
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radiolabelling of nanomaterials for medical imaging and therapy
Juan
Pellico†
 ,
Peter J.
Gawne†
and
Rafael
T. M. de Rosales
*
,
Peter J.
Gawne†
and
Rafael
T. M. de Rosales
*
School of Biomedical Engineering & Imaging Sciences, King's College London, St. Thomas’ Hospital, London SE1 7EH, UK. E-mail: rafael.torres@kcl.ac.uk
First published on 25th January 2021
Abstract
Nanomaterials offer unique physical, chemical and biological properties of interest for medical imaging and therapy. Over the last two decades, there has been an increasing effort to translate nanomaterial-based medicinal products (so-called nanomedicines) into clinical practice and, although multiple nanoparticle-based formulations are clinically available, there is still a disparity between the number of pre-clinical products and those that reach clinical approval. To facilitate the efficient clinical translation of nanomedicinal-drugs, it is important to study their whole-body biodistribution and pharmacokinetics from the early stages of their development. Integrating this knowledge with that of their therapeutic profile and/or toxicity should provide a powerful combination to efficiently inform nanomedicine trials and allow early selection of the most promising candidates. In this context, radiolabelling nanomaterials allows whole-body and non-invasive in vivo tracking by the sensitive clinical imaging techniques positron emission tomography (PET), and single photon emission computed tomography (SPECT). Furthermore, certain radionuclides with specific nuclear emissions can elicit therapeutic effects by themselves, leading to radionuclide-based therapy. To ensure robust information during the development of nanomaterials for PET/SPECT imaging and/or radionuclide therapy, selection of the most appropriate radiolabelling method and knowledge of its limitations are critical. Different radiolabelling strategies are available depending on the type of material, the radionuclide and/or the final application. In this review we describe the different radiolabelling strategies currently available, with a critical vision over their advantages and disadvantages. The final aim is to review the most relevant and up-to-date knowledge available in this field, and support the efficient clinical translation of future nanomedicinal products for in vivo imaging and/or therapy.
 Juan Pellico | Juan Pellico Sáez obtained his PhD degree in Chemistry from the Complutense University of Madrid (UCM) in 2016. He then obtained a grant to conduct postdoctoral research in the Spanish Centre for Cardiovascular Research (CNIC). In 2018, he moved to the University of Oxford as a Postdoctoral Research Associate (PDRA). He joined to the group of Dr Rafael T. M. de Rosales at King's College London in 2019 as a PDRA. His main area of interest combines novel particulate PET tracers with the application of nanotechnology in biomedicine to develop a new generation of imaging agents for multimodal molecular imaging applications. |
 Peter J. Gawne | Peter Gawne received his Masters in Chemistry from the University of Hull, before joining the Medical Imaging CDT at King's College London and Imperial College London in 2015; obtaining a Masters of Research in Medical Imaging Science, followed by his PhD in Radiochemistry at King's College London – under the supervision of Dr Rafael T. M. de Rosales. He is currently continuing his work as a Postdoctoral Research Associate focusing on the radiolabelling of cells and nanomedicines. |
 Rafael T. M de Rosales | Rafael T. M de Rosales obtained his BSc in Chemistry from the University of Granada (Spain), and a PhD in Bioinorganic Chemistry at the University of Edinburgh (UK) in 2004. After a Marie Curie Postdoctoral Fellowship in Naples (Italy), and a postdoctoral research position in bio-inspired inorganic catalysis at Imperial College London (UK), he moved to the School of Biomedical Engineering & Medical Imaging at King’s College London in 2007, where he is now Reader in Imaging Chemistry. His main interest is the development and application of radiochemical tools to investigate the in vivo behaviour of drug delivery systems and cell therapies. |
1 Introduction
Materials at the nanometric scale (i.e. with at least one dimension below 100 nm) have emerged in the last 20 years as tools with several unique applications in imaging, diagnosis and treatment in medicine. Since then, the use of nanomaterials in medicine (nanomedicine) has evolved tremendously, with an increasing number of examples that overcome previously unmet medical needs (Fig. 1).1 The size-dependent optical, magnetic, and/or electronic properties of nanomaterials offer multiple possibilities in different fields of application. In addition, the tuneable nature of their physicochemical properties, pharmacokinetics and biodistribution has allowed the development of improved drug delivery systems, where the formulation is mainly driven towards the malignant areas rather than healthy areas, decreasing undesirable side effects and boosting therapeutic efficacy.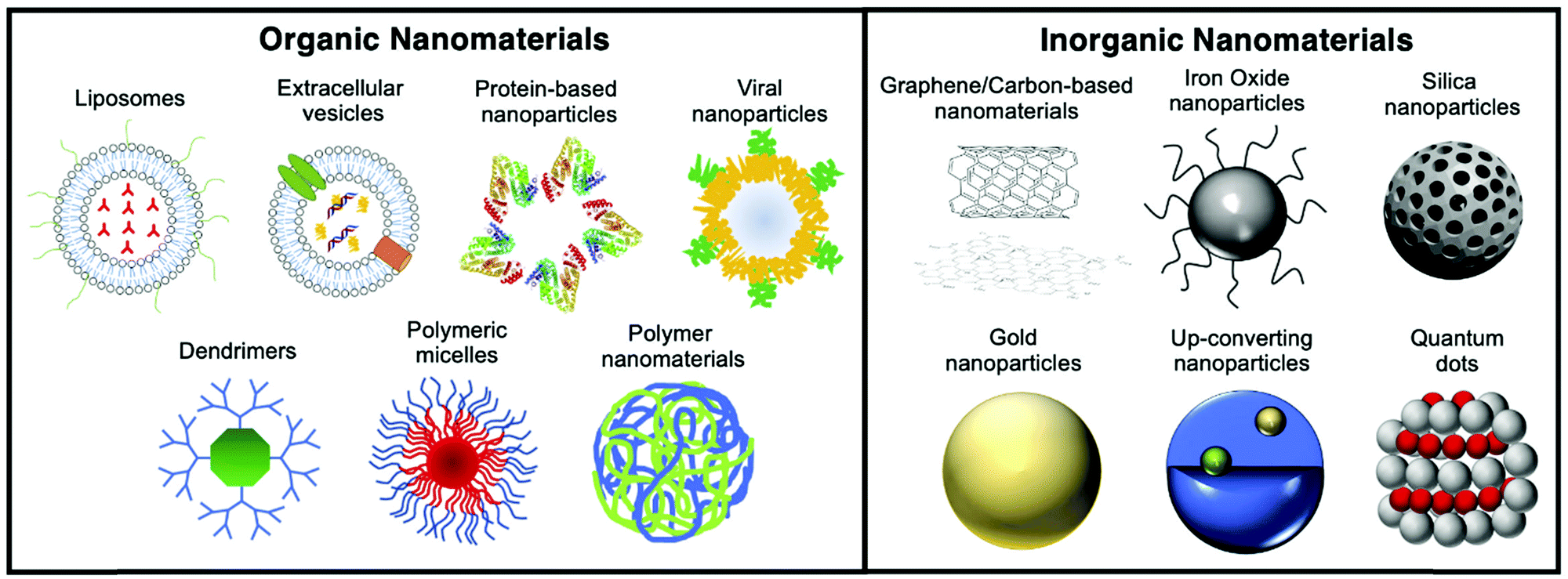 | ||
| Fig. 1 Schematic showing the various organic and inorganic nanomaterials discussed in this review. | ||
Since the approval in 1989 of Diprivan (a liposomal-based formulation used as anaesthetic) by the Food and Drug Agency (FDA), the number of clinically-approved nanomedicines has grown remarkably.2 One of the most notable early examples is the cancer nanomedicine Doxil/Caelyx (PEGylated liposomal doxorubicin), approved in 1995 and still widely used today in ovarian cancer, HIV-associated Kaposi's sarcoma and multiple myeloma.3 Several nanomedicines have since been approved by the FDA and/or the European Medicines Agency (EMA) for different purposes such as cancer therapy, iron-replacement, vaccines, anaesthetics, fungal treatments, muscular degeneration, or imaging.4 In 2015, PEGylated liposomal irinotecan (Onivyde MM-398) was approved for metastatic pancreatic cancer.5 Moreover, liposome technology has been applied to improve vaccines (Epaxal, Inflexal V), treatments for macular degeneration (Visudyne) and fungal infections (AmBisome), among other applications.6,7 Besides liposomes, several iron oxide NP formulations are being utilised as treatment for iron deficient anaemia (Venofer, Ferrlixit, Ferinject, Feraheme).8 Although the benefits of nanomedicinal formulations are well reported – with many preclinical examples supporting their effectiveness – their translation into the clinics is still an arduous, lengthy and costly pathway with multiple issues to be addressed.9 This is clearly evidenced by the relatively few examples of pre-clinical research that have translated into clinical applications.
In preclinical research, the use of NPs is still being widely explored for both imaging and therapeutic applications. Different imaging agents based on NPs can be found for several medical imaging techniques; providing anatomical and functional information with increased sensitivity and specificity.10 From the use of NPs to simply generate contrast in imaging techniques, work in this area has evolved towards more sophisticated formulations (“smart” NPs) capable of responding to external stimuli, biological targets or microenvironmental conditions in a specific manner relevant to the diagnostic and/or treatment of a disease.11
Current medical non-invasive imaging techniques include computed tomography (CT), magnetic resonance imaging (MRI), optical imaging techniques (OI) and nuclear imaging techniques – such as single photon emission computed tomography (SPECT) and positron emission tomography (PET). Each technique has advantages and drawbacks (see Section 2); and the choice of which imaging method is most appropriate must be carefully considered based on the clinical problem being addressed. In particular, radionuclide imaging techniques offer high sensitivity (defined as the concentration of tracer needed for contrast) and the ability to provide functional/metabolic information at the molecular level. These techniques require the use of exogenous compounds containing radioisotopes (radiotracers), to provide imaging contrast. Radiotracers usually consist of biologically active organic molecules previously modified (radiolabelled) with a SPECT or PET radionuclide (see Section 3). For instance, one of the most clinically used radiotracers for PET is 18F-fluorodeoxyglucose ([18F]-FDG) formed by a deoxyglucose molecule radiolabelled with the radionuclide fluorine-18 (18F). Considering the role of deoxyglucose in metabolic glycolytic pathways, many clinical studies are conducted daily to detect the increased level of glycolysis found in patients with cancer and other diseases.12 Besides small molecules, nanomaterials are also being explored as radiotracers that combine the size-dependent properties of nanomaterials with the high sensitivity provided by radionuclides. Although radiolabelled nanomaterials are not applied routinely in clinics, they could find applications thanks to specific properties such as the ability to incorporate multiple radionuclides per NP (leading to high sensitivity), vector ligands (leading to high target affinity), or therapeutic components in a single platform.13 This concept, known as multifunctionality, has generated new possibilities in the application of radiolabelled nanomaterials, not only for standard or multimodal molecular imaging but also for combined diagnosis and therapy – known as ‘theranostics’.
The term theranostics was introduced in 1998 by J. Funkhouser referring to “the ability to affect therapy or treatment of a disease state”.14 Being able to perform therapy and diagnosis with the same vector is an important step forward towards personalised medicine where the safety and effectiveness of a treatment can be predicted and monitored by medical imaging techniques. With a slow evolution during the first years, the use of nanomedicines as theranostics platforms – known as nanotheranostics – has arguably had a large impact on the field. Different nanoparticle-based treatments such as those based on chemotherapy, gene therapy, immunotherapy, radiotherapy, photothermal therapy or photodynamic therapy have been developed in combination with the imaging modalities mentioned above.15–17 The ability to image nanoparticle-based therapeutics non-invasively can provide information on target uptake of the nanomedicines – as well as potentially predict the therapeutic response. Hence, nanotheranostic platforms can potentially guide treatment regimens on a patient-to-patient basis. Additionally, the combination of nuclear imaging modalities with radiotherapies is especially attractive.18
One of the key aspects to consider when radiolabelling nanomaterials is the selection of the radionuclide. Different properties such as half-life, decay mode and biological response must be considered in advance (see Section 3). The chemistries available to integrate the radionuclide into the nanomaterial must be then considered; with special attention given to the type of material and their potential effects on their physicochemical properties, as well as the expected in vivo stabilities. (see Section 4). These considerations are essential to avoid time-consuming and inefficient protocols that could give misleading or unusable results. The interaction between the radionuclide and the nanomaterial, the level of loading/chemical modifications and the stability of the final formulation in physiological media are key properties that will influence the pharmacokinetics and pharmacodynamics of the radiolabelled nanomaterial.
The strategies used during early nanoparticulate radiolabelling studies were primarily based on the application of standard radiochemistry protocols for lower-molecular weight compounds. With the evolution of the field, novel advanced radiolabelling methods specifically designed for the radiolabelling of nanomaterials are continuously emerging. Whether a radiolabelling method is adequate or not is affected by multiple factors that need to be carefully addressed. This review aims to discuss all these factors and provide a thorough summary and critical review of the different strategies available to label nanomaterials with radionuclides, from traditional to recent innovative methods. Ultimately, we hope that this document will guide the reader to select the best strategy for developing efficiently radiolabelled nanomaterials for innovative imaging and/or therapeutic purposes.
2 Medical imaging techniques: focus on nuclear imaging and radionuclide therapy
2.1 Medical imaging
Medical imaging refers to the use of imaging scanners to non-invasively obtain in vivo information of living subjects – as opposed to ex vivo invasive medical procedures (e.g. biopsy). Patients/subjects are placed within a medical imaging scanner which provides information, based on image contrast achieved by an intrinsic mechanism of the imaging technique (US, MRI, CT). Alternatively, image contrast can be attenuated/boosted by exogenous ‘contrast agents’; which require pre- and post-contrast imaging allowing signal quantification (US, MRI, CT). Finally, imaging agents which have an inherent signal can be administered for ‘hot-spot’ imaging (e.g.19F-MRI, radioactive agents and fluorescent dyes). Depending on the technique, anatomical information and/or data on real-time biochemical processes (i.e. molecular imaging)19 can be obtained. The medical imaging modalities available have important differences in their properties (Table 1), including: imaging field of view (FOV), spatial and temporal resolution, sensitivity, and tissue depth limitation of the imaging signal. Multimodal imaging, in which two or more imaging modalities are combined into a single instrument, is often used to overcome some of the drawbacks associated with any imaging technique by providing synergistic information. In this review we focus on radionuclide-based imaging methods, however, to gain a good understanding of the pros and cons of these techniques for imaging NPs, we will provide a brief overview of other non-radionuclide based imaging modalities.| Imaging technique | Spatial resolution | Depth penetration | Sensitivity | Relative cost |
|---|---|---|---|---|
| MRI | ≤0.1 mm (PC) | No limit | μM–mM | €€€ |
| 1–2 mm (C) | ||||
| CT | ≤0.2 mm (PC) | No limit | mM | € |
| 0.5–1 (C) | ||||
| US | 1–2 mm (PC) | Several cm | ∼μM | € |
| ≤0.1 mm (C) | ||||
| OI | 5 mm | mm–cm | pM–nM | €–€€€ |
| PAI | ≤0.1 mm | Several cm | pM | € |
| SPECT | 0.5–2 mm (PC) | No limit | <pM | €€ |
| 5–12 mm (C) | ||||
| PET | 1–2 mm (PC) | No limit | fM | €€€ |
| 3–6 mm (C) |
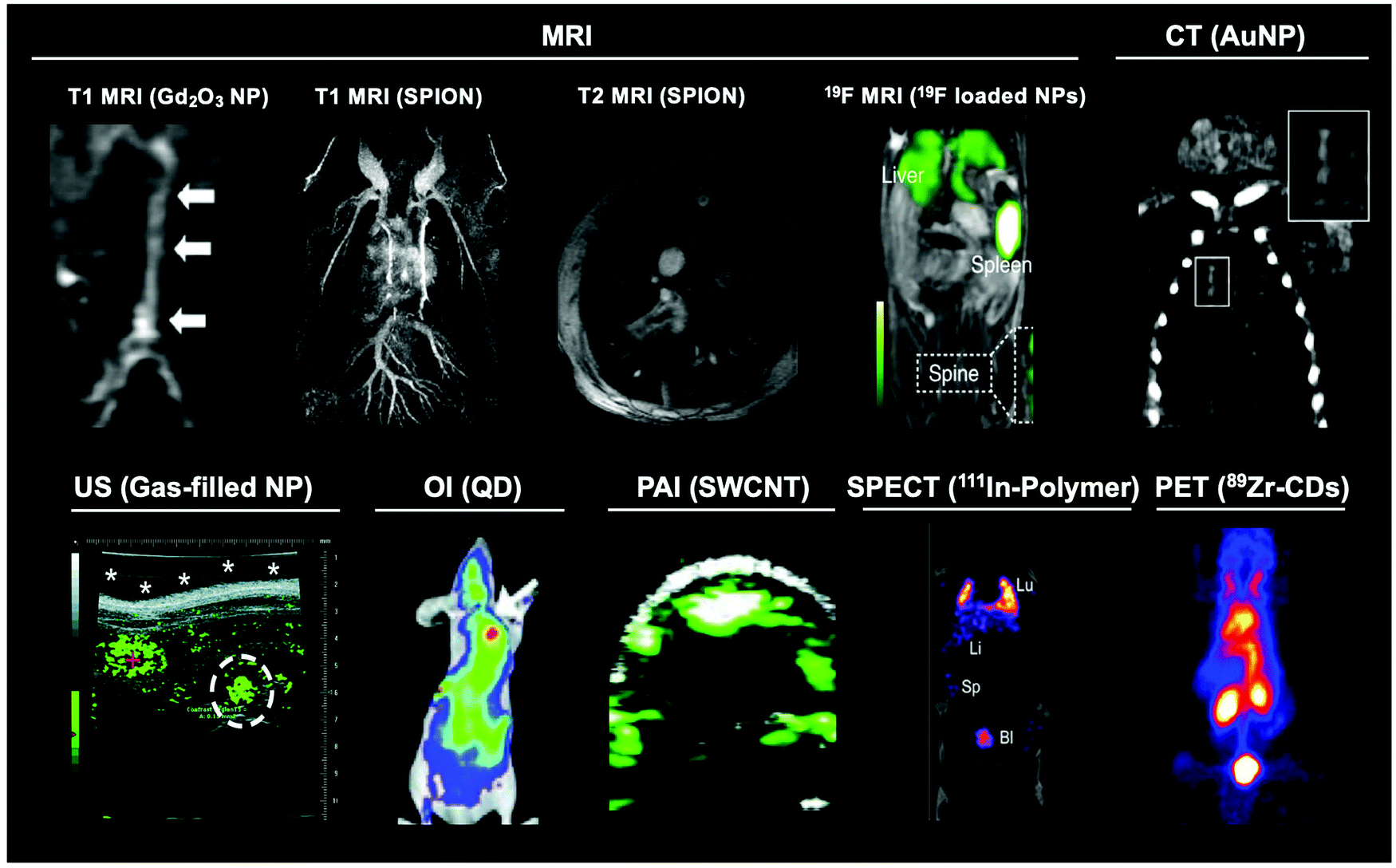 | ||
| Fig. 2 Representative images of the main modalities used to image different nanomaterials. Gd2O3 nanoparticle MR image adapted from Park et al.26T1 and T2 SPION MRI image adapted from Pellico et al.2719F MRI image adapted from Senders et al.28 CT image adapted from Chhour et al.29 US image adapted from Peyman et al.30 OI image adapted from Gao et al.31 PAI image adapted with permission from de la Zerda et al.32 Copyright (2010) American Chemical Society. SPECT image adapted from Imlimthan et al.33 PET image adapted from Cheng et al.34 | ||
2.2 Radionuclide imaging
Radionuclide or nuclear imaging refers to two main imaging techniques: single-photon emission computed tomography (SPECT, Fig. 3A) or positron emission tomography (PET, Fig. 3B). Both of these techniques rely on the detection of radioactive nuclides (radionuclides). Thus, tracking NPs using PET/SPECT requires their ‘tagging’ or ‘labelling’ with radionuclides (radiolabelling) allowing non-invasive in vivo imaging via the radioactive decay emissions of the radionuclide – using the appropriate scanner. Both techniques, however, differ in the detection method, leading to significant differences that are worth discussing below.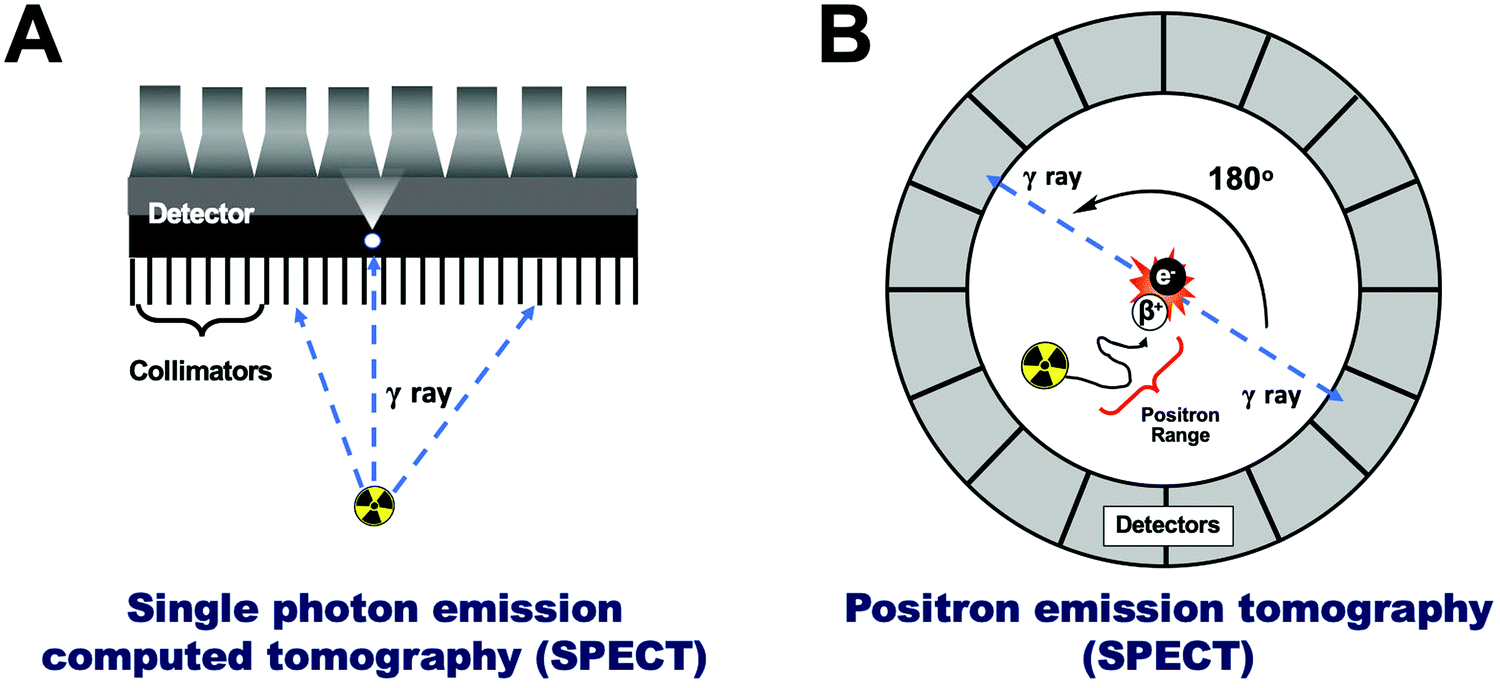 | ||
| Fig. 3 (A) Schematic representation of single photon emission computed tomography (SPECT), (B) schematic representation of positron emission tomography (PET). | ||
2.3 Radionuclide therapy
The decay properties of certain radionuclides allow their use as therapeutics, adding the possibility of using NPs as radionuclide therapy agents. These radionuclides emit α (alpha), β− (beta) particles or Auger electrons that are capable of depositing a substantial amount of energy, and hence damage, to tissues. These therapeutic radionuclides can be incorporated in high concentrations into nanomaterials with the aim of delivering their radio-emission ‘payload’ to specific tissues (e.g. tumours).46,47 For maximum therapeutic efficacy, the radionuclide decay type, range, and the energy deposited over that distance – the linear energy transfer (LET) – must be carefully considered and matched to the biological target.48 The three emission types for radionuclide therapy will be briefly summarised below.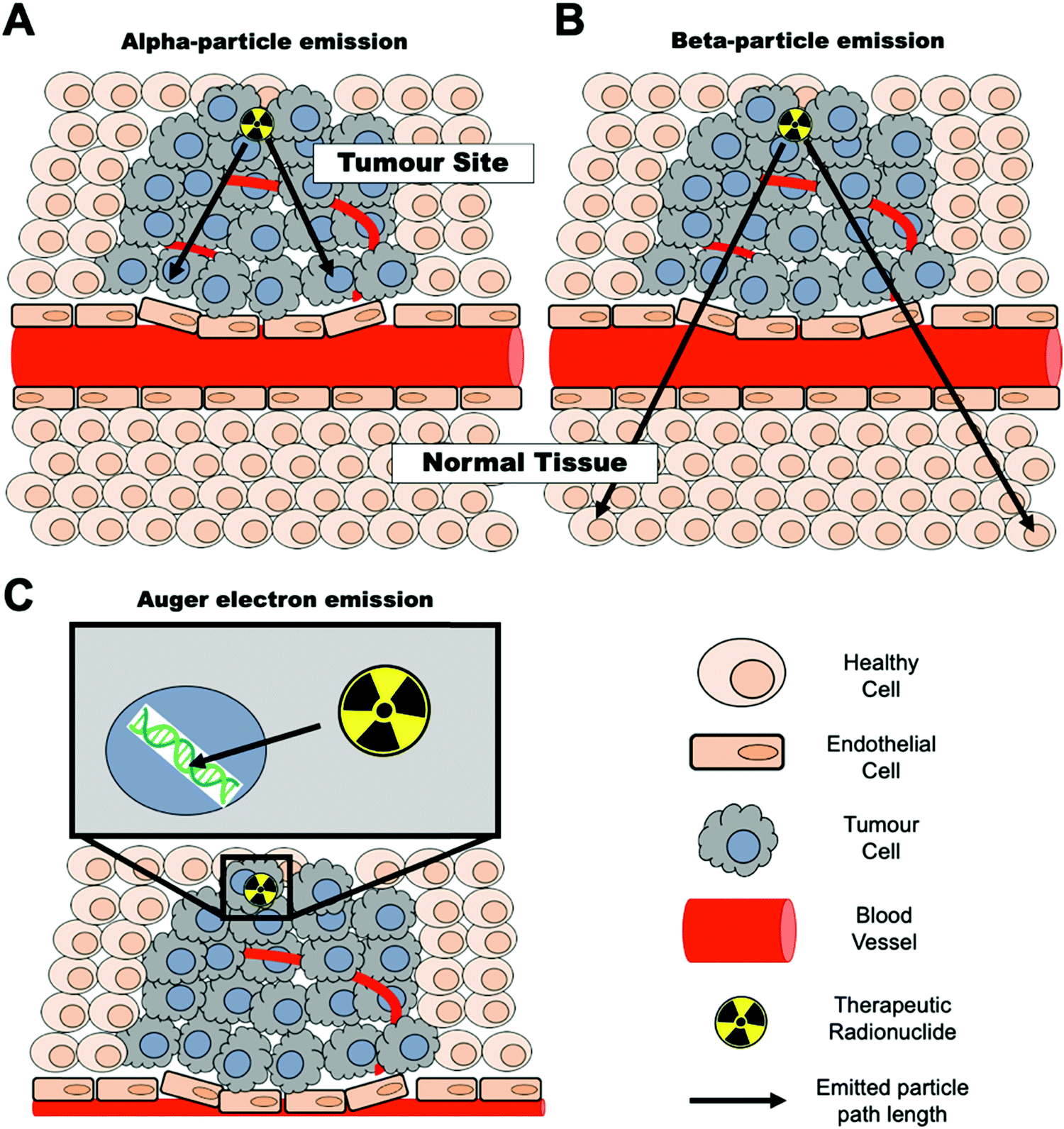 | ||
| Fig. 4 Radionuclide therapy mechanisms – representation of (A) alpha-particle emission, (B) beta-particle emission and (C) Auger electron emission. Black arrows represent the approximate path length of each emitted particle. | ||
3 Radionuclides
3.1 Production of radionuclides
Traditionally, the production of radionuclides for medical imaging and therapy has been associated with costly facilities and time-consuming protocols. Nevertheless, the optimisation of production processes and the modernisation of production technologies has facilitated their increased use in the clinical and preclinical settings. Four methods are currently applied for radionuclide production: fission, neutron activation, cyclotron and generator. These will be briefly described below.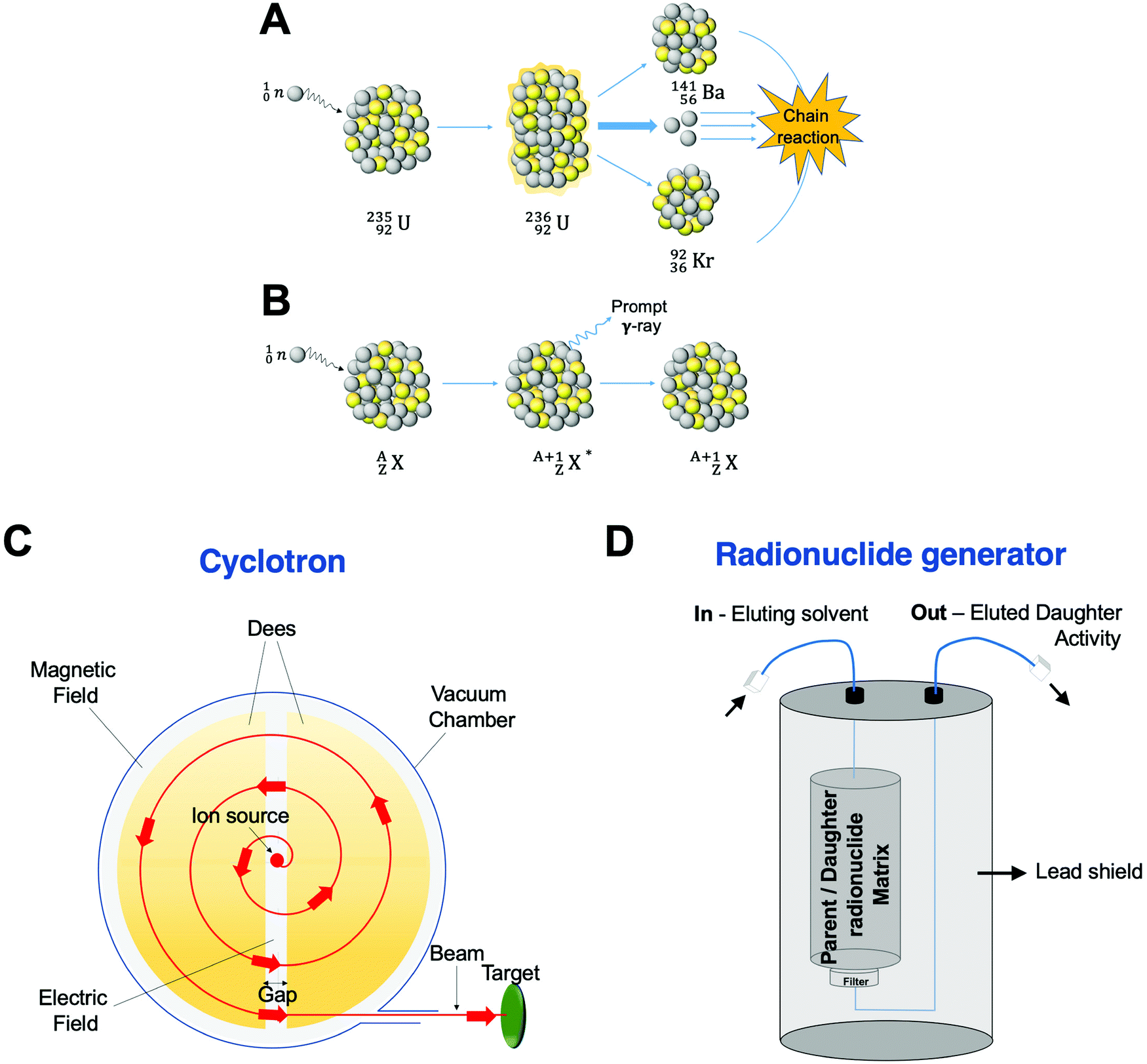 | ||
| Fig. 5 Production of radionuclides. Schematic representation of (A) nuclear fission of a 235U atom, (B) a (n,γ) neutron activation process, (C) cyclotron, and (D) standard radionuclide generator. | ||
Neutron activation is the other process carried out in a nuclear reactor. Here, the neutrons generated during the fission reaction are directed to a target with a stable nuclide, ZAX, giving an excited product nucleus, ZA+1X*. This excited nucleus then undergoes de-excitation to a ground state emitting a prompt γ photon, yielding a radioactive isotope of the same element, ZA+1X (Fig. 5B). Although the (n,γ) reaction is the most common in neutron activation, (n,p) reactions can also occur by emission of a proton, p. In this case, the starting target and the obtained product are different elements with the reaction represented as ZAX(n,p)Z−1AY.
| Radionuclide | Half-life | Max. energy (keV) | Decay | Production | Common production reaction |
|---|---|---|---|---|---|
| Au-198 | 2.7 d | 960 | β−, γ | Cyclotron | 197Au(n,γ)198Au |
| Au-199 | 3.1 d | 452.6 | β−, γ | Cyclotron | 198Au(n,γ)199Au |
| Co-57 | 270 d | 692 | EC, γ | Cyclotron | 56Fe(d,n)57Co |
| Fe-59 | 44.5 d | 1291 | β−, γ | Cyclotron | 59Co(p,n)59Fe |
| Ga-67 | 78.3 h | 300 | Auger e−, γ | Cyclotron | 68Zn(p,2n)67Ga |
| Gd-153 | 240.4 d | 103 | EC, γ | Cyclotron | 152Gd(n,γ)153Gd |
| In-111 | 2.81 d | 245 | γ | Cyclotron | 111Cd(p,n)111In |
| I-123 | 13.3 h | 159 | Auger e−, γ | Cyclotron | 127I(p,5n)123Xe |
| Re-186 | 91 h | 1080 | β−, γ | Cyclotron | 186W(p,n)186Re |
| Tc-99m | 6.0 h | 140 | γ | Generator | 99Mo/99mTc |
| Tl-201 | 3.0 d | 71 | γ | Cyclotron | 203Tl(p,3n)201Pb |
| Radionuclide | Half-life | Max. energy (keV) | Decay | Production | Common production reaction |
|---|---|---|---|---|---|
| As-72 | 25.9 h | 3320 | β+ | Cyclotron | 72Ge(p,n)72As |
| Br-76 | 16 h | 3980 | β+ | Cyclotron | 76Se(p,n)76Br |
| C-11 | 20.4 min | 961 | β+ | Cyclotron | 14N(p,α)11C |
| Cu-62 | 9.7 min | 2926 | β+ | Generator | 62Zn/62Cu |
| Cu-64 | 12.7 h | 656 | EC, β+, β− | Cyclotron | 64Ni(p,n)64Cu |
| F-18 | 109.7 min | 634 | EC, β+ | Cyclotron | 18F (F−): 18O(p,n)18F |
| Ga-68 | 67.6 min | 1899 | EC, β+ | Generator/cyclotron | 68Ge/68Ga |
| Ge-69 | 39.1 h | 1205 | β+ | Cyclotron | 69Ga(p,n)69Ge |
| I-124 | 4.2 d | 2100 | EC, β+ | Cyclotron | 124Te(p,n)124I |
| Mn-52 | 5.6 d | 1434 | β+ | Cyclotron | 52Cr(p,n)52Mn |
| N-13 | 9.9 min | 1199 | β+ | Cyclotron | 16O(p,α)13N |
| O-15 | 2.1 min | 1732 | β+ | Cyclotron | 15N(p,n)15O |
| Rb-82 | 1.3 min | 3378 | EC, β+ | Generator | 82Sr/82Rb |
| Y-86 | 14.7 h | 3150 | β+ | Cyclotron | 86Sr(p,n)86Y |
| Zr-89 | 78.4 h | 900 | EC, β+ | Cyclotron | 89Y(p,n)89Zr |
| Radionuclide | Half-life | Max. energy (keV) | Decay | Production | Max. particle range |
|---|---|---|---|---|---|
| β-Emission (LET ∼ 0.2 keV μm−1) | |||||
| Au-198 | 2.7 d | 960 | β−, γ | Cyclotron | 4 mm |
| Y-90 | 64.0 h | 2280 | β− | Generator | 12.0 mm |
| Lu-177 | 6.7 d | 500 | β−, γ | Cyclotron | 1.5 mm |
| I-131 | 8.0 d | 610 | β−, γ | Fission | 2.0 mm |
| Cu-67 | 62 h | 577 | β−, γ | Cyclotron | 1.8 mm |
| Re-186 | 91 h | 1080 | β−, γ | Cyclotron | 5.0 mm |
| Re-188 | 16.9 h | 2120 | β−, γ | Generator | 10.0 mm |
| α-Emission (LET ∼ 80 keV μm−1) | |||||
| At-211 | 7.2 h | 6000 | α | Cyclotron | 0.08 mm |
| Ac-225 | 10 d | 8000 | α, β− | Cyclotron | 0.1 mm |
| Bi-212 | 60.6 min | 6000 | α, β− | Cyclotron | 0.09 mm |
| Bi-213 | 46 min | 6000 | α, β− | Cyclotron | <0.1 mm |
| Ra-223 | 11.4 d | 7000 | α, β− | Cyclotron | <0.1 mm |
| Pb-212 | 10.6 h | 7800 | α, β− | Cyclotron | <0.1 mm |
| Tb-149 | 4.2 h | 400 | α | Cyclotron | <0.1 mm |
| Auger-emission (LET ∼ 4–26 keV μm−1) | |||||
| Ga-67 | 78.3 h | 300 | Auger e−, γ | Cyclotron | 10 nm |
| I-123 | 13.3 h | 159 | Auger e−, γ | Cyclotron | 10 nm |
| I-125 | 60.5 d | 27 | Auger e−, γ | Neutron ativation | 10 nm |
3.2 Radionuclides for SPECT
Radionuclides are mainly characterised by their decay modes, the energy emitted and the half-life of the products and sub-products generated until the stable isotope is reached.55,56 Gamma-emitters have been used since the beginning of nuclear medicine for γ-scintigraphy. With the development of SPECT – usually combined with CT – γ-emitting radionuclides are expanding the clinical imaging applications beyond the traditional γ-cameras. Nowadays, 99mTc is the most widely used radionuclide. This radionuclide combines a moderate short half-life (6 h), appropriate nuclear properties (89% of γ-rays abundance at 140 keV) and accessible generator production; making it a highly suitable choice for nuclear imaging studies.57 Due to its metallic character and several oxidation states available, radiolabelling with 99mTc is based on the formation of coordination complexes between the radionuclide (that needs to be reduced from Tc(VII) and a chelating ligand). Therefore, the versatility of 99mTc based radiolabelling, and that of other metallic radionuclides, is limited to coordination chemistry approaches (see Section 4.2).58 Other SPECT radionuclides, mainly iodine isotopes, are used for the formation of covalent bonds with carbon. In this regard, iodine radionuclides offer different isotopes to perform medium-term (123I, t1/2 = 13.3 h) or long-term imaging studies (125I, t1/2 = 60.5 d) and even radiotherapy (131I, t1/2 = 8 d, β−) with the same molecule.59 There is an extensive variety of useful SPECT radionuclides; not only for the radiolabelling of small molecules, peptides, proteins or antibodies, but also for the radiolabelling of nanomaterials (Table 2).3.3 Radionuclides for PET
Traditionally, clinical applications of PET have been mainly focused on four radionuclides: 11C, 18F, 13N and 15O.6018F is currently the main radionuclide used in clinical PET imaging, mostly due to its manageable half-life (t1/2 = 109.7 min), whereas that of 11C, 13N and 15O are very short (t1/2 = few minutes). Therefore, whereas having a cyclotron in close proximity and very fast radiolabelling protocols are required for 11C, 13N and 15O, this is not essential for 18F radiochemistry. Additionally, a substantial number of new drugs contain a F atom in their structure, increasing the interest of drug companies to use 18F-PET to study their in vivo properties.61 Furthermore, the half-life of 18F matches well with the pharmacokinetics of many small biomolecules.62 NPs, however, tend to have longer biological half-lives that are better matched by long-lived PET radionuclides.Metallic radionuclides elements are attractive candidates for PET applications, particularly for imaging NPs. 89Zr, with a long half-life of 3.3 days, has been attached to biomolecules with long circulation times, mainly antibodies for immuno-PET applications.6368Ga (t1/2 = 67.6 min), due to its generator-based production (Table 3), is increasingly being used for the radiolabelling of peptides and small molecules, making 68Ga the “PET version of 99mTc”.64 However, it has limited applications for in vivo NP imaging studies due to its short half-life. Several other radionuclides with different nuclear and chemistry properties have been also investigated for a variety of PET applications (Table 3).
3.4 Radionuclides for therapy
As discussed in the previous section, radionuclides with α, β− and Auger e− emissions have therapeutic applications (Table 4). The use of radionuclides for therapy is not a novel concept. The treatment of thyroid cancer and hyperthyroidism with thyroid-avid 131I-iodide was implemented more than 70 years ago.65 Other important therapeutic radionuclides used clinically is the bone-tropic 223Ra; with demonstrated effectiveness in bone related solid tumours and bone metastases in prostate cancer.66 Other emerging radionuclides for therapy are 177Lu and 225Ac, being investigated in different clinical trials for theranostics applications in neuroendocrine tumours and prostate cancer.67–69For therapeutic applications with antibodies (radioimmunotherapy), several formulations are also under evaluation using 90Y as a therapeutic radionuclide, with some of them already approved – such as 90Y-Ibritumomab tiuxetan (Zevalin®) used as treatment for non-Hodgkin's lymphoma.70 The integration of therapeutic radionuclides into nanomaterials has the potential of not only improving their therapeutic efficiency but also their theranostic capabilities with a broad variety of applications. However, the usual slow excretion of nanomaterials poses a significant barrier for this approach.
3.5 Theranostic pairs of radionuclides
Besides the use of individual radionuclides for imaging and/or therapy, certain combinations of radioisotopes can be used as theranostic pairs for both imaging and therapy. These combinations are formed by two radioisotopes of the same chemical element, one with the appropriate radio-physical properties to generate a signal for PET or SPECT detection, and the other isotope with suitable therapeutic properties. This is an interesting approach since both isotopes are radioisotopes of the same element and hence, only one chemical element is ultimately applied allowing both diagnosis and therapy.The first example of a theranostic pair application was described in 1993 by Herzog et al. where the pair 86Y/90Y was studied to evaluate, in a patient with bone metastases, the pharmacokinetics of the radiotracer 86Y-citrate as an analogue of the radiotherapeutic 90Y-citrate.71 Since then, different pairs have been proposed increasing the opportunities in personalised medicine. Theses pairs are formed by β+ or γ-emitters for PET or SPECT respectively, in combination with radionuclides with α, β− and Auger e− emissions for the therapeutic response. Some of the most important proposed pairs are: 72As/77As, 64Cu/67Cu, 68Ga/67Ga, 124I/131I, 110gIn/111In, 44gSc/47Sc, 83Sr/89Sr, 152Tb/161Tb, 152Tb/149Tb and 86Y/90Y.72
The nature of NPs offers unique possibilities in combination with theranostic radionuclide pairs, such as the ability of co-loading radionuclides and drugs with synergistic therapeutic properties. However, as mentioned in the previous section, the slow biological excretion profile of most nanomaterials represents a significant barrier towards the clinical translation of radionuclide-based therapeutic nanomaterials.
3.6 Biodistribution of free radionuclides
A key factor when in vivo studies are conducted with radiolabelled nanomaterials is the biodistribution of the “free” or unchelated radionuclide. Although this is often underestimated, the lack of consideration of this aspect can easily lead to misinterpreting imaging signal: wherein the biodistribution of the free radionuclide is wrongly attributed to the nanomaterial signal. On the contrary, knowledge of the radionuclide biodistribution can also aid the selection of the most appropriate radionuclide depending on the final application; to avoid, as far as possible, the overlapping between the signals of the free radionuclide and the radiolabelled nanomaterial. It is worth noting that this is mostly applicable when the radiolabeled NP releases its radionuclide in its ‘free’ form. When radionuclides are chelated to a well-suited small molecule-based ligand/chelator it is expected that release of this component from the NP structure will result in fast excretion via the renal excretion pathway, unless any biological process that may be involved in NP degradation affects the expected radiometal-chelator stability.Table 5 shows the biodistribution of the most important radionuclides used for the radiolabelling of nanomaterials. It is important to note that this table highlights the organs where an unchelated radionuclide can be found in a qualitative manner. The degree of uptake will depend on the type of specimen, experimental model and the biodistribution time. In addition, some radionuclides are often produced under different formulations (e.g.89Zr can be used as [89Zr]ZrCl4 or [89Zr]Zr-oxalate) with possible effects over the biodistribution, the chemical identity of the free radionuclide is defined in the table.
| Radionuclide | Qualitative biodistribution of “free” radionuclides | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blood | Liver | Kidneys | Heart | Spleen | Bone | Pancreas | Salivary glands | Thyroid | Stomach | Tumour | ||
| 111In (111InCl3) | ✓ | ✓ | ✓ | ✓ | 74 | |||||||
| 99mTc (99mTcO4) | ✓ | ✓ | 75 | |||||||||
| 198Au (198AuCl4) | ✓ | ✓ | ✓ | 76 | ||||||||
| 18F (Na18F) | ✓ | 77 | ||||||||||
| 67/68Ga (67Ga-citrate) | ✓ | ✓ | ✓ | ✓ | ✓ | 78 and 79 | ||||||
| radioI (NaradioI) | ✓ | ✓ | ✓ | 80 | ||||||||
| 64Cu (64CuCl2) | ✓ | ✓ | 81 and 82 | |||||||||
| 89Zr (89ZrCl4) | ✓ | 83 | ||||||||||
| 52Mn (52MnCl2) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 84 | |||||
| 90Y (90YCl3) | ✓ | ✓ | ✓ | 85 | ||||||||
| 177Lu (177LuCl3) | ✓ | 86 | ||||||||||
| 188Re (188ReO4) | ✓ | ✓ | ✓ | ✓ | ✓ | 87 | ||||||
| 223Ra (223RaCl2) | ✓ | ✓ | ✓ | 88 | ||||||||
| 225Ac (225AcCl3) | ✓ | ✓ | 89 | |||||||||
It is particularly worth highlighting that several radionuclides show high uptake in organs where nanomaterials commonly accumulate (e.g. liver), and this should be taken into account when analysing the images. In summary, there are different factors affecting the radionuclide choice. These involve the type of production, the radio-physicochemical properties and the biodistribution. The selection of the radionuclide usually delimits the type of radiolabelling method, although different methods for the same radionuclide can be applied as further described in the next sections.
4 Radiolabelling nanomaterials: basic concepts and methods
4.1 Basic concepts
In this section we will introduce and summarise basic radiochemical concepts which are widely applicable to any radiolabelling chemistry. However, we will place a particular emphasis on those aspects that are relevant to the radiochemistry of nanoparticles.4.2 Chelator-based radiolabelling
The labelling of compounds with non-metallic radionuclides (e.g. fluorine-18, carbon-11 and iodine-131, etc.) is achieved by direct covalent bond formation (see Section 4.4.3 for further details). However, radionuclides with metallic character (radiometals; e.g. copper-64, technetium-99m, zirconium-89) often require the use of chelators and hence coordination chemistry approaches to efficiently attach them to the NP of interest. The purpose of a chelator is to bind the radiometal ion through two or more bonds creating highly stable metal complexes and hence RCS. Due to the ‘always on’ nature of imaging contrast using nuclear imaging, any radiometals which are not stably bound may distribute differently in vivo causing misleading signal within the images. For this reason, the choice of chelator used with any particular radiometal is of paramount importance.Understanding the coordination chemistry of the chosen radiometal is essential to avoid the incorrect selection of a chelator. Firstly, the geometric preferences and coordination number will be affected by the atomic number, radii and charge. Additionally, the ‘hardness’ of the metal ion in terms of Pearson's acid–base concept must be assessed, with the chosen ligand having the appropriate hard/soft donor atoms and with the right electronic properties to improve the kinetic inertness of the complex. In terms of thermodynamic stabilities, polydentate ligands form stable complexes over their monodentate counterparts due to the “chelate effect”. This is, in a simplified way, due to the increase in entropy resulting from the complexation of a polydentate ligand and metal ion, as compared with multiple monodentate ligands. Polydentate ligands are usually split into two categories: acyclic/linear chelators and macrocyclic chelators. Acyclic or linear chelators often benefit from rapid radiometal complexation due to their lack of rigidity. This is in contrast to macrocyclic chelators, which have a relatively rigid and pre-organised structure resulting in higher complex stability (i.e. macrocyclic effect) but suffer from slow complexation kinetics, resulting in the need for high temperatures and long reaction times. This last requirement may be damaging for some heat-sensitive NP types (e.g. protein-based, exosomes). For this reason, the radiolabelling of heat-sensitive NPs with macrocyclic chelators is often done post-complexation via the use of bifunctional chelators (vide infra, Section 4.2.1).
Based on the above principles, an ideal chelator should allow rapid, quantitative complexation under mild conditions (aqueous solvent, room temperature and neutral pH), whilst demonstrating high kinetic inertness and thermodynamic stability in biologically relevant medium (i.e. serum). This stability should be for an appropriate amount of time to allow imaging and is usually based on the half-life of the radiometal and pharmacokinetics of the NP of interest. Several reviews have discussed optimised chelators for each radiometal in great detail, and are highly recommended for further reading.94–96Fig. 6 shows the chemical structures of all chelators used for the radiolabelling of NPs discussed in this review, with their corresponding radionuclide(s).
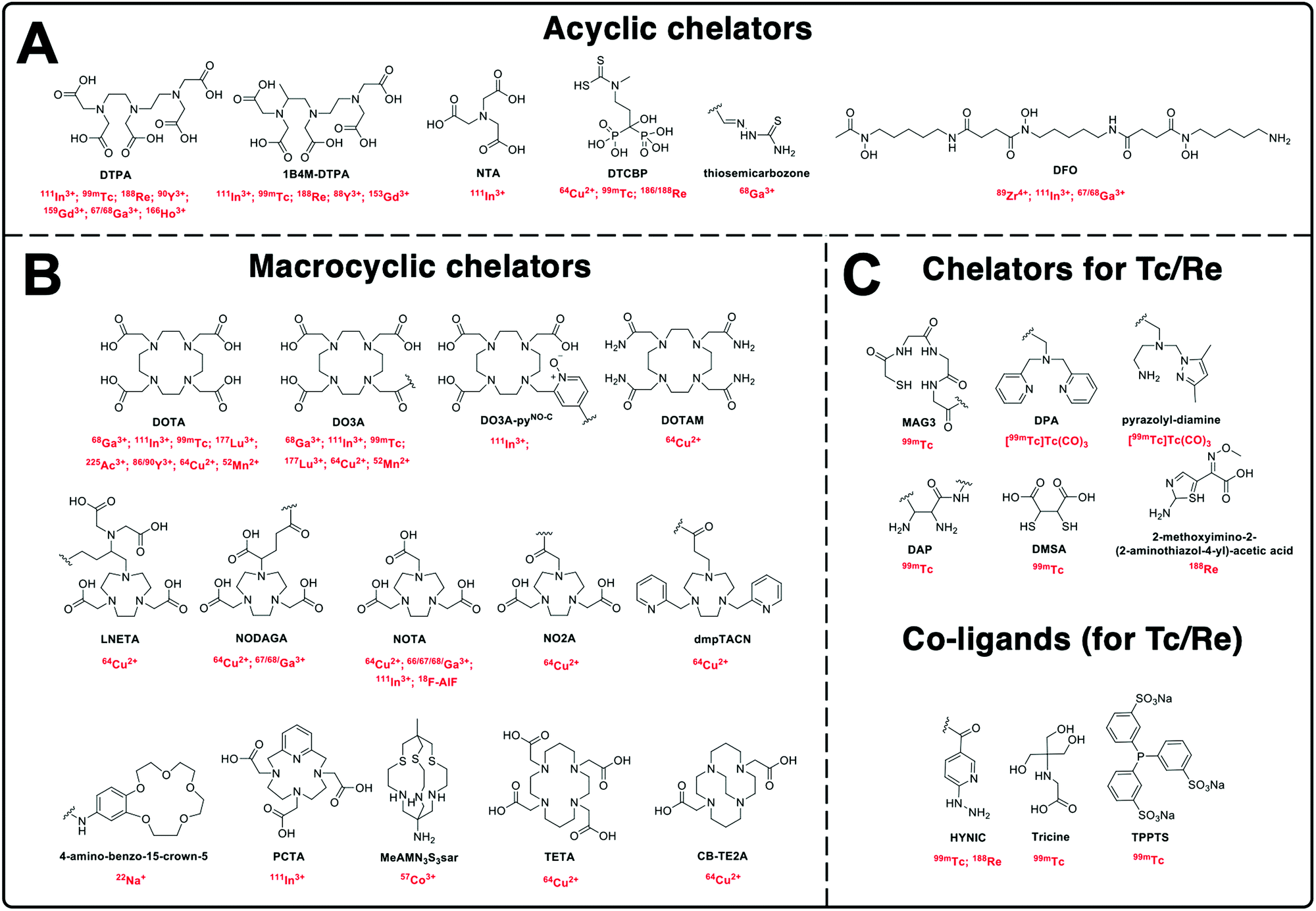 | ||
| Fig. 6 Chemical structures of the chelators used for radiolabelling nanomaterials described in this review with their corresponding radionuclide(s). | ||
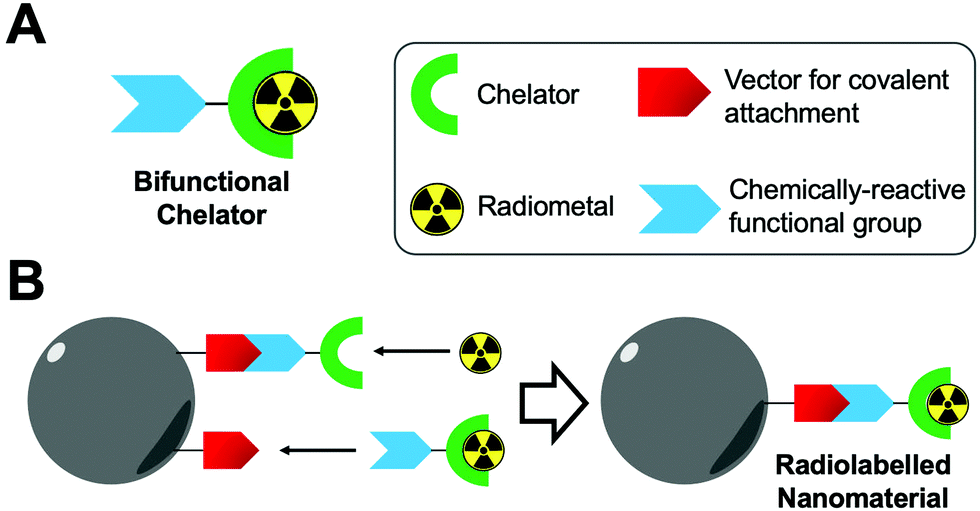 | ||
| Fig. 7 (A) Schematic representation of a bifunctional chelator. (B) Schematic representation of the radiolabelling of nanoparticles using bifunctional chelators. | ||
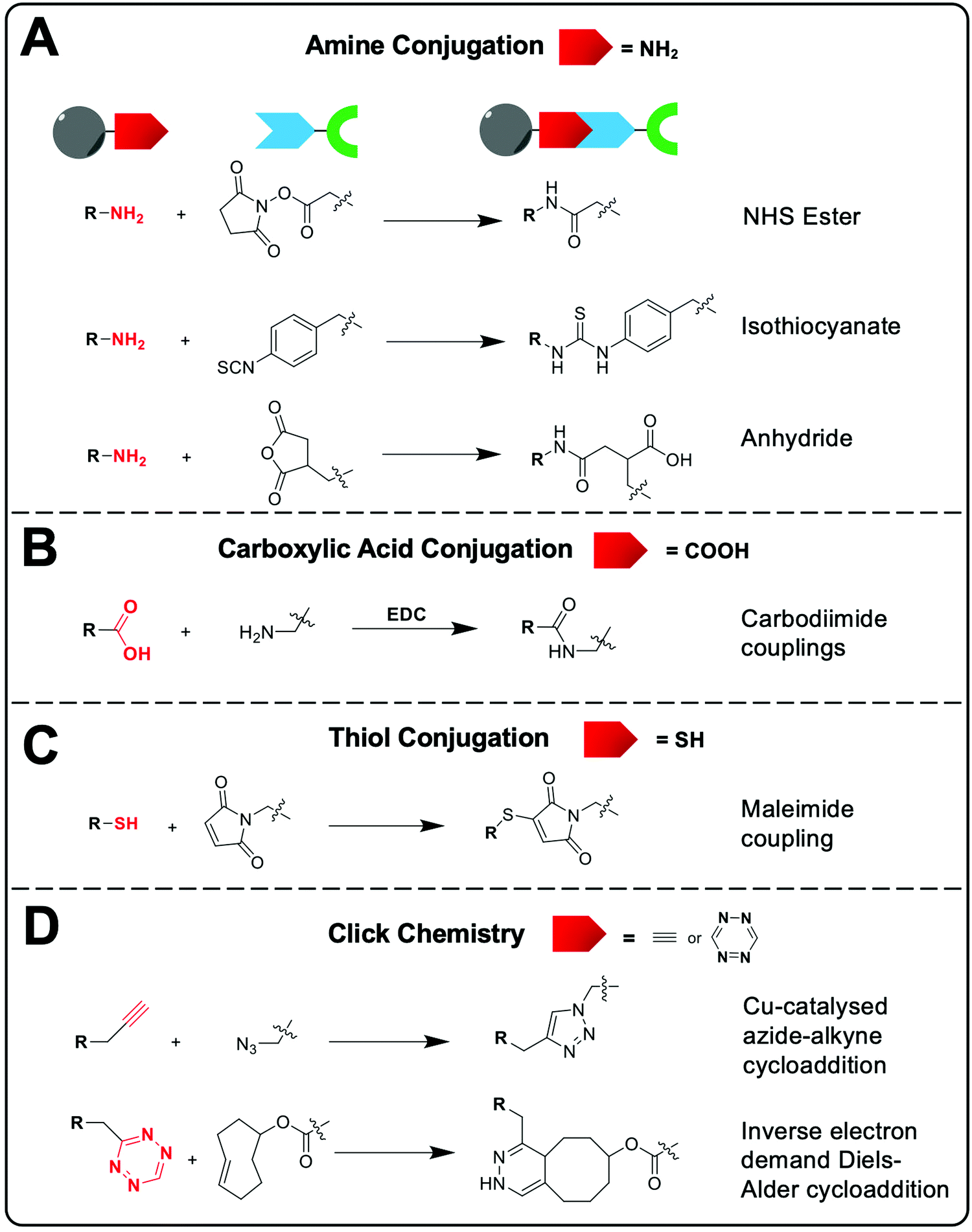 | ||
| Fig. 8 Common bioconjugation reactions that allow the attachment of bifunctional chelators to the nanomaterial surface. (A) Amine-based conjugation, (B) carboxylic acid-based conjugation, (C) thiol-based conjugation and (D) click chemistry conjugation. | ||
Common chemical functional groups present on the surface of nanomaterials can be radiolabelled using bifunctional chelators. For example, amines can be reacted with chelators containing NHS ester groups or cyclic anhydrides to form amide bonds, or with aryl isothiocyanate groups to form isothioureas (Fig. 8A). Carboxylate functionalised NPs can be reacted with amines via the use of carbodiimide coupling reagents, such as EDC, and free thiols can be conjugated using maleimides (Fig. 8B and C).98 Finally, click chemistry is often used due to its rapid, high yielding reactions. Two commonly used reactions are the copper-catalysed azide–alkyne (CuAAC) and inverse electron demand Diels–Alder cycloaddition between a tetrazine and trans-cyclooctene (Fig. 8D).99 These reactions have previously been discussed in the context of bifunctional chelators for radionuclide imaging in reviews that are highly recommended for further reading.99–101
The selection of the appropriate bioconjugation reaction may be often dictated by the nanomaterial of interest. For example, poly(amidoamine) (PAMAM) dendrimers or lipids used to formulate vesicles will often contain free amine groups capable of easily being reacted with appropriate functional groups. Additionally, polymer-based or polymer-coated and protein-based NPs will often intrinsically contain functional groups for bioconjugation (e.g. carboxylate groups on dextran or aspartic/glutamic amino acids). However, whilst the target vector for conjugation is often intrinsic to the NP formulation, the NP can also be modified to facilitate conjugation of the bifunctional chelator if need be.
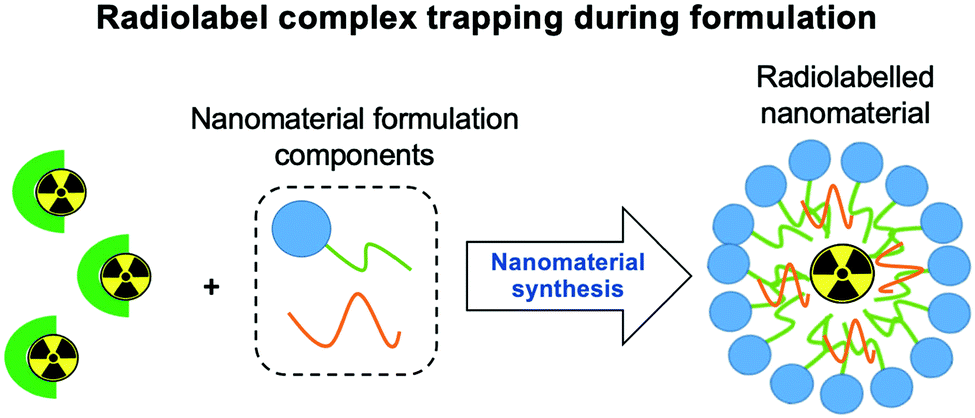 | ||
| Fig. 9 Schematic representation of the radiometal complex-trapping radiolabelling strategy. Radiometal complexes are added to the mixture during the formation of the nanomaterial and are then subsequently incorporated into the nanoparticle and become trapped. | ||
4.3 Ionophore-based radiolabelling
Whilst technically involving chelators, ionophore-based methods are distinct enough from the classic chelator-based methodologies described previously (Section 4.2). Although the following radiolabelling methods are only relevant for vesicle-based NPs (e.g. liposomes, exosomes) containing lipid membranes, they represent a significant portion of the NP literature. Hence, for the sake of clarity, we have separated these methods from the chelator-based methods described above. Fig. 10 summarises the strategies used for ionophore-based NP radiolabelling, that are discussed below.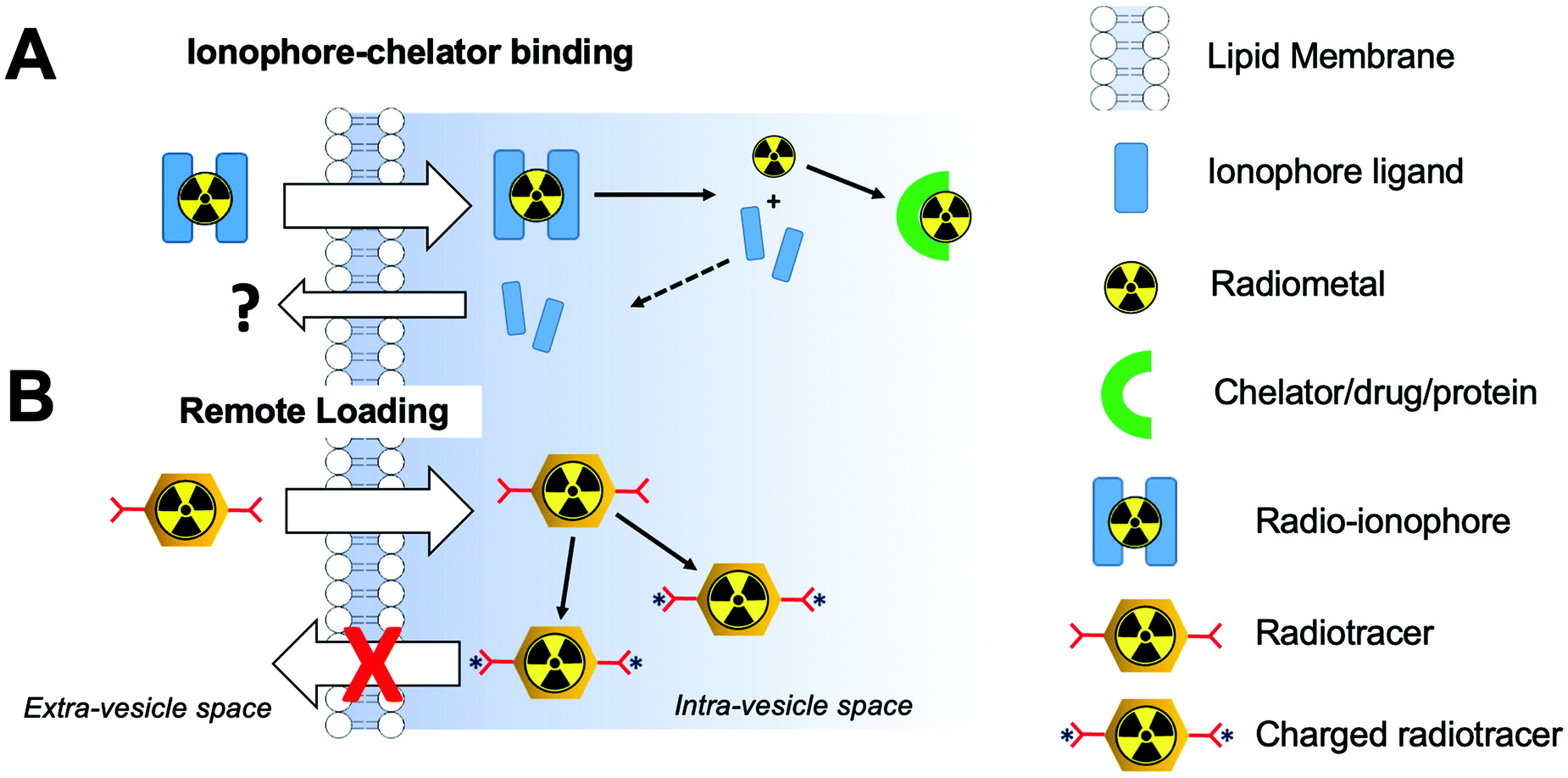 | ||
| Fig. 10 Schematic representation of (A) ionophore-based radiolabelling strategies and (B) remote loading radiolabelling. | ||
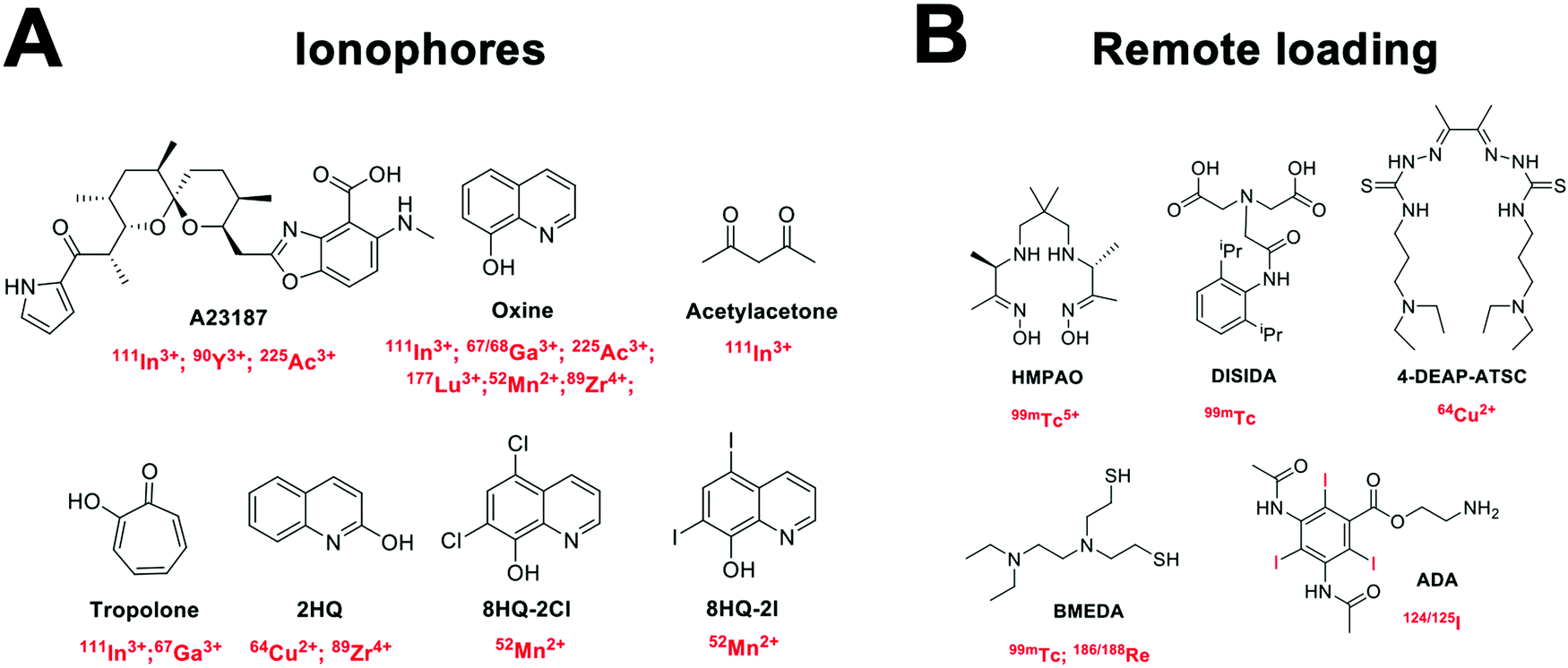 | ||
| Fig. 11 Chemical structures of (A) common ionophores used in ionophore-chelate vesicle radiolabelling with the corresponding radionuclides; and (B) chemical structures of compounds used for remote loading with their corresponding radionuclides. | ||
4.4 Non-chelator radiolabelling
Non-chelator based strategies involve the direct incorporation of radionuclides into the core and/or surface of nanomaterials, circumventing the need for external chelating agents. Hence, these methods are usually more straightforward and less time-consuming than chelator-based methods – though this is dependent on the type of nanomaterial and radionuclide being used. Removing the use of chelators will often decrease the number of reaction steps and most importantly, preserve the integrity of the nanomaterial by avoiding the bulky chelator molecule that could affect the in vivo behaviour.103 Non-chelator based strategies adapt a variety of common radiolabelling reactions, as well as implementing bespoke radiolabelling methods specifically designed for the integration of radionuclides into nanomaterials (Fig. 12).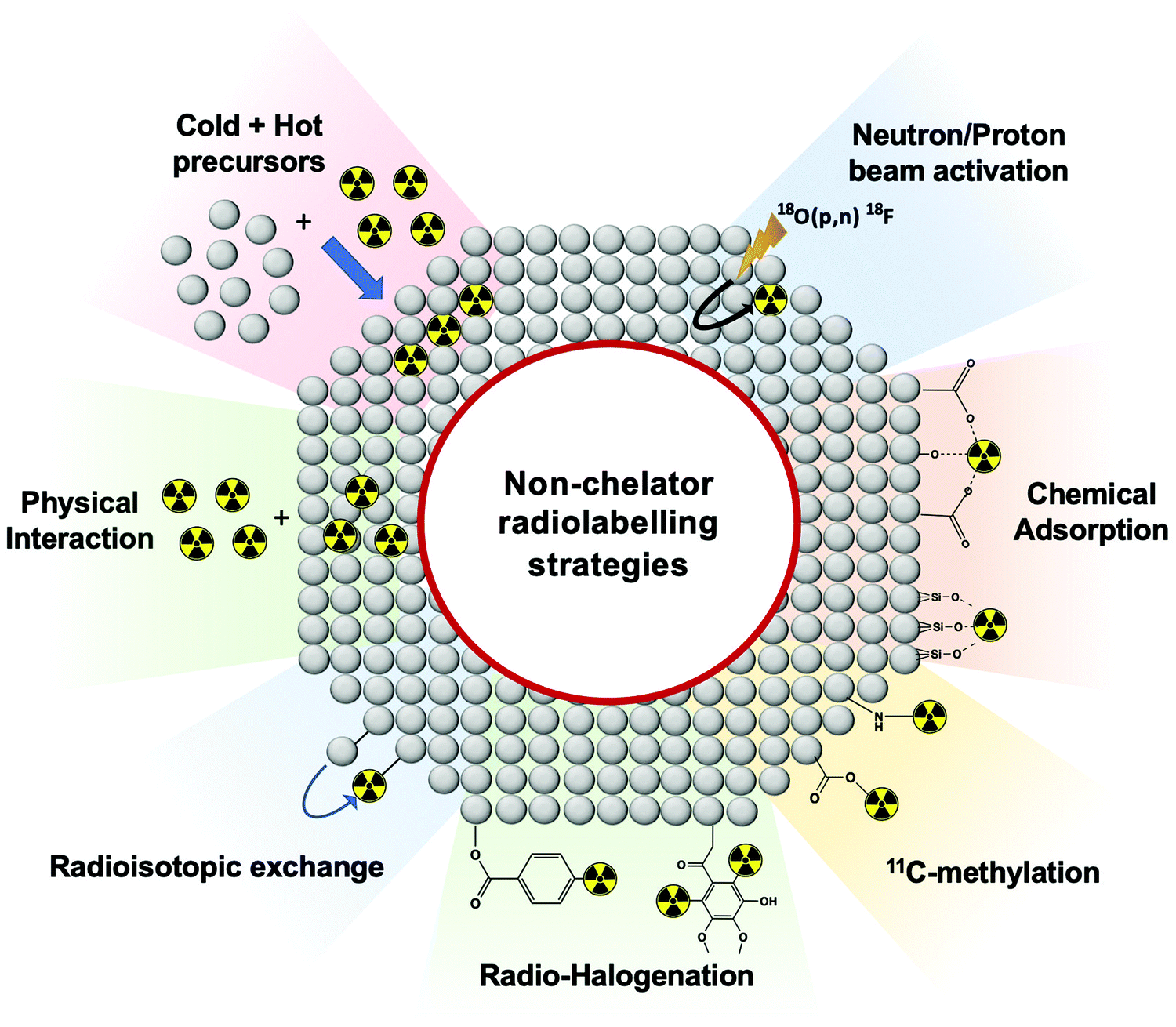 | ||
| Fig. 12 Schematic representation of non-chelator based radiolabelling methods. | ||
Traditional radiochemical reactions such as radio-halogenations, 11C-methylations or chemical adsorptions are often used. In addition, reactions such as the use of hot + cold NP precursors or proton beam activation of materials are specific for nanomaterials. Other non-standard radiochemical labelling methods such as those based on radioisotopic exchange or physical interactions take the advantage of the physicochemical properties of certain nanomaterials to facilitate radiolabelling. Each of the non-chelator based NP radiolabelling methods will now be discussed in detail.
 | ||
| Fig. 13 Representation of hot + cold precursors radiolabelling strategy. | ||
This strategy, exclusive for inorganic nanomaterials, is often straightforward with fast protocols, making this method the most widely used of the non-chelator NP radiolabelling methods. From a chemical point of view, this method is based on the radiochemical doping of the nanomaterial during synthesis. The radionuclide (hot precursor) is added in trace levels to the nanomaterial precursors (carrier-added) triggering a co-precipitation that leads to the incorporation of the radionuclide into the crystal lattice of the nanostructures.104 The ‘doping’ represents the main advantage of this strategy, as it maintains the nanomaterial's structural integrity, whilst allowing strong radiochemical stabilities. This is particularly the case with homo-radionuclide doping, (i.e. the nanomaterial core contains the same element as the radionuclide dopant) which allows imaging of the in vivo fate of some nanomaterials without modifications to the NP structure. For instance, diverse gold NPs have been doped with 195Au, 198Au or 199Au or iron oxide NPs with 59Fe for similar purposes.105–111 For example, Zhao et al. doped Au NPs with 199Au to study the biodistribution in tumour-bearing mice model after conjugation with D-Ala1-peptide T-amide (DAPTA) (Fig. 14A). SPECT/CT imaging experiments revealed the elimination of the [199Au]AuNPs by liver and spleen with specific accumulation in the tumour due to the DAPTA vectorisation (Fig. 14B).106
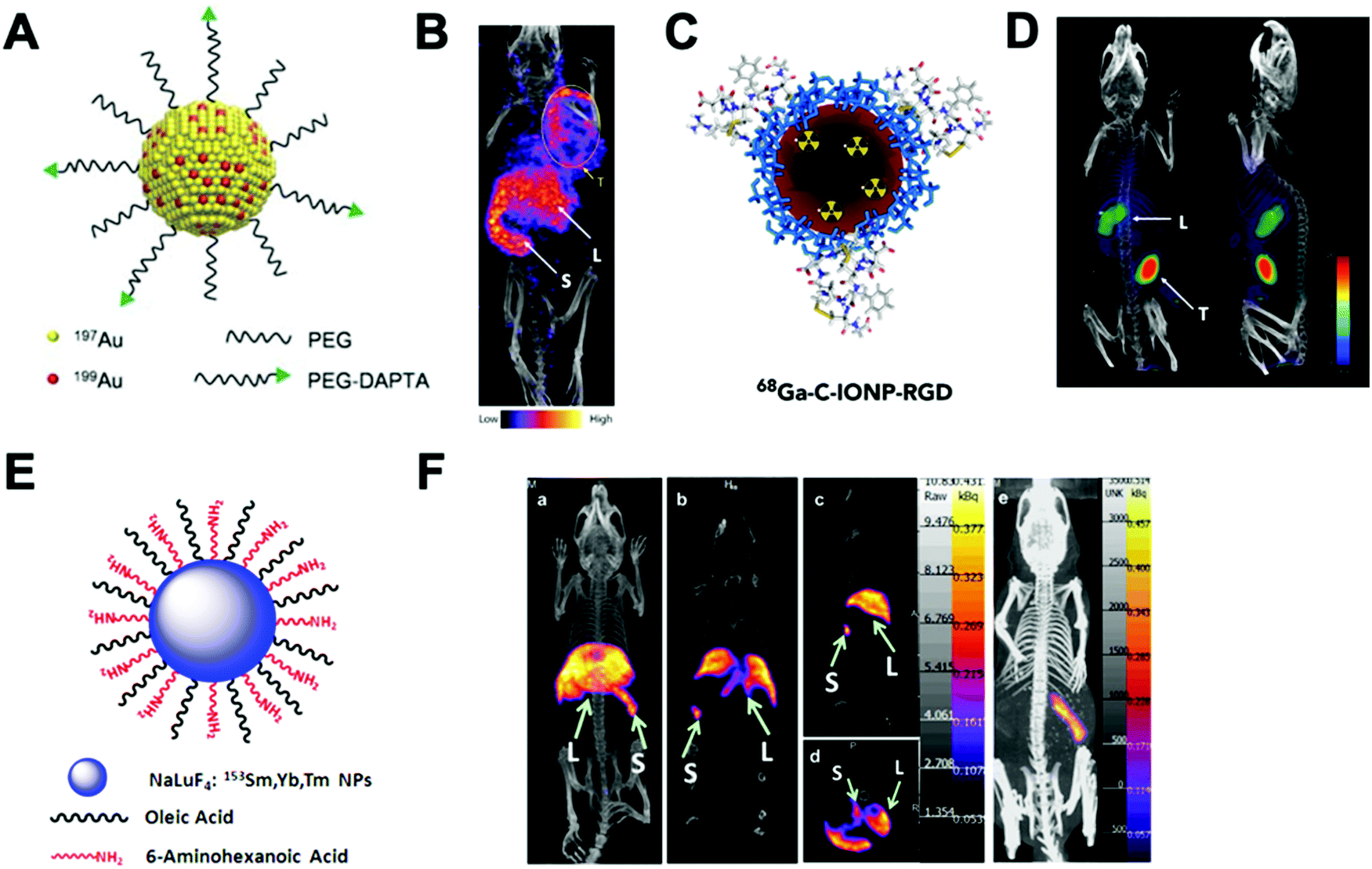 | ||
| Fig. 14 (A) Radioactive Au nanoparticles doped with 199Au atoms conjugated with D-Ala1-peptide T-amide (DAPTA), (B) NanoSPECT/CT image of a 4T1 tumour-bearing mouse 24 h post injection of the 5 nm 199Au–AuNP–DAPTA probe (the tumour is labelled by an ellipse in yellow colour. T: tumour, L: liver, S: spleen), (C) 68Ga core-doped iron oxide nanoparticles functionalised with RGD peptide (68Ga-C-IONP-RGD), (D) PET/CT imaging of tumour-bearing mice 1 hour after injection of 68Ga-C-IONP-RGD, showing strong activity in the tumour (T: tumour, L: liver), (E) the schematic diagram of the NaLuF4:153Sm,Yb,Tm nanoparticles, (F) in vivo SPECT images after intravenous injection of 153Sm–UCNPs. (a) Whole-body three-dimensional projection, (b) coronal, (c) sagittal and (d) transversal images acquired at 1 h and (e) whole-body three-dimensional projection images acquired at 24 h are shown respectively. The arrows inset point to the liver (L) and spleen (S). Adapted and reproduced with permission from ref. 112–114. | ||
There are some considerations in order to achieve high RCYs with this strategy. A high solubility between both, cold and hot, precursors is required. Considering most of radionuclides are delivered in aqueous solutions, this strategy is then limited to reactions conducted in water. It is also important to control the ionic strength of the reaction media to allow the nucleation and growth of the nanomaterial. The physicochemical properties of the radionuclide also play a key role. The ionic radius of the radionuclide and its corresponding non-radioactive ion should be similar. In addition, the radionuclide should have the same ionic charge, in order to coordinate with the intermediate complex formed by the cold precursors before nucleation. Considering this, mainly metallic cations can be integrated into NPs using this strategy with few suitable radionuclide–NP pair choices (Table 6). For instance, IONPs were doped with 68Ga for tumour imaging driven by the functionalisation with an RGD peptide (68Ga-C-IONP-RGD, Fig. 14C). PET/CT imaging showed high accumulation in the tumour 1 h after the injection of the 68Ga-C-IONP-RGD with no signals of free 68Ga3+ confirming the high stability of the radiolabelling (Fig. 14D).115 Other successful combination, reported by Yang et al., is the use of 153Sm as hot precursor for the formation of NaLuF4 UCNPs (Fig. 14E). The biodistribution of 153Sm–UCNPs was easily addressed by in vivo SPECT/CT imaging revealing a rapid clearance to the liver and spleen 1 h after the i.v. injection and main accumulation into the spleen after 24 h (Fig. 14F).
| Nanoparticle type | Radionuclide | Ref. |
|---|---|---|
| Iron oxide NPs | 225Ac, 64Cu, 59Fe, 68Ga, 111In | 109–111 and 115–119 |
| Gold NPs | 195Au, 198Au, 199Au | 105–108 |
| Up-converting NPs | 153Sm, 90Y | 112 and 120 |
| Quantum dots | 109Cd, 64Cu, 125mTe | 121–123 |
| Cerium oxide NPs | 141Ce, 65Zn | 124 |
| Silver NPs | 131I | 125 |
Finally, it is worth highlighting that if the radionuclide and the coating molecule are oppositely charged, the labelling may be conducted by chemical adsorption (see Section 4.4.5) rather than by the radioactive co-precipitation – with possible implications for the radiochemical stability. Being a convenient strategy for pre-clinical purposes, it presents a main limitation for clinical applications since the radionuclide is integrated from the beginning, demanding fast and effective purification protocols to reduce the radioactive exposition to the operator. The potential lack of reproducibility between independent batches could also be a limitation of this strategy which requires extremely reproducible synthetic protocols.
 | ||
| Fig. 15 Schematic representation of neutron/proton beam activation radiolabelling strategy. | ||
A high control over the radiolabelling location represents the main advantage of this method, as only specific atoms can undergo the nuclear reaction, with a consequently high RCS. However, this method has a key drawback in the requirement of a proton/neutron beam source, which involves the use of complex instruments that are not widely available. Additionally, the high energies used in these nuclear reactions may affect the integrity of sensitive biological species that may be attached to the nanomaterial, limiting the applications to purely inorganic nanomaterials.
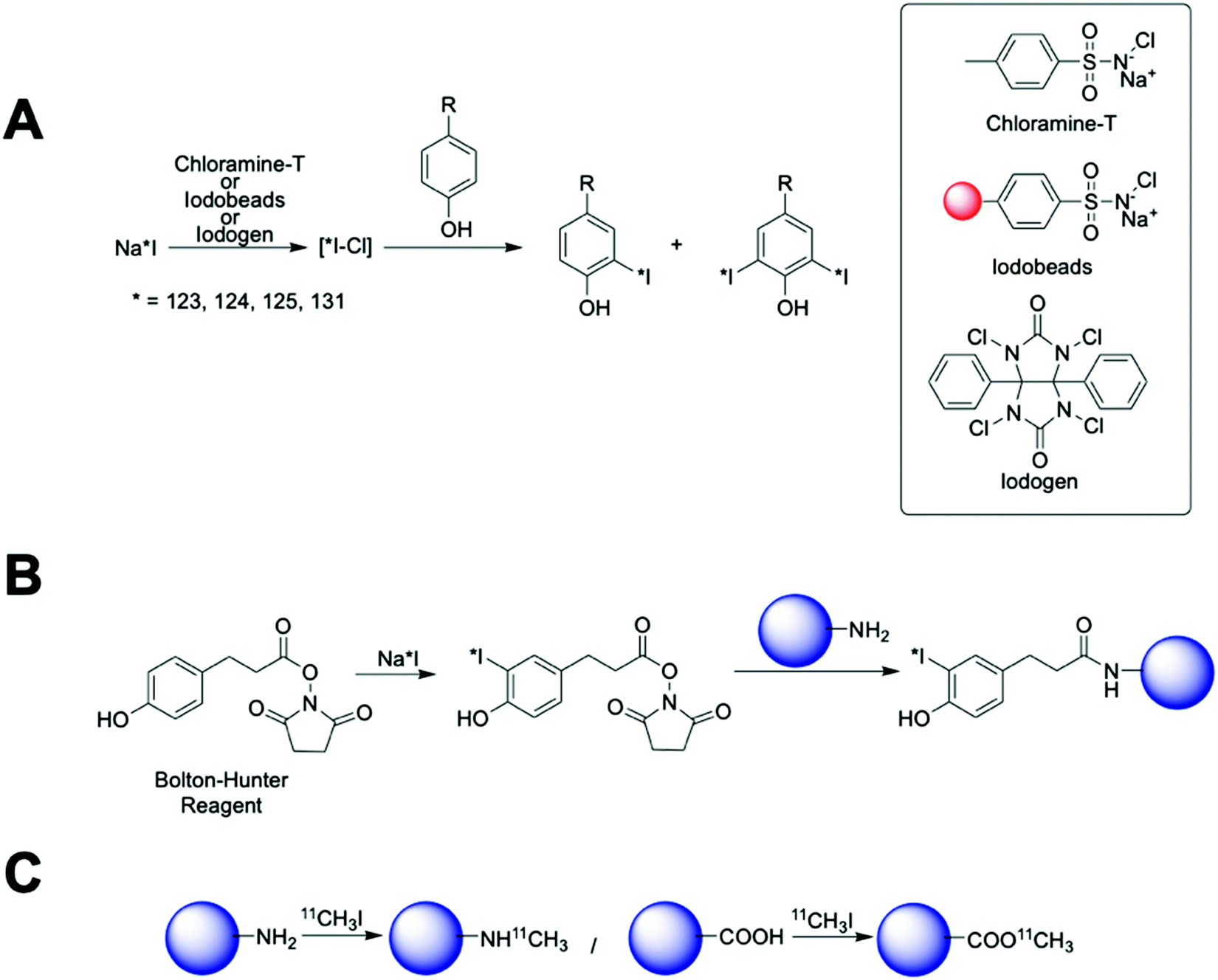 | ||
| Fig. 16 Radioiodination and 11C-methylation. (A) Scheme of radioiodination of tyrosine residues mediated by chloramine-T, iodobeads or iodogen, (B) scheme showing the radioiodination of amine-terminated nanoparticles by Bolton–Hunter reagent and (C) 11C methylation of amine and carboxylate-functionalised nanoparticles. | ||
These methods are quick, with the radioiodination occurring in seconds to a few minutes and usually in high radiolabelling yields. Chloramine-T is used in solution generating a strong oxidising environment that triggers the radioiodination in just 30 s. Then, subsequent quenching with a reducing agent (usually sodium metabisulfite) is required. Although the chloramine-T method is fast, cheap and reproducible, active biomolecules can be affected by the oxidant and/or the reducing agent. To overcome this limitation, the chloramine-T is immobilised in a polystyrene bead (Iodobead) where the reactivity is controlled under mild conditions without the need of reducing agents.134 Iodogen also facilitates radioiodination reactions under mild conditions. In this case, iodogen is dissolved in organic solvents and evaporated, to fix the molecule on the walls of the reaction vessel, preventing dissolution in water and direct contact with the biomolecule/NP. All these iodine radiolabelling mediators are generally limited to the presence of tyrosine or histidine moieties in the surface of the nanomaterials. The Bolton–Hunter reagent, a radioiodination mediator based on a pre-radiolabelled N-hydroxysuccinimide group, is frequently used for the radiolabelling of nanomaterials with free amino groups on the surface – extending the flexibility of the nanomaterial radioiodination protocols (Fig. 16B). With advantages and drawbacks, these radiolabelling mediators have been applied to the radiolabelling with 124I, 125I or 131I of a vast number of nanomaterials (Table 7). Generally, these protocols rendered high radiochemical yields; although, in some examples, poor radiochemical stability were reported giving an undesirable accumulation in the thyroid glands due to the iodine detachment from the nanomaterial.135–137 This situation has been previously attributed to an enzymatic-driven cleavage of the C–I bond in some molecules.138,139
| Nanoparticle type | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Iron oxide NP | 125I | Chloramine-T | 140 and 141 |
| Bolton–Hunter reagent | 142 | ||
| Silica NP | 124I | Bolton–Hunter reagent | 143 |
| 125I | Bolton–Hunter reagent | 144 | |
| Gold NP | 124I | Chloramine-T | 145 and 146 |
| 125I | Iodogen | 136 | |
| 131I | HPAO/chloramine-T | 147 and 148 | |
| UCNPs | 124I | Iodobeads | 149 |
| 125I | Bolton–Hunter reagent | 135 | |
| Q dots | 125I | Bolton–Hunter reagent | 150 |
| Silver NP | 131I | Chloramine-T | 151 |
| Dendrimers | 125I | Bolton–Hunter reagent | 152–156 |
| Chloramine-T | 157–160 | ||
| 131I | Iodogen | 161 | |
| Caprolactone polymeric NP | 125I | Chloramine-T | 162 |
| Graphene oxide/carbon NPs | 125I | Chloramine-T | 163 and 164 |
| 131I | Chloramine-T | 165 | |
| Chitosan NPs | 125I | Bolton–Hunter reagent | 166 |
| HPMA copolymer NP | 125I | Chloramine-T/iodogen | 167 |
| 131I | Chloramine-T | 168 | |
| 125I | Bolton–Hunter reagent | 155 | |
| Nanogel | 125I | Chloramine-T | 169 |
| Polymeric micelles | 125I | Chloramine T | 170 |
| Iodogen | 171 and 172 | ||
| 131I | Chloramine-T | 173 | |
| Poly(maleic anhydride-alt-1-octadecene) NP | 125I | Iodogen | 174 |
| 131I | Iodogen | 174 | |
| Poly(γ-glutamic acid) NP | 125I | Iodogen | 175 |
| PPP-type copolymers | 125I | Iodogen | 176 |
| Polyester-based NPs | 125I | Iodobeads | 177 |
| PLGA NPs | 125I | Iodobeads | 178 |
| PVP NPs | 125I | Chloramine-T | 179 |
| Iodination beads | 180 | ||
| 124I | Iodination beads | 180 | |
| PDPA NPs | 131I | Chloramine-T | 181 |
| Protein-based NPs | 125I | Iodogen | 182 |
| 131I | Iodogen | 182 | |
| Chloramine-T | 183 |
Besides iodine radionuclides, chloramine-T has been also used as mediator for efficient radiolabelling of dendrimers and polymeric NPs with 76Br providing RCYs greater than 95%.184,185 Other radio-halogenation reactions such as traditional nucleophilic or electrophilic substitutions have also been applied to nanomaterials for iodine radiolabelling as well as for radio-fluorination.186–189 Interestingly, as with chelator based methods, click-chemistry or biorthogonal reactions (Fig. 8D) have also recently been explored for the radio-halogenation of nanomaterials. These reactions are frequently fast, specific to certain prosthetic groups and regioselective allowing rapid and controllable radio-halogenation with high yields and stabilities.190 With this purpose, chemoselective oxime formation, alkyne-nitrone, copper catalysed azide–alkyne and azide-DBCO cycloadditions have been used for 18F, 123I and 125I radiolabelling of both organic and inorganic nanomaterials.191–197 As a main drawback, biorthogonal reactions require the control over the synthesis, characterisation and reactivity of two independent species, complicating their potential clinical translation.
11C methylation reactions can be also applied for the radiolabelling of nanomaterials (Fig. 16C). Sharma et al. reported the radiolabelling of iron oxide NPs using [11C]CH3I as a precursor to conduct N- and O-methylation on the coating of the NPs with poor RCY, but high RCS.132 Although this study represented a good proof-of-concept, the very short half-life of the radionuclide (20.4 min) does not seem to be suitable for biodistribution studies on nanomaterials that commonly show prolonged biological half-lives.
 | ||
| Fig. 17 Schematic representation of the radioisotopic exchange mechanism. | ||
A key advantage of this radiolabelling approach is its simplicity; however, few combinations of NP–radionuclide are truly effective with only a few examples in the literature of nanomaterials being radiolabelled by these methods. Homogeneous radioisotopic exchange between 19F and 18F has been reported as an attractive strategy for the radiolabelling of up-converting NPs (UCNPs). Two types of UCNPs with NaYF4 and NaGdF4 cores doped with Yb3+ and Er3+ have been investigated. NaYF4 (Fig. 18A) particles showed higher RCYs (∼92%) than NaGdF4 with moderate radiochemical yields up to 43% when radiolabelling is conducted at room temperature for 1–10 min. Both formulations reported high RCPs (>95%) with fast clearance from blood to liver and spleen for NaYF4:Y,Er (Fig. 18B). Although the high bone accumulation (up to 12% ID per g, Fig. 18C) found during in vivo studies strongly suggest radiochemical instability ([18F]F-fluoride is known to accumulate in bone, Table 5).198–201 Heterogenous exchange has been used on the radiolabelling of iron oxide NPs (IONPs) with 68Ga, quantum dots (QDs) with 68Ga and 64Cu and UCNPs with 153Sm.202–205 The method provided radiolabelled NPs with high RCY and purity. The mild and fast radiolabelling conditions required for 68Ga–QDs (37 °C for 15 min) or 64Cu–QDs (60 °C for 1 h, Fig. 18D) suggest a facile heterogeneous exchange on QDs and therefore, an appropriate radiolabelling strategy. In addition, in vivo PET biodistribution of 64Cu–QDs in tumour-bearing mice revealed passive accumulation of the particles in the tumour by EPR effect with liver and spleen excretion (Fig. 18E).204 As this biodistribution profile could be attributed to free 64Cu2+, the authors further conducted ICP measurements on excised tissues after the injection of non-radioactive QDs. The results indicated a linear correlation between the ex vivo gamma counter quantification and the ICP measurements, confirming the 64Cu–QDs biodistribution of the PET imaging. On the other hand, the harsh conditions for 68Ga–IONPs and 153Sm–UCNPs (T = 100–300 °C for 1–4 h) suggest that milder radiolabelling strategies may be more appropriate, particularly if heating results in changes of the physicochemical properties of these NPs.
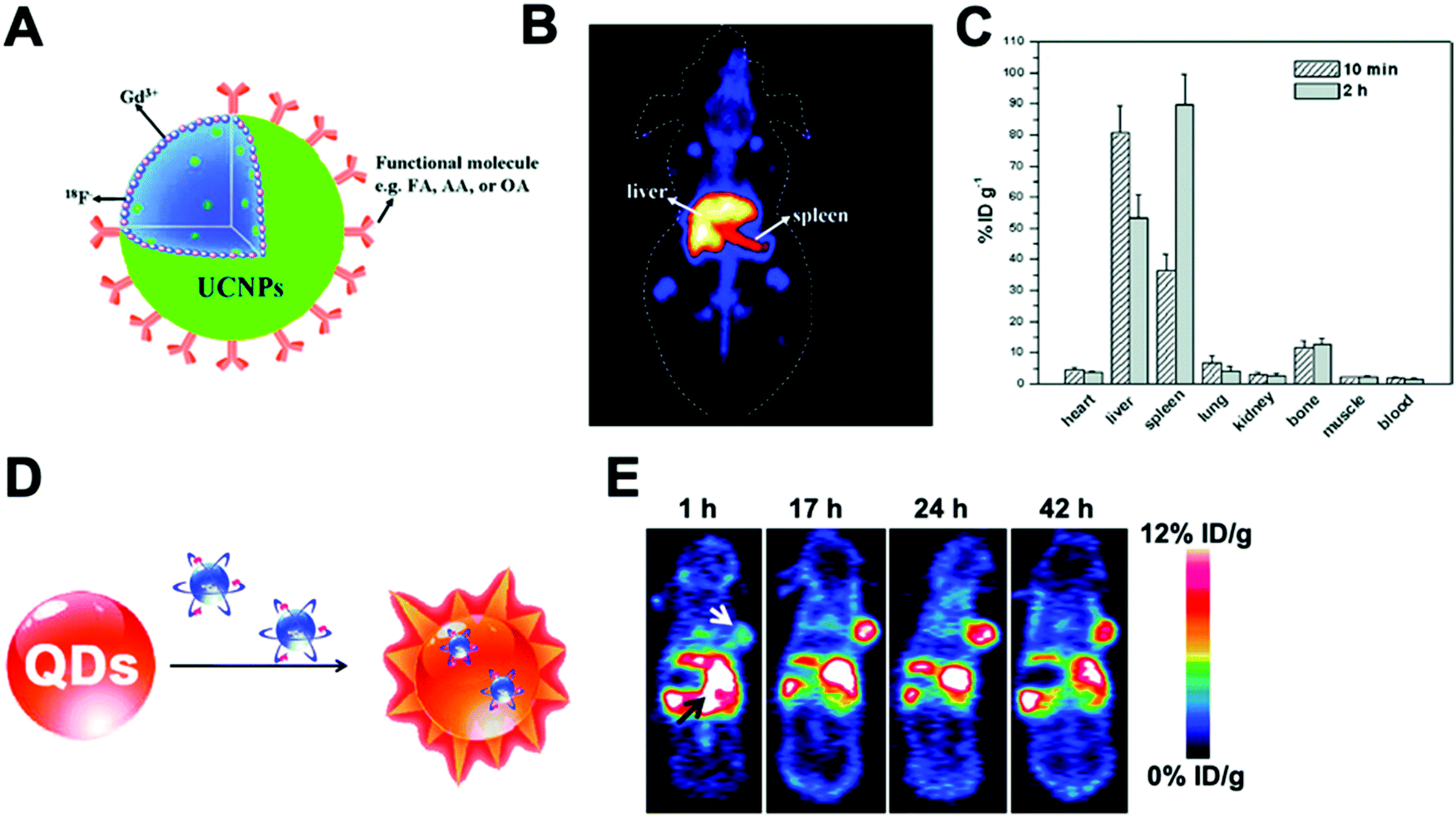 | ||
| Fig. 18 (A) Schematic of fluorine-18-labeled magnetic-upconversion functional nanocrystals. FA: folic acid; OA: oleic acid; AA: aminocaproic acid, (B) Kunming mice PET imaging 10 min postinjection of 18F-AA-Gd-UCNPs (200 μg mL−1), (C) biodistribution of 18F-AA-Gd-UCNPs at 10 min and 2 h postinjection; the data shown are based on five mice per group, (D) design of self-illuminating 64Cu-doped QDs, (E) representative whole-body coronal PET images of U87MG tumour-bearing mice at 1, 17, 24, and 42 h after intravenous injection of 250 μCi of 64Cu-doped QD580 (n = 3). White arrow, tumour area; black arrow, liver area. Adapted and reproduced with permission from ref. 198 and 204. | ||
 | ||
| Fig. 19 Schematic representation of the chemical adsorption strategy. | ||
This strategy, sometimes known as chemisorption, has been historically studied for other applications; mainly in catalysis and analytical chemistry to shed light on the mechanisms of interaction between metals and materials.206,207 Nevertheless, the first application for the radiolabelling of a nanomaterial appeared in 2013, where Cheng et al. described the chemical adsorption of various *As (* = 71, 72, 74, 76) radionuclides on the surface of a magnetite (IONP) NPs (Fig. 20A).208 In this case, the radiolabelling mechanism was attributed to the formation of stable As–Fe3O4 complexes where AsIIIO3 trigonal pyramids or AsVO4 tetrahedra may form on vacant tetrahedral spaces within the Fe3O4 octahedrally terminated (111) surface. The biodistribution of the *As–IONPs was studied by PET imaging in Balb/C mice after i.v. injection of free *As and *As–IONPs. The images showed a renal elimination for the free *As with high uptake in the bladder at 0.5 h and 3 h post-injection (Fig. 20B). Elimination through liver and spleen was observed for the *As–IONPs with significant signal in the bladder corresponding, most likely, to the in vivo desorption of *As from the NPs (Fig. 20C). After this work, several IONPs have been reported using the chemical adsorption strategy with a variety of other radionuclides. For example, feraheme/ferumoxytol NPs were successfully radiolabelled with different metallic radionuclides such as 89Zr, 64Cu and 111In with radiochemical yields between 66–93% and radiochemical purities greater than 98%.209 The greater RCY (93 ± 3%) was obtained using either [89Zr]Zr-oxalate or [89Zr]ZrCl4 at pH = 8 and 120 °C (Fig. 20D). With a RCS > 90% in human plasma, the biodistribution studies by PET/CT in wild-type B6C3F1/J mice revealed a circulation time in blood between 6–8 hours with final accumulation in liver, spleen and lymph nodes (high uptake in mesenteric lymph nodes) (Fig. 20E).
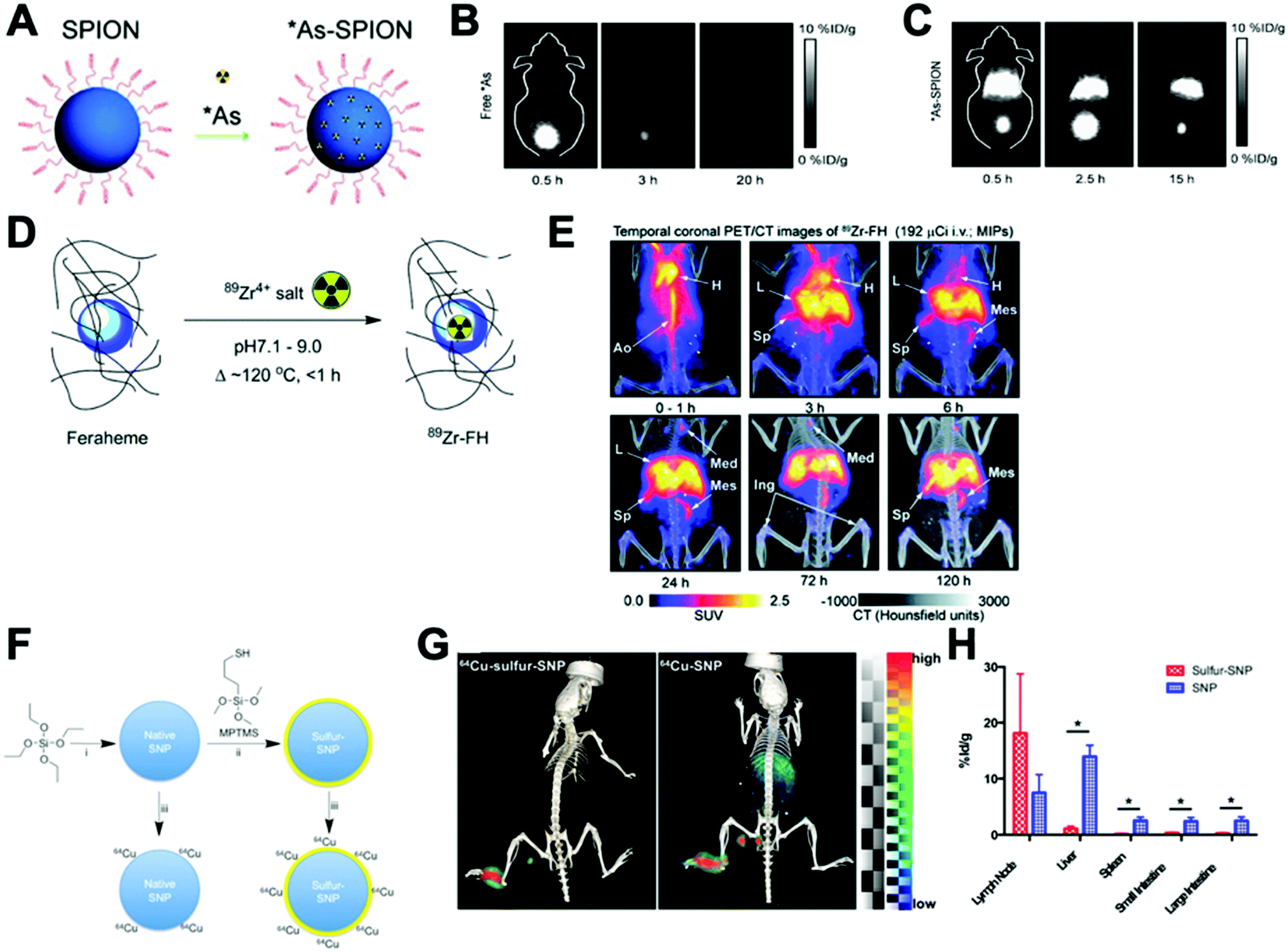 | ||
| Fig. 20 (A) Chelator-free synthesis of *As–SPIONs, (B) serial in vivo PET images of free *As at different time points after intravenous injection into mice, (C) serial in vivo PET images of *As–SPION at different time points after intravenous injection into mice, (D) reaction of FH with 89Zr4+ ion salts (oxalate or chloride) to give radiolabeled 89Zr–FH, (E) temporal PET/CT maximum intensity projection (MIP) images recorded between 0–120 h post-i.v. injection of 89Zr–FH in B6C3F1/J wild-type mice. Ao = aorta; H = heart; L = liver; Sp = spleen; Mes = mesenteric lymph node; Ing = inguinal lymph, (F) reaction schematic. Although native SNP (blue) stably bind hard oxophilic radiometals such as 89Zr and 68Ga, thiol-functionalization (yellow) of SNP allows stable retention of soft, sulfur-avid copper-64. (G) PET/CT and biodistribution of 64Cu–sulfur–SNP and 64Cu–SNP injected into the footpad allow lymph node imaging with little systemic uptake at 14 h post-injection, (H) quantitative ex vivo biodistribution values. Adapted and reproduced with permission from ref. 208–210. | ||
Additionally, silica NPs have showed particularly high affinities of oxophilic cations – such as 68Ga, 111In, 177Lu, 90Y and 89Zr – towards the silanol groups on the NP surface (see Section 4.6.3). This allows simple, fast and robust radiolabelling of silica-based nanomaterials.211 However, this work described a poor RCS for the radiolabelling with 64Cu. To overcome this limitation, the authors reported the functionalisation of the silica NPs (SNP) to introduce thiol groups on the surface (sulfur–SNP) (Fig. 20F).212 This brief modification increases the RCS from 34.9 ± 5.8% for SNP to 90.9 ± 5.8% for the sulfur–SNP. These results were confirmed by in vivo PET/CT studies in athymic mice injected in the footpad (Fig. 20G). Whilst SNP showed accumulation in liver, spleen and intestines due to the free 64Cu2+, sulfur–SNP were only observed in the footpad and draining lymph nodes as confirmed in the quantitative ex vivo biodistribution (Fig. 20H).
A key drawback of the chemical adsorption strategy is the high temperatures required for the radiochemical reactions, that can be limiting for heat-sensitive NP formulations. Additionally, the strength of the chemical interaction between the radionuclide and the nanomaterial surface must be carefully considered to avoid radiochemical stability issues, such as those reported in the radiolabelling of Fe3O4@Al(OH)3 NPs with 18F.213–215 In these studies, that relied on the formation of the theoretically strong Al–18F bonds, it was found that significant release of 18F-fluoride occurred in vivo, as evident from the increasing high signal from bone reported by Cui et al.215
 | ||
| Fig. 21 Schematic representation of the radiolabelling strategy involving physical interaction between materials and radionuclides. | ||
Although plausible, this methodology has not been widely explored for the radiolabelling of nanomaterials with only few examples reported in the literature. A key example of this strategy reported the encapsulation of 64Cu into the cavity of single-wall carbon nanotubes (SWCNTs).216 The radiolabelled SWCNTs presented quantitative RCP and high RCS in PBS after 24 h of incubation. However, the RCS decreased to 63% in 50% mouse serum confirming the poor stability of the radiolabelling. This is a good example on the application of the well-known loading capabilities of nanotubes to increase the specific molar activities of radionuclides in nanomaterials. Although the exploitation of physical properties of nanomaterials as the radiolabelling driven force is an interesting approach, it is currently not extensively used due to the hypothetical low radiochemical stability issues – as well as the lack of appropriate materials amenable to fully exploit these radiolabelling mechanisms.
4.5 Radiolabelling of organic nanomaterials
In the previous sections we have outlined the main methodologies of incorporating radionuclides into nanomaterials. We will now review the radiolabelling of specific types of nanomaterials, linking them with the different radiolabelling methods discussed above, and the potential benefits/drawbacks of each approach. This section will focus on organic-based nanomaterials and will be followed by inorganic nanomaterials in Section 4.6.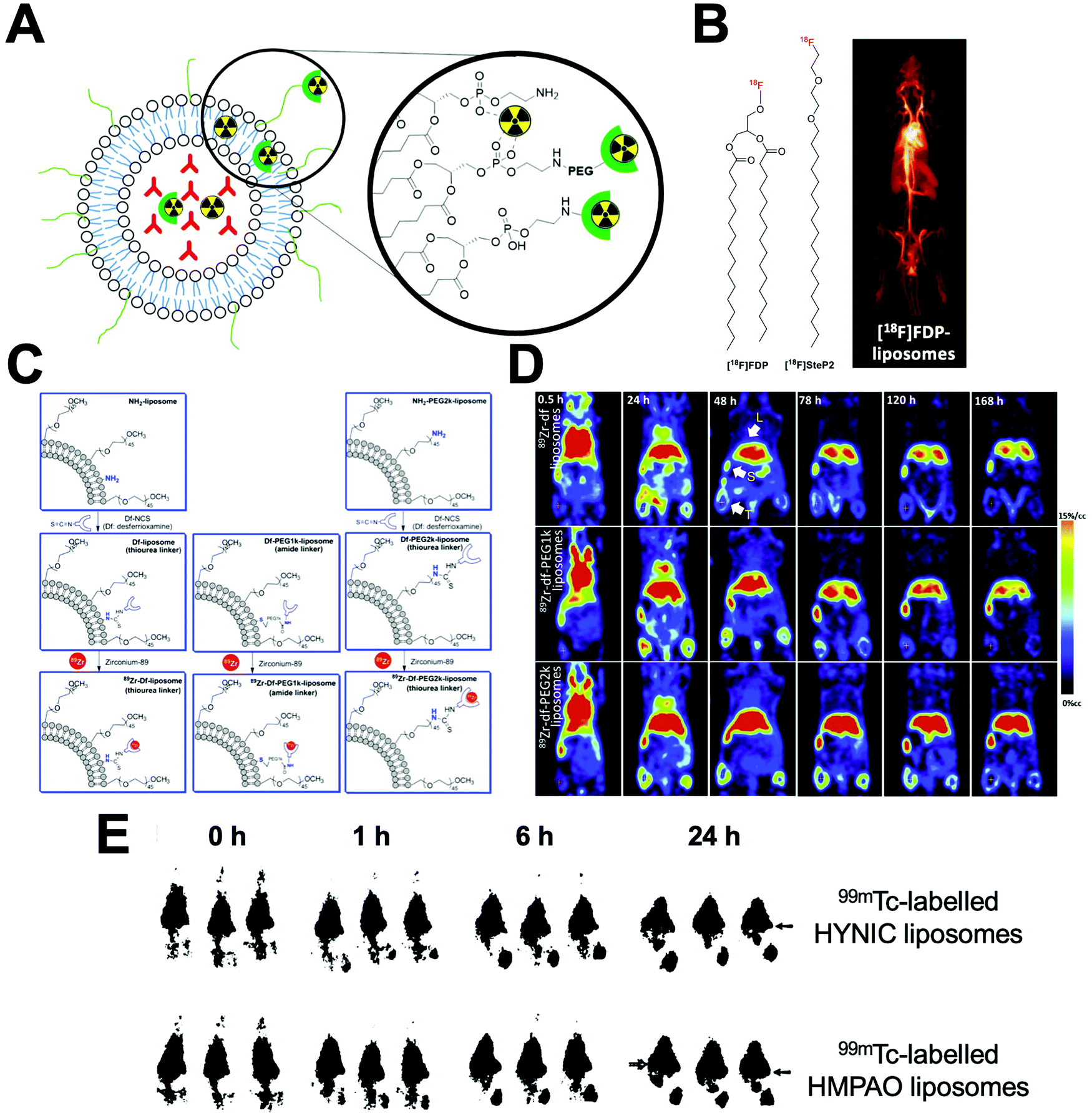 | ||
| Fig. 22 (A) Schematic representation of the different methods to radiolabel liposomes. Radionuclides can be bound to the surface using chelators or chelate-free or trapped intra-liposomally. (B) Chemical structures of [18F]FDP and [18F]SteP2. (left) PET image of [18F]FDP liposome in a rat model during a 90 min scan (right). Adapted with permission from Marik et al.218 (C) Schematic for the liposomal-labelling method with 89Zr, with different PEG chain lengths between the DFO chelator and liposome surface, used by Seo et al. (D) PET images at indicated time of 89Zr liposomes in mammary tumour bearing mice with no PEG chain (top), a 1k PEG chain (middle), and a 2k PEG chain (bottom) between the DFO chelator and liposomal surface. Clear differences in tumour and liver uptake can be observed. Adapted with permission from Seo et al.219 (E) Gamma camera images of 99mTc-labeled HYNIC liposomes (top row) and 99mTc-labelled HMPAO liposomes (bottom row) in rats with S. aureus abscess in calf muscle. Adapted from Laverman et al.220 | ||
| Radiolabelling method | Radioisotope | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Surface non-chelator | 99mTc | Direct labelling via SnCl2 reduction | 221–225 |
| 89Zr | Chelate free | 226 | |
| 18F | [18F]FDP | 218 and 227–229 | |
| [18F]-Fluorocholesteryl ether | 230 | ||
| [18F]SteP2 | 231–234 | ||
| CuAAC click reaction | 235 and 236 | ||
| Surface chelator-based | 99mTc | DTPA-sterylamine | 237 |
| DTPA | 238–244 | ||
| DTPA via99mTc-tricarbonyl | 244 | ||
| HYNIC + tricine co-ligand | 220 and 245–247 | ||
| 2-Iminothiolane via99mTc-tricarbonyl | 248 | ||
| 67Ga | DTPA-sterylamine | 237 | |
| 111In | DTPA | 244 and 249–255 | |
| 68Ga | DTPA | 244 | |
| NODAGA | 256 | ||
| 64Cu | BAT | 257–261 | |
| TETA | 262 and 263 | ||
| CB-TE2A | 262 | ||
| DOTA | 264 | ||
| DO3A | 265–267 | ||
| 52Mn | DO3A | 267 | |
| 177Lu | DTPA | 244 | |
| 90Y | DTPA | 268 | |
| 166Ho | DTPA | 269 | |
| 89Zr | DFO | 219 and 270–274 | |
| Complex trapping | 99mTc | DTPA complex during formulation | 275–278 |
| 111In | DTPA complex during formulation | 279 | |
| 159Gd | DTPA complex during formulation | 280 | |
| 225Ac | DOTA complex during formulation | 281 | |
| Ionophore-based (chelator binding) | 111In | A23187 (NTA) | 282 and 283 |
| Oxine (NTA) | 284 and 285 | ||
| Acetylacetone (NTA) | 286 | ||
| Tropolone (NTA) | 287 | ||
| Oxine (DFO) | 288 and 289 | ||
| Oxine (DTPA) | 254, 279 and 290–293 | ||
| 90Y | A23187 (DTPA) | 294 | |
| 67Ga | Oxine (DFO) | 295 and 296 | |
| Tropolone (DFO) | 295 and 296 | ||
| 177Lu | Oxine (DTPA) | 297 | |
| 64Cu | 2-HQ (DOTA) | 298–300 | |
| 52Mn | Oxine (DOTA) | 267 | |
| 8HQ-2Cl (DOTA) | |||
| 8HQ-2I (DOTA) | |||
| 225Ac | Oxine (DOTA) | 301 and 302 | |
| A23187 (DOTA) | |||
| 89Zr | Oxine (DFO) | 303 | |
| 2HQ (DFO) | |||
| Ionophore based (drug binding) | 89Zr | Oxine | 304 and 305 |
| 52Mn | Oxine | 304 and 306 | |
| 64Cu | 2HQ | 304 | |
| 111In | Oxine | 307 | |
| Unassisted loading (chelator binding) | 64Cu | DOTA | 308–311 |
| Remote loading | 99mTc | HMPAO | 220, 312 and 313 |
| DISIDA | 314 | ||
| BMEDA | 315–317 | ||
| 186Re | BMEDA | 318 and 319 | |
| 188Re | BMEDA | 320–323 | |
| 64Cu | 4-DEAP-ATSC | 324–327 | |
| 124I | Amino diatrizoic acid | 328 | |
| 125I | Amino diatrizoic acid | ||
The direct attachment of radionuclides to the surface of liposomes – without the use of chelators – was first described by Richardson et al. who showed that liposomes can be directly labelled with 99mTc after reduction of pertechnetate using stannous chloride (SnCl2) as a reducing agent.221–225 To the best of our knowledge, the exact binding site of 99mTc is not known; however, one likely possibility is chelation by the phosphonate groups on the liposome phospholipid surface. Labelling efficiencies (RCY) of >97% could be achieved after 15 min at room temp. However, there have been reports of in vivo instability of the radiolabel using this method.329 This direct labelling approach was also used by Abou et al. with 89Zr. However, this interaction was shown to be weak, resulting in low serum and in vivo stability.226
Non-chelator labelling of liposomes has also been achieved with radiofluorine-based agents. Several groups used 3-[18F]fluoro-1,2-dipalmitoylglycerol ([18F]FDP, Fig. 22B),218,227–229 which was added during liposomal preparation. Radiolabelled liposomes could be prepared in ca. 1 h with a RCY of 70%. In vivo stability was shown with no observable bone uptake (a consequence of defluorination).218 Alternatively, Urakami et al. synthesised an amphiphilic probe, 1-[18F]fluoro-3,6-dioxatetracosane ([18F]SteP2, Fig. 22B).231–234 The long alkyl chain on the probe allowed intercalation with the lipid bilayer on the liposome surface allowing a LE and stability in serum (after 30 min) of >80%.232
Chelator-based radiolabelling of liposomes is primarily performed by the attachment of a chelator onto the liposome surface, either to the phospholipid or PEG chains present on LCLs (Fig. 22A). Liposomes pre-formulated with DTPA conjugated to the phospholipid on the liposome have been widely used with several different radioisotopes; particularly with 99mTc – however, low serum and in vivo stability was observed using this method.238–240 Several reports have also shown that DTPA functionalised liposomes allow >95% RCY with 111In under mild conditions (25–37 °C, up to 1 h).244,249–255 Interestingly, Helbok et al. reported a direct comparison of the radiolabelling of DTPA-functionalised PEGylated liposomes with several different radionuclides.244 The liposomes were labelled with 99mTc (using both [99mTc][TcO4]− and [99mTc][Tc(CO)3]+ and 111In), with the latter showing the most favourable labelling (>95% LE). Labelling with [99mTc][TcO4]− was consistently lower (75%), and >80% RCY was achievable using [99mTc][Tc(CO)3]+ but only with 50-fold more liposomes. The authors also demonstrated radiolabelling with 68Ga and the therapeutic isotope 177Lu using the same formulation; achieving >95% and >80% RCY respectively – however higher concentrations of liposomes were necessary for the latter.244
A key consideration when radiolabelling liposomes via the surface is the biodistribution of these radiolabelled phospholipids in vivo, which may occur after tissue uptake/destruction of the liposomes. This was explored by Seo et al. who synthesised liposomes functionalised with the 64Cu-specific chelator TETA (Fig. 6).257–261 This allowed >80% LE after 1 h at room temp, with >90% stability in mouse serum for 48 h. Interestingly, the ex vivo biodistribution at 48 h of the liposomes compared to the 64Cu–PEG–lipid, showed liver uptake of the latter was roughly 3-fold higher than the liposomes.257 This uptake of lipids, that may arise as a result of in vivo liposome decomposition, should be carefully considered when tracking liposomes, as it may lead to misinterpretation of the amount of liposomes present in the liver.
Furthermore, several reports have shown that the biodistribution of radiolabelled liposomes can easily be altered solely based on the position of the radiocomplex, which could be viewed as a drawback to surface labelling of liposomes. Seo and collaborators looked at labelling using 64Cu complexes of 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid (TETA) and 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo-(6.6.2)hexadecane (CB-TE2A, Fig. 6).262 Intriguingly, the authors showed that attaching the complex to either PEG or non-PEGylated lipids altered the biodistribution, with 5% higher hepato-splenic uptake occurring after 48 h.262 This work was later expanded by Seo et al. who performed surface labelling with 89Zr using desferrioxamine (DFO) as a chelator, which allows radiolabelling at neutral pH with only mild heating.219,270–274 The authors compared the effect of increasing PEG-length between the liposomal surface and the 89Zr–DFO complex.219 Three formulations were prepared with DFO attached directly to the lipid or with a 1k or 2k PEG spacer (Fig. 22C). No significant differences in terms of % RCY, stability or blood half-life were observed. However, image-based analysis showed significantly higher tumour, liver and spleen uptake when using a 2k PEG spacer, over 7 d compared to the other two formulations (Fig. 22D). This highlights how small modifications in chelator position on the surface of radiolabelled liposomes can affect their biodistribution and pharmacokinetics.
Due to these potential drawbacks of chelator-based surface labelling, radiolabelling of liposomes is sometimes performed within the liposomal core. This approach can, in theory, increase the stability of the radiolabel as it is no longer present on the surface where it can interact with chelating compounds (e.g. serum proteins). However, the radiolabelling procedure can often become more complex; often involving the prior synthesis of a radiotracer to incorporate radionuclides inside the liposomes (see Section 4.3). Some of the earliest studies achieved this by simply encapsulating a radiometal complex with DTPA inside the liposomal core during formation of the liposomes (see Section 4.2.2). This was first done with 99mTc,275–278 and later with 111In279 and 159Gd–DTPA,280 as well as encapsulating the DOTA complex of 225Ac.281 One drawback of this method is the longer, more complicated radiosynthesis needed (especially relevant when using short-lived isotopes).
The most widely used ‘intra-liposomal’ radiolabelling method is the use of ionophores to transport radiometals across the lipid bilayer to encapsulated chelators (Fig. 10A and Section 4.3.1). The first example of this was reported by Gamble and collaborators who used the calcium ionophore A23187 (Fig. 11A) to transport 111In inside the liposomal core where it was chelated by encapsulated nitrilotriacetic acid (NTA, Fig. 6) allowing >90% RCY.282,283 Since then, several different ionophore and encapsulated chelator combinations have been reported (Table 8). A key study by Harrington and collaborators reported using the ionophore 8-hydroxyquinoline (oxine; Fig. 11A) to radiolabel liposomes containing DTPA with 111In, which allowed >90% LE after 15 min incubation and high serum stability for up to 10 days.290–292 An important study by Van der Geest et al. compared this ionophore-based radiolabelling with chelator-based surface labelling with 111In using DTPA–DSPE liposomes – along with the labelling of empty liposomes (without DTPA).254 Labelling efficiencies and serum stability (after 48 h) of >95% were reported using both radiolabelling methods, whereas the empty liposomes showed lower LE (62%) and serum stability (68%). Interestingly, when comparing the in vivo distribution of the formulations in mice, the surface-labelled liposomes showed significantly higher liver uptake over 72 h – compared to the oxine-DTPA liposomes.254 This may indicate that release of [111In]In–DTPA–DSPE from the liposomes is occurring, suggesting lower in vivo stability, as [111In]In–DTPA is rapidly cleared,291 whereas [111In]In–DTPA–DPSE (released from liposomes during degradation) will likely accumulate in the liver (vida supra).
A key consideration when using ionophore-based methods is the intra-liposomal pH; which can affect the rate of radiometal release, and subsequent transchelation. Petersen et al. used the ionophore 2-hydroxyquinoline (2HQ, Fig. 11A) to radiolabel DOTA-encapsulated liposomes with 64Cu, which had different intra-liposomal pHs.298–300 Liposome loading was >95% and 70% for pH 4 and 5.9 respectively, suggesting the complexation by DOTA was affected.298 This concept was explored further by Jensen et al. who used several oxine derivatives to load 52Mn into DOTA encapsulated liposomes.267 Labelling efficiencies above 90% could be achieved with an intraliposomal of pH 4 when using oxine and 5,7-dichloro-8-hydroxyquinoline (8HQ-2Cl, Fig. 11A), but increasing the pH to 7.8 led to a large reduction in labelling using oxine (ca. 30–70% LE) whereas this was not observed for 8HQ-2Cl. Therefore, the internal pH will not only affect the chelation by the internalised ligand, but also the dissociation of the ionophore complex used.
Our group showed that radiolabelling of liposomes is possible without the need for incorporated chelators and therefore without having to chemically modify the formulation.304–307 This is based on the metal-chelating properties of certain drug molecules (Fig. 23A and B), that are present in high concentrations inside the liposome, and able to bind the radionuclide after ionophore-mediated transport across the lipid bilayer (Fig. 23A). For example, manganese complexes of doxorubicin via hydroxyl and carbonyl groups on the doxorubicin backbone have been previously reported,330,331 and IR spectroscopy showed that Zr4+ interacted with the carboxylate present on methylprednisolone hemisuccinate.305 These interactions allowed us to radiolabel a variety of liposomal nanomedicines with 111In, 64Cu, 89Zr and 52Mn and image them longitudinally (Fig. 23C–F).304–307 This method overcomes the need to incorporate liposomes with a chelator which may limit its use to validate pre-formulated, commercially available liposomal nanomedicines. However, the extent of radiolabelling using this method will always be limited by the strength of interaction between the radiometal and the drug inside the liposomal formulation.304,305 Furthermore, the lack of a stable chelator means that release of the ‘free radiometal’ can occur after destruction of the liposomes. In particular, radioactive isotopes of endogenous metals, such as 52Mn and 64Cu, may be more susceptible to trafficking out of the tissues and into the bloodstream, resulting in secondary uptake in other organs (Fig. 23F). Specifically, in the case of 64Cu and 52Mn it may be difficult to distinguish between ‘free radiometal’ uptake from that of liposomal uptake in the liver and even in tumours.332,333 This has been shown to be less of an issue when labelling with 89Zr (a non-endogenous metal), which almost exclusively shows uptake in the bone (Fig. 23C and E).334
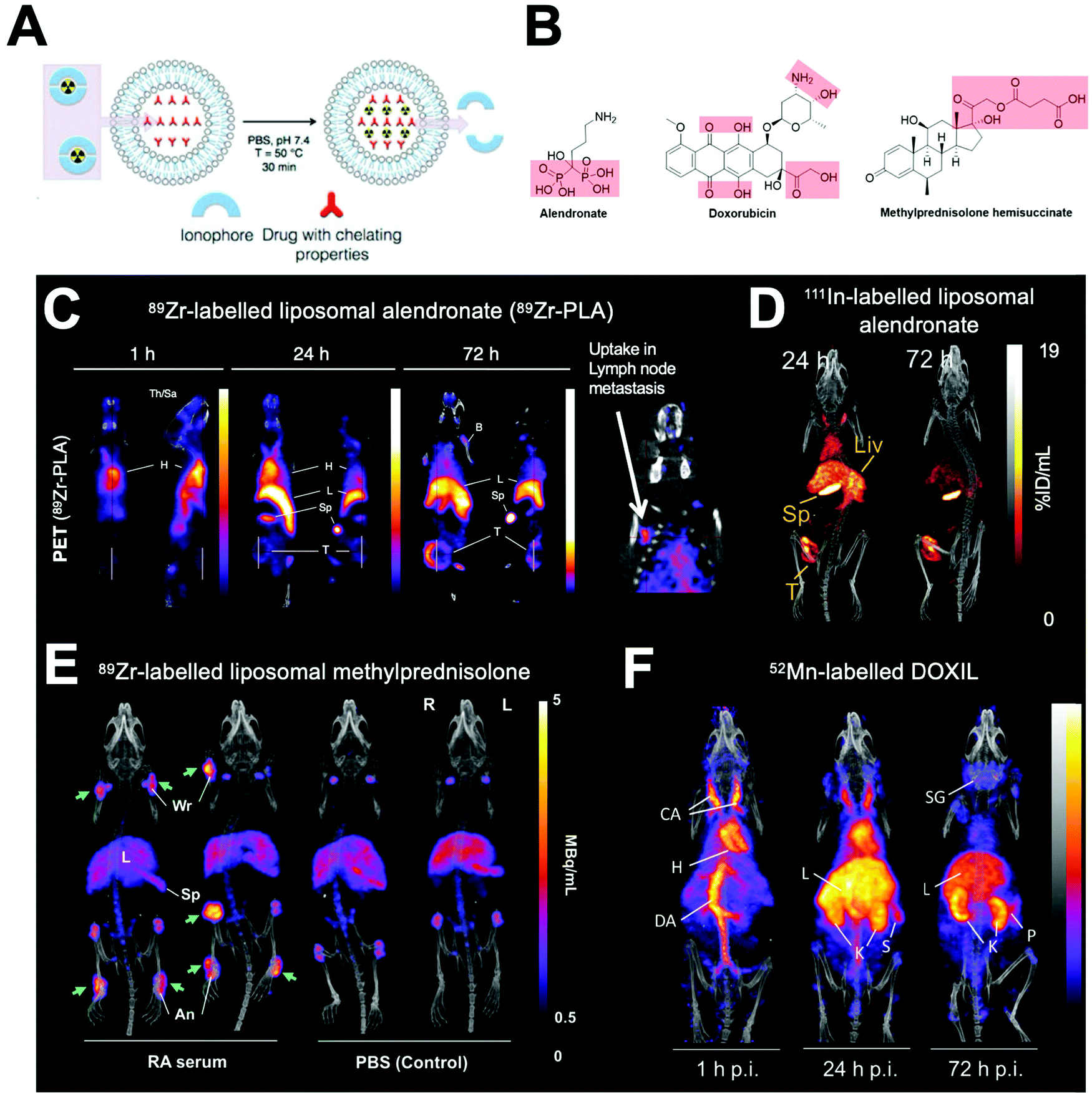 | ||
| Fig. 23 (A) Schematic showing the ionophore-based method for radiolabelling liposomes using the chelating properties of drugs. (B) Chemical structures of drugs incorporated inside liposomes capable of chelating radiometals. (C) PET/CT images of PEGylated liposomal alendronate (PLA) labelled with 89Zr in a mouse model of metastatic breast cancer, showing long circulation and gradual uptake in primary tumour (T) and lymph node metastasis. Adapted from Edmonds et al.304 (D) SPECT-CT images of 111In-labelled PEGylated liposomal alendronate in a breast cancer model. Adapted from Man et al.307 (E) PET/CT images of PEGylated liposomal methylpredinisolone hemisucinate labelled with 89Zr in a model of arthritis (left) and control animals (right). High uptake in arthritic joints denoted by green arrows. Adapted with permission from Gawne et al.305 (F) PET/CT images in healthy mice of DOXIL radiolabelled with 52Mn. Images at 72 h show release and redistribution of 52Mn from liposomes after tissue uptake. Adapted with permission from Gawne et al.306 | ||
Interestingly, Henriksen et al. showed that use of an ionophore to transport radiometals across the lipid bilayer of liposomes is not always necessary. They found that by incubating unchelated 64Cu2+ with liposomes containing a DOTA chelator allowed >90% RCY after 30–60 min at 55 °C.308–311 This ‘unassisted loading’ of 64Cu was proposed to occur due to the formation of a steep copper gradient, across the lipid membrane, by the chelation of non-radioactive copper inside the liposomal core by the DOTA chelator. This gradient then causes diffusion of 64Cu2+ into the liposome where it is trapped by chelation by the DOTA ligand. The increased simplicity of this technique is clearly beneficial, and additionally removes the need for ionophores, which are known to have a variety of biological activities.335 However, it may not be applicable to other radionuclides and more studies are required to fully understand the exact mechanism that allows charged hydrophilic ions such as Cu2+ to cross lipid bilayers.
The radiolabelling of liposomes can also be achieved by the remote loading of radiopharmaceuticals into the liposomal core (Section 4.3.2). Generally speaking, a neutrally charged, lipophilic, radiopharmaceutical crosses the lipid bilayer of liposomes into the aqueous core where it becomes protonated and trapped as a more hydrophilic form (Fig. 10B and Section 4.3.2). An early example of this was reported by Rudolph and collaborators who used 99mTc-labelled hexamethylpropyleneamine oxime (HMPAO, Fig. 11B) to radiolabel liposomes encapsulating with glutathione (GSH) which was necessary to allow high labelling of the liposomes (>90% LE).220,312,313 The authors postulated the complex would undergo reduction by interaction with glutathione, allowing trapping of the agent. Cao et al. also reported the GSH-dependent trapping in liposomes of the 99mTc complex of diisopropyl iminodiacetic acid (DISIDA, Fig. 11B).314 Laverman et al. later compared remote loading of [99mTc]Tc-HMPAO with surface radiolabelling using HYNIC conjugated to the lipid bilayer of liposomes.220 No difference in serum stability after 48 h, using the two methods was reported. However, in vivo tracking of the labelled liposomes. showed that kidney uptake was 3-fold higher after 24 h for HMPAO-labelled liposomes, suggesting release of [99mTc]Tc-HMPAO from the liposomes (Fig. 22E). A similar method was described by Bao and collaborators using the chelator N,N-bis(2-mercaptoethyl)-N′,N′-diethyl-ethylenediamine (BMEDA, Fig. 11B) for the remote-loading of liposomes with 99mTc,315–317186Re,318,319 and 188Re.320–323 Labelling efficiencies with the 99mTc complex were ca. 37% LE; with the presence of glutathione within the liposomal core allowing increased stability (>80%) in serum over 72 h compared with empty liposomes (<35% stability).315 However, in both of these examples, the need to encapsulate glutathione within liposomes to facilitate radiolabelling is a potential drawback, compared with other methods avoiding the need for modifications.
An excellent method by Lee et al. reported a 64Cu complex of diacetyl 4,4′-bis(3-(N,N-diethylamino)propyl)thiosemicarbazone (4-DEAP-ATSC, Fig. 11B) for the remote loading of liposomal nanomedicines without modification.324–327 [64Cu][Cu(4-DEAP-ATSC)] allowed >90% LE of several liposomal formulations after 10 min at 65 °C. The radiolabelled liposomes showed high serum stability >99% after 48 h. The authors compared the ex vivo biodistribution of the radiolabelled liposomes with the [64Cu][Cu(4-DEAP-ATSC)] complex and free 64Cu2+ and showed that the liposomes and complex had similar uptake in the liver – as well as that [64Cu][Cu(4-DEAP-ATSC)] and ‘free 64Cu’ had similar pharmacokinetics. Copper–bisthiosemicarbazone complexes are not stable in vivo,336 and thus any [64Cu][Cu(4-DEAP-ATSC)] released from the liposomes will likely decompose and release free 64Cu leading to accumulation in the liver 64Cu in its free form. Furthermore, small amounts (ca. 3% ID per g, 24 h p.i.) of tumour uptake of [64Cu][Cu(4-DEAP-ATSC)] was observed,324 which matches previous observations that 64Cu and its bisthiosemicarbazone complexes are known to accumulate in tumours.336,337 Hence, the release of the [64Cu][Cu(4-DEAP-ATSC)] complex – and indeed other remote loading complexes – from liposomes is a key consideration when using this method as it may distort tumour and liver uptake values of the radiolabelled liposomes. Finally, Engudar et al. reported a novel radioiodinated compound, amino diatrizoic acid (ADA, Fig. 11B), for the remote loading into liposomes.328 [125I]-ADA and [124I]-ADA could be incorporated into liposomes with >70% LE after 2 h. Good stability of the radiolabel in vivo was demonstrated by just 1% ID per g of the radioactivity in thyroid present after 72 h.
 | ||
| Fig. 24 (A) Schematic representation of the various methods used to radiolabel extracellular vesicles. Chelators can be attached to the surface via conjugation to phospholipids or protein residues, or radionuclides can be incorporated inside via ionophores. (B) Representative MIP PET/CT images of radiolabelled 89Zr-labelled PANC1 exosomes in healthy mice; showing signal in liver, spleen, several lymph nodes (arrowheads), and brain; adapted from Khan et al.340 (C) PET/MRI of 64Cu-labelled sEVs in healthy mice. Adapted from Banerjee et al.341 (D) PET-CT coronal images obtained at different time points of 124I-labelled EVs in healthy mice (top) and free [124I]NaI (bottom). Release of iodine can be seen at later timepoint, resulting in thyroid signal. Adapted from Royo et al.342 | ||
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 111In | DTPA | 343 |
| 64Cu | DOTA | 344 | |
| DO3A | 341 | ||
| Non-chelator | 125I | (3-125I-Iodobenzoyl)norbiotinamide | 345 and 346 |
| 124I | Iodination tube | 342 | |
| 131I | Iodination tube | 347 | |
| 99mTc | Via 99mTc tricarbonyl | 348 | |
| Direct labelling with SnCl2 reduction | 349–351 | ||
| Ionophore-based | 111In | Tropolone | 343 |
| Oxine | 352 | ||
| 89Zr | Oxine | 340 | |
| Remote loading | 99mTc | HMPAO | 353 |
Similarly to cells, a key consideration for radiolabelling cell-derived EVs, is that proteins on their surface can be utilised for functionalisation (Fig. 24A). Hence several groups have reported chelator-based labelling of the surface of exosomes. Shi et al. reported that the bifunctional chelator p-SCN-Bz-NOTA could be conjugated to the surface of EVs via free amines present on the surface membrane. This allowed >95% RCY and high serum stability (>80% at 24 h).344 Similarly, Banerjee et al. conjugated a DO3A-maleimide to the surface of EVs via free thiols present. However, RCYs were relatively low (ca. 16–25%), and in vivo PET imaging showed consistently high uptake in the bladder which peaked at 25% ID per g at 3 h; indicating that release of the bioconjugate from the EVs may have occurred (Fig. 24C).341 The presence of proteins on the EV surface also allows the direct radioiodination via electrophilic aromatic substitution on tyrosine residues (Fig. 24A).342,347 However, in each reported example uptake of radioactivity in the thyroid was observed, suggesting instability of the radiolabel (Fig. 24D). Hence, direct labelling of EVs with radioiodine may not be as appropriate compared with other methods. As with liposomes, several groups have reported the chelate-free direct labelling of different extracellular vesicles (EVs)/exosomes with reduced 99mTc.349–351 RCYs >95% were consistently reported with high serum stability (>90%) reported up to 48 h. In vivo SPECT/CT imaging showed that this labelling method was stable with minor uptake in the thyroid compared with pertechnetate.349 Taking into account the important role that surface proteins have in the biological behaviour of EVs, a potential disadvantage of targeting these proteins for radiolabelling EV surface proteins is the possibility of affecting their structure and function.
Due to their lipid bilayer, EVs are also capable of being radiolabelled using radio-ionophores, with the radiometal binding to proteins in the exosome core (Section 4.3.1 and Fig. 24A). This was first reported by Smyth et al. who used the [111In]In-oxine methodology to label PC3 and MCF-7 cell derived exosomes with labelling efficiencies between 67–81%.352 In a similar study, Faruqu et al. radiolabelled exosomes using the 111In-tropolone radio-ionophore complex, and compared this to labelling using a DTPA chelator conjugated to the surface.343 The radio-ionophore method was shown to be inferior to the surface labelling both with regards to radiolabelling and serum stability. 111In-tropolone labelled exosomes with just 4% LE and demonstrated only 14% serum stability after 24 h. This may be the result of the relatively high stability of 111In-tropolone that may have not dissociated inside the EVs. Finally, Hwang et al. reported the labelling of exosomes using remote loading with [99mTc]Tc-HMPAO which was facilitated by the presence of GSH inside the EVs.353 The radiolabelled EVs, had high (ca. >90% stability) in serum up to 5 h. However, in vivo SPECT/CT and ex vivo biodistribution of the labelled EVs showed uptake in the salivary glands (ca. 15% ID per g after 3 h), suggesting release of 99mTc from the EVs occurred.353
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. | |
|---|---|---|---|---|
| Protein-based nanoparticles | Non-chelator | 99mTc | SnCl2 reduction | 356 and 357 |
| Via 99mTc tricarbonyl | 358 | |||
| 125I | Iodogen | 182 | ||
| 131I | Iodogen | |||
| Chloramine-T | 183 | |||
| Chelator-based | 99mTc | MAG3 chelator | 359 | |
| Porphyrin | 360 and 361 | |||
| 111In | DTPA | 362 | ||
| 67Ga | NOTA | 363 | ||
| High-density lipoprotein (HDL) NPs | Chelator-based | 89Zr | DFO chelator | 28 and 364–370 |
| Viral nanoparticles (capsids) | Non-chelator | 125I | Iodogen | 371 |
| 124I | Iodogen | 372 | ||
| Bolton–Hunter reagent | ||||
| Chelator-based | 64Cu | NO2A dendrimer | 373 | |
| Bacteriophages | Non-chelator | 125I | Iodogen | 374 |
| Chelator-based | 111In | DOTA–biotin | 375 | |
| DTPA–avidin | 375 and 376 | |||
| 99Tc | MAG3 | 377 and 378 | ||
| HYNIC and tricine co-ligand | 379 and 380 | |||
| 64Cu | DO3A | 381 and 382 | ||
| DOTA | 381 | |||
| NOTA | ||||
| NO2A | ||||
A large portion of the radiolabelling of protein-based NPs has been carried out with serum albumin (SA) NPs. Jain et al. reported the direct radiolabelling of SA NPs using 99mTc via SnCl2 reduction which allowed 98% LE and 90% stability in PBS up to 24 h.356 A couple of groups reported the 99mTc labelling of the SA NPs conjugated with porphyrin photosensitising agents.360,361 Both studies demonstrated high labelling efficiencies >90%, however, no evidence was provided showing 99mTc was bound to the porphyrin, as opposed to directly to the albumin. The direct radioiodination (Fig. 25A) of SA particles has also been reported by Yi et al. using 125I and 131I for SPECT/CT and therapy respectively.182 However, no radiolabelling yields were reported, and SPECT/CT in mice showed large amounts of thyroid uptake at 3 d p.i. suggesting deiodination from the NPs (Fig. 25B).182 A chelator-based method for SA NP radiolabelling was reported by Woods et al. who conjugated albumin with p-BCS-Bz-DTPA which was then labelled with 111In. This was then used to synthesise the albumin NPs with an efficiency of 67%, showing >97% stability in serum for 48 h.362 Despite this excellent in vitro RCS, the long radiolabelling procedure involving the synthesis of labelled albumin followed by formulation of the particles (overnight) is a potential drawback.
 | ||
| Fig. 25 (A) Schematic representation of the methods used to radiolabel protein-based nanoparticles. Chelators can be conjugated to the nanoparticles via amine or thiol residues on the proteins, or tyrosine residues can be radiohalogenated. (B) SPECT/CT images of 125I-HSA in a mouse tumour model. Adapted from Yi et al.182 (C) CT (left) and PET/CT fusion (right) images of 89Zr-apoAI-DFO-HDL (top) and 89Zr-PL-DFO-HDL (bottom) in mice bearing orthotopic 4T1 tumour. Adapted from Pérez-Medina et al.364 | ||
Several other protein-based NPs have been radiolabelled. Yang et al. reported the radiolabelling of self-assembled protein NPs based on polypeptides designed to contain His-tags for labelling with 99mTc via the tricarbonyl core.358 Ferritin-based nanocages were radiolabelled by Liang et al using 99mTc using a MAG3 conjugated chelator via an NHS ester.359 Additionally, Gil et al reported the radiolabelling of casein NPs with 67Ga via the conjugation of p-SCN-Bn-NOTA.363 A highly robust method for radiolabelling high-density lipoprotein (HDL) based NPs was described by Mulder and collaborators who radiolabelled HDL NPs with 89Zr using the chelator DFO.28,364–370 Interestingly, a number of their studies showed that the placement of chelator had significant effects on in vitro stability and in vivo biodistribution of the particles.28,364,365 A key study compared the properties of radiolabelled HDL-NPs which were conjugated with p-SCN-DFO either to the phospholipid (PL-DFO NPs) layer or the HDL (apoA-I-DFO).364 The PL-DFO NPs showed lower RCYs than the apoA-I-DFO NPs (79% and 94% respectively). Large differences were observed in vivo (Fig. 25C); with PL-DFO NPs having a nearly 3-fold lower blood half-life compared with the apoA-I-DFO NPs, and had larger amounts of bone uptake (17% & 4% respectively) – indicating loss of the 89Zr. Additionally, apoA-I-DFO NPs showed that 28% of bone uptake of being associated with bone marrow, whereas this was only ca. 4% for the phospholipid labelled particles.364
4.5.3.1 Viral nanoparticles. Viral nanoparticles (VNPs) refers to several types of nanomaterials; such as plant viruses, bacteriophages and animal viruses. The application of VNPs, as well as virus-like NPs (which do not contain viral genomes), for drug delivery is of growing interest; due to their biocompatibility, ease of functionalisation and increased cellular uptake.383,384 Additionally, viral vectors are also being explored for gene delivery and therapy.385 VNPs can easily be modified to incorporate radiolabels, allowing their in vivo tracking using radionuclide imaging techniques. The radiolabelling methods used with VNPs are summarised in Table 10.
Wu et al. described the radioiodination of the viral NP tobacco mosaic virus, with 125I.371 This was performed using the iodogen method, with radiolabelling suggested to occur on tyrosine residues present on the VNPs (Fig. 26A).371 Similarly, Kothari et al. compared two different methods for the radioiodination of adeno-associated virus (AAV) capsids with 124I.372 Radiolabelling efficiencies were generally low, but higher with the iodogen method (10–18%) compared with 1.0–4.5% when using the Bolton–Hunter reagent to label protein amine residues. Chelator-based methods have also been used to radiolabel VNPs. In particular, Seo et al. developed a method to radiolabel AAV capsids using multimeric NO2A bioconjugate platforms.373 The multichelator systems contained eight NO2A rings attached to with either a transcyclooctene (TCO) or maleimide to allow conjugation through either a tetrazine-modified amine group or cysteine residue on the AAV surface (Fig. 26B and C). Both multimers allowed >99% RCYs and high molar activity compared to single chelator systems, but labelling efficiencies of the AAVs were low (2–7.5%) with both bioconjugate systems.373
 | ||
| Fig. 26 (A) Schematic representation of the methods used to radiolabel viral nanoparticles. Chelators can be conjugated to the nanoparticles via amine or thiol residues on the proteins, or radiohalogens can be attached via free amine or tyrosine residues. (B) Surface modification with multichelators (MC) on lysine residues in capsids (top) or the site-specific radiolabeling on cysteine residues in capsids via the multichelator-maleimide conjugate (bottom). (C) Representative PET/CT images of 64Cu-labelled viral capsids in healthy C57BL/6 mice. Adapted from Seo et al.373 | ||
Several reports have also investigated the radiolabelling of bacteriophages (Table 10).381 An interesting study by Holman et al. reported the radiolabelling of Pseudonamas bacteriophages with 99mTc using the HYNIC chelator.380 HYNIC was conjugated using an NHS ester derivative, but it was found that all but the briefest reaction (≤3 min) resulted in loss of infectivity of the phage. However, optimised conditions – which retained infectivity of the phage – radiolabelled the phage with 95% RCY using the co-ligand tricine.380 This highlights the need to ensure that the radiolabelling procedure of viral capsids does not affect their biological function, and how optimisation of the protocol can help mitigate this.
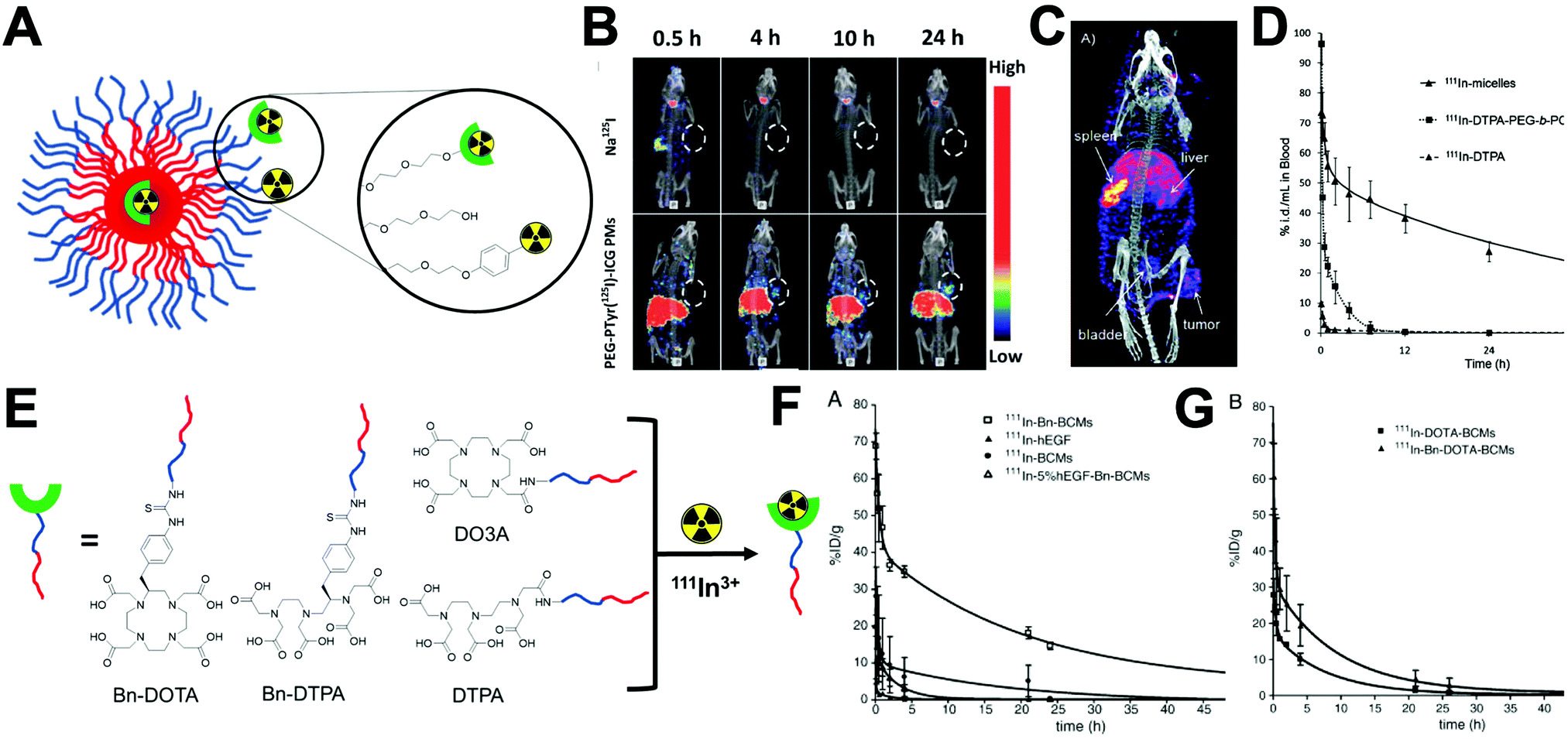 | ||
| Fig. 27 (A) Schematic representation of the methods of radiolabelling polymeric micelles. Chelators or radiohalogens can be attached to the surface or radionuclides can be trapped inside the micelle core. (B) Representative SPECT-CT images of 125I-radiolabelled polymeric micelles (bottom) along with free [125I]NaI at various timepoints in a tumour mouse model. Adapted from Yang et al.170 (C) MIP and sagittal image of tissue accumulation of 111In-micelles 48 h p.i. in a breast cancer tumour xenograft. Adapted from Hoang et al.388 (D) Comparison of the pharmacokinetics of 111In-labelled micelles with the free PEG-b-PCL polymer and the 111In-DTPA complex. Adapted from Hoang et al.388 (E) Graphical representation of the methodology deployed by Fonge et al. comparing the effect of different BFCs on the pharmacokinetics of radiolabelled micelles. (F) Blood pharmacokinetics of 60 nm 111In-labelled micelles in mice bearing human breast cancer xenografts labelled with 111In via p-SCN-Bn-DTPA (111In-Bn-BCMs) or DTPA bis-anhydride (111In-BCMs). Blood clearance curve of micelles containing 5 mol% hEGF targeted BCMs is also shown but not discussed. (G) Blood pharmacokinetics in the same model as above with micelles labelled with 111In via p-SCN-Bn-DOTA (111In-Bn-DOTA-BCMs) or NHS-DOTA (111In-DOTA-BCMs). Adapted from Fonge et al.389 | ||
| NP type | Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|---|
| Polymeric micelles | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 390–398 |
| 125I | Chloramine T | 170 | ||
| Iodogen | 171 and 172 | |||
| 131I | Chloramine-T | 173 | ||
| Chelator-based | 99mTc | Pyrazolyl-diamine chelator via99mTc(CO)3 | 399 | |
| DTPA | 400–402 | |||
| 111In | DTPA chelator | 388, 389 and 403–412 | ||
| DOTA chelator | 389 | |||
| DO3A chelator | 389 | |||
| 64Cu | LNETA chelator, followed by DBCO click chemistry | 413 | ||
| CB-TE2A | 414 | |||
| NOTA | 415 and 416 | |||
| 89Zr | DFO chelator, followed by DBCO click chemistry | 413 | ||
| Ionophore-based | 111In | Tropolone | 417 | |
| Complex trapping | 111In | Oxine complex added during formulation | 418 | |
| 68Ga | Oxine complex added during formulation | 418 | ||
| PEI/DNA polyplex micelles | Surface/polymer non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 419 |
| Napthalocyanine micelles | Surface/polymer non-chelator | 64Cu | Binds to napthalocyanines | 420 |
As with other NP types, several studies have reported the radiolabelling of polymeric micelles with 99mTc using the direct labelling method.390–398 Once again, this was shown to be a robust technique; with reported RCYs generally being >95% under optimised conditions and serum stability ca. 98% after 24 h.395–397 The radioiodination of polymeric micelles has also been reported. Kao et al. reported the radiolabelling of co-polymer based micelles containing a benzyl group allowing radiolabelling with 131I using the chloramine-T method (Fig. 27A).173 RCYs were 55%, but instability was observed in serum (53% after 48 h). Additionally, in vivo stability could not be assessed as the authors blocked thyroid/stomach uptake by the injection of non-radioactive iodide/perchlorate.173 A similar method was described by Yang et al. who synthesised micelles labelled with 125I, facilitated by binding to tyrosine residues via chloramine-T.170 This allowed RCYs of 77% and 98% stability in mouse plasma up to 48 h. Furthermore, in vivo stability also seemed high with little thyroid uptake observed by SPECT/CT imaging (Fig. 27B).170 One potential drawback of these reports was the need to radiolabel the polymer first, and then perform the synthesis of the micelles. This process can take long periods of time (>12 h in the study by Yang170) which could limit its adoption in clinical setups. Radiolabelling of already formulated micelles with radioiodine has been described. Zhan and collaborators reported using the iodogen method with 125I to label PEG–PLA micelles conjugated with targeting peptides.171,172
Alternatively, several groups have used chelator-based methods for radiolabelling polymeric micelles (Fig. 27A and Table 11). Hoang et al. highlighted a key consideration for radiolabelling micelles: the biodistribution of the ‘free’ copolymer.388 The authors compared the in vivo distribution of the 111In-labelled micelles with the single co-polymer. Whilst the pharmacokinetics were clearly distinct with hugely different blood half-lives (29 h and 2 h for the micelles and polymer, respectively; Fig. 27D), the ex vivo biodistribution showed near identical uptake in the liver (ca. 12% ID per g) after 48 h p.i.388 Hence, release of the polymer after micelle degradation may contribute to liver uptake observed (Fig. 27C). In another interesting study, Fonge et al. compared the effect of different BFCs on the pharmacokinetics of radiolabelled micelles using 111In.389 The co-polymers were conjugated with p-SCN-Bn-DTPA (DTPA-Bn), DTPA anhydride (DTPA), p-SCN-Bn-DTPA (DOTA-Bn) or with DOTA (DO3A, Fig. 27E), labelled with 111In and then used to formulate the polymeric micelles. Several differences were seen in vivo. Firstly, each formulation had different blood half-lives with DTPA-Bn having the highest (25 h) – the t1/2 for DTPA, DOTA-Bn and DO3A were 12 h, 9 h and 15 h respectively (Fig. 27F and G). Ex vivo biodistribution at 48 h in tumour-bearing mice also showed distinct differences in uptake. The DTPA-Bn had the highest uptake in all organs of interest with 32% ID per g in the liver, 15% ID per g in the spleen and 4% ID per g in the tumour. Compared with 18% ID per g, 1% ID per g and <1% ID per g for the DTPA labelled micelles in the liver, spleen and tumour respectively. Despite their differences in pharmacokinetics, the two DOTA based formulations had similar uptake at 48 h with ca. 10% ID per g, 2% ID per g and 2% ID per g in the liver, spleen and tumour respectively.389 These results highlight that chelator-based radiolabelling approaches can potentially have large effects on the in vivo behaviour of the radiolabelled particles.
Laan et al. reported the radiolabelling of micelles using the radio-ionophore [111In][In(tropolone)3] via either the trapping of the lipophilic complex during the formation of the NPs, or by labelling preformed micelles.417 The hypothesis was that the lipophilic complex would become trapped within the micellar core. Both methods showed relatively low labelling efficiency with 32% LE during micelle formation and 22% LE of preformed micelles. Incubation in serum showed ca. 20% loss of activity after 2 d.417 Similarly, de la Fuente et al. reported the radiolabelling of micelles via the addition of either [68Ga]Ga-oxine and [111In]In-oxine complexes during formulation.418 However, release of the oxine complexes was shown to occur rapidly in vivo; such that the ex vivo biodistribution of the labelled micelles and the administered oxine complexes as a control were nearly identical.
 | ||
| Fig. 28 (A) Schematic representation of methods used to radiolabel dendrimers. Chelators can be attached to the dendrimer core or to the outer layer of PAMAM dendrimers via the free amines. Alternatively, radiometals can be bound directly to free amines present. Finally, radiohalogens can also be attached to the outer layer. (B) Representative PET/CT of radiolabelled PSMA-targeted dendrimers in male NOD-SCID mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu tumour xenografts with 64Cu, adapted from Lesniak et al.422 (C) Scheme showing the different size dendrimers radiolabelled by Valliant and collaborators.423 (D) Chemical structures of the 18F-labelled NHS agents reported by Zhou et al. to radiolabel amine-functionalised PAMAM dendrimers.424 | ||
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 64Cu | dmpTACN | 425 |
| DO3A | 422 and 426 | ||
| 177Lu | DO3A | 427 and 428 | |
| DOTA | 429 and 430 | ||
| H4DO3A-pyNO-C | 431 | ||
| 111In | DTPA | 432–435 | |
| 1B4M DTPA | 436–438 | ||
| DO3A | 422 and 439 | ||
| H4DO3A-pyNO-C | 440 | ||
| 153Gd | 1B4M DTPA | 436 and 441 | |
| 88Y | 1B4M DTPA | 438 | |
| 99mTc | DPA and 99mTc(CO)3 | 423, 442 and 443 | |
| HYNIC and tricine co-ligand | 444 and 445 | ||
| 1B4M DTPA | 446–448 | ||
| MAG3 | 449 | ||
| 188Re | HYNIC and tricine co-ligand | 450 | |
| 1B4M DTPA | 451 | ||
| 89Zr | DFO | 452 | |
| 68Ga | DO3A | 453–455 | |
| NOTA | 456 | ||
| Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 457–461 |
| Via 99mTc(CO)3 | 462 | ||
| 64Cu | Direct labelling | 463 | |
| 125I | Bolton–Hunter reagent | 152–156 | |
| Chloramine-T | 157–160 | ||
| N-Succinimidyl 3-(123I) iodobenzoate conjugation | 464 | ||
| 123I | N-Succinimidyl 3-(123I) iodobenzoate conjugation | 465 | |
| 131I | Iodogen | 161 | |
| Chloramine-T | 466–468 | ||
| 76Br | Chloramine-T | 184 | |
| 18F | Isotopic exchange with trifluoroborate | 469 | |
| Conjugation of radiolabelled NHS agents | 424 | ||
The overwhelming majority of examples of dendrimer radiolabelling has been performed with poly(amidoamine) (PAMAM) based dendrimers. A common technique employed with this type of dendrimer is the conjugation of a chelator to amine residues present on the polymer (Fig. 28A). For example, several reports used DTPA-based conjugates to radiolabel dendrimers with 111In432–435 Sano et al. reported >99% RCYs using p-SCN-Bn-DTPA conjugated to PAMAM-based dendrimers.435 Furthermore, PAMAM dendrimers have been radiolabelled with the radiotherapeutic isotope 177Lu.427–431 using the amine reactive BFCs DOTA-NHS,428 and p-SCN-Bn-DOTA.429,430
The radiolabelling of PAMAM dendrimers with 64Cu has also been performed using the DOTA-NHS BFC system.422,426 This allowed RCYs of ca. 85% and >93% serum stability up to 20 h.426 Interestingly, Lesniak et al. compared the labelling and biodistribution of 64Cu-labelled and 111In-labelled dendrimers with this chelate system.422 Whilst RCYs with both radiometals were similar (80%), discrepancies in the biodistributions of the labelled dendrimers were found between the two. Liver uptake was 6–8 fold higher with 64Cu over 48 h compared with the 111In-labelled NPs, and spleen uptake at 1 h was 107% ID per g – decreasing to 68% ID per g at 48 h – for 64Cu-labelled dendrimers – whereas spleen uptake increased overtime for the 111In-labelled NPs (6.4% ID per g at 1 h to 34% ID per g at 48 h p.i.). Furthermore, the 111In-labelled dendrimer biodistribution matched more closely with that seen with dendrimers labelled with an optical probe.422 These results taken together suggest loss of 64Cu from the dendrimer. Indeed, it is well established the superiority of DOTA as a chelator for 111In, compared with 64Cu.94 Alternatively, NOTA has often been shown to be a more suitable chelator for 64Cu,94,470 This study highlights the stark differences in biodistribution that can occur from using different radiometals in the same system, and that chelators should be chosen appropriately to match with each radiometal.
A key study by Valliant and collaborators reported the radiolabelling of various dendrimers with 99mTc using DPA to chelate the [99mTc][Tc(CO)3]+ core (Fig. 28C).423,442,443 However, in this case the chelator was conjugated via an alkyl amine to the dendrimer core which had been functionalised with an NHS ester. RCYs of ca. 90% in just 5 min using a microwave synthesis unit were reported – albeit at high temperatures of 80–130 °C. Interestingly, the authors noted a reduction in RCY when radiolabelling larger dendrimers with G6 and G7 dendrimers having RCYs of 70% and 53%, respectively compared with ca. 90% observed with G5.423 This highlights a potential drawback with radiolabelling dendrimers via their core, as opposed to functional groups on the outer layers, wherein increasing dendrimer size potentially renders the chelator less accessible for radiolabelling (Fig. 28C).
The amine residues on PAMAM dendrimers also allow the radioiodination using Bolton–Hunter reagent,152–156 as well as another amine-reactive reagent, N-succinimidyl 3-iodobenzoate (Fig. 28A).464,465 The chloramine-T method was used by several groups to radiolabel dendrimers with 125I,157–160 and with the PET isotope 76Br,184 however the dendrimers usually had to be modified with either tyrosine or tyramine residues to allow radiolabelling. Alternatively, Zhou et al. reported several 18F-labelled NHS agents (Fig. 28D) to radiolabel amine-functionalised PAMAM dendrimers.424 RCYs under optimised conditions were 28%, 95% and 95% for [18F]4, [18F]7 and [18F]10 respectively after 5 min, with the RCY with [18F]4 increasing to 71% after 15 min. The higher labelling efficiency and faster reaction kinetics of [18F]7 and [18F]10 was hypothesised to occur due to electrostatic interactions of the NHS esters with the PAMAM dendrimers. [18F]7 and [18F]10 would become pre-localised to the dendrimers due to the interaction of the sulfonate and carboxylate groups respectively, with the positively charged amines on the dendrimers.424
Finally, the direct labelling of PAMAM dendrimers with radiometals has also been reported by several groups. In each case binding of the radiometals was proposed to occur via the amine groups on the dendrimer (Fig. 28A). Once again, the binding of [99mTc]TcO4 after SnCl2 reduction has been described by several groups,457–461 allowing high RCYs and serum stability. Interestingly, Tassano et al. found the [99mTc][Tc(CO)3]+ core could bind to PAMAM dendrimers with ca. 90% RCY.462 However, the complex was shown to be unstable in a competition assay with histidine; with over 50% of the activity being transchelated after just 1 h.462 The direct labelling of PAMAM dendrimers was also reported with 64Cu by Xu et al.463 Optimised conditions showed that ca. 95% RCYs could be achieved after just 15 min at room temp. and pH 7. Labelling at acidic conditions was reduced (30% at pH 3) suggesting that chelation was occurring via the amines on the PAMAM dendrimer, as these amine groups would become more protonated at lower pH. This interaction was shown to be relatively stable with ca. 80% stability in mouse plasma was after 24 h.463
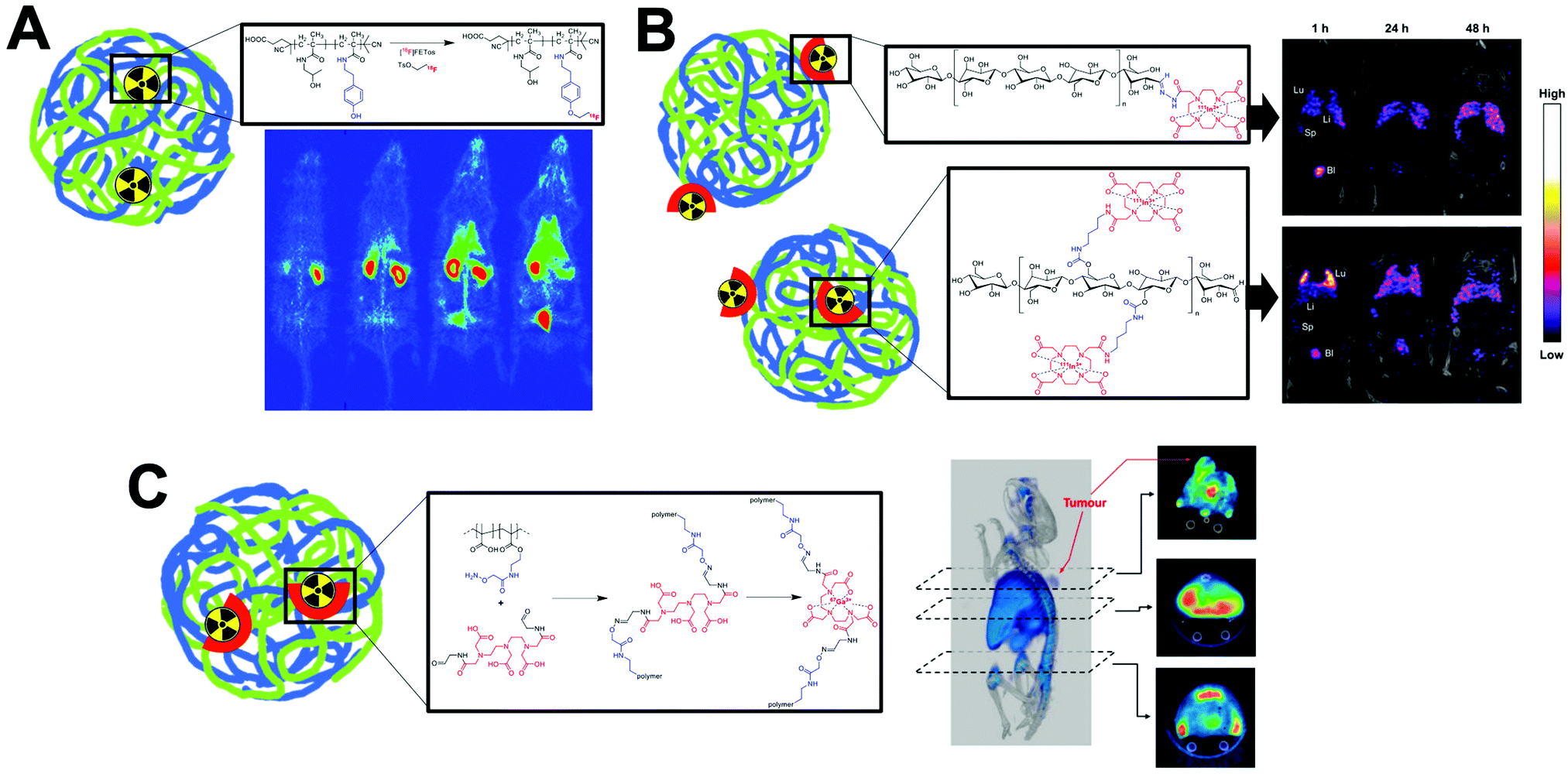 | ||
| Fig. 29 Figure showing the various ways of incorporating radionuclides into different polymer-based nanomaterials. (A) Radiohalogens can be attached to the polymer backbone. Schematic showing the radiolabelling of N-(2-hydroxypropyl)-methacrylamide (HPMA) with 2-[18F]fluoroethyl-1-tosylate ([18F]FETos) and PET images in healthy rats of the radiolabelled polymer reported by Allmeroth and collaborators. Adapted from Herth et al.471 (B) Chelators can be attached to the polymer backbone. Schematic showing the two different bioconjugate strategies used for the radiolabelling of cellulose-based nanoparticles with 111In reported by Imlimthan et al – and SPECT/CT in tumour-bearing mice. Adapted from ref. 33. (C) Chelators can be incorporated directly into the polymer structure. Schematic showing the functionalisation of single-chain poly-(methacrylic acid) with a DTPA derivative compound for radiolabelling with 67Ga along with SPECT/CT images in tumour bearing mice. Adapted from Benito et al.472 | ||
| NP type | Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|---|
| PLGA NPs | Complex trapping | 111In | Oxine complex added during formulation | 473 |
| Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 474–486 | |
| 125I | Iodobeads | 178 | ||
| 18F | Biotin conjugate after formulation | 487 | ||
| Chelator-based | 99mTc | DTPA | 488 | |
| 111In | DTPA chelator | 489 | ||
| 177Lu | DO3A chelator | 490 and 491 | ||
| Remote loading | 177Lu | DOTATATE complex | 492 | |
| HPMA copolymer NP | Non-chelator | 18F | Tosylate | 168, 471 and 493–495 |
| 131I | Chloramine-T | 168 | ||
| 125I | Chloramine-T | 167 | ||
| 72/74As | Thiol binding | 496 | ||
| Chitosan NPs | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 497–502 |
| 64Cu | Direct labelling | 503 | ||
| 89Zr | Direct labelling | 503 and 504 | ||
| Chelator-based | 64Cu | DO3A via DBCO click chem | 505 and 506 | |
| Chitosan/polyglutamic acid NPs | Chelator-based | 68Ga | NOTA | 507 |
| Quaternary ammonium palmitoyl glycol chitosan (GCPQ) NPs | Non-chelator | 125I | Bolton–Hunter reagent | 166 |
| Polyglucose NPs | Non-chelator | 18F | CuAAC click reaction | 508 |
| Dextran-based single chain polymer NPs | Chelator-based | 67Ga | NODAGA | 509 |
| Dextran NP | Chelator-based | 89Zr | DFO | 510 |
| Cellulose NPs | Chelator-based | 111In | DO3A | 33 |
| Shell cross-linked Knedel-like NPs | Chelator-based | 64Cu | TETA chelator | 511 |
| DOTA chelator | 512–515 | |||
| DO3A | 512 | |||
| Hyaluronan NPs | Chelator-based | 89Zr | DFO chelator | 516 |
| PEG chain | Chelator-based | 89Zr | DFO chelator | 517 |
| Nanogel | Non-chelator | 99mTc | SnCl2 reduction | 518 |
| Chelator-based | 68Ga | NOTA | 519 and 520 | |
| Polyoxazoline polymer | Non-chelator | 18F | Isotopic exchange using SiFA | 521 |
| Chelator-based | 111In | DOTA chelator | 522 and 523 | |
| 90Y | DOTA chelator | 523 | ||
| Caprolactone polymers | Non-chelator | 125I | Chloramine-T | 162 |
| Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 524 | |
| Poly(β-amino ester) NP | Ionophore-based | 111In | Oxine complex added during formulation | 525 |
| PEG–MA/MMA comb NP | Chelator-based | 64Cu | DO3A chelator | 526 and 527 |
| Poly(maleic anhydride-alt-1-octadecene) NP | Non-chelator | 125I | Iodogen | 174 |
| 131I | Iodogen | 174 | ||
| Poly(γ-glutamic acid) NPs | Non-chelator | 125I | Iodogen | 175 |
| Chelator-based | 111In | DTPA | 528 | |
| Polyester-based NPs | Non-chelator | 18F | 4-[18F]fluorobenzyl-2-bromoacetamide | 529 |
| 125I | Iodobeads | 177 | ||
| Polythiophene NPs | Chelator-based | 99mTc | HYNIC with co-ligands (TPPTS and tricine) | 530 |
| PLA NPs | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 531 and 532 |
| Encapsulated complex | 99mTc | HMPAO complex added during formulation | 533 and 534 | |
| Latex NPs | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 535 |
| Polyanhydride NPs | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 536 |
| Polyacrylamide NPs | Non-chelator | 125I | Chloroglycoluril | 185 |
| 76Br | Chloramine-T | 185 | ||
| Gelatin NPs | Non-chelator | 125I | Iodogen tube | 537 |
| Chelator-based | 111In | DTPA chelator | 538–540 | |
| Polyvinyl phenol NPs | Non-chelator | 125I | Iodination beads | 180 |
| Chloramine-T | 179 | |||
| 124I | Iodination beads | 180 | ||
| Polydopamine NPs | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 181 |
| 131I | Chloramine-T | 181 | ||
| p(BAEA-co-OEGA-co-VDM) polymeric nanostar | Chelator-based | 177Lu | DO3A chelator with DBCO click chem | 541 |
| 89Zr | DFO | 541 | ||
| Poly(methacrylic acid) (PMAAc)-based Single Chain Polymer NPs | Chelator-based | 67Ga | DTPA | 472 |
| Poly(n-butylcyano acrylate) (PBCA) NP | Non-chelator | 99mTc | Direct labelling with SnCl2 reduction | 542 |
| Melanin NPs | Non-chelator | 64Cu | Direct labelling to melanin | 543 and 544 |
| Silk fibroin NPs | Chelator-based | 111In | DTPA | 545 |
As with the other NP types discussed previously, several different polymer-based nanomaterials have been radiolabelled using the non-chelator direct labelling method with 99mTc. PLGA-based NPs (PLGA-NP),474–486 latex NPs,535 PLA NPs531,532 polydopamine (PDA) NPs,181 and poly(anhydride) NPs536 have all been radiolabelled in this way with high (>90%) RCYs generally reported. The radiolabelling of chitosan-based NPs with this method has also been reported by several groups, resulting in 85–98% RCYs.497–502 Binding of 99mTc is likely to occur via the free hydroxyl and amines present on the chitosan polymer, however this has not been characterised. The direct labelling of chitosan NPs with 64Cu and 89Zr has also been reported by Fairclough et al.503,504 RCYs were ca. 72% for both radiometals; with larger MW polymers (>190 kDa) resulting in higher LE for 89Zr over 64Cu (90% and 72%, respectively).503 Fan et al. showed that melanin NPs could be directly labelled with 64Cu, which was hypothesised to occur via free hydroxyl and carbonyl groups present on melanin.543,544 RCYs of 80% were achieved under mild condition (after 1 h at 40 °C) with ca. 90% stability in FBS after 24 h.
Several groups have used radiohalogenation-based reactions for the labelling of polymeric nanomaterials. However, these often involve the modification of the polymer structure first. For examples, Allmeroth and collaborators reported the labelling of N-(2-hydroxypropyl)-methacrylamide (HPMA) polymer structures; which were first modified with tyramine, which allowed subsequent reaction with 2-[18F]fluoroethyl-1-tosylate ([18F]FETos) (Fig. 29A).168,471,493–495 This allowed ca. 90% RCY under optimised conditions.471 Wagener et al. compared this method with the radioiodination with 131I using chloramine-T, which allowed ca. 50% RCY after 4 min.168 Interestingly, biodistribution studies of the two radiohalogenated derivatives showed stark differences in organ uptake after 2 h p.i. Liver uptake of the 131I-labelled polymer was 5-fold that of the 18F derivative, and was 12-fold higher in the spleen. Conversely, higher kidney uptake was observed for the 18F-labelled polymer than the 131I derivative (ca. 5.5% and 0.5% ID per g respectively). Additionally, thyroid uptake (ca. 22% ID per g) was observed for the 131I at 24 h p.i. strongly suggesting significant deiodination in vivo.168
The structures of certain polymers can however enable radioiodination reactions without the need for further modification. Rahmani et al. reported the radioiodination of PLGA–polymethylmethacrylate (PMMA) co-polymer NPs with 125I using iodobeads.178 Radiolabelling was facilitated by the phenol-containing PMMA, which allowed 95% RCP after synthesis and purification. The radiolabel was shown to be stable in vivo with <2% ID per g in the thyroid over 24 h p.i.178 Similarly, the phenol residues present on polyvinyl phenol (PVP)-based NPs allow the radioiodination.179,180 Simone et al. used iodination beads to radiolabel PVP NPs with both 125I and 124I, allowing ca. 90% RCY and >80% serum stability over 3 d. However, release of free iodide was observed in vivo with ca. 3% ID observed in the thyroid after 24 h.180 Similarly, Tang et al. radiolabelled PEGylated polyvinyl phenol NPs with 125I using the chloramine-T method which resulted in >90% RCYs.179 Stability of the radiolabel was high with >95% stability in human serum over 48 h, and <0.2% ID uptake in the thyroid of mice observed up to 4 d.179 Zhong et al. took advantage of the benzene rings present on polydopamine (PDA) NPs to radiolabel the particles with 131I using chloramine-T – resulting in 70% RCY.181
As well as their reaction with radiohalogens, polymer structures on NPs can also easily facilitate the bioconjugation of chelators for labelling with radiometals. For example, Gracia et al radiolabelled single-chain dextran based NPs with 67Ga by coupling the BFC NH2-NODAGA to carboxylate residues on the NPs which allowed RCYs of ca. 50% after.509 An interesting study by Imlimthan et al. radiolabelled cellulose-based NPs with 111In using two different bioconjugates.33 The cellulose polymers were functionalised with a DO3A chelator either via a terminal aldehyde group (with a DO3A-hydrazine BFC) or to one of the hydroxyl groups on the cellulose backbone (via an DO3A-amine BFC, Fig. 29B). RCYs for the aldehyde-conjugated NPs after reaction with 111In was much lower (7–18%) compared with the hydroxyl-conjugated NPs (54–65%) likely due to lower number of chelators present. Stability in human plasma was the same for both formulations with >90% stability over 72 h. Ex vivo biodistribution of the two formulations in mice showed similar uptake profiles with uptake in the liver and spleen. However, vastly different amounts of uptake over all time points were observed in the lung; with ca. 125% ID per g observed for the hydroxyl-conjugated NPs compared with ca. 12% ID per g for the aldehyde-conjugated NPs at 6 h p.i (Fig. 29B).33 This was attributed to the higher zeta-potential of the hydroxyl-conjugated NPs, and highlights once again how modification of NPs aimed at facilitating radiolabelling can lead to large differences in their properties and biodistribution in vivo.
A unique approach for chelator-based radiolabelling was reported by Benito et al. who radiolabelled single-chain poly-(methacrylic acid) NPs with 67Ga.472 Interestingly, this was performed by incorporating DTPA into the polymer chain by forming an aldehyde-functionalised DTPA derivative, which was reacted with an amine groups present on a modified polymer (Fig. 29C). This modification allowed ca. 65% labelling efficiencies, with >90% stability in saline solution over 48 h. One potential drawback of this method, however, is the potential reduction in the Ga complex stability, with only three carboxylates being available for reaction – due to the aldehyde functionalisation of the other two. Finally, Pereira et al. described the radiolabelling of PLA-based nanocapsules by the trapping of [99mTc]Tc-HMPAO during formulation of the NPs.533,534 The complex could be encapsulated with 50% LE, however >30% release of the complex was observed, suggesting the trapping of [99mTc]Tc-HMPAO a sub-optimal radiolabelling method.
4.6 Radiolabelling of inorganic nanomaterials
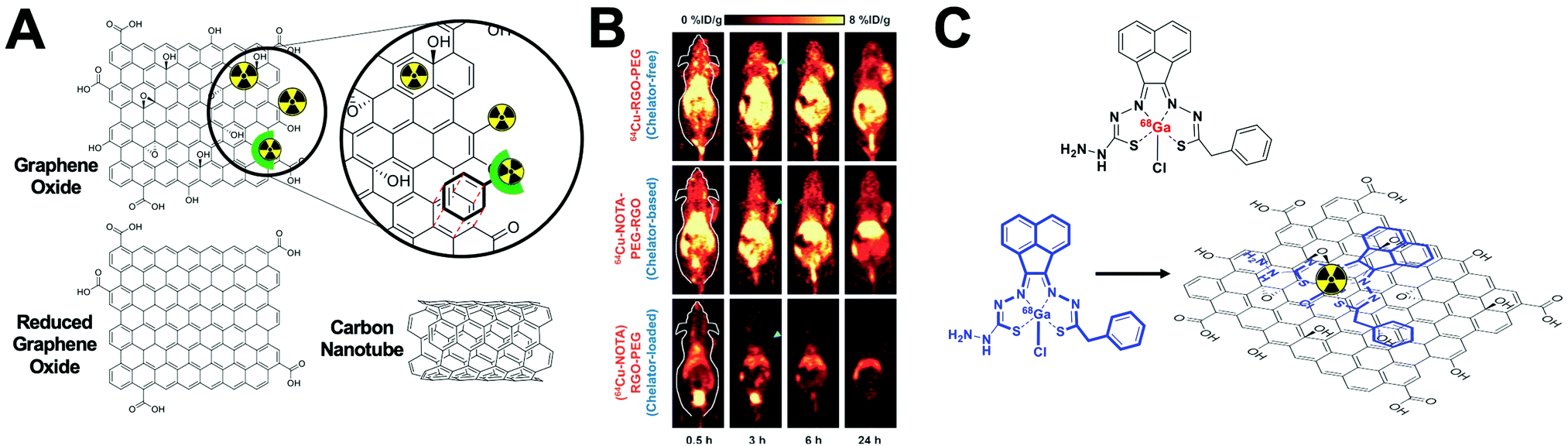 | ||
| Fig. 30 (A) Structures of graphene oxide (GO), reduced graphene oxide (rGO) and carbon nanotubes (CNTs) along with a schematic of the various ways to radiolabel these carbon-based structures. Radiometals and radiohalogens can bind directly to the graphene structures and chelators can be attached to the nanomaterials either via conjugation or by π–π interactions between BFCs and the graphene structures. (B) Representative PET images of graphene-oxide nanosheets radiolabelled with 64Cu in 4T1 tumor-bearing mice. Adapted from Shi et al.550 (C) Structure of a novel bissemithiocarbazonate 68Ga3+ complex for radiolabelling graphene oxide NPs. (top) and a schematic showing the proposed incorporation of the 68Ga3+ complex into the GO sheets (bottom). Reported by Sarpaki et al.551 | ||
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Non-chelator | 125I | Chloramine-T | 163, 164 and 552 |
| 131I | Chloramine-T | 165 | |
| 99mTc | Direct labelling with SnCl2 reduction | 553–558 | |
| Via 99mTc(CO)3 | 559 | ||
| 188Re | Direct labelling with SnCl2 reduction | 560–562 | |
| 64Cu | Direct labelling | 550 | |
| 68Ga | Direct labelling | 551 | |
| Chelator-based | 111In | DOTA chelator | 563–565 |
| DTPA chelator | 566–572 | ||
| 99mTc | DOTA chelator | 573 | |
| 64Cu | HPPH (porphyin PDT agent) chelator | 574 | |
| DO3A chelator | 575–577 | ||
| NOTA chelator | 550, 578 and 579 | ||
| DOTAM chelator | 580 | ||
| 66Ga | NOTA chelator | 581 | |
| 68Ga | NOTA chelator | 577 | |
| Intercalation of bissemithiocarbonato complex | 551 | ||
| DFO chelator | 582 | ||
| DO3A chelator | 577 | ||
| 86Y | DOTA chelator | 564 | |
| 89Zr | DFO chelator | 583 | |
| 225Ac | DOTA chelator | 583 | |
| 57Co | MeAMN3S3sar | 584 | |
| 177Lu | DOTA chelator | 585 | |
Several groups have reported the radiolabelling of nanographene oxide sheets with 125I using the chloramine-T method with RCYs of ca. 50–60%, with high in vitro serum and in vivo stability.163,164,552 Radioiodination was suggested to occur at the edges of the graphene sheets where defects exist. The non-chelator labelling of different types of graphene-based NPs with various radiometals has also been reported (Table 14). Zhan and co-workers radiolabelled multi-walled carbon nanotubes (MWCNTs) directly with 99mTc after SnCl2 reduction resulting in ca. 90% RCY.553,554 Direct labelling of graphene oxide nanomaterials with 99mTc has also been reported by several groups.555,556,559 Zhang et al. also used this direct labelling method for carbon NPs with 188Re.560–562 A slightly modified method was developed by Cao et al. who produced the [99mTc][Tc(CO)3]+ core, which was then reacted with PEGylated nanographene oxide.559 Radiolabelling peaked at 80% RCY after 5 min, but rapidly declined at later timepoints. The authors reported this as being due to reduction of carbonyl and hydroxyl groups on the GO layer by NaBH4 used to produce the [99mTc][Tc(CO)3]+ core. This was confirmed by the low labelling (8%) of GO reduced with NaBH4.559 However, this may also be due to the lack of appropriate binding sites on GO/rGO for [99mTc][Tc(CO)3]+ which prefers multidentate ligands, often containing aromatic amines.586 This work suggests this method may be inappropriate for use with GO NPs.
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 64Cu | DOTA | 587 and 588 |
| NOTA | 589–591 | ||
| Bis(dithiocarbamate)bisphosphonate | 592 | ||
| 68Ga | Thiosemicarbazone | 593 | |
| DOTA/NOTA | 594–596 | ||
| 89Zr | DFO | 597 | |
| 18F | 18F-AlF/NOTA | 598 | |
| 99mTc | Polyacrylic acid | 599 | |
| Bisphosphonate derivatives | 600–603 | ||
| DMSA | 604 and 605 | ||
| Lipoic acid based ligands | 606 | ||
| DTPA | 607–609 | ||
| 111In | PCTA | 116 | |
| DOTA | 610 | ||
| 90Y | Polyacrylic acid | 599 | |
| Imidodiphosphate (IDP) or Inositol hexaphosphate (IHP) | 611 | ||
| PEG600 diacid | 612 | ||
| 188Re | N2S4 | 613 | |
| (Z)-2-Methoxyimino-2-(2-aminothiazol-4-yl)-acetic acid | 614 | ||
| SnCl2 reduction | 615 | ||
| 177Lu | Polyacrylic acid | 599 | |
| Non-chelator | 64Cu | Chemical adsorption | 616 |
| Hot + cold precursors | 117 | ||
| 68Ga/67Ga | Chemical adsorption | 166, 592 and 617 | |
| Hot + cold precursors | 115 | ||
| 89Zr | Chemical adsorption | 209, 603, 618 and 619 | |
| 11C | [11C] methyl iodide | 132 | |
| 18F | 18FDG/chemoselective oxime formation | 197 | |
| Chemical adsorption/Al(OH)3 | 213–215 | ||
| 69Ge | Chemical adsorption | 620 | |
| 59Fe | Hot + cold precursors | 109–111 | |
| Core-doped/post-synthetic method | 621 | ||
| *As | Chemical adsorption | 208 | |
| 99mTc | SnCl2 reduction | 622 and 623 | |
| 111In | Chemical adsorption | 618 | |
| Cold + hot precursors | 119 | ||
| 125I | Chloramine-T | 140 and 141 | |
| Bolton–Hunter reagent | 142 | ||
| 90Y | Chemical adsorption | 616 | |
| Physisorption | 612 | ||
| 177Lu | Chemical adsorption | 616 | |
| 223Ra | Chemical adsorption | 624 | |
| 225Ac | Hot + cold precursors | 118 | |
As well as with Tc/Re, chelate-free labelling with other radiometals is also possible (Fig. 30A). Shi et al. reported a method to directly label GO and reduced GO with 64Cu2+.550 The binding of the copper ion was proposed to occur via an interaction with the π bond electrons on the graphene surface (Fig. 30A and B). This was supported by the increased RCY seen with reduced GO (60% RCY compared with ca. 20% for GO) which has higher abundance of π electrons. RGO also demonstrated higher serum stability with ca. 80% remaining on the GO after incubation in mouse serum for 24 h compared with ca. 50% for GO.550 Sarpaki et al. also reported that 68Ga3+ could also be attached to GO in a chelate-free method. This was again proposed to be based on interactions with the π bond electrons and also binding to oxygen donors on the GO surface. This interaction was shown to be highly favourable with RCYs and stability in human serum (up to 2 h) both >95%.551 The authors also reported a novel bis(semithiocarbazonate) 68Ga3+ complex (Fig. 30C) capable of radiolabelling GO. Radiolabelling with this complex allowed high RCY > 95% and high serum stability >95% up to 2 h.551 Characterisation using energy-dispersive X-ray spectroscopy (EDX) mapping the non-radioactive gallium complex suggested that the complex was incorporated non-covalently within the GO layers (Fig. 30C).
Several groups have used chelator-based methods for labelling graphene-based nanomaterials (Table 14); with a large number of reports using the chelator DTPA with 111In.566–572 Al-Jamal and collaborators reported RCYs varying between 8.0–85% with conjugation of the chelator occurring through one of the carboxylate groups on EDTA,566,567,569,571,572 whereas Zhang reported higher yields (up to 95%) when using an extended DTPA compound with an additional carboxylate for conjugation when using similar reaction conditions.570 Interestingly, Cornelissen et al. found that π-bond interactions between GO and the benzene ring (Fig. 30A) on the p-SCN-Bn-DTPA bifunctional chelator, could be strong enough to allow of the GO labelling with 111In.568 RCYs were high (>99%), with 95% serum stability up to 24 h. However, bladder uptake seen in early (1 h p.i.) SPECT/CT images suggests some of the 111In-DTPA is released from the GO and excreted.568 Similarly, Shi et al. reported that the p-SCN-Bn-NOTA bifunctional chelator also be non-specifically loaded onto RGO; which was hypothesised to be either from hydrophobic interactions or π-bond interactions between the RGO and the benzene ring on the.550 RGO loaded and conjugated with NOTA both showed high RCYs of ca. 90% with 64Cu. However, the NOTA-loaded RGO showed lower serum stability with only ca. 50% remaining after incubation in mouse serum for 24 h compared with >80% for the NOTA-conjugated RGO. Similar to the study by Cornelissen et al. in vivo bladder uptake for the NOTA-loaded RGO was observed suggesting the release and excretion of 64Cu-NOTA occurred (Fig. 30B).550
Initially, the main strategies for the radiolabelling of IONPs were based on chelating agents such as DTPA, DOTA and NOTA attached to the NP surface.625,627 In 2008, Jarret et al. introduced DOTA as a chelating agent for IONPs.587 Although with moderate RCY (up to 21%), this work revealed the benefit of incorporating 64Cu into DOTA before the conjugation with the NPs as well as the better performance of p-SCN-Bz-DOTA over p-NH2-Bz-DOTA during the conjugation – likely due to the decrease in the steric hindrance. Lee et al. reported a similar strategy not only for the 64Cu radiolabelling with DOTA but also for the conjugation with an RGD peptide through a PEG-maleimide linker (Fig. 31A). This formulation demonstrated high affinity towards integrins due to the RGD peptide allowing angiogenesis-targeted tumour PET/MRI detection (Fig. 31B).588 Among other chelating agents, bisphosphonate-based bifunctional chelators offer a versatile strategy for the radiolabelling of SPIOs with PET or SPECT radionuclides via direct binding to the Fe3O4 surface. Bisphosphonates have shown excellent properties as anchors to functionalise iron oxide and other nanomaterials based on metal oxides and calcium phosphates. For instance, bis(dithiocarbamatebisphosphonate) (dtcbp)2 was introduced as a chelating agent for 64Cu with instantaneous and quantitative complexation at room temperature. In combination with IONPs for 15 min at 100 °C, the attachment of [64Cu][Cu(dtcbp)2] provided 64Cu-labeled IONPs with 95% RCY, 100% RCP and quantitative RCS after 48 h of incubation with human serum at 37 °C, most likely a result of the protective action of the dextran coating.628 IONPs were also successfully PEGylated with high surface density and radiolabelled with 99mTc using bisphosphonate-functionalised PEG conjugates and dipicolylamine (DPA)-alendronate.600,601 The biodistribution of the PEG-bisphosphonate functionalised IONPs radiolabelled with 99mTc (Fig. 31C) was studied by SPECT/CT showing long circulation times, as expected from the high density of PEG, and high signal in the vasculature of Balb/C mice even at 200 min post i.v. injection (Fig. 31D).
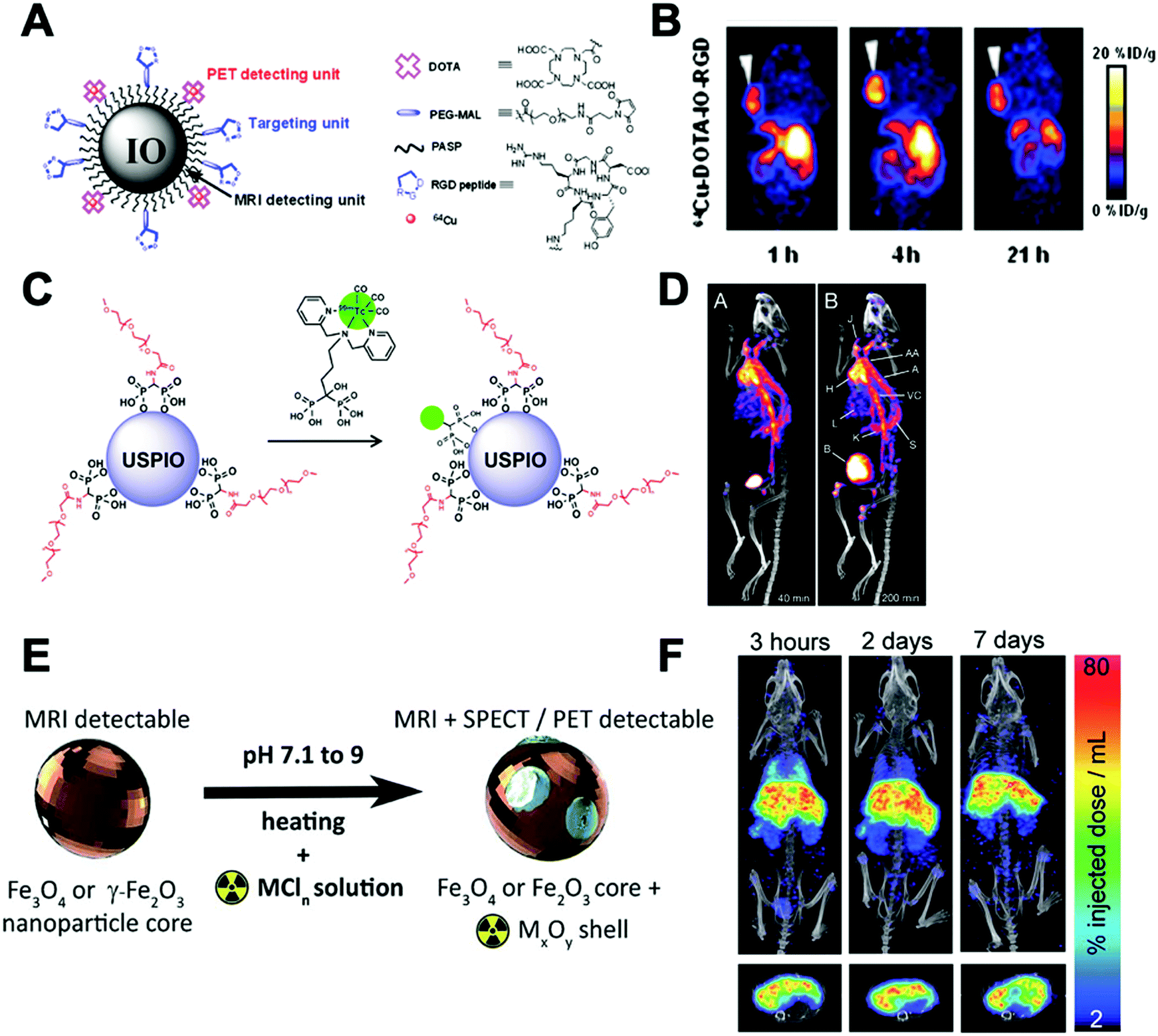 | ||
| Fig. 31 (A) Illustration of the 64Cu-DOTA-IO-RGD PET/MRI probe, (B) decay-corrected wholebody coronal PET images of nude mouse bearing human U87MG tumor at 1, 4, and 21 h after injection of 3.7 MBq of 64Cu-DOTA-IO-RGD, adapted with permission from ref. 588. (C) Radiolabeling of PEG(5)-BP-USPIOs with the radiolabelled bisphosphonate 99mTc-DPA-ale, (D) in vivo SPECT-CT studies with PEG(5)-BP-USPIO: (A and B) maximum intensity projection SPECT-CT images after i.v. injection of radiolabeled (99mTc) PEG(5)-BP-USPIO at the first (A, 40 min) and last (B, 200 min) time points (labels: H = heart, J = jugular vein, AA = aortic arch, A = aorta, VC = vena cava, L = liver, K = kidney, S = spleen, B = bladder), adapted with permission from ref. 601, (E) synthesis of radiolabelled IONPs using radiometal chloride salts (MCln) to form an oxidised radiometal coating, (F) maximum intensity projection 111In SPECT/CT at 3 h, 2 and 7 d post-injection confirms presence of labelled iron oxides in the liver, lung, kidneys, and spleen of C57BL/6 mice. Corresponding axial slices (bottom) show co-localisation of the radiolabelled IONPs and the liver, adapted with permission from ref. 618. | ||
Within the non-chelator based radiolabelling strategies described in Section 4.4, chemical adsorption and hot + cold precursors are the most widely reported methods to radiolabel of IONPs. In the chemical adsorption category, reported radiolabelling protocols showed the high affinity of the magnetite (Fe3O4) and maghemite (γ-Fe2O3) surface towards different metallic radionuclides as described in detail in Section 4.4.5. Recently, Patrick et al. introduced the concept of radio-mineralisation (SRM) to explain this affinity.618 In this work, 111In and 89Zr magnetite/maghemite NPs were synthesised by heat-induced chemical adsorption demonstrating the deposition of radionuclide metal oxides onto the surface of the IONPs (Fig. 31E). The reactions conducted at 90 °C and pH = 9 for 90 min provided RCYs between 79–85% for 111In-IONPs and 94% for 89Zr-IONPs. SPECT/CT biodistribution studies of 111In-IONPs in C57BL/6 mice showed main accumulation in liver and spleen with significant accumulation in the lungs in the first 3 hours post-injection (Fig. 31F).
The hot + cold precursors methodology has also been used for the integration of PET, SPECT and therapeutic radionuclides into the core of NPs. Dextran-coated IONPs doped with 64Cu or 68Ga were developed by fast microwave-driven protocols.115,117 Using FeCl3·6H2O, dextran and [68Ga]GaCl3 as starting reagents, a 10 min microwave protocol in water provided 68Ga doped IONPs with RCY greater than 90%, 100% RCP and quantitative RCS under different physiological media. The microwave synthesis was also successful when using citric acid as a coating (instead of dextran), demonstrating the versatility of this IONP radiolabelling method.629,630
Within the different radiolabelling methods for SiO2 NPs (Table 16), the chemical adsorption of radiometals appears to be the best choice due to its facile, fast and stable radiolabelling. Shaffer et al. evaluated the incorporation of 68Ga, 64Cu, 89Zr, 90Y, 111In and 177Lu into amorphous silica NPs at different temperatures, pH and reaction times.211 The results showed RCYs >99% with all radionuclides when radiolabelling is conducted at 70 °C and pH = 7.3 between 15 min and 1 h of incubation (Fig. 32A). No significant changes over the RCYs were observed at different pH whilst the RCY greatly increased with the temperature. Additionally, a competitive chelation protocol with EDTA was carried out showing stable radiolabelling only in samples heated at 70 °C. This suggests that high temperatures are needed to reach the required binding activation energy – rather than increase the radionuclide diffusion through the SiO2 NP. The RCS was also evaluated in 50% fetal bovine serum showing a clear relationship between the oxophilicity of the radionuclide and the RCS (Fig. 32A). This clearly highlights the affinity of the radionuclides towards the oxygen-rich matrix of the SiO2 NP. For ‘softer’ radiometals, such as 64Cu(II), the stability was very poor with 50% of radionuclide leaching after just 4 h (Fig. 32A). In vivo PET imaging demonstrated high RCS of 68Ga and 89Zr SiO2 NPs by showing high uptake in the elimination organs (liver and spleen), and different profiles compared to their respective free radionuclides (Fig. 32B and C). Interestingly, Cheng et al. compared this methodology for the 89Zr radiolabelling of ultrasmall cRGDY-conjugated fluorescent silica NPs (C′ dots) with the radiolabelling through a chelator-based protocol using DFO as chelating agent (Fig. 32D).34 Both NPs, with an average size of 6–7 nm were successfully radiolabelled with high RCY. To evaluate differences in RCS, both formulations were incubated in human serum at 37 °C obtaining high RCS of >99% in both cases. RCS and circulations half-lives were also studied by injecting both NPs in nude mice, finding a higher degradation of the non-chelator NPs (>25%) than chelator-based NPs (<2%) 48 h post-injection with similar circulation times of around 15 h. Biodistribution of both NPs was evaluated by dynamic PET during 60 min after the injection. Both formulations showed a similar trend with intense signal assigned to the circulation and most interestingly, the renal clearance due to the small size of the particles (Fig. 32E). Further ex vivo biodistribution studies revealed higher bone uptake in the non-chelator formulation attributed to the 89Zr detachment in agreement with the lower stability previously observed.
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | Cu-64 | NOTA | 634 |
| Zr-89 | DFO | 34, 635 and 636 | |
| Na-22 | 4-Amino-benzo-15-crown-5 | 637 | |
| Tc-99m | DTPA | 638–640 | |
| In-111 | DTPA | 641 and 642 | |
| DOTA | 643 | ||
| Lu-177 | DOTA | 644 | |
| Non-chelator | Cu-64 | Chemical adsorption | 211 |
| Chemical adsorption/thiol group | 212 | ||
| Ga-68 | Pyridine grafting | 645 | |
| Chemical adsorption | 211 and 646 | ||
| Zr-89 | Chemical adsorption | 34, 211, 647 and 648 | |
| F-18 | N-Succinimidyl 4-[18F]fluorobenzoate | 649 | |
| As* | Chemical adsorption/thiol group | 650 | |
| I-124 | Bolton–Hunter reagent | 143 | |
| Ti-45 | Chemical adsorption | 651 | |
| Tc-99m | SnCl2 reduction | 652 and 653 | |
| His-Tag | 654 | ||
| In-111 | Chemical adsorption | 211 | |
| I-125 | Bolton–Hunter reagent | 144 | |
| Y-90 | Chemical adsorption | 211 | |
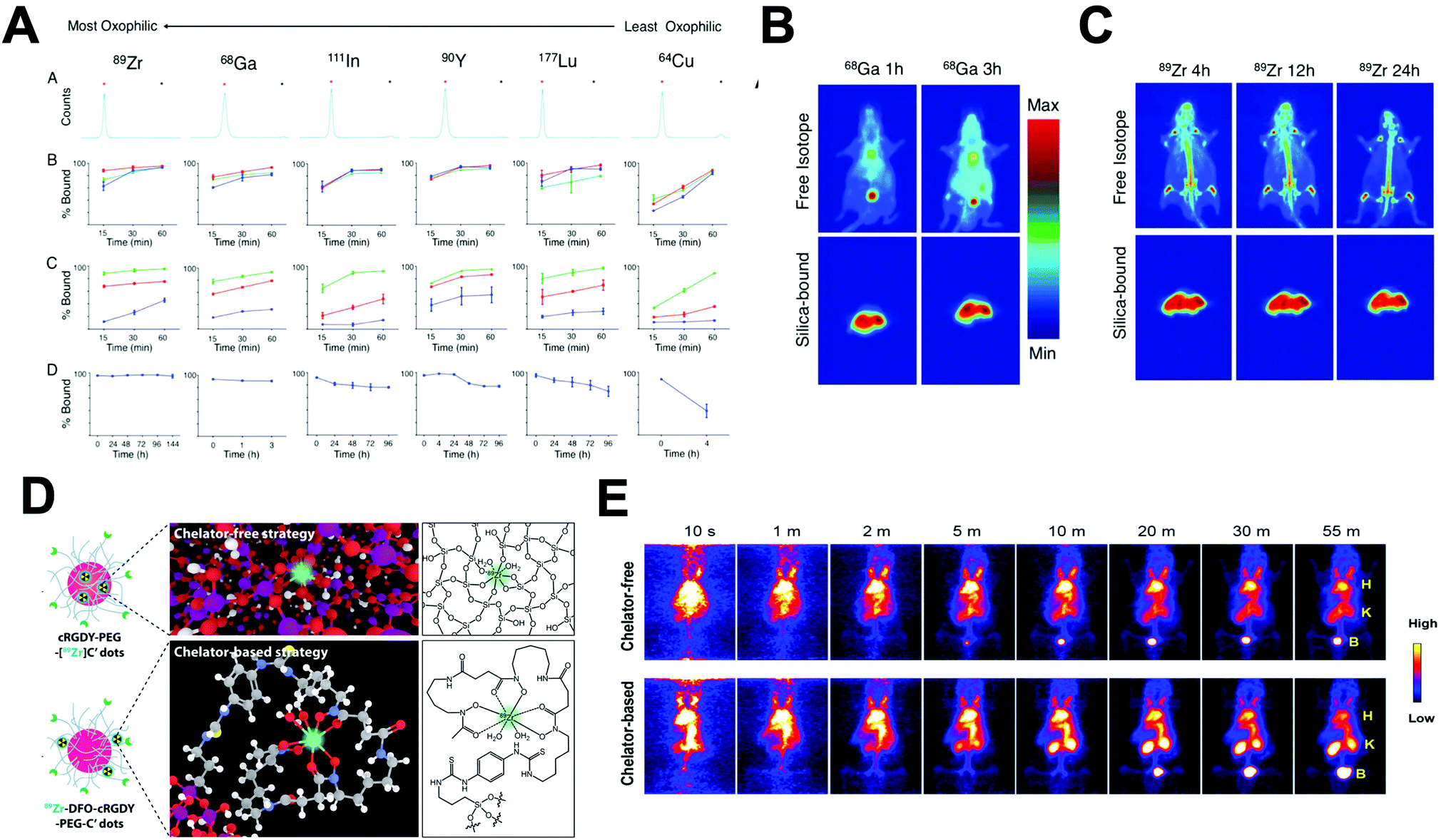 | ||
| Fig. 32 (A) Radiolabelling and serum stability of silica nanoparticles: (a) instant thin-layer chromatographs of radiolabelled silica nanoparticles. The red asterisk denotes the origin, where the nanoparticles remain, and the black asterisk denotes the solvent front, where the free activity would be located. Controls of buffer-only solutions (no particles) were performed with each condition with >95% signal at the free activity peak. (b) Percent radioisotope bound to silica nanoparticles as a function of time and pH. The blue, red, and green lines indicate radiolabelling at pH = 5.5, 7.3, and 8.8, respectively. (c) Percent radioisotope bound to silica nanoparticles as a function of time and temperature. The blue, red, and green lines indicate radiolabelling at 4, 37, and 70 °C, respectively. (d) Serum stability of silica nanoparticles radiolabelled at pH = 7.3 and 70 °C, then incubated in 50% FBS at 37 °C. (B) In vivo coronal PET maximum intensity projections (MIPs) of free (top) and silica-bound (bottom) 68Ga at 1 and 3 h post injection in athymic nude mice. (C) In vivo coronal PET maximum intensity projections (MIPs) of free (top) and silica-bound (bottom) 89Zr at 1 and 3 h post injection in athymic nude mice, (D) schematic representation of chelator-free cRGDY-PEG-[89Zr]C′ dots and chelator-based 89Zr-DFO-cRGDY-PEG-C′ dots, (E) comparison of dynamic PET imaging results in mice for chelator-free 89Zr-labeled cRGDY-PEG-C′ dots and chelator-based 89Zr-labeled cRGDY-PEG-C′ dots. H: heart; K: kidney; B: bladder. Adapted and reproduced with permission from ref. 34 and 211. | ||
As well as the above mentioned radionuclides, this strategy has also been evaluated to attach different radioarsenic isotopes, *As (* = 72, 76, 74, 71) and 45Ti demonstrating the high versatility of this radiolabelling method.650,651 Burke et al. used this strategy for the radiolabelling of iron oxide nanorods coated with silica. In this work, a series of nanorods conjugated with siloxane terminated DO3A chelator and a siloxane polyethylene glycol (PEG) derivative were radiolabelled with 68Ga at 90 °C for 15 min. The results showed quantitative RCY and 95% of RCS in human serum at 37 °C for 3 h in the sample without DO3A chelator showing the high affinity of the silica layer towards 68Ga.646
Similar to IONPs, it is difficult to highlight the most appropriate radiolabelling method for gold NPs due to the diversity of reported examples (Table 17) and each formulation should be considered individually. However, radiolabelling with 64Cu by both, chelator-based and chelator free methods and with 99mTc by chelator-based protocols make up a large portion of the literature. Moreover, due to the availability of different Au radionuclides, the radiolabelling by hot + cold precursors to incorporate 195Au, 198Au, or 199Au radionuclides into the crystal lattice of the particle is also a suitable strategy – as described in Section 4.4.1. Several radiolabelling strategies for gold NPs are based on the highly stable bonds that gold forms with sulfur. Campbell et al. reported the covalent attachment of thiolated gold NPs with DOTA-maleimide with further radiolabelling at different pH and temperatures (Fig. 33A). A high RCY of 96% was observed when the pH is increased from 5.5 to 8.8 without significant variations between room temperature and 60 °C reactions. RCS was also high at these conditions with a leaching of 4% of 64Cu after 24 hours of incubation in EDTA.659 PET/CT studies showed a rapid accumulation in liver and spleen when particles are administered intravenously. However, after oral administration, the images revealed a different pattern with initial accumulation in stomach and further uptake in small intestine, cecum, and large intestine with no presence of NPs after 24 h (Fig. 33B). Within the non-chelator strategies, a highly interesting report by Sun et al. described the reduction of 64Cu2+ over the surface of different PEGylated gold nanomaterials (Fig. 33C).660 In this radiolabelling reaction, 64Cu2+ is first reduced in hydrazine and exposed to the gold PEGylated nanomaterial in the presence of poly(acrylic acid) at room temperature. These conditions provided RCYs of near 100% after 1 h for gold NPs of different sizes (10, 20 and 80 nm) and most interestingly, with different shapes – such as spheres, rods and hexapods. In the absence of hydrazine, a decrease of the RCY to 30% was observed, highlighting the key role of the reducing agent. The RCS was also evaluated; with a 3% of 64Cu release after the incubation of the NPs for 24 h in PBS, further confirmed in vivo after an imaging biodistribution study showing different liver uptake profiles (Fig. 33D). Consequently, this technique clearly represents a highly versatile non-chelator method for the radiolabelling of AuNPs with 64Cu.
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 64Cu | DOTA | 659 and 661–665 |
| NODAGA | 666 | ||
| 68Ga/67Ga | DOTA | 667 and 668 | |
| 89Zr | DFO labelled antibody | 669 and 670 | |
| 99mTc | DTPA | 671 and 672 | |
| DOTA | 673 | ||
| HYNIC-TOC | 457 and 674 | ||
| HYNIC-GGC | 675–677 | ||
| 111In | DTPA | 136, 663, 678 and 679 | |
| DOTA | 680 | ||
| 177Lu | DOTA | 430, 674 and 681–683 | |
| 225Ac | DOTAGA | 684 | |
| Non-chelator | 64Cu | Reduction of [64Cu]Cu(0) onto the surface | 666 |
| Entrapment on a gold bilayer | 685 | ||
| Reduction of 64Cu on PEG surface | 660 | ||
| Hot + cold precursors/incorporation into crystal lattice | 686 and 687 | ||
| Hot + cold precursors/Au and Cu co-deposition | 688 | ||
| Hot + cold precursors/64Cu alloyed gold nanoclusters | 689 | ||
| 18F | Alkyne-nitrone cycloaddition | 196 | |
| [18F]SiFA-SH prosthetic group | 690 | ||
| [18F]-Fluorobenzoate | 189 | ||
| 124I | Isotopic exchange and anionic absorption | 691 | |
| Chloramine-T | 145 and 146 | ||
| 195Au | Hot + cold precursors/aerosol spark ignition | 105 | |
| 199Au | Hot + cold precursors/seed-mediated synthesis | 106 | |
| 198Au | Hot + cold precursors/S198AuNP | 107 | |
| Hot + cold precursors/(HAuCl4)–198Au precursor | 108 | ||
| 111In | Hot + cold precursors HAuCl4 + 111InCl3 | 692 | |
| 99mTc | SnCl2 reduction | 693 | |
| Doxorubicin/SnCl2 reduction | 694 | ||
| 123I | Azide–alkyne cycloaddition | 195 | |
| 125I | [125I]Azide-DBCO cycloaddition | 194 | |
| Chemisorption | 695 and 696 | ||
| Iodogen | 136 | ||
| 131I | HPAO/chloramine-T | 147 and 148 | |
| Chemisorption | 697 and 698 | ||
 | ||
| Fig. 33 (A) Schematic of Au–sulfur–maleimide–DOTA–64Cu NPs, (B) static microPET images at 2, 5, 24 and 48 h time points post IV or oral administration of radiolabelled SERS nanoparticles. Scale bar indicates % injected dose per gram (% ID per g) of tissue, (C) scheme of synthesis of chelator-free 64Cu-integrated Au NMs, (D) representative whole-body coronal PET images of mice at 2 and 24 h after intravenous injection of 130 μCi of [64Cu]80 nm Au (upper) as well as free 64Cu (lower), (E) design of the indium-111 labelled gold nanoparticles. Gold nanoparticles were synthesized with the particle core stably labelled with the γ-emitter indium-111 and the surface modified with linear and cyclic RGD ligands, (F) demonstrated higher uptake of RGD-modified indium-111 labeled gold nanoparticles in the M21 tumor (left) compared to the M21-L tumor (right). Adapted and reproduced with permission from ref. 659, 660 and 692. | ||
The hot + cold precursors method has also been extensively used not only for the radiolabelling with 64Cu, but also with 111In and different Au isotopes (195Au, 198Au and 199Au). Ng et al. synthesised Au NPs doped with 111In with further surface functionalisation with angiogenesis-targeting RGD peptides (Fig. 33E). The protocol rendered RCPs of 95% with high RCS after incubation in human plasma. In addition, SPECT/CT imaging allowed the identification of tumours in a manner relevant to integrin overexpression (Fig. 33F). Pang et al. reported a straightforward, one-step protocol where 199Au3+ is introduced in a low molar concentration during the growth step of the gold NPs (see Section 4.4.1).106 This synthesis – described as a seed-mediated synthesis due to the radionuclide doping during the growth step from a native Au seed – provided RCYs of 96%. This was shown to be reproducible irrespective of differences in specific activities, depending on the initial 199Au3+ concentration, and allowed quantitative RCP. This strategy seems as the most straightforward to incorporate Au radionuclides into the crystal lattice of gold NPs. Other reported strategies have utilised spark ignition to obtain gold NPs aerosols by a complex reaction set up, or used H198AuCl4 to prepare gold nanocages. However, these methods are highly complex and time-consuming.105,108
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 64Cu | DOTA | 702 |
| 99mTc | EDTA | 703 and 704 | |
| 2,3-Diaminopropionic acid (DAP) | 705 | ||
| Dithiocarbamate (DTC) derivatised bisbiotin ligand | 706 | ||
| Non-chelator | 64Cu | Radioisotopic exchange | 204 and 701 |
| Reduction by ascorbic acid | 707 | ||
| Hot + cold precursors/64CuCl2 starting reagent | 122 | ||
| 68Ga | Radioisotopic exchange | 701 | |
| MCM-41 thiol group | 708 | ||
| 99mTc | SnCl2 reduction | 709 | |
| 111In | Interface layer deposition | 710 | |
| 125I | Nucleophilic substitution Mannose triflate-cysteamine | 188 | |
| Bolton–Hunter reagent | 150 | ||
| Hytohemagglutinin-L (PHA-L)/iodogen | 711 | ||
| 131I | Nucleophilic substitution Mannose triflate-cysteamine | 188 | |
| 125mTe | Hot + cold precursors/mixture of 125mTe/124Te | 123 | |
| 109Cd | Hot + cold precursors | 121 | |
The hot + cold precursors method has been applied for the radiolabelling of QDS with 64Cu, 109Cd or 125mTe, which were successfully incorporated into the core of CuInS/ZnS, CdSe/CdZnS or CdTeSe/CdZnSe and CdTe QDs respectively.121–123 A key study by Guo et al. reported a one-pot synthesis of [64Cu]CIS/ZnS QDs from 64CuCl2/CuCl2, InCl3 and Na2S along with a ZnCl2 shell formation and then in situ capped with PEG and glutathione (GSH) (Fig. 34A).122 These QDs showed fluorescence in the near infrared (NIR) with a quantum yield (QY) of 25%. From the radiochemical point of view, this synthesis provided a RCY of ca. 100% and quantitative RCS in mouse serum. In vivo biodistribution studies in tumour bearing mice showed higher accumulation of the PEGylated GSH-[64Cu]CIS/ZnS in the tumour than GSH-[64Cu]CIS/ZnS and free 64CuCl2 due to the longer blood circulation time of the former that allows accumulation due to the EPR phenomenon. Despite the significant tumour uptake level differences, It is important to highlight the high tumour uptake of free 64CuCl2, that as we discussed above (Section 3.5 and Table 5) shows significant tumour uptake and similar biodistribution compared to both [64Cu]CIS/ZnS formulations, and may complicate image analysis. Interestingly, non-PEGylated GSH-[64Cu]CIS/ZnS showed significant bladder uptake due to the renal clearance of NPs that are smaller that 5.5 nm (Fig. 34B). These results highlight this method as suitable for the synthesis and radiolabelling of QDs in one step. Additionally, the protocol allowed the radiolabelling of QDs with 111In with the same favourable results, suggesting this protocol is a versatile and convenient strategy for QDs radiolabelling.
 | ||
| Fig. 34 (A) Illustration of the synthesis of intrinsically radioactive [64Cu]CIS/ZnS QDs, (B) representative whole-body coronal PET images of U87MG tumour-bearing mice at 2, 6, 18, 24, and 48 h after intravenous injection of 100 μL (50 μg, 300 μCi) of 64CuCl2, GSH-[64Cu]CIS/ZnS and PEGylated GSH-[64Cu]CIS/ZnS RQDs. Arrow indicates location of the tumour. Adapted and reproduced with permission from ref. 122. | ||
Radiolabelling examples of these nanomaterials are quite recent with the first reports in 2011 (Table 19). As discussed in Section 4.4.4, the isotopic exchange between natural 19F and 18F is the main protocol for effective and robust radiolabelling of UCNPs. Other methods have also demonstrated successful incorporation of radionuclides. Rieffel et al. reported a chelator-based protocol using a porphyrin–phospholipid as NP coating.714 The coating provided a high affinity for 64Cu by incubation at 37 °C and pH = 5.5 with RCYs greater than 80%. Interestingly, the combination of only two components, the porphyrin–phospholipid and the UCNP core, rendered excellent capabilities for six different imaging modalities (FL/PA/PET/CT/CL/UC). Alternatively, Yang et al. described a one-pot hydrothermal synthesis of NaLuF4:153Sm,Yb,Tm NPs.112 The use of [153Sm]SmCl3 in the starting reagents provided a 100% RCY with >99% RCS after incubation in FBS for 24 h. This high stability was expected since the 153Sm is incorporated in the crystal lattice of the particles.
| Radiolabelling method | Radionuclide | Radiolabelling mediator | Ref. |
|---|---|---|---|
| Chelator-based | 64Cu | NOTA | 715 |
| Porphyrin | 714 | ||
| Bisphosphonate | 716 | ||
| 68Ga | DOTA | 717 | |
| 99mTc | Bisphosphonate | 716 | |
| Non-chelator | 18F | Radioisotopic exchange | 198–201 |
| 124I | Iodo-beads | 149 | |
| 125I | Bolton–Hunter reagent | 135 | |
| 153Sm | Hot + cold precursors | 112 | |
| Radioisotopic exchange | 205 | ||
| 90Y | Hot + cold precursors | 120 | |
5 Applications of radiolabelled nanomaterials in imaging and therapy
The radiolabelling of nanomaterials can be performed for a variety of different applications with their use encompassing pre-clinical validation all the way to imaging in a clinical setting. In this section, the main applications for using radiolabelled NPs will be outlined, with important and/or interesting examples briefly discussed.5.1 Assessment of new formulations
One of the most widely used applications of radiolabelled nanomaterials is to assess the in vivo biodistribution of novel formulations. The sensitivity and quantitative nature of nuclear imaging easily allows elucidation of whole-body pharmacokinetics, biodistribution and target accumulation of different nanomaterials. A good example of this was reported by Tang et al. who created a library of lipoprotein-based NPs as candidates for atherosclerosis treatment. The leading candidates were radiolabelled with 89Zr, and their in vivo behaviour evaluated with PET imaging.366 This allowed comparison of their blood pharmacokinetics and uptake in organs of interest. As well as testing novel formulations, another key application is imaging the distribution of targeted NPs versus their non-targeted counterparts. For example, Yang et al. radiolabelled cRGD-functionalised SPIONs with 64Cu; demonstrating increased tumour accumulation in mice for the targeted NPs – compared with non-targeted SPIONs – which could be blocked via the administration of cRGD (Fig. 35A).718 On the other hand, Christensen et al. used PET imaging of folate-targeted liposomes labelled with 64Cu to show there was lower uptake in high folate-receptor expressing tumours, compared with non-targeted liposomes.719 This suggested that the functionalised used for targeting may result in reduction of EPR-mediated uptake of the liposomes. | ||
| Fig. 35 Summary of the five main applications of radiolabelled nanoparticles focused on in this review. (A) The radiolabelling of nanoparticles can allow the testing of new formulations and the assessment of NP targeting. PET images of U87MG tumor-bearing mice at various time points post-injection of 64Cu-labeled SPIO nanocarriers (cRGD-conjugated, cRGD-free, and cRGD-conjugated with a blocking dose of cRGD). Figure adapted from Yang et al.718 (B) Radiolabelled nanoparticles can aid the clinical translation of nanoparticles and assess target engagement in patients: personalised nanomedicines. Maximum intensity projection PET images of 2 patients with HER2-positive breast cancer injected with 64Cu-labelled HER2-targeted liposomes (top). Patient lesion deposition of the lowest uptake lesion within each patient from days 2 or 3 are shown and aligned in ascending order (bottom left). Patient PFS of the high versus low uptake patients is shown in a Kaplan–Meier curve (bottom right). Figures adapted from Lee et al.326 (C) Radiolabelled nanoparticles can be used for the diagnosis of disease, in this case by exploiting multimodal PET-MR imaging and 68Ga-labelled SPIOs to locate and identify metastatic lymph nodes. Figure adapted from Savolainen et al.720 (D) Radiolabelled nanoparticles can be used as cell labelling agents, allowing the in vivo tracking of cells. A single MDA-MB-231 cells is imaged with PET in the paw (left) and lung (right) of a mouse. Figure from Jung et al.721 (E) Nanoparticles can be used for radiotherapy. CT images for patients with esophageal cancer and lung metastases tumors: 1 month before (left column)and 4 months after (middle column) administration of 188Re-liposome injection, the metastatic lesions (green arrows) either decrease in size or show signs of cavitation (red arrows). SPECT/CT images (right column) show a high uptake and efficient targeting of 188Re-liposome in the corresponding tumor lesion (blue arrow). Figure adapted from Wang et al.722 | ||
5.2 Personalised nanomedicine
A key application of radiolabelled nanomaterials is for the assessment of target accumulation in the patients undergoing treatment with nanomedicines. The EPR mechanism that often drives the accumulation of nanomedicines in target tissues is highly heterogenous in humans.723–725 However, by imaging nanomedicines non-invasively within patients, they can be grouped into potential responders and non-responders allowing treatment stratification – a concept known as personalised nanomedicine.726 A key clinical study was reported by Lee et al. who radiolabelled HER2-targeted liposomal doxorubicin (MM-302) with 64Cu and performed PET imaging in patients with metastatic breast cancer.326 PET imaging showed heterogenous uptake of the liposomes in primary tumours and metastases; both from patient-to-patient and within lesions within the same patient. Despite this, a correlation was observed between tumour uptake of MM-302 and the patient's disease progression-free survival (Fig. 35B). As opposed to radiolabelling and imaging specific nanomedicines to assess their target accumulation, a potentially more robust method is to inject a ‘companion diagnostic’ which behaves similarly to or demonstrates EPR-mediated uptake. This concept has been previously demonstrated using the iron-oxide nanoparticle, Ferumoxytol.727 Both Perez-Medina et al.271 and Lee et al.327 developed radiolabelled nanoliposome platforms that could be injected both prior-to or with the injection of nanomedicines – allowing prediction of therapeutic response in preclinical cancer models.5.3 Diagnostics
Another application of radiolabelled nanomaterials is their use for diagnostics. The EPR-mediated uptake of NPs into tumours or sites of inflammation can clearly be taken advantage for diagnostic purposes. Mahakian et al. compared the diagnostic potential of long-circulating liposomes radiolabelled with 64Cu with [18F]FDG in a head and neck cancer mouse model. The tumour accumulation and signal to background ratios of the labelled liposomes were superior to [18F]FDG when imaging after 24 h.260 However, in a clinical setting, the requirement for delayed imaging may be limiting and the longer half-life of the radioisotope may also result in a higher absorbed radiation dose. Additionally, radiolabelled NPs can be used as multimodal systems; to take advantage of the sensitivity of nuclear imaging in conjunction with more anatomical-focused modalities – such as MRI and CT.139 However, the use of NPs as multimodal diagnostics should arguably provide benefits over lower MW tracers, and when using single modalities. An example of this approach was reported by Savolainen et al. who radiolabelled Sienna+/Magtrace® (a macrophage-targeted clinically-approved SPIO) with 68Ga, as a tool for PET-MRI guided sentinel lymph node (SLN) biopsies in metastatic cancer.720 In this approach, PET provides sensitive whole-body information of the location of the SLNs, while MRI provides information on their different macrophage levels and hence metastatic status (Fig. 35C).5.4 Cell tracking
Cell tracking describes the use of medical imaging techniques to non-invasively image the biodistribution and trafficking of cells in a living organism. This information can be beneficial for: disease diagnosis, the imaging of biological mechanisms, and evaluating the efficacy of treatments.728 More recently, cell tracking methods have been used for the development and evaluation of cellular therapies – such as CAR T-cell immunotherapies. To allow cell tracking, cells often have to labelled with a contrast agent. For example, for cell tracking with nuclear imaging, radionuclides have to be incorporated into cells; often by their attachment to cellular membranes, or internalisation via the use of radio-ionophores (as in Section 4.3.1). Alternatively, nanomaterials can enter cells through a variety of different mechanisms including such as phagocytosis, endocytosis or micropinocytosis.729 Hence these mechanisms can be taken advantage of to enable the radiolabelling of cells, to facilitate in vivo cell tracking. A variety of different cell types have been radiolabelled with chitosan-based NPs,503,504 gold NPs,730,731 SPIONs,213,610,732 and mesoporous silica NPs.721,733 The efficiency of this cell labelling process is exemplified by Jung et al. who used 68Ga-labelled MSNPs to radiolabel breast cancer cells with enough activity (ca. 30 Bq per cell) to allow the in vivo tracking of a single cell using PET (Fig. 35D).7215.5 Radionuclide therapy
Finally, nanomaterials can be labelled with therapeutic radionuclides (see Section 2.3) allowing them to be used for radionuclide therapy. As with their use as diagnostics, the accumulation of nanomaterials at tumour sites via the EPR effect or due to targeting can allow delivery of the radionuclide. However, the long-circulating properties of some nanomaterials may be considered a drawback in the context of radionuclide therapy due to the possible increase in radiation dose to the patient and non-target organs – such as the spleen. Wang et al. recently reported a phase 0 study of PEGylated liposomes radiolabelled with 188Re (beta emitter) in patients with metastatic cancer.722 A partial therapeutic effect was observed in some metastatic lesions, which also showed uptake of the radiolabelled liposomes (Fig. 35E). Despite this, dosimetry measurements showed the highest absorbed dose was in the spleen and liver. Whilst this work demonstrates clear progress in the use of nanomaterials for radiotherapy, the application of NPs for this purpose must show clear benefits – in terms of efficacy and vital organ dosimetry – over standard targeted radionuclide therapy methods (i.e. radiolabelled antibodies or small molecules).Besides radiotherapy applications, a novel therapeutic strategy based on Cerenkov luminescence (CL) is becoming a successful choice to induce a photodynamic therapy (PDT) response. This strategy, known as Cerenkov radiation-induced therapy (CRIT) leverage from the UV-blue light generated by the radionuclide decay that interacts with a photosensitive nanomaterial triggering the emission of long-wavelength photons that produce cytotoxic reactive oxygen species (ROS).734,735 Most of reported examples of CRIT applications are based on the combination of TiO2 nanoparticles as photosensitiser material with 68Ga, 64Cu, 18F or 89Zr radionuclides to generate the CL.736–739 Apart of TiO2 nanoparticles, other materials such as iron oxide NPs and porphyrins have shown a remarkable PDT response in combination with 89Zr,740,741 opening a very encouraging field within personalised nanomedicine due to the unique properties of the radiolabelled nanomaterials.
6 Conclusions and perspectives
In this review we have described and discussed the different radiochemical methods explored to date to radiolabel different nanomaterials of interest for medical applications, with applications in imaging and therapy. We believe there are several conclusions that we can draw from this work and should be taken into account when considering the best methodology for a specific NP-radiolabelling project. First, for each nanomaterial and formulation, each radiochemical approach available will have inherent advantages and disadvantages. Ultimately, the selection of methodology should be driven by its capabilities to provide the required information, but this is only achievable if we are aware of its pitfalls (known and potential). To facilitate this, we strongly believe it is important to incorporate as many control groups as required into any study involving the in vivo evaluation of radiolabelled NPs (e.g. radionuclide-only, radiochelate-only, radiolabelled component-only, etc.). Where relevant, it is also important to include ex vivo information to complement and support the in vivo results (e.g. histology methods). A particularly relevant aspect to take into account for in vivo studies is the fact that many radionuclides and radiochelates accumulate in, or are cleared by, the same organs as nanomaterials (e.g. liver, bone marrow, lungs, tumours), complicating image analysis in the absence of appropriate controls. Often, the selection of radiolabelling methodology will be limited by radionuclide/equipment availability, as this type of work requires special health and safety considerations and expensive facilities. However, we believe that the wide variety of methodologies and substantial knowledge already available for different radionuclides and nanomaterials, as described in this review, should allow ample choice for effective and informative radionuclide-based NP studies.The same properties of nanomaterials that make them attractive for biomedical applications, result in some important disadvantages when compared to other imaging/therapy platforms. Importantly, their relatively large size and surface area makes them easy targets for phagocytic cells, limiting their ability to reach their intended target. This can be in part addressed by the use of stealth coatings, but ultimately their excretion is likely to involve the mononuclear phagocyte system (MPS), which is slow compared to the renal excretion often found for smaller molecules. In addition, this results in low tissue penetration, limiting their potential as imaging agents and drug carriers when the target is not easily accessible from the vasculature. However, some of these disadvantages can be exploited for specific purposes, for example by using NPs as myeloid cell-targeting agents, with wide-ranging applications from inflammation imaging to novel therapeutic approaches such as those based on trained-immunity.742–744
In terms of future applications, it is likely that radiolabelled NPs will be increasingly used as tools to inform the clinical translation of nanoparticles as therapeutics, and potentially as theranostic agents. As mentioned above, radiolabelling allows an accurate and sensitive method to study their biodistribution and pharmacokinetics in both animals and humans. Hence, we strongly believe that integrating these techniques early in the development of therapeutic nanomedicines will significantly enhance their clinical translation potential, by allowing the selection of the best candidates and de-risking the process. If a clear association between the biodistribution of a therapeutic NP and their biodistribution can be made, radiolabelled NPs have significant potential as predictors of efficacy. The capacity of NPs to carry several radionuclides per particle also makes them attractive as radionuclide therapy agents, but the slow excretion and retention of NPs remains a significant barrier to overcome that limits their potential. Finally, new developments in PET technology – specifically total-body PET44,45 – provide exciting opportunities for NP-based biomedical applications. These are mainly driven by the increased sensitivity (allowing NP imaging with short-lived radionuclides and lower radiation doses). We believe this will facilitate more PET imaging-based clinical evaluation of novel nanomedicines in the future, increasing the impact of this technology in the effective clinical translation of novel nanomedicines.
Conflicts of interest
Research in the RTMR group has received funding by AstraZeneca plc (PhD studentship), GlaxoSmithKline plc (PhD studentship), Theragnostics Ltd (PhD studentship) and Lipomedix Pharmaceutical Ltd (in kind contributions).Acknowledgements
The authors are funded by EPSRC programme grants EP/S032789/1 (Probing Multiscale Complex Multiphase Flows with Positrons for Engineering and Biomedical Applications) and EP/R045046/1 (Next Generation Molecular Imaging and Therapy with Radionuclides). We also acknowledge support from the Wellcome/EPSRC Centre for Medical Engineering [WT/203148/Z/16/Z] and the KCL and UCL Comprehensive Cancer Imaging Centre funded by CRUK and EPSRC in association with the MRC and DoH (England). Radioanalytical equipment was funded by a Wellcome Trust Multiuser Equipment Grant: A multiuser radioanalytical facility for molecular imaging and radionuclide therapy research [212885/Z/18/Z]. The authors finally acknowledge support the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and KCL [grant number IS-BRC-1215-20006]. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.References
- B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopeček, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. Ling Lim, L. M. Liz-Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X.-E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 CrossRef.
- S. T. Duggan and G. M. Keating, Drugs, 2011, 71, 2531–2558 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 Search PubMed.
- F. C. Passero, D. Grapsa, K. N. Syrigos and M. W. Saif, Expert Rev. Anticancer Ther., 2016, 16, 697–703 CrossRef CAS.
- R. A. Schwendener, Ther. Adv. Vaccines, 2014, 2, 159–182 CrossRef CAS.
- H.-I. Chang and M.-K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- D. Girelli, S. Ugolini, F. Busti, G. Marchi and A. Castagna, Int. J. Hematol., 2018, 107, 16–30 CrossRef CAS.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS.
- S. Kunjachan, J. Ehling, G. Storm, F. Kiessling and T. Lammers, Chem. Rev., 2015, 115, 10907–10937 CrossRef CAS.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS.
- R. Bar-Shalom, A. Y. Valdivia and M. D. Blaufox, Semin. Nucl. Med., 2000, 30, 150–185 CrossRef CAS.
- E. C. Pratt, T. M. Shaffer and J. Grimm, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 872–890 CAS.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS.
- S. Mura and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 1394–1416 CrossRef CAS.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS.
- S. D. Jo, S. H. Ku, Y.-Y. Won, S. H. Kim and I. C. Kwon, Theranostics, 2016, 6, 1362–1377 CrossRef CAS.
- M. Hamoudeh, M. A. Kamleh, R. Diab and H. Fessi, Adv. Drug Delivery Rev., 2008, 60, 1329–1346 CrossRef CAS.
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897–965 CrossRef CAS.
- C. Pérez-Medina, A. J. P. Teunissen, E. Kluza, W. J. M. Mulder and R. van der Meel, Adv. Drug Delivery Rev., 2020, 154–155, 123–141 CrossRef.
- K. Heinzmann, L. M. Carter, J. S. Lewis and E. O. Aboagye, Nat. Biomed. Eng., 2017, 1, 697–713 CrossRef.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS.
- X. Han, K. Xu, O. Taratula and K. Farsad, Nanoscale, 2019, 11, 799–819 RSC.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- I. Fernández-Barahona, M. Muñoz-Hernando, J. Ruiz-Cabello, F. Herranz and J. Pellico, Inorganics, 2020, 8 Search PubMed.
- J. Y. Park, M. J. Baek, E. S. Choi, S. Woo, J. H. Kim, T. J. Kim, J. C. Jung, K. S. Chae, Y. Chang and G. H. Lee, ACS Nano, 2009, 3, 3663–3669 CrossRef CAS.
- J. Pellico, J. Ruiz-Cabello, I. Fernández-Barahona, L. Gutiérrez, A. V. Lechuga-Vieco, J. A. Enríquez, M. P. Morales and F. Herranz, Langmuir, 2017, 33, 10239–10247 CrossRef CAS.
- M. L. Senders, A. E. Meerwaldt, M. M. T. van Leent, B. L. Sanchez-Gaytan, J. C. van de Voort, Y. C. Toner, A. Maier, E. D. Klein, N. A. T. Sullivan, A. M. Sofias, H. Groenen, C. Faries, R. S. Oosterwijk, E. M. van Leeuwen, F. Fay, E. Chepurko, T. Reiner, R. Duivenvoorden, L. Zangi, R. M. Dijkhuizen, S. Hak, F. K. Swirski, M. Nahrendorf, C. Pérez-Medina, A. J. P. Teunissen, Z. A. Fayad, C. Calcagno, G. J. Strijkers and W. J. M. Mulder, Nat. Nanotechnol., 2020, 1–8 Search PubMed.
- P. Chhour, P. C. Naha, S. M. O’Neill, H. I. Litt, M. P. Reilly, V. A. Ferrari and D. P. Cormode, Biomaterials, 2016, 87, 93–103 CrossRef CAS.
- S. A. Peyman, J. R. McLaughlan, R. H. Abou-Saleh, G. Marston, B. R. G. Johnson, S. Freear, P. L. Coletta, A. F. Markham and S. D. Evans, Lab Chip, 2016, 16, 679–687 RSC.
- J. Gao, K. Chen, R. Luong, D. M. Bouley, H. Mao, T. Qiao, S. S. Gambhir and Z. Cheng, Nano Lett., 2012, 12, 281–286 CrossRef.
- A. de la Zerda, Z. Liu, S. Bodapati, R. Teed, S. Vaithilingam, B. T. Khuri-Yakub, X. Chen, H. Dai and S. S. Gambhir, Nano Lett., 2010, 10, 2168–2172 CrossRef CAS.
- S. Imlimthan, S. Otaru, O. Keinänen, A. Correia, K. Lintinen, H. A. Santos, A. J. Airaksinen, M. A. Kostiainen and M. Sarparanta, Biomacromolecules, 2019, 20, 674–683 CrossRef CAS.
- F. Chen, K. Ma, L. Zhang, B. Madajewski, P. Zanzonico, S. Sequeira, M. Gonen, U. Wiesner and M. S. Bradbury, Chem. Mater., 2017, 29, 8269–8281 CrossRef CAS.
- J. Kim, P. Chhour, J. Hsu, H. I. Litt, V. A. Ferrari, R. Popovtzer and D. P. Cormode, Bioconjugate Chem., 2017, 28, 1581–1597 CrossRef CAS.
- M. Shilo, T. Reuveni, M. Motiei and R. Popovtzer, Nanomedicine, 2012, 7, 257–269 CrossRef CAS.
- D. Xi, S. Dong, X. Meng, Q. Lu, L. Meng and J. Ye, RSC Adv., 2012, 2, 12515–12524 RSC.
- H.-D. Liang and M. J. K. Blomley, Br. J. Radiol., 2003, 76, S140–S150 CrossRef CAS.
- K. Christensen-Jeffries, O. Couture, P. A. Dayton, Y. C. Eldar, K. Hynynen, F. Kiessling, M. O’Reilly, G. F. Pinton, G. Schmitz, M. X. Tang, M. Tanter and R. J. G. van Sloun, Ultrasound Med. Biol., 2020, 46, 865–891 CrossRef.
- V. Ntziachristos and D. Razansky, Chem. Rev., 2010, 110, 2783–2794 CrossRef CAS.
- L. Zhou, H. Zhou and C. Wu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, e1510 Search PubMed.
- M. M. Khalil, J. L. Tremoleda, T. B. Bayomy and W. Gsell, Int. J. Mol. Imaging, 2011, 2011, 1–15 CrossRef.
- T. Fukuchi, T. Okauchi, M. Shigeta, S. Yamamoto, Y. Watanabe and S. Enomoto, Med. Phys., 2017, 44, 2257–2266 CrossRef CAS.
- S. R. Cherry, T. Jones, J. S. Karp, J. Qi, W. W. Moses and R. D. Badawi, J. Nucl. Med., 2018, 59, 3–12 CrossRef CAS.
- R. D. Badawi, H. Shi, P. Hu, S. Chen, T. Xu, P. M. Price, Y. Ding, B. A. Spencer, L. Nardo, W. Liu, J. Bao, T. Jones, H. Li and S. R. Cherry, J. Nucl. Med., 2019, 60, 299–303 CrossRef CAS.
- M. Silindir-Gunay, M. Karpuz and A. Y. Ozer, Cancer Biother. Radiopharm., 2020, 35(6), 446–458, DOI:10.1089/cbr.2019.3213.
- J. Jeon, Int. J. Mol. Sci., 2019, 20, 2323 CrossRef CAS.
- S. Poty, L. C. Francesconi, M. R. McDevitt, M. J. Morris and J. S. Lewis, J. Nucl. Med., 2018, 59, 878–884 CrossRef CAS.
- S. Aghevlian, A. J. Boyle and R. M. Reilly, Adv. Drug Delivery Rev., 2017, 109, 102–118 CrossRef CAS.
- K. E. Baidoo, K. Yong and M. W. Brechbiel, Clin. Cancer Res., 2013, 19, 530–537 CrossRef CAS.
- A. Ku, V. J. Facca, Z. Cai and R. M. Reilly, EJNMMI Radiopharm. Chem., 2019, 4, 27 CrossRef.
- K. P. Willowson, Eur. J. Phys., 2019, 40, 043001 CrossRef.
- K. Peach, P. Wilson and B. Jones, Br. J. Radiol., 2011, 84(1), S4–S10 CrossRef.
- A. Dash and R. Chakravarty, Am. J. Nucl. Med. Mol. Imaging, 2019, 9, 30–66 CAS.
- J. Magill and J. Galy, Radioactivity Radionuclides Radiation, Springer-Verlag, Berlin/Heidelberg, 2005 Search PubMed.
- K. F. Eckerman and A. Endo, MIRD: radionuclide data and decay schemes, Society of Nuclear Med, 2008 Search PubMed.
- A. Boschi, L. Uccelli and P. Martini, Appl. Sci., 2019, 9, 2526 CrossRef CAS.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC.
- G. Vaidyanathan and M. R. Zalutsky, Radiopharmaceutical Chemistry, Springer International Publishing, Cham, 2019, pp. 391–408 Search PubMed.
- J. Ródenas and E. Jabaloyas, J. Radioanal. Nucl. Chem., 2019, 322, 1691–1695 CrossRef.
- Fluorine and Health, ed. A. Tressaud and G. Haufe, Elsevier, 2008 Search PubMed.
- M. M. Alauddin, Am. J. Nucl. Med. Mol. Imaging, 2012, 2, 55–76 CAS.
- I. Verel, G. W. M. Visser, R. Boellaard, M. Stigter-van Walsum, G. B. Snow and G. A. M. S. van Dongen, J. Nucl. Med., 2003, 44, 1271–1281 CAS.
- J. Zhang-Yin, C. Provost, G. Cancel-Tassin, T. Rusu, M. Penent, C. Radulescu, E. Comperat, O. Cussenot, F. Montravers, R. Renard-Penna, J.-N. Talbot and A. Prignon, Nucl. Med. Biol., 2020, 84–85, 88–95 CrossRef CAS.
- C.-H. Yeong, M. Cheng and K.-H. Ng, J. Zhejiang Univ., Sci., B, 2014, 15, 845–863 CrossRef CAS.
- C. Parker, S. Nilsson, D. Heinrich, S. I. Helle, J. M. O’Sullivan, S. D. Fosså, A. Chodacki, P. Wiechno, J. Logue, M. Seke, A. Widmark, D. C. Johannessen, P. Hoskin, D. Bottomley, N. D. James, A. Solberg, I. Syndikus, J. Kliment, S. Wedel, S. Boehmer, M. Dall’Oglio, L. Franzén, R. Coleman, N. J. Vogelzang, C. G. O’Bryan-Tear, K. Staudacher, J. Garcia-Vargas, M. Shan, Ø. S. Bruland and O. Sartor, N. Engl. J. Med., 2013, 369, 213–223 CrossRef CAS.
- C. Kratochwil, F. Bruchertseifer, F. L. Giesel, M. Weis, F. A. Verburg, F. Mottaghy, K. Kopka, C. Apostolidis, U. Haberkorn and A. Morgenstern, J. Nucl. Med., 2016, 57, 1941–1944 CrossRef CAS.
- T. Langbein, W. A. Weber and M. Eiber, J. Nucl. Med., 2019, 60, 13S–19S CrossRef CAS.
- C. Kratochwil, U. Haberkorn and F. L. Giesel, Semin. Nucl. Med., 2020, 50, 133–140 CrossRef.
- T. E. Witzig, L. I. Gordon, F. Cabanillas, M. S. Czuczman, C. Emmanouilides, R. Joyce, B. L. Pohlman, N. L. Bartlett, G. A. Wiseman, N. Padre, A. J. Grillo-López, P. Multani and C. A. White, J. Clin. Oncol., 2002, 20, 2453–2463 CrossRef CAS.
- H. Herzog, F. Rösch, G. Stöcklin, C. Lueders, S. M. Qaim and L. E. Feinendegen, J. Nucl. Med., 1993, 34, 2222–2226 CAS.
- S. M. Qaim, B. Scholten and B. Neumaier, J. Radioanal. Nucl. Chem., 2018, 318, 1493–1509 CrossRef CAS.
- F. Man, P. J. Gawne and R. T. M. de Rosales, Adv. Drug Delivery Rev., 2019, 143, 134–160 CrossRef CAS.
- M. Quastel, A. Richter and J. Levy, Br. J. Cancer, 1990, 62, 885–890 CrossRef CAS.
- P. R. Franken, J. Guglielmi, C. Vanhove, M. Koulibaly, M. Defrise, J. Darcourt and T. Pourcher, Thyroid, 2010, 20, 519–526 CrossRef CAS.
- B. N. Bottenus, P. Kan, T. Jenkins, B. Ballard, T. L. Rold, C. Barnes, C. Cutler, T. J. Hoffman, M. A. Green and S. S. Jurisson, Nucl. Med. Biol., 2010, 37, 41–49 CrossRef CAS.
- J. Czernin, N. Satyamurthy and C. Schiepers, J. Nucl. Med., 2010, 51, 1826–1829 CrossRef CAS.
- R. G. Sephton, G. S. Hodgson, S. De Abrew and A. W. Harris, J. Nucl. Med., 1978, 19, 930–935 CAS.
- H. M. Chilton, R. L. Witcofski, N. E. Watson and C. M. Heise, J. Nucl. Med., 1981, 22, 1064–1068 CAS.
- J. Spetz, N. Rudqvist and E. Forssell-Aronsson, Cancer Biother. Radiopharm., 2013, 28, 657–664 CrossRef CAS.
- R. Chakravarty, S. Chakraborty and A. Dash, Mol. Pharmaceutics, 2016, 13, 3601–3612 CrossRef CAS.
- A. Piccardo, F. Paparo, M. Puntoni, S. Righi, G. Bottoni, L. Bacigalupo, S. Zanardi, A. DeCensi, G. Ferrarazzo, M. Gambaro, F. G. Ruggieri, F. Campodonico, L. Tomasello, L. Timossi, S. Sola, E. Lopci and M. Cabria, J. Nucl. Med., 2018, 59, 444–451 CrossRef CAS.
- D. S. Abou, T. Ku and P. M. Smith-Jones, Nucl. Med. Biol., 2011, 38, 675–681 CrossRef CAS.
- S. A. Graves, R. Hernandez, J. Fonslet, C. G. England, H. F. Valdovinos, P. A. Ellison, T. E. Barnhart, D. R. Elema, C. P. Theuer, W. Cai, R. J. Nickles and G. W. Severin, Bioconjugate Chem., 2015, 26, 2118–2124 CrossRef CAS.
- A. Vakili, A. R. Jalilian, K. Yavari, S. Shirvani-Arani, A. Khanchi, A. Bahrami-Samani, B. Salimi and A. Khorrami-Moghadam, J. Radioanal. Nucl. Chem., 2013, 296, 1287–1294 CrossRef CAS.
- W. A. Breeman, K. van der Wansem, B. F. Bernard, A. van Gameren, J. L. Erion, T. J. Visser, E. P. Krenning and M. de Jong, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 312–315 CrossRef CAS.
- E. Dadachova, B. Bouzahzah, L. Zuckier and R. Pestell, Nucl. Med. Biol., 2002, 29, 13–18 CrossRef CAS.
- S. Hanadate, K. Washiyama, M. Yoshimoto, H. Matsumoto, A. Tsuji, T. Higashi and Y. Yoshii, J. Nucl. Med., 2017, 58, 1030 Search PubMed.
- P. Sukthankar, L. A. Avila, S. K. Whitaker, T. Iwamoto, A. Morgenstern, C. Apostolidis, K. Liu, R. P. Hanzlik, E. Dadachova and J. M. Tomich, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2296–2305 CrossRef CAS.
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike and A. D. Windhorst, Ann. Nucl. Med., 2018, 32, 236–238 CrossRef.
- C. C. Wagner and O. Langer, Adv. Drug Delivery Rev., 2011, 63, 539–546 CrossRef CAS.
- J. J. M. de Goeij and M. L. Bonardi, J. Radioanal. Nucl. Chem., 2005, 263, 13–18 CrossRef CAS.
- S. E. Lapi and M. J. Welch, Nucl. Med. Biol., 2013, 40, 314–320 CrossRef CAS.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier Inc., 3rd edn, 2013 Search PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier, 2008, pp. 169–212 Search PubMed.
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS.
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720 RSC.
- B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168 RSC.
- A. Steinbrueck, A. C. Sedgwick, J. T. Brewster, K.-C. Yan, Y. Shang, D. M. Knoll, G. I. Vargas-Zúñiga, X.-P. He, H. Tian and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 3726–3747, 10.1039/C9CS00373H.
- S. Goel, F. Chen, E. B. Ehlerding and W. Cai, Small, 2014, 10, 3825–3830 CrossRef CAS.
- J. Lamb and J. P. Holland, J. Nucl. Med., 2018, 59, 382–389 CrossRef CAS.
- W. G. Kreyling, W. Möller, U. Holzwarth, S. Hirn, A. Wenk, C. Schleh, M. Schäffler, N. Haberl, N. Gibson and J. C. Schittny, ACS Nano, 2018, 12, 7771–7790 CrossRef CAS.
- Y. Zhao, B. Pang, H. Luehmann, L. Detering, X. Yang, D. Sultan, S. Harpstrite, V. Sharma, C. S. Cutler, Y. Xia and Y. Liu, Adv. Healthcare Mater., 2016, 5, 928–935 CrossRef CAS.
- N. Chanda, V. Kattumuri, R. Shukla, A. Zambre, K. Katti, A. Upendran, R. R. Kulkarni, P. Kan, G. M. Fent, S. W. Casteel, C. J. Smith, E. Boote, J. D. Robertson, C. Cutler, J. R. Lever, K. V. Katti and R. Kannan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8760–8765 CrossRef CAS.
- Y. Wang, Y. Liu, H. Luehmann, X. Xia, D. Wan, C. Cutler and Y. Xia, Nano Lett., 2013, 13, 581–585 CrossRef CAS.
- R. Weissleder, D. Stark, B. Engelstad, B. Bacon, C. Compton, D. White, P. Jacobs and J. Lewis, Am. J. Roentgenol., 1989, 152, 167–173 CrossRef CAS.
- C. Chouly, D. Pouliquen, I. Lucet, J. J. Jeune and P. Jallet, J. Microencapsulation, 1996, 13, 245–255 CrossRef CAS.
- D. Pouliquen, R. Perdrisot, A. Ermias, S. Akoka, P. Jallet and J. Le Jeune, Magn. Reson. Imaging, 1989, 7, 619–627 CrossRef CAS.
- Y. Yang, Y. Sun, T. Cao, J. Peng, Y. Liu, Y. Wu, W. Feng, Y. Zhang and F. Li, Biomaterials, 2013, 34, 774–783 CrossRef CAS.
- Y. Zhao, B. Pang, H. Luehmann, L. Detering, X. Yang, D. Sultan, S. Harpstrite, V. Sharma, C. S. Cutler, Y. Xia and Y. Liu, Adv. Healthcare Mater., 2016, 5, 928–935 CrossRef CAS.
- J. Pellico, J. Ruiz-Cabello, M. Saiz-Alía, G. del Rosario, S. Caja, M. Montoya, L. Fernández de Manuel, M. P. Morales, L. Gutiérrez, B. Galiana, J. A. Enríquez and F. Herranz, Contrast Media Mol. Imaging, 2016, 11, 203–210 CrossRef CAS.
- J. Pellico, J. Ruiz-Cabello, M. Saiz-Alía, G. del Rosario, S. Caja, M. Montoya, L. Fernández de Manuel, M. P. Morales, L. Gutiérrez, B. Galiana, J. A. Enríquez and F. Herranz, Contrast Media Mol. Imaging, 2016, 11, 203–210 CrossRef CAS.
- H. Zolata, H. Afarideh and F. A. Davani, Cancer Biother. Radiopharm., 2016, 31, 324–329 CrossRef CAS.
- R. M. Wong, D. A. Gilbert, K. Liu and A. Y. Louie, ACS Nano, 2012, 6, 3461–3467 CrossRef CAS.
- E. Cędrowska, M. Pruszyński, W. Gawęda, M. Żuk, P. Krysiński, F. Bruchertseifer, A. Morgenstern, M.-A. Karageorgou, P. Bouziotis and A. Bilewicz, Molecules, 2020, 25, 1025 CrossRef.
- J. Zeng, B. Jia, R. Qiao, C. Wang, L. Jing, F. Wang and M. Gao, Chem. Commun., 2014, 50, 2170 RSC.
- S. Najmr, T. Lu, A. W. Keller, M. Zhang, J. D. Lee, M. Makvandi, D. A. Pryma, C. R. Kagan and C. B. Murray, Nano Futures, 2018, 2, 025002 CrossRef.
- M. Sun, D. Hoffman, G. Sundaresan, L. Yang, N. Lamichhane and J. Zweit, Am. J. Nucl. Med. Mol. Imaging, 2012, 2, 122–135 CAS.
- W. Guo, X. Sun, O. Jacobson, X. Yan, K. Min, A. Srivatsan, G. Niu, D. O. Kiesewetter, J. Chang and X. Chen, ACS Nano, 2015, 9, 488–495 CrossRef CAS.
- S. J. Kennel, J. D. Woodward, A. J. Rondinone, J. Wall, Y. Huang and S. Mirzadeh, Nucl. Med. Biol., 2008, 35, 501–514 CrossRef CAS.
- L. Yang, G. Sundaresan, M. Sun, P. Jose, D. Hoffman, P. R. McDonagh, N. Lamichhane, C. S. Cutler, J. M. Perez and J. Zweit, J. Mater. Chem. B, 2013, 1, 1421 RSC.
- T. M. Sakr, O. M. Khowessah, M. A. Motaleb, A. Abd El-Bary, M. T. El-Kolaly and M. M. Swidan, Eur. J. Pharm. Sci., 2018, 122, 239–245 CrossRef CAS.
- X. Sun, W. Cai and X. Chen, Acc. Chem. Res., 2015, 48, 286–294 CrossRef CAS.
- C. Pérez-Campaña, V. Gómez-Vallejo, A. Martin, E. San Sebastián, S. E. Moya, T. Reese, R. F. Ziolo and J. Llop, Analyst, 2012, 137, 4902 RSC.
- C. Pérez-Campaña, V. Gómez-Vallejo, M. Puigivila, A. Martín, T. Calvo-Fernández, S. E. Moya, R. F. Ziolo, T. Reese and J. Llop, ACS Nano, 2013, 7, 3498–3505 CrossRef.
- I. Munaweera, Y. Shi, B. Koneru, R. Saez, A. Aliev, A. J. Di Pasqua and K. J. Balkus, Mol. Pharmaceutics, 2015, 12, 3588–3596 CrossRef CAS.
- W. M. da Silva, R. H. de Andrade Alves e Silva, M. F. Cipreste, G. F. Andrade, P. L. Gastelois, W. A. de Almeida Macedo and E. M. B. de Sousa, Appl. Radiat. Isot., 2020, 157, 109032 CrossRef.
- T. Tsoncheva and E. Sarkadi-Priboczki, Microporous Mesoporous Mater., 2012, 148, 1–7 CrossRef CAS.
- R. Sharma, Y. Xu, S. W. Kim, M. J. Schueller, D. Alexoff, S. D. Smith, W. Wang and D. Schlyer, Nanoscale, 2013, 5, 7476 RSC.
- M. Eisenhut and W. Mier, Handbook of Nuclear Chemistry, Springer US, Boston, MA, 2011, pp. 2121–2141 Search PubMed.
- H. H. Coenen, J. Mertens and B. Mazière, Radioionidation Reactions for Radio Pharmaceuticals, Springer Netherlands, Dordrecht, 2006, pp. 29–72 Search PubMed.
- U. Kostiv, V. Lobaz, J. Kučka, P. Švec, O. Sedláček, M. Hrubý, O. Janoušková, P. Francová, V. Kolářová, L. Šefc and D. Horák, Nanoscale, 2017, 9, 16680–16688 RSC.
- K. C. L. Black, W. J. Akers, G. Sudlow, B. Xu, R. Laforest and S. Achilefu, Nanoscale, 2015, 7, 440–444 RSC.
- K. C. L. Black, A. Ibricevic, S. P. Gunsten, J. A. Flores, T. P. Gustafson, J. E. Raymond, S. Samarajeewa, R. Shrestha, S. E. Felder, T. Cai, Y. Shen, A.-K. Löbs, N. Zhegalova, D. H. Sultan, M. Berezin, K. L. Wooley, Y. Liu and S. L. Brody, Biomaterials, 2016, 98, 53–63 CrossRef CAS.
- C. Pérez-Medina, N. Patel, M. Robson, M. F. Lythgoe and E. Årstad, Bioorg. Med. Chem. Lett., 2013, 23, 5170–5173 CrossRef.
- J. Ge, Q. Zhang, J. Zeng, Z. Gu and M. Gao, Biomaterials, 2020, 228, 119553 CrossRef CAS.
- H. Sun, B. Zhang, X. Jiang, H. Liu, S. Deng, Z. Li and H. Shi, Nanomedicine, 2019, 14, 5–17 CrossRef CAS.
- S. Deng, W. Zhang, B. Zhang, R. Hong, Q. Chen, J. Dong, Y. Chen, Z. Chen and Y. Wu, J. Nanopart. Res., 2015, 17, 19 CrossRef.
- A. L. B. de Barros, A.-M. Chacko, J. L. Mikitsh, A. Al Zaki, A. Salavati, B. Saboury, A. Tsourkas and A. Alavi, Mol. Imaging Biol., 2014, 16, 330–339, DOI:10.1007/s11307-013-0709-9.
- R. Kumar, I. Roy, T. Y. Ohulchanskky, L. A. Vathy, E. J. Bergey, M. Sajjad and P. N. Prasad, ACS Nano, 2010, 4, 699–708 CrossRef CAS.
- G. Xie, J. Sun, G. Zhong, L. Shi and D. Zhang, Arch. Toxicol., 2010, 84, 183–190 CrossRef CAS.
- S. B. Lee, S.-W. Lee, S. Y. Jeong, G. Yoon, S. J. Cho, S. K. Kim, I.-K. Lee, B.-C. Ahn, J. Lee and Y. H. Jeon, ACS Appl. Mater. Interfaces, 2017, 9, 8480–8489 CrossRef CAS.
- S. B. Lee, D. Kumar, Y. Li, I.-K. Lee, S. J. Cho, S. K. Kim, S.-W. Lee, S. Y. Jeong, J. Lee and Y. H. Jeon, J. Nanobiotechnol., 2018, 16, 41 CrossRef.
- L. Zhao, Y. Li, J. Zhu, N. Sun, N. Song, Y. Xing, H. Huang and J. Zhao, J. Nanobiotechnol., 2019, 17, 30 CrossRef.
- N. Sun, L. Zhao, J. Zhu, Y. Li, N. Song, Y. Xing, W. Qiao, H. Huang and J. Zhao, Int. J. Nanomed., 2019, 14, 4367–4381 CrossRef CAS.
- J. Lee, T. S. Lee, J. Ryu, S. Hong, M. Kang, K. Im, J. H. Kang, S. M. Lim, S. Park and R. Song, J. Nucl. Med., 2013, 54, 96–103 CrossRef CAS.
- J. Jun Park, T. Sup Lee, J. Hyun Kang, R. Song and G. Jeong Cheon, Appl. Radiat. Isot., 2011, 69, 56–62 CrossRef.
- H. Fayez, M. A. El-Motaleb and A. A. Selim, J. Labelled Compd. Radiopharm., 2020, 63, 25–32 CrossRef CAS.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS.
- W. Yang, R. F. Barth, D. M. Adams, M. J. Ciesielski, R. A. Fenstermaker, S. Shukla, W. Tjarks and M. A. Caligiuri, Cancer Res., 2002, 62, 6552–6558 CAS.
- G. Thiagarajan, S. Sadekar, K. Greish, A. Ray and H. Ghandehari, Mol. Pharmaceutics, 2013, 10, 988–998 CrossRef CAS.
- S. Sadekar, A. Ray, M. Janàt-Amsbury, C. M. Peterson and H. Ghandehari, Biomacromolecules, 2011, 12, 88–96 CrossRef CAS.
- R.-Q. Huang, Y.-H. Qu, W.-L. Ke, J.-H. Zhu, Y.-Y. Pei and C. Jiang, FASEB J., 2007, 21, 1117–1125 CrossRef CAS.
- K. de Winne, S. Vanderkerken, K. Hoste, P. Dubruel, E. Schacht, M. Jelinkova and B. Rihova, J. Bioact. Compat. Polym., 2004, 19, 367–382 CrossRef CAS.
- S. J. Guillaudeu, M. E. Fox, Y. M. Haidar, E. E. Dy, F. C. Szoka and J. M. J. Fréchet, Bioconjugate Chem., 2008, 19, 461–469 CrossRef CAS.
- J. Liu, J. Liu, L. Chu, L. Tong, H. Gao, C. Yang, D. Wang, L. Shi, D. Kong and Z. Li, J. Nanosci. Nanotechnol., 2014, 14, 3305–3312 CrossRef CAS.
- E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmaceutics, 2005, 2, 129–138 CrossRef CAS.
- R. He, H. Wang, Y. Su, C. Chen, L. Xie, L. Chen, J. Yu, Y. Toledo, G. S. Abayaweera, G. Zhu and S. H. Bossmann, RSC Adv., 2017, 7, 16181–16188 RSC.
- N. Nasongkia, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef.
- K. Yang, L. Feng, H. Hong, W. Cai and Z. Liu, Nat. Protoc., 2013, 8, 2392–2403 CrossRef CAS.
- T. Cao, P. You, X. Zhou, J. Luo, X. Xu, Z. Zhou, S. Yang, Y. Zhang, H. Yang and M. Wang, J. Mater. Chem. B, 2016, 4, 6446–6453 RSC.
- H. Zhao, Y. Chao, J. Liu, J. Huang, J. Pan, W. Guo, J. Wu, M. Sheng, K. Yang, J. Wang and Z. Liu, Theranostics, 2016, 6, 1833–1843 CrossRef CAS.
- A. Lalatsa, N. L. Garrett, T. Ferrarelli, J. Moger, A. G. Schätzlein and I. F. Uchegbu, Mol. Pharmaceutics, 2012, 9, 1764–1774 CrossRef CAS.
- O. Sedláček, J. Kučka and M. Hrubý, Appl. Radiat. Isot., 2015, 95, 129–134 CrossRef.
- K. Wagener, D. Moderegger, M. Allmeroth, A. Reibel, S. Kramer, B. Biesalski, N. Bausbacher, R. Zentel, O. Thews and F. Rösch, Nucl. Med. Biol., 2018, 58, 59–66 CrossRef CAS.
- B. Sadarani, A. Majumdar, S. Paradkar, A. Mathur, S. Sachdev, B. Mohanty and P. Chaudhari, Biomed. Pharmacother., 2019, 114, 108770 CrossRef CAS.
- L. Yang, C. Zhang, J. Liu, F. Huang, Y. Zhang, X. Liang and J. Liu, Adv. Healthcare Mater., 2020, 9, 1901616 CrossRef CAS.
- C. Zhan, B. Gu, C. Xie, J. Li, Y. Liu and W. Lu, J. Controlled Release, 2010, 143, 136–142 CrossRef CAS.
- J. Shen, C. Zhan, C. Xie, Q. Meng, B. Gu, C. Li, Y. Zhang and W. Lu, J. Drug Targeting, 2011, 19, 197–203 CrossRef CAS.
- H.-W. Kao, C.-J. Chan, Y.-C. Chang, Y.-H. Hsu, M. Lu, J. Shian-Jy Wang, Y.-Y. Lin, S.-J. Wang and H.-E. Wang, Appl. Radiat. Isot., 2013, 80, 88–94 CrossRef CAS.
- Y. Tao, P. Ren, X. Yi, H. Zhou, S. Xiong, F. Ge, L. Chen and K. Yang, Part. Part. Syst. Charact., 2019, 36, 1900018 CrossRef.
- R. Toita, Y. Kanai, H. Watabe, K. Nakao, S. Yamamoto, J. Hatazawa and M. Akashi, Bioorg. Med. Chem., 2013, 21, 5310–5315 CrossRef CAS.
- D. G. Colak, I. Cianga, D. O. Demirkol, O. Kozgus, E. I. Medine, S. Sakarya, P. Unak, S. Timur and Y. Yagci, J. Mater. Chem., 2012, 22, 9293–9300 RSC.
- P. P. Di Mauro, Int. J. Pharm., 2018, 553, 169–185 CrossRef CAS.
- S. Rahmani, C. H. Villa, A. F. Dishman, M. E. Grabowski, D. C. Pan, H. Durmaz, A. C. Misra, L. Colón-Meléndez, M. J. Solomon, V. R. Muzykantov and J. Lahann, J. Drug Targeting, 2015, 23, 750–758 CrossRef CAS.
- C. Tang, J. Edelstein, J. L. Mikitsh, E. Xiao, A. H. Hemphill, R. Pagels, A. M. Chacko and R. Prud’homme, J. Mater. Chem. B, 2016, 4, 2428–2434 RSC.
- E. A. Simone, B. J. Zern, A. M. Chacko, J. L. Mikitsh, E. R. Blankemeyer, S. Muro, R. V. Stan and V. R. Muzykantov, Biomaterials, 2012, 33, 5406–5413 CrossRef CAS.
- X. Zhong, K. Yang, Z. Dong, X. Yi, Y. Wang, C. Ge, Y. Zhao and Z. Liu, Adv. Funct. Mater., 2015, 25, 7327–7336 CrossRef CAS.
- X. Yi, H. Zhou, Z. Zhang, S. Xiong and K. Yang, Biomaterials, 2020, 233, 119764 CrossRef.
- A. Ji, Y. Zhang, G. Lv, J. Lin, N. Qi, F. Ji and M. Du, J. Labelled Compd. Radiopharm., 2018, 61, 362–369 CrossRef CAS.
- A. Almutairi, R. Rossin, M. Shokeen, A. Hagooly, A. Ananth, B. Capoccia, S. Guillaudeu, D. Abendschein, C. J. Anderson, M. J. Welch and J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 685–690 CrossRef CAS.
- Y. Liu, A. Ibricevic, J. A. Cohen, J. L. Cohen, S. P. Gunsten, J. M. J. Fréchet, M. J. Walter, M. J. Welch and S. L. Brody, Mol. Pharmaceutics, 2009, 6, 1891–1902 CrossRef CAS.
- P. P. Di Mauro, V. Gómez-Vallejo, Z. Baz Maldonado, J. Llop Roig and S. Borrós, Bioconjugate Chem., 2015, 26, 582–592 CrossRef CAS.
- E. M. El-Marakby, R. M. Hathout, I. Taha, S. Mansour and N. D. Mortada, Int. J. Pharm., 2017, 525, 123–138 CrossRef CAS.
- O. Akca, P. Unak, E. I. Medine, S. Sakarya, A. Yurt Kilcar, C. Ichedef, R. Bekis and S. Timur, Radiochim. Acta, 2014, 102(9), 849–859, DOI:10.1515/ract-2013-2152.
- S. Guerrero, J. R. Herance, S. Rojas, J. F. Mena, J. D. Gispert, G. A. Acosta, F. Albericio and M. J. Kogan, Bioconjugate Chem., 2012, 23, 399–408 CrossRef CAS.
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS.
- K. Wagener, M. Worm, S. Pektor, M. Schinnerer, R. Thiermann, M. Miederer, H. Frey and F. Rösch, Biomacromolecules, 2018, 19, 2506–2516 CrossRef CAS.
- A. T. Reibel, S. S. Müller, S. Pektor, N. Bausbacher, M. Miederer, H. Frey and F. Rösch, Biomacromolecules, 2015, 16, 842–851 CrossRef CAS.
- E. J. Keliher, Y.-X. Ye, G. R. Wojtkiewicz, A. D. Aguirre, B. Tricot, M. L. Senders, H. Groenen, F. Fay, C. Perez-Medina, C. Calcagno, G. Carlucci, T. Reiner, Y. Sun, G. Courties, Y. Iwamoto, H.-Y. Kim, C. Wang, J. W. Chen, F. K. Swirski, H.-Y. Wey, J. Hooker, Z. A. Fayad, W. J. M. Mulder, R. Weissleder and M. Nahrendorf, Nat. Commun., 2017, 8, 14064 CrossRef CAS.
- J. Jeon, J. A. Kang, H. E. Shim, Y. R. Nam, S. Yoon, H. R. Kim, D. E. Lee and S. H. Park, Bioorg. Med. Chem., 2015, 23, 3303–3308 CrossRef CAS.
- C.-Y. Wu, J.-J. Lin, W.-Y. Chang, C.-Y. Hsieh, C.-C. Wu, H.-S. Chen, H.-J. Hsu, A.-S. Yang, M.-H. Hsu and W.-Y. Kuo, Colloids Surf., B, 2019, 183, 110387 CrossRef CAS.
- S. Ghiassian, L. Yu, P. Gobbo, A. Nazemi, T. Romagnoli, W. Luo, L. G. Luyt and M. S. Workentin, ACS Omega, 2019, 4, 19106–19115 CrossRef CAS.
- M. De Simone, D. Panetta, E. Bramanti, C. Giordano, M. C. Salvatici, L. Gherardini, A. Menciassi, S. Burchielli, C. Cinti and P. A. Salvadori, Contrast Media Mol. Imaging, 2016, 11, 561–571 CrossRef CAS.
- Q. Liu, Y. Sun, C. Li, J. Zhou, C. Li, T. Yang, X. Zhang, T. Yi, D. Wu and F. Li, ACS Nano, 2011, 5, 3146–3157 CrossRef CAS.
- Y. Sun, M. Yu, S. Liang, Y. Zhang, C. Li, T. Mou, W. Yang, X. Zhang, B. Li, C. Huang and F. Li, Biomaterials, 2011, 32, 2999–3007 CrossRef CAS.
- J. Zhou, M. Yu, Y. Sun, X. Zhang, X. Zhu, Z. Wu, D. Wu and F. Li, Biomaterials, 2011, 32, 1148–1156 CrossRef CAS.
- S. Alonso-deCastro, E. Ruggiero, A. Lekuona Fernández, U. Cossío, Z. Baz, D. Otaegui, V. Gómez-Vallejo, D. Padro, J. Llop and L. Salassa, Inorganics, 2019, 7, 60 CrossRef CAS.
- L. L. Israel, F. Karimi, S. Bianchessi, E. Scanziani, L. Passoni, M. Matteoli, B. Langström and J.-P. Lellouche, Mater. Res. Express, 2015, 2, 095009 CrossRef.
- T. Tang, Y. Wei, Q. Yang, Y. Yang, M. J. Sailor and H.-B. Pang, Nanoscale, 2019, 11, 22248–22254 RSC.
- X. Sun, X. Huang, J. Guo, W. Zhu, Y. Ding, G. Niu, A. Wang, D. O. Kiesewetter, Z. L. Wang, S. Sun and X. Chen, J. Am. Chem. Soc., 2014, 136, 1706–1709 CrossRef CAS.
- Y. Sun, X. Zhu, J. Peng and F. Li, ACS Nano, 2013, 7, 11290–11300 CrossRef CAS.
- B. Roldan Cuenya and F. Behafarid, Surf. Sci. Rep., 2015, 70, 135–187 CrossRef CAS.
- M. Králik, Chem. Pap., 2014, 68, 1625–1638, DOI:10.2478/s11696-014-0624-9.
- F. Chen, P. A. Ellison, C. M. Lewis, H. Hong, Y. Zhang, S. Shi, R. Hernandez, M. E. Meyerand, T. E. Barnhart and W. Cai, Angew. Chem., Int. Ed., 2013, 52, 13319–13323 CrossRef CAS.
- E. Boros, A. M. Bowen, L. Josephson, N. Vasdev and J. P. Holland, Chem. Sci., 2015, 6, 225–236 RSC.
- T. M. Shaffer, S. Harmsen, E. Khwaja, M. F. Kircher, C. M. Drain and J. Grimm, Nano Lett., 2016, 16, 5601–5604 CrossRef CAS.
- T. M. Shaffer, M. A. Wall, S. Harmsen, V. A. Longo, C. M. Drain, M. F. Kircher and J. Grimm, Nano Lett., 2015, 15, 864–868 CrossRef CAS.
- T. M. Shaffer, S. Harmsen, E. Khwaja, M. F. Kircher, C. M. Drain and J. Grimm, Nano Lett., 2016, 16, 5601–5604 CrossRef CAS.
- S. Belderbos, M. A. González-Gómez, F. Cleeren, J. Wouters, Y. Piñeiro, C. M. Deroose, A. Coosemans, W. Gsell, G. Bormans, J. Rivas and U. Himmelreich, EJNMMI Res., 2020, 10, 73 CrossRef CAS.
- M. A. González-Gómez, S. Belderbos, S. Yañez-Vilar, Y. Piñeiro, F. Cleeren, G. Bormans, C. M. Deroose, W. Gsell, U. Himmelreich and J. Rivas, Nanomaterials, 2019, 9, 1626 CrossRef.
- X. Cui, S. Belo, D. Krüger, Y. Yan, R. T. M. de Rosales, M. Jauregui-Osoro, H. Ye, S. Su, D. Mathe, N. Kovács, I. Horváth, M. Semjeni, K. Sunassee, K. Szigeti, M. A. Green and P. J. Blower, Biomaterials, 2014, 35, 5840–5846 CrossRef CAS.
- B. T. Cisneros, J. J. Law, M. L. Matson, A. Azhdarinia, E. M. Sevick-Muraca and L. J. Wilson, Nanomedicine, 2014, 9, 2499–2509 CrossRef CAS.
- F. Man, P. J. Gawne and R. T. M. de Rosales, Adv. Drug Delivery Rev., 2019, 143, 134–160 CrossRef CAS.
- J. Marik, M. S. Tartis, H. Zhang, J. Y. Fung, A. Kheirolomoom, J. L. Sutcliffe and K. W. Ferrara, Nucl. Med. Biol., 2007, 34, 165–171 CrossRef CAS.
- J. W. Seo, L. M. Mahakian, S. Tam, S. Qin, E. S. Ingham, C. F. Meares and K. W. Ferrara, Nucl. Med. Biol., 2015, 42, 155–163 CrossRef CAS.
- P. Laverman, E. T. M. Dams, W. J. G. Oyen, G. Storm, E. B. Koenders, R. Prevost, J. W. M. van der Meer, F. H. M. Corstens and O. C. Boerman, J. Nucl. Med., 1999, 40, 192–197 CAS.
- V. J. Richardson, K. Jeyasingh, R. F. Jewkes, B. E. Ryman and M. H. Tattersall, Biochem. Soc. Trans., 1977, 5, 290–291 CrossRef CAS.
- V. J. Richardson, K. Jeyasingh, R. F. Jewkes, B. E. Ryman and M. H. Tattersall, J. Nucl. Med., 1978, 19, 1049–1054 CAS.
- V. J. Richardson, B. E. Ryman, R. F. Jewkes, M. H. Tattersall and E. S. Newlands, Int. J. Nucl. Med. Biol., 1978, 5, 118 CrossRef CAS , 121–122, 123.
- V. J. Richardson, B. E. Ryman, R. F. Jewkes, K. Jeyasingh, M. N. H. Tattersall, E. S. Newlands and S. B. Kaye, Br. J. Cancer, 1979, 40, 35–43 CrossRef CAS.
- M. P. Osborne, J. H. Payne, V. J. Richardson, V. R. McCready and B. E. Ryman, Br. J. Surg., 1983, 70, 141–144 CrossRef CAS.
- D. S. Abou, D. L. J. Thorek, N. N. Ramos, M. W. H. Pinkse, H. T. Wolterbeek, S. D. Carlin, B. J. Beattie and J. S. Lewis, Pharm. Res., 2013, 30, 878–888 CrossRef CAS.
- H. Zhang, J. Kusunose, A. Kheirolomoom, J. W. Seo, J. Qi, K. D. Watson, H. A. Lindfors, E. Ruoslahti, J. L. Sutcliffe and K. W. Ferrara, Biomaterials, 2008, 29, 1976–1988 CrossRef CAS.
- H. Zhang, N. Li, P. Sirish, L. Mahakian, E. Ingham, F.-R. Curry, S. Yamada, N. Chiamvimonvat and K. W. Ferrara, J. Controlled Release, 2012, 163, 10–17 CrossRef CAS.
- F. Emmetiere, C. Irwin, N. T. Viola-Villegas, V. Longo, S. M. Cheal, P. Zanzonico, N. Pillarsetty, W. A. Weber, J. S. Lewis and T. Reiner, Bioconjugate Chem., 2013, 24, 1784–1789 CrossRef CAS.
- A. T. I. Jensen, T. Binderup, T. L. Andresen, A. Kjær and P. H. Rasmussen, J. Liposome Res., 2012, 22, 295–305 CrossRef CAS.
- T. Urakami, S. Akai, Y. Katayama, N. Harada, H. Tsukada and N. Oku, J. Med. Chem., 2007, 50, 6454–6457 CrossRef CAS.
- T. Urakami, A. T. Kawaguchi, S. Akai, K. Hatanaka, H. Koide, K. Shimizu, T. Asai, D. Fukumoto, N. Harada, H. Tsukada and N. Oku, Artif. Organs, 2009, 33, 164–168 CrossRef CAS.
- N. Oku, M. Yamashita, Y. Katayama, T. Urakami, K. Hatanaka, K. Shimizu, T. Asai, H. Tsukada, S. Akai and H. Kanazawa, Int. J. Pharm., 2011, 403, 170–177 CrossRef CAS.
- T. Fukuta, T. Ishii, T. Asai, G. Nakamura, Y. Takeuchi, A. Sato, Y. Agato, K. Shimizu, S. Akai, D. Fukumoto, N. Harada, H. Tsukada, A. T. Kawaguchi and N. Oku, Artif. Organs, 2014, 38, 662–666 CrossRef CAS.
- A. T. Reibel, S. S. Müller, S. Pektor, N. Bausbacher, M. Miederer, H. Frey and F. Rösch, Biomacromolecules, 2015, 16, 842–851 CrossRef CAS.
- K. Wagener, M. Worm, S. Pektor, M. Schinnerer, R. Thiermann, M. Miederer, H. Frey and F. Rösch, Biomacromolecules, 2018, 19, 2506–2516 CrossRef CAS.
- D. J. Hnatowich, B. Friedman, B. Clancy and M. Novak, J. Nucl. Med., 1981, 22, 810–814 CAS.
- Q. F. Ahkong and C. Tilcock, Int. J. Radiat. Appl. Instrum., Part B, 1992, 19, 831–840 CrossRef CAS.
- C. Tilcock, Q. F. Ahkong and D. Fisher, Nucl. Med. Biol., 1994, 21, 89–96 CrossRef CAS.
- C. Tilcock, Q. F. Ahkong and D. Fisher, Biochim. Biophys. Acta, 1993, 1148, 77–84 CrossRef CAS.
- H. Hwang, H.-S. H.-J. Jeong, P.-S. Oh, M. Kim, T.-K. Lee, J. Kwon, H.-S. Kim, S. T. Lim, M.-H. Sohn and H.-S. H.-J. Jeong, Nucl. Med. Biol., 2016, 43, 552–558 CrossRef CAS.
- L. O. F. Monteiro, R. S. Fernandes, C. M. R. Oda, S. C. Lopes, D. M. Townsend, V. N. Cardoso, M. C. Oliveira, E. A. Leite, D. Rubello and A. L. B. de Barros, Biomed. Pharmacother., 2018, 97, 489–495 CrossRef CAS.
- A. T. M. e Silva, A. L. C. Maia, J. de Oliveira Silva, A. L. B. de Barros, D. C. F. Soares, M. T. Q. de Magalhães, R. José Alves and G. A. Ramaldes, Carbohydr. Res., 2018, 465, 52–57 CrossRef CAS.
- A. Helbok, C. Decristoforo, G. Dobrozemsky, C. Rangger, E. Diederen, B. Stark, R. Prassl and E. von Guggenberg, J. Liposome Res., 2010, 20, 219–227 CrossRef CAS.
- E. T. Dams, M. M. Reijnen, W. J. Oyen, O. C. Boerman, P. Laverman, G. Storm, J. W. van der Meer, F. H. Corstens and H. van Goor, Ann. Surg., 1999, 229, 551–557 CrossRef CAS.
- C. J. J. M. Sikkink, M. M. P. J. Reijnen, P. Laverman, W. J. G. Oyen and H. van Goor, J. Surg. Res., 2009, 154, 246–251 CrossRef CAS.
- A. Brouwers, D. De Jong, E. Dams, W. Oyen, O. Boerman, P. Laverman, T. Naber, G. Storm and F. Corstens, J. Drug Targeting, 2000, 8, 225–233 CrossRef CAS.
- Z. Varga, I. C. Szigyártó, I. Gyurkó, R. Dóczi, I. Horváth, D. Máthé and K. Szigeti, Contrast Media Mol. Imaging, 2017, 2017, 1–8 CrossRef.
- T. A. Elbayoumi and V. P. Torchilin, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1196–1205 CrossRef CAS.
- E. Jestin, M. Mougin-Degraef, A. Faivre-Chauvet, P. Remaud-Le Saëc, F. Hindre, J. P. Benoit, J. F. Chatal, J. Barbet and J. F. Gestin, Q. J. Nucl. Med. Mol. Imaging, 2007, 51, 51–60 CAS.
- M. Mougin-Degraef, C. Bourdeau, E. Jestin, C. Saï-Maurel, M. Bourgeois, P. R.-L. Saëc, P. Thédrez, J.-F. Gestin, J. Barbet and A. Faivre-Chauvet, Int. J. Pharm., 2007, 344, 110–117 CrossRef CAS.
- Y. Duan, L. Wei, J. Petryk and T. Ruddy, Int. J. Nanomed., 2016, 11, 5697–5708 CrossRef CAS.
- A. K. Iyer, Y. Su, J. Feng, X. Lan, X. Zhu, Y. Liu, D. Gao, Y. Seo, H. F. VanBrocklin, V. Courtney Broaddus, B. Liu and J. He, Biomaterials, 2011, 32, 2605–2613 CrossRef CAS.
- T. van der Geest, P. Laverman, D. Gerrits, G. M. Franssen, J. M. Metselaar, G. Storm and O. C. Boerman, J. Controlled Release, 2015, 220, 239–244 CrossRef CAS.
- E. D. Hood, C. F. Greineder, T. Shuvaeva, L. Walsh, C. H. Villa and V. R. Muzykantov, Bioconjugate Chem., 2018, 29, 3626–3637 CrossRef CAS.
- J. Malinge, B. Géraudie, P. Savel, V. Nataf, A. Prignon, C. Provost, Y. Zhang, P. Ou, K. Kerrou, J.-N. Talbot, J.-M. Siaugue, M. Sollogoub and C. Ménager, Mol. Pharmaceutics, 2017, 14, 406–414 CrossRef CAS.
- J. W. Seo, H. Zhang, D. L. Kukis, C. F. Meares and K. W. Ferrara, Bioconjugate Chem., 2008, 19, 2577–2584 CrossRef CAS.
- E. E. Paoli, D. E. Kruse, J. W. Seo, H. Zhang, A. Kheirolomoom, K. D. Watson, P. Chiu, H. Stahlberg and K. W. Ferrara, J. Controlled Release, 2010, 143, 13–22 CrossRef CAS.
- C. B. Rygh, S. Qin, J. W. Seo, L. M. Mahakian, H. Zhang, R. Adamson, J. Q. Chen, A. D. Borowsky, R. D. Cardiff, R. K. Reed, F.-R. E. Curry and K. W. Ferrara, Clin. Cancer Res., 2011, 17, 550–559 CrossRef CAS.
- L. M. Mahakian, D. G. Farwell, H. Zhang, J. W. Seo, B. Poirier, S. P. Tinling, A. M. Afify, E. M. Haynam, D. Shaye and K. W. Ferrara, Mol. Imaging Biol., 2014, 16, 284–292 CrossRef.
- A. W. Wong, E. Ormsby, H. Zhang, J. W. Seo, L. M. Mahakian, C. F. Caskey and K. W. Ferrara, Am. J. Nucl. Med. Mol. Imaging, 2013, 3, 32–43 CAS.
- J. W. Seo, L. M. Mahakian, A. Kheirolomoom, H. Zhang, C. F. Meares, R. Ferdani, C. J. Anderson and K. W. Ferrara, Bioconjugate Chem., 2010, 21, 1206–1215 CrossRef CAS.
- J. W. Seo, S. Qin, L. M. Mahakian, K. D. Watson, A. Kheirolomoom and K. W. Ferrara, J. Controlled Release, 2011, 151, 28–34 CrossRef CAS.
- S. Lee, K. Gangangari, T. M. Kalidindi, B. Punzalan, S. M. Larson and N. V. K. Pillarsetty, Nucl. Med. Biol., 2016, 43, 781–787 CrossRef CAS.
- N. Mitchell, T. L. Kalber, M. S. Cooper, K. Sunassee, S. L. Chalker, K. P. Shaw, K. L. Ordidge, A. Badar, S. M. Janes, P. J. Blower, M. F. Lythgoe, H. C. Hailes and A. B. Tabor, Biomaterials, 2013, 34, 1179–1192 CrossRef CAS.
- C. M. Kang, H. J. Koo, S. Lee, K. C. Lee, Y. K. Oh and Y. S. Choe, Nucl. Med. Biol., 2013, 40, 1018–1024 CrossRef CAS.
- A. I. Jensen, G. W. Severin, A. E. Hansen, F. P. Fliedner, R. Eliasen, L. Parhamifar, A. Kjær, T. L. Andresen and J. R. Henriksen, J. Controlled Release, 2018, 269, 100–109 CrossRef CAS.
- S. McQuarrie, J. Mercer, A. Syme, M. Suresh and G. Miller, J. Pharm. Pharm. Sci., 2005, 7, 29–34 Search PubMed.
- S. W. Zielhuis, J.-H. Seppenwoolde, V. A. P. Mateus, C. J. G. Bakker, G. C. Krijger, G. Storm, B. A. Zonnenberg, A. D. van het Schip, G. A. Koning and J. F. W. Nijsen, Cancer Biother. Radiopharm., 2006, 21, 520–527 CrossRef CAS.
- C. Perez-Medina, D. Abdel-Atti, Y. Zhang, V. A. Longo, C. P. Irwin, T. Binderup, J. Ruiz-Cabello, Z. A. Fayad, J. S. Lewis, W. J. M. Mulder and T. Reiner, J. Nucl. Med., 2014, 55, 1706–1711 CrossRef CAS.
- C. Pérez-Medina, D. Abdel-Atti, J. Tang, Y. Zhao, Z. A. Fayad, J. S. Lewis, W. J. M. Mulder and T. Reiner, Nat. Commun., 2016, 7, 11838 CrossRef.
- G. Srimathveeravalli, D. Abdel-Atti, C. Pérez-Medina, H. Takaki, S. B. Solomon, W. J. M. Mulder and T. Reiner, Mol. Imaging, 2018, 17 DOI:10.1177/1536012117749726.
- M. E. Lobatto, T. Binderup, P. M. Robson, L. F. P. Giesen, C. Calcagno, J. Witjes, F. Fay, S. Baxter, C. H. Wessel, M. Eldib, J. Bini, S. D. Carlin, E. S. G. Stroes, G. Storm, A. Kjaer, J. S. Lewis, T. Reiner, Z. A. Fayad, W. J. M. Mulder and C. Pérez-Medina, Bioconjugate Chem., 2020, 31, 360–368 CrossRef CAS.
- B. Yu, S. Goel, D. Ni, P. A. Ellison, C. M. Siamof, D. Jiang, L. Cheng, L. Kang, F. Yu, Z. Liu, T. E. Barnhart, Q. He, H. Zhang and W. Cai, Adv. Mater., 2018, 30, 1704934 CrossRef.
- V. J. Caride and B. L. Zaret, Science, 1977, 198, 735–738 CrossRef CAS.
- T. N. Palmer, V. J. Caride, L. A. Fernandez and J. Twickler, Biosci. Rep., 1981, 1, 337–344 CrossRef CAS.
- T. N. Palmer and J. P. Warner, Biosci. Rep., 1983, 3, 479–485 CrossRef CAS.
- N. Oku, Y. Namba, A. Takeda and S. Okada, Nucl. Med. Biol., 1993, 20, 407–412 CrossRef CAS.
- M. L. Corvo, O. C. Boerman, W. J. G. Oyen, J. C. S. Jorge, M. E. M. Cruz, D. J. A. Crommelin and G. Storm, Pharm. Res., 2000, 17, 600–606 CrossRef CAS.
- D. C. F. Soares, M. C. de Oliveira, R. G. dos Santos, M. S. Andrade, J. M. C. Vilela, V. N. Cardoso and G. A. Ramaldes, Eur. J. Pharm. Sci., 2011, 42, 462–469 CrossRef CAS.
- S. Sofou, J. L. Thomas, H. Lin, M. R. McDevitt, D. A. Scheinberg and G. Sgouros, J. Nucl. Med., 2004, 45, 253–260 CAS.
- M. R. Mauk and R. C. Gamble, Anal. Biochem., 1979, 94, 302–307 CrossRef CAS.
- R. T. Proffitt, L. E. Williams, C. A. Presant, G. W. Tin, J. A. Uliana, R. C. Gamble and J. D. Baldeschwieler, J. Nucl. Med., 1983, 24, 45–51 CAS.
- K. J. Hwang, J. Nucl. Med., 1978, 19, 1162–1170 CAS.
- P. L. Beaumier and K. J. Hwang, Biochim. Biophys. Acta, 1983, 731, 23–30 CrossRef CAS.
- P. L. Beaumier and K. J. Hwang, J. Nucl. Med., 1982, 23, 810–815 CAS.
- H. Essien and K. J. Hwang, Biochim. Biophys. Acta, 1988, 944, 329–336 CrossRef CAS.
- O. C. Boerman, G. Storm, W. J. G. Oyen, L. van Bloois, J. W. M. van der Meer, R. A. M. J. Claessens, D. J. A. Crommelin and F. H. M. Corstens, J. Nucl. Med., 1995, 36, 1639–1644 CAS.
- N. Postma, O. Boerman, W. J. Oyen, J. Zuidema and G. Storm, J. Controlled Release, 1999, 58, 51–60 CrossRef CAS.
- S. Stewart and K. J. Harrington, Oncology, 1997, 11, 33–37 Search PubMed.
- K. J. Harrington, G. Rowlinson-Busza, K. N. Syrigos, P. S. Uster, R. M. Abra and J. S. W. Stewart, Br. J. Cancer, 2000, 83, 232–238 CrossRef CAS.
- K. J. Harrington, G. Rowlinson-Busza, K. N. Syrigos, P. S. Uster, R. G. Vile and J. S. W. Stewart, Clin. Cancer Res., 2000, 6, 2528–2537 CAS.
- M. L. Corvo, O. C. Boerman, W. J. Oyen, L. Van Bloois, M. E. M. Cruz, D. J. Crommelin and G. Storm, Biochim. Biophys. Acta, Biomembr., 1999, 1419, 325–334 CrossRef CAS.
- D. Utkhede, V. Yeh, M. Szucs and C. Tilcock, J. Liposome Res., 1994, 4, 1049–1061 CrossRef CAS.
- A. Gabizon, J. Hliberty, R. M. Straubinger, D. C. Price and D. Papahadjopoulos, J. Liposome Res., 1988, 1, 123–135 CrossRef.
- A. Gabizon, D. C. Price, J. Huberty, R. S. Bresalier and D. Papahadjopoulos, Cancer Res., 1990, 50, 6371–6378 CAS.
- H.-E. Wang, H.-M. Yu, Y.-C. Lu, N.-N. Heish, Y.-L. Tseng, K.-L. Huang, K.-T. Chuang, C.-H. Chen, J.-J. Hwang, W.-J. Lin, S.-J. Wang, G. Ting, J. Whang-Peng and W.-P. Deng, Nucl. Instrum. Methods Phys. Res., Sect. A, 2006, 569, 533–537 CrossRef CAS.
- A. L. Petersen, T. Binderup, P. Rasmussen, J. R. Henriksen, D. R. Elema, A. Kjær and T. L. Andresen, Biomaterials, 2011, 32, 2334–2341 CrossRef CAS.
- A. L. Petersen, T. Binderup, R. I. Jølck, P. Rasmussen, J. R. Henriksen, A. K. Pfeifer, A. Kjær and T. L. Andresen, J. Controlled Release, 2012, 160, 254–263 CrossRef CAS.
- L. W. Locke, M. W. Mayo, A. D. Yoo, M. B. Williams and S. S. Berr, Biomaterials, 2012, 33, 7785–7793 CrossRef CAS.
- M.-Y. Chang, J. Seideman and S. Sofou, Bioconjugate Chem., 2008, 19, 1274–1282 CrossRef CAS.
- A. Bandekar, C. Zhu, R. Jindal, F. Bruchertseifer, A. Morgenstern and S. Sofou, J. Nucl. Med., 2014, 55, 107–114 CrossRef CAS.
- N. Li, Z. Yu, T. Pham, P. Blower and R. Yan, Int. J. Nanomed., 2017, 12, 3281–3294 CrossRef CAS.
- S. Edmonds, A. Volpe, H. Shmeeda, A. C. Parente-Pereira, R. Radia, J. Baguña-Torres, I. Szanda, G. W. Severin, L. Livieratos, P. J. Blower, J. Maher, G. O. Fruhwirth, A. Gabizon and R. T. M. de Rosales, ACS Nano, 2016, 10, 10294–10307 CrossRef CAS.
- P. J. Gawne, F. Clarke, K. Turjeman, A. P. Cope, N. J. Long, Y. Barenholz, S. Y. A. Terry and R. T. M. de Rosales, Theranostics, 2020, 10, 3867–3879 CrossRef CAS.
- P. Gawne, F. Man, J. Fonslet, R. Radia, J. Bordoloi, M. Cleveland, P. Jimenez-Royo, A. Gabizon, P. J. Blower, N. Long and R. T. M. De Rosales, Dalton Trans., 2018, 47, 9283–9293 RSC.
- F. Man, L. Lim, A. Volpe, A. Gabizon, H. Shmeeda, B. Draper, A. C. Parente-Pereira, J. Maher, P. J. Blower, G. O. Fruhwirth and R. T. M. de Rosales, Mol. Ther., 2019, 27, 219–229 CrossRef CAS.
- J. R. Henriksen, A. L. Petersen, A. E. Hansen, C. G. Frankær, P. Harris, D. R. Elema, A. T. Kristensen, A. Kjær and T. L. Andresen, ACS Appl. Mater. Interfaces, 2015, 7, 22796–22806 CrossRef CAS.
- A. E. Hansen, A. L. Petersen, J. R. Henriksen, B. Boerresen, P. Rasmussen, D. R. Elema, P. M. af Rosenschöld, A. T. Kristensen, A. Kjær and T. L. Andresen, ACS Nano, 2015, 9, 6985–6995 CrossRef CAS.
- B. Børresen, J. R. Henriksen, G. Clergeaud, J. S. Jørgensen, F. Melander, D. R. Elema, J. Szebeni, S. A. Engelholm, A. T. Kristensen, A. Kjær, T. L. Andresen and A. E. Hansen, ACS Nano, 2018, 12, 11386–11398 CrossRef.
- A. E. Hansen, F. P. Fliedner, J. R. Henriksen, J. T. Jørgensen, A. E. Clemmensen, B. Børresen, D. R. Elema, A. Kjær and T. L. Andresen, Nanomedicine, 2018, 14, 27–34 CrossRef CAS.
- A. S. Rudolph, R. W. Klipper, B. Goins and W. T. Phillips, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10976–10980 CrossRef CAS.
- W. T. Phillips, A. S. Rudolph, B. Goins, J. H. Timmons, R. Klipper and R. Blumhardt, Int. J. Radiat. Appl. Instrum., Part B, 1992, 19, 539–547 CrossRef CAS.
- M. Suresh and Y. Cao, J. Pharm. Pharm. Sci., 1998, 1, 31–37 CAS.
- A. Bao, B. Goins, R. Klipper, G. Negrete, M. Mahindaratne and W. T. Phillips, J. Pharm. Sci., 2003, 92, 1893–1904 CrossRef CAS.
- S. Li, B. Goins, W. T. Phillips and A. Bao, J. Liposome Res., 2011, 21, 17–27 CrossRef CAS.
- A. Bao, J. Pharmacol. Exp. Ther., 2003, 308, 419–425 CrossRef.
- A. Bao, B. Goins, R. Klipper, G. Negrete and W. T. Phillips, J. Nucl. Med., 2003, 44, 1992–1999 CAS.
- A. Soundararajan, A. Bao, W. T. Phillips, R. Perez and B. A. Goins, Nucl. Med. Biol., 2009, 36, 515–524 CrossRef CAS.
- L.-C. Chen, Y.-H. Wu, I.-H. Liu, C.-L. Ho, W.-C. Lee, C.-H. Chang, K.-L. Lan, G. Ting, T.-W. Lee and J.-H. Shien, Nucl. Med. Biol., 2012, 39, 35–43 CrossRef CAS.
- C.-W. Hsu, Y.-J. Chang, C.-H. Chang, L.-C. Chen, K.-L. Lan, G. Ting and T.-W. Lee, Cancer Biother. Radiopharm., 2012, 27, 481–489 CrossRef CAS.
- L.-T. Lin, C.-H. Chang, H.-L. Yu, R.-S. Liu, H.-E. Wang, S.-J. Chiu, F.-D. Chen, T.-W. Lee and Y.-J. Lee, J. Nucl. Med., 2014, 55, 1864–1870 CrossRef CAS.
- W.-C. Hsu, C.-N. Cheng, T.-W. Lee and J.-J. Hwang, Anticancer Res., 2015, 35, 4777–4788 CAS.
- H. Lee, J. Zheng, D. Gaddy, K. D. Orcutt, S. Leonard, E. Geretti, J. Hesterman, C. Harwell, J. Hoppin, D. A. Jaffray, T. Wickham, B. S. Hendriks and D. Kirpotin, Nanomedicine, 2015, 11, 155–165 CrossRef CAS.
- S. J. Blocker, K. A. Douglas, L. A. Polin, H. Lee, B. S. Hendriks, E. Lalo, W. Chen and A. F. Shields, Theranostics, 2017, 7, 4229–4239 CrossRef CAS.
- H. Lee, A. F. Shields, B. A. Siegel, K. D. Miller, I. Krop, C. X. Ma, P. M. LoRusso, P. N. Munster, K. Campbell, D. F. Gaddy, S. C. Leonard, E. Geretti, S. J. Blocker, D. B. Kirpotin, V. Moyo, T. J. Wickham and B. S. Hendriks, Clin. Cancer Res., 2017, 23, 4190–4202 CrossRef CAS.
- H. Lee, D. Gaddy, M. Ventura, N. Bernards, R. de Souza, D. Kirpotin, T. Wickham, J. Fitzgerald, J. Zheng and B. S. Hendriks, Theranostics, 2018, 8, 2300–2312 CrossRef CAS.
- G. Engudar, H. Schaarup-Jensen, F. P. Fliedner, A. E. Hansen, P. Kempen, R. I. Jølck, A. Kjæer, T. L. Andresen, M. H. Clausen, A. I. Jensen and J. R. Henriksen, Theranostics, 2018, 8, 5828–5841 CrossRef CAS.
- W. G. Love, N. Amos, B. D. Williams and I. W. Kellaway, J. Microencapsulation, 1989, 6, 105–113 CrossRef CAS.
- B. C. L. Cheung, T. H. T. Sun, J. M. Leenhouts and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1998, 1414, 205–216 CrossRef CAS.
- S. A. Abraham, K. Edwards, G. Karlsson, S. MacIntosh, L. D. Mayer, C. McKenzie and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2002, 1565, 41–54 CrossRef CAS.
- R. Chakravarty, S. Chakraborty and A. Dash, Mol. Pharmaceutics, 2016, 13, 3601–3612 CrossRef CAS.
- S. A. Graves, R. Hernandez, J. Fonslet, C. G. England, H. F. Valdovinos, P. A. Ellison, T. E. Barnhart, D. R. Elema, C. P. Theuer, W. Cai, R. J. Nickles and G. W. Severin, Bioconjugate Chem., 2015, 26, 2118–2124 CrossRef CAS.
- D. S. Abou, T. Ku and P. M. Smith-Jones, Nucl. Med. Biol., 2011, 38, 675–681 CrossRef CAS.
- V. Oliveri and G. Vecchio, Eur. J. Med. Chem., 2016, 120, 252–274 CrossRef CAS.
- R. Hueting, V. Kersemans, B. Cornelissen, M. Tredwell, K. Hussien, M. Christlieb, A. D. Gee, J. Passchier, S. C. Smart, J. R. Dilworth, V. Gouverneur and R. J. Muschel, J. Nucl. Med., 2014, 55, 128–134 CrossRef CAS.
- J. J. Bartnicka and P. J. Blower, J. Nucl. Med., 2018, 59, 1355–1359 CrossRef CAS.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS.
- O. M. Elsharkasy, J. Z. Nordin, D. W. Hagey, O. G. de Jong, R. M. Schiffelers, S. E. L. Andaloussi and P. Vader, Adv. Drug Delivery Rev., 2020, 159, 332–343, DOI:10.1016/j.addr.2020.04.004.
- A. Khan, F. Man, F. Faruqu, J. Kim, F. Al-Salemee, A. Volpe, G. O. Fruhwirth, K. T. Al-Jamal and R. T. M. de Rosales, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12730463.v1.
- A. Banerjee, V. Alves, T. Rondão, J. Sereno, Â. Neves, M. Lino, A. Ribeiro, A. J. Abrunhosa and L. S. Ferreira, Nanoscale, 2019, 11, 13243–13248 RSC.
- F. Royo, U. Cossío, A. Ruiz de Angulo, J. Llop and J. M. Falcon-Perez, Nanoscale, 2019, 11, 1531–1537 RSC.
- F. N. Faruqu, J. T. W. Wang, L. Xu, L. McNickle, E. M. Y. Chong, A. Walters, M. Gurney, A. Clayton, L. A. Smyth, R. Hider, J. Sosabowski and K. T. Al-Jamal, Theranostics, 2019, 9, 1666–1682 CrossRef CAS.
- S. Shi, T. Li, X. Wen, S. Y. Wu, C. Xiong, J. Zhao, V. R. Lincha, D. S. Chow, Y. Liu, A. K. Sood and C. Li, Bioconjugate Chem., 2019, 30(10), 2675–2683, DOI:10.1021/acs.bioconjchem.9b00587.
- M. Morishita, Y. Takahashi, M. Nishikawa, K. Sano, K. Kato, T. Yamashita, T. Imai, H. Saji and Y. Takakura, J. Pharm. Sci., 2015, 104, 705–713 CrossRef CAS.
- A. Matsumoto, Y. Takahashi, M. Nishikawa, K. Sano, M. Morishita, C. Charoenviriyakul, H. Saji and Y. Takakura, Cancer Sci., 2017, 108, 1803–1810 CrossRef CAS.
- M. H. Rashid, T. F. Borin, R. Ara, K. Angara, J. Cai, B. R. Achyut, Y. Liu and A. S. Arbab, Nanomedicine, 2019, 21, 102072 CrossRef CAS.
- Z. Varga, I. Gyurkó, K. Pálóczi, E. I. Buzás, I. Horváth, N. Hegedűs, D. Máthé and K. Szigeti, Cancer Biother. Radiopharm., 2016, 31, 168–173 CrossRef CAS.
- P. Gangadaran, C. M. Hong, J. M. Oh, R. L. Rajendran, S. Kalimuthu, S. H. Son, A. Gopal, L. Zhu, S. H. Baek, S. Y. Jeong, S.-W. Lee, J. Lee and B.-C. Ahn, Front. Pharmacol., 2018, 9, 817 CrossRef.
- S. H. Son, J. M. Oh, P. Gangadaran, H. D. Ji, H. W. Lee, R. L. Rajendran, S. H. Baek, A. Gopal, S. Kalimuthu, S. Y. Jeong, S. W. Lee, J. Lee and B. C. Ahn, Blood Cells, Mol., Dis., 2020, 80, 102375 CrossRef CAS.
- M. I. González, P. Martín-Duque, M. Desco and B. Salinas, Nanomaterials, 2020, 10, 1062 CrossRef.
- T. Smyth, M. Kullberg, N. Malik, P. Smith-Jones, M. W. Graner and T. J. Anchordoquy, J. Controlled Release, 2015, 199, 145–155 CrossRef CAS.
- D. W. Hwang, H. Choi, S. C. Jang, M. Y. Yoo, J. Y. Park, N. E. Choi, H. J. Oh, S. Ha, Y. S. Lee, J. M. Jeong, Y. S. Gho and D. S. Lee, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- W. Lohcharoenkal, L. Wang, Y. C. Chen and Y. Rojanasakul, BioMed Res. Int., 2014, 2014, 180549 Search PubMed.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS.
- S. Jain, R. Mathur, M. Das, N. K. Swarnakar and A. K. Mishra, Nanomedicine, 2011, 6, 1733–1754 CrossRef CAS.
- A. Bunschoten, T. Buckle, J. Kuil, G. D. Luker, K. E. Luker, O. E. Nieweg and F. W. B. van Leeuwen, Biomaterials, 2012, 33, 867–875 CrossRef CAS.
- Y. Yang, T. Neef, C. Mittelholzer, E. Garcia Garayoa, P. Bläuenstein, R. Schibli, U. Aebi and P. Burkhard, J. Nanobiotechnol., 2013, 11, 36 CrossRef.
- M. Liang, H. Tan, J. Zhou, T. Wang, D. Duan, K. Fan, J. He, D. Cheng, H. Shi, H. S. Choi and X. Yan, ACS Nano, 2018, 12, 9300–9308 CrossRef CAS.
- S. G. Yang, J. E. Chang, B. Shin, S. Park, K. Na and C. K. Shim, J. Mater. Chem., 2010, 20, 9042–9046 RSC.
- A. Ozgur, F. Y. Lambrecht, K. Ocakoglu, C. Gunduz and M. Yucebas, Int. J. Pharm., 2012, 422, 472–478 CrossRef CAS.
- A. Woods, A. Patel, D. Spina, Y. Riffo-Vasquez, A. Babin-Morgan, R. T. M. De Rosales, K. Sunassee, S. Clark, H. Collins, K. Bruce, L. A. Dailey and B. Forbes, J. Controlled Release, 2015, 210, 1–9 CrossRef CAS.
- A. G. Gil, J. M. Irache, I. Peñuelas, C. J. González Navarro and A. López de Cerain, Food Chem. Toxicol., 2017, 106, 477–486 CrossRef CAS.
- C. Perez-Medina, J. Tang, D. Abdel-Atti, B. Hogstad, M. Merad, E. A. Fisher, Z. A. Fayad, J. S. Lewis, W. J. M. Mulder and T. Reiner, J. Nucl. Med., 2015, 56, 1272–1277 CrossRef CAS.
- C. Pérez-Medina, T. Binderup, M. E. Lobatto, J. Tang, C. Calcagno, L. Giesen, C. H. Wessel, J. Witjes, S. Ishino, S. Baxter, Y. Zhao, S. Ramachandran, M. Eldib, B. L. Sánchez-Gaytán, P. M. Robson, J. Bini, J. F. Granada, K. M. Fish, E. S. G. Stroes, R. Duivenvoorden, S. Tsimikas, J. S. Lewis, T. Reiner, V. Fuster, A. Kjær, E. A. Fisher, Z. A. Fayad and W. J. M. Mulder, JACC: Cardiovasc. Imaging, 2016, 9, 950–961 Search PubMed.
- J. Tang, S. Baxter, A. Menon, A. Alaarg, B. L. Sanchez-Gaytan, F. Fay, Y. Zhao, M. Ouimet, M. S. Braza, V. A. Longo, D. Abdel-Atti, R. Duivenvoorden, C. Calcagno, G. Storm, S. Tsimikas, K. J. Moore, F. K. Swirski, M. Nahrendorf, E. A. Fisher, C. Pérez-Medina, Z. A. Fayad, T. Reiner and W. J. M. Mulder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6731–E6740 CrossRef CAS.
- K. H. Zheng, F. M. van der Valk, L. P. Smits, M. Sandberg, J. L. Dasseux, R. Baron, R. Barbaras, C. Keyserling, B. F. Coolen, A. J. Nederveen, H. J. Verberne, T. E. Nell, D. J. Vugts, R. Duivenvoorden, Z. A. Fayad, W. J. M. Mulder, G. A. M. S. van Dongen and E. S. G. Stroes, Atherosclerosis, 2016, 251, 381–388 CrossRef CAS.
- M. Lameijer, T. Binderup, M. M. T. Van Leent, M. L. Senders, F. Fay, J. Malkus, B. L. Sanchez-Gaytan, A. J. P. Teunissen, N. Karakatsanis, P. Robson, X. Zhou, Y. Ye, G. Wojtkiewicz, J. Tang, T. T. P. Seijkens, J. Kroon, E. S. G. Stroes, A. Kjaer, J. Ochando, T. Reiner, C. Pérez-Medina, C. Calcagno, E. A. Fischer, B. Zhang, R. E. Temel, F. K. Swirski, M. Nahrendorf, Z. A. Fayad, E. Lutgens, W. J. M. Mulder and R. Duivenvoorden, Nat. Biomed. Eng., 2018, 2, 279–292 CrossRef CAS.
- T. Binderup, R. Duivenvoorden, F. Fay, M. M. T. Van Leent, J. Malkus, S. Baxter, S. Ishino, Y. Zhao, B. Sanchez-Gaytan, A. J. P. Teunissen, Y. C. A. Frederico, J. Tang, G. Carlucci, S. Lyashchenko, C. Calcagno, N. Karakatsanis, G. Soultanidis, M. L. Senders, P. M. Robson, V. Mani, S. Ramachandran, M. E. Lobatto, B. A. Hutten, J. F. Granada, T. Reiner, F. K. Swirski, M. Nahrendorf, A. Kjaer, E. A. Fisher, Z. A. Fayad, C. Pérez-Medina and W. J. M. Mulder, Sci. Transl. Med., 2019, 11(506), eaaw7736, DOI:10.1126/scitranslmed.aaw7736.
- C. A. Mason, S. Kossatz, L. M. Carter, G. Pirovano, C. Brand, N. Guru, C. Pérez-Medina, J. S. Lewis, W. J. M. Mulder and T. Reiner, J. Nucl. Med., 2020, 61, 433–436 CrossRef CAS.
- M. Wu, J. Shi, D. Fan, Q. Zhou, F. Wang, Z. Niu and Y. Huang, Biomacromolecules, 2013, 14, 4032–4037 CrossRef CAS.
- P. Kothari, B. P. De, B. He, A. Chen, M. J. Chiuchiolo, D. Kim, A. Nikolopoulou, A. Amor-Coarasa, J. P. Dyke, H. U. Voss, S. M. Kaminsky, C. P. Foley, S. Vallabhajosula, B. Hu, S. G. DiMagno, D. Sondhi, R. G. Crystal, J. W. Babich and D. Ballon, Sci. Rep., 2017, 7, 39594 CrossRef CAS.
- J. W. Seo, E. S. Ingham, L. Mahakian, S. Tumbale, B. Wu, S. Aghevlian, S. Shams, M. Baikoghli, P. Jain, X. Ding, N. Goeden, T. Dobreva, N. C. Flytzanis, M. Chavez, K. Singhal, R. Leib, M. L. James, D. J. Segal, R. H. Cheng, E. A. Silva, V. Gradinaru and K. W. Ferrara, Nat. Commun., 2020, 11, 2102 CrossRef CAS.
- C. I. Øie, D. L. Wolfson, T. Yasunori, G. Dumitriu, K. K. Sørensen, P. A. McCourt, B. S. Ahluwalia and B. Smedsrød, Sci. Rep., 2020, 10, 1–9 CrossRef.
- J. R. Newton-Northup, S. D. Figueroa, T. P. Quinn and S. L. Deutscher, Nucl. Med. Biol., 2009, 36, 789–800 CrossRef CAS.
- J. R. Newton, Y. Miao, S. L. Deutscher and T. P. Quinn, J. Nucl. Med., 2007, 48, 429–436 CAS.
- M. Rusckowski, S. Gupta, G. Liu, S. Dou and D. J. Hnatowich, J. Nucl. Med., 2004, 45, 1201–1208 CAS.
- M. Rusckowski, S. Gupta, G. Liu, S. Dou and D. J. Hnatowich, Nucl. Med. Biol., 2008, 35, 433–440 CrossRef CAS.
- M. Elena Cardoso, L. Fernández, E. Tejería, P. Esperón and M. Terán, Curr. Radiopharm., 2016, 9, 137–142 CrossRef.
- D. Holman, M. P. Lungren, J. Hardy, C. Contag and F. Blankenberg, Bioconjugate Chem., 2017, 28, 2698–2706 CrossRef CAS.
- I. L. Aanei, A. M. Elsohly, M. E. Farkas, C. Netirojjanakul, M. Regan, S. Taylor Murphy, J. P. O’Neil, Y. Seo and M. B. Francis, Mol. Pharmaceutics, 2016, 13, 3764–3772 CrossRef CAS.
- M. E. Farkas, I. L. Aanei, C. R. Behrens, G. J. Tong, S. T. Murphy, J. P. O’Neil and M. B. Francis, Mol. Pharmaceutics, 2013, 10, 69–76 CrossRef CAS.
- Y. H. Chung, H. Cai and N. F. Steinmetz, Adv. Drug Delivery Rev., 2020, 156, 214–235, DOI:10.1016/j.addr.2020.06.024.
- M. Karimi, H. Mirshekari, S. M. Moosavi Basri, S. Bahrami, M. Moghoofei and M. R. Hamblin, Adv. Drug Delivery Rev., 2016, 106, 45–62 CrossRef CAS.
- C. Li and R. J. Samulski, Nat. Rev. Genet., 2020, 21, 255–272 CrossRef CAS.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS.
- M. Cagel, F. C. Tesan, E. Bernabeu, M. J. Salgueiro, M. B. Zubillaga, M. A. Moretton and D. A. Chiappetta, Eur. J. Pharm. Biopharm., 2017, 113, 211–228 CrossRef CAS.
- B. Hoang, H. Lee, R. M. Reilly and C. Allen, Mol. Pharmaceutics, 2009, 6, 581–592 CrossRef CAS.
- H. Fonge, H. Huang, D. Scollard, R. M. Reilly and C. Allen, J. Controlled Release, 2012, 157, 366–374 CrossRef CAS.
- R. Jain, S. Nabar, P. Dandekar, P. Hassan, V. Aswal, Y. Talmon, T. Shet, L. Borde, K. Ray and V. Patravale, Nanomedicine, 2010, 5, 575–587 CrossRef CAS.
- G. A. Abdelbary and M. I. Tadros, Int. J. Pharm., 2013, 452, 300–310 CrossRef CAS.
- S. A. Nour, N. S. Abdelmalak, M. J. Naguib, H. M. Rashed and A. B. Ibrahim, Drug Delivery, 2016, 23, 3681–3695 CrossRef CAS.
- F. Tesan, C. Cerqueira-Coutinho, J. Salgueiro, M. de Souza Albernaz, S. R. Pinto, S. R. Rezende Dos Reis, E. S. Bernardes, D. Chiapetta, M. Zubillaga and R. Santos-Oliveira, J. Drug Delivery Sci. Technol., 2016, 36, 95–98 CrossRef CAS.
- H. M. Rashed, R. N. Shamma and E. B. Basalious, Drug Delivery, 2017, 24, 181–187 CrossRef CAS.
- S. Dumoga, Y. Rai, A. N. Bhatt, A. K. Tiwari, S. Singh, A. K. Mishra and D. Kakkar, ACS Appl. Mater. Interfaces, 2017, 9, 22195–22211 CrossRef CAS.
- F. C. Tesan, M. G. Portillo, M. A. Moretton, E. Bernabeu, D. A. Chiappetta, M. J. Salgueiro and M. B. Zubillaga, Nucl. Med. Biol., 2017, 44, 62–68 CrossRef CAS.
- E. Grotz, N. L. Tateosian, J. Salgueiro, E. Bernabeu, L. Gonzalez, M. L. Manca, N. Amiano, D. Valenti, M. Manconi, V. García, M. A. Moretton and D. A. Chiappetta, J. Drug Delivery Sci. Technol., 2019, 53, 101170 CrossRef CAS.
- F. C. Tesan, M. B. Nicoud, M. Nuñez, V. A. Medina, D. A. Chiappetta and M. J. Salgueiro, Contrast Media Mol. Imaging, 2019, 2019, 4087895 Search PubMed.
- E. Ribeiro, I. Alho, F. Marques, L. Gano, I. Correia, J. D. G. Correia, S. Casimiro, L. Costa, I. Santos and C. Fernandes, Int. J. Pharm., 2016, 515, 692–701 CrossRef CAS.
- C. M. R. Oda, R. S. Fernandes, S. C. de Araújo Lopes, M. C. de Oliveira, V. N. Cardoso, D. M. Santos, A. M. de Castro Pimenta, A. Malachias, R. Paniago, D. M. Townsend, P. M. Colletti, D. Rubello, R. J. Alves, A. L. B. de Barros and E. A. Leite, Biomed. Pharmacother., 2017, 89, 268–275 CrossRef CAS.
- C. M. R. Oda, A. L. B. de Barros, R. S. Fernandes, S. E. M. Miranda, M. X. Teixeira, V. N. Cardoso, M. C. Oliveira and E. A. Leite, J. Drug Delivery Sci. Technol., 2018, 46, 182–187 CrossRef CAS.
- M. Silindir-Gunay and A. Y. Ozer, J. Drug Delivery Sci. Technol., 2020, 56, 101571 CrossRef CAS.
- V. S. Trubetskoy, M. D. Frank-Kamenetsky, K. R. Whiteman, G. L. Wolf and V. P. Torchilin, Acad. Radiol., 1996, 3, 232–238 CrossRef CAS.
- Z. Yang, S. Zheng, W. J. Harrison, J. Harder, X. Wen, J. G. Gelovani, A. Qiao and C. Li, Biomacromolecules, 2007, 8, 3422–3428 CrossRef CAS.
- A. Accardo, D. Tesauro, L. Aloj, L. Tarallo, C. Arra, G. Mangiapia, M. Vaccaro, C. Pedone, L. Paduano and G. Morelli, ChemMedChem, 2008, 3, 594–602 CrossRef CAS.
- R. R. Patil, J. Yu, S. R. Banerjee, Y. Ren, D. Leong, X. Jiang, M. Pomper, B. Tsui, D. L. Kraitchman and H. Q. Mao, Mol. Ther., 2011, 19, 1626–1635 CrossRef CAS.
- R. Zhang, C. Xiong, M. Huang, M. Zhou, Q. Huang, X. Wen, D. Liang and C. Li, Biomaterials, 2011, 32, 5872–5879 CrossRef CAS.
- B. Hoang, R. M. Reilly and C. Allen, Biomacromolecules, 2012, 13, 455–465 CrossRef CAS.
- B. Hoang, S. N. Ekdawi, R. M. Reilly and C. Allen, Mol. Pharmaceutics, 2013, 10, 4229–4241 CrossRef CAS.
- Y. Hong, H. Zhu, J. Hu, X. Lin, F. Wang, C. Li and Z. Yang, Bioorg. Med. Chem. Lett., 2014, 24, 2781–2785 CrossRef CAS.
- A. Arranja, O. Ivashchenko, A. G. Denkova, K. Morawska, S. Van Vlierberghe, P. Dubruel, G. Waton, F. J. Beekman, F. Schosseler and E. Mendes, Mol. Pharmaceutics, 2016, 13, 1158–1165 CrossRef CAS.
- A. Arranja, A. G. Denkova, K. Morawska, G. Waton, S. Van Vlierberghe, P. Dubruel, F. Schosseler and E. Mendes, J. Controlled Release, 2016, 224, 126–135 CrossRef CAS.
- J. Sun, L. Sun, J. Li, J. Xu, Z. Wan, Z. Ouyang, L. Liang, S. Li and D. Zeng, Acta Biomater., 2018, 75, 312–322 CrossRef CAS.
- A. I. Jensen, T. Binderup, P. K. Ek, C. E. Grandjean, P. H. Rasmussen, A. Kjaer and T. L. Andresen, J. Labelled Compd. Radiopharm., 2017, 60, 366–374 CrossRef CAS.
- J. Guo, H. Hong, G. Chen, S. Shi, Q. Zheng, Y. Zhang, C. P. Theuer, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2013, 34, 8323–8332 CrossRef CAS.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS.
- A. C. Laan, C. Santini, L. Jennings, M. de Jong, M. R. Bernsen and A. G. Denkova, EJNMMI Res., 2016, 6, 12 CrossRef.
- A. de la Fuente, S. Kramer, N. Mohr, S. Pektor, B. Klasen, N. Bausbacher, M. Miederer, R. Zentel and F. Rösch, Am. J. Nucl. Med. Mol. Imaging, 2019, 9, 67–83 CAS.
- A. Prasannan, T. A. Debele, H. C. Tsai, C. C. Chao, C. P. Lin and G. H. Hsiue, RSC Adv., 2015, 5, 107455–107465 RSC.
- Y. Zhang, M. Jeon, L. J. Rich, H. Hong, J. Geng, Y. Zhang, S. Shi, T. E. Barnhart, P. Alexandridis, J. D. Huizinga, M. Seshadri, W. Cai, C. Kim and J. F. Lovell, Nat. Nanotechnol., 2014, 9, 631–638 CrossRef CAS.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS.
- W. G. Lesniak, S. Boinapally, S. R. Banerjee, B. Behnam Azad, C. A. Foss, C. Shen, A. Lisok, B. Wharram, S. Nimmagadda and M. G. Pomper, Mol. Pharmaceutics, 2019, 16, 2590–2604 CrossRef CAS.
- M. C. Parrott, S. R. Benhabbour, C. Saab, J. A. Lemon, S. Parker, J. F. Valliant and A. Adronov, J. Am. Chem. Soc., 2009, 131, 2906–2916 CrossRef CAS.
- D. Zhou, S. H. Kim, V. M. Carroll, C. S. Dence and J. A. Katzenellenbn, Org. Biomol. Chem., 2014, 12, 8696–8701 RSC.
- K. Pant, D. Gröger, R. Bergmann, J. Pietzsch, J. Steinbach, B. Graham, L. Spiccia, F. Berthon, B. Czarny, L. Devel, V. Dive, H. Stephan and R. Haag, Bioconjugate Chem., 2015, 26, 906–918 CrossRef CAS.
- W. Ma, F. Fu, J. Zhu, R. Huang, Y. Zhu, Z. Liu, J. Wang, P. S. Conti, X. Shi and K. Chen, Nanoscale, 2018, 10, 6113–6124 RSC.
- T. S. Humani, S. Sutari, T. Triningsih, M. Ramli, R. Ritawidya and R. D. Haryuni, ITB J. Math. Fundam. Sci., 2017, 49, 258–268 CrossRef CAS.
- L. Kovacs, M. Tassano, M. Cabrera, C. B. Zamboni, M. Fernández, R. M. Anjos and P. Cabral, Cancer Biother. Radiopharm., 2015, 30, 405–410 CrossRef CAS.
- B. Gibbens-Bandala, E. Morales-Avila, G. Ferro-Flores, C. Santos-Cuevas, M. Luna-Gutiérrez, G. Ramírez-Nava and B. Ocampo-García, Polymers, 2019, 11, 1572 CrossRef CAS.
- H. Mendoza-Nava, G. Ferro-Flores, F. de, M. Ramírez, B. Ocampo-García, C. Santos-Cuevas, L. Aranda-Lara, E. Azorín-Vega, E. Morales-Avila and K. Isaac-Olivé, J. Nanomater., 2016, 2016, 1–11 CrossRef.
- A. Laznickova, V. Biricova, M. Laznicek and P. Hermann, Appl. Radiat. Isot., 2014, 84, 70–77 CrossRef CAS.
- C. Kojima, C. Regino, Y. Umeda, H. Kobayashi and K. Kono, Int. J. Pharm., 2010, 383, 293–296 CrossRef CAS.
- Y. Niki, M. Ogawa, R. Makiura, Y. Magata and C. Kojima, Nanomedicine, 2015, 11, 2119–2127 CrossRef CAS.
- H. Katsumi, M. Nishikawa, R. Hirosaki, T. Okuda, S. Kawakami, F. Yamashita, M. Hashida, T. Sakane and A. Yamamoto, Mol. Pharmaceutics, 2016, 13, 2867–2873 CrossRef CAS.
- K. Sano, Y. Iwamiya, T. Kurosaki, M. Ogawa, Y. Magata, H. Sasaki, T. Ohshima, M. Maeda and T. Mukai, J. Controlled Release, 2014, 194, 310–315 CrossRef CAS.
- H. Kobayashi, N. Sato, T. Saga, Y. Nakamoto, T. Ishimori, S. Toyama, K. Togashi, J. Konishi and M. W. Brechbiel, Eur. J. Nucl. Med., 2000, 27, 1334–1339 CrossRef CAS.
- M. Mamede, T. Saga, H. Kobayashi, T. Ishimori, T. Higashi, N. Sato, M. W. Brechbiel and J. Konishi, Clin. Cancer Res., 2003, 9, 3756–3762 CAS.
- H. Kobayashi, C. Wu, M. K. Kim, C. H. Paik, J. A. Carrasquillo and M. W. Brechbiel, Bioconjugate Chem., 1999, 10, 103–111 CrossRef CAS.
- A. Parat, D. Kryza, F. Degoul, J. Taleb, C. Viallard, M. Janier, A. Garofalo, P. Bonazza, L. Heinrich-Balard, R. Cohen, E. Miot-Noirault, J. M. Chezal, C. Billotey and D. Felder-Flesch, J. Mater. Chem. B, 2015, 3, 2560–2571 RSC.
- V. Biricová, A. Lázničková, M. Lázníček, M. Polášek and P. Hermann, J. Pharm. Biomed. Anal., 2011, 56, 505–512 CrossRef.
- H. Kobayashi, S. Kawamoto, T. Saga, N. Sato, T. Ishimori, J. Konishi, K. Ono, K. Togashi and M. W. Brechbiel, Bioconjugate Chem., 2001, 12, 587–593 CrossRef CAS.
- S. A. McNelles, S. D. Knight, N. Janzen, J. F. Valliant and A. Adronov, Biomacromolecules, 2015, 16, 3033–3041 CrossRef CAS.
- L. P. Sadowski, P. E. Edem, J. F. Valliant and A. Adronov, Macromol. Biosci., 2016, 16, 1475–1484 CrossRef CAS.
- M. Song, Z. Guo, M. Gao, C. Shi, D. Xu, L. You, X. Wu, X. Su, R. Zhuang, W. Pan, T. Liu and X. Zhang, Chem. Biol. Drug Des., 2017, 89, 755–761 CrossRef CAS.
- Z. Guo, L. You, C. Shi, M. Song, M. Gao, D. Xu, C. Peng, R. Zhuang, T. Liu, X. Su, J. Du and X. Zhang, Mol. Pharmaceutics, 2017, 14, 3780–3788 CrossRef CAS.
- Y. Zhang, Y. Sun, X. Xu, H. Zhu, L. Huang, X. Zhang, Y. Qi and Y. M. Shen, Bioorg. Med. Chem. Lett., 2010, 20, 927–931 CrossRef CAS.
- X. Xu, Y. Zhang, X. Wang, X. Guo, X. Zhang, Y. Qi and Y. M. Shen, Bioorg. Med. Chem., 2011, 19, 1643–1648 CrossRef CAS.
- Y. Zhang, Y. Sun, X. Xu, X. Zhang, H. Zhu, L. Huang, Y. Qi and Y. M. Shen, J. Med. Chem., 2010, 53, 3262–3272 CrossRef CAS.
- X. Chen, S. Dou, G. Liu, X. Liu, Y. Wang, L. Chen, M. Rusckowski and D. J. Hnatowich, Bioconjugate Chem., 2008, 19, 1518–1525 CrossRef CAS.
- M. Tassano, N. Oddone, M. Fernández, W. Porcal, M. F. García, W. Martínez-López, J. C. Benech and P. Cabral, Int. J. Radiat. Biol., 2018, 94, 664–670 CrossRef CAS.
- W. Cui, Y. Zhang, X. Xu and Y.-M. Shen, Med. Chem., 2012, 8, 727–731 CrossRef CAS.
- W. G. Lesniak, C. Chu, A. Jablonska, B. Behnam Azad, O. Zwaenepoel, M. Zawadzki, A. Lisok, M. G. Pomper, P. Walczak, J. Gettemans and M. Janowski, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1940–1951 CrossRef CAS.
- C. Wängler, S. Maschauer, O. Prante, M. Schäfer, R. Schirrmacher, P. Bartenstein, M. Eisenhut and B. Wängler, ChemBioChem, 2010, 11, 2168–2181 CrossRef.
- A. Ghai, B. Singh, P. Panwar Hazari, M. K. Schultz, A. Parmar, P. Kumar, S. Sharma, D. Dhawan and A. Kumar Mishra, Appl. Radiat. Isot., 2015, 105, 40–46 CrossRef CAS.
- G. Fischer, B. Wängler and C. Wängler, Molecules, 2014, 19, 6952–6974 CrossRef.
- S. Lindner, C. Michler, B. Wängler, P. Bartenstein, G. Fischer, R. Schirrmacher and C. Wängler, Bioconjugate Chem., 2014, 25, 489–500 CrossRef CAS.
- E. Orocio-Rodríguez, G. Ferro-Flores, C. L. Santos-Cuevas, F. de, M. Ramírez, B. E. Ocampo-García, E. Azorín-Vega and F. M. Sánchez-García, J. Nanosci. Nanotechnol., 2015, 15, 4159–4169 CrossRef.
- S. M. Ghoreishi, A. Khalaj, A. Bitarafan-Rajabi, A. D. Azar, M. S. Ardestani and A. Assadi, Drug Res., 2017, 67, 149–155 CAS.
- S. M. Ghoreishi, A. Khalaj, O. Sabzevari, L. Badrzadeh, P. Mohammadzadeh, S. S. Mousavi Motlagh, A. Bitarafan-Rajabi and M. Shafiee Ardestani, Int. J. Nanomed., 2018, 13, 4671–4683 CrossRef CAS.
- M. S. Ardestani, A. Bitarafan-Rajabi, P. Mohammadzadeh, S. Mortazavi-Derazkola, O. Sabzevari, A. D. Azar, S. Kazemi, S. R. Hosseini and S. M. Ghoreishi, Bioorg. Chem., 2020, 96, 103572 CrossRef CAS.
- A. G. Khosroshahi, M. Amanlou, O. Sabzevari, F. J. Daha, M. R. Aghasadeghi, M. Ghorbani, M. S. Ardestani, M. S. Alavidjeh, S. M. Sadat, M. H. Pouriayevali, L. Mousavi and S. E. S. Ebrahimi, Curr. Med. Chem., 2012, 20, 123–133 CrossRef.
- M. R. Tassano, P. F. Audicio, J. P. Gambini, M. Fernandez, J. P. Damian, M. Moreno, J. A. Chabalgoity, O. Alonso, J. C. Benech and P. Cabral, Bioorg. Med. Chem. Lett., 2011, 21, 5598–5601 CrossRef CAS.
- X. Xu, Y. Li, T. Cao, J. Cheng and Y. Zhang, J. Nanopart. Res., 2018, 20, 204 CrossRef.
- D. S. Wilbur, P. M. Pathare, D. K. Hamlin, K. R. Buhler and R. L. Vessella, Bioconjugate Chem., 1998, 9, 813–825 CrossRef CAS.
- Y. Kurokawa, H. Sone, T.-T. Win-Shwe, Y. Zeng, H. Kimura, Y. Koyama, Y. Yagi, Y. Matsui, M. Yamazaki and S. Hirano, Int. J. Nanomed., 2017, 12, 3967–3975 CrossRef CAS.
- Y. Cheng, J. Zhu, L. Zhao, Z. Xiong, Y. Tang, C. Liu, L. Guo, W. Qiao, X. Shi and J. Zhao, Nanomedicine, 2016, 11, 1253–1266 CrossRef CAS.
- N. Song, L. Zhao, X. Xu, M. Zhu, C. Liu, N. Sun, J. Yang, X. Shi and J. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 12395–12406 CrossRef.
- L. Zhao, J. Zhu, Y. Cheng, Z. Xiong, Y. Tang, L. Guo, X. Shi and J. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 19798–19808 CrossRef CAS.
- L. Trembleau, M. Simpson, R. W. Cheyne, I. Escofet, M. V. C. A. L. Appleyard, K. Murray, S. Sharp, A. M. Thompson and T. A. D. Smith, New J. Chem., 2011, 35, 2496–2502 RSC.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS.
- M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rösch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef CAS.
- A. B. Benito, M. K. Aiertza, M. Marradi, L. Gil-Iceta, T. Shekhter Zahavi, B. Szczupak, M. Jiménez-González, T. Reese, E. Scanziani, L. Passoni, M. Matteoli, M. De Maglie, A. Orenstein, M. Oron-Herman, G. Kostenich, L. Buzhansky, E. Gazit, H.-J. Grande, V. Gómez-Vallejo, J. Llop and I. Loinaz, Biomacromolecules, 2016, 17, 3213–3221 CrossRef CAS.
- S. E. Dunn, A. G. A. Coombes, M. C. Garnett, S. S. Davis, M. C. Davies and L. Illum, J. Controlled Release, 1997, 44, 65–76 CrossRef CAS.
- M. Snehalatha, K. Venugopal, R. N. Saha, A. K. Babbar and R. K. Sharma, Drug Delivery, 2008, 15, 277–287 CrossRef CAS.
- K. K. Halder, B. Mandal, M. C. Debnath, H. Bera, L. K. Ghosh and B. K. Gupta, J. Drug Targeting, 2008, 16, 311–320 CrossRef CAS.
- N. Mondal, K. K. Halder, M. M. Kamila, M. C. Debnath, T. K. Pal, S. K. Ghosal, B. R. Sarkar and S. Ganguly, Int. J. Pharm., 2010, 397, 194–200 CrossRef CAS.
- H. Gupta, M. Aqil, R. K. Khar, A. Ali, A. Bhatnagar and G. Mittal, Nanomedicine, 2010, 6, 324–333 CrossRef CAS.
- K. Ramanlal Chaudhari, A. Kumar, V. K. Megraj Khandelwal, M. Ukawala, A. S. Manjappa, A. K. Mishra, J. Monkkonen and R. S. Ramachandra Murthy, J. Controlled Release, 2012, 158, 470–478 CrossRef CAS.
- D. Sharma, D. Maheshwari, G. Philip, R. Rana, S. Bhatia, M. Singh, R. Gabrani, S. K. Sharma, J. Ali, R. K. Sharma and S. Dang, BioMed Res. Int., 2014, 2014, 156010 Search PubMed.
- P. J. Das, P. Paul, B. Mukherjee, B. Mazumder, L. Mondal, R. Baishya, M. C. Debnath and K. S. Dey, Mol. Pharmaceutics, 2015, 12, 2651–2664 CrossRef CAS.
- R. Baishya, D. K. Nayak, D. Kumar, S. Sinha, A. Gupta, S. Ganguly and M. C. Debnath, Pharm. Res., 2016, 33, 2691–2703 CrossRef CAS.
- Z. He, X. Zhang, J. Huang, Y. Wu, X. Huang, J. Chen, J. Xia, H. Jiang, J. Ma and J. Wu, Oncotarget, 2016, 7, 76635–76646 CrossRef.
- R. H. Gaonkar, S. Ganguly, S. Dewanjee, S. Sinha, A. Gupta, S. Ganguly, D. Chattopadhyay and M. Chatterjee Debnath, Sci. Rep., 2017, 7, 1–14 CrossRef CAS.
- I. Khan, A. Gothwal, A. Kaul, R. Mathur, A. K. Mishra and U. Gupta, Pharm. Res., 2018, 35, 1–11 CrossRef.
- K. Nigam, A. Kaur, A. Tyagi, K. Manda, R. Gabrani and S. Dang, Rejuvenation Res., 2019, 22, 235–245 CrossRef CAS.
- L. Mondal, B. Mukherjee, K. Das, S. Bhattacharya, D. Dutta, S. Chakraborty, M. M. Pal, R. H. Gaonkar and M. C. Debnath, Int. J. Nanomed., 2019, 14, 8073–8094 CrossRef CAS.
- R. W. Sirianni, M. Q. Zheng, T. R. Patel, T. Shafbauer, J. Zhou, W. M. Saltzman, R. E. Carson and Y. Huang, Bioconjugate Chem., 2014, 25, 2157–2165 CrossRef CAS.
- S. Subramanian, U. Pandey, D. Gugulothu, V. Patravale and G. Samuel, Cancer Biother. Radiopharm., 2013, 28, 598–606 CrossRef CAS.
- M. R. Gill, J. U. Menon, P. J. Jarman, J. Owen, I. Skaripa-Koukelli, S. Able, J. A. Thomas, R. Carlisle and K. A. Vallis, Nanoscale, 2018, 10, 10596–10608 RSC.
- R. M. Trujillo-Nolasco, E. Morales-Avila, B. E. Ocampo-García, G. Ferro-Flores, B. V. Gibbens-Bandala, A. Escudero-Castellanos and K. Isaac-Olive, Mater. Sci. Eng., C, 2019, 103, 109766 CrossRef CAS.
- B. Gibbens-Bandala, E. Morales-Avila, G. Ferro-Flores, C. Santos-Cuevas, L. Meléndez-Alafort, M. Trujillo-Nolasco and B. Ocampo-García, Mater. Sci. Eng., C, 2019, 105, 110043 CrossRef CAS.
- G. Arora, J. Shukla, S. Ghosh, S. K. Maulik, A. Malhotra and G. Bandopadhyaya, PLoS One, 2012, 7, e34019 CrossRef CAS.
- M. Allmeroth, D. Moderegger, B. Biesalski, K. Koynov, F. Rösch, O. Thews and R. Zentel, Biomacromolecules, 2011, 12, 2841–2849 CrossRef CAS.
- M. Allmeroth, D. Moderegger, D. Gündel, K. Koynov, H. G. Buchholz, K. Mohr, F. Rösch, R. Zentel and O. Thews, Biomacromolecules, 2013, 14, 3091–3101 CrossRef CAS.
- M. Allmeroth, D. Moderegger, D. Gündel, H. G. Buchholz, N. Mohr, K. Koynov, F. Rösch, O. Thews and R. Zentel, J. Controlled Release, 2013, 172, 77–85 CrossRef CAS.
- M. M. Herth, M. Barz, M. Jahn, R. Zentel and F. Rösch, Bioorg. Med. Chem. Lett., 2010, 20, 5454–5458 CrossRef CAS.
- M. C. Chen, H. S. Wong, K. J. Lin, H. L. Chen, S. P. Wey, K. Sonaje, Y. H. Lin, C. Y. Chu and H. W. Sung, Biomaterials, 2009, 30, 6629–6637 CrossRef CAS.
- A. Polyák, I. Hajdu, M. Bodnár, G. Trencsényi, Z. Pöstényi, V. Haász, G. Jánoki, G. A. Jánoki, L. Balogh and J. Borbély, Int. J. Pharm., 2013, 449, 10–17 CrossRef.
- A. Polyák, I. Hajdu, M. Bodnár, G. Dabasi, R. P. Jóba, J. Borbély and L. Balogh, Int. J. Pharm., 2014, 474, 91–94 CrossRef.
- H. Rajabi, S. Rasaneh and S. Salehi, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1450–1454 CrossRef CAS.
- I. S. Ahmed, H. M. Rashed, H. Fayez, F. Farouk and R. N. Shamma, Pharmaceutics, 2020, 12, 107 CrossRef CAS.
- J. R. Franca, L. L. Fuscaldi, T. G. Ribeiro, G. Foureaux, A. L. A. Cesar, R. O. Castilho, S. Cronemberger, A. J. Ferreira, S. O. A. Fernandes, V. N. Cardoso and A. A. G. Faraco, J. Biomed. Mater. Res., Part B, 2020, 108, 2227–2237 CrossRef CAS.
- M. Fairclough, C. Prenant, B. Ellis, H. Boutin, A. McMahon, G. Brown, P. Locatelli and A. K. P. Jones, J. Labelled Compd. Radiopharm., 2016, 59, 270–276 CrossRef CAS.
- M. Fairclough, B. Ellis, H. Boutin, A. K. P. Jones, A. McMahon, S. Alzabin, A. Gennari and C. Prenant, Appl. Radiat. Isot., 2017, 130, 7–12 CrossRef CAS.
- D. E. Lee, J. H. Na, S. Lee, C. M. Kang, H. N. Kim, S. J. Han, H. Kim, Y. S. Choe, K. H. Jung, K. C. Lee, K. Choi, I. C. Kwon, S. Y. Jeong, K. H. Lee and K. Kim, Mol. Pharmaceutics, 2013, 10, 2190–2198 CrossRef CAS.
- S. Lee, S. W. Kang, J. H. Ryu, J. H. Na, D. E. Lee, S. J. Han, C. M. Kang, Y. S. Choe, K. C. Lee, J. F. Leary, K. Choi, K. H. Lee and K. Kim, Bioconjugate Chem., 2014, 25, 601–610 CrossRef CAS.
- Z. Körhegyi, D. Rózsa, I. Hajdu, M. Bodnár, I. Kertész, K. Kerekes, S. Kun, J. Kollár, J. Varga, I. Garai, G. Trencsényi and J. Borbély, Anticancer Res., 2019, 39, 2415–2427 CrossRef.
- E. J. Keliher, Y.-X. Ye, G. R. Wojtkiewicz, A. D. Aguirre, B. Tricot, M. L. Senders, H. Groenen, F. Fay, C. Perez-Medina, C. Calcagno, G. Carlucci, T. Reiner, Y. Sun, G. Courties, Y. Iwamoto, H.-Y. Kim, C. Wang, J. W. Chen, F. K. Swirski, H.-Y. Wey, J. Hooker, Z. A. Fayad, W. J. M. Mulder, R. Weissleder and M. Nahrendorf, Nat. Commun., 2017, 8, 14064 CrossRef CAS.
- R. Gracia, M. Marradi, U. Cossío, A. Benito, A. Pérez-San Vicente, V. Gómez-Vallejo, H. J. Grande, J. Llop and I. Loinaz, J. Mater. Chem. B, 2017, 5, 1143–1147 RSC.
- M. D. Majmudar, J. Yoo, E. J. Keliher, J. J. Truelove, Y. Iwamoto, B. Sena, P. Dutta, A. Borodovsky, K. Fitzgerald, M. F. Di Carli, P. Libby, D. G. Anderson, F. K. Swirski, R. Weissleder and M. Nahrendorf, Circ. Res., 2013, 112, 755–761 CrossRef CAS.
- R. Rossin, D. Pan, K. Qi, J. L. Turner, X. Sun, K. L. Wooley and M. J. Welch, J. Nucl. Med., 2005, 46, 1210–1218 Search PubMed.
- J. Xu, G. Sun, R. Rossin, A. Hagooly, Z. Li, K. I. Fukukawa, B. W. Messmore, D. A. Moore, M. J. Welch, C. J. Hawker and K. L. Wooley, Macromolecules, 2007, 40, 2971–2973 CrossRef CAS.
- G. Sun, J. Xu, A. Hagooly, R. Rossin, Z. Li, D. A. Moore, C. J. Hawker, M. J. Welch and K. L. Wooley, Adv. Mater., 2007, 19, 3157–3162 CrossRef CAS.
- G. Sun, A. Hagooly, J. Xu, A. M. Nyström, Z. Li, R. Rossin, D. A. Moore, K. L. Wooley and M. J. Welch, Biomacromolecules, 2008, 9, 1997–2006 CrossRef CAS.
- A. Li, H. P. Luehmann, G. Sun, S. Samarajeewa, J. Zou, S. Zhang, F. Zhang, M. J. Welch, Y. Liu and K. L. Wooley, ACS Nano, 2012, 6, 8970–8982 CrossRef CAS.
- T. J. Beldman, M. L. Senders, A. Alaarg, C. Pérez-Medina, J. Tang, Y. Zhao, F. Fay, J. Deichmöller, B. Born, E. Desclos, N. N. Van Der Wel, R. A. Hoebe, F. Kohen, E. Kartvelishvily, M. Neeman, T. Reiner, C. Calcagno, Z. A. Fayad, M. P. J. De Winther, E. Lutgens, W. J. M. Mulder and E. Kluza, ACS Nano, 2017, 11, 5785–5799 CrossRef CAS.
- D. R. Beckford Vera, S. D. Fontaine, H. F. VanBrocklin, B. R. Hearn, R. Reid, G. W. Ashley and D. V. Santi, Mol. Cancer Ther., 2020, 19, 673–679 CrossRef.
- S. Soni, A. K. Babbar, R. K. Sharma and A. Maitra, J. Drug Targeting, 2006, 14, 87–95 CrossRef CAS.
- S. Singh, B. Bingöl, A. Morgenroth, F. M. Mottaghy, M. Möller and J. Schmaljohann, Macromol. Rapid Commun., 2013, 34, 562–567 CrossRef CAS.
- N. Drude, S. Singh, O. H. Winz, M. Möller, F. M. Mottaghy and A. Morgenroth, Biomacromolecules, 2017, 18, 2489–2498 CrossRef CAS.
- S. Berke, A. L. Kampmann, M. Wuest, J. J. Bailey, B. Glowacki, F. Wuest, K. Jurkschat, R. Weberskirch and R. Schirrmacher, Bioconjugate Chem., 2018, 29, 89–95 CrossRef CAS.
- K. Sano, Y. Kanada, K. Takahashi, N. Ding, K. Kanazaki, T. Mukai, M. Ono and H. Saji, Mol. Pharmaceutics, 2018, 15, 3997–4003 CrossRef CAS.
- K. Sano, Y. Kanada, K. Kanazaki, N. Ding, M. Ono and H. Saji, J. Nucl. Med., 2017, 58, 1380–1385 CrossRef CAS.
- S. R. R. dos Reis, S. R. Pinto, F. D. de Menezes, R. Martinez-Manez, E. Ricci-Junior, L. M. R. Alencar, E. Helal-Neto, A. O. da Silva de Barros, P. C. Lisboa and R. Santos-Oliveira, Pharm. Res., 2020, 37, 1–12 CrossRef.
- D. Shenoy, S. Little, R. Langer and M. Amiji, Pharm. Res., 2005, 22, 2107–2114 CrossRef CAS.
- P. K. Woodard, Y. Liu, E. D. Pressly, H. P. Luehmann, L. Detering, D. E. Sultan, R. Laforest, A. J. McGrath, R. J. Gropler and C. J. Hawker, Pharm. Res., 2016, 33, 2400–2410 CrossRef CAS.
- Y. Liu, H. P. Luehmann, L. Detering, E. D. Pressly, A. J. McGrath, D. Sultan, A. Nguyen, S. Grathwohl, M. Shokeen, M. Zayed, R. J. Gropler, D. Abendschein, C. J. Hawker and P. K. Woodard, ACS Appl. Mater. Interfaces, 2019, 11, 15316–15321 CrossRef CAS.
- R. Abellan-Pose, M. Rodríguez-Évora, S. Vicente, N. Csaba, C. Évora, M. J. Alonso and A. Delgado, Eur. J. Pharm. Biopharm., 2017, 112, 155–163 CrossRef CAS.
- P. P. Di Mauro, V. Gómez-Vallejo, Z. Baz Maldonado, J. Llop Roig and S. Borrós, Bioconjugate Chem., 2015, 26, 582–592 CrossRef CAS.
- B. Jia, X. Zhang, B. Wang, M. Chen, F. Lv, S. Wang and F. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 6646–6651 CrossRef CAS.
- S. Nascimento dos Santos, S. Rhaissa Rezende dos Reis, S. Rocha Pinto, C. Cerqueira-Coutinho, F. Nigro, T. Christina Barja-Fidalgo, N. Martins Alexandre Pinheiro, H. Affonso Paula Neto and R. Santos-Oliveira, J. Nanopart. Res., 2017, 19, 345, DOI:10.1007/s11051-017-4037-x.
- T. L. Braga, S. R. Pinto, S. R. R. dos Reis, F. L. Portilho, A. O. da Silva de Barros, E. S. Bernardes, S. N. dos Santos, L. M. R. Alencar, E. Ricci-Junior and R. Santos-Oliveira, Pharm. Res., 2019, 36, 1–8 CrossRef CAS.
- M. A. Pereira, V. C. F. Mosqueira, J. M. C. Vilela, M. S. Andrade, G. A. Ramaldes and V. N. Cardoso, Eur. J. Pharm. Sci., 2008, 33, 42–51 CrossRef CAS.
- M. A. Pereira, V. C. F. Mosqueira, V. A. S. Carmo, C. S. Ferrari, E. C. O. Reis, G. A. Ramaldes and V. N. Cardoso, Nucl. Med. Commun., 2009, 30, 749–755 CrossRef.
- G. Wunderlich, T. Grüning, B. R. Paulke, A. Lieske and J. Kotzerke, Nucl. Med. Biol., 2004, 31, 87–92 CrossRef CAS.
- P. Areses, M. T. Agüeros, G. Quincoces, M. Collantes, J. Á. Richter, L. M. López-Sánchez, M. Sánchez-Martínez, J. M. Irache and I. Penuelas, Mol. Imaging Biol., 2011, 13, 1215–1223 CrossRef.
- G. Kaul and M. Amiji, J. Drug Targeting, 2004, 12, 585–591 CrossRef CAS.
- S. Kommareddy and M. Amiji, J. Pharm. Sci., 2007, 96, 397–407 CrossRef CAS.
- M. D. Bhavsar and M. M. Amiji, AAPS PharmSciTech, 2008, 9, 288–294 CrossRef CAS.
- J. Xu, F. Gattacceca and M. Amiji, Mol. Pharmaceutics, 2013, 10, 2031–2044 CrossRef CAS.
- J. A. C. M. Goos, A. Cho, L. M. Carter, T. R. Dilling, M. Davydova, K. Mandleywala, S. Puttick, A. Gupta, W. S. Price, J. F. Quinn, M. R. Whittaker, J. S. Lewis and T. P. Davis, Theranostics, 2020, 10, 567–584 CrossRef CAS.
- K. R. Chaudhari, M. Ukawala, A. S. Manjappa, A. Kumar, P. K. Mundada, A. K. Mishra, R. Mathur, J. Mönkkönen and R. S. Ramchandra Murthy, Pharm. Res., 2012, 29, 53–68 CrossRef CAS.
- Q. Fan, K. Cheng, X. Hu, X. Ma, R. Zhang, M. Yang, X. Lu, L. Xing, W. Huang, S. S. Gambhir and Z. Cheng, J. Am. Chem. Soc., 2014, 136, 15185–15194 CrossRef CAS.
- H. Zhou, Q. Zhang, Y. Cheng, L. Xiang, G. Shen, X. Wu, H. Cai, D. Li, H. Zhu, R. Zhang, L. Li and Z. Cheng, Nanomedicine, 2020, 29, 102248 CrossRef CAS.
- T. Martínez Martínez, Á. García Aliaga, I. López-González, A. Abella Tarazona, M. J. Ibáñez Ibáñez, J. L. Cenis, L. Meseguer-Olmo and A. A. Lozano-Pérez, ACS Biomater. Sci. Eng., 2020, 6, 3299–3309 CrossRef.
- G. Shim, M. G. Kim, J. Y. Park and Y. K. Oh, Adv. Drug Delivery Rev., 2016, 105, 205–227 CrossRef CAS.
- B. S. Wong, S. L. Yoong, A. Jagusiak, T. Panczyk, H. K. Ho, W. H. Ang and G. Pastorin, Adv. Drug Delivery Rev., 2013, 65, 1964–2015 CrossRef CAS.
- H. Gong, R. Peng and Z. Liu, Adv. Drug Delivery Rev., 2013, 65, 1951–1963 CrossRef CAS.
- J. Lin, X. Chen and P. Huang, Adv. Drug Delivery Rev., 2016, 105, 242–254 CrossRef CAS.
- S. Shi, C. Xu, K. Yang, S. Goel, H. F. Valdovinos, H. Luo, E. B. Ehlerding, C. G. England, L. Cheng, F. Chen, R. J. Nickles, Z. Liu and W. Cai, Angew. Chem., Int. Ed., 2017, 56, 2889–2892 CrossRef CAS.
- S. Sarpaki, F. Cortezon-Tamarit, S. R. M. M. de Aguiar, R. M. Exner, D. Divall, R. L. Arrowsmith, H. Ge, F. J. Palomares, L. Carroll, D. G. Calatayud, S. J. Paisey, E. O. Aboagye and S. I. Pascu, Nanoscale, 2020, 12, 6603–6608 RSC.
- K. Yang, J. Wan, S. Zhang, Y. Zhang, S. T. Lee and Z. Liu, ACS Nano, 2011, 5, 516–522 CrossRef CAS.
- L. Zhan, G. Yanxia, Z. Xiaoyong, Q. Wei, F. Qiaohui, L. Yan, J. Zongxian, W. Jianjun, T. Yuqin, D. Xiaojiang and W. Wangsuo, J. Nanopart. Res., 2011, 13, 2939–2947 CrossRef.
- Q. Wei, L. Zhan, B. Juanjuan, W. Jing, W. Jianjun, S. Taoli, G. Yi’an and W. Wangsuo, Nanoscale Res. Lett., 2012, 7, 473 CrossRef.
- S. B. Challan and A. Massoud, J. Radioanal. Nucl. Chem., 2017, 314, 2189–2199 CrossRef CAS.
- F. Yurt, O. A. Ersöz, E. Harputlu and K. Ocakoglu, Chem. Biol. Drug Des., 2018, 91, 1094–1100 CrossRef CAS.
- M. Das, S. R. Datir, R. P. Singh and S. Jain, Mol. Pharmaceutics, 2013, 10, 2543–2557 CrossRef CAS.
- R. S. Fernandes, J. A. Lemos, A. L. Branco de Barros, V. Geraldo, E. Eleto da Silva, L. Alisaraie and D. C. Ferreira Soares, J. Drug Delivery Sci. Technol., 2019, 50, 266–277 CrossRef CAS.
- T. Cao, X. Zhou, Y. Zheng, Y. Sun, J. Zhang, W. Chen, J. Zhang, Z. Zhou, S. Yang, Y. Zhang, H. Yang and M. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 42612–42621 CrossRef CAS.
- X. Zhang, J. Yin, C. Kang, J. Li, Y. Zhu, W. Li, Q. Huang and Z. Zhu, Toxicol. Lett., 2010, 198, 237–243 CrossRef CAS.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Carbon, 2011, 49, 986–995 CrossRef CAS.
- X. Zhang, Y. Zhu, J. Li, Z. Zhu, J. Li, W. Li and Q. Huang, J. Nanopart. Res., 2011, 13, 6941–6952 CrossRef CAS.
- M. R. McDevitt, D. Chattopadhyay, B. J. Kappel, J. S. Jaggi, S. R. Schiffman, C. Antczak, J. T. Njardarson, R. Brentjens and D. A. Scheinberg, J. Nucl. Med., 2007, 48, 1180–1189 CrossRef CAS.
- M. R. McDevitt, D. Chattopadhyay, J. S. Jaggi, R. D. Finn, P. B. Zanzonico, C. Villa, D. Rey, J. Mendenhall, C. A. Batt, J. T. Njardarson and D. A. Scheinberg, PLoS One, 2007, 2, e907 CrossRef.
- C. H. Villa, M. R. McDevitt, F. E. Escorcia, D. A. Rey, M. Bergkvist, C. A. Batt and D. A. Scheinberg, Nano Lett., 2008, 8, 4221–4228 CrossRef CAS.
- L. Lacerda, A. Soundararajan, R. Singh, G. Pastorin, K. T. Al-Jamal, J. Turton, P. Frederik, M. A. Herrero, S. Li, A. Bao, D. Emfietzoglou, S. Mather, W. T. Phillips, M. Prato, A. Bianco, B. Goins and K. Kostarelos, Adv. Mater., 2008, 20, 225–230 CrossRef CAS.
- M. A. Herrero, L. Lacerda, A. Bianco, K. Kostarelos and M. Prato, Int. J. Nanotechnol., 2011, 8, 885–897 CrossRef CAS.
- B. Cornelissen, S. Able, V. Kersemans, P. A. Waghorn, S. Myhra, K. Jurkshat, A. Crossley and K. A. Vallis, Biomaterials, 2013, 34, 1146–1154 CrossRef CAS.
- H. Kafa, J. T. W. Wang, N. Rubio, K. Venner, G. Anderson, E. Pach, B. Ballesteros, J. E. Preston, N. J. Abbott and K. T. Al-Jamal, Biomaterials, 2015, 53, 437–452 CrossRef CAS.
- M. Zhang, D. Jasim, C. Ménard-Moyon, A. Nunes, S. Iijima, A. Bianco, M. Yudasaka and K. Kostarelos, Int. J. Nanomed., 2016, 11, 3317–3330 CrossRef CAS.
- H. Kafa, J. T. W. Wang, N. Rubio, R. Klippstein, P. M. Costa, H. A. F. M. Hassan, J. K. Sosabowski, S. S. Bansal, J. E. Preston, N. J. Abbott and K. T. Al-Jamal, J. Controlled Release, 2016, 225, 217–229 CrossRef CAS.
- J. T. W. Wang, N. Rubio, H. Kafa, E. Venturelli, C. Fabbro, C. Ménard-Moyon, T. Da Ros, J. K. Sosabowski, A. D. Lawson, M. K. Robinson, M. Prato, A. Bianco, F. Festy, J. E. Preston, K. Kostarelos and K. T. Al-Jamal, J. Controlled Release, 2016, 224, 22–32 CrossRef CAS.
- J. Da-wei, P. Cheng, S. Yan-Hong, J. Li-Na, L. Jian-Bo and Z. Lan, Nucl. Sci. Tech., 2015, 26(4), 040301, DOI:10.13538/j.1001-8042/nst.26.040301.
- P. Rong, K. Yang, A. Srivastan, D. O. Kiesewetter, X. Yue, F. Wang, L. Nie, A. Bhirde, Z. Wang, Z. Liu, G. Niu, W. Wang and X. Chen, Theranostics, 2014, 4, 229–239 CrossRef.
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS.
- Z. Liu, S. M. Tabakman, Z. Chen and H. Dai, Nat. Protoc., 2009, 4, 1372–1382 CrossRef CAS.
- G. Biagiotti, F. Pisaneschi, S. T. Gammon, F. Machetti, M. C. Ligi, G. Giambastiani, G. Tuci, E. Powell, H. Piwnica-Worms, E. Pranzini, P. Paoli, S. Cicchi and D. Piwnica-Worms, J. Mater. Chem. B, 2019, 7, 2678–2687 RSC.
- H. Hong, K. Yang, Y. Zhang, J. W. Engle, L. Feng, Y. Yang, T. R. Nayak, S. Goel, J. Bean, C. P. Theuer, T. E. Barnhart, Z. Liu and W. Cai, ACS Nano, 2012, 6, 2361–2370 CrossRef CAS.
- D. Yang, L. Feng, C. A. Dougherty, K. E. Luker, D. Chen, M. A. Cauble, M. M. Banaszak Holl, G. D. Luker, B. D. Ross, Z. Liu and H. Hong, Biomaterials, 2016, 104, 361–371 CrossRef CAS.
- J. Li, W. Yang, R. Cui, D. Wang, Y. Chang, W. Gu, W. Yin, X. Bai, K. Chen, L. Xia, H. Geng and G. Xing, Nanotechnology, 2016, 27, 155101 CrossRef.
- H. Hong, Y. Zhang, J. W. Engle, T. R. Nayak, C. P. Theuer, R. J. Nickles, T. E. Barnhart and W. Cai, Biomaterials, 2012, 33, 4147–4156 CrossRef CAS.
- J. Lamb, E. Fischer, M. Rosillo-Lopez, C. G. Salzmann and J. P. Holland, Chem. Sci., 2019, 10, 8880–8888 RSC.
- M. McDevitt, M. McDevitt, A. Ruggiero, C. H. Villa, J. P. Holland, S. R. Sprinkle, C. May, J. Lewis and D. Scheinberg, Int. J. Nanomed., 2010, 5, 783 CrossRef.
- R. W. Ormsby, T. McNally, C. A. Mitchell, A. Musumeci, T. Schiller, P. Halley, L. Gahan, D. Martin, S. V. Smith and N. J. Dunne, Acta Mater., 2014, 64, 54–61 CrossRef CAS.
- D. Satpati, A. Satpati, Y. Pamale, C. Kumar, R. Sharma, H. D. Sarma and S. Banerjee, RSC Adv., 2016, 6, 50761–50769 RSC.
- S. Liu, Chem. Soc. Rev., 2004, 33, 445–461 RSC.
- B. R. Jarrett, B. Gustafsson, D. L. Kukis and A. Y. Louie, Bioconjugate Chem., 2008, 19, 1496–1504 CrossRef CAS.
- H.-Y. Lee, Z. Li, K. Chen, A. R. Hsu, C. Xu, J. Xie, S. Sun and X. Chen, J. Nucl. Med., 2008, 49, 1371–1379 CrossRef CAS.
- M. Yang, K. Cheng, S. Qi, H. Liu, Y. Jiang, H. Jiang, J. Li, K. Chen, H. Zhang and Z. Cheng, Biomaterials, 2013, 34, 2796–2806 CrossRef CAS.
- C. G. England, H.-J. Im, L. Feng, F. Chen, S. A. Graves, R. Hernandez, H. Orbay, C. Xu, S. Y. Cho, R. J. Nickles, Z. Liu, D. S. Lee and W. Cai, Biomaterials, 2016, 100, 101–109 CrossRef CAS.
- J. Zhang, Y. Ma, W. Yang, J. Xue, Y. Ding, C. Xie, W. Luo, F. Gao, Z. Zhang, Y. Zhao, Z. Chai and X. He, Nanoscale, 2019, 11, 5909–5913 RSC.
- X. Cui, S. Belo, D. Krüger, Y. Yan, R. T. M. de Rosales, M. Jauregui-Osoro, H. Ye, S. Su, D. Mathe, N. Kovács, I. Horváth, M. Semjeni, K. Sunassee, K. Szigeti, M. A. Green and P. J. Blower, Biomaterials, 2014, 35, 5840–5846 CrossRef CAS.
- N. Gholipour, M. Akhlaghi, A. Mokhtari Kheirabadi, P. Geramifar and D. Beiki, Int. J. Biol. Macromol., 2020, 148, 932–941 CrossRef CAS.
- A. Lahooti, S. Shanehsazzadeh and S. Laurent, Nanotechnology, 2020, 31, 015102 CrossRef CAS.
- M. Hajiramezanali, F. Atyabi, M. Mosayebnia, M. Akhlaghi, P. Geramifar, A. R. Jalilian, S. M. Mazidi, H. Yousefnia, S. Shahhosseini and D. Beiki, Int. J. Nanomed., 2019, 14, 2591–2605 CrossRef CAS.
- S. Kim, M. K. Chae, M. S. Yim, I. H. Jeong, J. Cho, C. Lee and E. K. Ryu, Biomaterials, 2013, 34, 8114–8121 CrossRef CAS.
- P. Unak, V. Tekin, O. K. Guldu and O. Aras, Appl. Organomet. Chem., 2020, 34, e5616, DOI:10.1002/aoc.5616.
- Z. Sun, K. Cheng, F. Wu, H. Liu, X. Ma, X. Su, Y. Liu, L. Xia and Z. Cheng, Nanoscale, 2016, 8, 19644–19653 RSC.
- M. Ognjanović, M. Radović, M. Mirković, Ž. Prijović, M. del Puerto Morales, M. Čeh, S. Vranješ-Đurić and B. Antić, ACS Appl. Mater. Interfaces, 2019, 11, 41109–41117 CrossRef.
- R. Torres Martin de Rosales, R. Tavaré, A. Glaria, G. Varma, A. Protti and P. J. Blower, Bioconjugate Chem., 2011, 22, 455–465 CrossRef.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2013, 7, 500–512 CrossRef CAS.
- A. Adamiano, M. Iafisco, M. Sandri, M. Basini, P. Arosio, T. Canu, G. Sitia, A. Esposito, V. Iannotti, G. Ausanio, E. Fragogeorgi, M. Rouchota, G. Loudos, A. Lascialfari and A. Tampieri, Acta Biomater., 2018, 73, 458–469 CrossRef CAS.
- M. R. A. Abdollah, T. J. Carter, C. Jones, T. L. Kalber, V. Rajkumar, B. Tolner, C. Gruettner, M. Zaw-Thin, J. Baguña Torres, M. Ellis, M. Robson, R. B. Pedley, P. Mulholland, R. T. M. de Rosales and K. A. Chester, ACS Nano, 2018, 12, 1156–1169 CrossRef CAS.
- C. Tsoukalas, D. Psimadas, G. A. Kastis, V. Koutoulidis, A. L. Harris, M. Paravatou-Petsotas, M. Karageorgou, L. R. Furenlid, L. A. Moulopoulos, D. Stamopoulos and P. Bouziotis, Front. Chem., 2018, 6, 224, DOI:10.3389/fchem.2018.00224.
- S. Fatahian, D. Shahbazi-Gahrouei, M. Pouladian, M. H. Yousefi, G. R. Amiri and A. Noori, J. Radioanal. Nucl. Chem., 2012, 293, 915–921 CrossRef CAS.
- M. Felber and R. Alberto, Nanoscale, 2015, 7, 6653–6660 RSC.
- M. de Souza Albernaz, S. H. Toma, J. Clanton, K. Araki and R. Santos-Oliveira, Pharm. Res., 2018, 35, 24 CrossRef.
- Y. Zhao, Q. Yao, H. Tan, B. Wu, P. Hu, P. Wu, Y. Gu, C. Zhang, D. Cheng and H. Shi, J. Radioanal. Nucl. Chem., 2014, 299, 1273–1280 CrossRef CAS.
- C.-M. Lee, H.-J. Jeong, E.-M. Kim, D. W. Kim, S. T. Lim, H. T. Kim, I.-K. Park, Y. Y. Jeong, J. W. Kim and M.-H. Sohn, Magn. Reson. Med., 2009, 62, 1440–1446 CrossRef CAS.
- M. Zaw Thin, H. Allan, R. Bofinger, T. D. Kostelec, S. Guillaume, J. J. Connell, P. S. Patrick, H. C. Hailes, A. B. Tabor, M. F. Lythgoe, D. J. Stuckey and T. L. Kalber, Nanoscale, 2020, 12, 16570–16585, 10.1039/D0NR03237A.
- M. Radović, M. Mirković, M. Perić, D. Janković, A. Vukadinović, D. Stanković, Đ. Petrović, M. Bošković, B. Antić, M. Marković and S. Vranješ-Đurić, J. Mater. Chem. B, 2017, 5, 8738–8747 RSC.
- M. Radović, M. P. Calatayud, G. F. Goya, M. R. Ibarra, B. Antić, V. Spasojević, N. Nikolić, D. Janković, M. Mirković and S. Vranješ-Đurić, J. Biomed. Mater. Res., Part A, 2015, 103, 126–134 CrossRef.
- B. Azadbakht, H. Afarideh, M. Ghannadi-Maragheh, A. Bahrami-Samani and M. Asgari, Nucl. Med. Biol., 2017, 48, 26–30 CrossRef CAS.
- J. Cao, Y. Wang, J. Yu, J. Xia, C. Zhang, D. Yin and U. O. Häfeli, J. Magn. Magn. Mater., 2004, 277, 165–174 CrossRef CAS.
- S. Akbari-Karadeh, S. M. R. Aghamiri, P. Tajer-Mohammad-Ghazvini and S. Ghorbanzadeh-Mashkani, Appl. Biochem. Biotechnol., 2020, 190, 540–550 CrossRef CAS.
- Y. H. Gholami, L. Josephson, E. A. Akam, P. Caravan, M. Q. Wilks, X.-Z. Pan, R. Maschmeyer, A. Kolnick, G. El Fakhri, M. D. Normandin, Z. Kuncic and H. Yuan, Int. J. Nanomed., 2020, 15, 31–47 CrossRef CAS.
- A. Ruiz-de-Angulo, A. Zabaleta, V. Gómez-Vallejo, J. Llop and J. C. Mareque-Rivas, ACS Nano, 2016, 10, 1602–1618 CrossRef CAS.
- P. S. Patrick, L. K. Bogart, T. J. Macdonald, P. Southern, M. J. Powell, M. Zaw-Thin, N. H. Voelcker, I. P. Parkin, Q. A. Pankhurst, M. F. Lythgoe, T. L. Kalber and J. C. Bear, Chem. Sci., 2019, 10, 2592–2597 RSC.
- M. D. Normandin, H. Yuan, M. Q. Wilks, H. H. Chen, J. M. Kinsella, H. Cho, N. J. Guehl, N. Absi-Halabi, S. M. Hosseini, G. El Fakhri, D. E. Sosnovik and L. Josephson, Angew. Chem., Int. Ed., 2015, 54, 13002–13006 CrossRef CAS.
- R. Chakravarty, H. F. Valdovinos, F. Chen, C. M. Lewis, P. A. Ellison, H. Luo, M. E. Meyerand, R. J. Nickles and W. Cai, Adv. Mater., 2014, 26, 5119–5123 CrossRef CAS.
- B. Freund, U. I. Tromsdorf, O. T. Bruns, M. Heine, A. Giemsa, A. Bartelt, S. C. Salmen, N. Raabe, J. Heeren, H. Ittrich, R. Reimer, H. Hohenberg, U. Schumacher, H. Weller and P. Nielsen, ACS Nano, 2012, 6, 7318–7325 CrossRef CAS.
- R. Madru, P. Kjellman, F. Olsson, K. Wingardh, C. Ingvar, F. Stahlberg, J. Olsrud, J. Latt, S. Fredriksson, L. Knutsson and S.-E. Strand, J. Nucl. Med., 2012, 53, 459–463 CrossRef CAS.
- I. Tsiapa, E. K. Efthimiadou, E. Fragogeorgi, G. Loudos, A. D. Varvarigou, P. Bouziotis, G. C. Kordas, D. Mihailidis, G. C. Nikiforidis, S. Xanthopoulos, D. Psimadas, M. Paravatou-Petsotas, L. Palamaris, J. D. Hazle and G. C. Kagadis, J. Colloid Interface Sci., 2014, 433, 163–175 CrossRef CAS.
- O. Mokhodoeva, M. Vlk, E. Málková, E. Kukleva, P. Mičolová, K. Štamberg, M. Šlouf, R. Dzhenloda and J. Kozempel, J. Nanopart. Res., 2016, 18, 301 CrossRef.
- J. Pellico, J. Llop, I. Fernández-Barahona, R. Bhavesh, J. Ruiz-Cabello and F. Herranz, Contrast Media Mol. Imaging, 2017, 2017, 1–24 CrossRef.
- F. Ai, C. A. Ferreira, F. Chen and W. Cai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 619–630 CAS.
- F. Ai, C. A. Ferreira, F. Chen and W. Cai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 619–630 CAS.
- R. Torres Martin de Rosales, R. Tavaré, R. L. Paul, M. Jauregui-Osoro, A. Protti, A. Glaria, G. Varma, I. Szanda and P. J. Blower, Angew. Chem., Int. Ed., 2011, 50, 5509–5513 CrossRef.
- J. Pellico, I. Fernández-Barahona, M. Benito, Á. Gaitán-Simón, L. Gutiérrez, J. Ruiz-Cabello and F. Herranz, Nanomedicine, 2019, 17, 26–35 CrossRef CAS.
- J. Pellico, A. V. Lechuga-Vieco, E. Almarza, A. Hidalgo, C. Mesa-Nuñez, I. Fernández-Barahona, J. A. Quintana, J. Bueren, J. A. Enríquez, J. Ruiz-Cabello and F. Herranz, Sci. Rep., 2017, 7, 13242 CrossRef.
- P. G. Jeelani, P. Mulay, R. Venkat and C. Ramalingam, Silicon, 2020, 12, 1337–1354 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590 RSC.
- M. Manzano and M. Vallet-Regí, Adv. Funct. Mater., 2020, 30, 1902634 CrossRef CAS.
- R. Chakravarty, S. Goel, H. Hong, F. Chen, H. F. Valdovinos, R. Hernandez, T. E. Barnhart and W. Cai, Nanomedicine, 2015, 10, 1233–1246 CrossRef CAS.
- S. Nizzero, F. Li, G. Zhang, A. Venuta, C. Borsoi, J. Mai, H. Shen, J. Wolfram, Z. Li, E. Blanco and M. Ferrari, Acta Biomater., 2019, 97, 501–512 CrossRef CAS.
- L. Kramer, G. Winter, B. Baur, A. J. Kuntz, T. Kull, C. Solbach, A. J. Beer and M. Lindén, Nanoscale, 2017, 9, 9743–9753 RSC.
- A. Al Faraj, B. Alotaibi, A. Pasha Shaik, K. Shamma, I. Al Jammaz and J. Gerl, Int. J. Nanomed., 2015, 6293 CrossRef CAS.
- H. Gao, X. Liu, W. Tang, D. Niu, B. Zhou, H. Zhang, W. Liu, B. Gu, X. Zhou, Y. Zheng, Y. Sun, X. Jia and L. Zhou, Nanoscale, 2016, 8, 19573–19580 RSC.
- A. L. B. de Barros, K. S. de Oliveira Ferraz, T. C. S. Dantas, G. F. Andrade, V. N. Cardoso and E. M. B. de Sousa, Mater. Sci. Eng., C, 2015, 56, 181–188 CrossRef.
- G. F. Andrade, J. A. Q. A. Faria, D. A. Gomes, A. L. B. de Barros, R. S. Fernandes, A. C. S. Coelho, J. A. Takahashi, A. da Silva Cunha and E. M. B. de Sousa, J. Sol-Gel Sci. Technol., 2018, 85, 369–381 CrossRef CAS.
- P. Dogra, N. L. Adolphi, Z. Wang, Y.-S. Lin, K. S. Butler, P. N. Durfee, J. G. Croissant, A. Noureddine, E. N. Coker, E. L. Bearer, V. Cristini and C. J. Brinker, Nat. Commun., 2018, 9, 4551 CrossRef.
- A. Liberman, Z. Wu, C. V. Barback, R. Viveros, S. L. Blair, L. G. Ellies, D. R. Vera, R. F. Mattrey, A. C. Kummel and W. C. Trogler, ACS Nano, 2013, 7, 6367–6377 CrossRef CAS.
- S.-H. Cheng, D. Yu, H.-M. Tsai, R. A. Morshed, D. Kanojia, L.-W. Lo, L. Leoni, Y. Govind, L. Zhang, K. S. Aboody, M. S. Lesniak, C.-T. Chen and I. V. Balyasnikova, J. Nucl. Med., 2016, 57, 279–284 CrossRef CAS.
- X. Zhang, F. Chen, M. Z. Turker, K. Ma, P. Zanzonico, F. Gallazzi, M. A. Shah, A. R. Prater, U. Wiesner, M. S. Bradbury, M. R. McDevitt and T. P. Quinn, Biomaterials, 2020, 241, 119858 CrossRef CAS.
- Y. Fazaeli, M. A. Hosseini, M. Afrasyabi and P. Ashtari, Radiochim. Acta, 2019, 107, 157–164 CAS.
- B. P. Burke, N. Baghdadi, A. E. Kownacka, S. Nigam, G. S. Clemente, M. M. Al-Yassiry, J. Domarkas, M. Lorch, M. Pickles, P. Gibbs, R. Tripier, C. Cawthorne and S. J. Archibald, Nanoscale, 2015, 7, 14889–14896 RSC.
- F. Chen, S. Goel, S. Shi, T. E. Barnhart, X. Lan and W. Cai, Nano Res., 2018, 11, 4890–4904 CrossRef CAS.
- S. Goel, F. Chen, S. Luan, H. F. Valdovinos, S. Shi, S. A. Graves, F. Ai, T. E. Barnhart, C. P. Theuer and W. Cai, Adv. Sci., 2016, 3, 1600122 CrossRef.
- S. Rojas, J. D. Gispert, C. Menchón, H. G. Baldoví, M. Buaki-Sogo, M. Rocha, S. Abad, V. M. Victor, H. García and J. R. Herance, J. Nanopart. Res., 2015, 17, 131 CrossRef.
- P. A. Ellison, F. Chen, S. Goel, T. E. Barnhart, R. J. Nickles, O. T. DeJesus and W. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 6772–6781 CrossRef CAS.
- F. Chen, H. F. Valdovinos, R. Hernandez, S. Goel, T. E. Barnhart and W. Cai, Acta Pharmacol. Sin., 2017, 38, 907–913 CrossRef CAS.
- E. Helal-Neto, S. S. Cabezas, F. Sancenón, R. Martínez-Máñez and R. Santos-Oliveira, J. Pharm. Biomed. Anal., 2018, 153, 90–94 CrossRef CAS.
- L. T. M. Sá, S. Simmons, S. Missailidis, M. I. P. da Silva and R. Santos-Oliveira, J. Drug Targeting, 2013, 21, 427–434 CrossRef.
- P. Rainone, B. Riva, S. Belloli, F. Sudati, M. Ripamonti, P. Verderio, M. Colombo, B. Colzani, M. C. Gilardi, R. M. Moresco and D. Prosperi, Int. J. Nanomed., 2017, 12, 3447–3461 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759 RSC.
- J. Zhang, L. Mou and X. Jiang, Chem. Sci., 2020, 11, 923–936 RSC.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- S. Same, A. Aghanejad, S. Akbari Nakhjavani, J. Barar and Y. Omidi, Bioimpacts, 2016, 6, 169–181 CrossRef CAS.
- J. L. Campbell, E. D. SoRelle, O. Ilovich, O. Liba, M. L. James, Z. Qiu, V. Perez, C. T. Chan, A. de la Zerda and C. Zavaleta, Biomaterials, 2017, 135, 42–52 CrossRef CAS.
- X. Sun, X. Huang, X. Yan, Y. Wang, J. Guo, O. Jacobson, D. Liu, L. P. Szajek, W. Zhu, G. Niu, D. O. Kiesewetter, S. Sun and X. Chen, ACS Nano, 2014, 8, 8438–8446 CrossRef CAS.
- A. L. Tam, M. P. Melancon, M. Abdelsalam, T. A. Figueira, K. Dixon, A. McWatters, M. Zhou, Q. Huang, O. Mawlawi, K. Dunner, C. Li and S. Gupta, J. Biomed. Nanotechnol., 2016, 12, 296–307 CrossRef CAS.
- Y. Zhao, B. Pang, L. Detering, H. Luehmann, M. Yang, K. Black, D. Sultan, Y. Xia and Y. Liu, Mol. Imaging, 2018, 17, 153601211877582 CrossRef.
- H. Li, L. Diaz, D. Lee, L. Cui, X. Liang and Y. Cheng, Radiol. Med., 2014, 119, 269–276 CrossRef.
- M. Tian, W. Lu, R. Zhang, C. Xiong, J. Ensor, J. Nazario, J. Jackson, C. Shaw, K. A. Dixon, J. Miller, K. Wright, C. Li and S. Gupta, Mol. Imaging Biol., 2013, 15, 614–624 CrossRef.
- H. Xie, Z. J. Wang, A. Bao, B. Goins and W. T. Phillips, Int. J. Pharm., 2010, 395, 324–330 CrossRef CAS.
- M. Pretze, N. P. van der Meulen, C. Wängler, R. Schibli and B. Wängler, J. Labelled Compd. Radiopharm., 2019, 62, 471–482 CrossRef CAS.
- L. E. Chilug, R. A. Leonte, M. E. B. Patrascu, A. C. Ion, C. S. Tuta, A. Raicu, G. Manda and D. Niculae, J. Radioanal. Nucl. Chem., 2017, 311, 1485–1493 CrossRef CAS.
- F. Silva, A. Zambre, M. P. C. Campello, L. Gano, I. Santos, A. M. Ferraria, M. J. Ferreira, A. Singh, A. Upendran, A. Paulo and R. Kannan, Bioconjugate Chem., 2016, 27, 1153–1164 CrossRef CAS.
- L. Karmani, V. Bouchat, C. Bouzin, P. Levêque, D. Labar, A. Bol, G. Deumer, R. Marega, D. Bonifazi, V. Haufroid, C. Michiels, V. Grégoire, O. Feron, S. Lucas, T. Vander Borght and B. Gallez, Nanomedicine, 2014, 9, 1923–1937 CrossRef CAS.
- L. Karmani, D. Labar, V. Valembois, V. Bouchat, P. G. Nagaswaran, A. Bol, J. Gillart, P. Levêque, C. Bouzin, D. Bonifazi, C. Michiels, O. Feron, V. Grégoire, S. Lucas, T. Vander Borght and B. Gallez, Contrast Media Mol. Imaging, 2013, 8, 402–408 CrossRef CAS.
- Y. Li, L. Zhao, X. Xu, N. Sun, W. Qiao, Y. Xing, M. Shen, M. Zhu, X. Shi and J. Zhao, J. Biomed. Nanotechnol., 2019, 15, 1201–1212 CrossRef CAS.
- F. Silva, L. Gano, M. P. Cabral Campello, R. Marques, I. Prudêncio, A. Zambre, A. Upendran, A. Paulo and R. Kannan, Dalton Trans., 2017, 46, 14572–14583 RSC.
- M. Motiei, T. Dreifuss, T. Sadan, N. Omer, T. Blumenfeld-Katzir, E. Fragogeorgi, G. Loudos, R. Popovtzer and N. Ben-Eliezer, Chem. Lett., 2019, 48, 291–294 CrossRef CAS.
- N. Jiménez-Mancilla, G. Ferro-Flores, C. Santos-Cuevas, B. Ocampo-García, M. Luna-Gutiérrez, E. Azorín-Vega, K. Isaac-Olivé, M. Camacho-López and E. Torres-García, J. Labelled Compd. Radiopharm., 2013, 56, 663–671 CrossRef.
- B. E. Ocampo-García, F. de, M. Ramírez, G. Ferro-Flores, L. M. De León-Rodríguez, C. L. Santos-Cuevas, E. Morales-Avila, C. A. de Murphy, M. Pedraza-López, L. A. Medina and M. A. Camacho-López, Nucl. Med. Biol., 2011, 38, 1–11 CrossRef.
- O. J. Estudiante-Mariquez, A. Rodríguez-Galván, D. Ramírez-Hernández, F. F. Contreras-Torres and L. A. Medina, Molecules, 2020, 25, 1982 CrossRef CAS.
- E. Morales-Avila, G. Ferro-Flores, B. E. Ocampo-García, L. M. De León-Rodríguez, C. L. Santos-Cuevas, R. García-Becerra, L. A. Medina and L. Gómez-Oliván, Bioconjugate Chem., 2011, 22, 913–922 CrossRef CAS.
- A. Loiseau, J. Boudon, A. Oudot, M. Moreau, R. Boidot, R. Chassagnon, N. Mohamed Saïd, S. Roux, C. Mirjolet and N. Millot, Cancers, 2019, 11, 1962 CrossRef CAS.
- L. Song, S. Able, E. Johnson and K. A. Vallis, Nanotheranostics, 2017, 1, 232–243 CrossRef.
- H.-W. Kao, Y.-Y. Lin, C.-C. Chen, K.-H. Chi, D.-C. Tien, C.-C. Hsia, W.-J. Lin, F.-D. Chen, M.-H. Lin and H.-E. Wang, Nanotechnology, 2014, 25, 295102 CrossRef.
- H. Mendoza-Nava, G. Ferro-Flores, F. de, M. Ramírez, B. Ocampo-García, C. Santos-Cuevas, E. Azorín-Vega, N. Jiménez-Mancilla, M. Luna-Gutiérrez and K. Isaac-Olivé, Mol. Imaging, 2017, 16 DOI:10.1177/1536012117704768.
- G. Ferro-Flores, B. Ocampo-García, C. Santos-Cuevas, F. María Ramírez, E. Azorín-Vega and L. Meléndez-Alafort, Curr. Radiopharm., 2015, 8, 150–159 CrossRef CAS.
- A. Vilchis-Juárez, G. Ferro-Flores, C. Santos-Cuevas, E. Morales-Avila, B. Ocampo-García, L. Díaz-Nieto, M. Luna-Gutiérrez, N. Jiménez-Mancilla, M. Pedraza-López and L. Gómez-Oliván, J. Biomed. Nanotechnol., 2014, 10, 393–404 CrossRef.
- E.-A. Salvanou, D. Stellas, C. Tsoukalas, B. Mavroidi, M. Paravatou-Petsotas, N. Kalogeropoulos, S. Xanthopoulos, F. Denat, G. Laurent, R. Bazzi, S. Roux and P. Bouziotis, Pharmaceutics, 2020, 12, 188 CrossRef CAS.
- A. F. Frellsen, A. E. Hansen, R. I. Jølck, P. J. Kempen, G. W. Severin, P. H. Rasmussen, A. Kjær, A. T. I. Jensen and T. L. Andresen, ACS Nano, 2016, 10, 9887–9898 CrossRef CAS.
- B. Pang, Y. Zhao, H. Luehmann, X. Yang, L. Detering, M. You, C. Zhang, L. Zhang, Z.-Y. Li, Q. Ren, Y. Liu and Y. Xia, ACS Nano, 2016, 10, 3121–3131 CrossRef CAS.
- Y. Zhao, D. Sultan, L. Detering, S. Cho, G. Sun, R. Pierce, K. L. Wooley and Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 156–159 CrossRef CAS.
- M. Yang, D. Huo, K. D. Gilroy, X. Sun, D. Sultan, H. Luehmann, L. Detering, S. Li, D. Qin, Y. Liu and Y. Xia, ChemNanoMat, 2017, 3, 44–50 CrossRef CAS.
- Y. Zhao, D. Sultan, L. Detering, H. Luehmann and Y. Liu, Nanoscale, 2014, 6, 13501–13509 RSC.
- J. Zhu, J. Chin, C. Wängler, B. Wängler, R. B. Lennox and R. Schirrmacher, Bioconjugate Chem., 2014, 25, 1143–1150 CrossRef CAS.
- K. R. Pulagam, K. B. Gona, V. Gómez-Vallejo, J. Meijer, C. Zilberfain, I. Estrela-Lopis, Z. Baz, U. Cossío and J. Llop, Molecules, 2019, 24, 3609 CrossRef CAS.
- Q. K. T. Ng, C. I. Olariu, M. Yaffee, V. F. Taelman, N. Marincek, T. Krause, L. Meier and M. A. Walter, Biomaterials, 2014, 35, 7050–7057 CrossRef.
- R. Kamal, V. D. Chadha and D. K. Dhawan, Nanomedicine, 2018, 14, 1059–1071 CrossRef CAS.
- W. I. El-Ghareb, M. M. Swidan, I. T. Ibrahim, A. Abd El-Bary, M. I. Tadros and T. M. Sakr, Int. J. Pharm., 2020, 586, 119514 CrossRef CAS.
- X. Shao, A. Agarwal, J. R. Rajian, N. A. Kotov and X. Wang, Nanotechnology, 2011, 22, 135102 CrossRef.
- X. Shao, H. Zhang, J. R. Rajian, D. L. Chamberland, P. S. Sherman, C. A. Quesada, A. E. Koch, N. A. Kotov and X. Wang, ACS Nano, 2011, 5, 8967–8973 CrossRef CAS.
- Y. Zhang, Y. Zhang, L. Yin, X. Xia, F. Hu, Q. Liu, C. Qin and X. Lan, Contrast Media Mol. Imaging, 2017, 2017, 1–10 Search PubMed.
- N. Eskandari, K. Yavari, M. Outokesh, S. Sadjadi and S. J. Ahmadi, J. Labelled Compd. Radiopharm., 2013, 56, 12–16 CrossRef CAS.
- C. Matea, T. Mocan, F. Tabaran, T. Pop, O. Mosteanu, C. Puia, C. Iancu and L. Mocan, Int. J. Nanomed., 2017, 12, 5421–5431 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- T. Tang, Y. Wei, Q. Yang, Y. Yang, M. J. Sailor and H.-B. Pang, Nanoscale, 2019, 11, 22248–22254 RSC.
- C. Tu, X. Ma, A. House, S. M. Kauzlarich and A. Y. Louie, ACS Med. Chem. Lett., 2011, 2, 285–288 CrossRef CAS.
- R. Mathur, N. Bag, R. Varshney, F. Hussain, A. Kaul, N. Kumari, R. Chauhan, S. Singh, S. Singh and A. K. Mishra, RSC Adv., 2016, 6, 13562–13571 RSC.
- N. Bag, R. Mathur, F. Hussain, N. Indracanti, S. Singh, S. Singh, R. P. Chauhan, K. Chuttani and A. K. Mishra, Toxicol. Res., 2015, 4, 1416–1425 CrossRef CAS.
- M. Felber, M. Bauwens, J. M. Mateos, S. Imstepf, F. M. Mottaghy and R. Alberto, Chem. – Eur. J., 2015, 21, 6090–6099 CrossRef CAS.
- N. Bag, R. Mathur, S. Singh, F. Hussain, R. P. Chauhan, K. Chuttani and A. K. Mishra, MedChemComm, 2015, 6, 363–371 RSC.
- A. Riedinger, T. Avellini, A. Curcio, M. Asti, Y. Xie, R. Tu, S. Marras, A. Lorenzoni, S. Rubagotti, M. Iori, P. C. Capponi, A. Versari, L. Manna, E. Seregni and T. Pellegrino, J. Am. Chem. Soc., 2015, 137, 15145–15151 CrossRef CAS.
- A. Rasekholghol, Y. Fazaeli, S. Moradi Dehaghi and P. Ashtari, J. Radioanal. Nucl. Chem., 2020, 324, 599–608, DOI:10.1007/s10967-020-07102-y.
- H. S. Sezgin, F. Z. Biber Muftuler, C. Ichedef, A. Yurt Kilcar, S. Teksoz, P. Unak and C. Harmansah, J. Radioanal. Nucl. Chem., 2017, 313, 291–297 CrossRef CAS.
- M. Sun, G. Sundaresan, P. Jose, L. Yang, D. Hoffman, N. Lamichhane and J. Zweit, J. Mater. Chem. B, 2014, 2, 4456–4466 RSC.
- A. Kara, P. Ünak, C. Selçuki, Ö. Akça, E. İ. Medine and S. Sakarya, J. Radioanal. Nucl. Chem., 2014, 299, 807–813 CrossRef CAS.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef.
- G. Tessitore, G. A. Mandl, M. G. Brik, W. Park and J. A. Capobianco, Nanoscale, 2019, 11, 12015–12029 RSC.
- J. Rieffel, F. Chen, J. Kim, G. Chen, W. Shao, S. Shao, U. Chitgupi, R. Hernandez, S. A. Graves, R. J. Nickles, P. N. Prasad, C. Kim, W. Cai and J. F. Lovell, Adv. Mater., 2015, 27, 1785–1790 CrossRef CAS.
- H. J. Seo, S. H. Nam, H.-J. Im, J. Park, J. Y. Lee, B. Yoo, Y.-S. Lee, J. M. Jeong, T. Hyeon, J. Who Kim, J. S. Lee, I.-J. Jang, J.-Y. Cho, D. W. Hwang, Y. D. Suh and D. S. Lee, Sci. Rep., 2015, 5, 15685 CrossRef CAS.
- X. Cui, D. Mathe, N. Kovács, I. Horváth, M. Jauregui-Osoro, R. Torres Martin de Rosales, G. E. D. Mullen, W. Wong, Y. Yan, D. Krüger, A. N. Khlobystov, M. Gimenez-Lopez, M. Semjeni, K. Szigeti, D. S. Veres, H. Lu, I. Hernández, W. P. Gillin, A. Protti, K. K. Petik, M. A. Green and P. J. Blower, Bioconjugate Chem., 2016, 27, 319–328 CrossRef CAS.
- J. Gallo, I. S. Alam, J. Jin, Y.-J. Gu, E. O. Aboagye, W.-T. Wong and N. J. Long, Dalton Trans., 2014, 43, 5535 RSC.
- X. Yang, H. Hong, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, Y. Xiao, Y. Yang, Y. Zhang, R. J. Nickles, W. Cai, D. A. Steeber and S. Gong, Biomaterials, 2011, 32, 4151–4160 CrossRef CAS.
- E. Christensen, J. R. Henriksen, J. T. Jørgensen, Y. Amitay, H. Schmeeda, A. A. Gabizon, A. Kjær, T. L. Andresen and A. E. Hansen, Int. J. Nanomed., 2018, 13, 7647–7656 CrossRef CAS.
- H. Savolainen, A. Volpe, A. Phinikaridou, M. Douek, G. Fruhwirth and R. T. M. de Rosales, Nanotheranostics, 2019, 3, 255–265 CrossRef.
- K. O. Jung, T. J. Kim, J. H. Yu, S. Rhee, W. Zhao, B. Ha, K. Red-Horse, S. S. Gambhir and G. Pratx, Nat. Biomed. Eng., 2020, 1–10 Search PubMed.
- S. J. Wang, W. S. Huang, C. M. Chuang, C. H. Chang, T. W. Lee, G. Ting, M. H. Chen, P. M. H. Chang, T. C. Chao, H. W. Teng, Y. Chao, Y. M. Chen, T. P. Lin, Y. J. Chang, S. J. Chen, Y. R. Huang and K. L. Lan, EJNMMI Res., 2019, 9, 46 CrossRef.
- K. J. Harrington, S. Mohammadtaghi, P. S. Uster, D. Glass, A. M. Peters, R. G. Vile and J. S. W. Stewart, Clin. Cancer Res., 2001, 7, 243–254 CAS.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS.
- T. Lammers, L. Y. Rizzo, G. Storm and F. Kiessling, Clin. Cancer Res., 2012, 18, 4889–4894 CrossRef CAS.
- M. A. Miller, S. Arlauckas and R. Weissleder, Nanotheranostics, 2017, 1, 296–312 CrossRef.
- M. F. Kircher, S. S. Gambhir and J. Grimm, Nat. Rev. Clin. Oncol., 2011, 8, 677–688 CrossRef CAS.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- P. Bhatnagar, Z. Li, Y. Choi, J. Guo, F. Li, D. Y. Lee, M. Figliola, H. Huls, D. A. Lee, T. Zal, K. C. Li and L. J. N. Cooper, Integr. Biol., 2012, 5, 231–238 CrossRef.
- S. B. Lee, S. B. Ahn, S.-W. Lee, S. Y. Jeong, Y. Ghilsuk, B.-C. Ahn, E.-M. Kim, H.-J. Jeong, J. Lee, D.-K. Lim and Y. H. Jeon, NPG Asia Mater., 2016, 8, e281–e281 CrossRef CAS.
- P. Bhatnagar, M. Alauddin, J. A. Bankson, D. Kirui, P. Seifi, H. Huls, D. A. Lee, A. Babakhani, M. Ferrari, K. C. Li and L. J. N. Cooper, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- M. Yao, X. Shi, C. Zuo, M. Ma, L. Zhang, H. Zhang, X. Li, G.-Y. Yang, Y. Tang and R. Wu, ACS Appl. Mater. Interfaces, 2020, 12(34), 37885–37895, DOI:10.1021/acsami.0c10500.
- N. Kotagiri, M. L. Cooper, M. Rettig, C. Egbulefu, J. Prior, G. Cui, P. Karmakar, M. Zhou, X. Yang, G. Sudlow, L. Marsala, C. Chanswangphuwana, L. Lu, L. Habimana-Griffin, M. Shokeen, X. Xu, K. Weilbaecher, M. Tomasson, G. Lanza, J. F. DiPersio and S. Achilefu, Nat. Commun., 2018, 9, 275 CrossRef.
- T. M. Shaffer, E. C. Pratt and J. Grimm, Nat. Nanotechnol., 2017, 12, 106–117 CrossRef CAS.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS.
- D. Duan, H. Liu, Y. Xu, Y. Han, M. Xu, Z. Zhang and Z. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 5278–5286 CrossRef CAS.
- R. Tang, A. Zheleznyak, M. Mixdorf, A. Ghai, J. Prior, K. C. L. Black, M. Shokeen, N. Reed, P. Biswas and S. Achilefu, ACS Nano, 2020, 14, 4255–4264 CrossRef CAS.
- S. Kavadiya and P. Biswas, J. Nucl. Med., 2019, 60, 702–709 CrossRef CAS.
- D. Ni, C. A. Ferreira, T. E. Barnhart, V. Quach, B. Yu, D. Jiang, W. Wei, H. Liu, J. W. Engle, P. Hu and W. Cai, J. Am. Chem. Soc., 2018, 140, 14971–14979 CrossRef.
- B. Yu, D. Ni, Z. T. Rosenkrans, T. E. Barnhart, H. Wei, C. A. Ferreira, X. Lan, J. W. Engle, Q. He, F. Yu and W. Cai, Adv. Mater., 2019, 31, 1904894 CrossRef CAS.
- R. Weissleder, M. Nahrendorf and M. J. Pittet, Nat. Mater., 2014, 13, 125–138 CrossRef CAS.
- W. J. M. Mulder, J. Ochando, L. A. B. Joosten, Z. A. Fayad and M. G. Netea, Nat. Rev. Drug Discovery, 2019, 18, 553–566 CrossRef CAS.
- C. Wang, B. I. Leach, D. Lister, S. R. Adams, H. Xu, C. K. Hoh, P. McConville, J. Zhang, K. Messer and E. Ahrens, J. Nucl. Med., 2020 DOI:10.2967/jnumed.120.255273.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |