
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fundamentals and applications of photo-thermal catalysis†
Diego
Mateo
,
Jose Luis
Cerrillo
 ,
Sara
Durini
and
Jorge
Gascon
*
,
Sara
Durini
and
Jorge
Gascon
*
King Abdullah University of Science and Technology, KAUST Catalysis Center (KCC), Advanced Catalytic Materials, Thuwal 23955-6900, Saudi Arabia. E-mail: Jorge.gascon@kaust.edu.sa
First published on 18th December 2020
Abstract
Photo-thermal catalysis has recently emerged as an alternative route to drive chemical reactions using light as an energy source. Through the synergistic combination of photo- and thermo-chemical contributions of sunlight, photo-thermal catalysis has the potential to enhance reaction rates and to change selectivity patterns, even under moderate operation conditions. This review provides the fundamentals of localized surface plasmon resonance (LSPR) that explain the photo-thermal effect in plasmonic structures, describes the different mechanistic pathways underlying photo-thermal catalysis, suggests methodologies to disentangle the reaction mechanisms and proposes material design strategies to improve photo-thermal performance. Ultimately, the goal is to pave the way for the wide implementation of this promising technology in the production of synthetic fuels and chemicals.
 Diego Mateo | Diego Mateo received his bachelor's degree in Chemistry in 2009 and his bachelor's degree in Food Technology in 2011, both from the University of Valencia. After working for two years as a research technician at Complutense University of Madrid, in 2015 he started his PhD in the development of new graphene-based photocatalysts for the production of solar fuels in the group of Prof. Hermenegildo Garcia at the Institute of Chemical Technology (UPV-CSIC). After obtaining his PhD in 2019, he moved to the group of Prof. Jorge Gascon at King Abdullah University of Science and Technology (KAUST) as a postdoctoral fellow. His current research is focused on novel photo- and electro-catalytic routes for the production of fuels and chemicals. |
 Jose Luis Cerrillo | Jose Luis Cerrillo (1986) obtained his BSc in Chemistry in 2011 from the Universitat de València (Spain). After two years working as a R&D technician in a chemical company, he came back to academia and obtained his MSc in 2014 and his PhD in Sustainable Chemistry in 2019, both from Universitat Politècnica de València (Spain). His PhD research was funded by a Severo-Ochoa National scholarship and held at the Instituto de Tecnología Química (CSIC-UPV), focusing on the catalytic removal of water pollutants and on the study of novel biocide materials based on Ag-zeolites. Since October 2019, he has been doing postdoctoral research in Prof. Gascon's research group (Advanced Catalytic Materials) in King Abdullah University of Science and Technology (Saudi Arabia) where he investigates diverse heterogeneous catalytic processes related to the reutilization of CO2 to produce valued fuels and chemicals. |
 Sara Durini | Sara Durini obtained her bachelor's degree in Chemistry and Industrial Chemistry in 2009, her master's degree in Chemical Sciences in 2011, and her PhD in Chemistry in 2015 from the University of Insubria in Como (Italy). In 2016 she continued her work as a postdoctoral researcher at the University of Leipzig, where she worked on the synthesis of new luminescent metal–organic frameworks. In 2019 she joined Prof Gascon's group at King Abdullah University of Science and Technology (KAUST, Saudi Arabia) as a postdoctoral fellow, where she investigates porous materials for application in heterogeneous catalysis. |
 Jorge Gascon | Jorge Gascon (1977) received his MSc in Chemistry in 2002 and his PhD in Chemical Engineering in 2006, both from the University of Zaragoza (Spain). After his PhD he spent eleven years at TUDelft, the last four as Anthonie van Leeuwenhoek Professor of Catalysis Engineering. In 2018 he moved to King Abdullah University of Science and Technology (KAUST), where he is Professor of Chemical Engineering and the Director of the KAUST Catalysis Center. He has co-authored over 275 publications, several patents and books. His research group focuses on the design, development and demonstration of new heterogeneous catalysts and reactor engineering concepts. |
1. Introduction
Catalysis is an integral part of chemistry with large implications for the efficient production of everyday goods. Indeed, circa 95% of all chemical products are manufactured through catalytic routes. At the same time, many of these processes require an energy input that has, traditionally, been supplied by non-renewable sources.1,2 In the last few decades, with the discovery of heterogeneous semiconductor-based photocatalysis, the possibility of catalyzing chemical transformations using light as a source of energy has become a reality.In a semiconductor, upon absorption of a photon with equal or higher energy than the band gap, electron–hole pairs are generated. Eventually these charge carriers can migrate to the semiconductor surface and be transferred to adsorbed molecules, thereby initiating reduction or oxidation processes. Since the seminal paper of Fujishima and Honda in 1972, tremendous advances in the field of photocatalysis have been reported. In spite of this, the efficiency of most photocatalytic processes remains insufficiently low (typically in the range of hundreds of μmol g−1 h−1) mainly due to fast charge carrier recombination and low absorption and utilization of the solar spectrum by traditional wide band gap semiconductors.3–8
Next to charge separation upon light excitation, plasmon mediated processes have attracted a great interest. Hot charge carriers generated by means of the decay of localized surface plasmon resonance (LSPR) possess higher energies than those induced by direct photoexcitation.9,10 Furthermore, these energetic carriers can relax internally and dissipate their energy by local heating of the surroundings, causing a thermal effect on the material.11–13 This photo-thermal effect has been extensively applied in a large number of fields, including cancer therapy, degradation of pollutants, seawater desalination and water vaporization.14–19 It was therefore only a matter of time until similar concepts were applied to speed up chemical reactions. Indeed, photo-thermal catalysis combines photochemical and thermochemical contributions of sunlight and has emerged as a rapidly growing and exciting new field of research.20–31 Photo-thermal catalysis allows for a more effective harvesting of the solar spectrum, including low-energy visible and infrared photons that would be insufficient to promote photocatalytic reactions.30,32–34 Furthermore, due to the increase in the temperature of the catalytic active sites, photo-thermal catalysis renders outstanding production rates while operating under moderate conditions.35–37
Because of the rapidly growing nature of the field, we believe now is the time to write a holistic review on the field of photo-thermal catalysis focusing on the production of fuels and chemicals. As relatively newcomers, we make special emphasis on those topics that should facilitate the entrance of other new comers to this exciting field. We therefore pay special attention to the fundamentals of localized surface plasmon resonance and the underlying mechanisms behind photochemical and thermochemical processes that define the photo-thermal effect. Based on this analysis, we suggest characterization techniques and methodologies that should help assess the dominating pathway of reaction for a given catalytic system, and therefore gain a better insight. Next, we review in detail reactions driven by means of photo-thermal catalysis, gathering the most relevant information available in terms of efficiency, selectivity targets and mechanistic knowledge. Last but not least, we share our opinion on design strategies to further optimize catalytic performance and highlight potential future directions and challenges ahead.
2. Localized surface plasmon resonance and the photo-thermal effect
The localized surface plasmon resonance (LSPR) band in metallic nanoparticles (NPs) is an intense and broad absorption band along the UV-visible-NIR region of the electromagnetic spectrum. The physics behind this phenomenon has been traditionally explained by means of two theories: the Drude–Maxwell model and the theory developed by Gustav Mie in 1908.38,39 Although it is beyond the scope of this review to contribute to a deep and extensive discussion of these theories, these models can provide insightful information to describe the LSPR phenomenon and its application to catalysis, so a brief overview of them will be addressed in the ESI† of the present work.As per these theories, inside metal NPs conducting free electrons can move when guided by external incident irradiation. This motion is dampened by electron inelastic collisions and the restoring force on the electron cloud created by the accumulation of surface charges (Fig. 1).
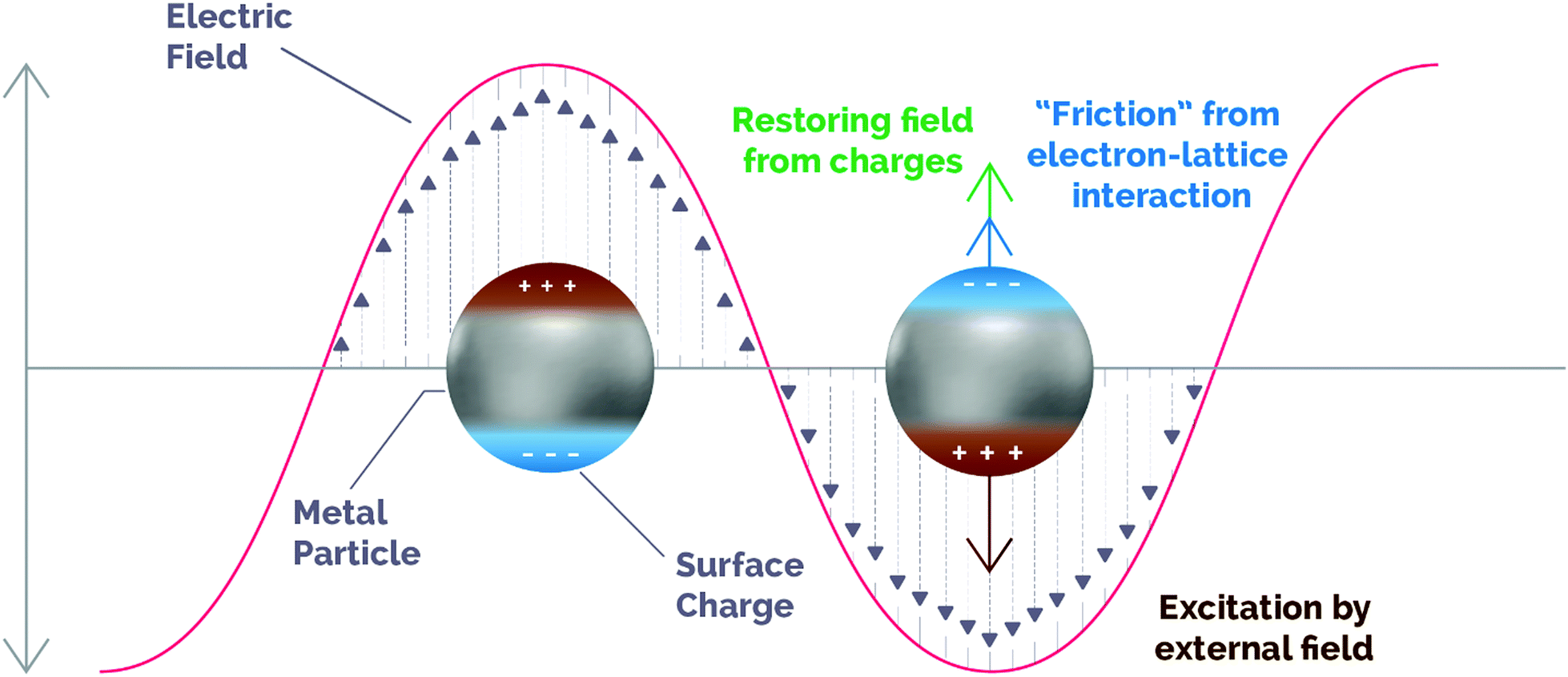 | ||
| Fig. 1 Schematic illustration of the dynamics of an excited plasmonic nanoparticle. The motion of carriers follows the changes induced by the incoming electromagnetic field (brown), while a restoring force is generated by the out-of-equilibrium surface charges (green) and the ionic network produces damping due to electronic collisions (blue). Adapted with permission from ref. 45. Copyright (2019) American Chemical Society. | ||
Under resonant conditions, both the incident electromagnetic wave and the resonant frequency of conduction electrons are in phase, thus maximizing the electric field in so-called “hot spots” on the surface of plasmonic NPs. Apart from the enhanced electric field, other relevant properties of LSPR arise from the different relaxation processes that occur within plasmonic structures. After excitation, the energy stored in surface plasmons can decay through several pathways, either radiative (as re-emitted photons) or non-radiative (electron–hole pair excitations and electron–electron collisions).40 Although for some specific purposes non-radiative losses are considered as a drawback for plasmonic performance, actually they can show potential scientific and technological applications.13,41 For instance, non-radiative decay may produce local heating as the electronic kinetic energy can be transmitted to metal lattice phonons (see Section 3.2). In spite of the fact that photo-thermal cancer therapy has traditionally benefited from this phenomenon, in the recent years the plasmon-induced heating effect has been also applied in a variety of research fields including environmental remediation, solar steam generation and, of more relevance to the scope of this review, catalytic processes.17,42,43
Besides, plasmon energy can be non-radiatively dissipated by absorption within the metal NPs and, eventually, generate energetic hot charge carriers in the plasmonic structure. This contribution to plasmon dephasing comes from electronic collisions with the surface of the plasmonic structure (surface scattering, also known as “Landau damping”) and is a pure quantum mechanical process in which the energy from a plasmon quantum is transferred on the timescale of femtoseconds (1–100 fs) into a single electron–hole pair excitation.40 This phenomenon is due to the non-conservation of the linear momentum of electrons near the surface and in hot spots, and enables an electron to absorb a photon quantum with energy ℏω.44 As we will describe in detail in the next section, hot carriers are able to escape from the plasmonic NPs and induce further chemical reactions.
However, before we delve into the mechanisms underlying photo-thermal catalysis, we would like to clarify some terms commonly used in the literature that, due to the lack of a strict definition, may cause misinterpretations in this field. Plasmon-enhanced catalysis derives from the synergistic combination of three properties arising from LSPR: hot carrier generation, local heating effect and optical near-field enhancement.45 When it comes to photo-thermal catalysis, Ozin et al. determined that hot carriers and the thermal effect contribute synergistically to the enhanced overall reaction rate, with both mechanisms playing a role to various degrees depending on the operating system.30,46 Therefore, in the case of plasmonic materials the photo-thermal effect arises from the combination of both thermal and photochemical contributions of non-radiative plasmon decay, as we will illustrate in subsequent sections. It should be emphasized, however, that the photo-thermal effect is not an exclusive phenomenon of plasmonic NPs. In fact, semiconductor oxides, chalcogenides, metal organic frameworks and other carbon-based materials such as graphene or carbon nanotubes (CNTs) can display photo-thermal performance as well, although in these cases the operating mechanisms have been less studied.29,47 For these reasons, in the next section we will also give a brief overview of these non-plasmonic approaches to photo-thermal catalysis.
3. Photo-thermal enhanced catalysis
3.1. Photochemical enhancement in plasmonic structures
As mentioned above, the excitation of plasmonic NPs can generate hot carriers (i.e. electrons and holes) via electronic intraband and/or interband transitions through non-radiative Landau damping.48 The absorption of a photon with energy hν promotes hot electrons with energies above the Fermi level (EF + hν) that, upon interaction with other species possessing electron-accepting orbitals, may be injected from the metal nanoparticle to these species.48,49 The kinetic energy of hot electrons excited during plasmon decay is transferred to the adsorbates, that are chemically activated via vibrational or electronic transitions. Ultimately, the promotion of high-energy electrons to antibonding orbitals of adsorbed molecules can result in the cleavage of molecular bonds, triggering subsequent chemical transformations.These above-mentioned hot electrons can be exploited for catalytic applications in four main pathways, depending on the existence of a single-component or a heterostructured plasmonic photocatalyst. The first two pathways make use of hot electrons of single plasmonic NPs interacting with adsorbates via indirect or direct electron transfer. The last two pathways are related to interactions of supported plasmonic metal nanostructures with semiconductors via an indirect injection mechanism to an acceptor or through direct promotion of the carrier into the conduction band (CB).50,51
This indirect mechanism for electron transfer has been evidenced in the plasmon-mediated activation of small molecules such as molecular hydrogen (H2) and oxygen (O2).49,55–58 Recently, Halas et al. using both DFT calculations and H/D exchange experiments proved that hot electrons generated on Au NPs could transfer via an indirect pathway to the antibonding orbital of molecular H2, thereby creating a negatively charged H2δ− species. Eventually, electrons transferred back to Au and H2 returned to their electronic ground state with accumulated vibrational energy that led to ultimate H2 dissociation.55 Linic and co-workers also reported the plasmon-induced partial oxidation of ethylene using Ag nanocubes. In this study, hot electrons generated on the surface of Ag nanocubes were transferred to antibonding states of O2 by an indirect mechanism, leading to the generation of O2−. In the same line as in the case of H2 activation, the accumulated vibrational energy overcomes the activation energy barrier and causes O2 dissociation.49
Interestingly, Linic's group observed that the photo-degradation of methylene blue by Ag nanocubes was driven via direct electron transfer.53,60 Although Ag nanocubes showed two plasmon absorption bands centered at 532 and 785 nm, the authors reported a higher degradation yield of methylene blue under irradiation at 785 nm. These results indicated that the longest wavelength, and so the lowest in energy, could transfer hot electrons more efficiently, thus suggesting a one-step process (direct electron transfer) rather than a two-step pathway (indirect mechanism). Based on their hypothesis, a strong hybridization between methylene blue and the Ag interface was established, generating electron-acceptor orbitals (LUMO) centered on methylene blue and electron-donor orbitals (HOMO) centered on Ag.53 However, for the LSPR decay of Ag to directly generated hot electrons in the electron-acceptor states, the energy gap of the HOMO–LUMO transition had to be resonant with the Ag plasmon band at 785 nm. Under this condition, the Ag LSPR decay allowed the direct electron promotion to the excited hybridized states.53,60 These results demonstrated that the direct electron transfer pathway can be a powerful strategy to selectively activate reactants only by choosing the wavelength that is resonant with the transition between the hybridized states.50
In a similar way to the indirect mechanism for non-supported plasmonic NPs, the most common mechanism for hot electron transfer in heterostructures takes place via a two-step pathway that involves hot carrier generation within the plasmonic nanoparticle in the time scale of femtoseconds followed by electron transfer through the metal/semiconductor interface (Fig. 2).50
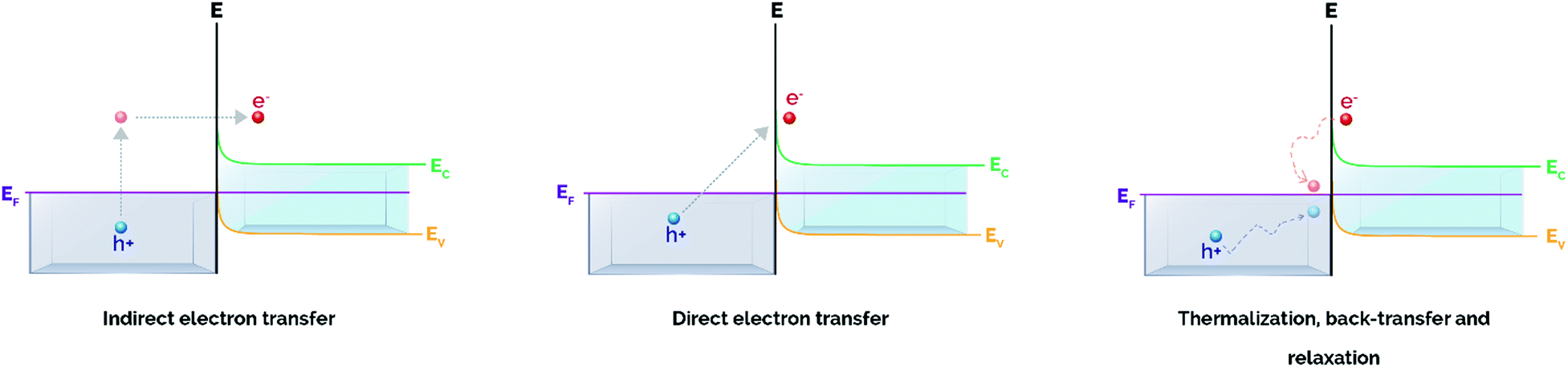 | ||
| Fig. 2 Plasmon-induced hot carrier generation and hot electron transfer/back-transfer processes in metal/semiconductor heterostructures. Adapted with permission from ref. 50. Copyright (2017) American Chemical Society. | ||
Upon the formation of a junction between a semiconductor and a metal nanoparticle, the Fermi values of both components align to favor the redistribution of charge and to ensure equilibrium in the system (Fig. 3).65 This gives rise to bending of both the valence and conduction bands of the semiconductor, thus creating a Schottky barrier at the semiconductor/metal interface whose energy (φSB) corresponds to the difference between the Fermi level of the metal and the interfacial CB edge.52,66
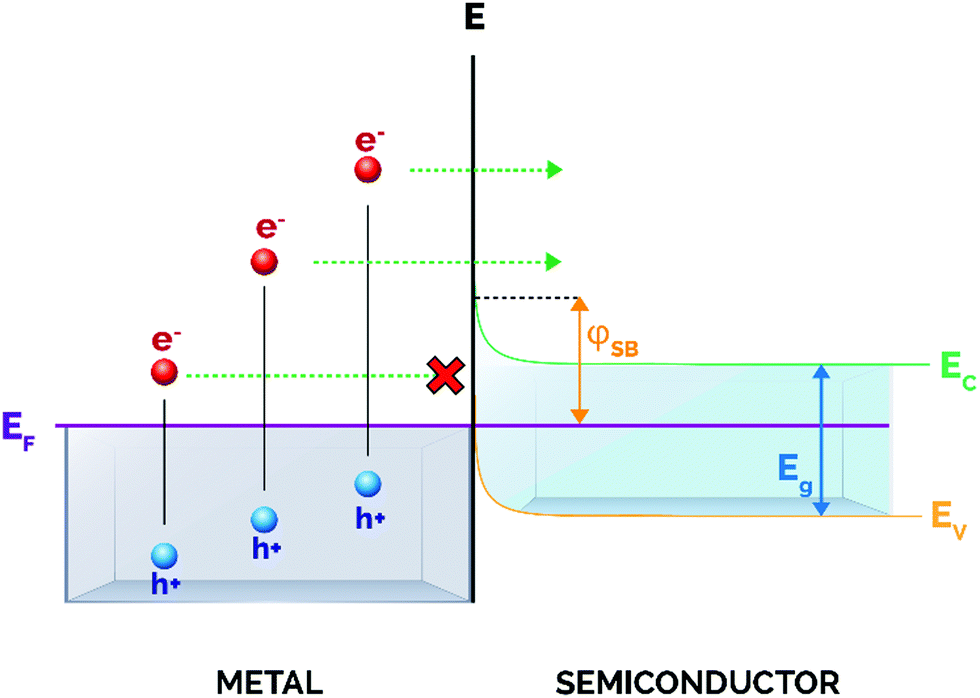 | ||
| Fig. 3 Schematic illustration of the metal/semiconductor Schottky barrier. Upon excitation, only hot electrons with enough energy can surpass the Schottky barrier (φSB) and transfer into the conduction band of the semiconductor. | ||
Importantly for catalytic applications, only hot electrons with energies above the energy threshold settled by the Schottky barrier will inject into the CB and will be available for subsequent electron-induced reactions at the semiconductor surface. In this regard, a low φSB will allow the promotion of a larger number of carriers at energies sufficiently high to traverse the barrier, and hence available for participating in chemical transformations. However, it is also of pivotal importance that the conduction band bending at the interface is high enough to prevent the back-transfer of electrons to the metal nanoparticle (Fig. 2), thereby guaranteeing the spatial separation of electron–hole pairs and increasing the average lifetime of charge carriers.45 For these reasons, an adequate balance between these two effects has to be taken into account when designing metal/semiconductor heterojunctions.
Typically, the magnitude of the Schottky barrier ranges between 0.5 and 1.5 eV and clearly determines the efficiency of hot electron injection from the metal nanoparticle to the CB of the semiconductor.52 Most relevant, the height of the Schottky barrier is smaller than the band gap of many semiconductors, and this features one of the key advantages of these heterostructures, as excited electrons are not required to possess greater energy than the semiconductor band gap to be extracted.67 As a matter of fact, many studies have described so far this metal–semiconductor indirect electron transfer mechanism and its practical application in a vast number of photocatalytic reactions including water splitting, CO2 reduction or the degradation of pollutants.21,68–81
In a recent work, Lian et al. observed a broadened absorption peak in the UV-visible spectrum for a Au/CdSe nanorod composite, suggesting a strong orbital hybridization between both Au and CdSe components.89 Transient absorption spectroscopy data also revealed an electron transfer time scale in the order of tens of femtoseconds with an overall efficiency exceeding 24%.89 In 2017, Ratchford and co-workers also determined an outstanding hot electron injection efficiency up to 45% of Au NPs embedded in TiO2 films.85 All these results were inconsistent with the traditional indirect pathway, hence indicating the existence of a direct electron transfer mechanism.
The assessment of the dominating electron transfer mechanism in metal/semiconductor hybrid materials can be tricky, but the study of QY can be a useful tool to distinguish the reaction pathway. For example, recent works have reported a very low QY of electron transfer for Au/graphene (∼10%) and Au/CdS (∼2.75%) heterostructures, thus revealing the existence of an indirect electron transfer mechanism.90,91 Nevertheless, most of the current studies still have not developed a solid distinction of electron transfer mechanisms in metal/semiconductor heterojunctions.50
3.2. Thermal enhancement in plasmonic structures
In the previous sections we have described the relevant physical mechanisms underlying photo-thermal catalysis of ultrafast processes (below 100 fs) occurring upon plasmon decay (i.e. electron–hole pair excitations via Landau damping). At longer relaxation times, hot carriers decay and convert their energy into heat through inelastic coulombic electron–electron scattering over a period from 100 fs to 1 ps. In parallel with this process, low energy electrons couple with phonon modes via electron-lattice collisions and so increasing the temperature of the metal lattice with a timescale of several to some hundreds of ps. At a later stage, this heat is transferred from the metal lattice to the surroundings in the time scale from 100 ps to 10 ns through phonon–phonon scattering (Fig. 4).59,92,93 Therefore, plasmon-induced heat can result in energy transfer from the metal nanoparticle to surface adsorbates and subsequently trigger further chemical transformations following an Arrhenius dependence of the reaction rate. Ultimately, this implies that the use of plasmon heating to perform chemical reactions is similar to externally heating the system and does not offer a single pathway to control product selectivity.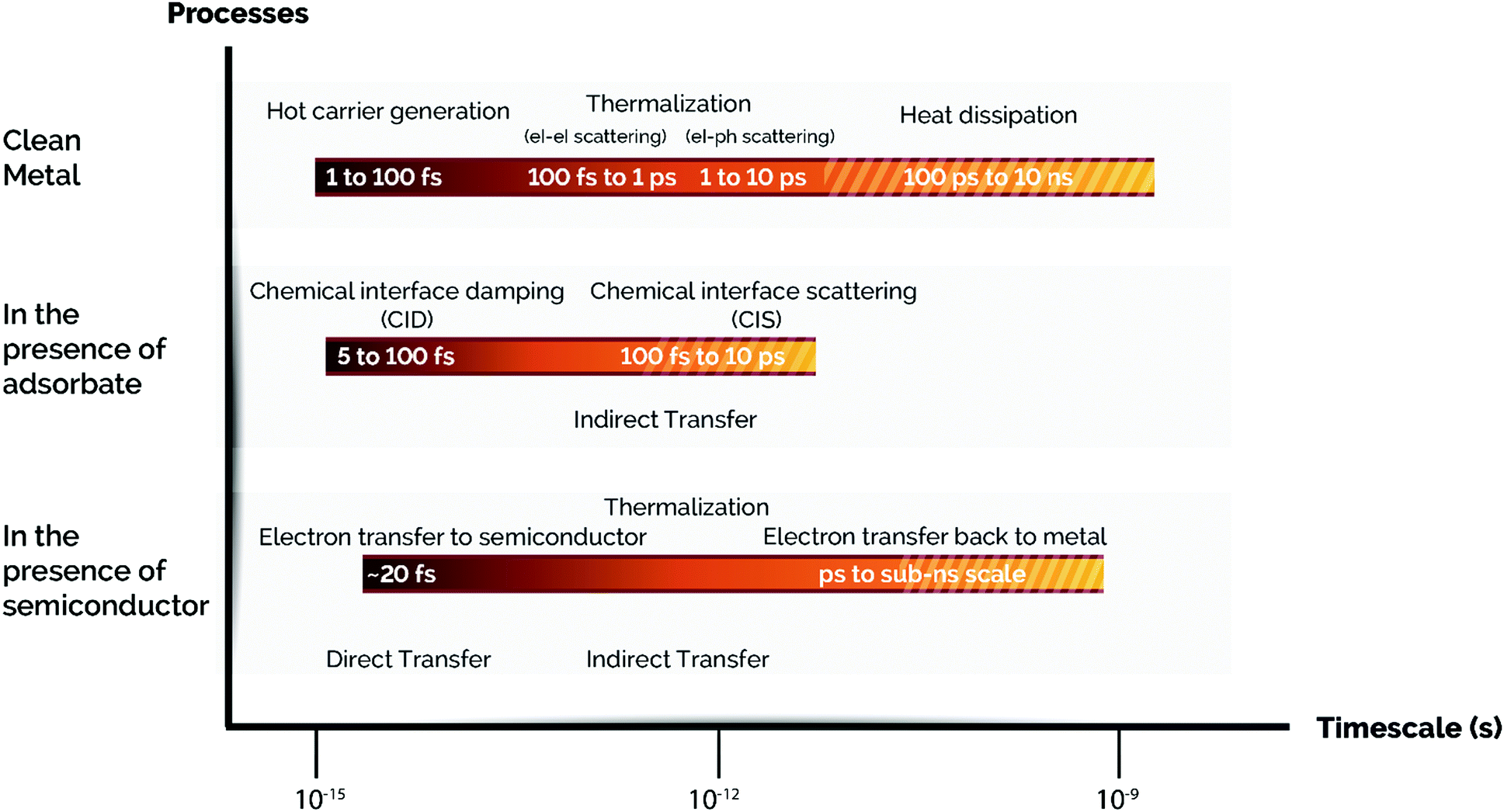 | ||
| Fig. 4 Time scales of plasmon-induced hot carrier generation, hot electron transfer, and thermalization processes with/without an adsorbate or a semiconductor. From top to bottom: A series of time scales corresponding to the fate of hot carriers in a clean metal NP, additional events in a metal NP capped with an adsorbate, and the processes involved in a metal NP loaded on a semiconductor support via a Schottky contact are shown. Note: el stands for electron and ph stands for phonon. Adapted with permission from ref. 50. Copyright (2017) American Chemical Society. | ||
In the particular case of a single spherical metal nanoparticle, the local temperature increase (ΔT) caused by the photo-thermal effect can be described by eqn (1):94
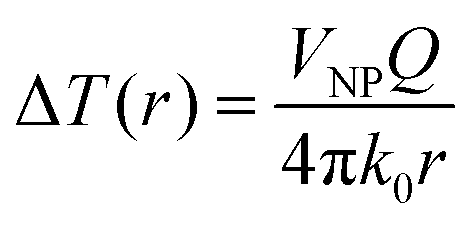 | (1) |
If we consider that the incident light wavelength is much larger than the nanoparticle radius, then the heat generation Q can be expressed as follows (eqn (2)):
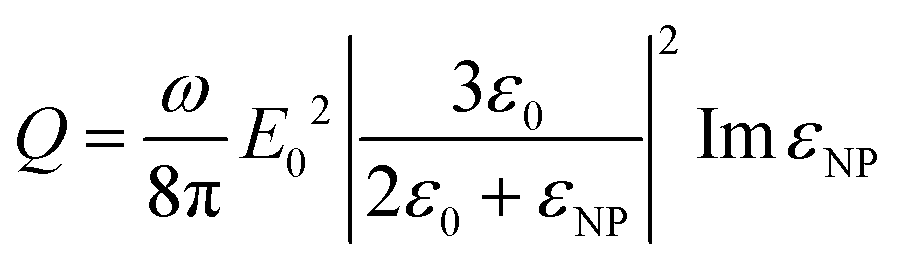 | (2) |
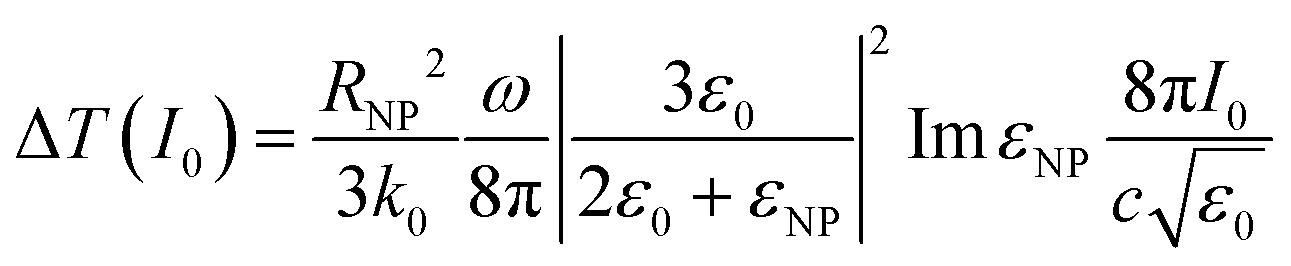 | (3) |
It is of great importance that from this equation it is possible to establish the dependence of temperature on the nanoparticle size:
| ΔT ∝ RNP2 |
Nevertheless, several experimental and theoretical studies suggest that the thermal contribution to photo-thermal catalysis represents a minority weight in the overall catalytic rate.49,94 An interesting work from Govorov et al. theorized that for light intensities similar to continuous-wave solar irradiance (around 100 mW cm−2) the temperature increment due to irradiation of a single gold NP became negligible (∼10−2 K). Only when the photon flux was about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more intense a significant temperature rise (in the order of a few K) was achieved.95 If we take into account that for commercial catalytic processes an increase of 10 K approximately doubles the reaction rate, it is difficult to imagine a scenario in which the light-induced heating plays a crucial role in the overall catalytic performance, at least under common solar irradiance operation conditions.96 It is worth reminding, however, that according to Govorov et al. the light-induced heating effect can be strongly enhanced in the presence of a large number of NPs owing to collective effects, so this opens up the possibility of achieving further temperature increases with appropriate material engineering.95 These observations are in line with those from Baffou et al. regarding significant collective temperature increases even in the presence of small amounts of plasmonic NPs.97
000 times more intense a significant temperature rise (in the order of a few K) was achieved.95 If we take into account that for commercial catalytic processes an increase of 10 K approximately doubles the reaction rate, it is difficult to imagine a scenario in which the light-induced heating plays a crucial role in the overall catalytic performance, at least under common solar irradiance operation conditions.96 It is worth reminding, however, that according to Govorov et al. the light-induced heating effect can be strongly enhanced in the presence of a large number of NPs owing to collective effects, so this opens up the possibility of achieving further temperature increases with appropriate material engineering.95 These observations are in line with those from Baffou et al. regarding significant collective temperature increases even in the presence of small amounts of plasmonic NPs.97
In fact, several groups have reported so far the use of plasmon-mediated heating in order to perform catalytic reactions. Boyd et al. described the catalytic steam reforming of ethanol using a flow-type microfluidic channel reactor with embedded 20 nm diameter Au NPs.98 Upon irradiation of a mixture of ethanol–water with a low-power laser at the resonant frequency of Au NPs, the authors could observe the formation of gaseous products corresponding to CO, CO2 and H2. These results demonstrated the possibility to drive endothermic reactions by plasmon-assisted heat generation.
More recently, Jiang and co-workers reported the first application in catalysis of the synergistic combination of the photo-thermal effect of plasmonic NPs with the advantageous properties of metal–organic frameworks (MOFs).99 In this work, the authors developed a Pd@ZIF-8 composite for the light-assisted selective hydrogenation of olefins at room temperature. Interestingly, the authors found that while Pd nanocube cores acted as nano-heater active sites, the ZIF-8 shell played multiple roles in the reaction mechanism. On the one hand, it accelerated the reaction rate due to its ability to act as a H2 gas reservoir. On the other hand, it favored Pd nanocube stabilization preventing them from migration/aggregation and acted as a molecular sieve for selective catalysis. As a result, the Pd@ZIF-8 composite effectively catalyzed the hydrogenation of olefins under UV-visible irradiation at room temperature, achieving a 66% conversion. Noticeably, the reaction yield was about 50% by heating at 50 °C under dark conditions, hence indicating the superior catalytic performance of the Pd@ZIF-8 composite upon light irradiation.
Similarly, Xu et al. also described a hybrid core–shell hierarchical nanostructure based on plasmonic NPs and MOFs for the photo-thermally catalyzed cyclocondensation reaction.35 Upon NIR irradiation, the plasmonic photo-thermal core of Cu7S4 generated a high temperature and efficiently transferred heat to the catalytic shell of ZIF-8 containing the acid–base Lewis active sites. This resulted in a 4-fold enhancement of the catalytic activity for cyclocondensation reaction compared to dark conditions.
It should be mentioned, however, that there is a lack of consensus across the literature when evaluating the thermal contribution to the overall photocatalytic rate in plasmon-mediated reactions.100 Consequently, it is still tricky to successfully distinguish the thermochemical pathway from the pure photochemical mechanism both underlying the photo-thermal effect. In Section 3.4, we will describe different methodologies that can help to identify which of the aforementioned mechanisms (i.e. photochemical and/or thermal) is preeminent in the photo-thermal reactions under study.
3.3. Photo-thermal catalysis in non-plasmonic structures
In a similar way to plasmonic structures, non-plasmonic elements can display the photo-thermal effect through direct intraband and/or interband electronic transitions. For instance, Sarina et al. demonstrated that non-plasmonic metal NPs supported on ZrO2 could catalyze cross-coupling reactions at low temperatures under visible light.101 According to the authors, upon irradiation with UV light electrons could shift to high-energy levels through interband transitions, and only those with enough energy could transfer to the LUMO of adsorbed molecules, just like in the case of plasmonic metal NPs. When excited with low-energy visible-IR light, electrons were not energetic enough to be injected into adsorbate states, thus contributing to the enhancement of the reaction rate by means of thermal effects.In the case of metal oxide non-plasmonic semiconductors, an effective charge separation is of pivotal importance to maximize the efficiency of the photochemical process. However, in these systems non-radiative relaxation processes compete with charge separation and occur in the form of either Auger or Shockley–Read–Hall recombination.30 It is of great importance that ultimately both processes are responsible for the emission of energy as lattice vibrations, hence providing an active mechanism for heat generation in this type of structures. Auger recombination takes place when an electron and hole recombine but, instead of primarily releasing the excess of energy in the form of light or heat, the energy is transferred to a third charge carrier that thermalizes back to the conduction band edge by emitting phonon vibrations. An alternative pathway for non-radiative relaxation is Shockley–Read–Hall recombination (also known as “trap-assisted recombination”). This mechanism usually takes place in semiconductors displaying a high degree of defects or vacancies that create mid-gap energy states. After excitation, electrons quickly move to these states releasing energy as phonons that eventually increase the temperature of the semiconductor. Altogether, photochemical and thermal effects contribute to the photo-thermal performance in non-plasmonic semiconductors. In 2019, Ye et al. reported an example of this approach for the photo-thermal reduction of CO2 using 2D In2O3−x as a catalyst.102 Compared with commercial In2O3, defective 2D In2O3−x displayed an outstanding photo-thermal CO production of 103.2 mmol g−1 h−1 by increasing the temperature of the system to 280 °C within 10 min. The authors rationalized that in this case oxygen vacancies in defective 2D In2O3−x had a dual function; in other words, they provided efficient local heat generation and active sites for CO2 adsorption that basically enhanced the catalytic performance.
Besides non-plasmonic metallic NPs and semiconductors, the photo-thermal effect has been also reported in a myriad of materials including MOFs, chalcogenides, MXenes and other carbon-based materials. In spite of the fact that in these cases the mechanisms underlying the photo-thermal effect are not completely understood, all of them offer promising applications not only in the field of catalysis, but also in photovoltaics and solar thermal energy.47
3.4. Methodologies to identify the reaction mechanisms in photo-thermal processes
The photo-thermal effect results from the combination of both photochemical and thermochemical contributions. Taking into account that the two mechanisms can coexist simultaneously, it is very challenging and not always evident to distinguish which pathway drives predominantly the system. For this reason, in this section we have summarized and listed different experimental and computational methodologies that can help to distinguish between hot-carrier and thermal effects in photo-thermal processes.Previous experimental and theoretical studies have shown that there is a linear relationship between the photon flux and the surface temperature increase of plasmonic NPs.94 However, if this linear relationship is combined with the Arrhenius equation for thermally driven reactions, the reaction rates arising from plasmon-induced heating exhibit an exponential dependence on light intensity.49 An example of this behavior was reported by Wang et al. when they used Au NPs supported on ZnO for the plasmon-assisted thermal hydrogenation of CO2 (Fig. 5a).103 The authors not only found an exponential dependence of the CO conversion rate on laser intensity, but they could also tune product distribution to either CO or CH4 by just varying the intensity of the laser. Therefore, an exponential dependence of the reaction rate on illumination intensity is a characteristic feature of thermally driven transformations.
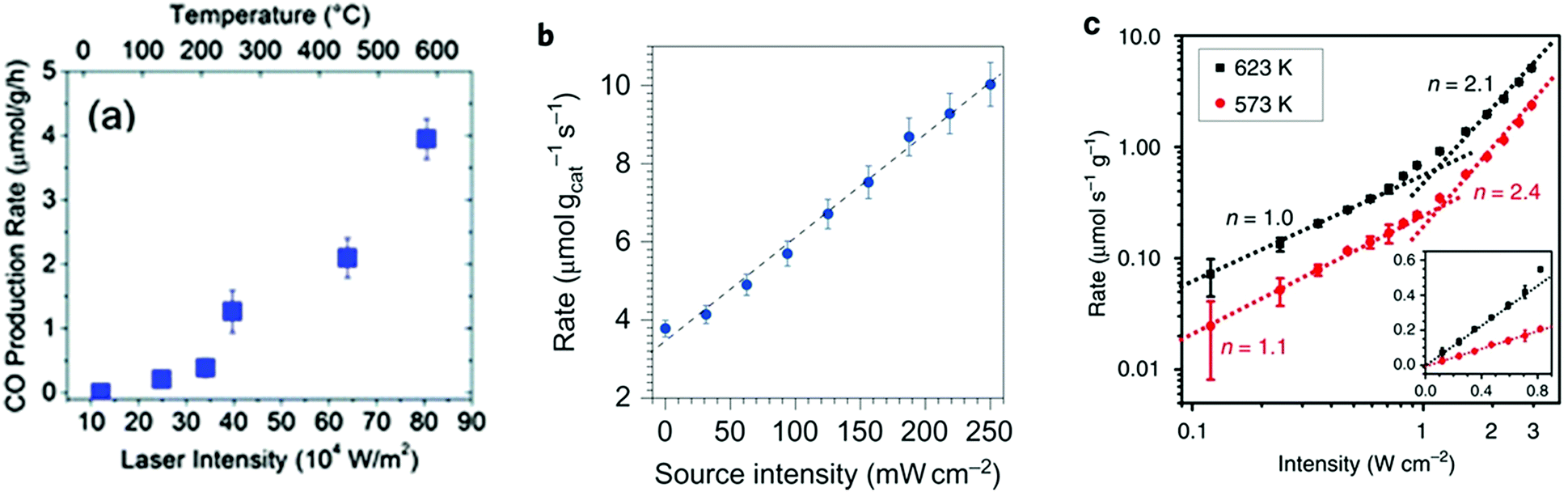 | ||
| Fig. 5 (a) CO production rate as a function of continuous wave laser intensity at 532 nm using ZnO-supported Au NPs. Reproduced with permission from ref. 103. Copyright (2013) Royal Chemical Society. (b) The rate of photo-thermal ethylene epoxidation as a function of light source intensity. Reproduced with permission from ref. 49. Copyright (2011) Springer Nature. (c) Rate of CH4 photo-production as a function of ultraviolet light intensity at 623 (black squares) and 573K (red circles). The intensity-dependent reaction rate shows a linear to super-linear transition with increasing light intensity. The inset shows the intensity-dependent reaction rate in the linear region. Reproduced with permission from ref. 108. Copyright (2017) Springer Nature. | ||
A linear dependence of the reaction rate on light intensity is, however, a signature of an electron-driven process and it is the most commonly reported mechanism for plasmonic reactions.49,104–106 A first order relationship between the photon flux and reaction rate implies that a single photon absorption induces the chemical reaction. It is worth mentioning that both linear (rate ∝ I) and superlinear (rate ∝ In, n > 1) regimes are distinctive features of electron-driven chemical transformations on metal surfaces, but the operation conditions are the key factor determining the prevalence of each of them. For instance, Linic et al. observed a linear dependence of the photocatalytic rate of ethylene epoxidation on light intensity up to 300 mW cm−2, while at higher source intensities the dependence turned into the superlinear regime (Fig. 5b).49,107 In a similar way, Zhang et al. described a linear dependence of the photocatalytic CO2 hydrogenation on Rh/Al2O3 heterostructures that became superlinear at the highest light intensities (Fig. 5c).108 In this case, the authors attributed the transition to superlinearity to multiple excitations of the vibrational modes of adsorbed species by plasmon-induced hot electrons.
The reaction rate dependence on the photon wavelength typically correlates absorption properties of the photocatalyst with its catalytic performance and is of great importance for this section; it can provide further evidence for electron-driven processes. In this respect, an agreement between the absorption spectrum of the plasmonic photocatalyst and the wavelength dependent reaction rates corroborates an electron-driven photochemical transformation.54 For instance, Ye and co-workers recently observed that the quantum efficiency for the photocatalytic steam reforming of CH4 using TiO2-supported Rh NPs was consistent with the optical absorption spectrum of the catalyst, thus suggesting that the hot electrons from interband transition in Rh NPs were responsible for the catalytic activity.109 In 2014, the same group reported the photo-thermal conversion of CO2 with H2 to produce methane (CH4) with group VIII nanocatalysts.110 However, in this case the electron-driven reaction pathway was completely excluded since the photo-action spectrum using monochromatic light showed no dependence on the irradiation wavelength. In view of these findings, the authors concluded that the photo-methanation of CO2 was mediated by a pure thermal effect. In the early 2020, Su and Xiao studied the visible light α-alkylation of ketones with alcohols using a titanium nitride (TiN) photocatalyst.111 The authors not only found that both action and optical absorption spectra were in good agreement, but also reported a positive relationship between reaction rate and light intensity. Altogether, the results certainly indicated that the reaction proceeded through an electron-driven mechanism operated by energetic charge carriers on TiN. The irradiation wavelength dependence of the reaction rate was also evaluated for O2 activation by different plasmonic Au-nanorods@Pd superstructures.112 In this work, the authors observed a clear matching between the photocatalytic performance and the visible-NIR spectrum of the photocatalyst, suggesting that plasmon-induced hot electrons drove the reaction mechanism by electron injection into the antibonding orbital of molecular oxygen. Furthermore, on the basis of theoretical calculations the thermal effect was found to be negligible, so the high photocatalytic activity was mainly ascribed to the large field-enhancement that maximized plasmon absorption and boosted hot electron generation in Au-nanorods@Pd heterostructures. In spite of the fact that all these studies are insufficient to distinguish between the direct and indirect electron transfer pathways, the combination of wavelength dependent experiments with a linear relationship of the reaction rate with light intensity provides convincing evidence of the hot electron-induced reaction mechanism.
In a recent review, Baffou and co-workers proposed a series of simple experimental procedures to discriminate and quantify the different thermal and non-thermal contributions of plasmon-driven chemical reactions.113 One of these methodologies is based on the variation of the light beam diameter using two approaches: a constant irradiance or a constant power. In the first case, the photon flux is proportional to the beam diameter, whereas in the second case the amount of photons remains constant. Both thermal and non-thermal contributions typically present a proportional dependence between reaction rate and the area of light beam under constant irradiance conditions. However, in the constant-power regime, the rate of electron-driven processes is independent of the beam diameter, while thermal-driven reactions show an inverse relationship between rate and beam radius. The study of these different behaviors by varying the illumination diameter appears to be an efficient means to elucidate the underlying reaction mechanism.
The activation energy (Ea) is a key parameter in catalysis that provides insightful information about the catalytic performance of a material and the mechanistic pathway driving the reaction.114 Because of that, a straightforward method to ascertain if photon-induced charge carriers contribute to the enhancement of the reaction rate consists in comparing activation barriers under both light and dark conditions at constant temperature. The reduction in the value of Ea under light irradiation compared to dark conditions is indeed typical for a hot carrier-driven mechanism.108,109,115,116
Another distinctive feature of electron-driven processes can arise from isotopic labelling experiments. Traditionally, the isotopic labelling of reactants has been a useful analytical probe to elucidate the reaction mechanisms in thermal catalysis through the measurement of the kinetic isotope effect (KIE). The KIE is obtained by measuring the change in the reaction rate due to the introduction of labelled reactants, as heavy isotopes require a higher energy input to reach the transition state. Interestingly, an electron-driven process displays a greater KIE under light irradiation than the thermally driven counterpart under dark conditions, so the comparison of both values can be a convenient methodology to differentiate between thermally and electron-driven reactions. For example, Ye et al. measured the KIE for photocatalytic CH4 steam reforming over a Rh/TiO2 catalyst by comparison of the H2 production rates using either CH4 or CD4 as a reactant.109 The authors found that the KIE under visible light irradiation (KIElight = 1.69) was larger than the one obtained under dark conditions (KIEdark = 1.55). Consequently, it was concluded that photo-induced hot electrons from interband transitions of Rh were responsible for the enhanced catalytic activity. In a similar way, for the photocatalytic ethylene epoxidation Linic and co-workers used 16O2 and isotopically labelled 18O2 to measure the KIE under both illumination and dark conditions.49 Experiments confirmed that the activation of O2 determined the overall reaction rate and the considerably larger KIE reported for the photocatalytic process suggested an electron-driven reaction mechanism.
Alternatively, the analysis of reaction selectivity can be another useful technique to assess the dominating reaction pathway. In thermally driven reactions, energy is provided to all available degrees of freedom including vibrational, rotational and translational states, leading to the simultaneous activation of a plethora of reaction pathways.117 In contrast, electron-driven processes offer the opportunity to modulate reaction yield towards the desired product by selectively injecting hot carriers to specific adsorbate–metal bonds.108 In this respect, Christopher and co-workers investigated the photocatalytic CO oxidation in a H2 rich stream over Al2O3-supported Pt NPs.118 The results showed that the resonant photoexcitation of Pt–CO bonds arising from the hybridization of adsorbate–metal states enhanced the selective oxidation of CO to produce CO2 over the competing H2 oxidation reaction. Accordingly, the authors proposed that the specific photoactivation of metal–adsorbate bonds could be a good methodology to control reaction selectivity in electron-driven plasmonic reactions. Cronin et al. studied the photoreduction of CO2 with H2O vapor using Au/TiO2 heterostructures and found that the product distribution was strongly dependent on the irradiation wavelength.74 While the excitation at 532 nm mostly produced CH4, the use of UV light of 254 nm yielded the generation of other products including ethane, methanol and formaldehyde. It was rationalized that depending on the irradiation wavelength it was possible to trigger different reaction pathways to drive the chemical reaction towards certain product distribution. Interestingly, any product was observed when the reaction was performed at 400 °C under dark conditions, thus excluding any thermal contribution to the reaction mechanism.
Transient absorption spectroscopy (TAS) is an effective methodology to study the dynamics of electron transfer processes. Consequently, many groups have used TAS to elucidate the mechanism of light-mediated reactions. For instance, Ye and co-workers determined an ultrafast electron transfer from Rh NPs to TiO2 under photoexcitation at 450 nm, attributable to the injection of hot electrons across the Schottky barrier at the metal–oxide interface (Fig. 6).109 Importantly, the authors found that the electron transfer event occurred in the time scale of hundreds of femtoseconds, while the transient signal showed a decay in the order of thousands of nanoseconds, thus indicating an effective charge carrier separation that prevented undesirable electron–hole recombination. In view of this, the authors concluded that the observed photo-enhancement of methane reforming using a Rh/TiO2 photocatalyst was due to excited hot carriers, rather than thermal effects. In the same way, Garcia and Corma performed transient spectroscopy measurements to study the reaction mechanism of photocatalytic CO2 hydrogenation to CH4 by Ni/SiO2·Al2O3.119 Experiments allowed the detection of transient signals under excitation at 355 and 532 nm that confirmed the presence of charge separation states. Since the reaction temperature was monitored at 150 °C, therefore insufficiently low to perform CO2 methanation, it was assumed that the reaction was driven by H2 activation through the formation of Ni–H species. A more recent work from the same group also used TAS measurements to demonstrate that NiO/Ni NPs supported on graphene could hydrogenate CO2 to CH4 by photo-excitation of electron–hole pairs.120
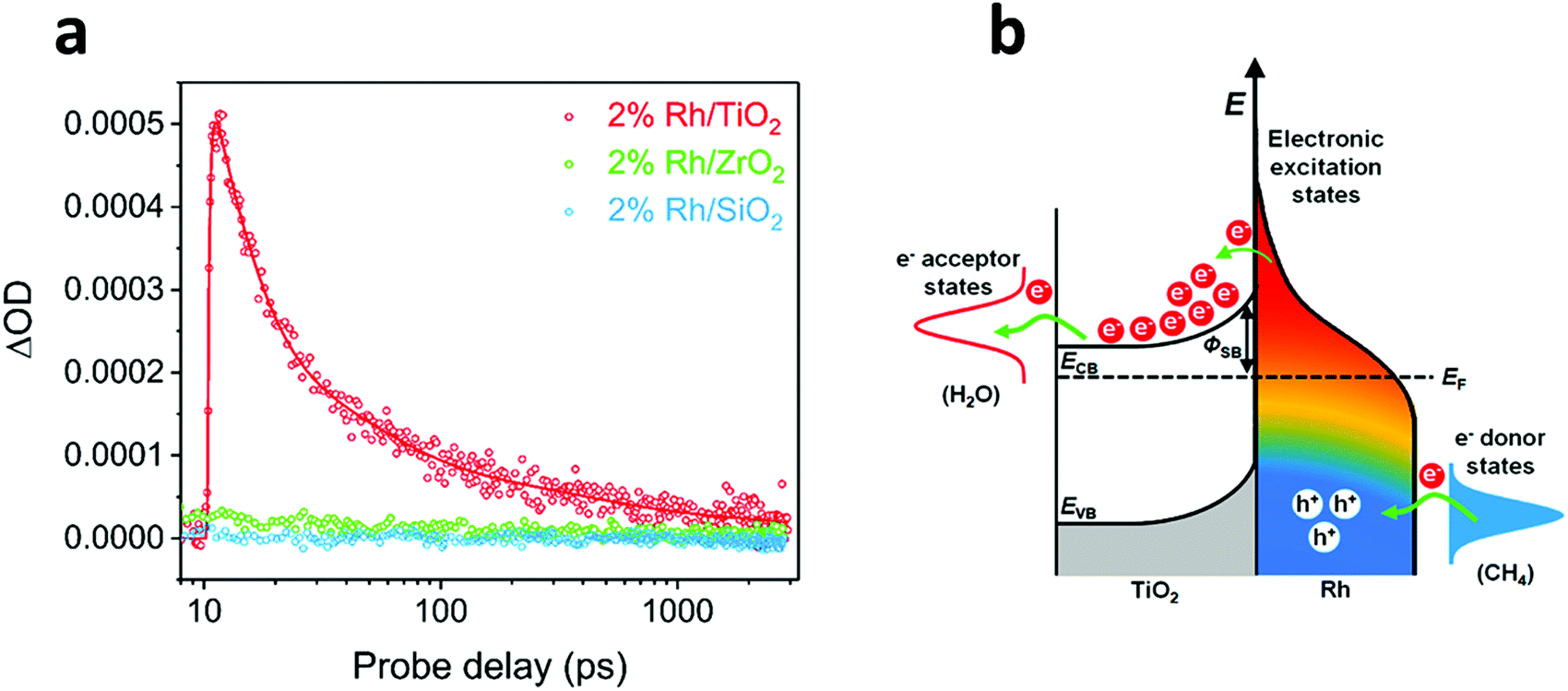 | ||
| Fig. 6 (a) Transient absorption kinetics at 5000 nm of Rh/TiO2, Rh/ZrO2 and Rh/SiO2 under laser excitation of 450 nm. (b) Mechanism of hot carrier-enhanced steam methane reforming. Visible light-excited hot carriers can be separated quickly at the Rh/TiO2 interface; the positively charged Rh surface favours C–H bond activation, whereas electron-rich states at the interface promote the reduction process. Reproduced with permission from ref. 109. Copyright (2018) American Chemical Society. | ||
3.4.2.1. Non-luminescence methods. Another approach to differentiate between the photochemical and the thermochemical contribution to photo-thermal reactions implies the direct temperature measurement at the surface active sites. Although carrying out this type of measurements is still very challenging, recent advances in metrology have allowed great improvements in nanoscale thermometry. For instance, scanning thermal microscopy equipped with nanoscale probe tips renders surface temperature measurements with spatial resolutions in the range of 10 nm and ∼10–50 mK precision.121,122 It is also possible to obtain local temperature maps with nanometer-scale resolution by means of the analysis of the temperature-dependent energy shift of plasmon peaks using scanning transmission electron microscopy (STEM).123 More recently in 2018, Idrobo et al. performed local temperature assessments in boron nitride nanoflakes by calculating the ratio between the gain and loss phonon peaks in the electron energy spectrum using monochromated aberration-corrected STEM.124
3.4.2.2. Luminescence methods. IR thermography is based on the Planck blackbody emission principle and estimates the temperature profile of a body in relation to its emitted energy. Although local and global temperatures can be obtained by thermal mapping with IR cameras in combination with bulk thermocouple measurements, the accuracy of these methodologies is still controversial due to different interpretations of the emissivity of catalysts and the parameter choice of thermal imaging cameras.100,125,126 It is also worth mentioning that under irradiation of thick catalyst beds, thermal gradients may exist due to the limited penetration ability of light and non-uniform heat diffusion. In these cases, the measurement from IR cameras only represents the surface temperature of the catalyst, that is commonly higher than the temperature inside the catalyst bed.125 For these reasons, IR camera measurements must be precisely recorded and attention has to be paid in order to avoid misinterpretations in the thermal contribution to photo-thermal systems.
Thermoreflectance and optical interferometry are luminescence techniques that use the reflection of light to obtain thermal measurements. Thermoreflectance relies on the relationship between the refractive index of a material and its temperature, giving rise to temperature profiles in the micrometer/submicrometer scale with high thermal and temporal resolutions typically in the order of 10−2 K and 10−1 μs, respectively.127 In spite of the fact that this technique has been widely implemented in microelectronics to study electrical self-heating, Wang et al. recently used different thermoreflectance techniques to evaluate the thermal enhancement of Au/Al2O3 plasmonic structures with outstanding spatial and temporal resolutions in the order of 100 nm and several nanoseconds.128 As for optical interferometry, it renders local temperature and thermal expansion measurements with very high temperature resolutions of ∼10 μK, but in this case the spatial resolutions are below the nanoscale (∼1 μm).127
Apart from the previously described techniques, tip-enhanced Raman spectroscopy (TERS) also enables mapping local temperatures at the nanometer scale by virtue of calculation of the ratio of the intensities of the anti-Stokes and Stokes Raman signals.129 In fact, the analysis of the observed linear shift in the position of Raman bands as a function of temperature can be a distinctive feature to characterize thermally driven photo-thermal reactions as well.130,131 Ozin and co-workers recently reported an appealing example of this effect using Pd NPs supported on Nb2O5 in the photocatalytic reduction of CO2 (Fig. 7).132 The authors observed a shift in the position of the niobium–oxygen phonon modes as a function of the excitation laser power, hence revealing a photo-thermal phenomenon. This heating effect was probed through a study of the Stokes and anti-Stokes Raman bands and ultimately allowed extracting the local temperature of the catalyst as high as 470 °C. According to the available data, Pd NPs acted as photo-thermal “nano-heaters” that effectively elevated the local temperature of the Nb2O5 support, which drove the CO2 hydrogenation reaction.
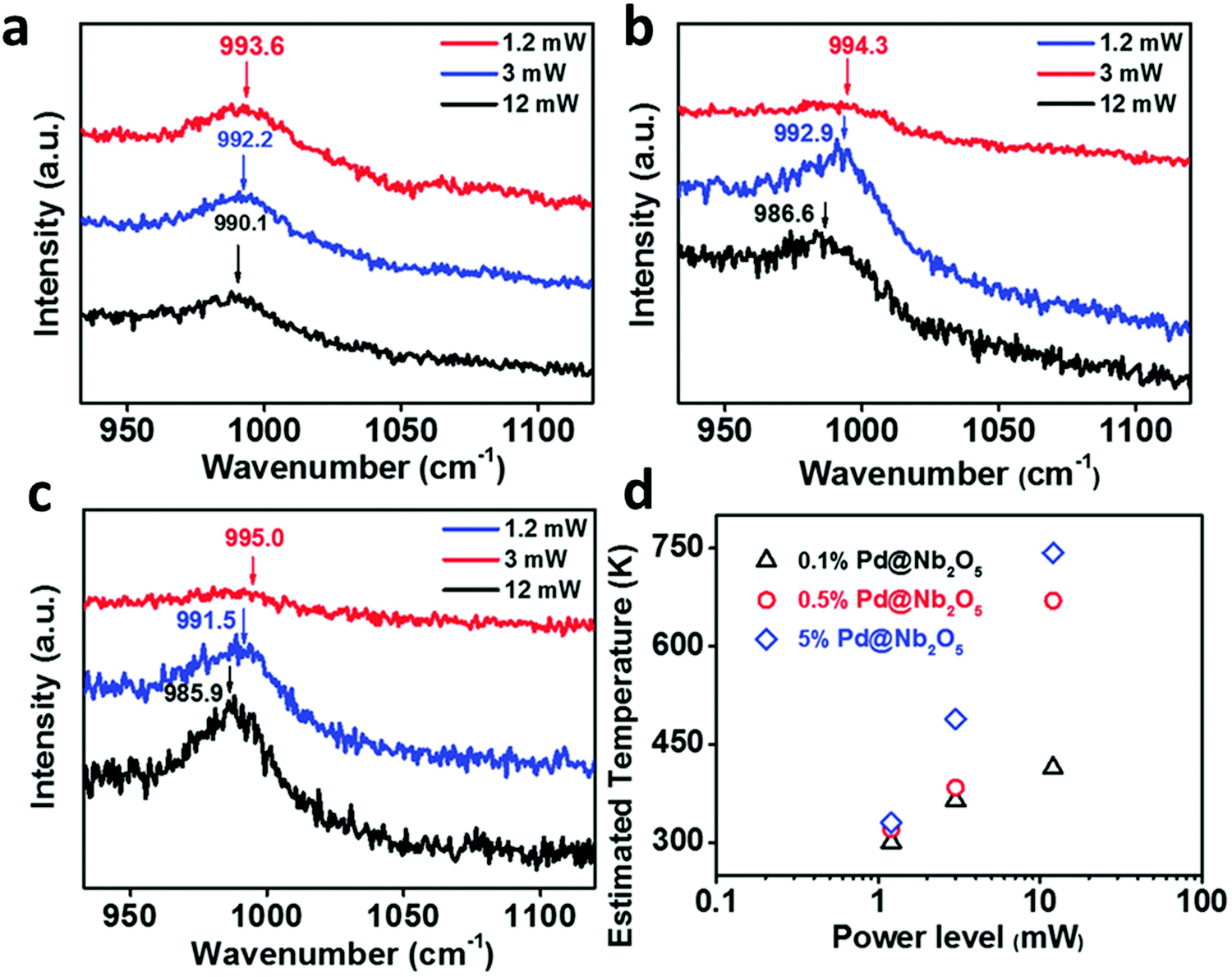 | ||
| Fig. 7 Dependence of the Raman frequency for Nb–O stretching vibrations of (a) 0.1% Pd@Nb2O5, (b) 0.5% Pd@Nb2O5, and (c) 5% Pd@Nb2O5 at different incident light power levels. (d) Estimated temperatures of 0.1%, 0.5%, and 5% Pd loaded onto Nb2O5 nanorods at different incident light power levels. Reproduced with permission from ref. 132. Copyright (2017) John Wiley and Sons. | ||
It is well established that the fluorescence signal strongly depends on temperature, since its intensity decreases and the peaks shift to longer wavelengths when the temperature increases. By taking advantage of this effect, nanoscale fluorescence thermometry can provide temperature measurements with nanometric spatial resolution using quantum dots (QDs) or organic dyes as temperature probes.133–136 For instance, Garcia et al. demonstrated the potential of this strategy to assess the reaction mechanism in the photocatalytic CO2 reduction by Cu2O NPs supported on graphene (Fig. 8).137 In this work, the authors used commercial core–shell CdSe@ZnS QDs as local thermometers and measured their emission lifetime as a function of temperature. Interestingly, the authors found that the irradiation of samples containing these QDs in contact with Cu2O NPs did not lead to significant changes in the QDs’ emission lifetime, thereby excluding a light-induced heating effect. In view of these findings, the authors concluded that the most likely reaction mechanism was the photo-generation of electron–hole pairs rather than a thermal activation. For further details regarding nanoscale thermal probing methodologies, the readers are referred to an excellent review by Wang et al.138 Carlos and Palacio's groups also performed a comprehensive revision of thermal measurement techniques at the nanoscale.127
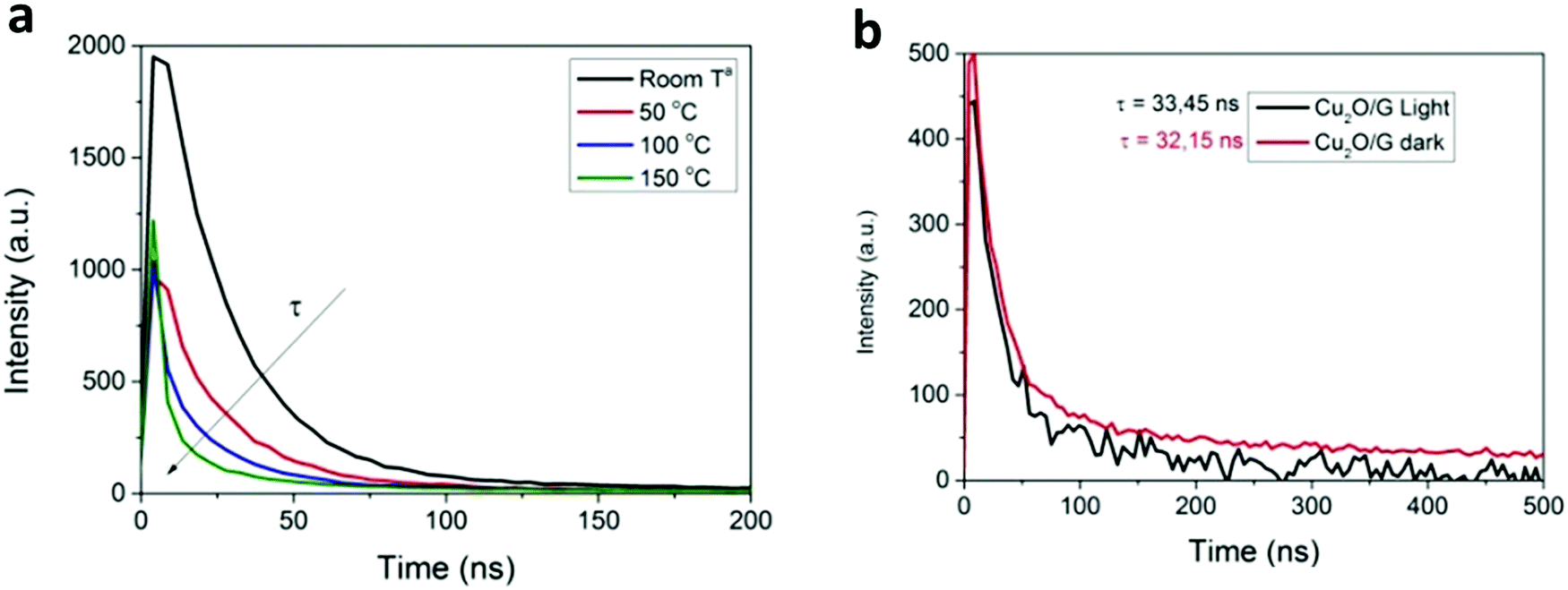 | ||
| Fig. 8 (a) Time correlated emission decay of commercial CdSe@ZnS QDs as a function of temperature. (b) Emission lifetime of thin films of CdSe@ZnS QDs supported on a Cu2O/graphene film under light and dark conditions. Reproduced with permission from ref. 137. Copyright (2017) Royal Society of Chemistry. | ||
3.4.2.3. Computational methods. Finally, besides the aforementioned experimental methodologies, computational methods can aid elucidation of the mechanisms underlying photo-thermal reactions. In this regard, the density functional theory (DFT) framework has proven to be a reliable model to study electronic structures in metal–semiconductor interfaces or adsorbed species in surface active sites.86,108,109,139 A remarkable example of the use of this model to assess the reaction pathway in Rh/TiO2 plasmonic nanostructures for CH4 activation was recently reported by Ye et al.109 Theoretical calculations suggested that under visible light excitation, hot electrons in Rh 4d orbitals could transfer to unoccupied 3d orbitals in Ti, so preventing them from recombination within the metal and favoring the generation of electron-deficient sites (Rhδ+) in the Rh surface. Consequently, these Rhδ+ sites were more prone to accept electrons from CH4, thus facilitating C–H bond cleavage and promoting CH4 activation. Liu and Everitt used DFT calculations as well to elucidate how hot electrons interacted with intermediate species in the photocatalytic CO2 hydrogenation by Rh nanocubes supported on Al2O3.108 This study revealed that anti-bonding orbitals from CHO intermediates could accept photo-generated electrons from Rh, thus weakening C–O bonds and assisting CH4 formation. Curiously, it was also found that the photo-injection of hot electrons into anti-bonding orbitals of the CO intermediate was far unlikely, somehow explaining the reaction selectivity towards CH4 production rather than CO formation.
It is worth reminding that all these methodologies can help to distinguish between photochemical and thermal contribution to photo-thermal conversion and, in the best-case scenario, determine which one is predominant in the system under study. Nonetheless, it is possible and very common indeed that both mechanisms coexist together and operate synergistically to enhance the overall photocatalytic rate. For example, Willets et al. quantified the relative contribution of hot carriers and heating to plasmon-mediated photoelectrochemical reactions based on scanning electrochemical microscopy (SECM) measurements.140 The authors determined that both mechanisms contributed to the photoelectrochemical performance, with a different dependence on excitation intensity. Another example of successful exploration and discrimination of thermal and non-thermal activities working synergistically in the photocatalytic activity of Rh NPs supported on TiO2 was recently provided by Zhang and co-workers.141 Based on both experimental measurements and theoretical models, it was found that heat and light worked not only simultaneously, but also cooperatively, and that a temperature increase gave rise to an improvement of the plasmon-enhanced catalysis. In a related precedent from the same group, the authors introduced a novel indirect illumination technique to distinguish between thermal and non-thermal effects on the photocatalytic CO2 methanation by Rh/TiO2.142 This technique makes use of an inactive layer of Ti2O3 (a well-known photo-thermal heater) on the top of the Rh/TiO2 catalyst. This Ti2O3 layer effectively absorbs light and produces heat, displaying a similar temperature profile to the one achieved from the direct illumination of the Rh/TiO2 catalyst. Under these indirect illumination conditions, the hot carrier contribution derived from light is completely extracted, hence allowing a solid distinction between thermal and non-thermal effects on the photocatalytic rate. In 2019, Zhan et al. studied the photoelectrochemical response of plasmonic Au electrodes and quantitatively disentangled the thermal from the hot carrier effect.143 To do this, the authors separated the overall plasmonic photocurrent into a rapid response current (RRC) and a slow response current (SRC) attributable to photo-electronic and photo-thermal effects, respectively. The results suggested that both effects influenced the reaction and the different timescales for hot carrier generation and heating effect served to assess each of the contributions to the overall reaction. For a deeper discussion of the synergy between thermal and non-thermal effects on plasmon-mediated photocatalysis the readers are referred to an excellent review by Liu and Everitt.144
4. Photo-thermal applications in catalysis
4.1. CO2 conversion
The possibility of producing synthetic fuels or chemicals through the reaction between CO2 and H2O (or indirectly between CO2 and H2 derived from H2O using green energy sources) is among the most promising alternatives to achieve CO2 neutrality in transportation and the chemical industry. In general, the reduction of CO2 can produce different chemicals and fuels, CO, CHOOH, CH3OH or CH4 among others, depending on the number of electrons that come into play. Over the last decade, the use of photothermal catalysis to speed up these processes has grown exponentially.Some of the most used semiconductor oxides for the photocatalytic CO2 reduction are TiO2, ZnO, CdS, FexOy or WO3 among others. All of them share properties such as low or non-toxicity, low cost and powerful redox properties. For the application of these photocatalysts in CO2 reduction processes, these materials should present high light harvesting, low charge recombination rates and easy activation of CO2. In order to improve some of these properties, the creation of superficial oxygen vacancies is one of the most suitable and used methods, usually by means of H2-treatments. The incorporation of these O-defects improves the adsorption of reagents and can also enhance the separation of photo-generated charges.152 Additionally, most of these semiconductor oxides are stable at high temperatures, permitting their adaptation to photo-thermal reaction conditions. To improve the photo-thermocatalysis, the same semiconductors have been functionalized by different metals or metallic oxide NPs. The use of these composites improves the yield of the process by combining the individual roles of each component in the overall reaction mechanism (e.g. photo-generated charges at TiO2 under UV light + H2 adsorption in metallic NPs such as Pd) or even, by means of cooperation between them (e.g. metallic NPs can reduce the recombination of photo-generated charges of semiconductors, by transfer of e−/h+). Moreover, most of the composites are designed in order to optimize the absorption of sunlight and exploit the full solar spectrum as a sustainable energy source.
Since the 70s, TiO2 has been one of the most used semiconductors in photocatalysis.153,154 Pure photocatalytic studies using this semiconductor with oxygen vacancies as a catalyst under artificial photosynthesis conditions have demonstrated a high CH4 selectivity. For example, Yin et al. produced 16.2 μmol CH4 gcat−1 h−1 and 2.7 μmol CH4 gcat−1 h−1, with a CH4 selectivity of 79 and 73% under solar light and visible light, respectively.155 However, the introduction of thermal energy in the photocatalytic process modifies the production rates and the main product. One of the first studies evaluating the use of TiO2 in photo-thermal catalysis was published in 1997.156 TiO2 was evaluated as an “artificial leaf” for the photocatalytic reduction of CO2 with H2O. The principal results showed CO, CH4 and C2H6 as the main products. However, the most relevant result was the increase of the CH4 rate when the temperature of the system was increased from 25 to 200 °C. The authors attributed this trend to the different physical adsorption/desorption capacities of the catalyst at each temperature and not to a clear cooperation between photo and thermocatalytic mechanisms. Anyway, this study can be considered as one of the first examples of photo-thermocatalysis.
DFT studies using supported MnOx NPs on TiO2 revealed that thermocatalysis aids photocatalysis by creating oxygen vacancies and modifying CO2 surface bindings, which results in an increased CO yield.157 Wang et al. employed TiO2 nanosheets alone and TiO2 nanosheets doped with CuS, for improving the light utilization efficiency, even in the IR, due to the narrow band gap energy of CuS. The catalytic results at room temperature using different light ranges revealed that the 2% CuS/TiO2 catalyst reduced CO2 to CO as the main product and the use of UV-vis-IR light achieved the best production rate (26 μmol CO gcat−1 h−1) increasing the temperature to 138 °C.158 The use of full spectrum light and cooling to 102 °C resulted in a reduction in the CO rate by almost 4 times, evidencing the photo-thermal cooperation. Similarly, metallic NPs (Ru and Au 1 wt%) were supported over TiO2 by an impregnation–reduction method.159 The UV-visible diffuse reflectance spectra uncovered that the incorporation of Au and Ru did not modify the absorption related to the band gap of the semiconductor, although Au-incorporation enhanced the absorption profile due to the LSPR at 570 nm. This LSPR absorption was shifted to 580 nm when Ru was incorporated, revealing a Au–Ru interaction. Photo-thermal experiments were carried out in a fixed-bed reactor at different temperatures (50–150 °C). The results demonstrated that, under temperature and light illumination, H2 and CH4 were formed simultaneously by water splitting coupled to CO2 reduction, obtaining the best results at 85 °C (0.3 μmol H2 gcat−1 h−1 and 27.1 μmol CH4 gcat−1 h−1). Control experiments without the catalyst or without irradiation did not produce any reduced product, concluding that the reaction is principally driven by photocatalysis. Briefly, the photocatalysis of water splitting reaction generates H2 that is able to react with CO2 in a thermocatalytic process over Au–Ru/TiO2. Besides, the rapid consumption of this generated H2 facilitates the migration of photo-generated electrons, minimizing the recombination of photo-induced carriers (Fig. 9).
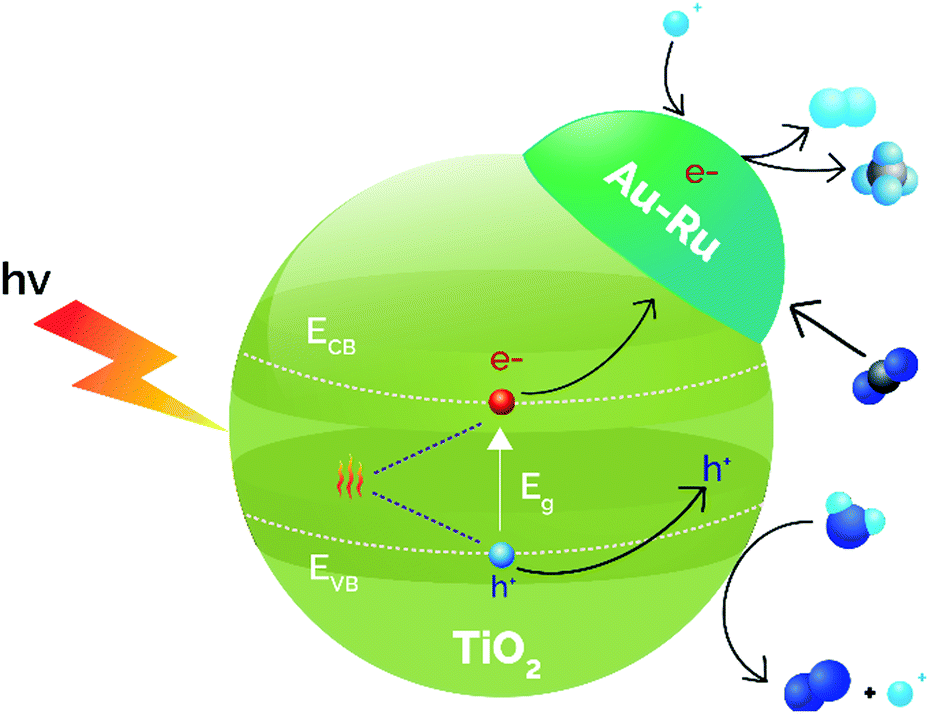 | ||
| Fig. 9 Synergistic effects between photocatalysis and thermocatalysis in the direct thermophotocatalytic reduction of CO2–H2O over Au–Ru/TiO2. Adapted with permission from ref. 159. Copyright (2017) John Wiley and Sons. | ||
As a way to create more sustainable processes, sunlight must be considered as the main energy source (photo and thermocatalytically). Solar light is the largest sustainable source of energy: renewable, clean, non-toxic and non-polluting, but also, almost 50% of the total sunlight is included in the range of visible light.160,161 This fact requires finding active photocatalysts that can efficiently harvest visible light.160 In this spirit, plasmonic metal NPs are one of the best options to enhance visible light absorption through plasmon resonance.162,163 Kumar et al. optimized a catalyst based on Au and Pt NPs on a SiO2 shell (Pt–AuNPs@SiO2) for the reduction of CO2 with H2O.164 The outcomes revealed an efficient, stable and recyclable photo-catalyst under visible-IR light at room temperature, with HCOOH as the main product along with traces of CH3OH. When the tests were carried out without temperature control, the temperature increased after 30 min from 25 to 85 °C, together with an increase of quantum yield up to 4.32%. These values confirmed the influence of the photo-thermal effect due to the irradiation of plasmonic nanomaterials using only visible and IR light. However, when comparing the formic acid yield at different irradiation wavelengths, the obtained profile was similar to the absorption spectrum of the catalyst. This equivalence is clear evidence of a mechanism directed mainly by the photo approach.
In the same direction, other studies focused on the incorporation of metallic NPs with LSPR into semiconductors, in order to generate hot charge carriers for CO2 photo-thermoreduction via plasmonic effects.74 This functionalization permits light absorption in a wide range of the spectrum, principally visible light, and also modifies the band gap of the semiconductor through the creation of a Schottky barrier. Recently, Hongjia Wang et al. studied the catalytic activity of Au/TiO2.165 This Au incorporation improved the harvest of light (LSPR absorption from Au at 550 nm) and increased about two times the CO rate (∼20 μmol gcat−1) and CH4 rate (∼4 μmol gcat−1) after 4 hours compared to the use of bare rutile-TiO2 under the full light spectrum. Moreover, the catalytic activity of Au/TiO2 under different irradiation conditions was proportional to the reached temperature: full spectrum (60 °C), IR cut-off filter (30 °C) and the one cooling down (15 °C). This similar trend suggests a temperature dependence of the catalytic process. Finally, two tests with Au/TiO2 at the same temperature were carried out using full spectra and IR cut-off irradiation + heating bath. The outcomes revealed a higher CO rate and a slightly increased CH4 rate in the full spectra experiment demonstrating that the reaction is boosted by IR (sunlight full spectra) and photo-thermal effects produced by LSPR of Au NPs.
Despite these results, the incorporation of temperature in a photocatalytic system produces some general alterations in the reaction: one benefit, the temperature enhances water splitting due to the endothermicity of this reaction; and two drawbacks, the recombination of hot carriers increases due to the thermal motion of photo-generated species and CO2 hydrogenation, being an exothermic reaction, is not thermodynamically favored. In order to minimize these drawbacks, disordered TiO2 was employed by Yu et al., reducing the charge carrier recombination and increasing the superficial oxygen defects. The disordered semiconductor was doped with Pt NPs, improving the visible light harvesting and increasing the dissociative adsorption of H2, aiding the reduction of CO2.166 The study always gave better results in the photo-thermocatalytic experiments (sunlight + 120 °C) than in photocatalytic tests (sunlight + 25 °C), giving the first evidence of the synergy between photo and thermal processes. The highest CH4 rate was achieved using Pt/disordered-TiO2 (17.1 μmol CH4 gcat−1 h−1) with a selectivity of 87.5%. Thermal H2 decomposition studies were carried out in dark conditions observing the highest H2-decomposition rate with the Pt/disordered-TiO2 catalyst, and then the best promotion of H2 splitting aiding the formation of CH4. In short, the incorporation of oxygen vacancies and disorder in the semiconductor, linked to the deposition of Pt NPs, are the most important variables to promote CH4 production, at least in the photo-thermocatalytic process.
Although TiO2 is the most studied, other semiconductors have also been evaluated for CO2 photo-thermocatalytic reduction using H2O. One of these examples is ZnO. Guo et al. evaluated initially the photocatalytic activity of CO2 artificial photosynthesis under UV and visible light at 25 and 200 °C using two different shapes of ZnO, rods and belts, alone or in layered-double-hydroxides (LDHs).167 The effects of the shape of the photocatalysts were observed and related to the electronic structure and surface defects, but the most important was the enhancement of production rates in all the evaluated photocatalysts when the temperature was increased to 200 °C. Besides, core–shell ZnO@LDHs (LDHs are 2D layered Cu–Zn–Al materials) were evaluated in the same photo-thermal conditions obtaining the best results with the belt shaped ZnO@LDHs (11.4 μmol CH4 gcat−1 h−1). The composite or heterojunction permitted an efficient transfer of photo-generated charges that minimized the recombination and also increased the photocatalyst surface area, improving the CO2 reduction. Again, the cooperation between light and heat increased the catalytic performance although the authors did not study the contribution of each of the energy sources to the overall reaction mechanism.
Other choice of a photo-thermocatalyst is the versatile, low cost, and non-toxic MoO3. This oxide was used by Li et al., tailoring it (MoO3−x) with extra superficial defects (O-vacancies).168 The introduction of O-defects extended the absorption of light permitting one to employ the whole spectrum of sunlight by means of the LSPR effect that narrowed the band gap of the material. Moreover, the increase of superficial defects improved the surface area and the available active sites, but also oxygen vacancies are responsible for decreasing the recombination of charge carriers. Catalytic tests using specific range of lights without any external heating system modified the activity as UV-vis-IR > IR > UV-vis, and also the reached temperature followed the same order from 160 to 105 °C, evidencing a high influence of thermal catalysis in the process. However, pure thermal reactions without irradiation were less active than experiments at the same temperature using light, and thus, photocatalysis played a role in the reaction too. Finally, synergistic photo-thermocatalytic artificial photosynthesis using MoO3−x produced the highest reaction rates, obtaining 41.2 μmol CO gcat−1 and 8.3 μmol CH4 gcat−1 using the full sunlight spectrum (UV-visible-IR).
Another example is WO3, a stable and non-toxic semiconductor able to harvest 12% of sunlight while offering a low conduction band that facilitates recombination. Wang et al. synthesized this semiconductor over a mesoporous template (KIT-6) and this was employed as a catalyst to reduce CO2 with H2O under visible light and/or temperature (250 °C).169 The mesoporous catalysts were pre-reduced under different temperatures controlling the amount of oxygen vacancies (related to the presence of W5+) and increasing lattice expansion and surface area compared to commercial WO3. The characterization of the diverse mesoporous WO3 (m-WO3) revealed that oxygen vacancies improved the visible-light absorption as well as narrowed the band gap. Accordingly, the higher the amount of oxygen vacancies, the higher the catalytic performance. The principal product was CH4 regardless of the used conditions; meanwhile CH3OH was formed only under thermal and photo-thermal catalysis conditions (<3 μmol CH3OH gcat−1). Oppositely, the photocatalytic route did not produce CH3OH due to the insufficient energy of hot electrons to reduce CO2 to the simplest carbon alcohol. Moreover, the different amount of oxygen defects at the catalysts using different reduction temperatures drove to different product distribution. The m-WO3 reduced at 550 °C presented the highest number of O-defects and also presented the highest CH4 production rate, specifically 0.15, 21.42 and 25.77 μmol CH4 gcat−1 after 12 h under photo, thermo and photo-thermal catalysis respectively. Examining these data, the extra 4.35 μmol CH4 gcat−1 after 12 h in the photo-thermal catalysis process compared to the thermocatalytic process was not just an addition of the other individual photo-processes. There was a clear cooperation between the two processes. The authors assigned this improvement to the fact that the temperature of the catalytic surface increased faster when the system was heated and irradiated. This intensified the photocatalytic performance because the electron excitation and relaxation were easier, increasing the production rate. Moreover, the m-WO3 reduced at 250 °C presented less oxygen vacancies and produced 6.0 μmol CH4 gcat−1 and almost 10 μmol CH3OH gcat−1 after 12 h under photo-thermal catalytic conditions. These outcomes disclosed that oxygen vacancies have an influence on the reaction mechanism and depending on the amount of these defects, the product distribution varies.
The same research group incorporated Mo NPs into the mesoporous WO3 (m-Mo/WO3), as a strategy to improve the photo-thermocatalytic performance by enhancing light harvesting, reducing the recombination of photo-generated charges and/or incorporating new catalytic active sites.170 Under the same experimental conditions, CH4 and CH3OH were the only products and the production rates were significant only when photo-thermal catalysis was carried out using m-Mo/WO3 after 12 h (5.96 μmol CH4 gcat−1 and 13.80 μmol CH3OH gcat−1). The authors reported a photo-thermal effect due to the better production rate when heating and irradiation were combined. The results highlighted how mesoporosity and Mo-doping increase the photocatalytic behavior by the reduction of charge carrier recombination due to the high crystallinity and ordered porosity together with the increase of the separation of photo-generated charges when Mo acts as cocatalyst.169 Anyway, more experiments at different temperatures or with different lights should be performed to analyze the effect of each energetic source.
Bai et al. have evaluated Bi NPs as a co-catalyst (substituting the usual noble metals) supported on the Bi4O5I2 semiconductor under sunlight irradiation.171 The study demonstrated photo-thermocatalytic reduction of CO2 without any external heating. Experiments were carried out with different lights, showing how the full spectrum (UV-vis-IR) yielded the best rates, 40.02 μmol CO gcat−1 h−1 and 7.19 μmol CH4 gcat−1 h−1. The temperature of the system using the full spectrum or just IR was close to 75 °C, although the catalytic performance was relevant only in the case of full irradiation. Finally, other tests were performed only under UV-vis light and external heating up to 70 °C, showing a higher catalytic behavior than the same experiment at room temperature. Based on these outcomes, the semiconductor was excited by UV-vis light and electrons can be transferred to the Bi surface (co-catalyst). Simultaneously, Bi NPs absorbed IR light increasing the temperature of the active sites and facilitating the reduction of CO2. In conclusion, the reduction mechanism is a photo-thermocatalytic process without external heating and using the whole range of sunlight and it is described in Fig. 10.
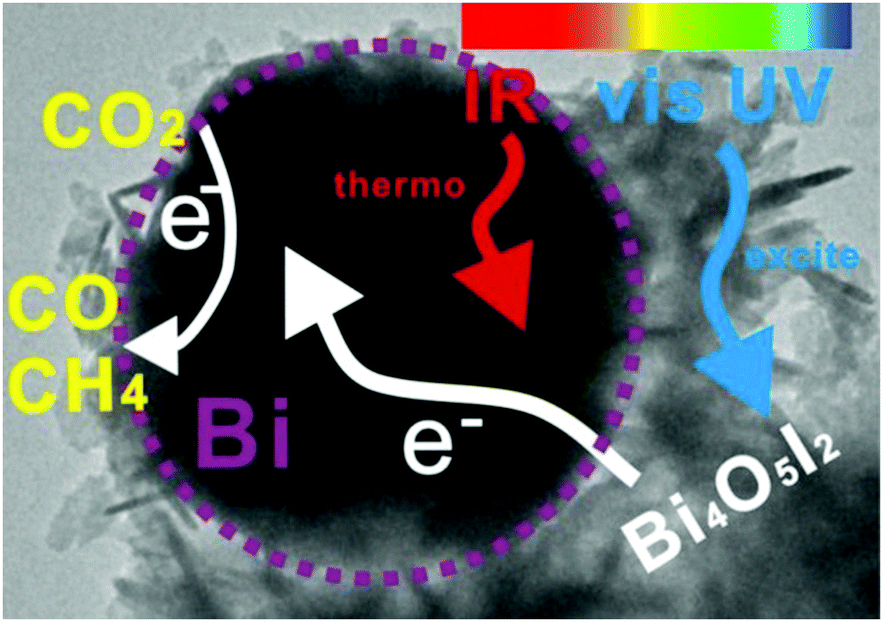 | ||
| Fig. 10 Scheme of the photo-thermocatalysis mechanism of semimetal Bi decorated Bi4O5I2 nanosheets under UV-vis-IR light. Reproduced with permission from ref. 171. Copyright (2018) Elsevier. | ||
Different research groups have studied the use of materials based on carbon or including carbon in their composition as photocatalysts for the CO2 reduction. Under UV irradiation a triple-catalyst based on carbon dots (0.9 wt%), TiO2 and Cu (0.9 wt%) was employed in the artificial photosynthesis reaction (Cu/TiO2–C) using a fixed bed flow reactor under atmospheric pressure.172 Characterization analysis determined that Cu species were dispersed homogeneously in the semiconductor, as well as carbon dots. XPS analyses highlighted carbon dots as electron savers, permitting the delivery of electrons during the reactions due to the excellent separation of charges. Regarding Cu, XPS revealed Cu(I) as a stable species during several treatments, with the stability of this species being one responsible factor for the higher activity of this triple catalyst. The catalytic tests were performed with the triple catalyst and compared to the same catalyst without carbon dots. Cu/TiO2 catalysts did not produce CH4 in dark conditions at any temperature, although with UV-irradiation the best results were 8 μmol CH4 gcat−1 h−1 at 250 °C. On the other hand, the Cu/TiO2–C catalyst showed significant thermocatalytic results at temperatures higher than 150 °C in dark conditions which demonstrates that carbon dots promoted the thermocatalysis of the reaction. Similarly, when UV light was involved, the CH4 production rate increased significantly, giving the best results of 60 μmol CH4 gcat−1 h−1 at 250 °C. Moreover, the results with UV light at low temperatures and highest temperatures in dark conditions were very similar, proving that the activity is not ruled by only the photo or thermal effect, revealing a synergetic photo-thermal effect. Finally, a specific mechanism under UV irradiation was described which included the reduction of CO2 to CO followed by hydrogenation to CH4via the *HCOO intermediate, along with a WGS reaction for the production of H2 from H2O which occurred at the same time. This reaction pathway is based on the reduction of CuO to Cu2O by electrons from TiO2 and the in situ regeneration of Cu2O using more electrons from TiO2 or the stored electrons in carbon dots, creating an excellent CuO/Cu2O cycle to ensure a continuous reaction.
In a similar approach, Xu et al. combined a traditional photocatalyst (TiO2) with graphene achieving a CH4 productivity of 26.7 μmol CH4 gcat−1 h−1 at 117 °C without any external heating.173 Further investigations should be carried out to elucidate the action of photo and thermocatalysis and to determine which one is the dominant pathway in the reaction mechanism.
Other research groups have also studied photonic crystals as photo-thermal catalysts. Low et al. prepared photonic crystals based on TiO2via an anodization-calcination method.174 The light-absorption characteristics of the catalyst showed a peak at 378 nm related to the band gap of anatase (increasing light utilization) and an absorption range between 400 and 800 nm due to the layered photonic crystal structure, allowing the use of IR light. The absorption in the IR region can produce localized surface photo-thermal (LSPT) effects raising the temperature of the catalyst. The increase of temperature should enhance the photo-conversion as well as catalytic efficiency, favoring the reactant adsorption and/or desorption. The CO2 reduction using chemically produced CO2 and H2O as reagents and irradiation with a Xe lamp produced 35 μmol CH4 h−1 m−2, which is 15 times higher than that achieved with TiO2-P25, and some CH3OH as a secondary product. The authors base this enhancement on the optical abilities of TiO2 in combination with the LSPT effect of the photonic crystalline structure, which permits absorption of heat radiation of IR light and accelerates the transfer and generation of photo-generated electron–hole pairs, in brief, improving the carbon dioxide reduction by means of photo-thermal effects. Moreover, the authors describe the concept of slow photon as another reason for the best catalytic performance. This concept is related to the position of the photonic band gap, which matches with the absorption range of TiO2 and lowers the propagation speed of light. Consequently, photons can be easily absorbed and employed for the photo-generation of hot carriers.
Perovskite-type materials are semiconductors with good stability and flexibility.175 Many of these compounds have been widely studied as photocatalysts for the production of H2 through water splitting.176–178
In 2017, Xu et al. reported a modified LaFeO3 perovskite oxide doped with Co as a catalyst for the CO2 reduction with H2O, using visible-IR light (λ > 420 nm).179 The photo-thermal catalytic activities were evaluated at 350 °C after 6 h of irradiation obtaining CH4 (437.28 μmol gcat−1) and CH3OH (13.75 μmol gcat−1) as unique products, with CH4 selectivity being higher than 96% in all cases. The results revealed that the modified-perovskite, LaCo0.6Fe0.4O3, generated 3.2 and 4.0 times more CH4 and CH3OH respectively than the unmodified perovskites (LaFeO3 or LaCoO3). This improvement is partially due to the narrower band gap of this perovskite (1.68 eV), making easier the formation of charge carriers (e−/h+). Besides, XPS analysis revealed that the substitution of Fe by Co created more oxygen vacancies, and the promotion of these defects, also adsorption sites, enhanced the catalytic behavior. Other works have evaluated similar perovskites.180 Specifically, using LaNi0.4Co0.6O3 under visible light at 350 °C it is possible to obtain 678.57 μmol CH4 gcat−1 and 20.83 μmol CH3OH gcat−1 after 6 h, the best results at artificial photosynthesis based on perovskites. As well as the previous work, the substitution of Ni by Co (x = 0.4) produced a higher amount of oxygen vacancies (67.90%), hence facilitating the adsorption and dissociation of CO2 and H2O. This substitution also modulated the positions of conduction and valence bands and the energy band gap (1.42 eV) of this perovskite to better promote both reactions involved in artificial photosynthesis. Finally, both studies shared that CH4 should be produced mainly by photocatalytic reduction of CO2, as well as water splitting. Meanwhile, CH3OH should be produced through a thermocatalytic route because the conduction band of the modified-perovskite is not negative enough to accomplish the reduction of CO2 to CH3OH. Nevertheless, the mechanisms are not completely clear and there is a lack of evidence to evaluate the synergy of photo and thermal catalysis to clear up which is the main pathway or how thermal and/or photo effects act in each step of the reaction.
Double perovskites prepared by combustion with the formula La1−xSrxCo1−yFeyO3−δ have also been studied alone (LSCF) or synthesized over a mesoporous template (3DOM-LSCF) for the artificial photosynthesis combined reaction.181 The photo-thermal reaction (UV-vis light + 350 °C) exhibited better results using both types of double perovskites than only thermocatalytic experiments, obtaining CH4 as the only product in all tests. Light absorption experiments demonstrated that these perovskites can absorb UV and visible light, photo-generating electron–hole pairs that improve the reduction of CO2 with H2O by means of photocatalysis. In accordance with the above results, the catalytic performance depends on the presence of oxygen vacancies and the smallest crystal size that increase the accessibility to active sites, with both properties being more related to thermocatalysis. On the other hand, although the band gaps of both double perovskites were very similar, the incorporation of the mesoporous structure in the 3DOM-LSCF catalyst influences the position of the conduction and valence bands, being more suitable in the case of the mesostructured material. Moreover, this specific structure also avoids the recombination of the photo-generated e−/h+, and thus, the photocatalytic process was improved. This was evidenced in the production rates which were 1.6 times higher when using 3DOM-LSCF (560 μmol CH4 gcat−1 h−1) than LSCF (350 μmol CH4 gcat−1 h−1). Concluding, this study demonstrates a photo-thermal coupling effect and both photo and thermocatalytic mechanisms are shown in the study. Nevertheless, more studies must be performed to elucidate which is the dominant mechanism.
Additionally, the use of perovskites as supports for different metallic NPs has drawn special attention. A triple catalytic system based on the CaTiO3 perovskite (70 wt%) as a semiconductor and support for Ni NPs (30 wt% NiO) and Pt NPs (1.0 wt%) has been applied as an “artificial leaf”.151 Each one of the components holds specific functions: perovskites are not only good light-harvesters but, according to CO2-TPD experiments, good CO2 adsorbents; Ni is the best metal to achieve the direct hydrogenation of CO2 to CH4, and also interacts with the perovskite generating oxygen vacancies demonstrated by XPS characterization; and finally small amounts of Pt lend efficient photo and thermal H2 activation, avoid the recombination of photo-generated charges and improve the formation of hot charges and local heating by means of LSPR. This work reveals unique reaction conditions because the authors employed both H2 and H2O as an effective H2 source or reducing agent (CO2:H2:H2O ratio of 1:2:2). Moreover, the catalysts were irradiated with a UV lamp in the temperature range of 40–240 °C in a fixed bed quartz reactor, and four catalytic conditions were tested modifying the use of the reduced or not-reduced catalyst and the energy sources (photo, thermo and photo-thermo). The photo-thermal results of the reduced catalyst showed the best catalytic performance at 180 °C reaching 46.48% CO2 conversion and 99.5% CH4 selectivity (35 μL CH4). Compared with thermal-methanation in the literature for which at least 200 °C is necessary, the photo-thermal system mixing H2O and H2 boosts the reaction at lower temperatures. Otherwise, the photo-results after 10 hours (not fixed reactor) displayed very low CO2 conversion although again the catalysts with Ni and Pt raised CH4 selectivity close to 100%. With respect to the thermal system without any irradiation, it showed similar trends to the photo-thermal system but with a lower production rate (26 μL CH4) and similar conversions (44.5%). Finally, the photo-thermal study was repeated with the non-reduced catalysts presenting the lowest results when heating is involved. This unlikeness is related to the better H2 adsorption in metallic components or alloys and the lesser amount of oxygen vacancies in perovskites that are usually promoted in the presence of metallic NPs. Summarizing, cooperation between photo and thermal approaches allows a successful catalytic reduction of CO2 using a mixture of H2 and H2O as a reducing agent.
As it has already been mentioned in the previous sections of this review, to optimize photo-thermal performance, photocatalysts should display a strong light absorption across the solar spectrum, low charge-carrier recombination rates and a good capability for heat transfer and/or generation. In the particular case of CO2 reduction, a high adsorption of CO2 is also desirable. A huge variety of catalysts have been studied with very diverse properties that permit tuning the products of the reaction or even working at different temperatures and with different ranges of light. In the specific case of artificial photosynthesis, two reactions must be carried out simultaneously, complicating the search of an active, stable, inexpensive and environmentally friendly catalyst.182 It seems that perovskite supports combined with plasmonic nanostructures could be especially convenient composite materials to perform efficient photo-thermal CO2 conversion.
+ H2), which via Fischer–Tropsch synthesis allows for the synthesis of important hydrocarbons, such as alkanes, olefins, alcohols, dimethyl ether or gasoline.183,184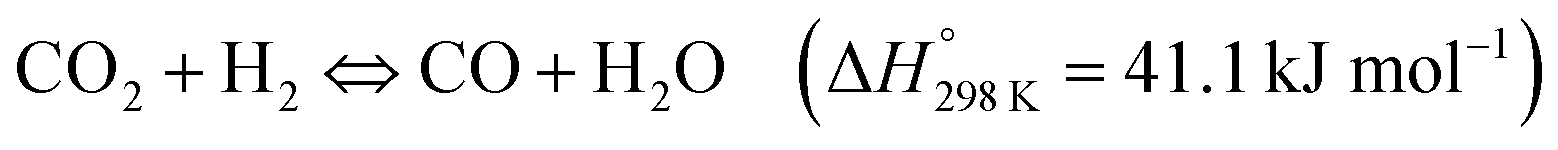 | (4) |
Ozin et al. described hydride-terminated silicon nanocrystals as potential photo-thermal catalysts for the reduction of CO2 to CO, according to the thermal and optical properties of silicon materials. Only-CO2 experiments using a metal halide lamp as a unique energy source at fixed 150 °C produced 4.5 μmol CO gcat−1 h−1, but the reaction rate decreased after each run.186
Many more examples can be found in the literature on the production of CO using H2 as a reducing agent. The most interesting works involve catalysts based on the incorporation of metals on photoactive supports or semiconductors. Many works have studied the efficiency of Au NPs of different shapes and sizes in different photocatalytic reactions.187–189 Using a Au/ZnO catalyst with a characteristic plasmon resonance around 538 nm, the authors were able to produce CO (4.22 μmol CO gcat−1 h−1) and small amounts of CH4 after 5 hours of irradiation with a continuous wave 532 nm laser, reaching 600 °C in a gas-tight photocatalysis cell with a H2:CO2 ratio of 3:1.103 Thermal catalytic tests in dark conditions revealed a different temperature profile than that under irradiation, offering evidence of photo-generated heat that improved the catalytic performance. Other catalysts based on Au NPs supported on TiO2, CeO2 and Al2O3 by deposition–precipitation have been evaluated in photo-thermal CO2 reduction.190 The use of Au/TiO2 with a metal loading of 1.4 wt% resulted in a CO2 consumption rate of 2.7 mmol gcat−1 h−1 at 400 °C under visible light irradiation. Lu et al. described a photocatalytic deposition method to produce Au/CeO2 nanorods.191 CO2 reduction under thermal conditions was not significant (<4% CO2 conversion). Nevertheless, the photo-thermal experiments achieving the same temperature as the thermal reactions just with irradiation (varying intensity) were much more active.
Another example is the use of Pd NPs below 10 nm size supported on the nanorods of Nb2O5 semiconductor.34 This catalyst was studied to elucidate the photo-thermocatalytic reduction of CO2, using a batch reactor pressurized up to 1.8 atm with a CO2:H2 ratio of 1, during 3 h under three different situations: no light and no heating, just irradiation with a Xe lamp and 160 °C without irradiation. In this way, the best results were obtained under light conditions (4.9 mmol CO gcat−1 h−1) followed by the externally heated system (1 mmol CO gcat−1 h−1); meanwhile no significant CO2 conversion was observed when neither light nor temperature was used. Focusing on the photocatalytic conditions, several experiments were carried out cutting-off the light by filters. The results demonstrated a clear influence of the range of irradiation; for instance, the CO reaction rate decreased until 1.8 mmol CO gcat−1 h−1, when UV-light was cut-off (only visible and IR light). According to the energy band gap of the semiconductor and the LSPR of Pd NPs, photoexcitation was not possible without UV-light. Therefore, the photons from visible and IR, not energetic enough to produce charge carriers, provided just thermal energy to the system by means of intra or interband transitions and non-radiative relaxation. In other words, when using visible and IR light, CO2 reduction to CO was possible by means of a photo-induced thermocatalysis. The most important conclusion was obtained when the authors compared the influence of both wavelength and light intensity. Both sets of experiments resulted in approximately the same exponential profile (Fig. 11), evidencing that the photocatalytic pathway was not the dominating one.
 | ||
| Fig. 11 Photo-thermal catalytic performance of the nanostructured Pd@Nb2O5 samples. The RWGS reaction rates plotted as a function of absorbed power for the series of batch reactions with different cut-off filters A–F (red line) and diverse intensities I–V (black line). Reproduced with permission from ref. 34. Copyright (2016) John Wiley and Sons. | ||
Using the same catalyst and conditions, the selectivity towards RWGS (CO) or Sabatier reaction (CH4) was tuned by varying the Pd loading.132 The 0.1% Pd/Nb2O5 catalyst produced the highest CO production rate (18.8 mol CO gPd−1 h−1) with a CO selectivity of 99.5%, meanwhile 10% Pd/Nb2O5 significantly increased the CH4 selectivity by 24 times, decreasing the CO production rate (∼1 mol CO gPd−1 h−1). The results normalized by Pd content pointed out an influence of the Pd-NP size on the activity and selectivity of the studied reaction; higher NP sizes produced higher CH4. Furthermore, different amounts of 10% Pd/Nb2O5 were evaluated obtaining a dependence between the mass of the catalyst and the CH4 production, and thus it was confirmed that selectivity was not related to just photocatalysis, but photo-thermal effects seemed to drive the reaction (light-to-heat). Ultimately, the characterization of the catalysts also demonstrated that oxygen vacancies and surface reduced Nb species are formed during the photo-thermal process by light irradiation, and this could improve the visible light absorption, avoid the recombination of e−/h+ and besides, increase the CO2 affinity.
Other metals with well-known hydrogenation properties have been analyzed. Anatase-TiO2 and γ-Al2O3 were doped with Pt (2 wt%) to obtain two similar catalysts for the reduction of CO2 with H2 using a ratio of 1:6.192 The experiments were carried out at different temperatures from 80 to 400 °C, under dark and irradiation conditions (Schott KL 2500 LCD visible-light). For both catalysts, the addition of light irradiation (photo-thermal process) promoted CO2 conversion and increased the CO production rate at every temperature, proving the photo-thermal effect. Specifically, for Pt/Al2O3, the increase was 9 times higher at 80 °C when the system was illuminated. However, better results were obtained with Pt/TiO2 (110 μmol CO gcat−1 h−1 at 400 °C) probably due to the strong interaction in Pt-TiO2. According to the presence of other products such as CH3OH, CHOOH or CH4, thermodynamic studies and operando DRIFT analysis, a possible mechanism was described based on thermocatalysis using Pt/Al2O3. Consequently, CO desorption was found to be the limiting step, which was enhanced by both heating and light irradiation, weakening the Pt–CO interaction.
Ru catalysts have also been described as efficient photo-thermal materials for CO2 reduction. Through an ion-exchange method Ru was incorporated in a Mg(OH)2 support with a three-dimensional nanoflower structure followed by 150 °C H2-reduction to create the metallic Ru.193 Using a batch reactor at atmospheric pressure irradiated with a Xe arc lamp (300–2500 nm and 18 suns) and a mixture of CO2 and H2 (1:1), the best CO2 conversion rate was 545 mmol gRu−1 h−1 using the 8.3% Ru/Mg(OH)2 catalyst. A clear exponential dependence between light intensity and photocatalytic activity was demonstrated (1413 mmol gRu−1 h−1 at 20 suns), therefore suggesting that thermocatalysis rules the photo-thermal reaction. Anyway, deeper investigations must be done to explain the photo-thermal effects and mechanisms but it is obvious that a suitable amount and dispersion of Ru increase the light absorption and can modify the product distribution, defining these variables as key factors in photo-thermal CO2 reduction.
The use of noble metals entails a significant cost. In order to reduce the price of the catalysts, non-noble transition metals have also been studied as part of catalysts in photo-thermocatalytic reactions as a more economical option. Ning et al. synthesized a hybrid carbon-based catalyst with embedded Co NPs together with dispersed Co–N species, denoted as Co@CoN & C.194 The reactions were carried out at 55 kPa, using an equal amount of CO2 and H2 (1:1) and a Xe lamp in a batch reactor. Initially, the photoinduced thermal-effect was demonstrated as evidenced by the increase of temperature of the catalyst surface during the reaction, which reached 518 °C, while the plain Co NP system increased only up to 300 °C. After 30 min of irradiation, the optimized catalyst (ratio of CN:Co(OH)2 of 1) showed 41% CO2 conversion and a CO selectivity of 91% (132 mmol CO gcat−1 h−1), meanwhile the unsupported Co NPs showed 43% CO selectivity. The high reaction rate of the hybrid catalyst was attributed to the significant photo-thermal effect, and the CO selectivity was improved by the special characteristics provided by the Co–N shell in comparison with plain Co NPs.
The utilization of metal organic frameworks (MOFs) as a sacrificing template for the manufacture of catalysts has also been applied in photo-thermal catalysis. A core–shell catalyst (Fe@C) was prepared by a double step pyrolysis of MIL-101(Fe).195 This catalyst was irradiated in a batch reactor using a Xe lamp and a CO2:H2 molar ratio of 1:1, presenting a CO2 reaction rate of 26.12 mmol gcat−1 h−1 with almost 100% CO selectivity. Meanwhile, other catalysts based on naked Fe NPs (Fe/SiO2 and Fe/CNT) also presented catalytic activity but never as active as Fe@C. Moreover, the selectivity to CO was a bit lower for the naked Fe-catalysts and for longer reaction times total selectivity was reduced, improving the selectivity to CH4 and even hydrocarbons such as C2H4, C2H6, C3H6 or C3H8. The same catalysts were employed under identical conditions in a fixed-bed reactor, producing 55.75 mmol CO min−1 as the highest reaction rate using Fe@C. These photo-thermal experiments presented the best results compared to traditional photocatalytic and thermocatalytic experiments, an evidence of photo-thermal effects. In addition, catalytic tests at diverse wavelengths showed optimum results using UV light, probably due to the plasmonic activity of Fe NPs. According to the characteristics of Fe@C and diverse catalytic outcomes, the absorption of light by Fe (LSPR) created local heating which was able to activate CO2 at high temperatures. It should be remarked that when carbon materials are used as supports, it is very important to demonstrate that all carbon products are derived from the fed CO2 and not from the direct hydrogenation of the support.
The hydrogenation of In2O3 at 400 °C results in the formation of oxygen vacancies (In2O3−x/In2O3),196 and in a heterostructure with a lower bandgap (2.36 eV) that improves solar light harvesting and absorption. This photo-enhancement (In2O3−x/In2O3 catalyst) was correlated to the best catalytic performance via RWGS, obtaining CO as the only product (19.64 μmol CO gcat−1 h−1) in a batch reactor system irradiated with a Xe lamp (H2:CO2 of 1). The increase of the temperature up to 160 °C under light irradiation evidenced a photo-thermal effect. Moreover, the authors evaluated the catalysts using a gas-phase flow reactor irradiated with the same lamp and with external heating. Compared to dark tests, the photo-experiments presented a 417% greater CO rate at 300 °C. Other researchers have studied the same semiconductor but using novel synthesis methods to obtain ultrathin 2D black In2O3−x nanosheets with photoinduced oxygen vacancies.102 This catalyst was compared to traditional In systems (bulk, hydroxide, defective oxide). The 2D structure improved the photoactivity (more electrons were trapped at oxygen defects) and the oxygen vacancies activated CO2 molecules providing more active sites for the photo-thermal CO2 reduction.
The good results of modified In2O3 as a semiconductor in photocatalytic studies motivated Hoch et al. to impregnate silicon nanowires (NWs) with In2O3−x(OH)x NPs. This catalyst was employed to reduce CO2 to CO at a rate of 22 μmol gcat−1 h−1 under irradiation with a Xe lamp (20 suns) without external heating.197 The same experiments using only λ > 495 nm (photons without the possibility to create charge separation in In2O3−x(OH)x) presented a CO rate 20 times lower. Most of the photons with wavelengths higher than 495 nm did not participate in the reaction in a radiative way. Instead, these photons were trapped by SiNWs and increased the temperature, facilitating the release of produced H2O and improving the thermocatalytic reaction. On the other hand, more energetic photons (<495 nm) contributed to the photocatalytic reduction of CO2 to CO.
Hydride-terminated silicon nanocrystals were also studied as catalysts for the reduction of CO2 using H2.186 Comparing experiments under dark and light conditions at the same temperature, the CO production rate is higher when the catalytic system is heated and irradiated (∼240 μmol CO gcat−1 h−1).
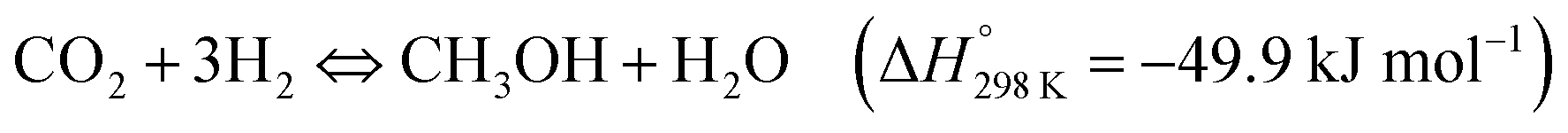 | (5) |
:3 CO2:H2 mixtures at 30 bar under irradiation (Xe lamp) in batch mode.36 While the Au nanocages did not show any conversion, Pt nanocubes after 3 hours produced small amounts of CH3OH at 150 °C with high selectivity (96%), both under dark and under illumination. The incorporation of Pt-nanocubes in ZIF-8 increased slightly the methanol production up to 0.7 mmol CH3OH, with no difference between dark and illumination conditions. In contrast, the 3-component catalyst consisting of Pt@Au@ZIF-8 showed an enhanced methanol productivity under light irradiation. It was concluded that (i) Pt nanocubes act as active sites for the transformation of CO2 to CH3OH; (ii) the addition of Au results in localized heating of the active sites by plasmonic photo-thermal effects (IR adsorption), and (iii) ZIF-8 acts as a heat insulator.
Pd–Zn catalysts have also been tested under photo-thermal conditions.201 Experiments at different temperatures (190–270 °C) using a 3:1 H2:CO2 ratio presented the best CO2 conversion and CH3OH production rates when the system was irradiated with UV light (Hg lamp). More importantly, the use of only visible light also resulted in an important enhancement in catalytic activity compared to experiments performed in the dark. Diffuse reflectance spectroscopy confirmed a shift to the visible range of the LSPR of Pd when alloyed with Zn. In view of these results, the authors speculate that hot-electrons generated upon light irradiation migrate to the active sites (Pd–Zn alloy), increasing in this way the electron density and therefore catalytic activity via electron injection to the antibonding orbital of CO2. We find that these results are very encouraging.
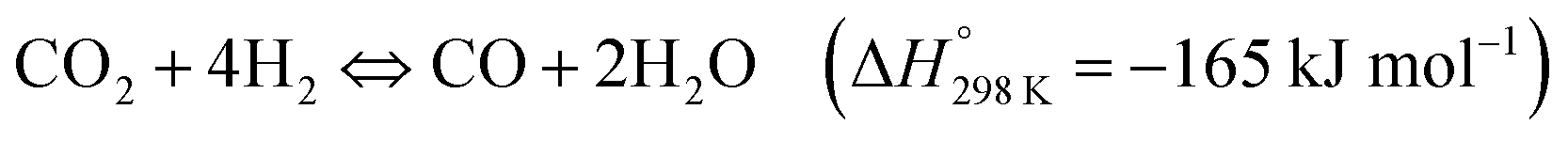 | (6) |
Incorporation of Ni in Y2O3 nanosheets allowed CO2 methanation under sunlight.207 Under 1 sun irradiation the catalyst with Ni NPs did not present good catalytic results (11% CO2 conversion and total CH4 selectivity), probably because the catalytic system reached only 78 °C temperature, not enough for the activation of the Sabatier reaction. To improve this catalytic activity, Ni was incorporated as single Ni atom species instead of Ni NPs, achieving 87% CO2 conversion and a temperature up to 240 °C. Finally, in order to obtain a photo-thermal catalytic system, the single atom Ni/Y2O3 catalyst was covered by a selective light absorber (AlNx/Al). This new system permitted elevating the temperature of the catalysts to 288 °C under just irradiation of 1 sun describing a reaction mechanism mainly driven by thermocatalytic effects. Using natural sunlight, a CH4 production rate of 7.5 L m−2 h−1 could be achieved (Fig. 12).
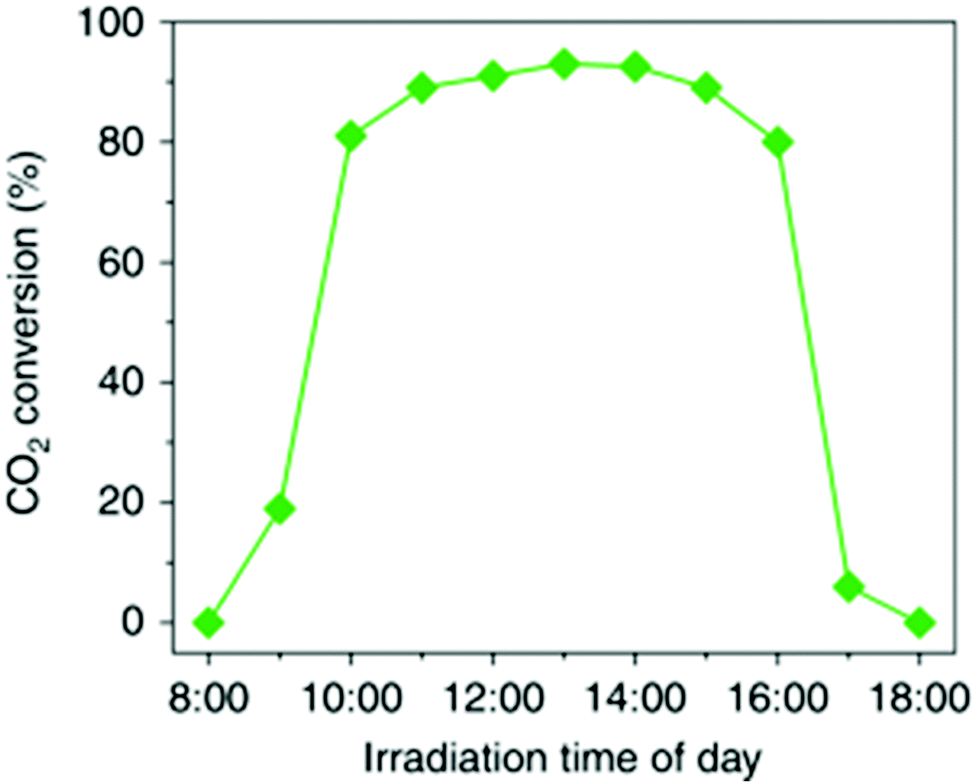 | ||
| Fig. 12 Photo-thermal CO2 conversion using SA Ni/Y2O3 nanosheets with a selective light absorber-assisted photo-thermal system as a function of time. Experiment carried out on June 30, 2018 from 8:00 to 18:00 in Baoding, Hebei, China. Reproduced with permission from ref. 207. Copyright (2019) Springer Nature. | ||
Since 1987, different researchers have employed Ru-based catalysts for photo and thermocatalytic CO2 reduction.32,208–210 Incorporation of Ru in TiO2, according to Wang et al., can narrow the energy band gap of the semiconductor (2.69 eV).211 Diverse catalytic tests were carrying out in a fixed bed reactor with a CO2:H2 ratio of 1:3. However, catalytic tests using sunlight (equivalent to 1 sun) at 150 °C did not show any difference with experiments performed in the dark at the same temperature. On the contrary, when increasing the temperature to 300 °C, the catalytic activity was three times higher under light illumination (69 mmol CH4 gcat−1 h−1) than in dark conditions. Nitrogen doping of TiO2 in similar systems can further enhance the photo-thermal behavior.212
Multimetallic systems such as Ru and Au supported on TiO2 have also been studied,159 once again confirming that the addition of Au greatly improves heat effects.108
When it comes to layered double hydroxides (LDHs) as catalytic supports,213,214 Ren et al. supported Ru NPs on ultrathin Mg–Al LDHs and tested the resulting catalysts under flow and UV illumination (Xe lamp).215 Under these conditions, the photo-thermal catalysts could reach up to 350 °C and showed similar activities to those obtained when the reaction is performed in the dark at the same temperature, highlighting the important thermal and the negligible photo effect for these specific catalysts. In the same spirit, Ru has been supported on many other semi-conductors such as silicon nanowires,32 silica opal and inverted silicon opal photonic crystals,203 planar silicon wafers26 or strontium titanate.216
Similar photo-thermal effects have been found when using Rh as the active phase and different semiconductors as supports,217 with very interesting studies on the effect of light intensity on catalyst activity (Fig. 13).141
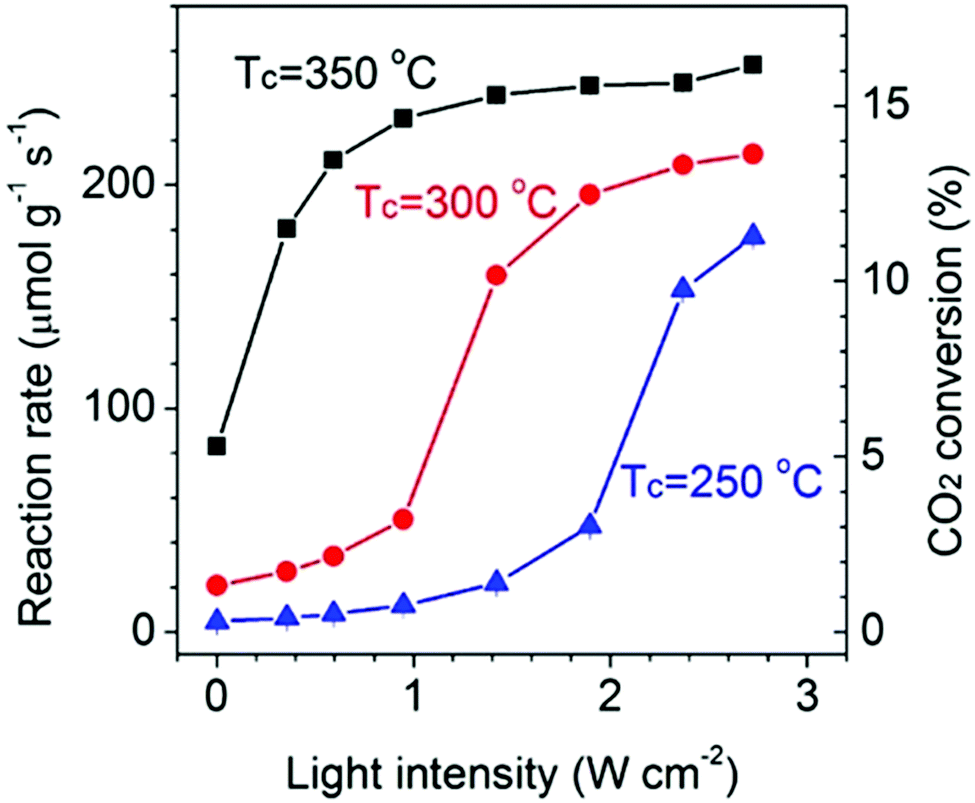 | ||
| Fig. 13 Total reaction rate and CO2 conversion at 350 °C (black squares), 300 °C (red circles) and 250 °C (blue triangles) as a function of light intensity. Reproduced with permission from ref. 141. Copyright (2018) American Chemical Society. | ||
As it can be concluded from the above, group VIII metals have attracted a great deal of attention. This is mostly due to an effective absorption in the full range of the solar spectrum (covering UV, visible and IR light) together with a high H2 activation ability. In a comparative study, Ye et al. impregnated an inert Al2O3 with 2–3 wt% group VIII metals (Ru, Rh, Ni, Co, Pd, Pt, Ir and Fe) and studied the catalytic properties of the resulting catalysts in a batch reactor with a gas mixture of CO2 and H2 (1:4) under Xe lamp irradiation.110 As a result of the strong increase in temperature (going up to 300–400 °C), CH4 production rates measured under these conditions were six orders of magnitude higher than under only photocatalytic conditions, highlighting, once again, the importance of photo-thermal processes in the final outcome of the reaction. Further studies on similar metals supported on SiO2218 revealed a linear relation between light intensity and CO2 conversion rate, demonstrating that the reaction is driven by hot electrons from the metal.
:H2 ratios close to 1) into longer hydrocarbons, is carried out at 20–30 bar and moderate temperature.
Surprisingly, following a photo-thermal approach, several groups have been able to demonstrate the formation of C2+ hydrocarbons from CO2:H2 mixtures at atmospheric pressure. In 2018, Zhang and co-workers, using an alumina-supported CoFe alloy reduced at 650 °C, were able to achieve 37% C2+ selectivity and a total CO2 conversion of 78.6% under irradiation with a Xe lamp at 5.2 W cm−2 irradiance and with no external heating.219 In order to elucidate if pure photochemical or thermal effects drove the CO2 hydrogenation, the authors monitored the temperature of the catalyst using a thermocouple in intimate contact with the catalyst bed. According to these experiments, under UV-light irradiation, the catalyst achieved temperatures as high as 310 °C, while in the absence of catalyst the reactor temperature was only 120 °C, proving the existence of a strong photo-thermal effect. To gain further evidence of this photo-thermal effect, CO2 conversion was also monitored as a function of temperature under both light and dark conditions. Interestingly, CO2 conversion profiles were almost identical in both cases, proving that the reaction was driven photo-thermally rather than photochemically. Computational calculations showed that the CoFe bimetallic alloy promoted C–C coupling reactions during CO2 hydrogenation, thus explaining the higher selectivity towards C2+ products.
More recently, Garcia and Corma reported a sodium-promoted Co@C (Na–Co@C) nanocomposite for the photo-thermal hydrogenation of CO2 to ethanol and C2+ hydrocarbons.220 Using this catalyst under photo-thermal conditions (235 °C and 24 kW m−2 power irradiance), the selectivity to ethanol and C2+ hydrocarbons was 7 and 36%, respectively, at a CO2 conversion of 37%. A blank experiment under dark conditions at the same temperature and CO2 conversion led to markedly lower selectivity, thus indicating that light irradiation had a strong influence on product selectivity. In order to further demonstrate the role of light in the catalytic performance, the authors conducted wavelength-dependent studies using monochromatic light. The results showed that UV light was mainly responsible for CO2 hydrogenation, and photo-action experiments had a clear match with the absorption spectrum of the material. Ultimately, this was clear evidence that the reaction mechanism was driven predominantly by the photo-generation of charge carriers. Additional near-ambient pressure X-ray photoelectron spectroscopy (AP-XPS) and in situ Raman mechanistic studies not only demonstrated that photo-induced electrons on Na–Co@C promoted CO2 dissociation to CO, but also that light irradiation stabilized CO on the catalyst surface, therefore contributing to a higher selectivity towards ethanol by a CO insertion mechanism. Furthermore, the authors also observed that UV light had an evident effect on the chain growth process according to the different surface intermediates compared to dark experiments, a fact that explains the difference in the reported selectivity. Altogether, these results evidenced the potential of light to selectively activate different reaction pathways in CO2 hydrogenation.
4.2. Fischer–Tropsch chemistry
In 2015, two research groups reported interesting results in the photo-thermally catalyzed Fischer–Tropsch (PFT) process.221,222 Guo and co-workers, using a worm-like ruthenium nanostructure dispersed on graphene sheets, reported the efficient formation of C20+ hydrocarbons from CO:H2 (1:1) mixtures at 20 bar, under irradiation from 400 nm to 800 nm using a 300 W Xe lamp.221 The catalytic activity in these conditions is 14.4 mol CO molRu−1 h−1, while the dark reaction, i.e. conducted without light irradiation, shows a halved activity value, 7.8 mol CO molRu−1 h−1. Moreover, this last value is comparable with the one reported by Kou and co-workers for another Ru nanocluster-based catalyst applied in FT synthesis, and this confirms that light irradiation plays indeed a key role.223 Guo's group attributes these improved FT reaction features to the formation of hot electrons in graphene; these high energy electrons can transfer to Ru, which also generates energetic electrons due to light absorption and initiates the process by activating CO. The authors confirmed this hypothesis proving distinctive features of the hot-electron mechanism: a linear relationship between reaction rate and light intensity, as shown in Fig. 14a, a lower activation energy under light irradiation compared to dark conditions, as shown in Fig. 14b, and a correlation between catalytic activity and irradiation wavelength, as shown in Fig. 14c and d.
 | ||
| Fig. 14 (a) Dependence of the photocatalytic activity of Ru/graphene on light intensity. (b) Arrhenius plots of photo-thermal Fischer–Tropsch reactions under light and dark conditions. (c) Dependence of the photocatalytic activity of Ru/graphene on wavelength regions. (d) Dependence of the photocatalytic activity of Ru/graphene on specific wavelength. Reproduced with permission from ref. 221. Copyright (2015) American Chemical Society. | ||
Yu et al. presented a series of iron-based catalysts supported on mesoporous titania (TiO2) at different Fe/Ti ratios, and they tested the catalytic performance of these materials in the photo-thermal Fischer–Tropsch process to convert coal to FT products.222 After a thorough characterization of five catalytic systems having 5%, 10%, 15%, 20%, and 30% Fe/Ti ratio, the researchers performed PFT tests at atmospheric pressure, at a temperature of 220 °C and with a H2/CO ratio of 1:2. Although the selectivity results did not follow a clear trend in correspondence with the amount of Fe in the catalyst, and the authors did not report a clear reaction mechanism, the study shows that the investigated catalytic systems are active in PFT and that light irradiation allows one to carry out PFT synthesis using mild conditions.
In recent years, more research groups have put their efforts to investigate deeper this topic; specifically they have tried to address the problem of increasing the selectivity towards light olefins. Su and co-workers designed a catalytic system combining a thermo-active part and a photosensitive support.224 They prepared via impregnation two TiO2 nanotube supported Co catalysts (20% and 30% Co/TiO2), and studied them for the FT reaction (CO/H2 = 1:2, pressure 20 bar) under photo-thermal conditions using UV light, thermal only condition and UV irradiation only condition. The results were compared after 40 hours on stream, and the data clearly showed that UV irradiation alone is not enough to overcome the energy barrier to catalyze CO hydrogenation. The 20% Co/TiO2 catalyst shows the most interesting result in terms of CO conversion; in fact, although the CO conversion value for the thermocatalytic process is 9.2%, it increases significantly to 63.9% in the presence of UV irradiation under the same temperature and pressure conditions. As Guo et al. already proposed, the support, graphene in their work and TiO2 in Su and co-workers’ paper, plays a key role as a photococatalyst.221 As a consequence of UV light irradiation, hot carriers (electron–hole pairs) are generated in TiO2: electrons can be collected on Co surface sites and transferred to CO to initiate the dissociation of C–O bonds; holes can be intercepted by the available reduced species. To prove that UV irradiation is the reason for the higher CO conversion because it activates CO dissociation, Su's group performed in situ Raman spectroscopy. This technique confirmed the adsorption of CO on the metal center and consequently the weakening of the C–O bond. Furthermore, for both catalysts the product distribution showed that the CO2 selectivity decreases under photo-thermal conditions. As far as the hydrocarbon selectivity is concerned, the quantity of CH4 is enhanced under UV light, due to the photohydrogenolysis of hydrocarbons with longer chains and the formation of hydrogen atoms from H2 dissociation. At the same time, in the C2–C4 fraction the photo-thermal process shifts the selectivity towards alkanes with a remarkable value of 98.6% versus 89.0% for the dark reaction. Overall, Su and co-workers also showed that the same CO conversion obtained with conventional thermo catalysis at 300 °C could be achieved at 220 °C using photo-thermal catalysis, when performing a more environmentally friendly Fischer–Tropsch synthesis.
In the same year Zhang and co-workers published a paper in which they investigated the photo-thermal Fischer–Tropsch process using a cobalt based catalytic system.225 They fabricated a series of catalysts consisting of Co and Co3O4 NPs supported on zinc oxide–alumina (ZnO–Al2O3), and they used them for the photo-thermal Fischer–Tropsch to olefins (FTO) synthesis. A detailed characterization of the obtained materials showed that, depending on the reduction temperature at which the catalysts are prepared, the presence at the surface of cobalt oxides and metallic cobalt can be tuned. The different catalysts were tested under the same reaction conditions: no external heating, 1.8 bar, 300 W Xe light (200–800 nm), 1 hour irradiation time, and the outcomes showed that the different surface composition strongly influences the CO conversion as well as the product selectivity. In particular, the most notable results were achieved using the catalyst prepared when the reduction temperature was 450 °C (Co-450), yielding 15.4% CO conversion and good olefins selectivity (36%) with a olefins to paraffins ratio of 6.1. In order to have a deeper insight into the process occurring, Zhang's group performed catalytic tests over Co-450 in different conditions: by monitoring the temperature under UV-irradiation; by using electric heating only; and by controlling light irradiation with monochromatic filters. Overall, they proved that the photo-thermal effect is the driving phenomenon for CO hydrogenation over this new cobalt-based catalyst rather than a pure photochemical route.
Another research group in 2018 investigated photo-thermal Fischer–Tropsch as a way to convert syngas into lower olefins. Ma and co-workers designed an iron-based catalyst, Fe5C2, whose surface was spontaneously decorated with oxygen atoms in situ when irradiated with UV light.226 After preparation, the catalyst was analyzed via energy dispersive X-ray (EDX) spectroscopy and powder X-ray diffraction (PXRD) to determine the elemental distribution and the phase purity. The researchers tested Fe5C2 as a catalyst for PFT performed at atmospheric pressure, molar ratio of CO/H2 = 1:2, 300 W Xe light (200–1100 nm), 0.5 hour irradiation time, and the results were quite surprising: CO conversion was 49.5%, selectivity toward CO2 was 18.9%, selectivity toward C2–4 olefins was 55.5 and selectivity toward C2–4 paraffins was 5.1% (o/p ratio 10.9). The temperature of the reaction was also monitored, and the catalyst bed reached 200 °C after 60 seconds under irradiation, while after 28 minutes the temperature rose to 490 °C. This behavior, supported by 3D excitation emission photoluminescence analysis, shows that the catalytic system undergoes photo-induced heat conversion, corroborating the hypothesis of the photo-thermal catalytic route. Additional investigations were performed to understand the reason behind the promoted production of olefins. X-ray photoelectron spectroscopy performed on fresh and used catalysts showed that photoirradiation leads to the formation of iron oxide on the Fe5C2 surface, in a significantly minor amount compared to the formation of iron oxide observed in the thermal FT process. This finding, integrated with density functional theory (DFT) calculations on Fe5C2 and oxygen-decorated Fe5C2, allowed Gao et al. to correlate the presence of a suitable amount of oxygen on the catalyst surface with the favorable production of olefins.
Zhang's research group expanded the investigation field on the PFT process and reported a new class of Co-based catalysts prepared via hydrogen reduction performed at different temperatures (from 300 °C until 700 °C) of CoAl layered-double-hydroxide (LDH) nanosheets.227 These materials perform better than those containing the same metal that the research group presented in a previous paper discussed above.225 The different catalysts were tested for CO hydrogenation at 1.8 bar and irradiation with a 300 W Xe light (200–800 nm) for 1 hour, without external heating, and an improving trend was observed with the increase of the reduction temperature used for the preparation of the catalyst. The material prepared at 700 °C, namely Co-700, gave the best catalytic performance yielding a CO conversion of 35.4% and a selectivity to higher hydrocarbons of 65%; therefore it was selected for additional catalytic tests using different conditions in order to optimize the procedure. Finally, Li et al. conducted a series of tests modifying the light source while monitoring the temperature, and they were able to prove that the investigated CO hydrogenation is a process driven by a photo-thermal route rather than a non-thermal photoexcitation process.
Very recently another paper from Zhang and co-workers was published, in which they reported the use of an unusual metal for the Fischer–Tropsch process: nickel.228,229 Wang et al. fabricated a novel class of catalysts, consisting of Ni NPs supported on manganese oxide (MnO), labelled Ni-x, where “x” represents the temperature at which the Ni–Mn mixed metal oxide was reduced to obtain the target materials. This metal–support combination was a winning strategy to optimize the hydrogenation properties of Ni to make it suitable for light olefins production via PFT synthesis, performed at 1.8 bar and irradiation with a 300 W Xe light (200–800 nm) for 1 hour, without external heating. Particularly, the performance of Ni-500 was the best compared to the other catalysts of the series. Ni-500 showed the highest value of selectivity to light olefins (33.0%) and a really low CO2 selectivity (0.2%). The Zhang group performed catalytic tests monitoring the temperature under UV irradiation, as well as dark tests with direct heating, and they confirmed that the studied Fischer–Tropsch process follows a photo-thermal route. Furthermore, they were able to clarify the role of MnO and Ni in the process. Ni K-edge X-ray-absorption near edge spectroscopy (XANES) showed that the properties of the support change the electronic features of metallic Ni NPs, and X-ray photoelectron spectroscopy (XPS) proved that charge transfer from MnO to Ni NPs occurs. These phenomena reduce the hydrogenation activity of Ni, and clarify the unexpectedly high selectivity of Ni-500 towards olefins.
4.3. Methane activation
In the last years, scientists have made significant efforts to develop new catalytic approaches for one of the biggest challenges in the chemical community: performing methane conversion to upgraded fuels and commodity chemicals under milder conditions. Photo-thermal-driven methane activation is a recent strategy that several research groups are exploring, because it allows exploring new routes, which require lower activation energies and, consequently, a more environmentally friendly approach.In 2019, two interesting reviews were published about sustainable conversion of methane driven by solar energy, and about the role of solar energy in obtaining fuels from catalytic C1 chemistry.230,231 Both publications contain a substantial section that revises a series of photo-thermal catalytic systems, which were tested for dry reforming of methane (DRM) and steam reforming of methane (SRM), reported until then.109,116,232–243
In addition to SRM and DRM, the partial oxidation of methane (POM) in the presence of oxygen has also been explored by photo-thermal means. In 2019, Miyauchi and co-workers used Pd NPs supported on strontium tantalate (Sr2Ta2O7) to perform the reaction under UV irradiation at temperatures lower than those conventionally required.244 They observed that, in contrast to any other thermal process reported, UV light initiates the partial oxidation at 150 °C, while in dark conditions the conversion of CH4 and O2 begins only above 350 °C. In addition, at this temperature, the conversion of CH4 goes from 21% in the dark to 60% under irradiation, and the yield of CO and H2 goes from 2% and 2% to 47% and 53%. The Arrhenius plot (logarithm of formation rate of CO versus the inverse temperature) allows calculating the activation energy (Ea) for the reaction under UV light (11.4 kJ mol−1) and in dark conditions (93.6 kJ mol−1), and the difference between the two shows that the light-induced energy plays a major role in the high performance at lower temperatures. The authors investigated the reaction mechanism and they proved that hot carriers are generated by the d to s interband excitation of Pd NPs via UV light irradiation. These hot carriers are transferred into the conductive band (CB) of Sr2Ta2O7, and reduce O2 to radical species. In parallel, hot holes in the Pd d-band activate CH4 for cracking. Finally, the authors found that the ultrawide-bandgap (UWBG) of the semiconductor, Sr2Ta2O7, is crucial to drive the photo-thermal reaction towards the desired product.
Very recently, the research group of Miyauchi expanded the investigation in photo-thermal DRM with two papers, in which the researchers studied the role of Rh as a plasmonic metal with two different supports, namely SrTiO3 and TaON.245,246 Surprisingly, in both cases the catalytic system tested exceeded the theoretical thermal limit calculated by using thermodynamic simulation software. Shoji et al. observed that the Rh based catalytic system supported on strontium titanate (Rh/STO) irradiated with a 150 W Hg–Xe lamp converts CH4 and CO2 giving H2 with a yield higher than 50% even at a temperature of 200 °C, while the performance in dark conditions was less than half even at 450 °C.245 In addition, they tested six different semiconductors as a support for Rh NPs and Ni/Al2O3 as catalytic systems only with photoirradiation, and they found that the band gap and the conduction band position of STO are optimal to drive CH4 and CO2 conversion. To understand thermal and photophysical contribution in this process, the authors performed the reaction under light irradiation while monitoring the working temperature, i.e. the temperature at the surface of the catalyst. They observed indeed that the temperature reached 300 °C; however this thermal energy is quite low to justify the high yield. In situ electron spin resonance (ESR) measurements conducted under light irradiation and at −173 °C proved that trapped electrons and trapped holes are generated in the valence band, that Rh NPs accept electrons in the DRM process, and that CH4 interacts with the holes while CO2 does not. In addition, Miyauchi's research group performed several experiments to elucidate the mechanism involved in this process: Kelvin probe force microscopy (KPFM) shows that the charge transfer process occurs at the semiconductor–metal interface; the correlation between light intensity and hydrogen generation rate shows that photoexcited charge carriers play the main role to drive the reaction; the results of the action spectrum obtained below 380 nm, corresponding to the bandgap energy of STO (3.2 eV), shows that the promotion of electrons from the valence band to the conductive band is also a driving force of the reaction. Catalytic tests conducted in the presence of a single gas, and the use of isotopic labeling allowed one to understand in detail the formation of CO. All this evidence allowed the authors to present a detailed mechanism for this photo-thermal DRM process (Fig. 15).
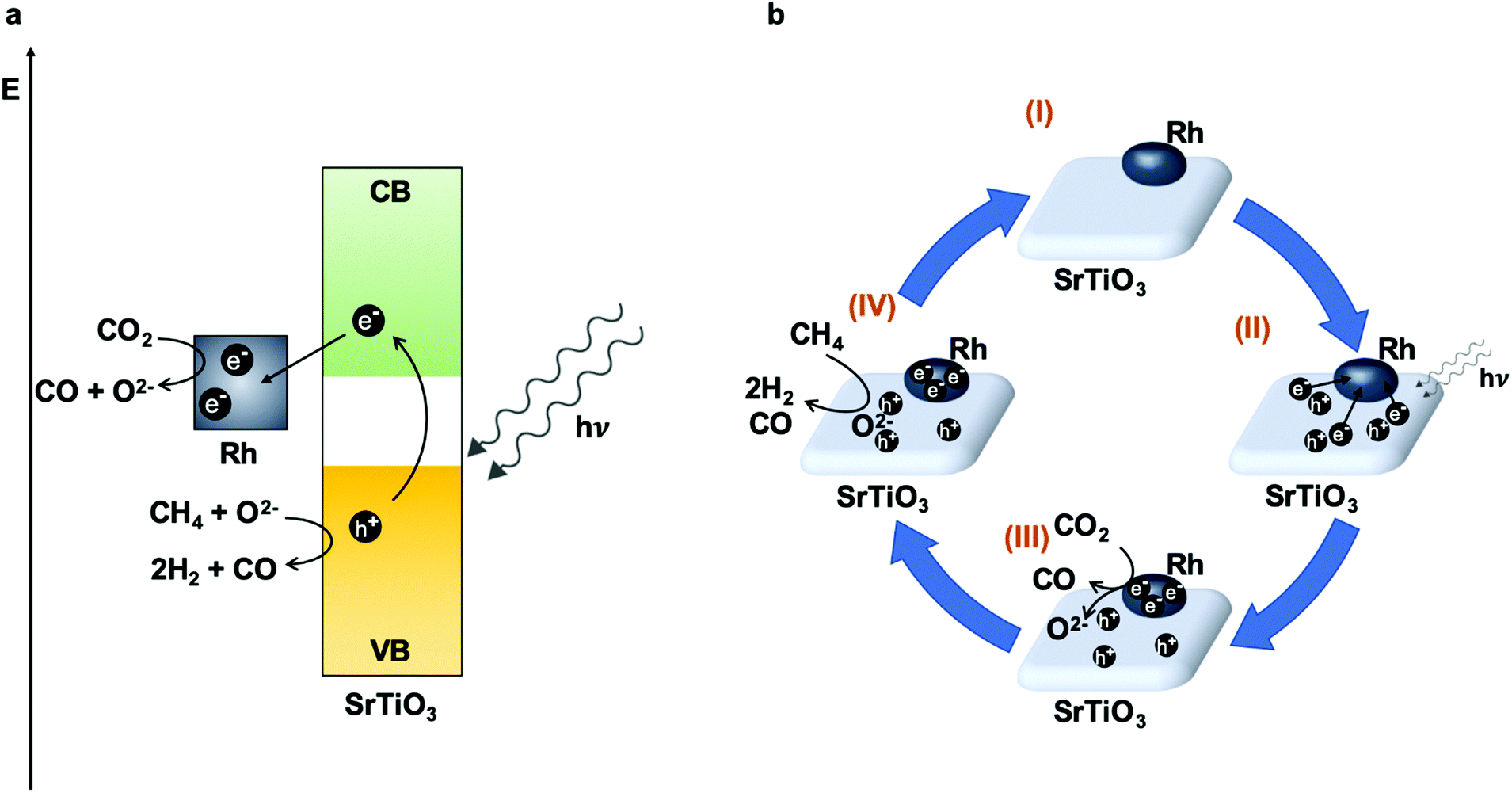 | ||
| Fig. 15 Mechanism of the photocatalytic DRM by Rh/STO. (a) Band diagram showing generation of electron–hole pairs in STO and expected redox reactions. (b) Schematic dynamics of charge carriers and oxygen ions. Adapted with permission from ref. 245. Copyright (2020) Springer Nature. | ||
Cho et al. studied the catalytic activity of rhodium-loaded tantalum oxynitride (Rh/TaON).246 In this case, the H2 yield at 450 °C under visible-light irradiation is 50.3%, a value that is significantly higher than the one obtained in dark conditions (15.6%), and higher than the theoretical thermal limit calculated by using thermodynamic simulation software (36.3%). The authors extended their studies to different combinations of metal and semiconductor support, and they confirmed that the interaction between these two components is pivotal to achieve the best results. Finally, they performed different experiments, in which they could monitor the activity of Rh/TaON with respect to the temperature generated by either light irradiation or external heating, and they proved that the high performance is not only due to the photogenerated thermal process, but it also involves the contribution of the plasmonic effect on Rh NPs and of hot electron–hole pairs in the support.
Inspired by the works of Ye's group on bimetallic-alloy plasmonic photocatalysts, Halas and co-workers prepared a series of Cu nanoparticle–Ru single-atom catalysts supported onto a MgO–Al2O3 composite (CuxRuy) for photo-thermal DRM.116,235,247 Cu19.9Ru0.1 and Cu19.8Ru0.2 showed the best stability due to coking resistance, with a minimal decrease in activity only after 50 hours, and Cu19.5Ru0.5 instead showed higher catalytic performance, but with a decrease in activity after 5 hours. Cu19.9Ru0.1 and Cu19.8Ru0.2 also showed higher selectivity compared to Cu19.5Ru0.5. The authors were able to prove that Ru atoms can be monodispersed on the Cu NPs in Cu19.9Ru0.1 and Cu19.8Ru0.2, and they attributed their superior performances to the presence of these atomically dispersed Ru sites. The researchers investigated the mechanism comparing the activity of the reaction under light irradiation and in dark conditions adding external heating, and they found that the DRM process is mainly driven by the generation of hot carriers, which promote the C–H activation on Ru sites and the release of H2. Finally, the study of the energy efficiency, expressed as the percentage of methane converted, proved even in this case that the photo-thermal induced process performs better than the thermal process having the same surface temperature.
4.4. NH3 synthesis/decomposition
The Haber–Bosch catalytic process is considered as one of the greatest game changer discoveries of the 20th century.248 When this synthesis to produce ammonia starting from its elements, hydrogen and nitrogen, was performed in BASF's laboratories, the operative conditions were really extreme. Since then, this century old process has been optimized in many aspects and is still applied worldwide to provide ammonia, a key chemical for the world's agricultural industry.249 However, this reaction is severely hampered by thermodynamics, and to achieve competitive industrial performance, the reactors need to operate at high temperatures (400 °C to 500 °C) and pressures (100 bar to 200 bar).In 2018, Zhang and co-workers presented a new strategy to achieve a Haber–Bosch comparable process based on sunlight only as the energy source.250 They prepared a catalytic system where K promoted Ru is supported on disordered TiO2−xHx (K/Ru/TiO2−xHx). This catalyst could generate ammonia simply by irradiation with sunlight at atmospheric pressure, without the need of external heating. K/Ru/TiO2−xHx is able to absorb in the UV-vis-NIR region of sunlight, and it can convert part of it to thermal energy; in fact, after 13 minutes of irradiation with a 300 W xenon lamp, the reactor reached a temperature of 360 °C. As a result, the process yields an ammonia generation rate of 112.6 μmol g−1 h−1 (gas flow rate of 6 mL min−1, TOF 3.9 × 10−4 s−1), which is surprisingly high when compared with the same and similar Ru based catalyst used under thermal conditions. In their work, Mao et al. highlight the importance of the surface disorder of TiO2−xHx to avoid H2 poisoning of Ru. In addition, they propose a mechanism, which involves the localized surface plasmon resonance (LSPR) effect of Ru facilitated by coupling with the support, TiO2−xHx, and which lowers activation barriers.
In the same year, Halas et al. presented an interesting study distinguishing the role of thermal and photochemical effects in reducing the activation barrier for ammonia decomposition.115 Particularly, the researchers tested a plasmonic Cu–Ru antenna-reactor (Cu–Ru-AR) as a photocatalyst, and they found out that the reaction rate was directly proportional to light irradiation. They were able to differentiate the plasmon-induced hot carriers and the photo-thermal heating contribution, and to mainly attribute to plasmon-induced hot carriers the ability of catalyzing NH3 decomposition. In addition, they calculated the activation energy (Ea) as a function of the irradiation wavelength and intensity, and they plotted the different values, obtained using the Arrhenius equation, obtaining a three-dimensional map, which shows that under optimized illumination conditions Ea can be reduced to 0.27 eV. This mapping proved to be a useful tool that provides information to predict and optimize catalytic performance.
One year later, Halas and co-workers published another paper in the same field, and they demonstrated an innovative route to produce ammonia using titanium nitride NPs as plasmonic antennas, which can heat magnesium-based nanomaterials to undergo chemical looping as active materials.251 The researchers manufactured a customized low-pressure reaction chamber to perform the three-step chemical looping process and to monitor the product formation in situ via isotopic labelling and rotational spectroscopy. During the experiment, the catalytic system was irradiated with a Ti:sapphire laser (808 nm wavelength, 150 fs pulses, 80 MHz repetition rate) with different timing for each step. Swearer et al. studied each stage of the looping process, and they proved that TiN plasmonic NPs play a crucial role in photo-thermal heating, which led to the increase of the surface temperature of the catalytic system above 550 °C. Furthermore, they succeeded in detecting ammonia evolution in the final step of the photo-thermal-promoted chemical looping. As the authors highlight in the conclusion, this process needs further improvements; however its advantages compared to classical strategies are evident, making this approach a promising green option for ammonia production.
Another innovative approach to produce ammonia without using thermal energy or elevated pressures was proposed by Liu and co-workers in 2019.252 Their strategy exploits photo-thermal heating in order to create and control a thermal gradient, which allows achieving high reaction rates and good conversion yields. For their investigation, Li et al. built a custom-made fixed-bed reactor equipped with a quartz window and two embedded thermocouples to monitor the temperature of the catalyst on the top-surface (T1) and on the bottom-surface (T2). The catalytic system consists of cesium-promoted ruthenium NPs supported on magnesium oxide (Ru–Cs/MgO); the powdered catalyst is loosely packed in the reactor, and it has a thickness of 3 mm. After reduction of the Ru catalyst, N2 and H2 were introduced in the reactor with a 1:3 ratio and a flow rate of 75 mL min−1. NH3 was produced giving the best results under irradiation from the top using UV and visible LEDs, at atmospheric pressure without adding external heating. The researchers observed that in these conditions photo-thermal heating forms a negative thermal gradient, meaning that T1 is higher than T2, within the vertical temperature profile of the packed catalyst. They demonstrated that this negative thermal gradient plays a crucial role in improving the reaction rate and the product yield, because it acts as a thermodynamic pump, which is able to shift the equilibrium towards the products. The hotter temperature on the top of the catalyst accelerates nitrogen scission, the rate-limiting step, while the product is pushed to the coldest region due to thermophoretic forces, and NH3 does not undergo decomposition reaction.253
Concluding this section, we consider worth mentioning three publications in the field of plasmonic assisted ammonia synthesis, although the authors do not discuss thermal contribution.254–256 The work presented in 2016 by Misawa and co-workers describes the production of ammonia and oxygen starting from nitrogen in water under visible light irradiation promoted by plasmonic induced charge separation.254 The researchers coated a niobium-doped strontium titanate photoelectrode with a thin layer composed of gold NPs, as a plasmonic metal, and with zirconium/zirconium oxide as a cocatalyst (AuNP/Nb–SrTiO3/Zr). The catalytic reaction was performed under ambient conditions, and the photoelectrode was irradiated using a xenon lamp using arbitrary light intensity and wavelength. Then the quantity of ammonia formed was quantitatively analyzed by GC-MS, and expressed as apparent quantum yield (AQY, %), i.e. the percentage of the ratio between reaction rate of NH3 formation and the flux of incident photon. Although the efficiency of this method is still not competitive for industrial applications, it proved to be highly selective and highly responsive to visible light. These aspects give high expectations for further development of sunlight-driven ammonia synthesis.
Gong and co-workers published in 2018 a work, where they described surface oxygen vacancies (Ovac) together with plasmon generation as a winning strategy for N2 fixation based on environmentally friendly methods.255 As in the previously reported example, the plasmonic metal of interest is gold, because it can broaden the light absorption range of the metal oxide support, TiO2. The optimized catalytic system consists of Au nanorods (NR) supported on rutile TiO2 modified on the surface by atomic layer deposition of amorphous TiO2 (TiO2/Au/a-TiO2). This modification introduces more defects, or Ovac, that promote the adsorption of N2 and its activation via hot electrons, without modifying the bulk properties. This was proved by testing the catalytic reaction by immersing different electrodes in water with continuous nitrogen bubbling under UV-visible light irradiation. The NH3 production rate increased with the enhancing of the surface modification of the electrodes: bare TiO2 < TiO2/AuNRs < TiO2/a-TiO2 < TiO2/Au/a-TiO2. With this paper Li et al. suggest a promising method to combine electrochemical ammonia synthesis with photovoltaic energy, exploiting a plasmon-enhanced reaction pathway.
Xiong and co-workers published very recently a paper about nitrogen fixation, where LSPR promotes nitrogen fixation through a N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N dissociative mechanism.256 This reaction pathway is normally unusual, since the nitrogen triple bond has a high dissociation energy. The series of catalysts that Hu et al. prepared, AuRux (x = 0.14, 0.23, 0.31, 0.39; it represents the percentage of Ru incorporated into Au NPs), contain Au in the core, because it absorbs efficiently a wide region of light, and Ru on the surface, because it facilitates N2 dissociation. AuRu0.31 was the catalyst that gave the best performance achieving a production rate of 101.4 μmol g−1 h−1 at room temperature and 2 bar of N2 pressure, under full-spectrum light irradiation (300 W Xe lamp, 400 mW cm−2). In addition, they performed electron spin resonance (ESR) to detect the reactive oxygen species, while catalytic N2 reduction by altering photon flux and wavelength-dependent apparent quantum efficiencies (AQEs) proved the plasmonic-driven mechanism. The researchers employed several techniques to follow carefully the different steps that N2 undergoes to generate NH3. They used 15N2 to verify the nitrogen source, they performed low-temperature Fourier-transform infrared (LT-FTIR) spectroscopy to identify the configuration of the adsorbed N2, and they brought evidence of the surface plasmon-driven N2 dissociation mechanism owing to in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) and in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS).
N dissociative mechanism.256 This reaction pathway is normally unusual, since the nitrogen triple bond has a high dissociation energy. The series of catalysts that Hu et al. prepared, AuRux (x = 0.14, 0.23, 0.31, 0.39; it represents the percentage of Ru incorporated into Au NPs), contain Au in the core, because it absorbs efficiently a wide region of light, and Ru on the surface, because it facilitates N2 dissociation. AuRu0.31 was the catalyst that gave the best performance achieving a production rate of 101.4 μmol g−1 h−1 at room temperature and 2 bar of N2 pressure, under full-spectrum light irradiation (300 W Xe lamp, 400 mW cm−2). In addition, they performed electron spin resonance (ESR) to detect the reactive oxygen species, while catalytic N2 reduction by altering photon flux and wavelength-dependent apparent quantum efficiencies (AQEs) proved the plasmonic-driven mechanism. The researchers employed several techniques to follow carefully the different steps that N2 undergoes to generate NH3. They used 15N2 to verify the nitrogen source, they performed low-temperature Fourier-transform infrared (LT-FTIR) spectroscopy to identify the configuration of the adsorbed N2, and they brought evidence of the surface plasmon-driven N2 dissociation mechanism owing to in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) and in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS).
4.5. Water gas shift reaction
Water gas shift reaction (WGS) is a key process for H2 production, as well as for ammonia or methanol synthesis, Fischer–Tropsch process, reforming systems for fuel cells or even one of the key steps in the automobile exhaust processes, producing H2 for the removal of toxic NOx gases.257,258 This reaction was discovered by Felice Fontana in 1780 and it is considered as one of the most important reactions for industry and in the history of Chemistry.259The reaction is a reversible and moderately exothermic process that permits, simultaneously, CO conversion into CO2 and H2 production using water as the H source (eqn (7)):
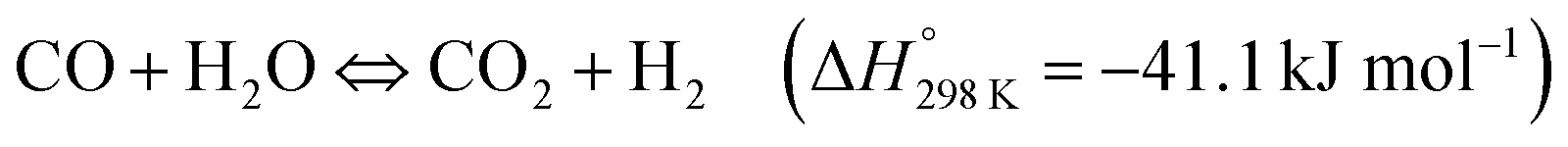 | (7) |
:0.6:0.4 Cu:Zn:Al (4.0 mmol H2 h−1). However, these results did not improve the catalytic performance of the catalysts with pure ZnO as a support (Cu/ZnO) that yielded a hydrogen production of 4.75 mmol H2 h−1 and a CO conversion of 85.6%. On the other hand, the pure Al2O3 catalyst (Cu/Al2O3) was less active (2.1 mmol H2 h−1). Furthermore, the study evaluated the time-dependent temperature increase revealing higher temperatures than 300 °C after three minutes of irradiation when using Cu/ZnO, Cu/Zn0.6Al0.4, Cu/Al2O3 and pure Cu NPs; meanwhile bare supports did not present higher temperatures than 150 °C, probably due to the absence of metallic Cu NPs. It is worth mentioning that Cu/Al2O3 reached the highest temperature (367 °C) with the highest photon absorption together with the plasmonic effect of metallic Cu NPs. The study, however, needs a deeper analysis of the photo and thermal approaches due to the existence of controversial results. The temperature study displayed the LSPR effect from plasmonic Cu, but the temperature was not indeed described as the dominant variable in the catalytic performance as the three catalysts presented higher temperatures than 250 °C (usual for WGS reaction at low temperatures), and the catalyst which reached higher temperatures was not the most active one. As more Cu is detected in Cu/ZnO and CuZn0.6Al0.4 catalysts than in Cu/Al2O3, it seems that the presence of ZnO improves the dispersion of Cu, offering a higher amount of active plasmonic sites. Finally, the authors also studied different reducing pretreatments for several samples obtaining as a better option the photo-reduction method, using also light for the reduction of the catalysts prior to their use in the photo-thermal reaction.
In the same line, Cu supported on Al2O3 (CuOx/Al2O3) was compared with Pd and Au supported on the same material as catalysts for WGS reaction in a closed-circulated system under light irradiation (UV-visible-IR).261 The catalysts with 2 wt.% loading of each metal performed different, ordered as Cu > Pt > Au (24.6, 6.3 and 3.5 μmol H2 gcat−1 s−1 respectively). Focusing on the most active and also cheapest metal, the loading of Cu was optimized up to 19 wt% revealing four times more catalytic activity, an improvement up to 122 μmol H2 gcat−1 s−1. The characterization of this Cu-based catalyst (XRD and HRTEM) showed mostly metallic Cu NPs (well-dispersed) and some Cu2O after the WGS reaction. The temperature reached during photoreaction was 285 °C offering twice the activity of usual thermal catalysis which occurs at 300 °C. This comparison demonstrated additional effects apart from the just heating of the catalyst (not only thermocatalysis). To elucidate the additional effects, several catalytic tests were carried out using light-filters: those filtering IR radiation and reducing the light-to-thermal conversion (only up to 60 °C) presented 54.6% less catalytic activity after 30 min; the other filters removed UV contribution and the catalytic activity was decreased to 48.4% achieving the same temperature as in full-spectrum experiments. Summarizing, these results revealed the photo-thermal oxidation of CO and reduction of H2O as a coupled and synergetic process, a cooperative action between photo-generated carriers and photoinduced heat.
4.6. H2 evolution and other processes driven by photo-thermal catalysis
The approach of using solar energy to improve reactions that normally require high temperature and pressure has been exploited successfully in several catalytic processes. Hydrogen evolution generated via water splitting reaction is indeed among these processes, and the first paper in this field was published in 2008.262 Li and Lu reported the formation of H2 from water steam splitting over titanium disilicide (TiSi2) and Pt supported on titanium oxide (Pt/TiO2) under visible light irradiation, and they studied the influence of pressure and temperature on the catalytic activity. The researchers carried out the reaction in a Pyrex flask, which was irradiated with a 300 W tungsten halogen lamp. They observed that for both catalysts the increase of pressure is essential to generate H2 at 190 °C with good yields.This topic gained attention again last year; in fact three different research groups studied water splitting reaction for hydrogen generation catalyzed by nickel-based materials.263–265 Zhang and co-workers combined the photo-thermal effect with electrocatalysis for hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).263 They prepared Ni NPs supported on reduced graphene oxide nanosheets (Ni/RGO) via hydrothermal synthesis and reduction in a H2/Ar atmosphere at 500 °C. The researchers compared the performance of the catalytic system under irradiation and in dark conditions, and they observed that light improves both the thermodynamics and the kinetics of the reaction, yielding better HER and OER activity. They explained this behavior with the ability of the catalyst to generate hot active species after light absorption, which increase the system's temperature to 50 °C in less than 5 minutes. Guo and co-workers observed a similar behavior, when they investigated nickel–tungsten–boride nanoneedles (Ni–W–B) supported on carbon cloth (CC) for photo-thermal catalysis in electrochemical water splitting.264 They tested the catalytic system for HER with and without irradiation, and they also compared the results with those obtained from bare CC and Pt–C deposited on CC. As expected, HER activity is facilitated by light irradiation, without an increase of the electrolyte temperature. In addition, the authors performed the catalytic process at different temperatures, and they observed that the performance of the photo-thermal HER was similar to the performance achieved at 48 °C. As far as the OER is concerned, the activity of Ni–W–B/CC was compared with bare CC and IrO2 supported on CC, and even in this case the Ni-based photo-thermal catalyst showed the best results. Finally, the researchers assembled an electrolyzer using Ni–W–B/CC as the cathode and anode, and they demonstrated its superior overall photo-thermal-assisted water splitting performance among the reported bifunctional catalysts. Even Liu and co-workers used abundant and cheap nickel to assemble a novel photo-thermal-enhanced electrochemical device for water splitting.265 They prepared a Ni nanosheets array supported on an Al2O3 ceramic chip, and they characterized thoroughly its electrochemical properties by performing electrocatalytic HER activity, electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV) and durability tests. Then, the researchers assembled a two-electrode device to investigate the overall water splitting during exposure to simulated sunlight, using the supported Ni nanosheets as the cathode, for HER, and NiFe hydroxide film on CC as the anode, for OER. The good production rates of hydrogen and oxygen make this device promising, although the authors suggest that they aim to improve the process with modifications in order to avoid thermal diffusion between the two ends of the device.
An alternative process to generate hydrogen proposed in this current year by Dai and by Naldoni is the photo-thermally enhanced hydrolysis of ammonia borane, whose hydrogen content is 19.6%.266–268 Wu et al. prepared a series of urchin-like sodium titanate microspheres supported on the reduced graphene oxide (RGO/Na2Ti3O7) photo-thermal catalyst with different graphene oxide content (0, 33, 50, 60, 66 wt%).266 Particularly, they focused on the challenging aspect of synthesis optimization, elucidating the role of graphene oxide sheets in the growth mechanism of Na2Ti3O7 microspheres. Also, they found that the photoelectrochemical performance of the prepared materials was related to the structural organization of the catalyst and the surface features of the Na2Ti3O7 microspheres. In fact, the branches of the urchin-like microspheres are crucial because they provide plenty of active sites, and they facilitate electron transfer at the interfaces. Then the authors studied the photo-thermal effect and its influence on the catalytic hydrolysis of ammonia borane. They observed that the high results in H2 evolution are a combination of photo-thermal properties of RGO and the ability of Na2Ti3O7 to generate hot spots. Finally, the researchers proved that the morphology of Na2Ti3O7 is also important, since they observed that RGO/Na2Ti3O7 microspheres have almost 2 times higher activity than RGO/Na2Ti3O7 nanowires. The synergistic effect between hot carriers and photo-thermal properties is also the driving mechanism which justifies the results reported by Naldoni and co-workers.267 They developed a procedure to decorate TiN nanocubes with Pt nanocrystals (TiN–Pt nanohybrids); they fully characterized the obtained materials, and they evaluated their catalytic performance for photo-thermal hydrogen evolution from ammonia borane dehydrogenation. As expected, visible-NIR light irradiation increases hydrogen production, compared to the results obtained in dark conditions, due to plasmonic generation of hot electrons. When the irradiation wavelength matches the LSPR, i.e. 700 nm, the value of apparent quantum yield (AQY) reaches 120%. This high value is explained with the high absorption ability of the catalysts and the low energy barrier of the dehydrogenation reaction, aspects that allow both hot electrons and low energy hot electrons (those which underwent inelastic scattering) to react efficiently with ammonia borane. In addition, the authors were able to detect with good approximation the temperature at the surface of the nanoparticles in the liquid medium, and to evaluate the photo-thermal heating accordingly. Finally, they demonstrated by performing theoretical COMSOL simulations the contribution of photo-thermal heating to the reaction rate, and they elucidated in detail the mechanism for the cleavage of the O–H bond, the rate determining step of the reaction, by hot electrons.
To conclude this part of the review, we believe that there are other noteworthy works, which did not fit in the previous sections. In fact, in the last few decades photo-thermal catalysis has contributed to the development of catalysis field bringing a different perspective to many catalytic reactions. One of the first examples was reported in 1998 by Kennedy and Datye, who described how the combination of a typical thermal oxidation catalyst, Pt, with a photocatalyst, TiO2, can improve the efficiency of ethanol oxidation.269 Almost twenty years later, Amal and co-workers investigated the mechanism of the same reaction using a TiO2-supported Au catalyst, and they reported an almost doubled catalytic activity in the photo-thermal regime.270 In the same year, Yang and co-workers reported the synthesis of AgPt bimetallic hollow nanoparticles, and they tested this material for LSPR-enhanced methanol oxidation.271 Linic and co-workers prepared another photo-thermal catalyst consisting of silver nanocubes supported on Al2O3, and they tested it for commercially relevant catalytic oxidations, which are normally performed at high temperatures and over expensive platinum-group metals.49 Zhang and co-workers investigated the effect and the mechanism of the photo-thermal enhanced catalytic oxidation of formaldehyde promoted by a graphene-based MnO2 composite.272 Another important application of photo-thermal catalysis is removal of VOCs (volatile organic compounds) via catalytic oxidation, as reported by Jia and coworkers.273 They tested four ACo2O4 (A = Ni, Cu, Fe, Mn) type spinels for photo-thermal degradation of toluene, and they observed the superior activity of NiCo2O4 due to better structural and photophysical features. Xu and co-workers reported the synthesis of a SmCoO3/SBA-15 composite via an impregnation method, which confers to the material an outstanding surface area.37 This feature, together with the good photo-thermal conversion, makes SmCoO3/SBA-15 a promising candidate for photo-thermal degradation of propane.
Finally, to have a more comprehensive overview on the advantages of plasmon-driven processes, we suggest three papers, which focus on plasmon induced crystal growth and material transformation.274–276 Xu and co-workers give a closer look at the different transformation processes that can occur among molecules when they are in the proximity of a plasmonic electromagnetic field, hot carriers, and heat.274 In their work, they explain the different paths that allow the energy transfer that induce the chemical transformation, as well as a significant contribution to plan efficient and tunable plasmonic catalytic systems. Zheng and his research group presented a nice example of an in situ transformation mediated by Au NPs.275 A polycrystalline material, NaYF4:Eu3+, can be converted into a single crystal luminescent material, Y2O3:Eu3+, in a rapid and controlled way. Sasaki and co-workers demonstrated that they were able to control the synthesis of a ZnO based plasmonic hybrid nanostructure in a targeted position.276 They realized a nanobutterfly gold structure, which directs plasmonic hotspots in a precise location, according to the direction of the incident light polarization. Thanks to this procedure, ZnO layers can selectively grow via hydrothermal synthesis where required, leaving the surrounding gold particles uncoated, and yielding the desired plasmonic hybrid nanostructure.
5. Design strategies for photo-thermal catalysts
For an optimal exploitation of the photo-thermal effect in catalytic processes, photocatalysts must meet a series of requirements, including an intense light absorption across the solar spectrum, an efficient charge carrier generation and a high capacity for heat production and/or heat transfer.30 In the subsequent sections, we outline different strategies for materials’ design and engineering in order to maximize photo-thermal effects and therefore efficiency for a given mechanism, i.e. hot electron or local heating effects.5.1. Size and shape effects
In plasmonic materials, size and shape are key factors determining the LSPR phenomenon (i.e. ratio of absorption to scattering, number of LSPR modes and position of the LSPR peak) and can be adjusted appropriately in order to match the plasmon band with the solar spectrum and thereby improve catalytic performance.277,278 In fact, both parameters have shown a strong influence not only on the capability for charge carrier generation, but also on the thermal properties of a material.Theoretical calculations have demonstrated that larger plasmonic NPs provide a larger amount of hot carriers; however, they display energies close to the Fermi level, since most of the absorbed energy is dissipated either by scattering or by heating.9 In contrast, smaller NPs render less electron–hole pairs than their counterparts, but they exhibit higher energies but showing very short lifetimes in the order of femtoseconds.279 This is of crucial importance in photo-thermal catalysis, as the higher the energy of hot electrons, the higher the probability for them to populate acceptor states or overcome Schottky barriers. Therefore, it seems that smaller NPs are especially convenient for hot electron induced catalysis. However, smaller NPs do not have to necessarily enhance the overall photo-thermal performance as the reduction of particle size can lead to inefficient charge carrier separation due to spatial confinement.280 In addition, plasmonic NPs have shown a red shift in their resonant frequency as the particle size increases, thus allowing a more efficient harvesting of the visible and IR regions of sunlight compared to their smaller equivalents.281,282 Given these circumstances, a precise balance between these effects should be considered when developing photo-thermal catalysts.
Morphology is another key parameter determining the optical properties of nanostructures, especially those featuring the LSPR effect. In this regard, certain shapes such as bars, bipyramids or branched structures have shown a red-shift in the position of their main LSPR absorption peaks, therefore being more appropriate to take advantage of the low energy regions of the solar spectrum.283–285 Theoretical studies from Govorov and co-workers pointed out that the ability for the generation of hot carriers was also closely related to the shape of plasmonic structures. In these works, the authors established that geometries displaying strong and inhomogeneous electric fields together with important confinement effects could generate high-energy hot electrons and holes. In contrast, those geometries without defects or featuring uniform electric fields like thin films or nanospheres afforded a less efficient production of hot carriers.286,287
The thermal conductivity of nanostructures can also be optimized by modifying phonon transport properties, by either size or shape engineering. For instance, it has been demonstrated that the thermal conductivity of a material can be drastically reduced by introducing a high density of grain boundaries which intensifies phonon scattering.288 In line with this, increasing the surface roughness has also proven to slow down phonon velocity.289 Then, decreasing the size of certain components of the photo-thermal system and maximizing its surface roughness contributes to the reduction of the thermal conductivity of the material, consequently favoring heat generation close to catalytic active sites.30
It should be also emphasized that size has direct consequences on the heating capability of plasmonic structures. In fact, as mentioned in the previous sections, the plasmon-heating effect in metallic nanospheres scales quadratically with the NP radius. However, larger plasmonic NPs are more prone to decay radiatively, thus reducing the amount of locally dissipated heat.290 For these reasons, an accurate balance between these two opposite effects has to be pondered when tailoring new photo-thermal catalysts. In addition, according to theoretical works developed by Baffou and co-workers, morphology also seems to play a key role when evaluating the heat production in plasmonic nanostructures.291 For example, the authors found that sharp, flat or elongated geometries exhibited a more intense heat generation than spheres. In the case of nanospheres, only the outermost part of the structure effectively faced the incoming light, while in the rest of the cases the whole volume of the structure participated in the heating process. Apart from this, heat production also benefited from the presence of corners and edges that enhanced electric fields locally.292
5.2. Hybrid materials
A very common and straightforward strategy to improve photo-thermal performance is the design of hybrid structures. In these composite materials, the combination of two or more components with different optical, electronic and thermal properties works synergistically to enhance catalytic rates. Typically, photo-thermal systems are comprised of an inorganic support or host (in most of the cases metal oxides) functionalized by either plasmonic or non-plasmonic metallic NPs. It is worth mentioning that each of the components is not restricted to a single function inside the photo-thermal structure and that, depending on the nature of each system, only one or more elements can act as catalytic active sites in the reaction.Recently, Jiang et al. described for the first time the preparation of a composite catalyst based on a porphyrinic MOF displaying the photo-thermal effect.296 The authors stabilized Pt nanocrystals on the MOF structure and the composite photocatalyst was tested for the oxidation of benzyl alcohol with molecular O2. Interestingly, according to the catalytic results a synergy between the photo-thermal effect of both the MOF and supported Pt nanocrystals was established. This cooperative work enhanced the photocatalytic rate of the composite compared to the individual contributions of each component separately.
More recently in 2018, Maspoch and co-workers studied the photo-thermal effect of a series of MOFs representing the major subfamilies of this type of porous materials and calculated the photo-thermal transduction efficiency for each of the MOFs under study.297 For example, UiO-66-NH2 and CPO-27-Ni reached 149 and 167 °C respectively under 30 min of irradiation, both showing photo-thermal efficiencies above 55%. In contrast, UiO-66 and ZIF-8 under the same experimental conditions were heated to 57 and 70 °C respectively, with photo-thermal efficiencies far below 10%. These results indicated that the observed photo-thermal effect was highly dependent on the optical absorption band of the MOFs. In other words, MOFs displaying absorption bands in the range of 300–600 nm featured a more pronounced photo-thermal effect than those that did not exhibit intense absorption in this region.
Some groups, however, have taken advantage of the heat insulating properties of the MOFs to create hybrid systems by combining them with active photo-thermal metallic species. In a work referenced in the previous section of this review (see Section 4.1.3), Zeng et al. designed a three-component heterostructure for the photocatalytic hydrogenation of CO2 based on Pt nanocubes and Au nanocages encapsulated in a zeolitic imidazole framework (ZIF).36 In this system, Pt nanocubes acted as catalytic sites facilitating CO2 reduction, while plasmonic Au nanocages effectively transformed light into heat. Particularly, ZIF served as a thermal insulator preventing heat dissipation, thus confining heat inside the structure and enhancing the catalytic activity of Pt nanocubes. This approach offers obvious advantages, as photo-thermal catalysis benefits from low thermal conductivity to enhance local heat generation, and opens up the possibility for further advances by proper material engineering in hybrid architectures.
Although much less studied, other porous materials such as zeolites have also shown potential applications as active components in photo-thermal catalysis. Zhu et al. evaluated the photocatalytic activity for the visible light-mediated oxidation of benzyl alcohol and derivatives by Au NPs supported on various zeolites.298 On the basis of experimental data, the authors proposed a reaction mechanism in which zeolitic supports effectively adsorbed reagent alcohols while Au NPs activated O2 molecules by transferring plasmon-induced hot electrons. The resulting O2− species then triggered the oxidation reaction towards the corresponding aldehydes. From the kinetic study of the reaction, it was also possible to calculate the activation energy under both dark and visible light irradiation conditions. Surprisingly, it was found that the plasmon contribution decreased the activation barrier by 40%.
For instance, a recent work from Kumar et al. described the preparation of Au plasmonic NPs coated with TiO2 and Pt NPs creating a core–shell structure (Pt@TiO2–AuNPs) and its application in CO2 photoreduction.300 It was demonstrated that the presence of a thin layer of TiO2 increased the quantum yield of the reaction compared to bare Au NPs due to a combination of positive effects including an enlargement of the surface area, a red-shift broadening of the LSPR peak and an increase in the light absorption. Furthermore, the deposition of Pt NPs assisted the trapping of hot electrons from the CB of TiO2, thus preventing electron–hole recombination and enhancing the overall reaction rate. The successful combination of these three components rendered a high efficiency (up to 70%) and selectivity for the photocatalytic reduction of CO2 to formic acid under NIR laser irradiation.
In a related precedent, Xiong and co-workers demonstrated that a precise tuning of the Pd shell thickness in the Au nanorods–Pd core–shell nanostructure could control not only the photo-thermal local heating but also the hot electron injection process in the photocatalytic styrene hydrogenation.301 By virtue of ultrafast TAS measurements, the authors could obtain the time constants of electron–phonon scattering (attributable to the photo-thermal effect) and the charge recombination process (attributable to hot electron lifetime). Interestingly, the Pd shell thickness appeared to be a key parameter to determine the preeminent surface plasmon decay pathway, either local heating or hot electron transfer.
Lastly, Yang et al. developed AuCu–CuS core–shell heterostructures immobilized on TiO2.302 This alloy–chalcogenide combination led to a synergistic effect between LSPR and photo-thermal heating that enhanced the photocatalytic glycerol oxidation at room temperature. Upon light irradiation, hot electrons from the plasmonic AuCu nanoalloy effectively transfer into the CB of TiO2, where they react with molecular oxygen to produce H2O2 or reactive superoxide ions (O2−) that drive the oxidation of glycerol. Meanwhile, the non-radiative recombination of electron–hole pairs in the plasmonic CuS shell induces thermal heating that provides an additional driving force for the overall catalytic process. In this work, the rational design of an effective bifunctional core–shell architecture proved to be a successful approach to perform photo-oxidation reactions under mild conditions.
6. Conclusions
In this review, we have succinctly explained the physics behind the LPSR phenomenon and its direct influence on the photo-thermal effect displayed by plasmonic NPs. Upon light irradiation, non-radiative plasmon decay leads to the local heat generation and/or hot carrier formation in plasmonic structures. Individually or synergistically working together, thermal and photochemical contributions of the photo-thermal effect in plasmonic or non-plasmonic nanostructures can be exploited to promote catalytic chemical transformations. A comprehensive analysis of the contribution of both approaches to the overall reaction rate is, however, crucial to further understand the reaction mechanisms, so an extensive revision of straightforward methodologies to successfully distinguish the dominant reaction pathway has been included in this review. Indeed, photo-thermal catalysis has proven to be an efficient strategy to perform a wide variety of high energy-demanding catalytic processes including CO2 hydrogenation, Fischer–Tropsch reaction, ammonia synthesis, methane activation or H2 production. In order to improve catalytic performances and selectivity, a suitable material engineering is also advisable. In the last section of this review, we have included strategies for catalyst design in order to maximize the efficiency for a given photo-thermal-mediated pathway. The vast number of catalytic applications together with the possibility of tailoring the thermal, optical and electronic properties of photocatalysts demonstrates the huge versatility and potential of photo-thermal catalysis.7. Outlook
Despite its afore-mentioned promising applications, the wide implementation of photo-thermal catalysis has still to compete with traditional thermal processes. In this regard, one of the main advantages of the photo-thermal approach is the possibility of increasing selectivity to target products by choosing adequate excitation wavelengths or light intensities. Hot-carrier chemistry has proved to be particularly efficient to trigger certain reaction mechanisms by selectively activating molecular bonds of adsorbed species, hence resulting in better selectivity than the thermochemical pathway. Photo-thermal catalysis also provides direct local heating at the nanoscale, thus concentrating heat at the surface of the active sites and avoiding unnecessary heating of the whole reactor system. Compared to traditional thermal catalysis, where fossil fuels are commonly used to achieve harsh reaction conditions, the photo-thermal approach renders a high productivity (in the order of mmol gcat−1) under mild conditions employing sunlight as a unique energy source. This offers obvious benefits not only in terms of sustainability, but also in terms of recyclability and stability of catalysts. Furthermore, the photo-thermal effect allows a much faster cooling and this opens up the possibility to apply this strategy to discontinuous operation conditions.Regardless of its outstanding performances presented here, there is still plenty of room for the improvement of catalytic efficiency in the field of photo-thermal catalysis. Taking into account that the photo-thermal effect derives from the combination of both photochemical and thermochemical pathways, different approaches can be followed depending on the dominating mechanism. For instance, when it comes to the photochemical process, photocatalysts with a strong light absorption, high population of charge carriers, long charge carrier lifetimes and low radiative emission are advisable.30 As we have previously commented in Section 5, size, morphology or surface defects clearly determine the efficiency of hot electron generation in plasmonic nanostructures. However, besides hot carrier generation efficiency, charge transfer efficiency is another key parameter that defines the photochemical contribution to the overall photo-thermal process. For example, in metal/semiconductor junctions only high-energy electrons are able to surpass the Schottky barrier at the metal/semiconductor interface, so the efficiency of the charge transfer will be dictated not only by the energy of hot carriers, but also the height of the Schottky barrier. Thus, a possible strategy to enhance charge transfer efficiency in this type of systems is to tailor the energy levels of the semiconductor by doping or formation of vacancies/defects in the structure. The spatial organization of the heterostructure or metal loading also determines the hot carrier transfer efficiency through the interface and, for that reason, the synergistic combination of all these parameters has to be pondered when designing photo-thermal nanostructures. In addition, it has been shown that small NPs shorten the pathway of hot electrons, hence favoring their injection to adsorbates. Last but not least, increasing the adsorption of intermediates is also a useful approach to extend their residence time on the surface of the active sites, which eventually enhances the hot electron transfer efficiency to these species. When it comes to the thermochemical process, materials with a high optical absorbance, low radiative emission and low thermal transport tend to amplify the temperature of the active sites, thus enhancing the catalytic performance. In this regard, non-stoichiometric black materials or materials with a high degree of defects in their structure are promising examples of photo-thermal catalysts. Overall, an adequate material engineering to improve both photochemical and thermochemical mechanisms can result in an extraordinary cooperation to boost photo-thermal performance in a vast number of chemical processes, as we have shown in this review.
Nevertheless, if photo-thermal catalysis aims to become a genuine source of solar fuels and chemicals it must overcome a series of limitations:
(a) In spite of the fact that photo-thermal catalysis does not require scaling considerations due to its close relationship with traditional catalytic heterogeneous systems, its main challenge is the incorporation of light into the processes. In contrast to other technologies, the photo-thermal approach does not demand extremely high temperatures but adequate photoreactors capable of harvesting and utilizing sunlight. Most of the works reported so far have been performed under batch conditions; however, the use of continuous flow systems appears to be more convenient for industrial-scale applications. For these reasons, research and development in reactor design and configuration are necessary for the wide implementation of this technique.
(b) Only a few studies among the vast number of works regarding photo-thermal catalysis have clarified the dominating pathway that rules the reaction mechanism. More fundamental studies in this direction are necessary.
(c) Although in the last few years a higher number of species apart from noble plasmonic metals (i.e. Au, Ag, Pd, Pt) have been studied, it is still advisable to move towards abundant and cost-efficient materials. Chalcogenides, nitrides or pnictogenides have recently appeared as promising candidates for photo-thermal catalysis owing to their capability to transform light into heat.303–305
(d) The investigation of long-term stability of photocatalysts remains insufficient, as most of the stability tests are limited to several hours. For an adequate scale-up to commercial plants, feasible and stable photocatalysts are required.
(e) Particularly in the case of CO2 conversion, CO and CH4 are the major products in most of the reported works. However, it would be more convenient to obtain high-value-added C2+ products, namely ethylene, propylene and other olefins. Selectivity control is one of the most attractive features of photo-thermal catalysis, so more work should be dedicated to the design of catalytic systems with remarkable selectivity. Furthermore, and especially in the case of carbon-based photocatalysts, experiments with isotopically labelled CO2 are necessary in order to demonstrate that all carbon based products from these processes are actually derived from CO2 and not from the support itself.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by King Abdullah University of Science and Technology (KAUST). The authors would like to thank Mrs Sandra Ramirez for her contribution to the graphical abstract and to the rest of the artwork in this manuscript.References
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef PubMed.
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Paik, M. Cargnello, T. R. Gordon, S. Zhang, H. Yun, J. D. Lee, H. Y. Woo, S. J. Oh, C. R. Kagan and P. Fornasiero, ACS Energy Lett., 2018, 3, 1904–1910 CrossRef CAS.
- H. Liu, P. Wu, H. Li, Z. Chen, L. Wang, X. Zeng, Y. Zhu, Y. Jiang, X. Liao and B. S. Haynes, Appl. Catal., B, 2019, 259, 118026 CrossRef CAS.
- C. Xiao, H. Wang, L. Zhang, S. Sun and W. Wang, ChemCatChem, 2019, 11, 6467–6472 CrossRef CAS.
- D. Li, S. Ouyang, H. Xu, D. Lu, M. Zhao, X. Zhang and J. Ye, Chem. Commun., 2016, 52, 5989–5992 RSC.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- B. Y. Zheng, H. Zhao, A. Manjavacas, M. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- H. H. Richardson, Z. N. Hickman, A. O. Govorov, A. C. Thomas, W. Zhang and M. E. Kordesch, Nano Lett., 2006, 6, 783–788 CrossRef CAS PubMed.
- H. H. Richardson, M. T. Carlson, P. J. Tandler, P. Hernandez and A. O. Govorov, Nano Lett., 2009, 9, 1139–1146 CrossRef CAS PubMed.
- G. Baffou and R. Quidant, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- W. Tao, X. Ji, X. Xu, M. A. Islam, Z. Li, S. Chen, P. E. Saw, H. Zhang, Z. Bharwani and Z. Guo, Angew. Chem., Int. Ed., 2017, 56, 11896–11900 CrossRef CAS PubMed.
- S. Lu, F. Liu, P. Qiu, M. Qiao, Y. Li, Z. Cheng, N. Xue, X. Hou, C. Xu and Y. Xiang, Chem. Eng. J., 2020, 379, 122382 CrossRef CAS.
- K.-K. Liu, Q. Jiang, S. Tadepalli, R. Raliya, P. Biswas, R. R. Naik and S. Singamaneni, ACS Appl. Mater. Interfaces, 2017, 9, 7675–7681 CrossRef CAS PubMed.
- A. Politano, P. Argurio, G. Di Profio, V. Sanna, A. Cupolillo, S. Chakraborty, H. A. Arafat and E. Curcio, Adv. Mater., 2017, 29, 1603504 CrossRef PubMed.
- Y. Shao, Z. Jiang, Y. Zhang, T. Wang, P. Zhao, Z. Zhang, J. Yuan and H. Wang, ACS Nano, 2018, 12, 11704–11710 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014–5019 CrossRef CAS PubMed.
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and C. Y. Jimmy, J. Mater. Chem. A, 2013, 1, 5790–5805 RSC.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed.
- S. Li, P. Miao, Y. Zhang, J. Wu, B. Zhang, Y. Du, X. Han, J. Sun and P. Xu, Adv. Mater., 2020, 2000086 Search PubMed.
- A. A. Jelle, K. K. Ghuman, P. G. O'Brien, M. Hmadeh, A. Sandhel, D. D. Perovic, C. V. Singh, C. A. Mims and G. A. Ozin, Adv. Energy Mater., 2018, 8, 1702277 CrossRef.
- S. Tang, J. Sun, H. Hong and Q. Liu, Front. Energy, 2017, 11, 437–451 CrossRef.
- L. Zhu, M. Gao, C. K. N. Peh and G. W. Ho, Mater. Horiz., 2018, 5, 323–343 RSC.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2018, 52, 356–366 CrossRef PubMed.
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC.
- Z. j. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016–8035 CrossRef CAS PubMed.
- P. G. O'Brien, A. Sandhel, T. E. Wood, A. A. Jelle, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400001 CrossRef PubMed.
- S. I. Nikitenko, T. Chave, C. Cau, H.-P. Brau and V. R. Flaud, ACS Catal., 2015, 5, 4790–4795 CrossRef CAS.
- J. Jia, P. G. O'Brien, L. He, Q. Qiao, T. Fei, L. M. Reyes, T. E. Burrow, Y. Dong, K. Liao and M. Varela, Adv. Sci., 2016, 3, 1600189 CrossRef PubMed.
- F. Wang, Y. Huang, Z. Chai, M. Zeng, Q. Li, Y. Wang and D. Xu, Chem. Sci., 2016, 7, 6887–6893 RSC.
- W. Zhang, L. Wang, K. Wang, M. U. Khan, M. Wang, H. Li and J. Zeng, Small, 2017, 13, 1602583 CrossRef PubMed.
- R. Zhang, J. Xu, C. Lu and Z. Xu, Mater. Lett., 2018, 228, 199–202 CrossRef CAS.
- P. Drude, Ann. Phys., 1900, 306, 566–613 CrossRef.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25 CrossRef CAS PubMed.
- A. Gellé and A. Moores, Curr. Opin. Green Sustainable Chem., 2019, 15, 60–66 CrossRef.
- L. Hu, S. Gao, X. Ding, D. Wang, J. Jiang, J. Jin and L. Jiang, ACS Nano, 2015, 9, 4835–4842 CrossRef CAS PubMed.
- J. Qiu and W. D. Wei, J. Phys. Chem. C, 2014, 118, 20735–20749 CrossRef CAS.
- L. V. Besteiro, X.-T. Kong, Z. Wang, G. Hartland and A. O. Govorov, ACS Photonics, 2017, 4, 2759–2781 CrossRef CAS.
- A. Gellé, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2019, 120, 986–1041 CrossRef PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu and E. E. Storey, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- Y. Zhao, A. Dunn, J. Lin and D. Shi, Novel Nanomaterials for Biomedical, Environmental and Energy Applications, Elsevier, 2019, pp. 415–434 Search PubMed.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467 CrossRef CAS PubMed.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2017, 118, 2927–2954 CrossRef PubMed.
- G. V. Hartland, L. V. Besteiro, P. Johns and A. O. Govorov, ACS Energy Lett., 2017, 2, 1641–1653 CrossRef CAS.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 CrossRef CAS PubMed.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- S. Sarina, E. Jaatinen, Q. Xiao, Y. M. Huang, P. Christopher, J. C. Zhao and H. Y. Zhu, J. Phys. Chem. Lett., 2017, 8, 2526–2534 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS PubMed.
- L. Zhou, C. Zhang, M. J. McClain, A. Manjavacas, C. M. Krauter, S. Tian, F. Berg, H. O. Everitt, E. A. Carter and P. Nordlander, Nano Lett., 2016, 16, 1478–1484 CrossRef CAS PubMed.
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 339, 1590–1593 CrossRef CAS PubMed.
- C. Bauer, J.-P. Abid, D. Fermin and H. H. Girault, J. Chem. Phys., 2004, 120, 9302–9315 CrossRef CAS PubMed.
- C. Boerigter, U. Aslam and S. Linic, ACS Nano, 2016, 10, 6108–6115 CrossRef CAS PubMed.
- B. Foerster, A. Joplin, K. Kaefer, S. Celiksoy, S. Link and C. Sönnichsen, ACS Nano, 2017, 11, 2886–2893 CrossRef CAS PubMed.
- M. Wang, M. Ye, J. Iocozzia, C. Lin and Z. Lin, Adv. Sci., 2016, 3, 1600024 CrossRef PubMed.
- H. G. Baldoví, S. T. Neaţu, A. Khan, A. M. Asiri, S. A. Kosa and H. Garcia, J. Phys. Chem. C, 2015, 119, 6819–6827 CrossRef.
- J. S. DuChene, B. C. Sweeny, A. C. Johnston-Peck, D. Su, E. A. Stach and W. D. Wei, Angew. Chem., Int. Ed., 2014, 53, 7887–7891 CrossRef CAS PubMed.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. Chowdhury and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- K. Marchuk and K. A. Willets, Chem. Phys., 2014, 445, 95–104 CrossRef CAS.
- P. J. Schuck, Nat. Nanotechnol., 2013, 8, 799 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- D. B. Ingram and S. Linic, J. Am. Chem. Soc., 2011, 133, 5202–5205 CrossRef CAS PubMed.
- C. Gomes Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, M. N. Hedhili, H. Zhang and P. Wang, Nano Lett., 2013, 13, 14–20 CrossRef CAS PubMed.
- Y. Zhu, Z. Xu, W. Jiang, W. Yin, S. Zhong, P. Gong, R. Qiao, Z. Li and S. Bai, RSC Adv., 2016, 6, 56800–56806 RSC.
- M.-Z. Ge, C.-Y. Cao, S.-H. Li, Y.-X. Tang, L.-N. Wang, N. Qi, J.-Y. Huang, K.-Q. Zhang, S. Al-Deyab and Y.-K. Lai, Nanoscale, 2016, 8, 5226–5234 RSC.
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- S. Bera, J. E. Lee, S. B. Rawal and W. I. Lee, Appl. Catal., B, 2016, 199, 55–63 CrossRef CAS.
- X. Cheng, P. Dong, Z. Huang, Y. Zhang, Y. Chen, X. Nie and X. Zhang, J. CO2 Util., 2017, 20, 200–207 CrossRef CAS.
- J. Y. Do, R. K. Chava, K. K. Mandari, N.-K. Park, H.-J. Ryu, M. W. Seo, D. Lee, T. Senthil and M. Kang, Appl. Catal., B, 2018, 237, 895–910 CrossRef CAS.
- P. Christopher, D. B. Ingram and S. Linic, J. Phys. Chem. C, 2010, 114, 9173–9177 CrossRef CAS.
- X. Chen, Z. Zheng, X. Ke, E. Jaatinen, T. Xie, D. Wang, C. Guo, J. Zhao and H. Zhu, Green Chem., 2010, 12, 414–419 RSC.
- H. Zhang, X. Fan, X. Quan, S. Chen and H. Yu, Environ. Sci. Technol., 2011, 45, 5731–5736 CrossRef CAS PubMed.
- M. Misra, N. Singh and R. K. Gupta, Catal. Sci. Technol., 2017, 7, 570–580 RSC.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS PubMed.
- T. P. White and K. R. Catchpole, Appl. Phys. Lett., 2012, 101, 073905 CrossRef.
- A. Naldoni, T. Montini, F. Malara, M. M. Mróz, A. Beltram, T. Virgili, C. L. Boldrini, M. Marelli, I. Romero-Ocaña and J. J. Delgado, ACS Catal., 2017, 7, 1270–1278 CrossRef CAS.
- D. C. Ratchford, A. D. Dunkelberger, I. Vurgaftman, J. C. Owrutsky and P. E. Pehrsson, Nano Lett., 2017, 17, 6047–6055 CrossRef CAS PubMed.
- S. Tan, A. Argondizzo, J. Ren, L. Liu, J. Zhao and H. Petek, Nat. Photonics, 2017, 11, 806–812 CrossRef CAS.
- Y. Yu, Z. Ji, S. Zu, B. Du, Y. Kang, Z. Li, Z. Zhou, K. Shi and Z. Fang, Adv. Funct. Mater., 2016, 26, 6394–6401 CrossRef CAS.
- J.-Y. Bigot, V. Halté, J.-C. Merle and A. Daunois, Chem. Phys., 2000, 251, 181–203 CrossRef CAS.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632–635 CrossRef CAS PubMed.
- A. Hoggard, L.-Y. Wang, L. Ma, Y. Fang, G. You, J. Olson, Z. Liu, W.-S. Chang, P. M. Ajayan and S. Link, ACS Nano, 2013, 7, 11209–11217 CrossRef CAS PubMed.
- K. Wu, W. E. Rodríguez-Córdoba, Y. Yang and T. Lian, Nano Lett., 2013, 13, 5255–5263 CrossRef CAS.
- K. Watanabe, D. Menzel, N. Nilius and H.-J. Freund, Chem. Rev., 2006, 106, 4301–4320 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef.
- A. O. Govorov, W. Zhang, T. Skeini, H. Richardson, J. Lee and N. A. Kotov, Nanoscale Res. Lett., 2006, 1, 84 CrossRef.
- H. Fogler, Essentials of Chemical Reaction Engineering, 2011 Search PubMed.
- G. Baffou, P. Berto, E. Bermúdez Ureña, R. Quidant, S. Monneret, J. Polleux and H. Rigneault, ACS Nano, 2013, 7, 6478–6488 CrossRef CAS PubMed.
- J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Nano Lett., 2009, 9, 4417–4423 CrossRef CAS PubMed.
- Q. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed.
- Y. Sivan, J. H. Baraban and Y. Dubi, OSA Continuum, 2020, 3, 483–497 CrossRef.
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., 2014, 126, 2979–2984 CrossRef.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2019, 1903915 Search PubMed.
- C. Wang, O. Ranasingha, S. Natesakhawat, P. R. Ohodnicki, M. Andio, J. P. Lewis and C. Matranga, Nanoscale, 2013, 5, 6968–6974 RSC.
- K. H. Kim, K. Watanabe, D. Mulugeta, H.-J. Freund and D. Menzel, Phys. Rev. Lett., 2011, 107, 047401 CrossRef PubMed.
- S. Navalon, M. de Miguel, R. Martin, M. Alvaro and H. Garcia, J. Am. Chem. Soc., 2011, 133, 2218–2226 CrossRef CAS PubMed.
- M. J. Landry, A. Gellé, B. Y. Meng, C. J. Barrett and A. Moores, ACS Catal., 2017, 7, 6128–6133 CrossRef CAS.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- H. Song, X. Meng, Z.-J. Wang, Z. Wang, H. Chen, Y. Weng, F. Ichihara, M. Oshikiri, T. Kako and J. Ye, ACS Catal., 2018, 8, 7556–7565 CrossRef CAS.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- P. Li, G. Xiao, Y. Zhao and H. Su, ACS Catal., 2020, 10, 3640–3649 CrossRef CAS.
- J. Guo, Y. Zhang, L. Shi, Y. Zhu, M. F. Mideksa, K. Hou, W. Zhao, D. Wang, M. Zhao and X. Zhang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 1–16 CrossRef PubMed.
- Y. Kim, D. Dumett Torres and P. K. Jain, Nano Lett., 2016, 16, 3399–3407 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter and P. Nordlander, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- H. Song, X. Meng, T. D. Dao, W. Zhou, H. Liu, L. Shi, H. Zhang, T. Nagao, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 408–416 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- M. J. Kale, T. Avanesian, H. Xin, J. Yan and P. Christopher, Nano Lett., 2014, 14, 5405–5412 CrossRef CAS PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. Garcia, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- D. Mateo, J. Albero and H. García, Appl. Catal., B, 2018, 224, 563–571 CrossRef CAS.
- D. G. Cahill, P. V. Braun, G. Chen, D. R. Clarke, S. Fan, K. E. Goodson, P. Keblinski, W. P. King, G. D. Mahan and A. Majumdar, Appl. Phys. Rev., 2014, 1, 011305 Search PubMed.
- F. Menges, P. Mensch, H. Schmid, H. Riel, A. Stemmer and B. Gotsmann, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- M. Mecklenburg, W. A. Hubbard, E. White, R. Dhall, S. B. Cronin, S. Aloni and B. Regan, Science, 2015, 347, 629–632 CrossRef CAS PubMed.
- J. C. Idrobo, A. R. Lupini, T. Feng, R. R. Unocic, F. S. Walden, D. S. Gardiner, T. C. Lovejoy, N. Dellby, S. T. Pantelides and O. L. Krivanek, Phys. Rev. Lett., 2018, 120, 095901 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, H. Robatjazi, A. Alabastri, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2019, 364, eaaw9545 CrossRef CAS PubMed.
- Y. Sivan, J. Baraban, I. W. Un and Y. Dubi, Science, 2019, 364, eaaw9367 CrossRef CAS PubMed.
- C. D. Brites, P. P. Lima, N. J. Silva, A. Millán, V. S. Amaral, F. Palacio and L. D. Carlos, Nanoscale, 2012, 4, 4799–4829 RSC.
- D. Wang, Y. R. Koh, Z. A. Kudyshev, K. Maize, A. V. Kildishev, A. Boltasseva, V. M. Shalaev and A. Shakouri, Nano Lett., 2019, 19, 3796–3803 CrossRef CAS PubMed.
- M. V. Balois, N. Hayazawa, F. C. Catalan, S. Kawata, T.-A. Yano and T. Hayashi, Anal. Bioanal. Chem., 2015, 407, 8205–8213 CrossRef CAS PubMed.
- S. Xie, E. Iglesia and A. T. Bell, J. Phys. Chem. B, 2001, 105, 5144–5152 CrossRef CAS.
- W. Sun, G. Zhong, C. Kübel, A. A. Jelle, C. Qian, L. Wang, M. Ebrahimi, L. M. Reyes, A. S. Helmy and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 6329–6334 CrossRef CAS PubMed.
- J. Jia, H. Wang, Z. Lu, P. G. O'Brien, M. Ghoussoub, P. Duchesne, Z. Zheng, P. Li, Q. Qiao and L. Wang, Adv. Sci., 2017, 4, 1700252 CrossRef PubMed.
- G. W. Walker, V. C. Sundar, C. M. Rudzinski, A. W. Wun, M. G. Bawendi and D. G. Nocera, Appl. Phys. Lett., 2003, 83, 3555–3557 CrossRef CAS.
- S. Li, K. Zhang, J.-M. Yang, L. Lin and H. Yang, Nano Lett., 2007, 7, 3102–3105 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS.
- P. Löw, B. Kim, N. Takama and C. Bergaud, Small, 2008, 4, 908–914 CrossRef PubMed.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
- Y. Yue and X. Wang, Nano Rev., 2012, 3, 11586 CrossRef PubMed.
- V. A. Spata and E. A. Carter, ACS Nano, 2018, 12, 3512–3522 CrossRef CAS PubMed.
- Y. Yu, V. Sundaresan and K. A. Willets, J. Phys. Chem. C, 2018, 122, 5040–5048 CrossRef CAS.
- X. Zhang, X. Li, M. E. Reish, D. Zhang, N. Q. Su, Y. Gutiérrez, F. Moreno, W. Yang, H. O. Everitt and J. Liu, Nano Lett., 2018, 18, 1714–1723 CrossRef CAS PubMed.
- X. Li, H. O. Everitt and J. Liu, Nano Res., 2019, 12, 1906–1911 CrossRef CAS.
- C. Zhan, B.-W. Liu, Y.-F. Huang, S. Hu, B. Ren, M. Moskovits and Z.-Q. Tian, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- X. Li, H. O. Everitt and J. Liu, Nano Res., 2020, 1–13 Search PubMed.
- M. Najafpour, Artificial photosynthesis, BoD–Books on Demand, 2012 Search PubMed.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef CAS.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu and L.-C. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- A. Nawaz, A. Kuila, A. Rani, N. S. Mishra, L. C. Sim, K. H. Leong and P. Saravanan, Industrial Applications of Nanomaterials, Elsevier, 2019, pp. 151–179 Search PubMed.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS PubMed.
- R. Poudyal, I. Tiwari, A. Koirala, H. Masukawa, K. Inoue, T. Tomo, M. Najafpour, S. Allakhverdiev and T. Veziroğlu, Compendium of Hydrogen Energy, Elsevier, 2015, pp. 289–317 Search PubMed.
- J. H. Lee, J. Y. Do, N.-K. Park, M. W. Seo, H.-J. Ryu, J.-P. Hong, Y. S. Kim, S. K. Kim and M. Kang, J. Photochem. Photobiol., A, 2018, 364, 219–232 CrossRef CAS.
- Y. Li, C. Wang, M. Song, D. Li, X. Zhang and Y. Liu, Appl. Catal., B, 2019, 243, 760–770 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- J. Low, B. Cheng and J. Yu, Appl. Surf. Sci., 2017, 392, 658–686 CrossRef CAS.
- G. Yin, X. Huang, T. Chen, W. Zhao, Q. Bi, J. Xu, Y. Han and F. Huang, ACS Catal., 2018, 8, 1009–1017 CrossRef CAS.
- F. Saladin and I. Alxneit, J. Chem. Soc., Faraday Trans., 1997, 93, 4159–4163 RSC.
- K. Schwartzenberg, J. Hamilton, A. K. Lucid, E. Weitz, J. Notestein, M. Nolan, J. A. Byrne and K. A. Gray, Catal. Today, 2017, 280, 65–73 CrossRef CAS.
- L. Wang, X. Liu, Y. Dang, H. Xie, Q. Zhao and L. Ye, Solid State Sci., 2019, 89, 67–73 CrossRef CAS.
- L. Zhang, G. Kong, Y. Meng, J. Tian, L. Zhang, S. Wan, J. Lin and Y. Wang, ChemSusChem, 2017, 10, 4709–4714 CrossRef CAS PubMed.
- M. Sun, W. Hao, C. Wang and T. Wang, Chem. Phys. Lett., 2007, 443, 342–346 CrossRef CAS.
- A. Tang, Y. Jia, S. Zhang, Q. Yu and X. Zhang, Catal. Commun., 2014, 50, 1–4 CrossRef CAS.
- L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2006, 6, 2060–2065 CrossRef CAS PubMed.
- Z. Zhang, S. Wang, T. Yu and T. Wu, J. Phys. Chem. C, 2007, 111, 17500–17505 CrossRef CAS.
- D. Kumar, C. H. Park and C. S. Kim, J. Mater. Chem. A, 2020, 8, 5734–5743 RSC.
- H. Wang, Y. Wang, L. Guo, X. Zhang, C. Ribeiro and T. He, Chin. J. Catal., 2020, 41, 131–139 CrossRef CAS.
- F. Yu, C. Wang, H. Ma, M. Song, D. Li, Y. Li, S. Li, X. Zhang and Y. Liu, Nanoscale, 2020, 12, 7000–7010 RSC.
- Q. Guo, Q. Zhang, H. Wang, Z. Liu and Z. Zhao, Catal. Commun., 2016, 77, 118–122 CrossRef CAS.
- J. Li, Y. Ye, L. Ye, F. Su, Z. Ma, J. Huang, H. Xie, D. E. Doronkin, A. Zimina and J.-D. Grunwaldt, J. Mater. Chem. A, 2019, 7, 2821–2830 RSC.
- L. Wang, Y. Wang, Y. Cheng, Z. Liu, Q. Guo, M. N. Ha and Z. Zhao, J. Mater. Chem. A, 2016, 4, 5314–5322 RSC.
- L. Wang, M. N. Ha, Z. Liu and Z. Zhao, Integr. Ferroelectr., 2016, 172, 97–108 CrossRef CAS.
- Y. Bai, P. Yang, P. Wang, H. Xie, H. Dang and L. Ye, J. CO2 Util., 2018, 23, 51–60 CrossRef CAS.
- K. Wang, R. Jiang, T. Peng, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2019, 256, 117780 CrossRef.
- M. Xu, X. Hu, S. Wang, J. Yu, D. Zhu and J. Wang, J. Catal., 2019, 377, 652–661 CrossRef CAS.
- J. Low, L. Zhang, B. Zhu, Z. Liu and J. Yu, ACS Sustainable Chem. Eng., 2018, 6, 15653–15661 CrossRef CAS.
- G. Zhang, G. Liu, L. Wang and J. T. Irvine, Chem. Soc. Rev., 2016, 45, 5951–5984 RSC.
- D. W. Hwang, H. G. Kim, J. Kim, K. Y. Cha, Y. G. Kim and J. S. Lee, J. Catal., 2000, 193, 40–48 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- X. Sun, Y. Mi, F. Jiao and X. Xu, ACS Catal., 2018, 8, 3209–3221 CrossRef CAS.
- L. Xu, M. N. Ha, Q. Guo, L. Wang, Y. Ren, N. Sha and Z. Zhao, RSC Adv., 2017, 7, 45949–45959 RSC.
- G. Wei, D. Zheng, L. Xu, Q. Guo, J. Hu, N. Sha and Z. Zhao, Mater. Res. Express, 2019, 6, 086221 CrossRef CAS.
- M. N. Ha, G. Lu, Z. Liu, L. Wang and Z. Zhao, J. Mater. Chem. A, 2016, 4, 13155–13165 RSC.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 1–12 CrossRef CAS.
- L. Pastor-Pérez, F. Baibars, E. Le Sache, H. Arellano-García, S. Gu and T. R. Reina, J. CO2 Util., 2017, 21, 423–428 CrossRef.
- X. Yang, X. Su, X. Chen, H. Duan, B. Liang, Q. Liu, X. Liu, Y. Ren, Y. Huang and T. Zhang, Appl. Catal., B, 2017, 216, 95–105 CrossRef CAS.
- C. Xu, W. Huang, Z. Li, B. Deng, Y. Zhang, M. Ni and K. Cen, ACS Catal., 2018, 8, 6582–6593 CrossRef CAS.
- W. Sun, C. Qian, L. He, K. K. Ghuman, A. P. Wong, J. Jia, A. A. Jelle, P. G. O’Brien, L. M. Reyes and T. E. Wood, Nat. Commun., 2016, 7, 1–9 CrossRef.
- R. Costi, A. E. Saunders, E. Elmalem, A. Salant and U. Banin, Nano Lett., 2008, 8, 637–641 CrossRef CAS PubMed.
- R. Kaur and B. Pal, J. Mol. Catal. A: Chem., 2012, 355, 39–43 CrossRef CAS.
- I. Tanabe, T. Ryoki and Y. Ozaki, RSC Adv., 2015, 5, 13648–13652 RSC.
- A. A. Upadhye, I. Ro, X. Zeng, H. J. Kim, I. Tejedor, M. A. Anderson, J. A. Dumesic and G. W. Huber, Catal. Sci. Technol., 2015, 5, 2590–2601 RSC.
- B. Lu, F. Quan, Z. Sun, F. Jia and L. Zhang, Catal. Commun., 2019, 129, 105724 CrossRef CAS.
- Z. Zhao, D. E. Doronkin, Y. Ye, J.-D. Grunwaldt, Z. Huang and Y. Zhou, Chin. J. Catal., 2020, 41, 286–293 CrossRef CAS.
- N. Kong, B. Han, Z. Li, Y. Fang, K. Feng, Z. Wu, S. Wang, A.-B. Xu, Y. Yu, C. Li, Z. Lin and L. He, ACS Appl. Nano Mater., 2020, 3, 3028–3033 CrossRef CAS.
- S. Ning, H. Xu, Y. Qi, L. Song, Q. Zhang, S. Ouyang and J. Ye, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS.
- H. Zhang, T. Wang, J. Wang, H. Liu, T. D. Dao, M. Li, G. Liu, X. Meng, K. Chang, L. Shi, T. Nagao and J. Ye, Adv. Mater., 2016, 28, 3703–3710 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- L. B. Hoch, P. G. O’Brien, A. Jelle, A. Sandhel, D. D. Perovic, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 9017–9025 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- D. Wu, K. Deng, B. Hu, Q. Lu, G. Liu and X. Hong, ChemCatChem, 2019, 11, 1598–1601 CrossRef CAS.
- E. T. Kho, T. H. Tan, E. Lovell, R. J. Wong, J. Scott and R. Amal, Green Energy Environ., 2017, 2, 204–217 CrossRef.
- P. G. O’Brien, K. K. Ghuman, A. A. Jelle, A. Sandhel, T. E. Wood, J. Y. Y. Loh, J. Jia, D. Perovic, C. V. Singh, N. P. Kherani, C. A. Mims and G. A. Ozin, Energy Environ. Sci., 2018, 11, 3443–3451 RSC.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- E. T. Kho, S. Jantarang, Z. Zheng, J. Scott and R. Amal, Engineering, 2017, 3, 393–401 CrossRef.
- S. Jantarang, E. C. Lovell, T. H. Tan, J. Scott and R. Amal, Prog. Nat. Sci.: Mater. Int., 2018, 28, 168–177 CrossRef CAS.
- Y. Li, J. Hao, H. Song, F. Zhang, X. Bai, X. Meng, H. Zhang, S. Wang, Y. Hu and J. Ye, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- K. R. Thampi, J. Kiwi and M. Graetzel, Nature, 1987, 327, 506–508 CrossRef CAS.
- K. K. Bando, H. Arakawa and N. Ichikuni, Catal. Lett., 1999, 60, 125–132 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- C. Wang, S. Fang, S. Xie, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 7390–7394 RSC.
- L. Lin, K. Wang, K. Yang, X. Chen, X. Fu and W. Dai, Appl. Catal., B, 2017, 204, 440–455 CrossRef CAS.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- J. Ren, S. Ouyang, H. Xu, X. Meng, T. Wang, D. Wang and J. Ye, Adv. Energy Mater., 2017, 7, 1601657 CrossRef.
- D. Mateo, J. Albero and H. García, Joule, 2019, 3, 1949–1962 CrossRef CAS.
- X. Li, H. O. Everitt and J. Liu, Nano Res., 2019, 12, 1906–1911 CrossRef CAS.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Nat. Commun., 2018, 9, 3027 CrossRef PubMed.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- L. Liu, A. V. Puga, J. Cored, P. Concepcion, V. Perez-Dieste, H. Garcia and A. Corma, Appl. Catal., B, 2018, 235, 186–196 CrossRef CAS.
- X.-N. Guo, Z.-F. Jiao, G.-Q. Jin and X.-Y. Guo, ACS Catal., 2015, 5, 3836–3840 CrossRef CAS.
- S. Yu, T. Zhang, Y. Xie, Q. Wang, X. Gao, R. Zhang, Y. Zhang and H. Su, Int. J. Hydrogen Energy, 2015, 40, 870–877 CrossRef CAS.
- C.-x. Xiao, Z.-p. Cai, T. Wang, Y. Kou and N. Yan, Angew. Chem., Int. Ed., 2008, 47, 746–749 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, X. Gu, Y. Zhang and H. Su, Catal. Sci. Technol., 2018, 8, 601–610 RSC.
- Z. Li, J. Liu, Y. Zhao, G. I. Waterhouse, G. Chen, R. Shi, X. Zhang, X. Liu, Y. Wei and X. D. Wen, Adv. Mater., 2018, 30, 1800527 CrossRef PubMed.
- W. Gao, R. Gao, Y. Zhao, M. Peng, C. Song, M. Li, S. Li, J. Liu, W. Li, Y. Deng, M. Zhang, J. Xie, G. Hu, Z. Zhang, R. Long, X.-D. Wen and D. Ma, Chem, 2018, 4, 2917–2928 CAS.
- Z. Li, J. Liu, Y. Zhao, R. Shi, G. I. Waterhouse, Y. Wang, L.-Z. Wu, C.-H. Tung and T. Zhang, Nano Energy, 2019, 60, 467–475 CrossRef CAS.
- Y. Wang, Y. Zhao, J. Liu, Z. Li, G. I. Waterhouse, R. Shi, X. Wen and T. Zhang, Adv. Energy Mater., 2020, 10, 1902860 CrossRef CAS.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- H. Song, X. Meng, Z.-J. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L.-Z. Wu, C.-H. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- H. Liu, X. Meng, T. D. Dao, H. Zhang, P. Li, K. Chang, T. Wang, M. Li, T. Nagao and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 11545–11549 CrossRef CAS PubMed.
- B. László, K. Baán, E. Varga, A. Oszkó, A. Erdőhelyi, Z. Kónya and J. Kiss, Appl. Catal., B, 2016, 199, 473–484 CrossRef.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494–497 CrossRef CAS.
- H. Liu, M. Li, T. D. Dao, Y. Liu, W. Zhou, L. Liu, X. Meng, T. Nagao and J. Ye, Nano Energy, 2016, 26, 398–404 CrossRef CAS.
- H. Liu, T. D. Dao, L. Liu, X. Meng, T. Nagao and J. Ye, Appl. Catal., B, 2017, 209, 183–189 CrossRef CAS.
- H. Liu, X. Meng, T. D. Dao, L. Liu, P. Li, G. Zhao, T. Nagao, L. Yang and J. Ye, J. Mater. Chem. A, 2017, 5, 10567–10573 RSC.
- Q. Zhang, M. Mao, Y. Li, Y. Yang, H. Huang, Z. Jiang, Q. Hu, S. Wu and X. Zhao, Appl. Catal., B, 2018, 239, 555–564 CrossRef CAS.
- H. Huang, M. Mao, Q. Zhang, Y. Li, J. Bai, Y. Yang, M. Zeng and X. Zhao, Adv. Energy Mater., 2018, 8, 1702472 CrossRef.
- H. Liu, H. Song, W. Zhou, X. Meng and J. Ye, Angew. Chem., Int. Ed., 2018, 57, 16781–16784 CrossRef CAS PubMed.
- M. Mao, Q. Zhang, Y. Yang, Y. Li, H. Huang, Z. Jiang, Q. Hu and X. Zhao, Green Chem., 2018, 20, 2857–2869 RSC.
- F. Pan, X. Xiang, W. Deng, H. Zhao, X. Feng and Y. Li, ChemCatChem, 2018, 10, 940–945 CrossRef CAS.
- Z. Jiang, Y. Li, Q. Zhang, Y. Yang, S. Wu, J. Wu and X. Zhao, J. Mater. Chem. A, 2019, 7, 4881–4892 RSC.
- H. Jiang, X. Peng, A. Yamaguchi, S. Ueda, T. Fujita, H. Abe and M. Miyauchi, Sol. RRL, 2019, 3, 1900076 CrossRef.
- S. Shoji, X. Peng, A. Yamaguchi, R. Watanabe, C. Fukuhara, Y. Cho, T. Yamamoto, S. Matsumura, M.-W. Yu and S. Ishii, Nat. Catal., 2020, 3, 148–153 CrossRef CAS.
- Y. Cho, S. Shoji, A. Yamaguchi, T. Hoshina, T. Fujita, H. Abe and M. Miyauchi, Chem. Commun., 2020, 56, 4611–4614 RSC.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- C. Mao, L. Yu, J. Li, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 224, 612–620 CrossRef CAS.
- D. F. Swearer, N. R. Knowles, H. O. Everitt and N. J. Halas, ACS Energy Lett., 2019, 4, 1505–1512 CrossRef CAS.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706–1711 CrossRef CAS PubMed.
- S. Duhr and D. Braun, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19678 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- C. Li, T. Wang, Z.-J. Zhao, W. Yang, J.-F. Li, A. Li, Z. Yang, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 5278–5282 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- W.-H. Chen and C.-Y. Chen, Appl. Energy, 2020, 258, 114078 CrossRef CAS.
- Y. Choi and H. G. Stenger, J. Power Sources, 2003, 124, 432–439 CrossRef CAS.
- F. Liu, L. Song, S. Ouyang and H. Xu, Catal. Sci. Technol., 2019, 9, 2125–2131 RSC.
- L. Zhao, Y. Qi, L. Song, S. Ning, S. Ouyang, H. Xu and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 7708–7712 CrossRef CAS PubMed.
- Q. Li and G. Lu, Catal. Lett., 2008, 125, 376–379 CrossRef CAS.
- L. Gu, C. Zhang, Y. Guo, J. Gao, Y. Yu and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3710–3714 CrossRef CAS.
- W. Hao, R. Wu, H. Yang and Y. Guo, J. Mater. Chem. A, 2019, 7, 12440–12445 RSC.
- L. Zhao, Z. Yang, Q. Cao, L. Yang, X. Zhang, J. Jia, Y. Sang, H.-J. Wu, W. Zhou and H. Liu, Nano Energy, 2019, 56, 563–570 CrossRef CAS.
- Y. Wu, Y. Sun, W. Fu, X. Meng, M. Zhu, S. Ramakrishna and Y. Dai, ACS Appl. Nano Mater., 2020, 3, 2713–2722 CrossRef CAS.
- S. Rej, L. Mascaretti, E. Y. Santiago, O. Tomanec, S. Kment, Z. Wang, R. Zbořil, P. Fornasiero, A. O. Govorov and A. Naldoni, ACS Catal., 2020, 10, 5261–5271 CrossRef CAS.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- J. C. Kennedy III and A. K. Datye, J. Catal., 1998, 179, 375–389 CrossRef.
- T. H. Tan, J. Scott, Y. H. Ng, R. A. Taylor, K.-F. Aguey-Zinsou and R. Amal, ACS Catal., 2016, 6, 1870–1879 CrossRef CAS.
- J. Bi, H. Cai, B. Wang, C. Kong and S. Yang, Chem. Commun., 2019, 55, 3943–3946 RSC.
- J. Wang, G. Zhang and P. Zhang, Appl. Catal., B, 2018, 239, 77–85 CrossRef CAS.
- X. Chen, S. Cai, E. Yu, J. Li, J. Chen and H. Jia, Appl. Surf. Sci., 2019, 484, 479–488 CrossRef CAS.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS PubMed.
- C. Zhang, J. Lu, N. Jin, L. Dong, Z. Fu, Z. Zhang and H. Zheng, Small, 2019, 15, 1901286 CrossRef PubMed.
- H. Fujiwara, T. Suzuki, C. Pin and K. Sasaki, Nano Lett., 2019, 20, 389–394 CrossRef PubMed.
- J. Mock, M. Barbic, D. Smith, D. Schultz and S. Schultz, J. Chem. Phys., 2002, 116, 6755–6759 CrossRef CAS.
- J. Zhang, H. Liu, Z. Wang and N. Ming, Adv. Funct. Mater., 2007, 17, 3295–3303 CrossRef CAS.
- S. Dal Forno, L. Ranno and J. Lischner, J. Phys. Chem. C, 2018, 122, 8517–8527 CrossRef CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- Q. Zhang, W. Li, C. Moran, J. Zeng, J. Chen, L.-P. Wen and Y. Xia, J. Am. Chem. Soc., 2010, 132, 11372–11378 CrossRef CAS PubMed.
- B. J. Wiley, Y. Chen, J. M. McLellan, Y. Xiong, Z.-Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS PubMed.
- B. J. Wiley, Y. Xiong, Z.-Y. Li, Y. Yin and Y. Xia, Nano Lett., 2006, 6, 765–768 CrossRef CAS PubMed.
- M. J. Mulvihill, X. Y. Ling, J. Henzie and P. Yang, J. Am. Chem. Soc., 2010, 132, 268–274 CrossRef CAS PubMed.
- H. Zhang and A. O. Govorov, J. Phys. Chem. C, 2014, 118, 7606–7614 CrossRef CAS.
- L. V. Besteiro and A. O. Govorov, J. Phys. Chem. C, 2016, 120, 19329–19339 CrossRef CAS.
- H. Wu, J. Carrete, Z. Zhang, Y. Qu, X. Shen, Z. Wang, L.-D. Zhao and J. He, NPG Asia Mater., 2014, 6, e108–e108 CrossRef CAS.
- J. Lim, K. Hippalgaonkar, S. C. Andrews, A. Majumdar and P. Yang, Nano Lett., 2012, 12, 2475–2482 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- G. Baffou, R. Quidant and F. J. García de Abajo, ACS Nano, 2010, 4, 709–716 CrossRef CAS PubMed.
- G. Baffou, R. Quidant and C. Girard, Appl. Phys. Lett., 2009, 94, 153109 CrossRef.
- K. Manthiram and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 3995–3998 CrossRef CAS PubMed.
- Z. Fang, S. Jiao, Y. Kang, G. Pang and S. Feng, ChemistryOpen, 2017, 6, 261–265 CrossRef CAS PubMed.
- R. Du, W. Liu, H. Bai, H. Wang and G. Xi, RSC Adv., 2020, 10, 2075–2084 RSC.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- J. Espín, L. Garzón-Tovar, A. Carné-Sánchez, I. Imaz and D. Maspoch, ACS Appl. Mater. Interfaces, 2018, 10, 9555–9562 CrossRef PubMed.
- X. Zhang, X. Ke and H. Zhu, Chem. – Eur. J., 2012, 18, 8048–8056 CrossRef CAS PubMed.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- D. Kumar, C. H. Park and C. S. Kim, ACS Sustainable Chem. Eng., 2018, 6, 8604–8614 CrossRef CAS.
- H. Huang, L. Zhang, Z. Lv, R. Long, C. Zhang, Y. Lin, K. Wei, C. Wang, L. Chen and Z.-Y. Li, J. Am. Chem. Soc., 2016, 138, 6822–6828 CrossRef CAS PubMed.
- L. Guo, Q. Sun, K. Marcus, Y. Hao, J. Deng, K. Bi and Y. Yang, J. Mater. Chem. A, 2018, 6, 22005–22012 RSC.
- S. Okeil, S. Yadav, M. Bruns, A. Zintler, L. Molina-Luna and J. J. Schneider, Dalton Trans., 2020, 49, 1032–1047 RSC.
- P. Ren and X. Yang, Sol. RRL, 2018, 2, 1700233 CrossRef.
- X. Niu, Y. Li, Y. Zhang, Z. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 17987–17993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00357c |
| This journal is © The Royal Society of Chemistry 2021 |