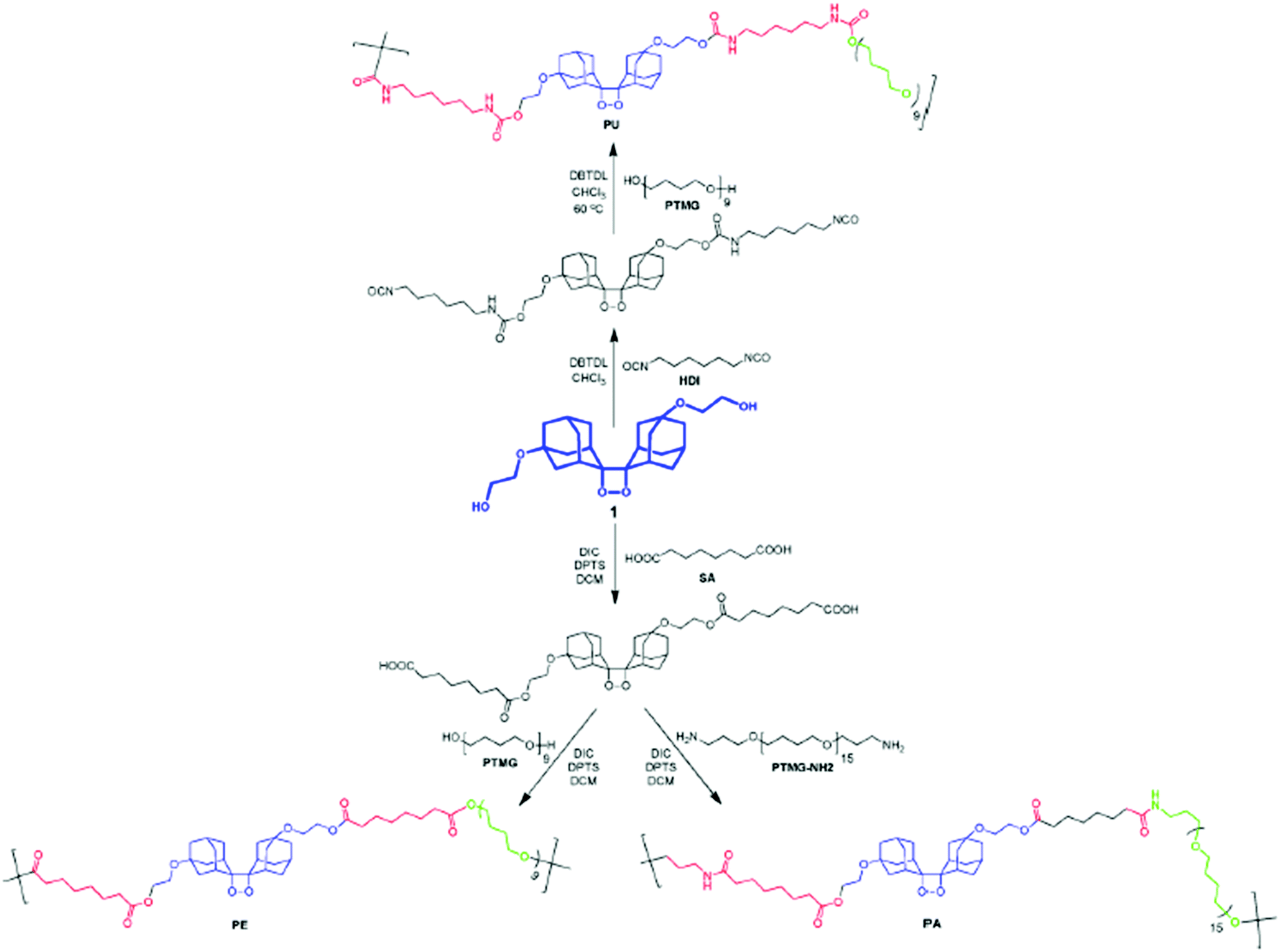 | ||
| Fig. 19 Synthetic scheme for SP-containing thermoplastic elastomers.148Fig. 19 is reprinted with permission from ref. 148, Copyright 2014 American Chemical Society. | ||
3.1.1.3 Coupling. Some studies report the incorporation of mechanophores in the polymer structure; not directly during the polymerization process but, rather, in a post-modification step.58,89,98,102,104,113,115,161–166 In Moore's group,162 benzocyclobutene (BCB) was incorporated into poly(ethylene glycol) (PEG) chains by coupling a difunctional carboxylic acid-terminated BCB with two end-functionalized α-methoxy-ω-amino-PEG resulting in mechanoresponsive PEG of various molar masses (Mw = 4 to 60 kDa) with one BCB moiety in the center of each chain. Alternatively, controlled/living radical polymerization techniques can be used to synthesize well-defined polymer chains bearing a reactive terminal group, which can then be used to incorporate the mechanophore in the center of the chain by employing well-known chemical strategies. This end-functionalization and coupling method opens the way to more complex, efficient, and sophisticated architectures. Verstraeten et al.104 used this strategy to incorporate highly reactive HABI in PMA chains by a simple oxidative coupling of triphenylimidazolyl-terminated PMA, prepared by SET-LRP, in presence of PbO2. Li et al.58,165 described the design of mechanoactive block copolymer micelles putting a maleimide–anthracene-Diels–Alder adduct at the junction of the hydrophilic block (poly(acrylic acid) (PAA)) and the hydrophobic block (PnBA or PS). Each polymer block was synthesized by controlled/living radical polymerization techniques with either a maleimide- or anthracene-functionalized initiator, and then coupled through a Diels–Alder reaction to obtain a diblock copolymer with a Diels–Alder adduct at the junction between blocks (Fig. 20a). By combining two polymerization steps, mechanophores were incorporated into other block copolymer architectures such as ABA triblock copolymers,167,168 or graft copolymers.163 Similarly, Oka et al.102 used controlled/living radical polymerization techniques to synthesize various azide-terminated PS which were then coupled to a DABBF-dialkyne derivative to obtain polymers with tunable architectures: linear, four-and eight-arm star PS. For systems based on metal-N-heterocyclic (NHC) complexes,89,113,115,161,166 a post-modification strategy is generally also required: the polymer synthesis (through cationic ROP, atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT), etc.) is followed by an ion exchange step and metal complexation (Fig. 20b).
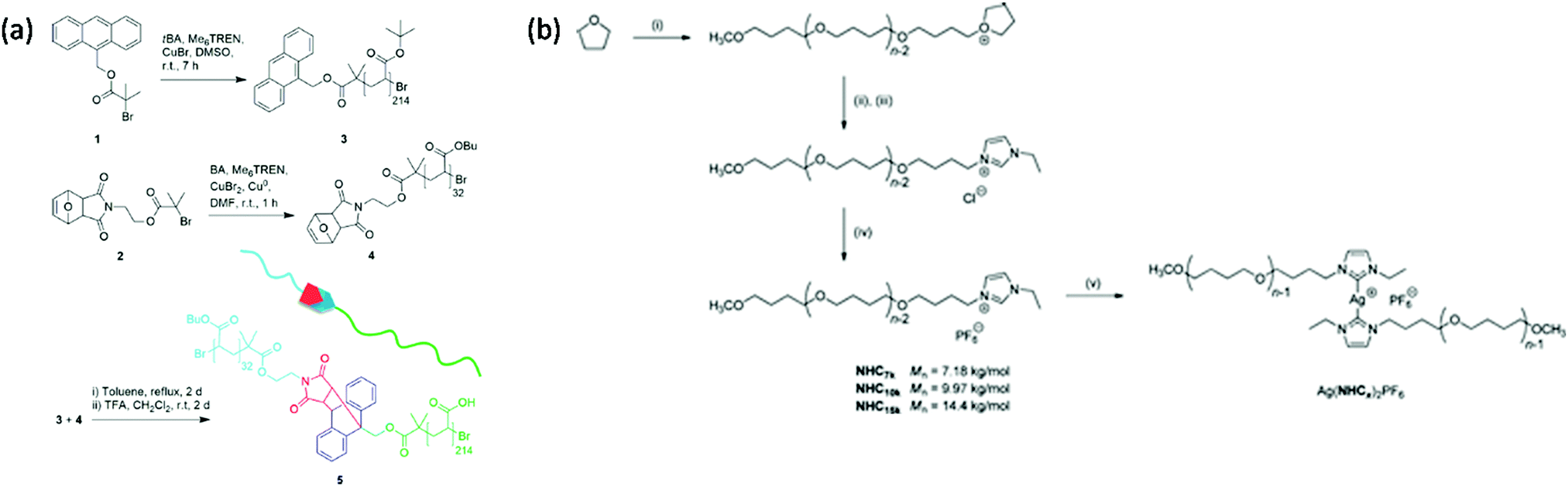 | ||
| Fig. 20 Synthetic route for the preparation of (a) PnBA-b-PAA containing a maleimide–anthracene Diels–Alder adduct mechanophore at the block junction58 and (b) Ag–NHC polymer complexes.113 (a) is reprinted with permission from ref. 58, Copyright 2016 American Chemical Society. (b) is reprinted with permission from ref. 113, Copyright 2011 American Chemical Society. | ||
3.1.1.4 High mechanophore content systems. Most of the described systems are based on incorporating a single mechanophore per chain or a low concentration of mechanophores (≤1 mol% relative to the monomer) into the chain or architecture, which may limit their use in stress-responsive applications in particular for the quantification of mechanically-induced damage. In this context, the interest for polymer chains containing multiple mechanophores, or for more complex polymer architectures, has grown.142,169 Preparation methods are quite similar to those described for structures containing a dilute mechanophore content: controlled/living polymerization techniques, post-modification steps, polycondensation, ring-opening metathesis, etc. In order to investigate the mechanical strength of scissile bonds, Craig's group designed multimechanophore-containing polybutadiene polymers:123,170 controlled contents of gem-dichlorocyclopropane (gDCC) units and weak bonds were incorporated into the backbone of the polymer using a ring-opening metathesis polymerization (ROMP) strategy. Very recently, they used a similar strategy to introduce a coumarin dimer mechanophore along the backbone of polybutadiene-based polymers.68 This new platform was then incorporated into silica-filled styrene-butadiene rubber using usual preparation methods for the synthesis of mechanophore-containing polymer nanocomposites (i.e. curing strategy, see Section 3.1.2.4 for details)
The same group reported a straightforward method based on RAFT polymerization for the preparation of a poly-mechanophore polymer system.171 Various copolymers made from cyclobutene carboxylates (CBCs) and nBA were synthesized with Mn varying from 8 to 127 kDa while maintaining a relatively low molecular weight dispersity (Đ ≤ 1.7).
3.1.2.1 Free radical polymerization. In some systems, the mechanophore is covalently incorporated into a polymer network. In this case, the typical strategy is based on the use of the mechanophore as a crosslinker. In 2009, the group of Moore incorporated SP in PMMA by bi-functionalizing the mechanophore with methacrylate moieties and copolymerizing it with the monomer in a free radical aqueous suspension polymerization process to obtain SP crosslinked PMMA beads.35 Thereafter, numerous studies on the introduction of SP in PMMA were reported,172–178 and nowadays, the incorporation of mechanophores in poly(meth-)acrylate networks is well-mastered.52,84,86,142,172–181 The network, containing generally 1 mol% of crosslinker relative to the monomer amount, can be made of either the mechanophore as unique crosslinker or with two different crosslinkers. In this latter case, the network architecture is designed by the primary cross-linker and only a small amount (0.018 mol% to 0.05 mol% relative to the monomer content) of mechanophore is introduced in the network.172–179 As illustrated in Fig. 21, the preparation method of the mechanophore-containing poly(meth-)acrylates networks is straightforward and highly versatile: the architecture and properties of the network are easily tuned by changing the nature,177 or quantity178 of the primary crosslinker; and the polymerization is generally performed in the bulk, directly in the mold. The method was also adapted to solution,52 and emulsion conditions.182 Apart from an inert atmosphere (for free radical polymerization) and good mixing of the reactants, no particular synthetic conditions are required. Using this strategy, and provided that the reactivity of the crosslinkers and monomers are not too different, a statistical incorporation of the mechanophore is expected and the mechanophore should be thus homogeneously dispersed in the network. The comparison of the mechanical properties of networks with and without mechanophores generally provides an indirect proof of the successful incorporation of the mechanophore in the network, without significantly impacting its material properties.86,175,177 To the best of our knowledge there is not yet a study investigating the kinetic aspects of the synthesis. It is well-known that free radical copolymerization of multifunctional monomers can result in different polymerization mechanisms; namely linear chain growth, cyclization as well as intra- and inter-crosslinking processes.183,184 Numerous parameters are involved in the kinetic control of these reactions:183–186 the initiator, the solvent, the monomer, the crosslinker, etc. The reactivity ratio is clearly specific to each monomer/crosslinker system and one can thus imagine that the nature and the amount of mechanophore crosslinker being used may impact the final labelled material. Unlike polymer solution or melts, which are at thermodynamic equilibrium, networks are frozen in the configuration obtained from the synthesis process.
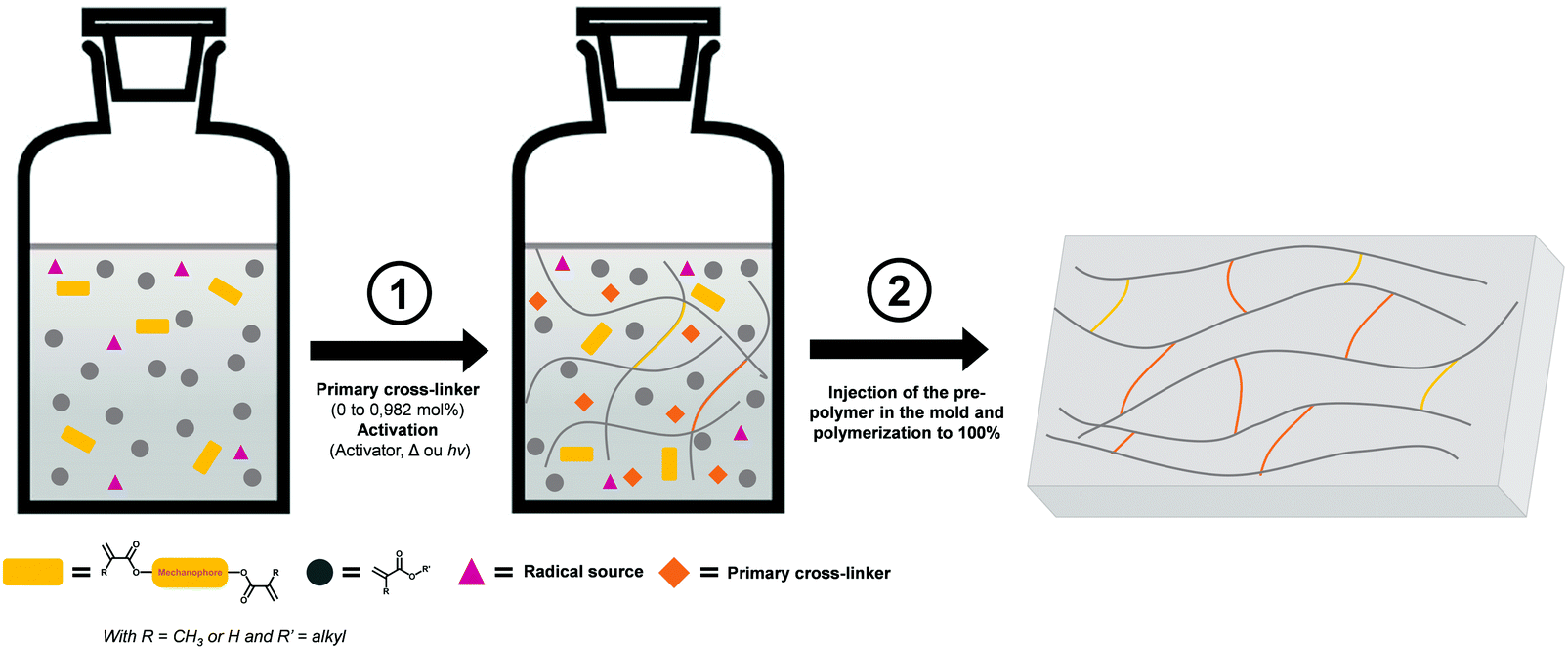 | ||
| Fig. 21 Illustration of the incorporation of mechanophores into a polymer network through a bulk radical polymerization process, directly in the mold (1 mol% of cross-linker). Notes: without primary crosslinker, the crosslinks are purely composed of mechanophore crosslinkers (in yellow); the procedure can be performed without the pre-polymer step via the direct incorporation of all the reactants in the mold as described by Clough and coworkers.179 | ||
This preparation method was extended to the preparation of other polymeric networks. In particular, it was applied to polyacrylamides: recently, Kabb et al.57 reported the incorporation of maleimide–anthracene Diels–Alder adducts in a poly(N,N-dimethylacrylamide) (PDMAc) network by copolymerizing DMAc and the mechanophore crosslinker by bulk free radical photopolymerization. The network properties were tuned by adding di(ethylene glycol) diacrylate (DEGDA) as primary crosslinker to vary the crosslink density.57 Similarly, Stratigaki et al.187 incorporated Diels–Alder adducts of π-extended anthracenes into poly(N-isopropyl acrylamide) (PNIPAAm) hydrogel networks to study the bond scissions in the network using confocal laser scanning microscopy.
Not only is the chemical nature of the network tunable, but also its architecture. More complex structures were achieved through sequential free radical polymerization steps: the multiple networks elastomers,52,86 which are known to be particularly tough due to the presence of sacrificial bonds. To investigate the reinforcement mechanism based on these sacrificial bonds, Ducrot et al.86 incorporated a bis(adamantyl)-1,2-dioxetane bisacrylate crosslinker into a single network, and in the first network of double and triple networks made of EA.
3.1.2.2 Hydrosilylation reaction. Because of its easy room temperature crosslinking or end-linking chemistry in the bulk and easy availability, poly(dimethylsiloxane) (PDMS) networks are among the most studied elastomers in research labs. As an example, Holder's group reported the incorporation of pyrene (Py) units in PDMS using two different strategies:188,189 Py-labelled precursors were prepared by the conjugation of allyl-functional Py with either PDMS oligomers, or triethoxysilane through hydrosilylation.189 Py-labelled PDMS was then obtained via the tin-catalyzed condensation of the precursor and hydroxyl-terminated PDMS, or via the platinum(Pt)-catalyzed hydrosilylation of the precursor and vinyl-terminated PDMS. In both cases, they showed that the incorporation of Py in the network did not impact the thermal and the mechanical properties of the elastomers.188,189
From a global point of view, the crosslinking process, generally via a Pt catalyzed hydrosilylation reaction, is the typical strategy to covalently and randomly incorporate mechanophores in silicone elastomers.44,48,158,190–196 Based on this strategy, Craig's group reported the introduction of SP in commercial PDMS:44,194–196 SP was functionalized with two alkene groups and covalently incorporated as a crosslinker in the network via the Pt catalyzed hydrosilylation reaction (Fig. 22).
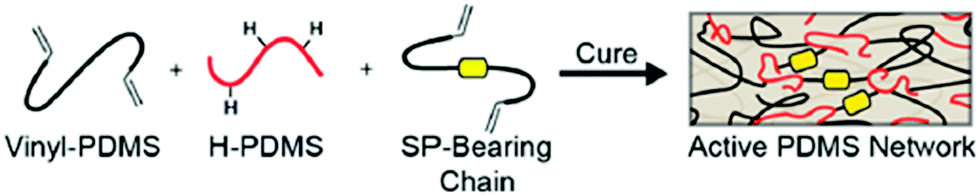 | ||
| Fig. 22 Schematic representation of SP-containing PDMS network: synthesis route.44Fig. 22 is reprinted with permission from ref. 44, Copyright 2014 American Chemical Society. | ||
Using a similar strategy, other mechanophores can be covalently incorporated in PDMS.48,190,191 As an example, to investigate the regiochemistry dependence of the mechanoresponsiveness, Robb et al.48 described the preparation of three naphthopyran regioisomers-containing PDMS networks: the mechanophore was first functionalized with 4-pentenoic anhydride, then mixed with the prepolymer solution and covalently incorporated during Pt cure hydrosilylation.
3.1.2.3 Other strategies. Other exotic strategies exist in parallel to the typical curing processes, as shown by Li et al.197 who prepared SP-doped PDMS films by mixing PDMS and 1,3,3-trimethylindolino-6-hydroxybenzopyrylospiran and pouring it onto fresh mulberry leaf. They studied in particular the effect of the SP mechanically-induced self-assembly on the film structure. Using a radical polymerization strategy, Kister and coworkers reported the preparation of a mechanoresponsive network made of SP, MMA and polybutadiene.198 Classic strategies used to incorporate mechanophores in the polymer chain were also used to incorporate the mechanophores as crosslinks in polymeric networks.199,200 For example, Yoshie et al.200 reported the preparation of a self-recovering network using maleimide–anthracene Diels–Alder adducts as crosslinks. In this work, the mechanophore is incorporated in the network by the Diels–Alder reaction of anthryl-telechelic poly(ethylene adipate) (PEAA2) and tris-maleimide (M3).
3.1.2.4 Polymer composites. Polymer composites, whether with particles or fibers, are ubiquitous in the engineering materials field due to their interesting combination of higher stiffness and/or better toughness. In particular, particle-filled polymer networks demonstrated a great enhancement of the toughness as well as a high mechanosensitivity, and were thus investigated with polymer mechanochemistry.172,173,190,201–209 One can distinguish two types of filled polymer networks: either a soft matrix reinforced with a hard filler, or a hard matrix toughened with soft filler. In both cases, the synthesis strategy is quite similar to the preparation of unfilled polymer networks, as described by Celestine and coworkers who reported the incorporation of SP in a toughened PMMA network.172,173 The crosslinked network was prepared classically by free radical polymerization, using SP as a secondary crosslinker; and the network was filled with rubber core–shell particles made of a butadiene-styrene core and a PMMA shell by simply dispersing the particles in the network. Clough et al.190 prepared mechanoluminescent silica-filled PDMS networks using a classic Pt catalyzed hydrosilylation reaction in presence of vinyl-functionalized silica nanoparticles.
In the field of mechanochemistry of polymer composites, epoxy-based thermoset networks are also well-known.201–206,209 In these systems, the fillers are mechanophore nanoparticles which bring mechanosensitive properties to the matrix rather than reinforcement. To incorporate a dimeric anthracene-based mechanophore in an epoxy matrix, Koo et al.203 mixed the carboxylic acid functional dimer particles with epoxy resin and the hardener. Then, the homogenized mixture was poured into a mold and allowed to cure at room temperature. This simple curing process (with possible adjustments) is typical for the preparation of such thermoset composites networks.
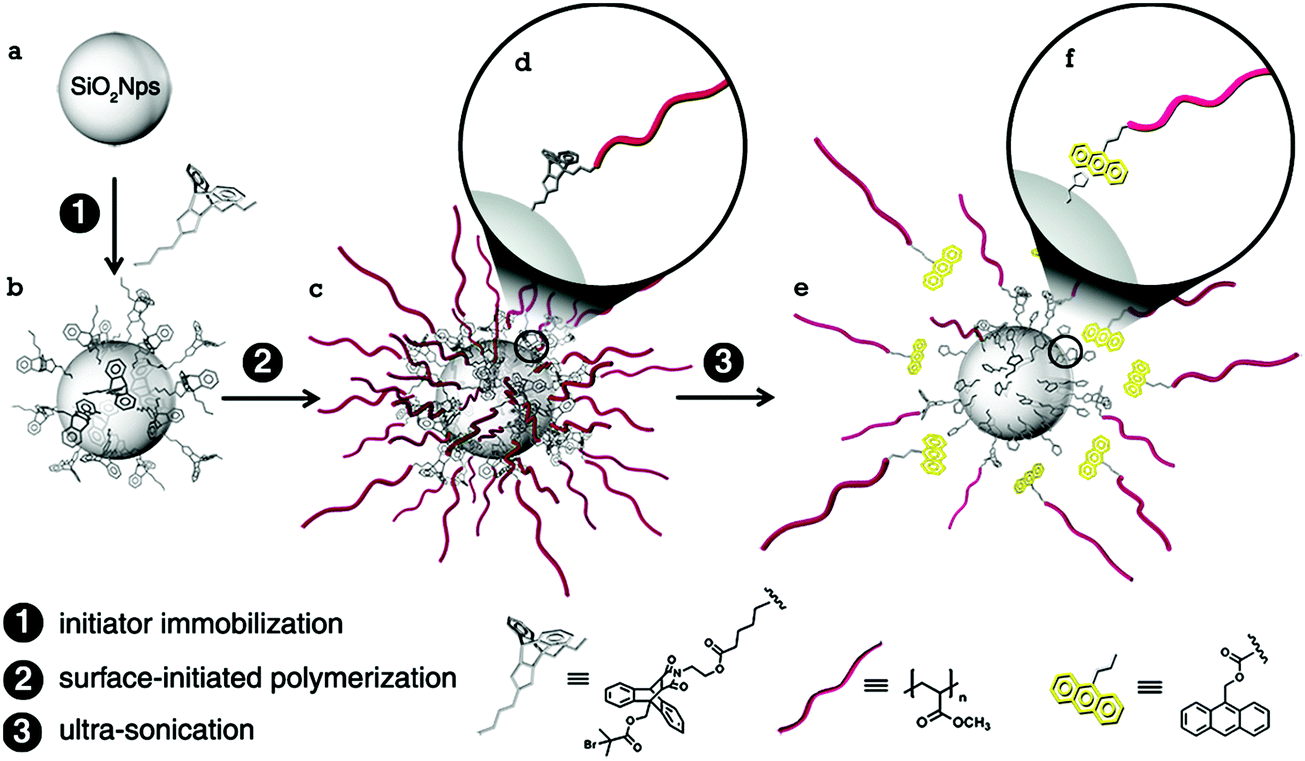 | ||
| Fig. 23 Illustration of the preparation method of maleimide–anthracene Diels–Alder adduct mechanophore-anchored PMA brush – grafted nanoparticles.59Fig. 23 is reprinted with permission from ref. 59, Copyright 2014 American Chemical Society. | ||
Other strategies were described to incorporate mechanophores at heterointerfaces and design new composite materials. To prepare highly mechanosensitive composites, Kosuge et al.211 incorporated DABBF in rigid networks using a sol–gel method. Sagara et al.212 proposed a straightforward strategy based on the covalent attachment of Py derivative micelles on glass beads, polymer beads, or living cells to prepare various mechanoluminescent materials. Woodcock et al.213 introduced a silk fiber in an epoxy matrix putting Rhodamine at the heterointerface.
| General characteristics | Polymers | ||
|---|---|---|---|
| Incorporation in the polymer chain | Chain-growth polym. | Process in solution | PMA,35,56,62,63,67,84,137–142 PMMA,143 PEA,144 PnBA,144 PiBA,144 PtBA,144 PS,145,167 copolymers,167,168,171 PCL,146,147 polybutadiene68,123,170 |
| Controlled/living polym. (SET-LRP, ATRP, ROP, ROMP, RAFT, etc.…) | |||
| Bifunctional mechanophore used as an initiator | |||
| Well-defined polymers with controlled molar masses (9–300 kDa), tunable architectures, low dispersities) | |||
| Step-growth polym. | Process in solution | PU,43,50,74,92,104,148–160 PES,43,70,148 PA148 | |
| Bifunctional mechanophore used as monomer (functionalization with OH, NCO or COOH) | |||
| Multiple mechanosensitive units throughout the backbone | |||
| Versatile method | |||
| Coupling | Synthesis of a functional polymers (generally using controlled/living polym.) | PEG,162 PMA,104 PS,98,102,164 copolymers,58,163,165 metal–NHC polymers complexes: PTHF;89,113,115,161,166 PS;89,113,115,161,166 PIB89,113,115,161,166 | |
| Post-polymerization coupling steps (amidification, oxidation, Diels–Alder reaction, etc.…) | |||
| Complex architectures | |||
| Incorporation in a network | Free radical polym. | Process in bulk, solution or dispersed medium | PMMA,35,172–179 PMA,84,181 PHMA,142 PEA,52,86,181 PDEA,180 PDMAc,57 PNIPAAm,187 copolymers,182 |
| Bifunctional mechanophore used as crosslinker (unique or secondary in a dual crosslinker system) | |||
| Straightforward synthetic conditions | |||
| Hydrosilylation react. | Bis-alkene functionalized mecanophore | PDMS44,48,158,188–196 | |
| Pt-catalyzed hydrosilylation | |||
| Typical strategy for the preparation of thermoset polymers | |||
| Other strategies | Tin-catalyzed condensation, curing in mulberry leaf, Diels–Alder reaction, curing, etc… | PDMS,189,197 PEAA2M3,200 Epoxy201–206,209 | |
| Incorporation at the interface | Surface funct. approach | Immobilization of the functional mechanophore on the surface | PMA59,61,64,210 |
| Growing of the polymer chain using the immobilized mechanophore as initiator | |||
| Other strategies | Sol–gel method, covalent attachment of dye micelles, etc.… | Rigid networks,211 PLA,212 Epoxy213 | |
3.2 Activation of the mechanophore in solution and in polymer materials
Based on this knowledge, the technique of ultrasonication has been widely employed in the field of polymer mechanochemistry to validate the activation of the molecule by force. In some particular cases, the method of activation is varied to be more suitable to the system; for example, the application of a vortex,212 and CO2-breathing activation.180 Depending on the chemistry of the mechanosensitive unit, various mechanical responses can be detected.
3.2.1.1 Type of activation. One can distinguish two main categories of mechanochemical response: the scission and the non-scission events. While in the second case the force generates isomerization reactions, in the first case, the force induces the cleavage of a covalent bond.215 In 2005, Moore's group introduced an azo moiety in a PEG chain and reported for the first time the site-specific bond cleavage of the polymer induced by ultrasound.103 They demonstrated in particular that the introduction of a weak bond in the polymer backbone favored the scission at that specific location resulting in a high degree of bond specificity that is revealed by a low dispersity of the polymer fragments.103 Similarly, the anthracene–maleimide Diels–Alder adduct,56,63 and coumarin,67 are scission-type mechanophores with a high site selectivity. They were incorporated into linear PMA chains, and the subsequent sonication of the polymer solutions revealed preferential bond cleavages due to cycloelimination reactions at the mechanophore location. Mechanophores based on scission reactions have also been well-studied for catalysis as reported by Groote et al.161 who investigated the mechanocatalytic activation of polymeric silver(I)–NHC complexes in solution for the transesterification of vinyl acetate with benzyl alcohol. Wei et al.141 reported the introduction of platinum–acetylide complex in bulk PMA and demonstrated its efficiency in the catalysis of olefin hydrosilylation reaction. Verstraeten et al.104 exploited the mechanical activation of HABI in PMA chains (which generates radicals) to study stress-induced crosslinking.
Scission reactions are generally not reversible (or hardly reversible), while isomerization reactions are generally fully reversible. This reversibility, and the generally better sensitivity to lower forces, is the reason why non-scissile-type mechanophores are used in polymer mechanochemistry. The best known and most efficient mechanophore based on an isomerization reaction is the SP. In 2007, Moore's group studied the ultrasound-induced ring-opening of SP in solution. Upon sonication, the colorless SP-centered PMA solution turned pink, revealing the SP-to-MC isomerization.138 The same group also reported the ultrasound-induced ring-opening reaction of BCB in PMA,138 and in PEG.162 The mechanically-induced ring-opening of STP was exploited by Zhang and coworkers, who incorporated the mechanophore in PES and reported load-induced addition reactions upon sonication of the solution in presence of N-ethylmaleimiede.43 The group of Craig used the non-scissile properties of gDCC mechanophore to quantify the strength of weak bonds.123,170 They designed a multimechanophore system in which gDCC and multiple scissile bonds were embedded in a polymer chain. Their approach relied on the competition between the ring-opening reaction of gDCC and the weak bonds scissions within the polymer chain triggered by ultrasound.
3.2.1.2 Role of intrinsic and extrinsic parameters on the activation. Besides the mechanophore chemistry (isomerization or bond scission reaction) and structure – which both play an important role62 – various parameters may influence the mechanoactivation. For individual polymer chains in solution, the main parameter to be considered is the polymer molar mass. A minimal chain length is required to allow force transduction to the mechanophore, and thus its mechanoactivation.103,138,141,161,162,167,216,217 The threshold value of molecular weight below which no activation is observed, generally estimated above 20 kDa, is dependent on the mechanophore structure (and on its activation force),217 and should be determined for each system. However, given the different published studies on the dependence of the degree of activation on polymer molecular weight a general tendency is clear: above the threshold value of molecular weight, the level of mechanoactivation increases with increasing molecular weight.103,138,141,161,162,167,216,217 This is an expected result, since the force on the covalent bonds depends on the relaxation time of the chain, which in turn varies with the square of the chain length in dilute solution.218 For flexible chains this force also depends on the position along the chain and is maximum in the center. The molecular weight dependence of the scission force has been investigated in detail by Nguyen and coworkers.218
Some studies suggested other important parameters to describe mechanical responsiveness; namely, the polydispersity index (PDI),67 and the degree of polymerization.144 Craig's group investigated the mechanoactivation of coumarin-centered PMA upon sonication and highlighted that higher PDI promotes “off-center” mechanophore structure leading to less selective bond scission.67 By comparing five SP-centered polyacrylates (namely, PMA, PEA, PnBA, PiBA and PtBA), Moore's group investigated the impact of the polymer composition, the side-chain nature, and the chain length on the mechanical response.144 Interestingly, they demonstrated that the mechanical transduction kinetics are governed by the degree of polymerization, i.e. the number of C–C bonds, rather than the molecular weight (Fig. 24).
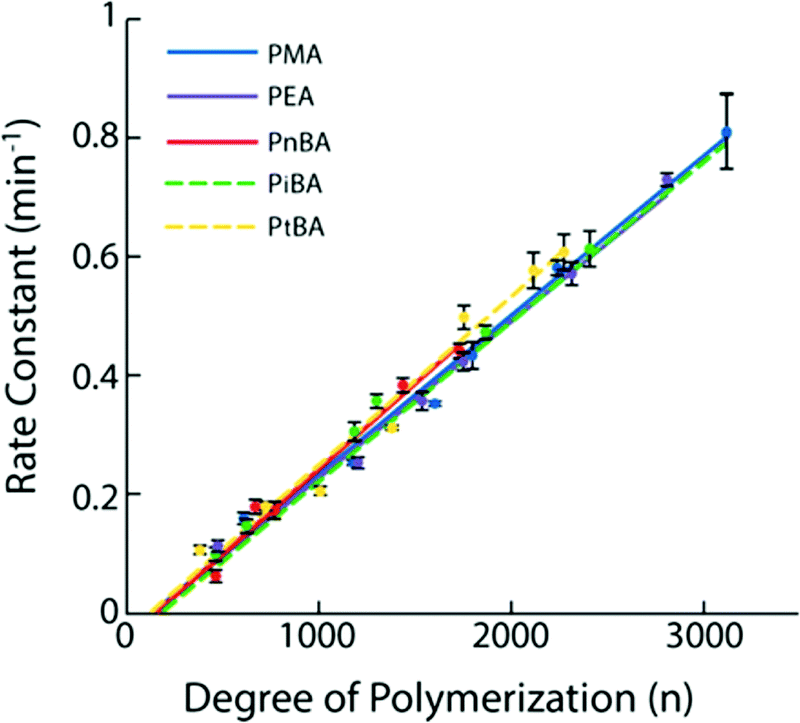 | ||
| Fig. 24 Rate constant of the SP activation as a function of degree of polymerization for various SP-containing polyacrylates.144Fig. 24 is reprinted with permission from ref. 144, Copyright 2016 American Chemical Society. | ||
The impact of the polymer architecture on the mechanical response was studied by Church et al.,56 who compared the bond scission mechanism of a linear PMA containing an anthracene–maleimide Diels–Alder adduct moiety and its three-arm counterpart (Fig. 25a). Both architectures demonstrated a high selectivity of the scission events; although, differences occurred in the chain scission rate. The study showed that the rate constant is defined by the number-average molar mass per arm rather than the global molar mass and the architecture. The specific case of the micellar structure was investigated by Wang et al.168 They demonstrated an increase by 5 of the reactivity of the SP-centered triblock copolymer made of tBA and NIPAAm. It self-assembled into micelles, in contrast to its linear counterpart (Fig. 25b). This enhancement was explained by the swelling of the micelle core and by the enhancement of the dielectric constant near SP units. Li et al.58 investigated the mechanical reactivity of the maleimide–anthracene Diels–Alder adduct mechanophores put at the interface between the hydrophobic core (PnBA) and the hydrophilic shell (PAA) of micelles. The study showed the scission of the centrally located mechanophore upon sonication when it was located at the interface between the hydrophobic core and hydrophilic shell in a micelle, but not when the polymer was in solution where the viscous drag is too low to transfer a sufficient force (Fig. 26).
 | ||
| Fig. 25 (a) Sonochemical activation of a linear PMA containing an anthracene–maleimide Diels–Alder adduct moiety and its three-arm counterpart;56 (b) UV absorbance vs. ultrasonication duration for SP-centered triblock copolymers made of tBA and NIPAAm, self-assembled into micelles and its linear counterpart.168 (a) is reprinted with permission from ref. 56, Copyright 2014 American Chemical Society. (b) is reprinted with permission from ref. 168, Copyright 2015 American Chemical Society. | ||
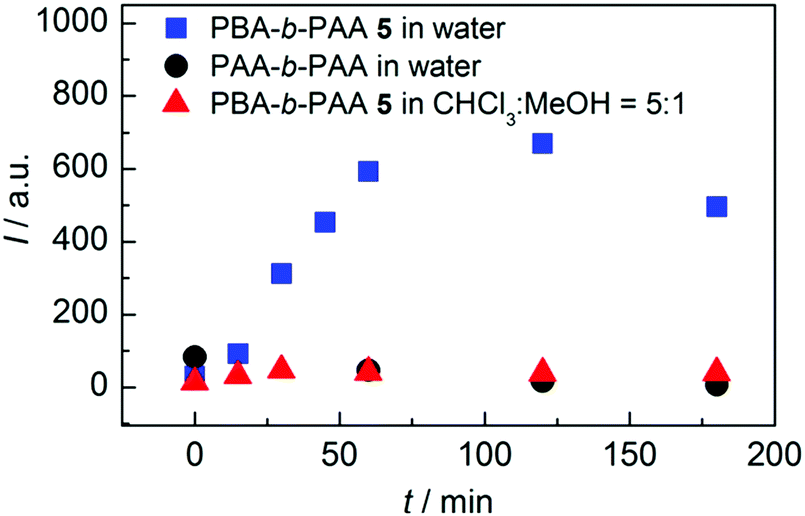 | ||
| Fig. 26 Evolution of the emission intensity at λem = 420 nm as a function of sonication time of PBA-b-PAA dispersed in water (square) or in solution (triangle) and PAA-b-PAA in aqueous solution (circle).58Fig. 26 is reprinted with permission from ref. 58, Copyright 2016 American Chemical Society. | ||
In addition to intrinsic parameters, external parameters can also be important for the mechanically induced response. In particular, the concentration of mechanophore is a key point, as shown by Groote and coworkers who demonstrated an increase of the mechanocatalyst activity with concentration.161 One can imagine that the process used for the activation is determining the mechanosensitive response; but interestingly, Surampudi et al.139 showed comparable isomerization conversion and polymer chain scission rate for an azobenzene-centered PMA exposed to continuous or pulsed sonication. By comparing the mechanochemical response of waterborne PU in solution, dispersion, and emulsion, Zhang and coworkers pointed out the role of the environment.160 While no activation was observed in emulsion, mechanoluminescence was detected in solution and dispersion with an optimal water content estimated at 15 v%. The lack of activation in emulsion was explained by the aggregation state of PU chains: the strong coiling of the chains hinders the force transduction. The key question is then how is the force applied to the chemical bond to trigger activation?
3.2.2.1 First proofs of concepts. There are myriad methods of activation and detection of mechanophores in materials.28 Due to its many advantages, the use of optical detection is generally favored. Indeed, optical detection methods are non-invasive, and the activation of the mechanophore in the material can be mapped within optical resolution. The use of optical detection techniques relies on the ability of the mechanosensitive molecule to change its optical properties when exposed to a force.134 The physicochemical mechanism involved in the optical response is directly related to the chemistry of the mechanosensitive molecule; in particular, one can distinguish the change of light absorption in the visible range, the fluorescence, and the chemiluminescence. For more information, the reader is invited to refer to the detailed overview on the subject by Göstl and coworkers.134
The activation of the mechanophore inside the material requires the application of a macroscopic force on the sample that may (or may not) activate molecules inside the material. By far the most common way to do this are uniaxial tensile or compressive tests. If the sample is stretchable and fracture in the grips can be avoided, tensile tests are more severe and more representative of the actual toughness of the sample. In this case a strip or a dogbone-shaped sample (e.g.Fig. 27) is typically fixed into a standard tensile tester and stretched while an optical observation setup of the sample is used while it is being deformed. If tensile tests are not practical or the sample is brittle, compression tests are a viable alternative for a proof of concept but the friction between sample and plate exerting the compression is unknown so local stress is actually much more difficult to determine than in tension.
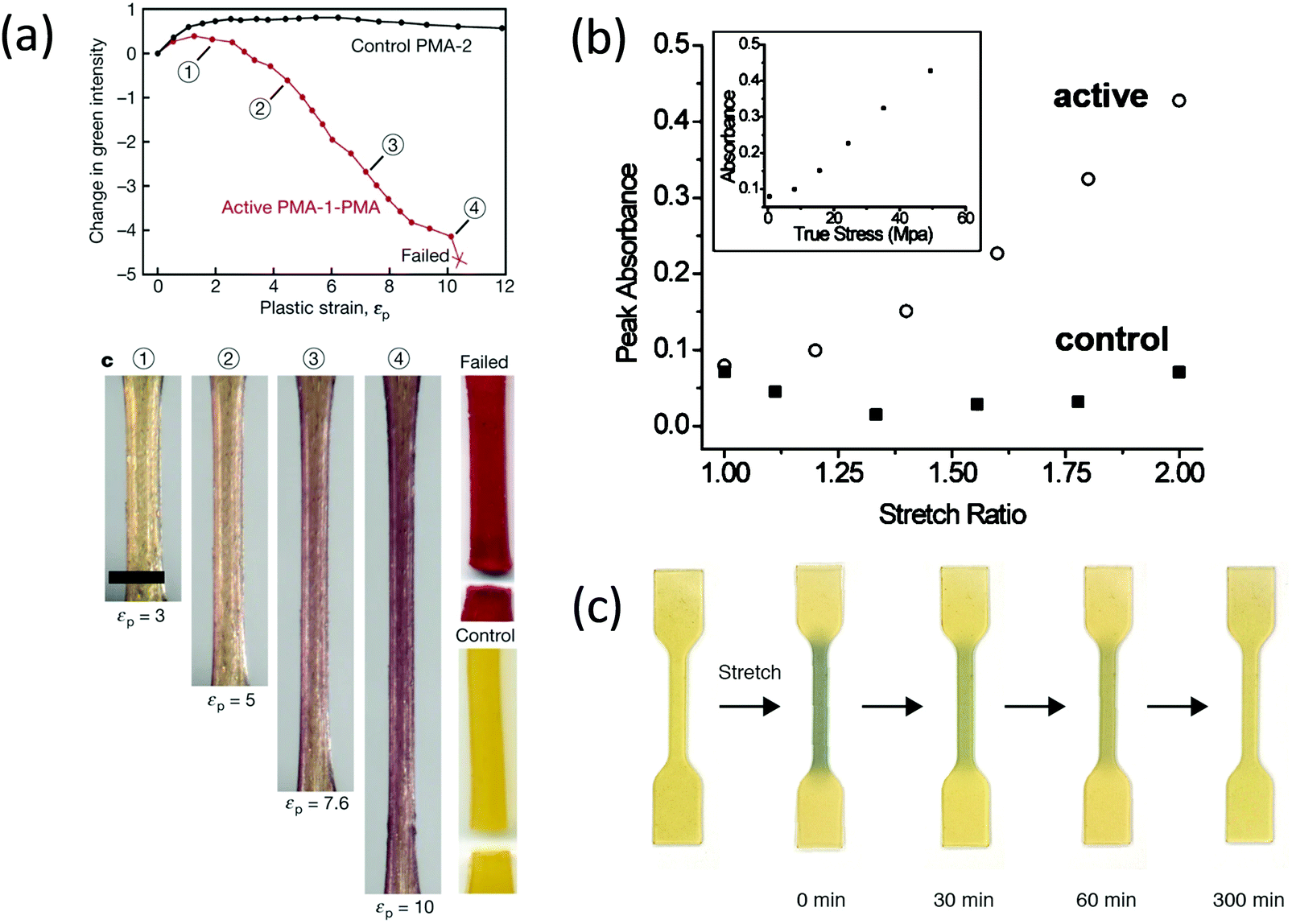 | ||
| Fig. 27 (a) Change in green intensity for SP-containing PMA and its inactive counterpart (control) as a function of plastic strain during cyclic testing.35 (b) Peak absorbance of SP-containing PU and its inactive counterpart (control) as a function of stretch ratio.154 (c) Optical images of DABBF-containing PU samples before and after being stretched manually.92 (a) is reprinted with permission from ref. 35, Copyright 2009 Springer Nature. (b) is reprinted with permission from ref. 154, Copyright 2010 American Chemical Society. (c) is reprinted with permission from ref. 92, Copyright 2015 American Chemical Society. | ||
Some early activation results were obtained by Rubner, who used visible spectrometry to investigate the optical properties of soft segmented PU containing reactive diacetylene groups in the hard segment.156 A shift and changes in shape of the absorption band were observed upon stretching a partially cross-polymerized thin film. In particular, the changes in the absorption spectrum highlighted the mechanisms involved in the deformation of the elastomer. Later, Kim and Reneker used UV-visible spectrometry to monitor the activation of the azo moiety in polyurethane copolymers upon uniaxial cyclic stretching.152 While no change in the UV-visible absorption spectrum was observed after three 100% tensile stretches, a relative increase of the amount of the trans-form was observed after three successive 200% tensile stretches. After three 300% tensile stretches, a quasi-total isomerization of the cis-form into the trans-form was observed. In a seminal paper in 2009, the group of Moore systematically studied the mechanical transduction of SP to MC in a polymer melt containing center-labeled PMA chains (Tg ∼ 15 °C) at room temperature during monotonic and cyclic tensile loading tests. A significant color change was observed upon plastically deforming these polymer melts up to failure, proving the SP to MC isomerization. Cyclic tests provided additional information: a red-green-blue (RGB) analysis of the digital images recorded by the color camera during the experiment showed a progressive decrease of the green intensity (i.e. increase of the activation) after reaching a plastic strain level of 200% until failure (Fig. 27a).35 By changing the polymer nature to a classical PU formulation, similar results were observed with a physically-crosslinked soft polymer (i.e. the increase of the SP to MC isomerization with stress (Fig. 27b).154 Activation kinetics within PU were studied using fluorescence imaging, showing in particular that the reversion from MC to SP after unloading the PU occurred in an hour, while no reversion was observed under constant strain.
The group of Otsuka introduced reversible scissile DABBF linkages in PU, and studied the time-dependent breakage of the bonds under tensile loading with EPR spectroscopy. Upon stretching, a blue coloration of the sample due to bond scissions occurred from 50% strain up, followed by a progressive increase of the cleavages with strain. After 5 h at RT, the linkages had recombined and the sample had almost recovered its shape, demonstrating the reversibility of the activation (Fig. 27c).92
In these uniaxial tensile tests, the degree of activation of the mechanophore typically increases monotonously with macroscopic uniaxial deformation (or stress), independent of the nature of the mechanophore and of the details of its incorporation (in linear chains,35,84 networks,84 physically cross-linked linear chains,92,152,156 or coatings made from emulsion);182 and activation starts to be detected at a threshold value of strain (dependent on the system properties).
These first activation studies in materials were clearly designed to address whether the molecule incorporated as a label in the material can be activated by a macroscopic stretch or compression, and concerned a variety of different polymer-based materials, including chemically-crosslinked elastomers52,84,86,142,190 and gels,180,184,187,195,219,220 glassy polymers,57,166,176–179 and physically crosslinked thermoplastic elastomers like polyurethanes,148–151,159 or triblock copolymers.167,168
3.2.2.2 Activation in elastomers and gels vs. activation in polymer glasses. Using uniaxial testing, Jiang et al.167 studied the mechanical activation of SP introduced in the center of triblock copolymers made of PS and PnBA as a function of the copolymer composition. By varying the PS content of the copolymer, the mechanical properties of the material were tuned from a soft elastomer (low PS content) to thermoplastic elastomers up to glassy-like polymer (high PS content). Consequently, the mechanoactivation under stretching at room temperature of SP varied in the corresponding manner: from weak homogeneous activation to an intense, but still homogeneous, color change, and up to a localized color change only once the fracture point had been reached. The enhancement of the activation with increasing PS content is related to a microphase separation structure induced by the formation of PS self-assembled domains playing the role of physical crosslinks. This example highlights the role of the polymeric matrix in the relationship between the macroscopic force applied to the material and the force detected by the mechanophore. The force transduction mechanism is clearly different in elastomers and glassy polymers.
In elastomeric polymers and gels, the forces along the backbone of the polymer chains can be orders of magnitude higher than between polymer chains. As a result, the force detected by the mechanophore can be quantitatively related to the macroscopic properties: stress, strain, or bond scission. However, in order to see widespread activation in the bulk, the material should not be too brittle. Therefore, unfilled, well-crosslinked elastomers far from their Tg were not used in these early studies. The two simple options were (1) the physically-crosslinked PU family of materials, where hydrogen bonds (hard segments) can dissipate energy and delay the nucleation and propagation of a crack; and (2) lightly crosslinked PMA with a Tg slightly below room temperature, where a high level of viscoelasticity also favors delayed crack propagation.221,222
In glassy polymers, the situation is very different since the chains are frozen in place, and the elastic properties are controlled by the van der Waals bonds between chains, i.e. enthalpic elasticity. In these conditions, it is very unlikely that any covalent bond activation occurs before actual yielding, i.e. before the van der Waals bonds start to fail. Hence, mechanophore activation can be used to detect localized yielding or bond scission after yielding, but is not a good measure of macroscopic stresses in the elastic regime, since the molecular forces are distributed over many more bonds and not only the backbone of the chain. This reasoning is key to the understanding and interpretation of all intermediate cases where the material is neither fully glassy nor fully elastomeric, such as in particle-filled composites, or polymers close to their Tg such as in some paints and coatings. In the following, we review in more detail the main findings obtained from this activation of mechanophores inside materials.
3.2.2.3 Activation of the mechanophores in elastomers and gels. The following section will highlight the role of material design (architecture, molar mass, crosslinking density, etc.) in the activation of mechanophores in soft elastic and viscoelastic materials, and discuss some of the interpretations.
Molecular weight. In soft linear polyurethane materials, the degree of activation shows similar trends in the bulk material and in solution, as shown by Chen and Sijbesma who studied the failure of mechanoluminescent PU.148 By varying the Mn of the PU from 9 to 43 kDa, they demonstrated a clear shift in behavior with Mn, with almost no mechanoluminescence detected for low molecular weight PU (Mn = 9–12 kDa), and a much more significant activation with increasing strain for Mn = 38–43 kDa.148 This trend is a general feature for mechanoresponsive linear polymers, and immediately demonstrates that even if the stress is macroscopically homogeneous, the forces on the bonds of the polymer chains are not.
Linear vs. cross-linked elastomers. Sijbesma's group compared the mechanically-activated luminescence of the bis(adamantyl)-1,2-dioxetane, incorporated either into a PMA chain or in a network.84 In the case of the network, a luminescence was detected from low stress during stretching; and a strong increase of the luminescence was observed with increasing stress until the sample fractured, at which point the luminescence turned off. In comparison, in the linear PMA, a weaker luminescence was detected during the test, revealing fewer bond scissions. To afford a quantitative analysis, the use of the Förster resonance energy transfer (FRET)223 was required. The stress-time curve showed higher stress values for the crosslinked PMA compared to the linear one, justiying the higher level of bond scissions detected in the network.
In their study on mechanoresponsive elastomeric acrylic latex coatings made of BA and MMA, Li et al.182 used vinyltriethoxysilane (VTES) as an interparticle crosslinker, and SP as an intra-particle crosslinker, and investigated the effect of intra- and interparticle crosslink density on the SP activation. They observed a higher stress sensitivity and lower stress activation threshold when increasing the intraparticle crosslink density (i.e. increasing the SP content); while the increase of the interparticle density did not impact the sensitivity but increased the activation threshold. This clear result suggests that the activation intensity is related to the concentration of activated SP molecules while the relation between macroscopic stress and concentration of activated molecules can be influenced by the material architecture. In other words, the interparticle crosslinking of the silane sustains a significant part of the stress and partially unloads the SP containing chains. As a result, activation of SP occurs at a higher macroscopic stress.
Composition. The mechanoactivation profile is determined by the nature, ratio, or compatibility of the monomer(s) that compose the material. As an example, by comparing the kinetics of the reversion of MC to SP under a white light exposure in different linear polymers, Moore's group showed the importance of the local environment and pointed in particular to the determining role of the polymer Tg: the duration of fluorescent room light exposure required to obtain the full reversion of MC back into SP increases with the Tg of the polymer, from 1 h for PU (Tg ≈ −60 °C) to 6 h for PMA (Tg ≈ 10 °C) up to several weeks for PMMA (Tg ≈ −105 °C).35,154
The introduction of physical crosslinks in the structure is a good way to improve mechanical activation of mechanophores in polymers.149–151,159,220 Weng's group incorporated a quadruple hydrogen bonding non-covalent 2-ureido-4-pyrimidone (UPy) motif into the SP-containing PU, thereby creating physical crosslinking through H-bonding between UPy.149,150 Upon stretching, organization in microdomains occurs and the fragmentation of the hard domains, as well as the dissociation of the UPy stacks and dimers, induced energy dissipation and delayed crack propagation.150 Further investigations showed that supramolecular interactions between UPy units promote chain orientation and strain-induced crystallization, which explained the improvement of the mechanical activation of SP (Fig. 28).149 Weng's group also reported the sensitive mechanoactivation of SP introduced into doubly-crosslinked PU: oligo-poly(lactic acid)-capped pentaerythritol (PTT–PLA) was chosen as a covalent cross-linker, and UPy dimer units played the role of physical crosslinks.159 Chen and Sijbesma highlighted the effect of the type of H-bonding on the activation by comparing rubbery materials made of segmented dioxetane-containing PU, PES, and PA. All dioxetane containing PU possessed similar molar mass (12 kDa) and were incorporated into PU of higher molar masses PU (42 kDa). A more densely-packed H-bond network favors the transduction of the force to the mechanophore (as revealed by a more intense light emission).148 In other words, the level of activation of the mechanophore can provide information on the internal relaxation of the polymer chains inside the material.
 | ||
| Fig. 28 Mechanism of SP mechanical activation enhanced by H-bonding in SP-containing PU, proposed by Weng's group. The stretching axis is vertical. (a) Before stretching, (b) within strain-hardening region, (c) SIC of PTHF segments, and (d) mechanical activation of SP to MC.149Fig. 28 is reprinted with permission from ref. 149, Copyright 2014 American Chemical Society. | ||
Orientation. Moore's group investigated the role of the mechanophore orientation in the mechanochemical response of the material. Using fluorescence polarization analyses on SP containing linear PMA and PMMA (plasticized or tested at high temperature), they showed that mechanophores oriented in the direction of the applied stress were preferentially-activated.219 Later, they proposed another method, based on photoelasticity and fluorescence measurements, to quantify the impact of polymer chain orientation on the activation of the mechanophore137 By comparing the mechanochemical response of SP, incorporated either in the soft or hard phase of a segmented PU, they highlighted the link between chain mobility, mechanophore orientation and activation.153 The high mobility in the soft segment promotes the alignment of the mechanophore; whereas the glassier state of the hard segment hinders the alignment under stress. The degree of activation in both the soft and hard phases is similar when SP orientation is parallel to the force direction, but SP in the hard segment can be activated at much lower alignment levels. In the soft segment, only the SP aligned with the force can be activated (force is translated into simultaneous rotating). In the hard segment, the activation is probably dominated by intermolecular chain forces, allowing the activation of molecules that are not completely parallel.
Regiochemistry. By comparing three naphthopyran regioisomers incorporated in PDMS, Moore's group reported the regiochemistry-dependence of the mechanical reactivity.48 In this study, only one regioisomer exhibited a mechanical response. In contrast, they studied the mechanoactivation of two SP mechanophores in a PDMS network; and in this case, comparable mechanoactivity was observed for both mechanophores.192 Similarly, Craig's group investigated the regiochemical effect on the mechanophore activation in PDMS by comparing three SP regioisomers.193 Interestingly, the study revealed differences in the relative colorometric response of the three regioisomers, but an identical detectable activation onset.
Besides material design, external parameters like temperature,182 solvent,74 strain rate,148,159,182,224 and laser excitation140 impact the force at which the mechanophore activates; presumably by modifying activation barriers for the reaction. Such sensitivity to the environment should be kept in mind if a quantitative interpretation of the mechanochemical response is desired. An important example is the activation of mechanophores in hydrogels. Wang and coworkers incorporated a rhodamine-based mechanophore into a micellar hydrogel. As the gel is stretched to large strains, the micelles are deformed and exert a force on the mechanophore that activates optical visibility.225
As described above, the probability of mechanical activation of mechanophores in a polymer backbone is affected by several factors from the molecular structure of mechanophores to the structure of polymer materials themselves. In Section 1, we reported values of the threshold force for activation measured or calculated for specific mechanophores by SMFS or CoGEF. These values are for individual molecules. However, the stress to activate the mechanophores embedded in polymer materials reflects an average concentration of activated molecules inside the material and depends also on the molecule's environment. For polymer melts it may depend on molecular weight and strain rate, for networks it may depend on the composition, orientation, regiochemistry of the polymers, network architecture and of course temperature relative to the glass transition temperature. This last point is essential since It determines the elastic modulus and hence the share of the total load carried by the mechanophore relative to the rest of the bonds.
For example, in soft hydrogels the covalent bonds carry a much larger share of the load than in polymer glasses and this affects the relationship between macroscopic stress and mechanophore activation.
To put the activity of mechanophores within each respective polymer matrix in prospective, we list in Table 2 the critical stress of mechanophore activation, the Young's modulus of polymer materials and the glass transition temperature when such information is available from the original publication.
| Matrix | Molar mass (kDa) | Young's modulus (MPa) | Critical stress of mechanophores (MPa) | T g (°C) | Ref. |
|---|---|---|---|---|---|
| a Estimated using stress–strain curve (tensile test experiment at RT). b Estimated using stress–strain curve (compression loading experiment at RT).Polymers: PMA: poly(methyl acrylate); PMMA: poly(methyl methacrylate); PU: polyurethane; PnBA: poly(n-butyl acrylate); PS: polystyrene; PDMS: polydimethylsiloxane; PEVA: poly(ethylene-vinyl acetate); PEOC: poly(ethylene octene copolymer); PEA: poly(ethyl acrylate); PHMA: poly(n-hexyl methacrylate); PDMA: poly(N,N-dimethylacrylamide). SN: single network; DN: double network elastomers; TN: triple network elastomers. | |||||
| Linear PMA | 18 to 287 | — | 10a (dioxetane) 1.5a (SP) | 12–15 | 35, 84 and 140 |
| PMA network | — | — | 100b (compound 24) | 10–15 | 84, 117, 119 and 120 |
| 150b (compound 23) | |||||
| Linear PMMA | 260 | 800a | 128 | 35, 143 and 219 | |
| Linear plasticized PMMA | 260 | 400a | 15a (SP at 90 °C) | Below room temperature | 143 |
| Cross-linked PMMA beads | — | 300b | 50b (SP) | — | 35 |
| PU | 50–70 | 2–13 | 40b (dioxetane) | −70 (soft segments) | 74, 92, 101, 148–150, 153, 154, 156, 159 and 160 |
| 5a (SP) | |||||
| 8a (rotaxane) | |||||
| 6a (BABBF) | |||||
| PS–PnBA–PS triblock copolymers | 37–51 | 0.32 (24 wt% PS) to 240 (45 wt% PS) | −40 (PnBA); 80 (PS) | 167 | |
| P(nBA-co-MMA) film (from crosslinked particles) | — | 2a | 2a (SP) | −4 (46 wt% BA) to 30 °C (51 wt% MMA) | 182 |
| PDMS | — | 1.4 | 2a (SP) | −125 | 40, 48, 190, 192 and 193 |
| 2a (dioxetane) | |||||
| PEVA, PEOC | — | 5a | −90 (semi-crystalline at room T) | 224 | |
| PEA (multiple networks) | — | 0.9 to 2.1 (SN) | 1.5a (SP in TN) | −20 | 41, 52 and 86 |
| 1.2 to 2.3 (DN) | 1.5a (rhodamine in TN) | ||||
| 1.7 to 5.4 (TN) | 2.5a (rhodamine in DN) | ||||
| 2.5 a (dioxetane in TN) | |||||
| 5a (dioxetane in DN) | |||||
| PHMA | — | — | 0.6 (TASN) | 0 | 100 |
| PDMA | — | 1.5 (1 mol% cross-linker) | 150b (compound 13) | 120 | 57 |
| 6 (5 mol% cross-linker) | |||||
| 9 (10 mol% cross-linker) | |||||
3.2.2.4 Activation of the mechanophore in glassy polymers. The material design plays a crucial role in the activation of mechanophores in glassy polymers as well. However, as discussed earlier, the activation of mechanophores in glassy polymers is closely related to the plastic deformation; and as such, is more sensitive to the monomer composition than to the crosslinking structure. Comparing, for example, the mechanical response of linear143 and cross-linked176 SP-containing PMMA with similar Tg (127 °C and 118 °C, respectively) under tensile load at 90 °C, similar mechanoactivation profiles were observed: a visible, mechanically-induced activation as the polymer yields, with an increase of intensity with plastic strain for crosslinked and uncrosslinked samples. The effect of the crosslink density was also investigated by Kabb and coworkers.57 The authors assumed that increasing the crosslink density would shorten the length of the chains between the crosslink junctions, therefore enhancing the mechanoactivation. However, by varying the crosslinker amount from 1 mol% to 10 mol% in a PDMA network containing maleimide-anthracene Diels–Alder adducts, no significant difference in the activation profile was noticed (Fig. 29). This expected result confirms that the mechanophore activation is more related to the onset of plasticity. On yet another glassy system, Moore's group evaluated the effect of the network structure on the mechanoactivation by comparing the shear-induced activation of SP in three cross-linked PMMA samples based on primary cross-linkers of different lengths: EGMA, PEG550 and PEG750. At the lowest strain rate, the onset of activation occurred at exactly the same strain.177
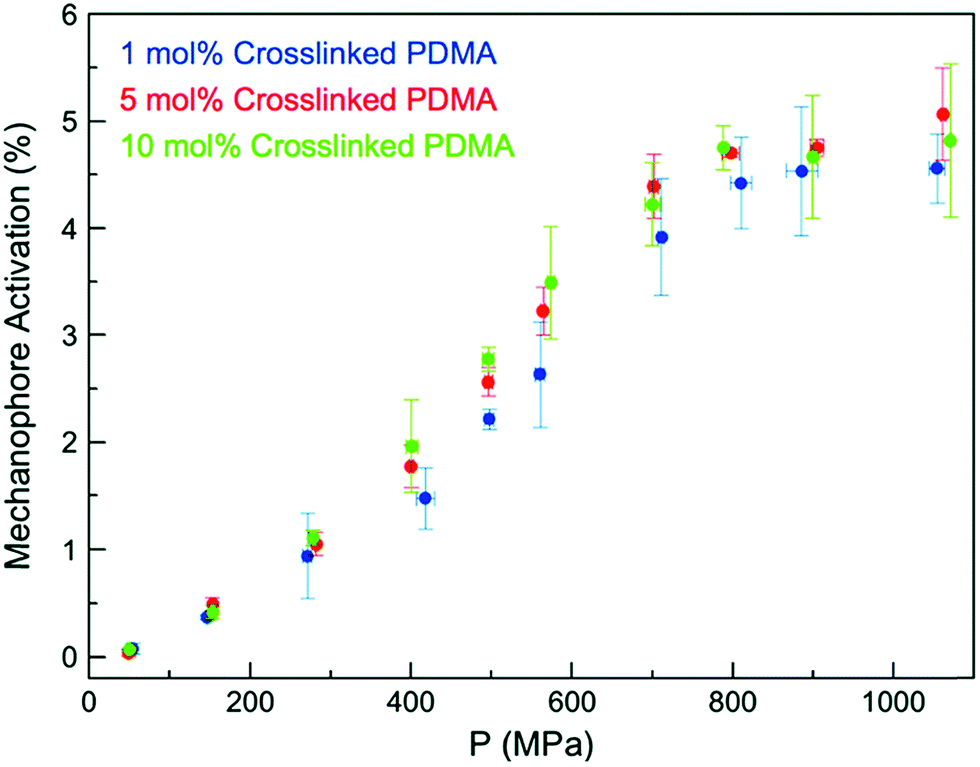 | ||
| Fig. 29 Activation of the mechanophore in maleimide–anthracene Diels–Alder adduct as a function of the force of compression for different crosslink densities.57Fig. 29 is reprinted with permission from ref. 57, Published by The Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). | ||
Another experiment that was carried out is the activation by solvent swelling. Since swelling by a solvent may cause polymer chain stretching, it is reasonable to believe that activation of a mechanophore would occur as a result. In this case different mechanophores reveal different features for the same experiment. The experiments of Clough et al. with the chemoluminescent bis(adamantyl)-1,2-dioxetane mechanophore reveal the early stages of PMMA swelling very well; where the solvent causes localized crazes, and covalent bonds are broken locally.179 The experiments of Lee et al. on SP cross-linked PMMA networks that focused on equilibrium swelling pointed to a crosslink density effect: an increase of the crosslinking density induces a longer equilibrium period and less activation (due to a lower degree of swelling and, presumably, of chain stretching).178
Linear vs. complex architectures. In order to investigate the effect of the architecture on the mechanoactivation, Oka et al.102 designed linear, and star (four- and eight-arm) polystyrenes that contained a DABBF moiety in the center of the structure. By performing grinding tests on bulk polymers, they showed an increase of DABBF dissociation (i.e. mechanoactivation) with molecular weight for linear polymers. Additionally, a comparison of linear and star polymers with similar molecular weights showed that the DABBF dissociation ratio was significantly higher for star polymers (Fig. 30a). Moreover, increasing the number of arms of the star polymers led to a drastic increase of the mechanoactivation, proving an efficient load transfer to the mechanophore in the star architecture (Fig. 30a). Interestingly, this study differs from previous ones which reported a decrease of reactivity for star polymers in solution by increasing the number of arms.56 This result was related to a better shear stability of star polymers in solution.56,218,225 Oka and coworkers explained the observed differences between bulk and solution behaviors by the increase of intermolecular entanglement in the bulk state relative to the solution, due to the multiple arms of the star architecture.102
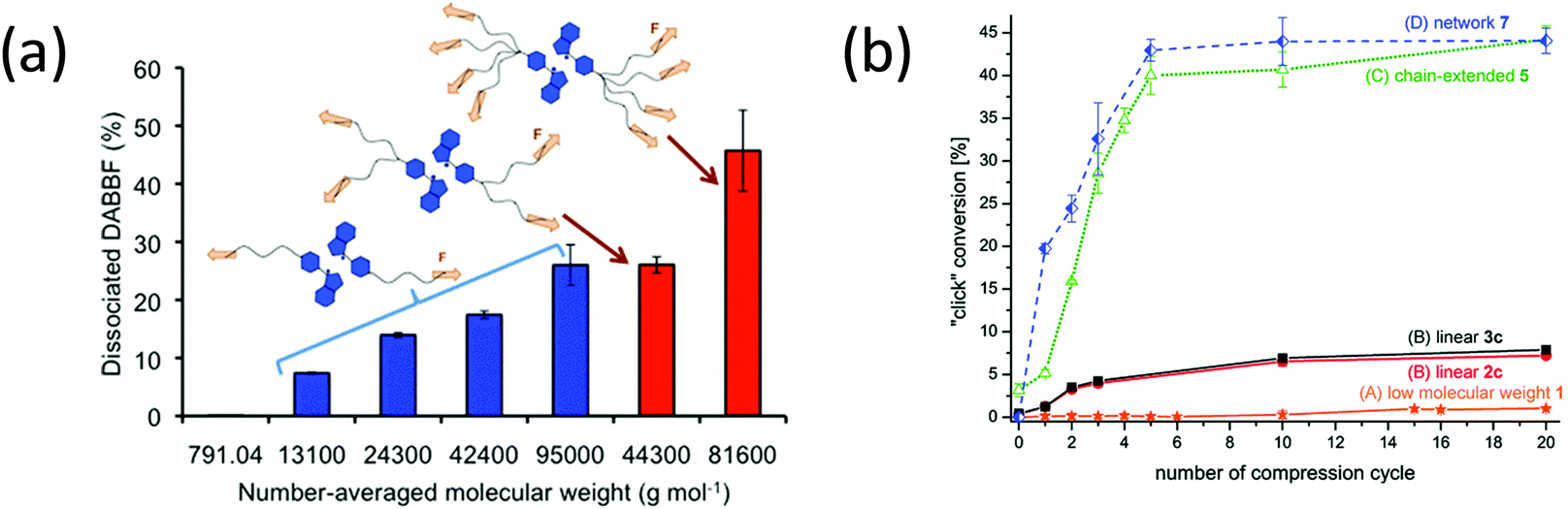 | ||
| Fig. 30 (a) Amount of dissociated DABBF as a function of the molecular weight for linear (blue bars) four- and eight-arm star (red bars) DABBF-centered PS.102 (b) “click” reaction conversion as a function of compression cycle for different Cu(I) bis(NHC) polymer complexes mechanocatalysts.166 (a) is reprinted with permission from ref. 102, Copyright 2016 American Chemical Society. (b) is reprinted with permission from ref. 166, Copyright 2018 Wiley-VCH. | ||
Michael et al.166 designed four mechanocatalysts based on Cu(I) bis(NHC) polymers of different architectures: a low molecular weight complex, linear complexes bearing either flexible PIB or stiffer PS chains (different chain length were explored), a chain-extended PS-based complex with multiple Cu(I) bis(NHC) moieties along the polymer backbone, and networks containing a number of Cu(I) bis(NHC)s as crosslinking points. The mechanically-induced catalysis of the fluorogenic “click” reaction of 3-azido-7-hydroxy-coumarin and phenylacetylene was investigated directly in bulk by compression test. The study highlighted that a chain-extended structure (multiple-mechanophore material) and the network architecture exhibited the highest mechanically-induced catalytic activation (Fig. 30b).
Testing parameters. It is known that external parameters such as temperature, strain rate, testing method, and solvent directly affect the mechanical response of polymers. In the case of polymer glasses, one may wonder how these parameters can be varied to influence mechanophore activation.
Independent of the structure (linear, chain, or network), increasing the temperature modifies the mechanical behavior of the glassy polymer; and the activation of the mechanophore reveals the change in the forces felt by the backbone chains in the polymer. Beiermann et al.143 tested an SP-labeled PMMA at different temperatures and in the presence or absence of solvent. The PMMA is glassy at room temperature and at 80 °C; and the SP only sees the load when plastic deformation is activated. In those conditions, activation only occurs very close to the propagating crack where a plastic zone exists,226 and was not detected by the authors because of lack of spatial resolution. At 90–105 °C, the PMMA is no longer brittle and plasticity occurs in the bulk, causing a spatially-homogeneous activation of the SP at an engineering stress of around 15–20 MPa. Finally, at 120 °C the linear chains of PMA relax too fast, and forces on the backbone never reach the value necessary for activation. Similarly, adding plasticizer (15–20 wt% of MeOH) to SP-containing PMMA network allowed a mechanical response at RT similar to those at 90 °C.
The solvent polarity also affects the mechanoactivation; in particular, in swelling-induced activation systems. Lee et al.178 showed that a solvent with an intermediate polarity is required to observe swelling-induced activation of SP in PMMA network. This may also be connected to the ability of the solvent to swell the network and hence to stretch the backbone chains.
The testing method, specifically the way the force is applied (stretching, compression, shear, torsional creep), is also important in the details of the mechanophore response. The comparison of three studies made on SP-containing crosslinked PMMA illustrates this idea. Davis et al.35 reported a quite constant threshold activation strain value (around 20%) with strain rate in uniaxial compression (Fig. 31a). Later, Kim et al.176 compared the mechanical response of crosslinked PMMA under compression and tension. The study showed that the threshold strain for activation was dependent on the strain rate; and although comparable trends are observed for both testing geometries, different threshold values were obtained (Fig. 31b). Shear experiments highlighted a near-constant threshold strain value at all tested shear rates, with a threshold value estimated around 45% strain (Fig. 31c).177 Consistent results were obtained by comparing monotonic and creep torsion experiments, revealing that large-scale polymer deformation and onset of flow and stress-induced mobility are needed to permit force transduction.174
 | ||
| Fig. 31 Activation (strain or stress) threshold for SP-linked crosslinked PMMA as a function of strain rate: (a) Threshold (color) stress, yield stress and threshold strain as a function of strain rate reported by Davis et al.;35 (b) Yield- and activation strain as a function of strain rate in tension test and compression test as described by Kim et al.;176 (c) Activation strain as a function of shear rate, plotted using the values reported by Kingsbury et al.177 (a) is reprinted with permission from ref. 35, Copyright 2009 Springer Nature. (b) is reprinted with permission from ref. 176, Copyright 2015 American Chemical Society. (c) is reprinted with permission from ref. 177, Published by The Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). | ||
Performing mechanical tests at very high strain rate (shockwave conditions) has been tested to promote mechanophore activation by limiting relaxation.145,175 using a high spatial resolution technique, Hemmer and coworkers proposed the following high strain rate induced activation scenario: a fast loading and fast fracture induces plastic heating and subsequent thermal activation and creates localized craze zones, in which strong activation is detected175
3.2.2.5 Activation at the interface in nanocomposites. Although the vast majority of studies use mechanophores incorporated within the bulk of the polymer (either in the main chain or as a crosslinker), some studies report the incorporation of mechanophore at organic–inorganic interfaces. For example, in 2014 Moore's group proposed a model system based on the maleimide–anthracene Diels–Alder adduct mechanophore grafted at the interface between silica nanoparticles and PMA linear chains to investigate the force-induced activation of mechanophores at the heterointerface.59,64 Upon sonication, the selective activation of the mechanophore was observed and the study revealed that activation characteristics at the heterointerface are comparable to those of mechanophore in polymers: in particular, a minimal chain length is required to allow force transduction and from this threshold Mn, the activation rate increases with Mn.59 Grafting density also affects the mechanophore activation, in that a low grafting density favors the mechanophore activation.64
The same interfacial activation has been studied in nanocomposites. In this case, because stresses are typically higher at soft–hard interfaces than in the bulk, the mechanophore is highly selective and reveals the heterogeneity of the composite.59,61 Activation can give detailed insight into molecular damage mechanisms in these more complex materials; but the particular structure of nanocomposites may also enhance the mechanophore activation, as was reported by Kim et al.210 who demonstrated a reduction of the activation threshold strain for SP at the heterointerface compared to one in bulk for silica-filled PMA materials. Otsuka's group studied the force-induced dissociation of DABBF in a complex polymer–inorganic composite structure comprised of a rigid silica network obtained by sol–gel, and embedded within a soft PBA matrix. They showed a significant increase of the sensitivity and a higher dissociation ratio of the mechanophore with increasing silica content. Moreover, the rigidity of the silica network (containing the grafted DABBF) hinders the deactivation of the mechanophore by recombination of the DA adduct.211 Generally speaking, if the mechanophore in a nanocomposite is grafted at the particle/matrix interface, it can be activated at a much lower macroscopic strain than the same mechanophore incorporated within the bulk of an unfilled soft polymer, which makes it possible to activate fluorescence or a change in absorption at a lower macroscopic strain and have a more sensitive strain probe.201,204,205,213
From a more general point of view, several parameters are important to enhance the sensitivity of the mechanophore to macroscopic strain, such as porosity of the structure,207 crosslink density and composition in polyurethane–silicone blends,227 strain rate,227 and pre-alignment of the mechanophore in the tensile direction.208
4. Applications and functions of mechanoresponsive polymer materials
Mechanoresponsive polymer materials, obtained by incorporating mechanophores into a polymer matrix, display a range of responses, including color change, luminescent and fluorescent emission, release of small molecules, and activation of a catalytic center. These responses have been used in a number of ways, varying from understanding polymer mechanics to warning against imminent damage. A comprehensive overview of these applications is presented in this section (Scheme 1).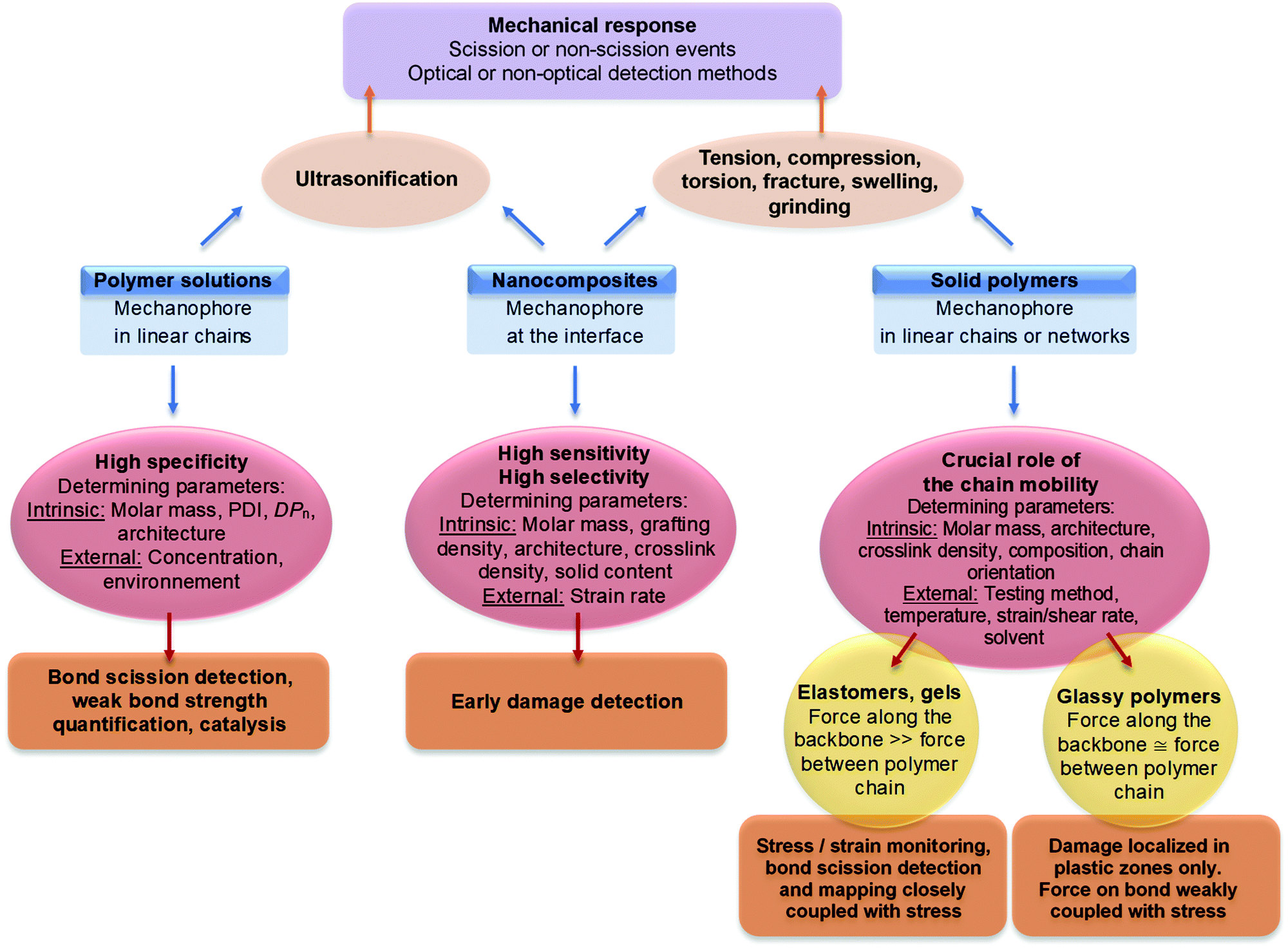 | ||
| Scheme 1 Schematic summary of the mechanophore activation in polymer materials and its interpretation. | ||
4.1 Using mechanophores to understand polymer mechanics
Mechanophores can be used as molecular indicators, and can be incorporated into different polymer systems. The color change, fluorescence, or luminescence obtained after mechanical activation can then be used to quantify and map the stress or strain, especially when the stress is heterogeneous; and in some cases, to obtain time- and space-resolved information on activation. These results are invaluable to develop mechanical models at the molecular model of material damage and fracture.As already discussed previously for PMMA at room temperature, the crux of the matter is the localization of high stress regions leading to the propagation of a crack. A brittle material does not fail by plastic flow but by the propagation of a crack nucleated by an inherent flaw in the material. This propagation can be localized at the nm level (as in inorganic glass), or very delocalized at the 100 μm-level, as in the tearing of rubber. While modern tools of mechanics (such as digital image correlation) can be used to visualize propagating cracks and map the strains near a crack, the stresses and, even more so, the extent of molecular damage were impossible to detect until recently.
As an example of the use of mechanophores in mechanics, Craig's group reported the incorporation of weak, fast-exchanging supramolecular crosslinks, and was able to increase the maximum-achievable strain of an organogel made of P4VP under compression.119 To investigate the mechanism involved in the enhancement of the mechanical properties of the organogel bis(adamantyl)-1,2-dioxetane, a scissile chemoluminescent crosslinker was introduced into the network. During a compression test, the activation of the mechanophore occurred in a very localized way, and only upon rupture. By varying the content of supramolecular units, the onset of activation could be shifted with the strain at break, while keeping a constant intensity (Fig. 32). This suggested that the reversible interactions delay the nucleation and propagation of a crack to higher strains, but do not influence the extent of bond scission once the crack propagates.
 | ||
| Fig. 32 Representative stress–strain curve and the corresponding emission intensity–strain plot of mechanoluminescent PN2·PdEt gels (P4VP networks with PdEt as physical crosslinker) as a function of [PdEt].220Fig. 32 is reprinted with permission from ref. 220, Copyright 2014 Wiley-VCH. | ||
In another example of space-resolved mapping, the color change of colorless SP into blue MC was used to map the stress distribution around crack tip in a polyurethane material by Weng's group.159 The SP was activated around the crack tip and revealed a loaded region via the blue color; whereas, due to the isomerization of MC, a second color switch (from blue to purple) was observed in the unloaded regions (Fig. 33a). In a different example, Craig et al.196 used the color change of SP to measure the continuous 3D spatial strain distribution in PDMS soft materials when they are hit by a projectile (Fig. 33d), and argued that this type of experiment could be used to simulate brain strain for traumatic brain injury applications. The fluorescence of MC was also used to map and localize the high stress distribution around the crack by Sprakel's,228 and Moore's group (as shown in Fig. 33b and c).172
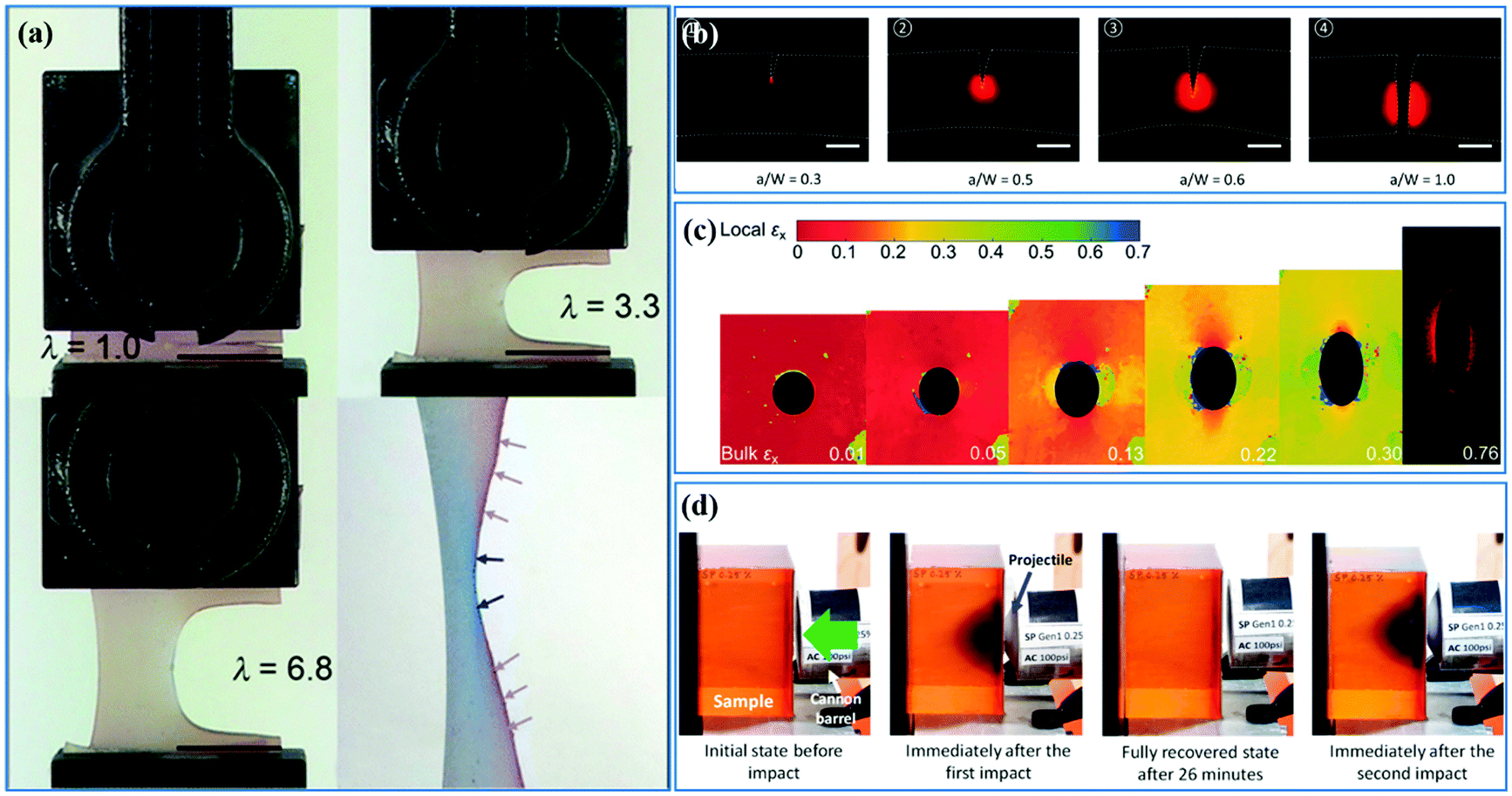 | ||
| Fig. 33 SP was used to detect and map high stress regions. (a) SP was incorporated into polyurethane and the stress distribution around the crack tip was mapped from the color change before and after crack propagation.159 (b) SP was covalently crosslinked into poly(methyl methacrylate) network and was used as an indicator to measure the plastic deformation around crack tip by fluorescent feature of MC.172 (c) The strain distribution around the defect of a hole was mapped by photonic array reporting. The fluorescent region indicated by SP matched with the high strain distribution measured by photonic array.228 (d) The color change after SP activation was applied for mapping the strain deriving from the impact of projectile in PDMS.196 (a) is reprinted with permission from ref. 159, Copyright 2014 American Chemical Society. (b) is reprinted with permission from ref. 172, Copyright 2014 Elsevier. (c) is reprinted with permission from ref. 228, Copyright 2020 Wiley-VCH. (d) is reprinted with permission from ref. 196, Published by MDPI (https://creativecommons.org/licenses/by/4.0/). | ||
While in all these previous examples the degree of mechanophore activation could be measured by either fluorescence intensity or color change, the absolute value of the average stress (an important quantity in mechanics) could not be directly measured for lack of a calibration method that would work across sample geometries. A first attempt in that sense was made by Chen et al.42 who carefully calibrated the color change in uniaxial tension, and used the calibration data to directly obtain absolute values of engineering stress around an opening crack. The results were compared with a finite element simulation carried out without any adjustable parameters. The correspondence was remarkable as can be seen in Fig. 34.
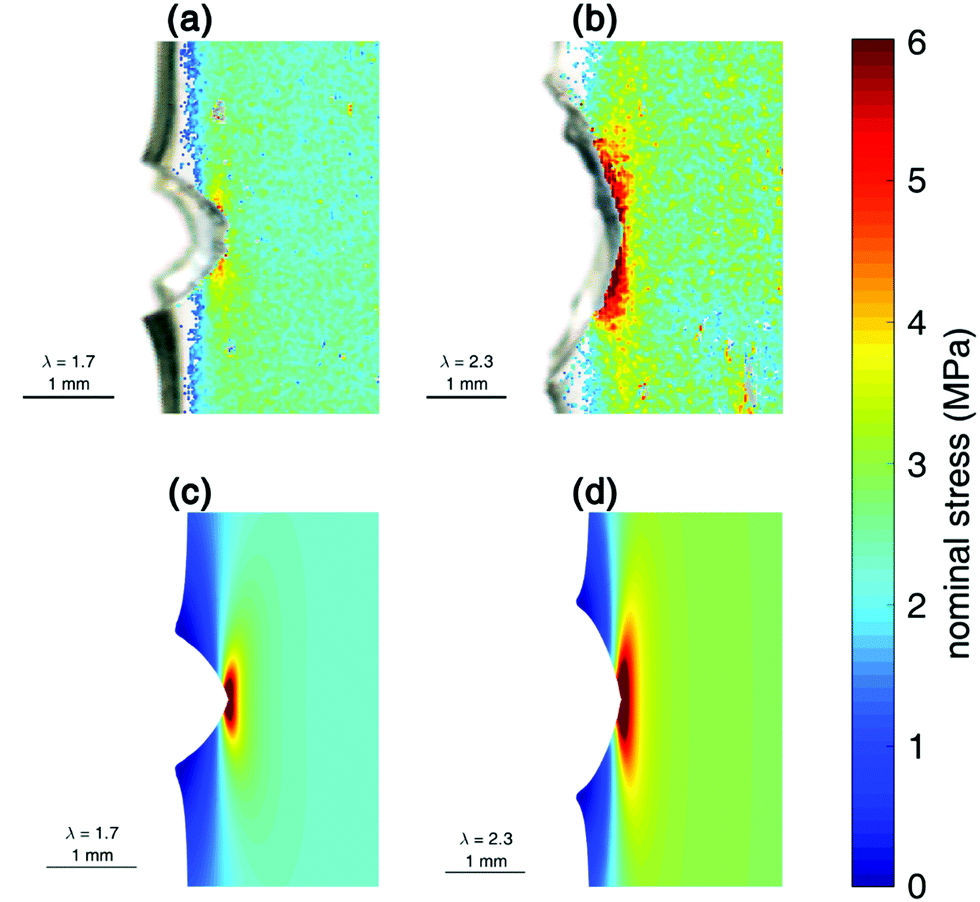 | ||
| Fig. 34 Stress maps around crack tip are quantified by the color change of spiropyran and the simulation results by finite element simulation. (a) Experiment results of stress map for stiff materials; (b) experiment results of stress map for soft materials; (c) simulated stress map for stiff materials; (d) simulated stress map for stiff materials for soft materials.42Fig. 34 is reprinted with permission from ref. 42, (published under a Creative Commons license, CC BY-NC), Copyright 2020 AAAS. | ||
While spatial mapping of stress is clearly a useful tool to identify the highly loaded regions in the sample, of even greater use is the extent of molecular damage caused by the fracture. This has been done with two types of molecules.
Mechanoluminescent 1,2-dioxetane mechanophore was used as a time-resolved molecular damage probe to map the damage distribution in multiple network elastomers by Creton's and Sijbesma's groups.86 A 1,2-dioxetane derivative was crosslinked in the filler network of multiple network elastomers; and in fracture tests, single, double, and triple network elastomers showed a very different scale of molecular damage upon crack propagation, as is revealed by luminescence due to the bond scission of 1,2-dioxetane that is shown in Fig. 35a. The blue light regions gradually increased with the numbers of networks in elastomers, consistent with the increase of toughness of multiple network elastomers. The same molecule was also used as a damage sensor190 in a commercial PDMS elastomer containing nanoparticles. The luminescence resulting from the 1,2-dioxetane scission was quantified in step-cyclic tests, and showed unambiguously the early molecular damage taking place directionally in nanocomposites as a function of strain. Interestingly, the time-resolved information showed that some damage also took place upon unloading (Fig. 35b).
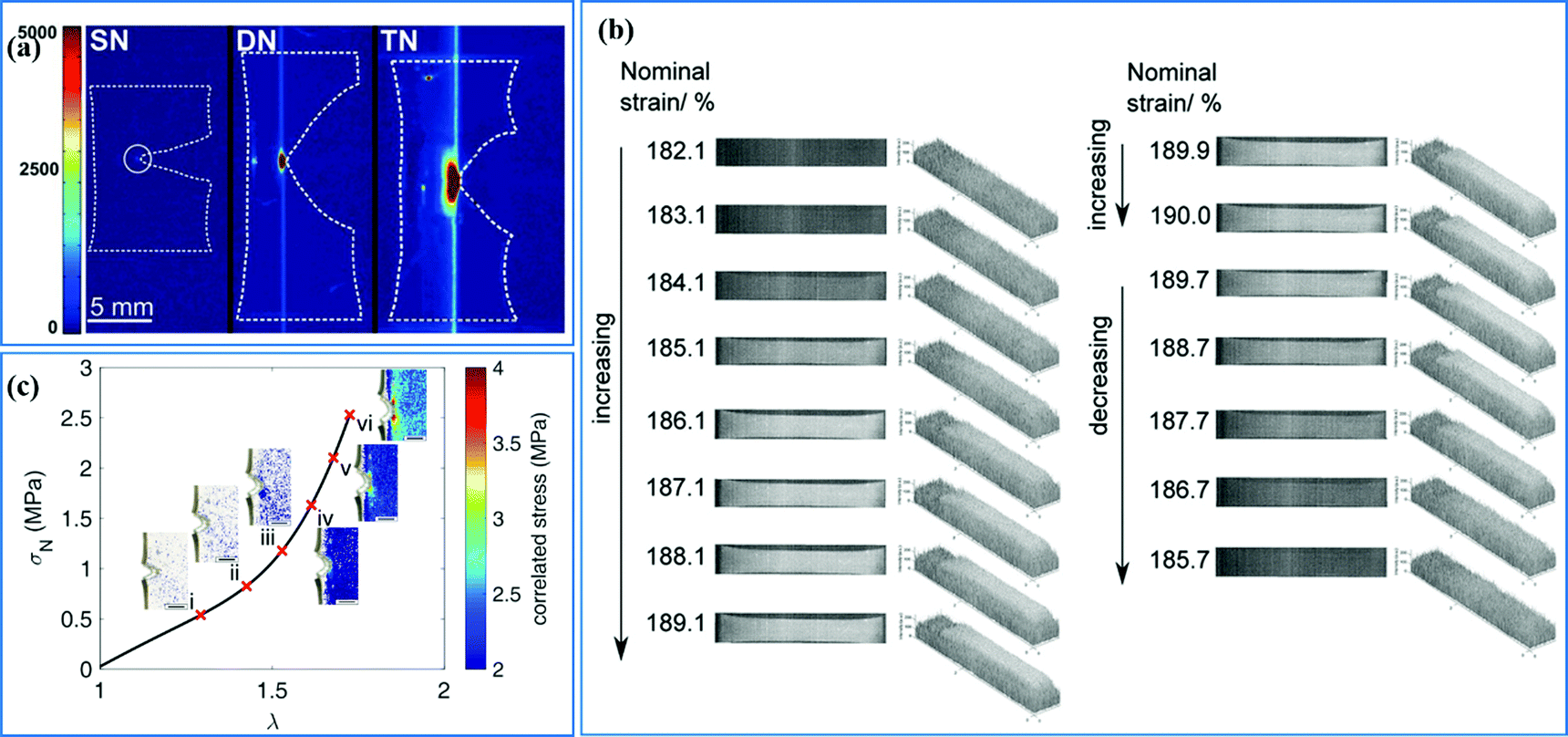 | ||
| Fig. 35 (a) 1,2-Dioxetane mechanophore as damage indicator showed the damaged region around crack tip for multiple network elastomers.86 (b) 1,2-Dioxetane mechanophore was integrated in polymer composite and the images show the luminescent intensity increase with strain and in relaxation luminescence gradually decrease.190 (c) SP was used as stress sensor to quantify the stress distribution around crack tip in multiple network elastomers. The regions and magnitude of stress around crack tip increase significantly with deformation.42 (a) is reprinted with permission from ref. 86, (published under a Creative Commons license, CC BY-NC), Copyright 2014 AAAS. (b) is reprinted with permission from ref. 190, Copyright 2016 Wiley-VCH and (c) is reprinted with permission from ref. 42, (published under a Creative Commons license, CC BY-NC), Copyright 2020 AAAS. | ||
Finally, it is important to mention the applications of a recently developed mechanofluorophore for damage detection. The Diels–Alder (DA) adduct of π-extended anthracene developed by Goestl and Sijbesma,63,142 emits a stable and high-yield fluorescent signal after scission of the DA bond. This new mechanofluorophore was used in particular to detect bond scission in hydrogels,187 and was used to devise a method to quantify bond scission in absolute terms by Slootman et al.181 By carrying out a series of fracture tests at different temperatures and stretch rates on pre-notched samples, they could demonstrate, for the first time, that bond scission during elastomer fracture increases up to 50 times with increasing stretch rate or decreasing temperature; it occurs over tens of microns near the crack tip, even in the absence of any sacrificial bonds; and it contributes significantly to the measured fracture energy, even at high strain rates, contradicting existing theories.229 An important point to validate the quantification of bond scission is the representativity of the mechanophore signal. In other words, although the mechanophore is a weaker bond than the normal C–C covalent bonds of the polymer backbone, is the fraction of broken DA bonds in the material equal to the fraction of broken strands? There is no absolute answer to this; but Slootman et al.181 compared the activation of a set of elastomers with the same total crosslinker concentration (and the same mechanical properties) but at different concentrations of mechanophores, and the activation was nearly proportional to the added concentration, as can be seen in Fig. 36.
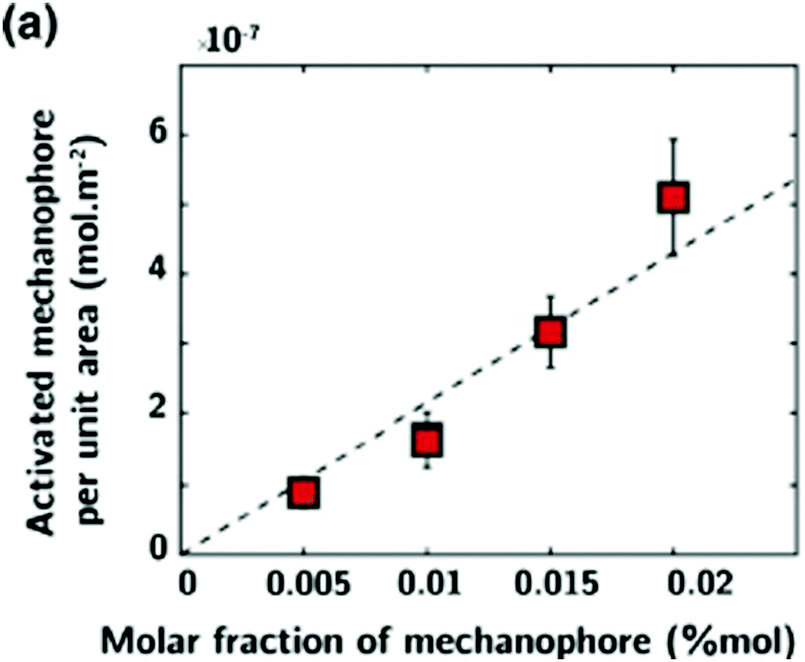 | ||
| Fig. 36 Linear dependence of fluorescence intensity with mechanophore concentration in the same material.181Fig. 36 is reprinted with permission from ref. 181, Copyright 2020 American Physical Society. | ||
4.2 Directed polymer degradation
In conventional degradable polymers, cleavable groups are normally located on the backbone of the polymer, and the degradable moieties degrade over time in an uncontrolled way in ambient conditions, making it difficult to have a polymer that is both durable and degradable. Mechanophores can be used here as mechanical triggers to protect the degradable motifs. When mechanophores are activated by an external force, they can introduce a degradable moiety into the backbone. In this way, the degradation of the polymer can be achieved on demand via mechanochemistry. Two similar cases were reported by the groups of Wang230 and Craig.231 They incorporated cyclobutene derivatives into the backbone, replacing the original degradable moieties. The cyclobutene derivatives – combined with suitable degradable moieties of lactone and the cyclobutene – acted as a ‘gate’ or ‘locker’, protecting the polymer backbone as shown in Fig. 37a. When the lactone hydrolyzed under a strong base, the polymer backbone stayed intact. However, when the ‘gate’ was opened by mechanical stimulus, the polymer materials could then be degraded in a basic solution (Fig. 37b). In this way, a mechanically-responsive, degradable polymer material was achieved.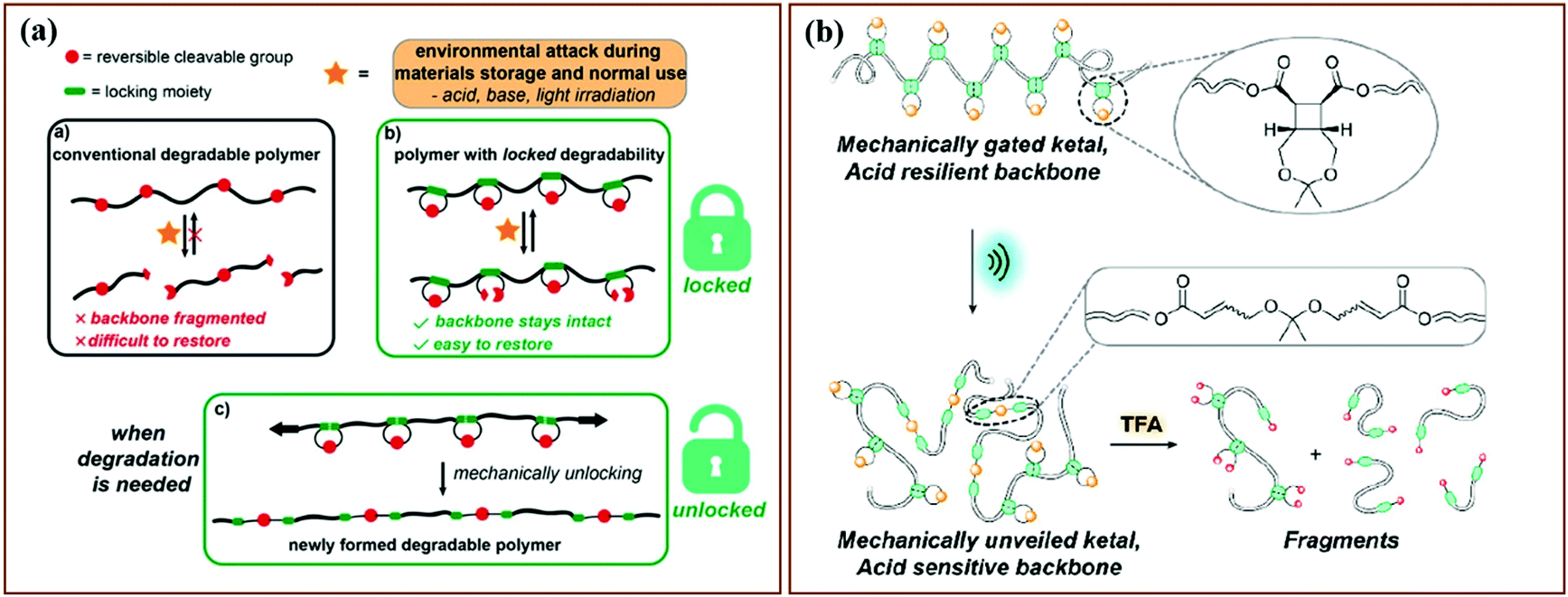 | ||
| Fig. 37 Mechanophores used as “locks” were integrated into the backbone of degradable polymer materials to control degradation of polymer materials. (a) The scheme of degradable polymer materials controlled by mechanophore activation.230 (b) The sequential degradation of polymer was initiated by the activation of mechanophores via sonication.231 (a) is reprinted with permission from ref. 230, Copyright 2020 American Chemical Society. (c) is reprinted with permission from ref. 231, Copyright 2020 American Chemical Society. | ||
4.3 Directed trigger/indicator or catalyst
Some mechanophores produce radicals or a catalytically-active site in response to a mechanical stimulus, as was described in Section 2; and the radicals or active sites can, in turn, initiate the polymerization of monomers at the damaged position. For instance, the polymer ruthenium(II)–NHC complex catalyzes the ring opening metathesis polymerization of the cyclooctene monomer.114 Another example is that of the mechanochromic HABI moiety, which can, under the application of a force, dissociate into two stable TPI radicals that act as initiators in the free radical polymerization of acrylates mediated by thiol-based chain transfer agents.104 The ability to trigger a polymerization reaction opens the possibility to design self-healing or self-reinforcement materials.Recently, Matsuda et al.99 achieved just that for double network hydrogels. They prepared tough double network hydrogels containing (in the water phase of the gel) ferrous ions, additional AMPS monomer, and crosslinker (Fig. 38c). Upon stretching the gel in an argon atmosphere, the scission of covalent bonds in the sacrificial network created radicals that, on the one hand, were detected by reacting with water to form hydrogen peroxide, which oxidized ferrous ions into ferric ions and changed the color of the solution to brown (Fig. 38b). On the other hand, when enough monomers were present in the water and monomers and crosslinkers were present in an equal ratio, the radicals formed upon scission triggered the polymerization and crosslinking, and reformed a sacrificial network that increased both stiffness (modulus) and stress at break (Fig. 38c). Despite the limitation of the argon atmosphere and relatively long polymerization time (12–24 hours), this clever proof-of-principle shows the potential of polymer mechanochemistry to repair even sophisticated materials, such as interpenetrated networks.
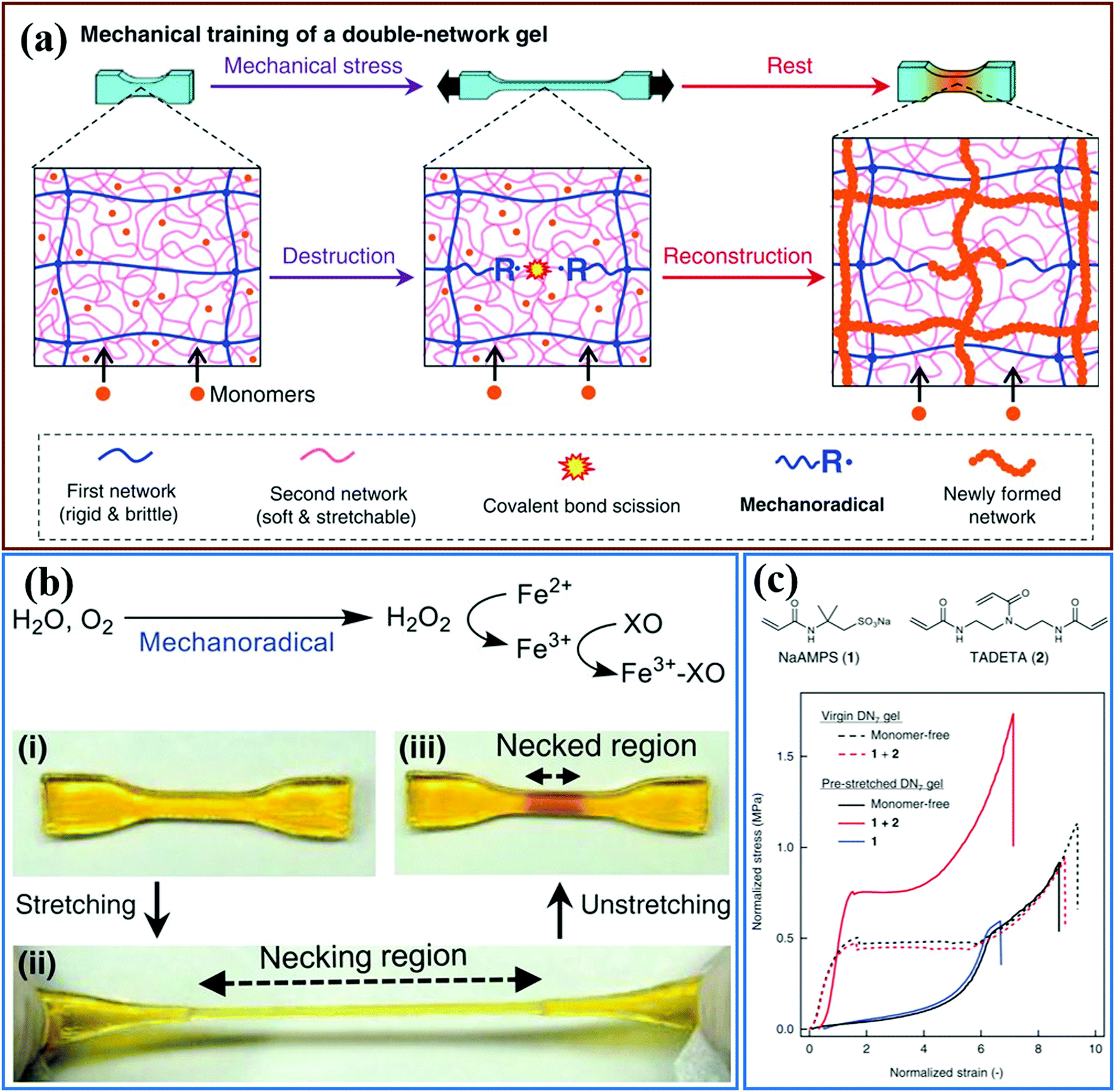 | ||
| Fig. 38 The radicals deriving from the bond scission were used to self-heal or strengthen the polymer materials. (a) The scheme of double network hydrogels strengthening via a mechanoradical initiated polymerization reaction due to bond scission. (b) The presence of mechanoradicals is indicated by a color change. Image (i to iii) shows the generation of mechanoradicals after stretching in the necked region. (c) Due to a new polymerization and crosslinking initiated by the mechanoradical, pre-stretched double network hydrogels reformed a sacrificial network and showed a significant strengthening of the mechanical properties.99Fig. 38 is adapted from ref. 99 (published under a Creative Commons license, CC BY-NC), Copyright 2019 AAAS. | ||
4.4 Force sensors
Depending on the specific design of the material and how the mechanophore is incorporated, the unique capability of mechanophores to elicit a change in optical properties upon application of a force on the molecular bond opens many possibilities in terms of sensing and mapping a higher level of deformation or local stress; and several examples have been reported.16,81,87–91,207 However, in polymeric networks the force on main chain bonds only becomes significant when the chain is highly stretched. In a random network of polymer chains, this happens only very close to the fracture point; and in polymer melts, it only occurs at very high strain rates and at temperatures close to Tg.25 Several tricks can, however, be played with the material architecture to reduce the overall stress and – to a greater extent – the overall strain at which mechanophores give an optical signal. They are all based on heterogeneities. Multiple interpenetrated networks create a dilute population of highly-stretched chains inside the material, so that if these chains are labeled with mechanophores, activation occurs at relatively low strains and well before macroscopic fracture.42,232 Nanofillers are another way to introduce heterogeneities, and hence cause some chains to be overstretched. Pores can also bring the same function. The work of Raisch et al.208 is a good example, where electrospun fibers containing SP in the main chain cause activation at 5% deformation of a PDMS/fiber composite.If the material is designed to be sensitive, the next hurdle is the calibration. Typically, uniaxial tension tests are carried out to match the optical signal with a value of stress or strain. This calibration curve can then in principle be used for an arbitrary geometry. There are however two main challenges:
– The relationship between macroscopic stress and fraction of activated molecules is going to depend on the 3D spatial organization of the mechanophore-containing polymer chains in the material, and will need to be established for any new material.
– Mechanophores can report on the intensity of the optical signal, reflecting the local concentration of activated molecules, but not on directionality. Yet, stress or strain are both tensors. Chen et al. have discussed this point, and proposed to use the maximum principal nominal stress as the activation criterion.40 This methodology makes it possible to apply the calibration curve obtained from uniaxial tension to more complex loading situations, and compare results with simulations.
Despite these difficulties mechanophores that activate with an optically-visible change in color may find many interesting applications and some examples are given below.
Mechanically-responsive polymer materials can also be used as a smart skin for soft robots207 or robot arms in order to detect the pressure or stress distribution in the operating process. In this case, a mechanochromic nanocomposite lowers the activation threshold in terms of strain; and the color change that occurs due to the mechanical response can then be used as input for other functions.
An interesting example is the application of mechanochromic polymer materials as an overcoat for a touch screen,233 combined with cross-aligned silver nanowire transparent conductive electrodes (AgNW TCE) layers, as shown in Fig. 39a. The SP functionalized PDMS overcoat changes color as a result of the application of a local pressure and the AnNW TCE layers detect the location of dynamic touch. Since different writing pressures lead to the different color intensities on the screen (as shown in Fig. 39a), the writing style can be quantified much more precisely by analyzing, with a spectrophotometer, the intensity of the color change. The local pressure can then be extracted from the representation of the color coordinating on the CIE 1931 color space. This technology is promising for personal information safety, since persons typically possess a unique writing habit and applied pressure on a pencil.
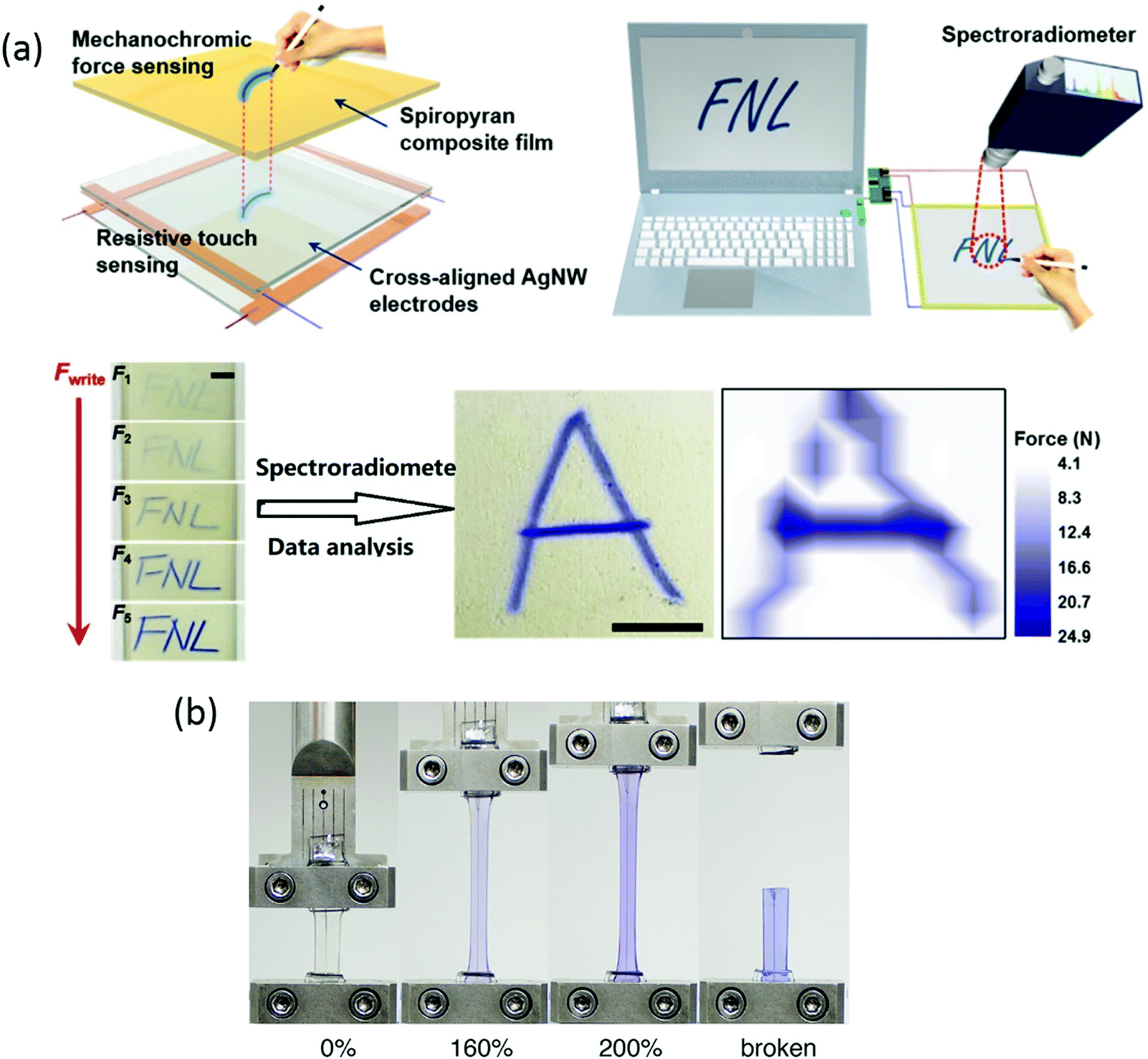 | ||
| Fig. 39 Mechanically responsive polymeric materials applied in electronic devices. (a) SP-containing PDMS was used as a tough screen where the writing force can be quantified by an optical technology.233 (b) A liquid metal wire was cast into the centre of an SP-containing PDMS elastomer and the material changed color in uniaxial tensile tests.234 (a) is adapted with permission from ref. 233, Copyright 2017 American Chemical Society. (b) is reprinted with permission from ref. 234, Copyright 2018 American Chemical Society. | ||
Another interesting example also came from Dickey and Craig's group.234 Liquid metal wires composed of a eutectic gallium indium alloy were embedded into the central section of the labelled PDMS materials as shown in Fig. 39b. Since the onset of color change can be tuned by adjusting the modulus of the silicone layering in the PDMS materials, the onset strain of color change can be matched with the maximum allowable deformation of the metal wires. This, therefore, gives the user a visual warning of the limiting stretch of these flexible electronics, as shown in Fig. 39b.
The SP-functionalized PDMS were also cured into a molded soft robot walker and gripper.235 In Fig. 40a, the color distribution reveals the spatial distribution of the maximum force during the actuation process of the gripper or walker – useful information for engineering design. Alternatively, rhodamine was integrated as a stress sensor into the filler network of multiple-network elastomers, and the materials used as robotic skin.52 The labelled elastomeric skin stuck on a finger, showed different colors during the action of making a fist (Fig. 40b). The skin became fluorescent when the finger bent, switching from blue to red, and then turning yellow as the finger straightened. These optical variations provide feedback loops, and open up a possibility of a feedback loop that is based on the optical information. In addition, similar mechanochromic polymer materials were also applied in biomimetic materials or devices by Craig,194 and Sprakel et al.228 For example, a biomimetic device was designed via electro-mechanochemically responsive elastomers to imitate the behavior of cephalopods, which can show various color patterns by selectively contracting muscles to reversibly activate chromophores (Fig. 40c).
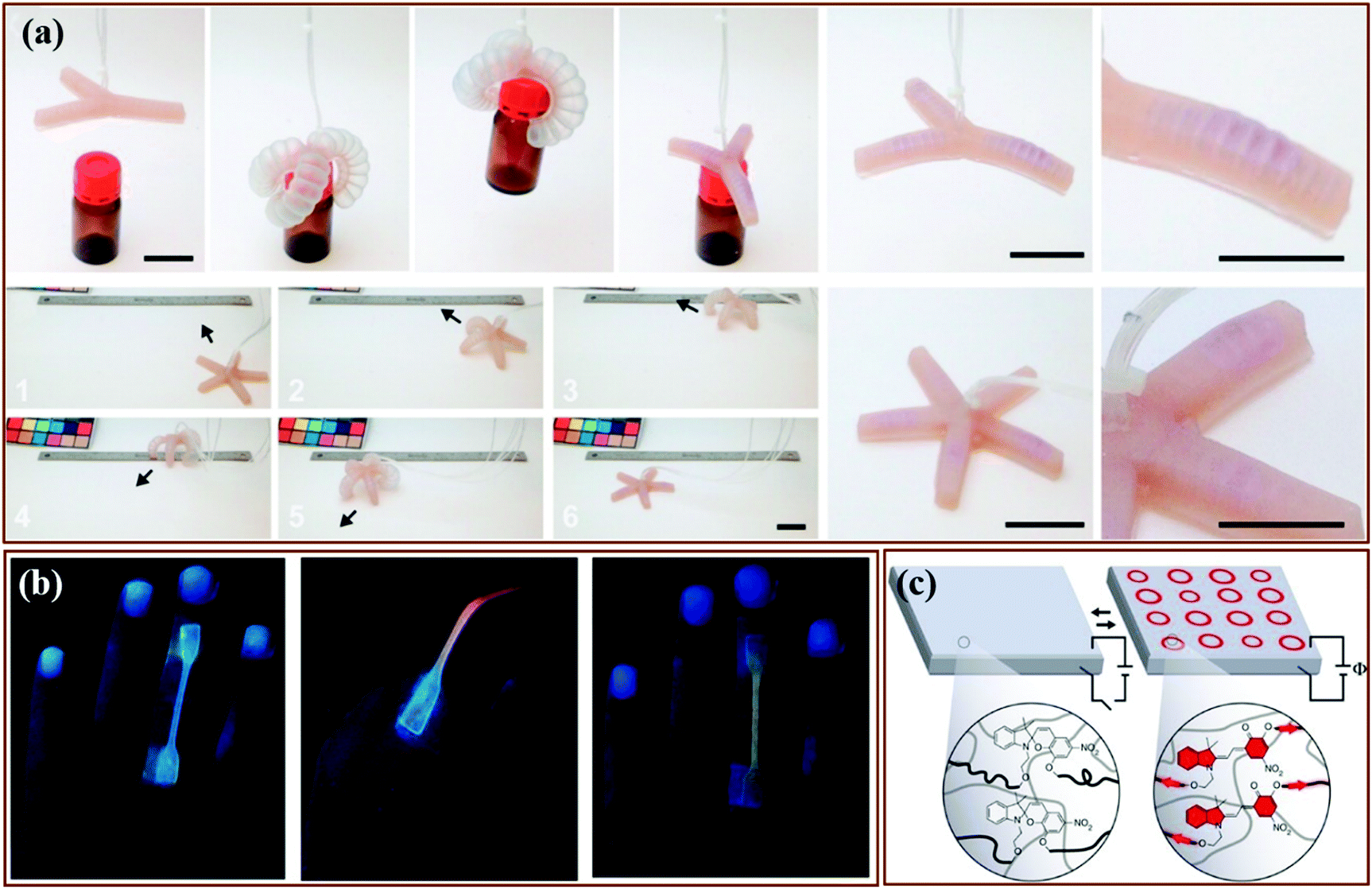 | ||
| Fig. 40 Mechanical responsive polymer materials are applied in soft robots and biomimetic device. (a) SP-containing PDMS was casted into a gripper and worker. The two robots showed color change in actuation process and indicated the high stress location after actuation.235 (b) Multiple network elastomers coupled of rhodamine acted as skin of finger and exhibited different fluorescent colors during the bending and straightening of finger.52 (c) SP was incorporated into PDMS network. PDMS materials combining with other electronic device was assembled to be an electro-mechanochemically responsive elastomers. The schematic shows the mechanism of the elastomers actuation.194 (a) is reprinted with permission from ref. 235, Copyright 2015 American Chemical Society. (b) is reprinted with permission from ref. 52, Copyright 2017 American Chemical Society. (c) is reprinted with permission from ref. 194, Copyright 2014 Springer Nature. | ||
5. Conclusion and outlook
The surge of research activities in polymer mechanochemistry over the last 15 years has led to remarkable progress. Important new insights have been obtained in the physical chemical background of bond activation by mechanical force. Mechanophores with a large range of optical responses have been introduced with high sensitivities for the detection of small forces in a small number of chains; and these mechanophores have been used to understand failure mechanisms in a wide range of polymeric materials, while other optical mechanophores show promise as force sensors. Mechanophores with chemical responses have been able to direct polymer degradation, or to lead reinforcement of polymer materials, by initiating a polymerization reaction. As a conclusion to this review, we would like to pinpoint a few areas where, in our personal view, this flourishing field has the most potential to contribute to future insights and applications.– With the development of extreme molecular force sensors, the road is now open towards application of synthetic mechanophores to the study of biological phenomena in vivo, such as cell adhesion, mechanotransduction and subcellular viscosity patterns.236 In mechanobiology, much progress can be made by combining the specificity of biological force sensors with the smaller size and versatility of fully synthetic mechanophores. The capability to release small molecules with a mechanical stimulus promises applications for targeted drug release.
– Optical mechanophores find increasing use in the study of failure of polymer materials. Although we reported multiple examples of mapping strains or bond scission in a given material, the main challenge now ahead is to be quantitative in an absolute way in order to be able to compare activation in different materials and compare experimental data with molecular models or simulations. Along this line there are two main challenges: absolute calibration of the signal with a reference sample as discussed in a recent work of Slootman et al.,181 and representativity of the data. Since mechanophores are typically weaker bonds than C–C covalent bonds, two questions need to be addressed: (1) is the mechanophore altering the properties of the material into which it is incorporated? and (2) is the mechanophore correctly reporting what is happening to C–C bonds? A second important current limitation is that optical mechanophores require transparent or translucent materials while many engineering polymeric materials are opaque. Infrared dyes, or the detection of radicals could be interesting options for the development of probes for opaque materials.
– A more sustainable economy requires durable and recyclable materials. There are ample opportunities for mechanochemistry to play a role in this area. Stress and damage sensors can help extend the service lifetime of polymer materials, provided that the sensors are robust and have a simple readout. Another exciting prospect is the use of mechanocatalysts and mechanoradical initiators to promote polymerization and crosslinking in self-healing materials. While proof-of-concept has been demonstrated with catalysts as well as radical initiators, much work needs to be done before such self-healing schemes will be practical for commercial materials.
Making these opportunities a reality will not only require creativity and imagination – characteristics that our colleagues in the field of polymer mechanochemistry have demonstrated to possess in abundance – but also sustained interdisciplinary work and close collaborations between organic chemists, physicists, and solid mechanics experts. We thank our community for the inspiring work that has greatly enriched the scientific literature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. P. Sijbesma, Y. Chen and D. van Luijk have received funding from the Dutch research council (NWO), project numbers 024.001.035 and 731.016.202. C. Creton and G. Mellot have received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement AdG No 695351 and grant agreement ERC Proof of Concept 2019 N° 899555.References
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS.
- S. Garcia-Manyes and A. E. M. Beedle, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- T. Stauch and A. Dreuw, Chem. Rev., 2016, 116, 14137–14180 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 18–33, DOI:10.1002/ejoc.201700961.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS.
- G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- B. Cheng and S. Cui, in Polymer Mechanochemistry, ed. R. Boulatov, Springer International Publishing, Berlin, 2015, pp. 97–134, DOI:10.1007/128_2015_628.
- J. Liang and J. M. Fernandez, ACS Nano, 2009, 3, 1628–1645 CrossRef CAS.
- Y. Liu and G. J. Vancso, Prog. Polym. Sci., 2020, 104, 101232 CrossRef CAS.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS.
- Y. Li and S. S. Sheiko, in Polymer Mechanochemistry, ed. R. Boulatov, Springer International Publishing, Berlin, 2015, pp. 1–36, DOI:10.1007/128_2015_627.
- J. Ribas-Arino and D. Marx, Chem. Rev., 2012, 112, 5412–5487 CrossRef CAS.
- Q. M. Zhang and M. J. Serpe, in Polymer Mechanochemistry, ed. R. Boulatov, Springer International Publishing, Berlin, 2015, pp. 377–424, DOI:10.1007/128_2015_626.
- A. L. Black, J. M. Lenhardt and S. L. Craig, J. Mater. Chem., 2011, 21, 1655–1663 RSC.
- J. M. Clough, A. Balan and R. P. Sijbesma, in Polymer Mechanochemistry, ed. R. Boulatov, Springer International Publishing, Berlin, 2015, pp. 209–238, DOI:10.1007/128_2015_641.
- M. Li, Q. Zhang, Y.-N. Zhou and S. Zhu, Prog. Polym. Sci., 2018, 79, 26–39 CrossRef CAS.
- Y. Yuan and Y.-l. Chen, Chin. J. Polym. Sci., 2017, 35, 1315–1327 CrossRef CAS.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- G. Chen, Y. Cui and X. Chen, Chem. Soc. Rev., 2019, 48, 1434–1447 RSC.
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, Polym. Chem., 2013, 4, 4846–4859 RSC.
- L. Anderson and R. Boulatov, Advances in Physical Organic Chemistry, 2018, vol. 52, pp. 87–143 Search PubMed.
- C. L. Brown and S. L. Craig, Chem. Sci., 2015, 6, 2158–2165 RSC.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS.
- N. Deneke, M. L. Rencheck and C. S. Davis, Soft Matter, 2020, 16, 6230–6252 RSC.
- M. Stratigaki and R. Gostl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS.
- K. M. Wiggins, J. N. Brantley and C. W. Bielawski, Chem. Soc. Rev., 2013, 42, 7130–7147 RSC.
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Chem, 2018, 4, 2499–2537 CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109–1112 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS.
- H. J. Kulik, in Reviews in Computational Chemistry, ed. A. L. Parrill and K. B. Lipkowitz, John Wiley & Sons, Inc., 2019, ch. 266, pp. 266–311 Search PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS.
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 6148–6151 CrossRef CAS.
- G. O'Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594–1600 CrossRef.
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 6148–6151 CrossRef CAS.
- M. H. Barbee, T. Kouznetsova, S. L. Barrett, G. R. Gossweiler, Y. Lin, S. K. Rastogi, W. J. Brittain and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 12746–12750 CrossRef CAS.
- Y. Lin, M. H. Barbee, C.-C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS.
- M. H. Barbee, T. Kouznetsova, S. L. Barrett, G. R. Gossweiler, Y. Lin, S. K. Rastogi, W. J. Brittain and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 12746–12750 CrossRef CAS.
- Y. Chen, J. Yeh, Y. Qi, R. Long and C. Creton, Sci. Adv., 2020, 6, eaaz5093 CrossRef CAS.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142(38), 16364–16381 CrossRef CAS.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS.
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS.
- H. Montenegro, M. Di Paolo, D. Capdevila, P. F. Aramendía and M. L. Bossi, Photochem. Photobiol. Sci., 2012, 11, 1081–1086 RSC.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS.
- T. Wang, N. Zhang, Y. Ge, C. Wang, Z. Hang and Z. Zhang, Macromol. Chem. Phys., 2020, 221, 1900463 CrossRef CAS.
- T. van de Laar, H. Schuurman, P. van der Scheer, J. Maarten van Doorn, J. van der Gucht and J. Sprakel, Chem, 2018, 4, 269–284 CAS.
- Y.-K. Song, K.-H. Lee, W.-S. Hong, S.-Y. Cho, H.-C. Yu and C.-M. Chung, J. Mater. Chem., 2012, 22, 1380–1386 RSC.
- D. C. Church, G. I. Peterson and A. J. Boydston, ACS Macro Lett., 2014, 3, 648–651 CrossRef CAS.
- C. P. Kabb, C. S. O'Bryan, C. D. Morley, T. E. Angelini and B. S. Sumerlin, Chem. Sci., 2019, 10, 7702–7708 RSC.
- H. Li, R. Göstl, M. Delgove, J. Sweeck, Q. Zhang, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2016, 5, 995–998 CrossRef CAS.
- J. Li, T. Shiraki, B. Hu, R. A. Wright, B. Zhao and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 15925–15928 CrossRef CAS.
- G. I. Peterson, J. Lee and T.-L. Choi, Macromolecules, 2019, 52, 9561–9568 CrossRef CAS.
- A. R. Sulkanen, J. Sung, M. J. Robb, J. S. Moore, N. R. Sottos and G. Y. Liu, J. Am. Chem. Soc., 2019, 141, 4080–4085 CrossRef CAS.
- S. S. M. Konda, J. N. Brantley, B. T. Varghese, K. M. Wiggins, C. W. Bielawski and D. E. Makarov, J. Am. Chem. Soc., 2013, 135, 12722–12729 CrossRef CAS.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC.
- J. Li, B. Hu, K. Yang, B. Zhao and J. S. Moore, ACS Macro Lett., 2016, 5, 819–822 CrossRef CAS.
- Y. Min, S. Huang, Y. Wang, Z. Zhang, B. Du, X. Zhang and Z. Fan, Macromolecules, 2015, 48, 316–322 CrossRef CAS.
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS.
- Z. S. Kean, G. R. Gossweiler, T. B. Kouznetsova, G. B. Hewage and S. L. Craig, Chem. Commun., 2015, 51, 9157–9160 RSC.
- Y. Zhang, E. Lund, G. R. Gossweiler, B. Lee, Z. Niu, C. Khripin, E. Munch, M. Couty and S. L. Craig, Macromol. Rapid Commun., 2020, 2000359, DOI:10.1002/marc.202000359.
- M. Karman, E. Verde-Sesto, C. Weder and Y. C. Simon, ACS Macro Lett., 2018, 7, 1099–1104 CrossRef CAS.
- M. Li, H. Zhang, F. Gao, Z. Tang, D. Zeng, Y. Pan, P. Su, Y. Ruan, Y. Xu and W. Weng, Polym. Chem., 2019, 10, 905–910 RSC.
- M. Karman, E. Verde-Sesto and C. Weder, ACS Macro Lett., 2018, 7, 1028–1033 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS.
- Z. a. Li, R. Toivola, F. Ding, J. Yang, P.-N. Lai, T. Howie, G. Georgeson, S.-H. Jang, X. Li, B. D. Flinn and A. K. Y. Jen, Adv. Mater., 2016, 28, 6592–6597 CrossRef CAS.
- T. Muramatsu, Y. Sagara, H. Traeger, N. Tamaoki and C. Weder, ACS Appl. Mater. Interfaces, 2019, 11, 24571–24576 CrossRef CAS.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874–881 CAS.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS.
- K. Imato, R. Yamanaka, H. Nakajima and N. Takeda, Chem. Commun., 2020, 56, 7937–7940 RSC.
- A. Aerts, S. J. D. Lugger, J. P. A. Heuts and R. P. Sijbesma, Macromol. Rapid Commun., 2020, 2000476, DOI:10.1002/marc.202000476.
- W. Yang, S. Wang, R. Li, J. Xu and W. Hao, React. Funct. Polym., 2018, 133, 57–65 CrossRef CAS.
- W. Zhang Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 560–574 CrossRef.
- G. A. Filonenko and J. R. Khusnutdinova, Adv. Mater., 2017, 29, 1700563 CrossRef.
- G. A. Filonenko, J. A. M. Lugger, C. Liu, E. P. A. van Heeswijk, M. M. R. M. Hendrix, M. Weber, C. Müller, E. J. M. Hensen, R. P. Sijbesma and E. A. Pidko, Angew. Chem., Int. Ed., 2018, 57, 16385–16390 CrossRef CAS.
- G. A. Filonenko, D. Sun, M. Weber, C. Müller and E. A. Pidko, J. Am. Chem. Soc., 2019, 141, 9687–9692 CrossRef CAS.
- Y. Chen, A. J. Spiering, S. Karthikeyan, G. W. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS.
- N. J. Turro, P. Lechtken, N. E. Schore, G. Schuster, H.-C. Steinmetzer and A. Yekta, Acc. Chem. Res., 1974, 7, 97–105 CrossRef CAS.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS.
- G. B. Schuster, N. J. Turro, H.-C. Steinmetzer, A. P. Schaap, G. Paler, W. Adam and J. C. Liu, J. Am. Chem. Soc., 1975, 97, 7110–7118 CrossRef CAS.
- P. Millereau, E. Ducrot, J. M. Clough, M. E. Wiseman, H. R. Brown, R. P. Sijbesma and C. Creton, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9110–9115 CrossRef CAS.
- J. M. Clough, A. Balan, T. L. J. van Daal and R. P. Sijbesma, Angew. Chem., Int. Ed., 2016, 55, 1445–1449 CrossRef CAS.
- H. Lebel, M. K. Janes, A. B. Charette and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5046–5047 CrossRef CAS.
- M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579–2582 CrossRef CAS.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS.
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC.
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1087–1091 CrossRef CAS.
- Y. Mori, N. Yamada, M. Kanazawa, Y. Horikoshi, Y. Watanabe and K. Maeda, Bull. Chem. Soc. Jpn., 1996, 69, 2355–2359 CrossRef CAS.
- K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 556–560 CrossRef CAS.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem. – Eur. J., 2018, 24, 3170–3173 CrossRef CAS.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS.
- H. Sakai, D. Aoki, K. Seshimo, K. Mayumi, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, ACS Macro Lett., 2020, 9, 1108–1113 CrossRef CAS.
- K. Imato, T. Kanehara, S. Nojima, T. Ohishi, Y. Higaki, A. Takahara and H. Otsuka, Chem. Commun., 2016, 52, 10482–10485 RSC.
- H. Oka, K. Imato, T. Sato, T. Ohishi, R. Goseki and H. Otsuka, ACS Macro Lett., 2016, 5, 1124–1127 CrossRef CAS.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- F. Verstraeten, R. Gostl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- A. Dowon, PhD thesis, University of Michigan, 2017.
- Q. Wu, Y. Yuan, F. Chen, C. Sun, H. Xu and Y. Chen, ACS Macro Lett., 2020, 9, 1547–1551 CrossRef CAS.
- J. Xia, P. Zhao, S. Pan and H. Xu, ACS Macro Lett., 2019, 8, 629–633 CrossRef.
- A. P. Wiita, S. R. K. Ainavarapu, H. H. Huang and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7222–7227 CrossRef CAS.
- P. Dopieralski, J. Ribas–Arino, P. Anjukandi, M. Krupicka and D. Marx, Nat. Chem., 2016, 9, 164–170 CrossRef.
- W. Li and F. Gräter, J. Am. Chem. Soc., 2010, 132, 16790–16795 CrossRef CAS.
- F. Wang, M. Burck and C. E. Diesendruck, ACS Macro Lett., 2017, 6, 42–45 CrossRef CAS.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS.
- R. Groote, B. M. Szyja, E. A. Pidko, E. J. M. Hensen and R. P. Sijbesma, Macromolecules, 2011, 44, 9187–9195 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS.
- P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS.
- P. Michael, S. K. Sheidaee Mehr and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3893–3907 CrossRef CAS.
- C. E. Diesendruck, B. D. Steinberg, N. Sugai, M. N. Silberstein, N. R. Sottos, S. R. White, P. V. Braun and J. S. Moore, J. Am. Chem. Soc., 2012, 134, 12446–12449 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 99–103 CrossRef CAS.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2014, 136, 1276–1279 CrossRef CAS.
- Y. Sha, Y. Zhang, E. Xu, Z. Wang, T. Zhu, S. L. Craig and C. Tang, ACS Macro Lett., 2018, 7, 1174–1179 CrossRef CAS.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS.
- B. Lee, Z. Niu, J. Wang, C. Slebodnick and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 10826–10832 CrossRef CAS.
- A. L. B. Ramirez, Z. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Nat. Chem., 2013, 5, 757–761 CrossRef CAS.
- J. Wang, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 11554–11557 CrossRef CAS.
- J. Wang, T. B. Kouznetsova, R. Boulatov and S. L. Craig, Nat. Commun., 2016, 7, 13433 CrossRef.
- Y. Lin, T. B. Kouznetsova, C.-C. Chang and S. L. Craig, Nat. Commun., 2020, 11, 4987 CrossRef CAS.
- M. H. Barbee, J. Wang, T. Kouznetsova, M. Lu and S. L. Craig, Macromolecules, 2019, 52, 6234–6240 CrossRef CAS.
- H. Zhang, X. Li, Y. Lin, F. Gao, Z. Tang, P. Su, W. Zhang, Y. Xu, W. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS.
- C. Calvino, L. Neumann, C. Weder and S. Schrettl, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 640–652 CrossRef CAS.
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC.
- R. Göstl, J. M. Clough and R. P. Sijbesma, in Mechanochemistry in Materials, ed. C. Y. Simon and L. S. Craig, Polymer Chemistry Series, Royal Society of Chemistry, 2018, no. 26, ch. 3, pp. 53–75 Search PubMed.
- L. W. McKeen, The Effect of UV Light and Weather on Plastics and Elastomers, Elsevier, 2019, ch. 1, pp. 1–20 DOI:10.1016/b978-0-12-816457-0.00001-0.
- M. Beija, M.-T. Charreyre and J. M. G. Martinho, Prog. Polym. Sci., 2011, 36, 568–602 CrossRef CAS.
- S. Kramer, B. Beiermann, D. Davis, N. Sottos, S. White and J. Moore, presented in part at the Proceedings of the SEM Annual Conference, Indianapolis, Indiana USA, 2013.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS.
- S. K. Surampudi, H. R. Patel, G. Nagarjuna and D. Venkataraman, Chem. Commun., 2013, 49, 7519–7521 RSC.
- M. van Horn, P. Smith, B. P. Mason, J. R. Hemmer, J. Read de Alaniz, J. P. Hooper and S. Osswald, J. Appl. Phys., 2015, 117, 043103 CrossRef.
- K. Wei, Z. Gao, H. Liu, X. Wu, F. Wang and H. Xu, ACS Macro Lett., 2017, 6, 1146–1150 CrossRef CAS.
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Göstl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS.
- B. A. Beiermann, D. A. Davis, S. L. B. Kramer, J. S. Moore, N. R. Sottos and S. R. White, J. Mater. Chem., 2011, 21, 8443–8447 RSC.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS.
- M. E. Grady, B. A. Beiermann, J. S. Moore and N. R. Sottos, ACS Appl. Mater. Interfaces, 2014, 6, 5350–5355 CrossRef CAS.
- G. O’Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594–1600 CrossRef.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS.
- Y. Chen and R. P. Sijbesma, Macromolecules, 2014, 47, 3797–3805 CrossRef CAS.
- Y. Chen, H. Zhang, X. Fang, Y. Lin, Y. Xu and W. Weng, ACS Macro Lett., 2014, 3, 141–145 CrossRef CAS.
- X. Fang, H. Zhang, Y. Chen, Y. Lin, Y. Xu and W. Weng, Macromolecules, 2013, 46, 6566–6574 CrossRef CAS.
- G. Hong, H. Zhang, Y. Lin, Y. Chen, Y. Xu, W. Weng and H. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
- S.-J. Kim and D. H. Reneker, Polym. Bull., 1993, 31, 367–374 CrossRef CAS.
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746–3752 CrossRef CAS.
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef CAS.
- M. F. Rubner, Macromolecules, 1986, 19, 2114–2128 CrossRef CAS.
- M. F. Rubner, Macromolecules, 1986, 19, 2129–2138 CrossRef CAS.
- E. Seo, J. Choi, B. Lee, Y.-A. Son and K. J. Lee, Sci. Rep., 2019, 9, 18648 CrossRef CAS.
- M. Torelli, F. Terenziani, A. Pedrini, F. Guagnini, I. Domenichelli, C. Massera and E. Dalcanale, ChemistryOpen, 2020, 9, 261–268 CrossRef CAS.
- H. Zhang, Y. Chen, Y. Lin, X. Fang, Y. Xu, Y. Ruan and W. Weng, Macromolecules, 2014, 47, 6783–6790 CrossRef CAS.
- Q. Zhang, Y. Wang, C. Xing, Y. Cai, K. Xi and X. Jia, RSC Adv., 2017, 7, 12682–12689 RSC.
- R. Groote, L. van Haandel and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4929–4935 CrossRef CAS.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS.
- Y. Jia, W.-J. Wang, B.-G. Li and S. Zhu, Macromol. Mater. Eng., 2018, 303, 1800154 CrossRef.
- K. Kawasaki, D. Aoki and H. Otsuka, Macromol. Rapid Commun., 2020, 41, 1900460 CrossRef CAS.
- H. Li, Y. Zhang, Y. Liu, R. P. Sijbesma, J. P. A. Heuts and Q. Zhang, Polym. Chem., 2017, 8, 3971–3976 RSC.
- P. Michael, M. Biewend and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1800376 CrossRef.
- S. Jiang, L. Zhang, T. Xie, Y. Lin, H. Zhang, Y. Xu, W. Weng and L. Dai, ACS Macro Lett., 2013, 2, 705–709 CrossRef CAS.
- L.-J. Wang, X.-J. Zhou, X.-H. Zhang and B.-Y. Du, Macromolecules, 2016, 49, 98–104 CrossRef CAS.
- B. H. Bowser and S. L. Craig, Polym. Chem., 2018, 9, 3583–3593 RSC.
- J. M. Lenhardt, A. L. Black Ramirez, B. Lee, T. B. Kouznetsova and S. L. Craig, Macromolecules, 2015, 48, 6396–6403 CrossRef CAS.
- B. H. Bowser, C.-H. Ho and S. L. Craig, Macromolecules, 2019, 52, 9032–9038 CrossRef CAS.
- A.-D. N. Celestine, B. A. Beiermann, P. A. May, J. S. Moore, N. R. Sottos and S. R. White, Polymer, 2014, 55, 4164–4171 CrossRef CAS.
- A. D. N. Celestine, N. R. Sottos and S. R. White, Strain, 2019, 55, e12310 CrossRef.
- C. M. Degen, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2013, 46, 8917–8921 CrossRef CAS.
- J. R. Hemmer, P. D. Smith, M. van Horn, S. Alnemrat, B. P. Mason, J. R. de Alaniz, S. Osswald and J. P. Hooper, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1347–1356 CrossRef CAS.
- J. W. Kim, Y. Jung, G. W. Coates and M. N. Silberstein, Macromolecules, 2015, 48, 1335–1342 CrossRef CAS.
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, J. Mater. Chem., 2011, 21, 8381–8388 RSC.
- C. K. Lee, C. E. Diesendruck, E. Lu, A. N. Pickett, P. A. May, J. S. Moore and P. V. Braun, Macromolecules, 2014, 47, 2690–2694 CrossRef CAS.
- J. M. Clough, J. van der Gucht and R. P. Sijbesma, Macromolecules, 2017, 50, 2043–2053 CrossRef CAS.
- M. Li, L. Lei, Q. Zhang and S. Zhu, Macromol. Rapid Commun., 2016, 37, 957–962 CrossRef CAS.
- J. Slootman, V. Waltz, C. J. Yeh, C. Baumann, R. Göstl, J. Comtet and C. Creton, In ArXiv, ArXiv cond-mat.soft, 2020.
- M. Li, W. Liu, Q. Zhang and S. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15156–15163 CrossRef CAS.
- S. V. Kurmaz, M. L. Bubnova, E. O. Perepelitsina and G. A. Estrina, Polym. Sci., Ser. A, 2006, 48, 696–706 CrossRef.
- H. J. Naghash and S. Mallakpour, Iran. Polym. J., 2004, 13, 287–295 CAS.
- R. Mao, Y. Liu, M. B. Huglin and P. A. Holmes, Macromolecules, 1995, 28, 6739–6744 CrossRef CAS.
- A. K. Tripathi, M. L. Neenan, D. C. Sundberg and J. G. Tsavalas, Polymer, 2016, 96, 130–145 CrossRef CAS.
- M. Stratigaki, C. Baumann, L. C. A. van Breemen, J. P. A. Heuts, R. P. Sijbesma and R. Göstl, Polym. Chem., 2020, 11, 358–366 RSC.
- D. R. T. Roberts, M. Patel, J. J. Murphy and S. J. Holder, Sens. Actuators, B, 2012, 162, 43–56 CrossRef CAS.
- N. A. A. Rossi, E. J. Duplock, J. Meegan, D. R. T. Roberts, J. J. Murphy, M. Patel and S. J. Holder, J. Mater. Chem., 2009, 19, 7674–7686 RSC.
- J. M. Clough, C. Creton, S. L. Craig and R. P. Sijbesma, Adv. Funct. Mater., 2016, 26, 9063–9074 CrossRef CAS.
- A. E. Früh, F. Artoni, R. Brighenti and E. Dalcanale, Chem. Mater., 2017, 29, 7450–7457 CrossRef.
- T. A. Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2018, 51, 9177–9183 CrossRef CAS.
- Y. Lin, M. H. Barbee, C. C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, Nat. Commun., 2014, 5, 4899 CrossRef.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, J. Mech. Phys. Solids, 2015, 82, 320–344 CrossRef CAS.
- Z. Xia, V. D. Alphonse, D. B. Trigg, T. P. Harrigan, J. M. Paulson, Q. T. Luong, E. P. Lloyd, M. H. Barbee and S. L. Craig, Molecules, 2019, 24, 542 CrossRef.
- L. Li, C. Peng, Q. Xu, S. Chen, Y. He, W. Xu and J. Jiang, Mater. Lett., 2018, 214, 150–153 CrossRef CAS.
- G. Kister, M. Moniruzzaman, M. Khan and S. Debnath, Dyes Pigm., 2019, 162, 309–314 CrossRef CAS.
- K. Imato and H. Otsuka, Polymer, 2018, 137, 395–413 CrossRef CAS.
- N. Yoshie, S. Saito and N. Oya, Polymer, 2011, 52, 6074–6079 CrossRef CAS.
- R. Gunckel, E. Nofen, J. Hansen-Staggs, S. Babcock, B. Koo, A. Chattopadhyay and L. L. Dai, Mater. Res. Express, 2017, 4, 075014 CrossRef.
- B. Koo, J. Miller, R. Gunckel, A. Hall, L. Dai and A. Chattopadhyay, Smart Mater. Struct., 2019, 28, 115035 CrossRef CAS.
- B. Koo, E. Nofen, A. Chattopadhyay and L. Dai, Comput. Mater. Sci., 2017, 133, 167–174 CrossRef CAS.
- E. M. Nofen, A. Dasgupta, N. Zimmer, R. Gunckel, B. Koo, A. Chattopadhyay and L. L. Dai, Polym. Eng. Sci., 2017, 57, 901–909 CrossRef CAS.
- E. M. Nofen, J. Wickham, B. Koo, A. Chattopadhyay and L. L. Dai, Mater. Res. Express, 2016, 3, 035701 CrossRef.
- E. M. Nofen, N. Zimmer, A. Dasgupta, R. Gunckel, B. Koo, A. Chattopadhyay and L. L. Dai, Polym. Chem., 2016, 7, 7249–7259 RSC.
- J. Park, Y. Lee, M. H. Barbee, S. Cho, S. Cho, R. Shanker, J. Kim, J. Myoung, M. P. Kim, C. Baig, S. L. Craig and H. Ko, Adv. Mater., 2019, 31, 1808148 CrossRef.
- M. Raisch, D. Genovese, N. Zaccheroni, S. B. Schmidt, M. L. Focarete, M. Sommer and C. Gualandi, Adv. Mater., 2018, 30, 1802813 CrossRef.
- J. Zou, Y. Liu, B. Shan, A. Chattopadhyay and L. L. Dai, Smart Mater. Struct., 2014, 23, 095038 CrossRef.
- T. A. Kim, C. Lamuta, H. Kim, C. Leal and N. R. Sottos, Adv. Sci., 2020, 1903464, DOI:10.1002/advs.201903464.
- T. Kosuge, K. Imato, R. Goseki and H. Otsuka, Macromolecules, 2016, 49, 5903–5911 CrossRef CAS.
- Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, J. Am. Chem. Soc., 2014, 136, 4273–4280 CrossRef CAS.
- J. W. Woodcock, R. Beams, C. S. Davis, N. Chen, S. J. Stranick, D. U. Shah, F. Vollrath and J. W. Gilman, Adv. Mater. Interfaces, 2017, 4, 1601018 CrossRef.
- G. J. Price and P. F. Smith, Polymer, 1993, 34, 4111–4117 CrossRef CAS.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Polym. Int., 2013, 62, 2–12 CrossRef CAS.
- Z. S. Kean, A. L. Black Ramirez, Y. Yan and S. L. Craig, J. Am. Chem. Soc., 2012, 134, 12939–12942 CrossRef CAS.
- M. J. Kryger, A. M. Munaretto and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 18992–18998 CrossRef CAS.
- L. Xue, U. S. Agarwal and J. P. Lemstra, Macromolecules, 2005, 38, 8825–8832 CrossRef CAS.
- B. A. Beiermann, S. L. B. Kramer, J. S. Moore, S. R. White and N. R. Sottos, ACS Macro Lett., 2012, 1, 163–166 CrossRef CAS.
- Z. S. Kean, J. L. Hawk, S. Lin, X. Zhao, R. P. Sijbesma and S. L. Craig, Adv. Mater., 2014, 26, 6013–6018 CrossRef CAS.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- A. N. Gent, Langmuir, 1996, 12, 4492–4496 CrossRef CAS.
- C. Li, Y. Zhang, J. Hu, J. Cheng and S. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS.
- M. Li, W. Liu and S. Zhu, Polymer, 2017, 112, 219–227 CrossRef CAS.
- A. M. Striegel, J. Biochem. Biophys. Methods, 2003, 56, 117–139 CrossRef CAS.
- W. Döll and L. Könczöl, in Advances in Polymer Science, ed. H.-H. Kausch, Crazing in Polymers, Springer, Berlin, Heidelberg, 1990, vol. 2, pp. 137–214 Search PubMed.
- N. R. Sottos, Nat. Chem., 2014, 6, 381–383 CrossRef CAS.
- J. M. Clough, J. Gucht, T. E. Kodger and J. Sprakel, Adv. Funct. Mater., 2020, 30, 2002716 CrossRef CAS.
- B. N. J. Persson, O. Albohr, G. Heinrich and H. Ueba, J. Phys.: Condens. Matter, 2005, 17, R1071–R1142 CrossRef CAS.
- T.-G. Hsu, J. Zhou, H.-W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, J. Am. Chem. Soc., 2020, 142, 2100–2104 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS.
- W. Qiu, P. A. Gurr and G. G. Qiao, Macromolecules, 2020, 53, 4090–4098 CrossRef CAS.
- S. Cho, S. Kang, A. Pandya, R. Shanker, Z. Khan, Y. Lee, J. Park, S. L. Craig and H. Ko, ACS Nano, 2017, 11, 4346–4357 CrossRef CAS.
- M. H. Barbee, K. Mondal, J. Z. Deng, V. Bharambe, T. V. Neumann, J. J. Adams, N. Boechler, M. D. Dickey and S. L. Craig, ACS Appl. Mater. Interfaces, 2018, 10, 29918–29924 CrossRef CAS.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, ACS Appl. Mater. Interfaces, 2015, 7, 22431–22435 CrossRef CAS.
- L. Michels, V. Gorelova, Y. Harnvanichvech, J. W. Borst, B. Albada, D. Weijers and J. Sprakel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18110–18118 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |