
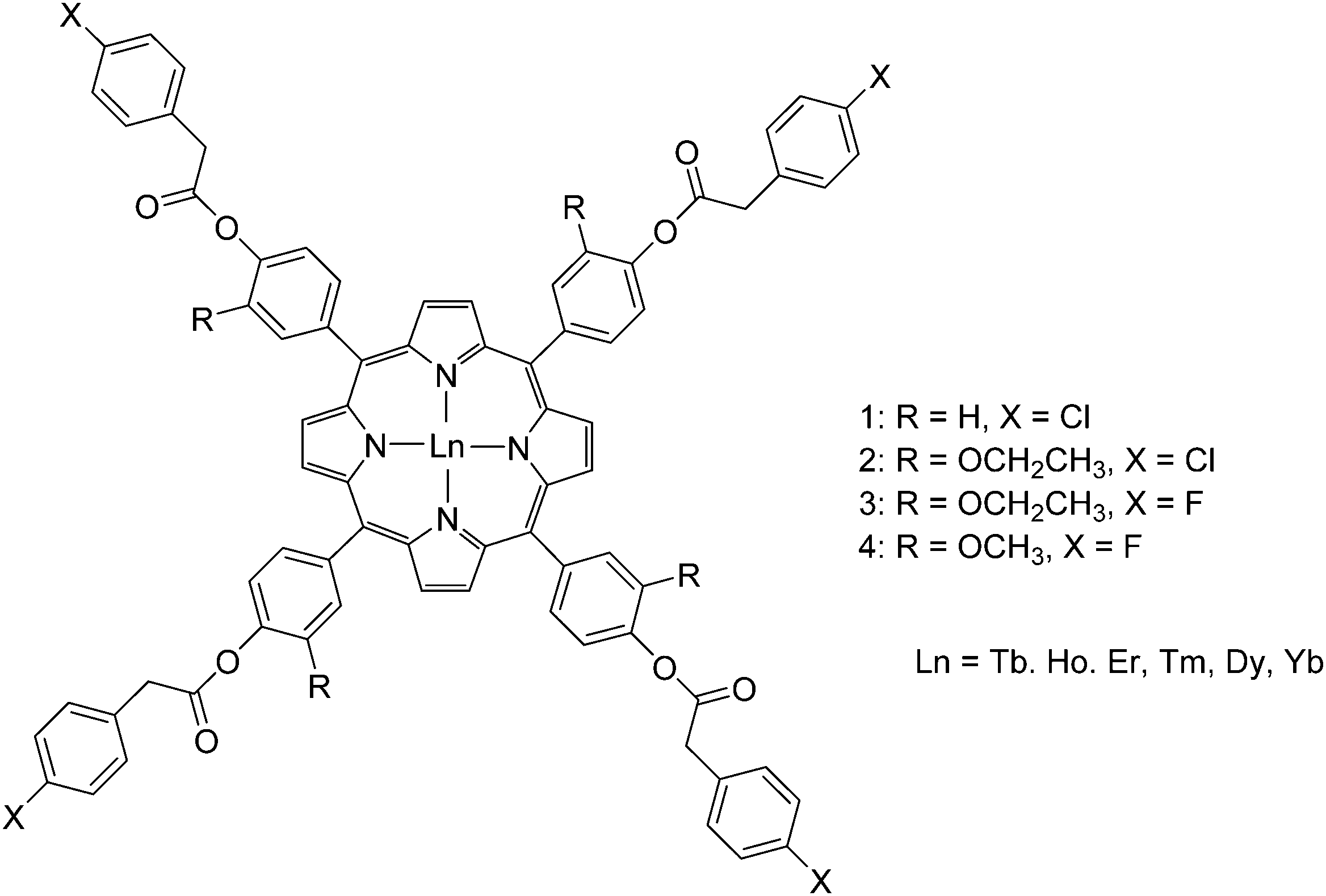
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide–tetrapyrrole complexes: synthesis, redox chemistry, photophysical properties, and photonic applications
Wai-Lun
Chan
 ab,
Chen
Xie
a,
Wai-Sum
Lo
b,
Jean-Claude G.
Bünzli
*ac,
Wai-Kwok
Wong
*a and
Ka-Leung
Wong
*a
ab,
Chen
Xie
a,
Wai-Sum
Lo
b,
Jean-Claude G.
Bünzli
*ac,
Wai-Kwok
Wong
*a and
Ka-Leung
Wong
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China. E-mail: wkwong@hkbu.edu.hk; klwong@hkbu.edu.hk
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China
cInstitute of Chemical Sciences & Engineering, Swiss Federal Institute of Technology, Lausanne (EPFL), Switzerland. E-mail: jean-claude.bunzli@epfl.ch
First published on 23rd September 2021
Abstract
Tetrapyrrole derivatives such as porphyrins, phthalocyanines, naphthalocyanines, and porpholactones, are highly stable macrocyclic compounds that play important roles in many phenomena linked to the development of life. Their complexes with lanthanides are known for more than 60 years and present breath-taking properties such as a range of easily accessible redox states leading to photo- and electro-chromism, paramagnetism, large non-linear optical parameters, and remarkable light emission in the visible and near-infrared (NIR) ranges. They are at the centre of many applications with an increasing focus on their ability to generate singlet oxygen for photodynamic therapy coupled with bioimaging and biosensing properties. This review first describes the synthetic paths leading to lanthanide–tetrapyrrole complexes together with their structures. The initial synthetic protocols were plagued by low yields and long reaction times; they have now been replaced with much more efficient and faster routes, thanks to the stunning advances in synthetic organic chemistry, so that quite complex multinuclear edifices are presently routinely obtained. Aspects such as redox properties, sensitization of NIR-emitting lanthanide ions, and non-linear optical properties are then presented. The spectacular improvements in the quantum yield and brightness of YbIII-containing tetrapyrrole complexes achieved in the past five years are representative of the vitality of the field and open welcome opportunities for the bio-applications described in the last section. Perspectives for the field are vast and exciting as new derivatizations of the macrocycles may lead to sensitization of other LnIII NIR-emitting ions with luminescence in the NIR-II and NIR-III biological windows, while conjugation with peptides and aptamers opens the way for lanthanide–tetrapyrrole theranostics.
 Wai-Lun Chan | Wai-Lun Chan received a BSc (Hons.) in Chemistry (2014) under the supervision of Prof. Ka-Leung (Gary) Wong and a MA in Communication (2015) from Hong Kong Baptist University, followed by a PhD in Chemistry (2019) from the National University of Singapore under the supervision of Prof. Yixin Lu. After a postdoctoral visit (2020) with Prof. Markus Zweckstetter at Max Planck Institute for Biophysical Chemistry and German Center for Neurodegenerative Diseases, he was appointed as a research assistant professor (2021) at the Department of Applied Biology and Chemical Technology, Hong Kong Polytechnic University. His research interests include lanthanide spectroscopy, asymmetric organocatalysis, natural product/peptide synthesis, bioimaging, and targeted protein degradation. |
 Chen Xie | Chen Xie received a MSc from Hong Kong Baptist University in 2017 and is now a PhD candidate in the research group of Prof. Ka-Leung (Gary) Wong. His current research focuses on the design and preparation of lanthanide-based complexes and the investigation of their photophysical properties and biological applications. |
 Wai-Sum Lo | Wai Sum Lo obtained a BSc in Applied Chemistry from Hong Kong Baptist University in 2010. In 2016, he received a PhD in Inorganic Chemistry from The Hong Kong Polytechnic University (PolyU), where his thesis focused on the synthesis and energy transfer studies of luminescent lanthanide complexes. After completing his postdoctoral research at PolyU on the photophysical studies of lanthanide complexes with potential bio-applications, he has started working as a Research Fellow at the same institute with fervent interest in understanding circularly polarized luminescence from lanthanide materials. |
 Jean-Claude G. Bünzli | Prof. Bünzli received his PhD degree from the Swiss Federal Institute of Technology, Lausanne (EPFL) in 1971. After post-doctoral stays at UBC, Vancouver, and ETHZ, Zürich, he returned to Lausanne in 1974 as an assistant professor at the University; he was promoted to Chair Professor in 1980; he served as Dean of the Faculty of Sciences (1990–1991) and Vice-President of the University (1991–1995) before transferring to EPFL in 2001. His research interests are focused on coordination, macrocyclic, and supramolecular chemistry of rare earths with main emphasis on the development of lanthanide-based luminescent materials and bioprobes. |
 Wai-Kwok Wong | Prof. Wai-Kwok Wong, a native of Hong Kong, received his PhD degree in Chemistry from the University of Wisconsin, Madison in 1978. He then spent a year at UCLA as a post-doctoral researcher with Prof. John Gladysz before moving on to work with the late Prof. Sir Geoffrey Wilkinson at Imperial College, London in 1979. He returned to Hong Kong in 1984 and took up a lecturer position at the Hong Kong Polytechnic University (then Hong Kong Polytechnic). In 1989, he joined the Chemistry Department of Hong Kong Baptist University (then Hong Kong Baptist College) where he stayed ever since. He was promoted to Chair Professor of Chemistry in 2005. He was Dean of Science (2002–2010) and the Vice-President (Research and Development) (2011–2019). He has published more than 260 papers. His main research interests include organometallic synthesis, bioactivity of inorganic/organometallic compounds, luminescent materials and homogeneous catalysis. |
 Ka-Leung Wong | Ka-Leung (Gary) Wong is the Dr Mok Man Hung Endowed Professor of Chemistry and Head of the Department at Hong Kong Baptist University. His undergraduate and graduate training was undertaken at Hong Kong University where he worked with Prof. Wing-Tak Wong, followed by post-doctoral periods at City University Hong Kong and Durham University (Royal Society post-doctoral fellowship with Prof. David Parker). His research interests span bioinorganic chemistry, the study of responsive luminescent materials as well as numerous applications of lanthanide science and optical spectroscopy. Recent work embraces the development of new probes and drugs for cancer diagnostics and photodynamic therapy. |
1 Introduction
Tetrapyrrole derivatives such as porphyrins or phthalocyanines are highly chemically stable macrocycles and considered as essential constituents in the emergence of life. Porphyrins are known since the seminal work of Hoppe-Seyler in the 1870s, the founder of physiological chemistry1 who studied various body fluids, including blood, and measured the electronic spectra of haemoglobin and chlorophyll, among others. The simplest compound is porphine (H2Por, Scheme 1) that does not occur in nature, but some substituted porphines do, such as protoporphyrin IX (H2ProP9, Scheme 1), a precursor to haem and haemoglobin.2 Common synthetic mimics of protoporphyrin IX used in coordination chemistry of transition metal ions and lanthanides are octaethylporphyrin (H2OEP), tetrabenzoporphyrin (H2TBP), and tetraphenylporphyrin (H2TPP) (Scheme 1). The highly delocalized π-system of porphyrins (highlighted in red in Scheme 1) confers them vivid colours and a wide variety of genuine optical, electric, and magnetic properties with applications ranging from catalysis to electronic materials, photonics, biomedical analysis, imaging, and therapy.3–5 Partial hydrogenation of H2Por results in highly photosensitive macrocycles termed chlorins (H2cl; bacteriochlorin H2Bcl; isobacteriochlorin H2iBcl, see Scheme 1) that are commonly used as singlet oxygen generators in photodynamic therapy (PDT) of cancer.6,7 When the methine bridges between the pyrrole moieties of porphyrins are replaced with methylene bridges, the corresponding hydrogenated macrocycles are named porphyrinogens (H4Pg). These compounds occur in living organisms, substituted with various side chains, and are reaction intermediates in the biosynthesis of several strategic life-indispensable molecules such as haemoglobins, cytochromes, or chlorophylls; a typical example is protoporphyrinogen IX (H4ProPg9, Scheme 1), the precursor to protoporphyrin IX. The meso-octasubstituted porphyrinogens constitute a class of macrocycles known since the 19th century, called calix[4]pyrroles (H4Cx[4]Py), and were put in front of the chemical scene in the 1990s by Floriani et al.8 and Sessler et al.,9 mainly in view of their host–guest properties and small-molecule activation capability. | ||
| Scheme 1 (A) Chemical structures and formulae of porphine (with the IUPAC numbering scheme10), porphyrins, chlorins, porphyrinogens, and phthalocyanines discussed in this review. In green: naturally occurring macrocycles; in red: 18 π-electron system of H2Por and H2Pc. (B) Chemical structures and formulae of common reagents involved in the syntheses of tetrapyrroles. | ||
Contrary to porphyrinogens that are usually colourless due to the lack of a delocalized π-electron system, phthalocyanines (H2Pc) are intensively coloured derivatives of tetraazaporphyrin (H2N4Por, Scheme 1), in which the methine bridges are replaced with imine nitrogens, with the pyrrole units being additionally fused with benzene rings. Such intensively blue-green-coloured synthetic macrocycles were apparently isolated in 1907 from a mixture resulting from the reaction of acetic anhydride with phthalamide11 and found to be identical to a product discovered by Posner in 1897 upon heating o-cyanobenzamide at an elevated temperature.12 These compounds can also be thought of as having an aza-annulene core. The metal derivatives of tetraazaporphyrin are not natural products, but due to their extended π-electron system, they have many applications as pigments—in particular the important phthalocyanine blue, [Cu(Pc)], electrochromic materials, redox catalysts, agents for photodynamic treatment of cancer or flow-injection analysis.13 Fusion of H2N4Por with anthracene rings results in naphthalocyanine (H2Nc, Scheme 1) which made the highlights in 2012 when scientists used it to visualize charge distribution in a single molecule by a combination of advanced microscopy techniques.14
From a historical perspective, lanthanide complexes with porphyrins were first reported by Horrocks et al., [Ln(TPP)L] (L is a β-diketonate, often acetylacetonate, acac−), in the search for efficient magnetic resonance dipolar probes,15 while in the same year Tsvirko et al. demonstrated energy transfer from TPP to YbIII and reported a quantum yield in solution of 0.5% for the metal-centred NIR luminescence.16 These 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes are usually prepared by reacting [Ln(acac)3(H2O)n] with the ligand in trichlorobenzene. By starting with [Ce(acac)3(H2O)3] and prolonging the refluxing time, Buchler et al. surprisingly isolated in 1983 the first neutral and diamagnetic bis(porphyrinato) complex [CeIV(TPP)2], followed by [PrIIIH(TPP)2].17 Then, the triple-decker complex [CeIII2(OEP)3] was obtained simultaneously with [CeIV(OEP)2] by a competitive condensation/autoxidation reaction.18 Regarding phthalocyanines, they were introduced as pigments on the market in the 1930s, consisting of either un-metalated H2Pc (blue-green) or [Cu(Pc)] (blue). When the benzene rings are fully chlorinated the corresponding CuII pigment is green; many colour variations can be obtained by modifying the ring substituents. When it comes to lanthanides, the first tetrapyrrole “sandwich” complexes were isolated by Kirin et al. in the 1960s, [LnH(Pc)2] (Ln = Pr, Nd, Er, Lu),19 who subsequently demonstrated their electrochromism. In fact the green LnIII–phthalocyanine sandwich complexes have a radical nature, which was established by EPR spectroscopy for the LuIII compound: one macrocycle is a di-anion but the other is a radical mono-anion so that the correct chemical formula is [Ln(Pc2−)(HPc−˙)],20 the structure responsible for their particular properties.21
:1 complexes are usually prepared by reacting [Ln(acac)3(H2O)n] with the ligand in trichlorobenzene. By starting with [Ce(acac)3(H2O)3] and prolonging the refluxing time, Buchler et al. surprisingly isolated in 1983 the first neutral and diamagnetic bis(porphyrinato) complex [CeIV(TPP)2], followed by [PrIIIH(TPP)2].17 Then, the triple-decker complex [CeIII2(OEP)3] was obtained simultaneously with [CeIV(OEP)2] by a competitive condensation/autoxidation reaction.18 Regarding phthalocyanines, they were introduced as pigments on the market in the 1930s, consisting of either un-metalated H2Pc (blue-green) or [Cu(Pc)] (blue). When the benzene rings are fully chlorinated the corresponding CuII pigment is green; many colour variations can be obtained by modifying the ring substituents. When it comes to lanthanides, the first tetrapyrrole “sandwich” complexes were isolated by Kirin et al. in the 1960s, [LnH(Pc)2] (Ln = Pr, Nd, Er, Lu),19 who subsequently demonstrated their electrochromism. In fact the green LnIII–phthalocyanine sandwich complexes have a radical nature, which was established by EPR spectroscopy for the LuIII compound: one macrocycle is a di-anion but the other is a radical mono-anion so that the correct chemical formula is [Ln(Pc2−)(HPc−˙)],20 the structure responsible for their particular properties.21
The diameters of lanthanide ions are larger than the cavity diameter of Por and Pc ligands so that the metal centres in the complexes lie above the planar macrocycles. The main stoichiometries and structures encountered in the coordination of lanthanides with tetrapyrrole ligands are illustrated in Fig. 1. The 1:1 and 1:2 (Ln:L) geometries are the commonest, followed by triple deckers ideally suited for the study of magnetic interactions since the Ln–Ln distance is rather short (∼0.35 nm); although less observed, the 2:2 stoichiometry also yields interesting f–f assemblies.22–24 More elaborate structures such as quadruple-decker 2:4 complexes25 in which two double-decker units are linked by a diethoxybenzyl unit26 have also been explored, as well as 4:5 bimetallic 4d–4f quintuple deckers.27
 | ||
| Fig. 1 Top: Schematic representation of some of the various structures encountered in lanthanide–tetrapyrrole Ln:L complexes; 1:1 and 2:2 complexes may be 7- or 8-coordinated. Bottom: Crystal structures of (left) double-decker [CeIV(OEP)2], (middle) triple-decker [CeIII2(OEP)3]. Redrawn from data in ref. 18; and (right) [TbIII2CdII2(OBPc)5], OBPc = octabutoxy-phthalocyanate, redrawn from data in ref. 27. | ||
Although lanthanide tetrapyrrole complexes are regularly mentioned in review articles dealing with one or another aspect of 4f-element coordination chemistry,28 supramolecular chemistry,29,30 photo- and spectro-chemistry,31–34 liquid crystals,35 analytical10,36 or bio-applications,4,37 non-linear optical properties,38 and magnetism,39 the field itself has seen a few comprehensive reviews during the past 20 years, since the publication of the Porphyrin Handbook.40 In addition, most of these reviews are focused onto a particular class of compounds, mononuclear complexes,41,42 sandwich complexes,43 porphyrin complexes,41 or phthalocyanine complexes.21
In this review, we therefore aim at giving an updated compendium of lanthanide–tetrapyrrole complexes, from their synthesis and structure to spectroscopy and reactivity, including the fast-developing fields of photophysical properties, non-linear optics and theranostics. Excluded herein are tetrapyrrole-attached/conjugated lanthanide nanomaterials/complexes, and single-molecule magnetism. Applications will be presented, including optical limiting materials, NIR-emitting sensors, bioprobes, and singlet oxygen generators. The literature is covered up to October 2020, with a particular emphasis on the period spanning the past 20 years.
2 Structure and synthesis
2.1 Lanthanide mono-tetrapyrrole complexes
(a) Porphyrins. One cannot discuss porphyrin synthesis without mentioning Rothemund's synthesis, first reported in 1936,44 which was achieved by reacting formaldehyde and pyrrole in an acid medium at high temperatures for 24 h. In 1967, a “simplified version” of Rothemund's synthetic procedures was published for the preparation of meso-substituted porphyrins, which is now commonly known as the Adler–Longo methodology (Scheme 2).45 Adler and Longo claimed that while their procedure does not necessarily lead to the highest synthetic yield (∼20%), it represents a convenient (in open air), fast (∼30 min), and reproducible synthesis of highly pure porphyrins. In general, equimolar amounts of freshly distilled pyrrole and aldehyde are added into propionic acid and refluxed for 30 min. The reaction mixture is then cooled and filtered; the filter cake is washed with methanol and hot water, followed by the removal of residual acid in vacuo. The substituted porphyrin is obtained as coloured crystals. Lindsey et al.46 further modified the procedure, putting emphasis on improving the synthetic yields and using milder reaction conditions, for instance, lowering the “unnecessarily high” reaction temperature, while broadening the choice of accessible benzaldehydes. The generic synthesis starts with adding equimolar amounts of pyrrole, aldehyde and triethyl orthoacetate into dry CH2Cl2 under N2. An aliquot of boron trifluoride etherate (Lewis acid) is also added and the reactants are mixed at room temperature for 1 h. Then, 2,3,5,6-tetrachlorobenzoquinone (chloranil, Scheme 1) is added as the oxidant and the reaction mixture is refluxed for 1 h. Removing the solvent in vacuo and further purification by column chromatography give the product in ∼45–50% isolated yield. The authors noted that, after systematic studies, the synthetic yields were found to be dependent on various conditions such as the nature of the acid catalyst, the choice of oxidant, the reaction time, the concentrations of the reactants, as well as on the solvent dryness.
 | ||
| Scheme 2 Classical Rothemund's and Adler–Longo synthetic approaches to porphyrins.44,45 | ||
In 1998, Wong et al. investigated the synthesis of cationic lanthanide porphyrin complexes, where the free base porphyrins in use, 5,10,15,20-tetrakis(p-tolyl)porphyrin (H2TTP, Scheme 1) and 5,10,15,20-tetrakis-(p-methoxyphenyl)porphyrin (H2TMOPP, Scheme 1), were conveniently synthesized according to the Adler–Longo method.47 Yu's group also obtained H2TPP with the same methodology for the solid state synthesis of [LnTPP(acac)] complexes.48 Both the Adler–Longo and Lindsey syntheses remained popular in the early 2000s with Asano-Someda's group,49 Tsukube's group,50 Wong's group,51,52 He's group53–56 and Zhang's group,57 all employing similar procedures for the synthesis of lanthanide-monoporphyrinates with different functional groups (Fig. 2), including H2OEP, crown ether-containing 5-(3-aminophenyl)-10,15,20-triphenylporphyrin (m-H2APTPP, Scheme 1) (see Section 2.1.8 for a specific discussion on crown-porphyrins), 5,10,15,20-tetrakis[3,4,5-tri(methoxy)phenyl]porphyrinate (H2TMO3,4,5PP), H2TMOPP and H2TTP, 5,10,15,20-tetrakis(p-chlorophenyl)porphyrin (H2TCPP), 5,10,15,20-tetrakis(p-bromophenyl)porphyrin (H2TBPP), 5,10,15,20-tetrakis(p-fluorophenyl)porphyrin (H2TFPP) and 5,10,15,20-tetrakis(2,4,6-trimethoxyphenyl)porphyrin (H2TMO2,4,6PP), see Scheme 1. As such, the Adler–Longo and Lindsey protocols have regularly underpinned the synthesis of porphyrins in recent decades.
 | ||
| Fig. 2 Examples of lanthanide monoporphyrinates synthesized using Alder and Longo's method.48,50–53,57 | ||
Lindsey later became interested in improving the synthesis of ortho-substituted tetraphenylporphyrins, especially those that are sterically hindered and are difficult to synthesize under the established mild conditions. He and Wagner discovered that, by adding a boron trifluoride–ethanol co-catalyst, the reaction yields of 2-alkyl-, 2-alkoxy- and 2,6-dialkoxybenzaldehydes increased, whereas the catalytic system did not improve the reaction involving six ortho-halogen-substituted benzaldehydes.58 In Wong's report of the first series of monoporphyrinato lanthanide complexes with large optical-limiting capabilities,59 the porphyrin free base 5,10,15,20-tetrakis(p-diphenylaminophenyl)-21H,23H-porphine (H2TDPAPP) was prepared by the condensation of pyrrole and 4-diphenylaminobenzaldehyde referencing to Cheng's synthesis,60 itself inspired by Lindsey's method.
When it comes to novel porphyrin-based electronic push–pull, bioconjugated framework systems for supramolecular photonic and biomedical applications, unsymmetrically substituted porphyrinoids have been foregrounded, in which the tetraphenylporphyrins comprise different meso-phenyl functionalities (A, B, C, and D), giving rise to A3B–Por, cis/trans-A2BC–Por, cis/trans-A2B2–Por and ABCD–Por combinations. To access them, three general approaches are employed: (i) statistical mixed condensation of pyrrole/dipyrromethene with different aldehydes,61 (ii) late-stage core/peripheral functionalization with organometallics,62 and (iii) cross-coupling of pre-formed pyrrolic precursors via the “2 + 2” (for the A2B2-type) and “3 + 1” (for the A3B-type) additions.63 All their synthetic details are well documented in the literature.
(b) Phthalocyanines. Phthalocyanines have structural similarity with porphyrins, with four isoindole units linked by nitrogen atoms in an azo position instead of pyrrole units linked by carbon bridges. They can be synthesized by various cyclotetramerization methods starting from a catalogue of phthalic acid derivatives, e.g. phthalonitrile, phthalic anhydride, phthalimide, and diiminoisoindoline (Fig. 3), using the seminal Linstead procedure.64 Two well-established routes are (i) metal-templated synthesis, by refluxing phthalonitrile and lithium metal in pentanol to form di-lithium phthalocyanines, followed by the removal of the lithium content under acidic conditions; (ii) metal-free synthesis, upon heating phthalonitrile either in a melt with hydroquinone as a reductant, or in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), or 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) (Scheme 1), in high-boiling pentanol. Subsequently, the insoluble crude product can be purified by sublimation and/or washing with various solvents under Soxhlet extraction or centrifugation. Microwave irradiation in an optimized solvent is an alternative for preparing both metal-free and metallo-phthalocyanines in high yields and in a very short period of time, e.g., in a few minutes.
 | ||
| Fig. 3 Common synthetic precursors for phthalocyanines. | ||
As far as less symmetrically substituted phthalocyanines are concerned, several synthetic methods have been developed. The first one is statistical mixed condensation in which equimolar quantities of dinitriles A & B produce a statistical mixture of A4 (8.33%), A3B (25%), cis-A2B2 (25%), trans-A2B2 (8.33%), AB3 (25%), and B4 (8.33%). The other ones are sterically driven cross condensation and polymer-supported exclusive preparation of the A3B-type congener. Interested readers are advised to consult the seminal review by Mack and Kobayashi.65
(a) Lanthanide porphyrinates. In 1974, Horrocks Jr.'s group reported a lanthanide complexation method with symmetrical free base porphyrins.66 In general, H2TPP and two equivalents of tris(2,4-pentanedionate)europium(III) were refluxed in TCB for 3–4 h and the crude product was then chromatographed to yield the desired complex. This method is still widely in use nowadays, as exemplified by Zhang's group in the 2010s (Scheme 3).67,68 An alternative method for preparing cationic lanthanide monoporphyrinates was reported by Wong's group in 1999 using lanthanide amide complexes as synthetic precursors.47 The lanthanide amide complex Ln[N(SiMe3)2]3·x[LiCl(THF)3] was first generated in situ in excess by reacting LnIII trichloride with lithium bis(trimethylsilyl)amide in THF. The free base porphyrin (H2TTP or H2TMPP) was subsequently added and the reaction was refluxed in a bis(2-methoxyethyl) ether–tetrahydrofuran mixture for 48 h to give the air-stable cationic [LnIII(TTP)(H2O)2(THF)]Cl and [LnIII(TMPP)(H2O)3]Cl complexes, respectively. Both purple crystals were then subjected to the following procedure: (i) extraction with CH2Cl2, filtration and then concentration to dryness, (ii) washing with n-hexane and diethyl ether, and (iii) extraction with THF and filtration before (iv) final crystallization. Since then, this synthetic procedure for cationic porphyrin complexes was continuously used by Wong's group (Fig. 4, left).69,70
 | ||
| Scheme 3 Lanthanide complexation to porphyrins by Zhang's group following the procedure developed by Horrocks Jr.'s group. Redrawn from ref. 67. | ||
 | ||
| Fig. 4 (Left) X-Ray crystal structure of the cationic Yb–porphyrinate complex. Reprinted with permission from ref. 69, copyright (2000) Elsevier B.V. (Right) Porphyrin–cyclen sandwich lanthanide complex. Redrawn from ref. 73. | ||
As mentioned above, in Yu's solid state synthesis,48 H2TPP and Ln(acac)3 were mixed in a 1:3 mass ratio and ground in a mortar, followed by transferring the mixture into a nitrogen atmosphere. The solids were slowly heated to 140–150 °C causing their melting and blackening. The temperature was then increased to 220–230 °C and maintained for 2 h to give a blue-purple reaction crude that was cooled and chromatographed on neutral Al2O3 with the use of a gradient CH2Cl2:MeOH eluent.
Boncella's group in 2002 reported the preparation of anhydrous lanthanide chloride complexes with tetraphenylporphyrin by a salt metathesis reaction of LnCl3 (Ln = Yb, Tm, Er, Ho) and an equimolar quantity of di-lithiotetraphenylporphyrin bis(dimethoxyethane) under reflux conditions (Scheme 4).71 Upon metathesis, the lithium chloride could be easily separated by hot filtration and the reaction yield reached 85%. The same group improved their method for the synthesis of Ln(TPP) complexes capped with multidentate ligands that had higher yields and could be scaled up to gram-scale synthesis.72
 | ||
| Scheme 4 Salt metathesis reaction for lanthanide complexation proposed by Boncella's group. Redrawn from ref. 71. | ||
Ishikawa et al. reported an interesting saturation of lanthanide coordination using two macrocycles.73,74 Freshly made hydrated lanthanide chloride was heated at high temperature with H2TPP in DMF containing DBU for 12–36 h. After simple work-up to obtain the monoporphyrinato intermediate complex, it was redissolved in methanol and 1,4,7,10-tetraazacyclododecane (cyclen) was added. The lanthanide centre in the final product [Ln(TPP)(cyclen)]Cl was sandwiched between the porphyrin and the cyclen, with each macrocycle contributing 4 nitrogen atoms for coordination (Fig. 4, right). Purification of the product was achieved by crystallization by liquid–liquid diffusion of hexane and CH2Cl2.
In 2008, He et al. disclosed another facile approach to prepare neutral monoporphyrinate lanthanide(III) complexes by, for example, reacting Yb(OAc)3·3H2O with H2TPP in TCB at 210 °C under N2 for 48 h, followed by stirring with methanol at 70 °C for 2 h. The desired product, seven-coordinated [Yb(TPP)(OOCCH3)(CH3OH)2], where acetate is monocoordinated, was exclusively obtained. It was used as a precursor for introducing bidentate ligands by exchange of the two labile methanol molecules, resulting in coordinatively saturated eight-coordinated monoporphyrinate YbIII complexes in which the acetate anion acts as a bidentate ligand after methanol substitution.75
(b) Lanthanide phthalocyaninates. Although the first examples of lanthanide–phthalocyanine complexes date back to as early as the 1960s,76 it was not until the 1980s that a large interest flourished in this research field. In 1983, through refluxing di-lithium phthalocyanine aggregate and tris(2,2,6,6-tetramethylheptane-3,5-dionato)LnIII in anhydrous THF under an argon atmosphere, that is, via the metal displacement strategy, Sugimoto et al. obtained a series of dinuclear [(μ-phthalocyaninato)(diketonato)4Ln2] complexes, where the phthalocyanine ring served as a bridging ligand (Fig. 5, left).77 Intriguingly, the authors found that the reaction gave the 1
:1 complexes instead in the presence of oxygen. Weiss et al. reported the synthesis and structure of [LuPc(OAc)(H2O)2]·H2O·2CH3OH, as well as of the sandwich [LuPc2]·CH2Cl2 in 1985, exploiting the metal-templating method, by refluxing Lu(OAc)3·xH2O, 1,2-dicyanobenzene, and DBU in boiling 1-hexanol.78 Adding a larger proportion of the lanthanide salt and shortening the heating time could slightly increase the yield of the monophthalocyaninate complex. With the same in situ generated Ln[N(SiMe3)2]3·x[LiCl(THF)3] reagent used for complexing the lanthanide ion, like in previous work,47 Wong et al. prepared a series of tripodal ligand-capped (monophthalocyaninato)lanthanide complexes in 2009.79 This procedure afforded green crystals of [Ln(t-Bu)4Pc(LOMe)], Ln = Yb and Er, with yields up to 65%.
 | ||
| Fig. 5 (Left) Crystal structure of dinuclear disamarium phthalocyaninate. Reproduced with permission from ref. 77, copyright (1983) The Royal Society of Chemistry. (Right) Magnified view of the STM image taken on a Ag(111) surface after subsequent deposition of a low coverage of gadolinium (0.06 monolayer) on the ADN monolayer, followed by annealing to 450 K, overlaid with a molecular model of Gd-SNPc. Reproduced with permission from ref. 82, copyright (2019) Nature Publishing Group. | ||
More recently, in 2015, Znoiko et al. demonstrated the successful synthesis of mixed 2-naphthoxy- and benzotriazolyl disubstituted lanthanide phthalocyaninates (NOBNPc, Scheme 1) with LnCl3·6H2O (Ln = Er, Yb) in the presence of urea at 190–195 °C, but the regioselectivity issue was not mentioned.80 The product was purified by, in sequence, (i) grinding in a mortar, (ii) washing with water and then acetone to remove the salt excess and unreacted phthalonitrile, respectively, and (iii) extraction with chloroform, before routine column chromatography (silica gel M 60, eluent: chloroform). In 2018, Dubinina et al. demonstrated that both templated and multi-step methods can be applied for efficiently synthesizing new lanthanide pyrazinoporphyrazine complexes with the assistance of both thermal and microwave irradiation techniques; the products were purified by simple precipitation from a MeOH:H2O (20:1 v/v) mixture, followed by washing with H2O, MeOH and acetone.81 In 2019, by taking advantage of the larger size of GdIII compared to FeII, Gottfried's group serendipitously obtained the template-controlled on-surface cyclopentamerization, rather than the expected formation of bis(phthalocyaninato) double deckers.78 The resulting gadolinium(III) supernaphthalocyanine Gd–SNPc could be observed via scanning tunnelling microscopy (STM) (Fig. 5, right).82
Most phthalocyanine-based lanthanide complexes and multi-deckers have been symmetrically designed, similarly prepared, and strategically studied for modern applications such as organic semiconductors, field-effect transistors, and single-molecule magnets.83 These aspects have been recently reviewed and are beyond the scope of this review so the following sub-sections will concentrate more on describing kaleidoscopes of lanthanide–porphyrin coordination compounds.
 | ||
| Fig. 6 Symmetrical porphyrins with extended planar meso-substituents. Redrawn from ref. 84–87. | ||
To unravel the effect of substituents on the photophysical properties of porphyrins, Wong's group systematically modified the electron-donating and electron-withdrawing substituents at the meso-position of the porphyrin phenyl rings (Fig. 7).52 Highly symmetric tetra-substituted porphyrin free bases with phenyl, cyanophenyl, pyridyl, pentafluorophenyl and 3,4,5-trimethoxyphenyl substituents were prepared by condensation of the respective benzaldehydes and pyrrole in propionic acid, following Lindsey's improved Rothemund's method.46,89 On the other hand, the less symmetric porphyrin free bases were obtained via the condensation of benzaldehyde and the respective dipyrromethene using trifluoroacetic acid or boron trifluoride diethyl etherate as acid catalysts (Fig. 8).90–92 These authors continued to employ the widely used complexation methodology starting from lanthanide amide precursors.47 Column chromatography was performed, followed by recrystallization via diffusion of methanol into CH2Cl2 solution.
 | ||
| Fig. 7 A series of Gd–monoporphyrinate complexes with various electron-donating and electron-withdrawing meso-substituents. Redrawn from ref. 52. | ||
 | ||
| Fig. 8 An example of a less symmetrical metalated monoporphyrinate complex. Redrawn from ref. 90. | ||
In 2012, Kim et al. reported a new series of porphyrins with four triazole groups at the ortho-positions of the phenyl rings of TPP with copper-mediated click chemistry (Fig. 9).93 The precursor ZnII porphyrin complex, with meta-alkynyl substituents, was prepared by first reacting meta-substituted benzaldehyde with pyrrole and trifluoracetic acid in CH2Cl2 and then mixing with zinc acetate after purification.94 Then, benzyl azide and the precursor complex—with the trimethylsilane group deprotected by tetrabutylammonium fluoride—were reacted according to an alkyne–azide click reaction catalysed by Cu(I). The desired product was obtained in good yield as the major product and was purified by column chromatography and recrystallization. The free base porphyrin was obtained by demetallation using trifluoroacetic acid and complexation was achieved according to Wong group's route using the lanthanide amide complex.
 | ||
| Fig. 9 Porphyrin ligand bearing triazole groups prepared by copper-mediated click chemistry. Redrawn from ref. 93. | ||
In 2013, Zhang et al. decided to introduce carbazole branches onto the porphyrin ring and imparted their intrinsic photophysical and redox properties to a lanthanide porphyrin complex.95 They reported the synthesis of three 5,10,15,20-tetrakis[alkylcarbazole]porphyrins with dodecyl, tetradecyl and hexadecyl alkyl chains. The porphyrins were synthesized by refluxing the bromoalkyl carbaldehyde with pyrrole in xylene using p-nitrobenzoic acid to catalyse the reaction. A hydroxy complex was prepared by reacting lanthanide trichloride with the porphyrin in molten imidazole under dry and inert conditions for 2 h (Fig. 10).
 | ||
| Fig. 10 Lanthanide monoporphyrinate complexes with carbazole branches exhibiting redox properties. Redrawn from ref. 95. | ||
 | ||
| Fig. 11 Monoporphyrinate YbIII complex with alkyl tails. Redrawn from ref. 97. | ||
 | ||
| Fig. 12 Examples of multi-halogenated lanthanide porphyrinates at various positions of the porphyrin backbone. Redrawn from ref. 99 and 100. | ||
Zhang and Gao's group designed and synthesized a series of YbIII complexes with octafluorinated porphyrins and obtained then-unprecedented in 2017, sizeable near-infrared luminescence quantum yields for this class of complexes as the C–H oscillators are completely replaced by C–F oscillators which minimizes luminescence quenching (Fig. 12, bottom right).100 The octafluoroporphyrins were prepared by Lindsey's method46via the boron trifluoride etherate-catalysed condensation of 3,4-difluoropyrrole and the respective benzaldehydes (pentafluorobenzaldehyde, 2,6-difluorobenzaldehyde and 4-trifluoromethylbenzaldehyde). Again, the desired products were isolated using silica gel chromatography and recrystallized from CH2Cl2/n-hexane.
 | ||
| Fig. 13 Examples of water-soluble porphyrins functionalized with hydroxamic derivatives. Redrawn from ref. 102 and 103. | ||
Wong's group also reported a systematic study on a series of water-soluble lanthanide porphyrinates with pyridyl substituents in the meso positions (Fig. 14).104 A convenient one-pot synthesis with pyrrole, benzaldehyde and 4-pyridinecarboxaldehyde in propionic acid yielded variously substituted and ionic porphyrins that were separated by column chromatography. The tricationic lanthanide porphyrinates were found to exhibit binding interactions with double-stranded DNA and moderate self-stacking was observed.
 | ||
| Fig. 14 Water-soluble capped lanthanide porphyrinates with pyridyl-substituents. Redrawn from ref. 104. | ||
In recent years, Zhang's group reported the synthesis of porphyrinates with carboxylate and cationic phosphonium groups for the preparation of water-soluble ytterbium complexes for conducting live cell NIR bioimaging. Based on their previous work on octafluorinated porphyrins,100 they modified the meso positions of the porphyrin phenyl rings with water-soluble moieties such as carboxyl, glycosyl and triphenylphosphonium using methyl p-formylbenzoate, 4-(2,3,4,6-tetraacetyl-glucopyranobenzyl)aldehyde, and p-chloromethylbenzaldehyde and reacted them with 2,6-difluoropyrrole for the porphyrin synthesis.105 The resultant porphyrins were then complexed by refluxing with lanthanide acetylacetonate in TCB overnight, followed by the addition of partially deuterated Kläui's tripodal ligand, sodium [(cyclopentadienyl)tris(di(methyl-d3)phosphito)cobaltate] (Na{(η5-C5H5)Co[P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OMeD)2]3}) or in short CoPCD3 (Fig. 15). The anionic carboxylate porphyrin required an additional deprotection of the ester, while the cationic triphenylphosphonium terminal was obtained by reaction with excess triphenylphosphine. The water-solubility of the anionic carboxylate porphyrin was even good enough (∼2 mM in water) to be utilized as an in vivo NIR lifetime probe.106
O)(OMeD)2]3}) or in short CoPCD3 (Fig. 15). The anionic carboxylate porphyrin required an additional deprotection of the ester, while the cationic triphenylphosphonium terminal was obtained by reaction with excess triphenylphosphine. The water-solubility of the anionic carboxylate porphyrin was even good enough (∼2 mM in water) to be utilized as an in vivo NIR lifetime probe.106
 O)(OMe)2]3} was subsequently added and the work-up gave the Yb–porphyrinate [Yb(p-OH-TPP)(CoPMe)]. Alternatively, 5-[p-(3-bromopropoxy)phenyl]-10,15,20-triphenylporphyrin (p-BrC3O-TPPH2) was prepared by reacting p-OH-TPPH2 with excess 1,3-dibromopropane in the presence of K2CO3; the bisporphyrin dyad was obtained by reacting p-BrC3O-TPPH2 with Yb(p-OH-TPP)(CoPMe) in dry THF and K2CO3 at 60 °C overnight, followed by chromatography on silica gel. In 2012, the same group reported similar rigid acetylene-bridged ZnII porphyrin–monophthalocyaninato YbIII hybrids, ZnPor–YbPc, through convergent Sonogashira coupling (in 58% yield) between meso-ethynyl ZnII–porphyrin and I(t-Bu)3Pc; selective ZnII demetallation using conc. HCl afforded H2Por–YbPc in 93% yield; then re-metalation with PdII acetate afforded PdPor–PcYb in 71% yield (Fig. 16, bottom).109
O)(OMe)2]3} was subsequently added and the work-up gave the Yb–porphyrinate [Yb(p-OH-TPP)(CoPMe)]. Alternatively, 5-[p-(3-bromopropoxy)phenyl]-10,15,20-triphenylporphyrin (p-BrC3O-TPPH2) was prepared by reacting p-OH-TPPH2 with excess 1,3-dibromopropane in the presence of K2CO3; the bisporphyrin dyad was obtained by reacting p-BrC3O-TPPH2 with Yb(p-OH-TPP)(CoPMe) in dry THF and K2CO3 at 60 °C overnight, followed by chromatography on silica gel. In 2012, the same group reported similar rigid acetylene-bridged ZnII porphyrin–monophthalocyaninato YbIII hybrids, ZnPor–YbPc, through convergent Sonogashira coupling (in 58% yield) between meso-ethynyl ZnII–porphyrin and I(t-Bu)3Pc; selective ZnII demetallation using conc. HCl afforded H2Por–YbPc in 93% yield; then re-metalation with PdII acetate afforded PdPor–PcYb in 71% yield (Fig. 16, bottom).109
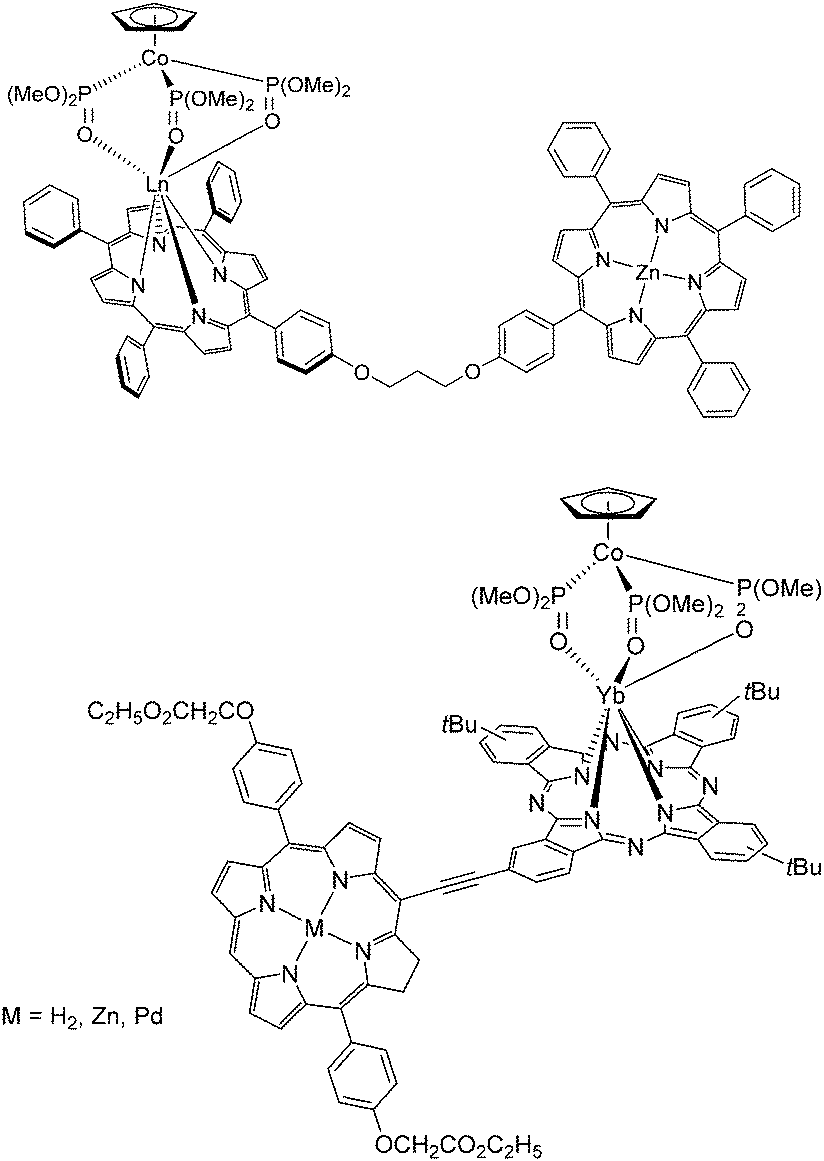 | ||
| Fig. 16 Heterodimetallic 3d–4f (top) bisporphyrin and (bottom) porphyrin–phthalocyanine lanthanide complexes designed and synthesized by the Wong group. Redrawn from ref. 107 and 108. | ||
In 2015, upon mixing and refluxing 4,5-bis-[4-tertbutyl phenoxy]-phthalonitrile, pre-synthesized 2,4,6-tris(3-thiopthalonitrile)-s-triazine, LnCl3·xH2O (Ln = Yb, Lu) and DBU in 1-pentanol in open air for 21 h, the Nyokong group isolated trimeric dendron-like lanthanide phthalocyanines in 69–77% yields; the products were purified by silica gel column chromatography by eluting first with a chloroform/hexane mixture (v/v = 1:1) and then with a chloroform/ethanol mixture (v/v = 3:1) (Fig. 17).108
 | ||
| Fig. 17 Dendron-like lanthanide triphthalocyaninate conjugates reported by the Nyokong group. Redrawn from ref. 109. | ||

 | ||
| Fig. 19 Structures of bimetallic lanthanide porphyrinates with EDTA and DTPA incorporated into the ligand design. Redrawn from ref. 112. | ||
 | ||
| Fig. 20 Corrole ligands conjugated with EDTA and DTPA synthesized with similar procedures as for porphyrin counterparts. Redrawn from ref. 115. | ||
The Wong group designed porphyrin with an appended β-diketonate which would help fulfil the LnIII coordination requirements in addition to the porphyrin N atoms (Fig. 21).116 The synthesis started with the condensation of pyrrole, benzaldehyde and o-hydroxybenzaldehyde in a molar ratio of 4:1:3 to give 5-(2-hydroxylphenyl)-10,15,20-triphenylporphyrin (o-OHTPPH2). Then, the porphyrin was mixed with 1,4-dibromobutane in dry DMF using K2CO3 as the base to give 5-(2-(4-bromobutyl)-phenyl)-10,15,20-triphenylporphyrin which was subsequently reacted with diethyl malonate in dry DMF in the presence of sodium methoxide to give 5-[2-(5,5′-ethoxycarbonyl)pentoxy]phenyl-10,15,20-triphenylporphyrin in high yield. Complexation was achieved with the reaction of a lanthanide amide complex precursor with the porphyrin free base, while purification was accomplished by silica gel column chromatography with chloroform:methanol (v/v = 5:1).
 | ||
| Fig. 21 Monometallic lanthanide porphyrinate complex with a coordinating β-diketonate moiety conjugated to the porphyrin backbone. Redrawn from ref. 116. | ||
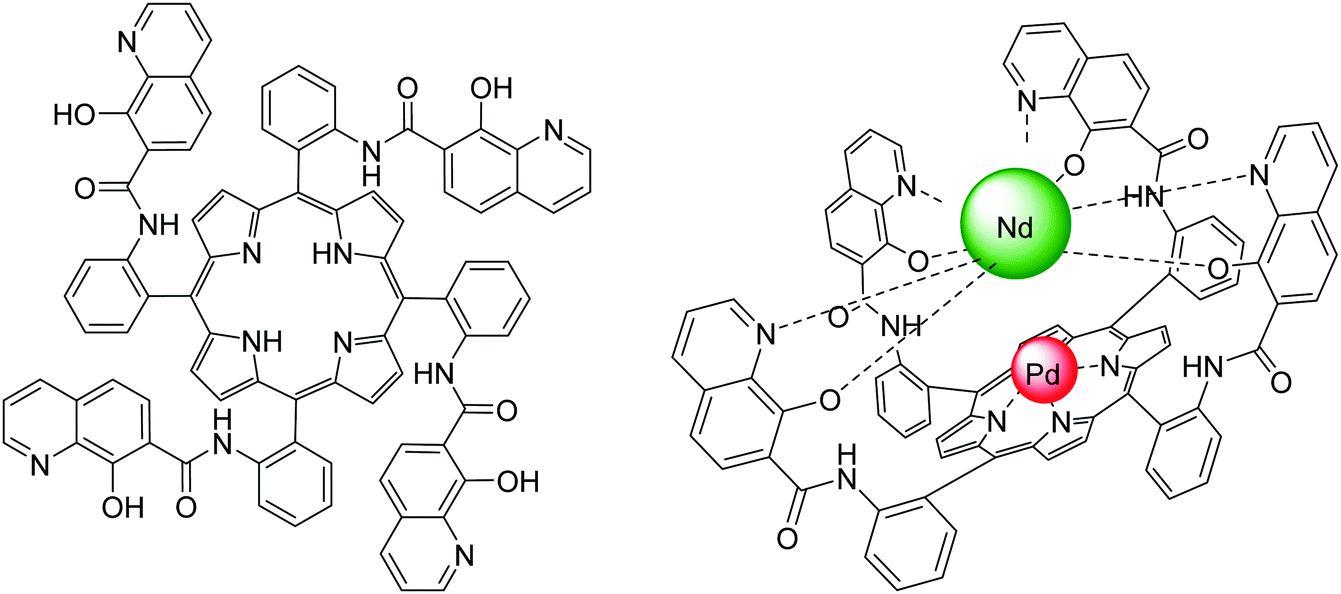 | ||
| Fig. 22 Structures of (left) the 8-hydroxyquinoline tetrafunctionalized porphyrin ligand and (right) the heterobimetallic PdII/NdIII porphyrinate; the steric hindrance and competition from 8-hydroxyquinolinyl units resulted in no lanthanide coordination in the porphyrin cavity. Redrawn from ref. 117 and 118. | ||
In 2011, the Wong group began designing porphyrin complexes appended to an external dye to explore the potential of photophysical properties of ytterbium porphyrinates (Fig. 23). They designed an unsymmetrical porphyrin with three pentafluorophenyl substituents at the meso position and attached one p-conjugated linker for covalent conjugation with a rhodamine B dye. The rhodamine B moiety was also functionalized with a short PEG chain to improve the overall water-solubility of the complex. The F15TPP “A3B” porphyrin was obtained via condensation of pentafluorobenzaldehyde, 4-[2-(trimethylsilyl)ethynyl]benzaldehyde and pyrrole using Lindsey's protocol.58,119 Complexation was performed via the established route of using a lanthanide amide complex precursor and the addition of a CoPMe anion to cap the YbIII porphyrinate. The trimethylsilyl (TMS) group was subsequently deprotected with tetrabutylammonium fluoride to give an alkynyl terminal group. The latter was subjected to a Sonogashira coupling reaction to produce an extended linker with a hydroxy terminal for conjugation with iodo-PEG-functionalized rhodamine B. The complex demonstrated its potential in a wide spectrum of applications, such as photophysical properties in aqueous solution,70 NIR imaging,120 photodynamic therapy,121 and cancer cell imaging.122 Furthermore, the synthetic flexibility offered by the linker for conjugation with various moieties allowed the properties of the porphyrinate complex to be modified relatively easily through the linker instead of on the porphyrin skeleton; it has allowed Wong's group to transform the rhodamine B moiety into a BODIPY-conjugate as a HgII-selective sensor,123 binding unit towards anionic phospholipids,124 or peptide-based integrin αvβ3 binding unit with NIR125 and MRI126 imaging capabilities. The purification of peptide-conjugated lanthanide–porphyrin systems requires several rounds of HPLC.


The same authors further modified the hydroxyl group on H2HPTPP by refluxing with 4-fluorobenzoyl chloride and triethylamine in benzene for 8 h to give 5-[p-(4-fluorobenzoyloxy)]-phenyl-10,15,20-triphenyl porphyrin (H2FBOPTPP, Fig. 24, top right).130 They also reacted H2HPTPP with myristyl chloride under similar experimental conditions to give 5-(4-myristyloxy)phenyl-10,15,20-triphenyl porphyrin (H2MPTPP, Fig. 24, bottom) and eventually its acac− complex.131 In 2011, Cui et al. reported an unsymmetrical 5-[p-(4-fluorobenzoyloxy)-m-ethyloxy]phenyl-10,15,20-triphenylporphyrin (H2FBOEPTPP, Fig. 24, top right) referencing to Liu's synthetic procedures for H2HPTPP as the precursor.132 The corresponding lanthanide complex was obtained by heating the free base porphyrin with lanthanide acetylacetonate in molten imidazole under dry nitrogen for 3.5 h, a procedure reported by Srivastava in 1978133 and also used by Che et al. in 2013.134
2.2 Complexation with N-confused porphyrins
Compared with normal porphyrins, N-confused porphyrins or “NC-P” have been discovered much more recently. The first NC-P was synthesized in 1994 as a new isomer of tetraphenylporphyrin by two different research groups, and different synthesis mechanisms were proposed.136,137 Looking at the structure of this macrocycle, one of the “confused” pyrrole rings is linked through its α–β′ axis rather than a common α–α one.138 Based on the traditional acid-catalysed condensation for preparing tetraphenylporphyrin, N-confused ones were synthesized with propionic acid, t-BuOH/CH2Cl2 (1:1), and concentrated HBr.136 Researchers kept investing efforts into exploring more derivatives of N-confused porphyrins and metalloporphyrins with novel synthetic methods during the past few decades,139–142 including the introduction of “neo-confused porphyrins” (Fig. 25).143 Although some comprehensive studies on the preparation of NC-Ps and meso-substituted NC-Ps have been published,144,145 there are only a few examples for metalation of NC-P or its derivatives, and even less involving lanthanide-based complexes because of their lability. In 2005, Wong et al. reported the first synthesis of lanthanide NC-P complexes146 using the tripodal anion CoPEt in view of its ability to stabilize labile trivalent lanthanide porphyrinates.51,69 The facile preparation of this first lanthanide-based N-confused-porphyrin complex consisted in adding an excess of Ln[(N(SiMe3)2]3·[LiCl(THF)3]x (Ln = Yb, Er) to N-confused tetraphenylporphyrin in refluxing toluene, followed by addition of an excess of NaCoPEt at room temperature. The crude products were purified by column chromatography, and the resulting air-stable green crystals were characterized as the final product in 75% yield. The researchers also found that no complex could be obtained if there was no addition of NaCoPEt to stabilize the intermediates. Unfortunately, there is little research on lanthanide complexes with N-confused porphyrins because the investigations on LnIII–NC-P did not show improved spectroscopic performance over regular porphyrinates.67
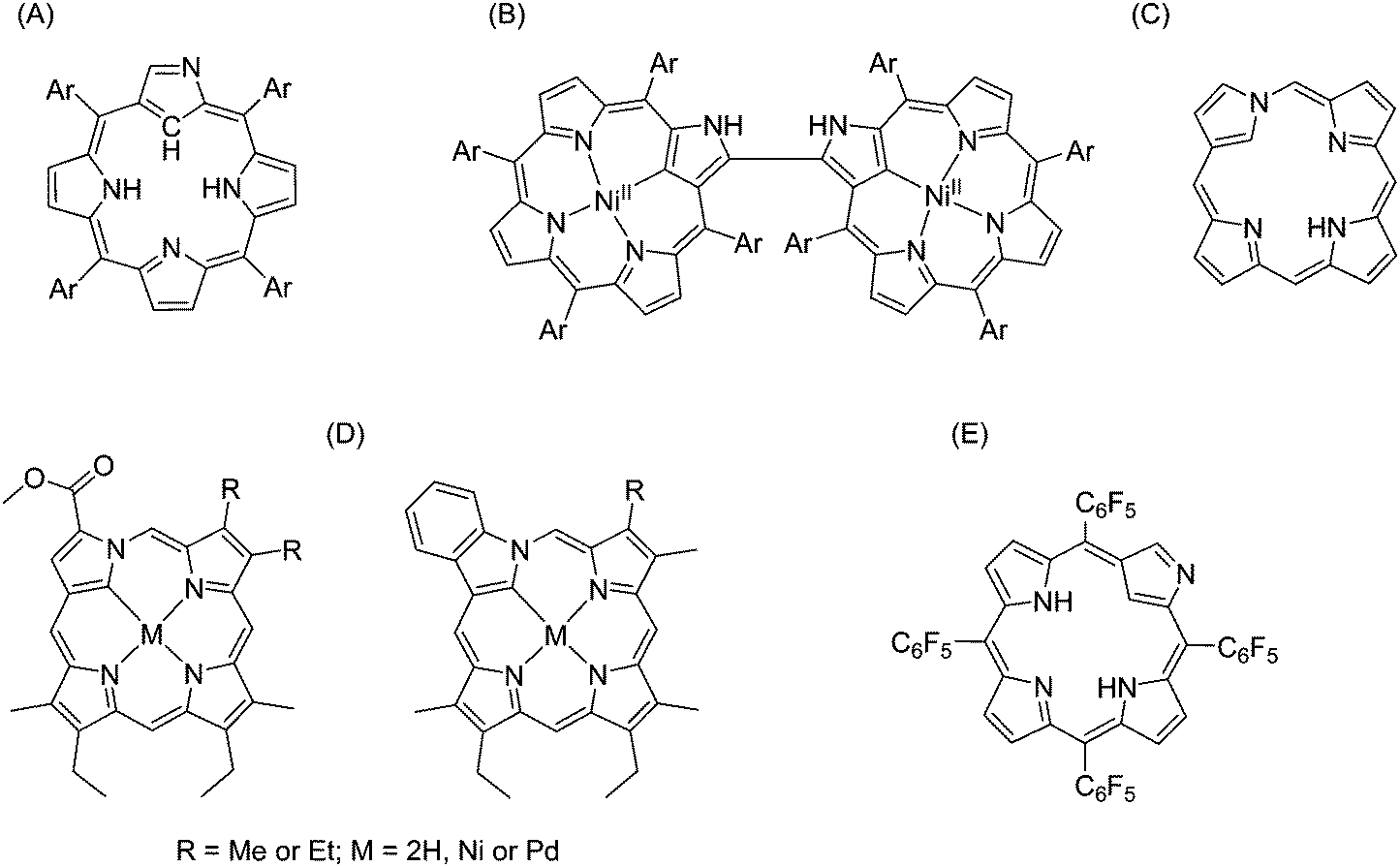 | ||
| Fig. 25 Examples of N-confused porphyrins. (A) N-Confused tetraphenylporphyrin;136 (B) N-confused porphyrin dimer;139 (C) neo-confused porphyrin;143 (D) metallo-neo-confused porphyrinates;141 (E) pentafluorophenyl-substituted N-confused porphyrins.142 | ||
2.3 Complexation with expanded porphyrins (texaphyrins)
Expanded porphyrins, as referred to by their name, are a group of porphyrinogen-like macrocycles containing more than four pyrrole units and therefore their cavities are larger than in regular porphyrins (Fig. 26). We note that some systems are sometimes considered as expanded porphyrins: although including larger polypyrrolic macrocycles, they neither have the typical carbon bridges as porphyrins nor show overall aromaticity.147 In principle, sapphyrin, which contains five pyrrole units directly linked by four methine groups, is considered being the first expanded porphyrin.148 Unfortunately, Bauer et al. only succeeded in isolating ZnII/CoII complexes, and there was no report describing lanthanide-based sapphyrin complexes until the 1990s.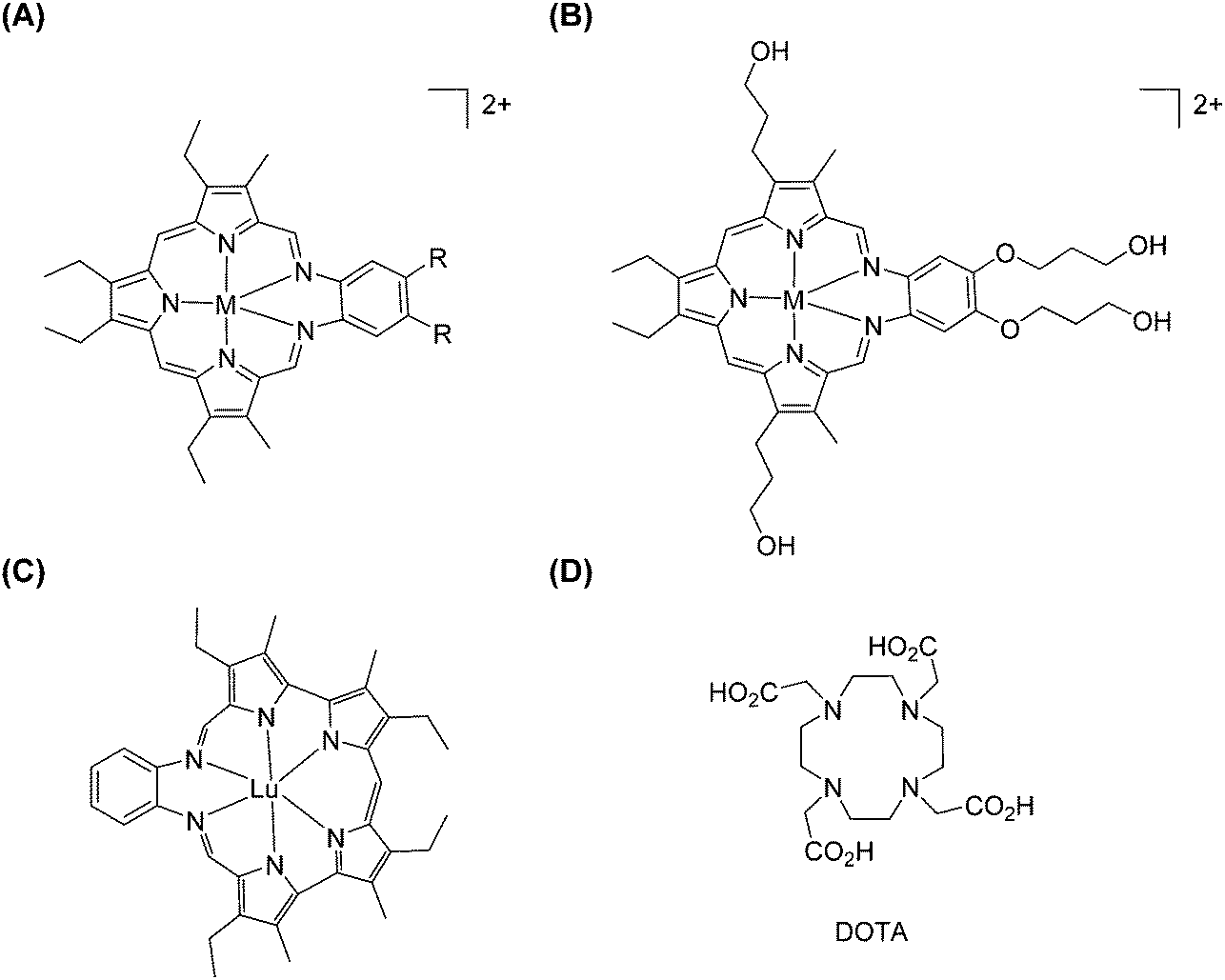 | ||
| Fig. 26 Examples of expanded porphyrinates: (A and B) LnIII texaphyrinates prepared by Sessler et al. (M = H or Ln, except Pm; R = CH3, OCH3 or O(CH2)3OH); (C) LuIII–grandephyrin. Redrawn from ref. 150 and 155. The structure of DOTA is given for comparison. | ||
The core ring of texaphyrin contains five nitrogen donor atoms and was reported by Sessler and co-workers in 1987.149 The first lanthanide-metalation of texaphyrin was described five years later by the same research group.150 They prepared this new class of tripyrrane-containing macrocycles in 44% yield by direct acid-catalysed condensation between diformyltripyrrane and o-phenylenediamine in the presence of concentrated HCl. It is worth noting that a metal template can facilitate this condensation because the addition of Pb(SCN)2 or UO2Cl2 improved the overall yields to 69% and 61%, respectively. In these three preparation methods, the pure products can be crystallized from dichloromethane, followed by washing with hexane.
Gadolinium(III)-based chelates are known since the 1980s for their clinical applications in imaging while coordinated to polyaminocarboxylate ligands, including DTPA or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, Fig. 26), for instance. Therefore, Sessler and co-workers tried to put trivalent lanthanide ions into alkyl-substituted texaphyrins (Fig. 26).150 A comprehensive study on a series of lanthanides embedded into expanded porphyrins has been performed: lanthanide salts and proton sponge (N,N′′,N′′,N′′′-tetramethyl-1,8-diaminonaphthalene) were reacted with texaphyrin in a chloroform/methanol mixed solvent at room temperature.151 Pure complexes were difficult to obtain in reasonable yields by direct chromatography on either silica gel or lipophilic Sephadex. A better optimized method made use of oxygenated methanol as the solvent into which a mixture of Ln(NO3)3·xH2O and triethylamine was heated. The yields increased from 25% to 55–70% for heavier lanthanide texaphyrin complexes (Nd–Lu, except Pm), while it remains around 30% for lighter ones (La–Pr). A water-soluble texaphyrin was also specifically designed for complexation with GdIII for potential application as a MRI contrast agent.152 The diamine portion of the macrocycle—a source of the fourth and fifth N atoms—was synthesized by nitration of o-bis-((3-hydroxypropyl)oxy)benzene to give a dinitro diacid intermediate, which was then reduced with borane–THF to give terminal hydroxy groups and further reduced. Then, condensation between the diamine and 2,5-bis((3-ethyl-5-formyl-4-methylpyrrol-2-yl)methyl)-3,4-diethylpyrrole yielded the targeted texaphyrin. However, the poor water-solubility of the complexes prompted modifications into a tetrahydroxylated texaphyrin. The presence of the four peripheral hydroxyl groups was vital to increasing the water-solubility of the texaphyrin complexes. Two lanthanide complexes with texaphyrin have been tested for medical applications: a GdIII complex, as a radiation sensitizer in radiotherapy, and a LuIII complex generating singlet oxygen for photodynamic treatment of cancer. Both complexes localize preferentially in tumour cells. For further information, the reader can refer to a recent review which covers the field comprehensively.153
Even though there are many other types of expanded porphyrins,153 only grandephyrin154 showed successful complexation with lutetium: bis(trimethylsilyl)amide of LuIII was added into the free-base macrocycle in THF solution and in the absence of water and oxygen. Unfortunately, the resulting LuIII–grandephyrin adduct was unstable and slowly decomposed upon exposure to air and moisture (Fig. 26).155
2.4 Complexation as double, triple and multiple deckers
Ever since the advent of metalloporphyrins, attention has been drawn to their stacked arrangements. The conventional construction of multiple-decker metalloporphyrins is either through the coordination of a metal ion sandwiched between two macrocycles or with the help of bridging ligands (see Fig. 1). In the case of lanthanides, most of the multi-sandwiched complexes reported are double or triple deckers and some of the provided ligands are shown in Fig. 27. | ||
| Fig. 27 Examples of functionalized porphyrins (top), of modification of H2OEP (bottom left and middle) and of N-confused porphyrins (bottom right) used in constructing homoleptic/heteroleptic multiple-decker lanthanide complexes. | ||
Weiss et al. reported lanthanide-based double/triple-decker arrangements without bridging ligands, for instance, CeIV bis(octaethylporphyrinate) [Ce(OEP)2] and dicerium(III) tris(octaethylporphyrinate) [Ce2(OEP)3], in the 1980s.18 The direct preparation of these complexes was to reflux Ce(acac)3·3H2O with H2(OEP) in TCB for 20 h. [Ce(OEP)2], [Ce2(OEP)3], and [Ce2(OEP)3]·2TCB were isolated by chromatography on an alumina column and eluted with toluene and methanol. The authors mentioned that two intermediates [Ce(OEP)(acac)] and [CeH(OEP)2] were formed, and they hypothesized that two pathways were responsible for the production of the final products: (i) condensation, [Ce(OEP)acac] losing H(acac), and (ii) autooxidation, [(CeH(OEP)2] being oxidized into [Ce(OEP)2]. In 1992, Jiang et al.156 reported the synthesis of another series of lanthanide–porphyrin sandwich complexes: bis(tetrapyridylporphyrinato)cerium(IV), [Ce(tptp)2], and bis(tetramethylpyridylporphyrinato)cerium(IV), [Ce(tmpyp)2]. They used Li2TPyP and Li2TMPyP obtained by the reaction between porphyrins and n-butyllithium to improve reactivity and solubility. In the following years, although other porphyrin/phthalocyanine-based homoleptic/heteroleptic complexes have been prepared,157,158 Chabach et al. were the pioneers in studying trivalent central metal ions in this context. They reported porphyrin–phthalocyanine sandwich-type complexes (Ln = La, Pr, Nd, Eu, Gd, Er, Lu, and Y),159 as well as mixed-metal triple-decker complexes with the porphyrin–phthalocyanine–porphyrin system.160 The smaller lanthanide ions (Er, Lu) were found to form double deckers with phthalocyanines more easily than the larger counterparts;159 their acetylacetonate salts were reacted with di-lithium phthalocyanine (Li2Pc) in boiling TCB for 3 h and then tetraphenylporphyrin was added. By contrast, the larger lanthanide ions (La–Gd) were heated first with H2TPP in TCB for 4 h and then with Li2Pc, since they coordinated more readily to porphyrin to form more stable sandwich complexes. The yields of neutral heteroleptic complexes with smaller lanthanide ions were around 85%, while those for the larger ions were in the range of 36–56%. All double deckers were neutral, except the one with LaIII which was first isolated in its protonated form, requiring further oxidation by phenoxathiinylium hexachloroantimonate into the neutral form. The first preparation of heteronuclear triple-decker complexes was achieved by the same research group: they reacted the metal acetylacetonate with metal-free porphyrin in refluxing TCB and then with the as-synthesized heteroleptic sandwich complexes. However, during the preparation of homonuclear porphyrin/phthalocyanine-based homoleptic/heteroleptic triple-decker complexes, Ng et al.161 mentioned that [Ln(Pc)TPP] (Ln = Er, Lu, and Y) were the sole products, and triple deckers can only be obtained if 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (H2TMPP) was used instead of H2TPP, yielding [Ln2(Pc)2(TMPP)]. Many other porphyrin derivatives were also tested using similar synthetic procedures. For instance, the Shinkai group162 studied CeIV-based double-deckers with tetrakis(4-pyridyl)porphyrin derivatives; the Ohta group163 investigated CeIV-based triple-decker complexes containing tetraalkoxylated terphenyl groups at the 5,15-positions of their parent porphyrin; the Lindsey group164 prepared two dyads by applying Sonogashira and Glaser coupling reactions on EuIII triple-deckers.
The usage of DBU in the preparation of sandwich-like lanthanide(III) complexes was reported in 1997.165 However, Ng's group only treated the corresponding phthalonitriles with Ln(acac)3·nH2O and DBU in amyl alcohol and obtained bis(phthalocyaninato)lanthanide(III) complexes, not porphyrinato complexes. The same group reported the one-pot preparation of [Ln(Nc)(TBPor)] complexes (Ln = La, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Y, Ho, Er, Tm; Nc = 2,3-naphthalocyanine; TBPor = 5,10,15,20-tetrakis(4-tert-butylphenyl)porphyrin, Scheme 1) by using DBU and n-octanol,166 and avoided the prior generation of the half-sandwich complexes [Ln(TBPor)(acac)] as in the conventional methods. Although the products [Eu[Nc(t-Bu)4]2] and [Eu[Nc(SC12H25)8]2] were unambiguously characterized by various methods, they contained traces of impurities that could not be removed by gel-permeation chromatography and recrystallization. They also further synthesized the first Nc-containing heteroleptic triple-decker complexes [Ln2(Nc)(OEP)2] (Ln = Nd, Eu) based on similar procedures.167 Again, the synthetic yields varied depending on the size of the lanthanide ion: complexes with smaller metal centres have increasing steric compression of the two macrocyclic ligands. Similar observations and explanations were also reported in previous researches.168,169 Microwave irradiation was tested to accelerate the reaction: Liu et al.170 provided a successful example in forming the [Lu(TBPor)Pc] double-decker in which all ligands and complexes were prepared in a microwave oven (240–560 W, 5–10 min) and the purification was performed via conventional column chromatography. Microwave-assisted synthesis is still a commonly used method: Jin et al. used microwave irradiation to overcome the low reactivity resulting from the steric hindrance from bulky peripheral groups of the lanthanide porphyrin–bis(phthalocyanine) triple-deckers.171
In 2014, Dumoulin's group studied and reported an expeditious one-step method to secure functionalized heteroleptic “A7B-type” lanthanide–phthalocyanine double-decker complexes via an optimized statistical method by changing the ratio of two different phthalonitriles (A = with hexylsulfanyl substitution; B = with long alkyl chains bearing polar OH, OMs and N3 tails).172
Once the standard procedures for preparing LnIII/IV-based multiple-deckers had been well established, researchers started to pay more attention to modifying the sidechains of the macrocycles and investigating their potential applications.173–176 Ohta et al.163,177 reported a series of phenyl-substituted porphyrins coordinating with CeIV and forming multiple deckers with long alkoxy sidechains; they emphasized that the chain length did not affect the reactivity. However, a longer reaction time gave improved yields, and the complexation time was usually longer when reacting with tetra-phenyl porphyrins than with bi-phenyl porphyrins because of steric hindrance. Also, as mentioned previously,164 Lindsey's group continued exploring more lanthanide triple-deckers including S-acetylthiomethyl groups178 or triallyl tripods179 incorporated on the porphyrinic moiety, while complexation procedures were still operated classically. It is worth noting that these examples164,178,180 proved that a coupling reaction could occur on the substituted phenyl groups of porphyrins without disrupting the as-formed double-decker structures. Besides, similar dyads (or triads) could also be formed in good yields by direct cross-condensation through an ester linkage between the metal-free 5-(4-acylchloride-phenyl)-10,15,20-tris(4-tert-butylphenyl)porphyrin and protonated YIII double-deckers (Fig. 28).181
 | ||
| Fig. 28 Examples of multiple-decker lanthanide porphyrinates with additional covalent attachments. Redrawn from ref. 178, 179 and 181. | ||
In order to explore more functionalities, other series of porphyrin derivatives were designed with sophisticated substituents at the interconnecting α-carbon. For example, porphyrins with four pyrenyl groups in the meso-positions were used to form EuIII multiple-decker complexes.182 Heteroleptic porphyrinate [Ln2(Pc)2(TCBP)] (TCBP = meso-tetrakis[3,4-(11,12,13,14-di(1′,2′-naphtho)-1,4,7,10,15,18-hexaoxacycloeicosa-2,11,13-triene)-phenyl]porphyrin, Fig. 27) featuring nonlinear optical activity were also prepared,183 where the α-carbon is derivatised with crown ether substituents. In heteroleptic porphyrin-containing multiple-decker complexes, it is common that one macrocycle is a crown-phthalocyanine instead of a porphyrin derivatised with crown ethers.184,185 As an alternative to coupling reactions for incorporating ferrocene onto a porphyrin ring through phenyl–ethynyl–phenyl links,180 ferrocenyl units can be directly attached to the α-carbon during the preparation of metal-free porphyrins (Fig. 27),186 and previously described experimental conditions can be applied for lanthanide complexation.
Finally, efforts have been put in the synthesis and characterization of homoleptic 5,15-diazaporphyrinato lanthanide sandwich complexes [Ln2DAB3] (DAB = 2,8,12,18-tetraethyl-3,7,13,17-tetramethyl-5,15-diazaporphyrinate, Scheme 1; Ln = Ce, Eu).187 The metal-free diazaporphyrin was simply reacted with [Ln(acac)3·H2O] in refluxing TCB, yielding the desired triple-decker in a one-pot synthesis. Other complexes with diazaporphyrin were isolated by Pushkarev et al.: mixing of 27-phenyl-29H,31H-tetrabenzo[b,g,l,q][5,10,15]-triazaporphyrin (H2PhTBTAP, Scheme 1) with Ln(OAc)3 or Ln(acac)3 to afford the monoporphyrinate and the homoleptic double-deckers [Ln(PhTBTAP)Ac] and [Ln(PhTBTAP)2], respectively (Ln = Eu, Lu). Heteroleptic double-decker compounds [Ln(PhTBTAP)Pc] were obtained upon the interaction of H2PhTBTAP with the corresponding monoporphyrinates.188 Unexpected formation of partially and completely de-phenylated side products was observed for both the monoporphyrinates and the double deckers. A triple-decker [Eu2(PhTBTAP)3] was also isolated with a very low yield of 4%.
N-Confused porphyrins were similarly tested for synthesizing lanthanide double-deckers. The first example came from the reaction between H2NTBPP (N-confused 5,10,15,20-tetrakis[(4-tert-butyl)phenyl]porphyrin, Fig. 27) or its N2-position methylated analogue H(CH3)NTBPP (Fig. 27) and half-sandwich complexes [LnIII(Pc)(acac)].189 With more comprehensive studies on lanthanide–porphyrin multiple-decker complexes progressing, it is also promising to modify the pyrrole units. Birin et al.190 demonstrated a new family of multiple-decker complexes bearing heterocycle-appended porphyrins.
Tetra-15-crown-5-phthalocyanines afforded a wide range of lanthanide mono-, double- and triple-decker homoleptic/heteroleptic complexes and pose long-standing synthetic challenges. Tsivadze et al. systematically screened different reaction conditions (e.g. solvent, reagent ratio, concentration and refluxing duration) and discovered that (i) monophthalocyaninato lanthanide complexes could be synthesized quantitatively in o-dichlorobenzene for some of the smaller lanthanides (i.e., Sm, Eu, Gd, Tb, Yb, Lu), the organic base used, like DBU, being then always coordinated as the axial ligand; (ii) 1-chloronaphthalene was the best solvent to obtain the double/triple-sandwich complexes (except for La and Nd).191 The remaining synthetic enigma with Er was finally resolved by changing the solvent and temperature, as well as via either direct metalation or metal-templated strategies.192
For more structurally novel multiple phthalocyaninato lanthanide complexes, Tomilova et al. documented the direct preparation of clamshell-type quadruple-decker dinuclear lanthanide phthalocyaninates by reacting their bisphthalocyanine-type clamshell ligand and LuIII monophthalocyaninate at 215 °C, in the mixed TCB/C16H33OH solvent and in the presence of the LiOMe base (Fig. 29, top).25 In 2020, Martynov et al. reported potassium-induced dimerization of heteroleptic triple-decker crown-phthalocyaninates [(15C5)4Pc]Ln(Pc)Ln(Pc) (Ln = Y, Tb; Fig. 29, middle).193 Cammidge's group disclosed a facile and controlled synthetic protocol for bridged and clamshell-type heteroleptic lanthanide porphyrin–phthalocyanine–porphyrin triple-decker assemblies, where the two porphyrins are conjugated with a flexible carbon-chain spacer (Fig. 29, bottom).194 Generally speaking, column chromatography is a common technique for purification of multiple-decker metalloporphyrins, and silica gel columns are suitable to remove unreacted ligands. However, repeated gel permeation chromatography is often applied afterward for better purification.
 | ||
| Fig. 29 Synthetic schemes for (top) clamshell-type quadruple-decker dinuclear lanthanide phthalocyaninates. Reprinted with permission from ref. 25, © 2018 Elsevier B.V.; (middle) self-assembled heteroleptic lanthanide crown-phthalocyaninates induced by potassium. Reprinted with permission from ref. 193, © 2020 The American Chemical Society; and (bottom) bigger clamshell-type heteroleptic lanthanide porphyrin–phthalocyanine–porphyrin triple decker. Reprinted with permission from ref. 194, copyright (2020) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
2.5 Complexation as oligomers and polymers
The large coordination numbers of lanthanide ions and the diversity of porphyrin macrocycles provide researchers with the chance of exploring unusual structures of lanthanide-containing materials. In addition to several classes of materials mentioned in the previous sections, the focus here will be on preparing polynuclear Ln–Por-based molecules, from dimeric ones to framework polymers.Years after the emerging lanthanide-based double-decker complexes, bridged single-decker complexes with two lanthanide cores were introduced. Jubb and Gambarotta195 used alkyl-derivatized porphyrinogen for nitrogen fixation. The encapsulation of dinitrogen was achieved by firstly mixing [SmCl3(THF)3] and Li4OEPg-(THF)4 (OEPg = octaethylporphyrinogen) in THF under an argon atmosphere and then reducing the product with metallic lithium, resulting in the precipitation of green crystals. The latter were recrystallized under a nitrogen atmosphere, affording orange-red crystals with formula [(THF2Li(OEPG)SmIII]2(N2Li4). Magnetic measurements confirmed the presence of SmIII, while the green crystals contained SmII; the crystal structure determination showed that the N–N distance is very long and compatible with a single bond, i.e. the last reaction step corresponds to a formal reduction of N2 into N24−. Campazzi et al. later reported two other meso-octaethylporphyrinogen–dinitrogen complexes containing PrIII and NdIII, respectively,196 where sodium was used instead of lithium to avoid possible reduction of N2 (Fig. 30A).
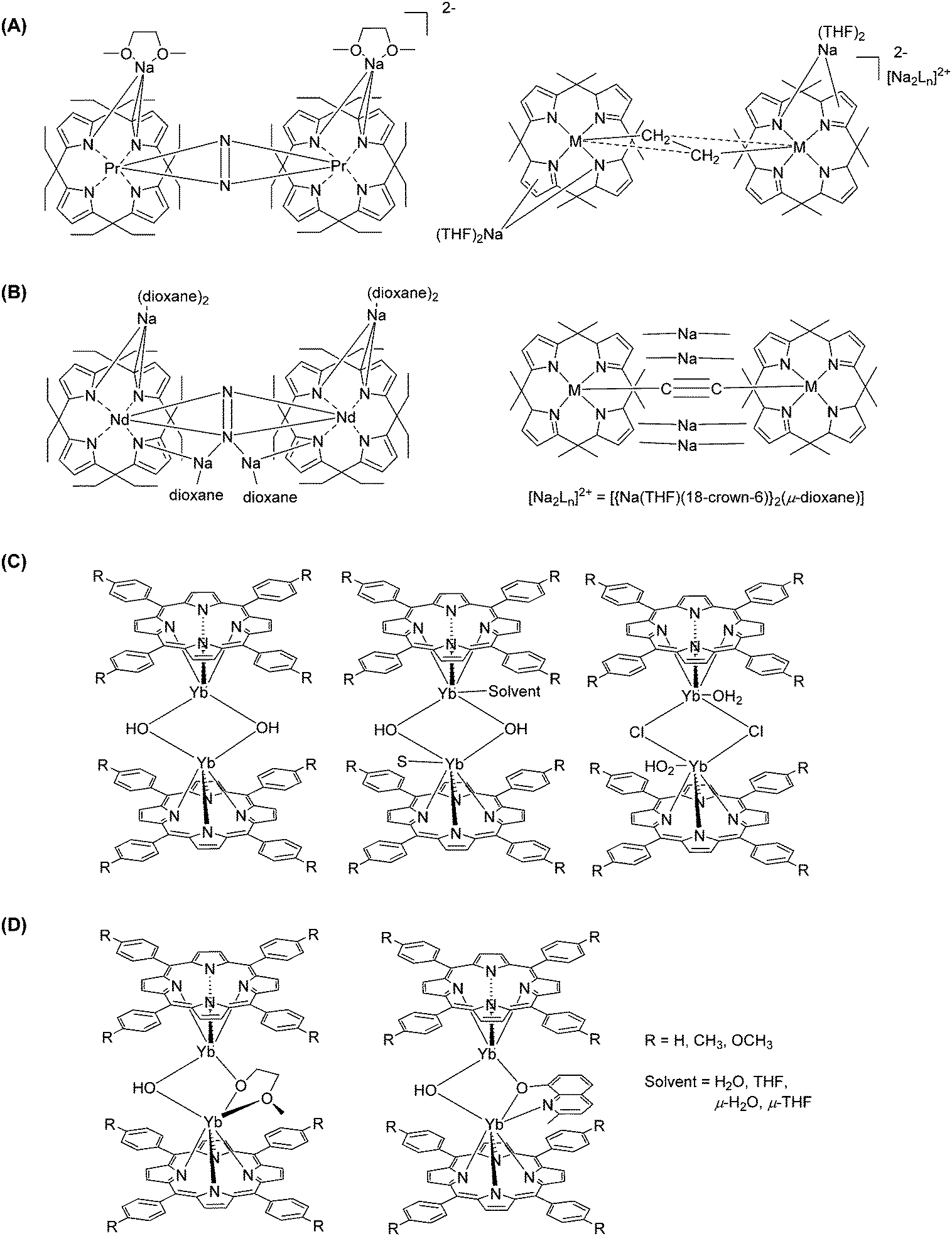 | ||
| Fig. 30 Examples of bridged lanthanide–porphyrin complexes. Redrawn from ref. 196 (A), ref. 197 (B), ref. 22 (C), and ref. 198 (D). | ||
The same half-sandwich compounds [{(η5:η1:η5:η1-Et8N4)Ln}Na(THF)2] (Ln = Pr, Nd) were used to establish other types of bridging (Fig. 30B).197 When the reaction was carried out with 18-crown-6, fixation of ethylene or acetylene was accomplished and the lanthanide cores were bridged by [C2H4]2− or [C2]2− anions. Wong et al.22 prepared other types of dinuclear LnIII porphyrinates having hydroxide bridging groups (Fig. 30C): [YbIII(NPh2)3·x[LiCl(THF)3] was generated from the mixture of YbCl3 and LiNPh2 in THF and then refluxed with H2TTP or H2TMPP in bis(2-methoxyethyl) ether, resulting in dinuclear bis(porphyrin) complexes {[YbIII(TMPP)(μ-OH)]2} and {YbIII(TTP)2(μ-OH)(μ-OCH2CH2OCH3)}. The purification steps were, in sequence, simple filtration, washing with ether, extraction with toluene, and another filtration. The final products were obtained as purple crystals from concentrated solutions. Although another starting material, Ln[N(SiMe3)2]3·x[LiCl(THF)3], could react with the same metal-free porphyrins and give the cationic LnIII monoporphyrinates in high yields,47 it could not form the bis(porphyrin) complexes because of the electric charges of the different intermediates. Comprehensive studies on the dimerization of these lanthanide monoporphyrinates, [Yb(Por)(H2O)3]Cl (Por = TPP2−, TTP2−, and TMPP2−), were performed by the same group, and different dimer bridges were formed under different conditions.198 TCB is not only widely applied in the preparation of multi-decker complexes, but it is also unexpectedly helps in the formation of the acetate-bridged dimer. He et al. documented a facile preparation method for the bridged dinuclear porphyrinates, in which ytterbium(III) acetylacetonate hydrate reacted with free base porphyrins in refluxing TCB (Fig. 30D).75 Besides internal bridging between two single-porphyrin complexes, polynuclear complexes were also produced by the direct linking between porphyrin rings through coupling reactions.178,179,199
Metal–organic frameworks (MOFs) as one important subclass of coordination polymers200 are also drawing much attention in the fields of lanthanide-based materials. Thanks to the large size and high coordination numbers of lanthanides, some researchers prefer using them rather than transition metals in building MOFs despite their lack of directionality. Porphyrins are common building blocks because of their tetradentate functionality and square-planar geometry. Goldberg's group had been putting efforts for years in preparing porphyrin frameworks assembled with the aid of lanthanide-bridging ions: the first example of hybrid architectures was obtained by hydrothermal techniques using free-base tetra(4-carboxyphenyl)porphyrin reacting with Ln2(oxalate)3 hydrated salts (Ln = Dy, Nd) (Fig. 31A and B).201 The second group of MOFs containing meso-tetra(carboxyphenyl)porphyrin species and polynuclear acetate/oxalate-bridged clusters was prepared by similar procedures where a dilute NaOH solution was used to enhance deprotonation of the carboxylic groups and a dilute HCl solution was provided to increase the reactivity of lanthanide salts (Fig. 31C and D).202 Later, another series of hybrid assemblies were synthesized, which were composed of meso-tetrapyridylporphyrin and various lanthanide complexes.203 Carbon dioxide fixation has been realized with a 2D bi-layered MOF consisting of 5,10,15,20-tetrakis(4-carboxylatophenyl)porphyrinato-zinc (ZnTCPP) linkers and [Ln2Na3(NO3)(H2O)3]8+ connecting clusters.204 Purification and isolation of the above-mentioned lanthanide–porphyrin polymers were achieved by crystallization where the reaction mixtures were heated and cooled down very slowly. Because of the different compositions of lanthanide–porphyrin MOFs, solvothermal methods used various solvents, water205–208 or organic solvents,203,204,209,210 as well as different reaction times and temperatures. Unfortunately, most lanthanide-based MOFs did not take advantage of the special spectroscopic properties of lanthanides, and the choice of lanthanides was not really targeted for a given property, but because most of these ions have similar size and coordination numbers.
 | ||
| Fig. 31 Examples of MOFs consisting of a Ln-bridged porphyrin layer. (A) 2:1 Pr:RPor;201 (B) 4:3 Dy:RPor (RPor = tetra(4-carboxyphenyl)porphyrin);201 (C) 1:1 La:R′Por;202 and (D) 2:1 Pr:R′Por (R′Por = tetra(m-carboxyphenyl)porphyrin).202 Reproduced with permission from ref. 201 and 202, copyright (2006 and 2007) The American Chemical Society. | ||
In addition to serving as structural clusters in the formation of MOFs, lanthanide ions can also play the role of core in porphyrin dendrons (Fig. 32). Several kinds of metalloporphyrin-containing ErIII-cored complexes are known.211,212 In these complexes, the porphyrins were first coordinated with other metals (PtII/ZnII), and then the core ErIII ion was coordinated by carboxyl groups from the porphyrin sidechains and by terpyridine. This novel synthetic method implies using a strong base, potassium hydride, for deprotonating the ligands, then followed by the addition of ErCl3. A dipicolinic acid (dpa) framework was also used to coordinate to a LnIII core.213 The lanthanide complexation was easily achieved in H2O/THF in which the corresponding salts were mixed with chlorin to form [Ln(dpa)2ChlPd]3− (Chl = chlorin).
 | ||
| Fig. 32 Examples of lanthanide-cored supramolecular complexes. (A) Redrawn from ref. 211; (B) reproduced with permission from ref. 212, copyright (2005) Elsevier B.V.; (C) redrawn from ref. 213. | ||
Lastly, some works focused on blending lanthanide–porphyrin complexes into conjugated or nonconjugated polymer hosts such as polystyrene, poly(methyl methacrylate), poly(n-butyl methacrylate), poly(bisphenol A–carbonate), poly(arylene–ethynylene), polytertbutylmethacrylate, and methylcellulose with routine fabrication methods.214–216 The lanthanide–porphyrin complexes used there were common and easy to prepare.
3 Redox properties
The extended π systems of metallo–porphyrinates and phthalocyaninates result in a rich redox chemistry itself generating fascinating electronic and spectroscopic properties. Indeed, the macrocyclic ligands can be easily oxidized or reduced and their electrical status, negatively charged, neutral, or positively charged, can be tuned, creating different electronic states and consequently largely different absorption spectra and colours. Electronic interactions between tetrapyrrole π-systems in close proximity play an essential role in photosynthetic bacteria, such as cyanobacteria, or in molecular organic conductors. Complexes with lanthanides are appealing models for understanding their redox and electrochromic properties. A good example is the historical [Lu(Pc2] double-decker coordination compound synthesized more than 50 years ago by Kirin et al.19 and which, according to EPR studies, could be formulated as [Lu(Pc)(Pc˙−)] since (i) the EPR spectrum displays a sharp signal at g = 2.0022217 (Fig. 33) and (ii) the magnetic susceptibility follows a Curie–Weiss behaviour with μeff = 1.76 BM, typical of a one-electron radical.218 In fact, modern analysis reveals that the unpaired electron is significantly delocalized on the two rings. Moreover, various forms of the complex exist depending on the oxidation state and have different colours, as usually determined on Langmuir–Blodgett (LB) films since there is enough space in the LB channels to facilitate charge-compensating ion transport during oxidation or reduction. For instance, oxidation of the green, paramagnetic [Lu(Pc)2] yields first a red-brown and diamagnetic form, [Lu(Pc)2]+, then a red and diamagnetic compound [Lu(Pc)2]2+. Reduction steps switch the green paramagnetic form [Lu(Pc)2] into a blue diamagnetic form [Lu(Pc)2]− and finally into a blue-violet and paramagnetic compound [Lu(Pc)2]2−. The complexes are sometimes written as their protonated forms,219 [Lu(Pc)2H]2+ (yellow-red), [Lu(Pc)2H]+ (green), [Lu(Pc)2H] (blue), and [Lu(Pc)2H]− (violet). The redox properties of complexes with tetrapyrrole macrocycles can be exploited in electrochromic devices (ECDs) such as rear-view mirrors, fitted in millions of cars, adjustable darkening windows in aircrafts, and displays; regarding the latter, organic liquid crystals are serious rivals but ECDs may be interesting for larger screens.220 Moreover, since the redox process is fairly reversible and can be cycled many times without altering the ECD component, another application is in molecular data storage.178 Finally, [Lu(Pc)2] behaves as an intrinsic molecular semiconductor and features high carrier mobility, making it a potential candidate for the design of field-effect transistors.221 | ||
| Fig. 33 X-Band EPR spectra of [Lu(Pc)2] (green, g = 2.0022) and [Lu(Pc)2]2− (violet, g = 2.0030) at room temperature (power: 5 mW, modulation: 0.63 G, gain: 2.5 × 105). Reproduced with permission from ref. 217, copyright (1979) The Electrochemical Society. | ||
In the following, we describe first the basic redox properties of lanthanide phthalocyaninates and some of the porphyrinates before giving a few hints on potential applications. It is noteworthy that investigations usually involve electrochemical studies coupled with absorption spectroscopy and magnetic measurements.
3.1 Mixed-ligand monophthalocyanine lanthanide complexes
In mono-phthalocyaninate complexes [Ln(Pc)(acac)] and [Ln(Pc)(acac)2]−, one oxidation and two reduction processes are seen; the half-wave potentials of oxidation and of the first reduction are quasi-insensitive to the nature of the Ln ion, while that of the second reduction step increases with increasing charge density on the metal ion.222 A study of similar complexes with other β-diketonate ancillary ligands confirms, by EPR measurements, the radical nature of the macrocycle and the various redox steps have been characterized by electronic absorption spectroscopy.2233.2 Homoleptic phthalocyaninato lanthanide sandwich complexes
Most of the reported works have focused on the redox properties of [Ln(Pc)2] double deckers, with main emphasis on seeking to determine how the nature of the lanthanide ion as well as that of the substituents on the periphery of the Pc framework influence the values of the redox potentials. The state of the sample – solid, thin film, or solution – and the exact experimental conditions, including the pH, also play important roles. It is worth noting that the purity of the final sample, which may depend on the synthetic method used, is crucial in such studies.Regarding the influence of the nature of the lanthanide ion on redox potentials, initial cyclic voltammetry studies in the 1980s reported variable results. For instance, the electrochemical properties of [Ln(Pc)2H] have been analysed in acidic (1 M HCl) and neutral aqueous media. The working electrode was a glass electrode, coated with 10 nm of sputtered platinum, on which the complexes were sublimed. The voltammograms are reversible and in acidic medium, during the second and subsequent cycles, oxidation potentials vs. saturated calomel electrode (Hg/Hg+, SCE) occur at +0.38 V and +0.6 V (orange-red form), while the reduction peak also occurs at +0.38 V (blue form), demonstrating chemical reversibility; the most reduced form (violet) appears at −0.2 V. The presence of oxygen and water influences the process, suggesting some interaction with the complexes, but apparently not the lanthanide ion (La, Nd, Eu, Gd, Dy, Er, Yb, Lu), consistent with the redox reactions occurring at the ligands. In neutral medium and for Eu, Gd, Dy, Er, and Yb, there is only one oxidation step at +0.7 V (orange-red) but two reduction steps at −0.6 V (blue form) and −1.4 V (violet form). However, no electrochemical behaviour is observed for La and Nd. A close look into the absorption spectra of these complexes reveals that they correspond to the reduced (blue) form [Ln(Pc)2]− which is electro-inactive. After a few weeks the solutions turn green with an electronic absorption spectrum identical to that of the free H2Pc; in addition, there is formation of a blue precipitate, pointing to the probable occurrence of a small amount of a triple-decker complex, [Ln2Pc3]. Apparently the presence of labile hydrogen is necessary for the complexes to display electrochemical behavior.224 The ambiguity noted above regarding the formulae of the lutetium bis(phthalocyanine) complexes, with or without “loosely bound” protons, arises from the fact that indeed, depending on the pH, acid–base reactions may also be an explanation to some observed colour changes.225
Half-wave potentials for the oxidation and reduction reactions of green NBu4[Ln(Pc)2] in o-dichlorobenzene are displayed in Fig. 34 for Ln = Y, Pr–Lu (except Tm).226 The reduction potentials show no dependence on the ionic radii, whereas oxidation potentials increase linearly with increasing ionic radii, with a slope of about 1.5 ± 0.2 V Å−1. In sandwich-type bis(tetrapyrrole) rare earth compounds the distance between the two isoindole 4N planes is very small; therefore, the proximity of the two conjugated π-systems in a face-to-face configuration induces splitting of the molecular orbitals. The dependence of the half-wave potentials upon ionic radii can then be reasonably explained by increasing ring–ring distance with increasing ionic radius and consequently decreasing π interaction between the aromatic rings, rendering the removal of electrons more difficult. A similar trend is observed for double-decker complexes with benzyloxy substituted phthalocyanines.227 Characteristic parameters often referred to are the differences between the two oxidation potentials, which is here ΔEo2–o1 = 0.45 V, and between the first oxidation and first reduction potentials, which is here ΔEo1–r1 = 1.19 V; the latter reflects the molecular electrochemical band gap, i.e., the energy difference between HOMO and LUMO orbitals.
 | ||
| Fig. 34 Redox potentials vs. Ag/AgCl (saturated KCl) of [Ln(Pc)2]− in o-dichlorobenzene as a function of ionic radii for the coordination number 8. Drawn from data reported in ref. 226. | ||
Peripheral octa-substitution by various alkyl groups, such as methyl, ethyl, n-butyl, t-butyl, or O-propyl,228–230 or mixed substitution231 can be used to modulate the redox potentials and therefore the colour of the complexes. A third reduction potential is found for some of the complexes with derivatized Pc, with E1/2 only slightly negative, which, like the oxidation potentials, varies slightly with the nature of the lanthanide ion. This is, for instance, the case for Pc derivatized with one benzyloxy and six t-butyl groups.231 This third reduction potential corresponds to the filling of the semi-occupied molecular orbital (SOMO) of the radial-anionic ligand.
Peripheral substitution of the macrocyclic ligand is used for several purposes, including increasing solubility in organic solvents or water, and sensing purposes, in which, for instance, the side chains, such as crown ethers, are complexation agents for metal ions.232,233 These substituents do not influence the basic structure of [Ln(Pc)2] with one macrocycle bearing a single negative charge; this is demonstrated by the EPR spectrum of a double-decker functionalized with 6-hydroxyhexylthio groups featuring a single signal with g = 2.001.234 However, redox properties are influenced, with respect to not only shifts in half-wave potentials but also the number of redox processes that can be triggered. For instance, the sandwich complexes with 6-hydroxyhexylthio-derivatised Pc display one oxidation step only but up to 5 reduction steps. An electrochemical study of double-decker complexes of the entire lanthanide series with Pc ligands derivatized with t-butyl, heptyl, butyloxy, pentyloxy, and octyloxy groups revealed five reduction steps and two oxidation steps, except for Ce. The first reduction potential is small and increases linearly from Lu to La (−0.02 to +0.27 V) in parallel with the first oxidation step (+0.40 to +0.61 V); the other reduction potentials have no Ln dependence and lie between −1.1 and −1.9 V. It is noteworthy that the latter potentials arise from the fact that the first and second LUMOs are close in energy and can accept up to four electrons. Experimentally, observation of several resolved redox processes is facilitated by the use of a glassy carbon disk resulting in less interaction between the electrochemically generated species and the electrode,235 as well as by complementing cyclic voltammetry measurements with differential pulse voltammetry data. The number of observed redox potentials evidently depends on the electrochemical window available under the given experimental conditions, where the solvent plays a crucial role.
A way to fine-tune the energy of the HOMO and LUMO orbitals of phthalocyaninates, i.e., tuning their redox potentials, is to play with the electron withdrawing or electron donating properties of the substituents. Jiang et al. have investigated a series of 17 metal-free phthalocyanines derivatized with alkoxy, alkylthio, alkyl, alkynyl, phenyloxy, and phenylthiol groups and concluded that the effects of peripheral and non-peripheral substitution on the values of half-wave potentials can be explained by considering the energy of the frontier orbitals. The tuning range is large, on the order of 0.45 V for the first three reduction potentials and between 0.6 and 0.8 V for the two oxidation potentials, while ΔEo1–r1 values span about 0.7 V.236 In addition to generating planned electronic effects, the choice of substituents is also guided by practical considerations, such as ease of processing thin films or mesophases. In an effort towards this goal, Bian et al. have introduced eight bulky 2,6-dimethylphenoxy groups on the periphery of Pc. The structure of the double-decker complexes with Pr, Sm, and Nd is similar to those of other homoleptic lanthanide phthalocyaninates, with the peculiar feature however that the Pc rings adopt a domed conformation towards the metal centres with dihedral angles being much larger than those observed for complexes with alkyl, alkylthio or phenoxy substituted Pc. This can be attributed to the presence of the bulky methyl groups on the substituents. With respect to unsubstituted [Ln(Pc)2], incorporation of these 2,6-dimethylphenoxy groups on the phthalocyanine ligands results in slightly more difficult oxidation and slightly easier reduction, consistent with the electron-withdrawing nature of the substituent and in accord with results obtained previously with phenoxy octasubstituted Pc.237
The search for practical electrochromic materials is a big drive for investigating various [Ln(Pc)2] complexes. For instance, double-deckers of Sm, Eu, Gd, Dy, and Lu with octa-2,2,3,3-tetrafluoropropoxy-substituted Pc have been evaluated toward this goal. Langmuir–Blodgett thin films of the complexes coated on ITO electrodes display three distinctive colours (green, orange, red); the Dy thin films were found to be ideal neutral and anodic colouring materials with short response times (4.3 s for green and 2.1 s for red, first cycle) and high optical stability.238
Naphthalocyanine (Nc) sandwich complexes with lanthanides behave similarly to phthalocyaninates with respect to their electrochromic properties (Fig. 35); however, interest for Nc compounds stems from their NIR-shifted absorptions by as much as 200 nm.239 The series of double deckers formed by tetra(t-but)-derivatized Nc (TBNc) with La–Lu, except Ce and Pm, display a rich electrochemistry, with 2–3 oxidation steps and 4–5 reduction processes, as shown by the differential pulse voltammogram for [Lu(TBNc)2] (Fig. 36, top). The dependence of the half-wave potentials on ionic radii is presented in Fig. 36 (bottom) and compared with that for similar double deckers with tetra(t-butyl)-derivatized Pc (TBPc). The three most negative reduction potentials have essentially no dependence on the radii, while the first reduction potential correlates linearly with these radii, as does the first oxidation potential, with values slightly increasing with increasing radii. The half-wave potentials for the second and third oxidation steps are also linearly dependent on the ionic radii of the metal ions but the slopes of the lines are opposite to those of the first oxidation and reduction potentials. When oxidation processes involve the same HOMO of the ligands, i.e., the 2nd and 3rd potentials, the slope of the potential dependence on ionic radii is the same. Comparison with [Ln(TBPc)2] reveals that the first two oxidation potentials and the first reduction potential of [Ln(TBNc)2] are less affected by the change in ionic radius, the corresponding gradients being about 40–50% less steep. The extension of the conjugated π system in going from Pc to Nc therefore attenuates the inter-ring separation effect on the redox potentials. In turn, the HOMO–LUMO energy separation is smaller; theoretical calculations give a 0.29 eV (∼2350 cm−1) difference for the un-complexed ligands, explaining why Nc compounds are more readily oxidized than Pc compounds.240
 | ||
| Fig. 35 Chemical formulae of pinene-substituted phthalocyanine H2Pc*, 2,3-naphthalocyanine H2Nc, and the C4v form of 1,2-naphthalocyanine H2Nc′. | ||
 | ||
| Fig. 36 (Top) Difference pulse voltammograms for [Lu(TBNc)2] in methylene chloride containing 0.1 M (NBu)4NClO4; the scan rate is 10 mV s−1. (Bottom) Dependence of half-wave potentials for [Ln(TBNc)2] and [Ln(TBPc)2] as a function of the ionic radii of the trivalent lanthanide ions. Reproduced with permission from ref. 240, copyright (2005) Society of Porphyrins and Phthalocyanines. | ||
In going from monophthalocyaninate [Ln(OPPc)acac] (H2OPPc is octaphenyl Pc, Scheme 1) to homoleptic double [Ln(OPPc)2] and triple [Ln2(OPPc)3] deckers, the number of redox processes increases from 4 to 6 and 10, respectively. For [Eu2(OPPc)3] in o-dichloro benzene, 6 reduction and 4 oxidation steps are observed and are completely reversible. The first oxidation (+0.67 V vs. +0.63 V) and the first reduction (+0.16 V vs. +0.16 V) half-wave potentials are very similar to those for the analogue double decker, with ΔEo1–r1 reaching 0.51 V compared with 0.47 V. Assignment of all half-wave potentials was performed by inspection of absorption spectra.239
Cerium double- and triple-deckers deserve special attention since, in addition to redox processes centred on the ligands, the metal ion could theoretically be switched between +3 and +4 oxidation states and therefore these compounds could display extra colours with respect to the other lanthanide derivatives. The reality appears to be subtle as the oxidation state of cerium depends on the ligand nature and/or the complex stoichiometry. For instance, cerium retains its trivalent oxidation state in a double decker with t-butyl-substituted naphthalocyanine for which no additional redox process was observed compared with other Ln complexes with the same ligand.241 On the other hand, in most other tetrapyrrole double deckers, cerium is predominantly tetravalent, such as in [CeIV(OEP)2] (H2OEP is octaethylporphyrin), with valence calculated from EXAFS data between 3.59 and 3.68. The first reduction wave at a slightly negative potential is then assigned to Cen/CeIII (n between 3 and 4).242 A Langmuir–Blodgett thin film made of cerium bis[tetra(15-crown-5)-phthalocyaninate] displays a similar behavior.243
3.3 Heteroleptic phthalocyaninato lanthanide sandwich complexes
Using two different tetrapyrrole ligands with different redox and optical properties to form dissymmetric lanthanide double deckers provides interesting information on the electronic structure of each ligand and, particularly, on charge delocalization in these macrocycles.43,244 Earlier studies focused on Por/Pc and Por/Por’ double deckers, demonstrating that, for instance, in [CeIV(TPP)(Pc)]+, the electronic hole (positive charge) resides mainly on the phthalocyanine ring, while in [CeIV(TPP)(OEP)]+ it is mainly delocalized on OEP. In general, the redox properties of mixed lanthanide double deckers feature several oxidation and reduction steps with dependences on the ionic radii similar to those evidenced for the homoleptic complexes.159,245,246In order to explore new electrochromic behaviours, novel tetrapyrrole rings have been synthesized. An example is given in Fig. 37 for the heteroleptic sandwich complex [Lu(Pc)(PTBTPor)] (H2PTBTPor is meso-phenyl-tetrabenzo-triazaporphyrin) which, like the homoleptic compound with PTBTPor, undergoes three quasi-reversible reductions and one quasi-reversible oxidation. The potential difference ΔEo1–r1 amounts to 0.45 V, which is at the higher end of the range usually observed for homoleptic Pc double deckers (0.35–0.45 V), meaning that the ring-to-ring interaction is larger compared with homoleptic Pc analogues.188,247
 | ||
| Fig. 37 (left) Structure of the heteroleptic lutetium complex with Pc and meso-phenyl-tetrabenzo-triazaporphyrin (H2PTBTPor). (right) Cyclic voltammograms (top) and square-wave voltammograms (bottom) of the complex in dichloromethane containing 0.15 M Bu4NBF4 (scan rate: 10 V s−1). Reproduced with permission from ref. 247, copyright (2013) The Royal Society of Chemistry. | ||
Owing to the remarkable redox properties of tetrathiafulvalene (TTF), such as oxidation at relatively low half-wave potentials converting its dithiolylidene rings into 6π-electron aromatic systems and use of its salts (e.g. [TTF]+Cl−) as semi-conductors or components of optoelectronic devices including sensitized solar cells, bis(thiopentyl)-derivatized TTF units have been fused with phthalocyanine rings. Heteroleptic double deckers [Eu(Pc)(PcTTFn)] (n = 1–4) have been isolated which display four reduction and two oxidation steps with some of them having half-wave potentials substantially shifted with respect to the homoleptic unsubstituted complex [Eu(Pc)2] for which two oxidation steps and three reduction processes are observed. All steps are reversible, except the second oxidation process. The shifts depend on the number of TTF substituents but in a nonlinear way. The first oxidation potential increases from 0.62 V (vs. SCE) to 0.65 V in going from the mono- to the tetra-substituted Pc, while it amounts to 0.54 V for the unsubstituted reference compound. The first reduction potential decreases from 0.17 V (n = 1) to 0.13 V (n = 4), the second increases from −1.06 V to −0.96 V, the third from −1.35 V to −1.27 V and the forth from −1.66 V to −1.59 V. The difference ΔEo1–r1 increases from 0.45 V to 0.52 V, compared with 0.43 V for the unsubstituted reference compound. These data reflect un-doubtful contribution of the TTF units to the electronic structure of the double deckers and represent a welcome tuning possibility for future electrochromic devices.248
As seen above, inter-ring π–π interactions in multidecker tetrapyrrole lanthanide complexes confer to these assemblies remarkable optical, electrical, and electrochemical properties. Researchers have consequently looked for ligands that may further enhance these properties. Good candidates are corroles (Cor) and their derivatives (Fig. 20). Corroles are contracted 18-π electron macrocycles containing a direct carbon–carbon single bond between two pyrrole rings; since they have three NH groups in the inner core, they act as trianionic ligands. The first lanthanide complexes with corroles were reported in 2013249 and the first heteroleptic lanthanide triple-deckers with tri-substituted corroles in 2015, with the formulae {Ln2[Pc(OBut)8]2[Cor(PhCl)3]}250 and {Eu2[Pc(OBut)8]2[Cor(Ph)n(PhNO2)3-n]} (n = 0–3).251 For the first series (Ln = Pr–Tb, except Tm), half-wave potentials for five oxidation and three reduction steps were determined. For Ln = Eu, the first oxidation and the three reduction potentials are similar to those observed for the homoleptic double-decker {Eu(Pc(OOct)8]2}, and so is ΔE01–r1 at 0.44 V versus 0.42 V for the reference; the other four oxidation potentials are more similar to those found for the triple-decker {Eu2[Pc(OOct)8]2(Pc)}. Replacing the butoxy groups with pentoxy or octaoxy substituents has little influence on the half-wave potentials. But the nature of the lanthanide ion has, as all potentials display a linear dependence on the ionic radii, usually increasing with increasing size, except for the 2nd oxidation potential which is fairly insensitive, while the 4th one displays an opposite trend (Fig. 38). In the second series of heteroleptic triple deckers, the nitrophenyl substituents are redox active, which leads to the observation of two additional reduction potentials between −1.19 and −1.33 V vs. SCE. All potentials, except the third oxidation one, vary with the number of nitrophenyl substituents. Regarding assignment, the first and fifth oxidation steps as well as the first reduction step arise from the corrole ligand, while the second and third reduction processes are localized on the nitrophenyl substituents and the other processes on the phthalocyanine ring.251 The analogue dysprosium triple deckers with pentoxy octasubstituted Pc and either fluoro or chloro trisubstituted Cor have electrochemical behavior similar to {Y2[Pc(OPent)8]2[Cor(PhCl)3]} with five oxidation steps and three reduction steps; ΔEo1–r1 = 0.40 V (fluoro) and 0.38 V (chloro), compared with 0.39 V for the yttrium compound (Fig. 39).252
 | ||
| Fig. 38 Half-wave potentials for {Ln2[Pc(OBut)8]2[Cor(PhCl)3]} (Ln = Pr–Tb, except Tm) in CH2Cl2 containing 0.1 M Bu4NClO4. Reproduced with permission from ref. 250, copyright (2015) American Chemical Society. | ||
 | ||
| Fig. 39 Cyclic voltammograms of {Dy2[Pc(OPent)8]2[Cor(PhX)3] (X = F, Cl) in CH2Cl2 containing 0.1 M Bu4NClO4. The top structure corresponds to the fluorinated complex. Reproduced with permission from ref. 252, copyright (2017) American Chemical Society. | ||
Heteroleptic complexes with phthalocyanine and naphthalocyanine can be obtained by a one-pot procedure and their redox and spectroscopic properties follow the same trends along the lanthanide series as porphyrin/phthalocyanine complexes, governed by the increased π-interaction between the aromatic rings. Double deckers {Ln(Nc)[Pc(α-OPent)4]} (Ln = Sm, Eu, Y) feature four oxidation and five reduction processes; ΔEo1–r1 is almost constant, 0.34–0.35 V, while ΔEr1–r2, which represents the HOMO–LUMO gap of the reduced forms {Ln(Nc)[Pc(α-OPent)4]}−, decreases from 1.21 V to 1.16 V and 1.09 V for Sm, Eu, and Y, respectively.253
3.4 Porphyrinato lanthanide complexes
The electrochemical properties of lanthanide porphyrinates have similarities with those described above for the phthalocyaninates so that we limit the description to a few examples. The europium double-decker complex with octaethylporphyrin [Eu(OEP)2] displays an EPR spectrum typical of an organic radical (g ∼ 2.0) meaning that one macrocycle is in the form of a radical mono anion. This and similar double deckers undergo several oxidation/reduction steps with concomitant colour changes. It is worth noting that, like the phthalocyaninates, protonation/deprotonation reactions also occur depending on the pH and sometimes lead to equilibria between two different species in solution. Electrochemical data are available for many complexes, including homoleptic and heteroleptic double deckers. For instance, octachloro-substituted tetraphenyl porphyrin (H2Cl8TPP) displays two oxidative and two reductive reversible one-electron processes in THF containing 0.1 M Bu4NClO4; the potentials do not vary much upon coordination to Tb in [Tb(Cl8TPP)(OAc)] but the complex displays a third oxidation step at a higher potential (1.63 vs. SCE); the ΔEo1–r1 difference is quite large, around 1.9 V. In principle, the electron withdrawing nature of the chloro substituents should result in porphyrins that are easier to reduce and harder to oxidize. This is however not the case and the reason is that the addition of eight relatively bulky groups on β-pyrrolic positions results in ring deformation leading to substantially modified redox properties due to a destabilization of the HOMO and to a macrocyclic ring that is easier to oxidize. The ring deformation depends on the number (4 or 8) and nature (chloro or bromo) of the substituents.99 The molecular structures of three oxidation forms of [Tb(TPP)2]n and [Tb(OEP)2]n (n = −1, 0, +1) have been determined by single-crystal X-ray diffraction and point to the redox state controlling the azimuthal rotation angle ϕ between the two porphyrin rings. For the TPP double decker this angle decreases with each oxidation step, from 44° to 34°, and 30°. Oxidation results in a shortening of the average Tb–N bond length, increasing the π–π interaction between the rings and the steric repulsion between the phenyl rings of one ligand and the β-hydrogen atoms on the other one. The eight ethyl groups on the porphyrin macrocycle of H2OEP seem to have a much smaller steric effect since the ϕ angle is equal to 45° for n = −1 and 0 and decreases to only 36° for n = +1.254In the series [Ln(Nc)(TBPP)], where H2TBPP is meso-tetra(4-t-butylphenyl)porphyrin, three quasi-reversible, one-electron oxidation and one reduction steps are revealed by cyclic voltammetry in methylene chloride at ambient temperature.166 A second reduction could not be detected owing to solvent breakdown. Linear correlations are observed between the measured potentials and the ionic radii of the trivalent lanthanide ions; the first oxidation and reduction steps have positive gradients, while the second and third oxidation potentials have negative gradients (Fig. 40). It is noteworthy that ΔEo1–r1 is large, with values in the range of 0.50 to 0.55 V, without a clear trend with respect to ionic radii. This is much larger than that in the bis(phthalocyaninate) analogues [Ln(Nc)2] (0.29–0.47 V) and slightly larger than that in the related [Ln(Pc)(TPP)] double deckers (0.45–0.48 V).
 | ||
| Fig. 40 (Top) Cyclic voltammogram of [Sm(Nc)(TBPP)] in CH2Cl2 containing 0.1 M Bu4NPF6 (scan rate: 100 mV s−1). (Bottom) Dependence of the redox potentials of [Ln(Nc)(TBPP)] on ionic radii. Reproduced with permission from ref. 166, copyright (2001) Wiley VCH Verlag GmbH. | ||
Both ferrocene and tetrapyrroles are extensively used in molecular electronics so that connecting them together has been tempted with the aim of tuning electronic coupling between ferrocene units. Two such ferrocene units have been grafted in syn configuration at the meso positions of the porphyrin framework (H2PorFc2), and heteroleptic complexes [Ln(Pc)(PorFc2)] (Ln = Eu, Y, Ho, Lu) and [Eu2(Pc)2(PorFc2)] have been prepared. The two tetrapyrrole ligands in the double deckers and the outer two macrocyclic ligands in the triple decker adopt a domed conformation, the degree of which depends on the nature of the metal ion, which enables the tuning of the ferrocene interaction. Indeed, the steric hindrance generated by the Pc ring in the sandwich complexes restricts the rotation of the ferrocene groups. Within the electrochemical window of CH2Cl2, all double-decker complexes exhibit three quasi-reversible one-electron oxidations and two quasi-reversible one-electron reductions (Fig. 41). Comparisons with reference compounds reveal that the first two reductions and the third oxidation involve the tetrapyrrole ligands, while the first two oxidations are clearly due to the ferrocenyl groups. By contrast, a single ferrocene-centred two-electron oxidation is detected in the free ligand H2PorFc2 pointing to the lack of an electronic interaction due to free rotation. The two ferrocene-centred oxidations occur between −42 (Eu) and −84 (Lu) mV and +39 (Eu) and +72 (Lu) mV, respectively, exhibiting the classical dependence on ionic radii. Their difference increases considerably, from 81 (Eu) to 156 (Lu) mV, with shrinking ionic radius, reflecting an increasing electronic interaction. This difference is much smaller (46 mV) for the triple-decker Eu complex, a consequence of the less domed conformation of the outer tetrapyrrole rings reducing the ferrocene interaction.186
 | ||
| Fig. 41 Molecular structure of [Eu(Pc)(PorFc2)] and differential pulse voltammetry of [Lu(Pc)(PorFc2)] in CH2Cl2 containing 0.1 M Bu4NClO4 (scan rate: 10 mV s−1). Reproduced with permission from ref. 186, copyright (2012) The American Chemical Society. | ||
4 NIR luminescence of tetrapyrrole complexes
The characteristics of trivalent lanthanide luminescence are well suited for essential photonic applications, including biomedical analyses, in view of narrow emission bands covering the entire spectroscopic range from UV to NIR—by suitable choice of the emitting ion, of long excited state lifetimes (μs to ms) enabling time-resolved detection, and of good resistance to photobleaching. However, since the implied transitions have very weak intensities owing to the Laporte's forbidden nature of electric dipole f–f transitions, a sensitization mechanism is needed. Photons are absorbed by the ligands surrounding the metal ion and energy is then transferred from a donor state, often a triplet state in coordination compounds, to the metal ion. The energy transfer is most efficient if the energy gap between the donor state of the ligands and the excited state of the emitting ion lies in the range of 2–5000 cm−1.255Applications in bioscience often require the use of NIR light in order to avoid too much absorption and scattering by the biological tissues. Looking into Fig. 42 which depicts schematically the main transitions of the NIR-emitting lanthanide ions, one realizes that, ideally, the donor states of the ligands should be located between 10000 and 15000 cm−1. Several classes of ligands are suitable for sensitizing the NIR luminescence of lanthanide ions256 and, among them, tetrapyrrole derivatives are especially well-adapted in view of their extended NIR spectroscopic properties.
 | ||
| Fig. 42 Partial energy diagram of the principal NIR-emitting trivalent lanthanide ions with main NIR transitions. The green rectangle indicates the ideal energy of the donor state of the ligand for efficient energy transfer. Adapted with permission from ref. 256, copyright (2007) Elsevier B.V. | ||
4.1 Electronic spectra of tetrapyrroles and their lanthanide complexes
The electronic spectra of simple metalloporphyrins, e.g. [MII(por)] with tetragonal symmetry, display one intense absorption with a maximum around 400–425 nm and logε = ∼4.5–5.5, corresponding to a S2 ← S0 transition and called the Soret (B) band; two other absorptions of lower intensities are detected in the spectral range of 500–650 nm, with logε ∼ 3–4 and assigned to two components of the S1 ← S0 transition, labelled Q(0,0) or Q0 and Q(1, 0) or Q1. When the tetragonal symmetry is broken as in the free ligand due to the presence of the NH protons, the Q bands are each split into two components labelled Qx0, Qx1, Qy0, and Qy1. A similar situation occurs for [LnIII(Por)L] complexes as shown in Fig. 43 for [Tb(TPP)(acac)] and H2TPP. Upon excitation in the Soret band, emission from S1 is observed at ∼650 and ∼714 nm for both coordinated and free TPP, corresponding to the Q(0,0) and Q(0,1) components, respectively.257 At 77 K, [Gd(TPP)(Ac)] (Ac is acetate) displays a structured emission band between 600 and 1200 nm corresponding to the phosphorescence of the porphyrin ring triplet state.258
 | ||
| Fig. 43 Absorption spectra (top) of [Tb(TPP)(acac)] (I) and H2TPP (II) in Nujol and emission spectra of the same compounds (10−5 M) in MeOH (bottom). Adapted with permission from ref. 257, copyright (2004) Elsevier B.V. | ||
In going from porphyrin to phthalocyanine, large spectroscopic changes occur. For instance, for [Ln(Pc*)2] (Pc* is tetra-pinene substituted Pc, Fig. 35; Ln = Eu, Er, Lu),259 the Soret band is blue-shifted to about 330–360 nm, while the Q band presents two components at 623 and 709 nm, the latter being as intense as the Soret band. In addition, two π-radical related weak bands are seen at 504–525 nm (blue valence band, BV – also called blue vibronic band) and 932–945 nm (red valence band, RV – also called red vibronic band) as well as a broad structured feature with maxima between 1620 and 1944 nm, assigned to the intervalence transition (IV) between the second-highest HOMO and the SOMO, corresponding to one electron hopping from one ring to the other.50 A partial molecular orbital diagram is shown in Fig. 44; the orbitals from which the Pc B bands originate are lower lying a2u and b2u and are not shown. Note that the intervalence band disappears when the two macrocycles bear the same 2− charge.
 | ||
| Fig. 44 Frontier molecular orbitals for the π system in [Lu(Pc)2]. Redrawn according to ref. 260. | ||
In the triple-decker series [(Pc)Ln(Pc(OEt)8)Ln(Pc)], the energies of the two Q bands shift linearly as a function of the ionic radii reflecting the increasing π-interaction between the macrocyclic rings along with the lanthanide contraction.261 Redox processes influence the absorption spectra, more particularly the intervalence band: when [(Pc2−)Tb(Pc˙−)]0 is reduced with 3 equivalents of Na[CpCo(CO)2] (Cp− is the cyclopentadienyl anion) to produce the di-anionic species [(Pc2−)Tb(Pc˙3−)]2−, the charge transfer (intervalence) band of the radical anions shifts from about 1600 nm to about 2130 nm, while the other transitions sustain little changes (Fig. 45). The reduced species can be isolated with two Na[cryptand(2,2,2)]+ as counter cations.262
 | ||
| Fig. 45 Absorption spectra of [(Pc2−)Tb(Pc˙−)]0 (1), [Tb(Pc2−)2]− (2), and [(Pc2−)Tb(Pc˙3−)]2− (3) in KBr pellets. Reproduced with permission from ref. 262, copyright (2019) The American Chemical Society. | ||
Absorption spectra of the naphthalocyaninate complexes retain the main features of their analogues with phthalocyaninates having, however, all absorption bands substantially red-shifted due to more extended π-conjugation. Dependences on the nature of the metal ion are also similar. For instance, in the double-decker series with tetra(t-butyl) substituted Nc, [Ln(NctBut4)2] (Ln = La–Tb, except Tm, Y, and Er), the Soret band sustains little changes, remaining in the range of 334–328 nm, while both components of the Q band (694–685 and 799–767 nm) as well as the (blue) π-radical anion band (648–593 nm) are blue-shifted with decreasing ionic radii of the metal ions. On the other hand, the red π-radical anion transition is red-shifted with decreasing ionic radii (1052–1084 nm). The spectra also display a broad NIR intervalence absorption, blue-shifted (2346–1818 nm) with the lanthanide contraction (Fig. 46, top).241 A comparable shift, ∼1400 cm−1 between Nd and Lu, is observed in double-decker complexes with tetra(hexylthio)-derivatized phthalocyanine (Fig. 46, bottom).263 A direct comparison between the spectra of [Er(OPPc)2] (OPPc is octaphenyl Pc) and [Eu(OPNc)2] (OPNc is octaphenyl Nc) is shown in Fig. 47, exemplifying an approximate bathochromic shift of 100 nm for the Q bands and up to 200 nm for the intervalence bands.239
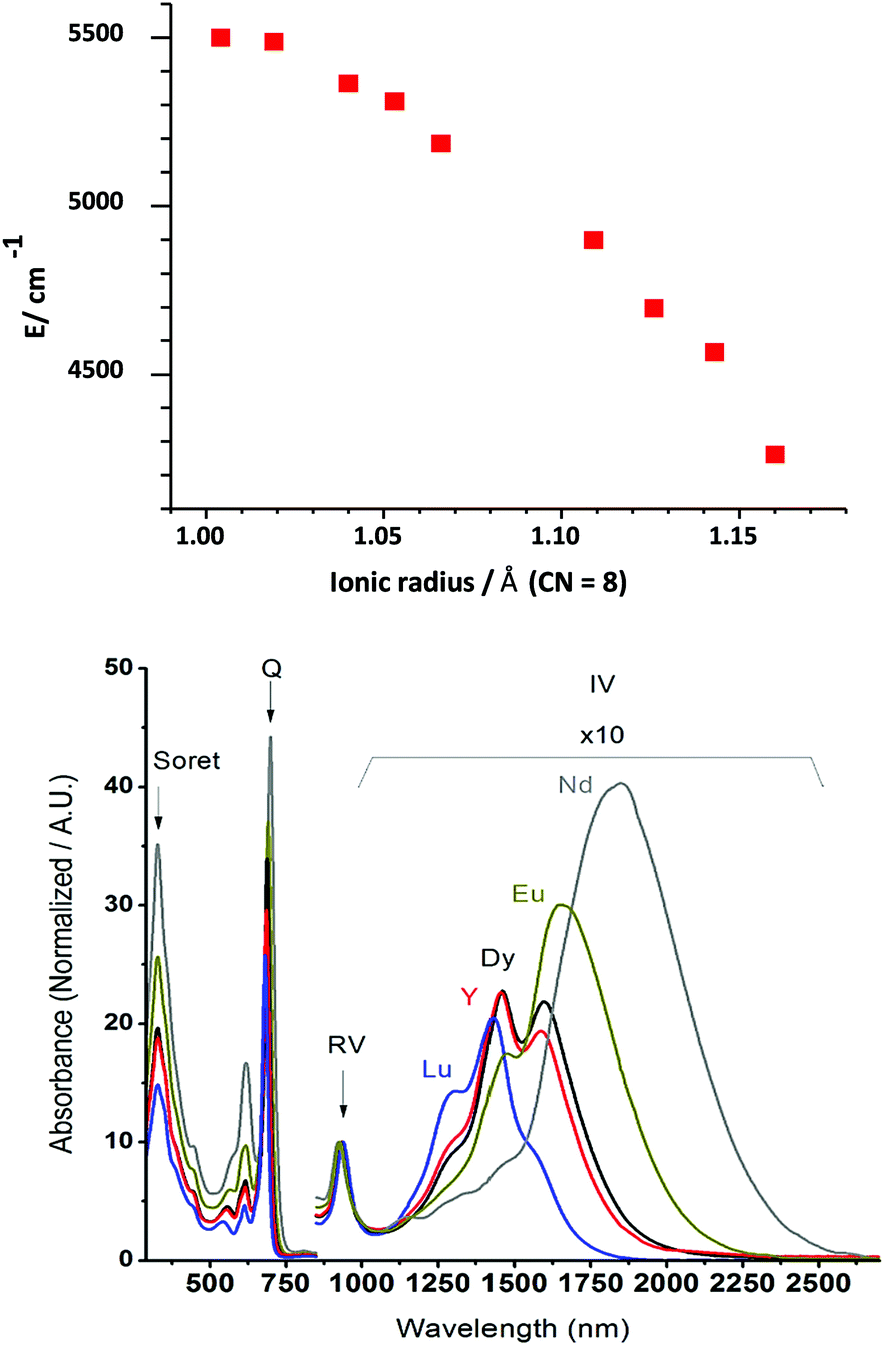 | ||
| Fig. 46 (Top) Dependence of the energy of the NIR intervalence band on ionic radii in the double-decker series [Ln(NctBut4)2] (Ln = La–Tb, except Pm, Y, and Er). Drawn from the data in ref. 241. (Bottom) Absorption spectra of oxidized [Ln(SHex4Pc˙−)2]+ showing the large blue shift of the IV band with decreasing ionic radius. Reproduced with permission from ref. 263, copyright (2014) The American Chemical Society. | ||
 | ||
| Fig. 47 Comparison between the absorption spectra of [Er(L)2] with L = OPPc and OPNc. Adapted with permission from ref. 239, copyright (2014) The Royal Society of Chemistry. | ||
As for the complexes with other corrole ligands, electronic spectra of the lanthanide naphthalocyaninates change with their oxidation state. An earlier study on double- and triple-decker complexes with 1,2-naphthalocyanine and 2,3-naphthalocyanine (Fig. 35) sheds light on this aspect by recording the absorption spectra of the double deckers [Lu(Nc′)2]n (n = −2, −1, 0, +1) and the triple deckers [Lu2(Nc′)3]n (n = −4, −3, −2, −1, 0, +1, and +2).264 Since only the first reduced and oxidized forms of these compounds are stable, spectra of more reduced or oxidized forms have been acquired under electrochemical conditions. The first reduction of neutral [Lu3+(Nc′)2−(Nc′)˙−] in benzonitrile does not influence much the Soret band but leads to a splitting of the Q band at 674 nm into two components (638 and 718 nm) for [Lu3+(Nc′)2−(Nc′)2−]− and, upon further reduction to [Lu3+(Nc′)2−(Nc′)˙2−]2−, into three components (∼590, ∼640, and ∼700 nm). The first reduction also suppresses the π-radical anion band at 509 nm. Compared with [Lu(Pc)2], the red shift of the Q band of [Lu(Nc′)2] is modest (18 nm), but considerable for [Lu(Nc)2] (102 nm). Oxidation of [Lu3+(Nc′)2−(Nc′)˙−] yielding [Lu3+(Nc′)˙−(Nc′)˙−]+ leads to a large splitting of the Q band into two components, at 550 nm and 710 nm, similarly to what is observed for [Lu(Pc)2] and [Lu(Nc)2]. Triple deckers display larger changes in their electronic spectra upon reduction and oxidation. Comparison between the double and triple deckers shows that stacking one [Lu(Nc′)2−]+ moiety over [Lu(Nc′)2−(Nc′)2−]− extends the delocalization over three rings and modifies significantly the electronic levels. In fact the observed spectrum in the region of the Q bands is very similar to a summation of the transitions of the neutral [Lu3+(Nc′)2−(Nc′)˙−] and reduced [Lu3+(Nc′)2−(Nc′)2−]− forms.
4.2 Sensitization of NIR-emitting lanthanide ions
The emissive properties of lanthanide ions are governed by the ease with which energy transfer from the donor ligands can be optimized and nonradiative deactivation paths minimized. Furthermore, selection rules forbidding electric dipole f–f transitions have to be somewhat relaxed. Important mechanisms are the J-mixing and the mixing with opposite-parity wavefunctions such as 5d orbitals, ligand orbitals, or charge transfer states. However, in view of the shielding of the 4f orbitals by the filled 5s25p6 subshells, the degree of mixing remains small. Coupling with vibrational modes is ambivalent; on the one hand it lowers the symmetry around the metal ion, resulting in more allowed transitions, but on the other hand it opens efficient non-radiative deactivation channels. While optimizing energy transfer from the surroundings to the emitting metal ion is relatively well understood and mastered despite its complexity involving several electronic levels and mechanisms,265 shutting down nonradiative deactivation mechanisms is much more difficult, particularly if the energy gap between the excited and ground states is small, which is the case for NIR emitting ions. This is predominantly true for coordination complexes with organic ligands which bring high-energy vibrations (e.g. O–H, N–H. C–H) in close proximity with the emitting ion. As a consequence, until recently, the quantum yields of NIR-emitting lanthanide coordination complexes with organic ligands were rather low, especially in the case of NdIII and ErIII. Ytterbium has a larger energy gap so that the quantum yields of a few percent have been routinely achieved.256 As will be described below, tetrapyrrole complexes have largely contributed to substantial improvements to this field.Sensitization of lanthanide luminescence is described by the following equations (Fig. 48). To quantify it, two different quantum yields have to be determined: the overall quantum yield QLLn obtained by excitation into the ligand levels (eqn (1)) and the intrinsic quantum yield QLnLn measured upon direct excitation into the excited states of the lanthanide ion (eqn (2)):
 | (1) |
 | (2) |
 | (3) |
 | ||
| Fig. 48 Simplified schematic energy diagram of the sensitization of lanthanide luminescence. A is absorption; E, emission; D, the ligand donor state; Ac and Em, the accepting and emitting states of the metal ion, respectively; ks are first order rate constants. | ||
Experimentally, measurement of the overall quantum yield is a commonly used procedure and, except for the necessary precautions to be exercised,267 is relatively straightforward even if experimental uncertainties are on the order of at least ±10%. Determination of QLnLn is more difficult because f–f transitions have faint cross sections and are often overlapped by intense and large absorption bands from the ligands. One way out is to consider the following equation:
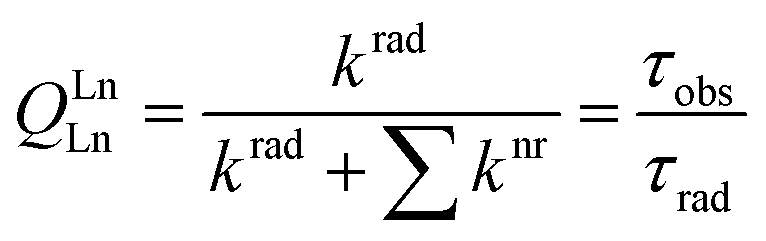 | (4) |
 :
: | (5) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) is the barycentre of the transition, n the refractive index, and DED and DMD the electric dipole and magnetic dipole oscillator strengths, respectively. Calculations are relatively involved since they necessitate the determination of Judd–Ofelt parameters.268 A more straightforward approach can be adopted if the corresponding absorption spectrum can be recorded, i.e., if the emissive transition terminates in the ground state:
is the barycentre of the transition, n the refractive index, and DED and DMD the electric dipole and magnetic dipole oscillator strengths, respectively. Calculations are relatively involved since they necessitate the determination of Judd–Ofelt parameters.268 A more straightforward approach can be adopted if the corresponding absorption spectrum can be recorded, i.e., if the emissive transition terminates in the ground state: | (6) |
) the absorption spectrum expressed in terms of molar absorption versus wavenumbers. In the case of EuIII, the situation simplifies because the excited 5D0 level has J = 0 and the 5D0 → 7F1 transition is purely magnetic dipolar. Then, τrad can be simply determined from the emission spectrum and the refractive index: | (7) |
 | (8) |
For NIR-emitting lanthanides, a major hurdle to highly luminescent materials is minimization of nonradiative processes. Indeed, the energy gap ΔE is small and can easily be bridged by vibrations and their overtones (see the right-hand side of Fig. 48). To put it simply, two models describe the situation. The first one, the “energy-gap law,” relates the non-radiative rate constant to the ratio of the energy gap divided by the most energetic vibrational mode in the molecule or matrix into which the luminescent ion is doped:
| kvibrnr = A·e−B(ΔE/hνmax) | (9) |
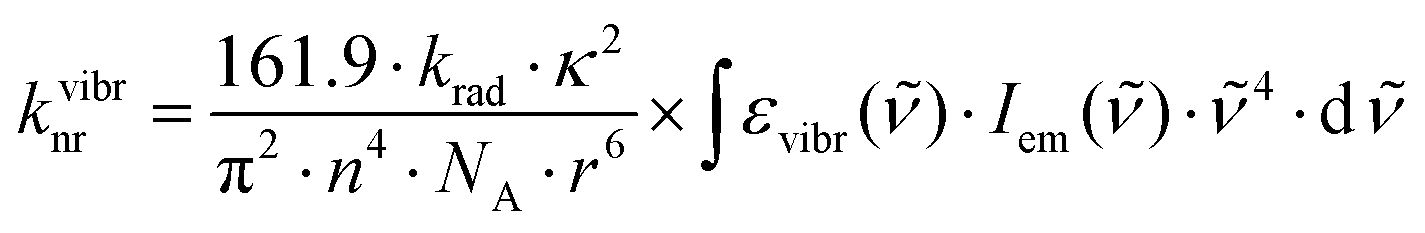 | (10) |
 | ||
| Fig. 49 Spectral overlap integrals between the second νC–D overtone and the first νC–H overtone with the 1D2 → 1G4 emission band of a PrIII cryptate. Reproduced with permission from ref. 269, copyright (2012) The American Chemical Society. | ||
As can be seen from eqn (4), shortening the radiative lifetime would be beneficial in that this would increase the intrinsic quantum yield. This can be done either by increasing the polarizability of the Ln–ligand bonds, not easy to achieve, or by modifying the refractive index: increasing the refractive index by doping the luminescent compound into a matrix, for instance, will decrease τrad, cf.eqn (5)–(7).
4.3 NIR-emissive lanthanide–porphyrin complexes
The combination of tetrapyrrole ligands with rare earth ions has been studied since the mid-1960s and this is also true for their luminescence properties, in particular when the ligand is a porphyrin. Indeed, due to low-energy electronic states, phthalocyanines and naphthalocyanines, particularly multiple-decker compounds, are less well suited for sensitizing NIR-emitting lanthanide ions. The field has been reviewed regularly since the turn of the century,32,256,260,270–274 including applications to bioanalysis and bioimaging.275–278 In some instances, polyaminocarboxylic acid groups115,279 or tris(dipicolinates)213 have been attached to porphyrins or to their transition metal complexes (M = Zn, Pd, Pt); the flexibility of the resulting assemblies is large enough to bring the LnIII ions close to the porphyrin core which then can sensitize their luminescence. Such work is not discussed in detail here.Porphyrin complexes with ytterbium are the most studied because the relatively large energy gap of this ion (∼10000 cm−1) limits nonradiative deactivations and because the triplet states of porphyrins (12–15000 cm−1) have adequate energy for efficient energy transfer onto YbIII. On the other hand, YbIII has only two spectroscopic levels, 2F7/2 (ground level) and 2F5/2, so that energy transfer is limited to the latter, contrary to other ions such as NdIII or ErIII which feature several higher energy levels (Fig. 42) rendering energy transfer processes easier. Since sensitization of YbIII luminescence by ligands with donor states of relatively high energy (20–25000 cm−1) has also been identified, alternative mechanisms have been proposed: either a phonon-assisted energy transfer or an electron-transfer path during which YbIII is first reduced to YbII producing a ligand radical cation which in turn re-oxidizes YbII producing excited YbIII.256
 | ||
| Fig. 50 Stabilization of a monoporphyrinate complex with Kläui's tripodal capping ligand. Drawn from data in ref. 51. | ||
Neutral bidentate ligands such as phenanthroline and its derivatives are equally efficient for replacing the solvent molecules in [Yb(TPP)(Ac)(Solv)2] and improving the photophysical properties, with the intrinsic quantum yield, calculated assuming τrad = 1.2 ms, increasing from 0.13% to about 1.5% in toluene.282
Influence of meso substitution of porphyrin by various alkyl (ethyl, n-propyl, n-butyl, n-nonyl and 2-, 3-, and 4-pyridinyl groups) on YbIII luminescence has been elucidated; the quantum yield and brightness (B = ε × QLLn) obtained by excitation in the Soret band are displayed schematically in Fig. 51 along with those for [Yb(TPP)(acac)] as a reference. The lowest quantum yields are obtained with the alkyl-substituted ligands and they decrease with increasing chain length, probably due to an increased number of C–H groups and to a larger flexibility of the alkyl chain leading to more vibrational quenching, reflected by a decrease in lifetime from 1.7 to 0.9 μs. Replacing the phenyl groups by pyridinyl ones results in a substantial increase of the quantum yield which reaches a maximum (1%) for 4-pyridinyl groups; this can be explained by better π-conjugation. The mixed substitution confirms these findings and exemplifies the positive effect of 4-pyridinyl groups on the quantum yield. Interestingly, the brightness of the monoporphyrinates with phenyl or pyridinyl substituents remains high (∼1000–1300 M−1 cm−1) despite the small quantum yields, owing to the large molar absorption coefficients (logε = 5.0–5.2). It is noteworthy that the best brightness, an important parameter for practical applications, does not necessarily correspond to the largest quantum yield. Finally, the influence of the additional ligand has also been tested. Among Cl−, Br−, acac−, ba− (benzoyl acetonate), and tta− (thenoyltrifluoroacetonate), acac− yields the best performance.283
 | ||
| Fig. 51 Quantum yield (black squares) and brightness (red circles) of [Yb(subst-Por)(acac)] complexes in methanol where Por is meso-substituted with different pyridinyl and alkyl groups. For alkyl groups, the numbering indicates the length of the carbon chain; for mixed substitution, the numbering refers to the number of pyridinyl/alkyl groups; there are two positional isomers for (2,2). Drawn from data reported in ref. 283. | ||
A relatively large intrinsic quantum yield has been reported for 8-coordinated [Yb(TPP)(acac)(4-MePhen)], 0.86% in methylene chloride based on τobs = 17.3 μs and by using τrad = 2 ms,75 which is too long. YbIII radiative lifetimes determined for 8-coordinated benzoxazole-substituted 8-hydroxyquinoline complexes in methylene chloride are in the range of 0.7–0.75 ms with an overall quantum yield of up to 2.6% and τobs of up to 20 μs,284 so that the intrinsic quantum yield of [Yb(TPP)(acac)(4-MePhen)] is more probably in the range of 2.1–2.4%.
Tetraphenylporphyrin has been unsymmetrized by adding one R substituent in position 7 (also designed as β-position). Among the substituents tested, chalcones gave the best photophysical results. For R = 1-methyl-prop-2-en-1-one and R = 1-phenyl-prop-2-en-1-one, the overall quantum yields of [Yb(TPPR)(acac)] in DMF solutions (5 × 10−5 M) amount to 1.3% and 4.7%, respectively; at the time of publication, the latter figure was one of the largest values reported for monoporphyrinates.285 One of the phenyl groups of TPP has also been derivatized in the 4-position by a polyaminocarboxylate group (EDTA or DTPA); mononuclear complexes (1 μM) in DMF show a low overall quantum yield, 0.11–0.14%, but when a second YbIII ion is bound to the polyaminocarboxylate moiety, this figure goes up to 0.28–0.32%.286
A decisive step was achieved by a systematic investigation of deuteration and fluorination effects. Tetrakis(pentafluorophenyl)porphyrin (F20TPP) has been β-substituted by deuterium or fluorine and the photophysical properties of the complexes with a non-deuterated or partially deuterated Kläui's tripodal ligand [Yb(F20TPPX)(CoPR)], where X = H, D, or F and R = CH3, CD3, C2D5 or C(CD3)2, have been determined in methylene chloride and compared.100 Deuteration of the methyl groups in Kläui's ligand increases the overall quantum yield from 5.1% for [Yb(F20TPPF)(CoPCH3)] to 25% for [Yb(F20TPPF)(CoPCD3)] (Fig. 52), while the lifetime increases from 49 to 285 μs. Replacing the deuterated methyl groups with deuterated ethyl or isopropyl groups has no influence so that subsequent measurements have been performed with the CD3-derivatized tripodal ligand. The influence of the β-substituents is also quite dramatic, the quantum yield increasing from 3.9% (H) to 15% (D) and 25% (F), with lifetime increasing from 54 to 180 and 285 μs, respectively. On the other hand, the hydrogen atoms of the phenyl substituents have less effect with the quantum yield of [Yb(TPPF)(CoPCD3)] remaining high at 20%, with a lifetime of 197 μs. As a comparison, [Yb(TPP)(CoPEt)] shows an overall quantum yield of only 2.4%. Therefore, the combined complete fluorination of the macrocycle and partial deuteration of the tripodal ligand improves the quantum yield by one order of magnitude! In deuterated methylene chloride the quantum yield of [Yb(F20TPPF)(CoPCD3)] reaches 63% with a lifetime of 714 μs.100 Until then the largest quantum yield reported for YbIII was 12% for a deuterated cryptate in deuterated methylene chloride.
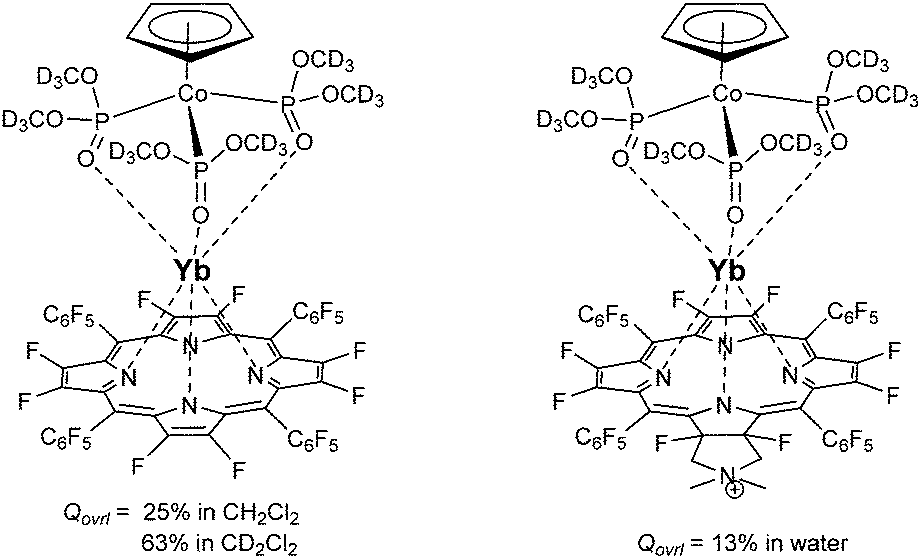 | ||
| Fig. 52 Ytterbium β-fluorinated tetrakis(pentafluorophenyl)porphyrinates with high overall quantum yields.100,105 | ||
Biomedical applications require water-soluble luminescent probes. A water-soluble ytterbium monoporphyrinate has been designed for this purpose. The porphyrin features three pentafluorophenyl groups, while the fourth phenyl group is para-substituted with a water-soluble moiety bearing either a quaternary amine (Lig–N) or a rhodamine B functionality (Lig–RhB). The ytterbium complex with Lig–N has a quantum yield of 2.7% in water and a lifetime of 12 μs upon excitation in the Soret band. It is efficient as a phospholipid marker and can differentiate between cancerous and normal cells in vitro.122 The other ligand has been designed for targeting mitochondria and the coordination is completed by the capping ligand CoPMe (Fig. 23). In water, the overall quantum yield of this bioprobe reaches 2.5% under excitation in the Soret band at 430 nm and the Yb(2F5/2) lifetime amounts to 18 μs. An additional feature of this probe is its ability to be excited at 860 nm by a two-photon process, with a sizable cross section of 375 GM.70 When one or two BODIPY chromophores are linked by ethynyl or phenylethynyl groups to the porphyrin framework, replacing phenyl group(s) or pentafluorophenyl groups(s) or triphenylamine substituents, the lifetime increases in the range of 37–43 μs and the corresponding [Yb(subst-Por)(CoPMe)] complex can be excited specifically in the BODIPY substituent at 528 nm; however, due to a relatively weak coupling with the porphyrin moiety the antenna effect of BODIPY remains modest, resulting in an overall quantum yield in the range of 0.35 to 0.73% only (Fig. 53, top).287 A useful aspect is that the two-photon cross section upon excitation at 800 nm (corresponding to excitation in the porphyrin Soret band) increases to 1000–2200 GM, so that these complexes are valuable NIR-to-NIR bioprobes. An attempt to introduce two BODIPY units as co-sensitizers of YbIII luminescence in [Yb(TPP)(Ac)(DBP-Phen)] (Fig. 53, bottom) resulted in a 2.4-fold increase in the intensity of the YbIII luminescence with respect to [Yb(TPP)(Ac)(Me-Phen)] and a broader excitation window, but in a somewhat smaller intrinsic quantum yield.288
 | ||
| Fig. 53 Molecular structures of porphyrin-based YbIII complexes conjugated to co-photosensitizers BODIPY and bis(BODIPY)phenanthroline. Redrawn from ref. 287 and 288. | ||
In other water-soluble bioprobes, the phenyl groups of TPP have been replaced with 1 to 4 pyridyl functions and, when at least two pyridyl groups are present, the Yb(2F5/2) lifetime amounts to 30 μs.104 As the literature demonstrates that fluorinating the C–H groups of organic ligands is beneficial to NIR luminescence, the β-positions of porphyrin have been fluorinated, whereas various water-soluble substituted phenyl groups have been grafted onto the meso positions. The complexes with a partially deuterated Kläui's tripodal ligand, [Yb(F8TPRP)(CoPCD3)], display sizeable overall quantum yields upon excitation in the Soret band: 5–13% in water and 9–23% in DMSO with lifetimes in the range of 56–173 μs in water and 84–249 μs in DMSO. The best performances are reached with F28TPPpyrrP, the completely fluorinated tetrakis(pentafluoro)porphyrin having one pyridinyl group fused with dimethylpyrrolidinium for water solubility. This porphyrinate has a remarkable photostability of over 3 h in water containing 0.1% DMSO upon 405 nm irradiation (0.2 W cm−2). In phosphate buffer saline (pH = 5), the same irradiation leads to a 10% decrease of the YbIII emission intensity after 4 h, while the corresponding non-β-fluorinated complex sustains 55% photobleaching.105 The ytterbium β-fluorinated porphyrinates are therefore adequate probes for NIR bioimaging277,289 or for the quantitative visualization of molecular events in vivo, such as pH determination.106
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(TMS)) revealed that sensitization of NIR luminescence is less efficient than in complexes with corresponding regular porphyrins, as shown by the Yb(2F5/2) lifetime in toluene remaining short, 0.19–0.38 μs.290
C(TMS)) revealed that sensitization of NIR luminescence is less efficient than in complexes with corresponding regular porphyrins, as shown by the Yb(2F5/2) lifetime in toluene remaining short, 0.19–0.38 μs.290
The luminescence of other NIR-emitting lanthanide ions is also sensitized in monoporphyrinates, but far less quantitative data are at hand. Overall quantum yields of 0.2% and 0.1% were reported for [Ln(TPP)(CoPEt)], Ln = Nd and Er, respectively; the corresponding values for [Ln(TPP)(TpH)] are 0.24% and 0.09%, respectively.72 A series of NdIII tetraphenyl monoporphyrinates have been studied, with the phenyl groups substituted with various functions (H, Me, OMe, F, Cl, Br), and intrinsic quantum yields in toluene were estimated to lie between 0.19 and 0.28% taking into account a literature radiative lifetime of 0.25 ms. The best sensitization was obtained for the bromo-substituted complex.54 With TpH as a capping ligand, intrinsic quantum yields between 0.1% (F) and 0.33% (H, Me, Cl) were obtained in the same solvent.281 8-Hydroxyquinoline is known to be a good sensitizer of lanthanide NIR luminescence and was attached to the α4-atropisomer of TPP. These groups bind NdIII, while the core of the porphyrin can be coordinated to a divalent transition metal ion such as PdII, which acts as a co-sensitizer of NdIII luminescence. The quantum yield upon excitation into the 8-hydroxyquinoline group amounts to 4.3 × 10−3% for the NdIII porphyrinate alone and to 9.9 × 10−3% when PdII is coordinated into the core, exemplifying the double sensitization process (Fig. 22).118
Because of the telecommunication applications of ErIII-doped materials and bio-applications of some ErIII complexes emitting in the second NIR biological window which are also tumour-selective probes,125 several ErIII porphyrinates were investigated; however, few quantitative data are reported often in view of too faint emission. An overall quantum yield value of 0.04% has been reported for [Er(TPP)(acac)] in chloroform; doping this complex in a film of the poly(arylene–ethynylene) polymer increases its NIR emission intensity in view of efficient Förster energy transfer from the polymer to the porphyrin ligand.215 A slight increase in quantum yield is observed upon fluorination of the phenyl groups (F20TPP), while the value approximately doubles when the β-positions are additionally fully brominated (Br8F20TPP).291 Hybridization of [Er(Cl4TPP)(acac)(R-Phen)] (R = 5-(N,N-bis(3-propyl)ureyl) results in a homogeneous and transparent material that could be useful, but the lifetime (2 μs in toluene, R = Me) is reduced 50–70%, pointing to quenching from the outer coordination sphere.56
750 cm−1 in F20TPP to ∼21450 cm−1 in F20TPPL (Fig. 54B). While the Soret band at ∼420 nm is hardly affected in going from the porphyrin to the porpholactone frameworks, the absorption spectra of the Q bands display more changes, particularly with respect to their intensity (Fig. 54C), and this is also true for the emission bands (Fig. 54D). On the other hand, the coordinative properties of the two macrocycles remain very comparable, so that porpholactones appear to be valuable alternatives to porphyrins for developing luminescent platforms for bio-applications.292
 | ||
| Fig. 54 Comparison between tetrakis(pentafluorophenyl)-substituted porphyrin and porpholactone: (A) chemical formulae; (B) molecular orbitals; and absorption (C) and emission (D) spectra. Adapted with permission from ref. 292, copyright (2019) The American Chemical Society. | ||
The ytterbium complexes [Yb(L)(acac)], L = F20TPP and F20TPPL, present similar spectral signatures, with a small red-shift of the Soret band (3 nm), larger red-shifts of the two Q bands (17 and 26 nm) and a much larger intensity of the more red-shifted Q band (8.3-fold). As a result, the overall quantum yield of [Yb(F20TPPL)(acac)] (4 × 10−6 M) in methylene chloride, under excitation of the Soret band, amounts to 3.3%, a 50% increase compared with [Yb(F20TPP)(acac)]. The Yb(2F5/2) lifetime is also slightly longer in the porpholactone complex, 26.5 μs versus 23.5 μs. The relative sensitization efficiency of the two macrocyclic ligands has been estimated, under the reasonable assumption that τrad is the same for the two complexes since they have the same coordination environment, and found to be 1.33, meaning that F20TPPL is a better sensitizer of YbIII luminescence.67 The influence of several aryl substituents has been tested: pentafluorophenyl (F5P), phenyl (P), chlorophenyl (ClP), trifluoromethylphenyl (F3CP), and 1,3,5-trimethylphenyl (Me3P) and the corresponding photophysical properties are presented in Table 1. The porpholactone derivatives are better than the porphyrin ones for all substituents; the best quantum yields (2.5% and 4.1%) are obtained with F3CP and this is also true for the highest brightness.
| Substituent | Porphyrin | Porpholactone | η O/ηy | B O/By | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| logε |
Q ovl/% | τ av/μs | B/M−1 cm−1 | logε |
Q ovl/% | τ av/μs | B/M−1 cm−1 | |||
| F5P | 5.62 | 2.2 | 23.5 | 9171 | 5.51 | 3.3 | 26.5 | 10679 |
1.33 | 1.16 |
| P | 5.62 | 1.6 | 24.5 | 6670 | 5.51 | 2.8 | 27.0 | 9061 | 1.59 | 1.36 |
| ClP | 5.62 | 1.5 | 26.3 | 6253 | 5.55 | 3.1 | 29.9 | 10999 |
1.82 | 1.76 |
| F3CP | 5.66 | 2.5 | 26.5 | 11427 |
5.52 | 4.1 | 25.5 | 13576 |
1.70 | 1.19 |
| Me3P | 5.67 | 1.1 | 23.4 | 5145 | 5.53 | 2.4 | 25.7 | 8132 | 1.99 | 1.58 |
When F20TPPL is combined with a partially deuterated Kläui's tripodal ligand, CoPCD3, and the resulting YbIII complex [Yb(F20TPPL)(CoPCD3)] dissolved into deuterated CD2Cl2, large increases in both the overall quantum yield (6.8%) and lifetime (75 μs) are observed, pointing to the benefit of replacing C–H vibrators with lower energy C–F ones. Therefore, in order to further minimize nonradiative deactivation induced by C–H vibrations, F20TPPL has been fully fluorinated (F28TPPL) and its complex with YbIII synthesized, [Yb(F28TPPL)(CoPCD3)]. The outcome is remarkable, with a lifetime of 525 μs and an overall quantum yield of 58%;293 these figures are comparable to those for the corresponding complex with fully fluorinated porphyrin (τ = 714 μs, QLLn = 63%).100 Experiments with two isomers of the porpholactol derivative of F28TPPL evidenced that, as expected, O–H vibrations quench more than C–H oscillators and that the formation of an intramolecular H-bond between the OH group and Kläui's tripodal ligand shortens the distance between the hydroxyl groups and the metal centre, thus leading to reduced NIR emission intensity.294 Furthermore, configurational isomerism in cis/trans-F20TPPDL porphodilactone (Fig. 55) not only red-shifts the absorption spectrum of the ligand but also results in a large difference in the NIR emission of YbIII in [Yb(F20TPPDL)(CoPMe)]: the overall quantum yield of the cis-isomer amounts to 3.3%, 8-fold larger than that of the trans-isomer (0.44%). The corresponding sensitization efficiencies estimated using τrad calculated from the absorption spectrum (0.79 ms) are 84% and 12%, respectively. Furthermore, the cis-isomer presents a useful oxygen-dependent thermosensitivity in air-saturated CH2Cl2 between 293 and 193 K (4% K−1) with a concomitant increase of the lifetime from 13.6 to 31 μs.295
 | ||
| Fig. 55 Effect of cis/trans isomerism in the absorption spectrum of F20TPPDL porphodilactone. Reproduced with permission from ref. 295, copyright (2017) The American Chemical Society. | ||
When a non-NIR luminescent lanthanide ion, such as GdIII or LuIII, is coordinated to porpholactones, ligand phosphorescence is observed in the range of 700–1200 nm, depending on the porpholactone. These complexes serve as efficient photosensitizers for the production of singlet oxygen68 or as triplet sensitizers for photon upconversion based on triplet–triplet annihilation.295
4.4 Lanthanide phthalocyaninates and naphthalocyaninates
Most of the reported complexes are with alkyl- or alkyloxy-β-substituted phthalocyanines but quantitative data are quasi-absent. Monophthalocyaninate complexes are usually stabilized with acac−296 or with Kläui's tripodal ligand.79 Sensitization of lanthanide NIR luminescence is weak because the triplet states are quite low in energy, ∼11000 cm−1 for Pc, and has essentially been observed in monophthalocyaninates, but not in double or triple deckers. Back energy transfer may be a reason.297 A few papers report NIR sensitization; for instance, ErIII luminescence is seen at 1530 nm for [Er(Pc(β-R4)acac] (R = t-but, O-C4H9) upon excitation into the Q band around 680 nm; the Er(4I11/2) lifetime amounts to 12.5–13 μs in solid state. When doped into PMMA, the resulting NIR luminescence increases up to 8 wt% concentration but then decreases due to resonant energy transfer296 In fact even ligand fluorescence and phosphorescence are weak; as an example, the three complexes [Lu(PcR4)(OAc)], [Dy(PcR4)2], and [Er(PcR4)Cl] with R4 = OPh have fluorescence quantum yields smaller than 1% and the triplet state lifetime could only be determined for the Lu complex (25 μs, in DMSO).298 Very weak NdIII sensitization was obtained for [Nd2(PcOR4)3] in DMSO.299 Metal-centred luminescence was observed upon excitation in the Soret band for [Yb(Pc)(OAc)] incorporated into PMMA and silica gel.300
5 Nonlinear optical applications
5.1 Definition
A nonlinear optical (NLO) medium is a dielectric medium in which electric polarization, expressing the density of induced electric dipoles, responds nonlinearly to the external electric field generated by light. This nonlinearity is observed for very high light intensities, i.e., usually produced by laser sources. There are two main phenomena associated with this situation: two- (or multiple-) photon absorption (TPA) and second (or higher) harmonic generation (SHG). The first one has been predicted in 1931 by the Nobel prize winner Maria Göppert–Mayer and observed in 1961 with the detection of two-photon excited luminescence in CaF2:EuII. In this process, two photons, of the same energy or different energies, are absorbed sequentially to reach an excited electronic state after transiting through a virtual state. The rate of transition differs from linear absorption in that it depends on the square of the light intensity and the cross section is expressed in units of Göppert–Mayer, 1 GM = 10−50 cm4 s photon−1. There are several applications of TPA, optical imaging – particularly bioimaging since TPA allows one to excite in the biologically friendly NIR range, optical power limiting to protect expensive optical sensors and/or human eyes of laser damages, 3D photopolymerization, or 3D optical data storage, among others. In the second process, SHG, two photons of the same energy interact with a nonlinear material, resulting in the production of a photon with double the frequency, so that the phenomenon is also termed frequency doubling. Adequate materials are crystals without an inversion centre and a common example is potassium dihydrogen phosphate (KH2PO4) used for doubling the frequency of the YAG:NdIII laser line at 1064 nm to obtain the popular green 532 nm line. Third harmonic generation, which depends on the third-order electric susceptibility χ(3), yields the 354 nm line. Both inorganic and metal complexes with organic ligands can present these phenomena and, in this respect, porphyrins, phthalocyanines and naphthalocyanines are particularly well-suited ligands.Regarding material requirements for efficient nonlinear optical properties, one has to consider the following equations expressing the dependence of the induced molecular dipole moment μ on the electric field, which is expressed as a series:301
| μ = α·E + β·EE + γ·EEE + … | (11) |
| P = ε0[χ(1)·E + χ(2)·EE + χ(3)·EEE +…] | (12) |
 | (13) |
The second-order effect is proportional to a change in dipole moment between the ground and excited states and to the square of the oscillator strength; conversely it is inversely proportional to the energy differences between the two states; moreover, a push–pull type of molecule with charge-transfer behaviour is favourable for large β values. For third harmonic generation, the requirement for a large χ(3) is mainly an extended delocalized π-system. It is noteworthy that χ(3) is a complex number and literature often reports only either the imaginary part or the bulk value:
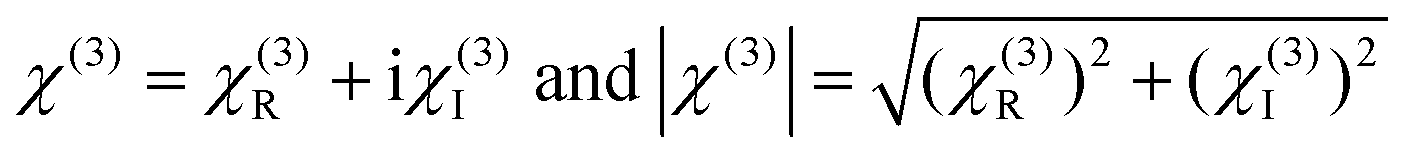 | (14) |
| n = n0 + n2·I = n0 + Δn | (15) |
 | (16) |
 | (17) |
There are several techniques for the determination of the nonlinear parameters defined above and of the nonlinear index of refraction n2. One technique is electric field-induced SHG (EFISHG) where a strong electric field is applied to the sample in solution, causing a bias in the average orientation of the molecules, partially removing isotropy, and allowing second-harmonic generation to occur. The product of the permanent dipole μ by β is gained from the intensity of the second-harmonic light; separate measurement of μ leads to the determination of β. A second, well-spread technique is the degenerate four-wave mixing (DFWM) for the characterization of third-order properties. In this process, three coherent waves are incident on a nonlinear medium, and a fourth wave (the phase conjugate wave) is generated with a strength depending on a coupling coefficient proportional to the effective χ(3). Henceforth, measurements of the phase conjugate intensity under various polarizations of the four beams yield the χ(3) tensor components of the medium. However only the moduli of these components can be determined, and extraction of the real and imaginary components requires additional techniques.
Another common technique is the so-called Z-scan where the transmittance of a nonlinear optical material is measured with a circular Gaussian laser beam in a tight focus geometry as a function of the position z of the sample. If the beam then goes through a finite aperture (Fig. 56, top) the sample behaves as a weak z-dependent lens and the detector identifies small distortions in the initial light beam; the technique is referred to as closed aperture Z-scan and this set-up is used to determine the real part of n2 using an appropriate model to interpret data. If the aperture is removed, the whole signal is measured by the detector, small distortions become unimportant, and the z-dependent signal is entirely due to the nonlinear absorption coefficient. The sample must be thin, namely thinner than Rayleigh length z0:
 | (18) |
 | (19) |
 | (20) |
 | (21) |
![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) the physical length of the sample.
the physical length of the sample.
 | ||
| Fig. 56 (Top left) Schematic set-up for a closed aperture Z-scan experiment: BS, beam-splitter; D1 and D2, detectors; L, lens; the dotted red lines show measurement of the entire beam with the open aperture set-up. (Bottom) Normalized transmission at a wavelength of 1064 nm (20 ps pulses) of [Eu(Pc)(Pyrenyl4Por)] in CHCl3versus z/z0 obtained from closed aperture (left) and open aperture (right) Z-scans. Adapted with permission from ref. 306, copyright (2012) Elsevier B.V. | ||
The two-photon absorption cross section σ2 is related to the two-photon absorption coefficient β by the following formula:
 | (22) |
| Parameter | Symbol | SI unit | cgs-esu unit | Conv. factor SI/cgs-esua |
|---|---|---|---|---|
| a Parameter (SI) = conversion factor × parameter (cgs-esu), e.g. σ (SI) = 10−4 × σ (cgs). b ε 0 = 8.85 × 10−12 F m−1 (SI); in the Gaussian (cgs-esu) system of equations, there is no ε0 (see eqn (16) and (17)). | ||||
| Linear absorption cross section | σ or σ0 | m2 | cm2 | 10−4 |
| Linear absorption coefficient | α or α0 | m−1 | cm−1 | 100 |
| Two-photon absorption cross section | σ 2 | 10−50 m4 s photon−1 | 1 GM = 10−50 cm4 s photon−1 | 10−16 |
| Two-photon (nonlinear) absorption coefficient | β | m W−1 | cm s erg−1 | 105 |
| Three-photon absorption coefficient | γ | m3 W−2 | cm3 s2 erg−2 | 108 |
| Third order susceptibility | χ (3) | m2 V−2 | esu = cm3 erg−1 | (4π/9) × 10−8 |
| Nonlinear refractive index | n 2 | m2 W−1 | cm2 s erg−1 | 103 |
| Vacuum permittivity | ε 0 | F m−1 = C V−1 m−1 | no | |
Metallo-porphyrinates and phthalocyaninates offer interesting possibilities for NLO performances. Indeed, there exists a correlation between the number of π-bonds in an organic π-delocalized system and χ(3), and tetracorroles and their complexes display extensive π-systems that even develop in 3D in double or triple deckers. Moreover, these compounds have large thermal stability, up to 400 °C, rendering the processing easy, and variation of both the central metal ion and ring peripheral substituents is a practical tool for modulating their electronic properties. Electron delocalization may also be extended by the formation of polymers, aggregation, or multiple-decker compounds.
5.2 Nonlinear optical parameters for some lanthanide tetrapyrrole complexes
Nonlinear optical parameters for lanthanide tetrapyrrole complexes have been reported by several authors (see for instance ref. 299, 302, and 306–315) and have been partially reviewed.271,301 Data are however not consistent from one study to the other in that experimental conditions are different and usually only a few lanthanides have been studied each time. | ||
| Fig. 57 Third-order hyperpolarizabilities at 1024 nm for LnPc2 in CHCl3. Redrawn from data reported in ref. 317. | ||
Comparisons between double- and triple-decker properties are available. One comparison deals with complexes with octa(t-but-phenoxy) substituted Pc, Pc(tbutPO)4. For 3.4 × 10−4 M solutions in CH2Cl2 and with a 532 nm laser as the excitation source, γ and χ(3)I increase slightly (+5%) in going from {Sm[Pc(tbutPO)4]2} to {Sm2[Pc(tbutPO)4]3}.318 Another study involves heteroleptic complexes with Pc and tetra(1-pyrenyl) substituted porphyrin, TPYRP. Replacing one phthalocyaninate ring in [EuPc2] with TPYRP to yield [Eu(Pc)(TPYRP)] leads to increases in γ from 22 to 104 × 10−32 esu and in χ(3) from 102 to 154 × 10−15 esu (0.72 × 10−4 M in CHCl3). On the other hand the triple-decker [Eu2(Pc)2(TPYRP)] (1.19 × 10−4 M) in CHCl3 has smaller nonlinear parameters, γ = 41 × 10−32 esu and χ(3) = 100 × 10−15 esu. The double decker with TPYRP has also a larger n2 value than the triple decker, 9.44 vs. 5.81 × 10−13 cm s· erg−1. The working wavelength was the same as that for [EuPc2] but the techniques were different, Z-scan vs. DFWM. That the double decker has larger nonlinear parameters than the triple decker is not usual and can be attributed to the fact that the intervalence band for the double decker around 1330 nm partly overlaps with the probe wavelength at 1064 nm resulting in enhanced nonlinear effects, while the triple decker has no intervalence band.306 Adding the TPP ligand to [SmPc2] to get the triple-decker [Sm2Pc2TPP] increases the value of χ(3) from 180 × 10−15 esu (10−4 M in DMF, 532 nm) to 210 × 10−15 esu (5 × 10−5 M in CHCl3, 830 nm); this value increases to 260 × 10−15 esu for [Sm2PcTPP2] (4.6 × 10−5 M in CHCl3, 830 nm). The authors attribute this large increase to the fact that the B band of TPP has its maximum at 415 nm, while the pump wavelength at 830 nm is exactly the double, so that part of the enhancement may arise from two-photon resonance.316 This shows that many factors influence nonlinear phenomena that are not always obvious to detect and therefore a detailed record of all experimental parameters is necessary when reporting such phenomena.
Within the frame of the work centred on developing efficient photodynamic procedures, hybrid systems were tested in which two macrocycles were linked by, for instance, acetylene bridges. One example is mixed M–porphyrin/Yb–phthalocyanine bimetallic complexes, with M = H2, Zn, and Pd (Fig. 16, bottom). Excitation at 416 nm in the Soret band results in singlet-oxygen quantum yields of 1.30, 1.38, and 1.07 for PH2–PcYb, PZn–PcYb, and PPd–PcYb, respectively. Quantum yields are larger than one because 1O2 can be produced by both the singlet and triplet states. When excitation is set at 675 nm, the quantum yields are less than 1. The two-photon absorption cross section σ2 is large for these systems: 2347, 3301, and 2365 GM, respectively.108 Boron dipyrromethene (BODIPY) is known for its light-harvesting and electron-acceptor properties. It has therefore been linked to phthalocyanine using phenyl-ethynyl bridges to form [Yb(Subst-Pc)CoPMe] with Pc bearing different substituents: 2 BODIPY and 2 C6F5 (Fig. 53, top), 1 BODIPY and 2 C6F5 (2), 1 BODIPY and 2 triphenylamine (TPA) (3), and 1 BODIPY and 2 phenyl (4). The measured σ2 cross sections of 10−4 M solutions in DMSO at 800 nm and acquired with the open-aperture Z-scan technique amount to 1417, 1048, 2226, and 1191 GM, respectively. The largest cross section is obtained with the strongest electron donor groups (TPA) because this conjugate has the most appropriate D–A push–pull structure. The length of the binding unit has some influence: replacing the phenylethynyl bridge with a shorter ethynyl bridge in (4) increases σ2 to 1446 GM (+21%). The power dependence of the two-photon absorption has been checked for conjugates 1 and 3 and the slopes of the log–log plots amount to 2.02 and 1.94, respectively, confirming the two-photon mechanism.287
In fact, despite the acetylene or phenylacetylene bridges, the electronic communication between the two entities, BODIPY and Pc, remains small so that attention has turned towards double deckers, with good success. Table 4 reports σ2 values for a series of [Ln(OOct8Pc)2] and [Ln(Pc)(OOct8Pc)] complexes. They are larger by factors of 10–60 compared with the monoporphyrinates. In the homoleptic series, the cross section increases 3.4 times between Ce and Lu, in line with increasing π–π interaction with decreasing ionic radius of the metal ion. Interestingly, the heteroleptic complexes have larger σ2 values than the homoleptic double deckers. This may be explained by the heteroleptic structure in that the hydrogen atoms on the unsubstituted Pc function as electron donors, while octyloxy groups of the substituted Pc are electron attractive so that the complex can be visualized as a D–A system connected by a large conjugated π system (D–π–A) and this type of configuration is known in organic chemistry for generating large nonlinear optical effects.310
5.3 Optical limiting properties
Optical limiters are materials that are transparent at normal light intensities but become opaque under larger light irradiation. They are essentially used for the protection of human eyes and sensitive optical components such as detectors when they are suddenly and, often, unexpectedly exposed to intense laser pulses. Therefore, these materials must possess fast response speeds. The present interest focuses on materials with small ground-state absorption cross sections and high excited-state cross sections, achieving optical limiting by reverse saturable absorption (RSA). Metalloporphyrins are ideally suited because they allow high transmission in a window between the Soret and Q bands, around 500–550 nm (see for instance Fig. 43), a range ideal for excitation with the 532 nm YAG:NdIII laser line. Nonlinear properties of many lanthanide tetrapyrrole complexes have been investigated with attention focused mainly on optical limiting properties and it was soon discovered that these complexes have often better characteristics than more classical materials such as C60 or transition metal phthalocyaninates. For instance, the erbium complex with tetrakis(p-diphenylaminophenyl)porphyrine (Scheme 1), [Er(TDPAPP)(CoPMe)], has optical limiting threshold Jlim = 0.093 J cm−2, compared with 0.15 and about 0.1 J cm−2 for C60 and MPc (M = Si, Ge), respectively.271A common model for interpreting optical limiting data is the so-called 5-level model depicted in Fig. 58.
 | ||
| Fig. 58 Five-level model corresponding to the RSA mechanism for optical limiting. σ's are absorption cross sections and ks are first-order rate constants reflecting the lifetime of various levels (τ = 1/k). | ||
A set of differential equations describe the population dynamics of the various levels:
 | (23) |
 | (24) |
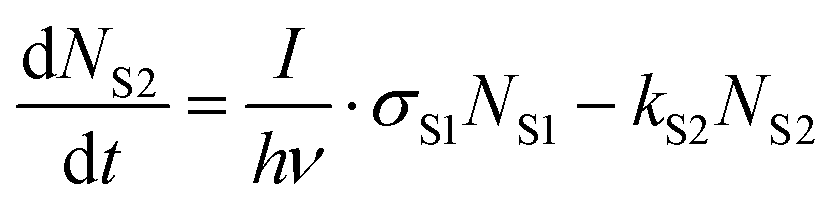 | (25) |
 | (26) |
 | (27) |
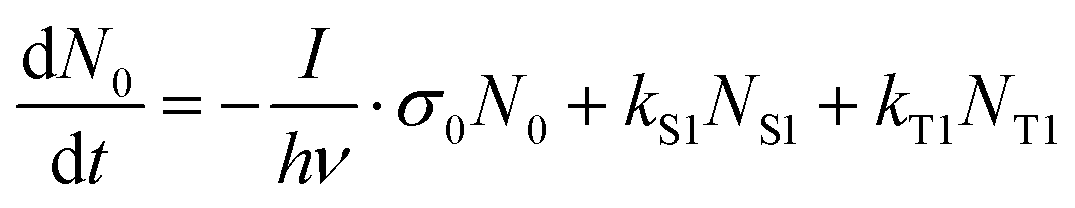 | (28) |
 | (29) |
 | (30) |
| Parameter | Units | Eu | Dy | Er | Lu |
|---|---|---|---|---|---|
| a Fixed and taken equal to the experimental value for [LuPc2]. | |||||
| σ 0 | 10−17 cm2 | 4.7 | 2.6 | 2.9 | 4.5 |
| σ S1 | 10−17 cm2 | 1.8 | 0.9 | 1.0 | 1.6 |
| σ T1 | 10−17 cm2 | 22.0 | 14.5 | 20.0 | 16.5 |
| σ T1/σ0 | — | 4.7 | 5.6 | 6.9 | 3.7 |
| k S1 | 108 s−1 | 5.0 | 5.0 | 5.0 | 5.0 |
| k isc | 108 s−1 | 3.85 | 3.45 | 3.33 | 3.85 |
Assuming steady-state approximation, i.e. that the populations of the states remain constant for the duration of the laser pulse, the intensity-dependent absorption coefficient can be written as
 | (31) |
Some recent data are listed in Table 6 for mono, double, triple, quadruple and sextuple phthalocyaninates. The optical limiting properties are reflected by Fsat values which are largely inferior to 1 J cm−2 and even reach values in the range of 0.05–0.25 J cm−2, while transmission can be reduced up to 80%. The reduced forms of the phthalocyaninates have the tendency to yield lower Fsat values.322 Introducing the optical limiting complexes into polymeric thin films is generally beneficial. For instance, when embedded into poly(bisphenol A carbonate) thin films (PBC), Fsat values of tris(monophthalocyaninate) complexes [Ln3Cl3L1] decrease from 0.31 to 0.11 J cm−2 (Yb) and from 0.79 to 0.26 J cm−2 (Lu).109 Similarly, PBC thin films of {Ln[Pc(tbutOPh)4]2} and their reduced forms {Ln[Pc(tbutOPh)4]2}− have much smaller Fsat values and transmission reduction can be as high as 88%.323 A very low Fsat value of 0.05 J cm−2 is reached for the PCB thin film incorporating [Nd(PcOPy4)2] and transmission attenuation reaches 73% instead of 39% for the solution.322
| Complex | Conditions | Ln | χ (3)I (10−10 esu) | F sat (J cm−2) | T reduction % | Ref. |
|---|---|---|---|---|---|---|
| a Absence of data means that 50% attenuation of transmittance is not achieved. b L1 = 2,4,6-tris[3-thio-9,10,16,17,23,24-hexa(4-terbutylphenoxy)phthalocyaninato]-1,3,5-triazin. c Counter-ion: Me3(C16H33)N+. d Counter-ion: [Nd(Ac)2]+. e Counter-ion: [Dy(Ac)2]+. | ||||||
| [Ln(PcOPy4)2] | 3.4 × 10−4 M, DMSO | La | 0.74 | 0.25 | — | 327 |
| Yb | 5.3 | 0.22 | — | |||
| [Ln(PcOPy4)2] | 4.6 × 10−5 M, CH2Cl2 | Nd | — | 0.24 | 39 | 322 |
| [Ln(PcOPy4)2]− | 4.6 × 10−5 M, CH2Cl2 | Nd | — | 0.15 | 64 | |
| {[Ln[Pc(OPhtbut)8]2} | CH2Cl2 | Ce | 2.55 | 0.08 | — | 318 |
| Gd | 2.68 | 0.07 | — | |||
| {[Ln2[Pc(OPhtbut)8]3} | CH2Cl2 | Ce | 2.12 | 0.09 | — | |
| [Ln3Cl3L1]b | 3.3 × 10−4 M, DMF | Yb | 1.4 | 0.31 | — | 109 |
| 3.4 × 10−4 M, DMF | Lu | 0.8 | 0.79 | — | ||
| [Ln(PcOBut6R2)2] | CH2Cl2 | Eu | 0.09 | — | — | 324 |
| [Ln2(PcOBut6R2)3] | CH2Cl2 | Eu | 0.41 | 0.57 | — | |
| [Ln(PcOBut8)2] | CH2Cl2 | Eu | 0.86 | — | — | |
| [Ln2(PcOBut8)3] | CH2Cl2 | Eu | 0.21 | 0.92 | — | |
| {Ln[Pc(OtbutPh)4]2} | CH2Cl2 | Nd | 3.77 | — | 48 | 323 |
| {Ln[Pc(OtbutPh)4]2}−c |
CH2Cl2 | Nd | 11.3 | 0.49 | 73 | |
| {Ln[Pc(OtbutPh)4]2}−d |
CH2Cl2 | Nd | 12.8 | 0.52 | 80 | |
| {Ln[Pc(OtbutPh)4]2}−e |
CH2Cl2 | Nd | 16.6 | 0.65 | 75 | |
| [Ln(Pc)2] | CH2Cl2 | Nd | 3.77 | — | 48 | 328 |
| [CdLn2(Pc)4] | CH2Cl2 | Nd | 20.0 | 0.34 | 66 | |
| [Cd2Ln4(Pc)6] | CH2Cl2 | Nd | 49.1 | 0.10 | 80 | |
In an effort to improve optical limiting properties, phthalocyaninates have been conjugated to various entities, e.g. quantum dots for the europium double and triple deckers with (PcOBut8) and (PcOBut6R2) leading to a decrease in Fsat up to 50%,324 or gold nanoparticles in the case of a completely chlorinated phthalocyanine, PcCl16. For the europium complex, the plasmonic effect of gold nanoparticles considerably increases reverse saturable absorption with the nonlinear absorption coefficient β increasing from 3.2 × 10−12 cm W−1 for the pure [Eu(PcCl16)2] compound to 11 and 18 × 10−12 cm W−1 for its conjugates with 20 and 30 nm gold nanoparticles, respectively.325 Finally, a bis(phthalocyaninate) di-neodymium complex has been conjugated with amino-functionalized multi-walled carbon nanotubes (MWCNT) with large decreases in β, from 3.78 to 0.70 × 10−8 m W−1, and in Fsat, from 0.42 to 0.055 J cm−2, which is one of the lowest values reported in the literature.326
5.4 Upconversion through triplet–triplet annihilation (TTA)
One mechanism of upconversion in organic compounds is triplet–triplet annihilation (TTA) which was first proposed in the 1960s to explain delayed fluorescence in anthracene. After initial excitation of the sensitizer molecules into their S1 states and subsequent intersystem crossing to T1 (ϕisc), these molecules transfer their triplet state energy to the emitter molecules (triplet–triplet transfer, ϕTTET). Triplet–triplet transfer then occurs between two emitter molecules (ϕTTA), one returning to its ground state and the other being promoted to a higher singlet state which finally de-excites by emitting delayed fluorescence (ϕF). The upconversion yield is given by| ϕUC-TTA = ϕisc × ϕTTET × ϕTTA × ϕF | (32) |
Although lanthanide ions have smaller spin–orbit constants than platinum or palladium for instance, gadolinium and lutetium porphyrinates display strong ligand-centred phosphorescence with a long lifetime, which is favourable for acting as a sensitizer in oxygen sensing or photodynamic therapy of cancer. Therefore Lu and Gd complexes with tetrakis(pentafluorophenyl)porphyrin (F20TPP), porpholactone (F20TPPL) and porphodilactone (F20TPPDL, cis and trans) have been recently tested as photosensitizers for TTA.329 The chosen sensitizers were 9,10-bis(2-phenylethynyl)anthracene (BPEA) for [M(F20TPP)CoPMe] (M = Lu, Gd, Pd) and [Lu(F20TPPL)CoPMe], and rubrene for [Lu(F20TPPDL)CoPMe]; measurements were conducted in degassed toluene. The Lu complexes with F20TPPDL have sensitization ability for rubrene comparable to that of previously reported Pd and Pt analogues, namely ϕUC-TTA = 0.9 and 0.5% for the cis and trans isomers, respectively. Interestingly, [Lu(F20TPPL)CoPMe] has a much larger ϕUC-TTA, 7.8%, while for complexes with F20TPP, ϕUC-TTA reaches 6.3% for Gd and 12.4% for Lu. As a comparison the Pd complex with the same ligand has ϕUC-TTA = 8.6%. Although [Lu(F20TPPL)CoPMe] has a smaller upconversion yield compared with [Lu(F20TPP)CoPMe], the incident wavelength can be shifted from 560 to just over 600 nm, which is an advantage for bio-applications. As a proof of concept, encapsulating BPEA and a Lu complex with F20TPP or F20TPPL into nanomicelles allows one to use the UC systems in aerated water and their imaging capability was tested successfully on HeLa cells.329
Triplet–triplet annihilation was recently invoked to explain anti-Stokes shifts in lanthanide double deckers “conjugated” with trifluoroacetic acid (TFA), [LnPc2(TFA)8], in which the protons of TFA interact with the meso N atoms of the phthalocyanine rings. Excitation in the Q band at 650 nm results in ligand fluorescence with modest anti-Stokes shifts of about 13–15 nm for some lanthanides (Nd, Sm, Eu, Gd) but not for the conjugates of Yb and Lu.330
5.5 Liquid crystals
When tetrapyrroles are fitted with long aliphatic and/or branched peripheral substituents, nematic phases can be obtained. As a matter of fact, the first lanthanide-containing metallomesogens were lutetium double deckers with phthalocyanines fitted with alkoxy chains containing 8-18 carbon atoms. In view of the nature of the ligands, most liquid crystalline phases are aligned columnar phases, where the molten alkyl chains act as insulating layers; therefore, these arrangements are a kind of molecular electric wires. The double deckers have large electrical conductivity and some, such as [Lu(Pc)2], behave like semiconductors. Lanthanidomesogens with tetrapyrrole ligands have been thoroughly reviewed,35,331 and most works in this field are devoted to characterizing the mesophases along with electrochemical properties. Therefore, we do not discuss these compounds further within the frame of this review.6 Sensing and biomedical applications
The perfect match between lanthanide(III) ions and macrocyclic tetrapyrrole ligands gave rise to kaleidoscopes of advanced functional materials which have been chemically, electronically, photonically, magnetically and even catalytically endowed, as showcased in the previous sections and other relevant reviews. Countless lanthanide–tetrapyrrole complexes and conjugates of fascinating architectures have, over the past few decades, spanned the fields of electrochromic and optoelectronic devices, photovoltaic cells, single-molecule magnets, Lewis acid/photo-catalysts, and even molecular rotors. In stark contrast, when it comes to sensing and biomedical applications, there had been distinctly scarcer examples in the early days, given (i) their underexplored biochemistry, (ii) the problematic availability of high-purity lanthanide elements, (iii) inefficient traditional synthetic and functionalization methods, and (iv) the intrinsic hydrophobicity of tetrapyrrolic macrocycles. This situation has dramatically changed since the turn of the century and more particularly during the recent decades, in that there have been fiery developments of lanthanide–tetrapyrrole complexes and materials in sensing and biomedical domains, thanks to continual and decisive advances in synthesis and spectroscopy. Regarding the former, one can cite smart ligation strategies on tetrapyrroles for global water solubility enhancements and selective bio-targeting achievements by tailored lanthanide complexes, while two-photon excitation and improved NIR detection systems have paved the way for sensitive biosensing and bioimaging. We have named this decade as the second golden period of development of lanthanide–tetrapyrrole chemistry. In this section, we are going to discuss in detail the design strategies and the structure–property relationship of the recently designed lanthanide–tetrapyrrole complexes for biomedical applications, hopefully casting more insights for more breakthroughs in the years to come!6.1 Chemical, biological and physical sensing
Lanthanide–tetrapyrrole systems have been widely and wisely employed in a diversity of chemical, biological and physical sensing—from various gases and metal ions, to chiral small molecules, biomolecules, and temperature. We have selected and categorized most of the related literature as follows.(a) NO2 & NH3. In 2009, Öztürk's et al. studied the NO2 and O3 sensing performances of a series of LuIII–phthalocyanine homoleptic double-decker complexes derivatized with various lengths of alkylthio chains in different concentration ranges (NO2 = 1–10 ppm; O3 = 50 ppb–1 ppm) and in the temperature range of 25–150 °C.335 The sandwich complexes exist in solid state at 25 °C and in the liquid crystalline phase at 150 °C; the response time and the sensor response were found to be highly dependent on the alkyl chain lengths. In 2012, Bouvet and Chen et al. prepared, by the quasi-Langmuir–Schäfer (QLS) method, solution-processed thin films of heteroleptic (phthalocyaninato)(porphyrinato) EuIII triple-decker complexes with different cis/trans-peripheral tert-butyl and hydroxyl groups to be used as NH3 sensors.336 Their supramolecular structures in nano-grains and nano-sheets were formed.
An ambipolar NO2 and NH3 sensor was developed by Chen et al. in 2007 based on QLS films of an amphiphilic and heteroleptic triple-decker [Eu2(TPyP)(Pc)2] complex; the sensor featured very good conductivity, crystallinity, and excellent linear sensitivity, 31.9% ppm−1 for NO2 but only 0.04% ppm−1 for NH3.337 In 2018, Jiang and Chen et al. discovered polymorphism (χ, β, and α phases) in the self-assembled nanostructures of new trifluoroethoxy-substituted homoleptic tris(phthalocyaninato) EuIII complexes of solvent-controlled nanomorphologies (nanobowls, nanobelts and nanoparticles) and confirmed the phase-dependent sensitivity towards NO2, phase χ > phase β > phase α; the largest response was recorded for phase χ, with a lowest detection limit of 20 ppb for room-temperature NO2 gas sensing.338
To further optimize the NO2 sensing behaviour of EuIII phthalocyaninato triple decker-based thin-film sensors, Jiang, Chen et al. proposed to extend the π-conjugation of the system by fusing two of them to form a quintuple-decker dimer sandwich (Fig. 59).339 Significant increases in the hole and electron mobilities, up to 0.07 and 0.06 cm2 V−1 s−1, respectively, and a much higher sensitivity, from 23.4% ppm−1 for the monomer to 46.7% ppm−1 for the quintuple decker were recorded for this OFET-based sensor. On the other hand, the authors had also attempted to substitute one of the middle-layer EuIII-layers with CdII and synthesized a novel sandwich-type EuIII–CdII tetrakis(phthalocyaninato) quadruple-decker.340 New records of ambipolar OFET properties of such a thin film, i.e., hole and electron mobilities of 5.6 × 10−3 and 1.2 × 10−3 cm2 V−1 s−1, respectively, and effective response towards NO2 concentrations ranging from 0.25 to 2.5 ppm, were obtained.
 | ||
| Fig. 59 Schematic molecular structures of {Eu2[Pc(SC6H13)8]3} (left) and {[Pc(SC6H13)8]2Eu2[BiPc(SC6H13)12] Eu2[Pc(SC6H13)8]2} (right) (EuIII = green, C = black, N = blue, S = yellow) with all the hydrogen atoms and n-hexyl chains omitted for clarity. The computed adsorption energy of the left compound towards NO2 is −34.3 kcal mol−1. Reproduced with permission from ref. 339, copyright (2019) Elsevier B.V. | ||
(b) O3 & O2. Differing from Öztürk et al.'s work using homoleptic bis[tetrakis(alkylthio)phthalocyaninato] double-decker LuIII complexes for sensing O3, Bouvet and Jiang et al. adopted the new QLS process in 2010 to fabricate amphiphilic heteroleptic tris(phthalocyaninato) EuIII triple-decker thin-film sensing transistors, in which two layers of the phthalocyanine sandwich were peripherally functionalized with crown ether moieties and the top layer with long alkyl chains.341 Outstanding sensitivity, and stable and reproducible responses towards O3 were reported. By loading monoporphyrinato LuIII and GdIII complexes into the thin films of copolymers of poly-tert-butylmethacrylate, polystyrene and methylcellulose, Ermolina et al. constructed an optical oxygen sensor by exploiting the O2 quenching effect on the room-temperature phosphorescence at 780 nm of the doped solid-state systems.216 The device response time and the detection limit of O2 were recorded as 1.0–1.2 s and 0.05%, respectively.
(c) SO2. In 2015, a structurally novel “molecular ball” composed of a LuIII–bis(phthalocyanine) complex as the core and four salen-type arch pillars was designed and synthesized by Bekaroğlu et al., which, in the form of thin film, electrically and reversibly responded to SO2 within 40 s (Fig. 60).342
 | ||
| Fig. 60 “Molecular ball”: LuIII–bis(phthalocyanine) sandwich complex chelated by four salen-type ligands capable of sensing SO2. Reproduced with permission from ref. 342, copyright (2015) Elsevier B.V. | ||
(d) Volatile organic compounds (VOCs). In 2001, Rodrìguez-Mendez and Saja et al. documented that the nature of the central lanthanide ion, the presence of tert-butyl groups, and the thickness of the LnPc2 (Ln = Pr, Lu) LB films could help modulate the device response of their sensor array, which could even discriminate between different types of olive oil aromas upon principal component analysis of the signals.343 In 2001, a wide range of VOCs, alcohols, aldehydes and esters, as well as gases, had been further examined by the authors,344 with the new possibility of NIR-emissive LB films of LnPc2 as gas/VOC-sensing materials in fibre-optic sensors being showcased. By octa-substituting the phthalocyanine with imidazole moieties, Bekaroğlu et al., in 2018, prepared more sensitive and semiconducting LB films of LnPc2 (Ln = EuIII, LuIII) for detecting organic vapours of toluene, ethanol, THF, n-hexane, chloroform, and xylene.345
 | ||
| Fig. 61 Scheme for chiral dicarboxylic acid sensing by sandwich CeIV-bisporphyrinates. Reproduced with permission from ref. 349, copyright (1999) The Royal Society of Chemistry. | ||
In 2001, Tsukube et al. constructed an ErIII porphyrinate/benzo-18-crown-6 conjugate which can synergistically recognize zwitterionic chiral amino acids and biogenic amines, actualizing induced CD-based chirality sensing of the tested biomolecules; in fact, just mixing the separate ErIII porphyrinate and the crown ether components failed to do so (Fig. 2).50 More intriguingly, the authors asserted that only the ErIII, but not the GdIII or YbIII, conjugates could afford such a CD-enhancement.
In 2003, Chudinova et al. developed a new donor–acceptor energy transfer and NIR-emissive marker pair based on (i) fluorescein isothiocyanate and (ii) YbIII–porphyrin–benzoyl trifluoroacetone–isothiocyanate; they applied it in a luminescence immunoassay for the detection of protein antigens on the surface of Langmuir–Blodgett films of antibodies; to reduce non-specific binding, a non-polar substrate consisting of layers of stearic acid was used as the substrate for the LB film.351 A high sensitivity (∼10−11 M) and a short assay time (6–8 min) were reported for the overall system.
In 2008, Basova et al. investigated the electrochromic properties of a spin-coated film of bis[octakis(hexylthio)phthalocyaninato] DyIII deposited on an indium tin oxide electrode. This film displays a single redox couple at E1/2 = 0.78 V and was used to determine nicotinamide adenine dinucleotide hydride (NADH) in aqueous solutions.352 The authors hypothesized that the oxidized form of the DyIII sandwich of the chemically and electrochemically modified film would be reduced to its neutral form, i.e., ([(C6H13S)8Pc]2Dy)+ → [(C6H13S)8Pc]2Dy, when interacting with the reducing NADH.
More recently, a new prototype of an albumin and nanosilica sensing system was proposed based on supramolecular films of polyvinyl pyrrolidone and polyvinyl alcohol comprising LuIII/YbIII diphthalocyanine double-decker complexes, highlighting the biomedical sensing potential of lanthanide sandwich complexes protected in biocompatible multicomponent host systems.353
In 2019, Nyokong's group reported new electrochemical sensors for detecting 4-chlorophenol, a toxic organic contaminant, in aqueous media by modifying a glassy carbon electrode (GCE) with two forms of blue and green diethylaminophenoxy-bearing thulium double-decker phthalocyaninates, capitalizing on their respective electrocatalytic effect arising predominantly from the redox cycles of the Pc rings. The blue form stands for [(Pc2−)TmIII(Pc2−)]− (i.e., both the HOMO and LUMO are fully filled), with its overall charge compensated by a hexadecyltrimethylammonium cation, and responds better to oxidizing analytes, while the green form [(Pc−1˙)TmIII(Pc2−)] with one electron hole prefers reducing analytes. The authors discovered that the blue-GCE demonstrated a lower peak potential of 0.88 V vs. Ag|AgCl, a higher catalytic current of 593 μA, and apparent sensitivity towards the presence of 4-chlorophenol with a lowest detection limit of 4.16 × 10−8 M.354
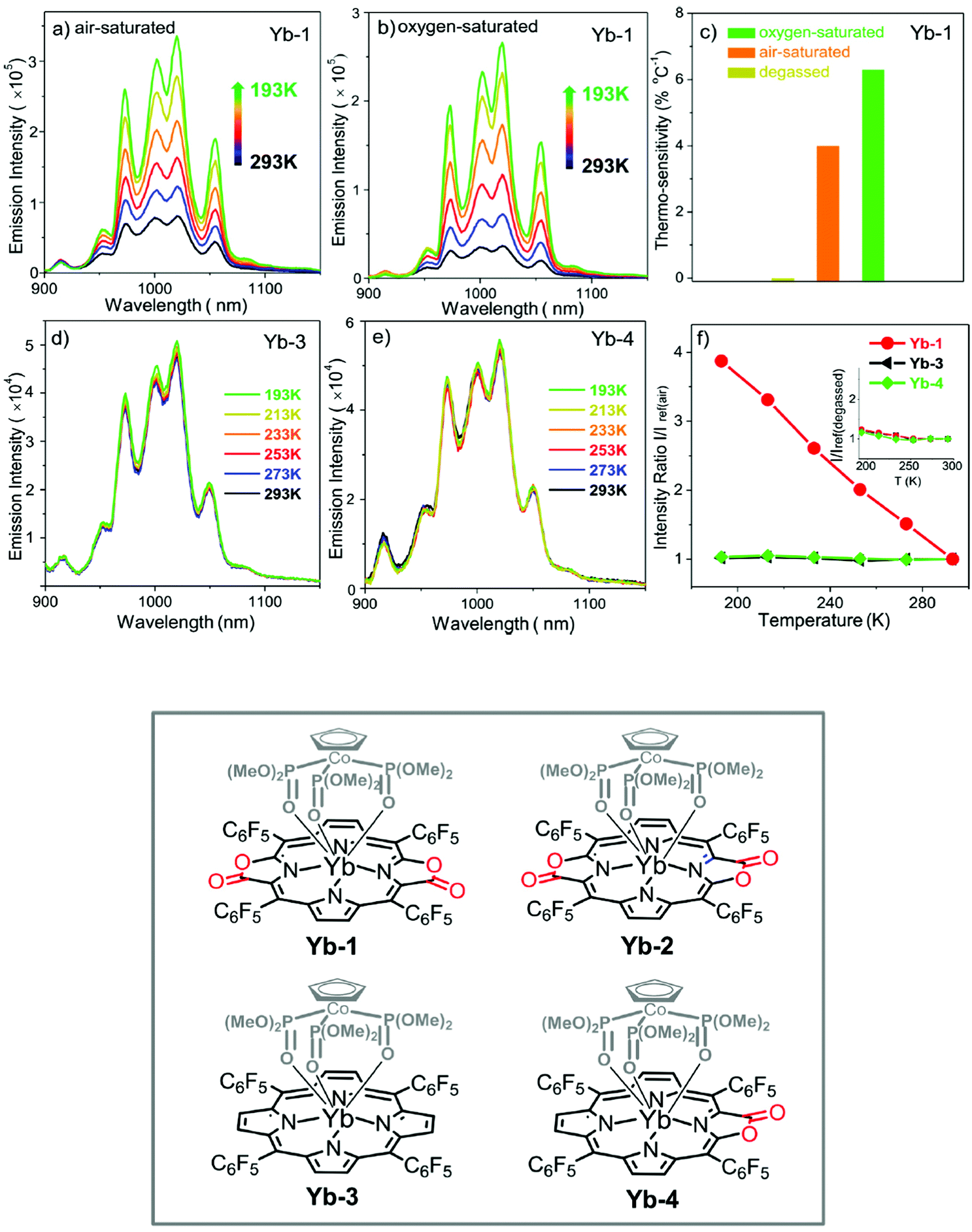 | ||
| Fig. 62 Temperature-dependent emission of Yb-1 in (a) air-saturated and (b) oxygen-saturated CH2Cl2 (293–193 K); (c) thermosensitivity of Yb-1 in different gas atmospheres in CH2Cl2 (293–193 K); temperature-independent emission of (d) Yb-3 and (e) Yb-4 in CH2Cl2 (293–193 K); (f) emission intensity ratio normalized according to that at 293 K of Yb-1 (red), Yb-3 (black), and Yb-4 (green) in air-saturated CH2Cl2 (Inset: Emission intensity ratio normalized according to that at 293 K in degassed CH2Cl2). Reproduced with permission from ref. 295, copyright (2017) The American Chemical Society. | ||
6.2 Time-resolved NIR imaging in vitro and in vivo
In the early 1990s, Guidak et al. employed their home-made fibre-laser spectrofluorometer and disclosed the first proof-of-concept of luminescence contrast-enhanced (i.e., autofluorescence-free) localization of tumours in vivo with NIR-emissive YbIII–porphyrin complexes.355 In parallel, Sessler et al. pioneered the use of gadolinium texaphyrin as a novel paramagnetic contrast agent in magnetic resonance imaging.356,357 These translational milestones laid the foundation of a new era for molecular lanthanide complexes in modern chemical biology and clinical radiology.One of the critical problems of commercially available organic-based imaging agents, e.g., fluorescein, is high background noises during microscopic studies, which are caused by biological samples’ autofluorescence and scattered light, as well as by the luminescence from the optical components and sample containers. Moreover, the poor photostability of most of them due to photobleaching is also a great concern. For this purpose, more attention has been focused on time-resolved luminescence techniques, with lanthanide complexes being new rising stars. The emission lifetimes of photostable lanthanide ions are in the micro- to milli-second scale, which is perfect for time-resolved detection. Moreover, some ions like NdIII, ErIII, or YbIII exhibit emission in the near-infrared II/III biological windows (1000–1700 nm), which enables higher tissue penetration. Several critical benchmarks have to be met when designing practical lanthanide/tetrapyrrole-based molecular in vitro/in vivo imaging probes: (i) high water solubility and biostability, (ii) adequate biocompatibility, (iii) impressive photophysical properties (i.e., high brightness and long lifetime), and (iv) target selectivity and specificity.
In the literature, when it comes to mainstream applications of lanthanide-based bioimaging and bioanalysis, visible-emissive EuIII and TbIII have been ubiquitous since they usually present large values of quantum yield and brightness, and long lifetimes. In contrast, before 2010, the highest NIR emission quantum yield and lifetime of YbIII with non-macrocyclic ligands were ca. 0.5% and <5 μs in water, respectively.358 In 2011, Wong et al. introduced the first water-soluble monoporphyrinato capped ytterbium(III) complex linked with rhodamine B (Yb–RhB) which showed mitochondria-specific subcellular localization and, at that time, record-high NIR emission quantum yield from YbIII (1% at λex = 340 nm; 2.5% at λex = 430 nm; both in water) (Fig. 23).70 By changing the vector from rhodamine B to ammonium salt, the same authors reported later another similar YbIII capped monoporphyrinate (Yb–N) which demonstrated strong binding to phosphatidylserine. The latter is an anionic phospholipid component appearing mainly in the interior of the cell membrane's lipid bilayer under normal conditions, but which is transported to the outer surface in the early and intermediate stages of apoptosis. Therefore, Yb–N differentiates the cancerous cells from the normal cells upon selective targeting of the more anionic phospholipid membrane of cancerous cells, as demonstrated by in vitro NIR imaging.122 Given that the extent and the rate of onset of induced targeted cancer apoptosis can serve as prognostic indicators of the treatment outcome, Yb–N holds promise as an NIR prognostic bioprobe.
To further ameliorate the NIR photophysical properties, water solubility and biocompatibility of YbIII porphyrin complexes, Zhang et al. introduced (i) fluorination at the meso-phenyl and β-peripheral positions of the porphyrin and (ii) deuteration of the Kläui ancillary ligand, to circumvent the quenching effect of the high-vibration X–H bonds (X = C, N and O); peripheral porphyrin β-cycloaddition and β-lactonization, and (iii) ligation with various hydrophilic side chains, were also attempted (Fig. 63).105 The resulting capped monoporphyrinato YbIII complexes gave rise to new records of NIR quantum yields (5–13% in water) and decay lifetimes (56–173 μs in water). Confocal live cell imaging and time-resolved fluorescence lifetime imaging (FLIM) in vitro verified the preferential localization of the NIR-emissive YbIII complex in the lysosome and high signal-to-noise ratios for effective autofluorescence discrimination. The authors subsequently employed the complexes in high-resolution non-invasive in vivo NIR-II imaging of small animals’ whole body, vasculature, and lymph nodes, and confirmed their safe metabolic clearance pathway through the hepatobiliary and renal systems like most organic fluorophores.289
 | ||
| Fig. 63 Top: NIR confocal images performed on living HeLa cells incubated with 10 mM Yb-4 for 12 h followed by 30 min incubation with 75 nM LysoTracker Green. (A) Bright field; (B) NIR signal arising from YbIII in channel 1 (λex, 620 nm; λem, 935, 170 nm bandpass); (C) visible signal arising from LysoTracker Green in channel 2 (λex, 470 nm; λem, 530, 43 nm bandpass); (D) merged b and c showing colocalization (P = 0.52). Scale bar: 10 μm. Bottom: NIR time-resolved images of living HeLa cells incubated with 10 mM Yb-4 and Yb-4c (λex, 408 nm; λem, 935, 170 nm bandpass; dwell time, 4 ms). Scale bar: 10 μm). Reproduced with permission from ref. 105, copyright (2018) The Royal Society of Chemistry. | ||
In 2010, Klapshina et al. adopted an encapsulation approach to prepare PEG-organized biocompatible silica-modified and disk-shaped nanoparticles doped with tricyanovinylbenzene-bridged bis(ytterbium(III) cyanoporphyrazine) complexes for in vivo imaging.359 Modification of the PEG nanoparticles with a hydrophobic organosilicon component led to uniform nanoparticles. The adopted sol–gel strategy rather than the commonly used water-in-oil method appears to be ideal for biomedical work since it avoids centrifugation and washing. Host protection helped in enhancing the water nano-suspension, biostability (for several weeks), and biocompatibility of the overall system. Protecting the lanthanide emitter from undesired interactions and shielding of living organisms from potentially toxic heavy metal effects were also achieved, in parallel with a large enhancement of NIR emission upon binding to biomolecules in physiological liquids.
6.3 Biomolecular interactions
Although free tetrapyrrolic macrocycles and their transition-metal complexes had been well-identified for their imaging and therapeutic potentials and for their interactions with biomolecules via intercalation, stacking, and binding,360,361 such information for their lanthanide counterparts had long been elusive. Liu and Wong et al. first spectroscopically investigated the thermodynamics of the binding between a lanthanide cationic porphyrin complex (YbTMPyP) and bovine serum albumin (BSA) in water, in 2007; they confirmed that such a process is enthalpy-driven and brings the reactants in close proximity so that molecular resonance energy transfer with tryptophan and tyrosine residues of BSA occurs.362 In 2011, Wong et al. synthesized a series of water-soluble meso-pyridyl-substituted LnIII porphyrinates with different substitution patterns and capped with a tripodal ligand, and examined their binding modes to and photocleavage of DNA. All the results of absorption and fluorescence titrations, induced circular dichroism measurements, plasmid DNA-unwinding assays and gel electrophoresis were complementary and supported the intercalative binding of the complex.104 For similar porphyrin systems, yet with a long protruding hydroxamic acid tail for external lanthanide coordination, Coutsolelos et al. observed different DNA-interaction modes depending on the nature of the DNA: intercalation for GC-rich DNAs, outside binding for AT-rich DNAs. They reported a positive correlation between the nuclease activity and the concentration and size of the lanthanide ion. Optimal conditions were found to correspond to a ratio of gadolinium/hydroxamic acid functionality of 2:1.363 A working model has been proposed (Fig. 64). In contrast, a negative correlation was found for interaction with RNAs, which was explained by the lanthanide interference with respect to the interaction between the porphyrin-hydroxamic acid derivatives and the open-chain RNAs.103 In 2017, Bağda et al. first studied the DNA-binding mechanism of a water-soluble LuIII double-decker phthalocyaninate, bearing bioactive quaternized 5-(3-pyridyl)-1,3,4-oxadiazole-2-thiol substituent groups, with calf thymus DNA; they found that the intercalative DNA binding process of such planar sandwich structure was highly favoured, being exoenergetic, and further favoured by an increase in viscosity.364
 | ||
| Fig. 64 Proposed model for the hydrolysis of a DNA ACG sequence by an intercalated porphyrin–hydroxamic acid–lanthanide ion adduct. Reproduced with permission from ref. 363, copyright (2001) World Scientific. | ||
6.4 Photosensitized singlet oxygen generation
Cancer has been the leading cause of human deaths for more than a century and research focusing on its treatment has always attracted huge attention. Radiotherapy and chemotherapy are the traditional methods for killing cancer cells effectively; however, their side effects can have deleterious effects on the patients. Therefore considerable efforts have been invested in designing efficient targeted cancer therapy while minimizing harmful side effects. Photodynamic therapy (PDT) has progressively become an attractive cornerstone for targeted cancer therapy, with tetrapyrrole-based photosensitisers being in the limelight.365–367 Indeed, cancer cells can be killed selectively through in situ photogeneration of cytotoxic reactive oxygen species (ROS). At the same time, specific targeting avoids undesired damage to normal cells. PDT proceeds in two main sequences: first, the administration of a photosensitizer into the patient's body and, second, the activation of this photosensitizer by non-thermal light of a specific wavelength, resulting in the production of singlet oxygen, which is a powerful oxidant causing serious cellular damages.7Laser is the frequently used light source for PDT as the light beam is coherent and monochromatic. It allows the light to be focused on a specific targeted area in order to bring less harm to the nearby tissue. Generally, lasers emitting in the near-infrared range are applied for PDT as NIR light has deeper tissue penetration ability compared to visible light, around 3 to 8 mm; in addition, shorter wavelength excitation may also result in strong light scattering and the strong absorption of light shorter than 580 nm by haemoglobin greatly interferes with in vivo treatments.6 There are two mechanisms for ROS generation (Fig. 65). In type I mechanism, radicals are produced through a series of electron-transfer processes. After activation of the photosensitizer by light, a mixture of oxygenated products is produced. Subsequent reactions between oxygen and the radicals produce ˙OH radicals and superoxide ˙O2−. In type II mechanism, singlet oxygen is produced from molecular oxygen (3O2) via a photochemical process. Since singlet oxygen has two valence electrons spin-paired in one π* orbital, it is more reactive than ground state molecular dioxygen.
 | ||
| Fig. 65 Type I and type II mechanisms of 1O2 generation. | ||
The photosensitizer determines the efficiency of PDT and several strategies can be used to enhance its efficiency: (i) introduction of heavy atoms such as halogens and metals in the proximity of the photosensitizer avoids the generation of free radicals and prevents interactions with the triplet excited state. (ii) Incorporation of a central metal into the photosensitizer prevents its aggregation; the latter results in a shorter lifetime of the triplet state which, in turn, reduces the 1O2 production. (iii) Expansion of the photosensitizer's conjugated system to increase its absorption efficiency at longer wavelengths for a better penetration depth in human tissue. (iv) Increasing the lipophilicity of the photosensitizer, by incorporating suitable functional groups, leads to better cellular uptake and improves its localization ability.
Since Sessler et al. reported that LuIII texaphyrin is an effective photosensitizer with an 1O2 quantum yield ΦΔ in water equal to 11%, and with high biostability and biocompatibility in photodynamic therapy,368–370 lanthanide–tetrapyrrole complexation systems have become attractive molecular platforms for singlet oxygen generation in vitro/in vivo. However, as discussed in Section 2, the intrinsic hydrophobicity renders water solubilization of most tetrapyrroles significantly challenging. In addition, there is still much room for improving the singlet oxygen generation efficiency, which is limited by their small molar absorption coefficients in the biological optical windows. New and smart molecular design strategies have recently emerged to overcome these bottlenecks.
The energies of the emissive 2F5/2 state of YbIII and of the T1 state of phthalocyanines are close so that YbIII emission is prevented due to back energy transfer, while 1O2 production is favoured, as seen from its phosphorescence in the NIR region. Taking this fact into consideration, Wong et al. developed potential one/two-photon photodynamic therapeutic agents and extended the π-conjugation of the tetrapyrrolic antenna ligand by developing acetylene bridged porphyrin–monophthalocyaninato YbIII hybrids (Fig. 16). The underlying reasons for such bridging are to (i) enlarge the two-photon absorption cross-sections (σ2) of the complexes by increasing the intramolecular charge-transfer and coherent structure domain and (ii) enhance the simultaneous 1O2 production with two conjugated photosensitizing moieties.108 The authors highlighted that the absorption spectra of the hybrids are not a superposition of the individual spectra of the porphyrin and the YbIII-phthalocyanine components. Singlet oxygen is efficiently produced in toluene upon one- or two-photon excitation either in the porphyrin Soret band or in the Q-band of the phthalocyanine: ΦΔ = 1.07–1.38, λex = 416 nm; ΦΔ = 0.73–0.79, λex = 675 nm. Hydrophilic modifications of the porphyrin chromophore to achieve better water solubility for the hybrids is highly feasible.
Apart from the π-conjugation extension strategy, Wong et al. continued to explore the impact of peripheral substitutions of their capped monophthalocyaninato YbIII complexes on the singlet oxygen quantum yields.371 In addition to the heavy-atom effect exerted by the lanthanide boosting the intersystem crossing (isc) from the ligand-centred singlet state to triplet state, the introduction of iodine and bulky 3,5-di-t-butylphenoxy substituents at the periphery further triggers isc and provides shielding from solvent-induced nonradiative quenching, leading to higher 1O2 yields (ΦΔ = 0.63–0.82 in toluene).
Considering the limited light penetration depth and irradiation efficiency of gadolinium–porphyrin complexes in biological medium, namely the weak Q band absorption at 500–600 nm, Zhang et al. proposed another π-conjugation modulation strategy to introduce a new type of macrocyclic tetrapyrrole-like antenna ligands—porpholactone derivatives (Fig. 66).68 In nature, macrocyclic pigments such as chlorins, bacteriochlorins, iso-bacteriochlorins and pyrrocorphins have all been biosynthesized by minor structural modifications to address various problems, water solubility, oxidation resistance and deep-red to NIR absorption tunability. Inspired by this, Zhang et al. replaced the porphyrin's β,β′-double bond with a lactone moiety, forming a series of porpholactones, cis- and trans-porphodilactones, and the corresponding capped GdIII complexes. Such replacement not only narrows the energy gap between the lowest triplet states of the ligand and the energy of the 1O2(1Δg → 3Σg transition) of 1O2, resulting in a higher yield of the reactive oxygen species, but also allows excitation in the visible to NIR spectral range. Quantum yields reach almost unity, ΦΔ = 0.76–0.99, in chloroform. To improve the water solubility of the new systems for further in vitro PDT studies, the authors glycosylated the four pentafluorophenyl rings at the methenyl bridges via nucleophilic aromatic substitution with 2,3,4,6-tetra-O-acetylglucosyl thioacetates, followed by removal of the acetyl protective group.
 | ||
| Fig. 66 Synthesis of Ln perfluorinated monoporphyrinates and porpholactone analogues for 1O2 production. (A) Typical EPR spectra of nitroxide radicals detected in DMSO solution containing TMPD (10 mm) and Gd–2 (0.01 mm), and irradiated (red) or not (black) with a mercury lamp (200 W, λ > 400 nm, 2 min) in the presence (or not) of NaN3 (10 mM). (B) Derived amount of 1O2 produced under the indicated conditions. (C) NIR emission of 1O2 at 1270 nm excited at 420 nm in CHCl3 (Inset: Relative intensity of 1O2 production versus absorption for Gd–1–4 and TPP at 420 nm). (D) 1O2 quantum yield measured upon excitation at 420 and 660 nm in CHCl3. Reproduced with permission from ref. 68, copyright (2016) Wiley-VCH Verlag GmbH & Co. | ||
In 2017, Cai et al. reported a water-soluble molecular dyad comprising two non-capped GdIII–sinoporphyrin sodium moieties, Gd–DVDMS; DVDMS is a porphyrin dimer combined with an ether bond isolated from Photofrin II and named sinoporphyrin by Fang.372 This system was proposed as a potential multifunctional theranostic agent, possibly accommodating two different metals.373 Gd–DVDMS exhibits large molar absorption coefficients because of the presence of two porphyrin rings and thus a reasonable 1O2 quantum yield (ΦΔ = 0.46 in methanol). The authors claimed another application of the dyad, namely phosphorescence-based oxygen sensing in PDT, by monitoring the intensity of the phosphorescence of the dyad at 712 nm, which is oxygen-sensitive.
More recently, in 2019, Wong et al. synthesized the first water-soluble and stable LnIII–porphyrin double-deckers, LnDD (Fig. 67), which have been a long-standing synthetic challenge. These double deckers displayed impressive NIR brightness and long lifetimes; more importantly, 1O2 generation was observed in aqueous solution, another long awaited property.374 At variance with their previous water-soluble polyethylene glycol (PEG) chain-conjugated, capped LnIII monoporphyrinates (YbN & GdN), the authors strategically fitted two optimally short hydrophilic methylated diethylene glycol chains to reach water solubility; moreover, the double-deckers sensitize efficiently the NIR luminescence of ytterbium by (i) having two antenna chromophores and (ii) minimizing the inner-sphere quenching effect by expelling bound water molecules from the inner sphere. These resulted in an NIR emission quantum yield of 2.8% for YbDD in water, slightly larger than that of YbN (ΦNIR = 2.7%); the singlet oxygen quantum yield of 46% in water was also better than that of YbN (ΦΔ = 42%). Although the 1O2 generation efficiency of GdDD is smaller than those of the FDA-approved commercially available photosensitizing agents Photofrin® (89%) and Levulan® (56%) (Fig. 68), GdDD has a much higher molar absorption coefficient, 223872 M−1 cm−1 at 412 nm and 52480 M−1 cm−1 at 580 nm compared to 3000 M−1 cm−1 at 632 nm for Photofrin® and 5000 M−1 cm−1 at 632 nm for Levulan®. More importantly, GdDD displayed a higher photodynamic therapeutic index against HeLa cells over the normal cells and performed similarly well under both normoxic and hypoxic conditions.
 | ||
| Fig. 67 (A) Structure of LnDD; (B) near-infrared 1O2 phosphorescence spectrum sensitized by GdDD, GdN, and the standard tetraphenylporphyrin H2TPP (in CHCl3. Absorbance = 0.05 at the excitation wavelength of 425nm); (C) photo-/dark cytotoxicity of GdDD, ErDD and GdN towards HeLa cancer and normal MRC5 cells under 1 J cm−2 light irradiation (λex = 430 nm); MTT assays were carried out after incubation at 37 °C for 24 h; (D) fluorescence intensity of emission at 525 nm of singlet oxygen sensor green of different samples in normoxic and hypoxic conditions (Exc.: NIR laser, 808 nm; 2 W cm−2; 1 min). Reproduced with permission from ref. 374, copyright (2019) Nature Publishing Group. | ||
 | ||
| Fig. 68 Structures of (A) Photofrin® (Porfimer sodium, a mixture of porphyrin oligomers formed by ether and ester linkages) and (B) Levulan® (5-aminolevulinic acid, a biosynthetic precursor to natural photosensitizer—protoporphyrin IX). | ||
6.5 Multimodal theranostics
As mentioned earlier, the past decade, from 2010 to 2020, can be landmarked as the second golden period of lanthanide–tetrapyrrole chemistry as many breakthroughs have been made, thanks to advances in synthetic chemistry: hydrophobic lanthanide–tetrapyrrole complexes have been strategically made water-soluble, biostable and biocompatible via novel tetrapyrrole ligand design such as peripherical perfluorination/lactonization, expanded/fused porphyrins, and PEGylation. Moreover, advanced synthesis allowed side-chain ligation and multiple-decker assembly. Such fundamental achievements pave the ways for further translational biomedical applications for these macrocyclic complexes, for instance, upon integrating their imaging capability with photodynamic therapy functionality—as novel multifunctional theranostic molecular platforms.In 2008, Liu and Wong et al. documented the first in vitro example of a combined agent for photodynamic therapy and NIR tumour-imaging based on an YbIII amphiphilic bis(porphyrin) complex.375 Porphyrin derivatives are likely to accumulate in tumours lacking lymphatic drainage due to their high hydrophobicity, vascular permeability and affinity towards proliferating endothelium/low-density lipoproteins. The authors took advantage of this passive bio-targeting/cellular uptake property by augmenting the molecular weight and hydrophobicity of the overall system, that is, connecting one more cationic porphyrin moiety to the YbIII monoporphyrinate complex, which resulted in a tumour-selective dual NIR-PDT functional probe. It should however be noted that the greater the hydrophobicity, the higher the cellular uptake efficiency, but the lower the efficiency of singlet oxygen generation, due to self-aggregation, and vice versa.
Later, Che et al. described a series of monomeric YbIII porphyrin complexes exhibiting potential anti-cancer activity, with IC50 values in the sub-micromolar range in DMSO/water.134 The best performing complex was an OEP complex which exists as a μ-OH dimer in CH2Cl2, {[Yb(OEP)]2(μ-OH)2}, and as a monomeric species in aqueous DMSO, [Yb(OEP)(DMSO)(H2O)(OH)]. The authors conducted comprehensive studies using transcriptomics, bioinformatics connectivity map analysis and biochemical experiments, and confirmed that the cancer-killing mechanism of this complex could be ascribed to the endoplasmic reticulum stress-induced apoptosis pathway. This is different from other anti-cancer non-macrocyclic lanthanide complexes targeting and photocleaving cellular DNA.
The same year, in continuation of the previous work on Yb–RhB as a monofunctional NIR imaging agent,70 Wong et al. replaced YbIII with ErIII and developed a new water-soluble, mitochondria-specific and one/two-photon-excitable porphyrin complex as a dual theranostic probe—NIR imaging and PDT (Fig. 23).120 Indeed, the energy difference between the porphyrin triplet state and the YbIII(2F5/2) state is small so that energy transfer is highly efficient, resulting in a poor singlet oxygen production (ΦΔ = 2% in chloroform). On the contrary, the larger energy gap between the porphyrin triplet state and the much lower ErIII(4I13/2) emissive level makes part of the excitation energy available for 1O2 production (ΦΔ = 10% in chloroform). Such an autofluorescence-suppressed optically traceable mild 1O2 generator selectively accumulated in the mitochondria of the cancer cells in vitro and enabled real-time longitudinal monitoring of the whole process for the first time.
A further improvement was disclosed by the same authors who presented a GdIII monoporphyrinate fitted with a short DEG ammonium chain (Gd–N) as the next-generation blueprint of selective in vivo cancer-tracking and a two-photon photodynamic therapeutic agent (Fig. 69).121 The complex recognises tumour cells by their anionic phosphatidylserine membrane. It also features a large 1O2 production with ΦΔ = 51% in chloroform upon excitation in the Soret band, i.e., comparable to the yield of H2TPP, in view of its highly energetic 6P7/2 emissive level, ∼32000 cm−1 compared with 23200, 15300 (singlets), and 12500 (triplet) cm−1 for the porphyrin ligand. Importantly, the PDT effect can be generated by 2-photon excitation in the NIR region (860 nm), which also results in exploitable NIR emission from the porphyrin for cell imaging. Gd–N-based photo-treatment at 860 nm not only efficaciously inhibits the growth of solid tumour but also reduces it by one half after 24 h in xenografted nude mice. A final advantage of this agent is its long residence time in the mice, around 8–9 h. The authors then continued to perform SILAC-based quantitative proteomics analysis and concluded that the newly developed Gd–N PDT led to (i) dysregulation of the expression of 485 proteins, phosphorylation of 106 proteins, and oxidation of 1050 proteins; (ii) the lysosome was discovered to be chiefly affected, along with the downregulation of most lysosomal acid hydrolases and the proton pump complex ATP6V/TCIRG1, where new insights into the cellular mechanism to oxidative stress and PDT effect were provided.124
 | ||
| Fig. 69 Left: (A) The molecular structure of the smart cancer specific photodynamic therapy agent (Gd–N) and their control analogues Yb–N and Gd–RhB. (B) The 3D in vitro imaging of Gd–N after 15 h incubation in HeLa cells. (C and D) The difference in subcellular localization of Gd–N in cancer cells (HeLa) and normal cells (WPMY-1). Right: In vivo studies of Gd–N as the cancer cell-specific PDT agent. (E) The representative gross images of tumours after PDT under 860 nm laser irradiation for four groups administered with different complexes: group 1: Yb–N; group 2: Gd–N; group 3: Yb–RhB; group 4: Gd–RhB. (F) Tumour volume in A. (G) In vivo biodistribution of Gd–N via ICP-MS studies. (H) Two-photon microscopic images of tumour samples in C. (I) In vivo tumour inhibition assays of Gd–N. (J) In vivo tumour inhibition via Gd–N and Gd–RhB–induced 1O2 through caudal vein injection. Reproduced with permission from ref. 121, copyright (2019) National Academy of Sciences of the United States of America. | ||
In a search for ultimate precision multimodal PDT treatment not only selectively targeting cancer cells over the normal cells, but accurately one cancer type over the others, Wong et al. proposed specifically modified lanthanide porphyrin complexes as in vitro NIR-PDT and in vivo NIR-MRI-PDT theranostic agents, respectively, taking bladder cancer as an example (Fig. 70).125,126 Various bladder cancer-specific molecules were developed via a combinatorial chemistry approach by targeting the integrin αvβ3 isoform, which is overexpressed in the neovasculature of bladder cancers; the peptides were conjugated to the previously developed porphyrin platforms. In addition to specificity for bladder cancer, this ensured enough water solubility and cell membrane permeability. Finally, a small hydrophilic peptide RrRK sequence was also conjugated to the hydrophobic bladder cancer-specific peptide sequences to make them amphiphilic. As previously demonstrated, singlet oxygen production varied depending on the lanthanide ion: GdIII (for MRI-PDT) ≫ ErIII (for NIR-PDT) ≫ YbIII (for NIR). Furthermore, for the in vivo GdIII agents, one complex, Gd-PEG-R3, featured (i) high photo-cytotoxicity (IC50 = 8.2 μM) against T24 bladder cancer cells while remaining non-cytotoxic in the dark (PTI = 199.0, 20-fold higher than 5-aminolevulinic acid) and in normal and HeLa cells; (ii) “off–on” responsive T1 relaxivity enhancement over 17 times upon αvβ3 binding; and (iii) a safe tumour-centred biodistribution profile without breaking the blood–brain barrier. This work substantiates the huge potential of lanthanide–tetrapyrrole systems for further clinical exploration.
 | ||
| Fig. 70 Left: (A) molecular structures of Gd-PEG-Rn (n = 1–3). (B) Displacement percentages of the vitronectin – αvβ3 integrin net binding at different concentrations of Gd-PEG-Rn. (C) Stability of 200 μM Gd-PEG-R3 in pH 7 and pH 5 PBS buffer solutions. Right: In vivo T1-weighted MR images of the mouse T24 tumour model after injection of (D) GdDOTA, (E) Gd-PEG-COOH, and (F) Gd-PEG-R3 at different time points. (G) In vivo biodistribution of Gd-PEG-Rn (n = 1–3) via ICP-MS studies, 3 samples in each group. Reproduced with permission from ref. 126, copyright (2019) Wiley VCH Verlag GmbH. | ||
7 Conclusion and outlook
The past decades have witnessed tremendous progress in the lanthanide–tetrapyrrole systems, and their unique properties and functionalities have been exploited in a range of applications from a wide spectrum of disciplines, from optical materials to biomedical agents277 and photocatalysis.376 Advanced synthetic and functionalization methodologies have been developed and exploited to realize various strategies for (1) conjugation extension in porphyrin/phthalocyanine rings for fine-tuning of their HOMO–LUMO energy gap, (2) modular lanthanide coordination and symmetry tailoring, (3) directed multiple-decker assembly with different twist angles, (4) better photosensitization for efficient Vis/NIR emission with high quantum yields and brightness, (5) improved solution-processability in organic solvents and water solubilization, and (6) enhanced biocompatibility, to impart lanthanide–tetrapyrrole complexes with novel biochemical properties, as well as for sensing, bio-targeting and multimodal theranostic applications. In this review, we briefly introduced the long and rich history of different lanthanide porphyrinates and phthalocyaninates, highlighting their syntheses and structure–property relationship, and surveying their applications in contemporary photonic, sensing and biomedical research. Despite the efforts and breakthroughs in this field in the past 20 years, there are still some challenges and opportunities lying ahead that we describe below.7.1 Hybrid organic–inorganic complexation to photosensitize less-explored luminescent lanthanides
NdIII and SmIII are luminescent lanthanide ions that are seldom employed with tetrapyrrolic ligands because of intrinsically weak quantum yields partly due to energy mismatch between the ligand donor states and the lanthanide excited states. Recently, however, organic–inorganic hybrid acetate-coordinated SmIII-based polyoxometalates have been reported with excitation-dependent reversible colour-tuneable photoluminescence.377 This approach enables reversible colour switching in the same material without adjusting the doping concentration of LnIII ions. On the other hand, Kögerler et al. disclosed new thermodynamically highly stable phthalocyanine–polyoxotungstate lanthanide double deckers, in which the redox-active polyoxometalates serve as versatile polydentate ligands;378 multiple decker-like phthalocyaninato lanthanide-ligated polyoxovanadate cages {[V12O32(Cl)](YbPc)x}n−5 (x = 1 or 2) have also been synthesized and characterized.379 Given the cornucopias of polyoxometalate structures, we believe that developing various functional lanthanide–tetrapyrrole–polyoxometalates with improved optical and redox properties and optimizing them for unparalleled photonic and biomedical applications will become an attractive research direction in this field.7.2 Photocatalytic transformations with lanthanide–tetrapyrrole complexes
Lanthanide–tetrapyrrole complexes, in particular phthalocyaninato lanthanide sandwiches, have been sporadically employed in photocatalysis, e.g., (i) photocatalytic degradation of 4-nitrophenol and 2-propanol upon impregnation onto TiO2,380,381 (ii) oxidative desulfurization of thiophene upon bonding on the surface of ZMS-5,382 and (iii) oxidative C–H bond functionalization of secondary or tertiary amines and oxygenation of cholesterol.68 In organic asymmetric synthesis, the concept of photoredox catalysis, operating through single-electron transfer, energy transfer and radical processes, has long become a research hotspot and gives rise to unprecedented reactivities and products.383 Given that eye-catching photocatalyst applications of lanthanides have recently come under the limelight,384,385 and that lanthanide–tetrapyrrole complexes possess unique photophysical and redox properties, we believe that new photocatalytic potentials of lanthanide–tetrapyrrole systems will soon be unleashed. One aspect is asymmetric synthesis, which is currently and actively pursued in our laboratory; on the other hand, further real-life photocatalytic applications of lanthanide–tetrapyrroles for environmental organic waste treatment and biomass conversion are also foreseeable.7.3 Towards peptide/aptamer-guided drug delivery and deep-tissue photoactivation of lanthanide–tetrapyrrole theranostic probes
To address the long-standing translational challenges of (i) poor water solubility, (ii) nonspecific toxicity, and (iii) lack of cancer selectivity of most lanthanide–tetrapyrrole-based PDT agents, different in vivo targeted delivery strategies have to be explored. For instance, both specific peptides and aptamers, which have been extensively surface-functionalized in nanomedicine for cancer targeting, can be conjugated similarly to the Ln–tetrapyrrole systems by optimization of their biostabilities and bioactivities. The systems can also be loaded onto proteins386 and attached to polymer carriers functionalized with specific vectors,387 or inserted into nanomaterials sensitive to tumour microenvironments (e.g., pH, presence of glutathione, etc.) and stimuli-activatable (e.g., through photoirradiation, ultrasound, etc.).388,389 This enables achieving safer and more specific targeting for multimodal imaging. Furthermore, X-ray and two-photon excitation can promisingly allow deep-tissue photodynamic therapy upon coupling to nanoscintillators,390 and this direction is under-explored with lanthanide–tetrapyrrole complexes. Hence, new multifunctional bioapplications of smart and biocompatible lanthanide–tetrapyrrole systems are conceivable.As a final remark, we would like to point out that developments in the field will need to be built on precise knowledge of all the crucial parameters relevant to each application. For instance, reports demonstrating the usefulness of lanthanide–tetrapyrrole complexes for bio-applications should not only describe their synthesis, structure, and imaging properties but also, and most crucially for biosafety, report their stability constants in vitro/in vivo. This has often been ignored, but is now being better acknowledged by the bioinorganic community. All photophysical parameters should also be quantitatively reported, interpreted and modelled. Providing such data in future work will give a more holistic view of the translational bioapplications of tetrapyrrole complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KLW acknowledges the support from Dr Mok Man Hung Endowed Professorship in Chemistry; this project was funded through grants RC-ICRS-18-19-01A from HKBU, RGC HKBU 12300318 and CAS-Croucher Funding Scheme for Joint Laboratories (CAS 18204) from Croucher Foundation. J.-C. G. B. thanks HKBU for a Dr Kennedy Wong Distinguished Visiting Professorship (2017–2019).References
- F. Hoppe-Seyler, Physiologische Chemie, A. Hirschwald Verlag, Berlin, 1877 Search PubMed.
- P. R. Ortiz de Montellano, Wiley Encycl. Chem. Biol., 2008, 1–10, DOI:10.1002/9780470048672.wecb221.
- S. Nardis, F. Mandoj, M. Stefanelli and R. Paolesse, Coord. Chem. Rev., 2019, 388, 360 CrossRef CAS.
- J. F. Longevial, S. Clément, J. A. Wytko, R. Ruppert, J. Weiss and S. Richeter, Chem. – Eur. J., 2018, 24, 15442 CrossRef CAS PubMed.
- X. D. Ren, X. Y. Hao, H. C. Li, M. R. Ke, B. Y. Zheng and J. D. Huang, Drug Discovery Today, 2018, 23, 1791 CrossRef CAS PubMed.
- S. B. Park, H. H. Jang, H. L. Lee, J. Kim, J. W. Nah, D. H. Kim, Y. I. Jeong and D. H. Kang, Bull. Korean Chem. Soc., 2019, 40, 439 CrossRef CAS.
- K. Yang, T. Niu, M. Luo, L. Tang and L. Kang, Photodiagn. Photodyn. Ther., 2018, 24, 332 CrossRef CAS PubMed.
- E. Campazzi, E. Solari, R. Scopelliti and C. Floriani, Chem. Commun., 1999, 1617 RSC.
- P. A. Gale, J. Anzenbacher and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57 CrossRef CAS.
- G. P. Moss, Eur. J. Biochem., 1988, 178, 277 CrossRef CAS PubMed.
- A. Braun and J. Tcherniac, Ber. Deutch. Chem. Ges., 1907, 40, 2709 CrossRef CAS.
- T. Posner, Ber. Deutch. Chem. Ges., 1897, 30, 1693 CrossRef CAS.
- J. K. F. van Staden, Talanta, 2015, 139, 75 CrossRef CAS PubMed.
- F. Mohn, L. Gross, N. Moll and G. Meyer, Nat. Nanotechnol., 2012, 7, 227 CrossRef CAS PubMed.
- C.-P. Wong, R. F. Venteicher and W. W. de Horrocks Jr., J. Am. Chem. Soc., 1974, 96, 7149 CrossRef CAS PubMed.
- T. F. Kachura, A. N. Sevchenko, K. N. Solov'ev and M. P. Tsvirko, Dokl. Acad. Nauk. SSSR, 1974, 217, 1121 CAS.
- J. W. Buchler, H. G. Kapellmann, M. Knoff, K. L. Lay and S. Pfeifer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1983, 38, 1339 CrossRef.
- J. W. Buchler, A. De Cian, J. Fischer, M. Kihn-Botulinski, H. Paulus and R. Weiss, J. Am. Chem. Soc., 1986, 108, 3652 CrossRef CAS.
- I. S. Kirin, P. N. Moskalev and Y. A. Makashev, Russ. J. Inorg. Chem., 1965, 10, 1065 Search PubMed.
- A. T. Chang and J.-C. Marchon, Inorg. Chim. Acta, 1981, 53, L241 CrossRef CAS.
- R. Weiss and J. Fischer, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Amsterdam, 2003, vol. 16, p. 171 Search PubMed.
- W. K. Wong, L. Zhang, F. Xue and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1999, 3053 RSC.
- W. K. Wong, L. L. Zhang, F. Xue and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 2000, 2245 RSC.
- X. Zhu, W.-K. Wong, J. P. Guo, W.-Y. Wong and J.-P. Zhang, Eur. J. Inorg. Chem., 2008, 3515 CrossRef CAS.
- Y. S. Korostei, V. G. Tarasova, V. E. Pushkarev, N. E. Borisova, A. K. Vorobiev and L. G. Tomilova, Dyes Pigm., 2018, 159, 573 CrossRef CAS.
- A. Y. Tolbin, V. E. Pushkarev, L. G. Tomilova and N. S. Zefirov, Mendeleev Commun., 2009, 19, 78 CrossRef CAS.
- Y. Horii, K. Katoh, N. Yasuda, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2015, 54, 3297 CrossRef CAS PubMed.
- F. T. Edelmann, New J. Chem., 2011, 35, 517 RSC.
- M. R. Sambrook and S. Notman, Chem. Soc. Rev., 2013, 42, 9251 RSC.
- C. Bazzicalupi, A. Bianchi, E. García-España and E. Delgado-Pinar, Inorg. Chim. Acta, 2014, 417, 3 CrossRef CAS.
- S. Comby and J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr., J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2007, ch. 235, vol. 37, p. 217 Search PubMed.
- V. Bulach, F. Sguerra and M. W. Hosseini, Coord. Chem. Rev., 2012, 256, 1468 CrossRef CAS.
- H. He, Coord. Chem. Rev., 2014, 273, 87 CrossRef.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100 CrossRef CAS PubMed.
- K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science, B.V., Amsterdam, 2013, ch. 254, vol. 43, p. 1 Search PubMed.
- S. Shinoda and H. Tsukube, Bunseki Kagaku, 2012, 61, 169 CrossRef CAS.
- O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550 RSC.
- M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. Ni Mhuircheartaigh, Adv. Mater., 2007, 19, 2737 CrossRef CAS.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press (Elsevier B.V.), Amsterdam, 1999 Search PubMed.
- W.-K. Wong, X. Zhu and W.-Y. Wong, Coord. Chem. Rev., 2007, 251, 2386 CrossRef CAS.
- D. K. P. Ng, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr., L. Eyring and G. H. Lander, Elsevier Science B.V., Amsterdam, 2001, ch. 210, vol. 32, p. 611 Search PubMed.
- J. Jiang and D. K. P. Ng, Acc. Chem. Res., 2008, 42, 79 CrossRef PubMed.
- P. Rothemund, J. Am. Chem. Soc., 1935, 57(10), 2010 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS.
- W.-K. Wong, L. Zhang, W.-T. Wong, F. Xue and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1999, 615 RSC.
- X.-B. Zhang, C.-C. Guo, J.-B. Xu, G.-L. Shen and R.-Q. Yu, Analyst, 2000, 125, 867 RSC.
- M. Asano-Someda and Y. Kaizu, J. Photochem. Photobiol., A, 2001, 139, 161 CrossRef CAS.
- H. Tsukube, M. Wada, S. Shinoda and H. Tamiaki, J. Alloys Compd., 2001, 323–324, 133 CrossRef CAS.
- W.-K. Wong, A. Hou, J. Guo, H. He, L. Zhang, W.-Y. Wong, K.-F. Li, K.-W. Cheah, F. Xue and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 2001, 3092 RSC.
- X.-J. Zhu, T. Zhang, S. Zhao, W.-K. Wong and W.-Y. Wong, Eur. J. Inorg. Chem., 2011, 3314 CrossRef CAS.
- H. He, W.-K. Wong, K.-F. Li and K.-W. Cheah, Synth. Met., 2004, 143, 81 CrossRef CAS.
- H. He, W.-K. Wong, J. Guo, K.-F. Li, W.-Y. Wong, W.-K. Lo and K.-W. Cheah, Inorg. Chim. Acta, 2004, 357, 4379 CrossRef CAS.
- H. He, J. Guo, Z. Zhao, W.-K. Wong, W.-Y. Wong, W.-K. Lo, K.-F. Li, L. Luo and K.-W. Cheah, Eur. J. Inorg. Chem., 2004, 837 CrossRef CAS.
- H. He, M. Dubey, A. G. Sykes and P. S. May, Dalton Trans., 2010, 39, 6466 RSC.
- J.-F. Zhang, H. Wong, A.-X. Hou, C.-F. Wang and H.-S. Zhang, J. Sep. Sci., 2004, 27, 1037 CrossRef CAS PubMed.
- J. S. Lindsey and R. W. Wagner, J. Org. Chem., 1989, 54, 828 CrossRef CAS.
- S. Fu, G. Zhou, X. Zhu, C. Ye, W.-K. Wong and Z. Li, Chem. Lett., 2006, 35, 802 CrossRef CAS.
- C.-W. Huang, K. Y. Chiu and S.-H. Cheng, Dalton Trans., 2005, 2417 RSC.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300 CrossRef CAS PubMed.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed.
- A. K. Burrell, D. L. Officer, P. G. Plieger and D. C. W. Reid, Chem. Rev., 2001, 101, 2751 CrossRef CAS PubMed.
- R. P. Linstead, J. Chem. Soc., 1934, 1016 RSC.
- J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281 CrossRef CAS PubMed.
- C.-P. Wong, R. F. Venteicher and W. D. Horrocks Jr., J. Am. Chem. Soc., 1974, 96, 7149 CrossRef CAS PubMed.
- X.-S. Ke, B.-Y. Yang, X. Cheng, S. L.-F. Chan and J.-L. Zhang, Chem. – Eur. J., 2014, 20, 4324 CrossRef CAS PubMed.
- X.-S. Ke, Y. Ning, J. Tang, J.-Y. Hu, H.-Y. Yin, G.-X. Wang, Z.-S. Yang, J. Jie, K. Liu, Z.-S. Meng, Z. Zhang, H. Su, C. Shu and J.-L. Zhang, Chem. – Eur. J., 2016, 22, 9676 CrossRef CAS PubMed.
- J. X. Meng, K. F. Li, J. Yuan, L. L. Zhang, W. K. Wong and K. W. Cheah, Chem. Phys. Lett., 2000, 332, 313 CrossRef CAS.
- T. Zhang, X. Zhu, C. C. W. Cheng, W.-M. Kwok, H.-L. Tam, J. Hao, D. W. J. Kwong, W.-K. Wong and K.-L. Wong, J. Am. Chem. Soc., 2011, 133, 20120 CrossRef CAS PubMed.
- T. J. Foley, K. A. Abboud and J. M. Boncella, Inorg. Chem., 2002, 41, 1704 CrossRef CAS PubMed.
- T. J. Foley, B. S. Harrison, A. S. Knefely, K. A. Abboud, J. R. Reynolds, K. S. Schanze and J. M. Boncella, Inorg. Chem., 2003, 42, 5023 CrossRef CAS PubMed.
- A. Santria, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Inorg. Chem., 2017, 56, 10625 CrossRef CAS PubMed.
- A. Santria, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Dalton Trans., 2019, 48, 7685 RSC.
- H. He and A. G. Sykes, Inorg. Chem. Commun., 2008, 11, 1304 CrossRef CAS.
- M. G. Gurevich and K. N. Solov'ev, Dokl. Acad. Nauk. SSSR, 1961, 5, 291–294 ( Chem. Abstr. , 1962 , 57 , 15948 ) CAS.
- H. Sugimoto, T. Higashi, A. Maeda, M. Mori, H. Masuda and T. Taga, J. Chem. Soc., Chem. Commun., 1983, 1234 RSC.
- A. De Cian, M. Moussavi, J. Fischer and R. Weiss, Inorg. Chem., 1985, 24, 3162 CrossRef CAS.
- H. Ke, W.-K. Wong, W.-Y. Wong, H.-L. Tam, C.-T. Poon and F. Jiang, Eur. J. Inorg. Chem., 2009, 1243 CrossRef CAS.
- S. A. Znoiko, O. N. Zubkova, A. V. Borisov, V. E. Maizlish and G. P. Shaposhnikov, Russ. J. Gen. Chem., 2015, 85, 2642 CrossRef CAS.
- A. D. Kosov, T. V. Dubinina, N. E. Borisova, A. V. Ivanov, K. A. Drozdov, S. A. Trashin, K. De Wael, M. S. Kotova and L. G. Tomilova, New J. Chem., 2019, 43, 3153 RSC.
- Q. Fan, J.-N. Luy, M. Liebold, K. Greulich, M. Zugermeier, J. Sundermeyer, R. Tonner and J. M. Gottfried, Nat. Commun., 2019, 10, 5049 CrossRef PubMed.
- R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728 CrossRef CAS PubMed.
- Z. X. Zhao, W. Liu, M. Jin and G. F. Liu, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 1747 CrossRef.
- Z.-X. Zhao, Q.-H. Xu, D.-M. Li, G.-F. Liu, L.-S. Li and R.-R. Xu, Solid State Sci., 2001, 3, 339 CrossRef CAS.
- Z. X. Zhao, T. F. Xie, D. M. Li, D. J. Wang and G. F. Liu, Synth. Met., 2001, 123, 33 CrossRef CAS.
- Z. X. Zhao and G. F. Liu, Synth. React. Inorg. Met.-Org. Chem., 2002, 32, 465 CrossRef CAS.
- M. Yu, G. Liu, Y. Cheng and W. Xu, Liq. Cryst., 2005, 32, 771 CrossRef CAS.
- J. S. Lindsey, K. A. MacCrum, J. S. Tyhonas and Y. Y. Chuang, J. Org. Chem., 1994, 59, 579 CrossRef CAS.
- K. Wada, T. Mizutani and S. Kitagawa, J. Org. Chem., 2003, 68, 5123 CrossRef CAS PubMed.
- T. P. Wijesekera, J. B. Paine III and D. Dolphin, J. Org. Chem., 1985, 50, 3832 CrossRef CAS.
- C.-H. Lee and L. S. Lindsey, Tetrahedron, 1994, 50, 11427 CrossRef CAS.
- E.-J. Kim, P. Kim, C.-H. Lee, J. Sung, H. Yoon, D. Kim and W.-D. Jang, Chem. Commun., 2012, 48, 5611 RSC.
- C.-H. Lee, S. Lee, H. Yoon and W.-D. Jang, Chem. – Eur. J., 2011, 17, 13898 CrossRef CAS PubMed.
- Y. J. Zhang, J. Shi, W. Liu and M. Yu, Synth. React. Inorg. Met.-Org. Chem., 2013, 43, 640 CrossRef CAS.
- C. Piechocki, J. Simon, J.-J. André, D. Guillon, P. Petit, A. Skoulios and P. Weber, Chem. Phys. Lett., 1985, 122, 124 CrossRef CAS.
- Z. Zhao and G. Liu, Liq. Cryst., 2002, 29, 1335 CrossRef CAS.
- T. Wijesekera, A. Matsumoto, D. Dolphin and D. Lexa, Angew. Chem., Int. Ed. Engl., 1990, 29, 1028 CrossRef.
- G. A. Spyroulias, A. P. Despotopoulos, C. P. Raptopoulou, A. Terzis, D. de Montauzon, R. Poilblanc and A. G. Coutsolelos, Inorg. Chem., 2002, 41, 2648 CrossRef CAS PubMed.
- J.-Y. Hu, Y. Ning, Y.-S. Meng, J. Zhang, Z.-Y. Wu, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 2702 RSC.
- J. Itoh, J. Liu and M. Komata, Talanta, 2006, 69, 61 CrossRef CAS PubMed.
- M.-C. Chalbot, L. Gryllos, K. Kefokeris, N. Manoussakis, C. Verchère-Béaur, M. Perrée-Fauvet and A. G. Coutsolelos, J. Porphyrins Phthalocyanines, 2011, 15, 704 CrossRef CAS.
- M. Marketaki, E. Touloupakis, G. Charalambidis, M.-C. Chalbot, D. F. Ghanotakis and A. G. Coutsolelos, J. Porphyrins Phthalocyanines, 2012, 16, 997 CrossRef CAS.
- X.-J. Zhu, P. Wang, H. W. C. Leung, W.-K. Wong, W.-Y. Wong and D. W. J. Kwong, Chem. – Eur. J., 2011, 17, 7041 CrossRef CAS PubMed.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742 RSC.
- Y. Ning, S. Cheng, J.-X. Wang, Y.-W. Liu, W. Feng, F. Li and J.-L. Zhang, Chem. Sci., 2019, 10, 4227 RSC.
- F.-L. Jiang, W.-K. Wong, X.-J. Zhu, G.-J. Zhou, W.-Y. Wong, P.-L. Wu, H.-L. Tam, K.-W. Cheah, C. Ye and Y. Liu, Eur. J. Inorg. Chem., 2007, 3365 CrossRef CAS.
- H. Ke, W. Li, T. Zhang, X. Zhu, H.-L. Tam, A. Hou, D. W. J. Kwong and W.-K. Wong, Dalton Trans., 2012, 41, 4536 RSC.
- K. E. Sekhosana, E. Amuhaya and T. Nyokong, Polyhedron, 2015, 85, 347 CrossRef CAS.
- Y. Korovin, Z. Zhilina, N. Rusakova, V. Kuz’min, S. Vodzinsky and Y. Ishkov, J. Porphyrins Phthalocyanines, 2001, 5, 481 CrossRef CAS.
- V. Thanabal and V. Krishnan, J. Am. Chem. Soc., 1982, 104, 3643 CrossRef CAS.
- N. N. Semenishyn, N. V. Rusakova, A. V. Mazepa and Y. V. Korovin, Macroheterocycles, 2009, 2, 57 CrossRef CAS.
- N. Rusakova, N. Semenishyn and Y. Korovin, J. Porphyrins Phthalocyanines, 2010, 14, 166 CrossRef CAS.
- N. N. Semenishyn, S. S. Smola, N. P. Efryushina and N. V. Rusakova, Theor. Exp. Chem., 2015, 51, 224 CrossRef CAS.
- N. N. Semenishyn and N. V. Rusakova, Macroheterocycles, 2016, 9, 163 CrossRef CAS.
- H.-S. He, Z.-X. Zhao, W.-K. Wong, K.-F. Lai, J.-X. Meng and K.-W. Cheah, Dalton Trans., 2003, 980 RSC.
- F. Eckes, V. Bulach, A. Guenet, C. A. Strassert, L. De Cola and M. W. Hosseini, Chem. Commun., 2010, 46, 619 RSC.
- A. Guenet, F. Eckes, V. Bulach, C. A. Strassert, L. De Cola and M. W. Hosseini, ChemPhysChem, 2012, 13, 3163 CrossRef CAS PubMed.
- J. Lindsey, J. Org. Chem., 1980, 45, 5215 CrossRef CAS.
- T. Zhang, C.-F. Chan, J. Hao, G.-L. Law, W.-K. Wong and K.-L. Wong, RSC Adv., 2013, 3, 382 RSC.
- T. Zhang, R. Lan, C.-F. Chan, G.-L. Law, W.-K. Wong and K.-L. Wong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5492 CrossRef CAS PubMed.
- T. Zhang, C.-F. Chan, R. Lan, H. Li, N.-K. Mak, W.-K. Wong and K.-L. Wong, Chem. Commun., 2013, 49, 7252 RSC.
- T. Zhang, C.-F. Chan, R. Lan, W.-K. Wong and K.-L. Wong, Chem. – Eur. J., 2014, 20, 970 CrossRef CAS PubMed.
- D. Qi, Q. Wang, H. Li, T. Zhang, R. Lan, D. W. J. Kwong, W.-K. Wong, K.-L. Wong, S. Li and F. Lu, Mol. BioSyst., 2015, 11, 3059 RSC.
- Y. Zhou, C.-F. Chan, D. W. J. Kwong, G.-L. Law, S. Cobb, W.-K. Wong and K.-L. Wong, Chem. Commun., 2017, 53, 557 RSC.
- C. Xie, H.-F. Chau, J.-X. Zhang, S. Tong, L. Jiang, W.-Y. Fok, H.-L. Lung, S. Zha, R. Zou, J. Jiao, C.-F. Ng, P. Ma, J. Zhang, J. Lin, K. K. Shiu, J.-C. G. Bünzli, W.-K. Wong, N. J. Long, G.-L. Law and K.-L. Wong, Adv. Ther., 2019, 1900068 CrossRef CAS.
- G. Liu, T. Shi and X. Liu, Polyhedron, 1994, 13, 2255 CrossRef CAS.
- D.-M. Li, Z.-X. Zhao, S.-Q. Liu, G.-F. Liu, T.-S. Shi and X.-X. Liu, Synth. Commun., 2000, 30, 4017 CrossRef CAS.
- E. Hasegawa, J.-I. Nemoto, T. Kanayama and E. Tsuchida, Eur. Polym. J., 1978, 14, 123 CrossRef CAS.
- X. L. Cui, M. Yu and G. F. Liu, Synth. React. Inorg. Met.-Org. Chem., 2005, 35, 785 CrossRef CAS.
- X. L. Cui, G. F. Liu and M. Yu, J. Coord. Chem., 2006, 59, 1361 CrossRef CAS.
- X. Cui, X. Shi and S. Li, Adv. Mater. Res., 2011, 308–310, 643 CAS.
- T. S. Srivastava, Bioinorg. Chem., 1978, 8, 61 CrossRef CAS PubMed.
- W.-L. Kwong, R. W.-Y. Sun, C.-N. Lok, F.-M. Siu, S.-Y. Wong, K.-H. Low and C.-M. Che, Chem. Sci., 2013, 4, 747 RSC.
- J. P. Collman, R. R. Gagne, C. A. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 87, 1427 CrossRef PubMed.
- P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779 CrossRef.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767 CrossRef CAS.
- K.-H. Schumacher and B. Franck, Angew. Chem., Int. Ed. Engl., 1989, 28, 1243 CrossRef.
- P. J. Chmielewski, M. Siczek and L. Szterenberg, Inorg. Chem., 2011, 50, 6719 CrossRef CAS PubMed.
- J. M. Fisher, V. K. Kensy and G. R. Geier, III, J. Org. Chem., 2017, 82, 4429 CrossRef CAS PubMed.
- R. Li, A. D. Lammer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2014, 79, 4078 CrossRef CAS PubMed.
- Q. Li, C. Li, J. Kim, M. Ishida, X. Li, T. Gu, X. Liang, W. Zhu, H. Agren, D. Kim, H. Furuta and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5294 CrossRef CAS PubMed.
- T. D. Lash, A. D. Lammer and G. M. Ferrence, Angew. Chem., Int. Ed., 2011, 50, 9718 CrossRef CAS PubMed.
- G. R. Geier, Y. Ciringh, F. Li, D. M. Haynes and J. S. Lindsey, Org. Lett., 2000, 2, 1745 CrossRef CAS PubMed.
- J. L. Shaw, S. A. Garrison, E. A. Alemán, C. J. Ziegler and D. A. Modarelli, J. Org. Chem., 2004, 69, 7423 CrossRef CAS PubMed.
- X. Zhu, W.-K. Wong, W.-K. Lo and W.-Y. Wong, Chem. Commun., 2005, 1022 RSC.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584 CrossRef CAS PubMed.
- V. J. Bauer, D. L. J. Clive, D. Dolphin, J. B. Paine III, F. L. Harris, M. M. King, J. Loder, S. W. Chien Wang and R. B. Woodward, J. Am. Chem. Soc., 1983, 105, 6429 CrossRef CAS.
- J. L. Sessler, M. R. Johnson and V. Lynch, J. Org. Chem., 1987, 52, 4394 CrossRef CAS.
- J. L. Sessler, T. D. Mody, G. W. Hemmi and V. Lynch, Inorg. Chem., 1993, 32, 3175 CrossRef CAS.
- J. L. Sessler, T. Murai and G. Hemmi, Inorg. Chem., 1989, 28, 3390 CrossRef CAS.
- J. Lisowski, J. L. Sessler, V. Lynch and T. D. Mody, J. Am. Chem. Soc., 1995, 117, 2273 CrossRef CAS.
- J. T. Brewster II, H. Zafar, H. D. Root, G. D. Thiabaud and J. L. Sessler, Inorg. Chem., 2020, 59, 32 CrossRef PubMed.
- S. M. Meyer, M. C. Hoehner, V. Lynch and J. L. Sessler, J. Porphyrins Phthalocyanines, 1999, 3, 148 CrossRef CAS.
- J. L. Sessler, P. J. Melfi, E. Tomat, W. Callaway, M. T. Huggins, P. L. Gordon, D. Webster Keogh, R. W. Date, D. W. Bruce and B. Donnio, J. Alloys Compd., 2006, 418, 171 CrossRef CAS.
- J. Jiang, K. Machida and G. Adachi, Bull. Chem. Soc. Jpn., 1992, 65, 1990 CrossRef CAS.
- J. W. Buchler, A. De Cian, J. Fischer, M. Kihn-Botulinski and R. Weiss, Inorg. Chem., 1988, 27, 339 CrossRef CAS.
- J. W. Buchler, A. De Cian, J. Fischer, P. Hammerschmitt, J. Löffler, B. Scharbert and R. Weiss, Chem. Ber., 1989, 122, 2219 CrossRef CAS.
- D. Chabach, M. Tahiri, A. De Cian, J. Fischer, R. Weiss and M. El Malouli Bibout, J. Am. Chem. Soc., 1995, 117, 8548 CrossRef CAS.
- D. Chabach, A. De Cian, J. Fischer, R. Weiss and M. El Malouli Bibout, Angew. Chem., Int. Ed. Engl., 1996, 35, 898 CrossRef CAS.
- J. Jiang, R. L. C. Lau, T. W. D. Chan, T. C. W. Mak and D. K. P. Ng, Inorg. Chim. Acta, 1997, 255, 59 CrossRef CAS.
- M. Takeuchi, T. Imada and S. Shinkai, Angew. Chem., Int. Ed., 1998, 37, 2096 CrossRef CAS PubMed.
- H. Miwa, N. Kobayashi, K. Ban and K. Ohta, Bull. Chem. Soc. Jpn., 1999, 72, 2719 CrossRef CAS.
- J. Li, D. Gryko, R. B. Dabke, J. R. Diers, D. F. Bocian, W. G. Kuhr and J. S. Lindsey, J. Org. Chem., 2000, 65, 7379 CrossRef CAS PubMed.
- J. Jiang, R. C. W. Liu, T. C. W. Mak, T. W. D. Chan and D. K. P. Ng, Polyhedron, 1997, 16, 515 CrossRef CAS.
- J. Jiang, W. Liu, K. L. Cheng, K. W. Poon and D. K. P. Ng, Eur. J. Inorg. Chem., 2001, 413 CrossRef CAS.
- J. Jiang, Y. Z. Bian, F. Furuya, W. Liu, M. T. M. Choi, N. Kobayashi, H. W. Li, Q. C. Yang, T. C. W. Mak and D. K. P. Ng, Chem. – Eur. J., 2001, 7, 5059 CrossRef CAS.
- J. W. Buchler, P. Hammerschmitt, I. Kaufeld and J. Löffler, Chem. Ber., 1991, 124, 2151 CrossRef CAS.
- W. Buchler, J. Huttermann and J. Löffler, Bull. Chem. Soc. Jpn., 1988, 61, 71 CrossRef.
- M. O. Liu and A. T. Hu, J. Organomet. Chem., 2004, 689, 2450 CrossRef CAS.
- H. G. Jin, X. Jiang, I. A. Kuhne, S. Clair, V. Monnier, C. Chendo, G. Novitchi, A. K. Powell, K. M. Kadish and T. S. Balaban, Inorg. Chem., 2017, 56, 4864 CrossRef CAS PubMed.
- S. Alpugan, Ü. İşci, F. Albrieux, C. Hirel, A. Gül Gürek, Y. Bretonnière, V. Ahsen and F. Dumoulin, Chem. Commun., 2014, 50, 7466 RSC.
- Y. Zhou, Y. Zhang, H. Wang, J. Jiang, Y. Bian, A. Muranaka and N. Kobayashi, Inorg. Chem., 2009, 48, 8925 CrossRef CAS PubMed.
- X. Wu, W. Lv, Q. Wang, H. Wang, X. Zhang and J. Jiang, Dalton Trans., 2011, 40, 107 RSC.
- Z. Valicsek, G. Eller and O. Horvath, Dalton Trans., 2012, 41, 13120 RSC.
- J. Kan, H. Wang, W. Sun, W. Cao, J. Tao and J. Jiang, Inorg. Chem., 2013, 52, 8505 CrossRef CAS PubMed.
- T. Nakai, K. Ban, K. Ohta and M. Kimura, J. Mater. Chem., 2002, 12, 844 RSC.
- K.-H. Schweikart, V. L. Malinovskii, J. R. Diers, A. A. Yasseri, D. F. Bocian, W. G. Kuhr and J. S. Lindsey, J. Mater. Chem., 2002, 12, 808 RSC.
- K. Padmaja, W. J. Youngblood, L. Wei, D. F. Bocian and J. S. Lindsey, Inorg. Chem., 2006, 45, 5479 CrossRef CAS PubMed.
- J. Otsuki, Y. Komatsu, D. Kobayashi, M. Asakawa and K. Miyake, J. Am. Chem. Soc., 2010, 132, 6870 CrossRef CAS PubMed.
- X. Zhang, Y. Li, D. Qi, J. Jiang, X. Yan and Y. Bian, J. Phys. Chem. B, 2010, 114, 13143 CrossRef CAS PubMed.
- N. Sheng, P. Zhu, C. Ma and J. Jiang, Dyes Pigm., 2009, 81, 91 CrossRef CAS.
- J. Lu, Y. Deng, X. Zhang, N. Kobayashi and J. Jiang, Inorg. Chem., 2011, 50, 2562 CrossRef CAS PubMed.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Magn. Reson. Chem., 2010, 48, 505 CrossRef CAS PubMed.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2012, 41, 9672 RSC.
- P. Zhu, X. Zhang, H. Wang, Y. Zhang, Y. Bian and J. Jiang, Inorg. Chem., 2012, 51, 5651 CrossRef CAS PubMed.
- N. Pan, Y. Bian, T. Fukuda, M. Yokoyama, R. Li, S. Neya, J. Jiang and N. Kobayashi, Inorg. Chem., 2004, 43, 8242 CrossRef CAS PubMed.
- V. E. Pushkarev, V. V. Kalashnikov, A. Y. Tolbin, S. A. Trashin, N. E. Borisova, S. V. Simonov, V. B. Rybakov, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2015, 44, 16553 RSC.
- W. Cao, H. Wang, X. Wang, H. K. Lee, D. K. Ng and J. Jiang, Inorg. Chem., 2012, 51, 9265 CrossRef CAS PubMed.
- K. P. Birin, I. A. Abdulaeva, A. I. Poddubnaya, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2020, 181, 108550 CrossRef CAS.
- Y. Gorbunova, L. A. Lapkina, A. G. Martynov, I. V. Biryukova and A. Y. Tsivadze, Russ. J. Coord. Chem., 2004, 30, 245 CrossRef CAS.
- A. A. Sinelshchikova, Y. G. Gorbunova, L. A. Lapkina, N. Y. Konstantinov and A. Y. Tsivadze, Russ. J. Inorg. Chem., 2011, 56, 1370 CrossRef CAS.
- A. G. Martynov, M. A. Polovkova, G. S. Berezhnoy, A. A. Sinelshchikova, F. M. Dolgushin, K. P. Birin, G. A. Kirakosyan, Y. G. Gorbunova and A. Y. Tsivadze, Inorg. Chem., 2020, 59, 9424 CrossRef CAS PubMed.
- D. González-Lucas, S. C. Soobrattee, D. L. Hughes, G. J. Tizzard, S. J. Coles and A. N. Cammidge, Chem. – Eur. J., 2020, 26, 10724 CrossRef PubMed.
- J. Jubb and S. Gambarotta, J. Am. Chem. Soc., 1994, 116, 4477 CrossRef CAS.
- E. Campazzi, E. Solari, C. Floriani and R. Scopelliti, Chem. Commun., 1998, 2603 RSC.
- E. Campazzi, E. Solari, R. Scopelliti and C. Floriani, Chem. Commun., 1999, 1617 RSC.
- H. He, X. Zhu, A. Hou, J. Guo, W.-K. Wong, W.-Y. Wong, K.-F. Li and K.-W. Cheah, Dalton Trans., 2004, 4064 RSC.
- J. J. Le Roy, J. Cremers, I. A. Thomlinson, M. Slota, W. K. Myers, P. H. Horton, S. J. Coles, H. L. Anderson and L. Bogani, Chem. Sci., 2018, 9, 8474 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, CrystEngComm, 2012, 14, 3001 RSC.
- S. George, S. Lipstman and I. Goldberg, Cryst. Growth Des., 2006, 6, 2651 CrossRef CAS.
- S. Muniappan, S. Lipstman, S. George and I. Goldberg, Inorg. Chem., 2007, 46, 5544 CrossRef CAS PubMed.
- S. Lipstman and I. Goldberg, Cryst. Growth Des., 2010, 10, 1823 CrossRef CAS.
- G. Nandi and I. Goldberg, Chem. Commun., 2014, 50, 13612 RSC.
- J. Demel, P. Kubat, F. Millange, J. Marrot, I. Cisarova and K. Lang, Inorg. Chem., 2013, 52, 2779 CrossRef CAS PubMed.
- Y.-P. Pei, J.-G. Huang, R.-H. Hu, Y.-X. Yang, J. Zhou and W.-T. Chen, J. Porphyrins Phthalocyanines, 2015, 19, 811 CrossRef CAS.
- X.-G. Yi, J.-G. Huang, R.-H. Hu, Z.-G. Luo, Y.-P. Pei and W.-T. Chen, J. Porphyrins Phthalocyanines, 2015, 19, 1072 CrossRef CAS.
- C. F. Pereira, F. Figueira, R. F. Mendes, J. Rocha, J. T. Hupp, O. K. Farha, M. M. Q. Simoes, J. P. C. Tome and F. A. A. Paz, Inorg. Chem., 2018, 57, 3855 CrossRef CAS PubMed.
- S. Lipstman and I. Goldberg, J. Mol. Struct., 2008, 890, 101 CrossRef CAS.
- L. Zhang, S. Yuan, L. Feng, B. Guo, J. S. Qin, B. Xu, C. Lollar, D. Sun and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5095 CrossRef CAS PubMed.
- J. B. Oh, K. L. Paik, J.-W. Ka, S.-G. Roh, M.-K. Nah and H. K. Kim, Mater. Sci. Eng., C, 2004, 24, 257 CrossRef.
- J. B. Oh, Y. H. Kim, M. K. Nah and H. K. Kim, J. Lumin., 2005, 111, 255 CrossRef CAS.
- R. Xiong, D. Mara, J. Liu, R. Van Deun and K. E. Borbas, J. Am. Chem. Soc., 2018, 140, 10975 CrossRef CAS PubMed.
- B. S. Harrison, T. J. Foley, A. S. Knefely, J. K. Mwaura, G. B. Cunningham, T.-S. Kang, M. Bouguettaya, J. M. Boncella, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2004, 16, 2938 CrossRef CAS.
- R. Pizzoferrato, L. Lagonigro, T. Ziller, A. Di Carlo, R. Paolesse, F. Mandoj, A. Ricci and C. Lo Sterzo, Chem. Phys., 2004, 300, 217 CrossRef CAS.
- E. G. Ermolina, R. T. Kuznetsova, T. A. Solodova, E. N. Tel'minov, T. N. Kopylova, G. V. Mayer, N. N. Semenishyn, N. V. Rusakova and Y. V. Korovin, Dyes Pigm., 2013, 97, 209 CrossRef CAS.
- G. A. Corker, B. Grant and N. J. Clecak, J. Electrochem. Soc., 1979, 126, 1339 CrossRef CAS.
- A. T. Chang and J.-C. Marchon, Inorg. Chim. Acta, 1981, 53, L241 CrossRef CAS.
- R. B. Daniels, G. L. Payne and J. Peterson, J. Coord. Chem., 1993, 28, 23 CrossRef CAS.
- P. M. S. Monk, R. J. Mortimer and D. A. Rosseinsky, Electrochromism and electrochromic devices, Cambridge University Press, Cambridge, 2007 Search PubMed.
- G. Guillaud, M. Al Sadoun, M. Maitrot, J. Simon and M. Bouvet, Chem. Phys. Lett., 1990, 167, 503 CrossRef CAS.
- A. Iwase and K. Tanaka, Electrochim. Acta, 1990, 35, 1707 CrossRef CAS.
- N. Nemykin, Y. Chernii and V. Volkov, J. Chem. Soc., Dalton Trans., 1998, 2995 RSC.
- M. M'Sadak, J. Roncali and F. Garnier, J. Electroanal. Chem. Interfacial Electrochem., 1985, 189, 99 CrossRef.
- R. B. Daniels, J. Peterson, W. C. Porter and Q. D. Wilson, J. Coord. Chem., 1993, 30, 357 CrossRef CAS.
- H. Konami, M. Hatano, N. Kobayashi and T. Osa, Chem. Phys. Lett., 1990, 165, 397 CrossRef CAS.
- I. P. Kalashnikova, I. V. Zhukov, L. G. Tomilova and N. S. Zefirov, Russ. Chem. Bull., 2005, 54, 2094 CrossRef CAS.
- M. L. Rodrìguez-Mendez, R. Aroca and J. A. Desaja, Chem. Mater., 1992, 4, 1017 CrossRef.
- Y. Liu, K. Shigehara, M. Hara and A. Yamada, J. Am. Chem. Soc., 1991, 113, 440 CrossRef CAS.
- I. V. Zhukov, V. E. Pushkarev, L. G. Tomilova and N. S. Zefirov, Russ. Chem. Bull., 2005, 54, 189 CrossRef CAS.
- V. E. Pushkarev, A. Y. Tolbin, N. E. Borisova, S. A. Trashin and L. G. Tomilova, Eur. J. Inorg. Chem., 2010, 5254 CrossRef CAS.
- I. V. Zhukov, L. A. Lapkina, Y. G. Gorbunova, V. E. Larchenko and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2005, 9, 1 CrossRef CAS.
- N. Sheng, R. Li, C.-F. Choi, W. Su, D. K. P. Ng, X. Cui, K. Yoshida, N. Kobayashi and J. Jiang, Inorg. Chem., 2006, 45, 3794 CrossRef CAS PubMed.
- M. N. Yarasir, M. Kandaz, A. Koca and B. Salih, J. Porphyrins Phthalocyanines, 2006, 10, 1022 CrossRef CAS.
- P. Zhu, F. Lu, N. Pan, D. Arnold, S. Zhang and J. Jiang, Eur. J. Inorg. Chem., 2004, 510 CrossRef CAS.
- R. Li, X. Zhang, P. Zhu, D. K. P. Ng, N. Kobayashi and J. Jiang, Inorg. Chem., 2006, 45, 2327 CrossRef CAS PubMed.
- X. Li, D. Qi, C. Chen, L. Yang, J. Sun, H. Wang, X. Li and Y. Bian, Dyes Pigm., 2014, 101, 179 CrossRef CAS.
- E. B. Orman, A. Koca, A. R. Özkaya, Ì. Gürol, M. Durmus and V. Ahsen, J. Electrochem. Soc., 2014, 161, H422 CrossRef CAS.
- T. V. Dubinina, K. V. Paramonova, S. A. Trashin, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2014, 43, 2799 RSC.
- R. Li, X. Zhang, N. Pan, P. Zhu, N. Kobayashi and J. Jiang, J. Porphyrins Phthalocyanines, 2005, 9, 40 CrossRef CAS.
- J. Jiang, W. Liu, K. W. Poon, D. Du, D. P. Arnold and D. K. P. Ng, Eur. J. Inorg. Chem., 2000, 205 CrossRef CAS.
- Y. Bian, J. Jiang, Y. Tao, M. T. M. Choi, R. Li, A. C. H. Ng, P. Zhu, N. Pan, X. Sun, D. P. Arnold, Z. Y. Zhou, H. W. Li, T. C. W. Mak and D. K. P. Ng, J. Am. Chem. Soc., 2003, 125, 12257 CrossRef CAS PubMed.
- S. L. Selector, V. V. Arslanov, Y. G. Gorbunova, O. A. Raitman, L. S. Sheinina, K. P. Birin and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2008, 12, 1154 CrossRef CAS.
- N. Ishikawa, J. Porphyrins Phthalocyanines, 2001, 5, 87 CrossRef CAS.
- T. H. Tran-Thi, T. A. Mattioli, D. Chabach, A. De Cian and R. Weiss, J. Phys. Chem., 1994, 98, 8279 CrossRef CAS.
- M. Tahiri, D. Chabach, M. Elmalouli-bibout, A. De Cian, J. Fischer and R. Weiss, Ann. Chim.-Sci. Mater., 1995, 20, 81 CAS.
- V. E. Pushkarev, V. V. Kalashnikov, S. A. Trashin, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2013, 42, 12083 RSC.
- H. Pan, L. Gong, W. Liu, C. Lin, Q. Ma, G. Lu, D. Qi, K. Wang and J. Jiang, Dyes Pigm., 2018, 156, 167 CrossRef CAS.
- H. L. Buckley, M. R. Anstey, D. T. Gryko and J. Arnold, Chem. Commun., 2013, 49, 3104 RSC.
- G. Lu, J. Li, S. Yan, W. Zhu, Z. Ou and K. M. Kadish, Inorg. Chem., 2015, 54, 5795 CrossRef CAS PubMed.
- G. Lu, J. Li, X. Jiang, Z. Ou and K. M. Kadish, Inorg. Chem., 2015, 54, 9211 CrossRef CAS PubMed.
- G. Lu, C. He, K. Wang, J. Sun, D. Qi, L. Gong, C. Wang, Z. Ou, S. Yan, S. Zeng and W. Zhu, Inorg. Chem., 2017, 56, 11503 CrossRef CAS PubMed.
- R. Wang, Y. Li, R. Li, D. Y. Y. Cheng, P. Zhu, D. K. P. Ng, M. Bao, X. Cui, N. Kobayashi and J. Jiang, Inorg. Chem., 2005, 44, 2114 CrossRef CAS PubMed.
- K. Yamashita, T. Yamanaka, N. Sakata and T. Ogawa, Chem. – Asian J., 2018, 13, 1692 CrossRef CAS PubMed.
- J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science, B.V., Amsterdam, 2016, ch. 287, vol. 50, p. 141 Search PubMed.
- S. Comby and J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr., J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2007, ch. 235, vol. 37, p. 217 Search PubMed.
- R. Wiglusz, J. Legendziewicz, A. Graczyk, S. Radzki, P. Gawryszewska and J. Sokolnicki, J. Alloys Compd., 2004, 380, 396 CrossRef CAS.
- G. E. Khalil, E. K. Thompson, M. Gouterman, J. B. Callis, L. R. Dalton, N. J. Turro and S. Jockusch, Chem. Phys. Lett., 2007, 435, 45 CrossRef CAS.
- W. Zheng, B.-B. Wang, J.-C. Lai, C.-Z. Wan, X.-R. Lu, C.-H. Li and X.-Z. You, J. Mater. Chem. C, 2015, 3, 3072 RSC.
- N. Kobayashi, Coord. Chem. Rev., 2002, 227, 129 CrossRef CAS.
- P. Zhu, N. Pan, R. Li, J. Dou, Y. Zhang, D. Y. Y. Cheng, D. Wang, D. K. P. Ng and J. Jiang, Chem. – Eur. J., 2005, 11, 1425 CrossRef CAS PubMed.
- D. V. Konarev, S. S. Khasanov, M. S. Batov, A. G. Martynov, I. V. Nefedova, Y. G. Gorbunova, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Inorg. Chem., 2019, 58, 5058 CrossRef CAS PubMed.
- M. M. Ayhan, A. Singh, E. Jeanneau, V. Ahsen, J. Zyss, I. Ledoux-Rak, A. G. Gürek, C. Hirel, Y. Bretonnière and C. Andraud, Inorg. Chem., 2014, 53, 4359 CrossRef CAS PubMed.
- F. Guyon, A. Pondaven, J. M. Kerbaol and M. L'Her, Inorg. Chem., 1998, 37, 569 CrossRef CAS PubMed.
- A. N. Carneiro Neto, E. E. S. Teotonio, G. F. de Sá, H. F. Brito, J. Legendziewicz, L. D. Carlos, M. C. Felinto, P. Gawryszewska, R. T. Moura, R. L. Longo, W. M. Faustino and O. L. Malta, in Handbook on the Physics and Chemistry of Rare Earths, Including Actinides, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier B.V., Amsterdam, 2019, ch. 310, vol. 56, p. 55 Search PubMed.
- K.-L. Wong, J.-C. G. Bünzli and P. A. Tanner, J. Lumin., 2020, 224, 117256 CrossRef CAS.
- H. Ishida, J.-C. G. Bünzli and A. Beeby, Pure Appl. Chem., 2016, 88, 701 CAS.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346 RSC.
- C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Güden-Silber, J. Lügger, C. Platas-Iglesias and M. Seitz, J. Am. Chem. Soc., 2012, 134, 16413 CrossRef CAS PubMed.
- Y. V. Korovin and N. V. Rusakova, Rev. Inorg. Chem., 2001, 21, 299 CAS.
- W.-K. Wong, X. Zhu and W.-Y. Wong, Coord. Chem. Rev., 2007, 251, 2386 CrossRef CAS.
- X. Zhu, W. K. Wong, W. Y. Wong and X. Yang, Eur. J. Inorg. Chem., 2011, 4651 CrossRef CAS.
- O. V. Snurnikova, A. P. Lukyanenko, E. A. Alekseeva, Y. V. Korovin and N. V. Rusakova, Macroheterocycles, 2011, 4, 93 CrossRef CAS.
- H. He, Coord. Chem. Rev., 2014, 273, 87 CrossRef.
- X. H. Chang, J. Zhang, L. H. Wu, Y. K. Peng, X. Y. Yang, X. L. Li, A. J. Ma, J. C. Ma and G. Q. Chen, Micromachines, 2019, 10, 422 CrossRef PubMed.
- X. X. Peng, X. F. Zhu and J.-L. Zhang, J. Inorg. Biochem., 2020, 209, 111118 CrossRef CAS PubMed.
- G. Q. Jin, Y. Ning, J.-X. Geng, Z.-F. Jiang, Y. Wang and J.-L. Zhang, Inorg. Chem. Front., 2020, 7, 289 RSC.
- Y. Ning, M. Zhu and J. L. Zhang, Coord. Chem. Rev., 2019, 399, 213028 CrossRef CAS.
- J. Y. Liew, J. J. Brown, E. G. Moore and M. Schwalbe, Chem. – Eur. J., 2016, 22, 16178 CrossRef CAS PubMed.
- H. He, P. S. May and D. Galipeau, Dalton Trans., 2009, 4766 RSC.
- H. He, W.-K. Wong, J. Guo, K.-F. Li, W.-Y. Wong, W.-K. Lo and K.-W. Cheah, Aust. J. Chem., 2004, 57, 803 CrossRef CAS.
- H. He, A. G. Sykes, P. S. May and G. He, Dalton Trans., 2009, 7454 RSC.
- N. V. Rusakova, Y. V. Korovin, Z. I. Zhilina, S. V. Vodzinskii and Y. V. Ishkov, J. Appl. Spectrosc., 2004, 506 CrossRef CAS.
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Inorg. Chem., 2009, 48, 7937 CrossRef CAS PubMed.
- S. Zhuravlyov, N. Rusakova and Y. Korovin, J. Alloys Compd., 2008, 451, 334 CrossRef CAS.
- N. N. Semenishyn, N. V. Rusakova, A. V. Mazepa and Y. V. Korovin, Macroheterocycles, 2009, 2, 57 CrossRef CAS.
- T. Zhang, X. Zhu, W.-K. Wong, H.-L. Tam and W.-Y. Wong, Chem. – Eur. J., 2013, 19, 739 CrossRef CAS PubMed.
- H. He, J. D. Bosonetta, K. A. Wheeler and S. P. May, Chem. Commun., 2017, 53, 10120 RSC.
- Y. Ning, S. Chen, H. Chen, J. X. Wang, S. He, Y. W. Liu, Z. Cheng and J.-L. Zhang, Inorg. Chem. Front., 2019, 6, 1962 RSC.
- X.-J. Zhu, F.-L. Jiang, C.-T. Poon, W.-K. Wong and W.-Y. Wong, Eur. J. Inorg. Chem., 2008, 3151 CrossRef CAS.
- R. Pizzoferrato, R. Francini, S. Pietrantoni, R. Paolesse, F. Mandoj, A. Monguzzi and F. Meinardi, J. Phys. Chem. A, 2010, 114, 4163 CrossRef CAS PubMed.
- Y. Ning, G. Q. Jin and J. L. Zhang, Acc. Chem. Res., 2019, 52, 2620 CrossRef CAS PubMed.
- J.-Y. Hu, Z.-Y. Wu, K. Chai, Z.-S. Yang, Y.-S. Meng, Y. Ning, J. Zhang and J.-L. Zhang, Inorg. Chem. Front., 2017, 4, 1539 RSC.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332 CrossRef CAS PubMed.
- Y. Ning, X.-S. Ke, J.-Y. Hu, Y.-W. Liu, F. Ma, H.-L. Sun and J.-L. Zhang, Inorg. Chem., 2017, 56, 1897 CrossRef CAS PubMed.
- S. Bo, J. Hu, Q. Wang, X. Liu and Z. Zhen, Photochem. Photobiol. Sci., 2008, 7, 474 CrossRef CAS PubMed.
- J. Hanuza, P. Godlewska, R. Lisiecki, W. Ryba-Romanowski, P. Kadlubanski, J. Lorenc, A. Lukowiak, L. Macalik, Y. Gerasymchuk and J. Legendziewicz, Spectrochim. Acta, Part A, 2018, 196, 202 CrossRef CAS PubMed.
- R. Zugle, C. Litwinski and T. Nyokong, Polyhedron, 2011, 30, 1612 CrossRef CAS.
- K. E. Sekhosana, E. Amuhaya, S. Khene and T. Nyokong, Inorg. Chim. Acta, 2015, 426, 221 CrossRef CAS.
- Y. Gerasymchuk, M. Guzik, R. Lisiecki, M. Sobczyk, J. Janski, A. Koll, G. Boulon and J. Legendziewicz, J. Lumin., 2018, 193, 84 CrossRef CAS.
- G. de la Torre, P. Vázquez, F. Aguillo-Lopez and T. Torres, Chem. Rev., 2004, 104, 3723 CrossRef CAS PubMed.
- R. L. Sutherland, D. G. McLean and S. Kirkpatrick, Handbook of Nonlinear Optics, Marcel Dekker, New York & Basel, 2003 Search PubMed.
- K. P. Unnikrishnan, J. Thomas, V. P. N. Nampoori and C. P. G. Vallabhan, Chem. Phys., 2002, 279, 209 CrossRef CAS.
- R. del Coso and J. Solis, J. Opt. Soc. Am. B, 2004, 21, 640 CrossRef CAS.
- R. A. Ganeev, A. I. Ryasnyansky, A. L. Stepanov and T. Usmanov, Phys. Status Solidi B, 2004, 241, 935 CrossRef CAS.
- N. Sheng, Z. Yuan, J. Wang, W. Chen, J. Sun and Y. Bian, Dyes Pigm., 2012, 95, 627 CrossRef CAS.
- K. P. Unnikrishnan, J. A. Y. A. Thomas, B. I. N. O. Paul, A. C. H. A. Kurian, P. R. A. M. Gopinath, V. P. N. Nampoori and C. P. G. Vallabhan, J. Nonlinear Opt. Phys. Mater., 2001, 10, 113 CrossRef CAS.
- K. E. Sekhosana, E. Amuhaya, J. Mack and T. Nyokong, J. Mater. Chem. C, 2014, 2, 5431 RSC.
- K. E. Sekhosana and T. Nyokong, Opt. Mater., 2014, 37, 139 CrossRef CAS.
- N. Sheng, D. Liu, B. Gu, J. He and Y. Cui, Dyes Pigm., 2015, 122, 346 CrossRef CAS.
- Z. Li, F. Gao, Z. Xiao, G. Ao, X. Wu, Y. Fang, Z. Nie, T. H. Wei, J. Yang, Y. Wang, X. Zhang, J. Zuo and Y. Song, Dyes Pigm., 2015, 119, 70 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Inorg. Chim. Acta, 2016, 450, 87 CrossRef CAS.
- B. Ren, N. Sheng, B. Gu, Y. Wan, G. Rui, C. Lv and Y. Cui, Dyes Pigm., 2017, 139, 788 CrossRef CAS.
- A. I. Plekhanov, T. V. Basova, R. G. Parkhomenko and A. G. Gürek, Opt. Mater., 2017, 64, 13 CrossRef CAS.
- Z. Li, F. Gao, Z. Xiao, X. Wu, J. Zuo and Y. Song, Opt. Laser Technol., 2018, 103, 42 CrossRef CAS.
- W. Huang, H. Xiang, Q. Gong, Y. Huang, C. Huang and J. Jiang, Chem. Phys. Lett., 2003, 374, 639 CrossRef CAS.
- A. Y. Tsivadze, G. V. Ionova and V. K. Mikhalko, Prot. Met. Phys. Chem. Surf., 2010, 46, 149 CrossRef CAS.
- K. E. Sekhosana, M. H. Manyeruke and T. Nyokong, J. Mol. Struct., 2016, 1121, 111 CrossRef CAS.
- N. Sheng, B. Gu, B. Ren, Y. Wang, L. Han, J. Wang, H. Cao, M. Guan, X. Zhai and J. Sha, Dyes Pigm., 2017, 136, 553 CrossRef CAS.
- S. M. O'Flaherty, S. V. Hold, M. J. Cook, T. Torres, Y. Chen, M. Hanack and W. J. Blau, Adv. Mater., 2003, 15, 19 CrossRef.
- A. B. Karpo, V. E. Pushkarev, V. I. Krasovskii and L. G. Tomilova, Chem. Phys. Lett., 2012, 554, 155 CrossRef CAS.
- K. E. Sekhosana, M. Shumba and T. Nyokong, Polyhedron, 2017, 138, 154 CrossRef CAS.
- K. E. Sekhosana, R. Nkhahle and T. Nyokong, ChemistrySelect, 2018, 3, 6671 CrossRef CAS.
- D. O. Oluwole, A. V. Yagodin, N. C. Mkhize, K. E. Sekhosana, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and T. Nyokong, Chem. – Eur. J., 2017, 23, 2820 CrossRef CAS PubMed.
- E. A. Kuzmina, T. V. Dubinina, A. V. Zasedatelev, A. V. Baranikov, M. I. Makedonskaya, T. B. Egorova and L. G. Tomilova, Polyhedron, 2017, 135, 41 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, J. Mol. Struct., 2016, 1117, 140 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Opt. Mater., 2015, 47, 211 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, RSC Adv., 2019, 934, 16223 RSC.
- Z.-S. Yang, Y. Ning, H.-Y. Yin and J.-L. Zhang, Inorg. Chem. Front., 2018, 5, 2291 RSC.
- G. Dyrda, M. Zakrzyk, M. A. Broda, T. Pedzinski, G. Mele and R. Slota, Molecules, 2020, 25, 3638 CrossRef CAS PubMed.
- K. Binnemans, Chem. Rev., 2002, 102, 2303 CrossRef CAS PubMed.
- B. Berno, A. Nazri and R. Aroca, Appl. Spectrosc., 1997, 51, 1525 CrossRef CAS.
- M. L. Rodrìguez-Mendez, Comments Inorg. Chem., 2000, 22, 227 CrossRef.
- X. Tang, Q. Liu, C. Wei, X. Lv, Z. Jin, Y. Chen and J. Jiang, J. Rare Earths, 2021, 39, 113 CrossRef CAS.
- N. Kılınç, D. Atilla, A. G. Gürek, Z. Z. Öztürk and V. Ahsen, Sens. Actuators, B, 2009, 142, 73 CrossRef.
- J. Gao, G. Lu, J. Kan, Y. Chen and M. Bouvet, Sens. Actuators, B, 2012, 166–167, 500 CrossRef CAS.
- K. Abdullah, Y. Wu, S. Zhao, X. Kong, X. Li and Y. Chen, Inorg. Chem. Commun., 2017, 86, 1 CrossRef CAS.
- Z. Dong, X. Kong, D. Qi, S. Zhao, X. Li, Y. Chen and J. Jiang, Org. Electron., 2018, 53, 127 CrossRef CAS.
- G. Lu, X. Kong, C. Wang, L. Zhao, D. Qi, Y. Jiang, S. Zhao, Y. Chen and J. Jiang, Dyes Pigm., 2019, 161, 240 CrossRef CAS.
- G. Lu, X. Kong, H. Wang, Y. Chen, C. Wei, Y. Chen and J. Jiang, New J. Chem., 2019, 43, 15763 RSC.
- Y. Chen, M. Bouvet, T. Sizun, Y. Gao, C. Plassard, E. Lesniewska and J. Jiang, Phys. Chem. Chem. Phys., 2010, 12, 12851 RSC.
- A. Yazıcıa, B. Sarıçiçek, A. Altındal, B. Salih and Ö. Bekaroğlu, Inorg. Chim. Acta, 2015, 428, 83 CrossRef.
- N. Gutierrez, M. L. Rodrìguez-Mendez and J. A. de Saja, Sens. Actuators, B, 2001, 77, 431 CrossRef.
- M. L. Rodrìguez-Mendez, Y. Gorbunova and J. A. de Saja, Langmuir, 2002, 18, 9560 CrossRef.
- E. Yabaş, M. Sülü, F. Dumludağ, B. Salih and Ö. Bekaroğlu, Polyhedron, 2018, 153, 51 CrossRef.
- M. Kandaza, A. T. Bilgicli and A. Altındal, Synth. Met., 2010, 160, 52 CrossRef.
- R. Boroujerdi, J. Fluoresc., 2016, 26, 781 CrossRef CAS PubMed.
- X. Chen, Y. Wang, X. Zhao, B. Liu, Y. Xu and Y. Wang, Microchim. Acta, 2019, 186, 63 CrossRef PubMed.
- A. Sugasaki, M. Ikeda, M. Takeuchi, A. Robertson and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1999, 3259 RSC.
- A. Sugasaki, M. Ikeda, M. Takeuchi, K. Koumoto and S. Shinkai, Tetrahedron, 2000, 56, 4717 CrossRef CAS.
- G. K. Chudinova, I. A. Nagovitsyn, R. E. Karpov and V. V. Savranskii, Quantum Electron., 2003, 33, 765 CrossRef CAS.
- T. Basova, I. Jushina, A. G. Gürek, V. Ahsen and A. K. Ray, J. R. Soc., Interface, 2008, 5, 801 CrossRef CAS PubMed.
- A. V. Lobanov, G. A. Gromova, Y. G. Gorbunova and A. Y. Tsivadze, Prot. Met. Phys. Chem. Surf., 2014, 50, 570 CrossRef CAS.
- K. E. Sekhosana, M. Shumba and T. Nyokong, ChemistrySelect, 2019, 4, 8434 CrossRef CAS.
- M. I. Guidak and V. V. Grigoryants, J. Photochem. Photobiol., B, 1990, 7, 15 CrossRef.
- J. L. Sessler, T. D. Mody, G. W. Hemmi, V. Lynch, S. W. Young and R. A. Miller, J. Am. Chem. Soc., 1993, 115, 10368 CrossRef CAS.
- S. W. Young, Q. Fan, A. Harriman, J. L. Sessler, W. C. Dow, T. D. Mody, G. Hemmi, Y. Hao and R. A. Miller, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6610 CrossRef CAS PubMed.
- Y. V. Korovin, N. V. Rusakova, Y. A. Popkov and V. P. Dotsenko, J. Appl. Spectrosc., 2002, 69, 841 CrossRef CAS.
- L. G. Klapshina, W. E. Douglas, I. S. Grigoryev, E. Y. Ladilina, M. V. Shirmanova, S. A. Mysyagin, I. V. Balalaeva and E. V. Zagaynova, Chem. Commun., 2010, 46, 8398 RSC.
- W. T. Morgan, A. Smith and P. Koskelo, Biochim. Biophys. Acta, 1980, 624, 271 CrossRef CAS.
- R. J. Fiel, J. Biomol. Struct. Dyn., 1989, 6, 1259 CrossRef CAS PubMed.
- P. Liu, Y. Liu, X. Li and W.-G. Huang, Chin. J. Chem., 2007, 25, 917 CrossRef CAS.
- M.-C. Chalbot, L. Gryllos, K. Kefokeris, N. Manoussakis, C. Verchère-Béaur, M. Perrée-Fauvet and A. G. Coutsolelos, J. Porphyrins Phthalocyanines, 2011, 15, 704 CrossRef CAS.
- E. Bağda, E. Yabaş and E. Bağda, Spectrochim. Acta, Part A, 2017, 172, 199 CrossRef PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041 RSC.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185 RSC.
- S. W. Young, K. W. Woodburn, M. Wright, T. D. Mody, Q. Fan, J. L. Sessler, W. C. Dow and R. A. Miller, Photochem. Photobiol., 1996, 63, 892 CrossRef CAS PubMed.
- J. L. Sessler, W. C. Dow, D. O’Connor, A. Harriman, G. Hemmi, T. D. Mody, R. A. Miller, F. Qing, S. Springs, K. Woodburn and S. T. Young, J. Alloys Compd., 1997, 249, 146 CrossRef CAS.
- L. I. Grossweiner, M. D. Bilgin, P. Berdusis and T. D. Mody, Photochem. Photobiol., 1999, 70, 138 CrossRef CAS.
- Q. Zhang, G. Cheng, H. Ke, X. Zhu, N. Zhu, W.-Y. Wong and W.-K. Wong, RSC Adv., 2015, 5, 22294 RSC.
- Q. C. Fang, Chin. J. New Drugs, 2014, 23, 1540 CAS.
- L. Zang, H. Zhao, J. Hua, F. Qin, Y. Zheng, Z. Zhang and W. Cao, Dyes Pigm., 2017, 142, 465 CrossRef CAS.
- J.-X. Zhang, W.-L. Chan, C. Xie, Y. Zhou, H.-F. Chau, P. Maity, G. T. Harrison, A. Amassian, O. F. Mohammed, P. A. Tanner, W.-K. Wong and K.-L. Wong, Light Sci. Appl., 2019, 8, 46 CrossRef PubMed.
- F.-L. Jiang, C.-T. Poon, W.-K. Wong, H.-K. Koon, N.-K. Mak, C. Y. Choi, D. W. J. Kwong and Y. Liu, ChemBioChem, 2008, 9, 1034 CrossRef CAS PubMed.
- Z. W. Jiang, Y. C. Zou, T. T. Zhao, S. J. Zhen, Y. F. Li and C. Z. Huang, Angew. Chem., Int. Ed., 2020, 59, 3300 CrossRef CAS PubMed.
- M. Zapata-Lizama, P. Hermosilla-Ibáñez, D. Venegas-Yazigi, G. Minguez Espallargas, L. J. Q. Maia, G. Gasparotto, R. C. D. Santana and W. Cañón-Mancisidor, Inorg. Chem. Front., 2020, 7, 3049 RSC.
- S. Sarwar, S. Sanz, J. van Leusen, G. S. Nichol, E. K. Brechin and P. Kögerler, Dalton Trans., 2020, 49, 16638 RSC.
- R. Pütt, X. Qiu, P. Kozłowski, H. Gildenast, O. Linnenberg, S. Zahn, R. C. Chiechi and K. Y. Monakhov, Chem. Commun., 2019, 55, 13554 RSC.
- G. Mele, E. Garcìa-Lòpez, L. Palmisano, G. Dyrda and R. Słota, J. Phys. Chem. C, 2007, 111, 6581 CrossRef CAS.
- G. Marcì, E. Garcìa-Lòpez, G. Mele, L. Palmisano, G. Dyrda and R. Słota, Catal. Today, 2009, 143, 203 CrossRef.
- Y. Zhang, D. Wang, R. Zhang, J. Zhao and Y. Zheng, Catal. Commun., 2012, 29, 21 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed.
- J. Ma, F. Schäfers, C. Daniliuc, K. Bergander, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 9639 CrossRef CAS PubMed.
- L. Zhou, T. Yang, J. Wang, Q. Wang, X. Lv, H. Ke, Z. Guo, J. Shen, Y. Wang, C. Xing and H. Chen, Theranostics, 2017, 7, 764 CrossRef CAS PubMed.
- B. Xiang, Y. Xue, Z. Liu, J. Tian, H. Frey, Y. Gao and W. Zhang, Polym. Chem., 2020, 11, 3913 RSC.
- C.-F. Chan, Y. Zhou, H. Guo, J. Zhang, L. Jiang, W. Chen, K.-K. Shiu, W.-K. Wong and K.-L. Wong, ChemPlusChem, 2016, 81, 535 CrossRef CAS PubMed.
- X.-F. Qiao, J.-C. Zhou, J.-W. Xiao, Y.-F. Wang, L.-D. Sun and C.-H. Yan, Nanoscale, 2012, 4, 4611 RSC.
- W. Sun, Z. Zhou, G. Pratx, X. Chen and H. Chen, Theranostics, 2020, 10, 1296 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |