
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic methods for amino acid modification
Thomas A.
King
 ,
Jiyan
Mandrup Kandemir
,
Stephen J.
Walsh
and
David R.
Spring
*
,
Jiyan
Mandrup Kandemir
,
Stephen J.
Walsh
and
David R.
Spring
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
First published on 11th November 2020
Abstract
Amino acid modification plays an important role across several fields, including synthetic organic chemistry, materials science, targeted drug delivery and the probing of biological function. Although a myriad of methods now exist for the modification of peptides or proteins, many of these target a handful of the most reactive proteinogenic amino acids. Photocatalysis has recently emerged as a mild approach for amino acid modification, generating a sizable toolbox of reactions capable of modifying almost all of the canonical amino acids. These reactions are characterised by their mild, physiologically compatible conditions, greatly enhancing their usefulness for amino acid modification. This review aims to introduce the field of photocatalytic amino acid modification and discusses the most recent advances.
 Thomas A. King | Thomas King received his Masters degree from the University of Cambridge in 2018, during which he conducted research in the group of Professor David Spring in the field of diversity-oriented synthesis. He began his PhD in the same group later that year. His current research focus is on the use of photoredox catalysis for amino acid modification. |
 Jiyan Mandrup Kandemir | Jiyan Mandrup Kandemir received his MSc in Medicinal Chemistry from the University of Copenhagen in 2019, having worked on the synthesis and evaluation of HDAC inhibitors in the laboratory of Professor Christian Adam Olsen. He is currently a PhD student in the laboratory of Professor David Spring at the University of Cambridge, where his current research focuses on the use of photoredox catalysis for the modification of amino acids. |
 Stephen J. Walsh | Stephen Walsh received his PhD in 2019 from the University of Cambridge, where he worked on bioconjugation methodology development in the laboratory of Professor David Spring. He is currently a postdoctoral research associate in the same group, where his work involves the development of new biotherapeutic modalities. |
 David R. Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
Key learning points(1) Amino acids are involved across a wide range of fields, from organic synthesis to biology to materials science.(2) Side chains of amino acids provide unique properties to peptides or proteins. (3) Side chains of amino acids can be selectively targeted to enable modification of the amino acid, peptide or protein. (4) Photocatalysis uses mild conditions to facilitate modification, making it appropriate for use on biological systems. (5) Appropriate selection of photocatalyst, based on redox potentials, can enable selective modification of most amino acids. |
1. Introduction
α-Amino acids are the building blocks of peptides and proteins synthesised by living cells.1 Containing a primary amine and a carboxylic acid, separated by a single carbon atom, amino acids are primed for amide, or peptide, bond formation (Fig. 1). Nature provides twenty common amino acids, with side chains displaying polar, aromatic or aliphatic groups. Within proteins, these groups provide interactions for stabilising the three-dimensional, tertiary, structure. On the surface, these groups provide unique environments for recognition of other biomolecules. Within proteins with catalytic function, enzymes, nearby side chains set the steric and electronic requirements for the binding of substrate molecules and can contribute to catalysis.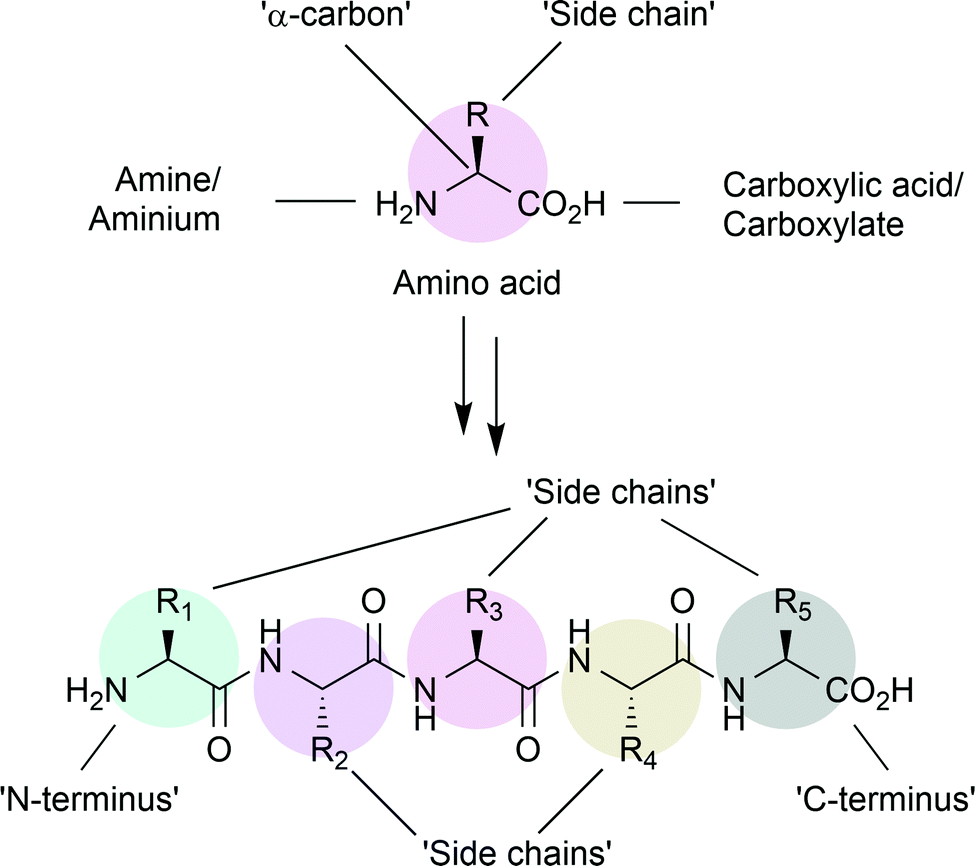 | ||
| Fig. 1 Basics of amino acid nomenclature. Amino acids, and peptides, contain an amino (N) and a carboxy (C) terminus. Each individual amino acid also has a side chain, of which there are 20 commonly found in nature, and which proffer specific properties to peptides. | ||
Researchers typically produce peptides or proteins for study in one of two ways. Peptides and short proteins may be chemically synthesised by solid-phase peptide synthesis (SPPS). This method can easily facilitate the introduction of non-canonical amino acids (ncAAs), and while this was previously limited to peptides of less than 50 AA, recent developments have drastically increased that limit.2 Alternatively, larger peptides and proteins may be produced by in vitro cellular expression, by introduction of the encoding DNA sequence into the expression host. While large proteins are more easily produced using this method, introduction of ncAAs in vitro requires the use of codon-suppression technology and addition of the ncAA to the cell suspension.3
Amino acid modification is relevant across multiple fields (Fig. 2). In nature itself, many amino acid residues have post-translational modifications (PTMs); phosphate groups, acetyl groups, sulfonate groups and methyl groups are all known to be added to amino acid side chains after the translation of an mRNA strand into a peptide to transiently control their activity and increase the structural diversity available from just 20 available building blocks.4 Mimicking these modifications can help researchers study their effects. Beyond simple PTMs, a range of other chemical moieties have been artificially added to peptides or proteins.5 For instance, addition of chemical moieties (e.g. fluorophores) to proteins of interest can aid in the elucidation of cellular processes (e.g. subcellular protein localisation). Alternatively, addition of a cytotoxic or modulatory drug to a targeting protein (e.g. a tumour-specific antibody) can produce a biotherapeutic with superior efficacy and tolerability compared to chemotherapy.6 As readily available chiral reagents, α-amino acids are used extensively in synthetic organic chemistry. Therefore, methods for the modification of the common 20 amino acids enhances their usefulness as building blocks. Beyond this, biocatalytic transformations are now widespread in organic synthesis. While separation of an enzyme from a reaction mixture can be challenging, modification of the enzyme such that it is attached to a solid support (at a residue which is not significant for enzyme activity), purification can be greatly simplified. Finally, the use of biomolecules in the development of materials is garnering increased interest.7
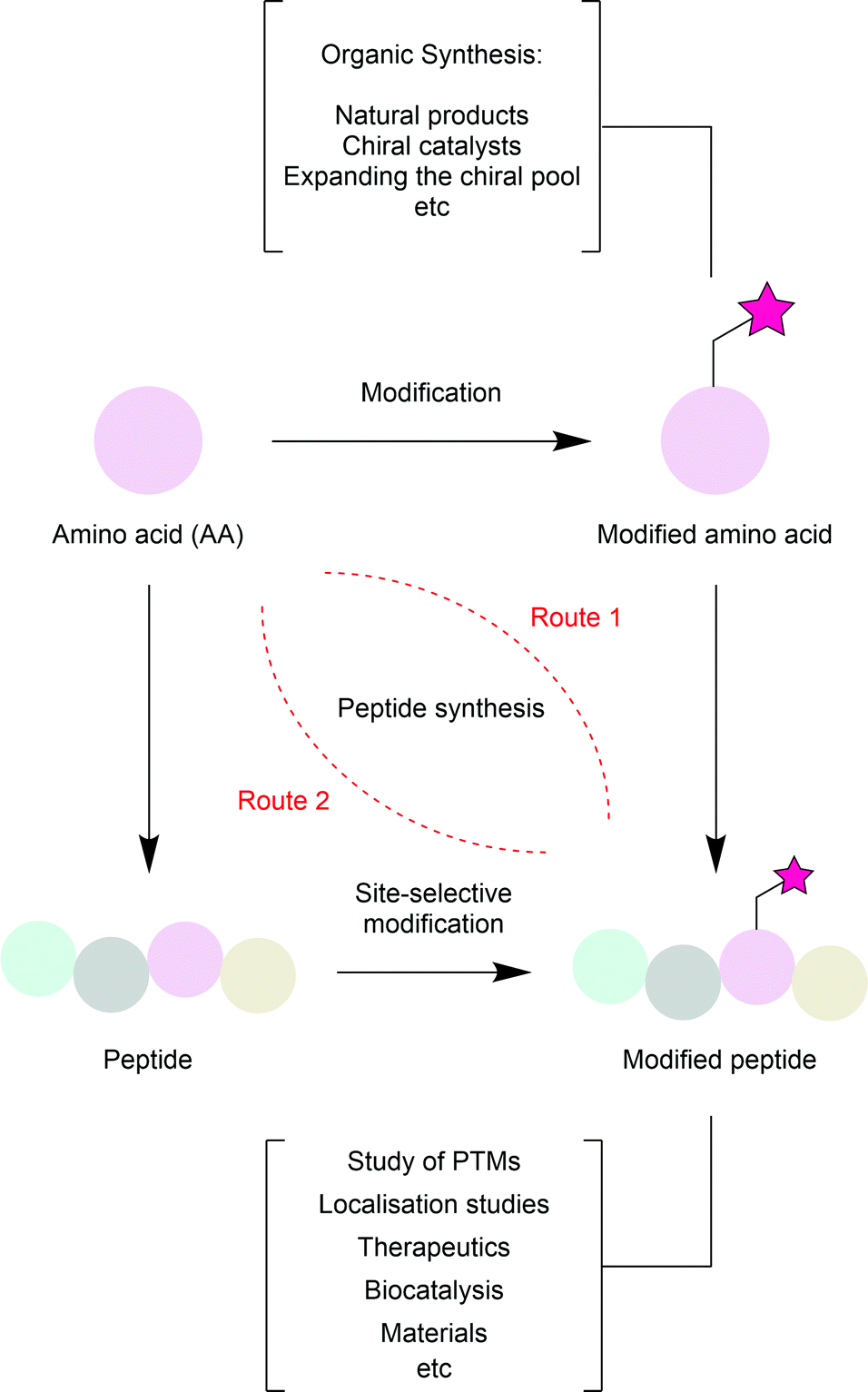 | ||
| Fig. 2 Routes to, and applications of, amino acid and peptide modification. Amino acids and their derivatives can be used in chemical synthesis as chiral reagents or building blocks to control or install stereochemistry. Amino acids can also be used to generate modified peptides via two distinct routes. Modification can be either made to an individual amino acid which is then incorporated during peptide synthesis (route 1) or achieved site-selectively after peptide synthesis (route 2). Modification of peptides can aid discovery across a wide variety of fields. | ||
The modification of an amino acid greatly expands its utility in chemical synthesis. Many synthetic strategies rely on naturally occurring chirality to control the stereoselectivity of reactions, either using chiral catalysts (reagent control), or through the influence of chiral centres within the substrate (substrate control). α-Amino acids constitute a significant portion of the so-called ‘chiral pool’ used by researchers to this end. There are 20 commonly available amino acids, which can provide unique functionality within a synthesis, either through incorporation into the substrate, or for the generation of catalysts. Methods for modifying an amino acid without epimerisation of the chiral centre would enable the generation of a far wider selection of reagents, thereby deepening the chiral pool. These alternative amino acids can also be used in peptide or protein synthesis as ncAAs.
The modification of an amino acid within a peptide or protein through the addition of a chemical moiety is commonly known as bioconjugation. Bioconjugation is also used more generally to describe the chemical addition of a moiety to any biological molecule. It has long been established that these added moieties can have a profound impact on the behaviour of the biomolecule of interest.
Any bioconjugation reaction must consider the implications of the chemistry on the substrate. Peptides and proteins are typically folded into an intricate secondary and often tertiary structure, formed in their native cellular environment. This can be disrupted by reagents (such as ureas), pH fluctuation, or heat, leading to a breakdown of structure and consequently function.1 Therefore, bioconjugation protocols must utilise exceptionally mild reaction conditions to maintain the structure of the biomolecule. Additionally, many peptides or proteins use particular side chains or groups of side chains to carry out their function. The purpose of the modification must therefore be carefully considered; to block or inhibit function through modification, an amino acid side chain which is required for activity should be targeted. Conversely, if retained function is desired, modification distal to the functional site or domain should be undertaken. An ‘ideal’ bioconjugation strategy can therefore be said to involve:
• Site-selectivity (chemo- and regioselective)
• Compatibility with physiological conditions (ca. 37 °C, aqueous solvent, neutral pH)
• Fast reaction kinetics
• Minimal, non-toxic by-products
Using traditional chemistry, the vast majority of bioconjugation tools have made use of the nucleophilicity of a handful of amino acid side chains, particularly lysine or cysteine, to install the modification.8 An alternative strategy which has been widely used in protein or peptide modification is the incorporation of non-canonical amino acids with side chains that contain bioorthogonally reactive functional groups (e.g. azides, ketones).9 These groups are then amenable to selective modification with the desired payload containing a complementary reactive handle. While effective, these strategies require genetic code expansion for ribosomal installation in a growing peptide chain and/or chemical synthesis of the complex ncAAs. In recent years, transition metal catalysis has also been applied to the modification of amino acids, peptides and proteins.10
Photocatalysis has recently emerged as a mild method for amino acid modification.11 Prominent in the attractive attributes of photocatalysis are the mild conditions often required and the potential modification of residues with otherwise inert side chains. Through photocatalysis, radical generation is simplified greatly – the need for high energy reagents or initiators, which are commonly required in traditional radical processes, is eliminated. Furthermore, visible-light-mediated methods for radical generation can typically be achieved at ambient temperature. For amino acid modification, these mild conditions reduce the chance of epimerisation of the α-carbon. While some photoredox-catalysed methods are only suitable for individual amino acid modification, the mild conditions can also allow for application of the chemistry to peptides, or even proteins.
This tutorial review provides an introduction to photoredox catalysis for amino acid modification, both individually and within larger peptide sequences. The underlying principles of photoredox catalysis are described, followed by implications for amino acid modification. For each amino acid, a brief history of its photocatalytic modification is provided, along with the most recent advances. Of particular note is the recent development of C–H functionalisation methods, which have enabled modification of amino acids for which there are currently limited alternatives. For discussions of other amino acid modification strategies please refer to other reviews of the field.8,12–16
2. Photoredox catalysis
The advent of visible-light photosensitisers has re-energised research into light-mediated transformations. Light has been used in chemistry for decades, for example to promote bond cleavage (e.g. initiators such as azobisisobutyronitrile (AIBN)) or enable thermally-prohibited transformations by promoting alkenes to their excited states (e.g. cycloadditions/pericyclic reactions). However, with the discovery of molecules which interact with visible light came the realisation that they can act as mediators for a plethora of chemical transformations. Using visible light, a photosensitiser (or photocatalyst) and a substrate with an appropriately matched redox potential, radical species can be generated under remarkably mild conditions, and subsequently harnessed to achieve chemical transformations.17,18The photocatalytic cycle for most photochemical processes discussed here involves electron transfer between excited catalyst and substrate, although energy transfer is also possible.19 Photoredox catalysis begins with the electronic excitation of an organic or inorganic compound (PC), through absorption of light (Fig. 3). These excited state molecules (PC*) then undergo quenching, typically through single electron transfer (SET), resulting in oxidation (blue) or reduction (red) of a substrate. The substrate is consequently activated for bond cleavage, atom abstraction, or nucleophilic or electrophilic attack. After quenching, the oxidised or reduced catalyst (PCox or PCred) regains, or loses, an electron to return to the starting ground state catalyst (PC).
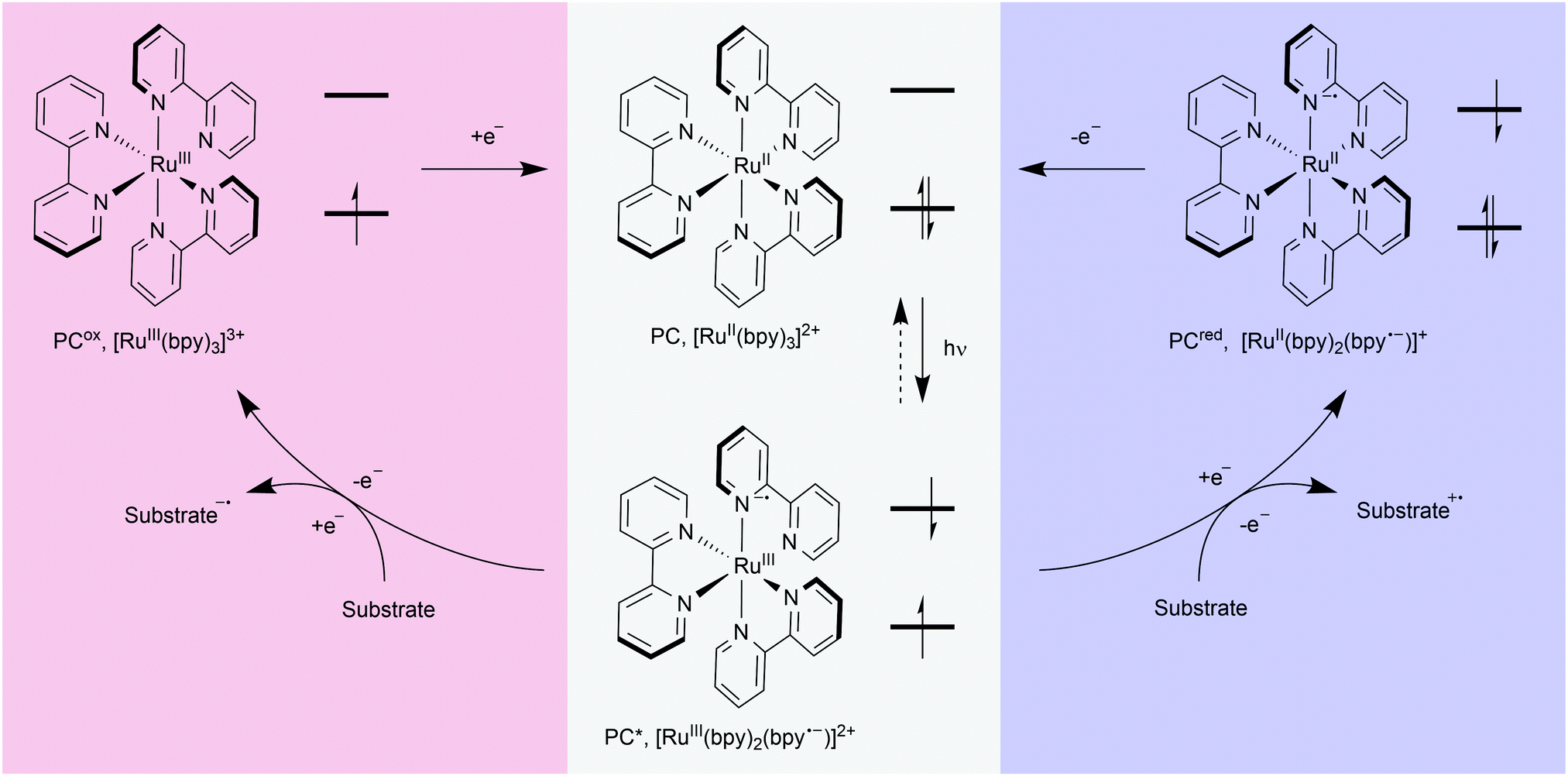 | ||
| Fig. 3 Visible-light-mediated activation of a photoredox catalyst: upon absorption of light of an appropriate wavelength, the photocatalyst (PC) becomes excited (usually denoted by an asterisk), with one of its electrons promoted to a higher energy orbital. This excited state is activated for electron (or energy) transfer. Substrates with a higher redox potential can accept an electron from the excited catalyst (red); substrates with a lower redox potential can lose an electron to the excited catalyst (blue). In this way, organic radicals are formed on the substrates, which can then undergo further reaction. Subsequently, electrons are returned to, or taken from, the intermediate catalyst species to regenerate the ground state (GS) catalyst and close the catalytic cycle. Bpy = 2,2′-bipyridyl. | ||
In developing a photocatalytic protocol, several key properties of the catalyst must be considered: redox potential, excited state lifetime, absorption maximum, and solubility. If oxidation of the substrate is desired, a photocatalyst which has an excited state reduction potential above that of the substrate is required. Conversely, if reduction is desired, the reverse is true. Photocatalysts have been discovered or developed, with a wide range of excited state and ground state redox potentials (most within ca. ±2 V vs. the standard hydrogen electrode, SHE), thus enabling selection of the most appropriate catalyst for a chosen substrate. Significantly, the lifetime of the excited state of the catalyst before spontaneous return to the ground state varies greatly across catalysts. Catalysts with longer lifetimes allow for a greater probability of reaction, as the probability of the reactive catalyst species associating with substrate molecules increases with increased excited state lifetime. Transition metal-based catalysts tend to have longer excited state lifetimes than purely organic photocatalysts (μs compared to ns). Finally, the wavelength at which the catalyst absorbs is also important. Matching the light source with the absorption maximum of the catalyst will greatly increase the proportion of catalyst molecules in the excited state, thereby increasing the rate of reaction. Most commonly used photocatalysts absorb in the blue region of the visible spectrum, although new catalysts are being developed which can absorb longer wavelength red light.20 Redox potentials, excited state lifetimes, and absorption maxima for commonly used photocatalysts are readily available.17,18
Practically, photocatalysis is simple to perform. After careful selection of an appropriate catalyst, a light source must be acquired. Despite calls for standardisation in recent years,21 most published reports detail the use of ‘shop-bought’ LED light sources. Provided the absorption maximum of your catalyst lies within the emitted wavelength of your source, any light should provide results. However, while household iridescent lightbulbs can provide the required photons, they often also emit large quantities of heat. LEDs, on the other hand, offer irradiation without excessive heating. Despite this, over time, minimal heating from the light source can significantly raise the reaction temperature. For this reason, a fan is often used to maintain constant reaction conditions.
3. Photocatalytic amino acid modification
Many side chains of amino acids, particularly those with heteroatoms, have appropriate oxidation potentials for quenching excited state photocatalysts. With careful control of oxidation potentials via selection of an appropriate catalyst, selective oxidation or reduction of, and therefore radical generation at, amino acid residues can be performed. These radicals can then be coupled, generally with electrophiles or other radicals, to install the desired modification.Recent years have witnessed a significant increase in the number of photocatalytic methods being reported. While the majority of these do not focus solely on amino acid modification, several document investigations on amino acid or peptide substrates to showcase the utility of the methodology. In the following sections, advances aimed specifically at amino acid or peptide modification, and those that demonstrate applicability to these, will be discussed. To aid the reader in understanding the application of each method, a colour code will be used. Procedures used in modification of individual amino acids will be marked red, those demonstrated on peptide substrates will be marked blue, and those demonstrated on protein substrates will be marked green.
Polar amino acid side chains
Polar amino acids (see Fig. 4), most notably cysteine and lysine, have received great interest from the scientific community in the development of ionic methods for amino acid modification.8 However, they are also amenable to modification via radical processes, and have thus been targeted through photocatalysis.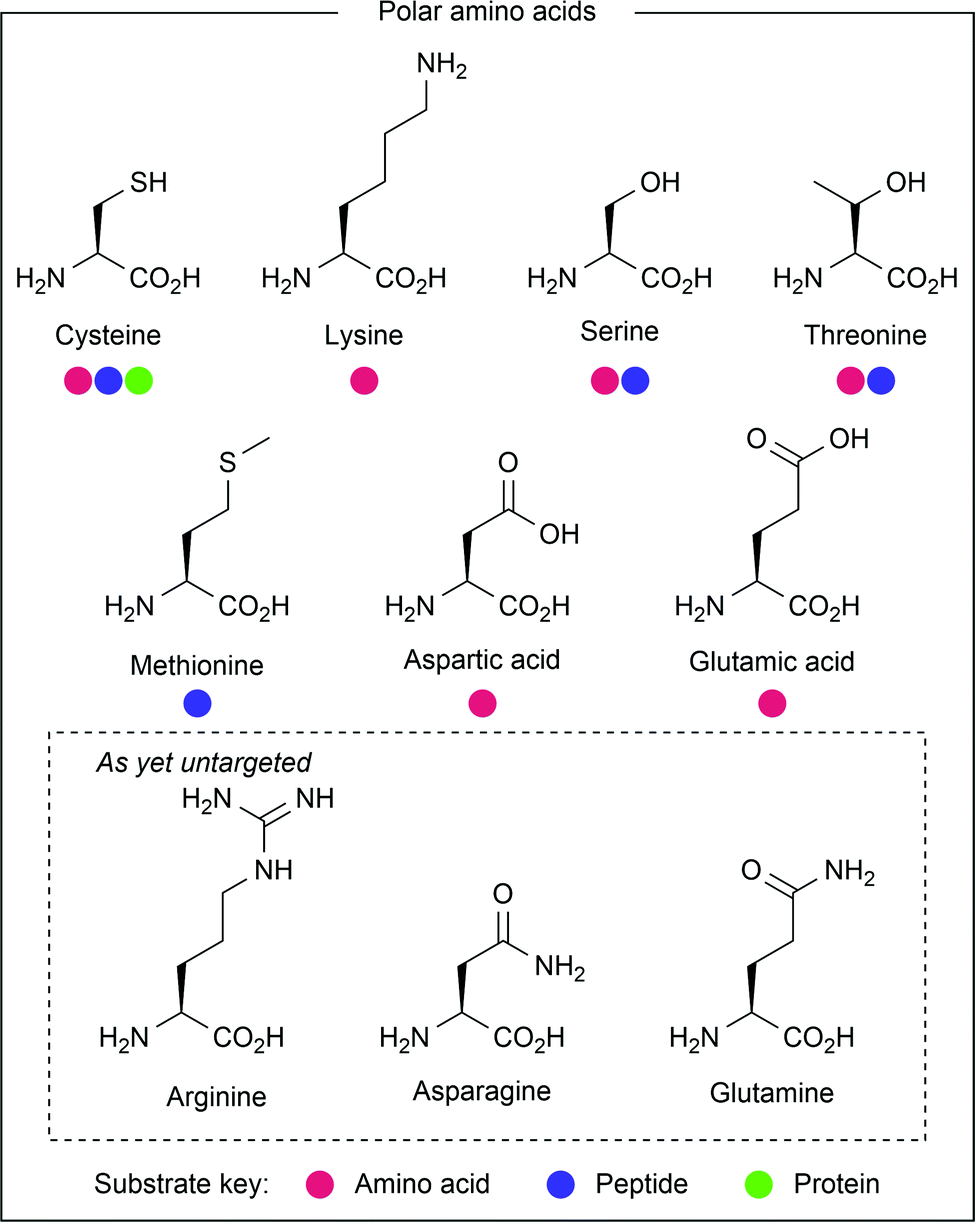 | ||
| Fig. 4 Amino acids containing polar side chains, or whose photochemical activity involves the presence of heteroatoms. | ||
Thiol–ene reactions, commonly achieved using chemical radical initiators or ultraviolet (UV) light, can also be achieved using visible light photocatalysis. In 2017, Wang and co-workers used allyl alcohols and amides as reagents for cysteine and thio-sugar modification (Fig. 5).22 An excited acridinium catalyst was used as the necessary oxidising agent, generating the thiyl radical after deprotonation. The alkene could then be attacked, and the product generated after a final hydrogen atom abstraction, possibly from unreacted thiol, thereby generating additional thiyl radical. Testing the reaction scope with benzyl mercaptan, allyl esters, amides, alcohols and silanes were shown to be competent coupling partners after irradiation for 6 hours (76–87%). Although few examples using cysteine were presented, those couplings were also achieved in high yields (75–90%), attaching a sugar, nucleic acid, or peptide to the residue. Of particular interest was the treatment of cysteine with the allyl ester of aspartic acid, which gave the cross-linked amino acids in high yield (85%), suggesting the potential utility of this methodology for peptide macrocyclisation.
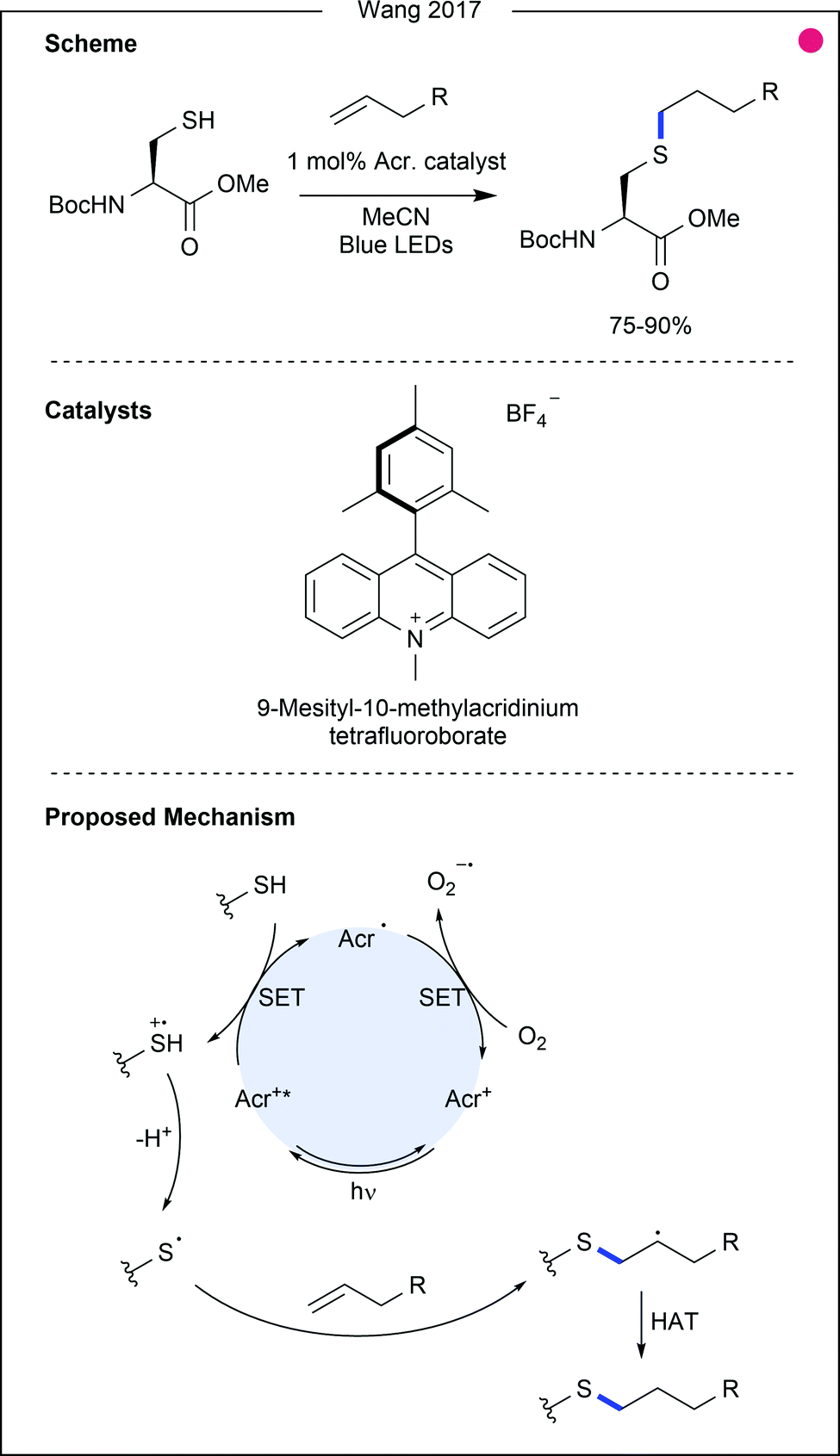 | ||
| Fig. 5 Thiol–ene reaction of cysteine. Using an acridinium catalyst to generate the thiyl radical, cysteine undergoes alkene addition. The resultant secondary radical persists for long enough for hydrogen atom transfer (HAT) to occur to generate the modification product. Acr. = 9-mesityl-10-methylacridinium. | ||
In 2018, Molander and co-workers developed a method for cysteine arylation using a ruthenium photocatalyst, nickel co-catalyst and a silicate-based hydrogen atom transfer reagent (Fig. 6).23 After excitation of the photocatalyst, oxidation of the silicate leads to the homolytic cleavage of the C–Si bond to produce a carbon-centred radical. This was able to abstract the proton from the cysteine thiol, generating a thiyl radical. Trapping of the thiyl radical with the nickel(0) species was then followed by oxidative addition of an aryl halide. Finally, reductive elimination from the nickel(III) complex produced the C(sp2)–S bond. The aryl substrate scope and amino acid compatibility was explored using glutathione (GSH) as substrate. Substituents on the aryl bromide greatly influenced the success of the reaction, as yields ranged from 14% (with a para-alcohol) to 83% (using 4-bromobenzyl boronic acid). Aryl bromides containing nitrile, halide, amide, sulfonamide, ether, alcohol, and carboxylate groups were all successful coupling partners. Disappointingly, a pendant azide group was not tolerated on the aryl bromide. Aryl bromide-containing chemical linkers, biologic probes and drug molecules were all used for the modification, producing GS-aryl conjugates in moderate to good yields (35–83%). The method was also successfully applied to the modification of a polypeptide (9 amino acids (AAs)) using 4-bromobenzonitrile (complete conversion was observed). Carboxylate functionality and aromatic amino acids were shown not to interfere with the reaction, whereas the presence of lysine, proline and arginine significantly hindered reactivity.
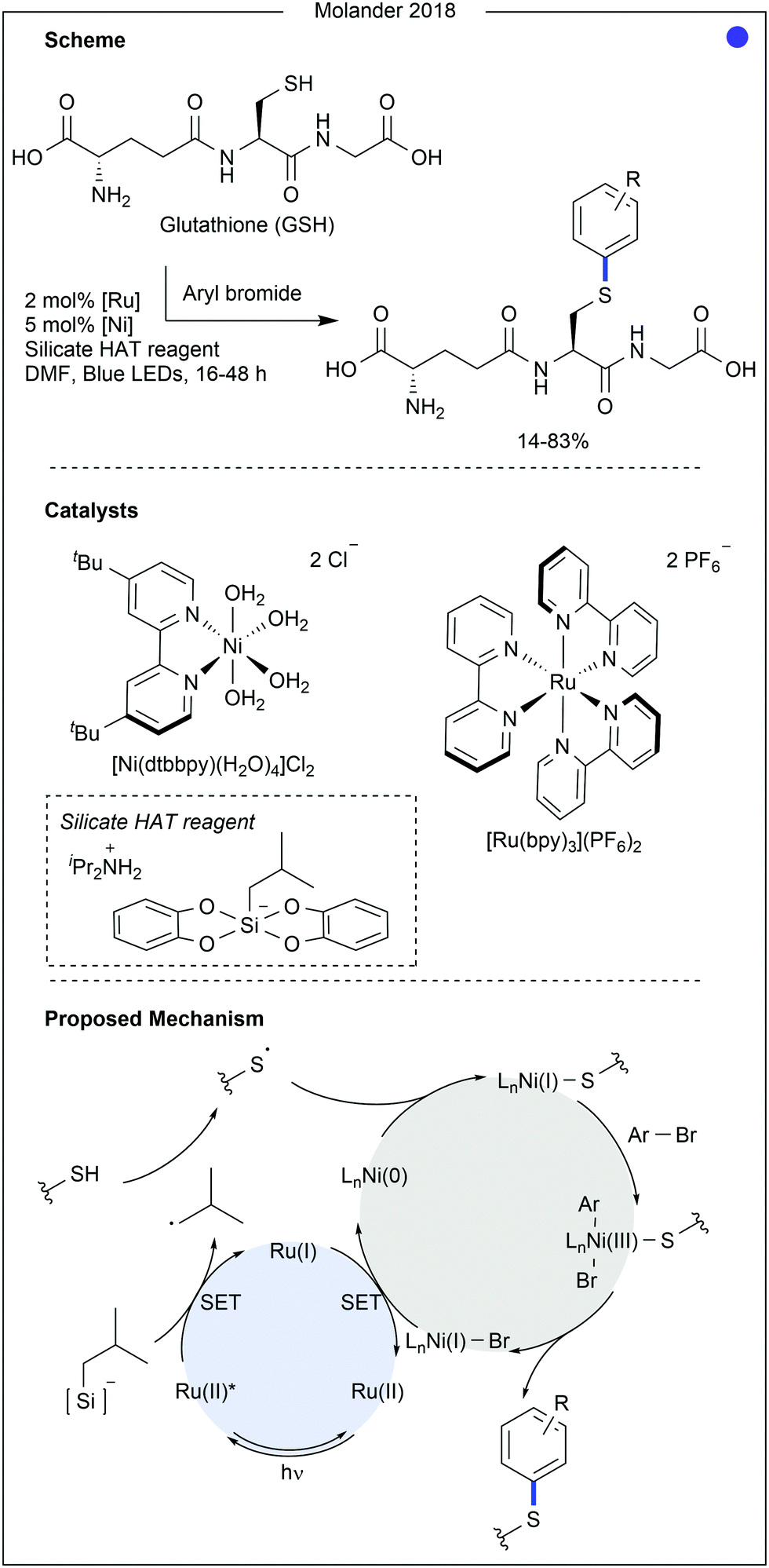 | ||
| Fig. 6 Cysteine arylation using dual nickel–ruthenium catalysis. A silicate reagent was proposed to generate the thiyl radical required in the nickel catalytic cycle through hydrogen atom transfer (HAT). Dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl. Bpy = 2,2′-bipyridyl. | ||
Bon, Wilson and co-workers recently demonstrated a ligand-directed approach to the selective labelling of a native cysteine-containing protein, and current anticancer target, human MCL-1 (Fig. 7).24 By appending a ruthenium photocatalyst to an oligopeptide (19 AAs) known to bind the target protein, the group were able to selectively oxidise and therefore modify a proximal cysteine residue (Cys286) on MCL-1 after irradiation with visible light. The thiyl radical formed after cysteine oxidation was trapped with aniline-based reagents similar to those used in previous work by Nakamura and co-workers for photocatalytic tyrosine modification (vide infra). While significant conversion was observed, increased irradiation time led to the generation of by-products, presumed to be due to oxidation of the MCL-1 protein by the photocatalyst. Attachment of a fluorophore or a biotin tag was achieved, the latter of which enabled purification of the target protein conjugate. Finally, competition experiments were performed using a mixture of MCL-1 and an analogous protein, BCl-xι to which the ligand peptide did not bind. Only limited labelling of the control protein was observed.
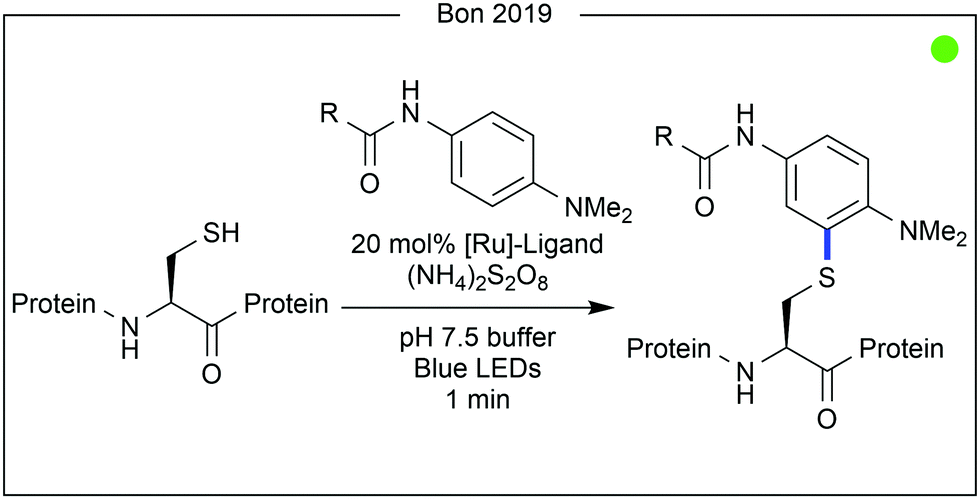 | ||
| Fig. 7 Ligand directed modification of cysteine on a protein surface. Using methodology similar to that used on tyrosine by Nakamura (vide infra), Wilson and co-workers were able to selectively label cysteine on the MCL-1 protein. | ||
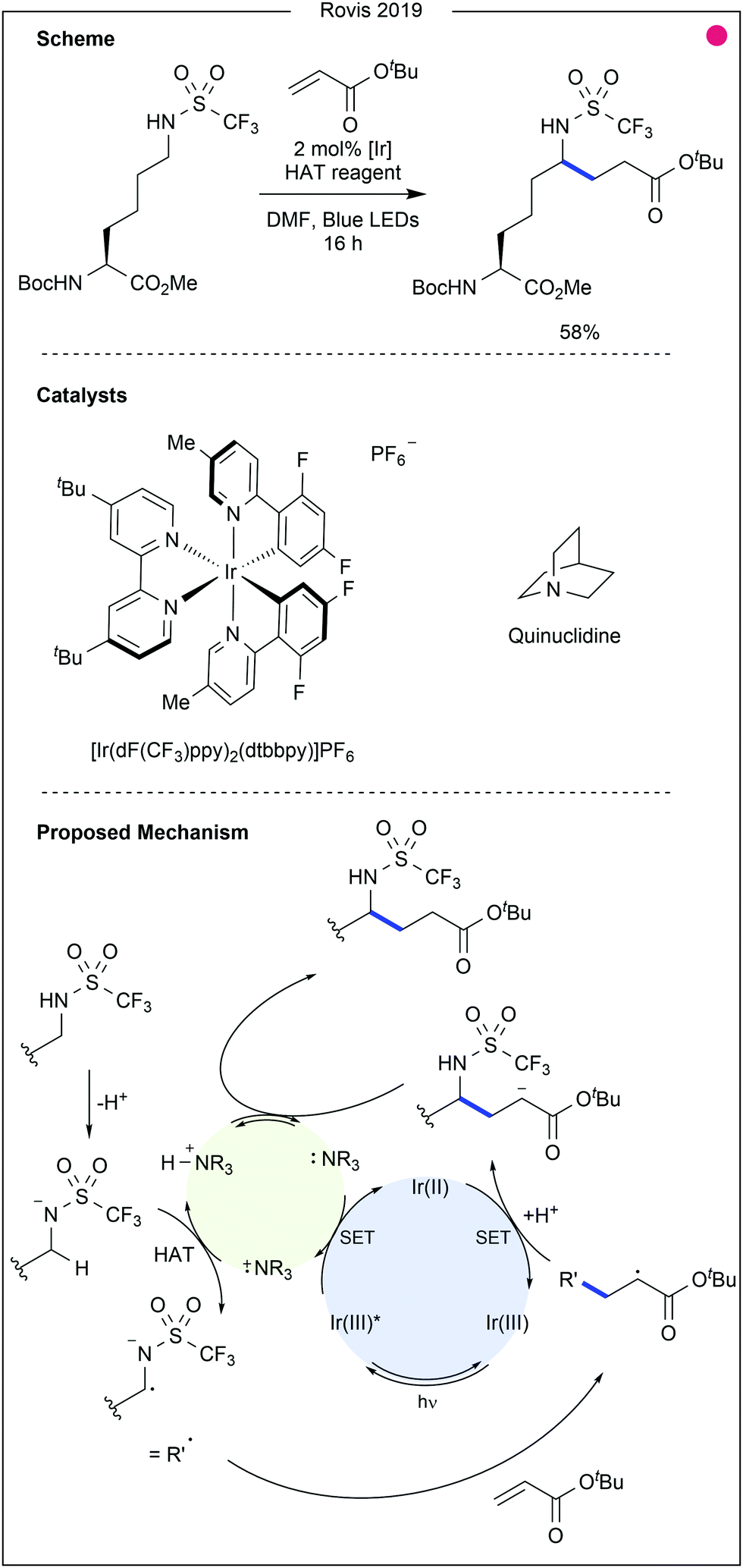 | ||
| Fig. 8 Triflamide-enabled photocatalytic modification of lysine residues. The use of a triflamide to acidify the side chain N–H encourages hydrogen abstraction from the adjacent carbon atom. Conjugate addition then occurs, and the resultant radical is reduced to an anion through single electron transfer (SET) from the photocatalyst. Protonation then provides the final product. Quinuclidine acts as an intermediary; after oxidation by the excited photocatalyst, it becomes primed for hydrogen atom transfer from the substrate. [Ir] = [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
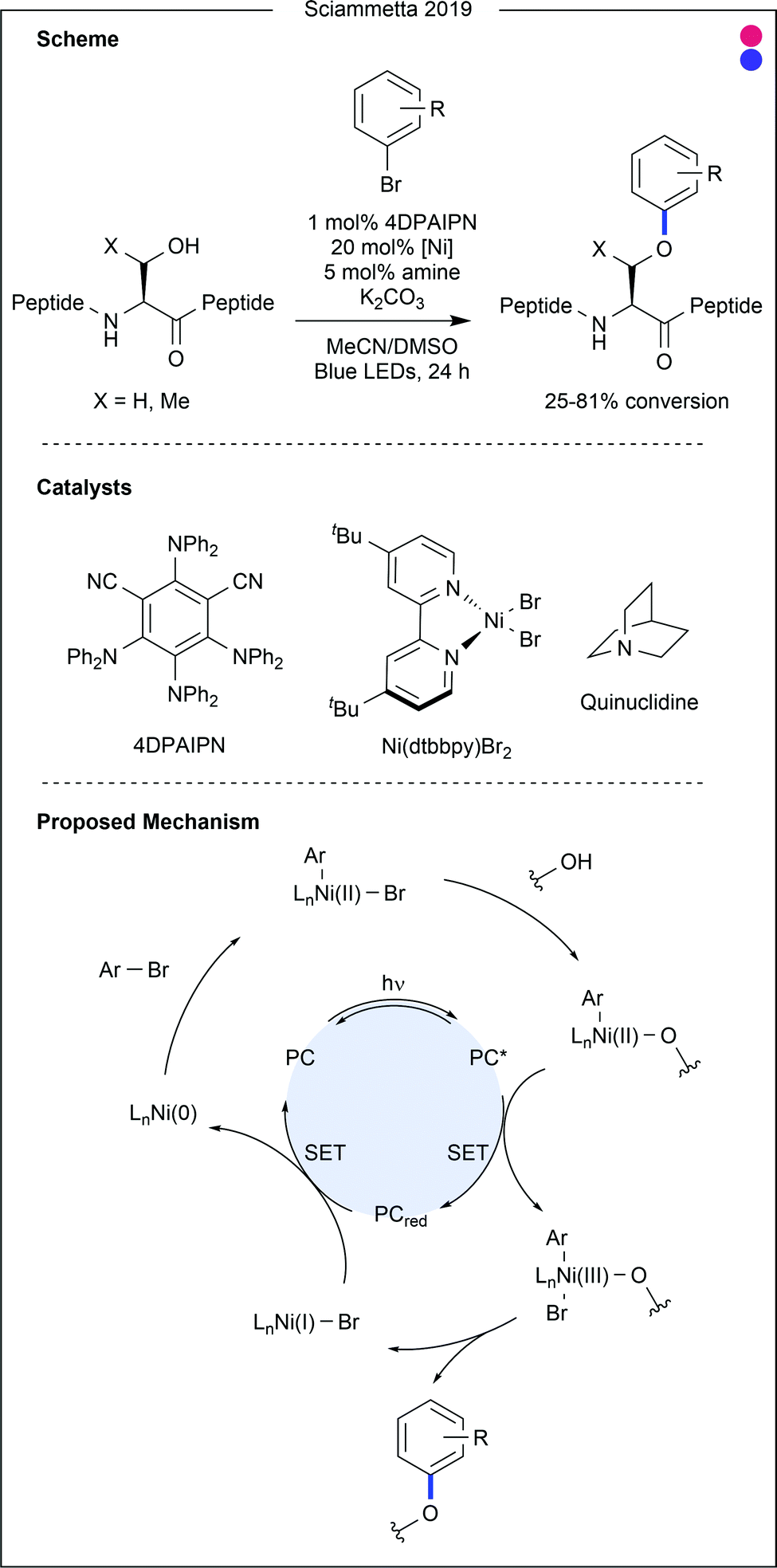 | ||
| Fig. 9 Serine cross-coupling with aryl halides using dual organophotocatalysis and nickel catalysis. Using a photocatalyst to modify the electronic state of nickel during the catalytic cycle enables efficient cross coupling of alcohols such as serine and threonine with aryl bromides. PC = photocatalyst (4DPAIPN). | ||
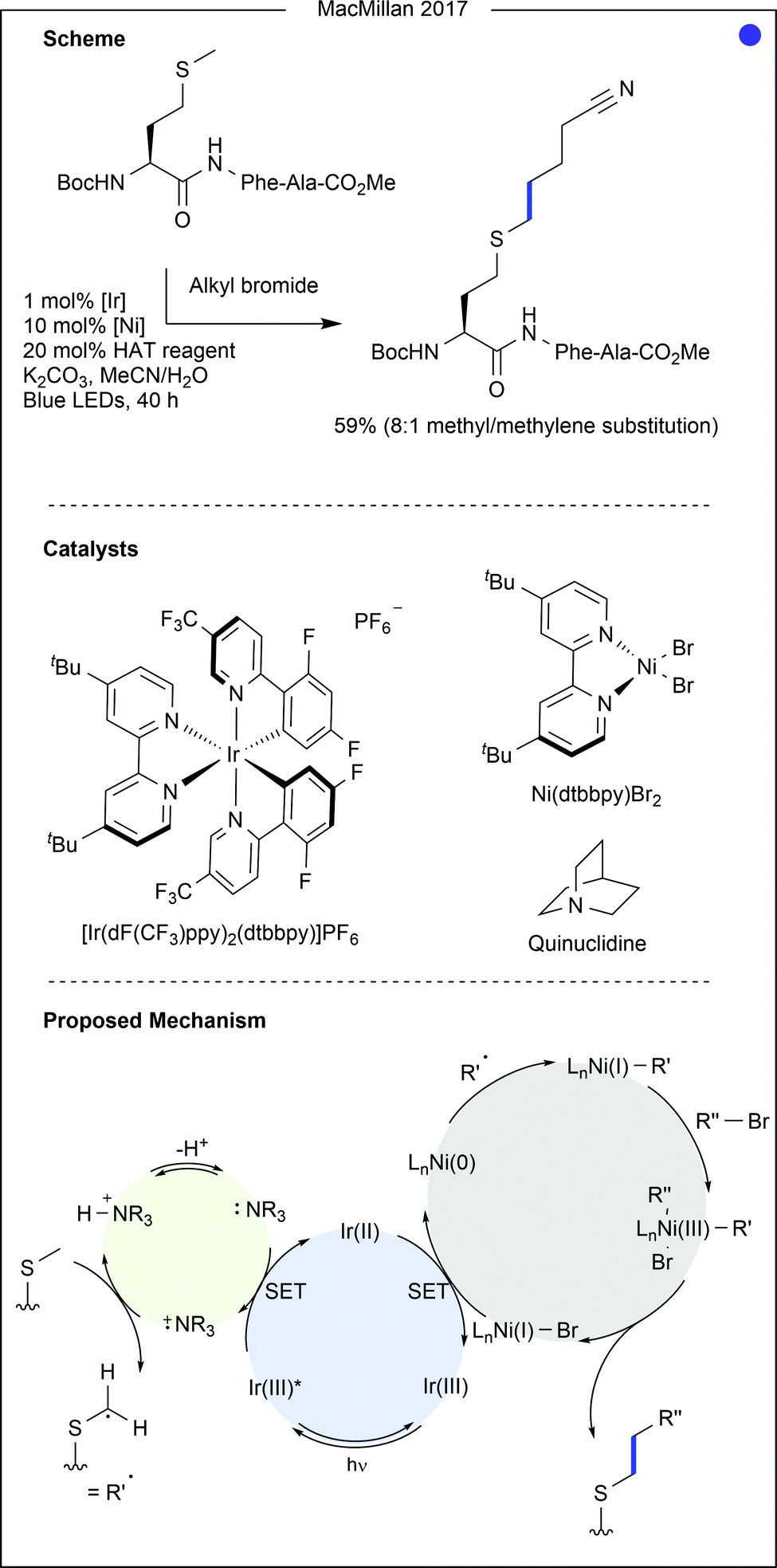 | ||
| Fig. 10 Recent photocatalytic modification of methionine using iridium/nickel/HAT triple catalysis. Quinuclidine acts as a mediator between the iridium photocatalyst and the methionine side chain. After oxidation by the excited photocatalyst, the amine is capable of hydrogen atom transfer (HAT) from the carbons adjacent to the sulfur atom. Ligation to the nickel centre, followed by an oxidative addition, produces a nickel(III) complex primed for reductive elimination to generate the product. [Ir] = [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. Dtbbpy = di-tert-butyl-bipyridyl. Phe = phenylalanine. Ala = alanine. | ||
While photocatalytic modification of native methionine within proteins remains limited, recent work from Gaunt and co-workers demonstrated site-selective addition of a reactive diazo handle to methionine, upon which photochemical modification was performed (Fig. 11).29 Using a C4-modified Hantzsch ester and a ruthenium photocatalyst, good conversion of the diazo group to a benzyl moiety was achieved (86%).
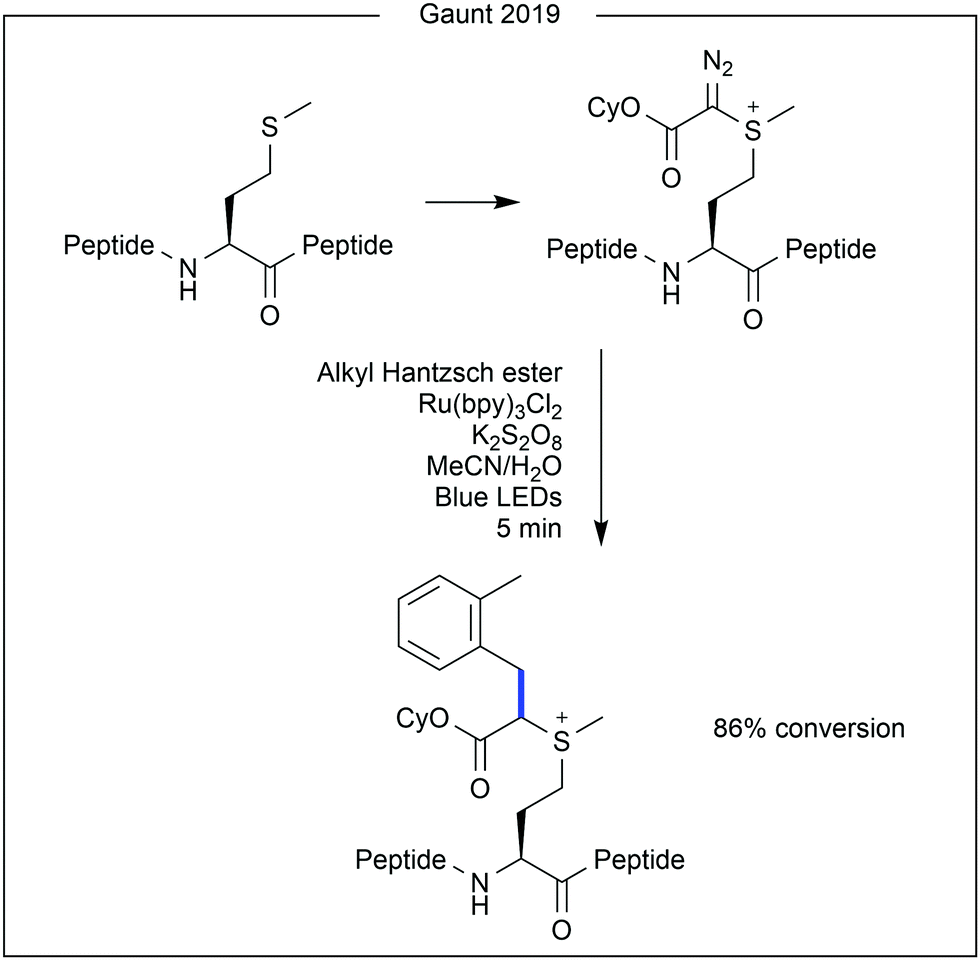 | ||
| Fig. 11 Photocatalysis for further modification of a protein after initial handle attachment to methionine. A C4-substituted Hantzsch ester releases a benzyl radical after photocatalytic oxidation. After cleavage of the pendant diazo group on the peptide, the two radicals can combine to form the further modified product. Bpy = 2,2′-bipyridyl. | ||
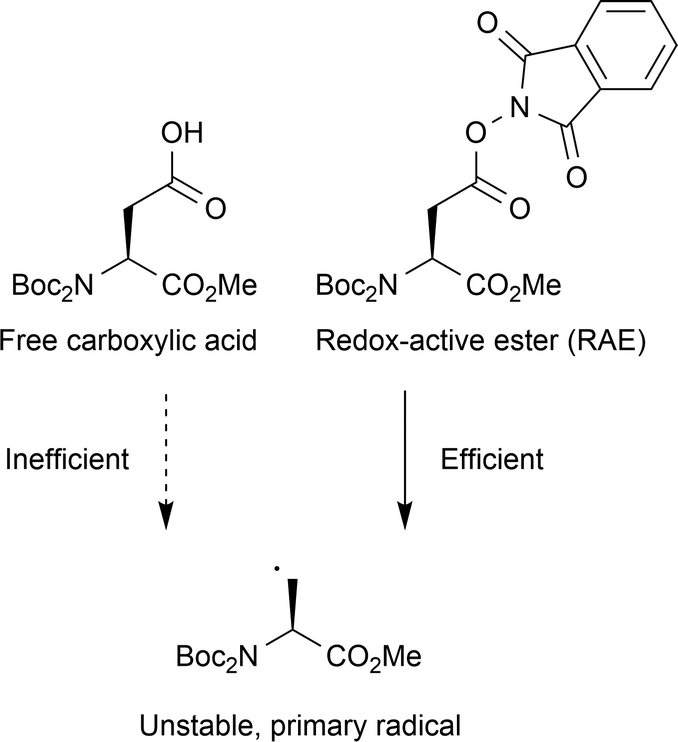 | ||
| Fig. 12 Primary radical generation on amino acids containing side chain carboxylates. Due to the relative instability of the resulting primary radical, photocatalytic decarboxylation is inefficient from the free carboxylic acid. Instead, the side chain must be activated for decarboxylation using a redox-active ester (RAE), which undergoes simple O–N homolytic fission after single electron reduction. This generates a carboxylate radical which will rapidly decarboxylate to provide the desired primary carbon radical. | ||
In this vein, Fu and co-workers generated 83 unnatural amino acids from either aspartic acid or glutamic acid building blocks (Fig. 13).30 Trapping of the primary radical was performed with a variety of α,β-unsaturated carbonyls (ketones, esters and amides, 52–92% yields), and alkynyl sulfones (62–85% yields), bearing aromatics with ether, halide and nitrile functional groups. These applications, whilst demonstrating significant utility for the modification of these two residues, involve additional steps to form the redox-active esters, thereby reducing the simplicity of the approach.
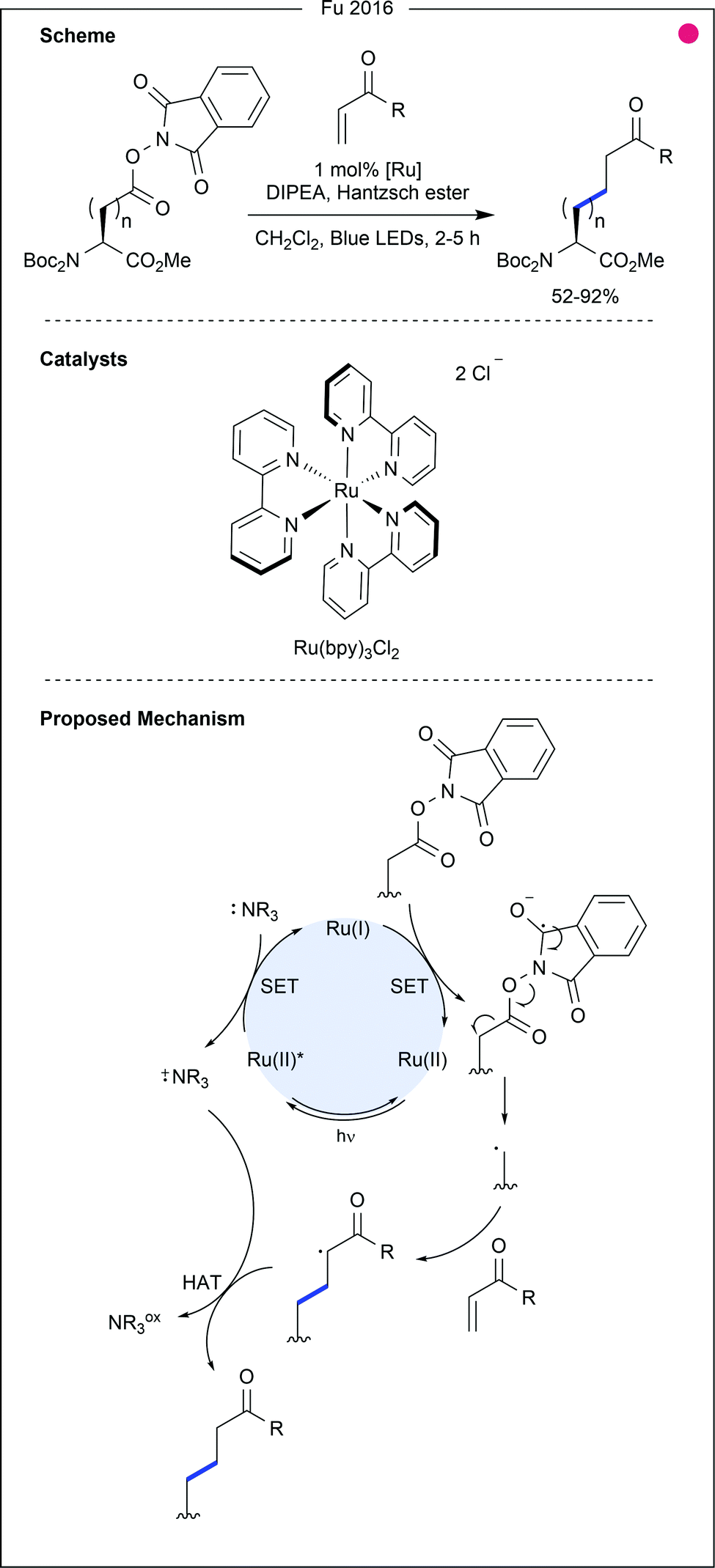 | ||
| Fig. 13 Alkylation of aspartic acid (n = 1) and glutamic acid (n = 2) redox-active esters (RAEs). Single electron reduction of the RAE provides an alkyl radical which can undergo conjugate addition. Stoichiometric amine acts to afford catalyst turnover and subsequently enables hydrogen atom transfer (HAT) to generate the final product. Bpy = 2,2′-bipyridyl. | ||
Aromatic amino acid side chains
The aromatic amino acids (see Fig. 14) offer great potential for photocatalysis as benzyl and heteroaryl rings have accessible redox potentials. In the context of bioconjugation, due to similarity across the aromatic amino acids, many traditional modification approaches fail to differentiate them. However, due to electronic differences, photocatalysis has been harnessed to enable residue-selective modification. | ||
| Fig. 14 Amino acids bearing aromatic side chain groups. | ||
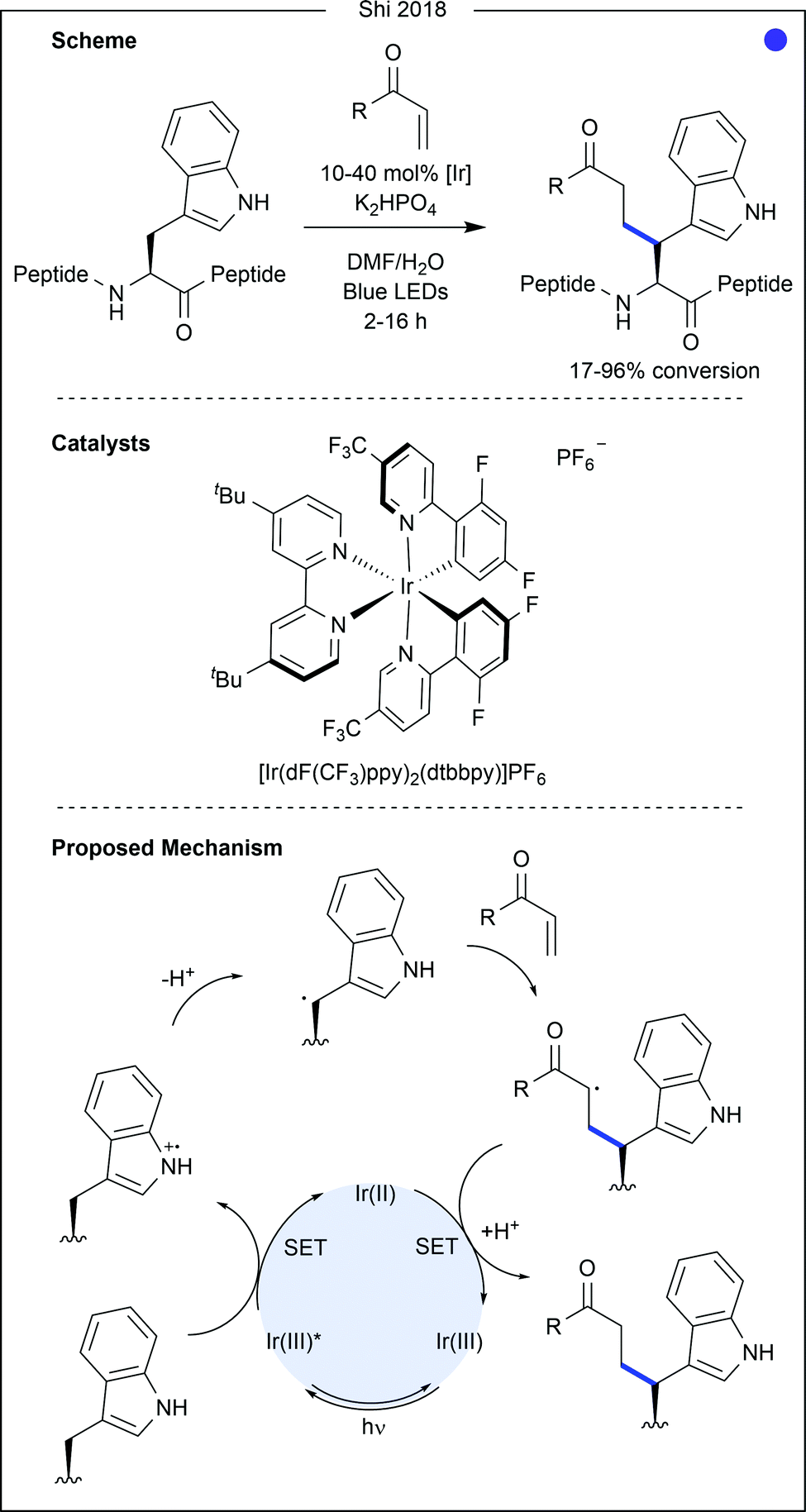 | ||
| Fig. 15 Photocatalytic modification of tryptophan at the methylene position. Single electron oxidation of the tryptophan nitrogen activates the methylene carbon for deprotonation. The resultant radical species, a stable secondary benzylic radical, can undergo conjugate addition. Single electron reduction, again by the photocatalyst itself, coupled with protonation generates the final product. [Ir] = [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
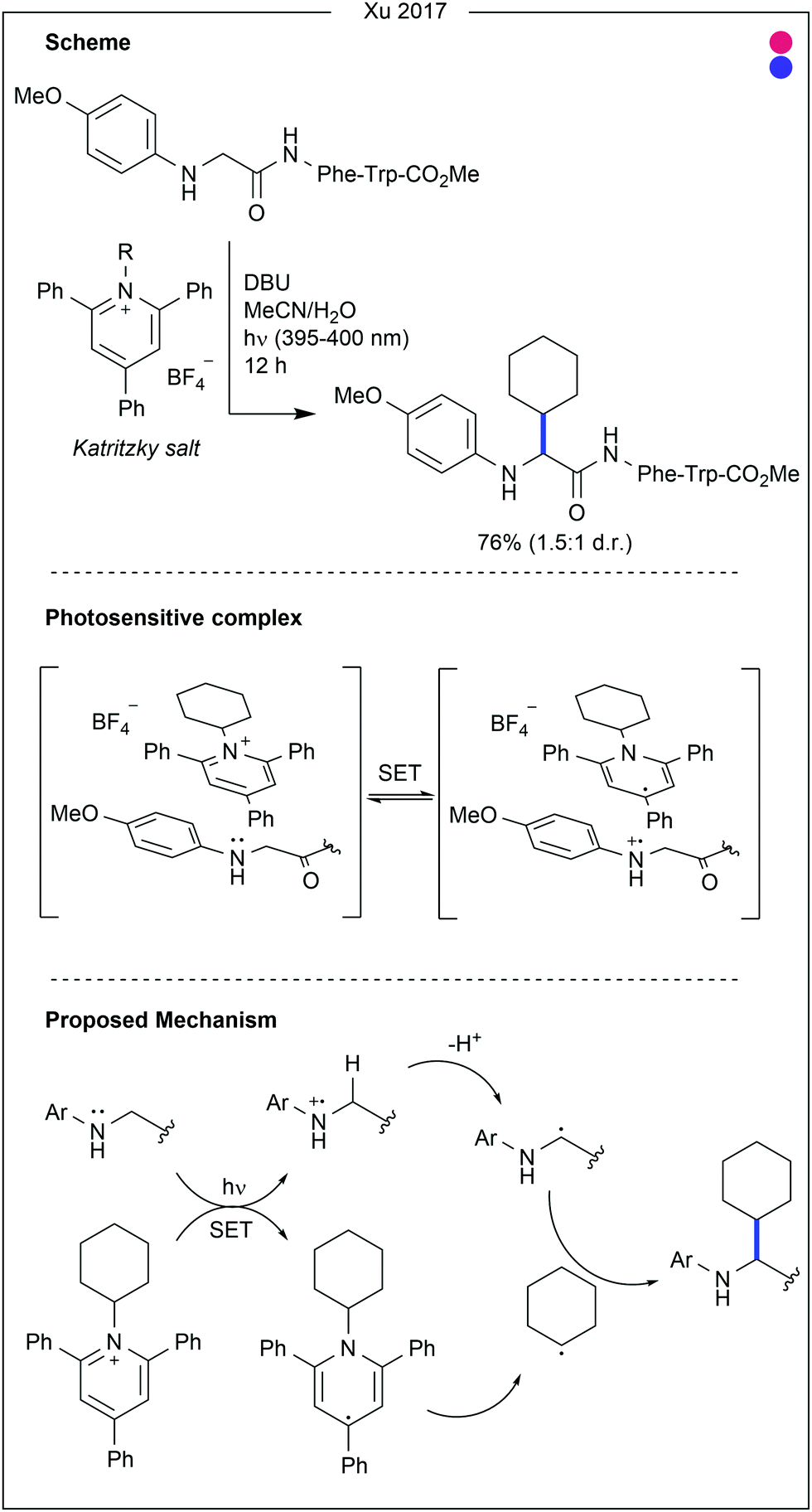
Contrastingly, Taylor and co-workers recently used photocatalysis to access modification of the C2-position of tryptophan (Fig. 16).32 By employing N-carbamoylpyridinium salts, the resulting charge transfer complex between the pyridinium and tryptophan indole could be excited by irradiation with ultraviolet (UV-B) light. Single electron transfer from tryptophan to the pyridinium leads to homolytic cleavage of the N–N bond, generating a radical intermediate. As this intermediate is generated proximal to the oxidised tryptophan residue, combination of the two to produce the product is favourable. A range of carbamoyl groups containing pendant functional groups such as ethers and carbamates, tags such as biotin, or reactive groups for further modification such as an alkyne could be transferred to tryptophan in this way. After demonstrating high reactivity on a short peptide (8 AAs, >90% conversion for all salts used), the chemistry was applied to larger peptide and protein substrates (1–14 kDa) achieving high (>85%) conversion in all cases. Notably, this protocol does not require any organic co-solvent, proceeding in aqueous buffer at near-neutral pH, and is fast, only requiring irradiation for 30–75 minutes to achieve high conversion.
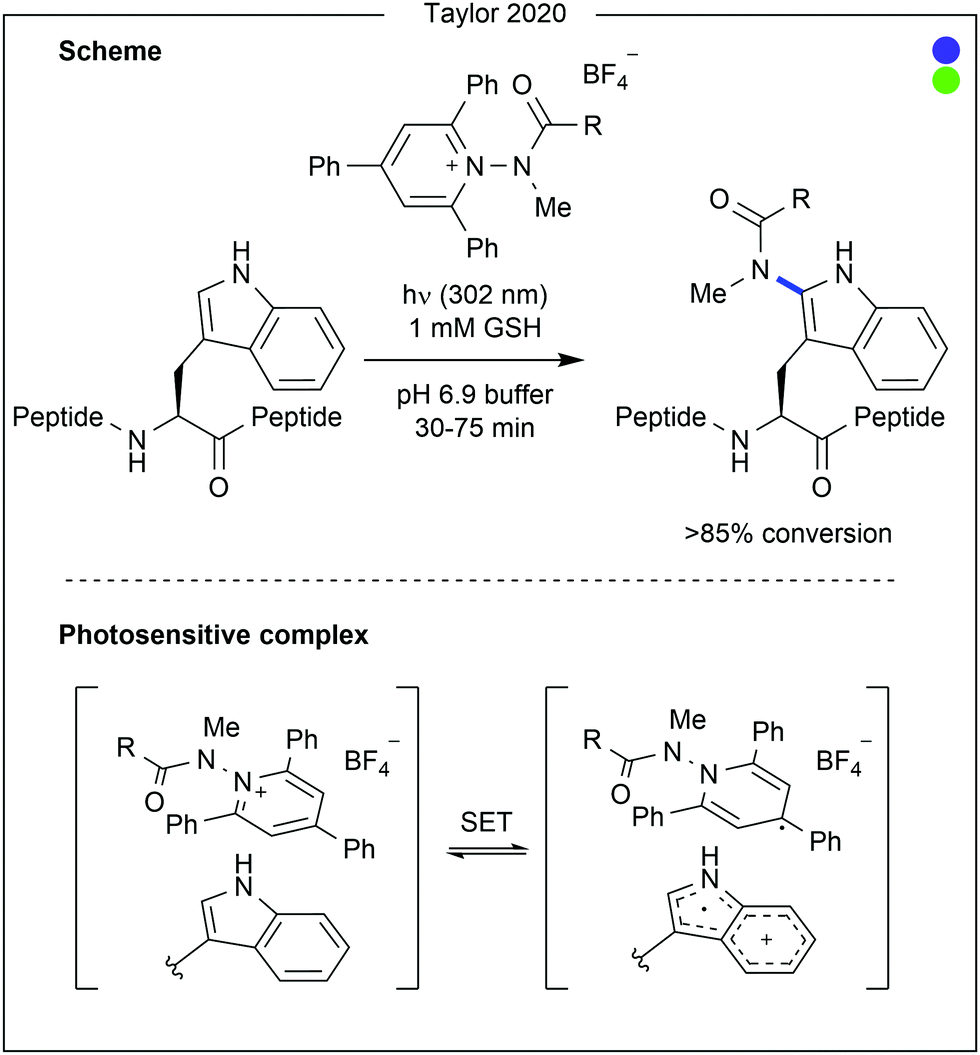 | ||
| Fig. 16 Photo-induced modification of tryptophan at the C2-position. The photosensitive complex undergoes single electron transfer when irradiated by ultraviolet light, leading to scission of the N–N bond in the Katritzky-type salt. Radical combination then generates the final product. GSH = glutathione. | ||
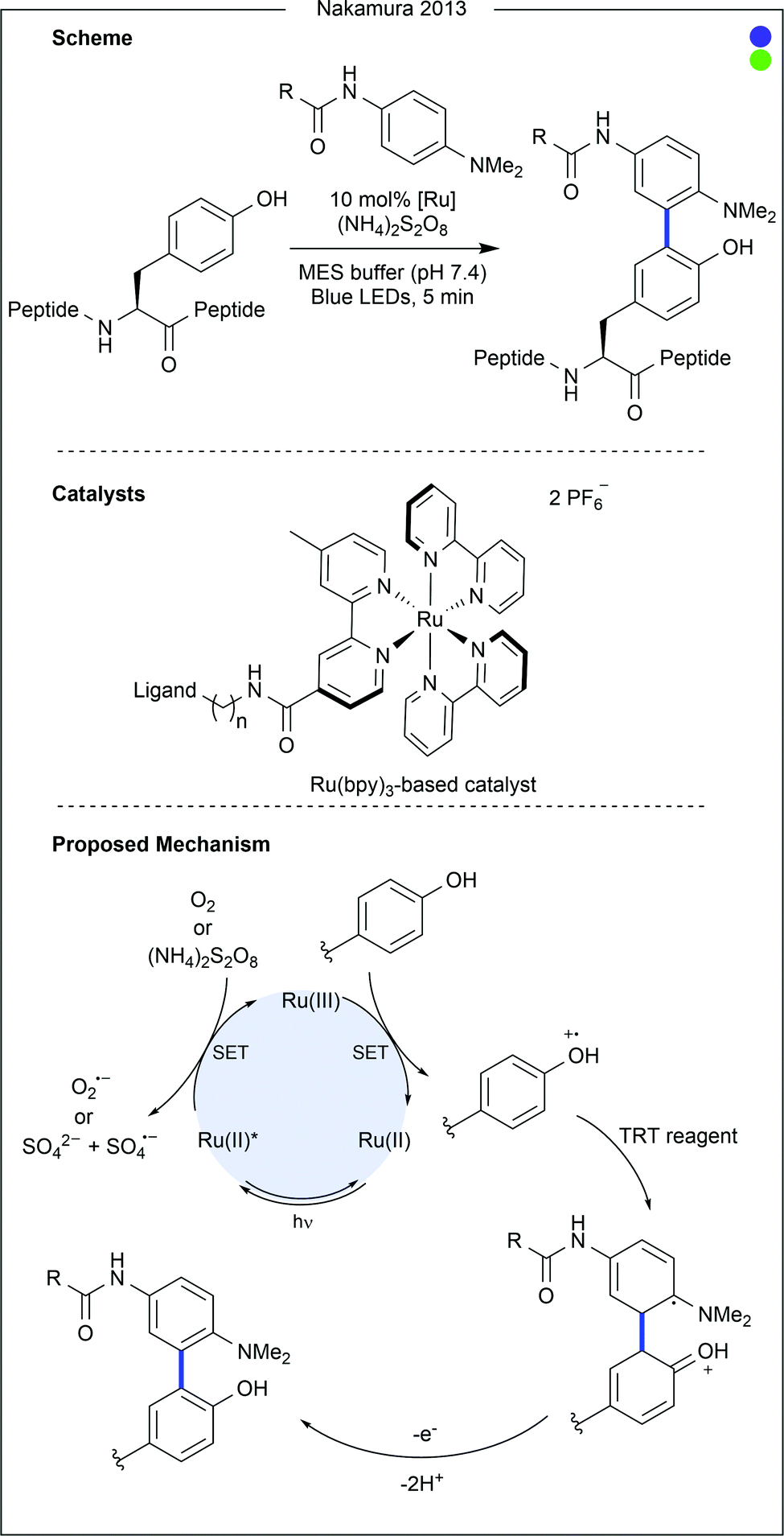 | ||
| Fig. 17 Ligand-directed photocatalytic modification of tyrosine. By attaching a ruthenium photocatalyst to a known ligand, selective modification of proximal tyrosine residues on the target protein was conducted. After irradiation, the excited ruthenium catalyst was proposed to reduce either oxygen or ammonium persulfate. The resultant ruthenium(III) species could then oxidise nearby tyrosine residues. A substituted aniline, a tyrosyl radical trapping (TRT) reagent, could then attack the tyrosyl radical, which after further oxidation led to the bi-aryl cross-coupled product. MES = 2-morpholinoethanesulfonic acid. Bpy = 2,2′-bipyridyl. | ||
Tyrosine-derived ethers have also been recently modified through photocatalytic oxidation of the tyrosine aryl ring. Work by Nicewicz and co-workers on novel SNAr chemistry highlighted methoxybenzenes as suitable reagents following initial oxidation by an excited acridinium photocatalyst (Fig. 18).35 The work demonstrated unique ipso-reactivity, whereby methanol was released after attack of a nucleophilic N-heterocycle. Among the substrate scope was tyrosine methyl ether, which was reacted with imidazole in moderate yield (38%). Tyrosine methyl ether was also reacted with a C- and N-terminally protected histidine residue to produce the coupled product, albeit in low yield (17%). This protocol makes use of unique tyrosine reactivity to enable the generation of unnatural amino acids. While initial results also suggest that the protocol could be used for amino acid cross-coupling or peptide macrocyclisation, further optimisation would be required to make this a viable tool in this respect.
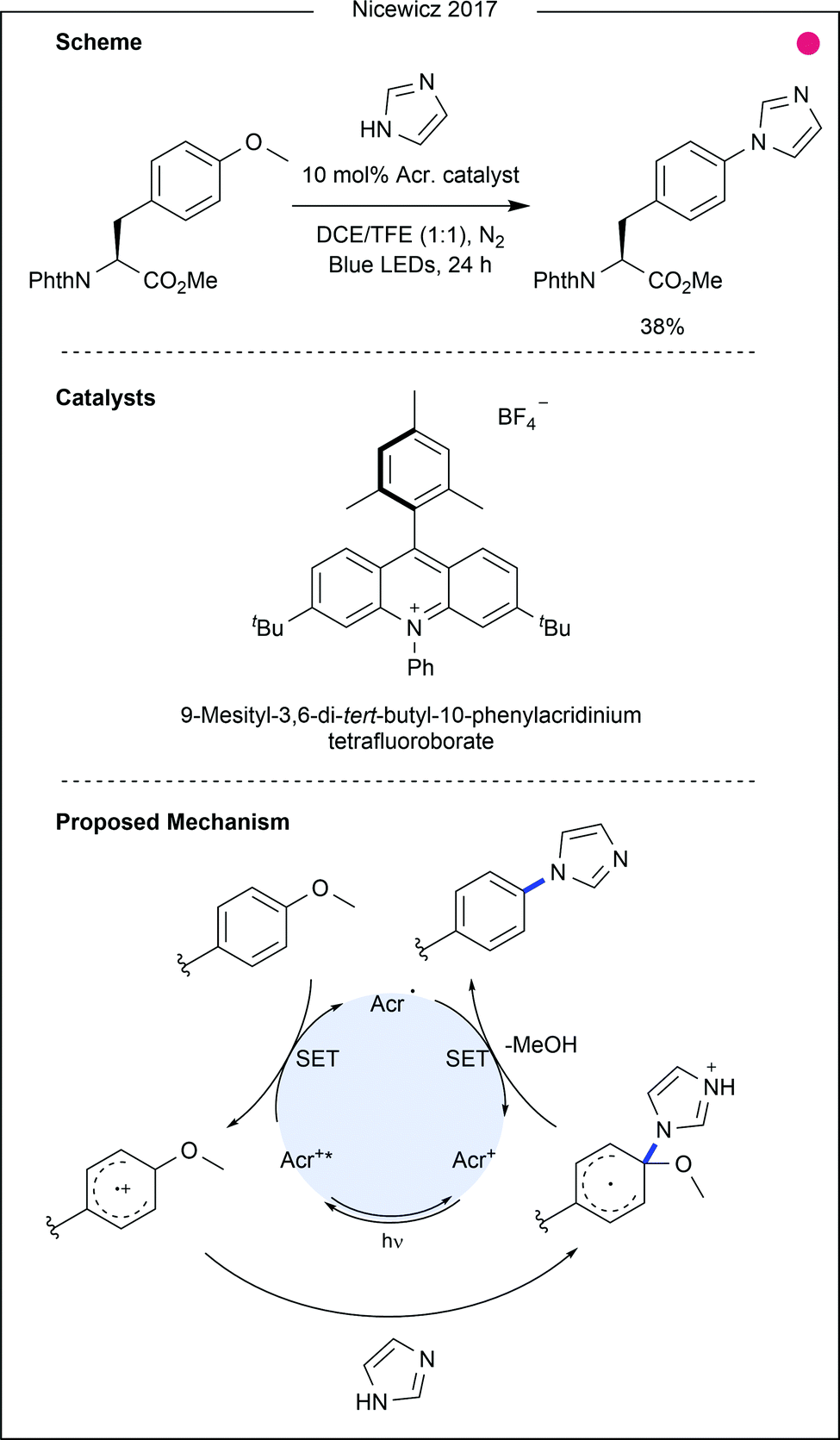 | ||
| Fig. 18 Tyrosine methyl ether activated for SNAr reactivity through the use of an acridinium photocatalyst. After oxidation of the tyrosine aryl ring by the photocatalyst, imidazole was shown to attack. Reduction occurred with loss of methanol to generate the product. Phth = phthaloyl. Acr. = 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate. DCE = dichloroethane. TFE = trifluoroethanol. | ||
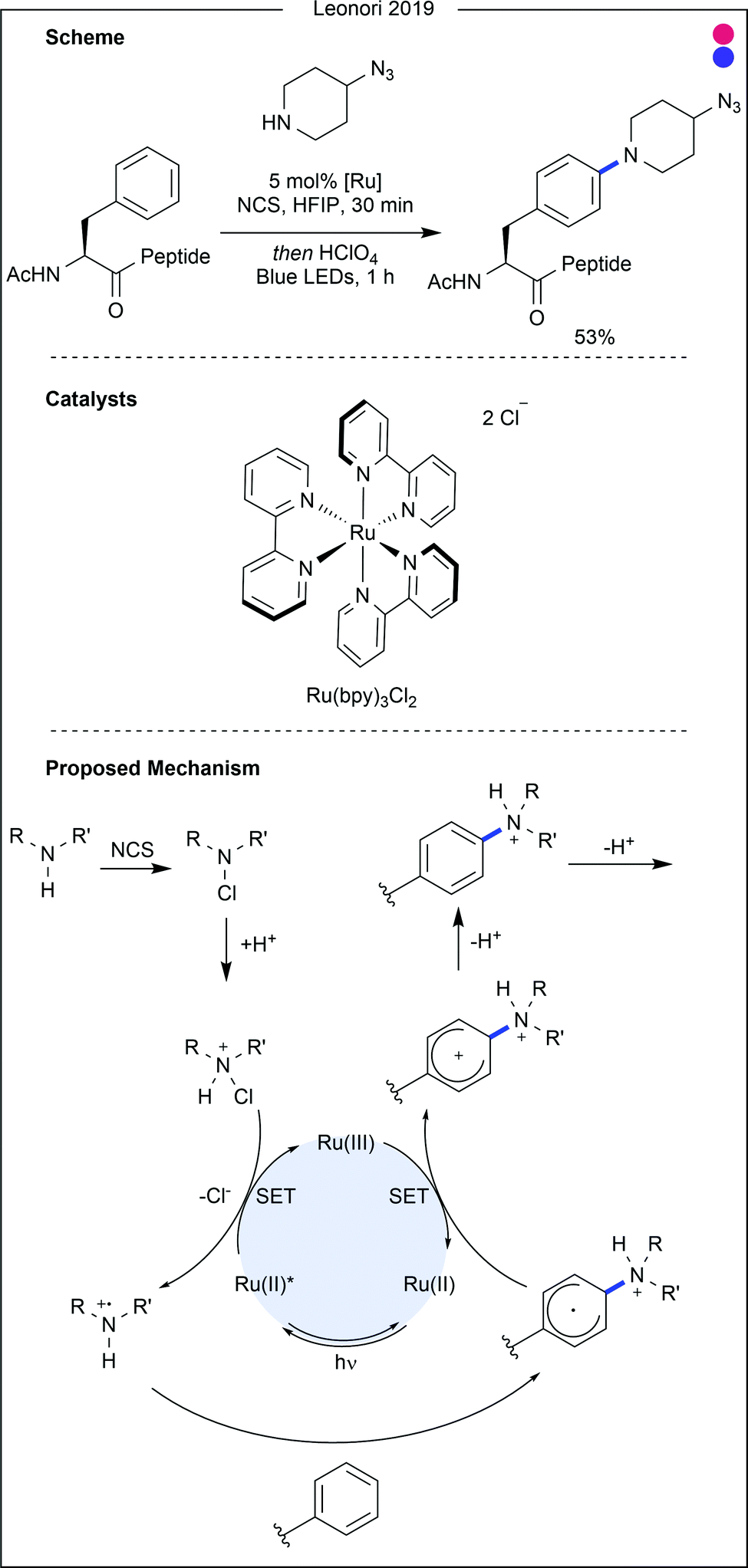 | ||
| Fig. 19 Recent photocatalytic modification of phenylalanine. An excited ruthenium catalyst was shown to reduce N-chloroammonium reagents to produce oxidised secondary amines. These species were sufficiently reactive to undergo radical attack from the aryl side chain of phenylalanine. After single electron oxidation by the photocatalyst, double deprotonation formed the aminated amino acid in moderate to high yields. Bpy = 2,2′-bipyridyl. NCS = N-chlorosuccinimide. HFIP = hexafluoroisopropanol. | ||
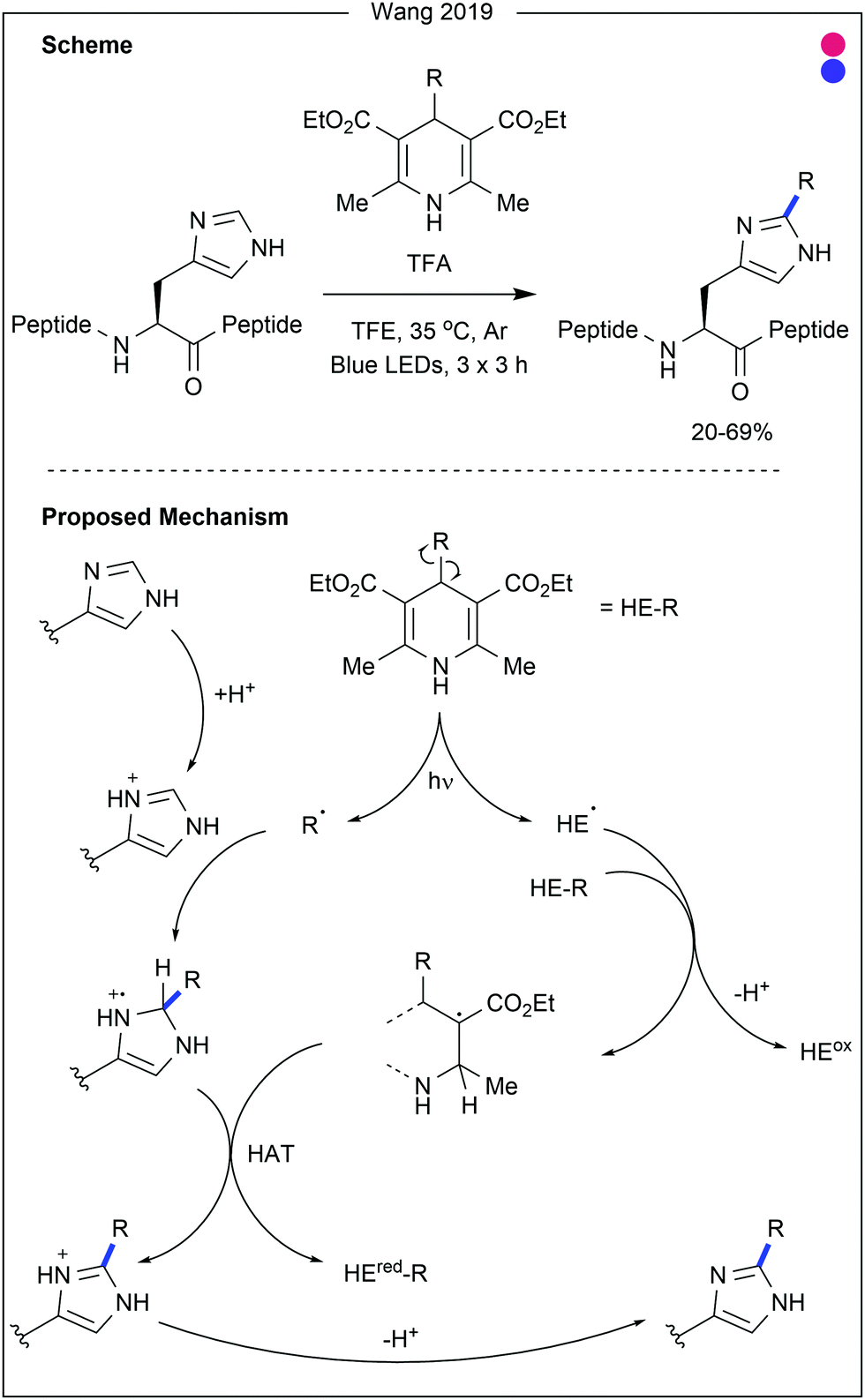 | ||
| Fig. 20 Photomediated histidine modification using alkylated Hantzsch esters (HE-R). After photomediated homolytic cleavage of the HE-R bond, the alkyl radical can undergo Minisci-type addition to protonated histidine residues. Hydrogen atom transfer (HAT) to a later Hatzsch ester derivative generates the final product. TFA = trifluoroacetic acid. TFE = trifluoroethanol. | ||
Aliphatic amino acid side chains
Aliphatic residues (see Fig. 21) have remained underexploited in the realm of amino acid and peptide modification, as their inherently inert side chains preclude modification using traditional methods. Palladium-catalysed C–H activation chemistry has been used to target these residues, but many procedures require biologically-incompatible conditions, most notably exceedingly high temperatures. In contrast, the milder conditions used in photocatalytic protocols are more suitable for bioconjugation purposes.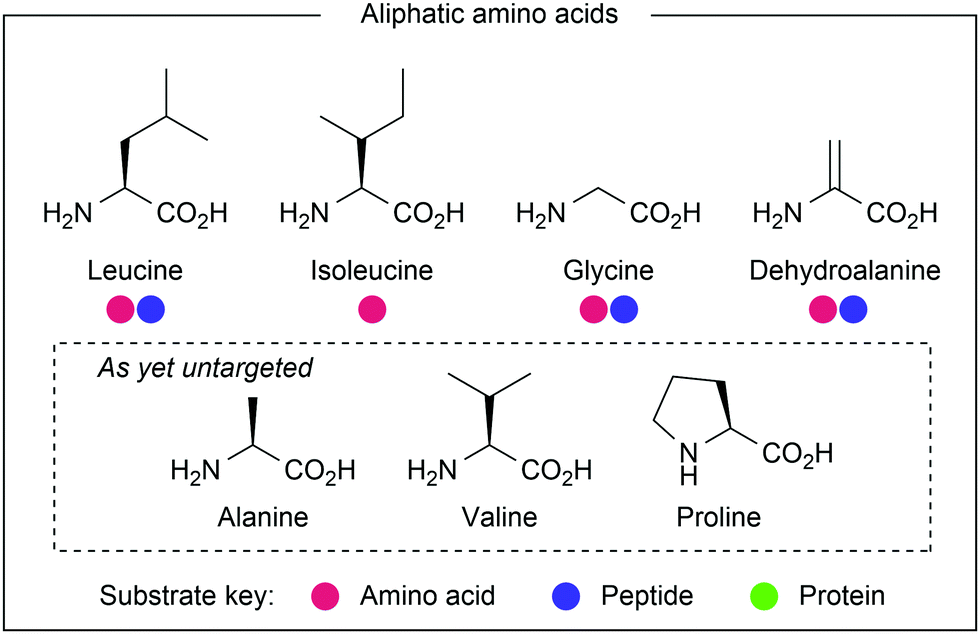 | ||
| Fig. 21 Amino acids with aliphatic side chains. | ||
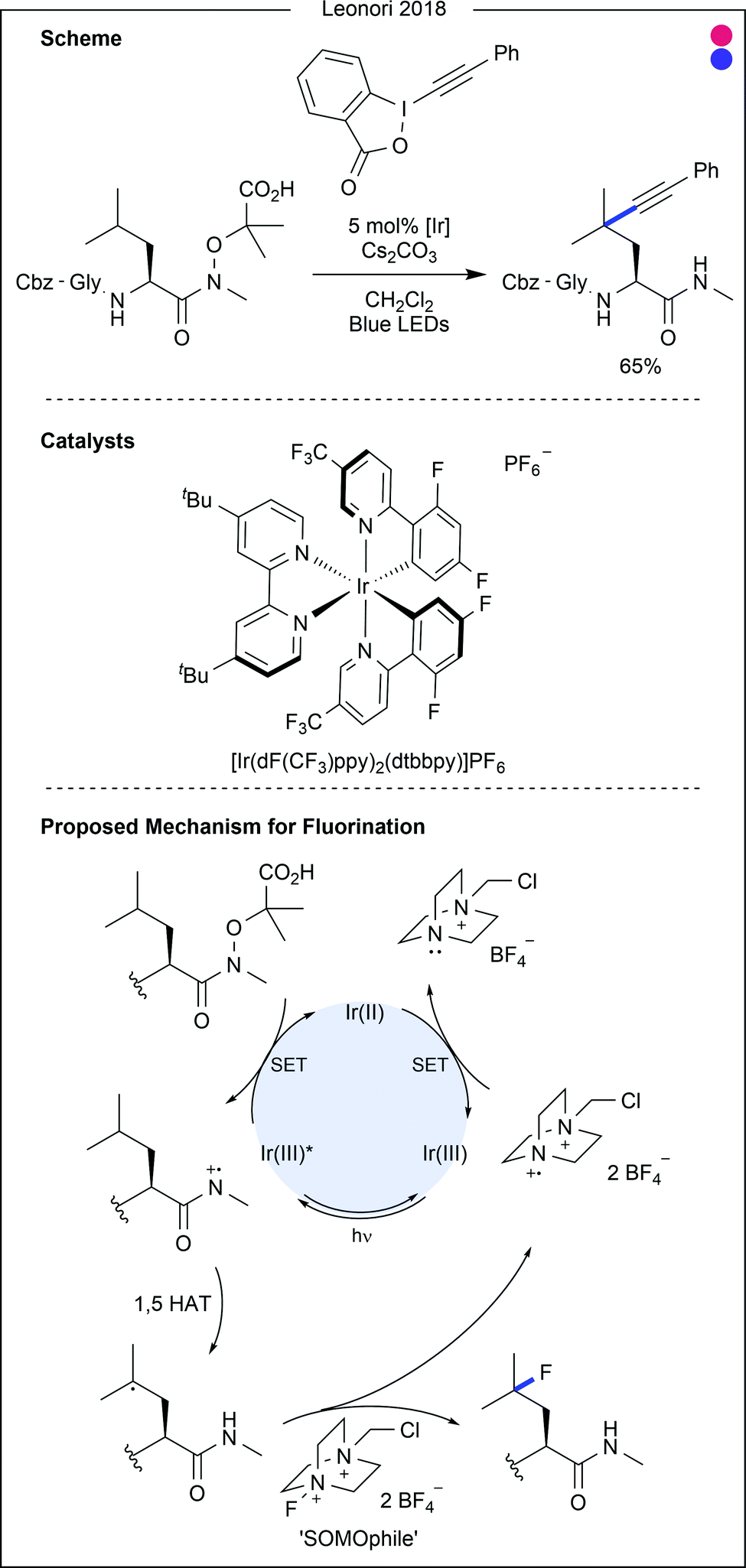 | ||
| Fig. 22 Photocatalytic modification of leucine and isoleucine using 1,5 hydrogen atom transfer (HAT). Using a photolabile group, after single electron oxidation an amidyl radical could be formed. Via a 6-membered cyclic transition state, hydrogen atom transfer from the side chain to the amide nitrogen could be achieved, generating an alkyl radical. Using radical reaction partners such as alkynyl hypervalent iodine or N-fluoro species, modified amino acids could be generated. Gly = glycine. Dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl. | ||
 | ||
| Fig. 23 Photo-enabled modification of glycine residues using Katritzky salts. An N-aryl glycine at the N-terminus of a short peptide sequence was shown to form an electron donor–acceptor (EDA) complex with Katritzky salts. Upon irradiation, single electron transfer produces both a glycine amino radical and a pyridinium radical. The pyridinium radical is able to collapse to generate the pyridine, with loss of an alkyl radical (R˙), while deprotonation with base moves the glycine radical to the α-carbon. Radical–radical combination then affords the modified product. Phe = phenylalanine. Trp = tryptophan. | ||
Dha has been used to generate unnatural amino acids, either independently or within a peptide chain (Fig. 24). Jui and co-workers used photocatalysis to generate a range of pyridyl radicals which performed radical conjugate addition into protected Dha, forming heteroaryl-containing amino acids after further reduction and protonation.42 Following single electron reduction of the halo-pyridine, mesolytic cleavage of the C–X bond occurs, releasing halide and forming a C(sp2)-centred radical. Hantzsch ester was used to provide terminal single electron reduction after conjugate addition. 2-, 3- and 4-halopyridines were successful coupling partners for N-Boc Dha methyl ester (34–97% yields). Alcohols, halides, amines and ethers were all compatible heteroaryl functional groups. Finally, Karady–Beckwith alkene, a chiral oxazolidinone derivative of Dha, was used to control the chirality at the α-carbon, inducing diastereoselective protonation of the enolate that is formed upon final reduction. Good yields (57–80%) were obtained using a small selection of heteroaromatic halides. Facile deprotection with hydrochloric acid yielded an example enantioenriched pyridylalanine product (97% ee).
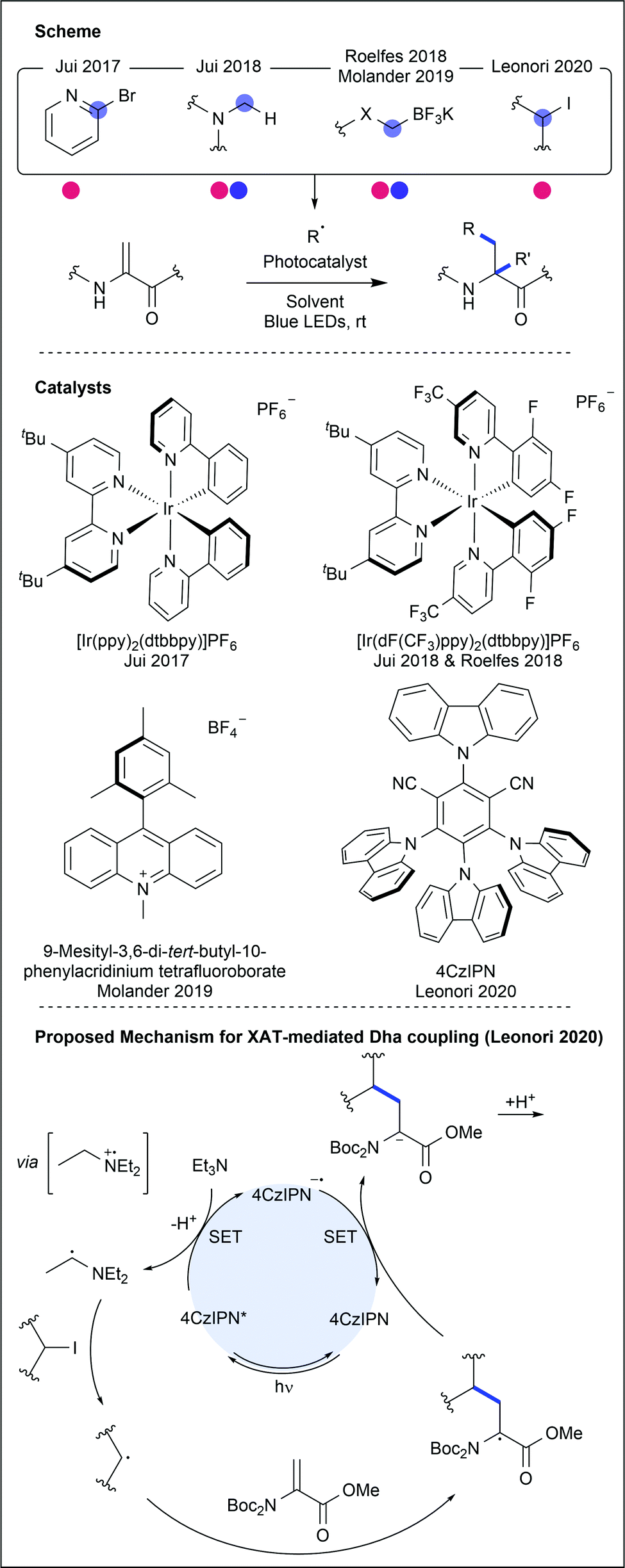 | ||
| Fig. 24 Recent developments in the modification of dehydroalanine (Dha). Photocatalytic methods for Dha modification generally involve alkyl or aryl carbon radicals, which undergo conjugate addition to the electron-deficient alkene. A variety of catalysts have been used, due to differing methods of radical formation. Most recently, alkyl and aryl halides have been shown to be suitable radical precursors, thereby opening up a wide, commercially available pool of potential substrates. | ||
A similar methodology was then reported using tertiary amines to generate α-amino radicals that also performed radical conjugate additions into Dha and the Karady–Beckwith oxazolidinone derivative.43 Single electron oxidation of a tertiary amine produces an amino radical cation, greatly acidifying the α-protons. Deprotonation then generates the required α-amino radical. Use of aryl and alkyl tertiary amines, containing aldehydes, halides, alkenes, acids and esters, as well as complex bioactive tertiary amines, generated addition products in good yields (62–91%). Modification using a propargylamine derivative was less successful (25% yield). Modification of Dha was then attempted within short peptide sequences (3–9 AAs), providing the conjugate products in good isolated yields (41–86%).
More recently, Roelfes and Molander have independently reported using trifluoroborate salts as radical precursors for Dha modification.44,45 When exposed to photocatalytic conditions, C–B cleavage within the salt generated a carbon-centred radical which could undergo radical conjugate addition. A variety of unnatural amino acids were generated, including fluorinated variants.
Dha was also recently used as a substrate to demonstrate work from Leonori and co-workers on α-amino-radical-promoted halogen atom transfer (XAT).46 The process, involving 4CzIPN as the photocatalyst, generates the α-radical of a tertiary amine through SET and deprotonation. This radical demonstrates efficient XAT from alkyl and aryl halides, producing the corresponding alkyl and aryl radicals. Assessing the substrate scope for the alkene coupling partners, the group generated 16 unnatural amino acids through coupling with Boc-protected Dha methyl ester, in moderate to excellent yields (22–99%). Halides containing amine, ether, alkyne, silane and boronic ester functionalities were all suitable coupling partners. The inclusion of both primary and secondary halides as coupling partners overcomes significant obstacles of other procedures, which often only enable the coupling of secondary carbon radicals to amino acids. It is, however, important to note that no control over stereochemistry could be achieved.
Reactivity at terminal amino acid residues
The C-terminal carboxylate of amino acids and peptides presents an opportunity to apply the plethora of photocatalysed decarboxylation methods to amino acid modification. Indeed, many protocols have been demonstrated to work on amino acids or their derivatives.47 However, modification of the C-terminal carboxylate of a lone amino acid precludes any subsequent products from being used in peptide synthesis. As these protocols would also not allow modification of a native peptide sequence, except at the C-terminus, most are beyond the scope of this review. There have, however, been a number of recent reports of C-terminal modification of peptides (Fig. 25).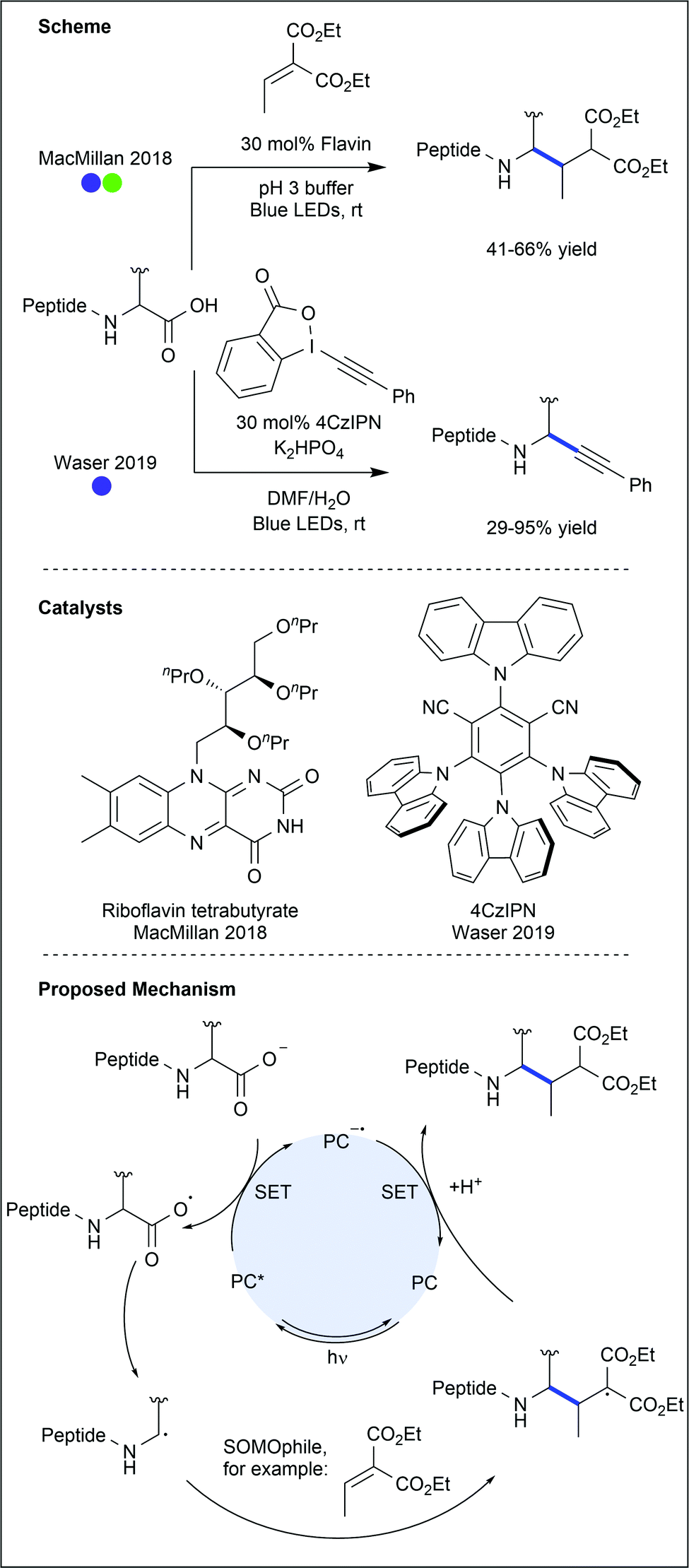 | ||
| Fig. 25 Decarboxylative C-terminal modification of short peptides. Modification strategies showed selectivity for the C-terminal carboxylate over internal carboxylate residues (glutamic acid and aspartic acid). Decarboxylation generates the alkyl radical which can react with SOMOphiles, such as α,β-unsaturated carbonyls or hypervalent iodine reagents. PC = photocatalyst. | ||
MacMillan and co-workers described the selective C-terminal decarboxylation of native peptides using flavin-based photocatalysts.48 Due to the significant difference in reduction potentials between the C-terminus carboxylate and those carboxylates found on amino acid side chains within the peptides, selective modification of the C-terminus could be achieved under oxidative conditions. C-Terminal decarboxylation results in a primary (if the amino acid is glycine) or secondary (if any other amino acid is used) α-amino carbon radical, which was trapped with α,β-unsaturated esters. Using a tetrapeptide containing a variable N-terminal residue, most polar and aliphatic amino acids were shown to be compatible with the reaction conditions. Tyrosine and histidine, however, significantly decreased conversion when present in the peptide. Eight larger peptides, ranging from 8–58 amino acids in length, were then successfully modified with moderate to good conversions (31–66%). While most of the substrates were trapped with an α,β-unsaturated diester, the potential for this chemistry to enable selective installation of an alkyne group on the A-chain C-terminus of human insulin was also demonstrated (41% conversion).
A decarboxylative alkynylation protocol has been developed by Waser and co-workers.49 Using donor–acceptor cyanoarene (DACA) photocatalysts and ethynyl benziodoxolone (EBX) reagents, selective modification of the C-terminus was achieved on di-, tetra- and hexapeptides (17–95% yields). While amino acids such as cysteine, tyrosine and tryptophan led to reduced yields when incorporated in the peptide sequence, protecting groups, or the use of a less oxidising catalyst were shown to revive reactivity. Furthermore, selectivity for the C-terminal carboxylate was observed in the presence of glutamic acid and aspartic acid. While other C-terminal decarboxylative alkynylations have been developed, most require chemical pre-activation of the carboxylate. This procedure brings alkynylation in line with arylation, reduction, cyanation and Giese-type coupling, all of which have previously been applied to free C-terminal carboxylates (see references within ref. 49).
Recent advances in decarboxylation chemistry have enabled azidation of amino acid C-termini, as reported by Leonori and co-workers (Fig. 26).50 Using a rhodamine-based photocatalyst, proline residues were decarboxylated to form the secondary alkyl radical, which was then trapped with an azide transfer reagent. The reaction proceeded in excellent yield when applied to N-Boc proline (87%), but the yields dropped when the procedure was attempted on dipeptides (54–68%). Azidation of proteins would allow for further modification through ‘click’ chemistry, but further progress on this azidation reaction is warranted for efficient protein modification.
 | ||
| Fig. 26 Developments in decarboxylative azidation, shown to be applicable to proline and peptide substrates containing a C-terminal proline residue. DCE = dichloroethane. | ||
4. Conclusions and outlook
Amino acid modification has been a key interest in recent years and has been applied across a wide variety of fields. Photocatalysis has offered new synthetic routes to amino acid modification. While lysine and cysteine have been widely targeted in bioconjugation through ionic methods, photocatalysis provides complementary methods to these, enabling modification with higher chemoselectivity. In addition to this, photocatalysis enables modification of amino acids for which there are either few or no other reported methods. An overview of the applicability of current photocatalytic methods for amino acid modification is given in Fig. 27.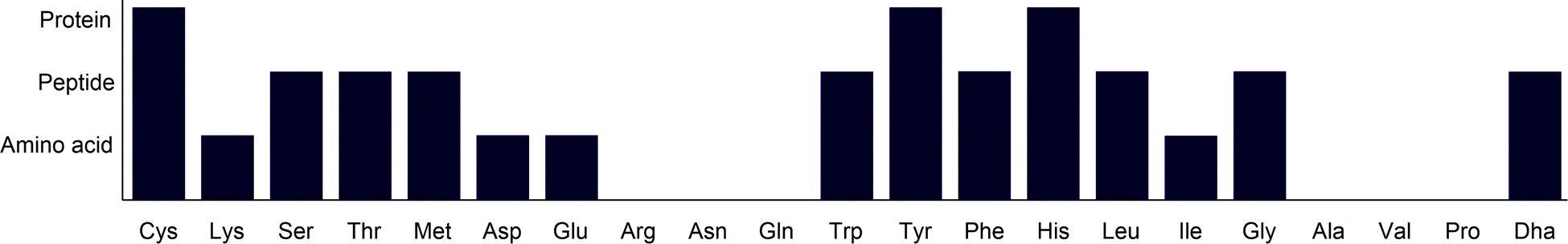 | ||
| Fig. 27 Overview of the complexity of substrates to which photocatalytic amino acid modification has been applied. Proteins are defined here as peptides containing over 50 amino acids. | ||
One area in which photocatalysis is a great advance on previous methods is C–H functionalisation. Whereas most previous protocols require high temperature, which to a protein would cause significant decomposition, photocatalytic processes are rarely performed above room temperature, leading to greater stability of the substrates. This has notably been applied to aliphatic residues such as leucine, isoleucine and glycine, which have been modified with simple electrophiles under ambient conditions. The emergence of C–H functionalisation protocols compatible with peptide substrates and capable of modifying aliphatic side chains shows great promise for the future of amino acid modification.
Despite great advances in the field of amino acid modification, significant challenges remain. Many of the exciting new protocols have been shown to be less effective when applied to larger peptide substrates. While amino acid modification is itself a significant goal, for these to be valuable tools beyond single residue modification, progress must be made in adapting and improving the procedures. In addition, many of the procedures detailed in recent years involve organic solvents which are themselves not suitable for solvating larger peptides or proteins. For these protocols to be more widely used, developments should be made to enable the transformations in aqueous solvent, preferably buffered at physiological pH.
The number of articles detailing amino acids among their substrate scopes has risen exponentially over the last two decades. As amino acid modification remains a rich area for discovery, this is unlikely to fall soon. Photocatalysis shows great potential in expanding the toolbox of methods for bioconjugation and amino acid modification.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Berg, J. L. Tymoczko, G. J. Gatto and L. Stryer, Biochemistry, New York, 9th edn, 2019 Search PubMed.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 CrossRef CAS.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- H. F. Lodish, Enzyme Microb. Technol., 1981, 3, 178–188 CrossRef CAS.
- N. Krall, F. P. Da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS.
- P. Akkapeddi, S. A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954–2963 RSC.
- A. J. Russell, S. L. Baker, C. M. Colina, C. A. Figg, J. L. Kaar, K. Matyjaszewski, A. Simakova and B. S. Sumerlin, AIChE J., 2018, 64, 3230–3245 CrossRef CAS.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS.
- J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176–6199 CrossRef CAS.
- J.-Q. Liu, A. Shatskiy, B. S. Matsuura and M. D. Kärkäs, Synthesis, 2019, 2759–2791 CAS.
- W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS.
- J. N. de Gruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS.
- L. R. Malins, Pept. Sci., 2018, 110, e24049 CrossRef.
- C. Bottecchia and T. Noël, Chem. – Eur. J., 2019, 25, 26–42 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS.
- C. C. Le, M. K. Wismer, Z. C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647–653 CrossRef CAS.
- G. Zhao, S. Kaur and T. Wang, Org. Lett., 2017, 19, 3291–3294 CrossRef CAS.
- B. A. Vara, X. Li, S. Berritt, C. R. Walters, E. J. Petersson and G. A. Molander, Chem. Sci., 2018, 9, 336–344 RSC.
- H. A. Beard, J. R. Hauser, M. Walko, R. M. George, A. J. Wilson and R. S. Bon, Chem. Commun., 2019, 2, 133 CrossRef CAS.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef CAS.
- H. Lee, N. C. Boyer, Q. Deng, H. Y. Kim, T. K. Sawyer and N. Sciammetta, Chem. Sci., 2019, 10, 5073–5078 RSC.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS.
- M. Jiang, Y. Jin, H. Yang and H. Fu, Sci. Rep., 2016, 6, 26161 CrossRef CAS.
- Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797–6800 CrossRef CAS.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS.
- S. Sato and H. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 8681–8684 CrossRef CAS.
- N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100–16104 CrossRef CAS.
- A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11, 426–433 CrossRef CAS.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS.
- S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS.
- C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 1–7 CrossRef.
- M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem. – Eur. J., 2019, 25, 8240–8244 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- R. A. Aycock, D. B. Vogt and N. T. Jui, Chem. Sci., 2017, 8, 7998–8003 RSC.
- R. A. Aycock, C. J. Pratt and N. T. Jui, ACS Catal., 2018, 8, 9115–9119 CrossRef CAS.
- A. D. de Bruijn and G. Roelfes, Chem. – Eur. J., 2018, 24, 11314–11318 CrossRef CAS.
- J. Sim, M. W. Campbell and G. A. Molander, ACS Catal., 2019, 9, 1558–1563 CrossRef CAS.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS.
- Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS.
- D. C. Marcote, R. Street-Jeakings, E. Dauncey, J. J. Douglas, A. Ruffoni and D. Leonori, Org. Biomol. Chem., 2019, 17, 1839–1842 RSC.
| This journal is © The Royal Society of Chemistry 2021 |