
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromaticity reversals and their effect on bonding in the low-lying electronic states of cyclooctatetraene†
Peter B.
Karadakov
 * and
Nicholas
Preston
* and
Nicholas
Preston
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: peter.karadakov@york.ac.uk
First published on 22nd October 2021
Abstract
Aromaticity reversals and their effect on chemical bonding in the low-lying electronic states of cyclooctatetraene (COT) are investigated through a visual approach which examines the variations in isotropic magnetic shielding in the space surrounding the molecule. The ground state (S0) of COT is shown to be strongly antiaromatic at the π-bond-shifting transition state (TS), a regular octagon of D8h symmetry; S0 antiaromaticity decreases at the D4h planar bond-alternating tub-to-tub ring-inversion TS but traces of it are shown to persist even at the tub-shaped D2d local minimum geometry. The lowest triplet (T1) and first singlet excited (S1) states of COT are found to have very similar D8h geometries and visually indistinguishable shielding distributions closely resembling that in benzene and indicating similarly high levels of aromaticity. Unexpectedly, COT diverges from its antiaromatic predecessor, cyclobutadiene, in the properties of the second singlet excited state (S2): In cyclobutadiene S2 is antiaromatic but in COT this state turns out to be strongly aromatic, with a shielding distribution closely following that around S2 benzene.
Introduction
Cyclooctatetraene (C8H8, COT) has been aptly described as “one of the decisive molecules in the history of chemistry”.1 It has been the subject of numerous experimental and theoretical studies; a comprehensive account of pre-2012 research has been provided by Schleyer and co-workers.1 The ground-state (S0) potential energy surface (PES) of COT has been examined in considerable detail2–8 and has been found to include symmetry-equivalent tub-shaped local minima of D2d symmetry connected through planar tub-to-tub ring-inversion transition states (TS) of D4h symmetry; these, in turn, are connected through a planar π-bond-shifting TS in the form of a regular octagon of D8h symmetry. In its electronic ground state D8h COT is a singlet diradical and its full-symmetry description requires at least a two-determinant wavefunction; the attempt to use the standard single closed-shell Slater determinant results in a “broken-symmetry” wavefunction which suggests energetically favourable distortions of the nuclear framework to one of the two bond-alternating D4h geometries. As shown by Hrovat and Borden,3 the ground-state PES of COT can be described with high accuracy using a complete active-space self-consistent field (CASSCF) wavefunction with “8 electrons in 8 orbitals”, CASSCF(8,8), accounting for the eight electrons involved in the bond rearrangements linking the D2d, D4h and D8h stationary points; CASSCF(8,8) wavefunctions have been also found to work well in studies of several low-lying electronic states of COT.4,6,9,10Hückel molecular orbital (HMO) theory depicts the ground electronic state of D8h COT as an antiaromatic triplet which is incorrect in more than one way: Firstly, at this geometry COT has an antiaromatic singlet ground electronic state which violates Hund's rule and secondly, the lowest triplet state (T1) of D8h COT has been shown to be aromatic.10–13D8h COT was used by Baird as an example of a molecule featuring an aromaticity reversal on excitation from S0 to T1;11 subsequent research revealed that D8h COT experiences a similar aromaticity reversal on excitation from S0 to the first singlet excited state (S1).10 Recent experimental work has demonstrated that cyclooctatetraene-fused acene dimers exhibit remarkable conformational flexibility associated with the change in aromaticity of the central COT moiety on excitation from S0 to S1; such “flapping” fluorophores (FLAP) provide a versatile platform for designing novel photofunctional systems.14–16
In this paper we investigate aromaticity and chemical bonding at all stationary points of the ground-state PES of COT, including the D2d tub-shaped, D4h bond-alternating and D8h geometries, as well as at the D8h geometries of the lowest triplet and first and second singlet (T1, S1 and S2) electronic excited states by analysing, for each geometry and electronic state, the changes in the off-nucleus isotropic shielding, σiso(r) = 1/3[σxx(r) + σyy(r) + σzz(r)], within the space surrounding the molecule. All σiso(r) calculations make use of state-specific CASSCF(8,8) wavefunctions constructed from gauge-including atomic orbitals (GIAOs). The aromaticities of the S0, T1 and S1 states of COT have been examined at the CASSCF(8,8)-GIAO level10 with several types of nucleus-independent chemical shifts (NICS),17–22 proton shieldings and magnetic susceptibilities. However, when applying NICS in order to assess the aromaticity of a larger ring such as COT, it is logical to ask to what extent we can trust predictions based on a single shielding value calculated at or above the centre of this ring. In contrast to NICS and other single-value aromaticity criteria, σiso(r) isosurfaces and contour plots provide somewhat more detailed and, arguably, more reliable information about aromaticity and its effect on chemical bonding.23–27 Representing off-nucleus magnetic isotropic shielding as a function of position in the space surrounding a molecule requires a very large number of closely spaced data points which addresses potential deficiencies of single-point NICS such as the need to choose, in a more or less arbitrary manner, locations at which these quantities are calculated (NICS can exhibit strong positional dependence and, in certain situations, standard choices can be inappropriate28,29), and the argument that a single number might not be sufficient to characterise all aspects of aromatic behaviour, supported by the observation that different ring current maps can produce nearly indistinguishable single-point NICS values.30,31 The application of this approach to the low-lying electronic states of the archetypal examples of aromatic and antiaromatic systems, benzene (C6H6) and square cyclobutadiene (C4H4)24 has shown that the profoundly different shielding distributions in the S0 states of C6H6 and C4H4 can be viewed as aromaticity and antiaromaticity “fingerprints” which are reproduced in other electronic states of the two molecules and allow classification of these states as aromatic (S0 and S2 for C6H6, T1 and S1 for C4H4) or antiaromatic (S0 and S2 for C4H4, T1 and S1 for C6H6); S2 C6H6 was predicted to be even more aromatic than S0 C6H6. Whether or not the low-lying electronic states of D8h COT follow a pattern similar to that observed in its antiaromatic cyclic conjugated predecessor with 4n π electrons, square cyclobutadiene, is one of the questions targeted through the research reported in this paper.
Computational procedure
The active spaces in the state-specific CASSCF(8,8) wavefunctions for the S0, T1, S1 and S2 states of D8h COT studied in this paper were formed from the π-orbital configuration (a2u)2(e1g)4(e2u)2 and two higher-energy π orbitals, a doubly-degenerate e3g and a b2u. As explained in ref. 10, the symmetries of the singlet and triplet states associated with the direct product e2u × e2u follow from the compositions of its symmetric and antisymmetric parts, [e2u × e2u] = a1g + b1g + b2g and {e2u × e2u} = a2g, respectively. Thus e2u × e2u gives rise to three singlet states, 11A1g (S1), 11B1g (S0) and 11B2g (S2), and one triplet state, 13A2g (T1). The largest subgroup of the D8h point group supported by one of the program packages we used for CASSCF(8,8) calculations, Dalton,32 is D2h, therefore in that subgroup the S0, S1, S2 and T1 states of D8h COT were treated as the 11B1g, 11Ag, 21Ag and 13B1g states, respectively. The 21Ag state was accessed by requesting the second root of the CASSCF(8,8) configuration interaction (CI) problem in that symmetry. In the CASSCF(8,8) calculations carried out with Gaussian1633 the S1 and S2 states were accessed by requesting the second and third roots, respectively, of the CASSCF(8,8) CI problem. The selection of active-space orbitals for the CASSCF(8,8) wavefunctions for the S0 states of D4h and D2d COT was, in each case, a straightforward task.The CASSCF(8,8)/6-31G** optimised geometries of the 11B1g state of D8h COT (TS for the π bond shift process on the S0 PES), the 11A1g state of D4h COT (TS for the ring inversion process on the S0 PES), the 11A1 state of D2d COT (local minimum on the S0 PES), the 13A2g state of D8h COT (local minimum on the T1 PES) and the 11A1g state of D8h COT (local minimum on the S1 PES) were taken from an earlier study of COT.10 Additionally, the geometry of the 11B2g state of D8h COT (local minimum on the S2 PES) was optimised at the same CASSCF(8,8)/6-31G** level, and then the geometries of all stationary points on the S0, T1, S1 and S2 PES of COT studied in this paper were reoptimised at the CASSCF(8,8)/cc-pVTZ level. As the differences between the CASSCF(8,8)/6-31G** and CASSCF(8,8)/cc-pVTZ optimised geometries were observed to be minor, all subsequent magnetic shielding calculations were carried out at the CASSCF(8,8)/6-31G** geometries; according to our experience with calculations of this type, off-nucleus shieldings are relatively insensitive to minor changes in molecular geometry. All of the additional geometry optimizations were carried out using Gaussian1633 and the local minimum or saddle point nature of each optimised geometry was verified through an analytic harmonic frequency calculation.
All CASSCF(8,8)-GIAO calculations were performed by means of the MCSCF-GIAO (multiconfigurational SCF with GIAOs) methodology34,35 that is implemented in the Dalton program package;32 the basis set used in these calculations was 6-311+G*. All reported total energies of various electronic states at CASSCF(8,8)/6-31G** geometries were also computed within the 6-311+G* basis, with both Gaussian16 and Dalton. Following previous work on NICS10,12,24,36 and ring currents37 in triplet systems, the CASSCF-GIAO isotropic shieldings in the lowest triplet electronic state of COT reported in this paper include only contributions that arise from the perturbation to the wavefunction. With this choice, the values reported for a triplet state can be compared directly with those for singlet states. For comparisons to experimental data when and if such data becomes available, one will need to take into account the large terms associated with the interaction between the electronic spin angular momentum and the magnetic field.38,39
The grids of points used in the construction of σiso(r) isosurfaces and contour plots for the S0, T1, S1 and S2 electronic states of COT studied in this paper are regular, with a spacing of 0.05 Å, in the shape of rectangular boxes centred at the origins of centre-of-mass right-handed Cartesian coordinate systems in which the z axes are oriented along the respective principal symmetry axes and the x and y axes are oriented along the C2′ axes (D2d geometry) or the C2′′ axes (D4h and D8h geometries). The grid for the S0 state of D2d COT consists of 1332 × 81 points, and the grids for all other states consist of 1612 × 101 points. In order to reduce computational effort, the σiso(r) values for each electronic state were calculated only at symmetry-unique grid points (1/4 of all points for D2d COT and 1/8 of all points for D4h and D8h COT, respectively) and data were then replicated by symmetry. For visualization purposes, all σiso(r) values obtained for the electronic states of COT were assembled into Gaussian cube files.40
Results and discussion
The carbon–carbon and carbon-hydrogen bond lengths in the S0, T1, S1 and S2 electronic states of COT from geometries optimised using state-specific CASSCF(8,8) wavefunctions in the 6-31G** and cc-pVTZ basis sets are shown in Table 1 together with the lowest harmonic frequencies and total energies of these states. The carbon–carbon and carbon–hydrogen bond lengths calculated in the cc-pVTZ basis set are slightly shorter; the largest difference between 6-31G** and cc-pVTZ bond lengths of 0.0052 Å is observed for the carbon–carbon “double” bond in D2d COT.| Geometry | State | Basis | R(CC) | R(CH) | υ 1 | Energy |
|---|---|---|---|---|---|---|
| D 2d | S0 (11A1) | 6-31G** | 1.3436, 1.4794 | 1.0791 | 179.8 (a1) | –307.691 721 |
| cc-pVTZ | 1.3384, 1.4767 | 1.0767 | 180.5 (a1) | –307.735 840 | ||
| D 4h | S0 (11A1g) | 6-31G** | 1.3510, 1.4718 | 1.0777 | 112.6i (b2u) | –307.674 646 |
| cc-pVTZ | 1.3454, 1.4705 | 1.0753 | 144.4i (b2u) | –307.718 937 | ||
| D 8h | S0 (11B1g) | 6-31G** | 1.4081 | 1.0774 | 2304.0i (b2g) | –307.663 411 |
| cc-pVTZ | 1.4044 | 1.0750 | 2595.3i (b2g) | –307.707 176 | ||
| T1 (13A2g) | 6-31G** | 1.4063 | 1.0773 | 159.5 (e2u) | –307.638 857 | |
| cc-pVTZ | 1.4026 | 1.0750 | 162.4 (e2u) | –307.682 884 | ||
| S1 (11A1g) | 6-31G** | 1.4074 | 1.0772 | 157.7 (e2u) | –307.612 618 | |
| cc-pVTZ | 1.4036 | 1.0748 | 160.1 (e2u) | –307.656 593 | ||
| S2 (11B2g) | 6-31G** | 1.3957 | 1.0773 | 162.8 (e2u) | –307.557 953 | |
| cc-pVTZ | 1.3927 | 1.0749 | 166.0 (e2u) | –307.603 528 |
The results for the electronic states of D8h COT obtained in the cc-pVTZ basis provide further indication that the antiaromatic S0 and aromatic T1 and S1 geometries can be expected to be very similar, with carbon–carbon bond lengths closer to that in benzene and significantly shorter than that in cyclobutadiene, 1.3961 Å and 1.4434 Å, respectively, from CASSCF(6,6)/6-31G** and CASSCF(4,4)/6-31G** S0 geometry optimizations of D6h benzene and D4h cyclobutadiene.10 The carbon–carbon bond length in the S2D8h COT geometry is almost identical to that in S0 benzene, with a difference of just 0.0004 Å (in the 6-31G** basis), which suggests an increase in aromaticity in comparison to T1 and S1 COT.
The variations in isotropic shielding around the S0, T1, S1 and S2 electronic states of COT at the CASSCF(8,8)/6-31G** geometries reported in Table 1 are illustrated in Fig. 1. The isovalues of σiso(r) = ±16 ppm and the molecular orientations were chosen so as to show reasonable levels of detail. Other orientations and isosurfaces for other isovalues can be examined using the Gaussian cube files with isotropic shielding values for the different geometries and electronic states of COT that are included in the ESI.† Further information about the behaviour of the isotropic shielding in the electronic states of COT at the planar D4h and D8h geometries is provided by contour plots in three planes: the molecular plane (Fig. 2), a plane 1 Å above it, and a plane perpendicular to the molecular plane (Fig. S1 and S2 in the ESI†).
 | ||
| Fig. 1 Isotropic shielding isosurfaces at σiso(r) = ±16 ppm (“ +” in blue, “−” in orange) for the electronic states of COT studied in this paper; additional σiso(r) = −1 ppm isosurface (yellow) included for S0 (D2d geometry). For details, see text. | ||
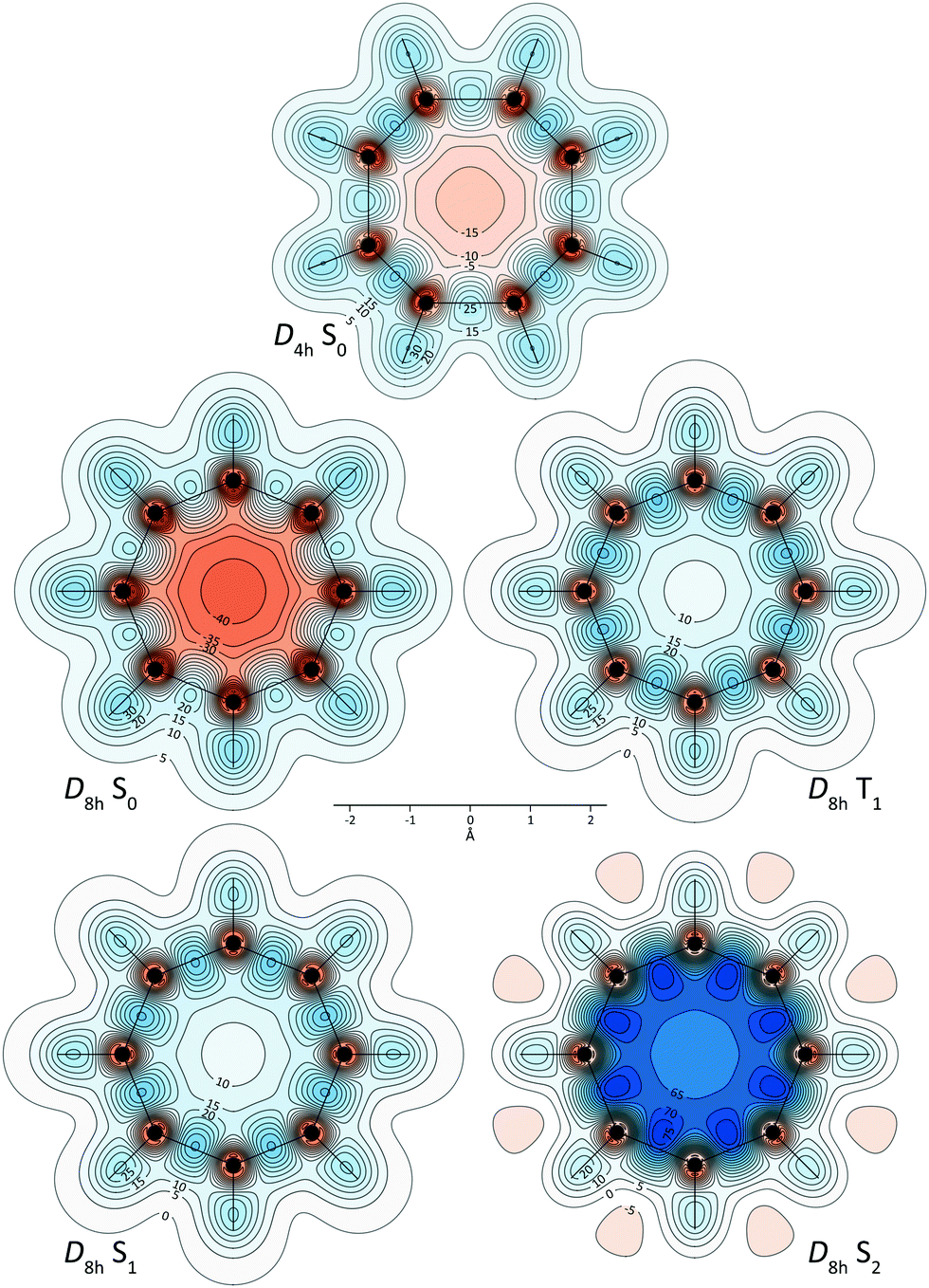 | ||
| Fig. 2 Isotropic shielding contour plots in the molecular (horizontal) planes for the S0 state of D4h COT and the S0, T1, S1 and S2 states of D8h COT. Contour levels at −75(5)75 ppm, orange (deshielded) to blue (shielded). | ||
The shielding along the carbon framework in the S0 electronic ground state at the D2d tub-shaped geometry outlines a sequence of alternating “single” and “double” carbon–carbon bonds similar to those reported in the magnetic shielding study of bonding in trans-1,3-butadiene.41 It is interesting to observe that this clearly non-aromatic picture retains a minor hint of antiaromaticity in the form of a small deshielded region surrounded by the yellow σiso(r) = –1 ppm isosurface in the centre of the ring. Note that other parts of this isosurface at –1 ppm can be seen as larger “halos” over the smaller orange “halos” at –16 ppm surrounding carbon atoms familiar from previous magnetic shielding studies of conjugated systems involving sp2 and sp hybridised carbon atoms and other sp2 hybridised first main row atoms.23,24,27,42,43 A recent study of magnetically induced currents in D2d COT reported a weak paratropic global molecular ring current,8 an indication of a very low level of antiaromaticity, which is in line with the findings of the current work. The shielding along the carbon framework does not show signs of additional conjugation over that in trans-1,3-butadiene41 which suggests that the interesting concept of “two-way” (double) hyperconjugation in D2d COT1 is likely to make only a minor contribution to bonding in this molecule.
The differences between the shielding around “single” and “double” carbon–carbon bonds are still obvious but somewhat less pronounced at the S0 planar geometry of D4h symmetry. There is some more evidence of antiaromaticity at the ring centre, in the form of a small almost spherical deshielded region with a boundary at –16 ppm. In fact, the D4h S0σiso(r) contour plot in Fig. 2 indicates that most of the interior of the carbon ring is moderately deshielded, including a sizeable region deshielded by more than 10 ppm. Antiaromaticity is also manifested by the weakened shielding around carbon–carbon bonds, the bulk of which is displaced away from the ring centre; this affects more the four “single” bonds.
The ground-state antiaromaticity of COT is most pronounced at the D8h planar geometry, at which the shielding picture is dominated by a large strongly deshielded dumbbell-shaped region in the centre of the molecule. The presence of this strongly deshielded region leads to further reduction of the shielding around carbon–carbon bonds and further displacement of this shielding towards the exterior of the ring. The variations in isotropic shielding around S0D8h COT resemble closely those observed for the paradigm of antiaromaticity, the electronic ground state of square cyclobutadiene.24 It has been suggested, on the basis of NICS values, that the S0 state of D8h COT is more antiaromatic than the respective state of square cyclobutadiene.10 Indeed, the deshielding of the ring interior shown in Fig. 1, 2 and Fig. S1, S2 (ESI†) is more intensive than that seen in analogous contour plots for square cyclobutadiene23,24 but, at the same time, in COT the extent to which the central deshielded region disrupts the shielding around carbon–carbon bonds is smaller. It would be correct to say that while the S0 state of D8h COT is undoubtedly strongly antiaromatic, antiaromaticity affects bonding along the carbon framework to a lesser degree than in the respective state of square cyclobutadiene. This is in line with the similarity between the antiaromatic S0 and aromatic T1 and S1D8h COT geometries illustrated by the data in Table 1.
Clearly, σiso(r) isosurfaces and contour plots differentiate between the levels of aromaticity of ground-state COT at its D8h, D4h and D2d geometries in a more nuanced way than modern valence bond (VB) theory: According to a spin-coupled generalised valence-bond (SCGVB) study,44 antiaromaticity at the D8h geometry follows from the singlet biradical character of the wavefunction (triplet pairs coupled to an overall singlet), whereas the absence of resonance within the wavefunctions at the D4h and D2d geometries was taken as a sign of non-aromatic behaviour.
It is difficult to tell apart the isotropic shielding pictures observed in the T1 and S1 electronic states of COT (Fig. 1, 2 and Fig. S1, S2, ESI†); both of these bear surprisingly close resemblance to the doughnut-shaped region of increased shielding enveloping the carbon framework in the electronic ground state of benzene,23,24 extended over a ring of eight rather than six carbons atoms. This observation provides compelling visual evidence that the T1 and S1 electronic states of COT exhibit levels of aromaticity very similar to that of the electronic ground state of benzene.
The negligible change in the level of aromaticity between the T1 and S1 electronic states of COT is one of the differences between the aromaticity characteristics of the low-lying excited states of COT and square cyclobutadiene: In square cyclobutadiene T1 is noticeably more aromatic than S1.24 A second, more important and somewhat unexpected difference is associated with the aromaticity of the second singlet excited state S2. In square cyclobutadiene S2 is antiaromatic but less so than S0.24 In stark contrast, in the S2 of COT the interior of the carbon ring is intensively shielded all over (Fig. 1, 2 and Fig. S1, S2, ESI†), in close resemblance to the shielding picture observed in the S2 state of benzene.24
The conclusion that can be drawn from this unexpected finding is that the S2 electronic state of COT, similarly to the corresponding state of benzene, exhibits a significant level of aromaticity, surpassing that of benzene in its electronic ground state.
Carbon and proton isotropic shieldings and various NICS indices for the S0, T1, S1 and S2 electronic states of COT, calculated at the same level of theory as the data for the shielding isosurfaces and contour plots, are collected in Table 2. The NICS indices include the original NICS index NICS(0) = −σiso(at ring centre),17 as well as NICS(1) = −σiso(at 1 Å above ring centre),18,19 NICS(0)zz = −σzz(at ring centre)20,21 and NICS(1)zz = −σzz(at 1 Å above ring centre).22 The position at which NICS(1) was evaluated for the non-planar D2d ground state of COT was 1 Å away from the ring centre along the S4 improper rotation axis. The data for states other than the S2 electronic state of COT were found to be identical to those reported earlier10 and have been included in order to facilitate comparison with the rather unexpected magnetic features of this state. According to the NICS(0), NICS(1), NICS(0)zz and NICS(1)zz values for the S2 electronic state of COT, this state of COT is decidedly more aromatic than the corresponding state of benzene.24 This conclusion is reinforced by the very significant proton deshielding observed in S2 COT: the corresponding σiso(1H) value is more than 8 ppm lower than that in the D2d ground state. Despite the fact that NICS carry much less information than the shielding isosurfaces and contour plots shown in Fig. 1, 2 and Fig. S1, S2 (ESI†), the data included in Table 2 demonstrates that they provide reasonably accurate assessments of the aromaticities of the S0, T1, S1 and S2 electronic states of COT. Of course, due to their single-point nature, NICS are unable to provide any of the insights into the influence of aromaticity and antiaromaticity on bonding in the S0, T1, S1 and S2 electronic states of COT that can be analysed and visualised through the respective isotropic shielding isosurfaces and contour plots.
| Geometry | State | σ iso(13C) | σ iso(1H) | NICS(0) | NICS(1) | NICS(0)zz | NICS(1)zz |
|---|---|---|---|---|---|---|---|
| D 2d | S0 (11A1) | 76.57 | 27.26 | 1.16 | −1.57 | 14.08 | 4.21 |
| D 4h | S0 (11A1g) | 79.48 | 28.91 | 16.10 | 11.98 | 55.40 | 37.62 |
| D 8h | S0 (11B1g) | 70.21 | 31.70 | 40.71 | 32.23 | 128.71 | 98.22 |
| T1 (13A2g) | 81.93 | 25.57 | −8.93 | −8.98 | −20.64 | −25.60 | |
| S1 (11A1g) | 80.01 | 25.43 | −8.87 | −9.02 | −21.52 | −26.34 | |
| S2 (11B2g) | 86.02 | 18.88 | −61.55 | −52.97 | −181.20 | −159.09 |
Conclusions
The analysis of the spatial variations in isotropic shielding, σiso(r), at all stationary points of the ground-state PES of COT, including the D2d tub-shaped, D4h bond-alternating and D8h geometries, as well as at the D8h geometries of the lowest triplet and first and second singlet (T1, S1 and S2) electronic excited states reveals both expected and unexpected features of bonding and aromaticity, some of which change dramatically between electronic states.As expected, the intense antiaromaticity of the electronic ground state at the D8h planar regular octagon geometry experiences rapid decrease on passing, through the lower-energy D4h planar geometry, to the lowest-energy D2d non-planar tub-shaped geometry. Interestingly, even at the D2d geometry, in which the alternating “single” and “double” carbon–carbon bonds are clearly outlined by the changes in isotropic shielding, ground-state COT retains a hint of antiaromaticity in the form of a small deshielded region surrounding the geometric centre of the molecule.
The features of the first and second singlet excited states of COT revealed by the current magnetic shielding study indicate that Hückel-antiaromatic rings with eight π electrons can be expected to behave differently from square cyclobutadiene in their first and second singlet excited states, with S1 becoming as aromatic as T1 and S2 turning out as strongly aromatic rather than antiaromatic.
The shielding isosurfaces and contour plots suggest that π bonding in the S2 state of COT (as well as in the S2 state of benzene24) is strengthened, which is associated with increased shielding within, above and below the ring; this increased shielding creates the perception of a substantially higher level of aromaticity. However, all of this is at the expense of the shielding over the σ framework and the strength of the σ bonds, hence the higher energy of this state. These findings can have important implications for the design of novel photofunctional materials involving flapping molecules14–16 and suggest that researchers in the area should be advised to examine not only the first singlet excited state of a flapping molecule but also the second singlet excited state—annulation can enhance the σ bond framework, decrease the energy of this state and make it more accessible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for support of this work by the University of York.Notes and references
- J. I. Wu, I. Fernández, Y. Mo and P. v. R. Schleyer, J. Chem. Theory Comput., 2012, 8, 1280–1287 CrossRef CAS PubMed.
- M. J. S. Dewar and K. M. Merz, Jr., J. Phys. Chem., 1985, 89, 4739–4744 CrossRef CAS.
- D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 1992, 114, 5879–5881 CrossRef CAS.
- P. G. Wenthold, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Science, 1996, 272, 1456–1459 CrossRef CAS PubMed.
- F.-G. Klärner, Angew. Chem., Int. Ed., 2001, 40, 3977–3981 CrossRef.
- J. L. Andrés, O. Castaño, A. Morreale, R. Palmeiro and R. Gomperts, J. Chem. Phys., 1998, 108, 203–207 CrossRef.
- A. Schild and B. Paulus, J. Comput. Chem., 2013, 34, 1393–1397 CrossRef CAS PubMed.
- R. J. F. Berger and A. Viel, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 327–339 CrossRef CAS.
- M. Garavelli, F. Bernardi, A. Cembran, O. Castaño, L. M. Frutos, M. Merchan and M. Olivucci, J. Am. Chem. Soc., 2002, 124, 13770 CrossRef CAS PubMed.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 12707–12713 CrossRef CAS PubMed.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- V. Gogonea, P. v. R. Schleyer and P. R. Schreiner, Angew. Chem., Int. Ed., 1998, 37, 1945–1948 CrossRef CAS.
- F. Feixas, J. Vandenbussche, P. Bultinck, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2011, 13, 20690–20703 RSC.
- T. Yamakado, S. Takahashi, K. Watanabe, Y. Matsumoto, A. Osuka and S. Saito, Angew. Chem., Int. Ed., 2018, 57, 5438–5443 CrossRef CAS PubMed.
- S. Saito, Flapping Molecules for Photofunctional Materials, in Molecular Technology: Materials Innovation, ed H. Yamamoto and T. Kato, Wiley-VCH, Weinheim, 2019, vol. 3, pp 17–51 Search PubMed.
- R. Kimura, H. Kuramochi, P. Liu, T. Yamakado, A. Osuka, T. Tahara and S. Saito, Angew. Chem., Int. Ed., 2020, 59, 16430–16435 CrossRef CAS PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- P. v. R. Schleyer, H. Jiao, N. J. R. van Eikema Hommes, V. G. Malkin and O. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- P. v. R. Schleyer, M. Manoharan, Z. X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. van Eikema Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- I. Cernusak, P. W. Fowler and E. Steiner, Mol. Phys., 2000, 98, 945–953 CrossRef CAS.
- E. Steiner, P. W. Fowler and L. W. Jenneskens, Angew. Chem., Int. Ed., 2001, 40, 362–366 CrossRef CAS PubMed.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- P. B. Karadakov and K. E. Horner, J. Phys. Chem. A, 2013, 117, 518–523 CrossRef CAS PubMed.
- P. B. Karadakov, P. Hearnshaw and K. E. Horner, J. Org. Chem., 2015, 81, 11346–11352 CrossRef PubMed.
- P. B. Karadakov, M. A. H. Al-Yassiri and D. L. Cooper, Chem. – Eur. J., 2018, 24, 16791–16803 CrossRef CAS PubMed.
- P. B. Karadakov, Org. Lett., 2020, 22, 8676–8680 CrossRef CAS PubMed.
- P. B. Karadakov, M. Di and D. L. Cooper, J. Phys. Chem. A, 2020, 124, 9611–9616 CrossRef CAS PubMed.
- C. Foroutan-Nejad, S. Shahbazian, F. Feixas, P. Rashidi-Ranjbar and M. Solà, J. Comput. Chem., 2011, 32, 2422–2431 CrossRef CAS PubMed.
- C. Foroutan-Nejad, Theor. Chem. Acc., 2015, 134(8), 1–9 Search PubMed.
- S. Fias, P. W. Fowler, J. L. Delgado, U. Hahn and P. Bultinck, Chem. – Eur. J., 2008, 14, 3093–3099 CrossRef CAS PubMed.
- S. Van Damme, G. Acke, R. W. A. Havenith and P. Bultinck, Phys. Chem. Chem. Phys., 2016, 18, 11746–11755 RSC.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, R. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS , Dalton, a molecular electronic structure program, Release Dalton2018.1, 2019, see http://daltonprogram.org.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. Ruud, T. Helgaker, R. Kobayashi, P. Jørgensen, K. L. Bak and H. J. A. Jensen, J. Chem. Phys., 1994, 100, 8178–8185 CrossRef CAS.
- K. Ruud, T. Helgaker, K. L. Bak, P. Jørgensen and J. Olsen, Chem. Phys., 1995, 195, 157–169 CrossRef CAS.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 7303–7309 CrossRef CAS PubMed.
- P. W. Fowler, E. Steiner and L. W. Jenneskens, Chem. Phys. Lett., 2003, 371, 719–723 CrossRef CAS.
- Z. Rinkevicius, J. Vaara, L. Telyatnyk and O. Vahtras, J. Chem. Phys., 2003, 118, 2550–2561 CrossRef CAS.
- J. Vaara, Phys. Chem. Chem. Phys., 2007, 9, 5399–5418 RSC.
- See https://gaussian.com/cubegen/(checked on 24 September 2021).
- P. B. Karadakov and K. E. Horner, J. Chem. Theory Comput., 2016, 12, 558–563 CrossRef CAS PubMed.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2013, 78, 8037–8043 CrossRef CAS PubMed.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2015, 80, 7150–7157 CrossRef CAS PubMed.
- P. B. Karadakov, J. Gerratt, D. L. Cooper and M. Raimondi, J. Phys. Chem., 1995, 99, 10186–10195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Isotropic shielding contour plots for the S0 (at D4h and D8h geometries), T1, S1 and S2 (at D8h geometries) electronic states of COT in planes 1 Å above the respective molecular planes and in planes perpendicular to the molecular planes; computational details and D2d S0 and D4h S0 optimised geometries; details of the compositions of the CASSCF wavefunctions for the S0, S1, S2 and T1 electronic states of D8h COT (PDF). Zip archive of Gaussian cube files with isotropic shielding values for the S0 (at D2d, D4h and D8h geometries), T1, S1 and S2 (at D8h geometries) electronic states of COT (ZIP). See DOI: 10.1039/d1cp04394c |
| This journal is © the Owner Societies 2021 |