
Optoelectronic processes in covalent organic frameworks
Niklas
Keller
 and
Thomas
Bein
*
and
Thomas
Bein
*
Department of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstraße 5-13, 81377 Munich, Germany. E-mail: bein@lmu.de
First published on 17th December 2020
Abstract
Covalent organic frameworks (COFs) are crystalline porous materials constructed from molecular building blocks using diverse linkage chemistries. Their modular construction system allows not only for tailor-made design but also for an immense variety of building blocks, opening the door to numerous different functionalities and potential applications. As a consequence, a large number of building blocks that can act as light-harvesters, semiconductors, ligands, binding sites or redox centers have recently been integrated into the scaffolds of COFs. This unique combination of reticular chemistry with the molecular control of intrinsic properties paves the way towards the design of new semiconducting materials for (opto-)electronic applications such as sensors, photocatalysts or -electrodes, supercapacitor and battery materials, solar-harvesting devices or light emitting diodes. With new developments regarding the linkage motif, highly stable but still tunable COFs have been developed for applications even under harsh conditions. Further, the molecular stacking modes and distances in the COFs have been investigated as a powerful means to control optical and electrical characteristics of these self-assembled frameworks. Advanced understanding of optoelectronic processes in COFs has enabled their implementation in optoelectronic devices with promising potential for real-world applications. This review highlights the key developments of design concepts for the synthesis of electro- and photoactive COFs as well as our understanding of optoelectronic processes in these frameworks, hence establishing a new paradigm for the rational construction of well-defined novel optoelectronic materials and devices.
 Niklas Keller | Niklas Keller received his MSc in Chemistry from the LMU Munich. He pursued his PhD in Physical Chemistry in the group of Thomas Bein and after graduating in 2020, he continued as a postdoctoral fellow at the LMU Munich. His research interests cover the synthesis and optoelectronic properties of novel Covalent Organic Frameworks. He recently started a position with Fraunhofer Society. |
 Thomas Bein | Thomas Bein received his PhD in Chemistry from the University of Hamburg (Germany) and the Catholic University Leuven (Belgium) in 1984. He continued his studies as Visiting Scientist at the DuPont Central Research and Development Department in Wilmington, DE (USA). From 1986 to 1991 he was Assistant Professor of Chemistry at the University of New Mexico in Albuquerque (USA). In 1991 he joined Purdue University (Indiana) as Associate Professor, and was promoted to Full Professor of Chemistry in 1995. In 1999 he was appointed Chair of Physical Chemistry at the University of Munich (LMU). |
Introduction
Covalent organic frameworks are created through the condensation of molecular building blocks, resulting in crystalline porous materials with widely tunable channel systems. They have attracted great scientific interest since the first reports of their promising potential appeared in 2005.1 Since then, thorough investigations have pointed towards a broad field of applications where covalent organic frameworks can be of great advantage. The porous and crystalline nature of these robust and stable materials combined with the large structural diversity and the ability to fine-tune at an atomic scale makes them attractive candidates for applications in gas storage,2–4 molecular separation5,6 and energy storage.7–9 Furthermore, their conductive properties and their optical behaviour in the visible and NIR region allow for applications as proton conducting materials,10–13 in photocatalysis14–17 or in optoelectronics, including sensors,18,19 light-emitting diodes,20,21 or photovoltaic devices.22–24Being organic materials, COFs are of great interest because they are made of sustainable and light-weight elements and exhibit low-energy processability. Additionally, their constituting elements are mostly non-toxic. One of the attractive features of covalent organic frameworks is the diversity of the linker chemistry in combination with the vast choice of organic building blocks. The linkage motif can be chosen from non-conjugated bonds such as boronate esters to sp2 carbon-conjugated frameworks.25–28 Hence, one can not only decide if the linkers communicate over a conjugated bond but also adjust the degree of conjugation between the building units. With a view towards optoelectronics, the tunability of covalent organic frameworks allows for tailor-made absorption and photoluminescence behaviour of the building blocks by choosing from a whole range of chromophores.29 Even sterically demanding side groups can be integrated into a framework by employing an asymmetric functionalization strategy.30 Another parameter that can be adjusted to some degree is the stacking distance in two-dimensional (2D) covalent organic frameworks, which controls the nature and strength of the electronic overlap of adjacent layers and can promote the formation of long-lived excited species.31
In this review, we will mostly discuss 2D COFs, which reflects the less developed field of 3D COFs and also points to the great potential of 3D COFs that still remains mostly undiscovered. We will discuss recent studies dealing with the optoelectronic properties of COFs, and explore their potential regarding key features such as efficient transport of excitons or charge carriers, suitable absorption bands for harvesting the solar spectrum, or tunable emission for light-emitting devices.
Structural aspects – impact of different COF structures on electronic interactions
Influence of the linker geometry and lattice symmetry
In general, the geometry of the linker molecules controls the geometry of the COF pores and the lattice symmetry which further determines the pore size. With hexagonal pore shapes leading to larger pores than square or triangular pores, the π-density of the COF can also be directly varied.Bredas and co-workers investigated the influence of symmetry of (trigonal and tetragonal) nodes on the resulting frameworks by applying DFT calculations. A 2D benzene-ethynylene COF was taken as an example for three-arm nodes, where benzene is connected in meta position by ethynylene moieties. The hexagonal pore system exhibits high symmetry with a kagome sublattice (honeycomb–kagome lattice) which leads to almost flat bands and no dispersion around the Fermi level. The authors derived this behavior from the cyclopropenyl species representing the triangular structure apparent in the kagome unit cell. These flat bands generally feature charge carriers with high effective masses. In order to achieve stronger band dispersion, it was calculated that the reduction of symmetry can be a promising method. Furthermore, tetratopic nodes such as zinc porphyrin and pyrene connected with diacetylene bridges form a (distorted) square lattice resulting in high band dispersion at the band edges. The 2D layers can be viewed as a Lieb lattice and the electronic band structure can be derived from allyl species serving as a molecular analogue for which the nonbonding levels are lower in energy.32 Based on the work above Thomas et al. extended their study to predict the electronic structure of four-arm nodes such as pyrene, porphyrin and zinc porphyrin nodes connected by diacetylene bridges leading to Pyr-, Por- and ZnPor–COFs, respectively. The high band dispersion of the square lattices resulted in light holes and electrons with stronger anisotropic character found for the Pyr–COF due to the distorted square framework. The authors state the importance of electron–phonon couplings when predicting charge carrier mobilities and point out that the calculated mobility values rank among the highest mobilities predicted for organic molecular semiconductors.33
Linkage motif as a key feature controlling electronic interactions (conjugation)
A powerful approach towards tuning or controlling electronic interactions within the COF is to vary the linkage motif with which the building blocks are coupled. The first linkage motif leading to the family of crystalline covalent organic frameworks was the B–O bond, specifically involving boronate esters and boroxine rings.1 This linkage motif is moderately stable and does not result in conjugated building blocks. It provides a suitable tool for generating non-conjugated heterojunctions, for example in a donor–acceptor COF.23 However, the stability issue and the missing conjugation has limited applications in other optoelectronic fields.Additional dynamic (slightly reversible) bond formation reactions have been implemented on the way towards new functional and stable COFs, such as the imine bond formed by the condensation of an aldehyde and an amine. In 2009, Yaghi and co-workers synthesized COF-300 from the condensation of tetra-(4-anilyl)methane and terephthalaldehyde, resulting in a 3D COF and the first COF using imine linkages as the connecting part.34 This motif leads to conjugation throughout the frameworks across the linkage bonds. This has a direct impact on absorption and photoluminescence properties of the resulting imine-linked COFs, leading to larger conjugated, connected systems and hence to red-shifted optical features. Even though the stability of these (often) highly crystalline frameworks exceeds that of the boronate ester-linked COFs, imine bonds are still sensitive towards hydrolysis. Additional stability was built into COFs by employing a keto–enol tautomerism occurring in building blocks with hydroxyl groups adjacent to the aldehyde function. The reversible condensation reaction of 1,3,5-triformylphloroglucinol and p-phenylenediamine or 2,5-dimethyl-p-phenylenediamine yielded the COFs TpPa-1 and TpPa-2, respectively. By means of an irreversible tautomerization, the COFs adopt a stable form that is prevented from undergoing the hydrolysis reaction in boiling water and acid, whereas the TpPa-2 even showed strong resistance against base.35
The combination of hydrazone- and boroxine-linked COFs could be applied to construct nanotubes by selectively hydrolyzing the boroxine bonds. The hydrazone-linked hexagons were connected via boroxine rings to form COF-OEt from the building blocks 3,5-diformylphenylboronic acid and 2,5-diethoxyterephthalohydrazide. The different linkage stabilities then enabled the selective hydrolysis of the boroxine rings leaving the hydrazone-linked nanotubes.36
In general, a rotation around the imine bond is possible and occurs within the frameworks. This rotation of the building units, however, weakens the conjugation via the linking bond. Searching for a way to overcome this undesired rotation and to introduce an additional stability factor, the groups of Lotsch and Yaghi developed two different strategies towards locking the imine bond. In one approach, the TTI–COF (made of triazine triphenyl aldehyde and the respective amine) was treated post-synthetically with elemental sulfur at elevated temperatures. Under such conditions, aromatic imines oxidize to form a thioamide and then cyclize into a thiazole ring. The resulting TTT–COF showed high electron beam stability and provided a perfect system to study the local structure by TEM (Fig. 1).37 The group of Yaghi chose a different approach towards creating the thiazole (and oxazole) linkage. Using a linker exchange reaction followed by oxidative cyclization, ILCOF-1 was first synthesized from 1,3,6,8-tetrakis(4-formylphenyl)pyrene (1) and 1,4-phenylenediamine and then the substitution was carried out with 2,5-diaminobenzene-1,4-dithiol dihydrochloride and 2,5-diaminohydroquinone dihydrochloride to yield COF-921 and LZU-192, respectively.38
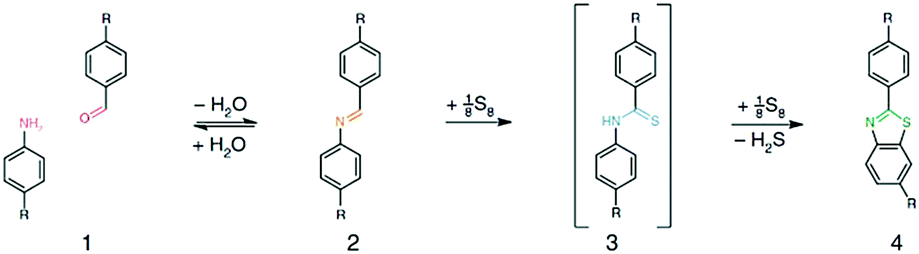 | ||
| Fig. 1 General scheme of the topochemical conversion of the imine bond into the thiazole-linkage in TTT-COF. Reproduced with permission.37 Copyright 2018, Nature Publishing Group. | ||
Another post-modification of the imine bond was established by a cyclization of a thiophene group and the adjacent imine bond in a B-COF-1 and T-COF-1. Both COFs consist of the linear bridge 2,5-bis(5-methylthiophen-2-yl)benzene-1,4-diamine and the trigonal nodes 1,3,5-tris(4-formylphenyl)benzene and 2,4,6-tris(4-formylphenyl)1,3,5-triazine, respectively. The resulting thieno[3,2-c]pyridine-linked COFs were highly stable in acids and bases at elevated temperatures and under electron beam irradiation.39 A long-standing aim of the COF research community has been to synthesize COFs purely based on sp2 carbon-conjugated bonds. In 2016, Feng and co-workers established the Knoevenagel condensation as C–C coupling reaction to deliver sp2 carbon-based COFs.40 Inspired by this report, robust sp2 carbon-based COFs are emerging and Jiang and co-workers were able to obtain the sp2c-COF in 2017. The sp2c-COF was synthesized by polycondensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene as a tetravalent node and 1,4-phenylenediacetonitrile under solvothermal reaction conditions (Fig. 2).25 Based on these findings, other COFs linked by carbon–carbon double bonds followed, already indicating that not only the stability is of great advantage for further applications but also that the COFs demonstrate promising optoelectronic features such as tunable light emission with high PL quantum yields and photocatalytic activity for hydrogen evolution.41,42
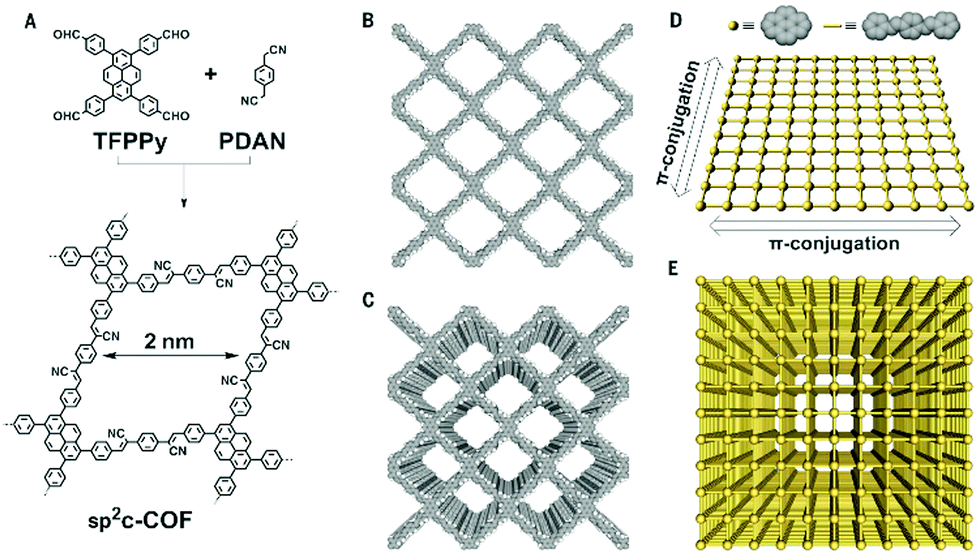 | ||
| Fig. 2 Scheme of sp2c-COF synthesis (A). Crystal structures of a single layer (B) and many layers (C) of the sp2c-COF exhibiting ordered pyrene π-stacks and 1D pores. Schematic overview of the intralayer π-conjugation of a single (D) and many layers (E). Reproduced with permission.25 Copyright 2017, American Association for the Advancement of Science. | ||
A different approach towards accessing completely olefin-linked COFs was developed by Yaghi and co-workers in 2019. 2,4,6-Trimethyl-1,3,5-triazine (TMT) and 4,4′-biphenyldicarbaldehyde undergo a trifluoroacetic acid catalyzed aldol condensation reaction to form COF-701 with high chemical robustness.43 Shortly after, Acharjya et al. investigated a base-catalyzed aldol condensation of TMT with terephthalaldehyde and 1,3,5-tris(4-formyl)phenyl benzene leading to V-COF-1 and V-COF-2, respectively.26
The above examples illustrate the variety of bond formation reactions that are already available for constructing COFs, hence offering many options for tuning and controlling conjugation and stability of tailor-made COFs in optoelectronic devices.
π-Stacking – tuning by proximity effects
The mode of stacking and the stacking distance of the π-systems embedded in COFs is another means of adjusting their electronic properties. In the first semiconducting COF, discovered by the Jiang group in 2008, the prominent π-stacking of the TP-COF synthesized from 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) and pyrene-2,7-diboronic acid (PDBA) was seen as a promising characteristic for a semiconducting COF material. The electrical conductivity was measured applying a two-probe method, showing increased current for the stacked, covalently connected building blocks in the COF compared to the neat monomers.44 Furthermore, Medina et al. showed that in thin films of a boronate ester-linked BDT-COF, consisting of benzo[1,2-b:4,5-b]dithiophene-2,6-diyldiboronic acid and 2,3,6,7,10,11-hexahydroxytriphenylene, significant hole-mobility along the stack can be observed (measured in a diode configuration). In contrast, in-plane charge transport seems to be decreased due to the non-conjugated boronate ester moieties.45 Recently, a pyrazine-linked COF condensed from 2,3,9,10,16,17,23,24-octaaminophthalocyaninato zinc(II) and tert-butylpyrene-tetraone was reported to exhibit a similar anisotropic behavior as observed above. With a mobility along the stack of almost 5 cm2 V−1 s−1 and DFT calculations revealing that there is almost no charge transport in-plane, stacking of the 2D COF sheets becomes a crucial factor that needs to be understood and controlled.46The above publications point to the importance of stacking in COFs for optoelectronic applications. In order to control the stacking of the COF layers and its effect on optoelectronic properties, many groups have investigated different approaches and designed concepts taking advantage of the modular system of COF synthesis. Chen et al. controlled the interlayer distance by inducing self-complementary π-electronic interactions. Partially replacing non-substituted arenes with fluoro-substituted arenes in a porphyrin-COF, the interlayer interactions and distances could be tuned (Fig. 3). Along with the increased π–π interactions, the crystallinity was enhanced and the HOMO–LUMO gap was reduced, suggesting an enhanced ability for electron transfer.47 Several concepts aiming at increased crystallinity by efficient and geometry-guided stacking were elaborated by our group. The studies highlight the significance of the interplay of propeller- and armchair-shaped tri- and tetradentate building blocks with their linear counterparts (1,1,2,2-tetrakis(4-formylphenyl)ethene (TPE) and pyrene, respectively).48,49 Here, the often used tetraphenyl pyrene linker was intensively studied owing to the fact that pyrene serves as an important building block for several optoelectronic COF materials. The synchronized offset induced by the chair conformation of the pyrene unit provides optimum conditions for the formation of several highly crystalline, densely packed COFs (Fig. 4).49 Considerable delocalization of excitations along the close-packed, slip-stacked pyrene stacks was observed and it was also shown that charge-transfer excitations are possible across the imine bonds. This design concept allows for a broad range of tailor-made 2D COFs with extended π-conjugated building blocks for applications in photocatalysis and optoelectronics.
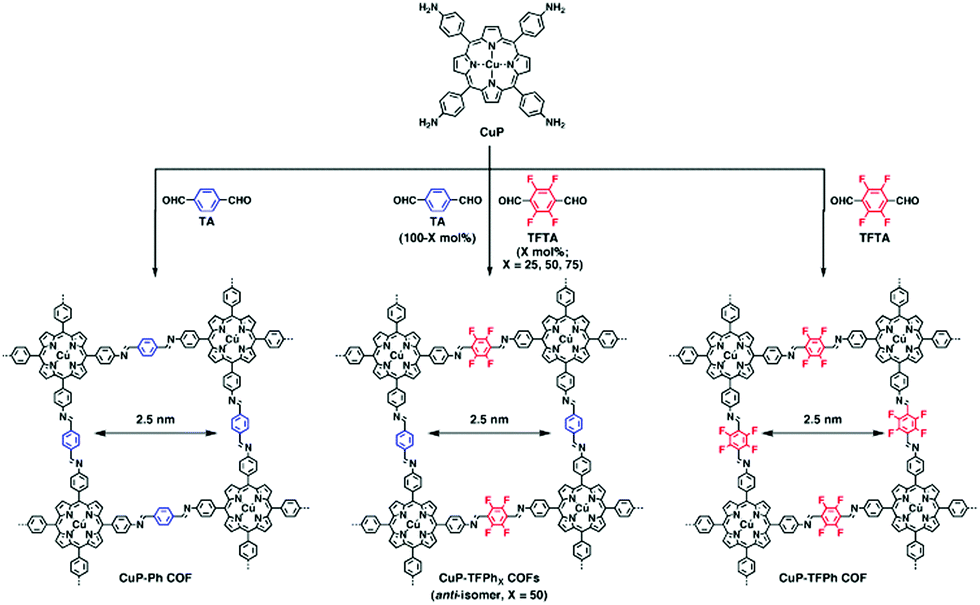 | ||
| Fig. 3 Scheme of a COF synthesis implementing self-complementary π-electronic interactions (TFPh50) in the middle and the resulting CuP-Ph COF and CuP-TFPh COF. Reproduced with permission.47 Copyright 2013, American Chemical Society. | ||
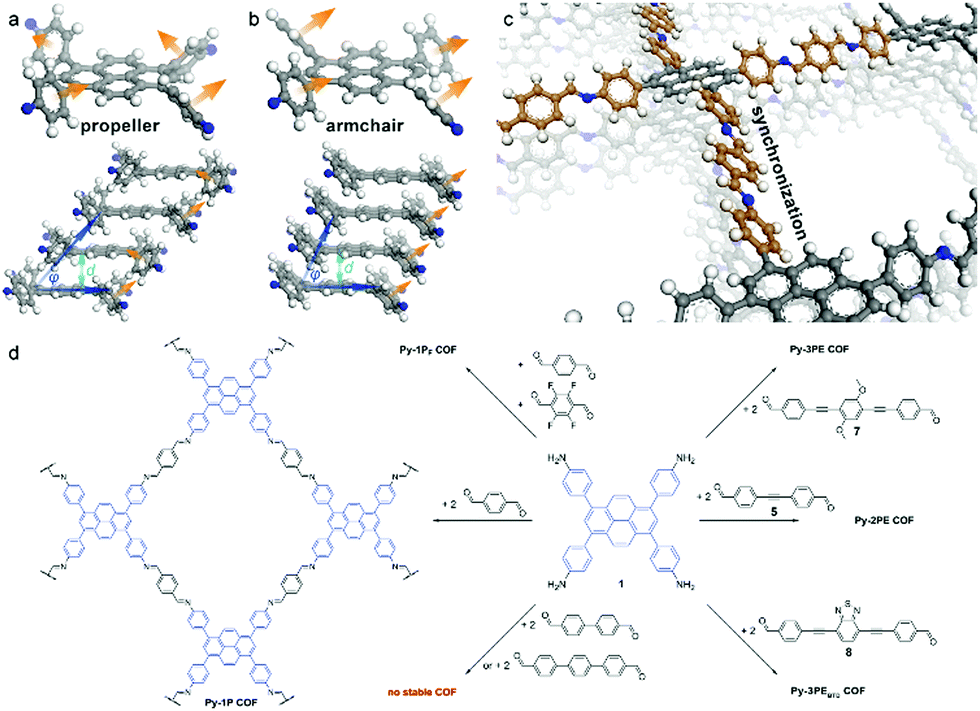 | ||
| Fig. 4 The structure of the different possible conformations of the pyrene molecule (a and b). The crystal structure of the Py-1P COF with pyrene in the armchair conformation allowing for closely stacked layers (c). Scheme of COF syntheses (d). Reproduced with permission.49 Copyright 2016, American Chemical Society. | ||
The group of Loh studied the photoluminescent properties of different 2D COFs and introduced approaches to overcome fluorescence quenching by strong π–π interactions and by locking-in imine bonds that could potentially rotate under excitation. In the first work,50 the authors proposed that the π–π interlayer interactions in the stack are reduced when pyrene is combined with a non-planar building block such as TPE. In conjunction with the fabrication of spherical COF nanoparticles in which a curvature-induced strain might lead to restricted imine rotations and decreased π–π interactions, the aggregation-caused quenching (ACQ) mechanism was shown to be disabled.50
In the second study of the Loh group,21 the imine bonds were locked-in by intramolecular hydrogen bonds to inhibit the rotationally labile linkage. Enabling a restriction of intramolecular bond rotations (RIR), the photoluminescence of the COFs could be enhanced. In addition, this RIR mechanism could be intensified by additional intermolecular hydrogen bonds that occur in this eclipsed COF structure (Fig. 5). Both kinds of hydrogen bonds could be used to tune the PL emission wavelength to obtain almost white emission. In comparison, related eclipsed-stacked COFs without hydrogen bonding exhibited significantly weaker fluorescence.21
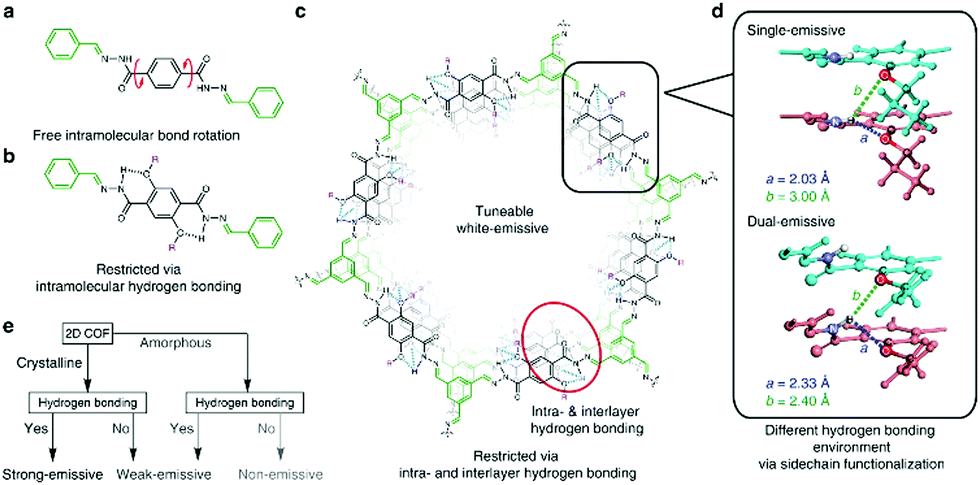 | ||
| Fig. 5 Intralayer hydrogen bonds restrict rotation leading to enhanced emission in the UV to violet range (a–c). The combination of intralayer and interlayer hydrogen bonds results in single- and dual emissive COFs (d) and the strategy to obtain strongly-emissive COFs. Reproduced with permission.21 Copyright 2018, Nature Publishing Group. | ||
Recent work of the group of Zamora showed the effect of different stacking modes on the photoluminescence behavior of the COFs. By modifying the trigonal aldehyde linker to obtain 1,3,5 benzenetricarboxaldehyde or 2,4,6-triformylphloroglucinol, it was possible to shift the stacking from the staggered to the eclipsed form, respectively (Fig. 6). As a consequence, this change turns on the fluorescence in the staggered-stacked IMDEA-COF-1 due to the decreased π–π interactions between the pyrene chromophores.51 Another way to influence the stacking mode is by steric tuning of the building blocks. Attaching alkyl chains to the core structure 1,3,5-tris(4-aminophenyl)benzene leads to different stacking behavior; whereas the absence of alkyl chains creates an AA stacking, ethyl and isopropyl chains induce an AB or ABC stacking. The study investigated binary, ternary and quaternary COFs with linear (terephthalaldehyde, 4,4′-biphenyldicarboxaldehyde and 2,2′-bipyridine-5,5′-dicarbaldehyde) and trigonal aldehydes (benzene-1,3,5-tricarbaldehyde and tris(4-formylphenyl)amine).52
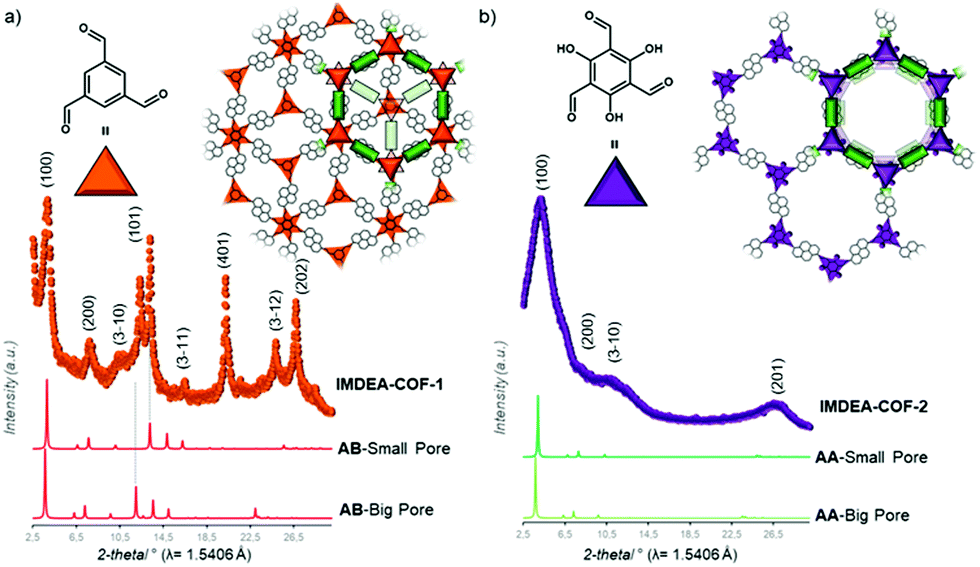 | ||
| Fig. 6 The crystal structure of IMDEA-COF-1 (a) and -2 (b) showing AB- and AA-stacking, respectively, and the experimental diffraction data. Reproduced with permission.51 Copyright 2018, American Chemical Society. | ||
In order to investigate the planarization of the well-known TPE building unit that adopts a propeller-like structure, recent work introduced a novel linker unit called dibenzo[g,p]chrysene (DBC). This chrysene unit features additional C–C bonds that cause a more planar and rigid tetradentate chromophore. The studies showed that the dibenzochrysene linker allows for closely packed docking, with the interlayer stacking distance reduced by almost 1 Å in comparison with analogous COFs embedding the TPE node.31,53 Furthermore, our group established the influence on the photoluminescence properties of the closer stacked DBC-COFs compared to the COFs using the propeller-shaped TPE. The enhanced PL lifetimes obtained in the DBC COF are postulated to be the consequence of a structurally induced stabilization of the photoexcited species across the DBC stacks.
Recently, Heine and co-workers calculated the effect of stacking 2D COF-5 sheets on their electronic features (Fig. 7). Flat bands with light electrons and heavy holes, and a direct bandgap of 2.7 eV were found for the COF-5 monolayer. However, upon stacking of the COF-5 layers in three different modes (eclipsed, inclined and serrated) a dispersion of the bands and an indirect bandgap were calculated. For the energetically unfavored eclipsed stacking, the indirect bandgap was obtained to be 1.3 eV, whereas for the inclined and serrated stacking mode indirect bandgaps of 2.2 eV were found. The band dispersion in the multilayer structures leads to light electrons and also light holes.54
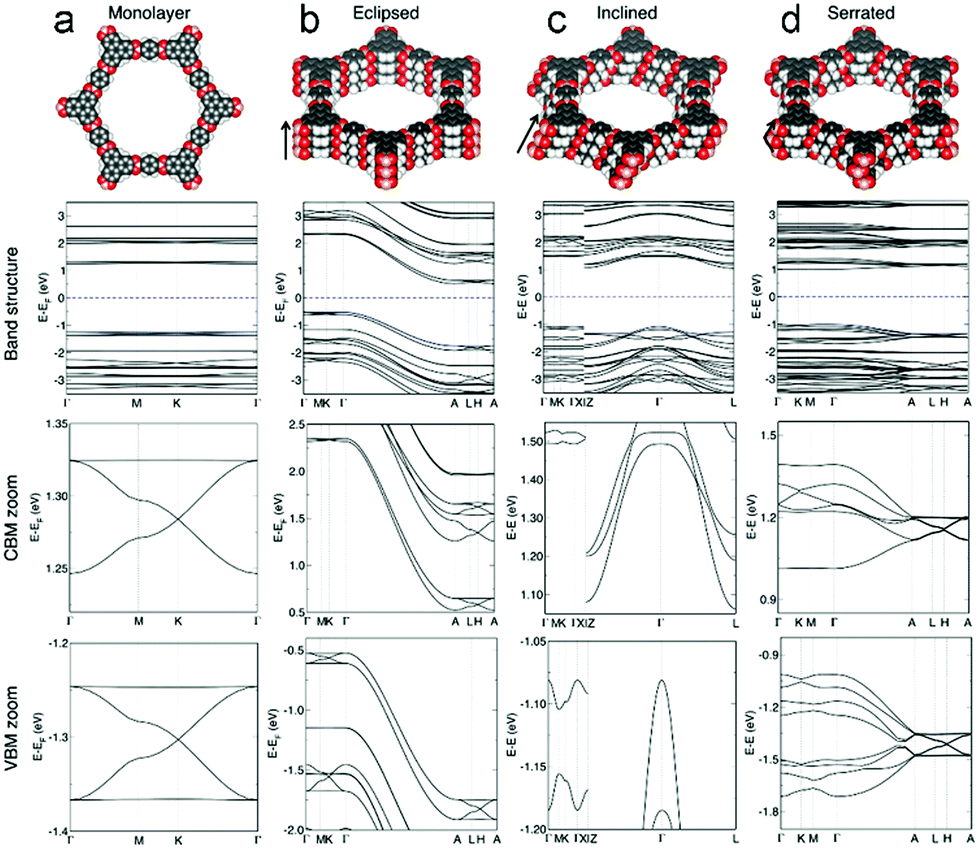 | ||
| Fig. 7 Structures of COF-5 with the corresponding calculated electronic band structures of a (a) monolayer, (b) eclipsed, (c) inclined and (d) serrated stackings of COF-5 sheets with the Fermi level depicted as blue dashed line. Reproduced with permission.54 Copyright 2020, John Wiley and Sons. | ||
Self-assembled H- and J-aggregates
When a specific spatial stacking arrangement of chromophores is present in the COF, they can form H- or J-type aggregates within the framework of the COF. The different arrangements of the (transition) dipoles occur by either distinctly eclipsed alignment (H-aggregate) or a shifted (in the extreme case head-to-tail) alignment (J-aggregate). As a result, the optical features change and get blue-shifted for the H-aggregate and red-shifted for the J-aggregate.29In the work of Ding et al., the authors synthesized a metallophthalocyanine-based COF where the stacked phthalocyanines are supposed to form an H-aggregate. Through the ordered, π-stacked columns of phthalocyanines the material exhibits enhanced intensity of the absorption bands in the visible and NIR light, is semiconducting and allows for an increased charge carrier transport in the COF.55 Yaghi and co-workers were able to tune chromophore absorption spectra by fabricating H-aggregates of porphyrin units in COF-66 and COF-366.56 Our group discovered that by the choice of the linear linker the nature of porphyrin aggregates could be switched to an extended J-aggregate (Fig. 8). The solid COF matrix enforced the packing of the porphyrin units into J-aggregate formation, which led to strongly enhanced absorption into the IR region and stabilized photoexcited species with slower decay dynamics.29
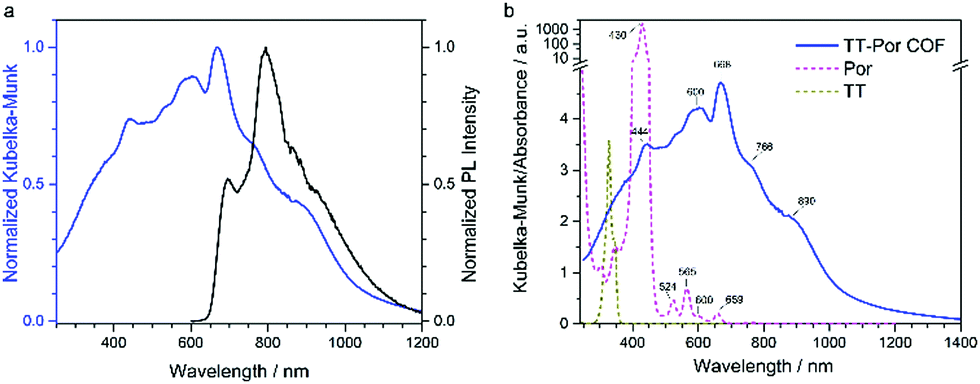 | ||
| Fig. 8 (a) Optical absorption (blue) spectrum of TT-Por COF measured as diffuse reflectance of the solid and converted with the Kubelka–Munk equation, and PL (λexc = 365 nm, black) spectrum of TT-Por COF. (b) Absorption spectra of the TT-Por COF (blue) compared to the monomers thienothiophene (TT) and porphyrin (Por) measured in diluted solution (50 μM, dioxane). Reproduced with permission.29 Copyright 2018, American Chemical Society. | ||
In another study of porphyrin-containing COFs, the influence of the different dimensionality in 2D and 3D frameworks on the photocatalytic performance was investigated. [5,10,15,20-Tetrakis(4-benzaldehyde)porphyrin]palladium was coupled with phenylenediamine or tetra(p-aminophenyl)methane to construct 2D-PdPor-COF and 3D-PdPor-COF, respectively. The aggregation mode of the Pd-porphyrin macrocycles in the 3D-COF led to extended triplet lifetimes and consequently to a better generation of superoxide radical anions resulting in higher yields of the photocatalytic selective oxidation of sulfides. The five-fold interpenetrated pts topology in the 3D-COF allowed for a strongly staggered stacking behavior.57 The formation of H-aggregates in boronate ester-based COFs was investigated by Spitler and Dichtel. They adapted a Lewis-acid catalysed procedure for protected catechols to overcome the issue of insoluble building blocks. This way, it was possible to synthesize a cofacially stacked phthalocyanine-based COF in which the phthalocyanine columns formed H-aggregates connected by 1,4-phenylenebis(boronic acid). This type of aggregation was indicated by the induced blue shift of the absorption maxima and a broadening into the NIR. Together with the non-emissive behaviour compared to the neat monomers, the H-aggregate stacking of the phthalocyanines could be confirmed.58 Another study investigating COFs interlinked by boronate ester formation (made of 4,4′-benzildiboronic acid (BZLBA) and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP)) was performed by Wang et al., who analysed the influence of the interlayer distance on the phosphorescent properties of the eclipsed COFs. The authors show that next to the RIR mechanism and the H-aggregate formation the stacking distance is an important factor for tuning the photoluminescence in COFs (Fig. 9).59
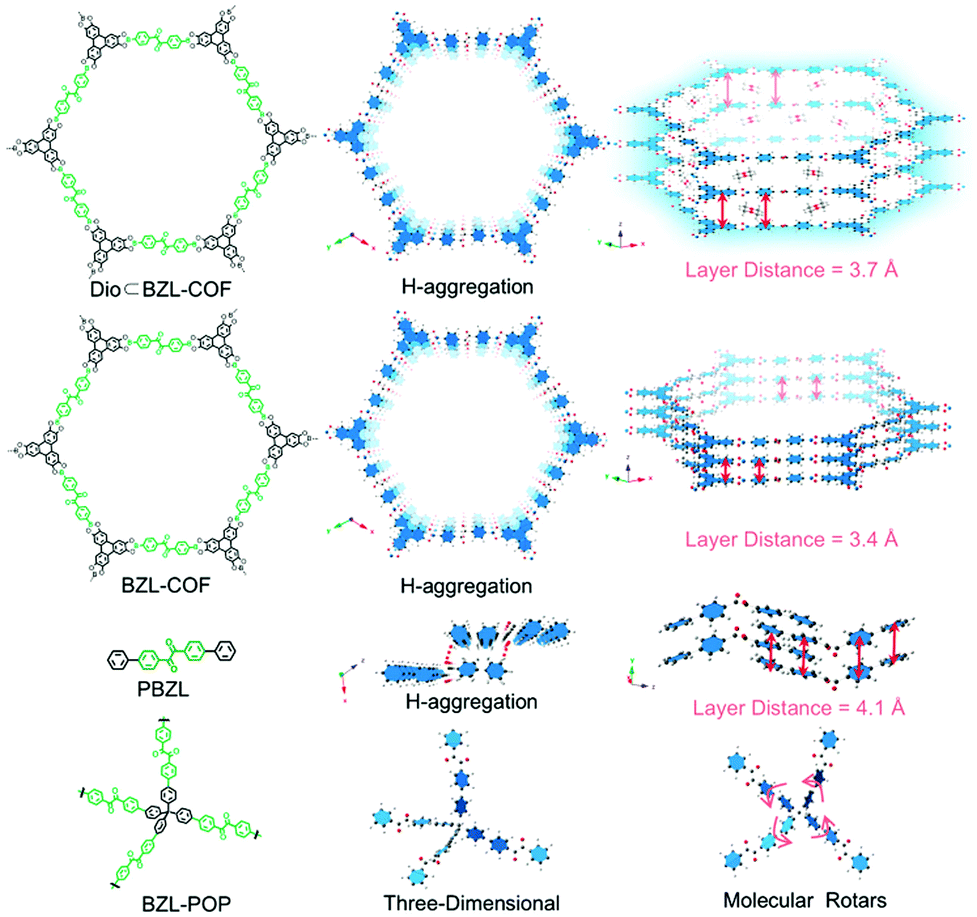 | ||
| Fig. 9 Structures of Dio BZL-COF, BZL-COF, PBZL and BZL POP (BZL: 4,4′-benzildiboronic acid; Dio: dioxane; PBZL: model compound 4,4′-diphenylbenzil) and the corresponding interlayer distance. Reproduced with permission.59 Copyright 2018, Royal Society of Chemistry. | ||
Theoretical insights on growth and stacking of COFs
First theoretical simulations based on DFT-B allowed for enhanced understanding of properties regarding structure and energy of several investigated boronate ester-based COFs. The COFs under study were obtained through a combination of linkers (1,4-phenylenediboronic acid (BDBA), [1,1′-biphenyl]-4,4′-diylboronic acid (BPDA), 5′-(4-boronophenyl)-[1,1′:3′,1′′-terphenyl]-4,4′′-diboronic acid (BTPA), benzene-1,3,5-triyltriboronic acid (BTBA) and pyrene-2,7-diylboronic acid (PDBA)) with connectors (2,3,6,7,10,11-hexahydroxytriphenylene (HHTP), hexahydroxybenzene (HHB), dodecahydroxycoronene (DHC) and tetrahydroxydiborane (THDB)). The stacking modes of the 2D COFs considered here were AA and AB stacking as well as low-symmetry versions such as serrated and inclined. It was found that the shifted layers such as AB, serrated and inclined lead to less repulsive interlayer interactions and subsequently to reduced stacking distances (Fig. 10). However, the simulated XRD patterns for the respective stacking forms revealed that the AB stacking shows a distinct disagreement with experimental XRD data. Furthermore, the bandgaps of all COFs were calculated to be between 1.7 eV (COF-9M built from DHC and BPDA) and 4.0 eV (COF-3M built from THDB and BTPA), indicating that these COFs are semiconductors. It is important to note that the stacking mode as well as the distance of the adjacent layers greatly influence the bandgap.60 These findings were supported by another study that found the serrated and inclined stacking behavior in hexagonal COFs (COF-1, -5, -6 and -8) to be the energy minimum.61 These results are in good agreement with the theoretical work of Spitler et al. showing that the hexagonal COF layers in HHTP-DPB COF (4,4′-diphenylbutadiynebis(boronic acid) (DPB)) are slightly offset by 1.7–1.8 Å.62 | ||
| Fig. 10 Calculated COF layer stackings AA, AB, serrated and inclined. Reproduced with permission.60 Copyright 2010, Beilstein-Institute. | ||
Another study showed that eclipsed stacking of triphenylenes leads to strong electronic coupling of the adjacent COF layers in HHTP-based COFs, thereby allowing for charge delocalization across the stack. The authors postulate band-like transport of delocalized charge carriers with promising mobility values for COFs.63
The effect of stacking was also investigated with regard to out-of-plane deformation and vacancy defects in the COF sheets. To investigate the influence of the stacking, the deformation and defects were studied for different numbers of layers in COF-5. It can be observed that the COF sheets in general exhibit large out-of-plane deformations in the range of several nm for isolated COF monolayers. If the COF layers are then stacked, the deformation decreases significantly and the role of defects diminishes. The defect concentration was found to be proportional to the out-of-plane deformation (Fig. 11).64
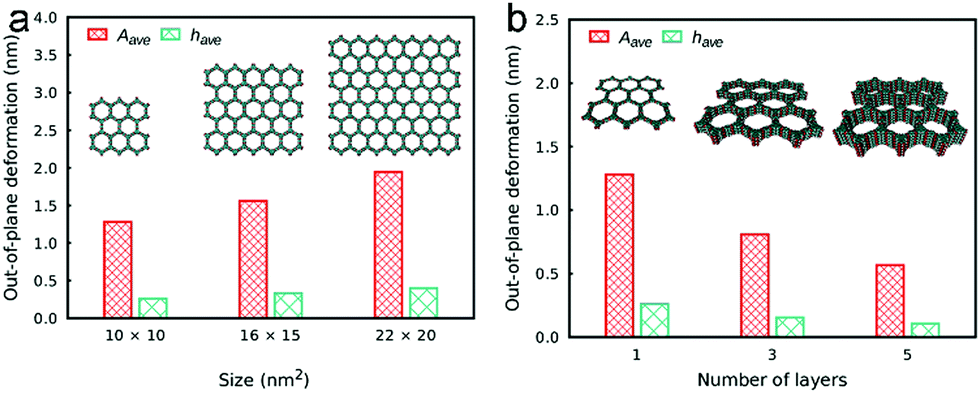 | ||
| Fig. 11 Out-of-plane deformation as a function of the COF sheet size (a) and of the number of stacked layers (b) in COF-5. Reproduced with permission.64 Copyright 2018, American Chemical Society. | ||
Furthermore, simulations of the crystallization process during the synthesis showed that enhanced stacking interactions of the building blocks in COF-5 lead to a rapid assembly of the monomers, accompanied by an increasing amount of defects causing poor crystallinity (Fig. 12).65 The authors suggest to use solvents that reduce the stacking interactions or nonplanar linker molecules with decreased stacking free energy.
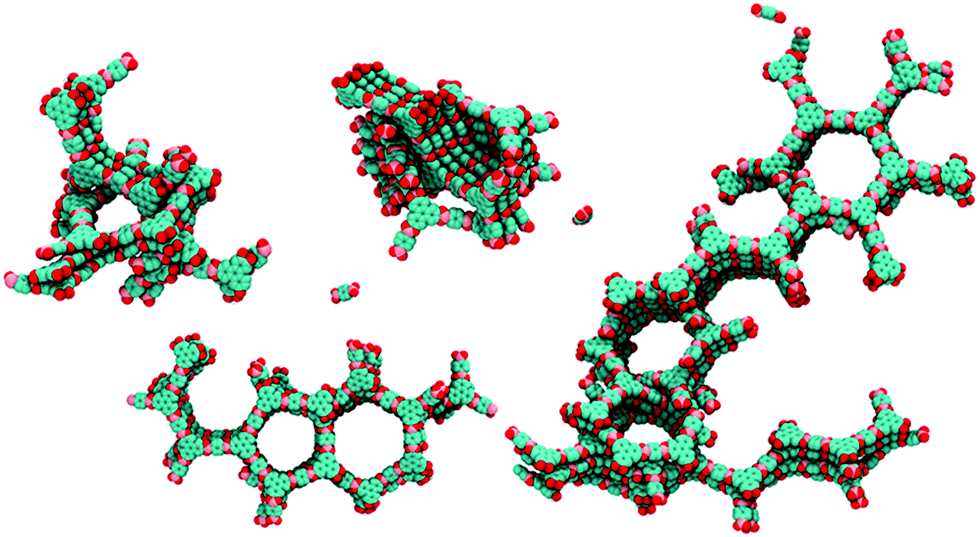 | ||
| Fig. 12 View of calculated clusters during synthesis with typical defects such as interpenetrating rings, five-membered rings and screw dislocations in COF-5. Reproduced with permission.65 Copyright 2018, American Chemical Society. | ||
Creating more complex COF stacking patterns
Since there is a large pool of building blocks to choose from, not only the bond formation, conjugation or stacking can be varied, but other features such as the electronic nature of the building blocks can be designed and controlled as well. The combination of electron donor and electron acceptor units in the COF can lead to the formation of an intrinsic heterojunction of spatially separated donor and acceptor columns. The implementation of heterojunctions in the COF material is an attractive synthetic target for potential applications in organic electronics such as organic photovoltaics. Conceptually, this can be achieved by either inserting acceptor/donor molecules into the pores of an appropriate COF, or by tailoring the pore walls (in 2D COFs) such that donor and acceptor stacks are embedded in the COF structure, enabling charge separation and transport.Hence, by modifying the fundamentally p-type NiPc COF made from 1,4-benzenediboronic acid and (2,3,9,10,16,17,23,24-octahydroxyphthalocyaninato)nickel(II)55 with electron deficient linear units such as benzothiadiazole yielded an electron-conducting material.66 This was elaborated further in the 2D D–A COF (2,1,3-benzothiadiazole-4,7-diboronic acid and HHTP) synthesized by Jiang and co-workers, where the triphenylene building block assumed the role of the donor and the benzothiadiazole building block represented the acceptor unit. This design allowed for significant charge carrier transport, electrons as well as holes, and photoconductivity.67 Further studies showed that the charge generation and separation in these D–A frameworks occurs in the femto- and picosecond regime and that it leads to long-lived charge separation with a lifetime of several microseconds (Fig. 13).68–70
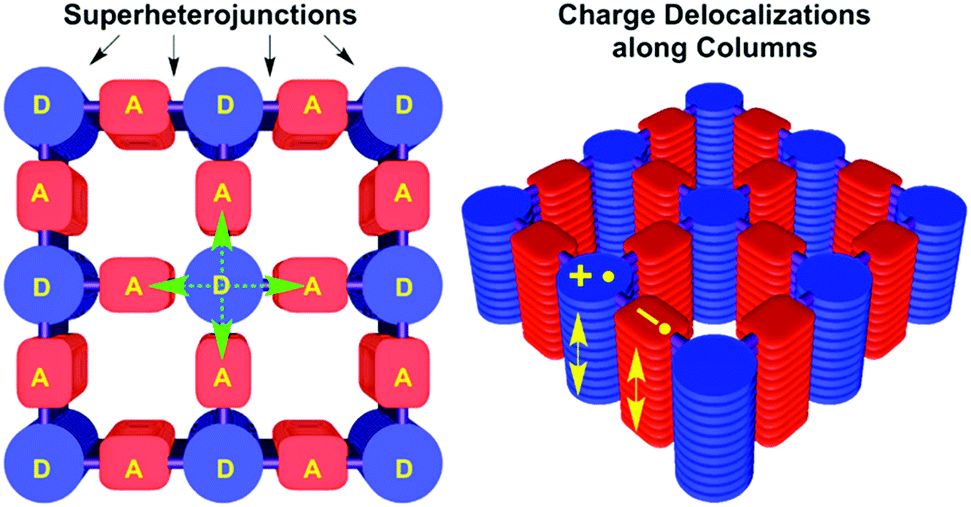 | ||
| Fig. 13 Illustration of segregated donor–acceptor columns in DMPc–ADI-COF. Photoinduced electron transfer occurs directly to the adjacent acceptor and also to diagonally disposed acceptors. Charge carrier migration is depicted as arrows along the columns. Reproduced with permission.69 Copyright 2015, American Chemical Society. | ||
The design of D–A heterojunctions in COFs leads to broadband light absorption and photoconductivity, which allows for the design of a metal-free photocatalyst COF-JLU22 for reductive dehalogenation71 or a photoinitiator TTT-DTDA and -BTDA COF for free radical polymerization.72 Even the introduction of non-linear optical behavior is possible in the Sa-TAPA COF containing a donor–acceptor dyad.73
Another approach to synthesize ordered donor–acceptor interfaces is by attaching azide groups to a fraction of the linear linkers in 2D COFs. These groups then operate as anchors in the pore walls for alkyne-modified fullerenes that can be attached by click chemistry. The electron-donating N3-ZnPc COF is then precisely connected to the electron-accepting fullerene units in the channels.74 Further, Crommie, Yaghi and Li developed a COF containing molecular heterojunctions consisting of the same chromophores. The condensation of 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (core A) and 5,10,15,20-tetrakis(4-formylphenyl)porphyrin (core B) took place in ultrahigh vacuum on a gold surface. Using thermal evaporation, first core B and then core A was deposited on the surface yielding the single-layer COF-420. Even though the COF consists solely of porphyrin-based building blocks, the donor–acceptor nature is induced by the asymmetric imine linkage bond causing the conduction band and valence band to localize onto different cores (Fig. 14).75
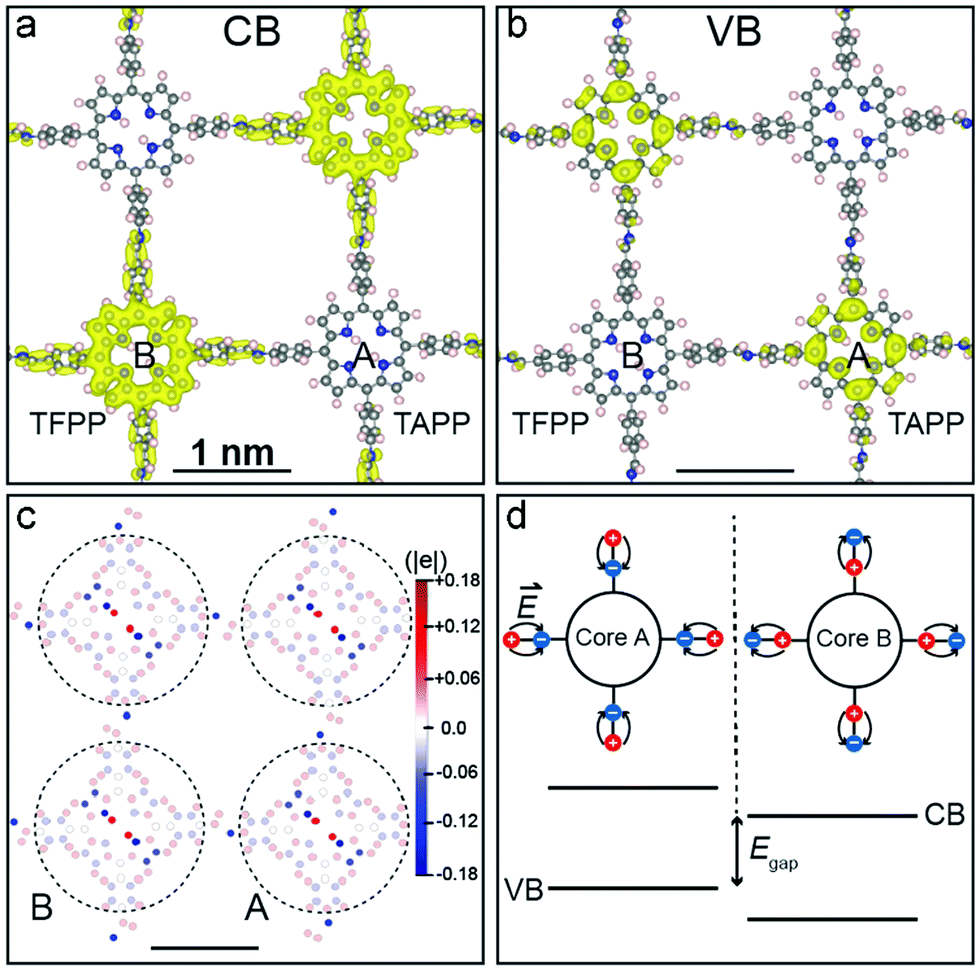 | ||
| Fig. 14 Charge density distribution for COF-420 at the conduction band minimum (a) and at the valence band maximum (b). (c) Hirshfeld population plot of COF-420 with local dipoles on nitrogen atoms (blue), and on carbon and hydrogen atoms (red). (d) Energy diagram with offset between core A and B. Reproduced with permission.75 Copyright 2019, John Wiley and Sons. | ||
The strong π–π interactions between a sterically demanding 2,5,8,11-tetra(formylphenyl)-perylene diimide (acceptor) and a simple perylene unit (donor) were used to construct alternating acceptor–donor columns that were connected in-plane with phenylenediamine. The authors assume that this stacking behavior led to an intercalated COF with 2D sheets of phenylenediamine-linked acceptor units spaced in the 3rd dimension by perylene donor moieties.76 With the two-in-one design strategy introduced by the group of Chen, an imine COF was synthesized by self-condensation of a pyrene bearing amine and aldehyde functions all in one building block (1,6-bis(4-formylphenyl)-3,8-bis(4-aminophenyl)pyrene (BFBAPy)). The resulting solvent-independent synthesis of the COF leads to simple handling and easy film synthesis. The films were investigated as a proof-of-principle in perovskite solar cells as hole transporting material.77
In a micelle-assisted approach, Zamora and co-workers could control the growth of sub-20 nm TAPB-BTCA COF particles created with 1,3,5-tris(4-aminophenyl)benzene (TAPB) and 1,3,5-triformylbenzene building blocks. The linkers were added to an aqueous micellar medium of cationic hexadecyltrimethylammonium bromide and anionic sodium dodecyl sulfate surfactants. Utilizing the resulting stable colloidal solution enabled easy processing of the imine-linked COF particles as ink for printing or molding.78
Interaction of light with COFs
Covalent organic frameworks can be designed to contain interconnected chromophore units that absorb visible light. Changing the size of the chromophore or introducing donor–acceptor features into the framework can enhance the absorption at lower energies. In addition, the degree of conjugation between the linked building units and the rotation of bonds within and between the linker molecules play an important role when it comes to absorbing visible light. With COFs, it is possible to tune these features and to synthesize strong absorbers not only in the visible but also in the (near) infrared (IR). The photoexcited states in the COF are then able to relax by emission or non-radiative decay. The emission can be of fluorescent or phosphorescent nature, depending on the spin relaxation pathways designed through the choice of linker molecules.Photoemitting frameworks
The ability of COFs to serve as photoemitters was first reported in 2008, leading to an increased focus on optical properties.44 Due to the aggregation-induced quenching effect of the π-stacked layers in 2D COFs, the PL emission is often reduced.58 Furthermore, the popular imine bond formed during condensation of amine/aldehyde building blocks is also known to quench photoluminescence by temperature-dependent bond rotation.79 These insights led to the development of first design principles for strongly emissive frameworks, by introducing building units that show aggregation-induced emission (AIE). For example, in a boronate ester-based COF propeller-shaped tetraphenylethylene (TPE) units were condensed with 1,2,4,5-tetrahydroxybenzene to obtain TPE-Ph COF. The PL properties of the resulting COF revealed fluorescence quantum yields of 32% in the solid COF material. When compared to a related model compound (tetraphenylethylenetetraboronic acid-pinacol ester), the PL quantum yield (QY) was enhanced in the COF implying that the stacking and interactions between the layers contribute to the higher PLQY values.80 Another design strategy taking advantage of the AIE mechanism was based on mechanically entangling 1D organic strands with metal-coordinating phenanthroline building blocks. The interlocked 3D ‘woven’ COF-500 was found to be non-emissive in the metalated form where the 4′,4′′′,4′′′′′,4′′′′′′′-(ethene-1,1,2,2-tetrayl)tetrakis([1,1′-biphenyl]-4-amine) (ETTBA) linker was spatially segregated from the adjacent linker (4,4′-(1,10-phenanthroline-2,9-diyl)dibenzaldehyde), thus enabling rotation of the phenyl rings. After demetalation, the free rotation was restricted due to the non-coordinating phenanthroline units spatially approaching the central ETTBA linker (Fig. 15). This restriction of bond rotation led to aggregation-induced emission.81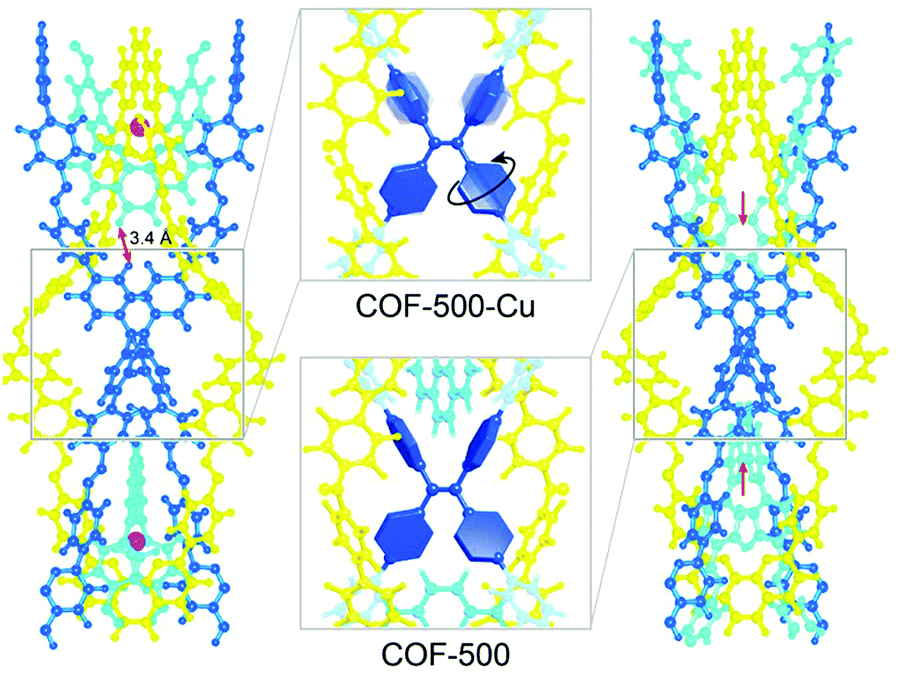 | ||
| Fig. 15 Motion of the phenylene groups of the tetraphenyleneethylene (TPE) node is restricted upon demetalation which enables the phenanthroline moieties (cyan) to move towards the TPE node in COF-500. Reproduced with permission.81 Copyright 2019, American Chemical Society. | ||
We have recently developed a strategy allowing the construction of a quaterthiophene-based COF series by an asymmetric functionalization of the building blocks. Using this approach, the steric repulsion was minimized enabling close face-on packing of the linear quaterthiophene (4T) units. The concept could also be transferred to optically interesting 4T-derived building blocks such as 3,3′′-dibutyl-[2,2′:5′,5′′:2′′,2′′′-quaterthiophene]-5,5′′′-dicarbaldehyde (a4T), 4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (TPD) and thieno[3,4-b]thiophene (TT) modified quaterthiophene (4TTPD, 4TTT). It was shown that the optical properties of such oligothiophene COFs can be precisely tuned and tailored, resulting in COFs with optical absorption and emission far into the NIR region (Fig. 16). Additionally, for the first time a charge transfer between the complementary pyrene node and the 4T units across the imine bonds was observed.30
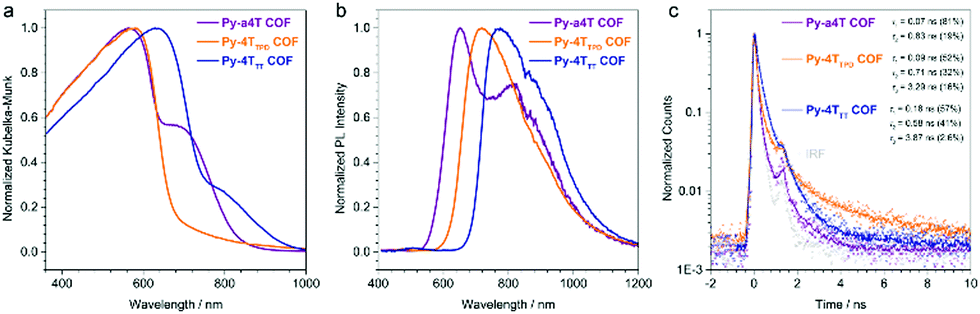 | ||
| Fig. 16 Optical properties of COFs built with quaterthiophene derivatives. Sterically demanding modifications on the backbone were enabled by an asymmetric functionalization approach. Reproduced with permission.30 Copyright 2017, American Chemical Society. | ||
Haldar et al. reported the first COF-LED with the IISERP-COF7 consisting of an anthracene and a triformyl resorcinol unit. The COF dispersed in N-methylpyrrolidone (NMP) showed a bright white emission. The source of the white light emission was attributed to the combination of the π-stacked anthracene units acting as a blue emitter and the resorcinol units contributing the red and green emission.82 Approaching the white light emission challenge from a different angle, the first report on COFs grafted with lanthanides showed similar emission behavior. The COF (built from 4,4′4′′-(1,3,5-triazine-2,4,6-triyl)trianiline and 2,6-diformylpyridine) was designed to bear coordination pockets for Eu3+ and Tb3+ that played an important role in complementing the blue emission feature of the COF with green and red emission from the lanthanides. The authors succeeded in synthesizing a solid-state white-light emitter, and by varying the excitation energy the emission could be tuned from orange over yellow to white light.83
Recently, a boroxine-linked COF series (self-condensed from 1,4-phenylenebis(boronic acid), 2,7-pyrenebisboronic acid, tetraphenylmethylboronic acid, and two previously unreported COFs condensed from 4,4′-biphenylbisboronic acid and diphenylbutadiynebisboronic acid) was synthesized in the form of stable colloidal suspensions. In this solution-based system, the emission of these highly photoluminescent COF colloids was suggested to originate from electronically communicating chromophores in the π-stacked COFs.84
Synthesizing COF single-crystals in sizes suitable for X-ray diffraction marked a big step forward for the structural and optical characterization of COFs (demonstrated for the imine-linked 3D COF-300, -303, LZU-79, and LZU-111 and for the boronate ester-based 2D COF-5 and COF-10).85,86 Next to structural insights, the COF-5 colloids were investigated with transient absorption (TA) spectroscopy to identify the excitation dynamics dependent of the size of the crystallite. It was observed that for high photon fluence the exciton decay lifetimes drop with decreasing crystallite sizes. This behavior was postulated to occur due to exciton–exciton annihilation. The probability of excitons generated on smaller colloids to collide is higher than on larger crystallites where the crystallite size exceeds the diffusion length by far.86
Ultrafast charge transfer in a D–A COF, condensed from 3,4,9,10-perylenetetracarboxylic acid diimide (PDI) and porphyrin building blocks, could be observed with femtosecond TA spectroscopy and further studied by non-adiabatic molecular dynamics calculations. Photoexcitation of the PDI unit of the COF leads to charge generation, followed by hole injection from the PDI chromophore to the porphyrin ring resulting in polarons within the crystal lattice of the framework. The charge transfer with a time constant of 124 fs was phonon-assisted by the scaffold, especially by the motion of the phenyl rings connected to the porphyrin core (Fig. 17).70
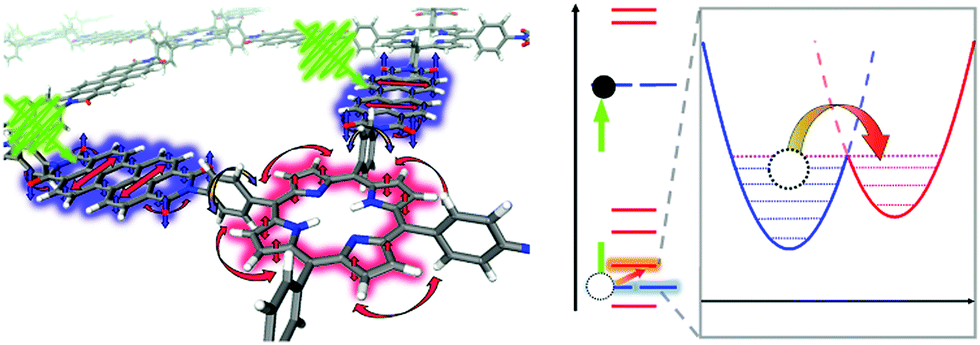 | ||
| Fig. 17 Schematic overview of phonon-assisted charge carrier dynamics in the photo-excited PDI-porphyrin COF. The rotation of the phenyl ring correlates with the π-interactions between the PDI and the porphyrin linkers, leading to ultrafast hole transfer from PDI to the porphyrin macrocycle. Reproduced with permission.70 Copyright 2019, Nature Publishing Group. | ||
To study the excited-state dynamics in COFs grown as oriented thin films, Jakowetz et al. investigated a series of imine-linked COFs with TA spectroscopy. The measurements revealed similar dynamics independent of the COF topology and the authors proposed a standard model in which singlet excitons diffuse through the framework and possibly collide. Through these singlet–singlet annihilation events, charges are generated with lifetimes of up to tens of microseconds, exceeding lifetimes of charges in classical polymer systems by orders of magnitude (Fig. 18).87
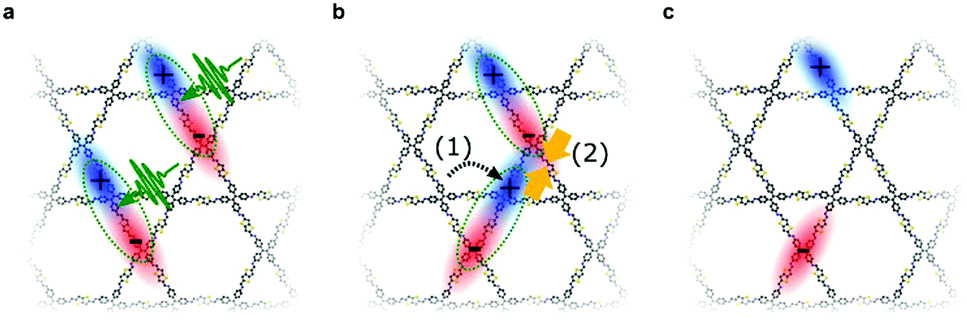 | ||
| Fig. 18 Illustration of the proposed photoinduced electronic processes in different imine-linked COFs (star-shaped COFs 4PE-TT and 4PE-BDT and hexagonal COF 3PB-BDT). (a) Photoexcitation leads to singlet excitons. (b) Diffusion and collision of generated singlets throughout the framework leads to singlet–singlet annihilation. (c) Isolated free charges with lifetimes of tens of microseconds. Reproduced with permission.87 Copyright 2019, American Chemical Society. | ||
By the introduction of an azobenzene functionalized linker, namely (E)-4-(4-(phenyldiazenyl)phenyl)pyridine-2,6-dicarbaldehyde, into COF TTA-AzoDFP, a photo-triggered cargo-release mechanism via the isomerization from trans to cis could be established. The imine-based hydrophobic COF, with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline serving as node, was able to load hydrophilic rhodamine B in its pores and release the dye when irradiated with UV light. The release was attributed to the motion of the azobenzene, which could be reversed by irradiation of light at 450 nm. The photoisomerization had only a slight impact on the degree of crystallinity of the COF sample.88
Just recently, the group of Jiang reported the growth of sp2 carbon-conjugated COFs via a template approach. Hereby, the topology of the imine-linked template COFs was imparted on the sp2c-COF series with hexagonal, tetragonal and kagome structures and the respective COFs were obtained upon hydrolysis of the template COF. The sp2c-COFs exhibited highly luminescent COF structures as well as nanosheets. Fluorescence anisotropy measurements revealed that the polarization is dependent on the topology, whereas the polarization P decreased in the order from hexagonal over tetragonal to kagome topology. The authors attributed this behavior to differences in exciton migration due to the different π electron couplings, which are more extended and efficient for kagome and tetragonal lattices than for the hexagonal lattice.89
In a communication from the group of Loh, the authors refer to the recently found antiparallel stacking behavior in hydrazone-linked COFs that exhibit interlayer hydrogen-bonds.90 For Tf-DHzDAll and Tf-DHzDPr, prepared from 1,3,5-triformylbenzene and 2,5-dialkyloxyl-terephthalohydrazide with allyl or propyl groups, respectively, experimental data suggest an antiparallel packing and further indicate that the strength of the interlayer hydrogen bond between the acyl oxygen and the side chain hydrogen controls the occurrence of dual or single emission. For Tf-DHzDAll the described interlayer hydrogen bond is stronger than the competing hydrogen bond between the acyl oxygen and the acyl hydrogen of the adjacent layer. DFT calculations suggest a proton shift in the excited state, namely excited-state interlayer proton shift (ESIPS) which then leads to a new conformer with a different fluorescence energy.91
Sensing ability
The ability to control the optical properties of COFs by various effects leading to different luminescence properties opens up the field of chemical sensing applications. The combination of the large surface area with pore channels accessible for small molecules and their delicate interplay with the photoluminescence properties of the framework makes COFs promising materials to sense and detect explosives, metal ions or even gases such as ammonia and HCl. The read-out of these COFs sensors occurs mostly via PL quenching of the material when interacting with guest molecules or ions.With this in mind, one design of COFs requires functional sites in the pore walls for interacting with and sensing intruding molecules. This can be achieved by using azine-linked luminescent COFs in which the azine bridge acts as a possible docking site for hydrogen bonding. Such interactions with a trinitrophenol-type explosive led to sensitive and selective quenching of the photoluminescence of a COF built from 1,3,6,8-tetrakis(4-formylphenyl)pyrene and hydrazine.92 Adding hydroxyl groups as active sites in the framework allows for the detection of Cu2+ ions that bind to the nitrogen atoms of the azine linkage and the hydroxyl groups in the trigonal linker (in a COF made from 1,3,5-tris(3′-tert-butyl-4′-hydroxy-5′-formylphenyl)benzene and hydrazine hydrate).93 The chelating effect of a hydrazine-linked COF was used for the selective and sensitive detection of Fe3+ ions in water with a quenching constant of 2.3 × 104 M−1. This method was enabled by the geometrically precise design of the pockets with the suitable linker choice of benzene-1,3,5-tricarbohydrazide with 2,5-dihydroxyterephthalaldehyde or 2,5-dimethoxyterephthalaldehyde.94 Using DFT and TDDFT calculations, the influence of hydrogen bonding of simple formaldehyde on the emission profile of a representative boronate ester-based COF (anthracene connected with phenylene unit) was elucidated. The analysis resulted in changes of the frontier molecular orbitals as well as electron configuration, which coincides with fluorescence quenching.95
Other docking sites for guest molecules can be thioether groups serving as receptor for the detection and removal of mercury. Again, the fluorescent COF (made from 2,5-bis(3-(ethylthio)propoxy)terephthalohydrazide and 1,3,5-triformylbenzene) strongly interacts through its thioether arms with penetrating Hg2+ ions, which leads to a sensitive chelation-enhanced quenching effect. Additionally, the strong binding between the sulfur atoms and Hg2+ ions enables the efficient removal from mercury-containing solutions.96 The same mechanism (using the COF condensed from 2,5-bis(2-(ethylthio)ethoxy) terephthalohydrazide and 1,3,5-triformylbenzene) was applied for detecting Au3+ ions in water and successfully recovering metallic gold powder after reduction and pyrolysis treatments.97
Guest molecules such as ammonia can also form Lewis acid–base pairs with the boronate ester linkage in COFs. Elaborating this approach with the TPE-Ph COF, highly sensitive detection of ammonia with a fluorescence quenching rate constant of up to 6.3 × 1014 M−1 s−1 was enabled in solutions with ammonia gas at concentrations below the ppm range.80 An alternative sensing mechanism for triacetone triperoxide (TATP) in β-ketoenamine COFs (3BD and 3′PD) is suggested to occur by oxidation of the enamine moiety by TATP.98
In a straightforward approach, a β-ketoenamine-based COF (made from (1,4-phenylene)bis(3-hydroxyprop-2-en-1-one) and 1,3,5-tris-(4-aminophenyl) triazine) known for its chemical stability was found to be applicable for HCl vapor detection. This sensing mechanism is based on the rapid change of UV-Vis absorption by protonation of the triazine ring. The induced electron enrichment in the triazine unit goes along with a conductivity increase by two orders of magnitude and PL quenching. The protonation can be reversed by deprotonation with ammonia or by physical treatments such as heating or vacuum.99
The dual emission of COF-4-OH (made from 1,3,5-triformylphloroglucinol and 9,9-dibutyl-2,7-diaminofluorene) induced by the keto–enol tautomerism leading to an excited-state induced proton transfer was used as a sensing concept for water content in organic solvents and as pH sensor. Furthermore, sterically demanding butyl chains were introduced to reduce aggregation-caused quenching (ACQ).100
Exfoliation of an imide-based COF (made from tetra(4-aminophenyl) porphyrin and perylenetetracarboxylic dianhydride) exhibited selective sensing of 2,4,6-trinitrophenol (TNP), whereas the fluorescence quenching constant was found to be higher than in the corresponding COF bulk material.101 High selectivity towards TNP was also observed in a dual-pore COF (made from 4′-(bis(4-formylphenyl)amino)-[1,1′-biphenyl]-3,5-dicarboxaldehyde with diaminobenzene/benzidine) where the sensing event was visible to the naked eye as a rapid color change.102 Organic pollutants and dyes could be detected with the exfoliated IMDEA-COF-1, a pyrene-containing imine-linked COF made from 1,6-diaminopyrene and benzene-1,3,5-tricarbaldehyde. The intercalated organic molecules were presumably responsible for fluorescence quenching, correlating with the surface area of the colloidal COF particles.103
By synthesizing highly fluorescent and chiral COF (CCOF) nanosheets from TPE and chiral 6,6′-dichloro-2,2′-diethoxy-1,1′-binaphthyl-4,4′-dialdehyde, it was possible to achieve enantioselective sensing of chiral vapors in solution as well as in membranes (Fig. 19). The selective fluorescence quenching was attributed to different binding energies of the enantiomers and a static enhancement due to the CCOF–vapor adduct, which may also lead to structural changes.104
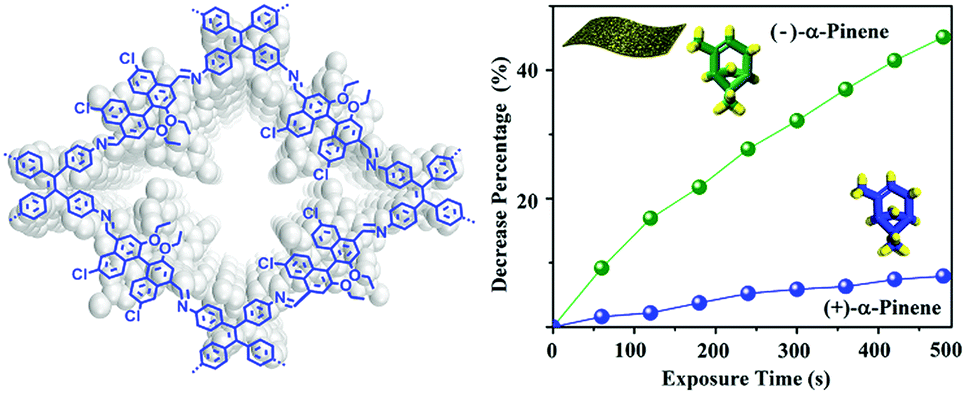 | ||
| Fig. 19 Structure of the chiral COF CCOF 7 and the observed selective fluorescence quenching of α-pinene. Reproduced with permission.104 Copyright 2019, American Chemical Society. | ||
Materials sensitive towards toxic pollutants such as perfluorooctane sulfonate (PFOS) are highly desirable. In this context, fluorescence quenching of a COF (made from 1,3,5-triformylbenzene and 1,4-phenylenediamine) coated lanthanide-doped nanocrystal upon exposure to PFOS can function as an ultrasensitive detector with a detection limit as low as 0.15 pM. The detection mechanism was attributed to hydrogen bond formation or electrostatic interactions of the PFOS in the COF pores.105
The detection of uranium in the nuclear industry is of high importance to detect leakages. This motivation led the group of Qiu to synthesize an sp2 carbon-conjugated COF consisting of 2,4,6-tris(4-formylphenyl)-1,3,5-triazine and 2,2′,2′′-(benzene-1,3,5-triyl)triacetonitrile. The free cyano groups of the resulting COF TFPT-BTAN could be post-synthetically functionalized to form amidoxime groups. These moieties work as highly selective and sensitive ligands for the detection of UO22+. The mechanism of detection is proposed to be based on the fact that the amidoxime group (amine and hydroxyl group) coordinates to UO22+ and leads to chemisorption of the target compound in the pores. Moreover, this behavior allowed also for the extraction of uranium by capturing the metal in the 1D pores, which led to more efficient UO22+ extraction compared to an analogous amorphous polymer.106
Photoconductive COFs
The ordered structure of (2D) covalent organic frameworks allows for the modular assembly of building blocks in closely stacked columns with conjugated in-plane connections. This framework architecture with its intrinsic pathways for excitons and charge carriers is a determining characteristic pointing to the great potential of COFs as semiconducting materials. Considering the combination of these structural features with the properties of chromophores serving as building blocks in photoabsorbing COFs harvesting a large fraction of the solar spectrum, the conceptual leap to photoconductive materials is evident. The foundation for this strategy was laid by Jiang's group with the report of the first COF showing semiconducting behavior due to its highly organized π-conjugated structure.44It was again Jiang's group who designed the first photoconductive COF. The requirement of strong absorbance in the visible spectrum was fulfilled by the choice of pyrenediboronic acid that self-condensed to a crystalline COF leading to the essential order of the structure. The exciton migration in this single-component COF was found to occur within the COF layer as well as in the direction of stacking. The rapid photoresponse led to photocurrents with an on–off ratio of over 8.0 × 104. The ordered structure seemed to be key since the neat components showed almost no response when illuminated. Iodine doping led to an increased current in the hole-conducting COF material.107 By utilizing Ni-phthalocyanine in an eclipsed framework coupled with BDBA, high hole mobility and strong light-harvesting resulted in a photocurrent of 3 μA and a photocarrier generation yield ϕ of 3.0 × 10−5. In this boronate ester-based COF the conducting species are holes with a reported mobility of 1.3 cm2 V−1 s−1.55 Switching from hole-conducting to an electron-conducting phthalocyanine-containing COF with the linear building unit 1,4-benzothiadiazole diboronic acid broadened the absorption bands far into the IR with a high sensitivity towards near IR photons. Charge carrier generation upon illumination led to an increased photocurrent of up to 15 μA with a photocarrier generation yield of 9.2 × 10−4 measured by time-of-flight.66 In order to harvest more light and to study the influence of the coordinated ions, the central metal in the phthalocyanine chromophore was exchanged by Co, Cu and Zn. The difference in electron density resulted in photocurrents ranging from 0.14 nA to 110 nA, which was attributed to a decrease of electron density in the metalated phthalocyanines.108
In order to increase the π-density of conjugated COF material at the expense of the pore size and volume, Jiang's group introduced two C6-symmetric linkers, namely hexaphenylbenzene and hexabenzocoronene, to form trigonal pores with a π-column density of 0.25 nm−2. Time-of-flight measurements revealed a photocarrier generation yield ϕ of 2.1 × 10−5 for the hexabenzocoronene COF with a hole mobility of 0.7 cm2 V−1 s−1.109
The orientation of COF films showed a significant difference regarding the generated photocurrent. The rapid photoresponse in oriented thin films varied by a factor of 5 compared to non-oriented COF films made from thieno[3,2-b]thiophene-2,5-diyldiboronic acid and the polyol 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP).110 Photocurrent could be greatly enhanced after iodine doping of an azodioxy-linked porphyrin-containing COF, whereas the non-grafted COF showed hardly any photoresponse.111
Upon implementation of photo-switchable molecules such as the dithienylethene moiety in COFs, it was possible to control the electrical conductivity by irradiation with UV or visible light. Upon UV irradiation at 365 nm the dithienylethene undergoes a ring closure which correlates with an increase in conductivity by the factor 200 to 2 × 10−5 S cm−1 compared to the open ring structure (Fig. 20). The process is reversible by irradiating the material with light in the visible above 550 nm and was attributed to a smaller bandgap in the closed state.112
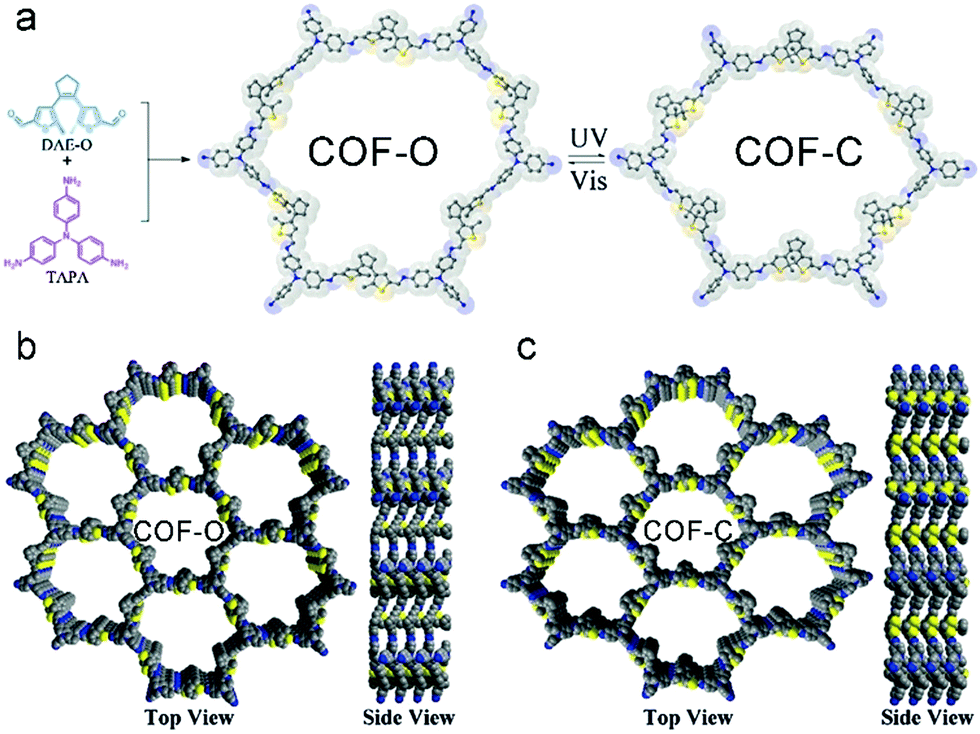 | ||
| Fig. 20 Synthesis and structure of COF-O and the corresponding photostimulated closed-ring state COF-C. Reproduced with permission.112 Copyright 2019, John Wiley and Sons. | ||
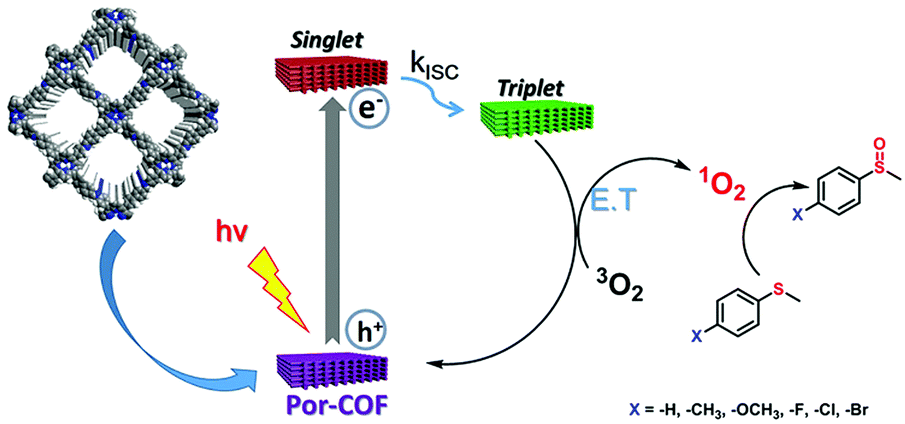
Since there is a great need for sustainable and effective energy sources, application in photocatalytic water splitting is an obvious step to take when considering the suitable characteristics of COFs. The photocatalytic activation of oxygen to singlet oxygen was already a first indication of the great potential. Jiang's group was able to implement a newly designed linkage motif for stable and conjugated COFs. Here, the squaraine-linked porphyrin-containing COF showed excellent stability under the tested conditions and a reduced bandgap for enhanced absorption at lower energies. The stacking of the COF building blocks seems to be crucial since the authors observed hardly any activity for the neat porphyrin monomer. The photoexcited states in the COF generate triplet states, which then activate molecular oxygen.113
Photocatalytic water splitting
In a further important development, Lotsch and co-workers reported the first COF applied as a photocatalyst for hydrogen evolution during the water splitting reaction. The authors designed a TFPT COF with hydrazone linkage for stability during catalytic operation and triazine units for achieving a bandgap suitable for light-harvesting as well as water splitting (Fig. 21). Using sodium ascorbate as sacrificial donor and Pt as proton reduction catalyst led to a hydrogen evolution rate of 230 μmol h−1 g−1 (under visible light illumination >420 nm), whereas using triethanolamine as sacrificial donor gave even higher values of 1970 μmol h−1 g−1. This concept showed that the COF can compete with other photocatalysts and laid the foundation for developing new types of organic materials for photocatalytic hydrogen evolution.114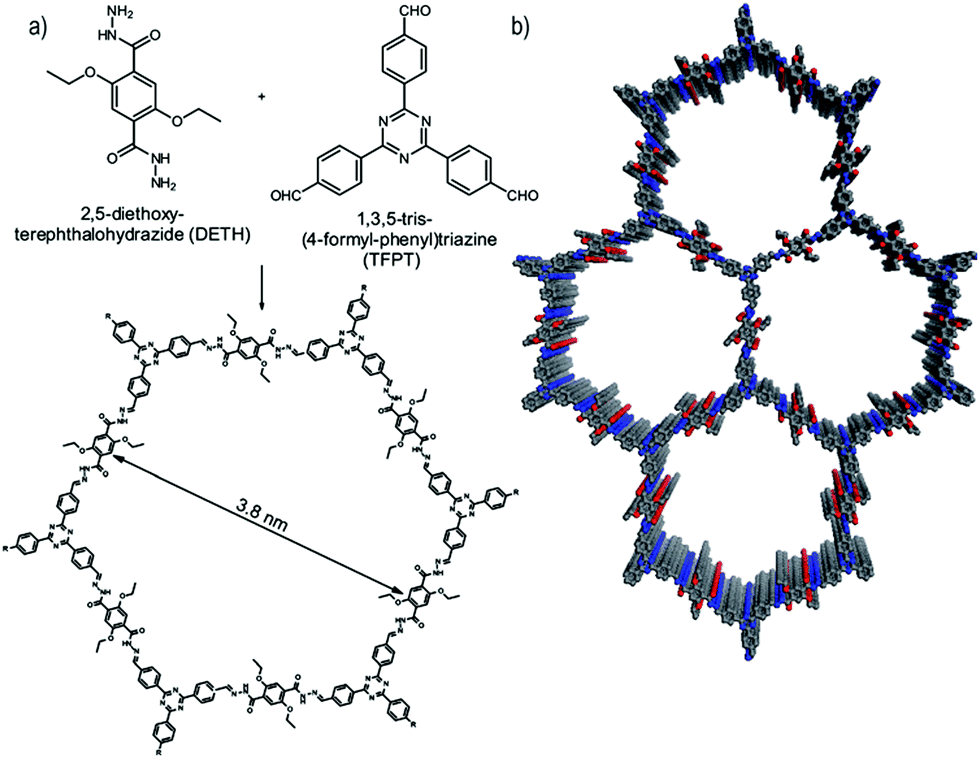 | ||
| Fig. 21 Hydrazone-linked TFPT-COF with a cofacial stacking of the layers exhibiting photocatalytic hydrogen evolution in the presence of a sacrificial agent and deposited Pt particles. Reproduced with permission.114 Copyright 2014, Royal Society of Chemistry. | ||
Further research on COFs as photocatalysts led to insights about the role of different linkers and functionalization. For example, an investigation of acetylene-functionalized COFs revealed the first operational COF photocatalyst without heteroaromatic building blocks. The catalytic activity was shown to be strongly dependent on the number of acetylene units in the COF. The TP-EDDA COF, made from 1,3,5-triformylphloroglucinol (TP) with 4,4′-(ethyne-1,2-diyl)dianiline (EDDA) with only one acetylene bridge in the linear linker exhibited a hydrogen evolution rate (in the presence of TEOA and photodeposited Pt particles) of 30 μmol h−1 g−1, which was enhanced compared to a COF with a linear linker of the same length but no acetylene moiety. Extending the acetylene bridge to a diacetylene bridge led to a much enhanced hydrogen evolution rate of 324 μmol h−1 g−1.115 This positive impact of acetylene moieties in the COF on the HER was also observed in related computational studies, pointing to an enhanced diffusion of photogenerated excitons along the polyyne moieties in the COF.116
Changing the focus to a pyrene-based tetravalent linker containing phenyl rings with various nitrogen contents (namely 1,3,6,8-tetrakis(4-ethynylbenzaldehyde)-pyrene (TEB PY), 1,3,6,8-tetrakis(6-ethynylnicotinaldehyde)-pyrene (TEN PY), and 1,3,6,8-tetrakis(2-ethynylpyrimidin-5-carbaldehyde)pyrene (TEP PY)) that are connected via acetylene bridges to the node by condensation with hydrazine, the impact of the nitrogen atoms in the ring on the photocatalytic hydrogen evolution was investigated. The experimental data in combination with quantum chemical calculations revealed A-TEB PY-COF with the lowest number of nitrogen atoms to be the most efficient in water splitting, due to the highest driving force for the hydrogen evolution reaction (HER) of the more electron-rich framework. The hydrogen evolution rate was found to be 98 μmol h−1 g−1 for the COF with pure phenyl rings without nitrogen atoms, compared to 22 and 6 μmol h−1 g−1 for the COFs with pyridine and pyrimidine rings, respectively.16
Examining the possible replacement of Pt as co-catalyst for COF-catalysed hydrogen evolution reactions, it was found that [Mo3S13]2− clusters in the pores of a COF made from ethidium bromide and 1,3,5-triformylphloroglucinol exhibited great potential, delivering hydrogen at a stable rate of up to 13.2 mmol h−1 g−1 for 18 h. The highest values were measured using Ru(bpy)3Cl2 as a photosensitizer, ascorbic acid as a sacrificial donor and the MoS clusters loaded into the COF pores.117 Alternatively, a combination of Eosin Y and triethanolamine (TEOA, sacrificial donor), with Pd0 as co-catalyst reduced on site with EtOH also showed significant hydrogen evolution activity of 10.4 mmol h−1 g−1 for at least 2 h with the COF TpPa-1 (made from 1,3,5-triformylphloroglucinol and p-phenylenediamine).118
Further research led to the substitution of Pt by Ni-based co-catalysts. The group of Lotsch reported the synthesis and successful application of a thiazolo[5,4-d]thiazole-bridged COF (with 1,3,5-triformylphloroglucinol nodes) as the photoactive material. In photocatalysis studies, a Ni-thiolate cluster was applied as co-catalyst and TEOA as a sacrificial electron donor (Fig. 22). The system was able to deliver a hydrogen evolution rate of up to 941 μmol h−1 g−1 for at least 70 h. The Ni-thiolate cluster is known for efficiently catalyzing the HER in combination with erythrosin B as sensitizer with rates up to 49.3 mmol h−1 g−1, but the catalyst fully degrades within 7 h resulting in a turnover number based on Ni that is 12 times lower than for the stable COF system.17
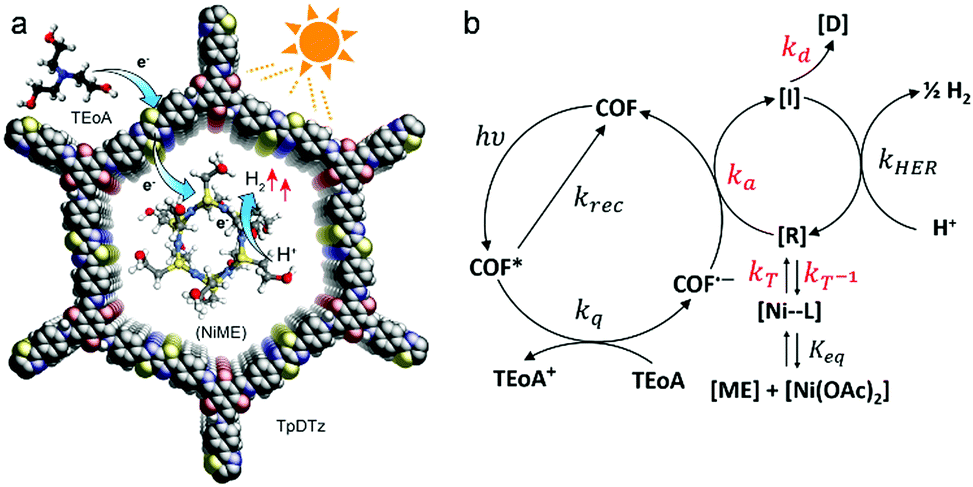 | ||
| Fig. 22 (a) Schematic representation of the proposed mechanism for the H2 evolution. (b) Key steps of the photocatalysis: [R] active catalyst; [D] deactivated species [I] intermediate reduced catalyst species. Reproduced with permission.17 Copyright 2019, American Chemical Society. | ||
Further research on noble metal alternatives showed that Ni(OH)2 was efficient as co-catalyst in a COF photocatalytic system (made from 1,3,5-triformylphloroglucinol and 2,5-dimethyl-p-phenylenediamine), resulting in a hydrogen production rate of up to 1896 μmol h−1 g−1 for at least 5 h. The sacrificial donor in this case was sodium ascorbate.119 Modifying a COF made from a triazine node and 1,3,5-triformylphloroglucinol with g-C3N4 (graphitic carbon nitride which was recently reported to show significant activity in photocatalytic water splitting120–122) to form CN–COF enhanced the photocatalytic performance to hydrogen evolution rates of up to 10.1 mmol h−1 g−1, superior to any other g-C3N4-based photocatalyst reported (Fig. 23).123
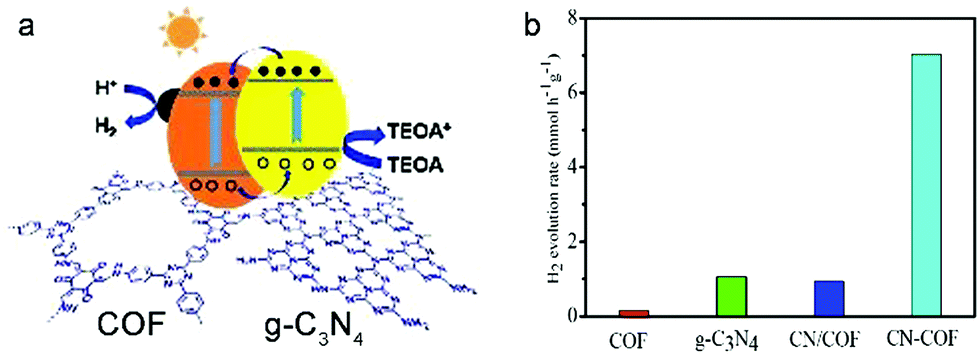 | ||
| Fig. 23 (a) Proposed photocatalytic mechanism for CN–COF. (b) H2 production rate, whereby CN/COF is the physical mixture of g-C3N4 and COF. Reproduced with permission.123 Copyright 2019, Royal Society of Chemistry. | ||
To use seawater as water source for photocatalytic watersplitting, it is important that the cocatalyst is strongly coordinated by the COF material to overcome competitive metal ion coordination, such as Na+, K+, Mg2+ and Ca2+. Therefore, the hydrazone-linked TTR-COF exhibiting thioether moieties was designed consisting of 1,3,5-tris(4-formylphenyl)triazine and 2,5-bis(2-(ethylthio)ethoxy)-terephthalohydrazide. The thioether shows high affinity towards Au cations that functioned as a cocatalyst in the photocatalytic hydrogen evolution with TEOA as sacrificial donor. The experiments in seawater showed a decreased production rate of 141 μmol h−1 g−1 compared to pure water but the rate remained stable over the course of 20 h. The decrease of production rate could be attributed to the chelation of Mg2+ ions. The significance of the thioether group could be shown by comparison with a COF containing no thioether. Here the catalytic activity decreased drastically since the Au particles agglomerated.124
So far only photocatalytic hydrogen evolution with COFs has been extensively studied, while oxygen evolution seemed to be out of reach since the band alignment is crucial and the valence band needs to be below the energy level of the water oxidation. However, in 2019 the group of Zhang synthesized the first COF (made from 3,5-dicyano-2,4,6-trimethylpyridine and 4,4′′-diformyl-p-terphenyl) that was reported to be able to perform both half-reactions of water splitting (Fig. 24). The sp2 carbon-conjugated g-C40N3–COF revealed hydrogen production rates of 4120 μmol h−1 g−1 with TEOA and Pt, and also exhibited an oxygen production rate of 50 μmol h−1 g−1 with silver nitrate as electron acceptor and La2O3 as a pH buffer.27
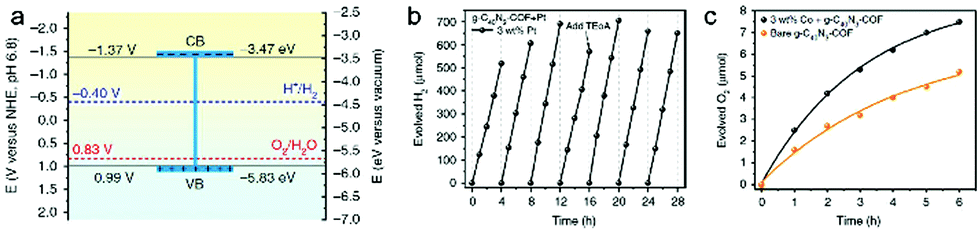 | ||
| Fig. 24 (a) Bandgap diagram with marked potential for HER and OER. (b) Hydrogen and (c) oxygen production during photocatalytic water splitting based on g-C40N3–COF. Reproduced with permission.27 Copyright 2019, Nature Publishing Group. | ||
With this work opening the field of COF-based photocatalytic OER, a first imine-linked COF constructed from TAPB nodes with a bipyridine bridge used to coordinate Co ions as co-catalyst showed oxygen evolving abilities, with an O2-production rate of 152 μmol h−1 g−1 with silver nitrate as electron acceptor.125
In a study evaluating the effect of different linkages, such as alkene, imide and imine, on the photocatalytic activity towards the hydrogen revolution reaction, three COFs were synthesized from triphenylbenzene nodes and phenyl bridges, namely COF-alkene, COF-imide and COF-imine connected with the indicated linkage motifs. Hereby the alkene-linked COF with its cyano moieties yielded the highest hydrogen production rate of 2330 μmol h−1 g−1. In the same design of experiment the compared COF-imide and COF-imine exhibited only rates of 34 and 12 μmol h−1 g−1. The experiments were performed with triethanolamine (TEOA) as sacrificial donor and 3% Pt as a cocatalyst. The authors attribute the exceeding activity of COF-alkene to the longer decay times of the photoexcited states and to the donor–acceptor character induced by the electron-withdrawing cyano moiety.126
Additional photoactive COFs – photocatalytic and photothermal materials
Converting solar energy with COFs into sustainable fuels can not only occur via photocatalytic hydrogen evolution but also by photocatalyzing CO2 reduction. Notably, without the use of additional sacrificial agents and co-catalysts, the azine-linked COFs ACOF-1 (made from 1,3,5-triformylbenzene and hydrazine hydrate) and N3–COF (made from 2,4,6-tris(4-formylphenyl)-1,3,5-triazine and hydrazine hydrate) could photocatalytically convert CO2 in water to methanol, with a yield of 13.7 μmol g−1 within 24 h for the N3–COF in a 100% CO2 atmosphere. When the CO2 concentration was reduced to 1%, the amount of methanol was still 9.9 μmol g−1 within 24 h for the N3–COF. The presence of only traces of H2 and O2 illustrates the photocatalytic selectivity, which is attributed to the high selectivity of the nitrogen-rich COF systems.127Enhancing the photocatalytic activity of the COFs can again be achieved by introducing metal-based co-catalysts into the framework. For example, when introducing bipyridine into a COF (connected with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline nodes), the two neighboring nitrogen sites can coordinate a Re complex using a simple post-synthetic procedure. Here, the reduction of CO2 yielded CO as a product at amounts of almost 15 mmol g−1 over the course of more than 20 h with the use of TEOA as sacrificial agent. The photocatalytic process is selective and shows recyclability for at least three cycles of 3 h each.128 It was found that the efficiency is strongly dependent on the ordered structure in the COF.129
Notably, the rhenium in the above COF could also be substituted by Ni(II) ions coordinated by the bipyridine units. Connected via 1,3,5-triformylphloroglucinol nodes, the photocatalytically selective CO2 reduction produced 811.4 μmol g−1 h−1 CO in the presence of TEOA as sacrificial electron donor. The synergistic combination of the COF and the single Ni-sites coordinated at the bipyridine linkers were experimentally and computationally found to be key for the high CO production rate.130
In a different study, several abundant metal ions such as Ni, Co and Zn coordinated to the quinone oxygens of adjacent layers in an anthraquinone-based COF (again linked with 1,3,5-triformylphloroglucinol) exhibited significant CO production rates, as high as 1020 μmol g−1 h−1 with TEOA for the Co-COF, whereas the Zn-coordinated COF selectively produced formic acid at a rate of 152.5 μmol g−1 h−1, also in the presence of TEOA.131
A bottom-up approach for the synthesis of ultrathin COF (made from porphyrin and TAPB nodes and terephthalaldehyde and 4,4′-biphenyldialdehyde bridges) nanosheets by adding 2,4,6-trimethylbenzaldehyde at high excess as modulator driving lateral growth due to steric hindrances between the layers led to promising photocatalytic performance for CO2 reduction. The photocatalysis showed a high selectivity of 78% for the CO evolution compared to H2 generation, with a CO production rate of 10.2 mmol g−1 h−1. The reaction was performed in CO2-saturated, 0.1 M KHCO3 aqueous solution, with [Ru(bpy)3]Cl2 as photosensitizer and ascorbic acid as sacrificial electron donor. The nanosheet structures exhibited a superior catalytic activity towards CO2 reduction than the bulk COF, showing a CO production rate of only 124 μmol g−1 h−1 and a selectivity of 13%.132
In a recent metal-free approach, CT-COF which was condensed from e−-donating 9-ethyl-9H-carbazole-2,7-dicarboxaldehyde and e−-accepting tris-(4-aminophenyl)triazine revealed a CO production rate of 102.7 μmol g−1 h−1 without a sacrificial donor or a co-catalyst. After 3 cycles the rate decreased to 95.5 μmol g−1 h−1. The authors attributed the efficient photocatalytic behavior based on DFT calculations to the active nitrogen in the triazine moieties. Further the authors suggest that charges from the electron-richer carbazole moiety transfer to the triazine node where CO2 is adsorbed. The authors did not comment on the use of a sacrificial donor.133
The high surface area and the tunable bandgap of COFs makes this new class of porous organic frameworks a promising metal-free platform for diverse photocatalytic applications beyond water splitting. One type of application involves the use of COFs as photocatalyst in selective organic transformations.
Introducing the benzoxazole linkage into COF systems not only increases their stability, chemically as well as physically, but also enhances their capability to harvest light at lower energies. Both characteristics made LZU-190, a COF condensed from 2,5-diamino-1,4-benzenediol dihydrochloride and 1,3,5-triformylbenzene, an excellent example of precise application-oriented tailoring of a photocatalytic COF. The COF achieved yields of up to 99% for the oxidative hydroxylation of arylboronic acids to phenols when illuminated and under air. The authors suggest a mechanism in which the photoexcited COF forms superoxide radical anions by single electron transfer. These events are followed by a rearrangement and hydrolysis step to yield the desired phenols.134
Another hydrazone-based COF made from 2,5-dimethoxyterephthalohydrazide and 1,3,5-triformylbenzene exhibited photoinduced catalytic activity with high yields for tandem radical addition–cyclization reactions of 2-aryl phenyl isocyanides. Mild conditions and recyclability of the COF as photosensitizer account for a metal-free and sustainable alternative for conventional catalysts. The proposed reaction mechanism includes the formation of radical anions and subsequent reduction of the COF to close the catalytic cycle.135
The design of donor–acceptor COFs with electron-deficient triazine units (4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline) connected with electron-rich thiophene-based building blocks (specifically, thieno[3,2-b]thiophene-2,5-dicarbaldehyde or [2,2′-bithiophene]-5,5′-dicarbaldehyde) led to applications as photoinitiators for free radical polymerizations to generate poly(methyl methacrylate). Upon photoexcitation of the COF, generated holes oxidize triethylamine to form radical cations. Reaction with a second amine leads to amine radicals initiating the polymerization, which is not the case for reference systems without the COF initiator (Fig. 25).72
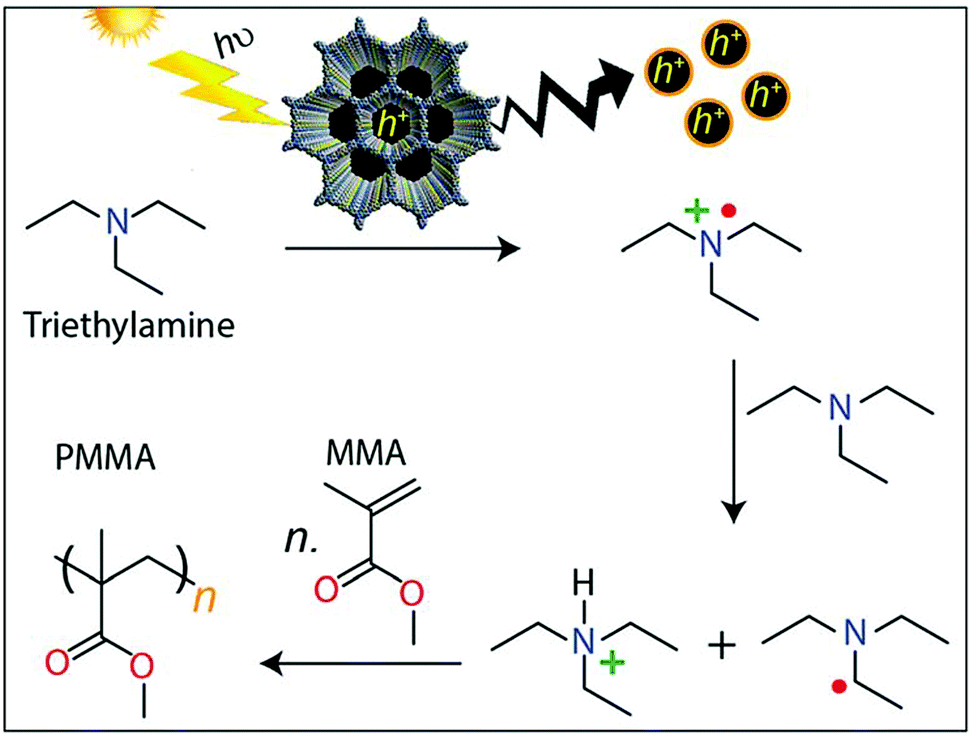 | ||
| Fig. 25 Illustration of proposed mechanism for the COF-initiated free radical polymerization of MMA to PMMA. Reproduced with permission.72 Copyright 2019, Royal Society of Chemistry. | ||
The two-in-one linkage strategy introduced by Chen and co-workers led to various COFs that can photocatalyze organic transformations with high selectivity and efficiency. On the one hand, the group developed a single component COF consisting of imine-linked porphyrin units, catalysing the oxidation of sulphides and Knoevenagel condensation reactions (Fig. 26).136 On the other hand, the group developed a benzoxazole-based COF (specifically, 4,4′-(2,6-bis(4-aminophenyl)benzo[1,2-d:4,5-d′]bis(oxazole)-4,8-diyl)dibenzaldehyde) with high photocatalytic performance for the oxidative hydroxylation of arylboronic acids.137 This two-in-one linkage strategy seems to be promising for the efficient synthesis of a platform of photocatalytically active COFs existing of only a single component with two different linkage functionalities.
 | ||
| Fig. 26 Illustration of proposed mechanism for the photocatalytic oxidation of thioanisole. Reproduced with permission.136 Copyright 2019, American Chemical Society. | ||
It is also possible to use MOF–COF hybrids covalently linked for the selective photocatalytic oxidation of alcohols. Here, TAPB-PDA (terephthalaldehyde, PDA) was grown on the MOF MIL-125 to form a hybrid structure. The proof-of-principle framework led to increased light absorption and showed superior photocatalytic activity compared to related MOF-only catalysts.138
The π-conjugation in sp2 carbon-conjugated COFs allows for broadened solar light harvesting and enhanced photocatalytic activity. For example, the ability to photocatalyze the regeneration of the coenzyme in an artificial photosystem I (a COF made from 2,4,6-tris(4-formylphenyl)-1,3,5-triazine (TFPT) and 2,2′-(1,4-phenylene)diacetonitrile (PDAN)) boosted the conversion of the NADH-consuming L-glutamate dehydrogenase to 97%, the highest value reported (Fig. 27). Even without a mediator, the high efficiency was retained and attributed to a charge transfer complex of the COF and NAD+ resulting from π–π interactions, amplifying electron transfer from the photoexcited COF.139,140
 | ||
| Fig. 27 Illustration of the artificial photosystem-I-induced coenzyme regeneration and photoenzymatic synthesis of glutamate by glutamate dehydrogenase (GDH) (sacrificial donor: triethanolamine (TEOA)). GDH structure image taken from the RCSB PDB (www.rcsb.org) of PDB ID 1HWX.140 Reproduced with permission.139 Copyright 2019, John Wiley and Sons. | ||
Benefitting from photothermal conversion, a porphyrin-containing chiral COF made from S-(+)-2-methylpiperazine and a copper 5,10,15,20-tetrakis(4-bromophenyl)porphyrin loaded with Pd or Au nanoparticles delivers the required thermal energy under visible light as well as the chiral confinement to drive and catalyze asymmetric Henry and A3-coupling reactions. The asymmetric catalysis showed not only high yields but also excellent enantiomeric excess.141
The application of COFs in the photoreduction of metal ions was demonstrated by Chen's group with a COF containing trigonal triazine units paired with benzothiadiazole via imine linkages. The authors were able to use the COF without additional sacrificial agent to achieve a photoreduction of Cr6+ to Cr3+ with 99% yield. The authors suggest an oxidation mechanism that is based on the oxidation of H2O and OH-groups which further oxidize the framework or form oxygen.142
The photothermal effect has also been studied with respect to photothermal in vitro therapy of cancer cells. A pyrene-containing COF (connected with [2,2′-bipyridine]-5,5′-dicarbaldehyde) containing a stable radical cation in the linear linker exhibited high photothermal conversion upon irradiation with an IR laser, leading to enhanced cytotoxicity of the COF particles against A549 cancer cells. The radical cation was suggested to be stabilized as π-radical across the stacked COF layers and the broadened optical absorption spectrum allowed for enhanced IR sensitivity. Photoacoustic imaging with the irradiation of the COF has been tested successfully in vivo.143 Biomedical applications of photoactive COFs have also been investigated by Lang and co-workers. For example, coatings of porphyrin-containing COFs on polystyrene showed singlet oxygen production under illumination, which was shown to inactivate bacteria.144
Recently, the group of Ma constructed a corrole-based COF keeping the porphyrin macrocycle but introducing an asymmetric character. The TPAPC-COF was condensed from 5,10,15-tris(p-aminophenyl)corrole and terephthalaldehyde and exhibited a desymmetric hcb topology with a staggered stacking mode. Strong absorption over the complete visible light spectrum and the ability of singlet oxygen generation together with the avoided self-quenching of excited states by the staggered corrole aggregates and easy diffusion in the porous framework led to an efficient photosensitizer for the photodynamic therapy.145
A combined approach of photodynamic and photothermal therapy was reported for a hybrid material of COF-LZU-1 (made from 1,3,5-triformylbenzene and 1,4-diaminobenzene) and Ag2Se nanoparticles. In vitro and in vivo studies showed that the combined generation of singlet oxygen and increase in temperature led to highly efficient anticancer therapy.146
Electronic transport in COFs
The designed columnar pathways and the intralayer conjugation network in (2D) COFs allows for the migration of excitons or charge carriers, both holes and electrons. Studies have shown that ordering the building blocks in these frameworks and interconnecting them in-plane by covalent bonds shows superior behavior in charge carrier transport and separation in comparison to their neat monomer units. Charges can be generated by photoexcited states such as singlet excitons that spatially segregate at donor–acceptor interfaces within the framework, through host–guest interactions, or by singlet–singlet annihilation where the migration and collision of adjacent singlets leads to charge generation. Photoexcitation and subsequent charge separation in COFs have motivated investigations of photovoltaic devices or of photocatalysts for various reactions such as the hydrogen evolution reaction. In this context, the intrinsic high surface area of COFs and adjustment of both bandgap and band alignment are crucial for realizing efficient applications.The theme of photoconductive COFs was already discussed in the previous chapter ‘Interaction of light with COFs’, and will now be extended to the whole field of conducting covalent organic frameworks.
Macroscopic and microscopic characterization techniques
Conductivity and mobility analysis techniques can differ regarding the observed parameters and the spatial extent of the relevant process, hence the meaning of the resulting data. Therefore, the results need careful consideration when compared to those obtained with other methods. For example, using a macroscopic technique, grain boundaries would strongly influence results, leading to reduced values compared to microscopic techniques where processes in single crystallites/domains may be probed.A simple measurement setup is the two-contact probe for which the sample is contacted with two electrodes, or sandwiched in between two electrodes. By applying a voltage (and possibly illuminating the sample) a typical I–V curve is obtained from which the electrical conductivity can be retrieved. This macroscopic technique reflects the average conductivity of the gap in-between the two contacts.147
Based on a similar principle of a two-contact probe, time of flight (ToF) analysis with pulsed laser excitation can be used to obtain transient photocurrents and to reveal data such as nature and number of the generated charge carriers, the photocarrier generation yield ϕ, and the mobility.148
Another macroscopic method is a four-electrode setup such as the van der Pauw method which can be applied to macroscopic COF samples such as thin films. The applied current between contact 1 and 2 induces a voltage which can be measured between contact 3 and 4 and converted to the resistivity (Fig. 28). Together with the sample thickness the elucidation of the conductivity is possible. To retrieve more data such as charge carrier concentration and mobility, an external magnetic field affecting the moving charge carriers needs to be applied.147
 | ||
| Fig. 28 Electrode pattern used for van der Pauw method with four electrodes (gold) on sample with random shape (grey). | ||
It is also possible to design devices in the form of a field-effect transistor with the COF serving as the active layer. By applying a gate voltage, the source-to-drain current initially increases linearly. This linear regime in the current vs. gate voltage plot is then used to calculate the mobility of the charge carriers on a macroscopic scale.149
Going to smaller dimensions, time-resolved microwave conductivity (TRMC) is a non-destructive and non-contact method where the sample is exposed to microwave radiation in the GHz range and the transmitted or reflected microwave power is detected. After photoexcitation with a laser, charge carriers are generated on the surface or in the bulk material depending on the wavelength. The charge carriers then cause a change in the detected signal that can be related to the conductivity, photocarrier generation yield, carrier lifetime and mobility, and also allows for the determination of the nature of the carriers. The high frequency of the microwave radiation implies that the motion of the charge carriers only extends over microscopic length scales.150
If the frequency is further increased, we reach the optical pump terahertz probe spectroscopy (OPTP), which is also a non-contact method. The sample is optically pumped by a pulsed laser and then probed with a THz pulse. The photoexcited electron–hole pairs interact with the electric field of the terahertz pulse, leading to the photoinduced conductivity. From correlation with frequency and time after the optical pump, doping densities, carrier mobilities and lifetimes can be extracted. These features are again probed on a microscopic scale with a temporal resolution in the femtosecond region.151
For comparison reasons, in the following the reported conductivity of COFs will be given in S cm−1.
Conducting COFs
In 2008, Jiang and co-workers reported the first semiconducting COF (made from 2,3,6,7,10,11-hexahydroxytriphenylene and pyrene-2,7-diboronic acid). The stacked triphenylene and pyrene building blocks in the TP-COF led to an increased electric current of 4.3 nA at 2 V bias voltage compared to the neat monomer mixture. The measurements were performed with a two-contact probe across a 10 μm Pt gap under ambient conditions. The current could be increased by oxidative iodine doping allowing for the conclusion that the TP-COF is a p-type semiconductor.44 It was again Jiang's group reporting the first n-type semiconducting COF, consisting of Ni-phthalocyanine units linked with electron-deficient benzothiadiazole bridges (Fig. 29d and e). Flash-photolysis (FP) TRMC revealed a ϕ∑μ of 5.8 × 10−4 cm2 V−1 s−1, with ϕ being the photocarrier generation yield and μ the carrier mobilities, for the 2D-NiPc-BTDA COF. In combination with time-of-flight measurements, a carrier mobility of 0.6 cm2 V−1 s−1 was derived, which was attributed to the eclipsed stacking of the chromophore units.66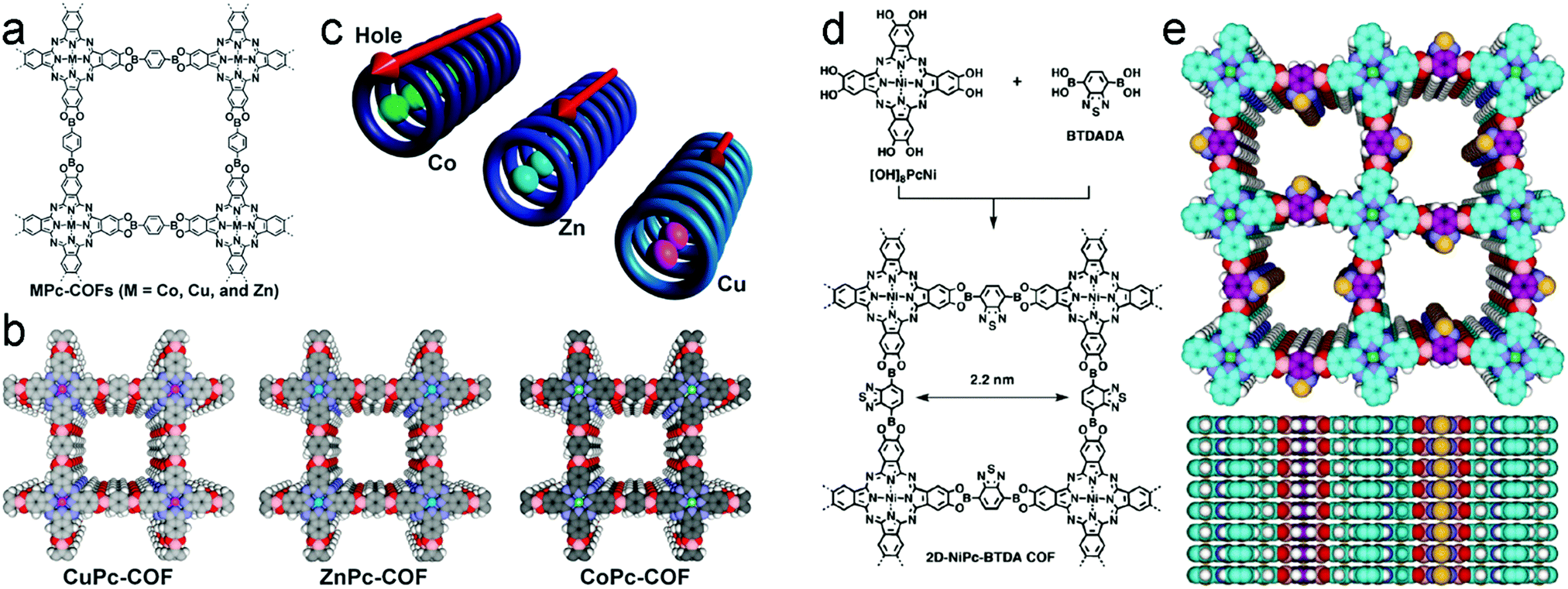 | ||
| Fig. 29 (a) Chemical structure of metalated phthalocyanine (MPc) COFs. (b) Slipped-AA stacking structure of MPc-COFs and (c) illustration of stacked phthalocyanine macrocycles allowing for hole transport. Reproduced with permission.108 Copyright 2012, Royal Society of Chemistry. (d) Synthesis of the first n-type semiconducting COF and (e) eclipsed stacking mode of the resulting 2D-NiPc-BTDA COF. Reproduced with permission.66 Copyright 2011, American Chemical Society. | ||
Yaghi and co-workers were then able to synthesize two porphyrin-containing COFs imine-linked by a phenyl ring (COF-366) and linked by boronate ester bonds with an anthracene unit (COF-66). Applying the FP-TRMC method, the associated COF/PMMA films revealed high charge carrier mobilities of 8.1 and 5.0 cm2 V−1 s−1 for COF-366 and COF-66, respectively, which was two orders of magnitude higher than the values obtained from the contact-probed TOF measurement. These mobilities exceed the values typical for many polymers and amorphous silicon and combined with the long carrier lifetime of around 80 μs, allow for promising charge carrier separation abilities of the two COFs.56
Notably, the nature of majority charge carriers in porphyrin COFs (made from tetrahydroxybenzene and 5,10,15,20-tetrakis[4(dihydroxyboryl)phenyl]porphine) could be switched by introducing different central divalent metal centers. The free-base porphyrin COF exhibited a hole-conducting behavior with high charge carrier mobilities of 3.5 cm2 V−1 s−1 measured by FP-TRMC. At the cost of reduced hole transport through the stacked macrocycles, the electron transport could be increased by inserting metal centers. Whereas Zn led to the first ambipolar conducting COF with mobility values μe and μh of 0.016 and 0.032 cm2 V−1 s−1, the insertion of Cu led to an electron-transporting material with a carrier mobility of 0.19 cm2 V−1 s−1. The combination of both transport channels, along the macrocycles and along the metals, also led to an increased photocurrent of 26.8 nA for the ZnP-COF with a high on–off ratio compared to 0.6 and 0.01 nA for the Cu-porphyrin and base-free porphyrin COF, respectively. The device structure was a COF film (25–30 μm) sandwiched between Al and ITO electrodes.152
Using the coordinating central metal in macrocycles such as porphyrins or phthalocyanines allows for tuning the electron density in the macrocycles, thus influencing the photoelectric behavior. This was further shown by Ding et al. in a study where the influence of the central metal on photoelectric properties was investigated using FP-TRMC and by the conventional two-contact probe method where the COF (made from (2,3,9,10,16,17,23,24-octahydroxyphthalocyaninato)cobalt(II)-M(II) and 1,4-benzene diboronic acid) was sandwiched between Al and Au electrodes. The TRMC results showed ϕ∑μ values consistent with the electron densities of the metallophthalocyanines (CuPc-COF < ZnPc-COF < CoPc-COF) of 1.4 × 10−4, 2.2 × 10−4 and 2.6 × 10−4 cm2 V−1 s−1, respectively (Fig. 29a–c). The two-probe photocurrents of 110, 0.6 and 0.14 nA, respectively, differ from the trend of the ϕ∑μ values and suggest the non-negligible influence of grain boundaries and morphologies of the COF material.108
Using femto- and nanosecond transient absorption spectroscopy, the charge carrier dynamics in a donor–acceptor (D–A) COF with Zn-phthalocyanine nodes connected with naphthalenediimide (NDI) bridges were investigated. The authors suggested fast light-induced charge separation within 1.4 ps between the D–A pairs. The delocalization along the π-stacked arrays allowed for long lifetimes of the separated charges of up to 10 ns in benzonitrile suspension. Investigations of the solid COF material with time-resolved electron spin resonance spectroscopy confirmed the formation of charged D–A columns, with the donor phthalocyanine and the acceptor NDI. The lifetimes of the separated charges were found to be 1.8 and 1500 μs at 280 and 80 K, respectively.68
Tetrathiafulvalene (TTF) is known for forming cation radicals and conducting charge-transfer salts. The introduction of the TTF unit into organic materials was therefore the focus of many studies. The charge carrier mobilities of phenyl- (TTF-Ph) and pyrene-containing (TTF-Py) TTF-COFs obtained with FP-TRMC were 0.2 and 0.08 cm2 V−1 s−1, respectively. The higher mobility of the TTF-Ph-COF was attributed to the slightly tighter stacking behavior compared to the TTF-Py-COF. An increase of up to 6 orders of magnitude in electrical current was observed when the TTF unit was oxidized with iodine leading to a charge-transfer complex of the TTF radical cation and iodide anion. The conductivity of this oxidized COF was measured on pellets with a thickness of 0.5 mm and resulted in values of 10−5 and 10−6 S cm−1 for TTF-Ph and TTF-Py COF, respectively.153 A comparable electroactive behavior was also found by Zhang and co-workers, pointing out that the oxidized TTF-Ph COF retained the crystalline structure and showed a similar conductivity of 1.8 × 10−6 S cm−1.154
The conductivity of the TTF-Ph COF could be even more enhanced in iodine-doped oriented thin films, where the high resulting values of 2.8 × 10−3 S cm−1 were attributed to the delocalization of the radical across the mixed-valence TTF stacks (Fig. 30).155
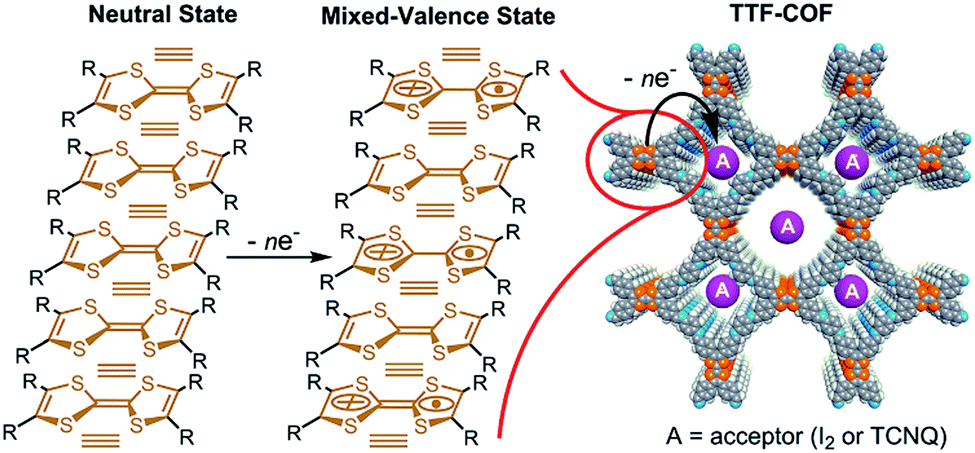 | ||
| Fig. 30 Scheme of the radical delocalization in the TTF-COF leading to mixed-valence state TTF species. Reproduced with permission.155 Copyright 2014, Royal Society of Chemistry. | ||
When aiming for high π-column density in COFs, the hexabenzocoronene (HBC) building block offers attractive features such as a large ‘graphitic’ coronene core, favorable stacking and multiple points for functionalization. A corresponding HBC-COF made from planar hexabenzocoronene and terephthalaldehyde shows a high hole mobility of 0.7 cm2 V−1 s−1 determined by FP-TRMC. The ϕ∑μ values of the HBC-COF were found to be 1.5 × 10−5 cm2 V−1 s−1 by the direct current integration method.109 Further FP-TRMC investigations of a boronate ester-based COF containing non-planar 2,3,10,11,18,19-hexahydroxy-cata-hexabenzocoronene as linker molecules connected with pyrene-2,7-diboronic acid showed that the sum ϕ∑μ of 0.6 × 10−4 cm2 V−1 s−1 is not affected by the core-twisted structure of the building units, which induce a wavy 2D structure in the obtained COF (Fig. 31).156
 | ||
| Fig. 31 Schematic illustration of core-twisted HBC node inducing a wavy structure in Marta-COF-1 and guiding the layer stacking of the COF. Reproduced with permission.156 Copyright 2019, American Chemical Society. | ||
In a single component COF in which protected tetrakis(arylhydroxylamine)porphyrin units are fused by azodioxy bridges, the electrical conductivity could be enhanced by three orders of magnitude upon iodine doping to reach 1.52 × 10−7 S cm−1. The increase is attributed to the decrease of the energy barrier for charge transport and to electron traps leading to better hole mobility due to the iodine doping.111
Our group studied directional charge transport in boronate ester-based COF thin films made from BDT and HHTP and showed a correlation between hole mobility and film thickness. The hole mobilities were measured for a hole-only device with MoOx serving as the hole-transporter and ITO and gold serving as the electrodes. For films with a thickness of around 80–90 nm, the hole mobility reached up to 3.0 × 10−7 cm2 V−1 s−1 and turned out to be dependent on illumination, which was attributed to photoexcited charge carriers filling up electronic trap states. Furthermore, we observed an in-plane conductivity of up to 5 × 10−7 S cm−1 in oriented thin film devices, which exceeded the conductivity of non-oriented, pressed COF pellets.157 For another boronate ester-based COF, the DPP2-HHTP-COF made from ((2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]-pyrrole-1,4-diyl)bis(thiophene-5,2-diyl))diboronic acid, we were able to induce diketopyrrolopyrrole stacks within the COF scaffold, which led to the highest conductivity (of up to 2.2 × 10−6 S cm−1) found for boronate ester-linked COF bulk materials. The measurements were performed with a four-point probe Hall measurement providing macroscopic conductivity values.158
The application of COFs as bulk doping agent for perovskite solar cells (PSC) was investigated with 3D COFs bearing spirobifluorene units well known for their potential in optoelectronic devices. The power conversion efficiency (PCE) of the SP-3D-COF-1 and -2 doped PSCs could be increased by up to 18% and the electron mobility in the COF-doped perovskite layer was almost doubled reaching 4.44 × 10−3 cm2 V−1 s−1. Based on computational studies, the mechanism could be attributed to the additional charge generation and the spatial separation of HOMO and LUMO in the COF preventing charge recombination.159
In a 3D-COF, a TTF-based building block was combined with an adamantane unit leading to a non-interpenetrated pts topology (Fig. 32). The open pores and high surface areas allowed for efficient iodine doping and tunable electrical conductivity. The values could be enhanced up to 2.7 × 10−4 S cm−1 upon iodine doping for 48 h at 25 °C, measured with a two-probe method using gold paste as electrodes. Increasing the temperature to 120 °C led to a conductivity of 1.4 × 10−2 S cm−1, which exceeds the value found for 2D TTF-COFs.160
 | ||
| Fig. 32 Synthesis of the 3D-TTF-COFs with a non-interpenetrated structure (f, JUC-518) and an interpenetrated structure (g, JUC-519). Reproduced with permission.160 Copyright 2019, American Chemical Society. | ||
To reach for even higher conductivity and mobility values, the conjugation across the bonds connecting the building blocks was increased by a pyrazine linkage motif. The hcc-COF consisting of pyrazine-linked benzene rings showed a conductivity, measured in a FET-type device of a COF film, of 4.0 × 10−3 S cm−1. We note that this value was found for the pristine COF without any added doping agents, and that the data do not allow conclusions regarding the orientation of the film. In the corresponding bulk material, conductivity values of up to 2.2 × 10−5 S cm−1 were obtained with a two-probe method.161
Mirica and co-workers used a pyrazine linkage to connect a Ni-phthalocyanine macrocycle with a pyrene unit to form a COF (reacting 2,3,9,10,16,17,23,24-octaaminophthalocyaninato nickel(II) and pyrenetetraone). Using a four-point probe method, the bulk conductivity of the COF was found to be 2.51 × 10−5 S cm−1. Upon iodine doping of the COF device with interdigitated gold electrodes, the current increased by a factor of 103. DFT calculations suggest an eclipsed stacking of the building blocks and thus facilitated charge transport along the stacked macrocycles.162
Changing the metal center of the phthalocyanine unit to Zn and Cu and coupling with a tert-butylpyrene-tetraone via a pyrazine-linkage produced COFs with Hall mobilities of 4.8 cm2 V−1 s−1 for the ZnPc-COF and 0.9 cm2 V−1 s−1 for the CuPc-COF and conductivities of around 5 × 10−5 S cm−1, representing the direct current limit on a macroscopic scale. Further investigation of these COFs with terahertz spectroscopy confirmed the above trend, revealing mobilities of 2.0 and 0.7 cm2 V−1 s−1 for the Zn- and Cu-version, respectively. Notably, DFT calculations suggested that an influence of the metal centers on the hole mobilities could be excluded and that differences in boundary scattering led to the change in mobility. The boundary scattering is affected by depletion regions between the crystallites. In-plane charge carrier transport was calculated to be very low compared with the out-of-plane mobility (Fig. 33).163
 | ||
| Fig. 33 Electronic band structures and projected density of states (PDOS). (a) Monolayer and (b) serrated AA stacked multilayers of ZnPc-pz. (c) Monolayer and (d) serrated AA stacked multilayers of CuPc-pz. Reproduced with permission.163 Copyright 2019, American Chemical Society. | ||
To further evolve the level of conjugation, one can move from pyrazine-linked COFs to sp2 carbon-conjugated linkage motifs to create a planar fully π-conjugated bridge between the aromatic building blocks. This is expected to allow for facilitated charge carrier transport, high stability and thus enhanced potential for application as semiconducting material. The group of Jiang was able to synthesize a fully sp2 carbon-conjugated framework (made from tetrakis(4-formylphenyl)pyrene and 1,4-phenylenediacetonitrile) that was identified as an insulator with a conductivity of 6.1 × 10−16 S cm−1. However, upon oxidation with iodine the conductivity could be immensely increased by a factor of 1012 to 7.1 × 10−4 S cm−1. Furthermore, the spins of the generated radicals exhibit paramagnetic behavior but undergo a ferromagnetic phase transition below 10 K.25 Based on computational analysis, a mechanism was proposed to explain the conducting behavior of the iodine-doped COF. Here, the large effective mass of carriers leads to low mobilities in the undoped framework whereas the iodine oxidation leads to increased mobility values. In combination with the high conductivity calculated for the I3− anion in the channels of the framework, the authors propose that the increase in conductivity is based not only on the increased charge carrier mobilities in the framework but also aided by high ionic conductivity.164
Zhang, Bi and co-workers further developed this approach to study the properties of semiconducting COFs and observed current densities of up to 45 μA cm−2 (with a COF made from 2,4,6-trimethyl-1,3,5-triazine and terephthalaldehyde) when applied as photoelectrodes in aqueous medium at 0.2 V vs. RHE under conditions of photocatalytic hydrogen generation.165
In a computational study, C–C-bonded single component COFs containing pyrene, porphyrin and Zn-porphyrin building blocks were investigated regarding their charge carrier mobilities. The calculations predicted a carrier mobility of 66 cm2 V−1 s−1 for holes and electrons in the pyrene COF and hole mobilities of 82 and 94 cm2 V−1 s−1 in porphyrin and Zn–porphyrin COFs, respectively. Subsequently, a synthesized single component ZnPor COF (made from 5,10,15,20-tetrakis(ethynyl)porphyrin-zinc(II)) with poor crystallinity revealed a mobility of 3.5 × 10−2 cm2 V−1 s−1 obtained from TRMC with ϕ∑μ of 3 × 10−5 cm2 V−1 s−1.33
Perepichka and co-workers designed an imine-linked COF with trioxaazatriangulene (TANG) as a trigonal node connected by 2,5-dihydroxyterephthalaldehyde. The large electron-rich aromatic system of TANG led to increased π-interactions of adjacent layers which consequently yielded strong absorption far into the near-infrared region. Upon iodine doping the small bandgap of 1.2 eV of the pristine TANG-COF was decreased to <0.2 eV along with an increase of conductivity from 1.6 × 10−5 S cm−1 to 2 × 10−3 S cm−1 measured with two-contact and four-contact probes. Under vacuum this value even increased to 1 × 10−2 S cm−1, positioning the TANG-COF in the class of highly conducting COFs. Furthermore, the amount of free spins in the doped poly(radical cation) TANG-COF was reported to reach 10% and the COF showed a paramagnetic behavior in the temperature range from 300–20 K.166
We note that COFs have also been used as precursors for carbon nanostructures, using pyrolysis or ionothermal transformation to increase the conductivity in the resulting materials.167,168
Energy storage with COFs
Their high surface area and the ability to combine a large variety of building units, including those with redox-active groups, makes covalent organic frameworks interesting for applications in energy storage, such as batteries, capacitors, supercapacitors or electrocatalysts.169 The first COF showing reversible redox processes was reported by Dichtel and co-workers. Specifically, the ketoenamine-linked DAAQ-TFP COF (made from 2,6-diaminoanthraquinone (DAAQ) and 1,3,5-triformylphloroglucinol (TFP)) bearing a redox-active 2,6-diaminoanthraquinone unit showed an initial capacitance of up to 48 F g−1 and even after 5000 cycles a stable capacitance of 40 F g−1 for COF bulk material mixed with carbon black. This work laid the foundation for future applications of COFs in pseudocapacitive energy storage.170 It was again Dichtel's group that demonstrated the effect of oriented COF thin films on electrochemical behavior. Starting from non-oriented bulk material of the DAAQ-TFP COF with a charge storage ability of 0.4 mF cm−2, this value was increased to 3 mF cm−2 by the enhanced access into the pores of oriented films, featuring high stability even after 5000 cycles.7Staying with the same framework of ketoenamine-linked anthraquinone building units, Mulzer et al. and later Wu et al. constructed ‘electron highways’ in the COF pores by polymerization of 3,4-ethylenedioxythiophene (EDOT) within the COF. The electropolymerization of EDOT occurred in COF thin films and led to a maximum capacitance of 350 F cm−3 but was limited to very low amounts of COF sample. The first step to scale up the amount of COF was achieved by a chemical polymerization (Fig. 34).171
 | ||
| Fig. 34 Schematic illustration of DAAQ-TFP films with incorporated PEDOT (a) and of corresponding oxidation and reduction of the DAAQ moieties in the COF. (b) Exemplary scheme of reduction and oxidation processes in the COF pores. Reproduced with permission.171 Copyright 2016, American Chemical Society. | ||
A further step was taken by Wu et al. with a solvent- and catalyst-free approach of solid-state polymerization of EDOT. With PEDOT strands winding through the pores, the COF composite reached a conductivity of up to 1.1 S cm−1 measured by two-probe and four-probe methods. Also, the specific capacitance increased to 1663 F g−1 compared to the pristine DAAQ-TFP COF (see work of Dichtel170 above), exhibiting high stability and fast charge and discharge rates.172
Banerjee and co-workers combined the redox-active anthraquinone units with anthracene linkers embedded in the COF (made from 2,6-diaminoanthracene (Da), 2,6-diaminoanthraquinone (Dq) and 1,3,5-triformylphloroglucinol (TP)). This strategy allowed for the synthesis of flexible yet electrochemically active sheets. Their use in quasi-solid-state supercapacitors was investigated comparing anthraquinone-only COF (DqTp) sheets and mechanically more flexible COF sheets blended one-to-one with anthraquinone and anthracene bridges (Dq1Da1Tp). The highest energy density was 0.43 and 0.30 μW h cm−2 for the DqTp and Dq1Da1Tp COFs, respectively. Considering the power densities, the values were determined to be 980 and 960 μW cm−2, respectively. The additional mechanical stability achieved by introducing the anthracene part led to a superior long-term stability compared to the DqTp COF.173
In order to further increase the electrochemical stability of the above COF, the hydroxy groups of 1,3,5-triformylphloroglucinol were substituted by methoxy groups to enable the formation of interlayer hydrogen bonds. As a result, instead of the ketoenamine linkage, imine bonds formed the COF in a condensation step with the anthraquinone moiety. Consequently, the COF sheets exhibited good performance when applied as supercapacitor in 3 M aqueous sulfuric acid with capacitance up to 1600 mF cm−2 and 169 F g−1. The fabrication of a symmetric solid-state supercapacitor yielded a device with a capacitance of 84 mF cm−2 and energy and power densities of 2.9 μW h cm−2 and 61.8 μW cm−2, respectively.174
The combination of the mechanically stable mixed one-to-one anthraquinone and anthracene COF (see work of Banerjee173 above) with carbon nanofibers increased the conductivity and led to outstanding performance in a quasi-solid-state supercapacitor with areal capacitance of 167 mF cm−2 (Fig. 35). The energy and power density were determined to be 5.8 μW h cm−2 and 125 μW cm−2, respectively. The supercapacitive capability of this device exceeds the values of MOF, COF and other carbon-based supercapacitors.175
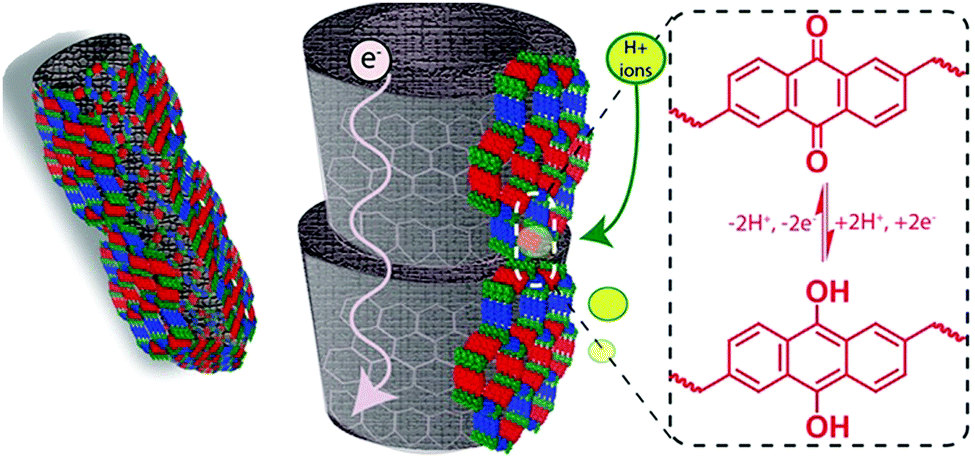 | ||
| Fig. 35 Illustration of supercapacitor material with carbon nanofiber as electron-conducting and COF as redox-active compound with the scheme of oxidation and reduction in the enlarged area. Reproduced with permission.175 Copyright 2019, American Chemical Society. | ||
Jiang and co-workers synthesized a COF consisting of a Ni-porphyrin framework connected by linear bridges with ethynyl groups attached (made from 2,5-bis(2-propynyloxy)terephthalaldehyde and 2,5-dimethoxyterephthalaldehyde at different molar ratios with nickel 5,10,15,20-tetrakis(4′-tetraphenylamino) porphyrin). By click reaction, 4-azido-2,2,6,6-tetramethyl-1-piperidinyloxy groups were covalently bound to the framework leading to a radical-containing COF. This strategy led to enhanced capacitance compared to the corresponding non-radical COF scaffold.176
The influence of the morphology on the performance of a COF-based supercapacitor was established in a study investigating carbazole-based COFs. Even though Car-TPT COF (made from 9-(4-aminophenyl)-carbazole-3,6-diamine and 2,4,6-tris(4-formylphenyl)triazine) showed the lowest BET surface area in this COF series and exhibited only a single redox-active site per building block, this COF featured the highest capacitance of 17.4 F g−1 due to its accessible microtubular structure.177
Recently, a series of pyridyl-functionalized COFs (for example, made from tripyridine-triazine and different phenolic trialdehydes) was shown to exhibit high power density and stability over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in supercapacitor applications. The redox-activity could be controlled by the number of hydroxyl groups attached to the trialdehyde linker. One attached hydroxyl group led to the largest BET surface and to a specific capacitance of 92 mF cm−2 (at 0.5 mA cm−2) and a power density of 98 μW cm−2.178
000 cycles in supercapacitor applications. The redox-activity could be controlled by the number of hydroxyl groups attached to the trialdehyde linker. One attached hydroxyl group led to the largest BET surface and to a specific capacitance of 92 mF cm−2 (at 0.5 mA cm−2) and a power density of 98 μW cm−2.178
The group of Kuo was able to synthesize the first COF based on tetraphenyl-p-phenylenediamine that led to the introduction of two redox-active triphenylamine sites per node and was coupled with 1,3,6,8-tetrakis(4-formylphenyl)pyrene or 1,1,2,2-tetrakis[4-formyl-(1,1′-biphenyl)]ethene to yield TPPDA-TPPyr and TPPDA-TPTPE COFs, respectively. Both COFs showed high specific capacitance of up to 237.1 F g−1 and capacity retention of above 85.6% after 5000 cycles.179
For applications of COFs in batteries, Jiang and co-workers proposed to combine COF material with carbon nanotubes. In their study, they investigated a boronate ester-based COF made from naphthalene diimide and triphenylene nodes. The resulting accessible open pore structure of the COF was viewed as a key factor for efficient Li ion diffusion, redox-active COF architecture and facilitated electron conduction through the carbon nanotubes.180 High Li ion conductivity of up to 0.26 mS cm−1 could be demonstrated in pressed COF samples inducing some orientation of the COF layers.181 In a 3D cyclodextrin-based anionic COF (connected with trimethyl borate), Li+ was chosen as a counterion resulting in Li channels along the framework. The Li ion conductivity at 30 °C was determined to be 2.7 mS cm−1, providing promising characteristics for Li-based batteries for energy storage.182
In order to trap polysulfides in COFs to make them applicable in Li-S batteries, COF-nets (COF-1, COF-5) were synthesized on a carbon nanotube net creating an interlayer between COF and carbon nanotube. Here, boronate ester bonds in the COF served as adsorption sites for both Li ions and Sx2−, showing promising results.183,184 Further investigations established COFs (for example, made from pyrene and terephthalaldehyde, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline and 2,3,5,6-tetrafluoroterephthalaldehyde, or TAPB and 2,5-divinylterephthalaldehyde) as potential cathode material in Li–S batteries with high capacity and cyclability.185–187 It was also shown that COFs (made from 1,3,5-tris(4-formylphenyl)benzene and DAAQ, or pyromellitic dianhydride and tris(4-aminophenyl)amine or TAPB) could serve as electrode material in Li ion batteries. Here, the Li-ion diffusion was facilitated by exfoliation of the COF material leading to few-layered COF sheets with superior performance compared to the bulk material, with promising capacities around 1000 mA h g−1.188–190
The approach of using exfoliated COF nanosheets was also transferred to other alkali ion batteries, such as sodium and potassium ion batteries in which the COF served as anode material. In a sodium ion battery, the use of TFPB-TAPT COF, which was synthesized from 1,3,5-tris(4-formyl phenyl)benzene and 1,3,5-tris(4-amino phenyl)-triazine, as anode material led to reversible capacities of up to 248 mA h g−1 and a capacity retention of 125 mA h g−1 after 500 cycles at 30 mA g−1. The performance of this material is based on the porous nature of the framework which allows for easy access of sodium ions into the channels.191
In a hybrid material consisting of the few-layered boronate ester-based COF-10 grown on carbon nanotubes (COF-10@CNT) the high potassium storage ability led to reversible capacities of 288 mA h g−1 after 500 cycles at 100 mA g−1. The pristine COF-10 (built from 4,4′-biphenyldiboronic acid and 2,3,6,7,10,11-hexahydroxytriphenylene) exhibited a capacity of 57 mA h g−1 with the same parameters as above. The authors suggest a mechanism where the potassium ions intercalate in-between the layers and reversibly interact with π-electrons from the aromatic benzene rings (Fig. 36). The superior performance of COF-10@CNT was suggested to arise from decreased diffusion pathways and thus faster insertion and extraction dynamics of K+. These features also allow for a capacity of 161 mA h g−1 after 4000 cycles at 1000 mA g−1, exceeding the performance of other organic and inorganic carbon anodes for potassium ion batteries.192
 | ||
| Fig. 36 Scheme of proposed potassium storage mechanism of COF-10. Reproduced with permission.192 Copyright 2019, American Chemical Society. | ||
Charge separation in COF heterojunctions
In the chapters ahead, we will discuss charge separation in the COF as a crucial requirement for the envisioned applications of COF-based heterojunctions. Already having provided an overview of charge generation, charge transport, recombination processes and also charge separation and extraction to evolve hydrogen, reduce metals, or harvest solar energy, this chapter will briefly describe the different approaches towards structuring heterojunctions in COF devices.Several sophisticated strategies for creating heterojunctions in COF materials have already been reported in the literature.
On the one hand, we can build up a heterojunction from molecular building blocks using the columnar arrays in the framework of 2D COFs in order to create separate donor and acceptor stacks (named integrated heterojunction). The holes and electrons generated upon absorption of light in principle can then be spatially segregated onto the π-stacked donor or acceptor columns along which they can move and from where they can be extracted. By means of this arrangement and given a favorable band alignment, the charges can be easily separated due to a short diffusion distance to the heterojunction interface leading to ultrafast separation and an ambipolar conducting framework.67
It was already shown that in favorable cases these separated charges can have long lifetimes in the framework due to the delocalization across the π-stack.193 This approach does not primarily depend on pore size or pore structure but on the energy levels of the COF building units. The energy alignment of the donor and acceptor is crucial for achieving an efficient driving force for separating the initially generated excitons. Following this principle, HOMO and LUMO of the COF structure should be spatially separated and located on different nodes serving as donor and acceptor.69 If the driving force is too low, for example when coupling a weaker electron acceptor to the donor, one can face a situation where no charge separation occurs, as with DTP–APyrDI-COF.194
Structuring the COF in this manner conceptually allows for its implementation in solar cell devices with ordered donor and acceptor columns. Challenges arising include loss mechanisms and conductivity issues occurring in the COF, thus impeding the extraction of charges.23
On the other hand, heterojunctions can be constructed by inserting an electron acceptor phase into the pore channels of a donor-type COF (or vice versa, inserting a donor phase into an acceptor-type COF), leading to an interpenetrated COF heterojunction. For this strategy to be realistic, the pore size needs to be in the mesoporous range to allow for the formation of a sufficiently large included phase consisting of guest molecules. This also requires good accessibility of the pores, whereas blocked pores by displaced COF layers or entrapped COF oligomers can significantly decrease the interface of the interpenetrated host–guest heterojunction.195 Compared to the abovementioned concept, the travel distance of charge carriers might then be increased due to pore blocking or inadequate filling. If the scaffold serves as a hole-conducting material, the acceptor phase within the pores is expected to transport electrons. For example, in the TT-COF (made from thienothiophene and HHTP moieties), charge extraction could be achieved by inserting the soluble C60 derivative PCBM into the pores of the photoconductive thienothiophene-containing framework (Fig. 37). The associated optoelectronic features allowed for the fabrication of a first heterojunction device based on the COF thin film grown on ITO and contacted with an aluminium top electrode. Under illumination, photovoltaic studies showed successful charge-transfer from the COF onto the PCBM, followed by charge separation and extraction.22 Transient absorption spectroscopy revealed longer lifetimes of polarons, which can be attributed to the suitable interface created where charges dissociate and form polarons.196
 | ||
| Fig. 37 Schematic overview of fullerene insertion into TT-COF pore and photo-induced electron transfer from the framework onto the electron-acceptor guest molecule. Reproduced with permission. Copyright 2013,22 John Wiley and Sons. | ||
Another elegant approach to create an ordered heterojunction with electron-accepting guest molecules includes the Huisgen cycloaddition between the azide-functionalized N3-ZnPc-COF (made from 1,4-phenylenediboronic acid (BDBA), 2,5-bis(azidomethyl)-1,4-phenylenediboronic acid and 2,3,9,10,16,17,23,24-octahydroxyphthalocyaninato) and fullerene units with an alkyne group attached (Fig. 38). This allowed for controlled filling of the pores with fullerenes that could be easily observed in IR spectra. ESR data showed that an increase of fullerene content in the COF enhanced the signal intensity correlated with the amount of radical species. This indicated photo-induced charge generation and electron transfer to the electron-accepting fullerene units clicked to the framework.74
 | ||
| Fig. 38 (a) Scheme of the synthetic route for a heterojunction COF and the subsequent functionalization of the pore walls with fullerene via Huisgen cycloaddition. (b) Top and side view of the resulting envisioned donor–acceptor structure. Reproduced with permission.74 Copyright 2014, American Chemical Society. | ||
A different approach towards creating a heterojunction was already elaborated in the photocatalysis part above (‘Interaction of light with COFs’), where the interface occurs between the liquid electrolyte in the COF pores and the solid COF itself.
Macroscopic devices based on COF architectures
With the ability to structure COF-based heterojunctions and other functional morphologies in various ways, they can be implemented in working devices requiring charge separation and other processes. In this context, it is worth noting that there are many ways to grow and structure films. Since COF film growth is not generic for any application, the synthetic procedure is chosen with regard to the final device use. Several reports in the literature have demonstrated the possibilities and ways to synthesize different kinds of films, e.g., oriented films, free-standing films at interfaces (air–water, organic-water, etc.), using a solid-state approach or 3D printing, exfoliated nanosheets, or hierarchical structures created by electrophoretic deposition.78,157,197–205 In the following we will discuss COF-based working devices utilizing some of the COF architectures mentioned above.Photovoltaic devices
In 2013, our group reported the construction of the first solar cell device with a COF acting as the photoactive layer. The oriented TT-COF thin film (see TT-COF discussed above) was spin-coated with a solution of PCBM serving as electron acceptor to achieve the desired heterojunction for charge separation. Sandwiched between ITO and aluminium, this photovoltaic device yielded a modest power conversion efficiency (PCE) of 0.053% with a VOC of 622 mV, a JSC of 0.213 mA cm−2, and a fill factor 40%. These findings prove the conceptual feasibility of COFs integrated in photovoltaic applications, but also expose challenges regarding light harvesting and loss mechanisms limiting charge extraction.22In related work, the group of Jiang chose a different approach with non-oriented COF bulk material, made from triphenylene hexaamine (TPHA) and tert-butylpyrene tetraone. To insert the PCBM, thermal sublimation/diffusion at 400 °C was performed leading to a COF with fullerene inside the pores but also fullerene aggregates between COF particles. The fullerene content was 50 wt% and also acted as a glue which was additionally added in the spin-coating process. This composite material was spin-coated on ITO and contacted from the top with aluminium. The devices showed an increased performance compared to the oriented TT-COF film device above, leading to a power conversion efficiency (PCE) of 0.9% with a VOC of 980 mV, a JSC of 1.7 mA cm−2, and a fill factor of 54%. While the photovoltaic features could be enhanced, it is evident that extracting the photogenerated charges is still a challenge.24
The above examples are based on interpenetrated COF heterojunctions utilizing the COF pores to insert electron acceptors. The alternative approach, relying on the combination of donor and acceptor building blocks to design an integrated COF heterojunction between the respective columns of donor and acceptor moieties, was reported by our group in 2014. In this Tp-Por COF made from linear 5,15-bis(4-boronophenyl)porphyrin and HHTP, the electron transfer from the triphenylene unit to the porphyrin could be observed in photoinduced absorption (PIA) spectroscopy and led to the first working photovoltaic device using the COF donor–acceptor interface for charge separation. The device showed superior (but still moderate) performance compared to a device constructed from the simply mixed neat building blocks, with a VOC of 312 mV and a JSC of 0.045 mA cm−2 (Fig. 39). The existence of severe loss processes was shown by the low external quantum efficiency of below 1%, which could be increased under reverse bias of 2 V to more than 30% at 350 nm.23
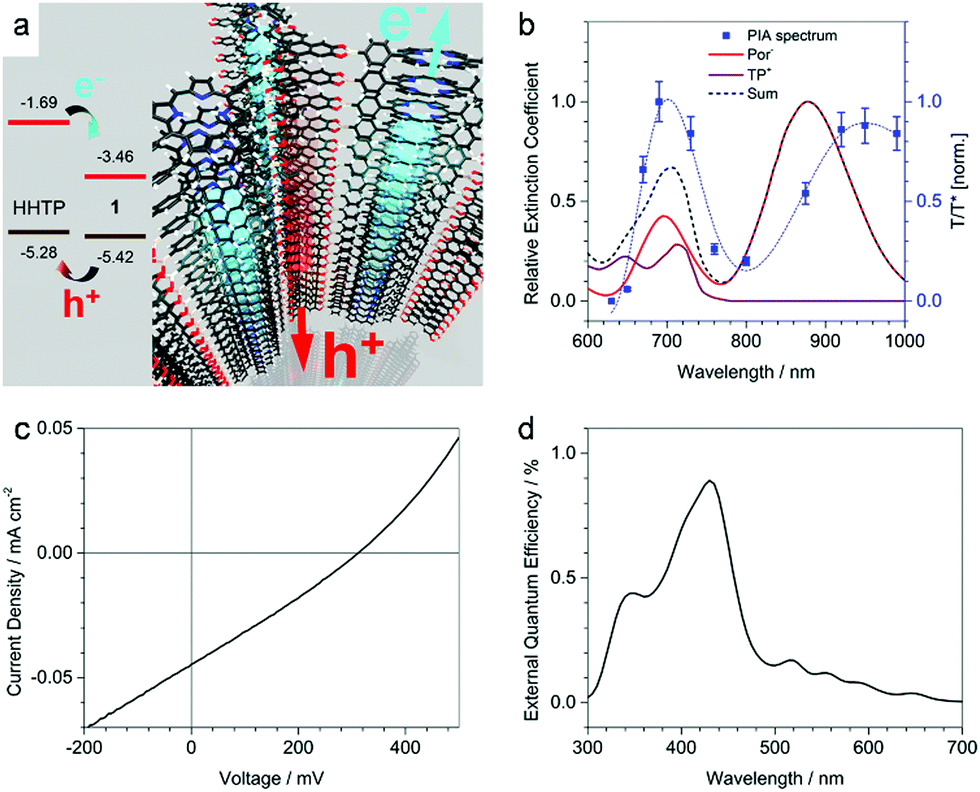 | ||
| Fig. 39 (a) Frontier orbital energies of both Tp-Por COF subunits and schematic illustration of the photogenerated charge separation. (b) PIA spectrum of the COF film together with radical ion absorption of the subunits. (c) J–V curve of the COF-based photovoltaic device and (d) the corresponding EQE spectrum. Reproduced with permission.23 Copyright 2014, American Chemical Society. | ||
The challenge regarding efficient light harvesting has been successfully addressed in recent years. We have reported several strategies to drastically increase absorption in the near IR (NIR) and IR. Connecting oligothiophene backbones with pyrene units led to tunable optical absorption bands in the IR-region up to 1000 nm.30 The absorption behavior of a thienoisoindigo-COF could be applied in a NIR-responsive photodetector. The small bandgap was created by integrating electron-deficient isoindigo derivatives (such as 6,6′-bis(4-formylphenyl)-N,N′-dibutyl-isoindigo, 5,5′-bis(4-formylphenyl)-N,N′-dibutyl-thienoisoindigo and 5,5′-bis(2-formylthiophen-5-yl)-N,N′-dibutyl-thienoisoindigo) with electron-rich pyrene nodes in a COF structure. Fabricating a heterojunction with PCBM as electron acceptor in the COF pores led to a spectrally switchable photodetector.18 The matrix of a 2D COF made of porphyrin nodes and linear thienothiophene enforced extended J-aggregate formation of the porphyrin macrocycles leading to enhanced light-harvesting in the near IR with strongly shifted peak ratios and much stronger absorption in the Q-bands.29
However, the extraction of the generated charges from the two different types of heterojunctions still remains a challenge and needs to be addressed for the design of efficient photovoltaic devices.
Photocatalytic water splitting
As mentioned in the chapter about the interactions of COFs with light, photocatalytic hydrogen evolution from water splitting reactions was first reported by the group of Lotsch in 2014. The respective COF (made from 1,3,5-tris-(4-formyl-phenyl)triazine and 2,5-diethoxy-terephthalohydrazide) was simply dispersed in water and in combination with platinum nanoparticles as proton reduction catalyst and triethanolamine as sacrificial donor the hydrogen evolution reached values as high as 1970 μmol h−1 g−1, but degraded after 5 hours.114This is a straightforward setup that can be safely applied since only one of the two half-reactions of water splitting occurs, otherwise the additional evolution of oxygen in the same chamber would create dangerous gas mixtures and limit scale-up. In subsequent work, the stability of COFs in photocatalytic systems was frequently ensured by the use of 1,3,5-triformylphloroglucinol as building block, leading to keto–enol tautomerism and stable linkages.
Copolymers containing dibenzo[b,d]thiophene sulfone moieties are known to be highly active, leading to significant hydrogen evolution rates. HER experiments with the polymer P7 (co-polymerized from 3,7-dibromodibenzo[b,d]thiophene 5,5-dioxide and 1,4-benzenediboronic acid) were performed in the presence of triethylamine and significant residues of Pd (from polymer synthesis).206 This knowledge was transferred to COFs in order to investigate the impact of ordered crystalline and porous materials on the photocatalytic behavior of these moieties. The hydrogen evolution reaction induced by S-COF (made from 3,7-diaminodibenzo[b,d]thiophene sulfone and 1,3,5-triformylphloroglucinol) with a single sulfone group in the linear bridge already led to increased rates of up to 4.44 mmol h−1 g−1. The related FS-COF bearing two sulfone moieties doubled the rate to 10.1 mmol h−1 g−1 with the sacrificial donor ascorbic acid and with deposited Pt nanoparticles (Fig. 40). By adding the dye WS5F ((E)-2-cyano-3-(5-(2-octyl-7-(4-(p-tolyl)-1,2,3,3a,4,8b-hexahydrocyclopenta[b]indol-7-yl)-2H-benzo[d][1,2,3]triazol-4-yl)furan-2-yl)acrylic acid) to enhance the optical absorption, the hydrogen evolution rate could be increased by another 60%, up to 16.3 mmol h−1 g−1. Compared to the related polymer P7 (see work of Sprick et al.206 above), the dye-sensitized COF sample exceeded the rates by one order of magnitude, showing the importance of crystallinity and accessible pore surface in these photocatalytic systems. A device of dropcast platinized FS-COF on glass exhibited a stable HER rate of 15.8 mmol h−1 m−2 under solar simulator irradiation (AM1.5G) which is comparable to carbon nitride films.207
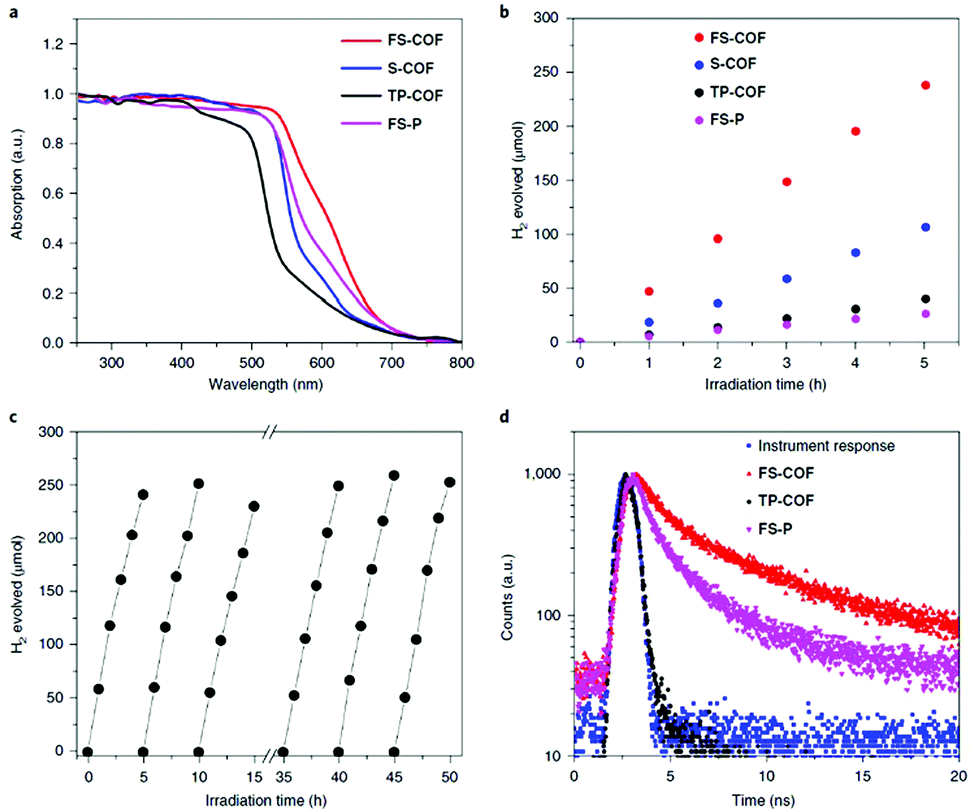 | ||
| Fig. 40 (a) Optical absorption and (b) photocatalytic H2 production as a function of time of the FS-COF, S-COF, TP-COF (reference without sulfone moiety) and the FS-P (analogous amorphous polymer). (c) Stability tests of the catalytic activity of FS-COF and (d) time-correlated single photon counting traces of the FS-COF, reference TP-COF and amorphous FS-P. Reproduced with permission.207 Copyright 2018, Nature Publishing Group. | ||
Photoelectrochemistry (PEC) separates the light-induced oxidation- and reduction half-reactions from each other such that they occur at different electrodes and in different compartments if so desired. This can be beneficial if reaction products need to be separated (as hydrogen and oxygen in water splitting reactions). In this context, our group reported the first COF-based photocathode applicable for the HER in water splitting. Oriented BDT-ETTA COF (condensed from propeller-shaped TPE (also called ETTA) and benzo[1,2-b:4,5-b′]dithiophene-2,6-dicarboxaldehyde) thin films were grown on ITO and photoelectrochemically investigated in an aqueous 0.1 M Na2SO4 electrolyte without the use of a sacrificial donor or a co-catalyst. The hydrogen reduction resulted in a photocurrent of 4.3 μA cm−2 at 0.3 V vs. RHE, being strongly dependent on the film thickness. An increase of the photocurrent by the factor of 4 could be achieved by depositing Pt nanoparticles as co-catalysts.14 The same COF system was used in a study addressing the importance of the COF morphology for catalytic applications. Using electrophoretic deposition (EPD), COF films can be easily prepared from COF suspensions within seconds, featuring textural porosity and thicknesses depending on the deposition time. Using such an EPD-based film of BDT-ETTA COF, the HER photocurrent density reached up to 21.1 μA cm−2 at 0.1 V vs. RHE under otherwise the same conditions with illumination at AM1.5G (Fig. 41). The EPD approach not only produces desirable textural porosity but also allows for the simultaneous co-deposition of a co-catalyst such as Pt nanoparticles. With such a co-catalyst, the photoelectrolysis of water resulted in a photocurrent density of 128.9 μA cm−2 at 0.1 V vs. RHE, showing the vital role of both morphology and co-catalysts in the photocatalysis of water.205
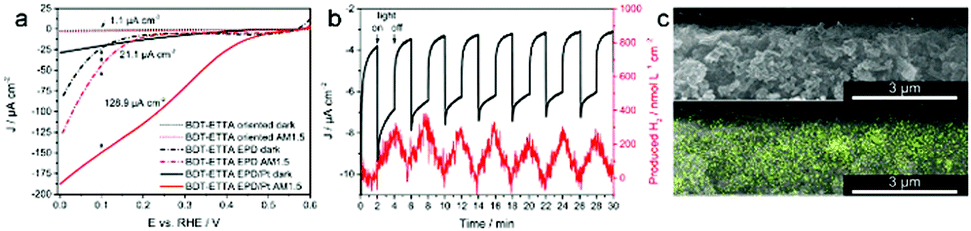 | ||
| Fig. 41 (a) PEC linear sweep voltammograms of electrodes coated with BDT-ETTA COF grown as an oriented film, a film prepared by EPD and a film with co-deposited Pt nanoparticles prepared by EPD. (b) Dynamic hydrogen evolution measurement under chopped illumination at 0.2 V vs. RHE. (c) SEM cross-section of COF/Pt hybrid film with EDX mapping of Pt. Reproduced with permission.205 Copyright 2019, American Chemical Society. | ||
Further research and implementation of unsubstituted olefin linkages led to sp2 carbon-conjugated g-C18N3–COF with triazine nodes (made from 2,4,6-trimethyl-1,3,5-triazine and terephthalaldehyde) allowing for bandgap tuning, high stability and accessibility through the pores. The n-type semiconductor exhibited a PEC current density of up to 45 μA cm−2 at 0.2 V vs. RHE when dropcast onto an ITO substrate and measured in 0.2 M Na2SO4. Photocatalytic hydrogen evolution with ascorbic acid as sacrificial donor and deposited Pt on the same COF material resulted in promising results of 292 μmol h−1 g−1.165
The above examples show the great potential of COFs as photocatalytic and photoelectrochemical systems for the generation of hydrogen and increasingly also oxygen from water. Undoubtedly, this research will be further driven by the increasing knowledge about stabilizing highly conjugated COFs, controlling morphology and diffusion pathways, and embedding catalytic functionality and co-catalysts into these well-defined systems.
Sensors and detectors
The optoelectronic properties of COFs can also inspire new concepts aiming at applications in sensor or detector devices. For example, a popular approach has focused on luminescent COFs where the signal output consists of fluorescence enhancement or quenching in the presence of the detected species. Engaging electronic interactions such as charge transfer, the optical properties change and can even be detected conveniently with the naked eye upon illumination. As a result, complex device structures have typically not been established yet. Nevertheless, the development is rapidly progressing in this field, leading to insights regarding mechanisms and new detection systems.The group of Banerjee reported highly emissive covalent organic nanosheets (CONs) exfoliated from TfpBDH bulk COF (made from 1,3,5-tris(4-formylphenyl)benzene and pyromellitic-N,N′-bisaminoimide), leading to the first turn-on/off sensing mechanism for nitroaromatics, especially 2,4,6-trinitrophenol (TNP). In solution, fluorescence quenching occurred by charge transfer from the nitroaromatic compound to the COF. In contrast, in the solid state the CON powders deposited on a paper strip exhibited fluorescence enhancement upon exposure to TNP. This effect was attributed to proton transfer from the TNP to the imine bond.208
A field-effect transistor (FET)-device for photo- and moisture-detection was built with an Lp-pi-COF thin film (made from TAPB and terephthalaldehyde), which was obtained by photon-assisted condensation of the building blocks on the water surface. The film was transferred to a SiO2 substrate, then chromium and gold contacts (5 and 20 nm thickness, respectively) were deposited on top by electron beam metal evaporation. A current change by three orders of magnitude could be measured with the Lp-pi-COF-based FET-device upon water vapor exposure (Fig. 42).209
 | ||
| Fig. 42 (a) I–V curve of the Lp-pi-COF film with tested device shown in photograph. (b) On–off switching behavior upon light irradiation of device from graph (a). (c) I–V curve of the Lp-pi-COF recorded in argon (black) and water vapor (blue) environment with tested device shown in photograph. (d) On–off switching behavior upon moisture exposure of device from graph (c). Reproduced with permission.209 Copyright 2018, American Chemical Society. | ||
Combining tetraphenylethylene monomers as photoactive component and triazine moieties as electron acceptor in a COF (4′,4′′′,4′′′′′,4′′′′′′′-(1,2-ethenediylidene)tetrakis[1,1′-biphenyl]-4-carboxaldehyde and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine), COF-based photodetectors with high photoresponsivity of 3.2 × 107 A W−1 and fast response times of 1.14 ms could be realized. The device for the photoelectric measurements of the COF film on graphene was constructed on top of gold electrodes deposited on a Si/SiO2 substrate. Due to the different electronic interactions with vapors of changing polarity, the photocurrent could be adjusted significantly.210
Solvatochromism is another promising concept for the design of chemical sensors. Accordingly, the solvatochromic Py-TT COF (made from 1,3,6,8-tetrakis(4-aminophenyl)pyrene and thieno-[3,2-b]thiophene-2,5-dicarboxaldehyde) proved to be a promising candidate for sensing of solvent or water vapors. The solvatochromism is based on electronic interactions between the solvent molecules and the COF, while excluding structural or chemical changes. In a sensor device, a COF thin film was grown, serving as an optical filter for an LED. As a result of the solvatochromic effects, the optical transmission of the COF at a specific wavelength was altered, thus inducing fast current changes in a photo-dependent resistor (Fig. 43). The response times of the transition of dry to humid and humid to dry atmosphere were 0.21 s and 0.15 s, respectively.19 In a related study, imine-linked perylene-based COFs (connected with the linear bridges phenylene-, naphthalene-, and pyrene-based dicarbaldehydes) were applied as acid vapor sensors based on the ability to protonate and deprotonate their imine bonds, resulting in optical changes with high sensitivity. Additional information could be gained about the acid strength and concentration in non-aqueous solutions, which is enabled by the stepwise protonation of the two imine bonds connecting the linear bridges in the COF.211
 | ||
| Fig. 43 (a) GIWAXS pattern of oriented Py-TT COF film on sapphire. (b) UV-Vis absorption of COF film at different moisture contents. (c) Humidity-induced absorbance changes. (d) UV-Vis absorption of COF film in saturated solvent atmospheres. (e) Solvatochromic response of COF film to changes from dry to H2O-saturated N2-streams and back. (f) TD-DFT calculated electron density difference upon the one-electron excitation from ground state to the first singlet excited state showing a distinct charge-transfer character. Reproduced with permission.19 Copyright 2018, Nature Publishing Group. | ||
Tautomerization in COFs leads to β-ketoenamine-linked highly stable COFs, but can also be applied in humidity sensing. The TAPB-PDA-OH COF, condensed from TAPB and 2,5-dihydroxyterephthaldehyde, underwent a reversible keto–enol tautomerism in the presence of water, resulting in the iminol/cis-ketoenamine COF. DFT calculations of the model compound BPH (2,5-bis((E)-(phenylimino)methyl)benzene-1,4-diol) revealed that the iminol/cis-ketoenamine COF is the preferred tautomer in the presence of water compared to the diiminol and diketoenamine form, suggesting the same behavior for the COF. A device made from an oriented COF film on fused silica exhibited an additional absorption band at longer wavelengths when exposed to saturated H2O vapor due to the tautomerism and the resulting charge-transfer-type excited state. The humidity sensing experiments revealed stability of up to two months and response times of 9 s and <1 s for the transition of dry to humid and humid to dry atmosphere.212
The sensing abilities of COFs were also applied in biomedical systems, enabling the sensitive detection of methylglyoxal (MGO) for diabetes mellitus diagnosis in serum systems. Notably, the TpPa-1 COF (made from triformylphloroglucinol and p-phenylenediamine) could be deprotonated at the N–H bond leading to a 70-fold increased PL emission. Upon exposure to MGO, the excited, deprotonated COF forms an excited-state charge-transfer complex with MGO, also called an exciplex. By the implementation of two logic gates, the red-shifted fluorescence upon MGO exposure can be directly traced as an output signal, leading to an easy read-out diagnostic device.213
The above examples illustrate the main strategies developed so far to demonstrate chemical sensing with COFs. Similar applications of COFs based on these principles have been reported for numerous additional analytes but will not be discussed here.
A boronate ester-linked 2D BECOF-PP film condensed from tetra-4-(dihydroxyborylphenyl)porphyrin and 1,2,4,5-tetrahydroxybenzene via a surfactant-monolayer assisted interfacial synthesis could be transferred as a free-standing film onto a silicon nanowire (SiNW) device to mimic neuronal memory processes. A COF/SiNW-based field-effect transistor was designed with contact electrodes of a titanium layer (50 nm) and a silver layer on top (200 nm). Upon applying a gate voltage (VG) charges accumulated at the interface of BECOF-PP and SiNW. These charges were trapped and maintained after the gate voltage was set to zero. This device was able to forget (VG = 0 V) or erase (VG = −5 V; defined by different discharge kinetics) the memory content. This memory behavior could open the road to potential application of COFs as logic and memory electronics.214
Light-emitting devices
The luminescent properties of COFs have been intensively studied. Various light-emitting COFs have been synthesized by implementing different strategies such as aggregation-induced emission (AIE) or restriction of intramolecular bond rotations (RIR) mechanisms.The first COF-LED reported derived its white light from the blue emission of π-stacked anthracene units and the red and green emission from the resorcinol moieties in a COF (2,4-dihydroxybenzene-1,3,5-tricarbaldehyde and anthracene-2,6-diamine), denoted as IISERP-COF7. It was even possible to fine tune the emission of this COF by the use of N-donor or O-donor solvents. Upon UV illumination, a film consisting of an IISERP-COF7-PMMA gel exhibited intense white light emission with a CIE coordinate of (0.35, 0.36). In a proof-of-principle COF-LED, poly(9-vinylcarbazole) served as the host for the same COF material, which was then implemented in an electroluminescent device (ITO, ZnO, polyethylenimine ethoxylated (PEIE), the hybrid material, MoO3 and gold, Fig. 44).82
 | ||
| Fig. 44 (A) PL lifetime traces of COF dispersion in NMP vs. COF film recorded in the blue, green and red region. (B) Variable temperature PL spectra of the COF film. (C) J–V plot of the COF-based LED device using IISERP-COF7 with scheme showing device structure. (D) UV lamp emission without and with COF coating as phosphor. Reproduced with permission.82 Copyright 2018, American Chemical Society. | ||
Applying the principle of aggregation-induced emission (AIE), tetraphenylethylene, a known AIEgen, was used as a building block in a 3D COF (made from tetra(p-aminophenyl)methane and 1,1,2,2-tetrakis(4-formyl-(1,1′-biphenyl)ethene)) that was found to be highly emissive with a CIE coordinate of (0.38, 0.57). This COF was utilized as a yellow-emitting phosphor for a blue LED by coating the latter with the COF powder, obtaining a white light emission with CIE coordinate (0.30, 0.35).20
Since lanthanides have already shown their luminescent potential in conventional LEDs, a recent study explored their combination with the luminescence from a COF. The blue-emitting COF made from 4,4′4′′-(1,3,5-triazine-2,4,6-triyl)trianiline and 2,6-diformylpyridine was hereby combined with the green and red emission from the lanthanides Eu3+ and Tb3+, respectively, coordinated into nitrogen-rich pockets in the pore wall.83 The combination with the COF resulted in a white light emitter upon excitation in the UV, with CIE coordinates (0.3636, 0.3355; 0.3636, 0.3355) for different ratios of the lanthanides.
Conclusions
In this review we have given an extensive overview about (opto)electronic properties and processes in covalent organic frameworks and their potential applications in various fields. Starting from luminescent and semiconducting frameworks, important insights into the impact of linkage motifs, stacking behavior, and functionalization on optoelectronic processes within the COF have been gathered over the last few years. The construction principles of COFs, based on the covalent coupling of molecular building blocks, allow for the design of diverse framework and pore topologies in combination with the properties of the building blocks themselves. Motivated by this potential, a large number of building blocks that can act as light-harvesters, semiconductors, ligands, binding sites or redox centers have been embedded in the frameworks of COFs. Coupled with the highly ordered structures and control of intrinsic and textural porosity, intriguing functionalities in the fields discussed in this review, i.e. defined donor–acceptor interfaces, batteries, supercapacitors, sensors, field-effect transistors, LEDs, photocatalysts and photovoltaic devices have recently been developed.The chemical stability of COFs – a critical feature for several envisioned applications – has been greatly enhanced over the years by introducing new linkage motifs such as sp2 carbon-conjugated bonds, intralayer/interlayer hydrogen bonds or the keto–enol tautomerism. It is anticipated that this progress will open access to applications where COFs need to overcome demanding conditions.
Controlling the stacking distances and stacking mode of 2D COF layers has been a powerful strategy to tune optical and electrical properties. However, challenges still remain regarding the rather modest electrical conductivity of these frameworks, leading to potential bottlenecks for some applications in optoelectronics, including photovoltaic devices. The processes of charge carrier generation, diffusion and recombination should therefore be further investigated to enable the development of design rules for further enhancing charge carrier mobilities and conductivity, for example via linker design or by creating suitable hybrid host–guest systems. In addition, the theoretical understanding and predictability of both structure and electronic properties needs to be further developed to enable precise tailoring of COFs. This understanding needs to cover both 2D and 3D COF families and should particularly catalyse research towards the less investigated 3D class of COFs, where issues of controlling crystallization, interpenetration and the formation of complex structures are some of the challenges ahead.
Embedding COFs into suitable device structures with morphologies optimised for desired applications will be key for the next generation of high-performance COFs. Therefore, highly-controlled thin film synthesis is expected to be a key factor among the synthetic challenges on the road towards reliable COF-based devices. For example, free-standing or surface-grown, oriented and highly crystalline thin films are expected to play an important role in the design of future devices. Processability is another key factor in thin film development – in this context free-standing or exfoliated transferable films could open new opportunities in scalable processing. Moreover, sustainable, low-cost linker materials that allow for scalable processing will be of great importance to achieve the transition from fundamental research to real-world applications. The solid foundation developed in 15 years of COF science is an excellent basis for future developments focusing on specific demands and intriguing applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via SPP 1613 and under Germany's Excellence Strategy – EXC 2089/1 – 390776260 and NIM (Excellence Cluster 4), and from the Free State of Bavaria (research networks ‘Solar Technologies Go Hybrid’ and UMWELTnanoTECH). Research leading to these results received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-3013)/ERC grant Agreement 321339.Notes and references
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed.
- H. Yang, S. Zhang, L. Han, Z. Zhang, Z. Xue, J. Gao, Y. Li, C. Huang, Y. Yi, H. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 5366–5375 CrossRef CAS PubMed.
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S.-h. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518–7525 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722 CrossRef CAS PubMed.
- Z. Meng, A. Aykanat and K. A. Mirica, Chem. Mater., 2019, 31, 819–825 CrossRef CAS.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. M. Evans, M. Döblinger, S. Herbert, K. Peters, D. Boehm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS PubMed.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- D. Bessinger, L. Ascherl, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 12035–12042 CrossRef CAS PubMed.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Nat. Commun., 2018, 9, 3802 CrossRef PubMed.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef CAS PubMed.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z.-H. Chen, H.-S. Xu, W. Tang, K. Leng, G.-H. Ning, J. Wu, Q.-H. Xu, S. Y. Quek, Y. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 CrossRef PubMed.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920–2924 CrossRef CAS PubMed.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- N. Keller, M. Calik, D. Sharapa, H. R. Soni, P. M. Zehetmaier, S. Rager, F. Auras, A. C. Jakowetz, A. Görling, T. Clark and T. Bein, J. Am. Chem. Soc., 2018, 140, 16544–16552 CrossRef CAS PubMed.
- N. Keller, D. Bessinger, S. Reuter, M. Calik, L. Ascherl, F. C. Hanusch, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 8194–8199 CrossRef CAS PubMed.
- N. Keller, T. Sick, N. N. Bach, A. Koszalkowski, J. M. Rotter, D. D. Medina and T. Bein, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9638633.v2.
- S. Thomas, H. Li, C. Zhong, M. Matsumoto, W. R. Dichtel and J.-L. Bredas, Chem. Mater., 2019, 31, 3051–3065 CrossRef CAS.
- S. Thomas, H. Li, R. R. Dasari, A. M. Evans, I. Castano, T. G. Allen, O. G. Reid, G. Rumbles, W. R. Dichtel, N. C. Gianneschi, S. R. Marder, V. Coropceanu and J.-L. Brédas, Mater. Horiz., 2019, 6, 1868–1876 RSC.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- R.-R. Liang, R.-H. A., S.-Q. Xu, Q.-Y. Qi and X. Zhao, J. Am. Chem. Soc., 2020, 142, 70–74 CrossRef CAS PubMed.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 9099–9103 CrossRef CAS PubMed.
- Y. Wang, H. Liu, Q. Pan, C. Wu, W. Hao, J. Xu, R. Chen, J. Liu, Z. Li and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 5958–5963 CrossRef CAS PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11(3), 2706–2713, DOI:10.1021/acsnano.6b07692.
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- X. Chen, M. Addicoat, S. Irle, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 546–549 CrossRef CAS PubMed.
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, J. Am. Chem. Soc., 2016, 138, 16703–16710 CrossRef CAS PubMed.
- Q. Gao, X. Li, G.-H. Ning, K. Leng, B. Tian, C. Liu, W. Tang, H.-S. Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349–2352 RSC.
- P. Albacete, J. I. Martínez, X. Li, A. López-Moreno, S. A. Mena-Hernando, A. E. Platero-Prats, C. Montoro, K. P. Loh, E. M. Pérez and F. Zamora, J. Am. Chem. Soc., 2018, 140, 12922–12929 CrossRef CAS PubMed.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS PubMed.
- A. Kuc, M. A. Springer, K. Batra, R. Juarez-Mosqueda, C. Wöll and T. Heine, Adv. Funct. Mater., 2020, 1908004 CrossRef CAS.
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- Y. Meng, Y. Luo, J. L. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed.
- S. Wang, L. Ma, Q. Wang, P. Shao, D. Ma, S. Yuan, P. Lei, P. Li, X. Feng and B. Wang, J. Mater. Chem. C, 2018, 6, 5369–5374 RSC.
- B. Lukose, A. Kuc, J. Frenzel and T. Heine, Beilstein J. Nanotechnol., 2010, 1, 60–70 CrossRef CAS PubMed.
- B. Lukose, A. Kuc and T. Heine, Chem. – Eur. J., 2011, 17, 2388–2392 CrossRef CAS PubMed.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416–19421 CrossRef CAS PubMed.
- S. Patwardhan, A. A. Kocherzhenko, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2011, 115, 11768–11772 CrossRef CAS.
- H. Li and J.-L. Brédas, J. Phys. Chem. Lett., 2018, 9, 4215–4220 CrossRef CAS PubMed.
- V. Nguyen and M. Grünwald, J. Am. Chem. Soc., 2018, 140, 3306–3311 CrossRef CAS PubMed.
- X. Ding, L. Chen, Y. Honsho, X. Feng, O. Saengsawang, J. Guo, A. Saeki, S. Seki, S. Irle, S. Nagase, V. Parasuk and D. Jiang, J. Am. Chem. Soc., 2011, 133, 14510–14513 CrossRef CAS PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS PubMed.
- T. W. Kim, S. Jun, Y. Ha, R. K. Yadav, A. Kumar, C.-Y. Yoo, I. Oh, H.-K. Lim, J. W. Shin, R. Ryoo, H. Kim, J. Kim, J.-O. Baeg and H. Ihee, Nat. Commun., 2019, 10, 1873 CrossRef PubMed.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334–342 CrossRef CAS.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- X. Li, Q. Gao, J. Aneesh, H.-S. Xu, Z. Chen, W. Tang, C. Liu, X. Shi, K. V. Adarsh, Y. Lu and K. P. Loh, Chem. Mater., 2018, 30, 5743–5749 CrossRef CAS.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- T. Joshi, C. Chen, H. Li, C. S. Diercks, G. Wang, P. J. Waller, H. Li, J. L. Bredas, O. M. Yaghi and M. F. Crommie, Adv. Mater., 2019, 31(3), 1805941 CrossRef PubMed.
- H. Li, P. Shao, S. Chen, G. Li, X. Feng, X. Chen, H.-J. Zhang, J. Lin and Y.-B. Jiang, J. Am. Chem. Soc., 2020, 142, 3712–3717 CrossRef CAS PubMed.
- Y. Li, Q. Chen, T. Xu, Z. Xie, J. Liu, X. Yu, S. Ma, T. Qin and L. Chen, J. Am. Chem. Soc., 2019, 141, 13822–13828 CrossRef CAS PubMed.
- C. Franco, D. Rodríguez-San-Miguel, A. Sorrenti, S. Sevim, R. Pons, A. E. Platero-Prats, M. Pavlovic, I. Szilágyi, M. L. Ruiz Gonzalez, J. M. González-Calbet, D. Bochicchio, L. Pesce, G. M. Pavan, I. Imaz, M. Cano-Sarabia, D. Maspoch, S. Pané, A. J. de Mello, F. Zamora and J. Puigmartí-Luis, J. Am. Chem. Soc., 2020, 142, 3540–3547 CrossRef CAS PubMed.
- S. Dufresne and W. G. Skene, J. Phys. Org. Chem., 2012, 25, 211–221 CrossRef CAS.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- Y. Liu, C. S. Diercks, Y. Ma, H. Lyu, C. Zhu, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 677–683 CrossRef CAS PubMed.
- S. Haldar, D. Chakraborty, B. Roy, G. Banappanavar, K. Rinku, D. Mullangi, P. Hazra, D. Kabra and R. Vaidhyanathan, J. Am. Chem. Soc., 2018, 140, 13367–13374 CrossRef CAS PubMed.
- C. Krishnaraj, A. M. Kaczmarek, H. S. Jena, K. Leus, N. Chaoui, J. Schmidt, R. Van Deun and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2019, 11, 27343–27352 CrossRef CAS PubMed.
- A. M. Evans, I. Castano, A. Brumberg, L. R. Parent, A. R. Corcos, R. L. Li, N. C. Flanders, D. J. Gosztola, N. C. Gianneschi, R. D. Schaller and W. R. Dichtel, J. Am. Chem. Soc., 2019, 141, 19728–19735 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- A. C. Jakowetz, T. F. Hinrichsen, L. Ascherl, T. Sick, M. Calik, F. Auras, D. D. Medina, R. H. Friend, A. Rao and T. Bein, J. Am. Chem. Soc., 2019, 141, 11565–11571 CrossRef CAS PubMed.
- G. Das, T. Prakasam, M. A. Addicoat, S. K. Sharma, F. Ravaux, R. Mathew, M. Baias, R. Jagannathan, M. A. Olson and A. Trabolsi, J. Am. Chem. Soc., 2019, 141, 19078–19087 CrossRef CAS PubMed.
- E. Jin, K. Geng, K. H. Lee, W. Jiang, J. Li, Q. Jiang, S. Irle and D. Jiang, Angew. Chem., Int. Ed., 2020, 59(29), 12162–12169, DOI:10.1002/anie.202004728.
- X. Li, J. Qiao, S. W. Chee, H.-S. Xu, X. Zhao, H. S. Choi, W. Yu, S. Y. Quek, U. Mirsaidov and K. P. Loh, J. Am. Chem. Soc., 2020, 142, 4932–4943 CrossRef CAS PubMed.
- X. Li, J. Qiao, S. Y. Quek and K. P. Loh, ACS Mater. Lett., 2020, 2, 654–657 CrossRef.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613–6616 RSC.
- G. Chen, H.-H. Lan, S.-L. Cai, B. Sun, X.-L. Li, Z.-H. He, S.-R. Zheng, J. Fan, Y. Liu and W.-G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830–12837 CrossRef CAS PubMed.
- X. Song, Z. Zhao, D. Si, X. Wang, F. Zhou, M. Zhang, Y. Shi and C. Hao, J. Mol. Model., 2019, 25, 248 CrossRef PubMed.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- Z. Zhou, W. Zhong, K. Cui, Z. Zhuang, L. Li, L. Li, J. Bi and Y. Yu, Chem. Commun., 2018, 54, 9977–9980 RSC.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- R. Kulkarni, Y. Noda, D. Kumar Barange, Y. S. Kochergin, P. Lyu, B. Balcarova, P. Nachtigall and M. J. Bojdys, Nat. Commun., 2019, 10, 3228 CrossRef PubMed.
- H.-Q. Yin, F. Yin and X.-B. Yin, Chem. Sci., 2019, 10, 11103–11109 RSC.
- C. Zhang, S. Zhang, Y. Yan, F. Xia, A. Huang and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- M.-W. Zhu, S.-Q. Xu, X.-Z. Wang, Y. Chen, L. Dai and X. Zhao, Chem. Commun., 2018, 54, 2308–2311 RSC.
- P. Albacete, A. López-Moreno, S. Mena-Hernando, A. E. Platero-Prats, E. M. Pérez and F. Zamora, Chem. Commun., 2019, 55, 1382–1385 RSC.
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS PubMed.
- J. Li, C. Zhang, M. Yin, Z. Zhang, Y. Chen, Q. Deng and S. Wang, ACS Omega, 2019, 4, 15947–15955 CrossRef CAS PubMed.
- W.-R. Cui, C.-R. Zhang, W. Jiang, F.-F. Li, R.-P. Liang, J. Liu and J.-D. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- X. Ding, X. Feng, A. Saeki, S. Seki, A. Nagai and D. Jiang, Chem. Commun., 2012, 48, 8952–8954 RSC.
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed.
- Y. Chen, H. Cui, J. Zhang, K. Zhao, D. Ding, J. Guo, L. Li, Z. Tian and Z. Tang, RSC Adv., 2015, 5, 92573–92576 RSC.
- B. Nath, W.-H. Li, J.-H. Huang, G.-E. Wang, Z.-h. Fu, M.-S. Yao and G. Xu, CrystEngComm, 2016, 18, 4259–4263 RSC.
- F. Yu, W. Liu, B. Li, D. Tian, J. L. Zuo and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 16101–16104 CrossRef CAS PubMed.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomacker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- H. Ma, M. Wei, F. Jin, T. Chen and Y. Ma, J. Phys. Chem. C, 2019, 123, 24626–24633 CrossRef CAS.
- Y.-J. Cheng, R. Wang, S. Wang, X.-J. Xi, L.-F. Ma and S.-Q. Zang, Chem. Commun., 2018, 54, 13563–13566 RSC.
- S.-Y. Ding, P.-L. Wang, G.-L. Yin, X. Zhang and G. Lu, Int. J. Hydrogen Energy, 2019, 44, 11872–11876 CrossRef CAS.
- H. Dong, X.-B. Meng, X. Zhang, H.-L. Tang, J.-W. Liu, J.-H. Wang, J.-Z. Wei, F.-M. Zhang, L.-L. Bai and X.-J. Sun, Chem. Eng. J., 2020, 379, 122342 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Chen, B. Lin, W. Yu, Y. Yang, S. M. Bashir, H. Wang, K. Takanabe, H. Idriss and J.-M. Basset, Chem. – Eur. J., 2015, 21, 10290–10295 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- M. Luo, Q. Yang, K. Liu, H. Cao and H. Yan, Chem. Commun., 2019, 55, 5829–5832 RSC.
- L. Li, Z. Zhou, L. Li, Z. Zhuang, J. Bi, J. Chen, Y. Yu and J. Yu, ACS Sustainable Chem. Eng., 2019, 7, 18574–18581 CrossRef CAS.
- J. Chen, X. Tao, C. Li, Y. Ma, L. Tao, D. Zheng, J. Zhu, H. Li, R. Li and Q. Yang, Appl. Catal., B, 2020, 262, 118271 CrossRef CAS.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Adv. Sci., 2020, 7, 1902988 CrossRef CAS PubMed.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- S.-Y. Li, S. Meng, X. Zou, M. El-Roz, I. Telegeev, O. Thili, T. X. Liu and G. Zhu, Microporous Mesoporous Mater., 2019, 285, 195–201 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- M. Lu, Q. Li, J. Liu, F.-M. Zhang, L. Zhang, J.-L. Wang, Z.-H. Kang and Y.-Q. Lan, Appl. Catal., B, 2019, 254, 624–633 CrossRef CAS.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- S. Liu, W. Pan, S. Wu, X. Bu, S. Xin, J. Yu, H. Xu and X. Yang, Green Chem., 2019, 21, 2905–2910 RSC.
- W. Hao, D. Chen, Y. Li, Z. Yang, G. Xing, J. Li and L. Chen, Chem. Mater., 2019, 31, 8100–8105 CrossRef CAS.
- X. Yan, H. Liu, Y. Li, W. Chen, T. Zhang, Z. Zhao, G. Xing and L. Chen, Macromolecules, 2019, 52, 7977–7983 CrossRef CAS.
- G. Lu, X. Huang, Y. Li, G. Zhao, G. Pang and G. Wang, J. Energy Chem., 2020, 43, 8–15 CrossRef.
- Y. Zhao, H. Liu, C. Wu, Z. Zhang, Q. Pan, F. Hu, R. Wang, P. Li, X. Huang and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 5376–5381 CrossRef CAS PubMed.
- J. Bailey, L. Powell, L. Sinanan, J. Neal, M. Li, T. Smith and E. Bell, FEBS J., 2011, 278, 3140–3151 CrossRef CAS PubMed.
- H.-C. Ma, C.-C. Zhao, G.-J. Chen and Y.-B. Dong, Nat. Commun., 2019, 10, 3368 CrossRef PubMed.
- W. Chen, Z. Yang, Z. Xie, Y. Li, X. Yu, F. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998–1004 RSC.
- Z. Mi, P. Yang, R. Wang, J. Unruangsri, W. Yang, C. Wang and J. Guo, J. Am. Chem. Soc., 2019, 141, 14433–14442 CrossRef CAS PubMed.
- J. Hynek, J. Zelenka, J. Rathouský, P. Kubát, T. Ruml, J. Demel and K. Lang, ACS Appl. Mater. Interfaces, 2018, 10, 8527–8535 CrossRef CAS PubMed.
- Y. Zhao, W. Dai, Y. Peng, Z. Niu, Q. Sun, C. Shan, H. Yang, L. Wojtas, D. Yuan, Z. Zhang, H. Dong, X. Zhang, B. Zhang, Y. Feng and S. Ma, Angew. Chem., Int. Ed., 2020, 59, 4354–4359 CrossRef CAS.
- C. Hu, L. Cai, S. Liu and M. Pang, Chem. Commun., 2019, 55, 9164–9167 RSC.
- Y. Singh, Int. J. Mod. Phys.: Conf. Ser., 2013, 22, 745–756 CAS.
- A. J. Mozer, N. S. Sariciftci, A. Pivrikas, R. Österbacka, G. Juška, L. Brassat and H. Bässler, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035214 CrossRef.
- L. Torsi, A. Dodabalapur, L. Sabbatini and P. G. Zambonin, Sens. Actuators, B, 2000, 67, 312–316 CrossRef CAS.
- G. Dicker, M. P. de Haas, L. D. A. Siebbeles and J. M. Warman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045203 CrossRef.
- J. L. Boland, A. Casadei, G. Tütüncüoglu, F. Matteini, C. L. Davies, F. Jabeen, H. J. Joyce, L. M. Herz, A. Fontcuberta i Morral and M. B. Johnston, ACS Nano, 2016, 10, 4219–4227 CrossRef CAS PubMed.
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS PubMed.
- S. Jin, T. Sakurai, T. Kowalczyk, S. Dalapati, F. Xu, H. Wei, X. Chen, J. Gao, S. Seki, S. Irle and D. Jiang, Chem. – Eur. J., 2014, 20, 14608–14613 CrossRef CAS PubMed.
- H. Ding, Y. Li, H. Hu, Y. Sun, J. Wang, C. Wang, C. Wang, G. Zhang, B. Wang, W. Xu and D. Zhang, Chem. – Eur. J., 2014, 20, 14614–14618 CrossRef CAS PubMed.
- S.-L. Cai, Y.-B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S.-R. Zheng, W.-G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- M. Martínez-Abadía, C. T. Stoppiello, K. Strutynski, B. Lerma-Berlanga, C. Martí-Gastaldo, A. Saeki, M. Melle-Franco, A. N. Khlobystov and A. Mateo-Alonso, J. Am. Chem. Soc., 2019, 141, 14403–14410 CrossRef.
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11, 2706–2713 CrossRef CAS PubMed.
- S. Rager, A. C. Jakowetz, B. Gole, F. Beuerle, D. D. Medina and T. Bein, Chem. Mater., 2019, 31, 2707–2712 CrossRef CAS PubMed.
- C. Wu, Y. Liu, H. Liu, C. Duan, Q. Pan, J. Zhu, F. Hu, X. Ma, T. Jiu, Z. Li and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 10016–10024 CrossRef CAS PubMed.
- H. Li, J. Chang, S. Li, X. Guan, D. Li, C. Li, L. Tang, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 13324–13329 CrossRef CAS PubMed.
- S. Kim and H. C. Choi, Commun. Chem., 2019, 2, 60 CrossRef.
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- Q. Zhang, M. Dai, H. Shao, Z. Tian, Y. Lin, L. Chen and X. C. Zeng, ACS Appl. Mater. Interfaces, 2018, 10, 43595–43602 CrossRef CAS PubMed.
- S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed.
- V. Lakshmi, C.-H. Liu, M. Rajeswara Rao, Y. Chen, Y. Fang, A. Dadvand, E. Hamzehpoor, Y. Sakai-Otsuka, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 2155–2160 CrossRef CAS PubMed.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, 1706330 CrossRef PubMed.
- H. Wei, J. Ning, X. Cao, X. Li and L. Hao, J. Am. Chem. Soc., 2018, 140, 11618–11622 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, J. M. Rotter, D. J. Fortman, D. dos S. Abreu, R. A. Timm, I. C. N. Diógenes, L. T. Kubota, H. D. Abruña and W. R. Dichtel, Angew. Chem., Int. Ed., 2015, 54, 13225–13229 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- Y. Wu, D. Yan, Z. Zhang, M. M. Matsushita and K. Awaga, ACS Appl. Mater. Interfaces, 2019, 11, 7661–7665 CrossRef CAS PubMed.
- A. Khayum M, V. Vijayakumar, S. Karak, S. Kandambeth, M. Bhadra, K. Suresh, N. Acharambath, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2018, 10, 28139–28146 CrossRef CAS PubMed.
- A. Halder, M. Ghosh, A. Khayum M, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- A. K. Mohammed, V. Vijayakumar, A. Halder, M. Ghosh, M. Addicoat, U. Bansode, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2019, 11, 30828–30837 CrossRef CAS PubMed.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed.
- A. F. M. El-Mahdy, C. Young, J. Kim, J. You, Y. Yamauchi and S.-W. Kuo, ACS Appl. Mater. Interfaces, 2019, 11, 9343–9354 CrossRef CAS PubMed.
- S. Haldar, R. Kushwaha, R. Maity and R. Vaidhyanathan, ACS Mater. Lett., 2019, 1, 490–497 CrossRef CAS.
- A. F. M. El-Mahdy, M. G. Mohamed, T. H. Mansoure, H.-H. Yu, T. Chen and S.-W. Kuo, Chem. Commun., 2019, 55, 14890–14893 RSC.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- D. A. Vazquez-Molina, G. S. Mohammad-Pour, C. Lee, M. W. Logan, X. Duan, J. K. Harper and F. J. Uribe-Romo, J. Am. Chem. Soc., 2016, 138, 9767–9770 CrossRef CAS PubMed.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS.
- J. Yoo, S.-J. Cho, G. Y. Jung, S. H. Kim, K.-H. Choi, J.-H. Kim, C. K. Lee, S. K. Kwak and S.-Y. Lee, Nano Lett., 2016, 16, 3292–3300 CrossRef CAS PubMed.
- Z. A. Ghazi, L. Zhu, H. Wang, A. Naeem, A. M. Khattak, B. Liang, N. A. Khan, Z. Wei, L. Li and Z. Tang, Adv. Energy Mater., 2016, 6(24), 1601250 CrossRef.
- Y. Meng, G. Lin, H. Ding, H. Liao and C. Wang, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC.
- D.-G. Wang, N. Li, Y. Hu, S. Wan, M. Song, G. Yu, Y. Jin, W. Wei, K. Han, G.-C. Kuang and W. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 42233–42240 CrossRef CAS PubMed.
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- Z. Wang, Y. Li, P. Liu, Q. Qi, F. Zhang, G. Lu, X. Zhao and X. Huang, Nanoscale, 2019, 11, 5330–5335 RSC.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31(29), 1901640 CrossRef PubMed.
- B. C. Patra, S. K. Das, A. Ghosh, A. Raj K, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC.
- X. Chen, H. Zhang, C. Ci, W. Sun and Y. Wang, ACS Nano, 2019, 13, 3600–3607 CrossRef CAS PubMed.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC.
- E. L. Spitler, J. W. Colson, F. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS PubMed.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS.
- H. Sahabudeen, H. Qi, M. Ballabio, M. Položij, S. Olthof, R. Shivhare, Y. Jing, S. Park, K. Liu, T. Zhang, J. Ma, B. Rellinghaus, S. Mannsfeld, T. Heine, M. Bonn, E. Cánovas, Z. Zheng, U. Kaiser, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 6028–6036 CrossRef CAS PubMed.
- K. Liu, H. Qi, R. Dong, R. Shivhare, M. Addicoat, T. Zhang, H. Sahabudeen, T. Heine, S. Mannsfeld, U. Kaiser, Z. Zheng and X. Feng, Nat. Chem., 2019, 11, 994–1000 CrossRef CAS PubMed.
- H. S. Sasmal, A. Halder, S. Kunjattu H, K. Dey, A. Nadol, T. G. Ajithkumar, P. Ravindra Bedadur and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 20371–20379 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139(37), 13083–13091, DOI:10.1021/jacs.7b06640.
- K. Dey, S. Kunjattu H, A. M. Chahande and R. Banerjee, Angew. Chem., Int. Ed., 2020, 59, 1161–1165 CrossRef CAS.
- M. Zhang, L. Li, Q. Lin, M. Tang, Y. Wu and C. Ke, J. Am. Chem. Soc., 2019, 141, 5154–5158 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- J. M. Rotter, S. Weinberger, J. Kampmann, T. Sick, M. Shalom, T. Bein and D. D. Medina, Chem. Mater., 2019, 31, 10008–10016 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- S. Kim, H. Lim, J. Lee and H. C. Choi, Langmuir, 2018, 34, 8731–8738 CrossRef CAS PubMed.
- Y. Xiong, Q. Liao, Z. Huang, X. Huang, C. Ke, H. Zhu, C. Dong, H. Wang, K. Xi, P. Zhan, F. Xu and Y. Lu, Adv. Mater., 2020, 32, 1907242 CrossRef CAS.
- L. Ascherl, E. W. Evans, J. Gorman, S. Orsborne, D. Bessinger, T. Bein, R. H. Friend and F. Auras, J. Am. Chem. Soc., 2019, 141, 15693–15699 CrossRef CAS PubMed.
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J.-L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS.
- J. Wang and B. Yan, Anal. Chem., 2019, 91, 13183–13190 CrossRef CAS.
- S. Park, Z. Liao, B. Ibarlucea, H. Qi, H.-H. Lin, D. Becker, J. Melidonie, T. Zhang, H. Sahabudeen, L. Baraban, C.-K. Baek, Z. Zheng, E. Zschech, A. Fery, T. Heine, U. Kaiser, G. Cuniberti, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 8218–8224 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |