
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational modelling of ammonia addition on partially reduced graphene oxide flakes†
Eszter
Makkos
 *,
Dominika
Bodrogi
and
Dénes
Szieberth
*
*,
Dominika
Bodrogi
and
Dénes
Szieberth
*
Budapest University of Technology and Economics, Budapest, Hungary. E-mail: eszter.makkos@edu.bme.hu
First published on 18th October 2021
Abstract
Density functional theory is employed to model the chemisorption of ammonia on epoxy-containing polycyclic aromatic hydrocarbons (PAHs) and understand the reaction mechanism of ammonia addition on partially reduced graphene oxide flakes. Coronene (C24H12) and ovalene (C32H14) based four-epoxy group containing molecules are used to mimic the RGO surface properties. The reaction mechanism changing effect of a second ammonia molecule as well as explicit water molecules is considered. The proposed reaction mechanism consists of two steps: the migration of one epoxy group out of the modelled four-epoxy group formation to a thermodynamically less stable one and the nucleophilic addition of the ammonia molecule. The second step involves forming an amine group and reducing an epoxy group to a hydroxyl one. Interestingly, the forming amine group bonds to the carbon atom with the smallest bond order among the available ones and not necessarily to the carbon atom of the opening epoxy ring. Incorporating a second ammonia molecule has a negligible effect on the overall reaction mechanism, while in the presence of one water molecule, the reaction goes through a different pathway involving a trimolecular state during the nucleophilic addition. Including more than one water molecule or applying an implicit solvent model does not cause further changes in the reaction.
1. Introduction
Graphene has initiated great research activities due to its beneficial properties, such as large surface area, high chemical stability, and novel electronic properties.1–6 An effective route to tune graphene's properties is chemical modification, which enables us to use it for potential applications, including graphene nanoribbon field-effect transistors,7,8 composite materials,9 adsorbents and sensors.10–14 Previous studies show that sp2 carbon-based nanomaterials and their derivatives could react with nitrogen-containing molecules such as NO2 and NH3, resulting in a local change in carrier concentration, after which physical properties are changing.15 Although only weak adsorption was found when they reacted with pristine graphene, a growing amount of defects and active sites on the surface effectively improved the adsorption.14,16 Graphene oxides (GOs), containing oxygen-based active sites, are essential precursors of graphene preparation. The thermal and chemical reduction of GO leads to reduced graphene oxides (RGOs) in which significant restoration of the sp2 carbon network takes place, although some oxygen-containing groups (mainly epoxy, ketone and carboxyl groups) remain which may enhance the interaction of molecules with RGO.17,18Experimental studies have been carried out to characterise the adsorption of ammonia and water on graphene and reduced graphene oxides.16,19–22 Measurements with Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy, and elemental analysis on graphene-oxides made by the Hummers' method show the formation of amine and hydroxyl groups during the reaction of NH3 and epoxy groups on the surface.20,23 In the case of various amounts of water within the interlayer space, it was found that water enhances the reaction via dissolving ammonia and promoting the dissociation of surface functional groups. Although above a certain amount, it reduces the accessibility of epoxy and –COOH groups, and thus limits the amount of ammonia adsorbed.21
Theoretical studies24–28 in connection with the physisorption of ammonia on pristine graphene found small binding energies independent of both the site and orientation of the molecule, while adsorption of NH3 on GO is generally stronger due to the presence of active sites.25 First-principles studies predict the reaction of NH3 with the hydroxyl and epoxy groups to be exothermic, with activation barriers being strongly dependent on the type and atomic arrangement of these groups. Hydroxyl groups interact with NH3 and NH4+ species via hydrogen bonds; therefore, only physisorption occurs between them and ammonia molecules, while epoxy groups and carbon vacancies are highly reactive centres for ammonia dissociation, creating a range of different chemisorbed species and causing a small level of charge transfer.26,27 Because the adsorption and dissociation of NH3 modify the GO's structural and electronic properties, it is likely to be partially responsible for removing oxygen-containing groups from graphene. The most considerable binding energy and the highest electron donor effect accompanied by a shift in Fermi energy is induced by the adsorption of NH3 on a surface with a single epoxy group.28 Theoretical investigations on the reactivity of epoxy and hydroxyl groups in the presence of water showed that an H2O molecule formed strong hydrogen bonds with the oxygen atoms of the functional groups. These findings agree with the experimental findings, such as the presence of water limits the amount of ammonia absorbed.21
The chemisorption of ammonia on graphene-oxide surfaces resembles nucleophilic substitution, a common reaction mechanism between an epoxy group and amines based on former studies.29–34 During nucleophilic substitution, the lone electron pair of the nitrogen atom in the amine initiates a nucleophilic attack on one of the carbon atoms connected to the oxygen atom of the epoxy group, which causes the disintegration of the O–C bond and consequently the formation of a compound containing a secondary amine and secondary alcohol (Fig. 1a).30,31 The activation energy of this reaction is around 12–14 kcal mol−1 in small molecular weight systems.30 Noncatalytic mechanism of the epoxy-amine reaction is believed to involve the formation of a trimolecular transition state. Alternatively, previous publications31–33 suggested a trimolecular transition state with an additional hydroxyl group involved in the reaction (Fig. 1b). A third mechanism was proposed34 based on a second, more reactive amine dimer (Fig. 1c). The effect of the formed aromatic ring adjacent to the epoxy group on trimolecular reaction mechanisms was also previously studied. It was shown that the reaction of a cyclic compound containing epoxy group is significantly slower than the reaction of an open-chain compound, presumably due to a higher energy barrier.35 Concerning the surface modification reactions of GO with amine or amine-containing molecules, Bourlinos et al. predicted that nucleophilic substitution reactions on the epoxy groups of GO proceed efficiently as the main insertion pathway.36
 | ||
| Fig. 1 The proposed mechanism (a) and possible transition states (b and c) of the amine and epoxy group reaction. | ||
The mentioned theoretical studies concentrated on the reaction mechanism of single ammonia adsorption on a surface model containing one or two oxygen-based functional groups. However, GO, and even RGO systems are known to have various oxygen-containing functional groups simultaneously. Besides, it is most likely that in experimental conditions, the ammonia adsorption takes place in the presence of additional ammonia or water molecules which allows a trimolecular nucleophilic substitution to take place and has not yet been studied with electronic structure calculations. Therefore, we apply a model molecule containing an epoxy group formation instead of a single oxygen-based functional group and concentrate on the assistance of explicit water and ammonia molecules in the close surrounding.
Today the most accepted model of graphene-oxide is the Lerf–Klinowski model, in which epoxy groups and hydroxyl groups are located on the surface, while carboxyl groups and lactones are on the edges of the GO sheet (Fig. 2).37 Regarding the location of epoxy groups, it is assumed that they form a rectangular grid on both sides of the GO sheet.38 Previous investigations showed that upon thermal treatment during the reduction of GO, desorption of hydroxyl groups occur at lower temperatures while epoxy groups need a higher temperature to leave.39 Recently, Uthaisar et al. reported that oxygen-containing groups are desorbing and migrating towards the edges until most functional groups disappear from the middle of the surfaces, i.e. upon partial reduction, epoxy groups are most likely found near to the edges.17 We applied finite-sized polycyclic aromatic hydrocarbons (PAHs) in our study to focus on the near-edge regions of PRGO systems, similarly to the area highlighted on the Lerf–Klinowski model of Fig. 2. Our research group had investigated the optimal structure of several adjacent epoxy groups on the surface of partially reduced graphene oxide with periodic boundary conditions and identified favourable group arrangements from 3.1% up to 18.8% epoxy group density (O/C%).40 The most stable four epoxy group arrangement was tested on PAH molecules and used further to study ammonia adsorption.
 | ||
| Fig. 2 The Lerf–Klinowski type structural model of graphene-oxide flakes37 blue dots are hydroxyl groups, while red dots are epoxy groups on the GO surface. (Up- and down-facing functional groups relative to the plane of the carbon grid are not differentiated.) The highlighted region represents the area we model with our PAH systems. | ||
The scope of this paper is to explore the energy profiles of the ammonia addition on a finite-sized partially reduced graphene oxide flake model with four epoxy groups and understand the effect of additional water or ammonia molecules on the reaction mechanism using density-functional theory calculations.
2. Computational methods
In the present work, the surface of PRGO was described with finite-sized molecular systems containing epoxy groups in a stable arrangement established with calculations using periodic boundary conditions.40,41 Calculations for each reaction step in the explored energy profiles included geometry optimisation and transition state searches. Frequency calculations were done to identify local minima and saddle points on the potential energy surface by their second derivatives. Natural population analysis (NPA)42 were applied to describe the electron distribution within the systems and determine atoms’ natural charges.Epoxy group arrangements were studied with the CRYSTAL09 code43,44 using periodic density functional methods. The Perdew–Burke–Ernzerhof (PBE)45 exchange–correlation functional was employed together with the following atom centred basis functions: Dovesi basis set for carbon (C: 6-21G*)46 and Muscat basis set for oxygen atoms (O: 8-411).47 The graphene sheet was modelled as a single carbon layer using experimental cell parameters and a 3 by 3 hexagonal unit cell containing 32 atoms, periodic in two dimensions. Only translational symmetry operators were set for the applied unit cells (with or without epoxy groups). Geometry optimisations were performed by sampling the reciprocal space with the Monkhorst–Pack grid48 using a shrinking factor of 8. The SCF energy convergence criteria were set to 10−11 a.u., while the Coulomb and exchange series was controlled with the following threshold values during the geometry optimisation: 10−7, 10−7, 10−7, 10−7 and 10−14 a.u.
All molecular structure calculations were performed with the Gaussian 09 program package.49 For the visualisation of the optimised structures, the MOLDEN program50 has been used. Minima, intermediates, and transition states were described by density functional theory using the B3LYP method, which consists of the three-parameter exchange functional of Becke51 and the correlation functional of Lee Yang and Parr.52 The molecules were described by the 6-31+G*53–55 basis set of double zeta quality with diffuse and d polarisation functions, and 6-31+G(2d,p), 6-311+G(2d,p), 6-311++G(2d,p)53,55–57 were used for further tests. Using larger basis sets did not change the energies substantially; therefore, we found 6-31+G* appropriate for this study. Test calculations were done to verify the applied level of theory for systems containing one ammonia molecule and a coronene or ovalene, by using B97D58 and ωB97XD59 functionals for geometry optimisations and by calculating single point energies with the following method/basis pairs: ωB97XD/6-311++G(2d,p), MP2/6-31+G(2d,p) and MP2/6-311++G(2d,p) at the ωB97XD/6-31+G(2d,p) level, B3LYP/6-31+G*//B3LYP-D/6-31+G* and B97/6-31+G*//B98/6-31+G*. We found that applying different functionals might change the activation barriers, but the reaction mechanism is unchanged. For more details, see the ESI.†
Thermodynamic potentials at room temperature were calculated for each reaction route. Gibbs free energies showed an insignificant difference in energy barrier heights but elevated the energy of the final products, therefore, changing the type of the reaction from exotherm to closely thermoneutral. Potentials and Gibbs free energies for the presented adsorption mechanisms are summarised in Table 1.
| Reaction energies (kcal mol−1) | |||||||
|---|---|---|---|---|---|---|---|
| Systems | 0 | TS0-1 | 1 | TS1-2 | 2 | TS2-3 | 3 |
| COR | 0.0 (0.0) | 31.4 (31.9) | 3.3 (4.2) | 30.4 (34.7) | −8.1 (−0.6) | — | — |
| (OV) | 0.0 (0.0) | 33.8 (33.8) | 10.6 (10.5) | 34.0 (37.5) | −8.4 (−1.3) | — | — |
| (OV)-H2O | 0.0 (0.0) | 29.2 (29.3) | 10.0 (10.1) | 29.8 (32.5) | 6.6 (13.9) | 8.5 (13.6) | −9.7 (−2.6) |
| (OV)-2NH3 | 0.0 (0.0) | 33.3 (33.0) | 11.2 (10.2) | 31.1 (35.2) | −6.3 (−2.5) | — | — |
Although additional water or ammonia molecules were explicitly added, the surrounding medium might also significantly affect the reaction mechanism. Consequently, water as a solvent was also considered using an implicit method. Single point calculations were made with the IEFPCM solvent model at B3LYP/6-31+G* level. Results showed that applying solvent correction did not cause any significant change in relative energies; the deviations were only 1–1.5 kcal mol−1. Considering that aqueous media caused only a slight decrease in the energy barrier of the migration of epoxy groups, the effect was small (see Table S3, ESI†). For the above reasons, we presented the results in gas phase without the IEFPCM model.
3. Results and discussion
A. Applied models
The interaction between ammonia and the partially reduced graphene oxide is investigated close to the side of the PRGO flakes. PAH molecules are good candidates to mimic edge behaviour due to their finite size and large surface/edge ratio.60,61 Moreover, molecular models have the advantage of analytical second derivatives of the wavefunction, which is essential to transition state searches. We limited ourselves to investigating the interaction between ammonia and one particular epoxy arrangement on the surface in the present work. At the same time, other types of functional groups which are typical at the edge (e.g. keto, carboxyl) will be the object of a future study.Although the distribution of the different functional sites within the more oxygen dense regions is described to be relatively random,38 some group unit formations are more stable than others. Epoxy groups tend to form a line on one side of the surface62,63 Using a 32 carbon atom containing unit cell and periodic boundary conditions for the graphene sheet; we optimised several possible configurations of epoxy groups on the surface with increasing epoxy group density. The oxygen concentration of reduced graphene-oxide surfaces produced with chemical reduction is between 8 and 16%;64,65 consequently, we limited our investigation in the region of 3.1% up to 18.8% (O/C%). We compared the relative stability of different configurations by calculating the formation energy. The complete account of considered formations and applied methods are included in the ESI,† while the most stable configurations are presented in Fig. 3. All Ef energies are positive, suggesting that the creations of epoxy groups on pure graphene lead to disturbance of the aromatic framework and destabilisation. However, they only vary within a small energy range (19–25 kcal mol−1), which shows that after breaking the delocalised electron net with one epoxy, the addition of other groups does not require significant energy investment.
 | ||
| Fig. 3 Unit cell sketches of the most stable epoxy group s for each studied epoxy group density and their formation energies (Ef, kcal mol−1) (dark red = up-facing, light red = down-facing epoxy groups). | ||
In periodic boundary conditions, the epoxy groups experience different environmental conditions than in small-sized PAHs. In the first case, the effect of neighbouring cells on the adsorption arises, while in the second case, the edge-effect of the finite-sized molecules. We aim to investigate the ammonia adsorption close to the edge of a PRGO flake. Besides, the reaction site can be viewed as an isolated activation centre in chemical reactions, which is better achieved with finite-sized models. Therefore we chose to describe the close-edge surface behaviour of PRGO on polycyclic aromatic hydrocarbons: C24H12 coronene (COR) is our smallest model, and C32H14 ovalene (OV) is the most stable 32 carbon PAH molecule (shown in Fig. 4). We chose the most stable epoxy group formation for 12.5% epoxy density because, in PRGO systems, there are sp2 carbon atoms available in the immediate surrounding of the oxygen-rich regions. Using the established four-epoxy group formation in ovalene leaves three sp2 carbons within the molecule, allowing more freedom during the reaction mechanism on the PRGO surface. There are two possible positions relative to the ovalene's geometry for the chosen four-epoxy group formation. We investigated both (see ESI†), from which only the geometry of OV on Fig. 4 had negative formation energy:‡ −22 kcal mol−1.
 | ||
| Fig. 4 Ball and stick representation of the used polycyclic aromatic hydrocarbons. Stable positions of the epoxy groups are numbered on the coronene (COR, left) and ovalene (OV, right). (red: oxygen, white: hydrogen, orange: carbon). | ||
We calculated relative stabilities for a series of four-epoxy group variations, differentiating between up and down-facing epoxy groups§ on the graphene/PAH plane (Fig. 5). The reference structures were the most stable 32 carbon unit cell with 12.5% epoxy density (PBC) and the ovalene molecule with the same epoxy group arrangement (OV). We note that due to the inherently different methodology, the two types of calculations cannot be compared quantitatively, but we can draw certain conclusions by plotting the two sets of relative energies next to each other. Nevertheless, the transferability of the epoxy group formations’ stability obtained with periodic boundary conditions onto PAH molecules is not evident, and their direct comparison is not unequivocal. Systems containing only up-facing epoxy groups (2, 6) tend to be less favourable under periodic boundary conditions, most likely due to differently treated morphological changes. While morphological stress introduced in the unit cell is multiplied in PBC calculations resulting in a wrinkling surface plane, the bending of small-sized PAH molecules is less energetically demanding up to a certain angle. However, the most stable structure is identical using both models.
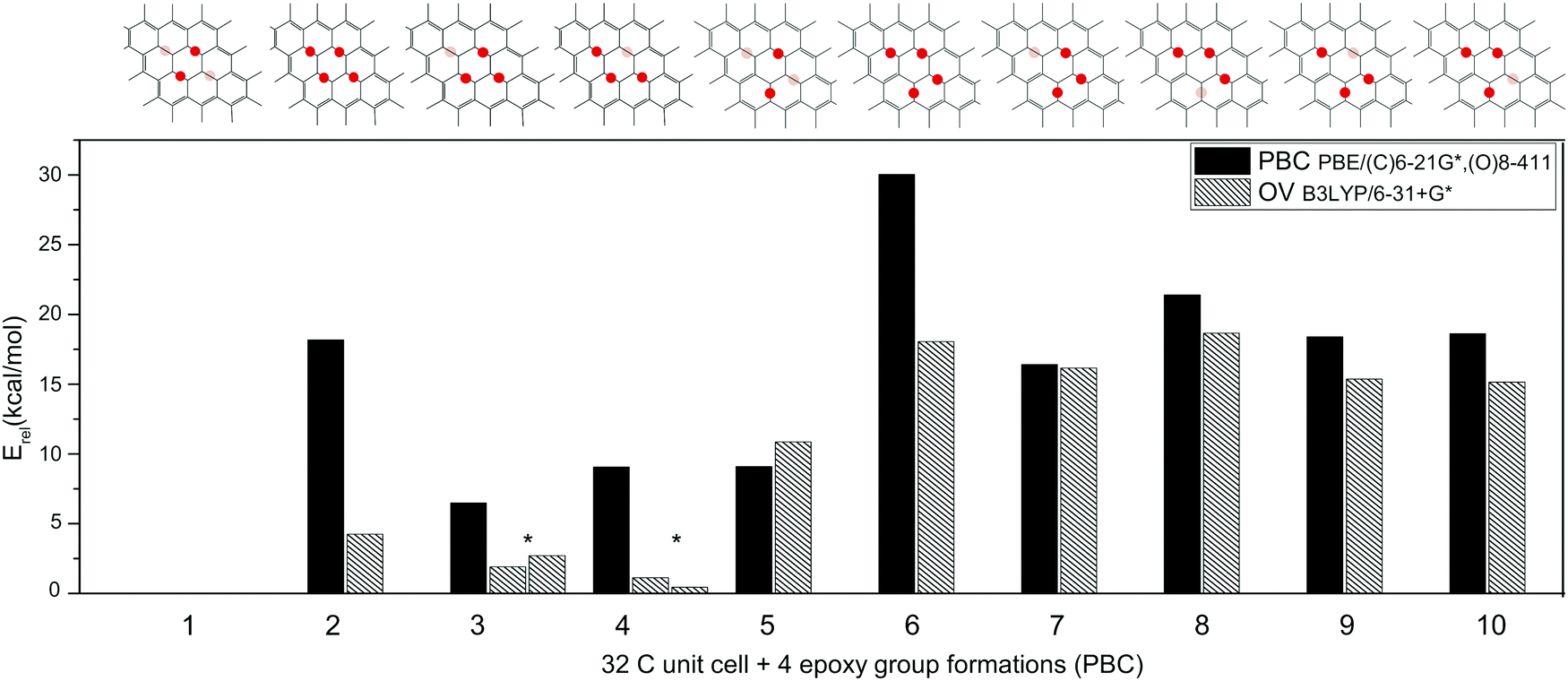 | ||
Fig. 5 Relative energies (Erel) of different four-epoxy group formations compared to the reference structures containing two up-, and two down-facing epoxy groups. Black columns represent 32C unit cells with four epoxy groups calculated using periodic boundary conditions on PBE/(C)6-21G*, (O)8-411 level of theory (PBC). Striped columns represent the oxidised ovalene molecules calculated on B3LYP/6-31+G* functional/basis pair (OV). Dark red = up-facing, light red = down-facing epoxy groups. (*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) We note that structures 3 and 4 have two variations within the ovalene because of the different relative positions of the epoxy groups within the molecule.) We note that structures 3 and 4 have two variations within the ovalene because of the different relative positions of the epoxy groups within the molecule.) | ||
The presented energy profiles are always calculated relative to the energy of the starting geometry of each model system. The following nomenclature is used in every section: 0, 1, 2 for the optimised stable geometries numbered in the order within the energy profile, and TS0-1, TS1-2 for the transitions states. Coronene models were differentiated by adding COR- before the numbers, while additional molecules within the system were noted afterwards, such as -H2O or -NH3.
B. Addition of ammonia on the PRGO models
Chemical intuition and results of the previous studies20,22,26,66 suggest that the ammonia molecule dissociates when it reacts with a PRGO surface and form an amine group at moderate temperatures (Fig. 6). Calculations based on coronene and ovalene hydrocarbon models showed the formation of such product to be 8 kcal mol−1 energetically more stable than a starting complex of an ammonia molecule bound loosely to the surface by a hydrogen bond. Initially, we assumed a one-step nucleophilic substitution mechanism for this reaction, as shown in Fig. 1a. That would involve a single transition state (TS) between the starting complex and the final product. However, after a thorough search for such a reaction barrier from different reaction coordinates and starting positions, we could not detect a transition state for this mechanism. Huang and Gubbins showed the formation of a hydroxylamine molecule through a one-step nucleophilic substitution of ammonia on the epoxy group.26 However, their model included a single epoxy group on the middle of the graphene sheet, which is known to be an energetically less stable40,41,67,68 than an epoxy group formation, therefore more likely to open for a reaction. | ||
| Fig. 6 Ball and stick representation of the optimised final product for ammonia addition on ovalene (2). The participating atoms in the reaction are labelled on the image (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
Electrostatic potential energy maps of the oxidised coronene and ovalene (see Fig. S6 in ESI†) show that the lone pairs of the epoxy oxygens face each other, affecting their reactivity. Previous investigations on partially reduced graphene oxides described epoxy group migration on the surface.17,63 Li et al. modelled the movement of an epoxy group on a PRGO coronene model and found an energy barrier of 0.83 or 1.1 eV depending on the new position of the migrated epoxy group.63 A sterically more favourable functional group arrangement may evolve for a nucleophilic attack through ammonia-assisted epoxy migration.
We studied the movement of the O1 oxygen both on COR and OV, with and without the presence of ammonia. Fig. 7 shows that the migration of the epoxy group on our PRGO model is an endotherm reaction with an energy difference of 3 or 10 kcal mol−1 depending on the used model size. The positive energy difference can be explained by the fact that in our case, the epoxy is required to move out from the more stable parallel position onto the neighbouring hexagonal ring. The new position of O1 is different on the COR and OV relative to the geometry of the molecules: while it moves towards the edge of the coronene, it migrates inward on the ovalene. We note that although the product is more stable using the smaller model (COR-1), the size of the coronene model limits the possible reaction sites (see ESI† for comparison). Without the presence of the ammonia molecule, the reaction can be activated with an energy barrier of cc. 33.3 or 36.8 kcal mol−1 in the COR and OV model (COR-TS0-1, TS0-1). Although these transition states are higher in energies than the values of Li et al.,63 this difference might be the result of the different starting formation since our four-epoxy group model is relatively more stable than a system with two groups.40,41,67 We found that in both cases, the presence of ammonia slightly decreased the height of the activation barrier (with 2–3 kcal mol−1) but did not affect the energy of the new epoxy arrangement.
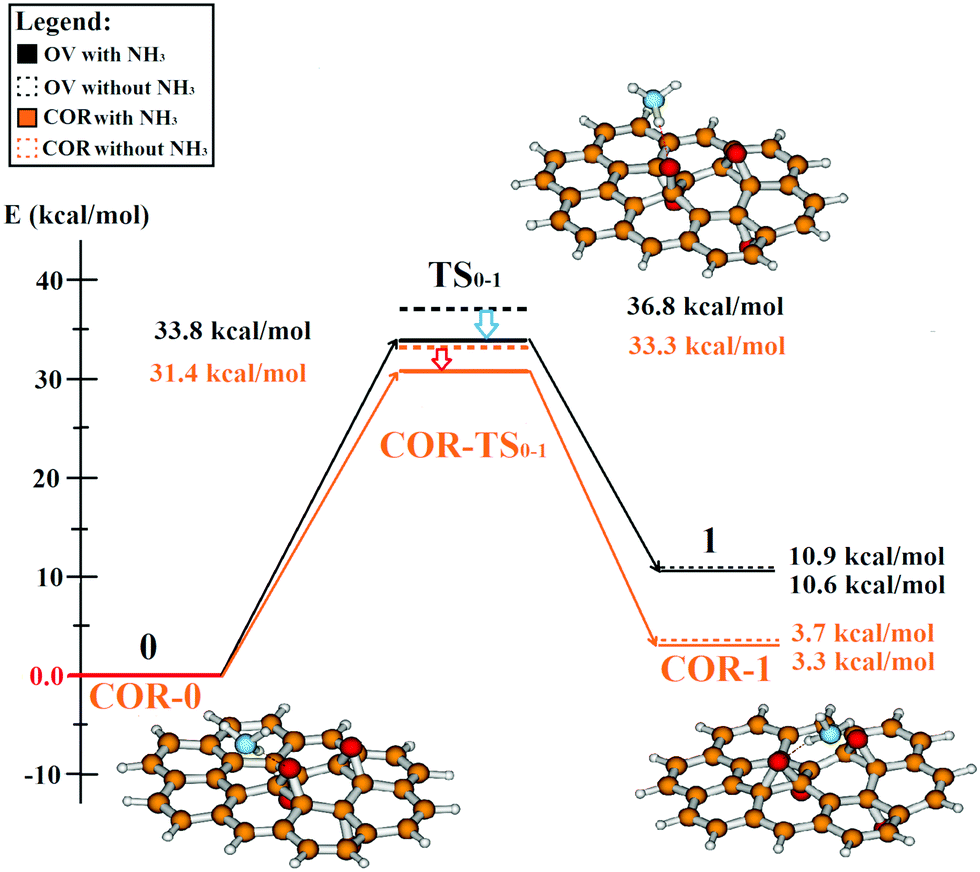 | ||
| Fig. 7 Energy profile (kcal mol−1) of an epoxy group migration with and without an ammonia molecule. The absolute energy of the starting complex is the basis of the energy comparison (0, COR-0). Black lines show energies calculated for the ovalene model, while the orange lines are related to coronene based results (COR-). The geometry of the starting formation (0), transition state (TS0-1) and migration product (1) using ovalene is represented with the ball and stick images (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
Although the product of the one-step epoxy migration is energetically less stable than the starting position, it can be considered an intermediate structure for the dissociation reaction of the ammonia molecule. Fig. 8 and Table 1 shows the energy profile of the whole two-step mechanism of ammonia addition. After the endothermic formation of a new epoxy arrangement (COR-1, 1) which allows the nucleophilic attack of the ammonia molecule on the migrated epoxy group, NH3 dissociates in an exothermic reaction; resulting in the previously established energetically stable product (shown for ovalene in Fig. 6). This second step (COR-TS1-2, TS1-2) requires the simultaneous protonation of the epoxy oxygen, the repeated opening of the epoxy group accompanied with the formation of the NH2 group. Furthermore, it has a similar energy barrier height to the epoxy migration (COR-TS0-1, TS0-1). The ammonia molecule adds on the PRGO model, seemingly with the previously established nucleophilic substitution (Fig. 1a). However, a major difference is that the C–N bond is formed with the C1 carbon atom instead of the carbon atom of the opening C–O bond.
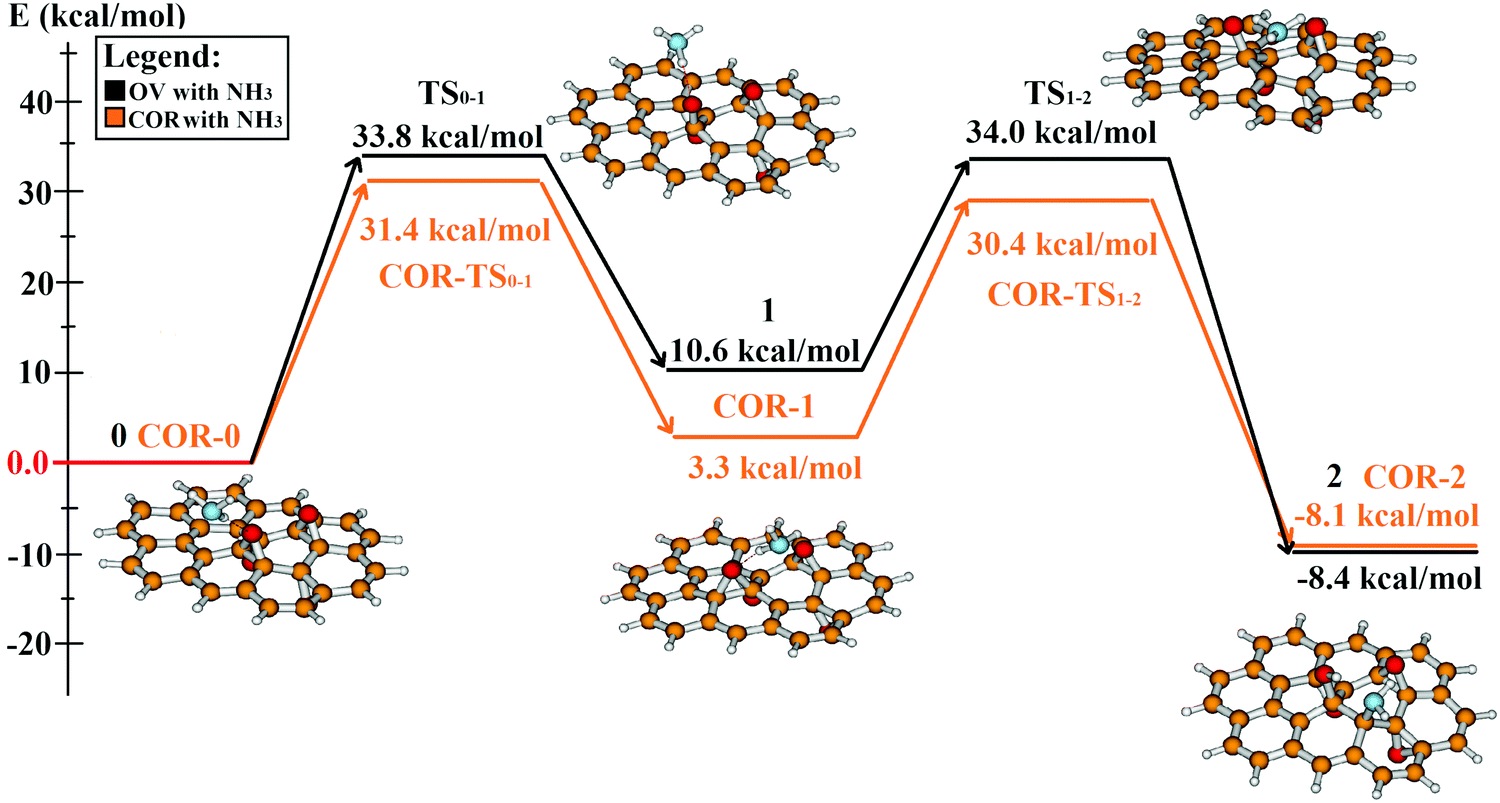 | ||
| Fig. 8 The whole reaction mechanism of ammonia addition on the used PRGO models. The absolute energy of the starting complex is the basis of the energy comparison (0). Black lines show energies calculated for the ovalene model, while the orange lines are the coronene based results. The optimised geometry of each reaction step using the ovalene model is shown with the ball and stick images (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
Our results are similar to the findings of Tang et al. for two (non-parallel) epoxy groups, in which the ammonia is bonded to the PRGO surface in a nucleophilic addition via the opening of the epoxy group.25
The two steps of the chemical reaction can be traced by the changes in the listed atomic distances and natural atomic charges calculated by natural bond order analysis (NBO). The relevant values for the ovalene model are summarised in Table 2. Since the reaction mechanism of coronene- and ovalene-based systems are qualitatively similar but energetically different from this point on, we decided to concentrate only on the ovalene results. However, all bond lengths, charges, and values for the COR model are in the ESI.† First, the ammonia interacts with the adjacent epoxy group via a hydrogen bond (1.966 Å N–H1⋯O1 at TS0-1). At the first transition state, the previously bonded C1–O1–C2 epoxy group opens as it is shown by the long O1–C1 distance (2.275 from 1.441 Å), the shortened O1–C2 bond length (1.389 from 1.443 Å) and the less positive partial charge on C1 (0.17 from 0.24 a.u.). The epoxy group is migrated to a new position at the intermediate step (C2–O1–C3), while C1 becomes a typical sp2 carbon without a notable partial charge.
| Atomic distances (Å) | Natural charges (a.u.) | |||||||
|---|---|---|---|---|---|---|---|---|
| Structures | N–C1 | N–H1 | O1–H1 | O1–C1 | O1–C2 | C1 | N | NH3 |
| 0 | 4.245 | 1.020 | 2.342 | 1.441 | 1.443 | 0.24 | −1.17 | 0.00 |
| TS0-1 | 5.005 | 1.028 | 1.966 | 2.275 | 1.389 | 0.17 | −1.18 | 0.00 |
| 1 | 3.808 | 1.020 | 2.314 | 2.491 | 1.444 | −0.02 | −1.17 | 0.00 |
| TS1-2 | 2.212 | 1.031 | 1.806 | 2.480 | 1.385 | 0.18 | −1.03 | 0.25 |
| 2 | 1.488 | 1.815 | 0.986 | 2.489 | 1.430 | 0.08 | −0.92 | — |
Interestingly, at the TS1-2 step, the ammonia attacks C1, a double-bonded carbon with a natural charge of −0.02 a.u., instead of other partially positive carbons of the epoxy group (0.23 a.u., see Table S9 in ESI†), as it would happen in a nucleophilic reaction. Therefore, the actual reaction between NH3 and the PRGO is an amine addition, accompanied by a repeated opening of the O1 containing epoxy group and a proton transfer between the ammonia and the O1 oxygen. To understand why a nucleophile reacts with a neutral carbon atom instead of the partially charged ones of the epoxy, we applied NBO bond order analysis and looked at total overlap-weighted NBO bond orders of the carbon atoms within system 1. We found that although between the sterically available carbon atoms, C1 is less positively charged, it has a lower bond order (cc. 3.24) than the neighbouring oxygen connected carbon atoms (cc. 3.44). This difference makes the C1 atom the preferable reaction site in its surroundings (all bond order values and natural charges of the carbon atoms are in Table S9, ESI†).
While the ammonia molecule is only weakly absorbed at the first part of the reaction mechanism via hydrogen bonding with one of the epoxy groups, at TS1-2, it is finally positioned above the C1 carbon atom with a relatively short distance (2.212 Å) by its hydrogens pointing towards the surface oxygens (see Fig. 9). While the usual H–N–H angle in an ammonia molecule is 107°, the interacting molecule flattens in the transition state with an average bond angle of 112°. The future protonation of O1 is predicted by the short H-bond of O1⋯H1 (1.806 from 2.314 Å). The ammonia starts a nucleophilic attack which is shown by the decreasing negative charge of the nitrogen (1.03 from 1.17 a.u.). The final charge transfer between the ammonia molecule and the PRGO surface is around 0.25 a.u.
 | ||
| Fig. 9 Ball and stick representation of the optimised transition state for nucleophilic attack (TS1-2). Atomic distances and bond lengths are given in Å (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
The final product (2) contains a primary amine group with a 1.488 Å N–C1 distance, which is slightly longer than it is in an aniline molecule (1.40 Å)69,70 but agrees with the calculated bond length of Tang et al. (1.49 Å).25 Similarly, the 1.430 Å C–O distance in the hydroxyl group is longer than in a typical phenol molecule (1.37 Å).71,72 In our model, the hydroxyl group points towards the direction of the amine group forming an H-bond between the two functional groups (shown by the N⋯H1–O1 distance of 1.815 Å).
C. Ammonia addition on PRGO models in the presence of water molecules
As it was presented in Fig. 1b, a proton transfer between an ammonia molecule and an epoxy group in a nucleophilic substitution reaction is also possible with the assistance of a hydroxyl group-containing proton donor, such as water.31–33 We considered this scenario by modelling the ammonia addition reaction on the ovalene based PRGO model in the presence of one or more water molecules.We initially assumed two possible starting positions for water depending on which side of the surface it is: same (Fig. 10a) or opposite side (Fig. 10b) as the ammonia molecule. However, as expected, calculations with the opposite side model confirmed that the water molecule on the other side of the PRGO model has no notable effect on the epoxy migration nor on the ammonia addition. The reaction goes as it was shown in the case of a single ammonia molecule (the calculated energies for this scenario are in Table S10, ESI†).
 | ||
| Fig. 10 Ball and stick representations of the optimised starting arrangements with a water molecule (a) coming from the same side (b) opposite side than the ammonia molecule. Relevant atomic positions are labelled on the images (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
Starting the investigation from Fig. 10a, we found a mechanism similar to the previous two-step reaction route: the migration of the O1 containing epoxy group is followed by the dissociation of the ammonia molecule. The energy profile of this reaction is shown in Fig. 11 and Table 1. Nevertheless, the water molecule plays a determining role in both parts of the reaction mechanism. First, it decreases the energy barrier of the epoxy group migration by 5 kcal mol−1 compared to the ammonia assisted migration (see Fig. 7).
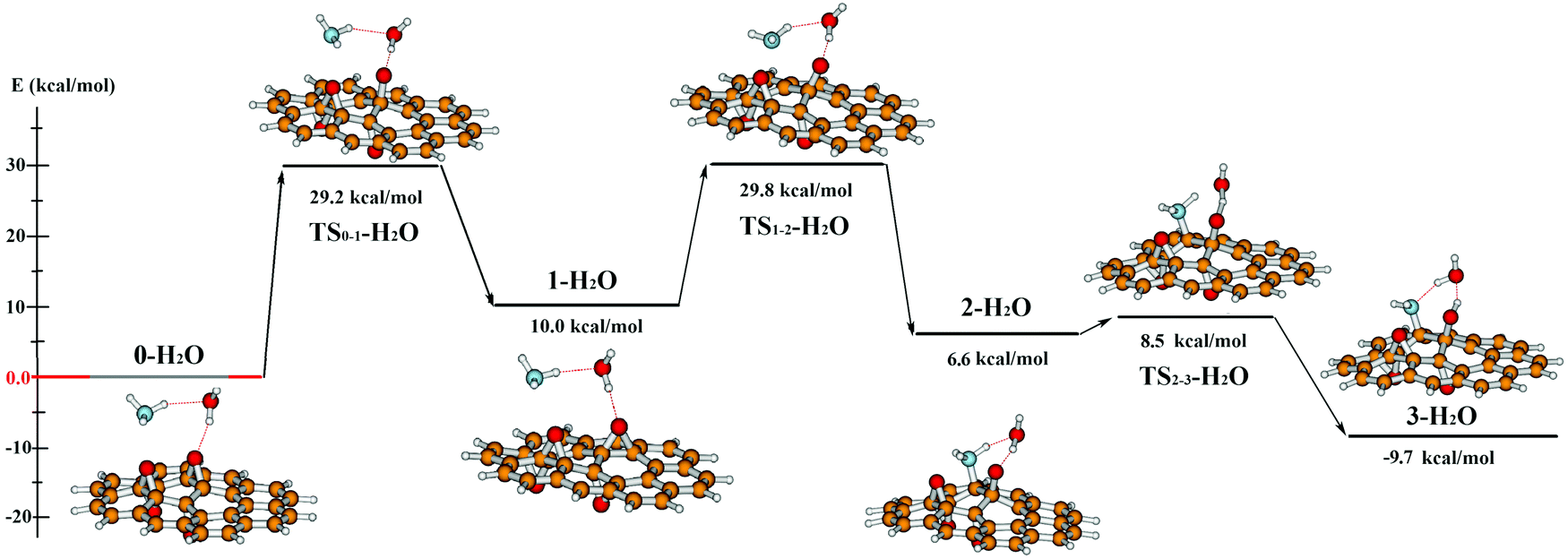 | ||
| Fig. 11 The whole reaction mechanism of ammonia addition on the used PRGO models in the presence of a water molecule. The absolute energy of the starting complex is the basis of the energy comparison (0-H2O). The optimised geometry of each reaction step is shown with the ball and stick images. (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
Second, in the presence of water, instead of the simultaneous addition and deprotonation of the ammonia, the addition mechanism happens in two steps: NH3 adds on a double-bonded carbon atom and forms an ionic intermediate (2-H2O),¶ then the stable final product (3-H2O) is formed by a simultaneous proton transfer between the NH3+ moiety and the water molecule, and between the water molecule and the oxygen of the opened epoxy group. The TS2-3-H2O activation barrier is small (only 2 kcal mol−1) and similar to the trimolecular transition state we expect based on the usual proton transfer in a nucleophilic substitution assisted by water.73
Bond lengths and natural charges (summarised in Table S11, ESI†) confirm the above described two differences in reaction routes. Compering the hydrogen bond lengths in the TS0-1 and TS0-1-H2O structures of Fig. 8 and 11, when H2O interacts with the oxygen of the epoxy group, the H-bond is shorter (O1–H4 is 1.733 Å), i.e. stronger, than between NH3 and the same oxygen (O1–H1 in Table 2 is 1.966 Å). Stronger interaction between the starting molecules lowers the energy of the ring-opening, i.e. the activation barrier of the epoxy migration.
Looking at the charge transfer between the ammonia molecule and the surface, we see that in TS1-2-H2O, the charge transfer is slightly smaller than in TS1-2 (0.16 a.u. vs. 0.25 a.u.). However, the intermediate structure of 2-H2O and the following transition state of TS2-3-H2O have a total charge of cc. 0.43 a.u. on the connected ammonia moiety, which confirms the partially charged state of these structures.
Previous studies suggest that the presence of excess water besides ammonia can prohibit the adsorption of the latter due to the strong interaction between water molecules and epoxy groups.21 To further investigate this, we increased the number of water molecules to two and three and studied their additional effect on the reaction barrier heights. Considering that the transition state of TS2-3-H2O is small compared to the other activation energies within the mechanism and expected to be low in every case, we focused only on the rate-determining barriers (TS0-1-H2O and TS1-2-H2O). Table 3 shows the calculated relative energies for each step of the whole reaction mechanism with two (-2H2O) and three water (-3H2O) molecules. Comparing the values to the one water results in Table 1, the energy level of the transition states stayed similar, while the energies of the final product somewhat increased (from −9.7 kcal mol−1 to −6.8 and −4.9 kcal mol−1, in the case of 2-2H2O and 2-3H2O respectively). In both cases, we optimised two possible intermediate structures (1-, 1′-), which are energetically different, but structurally differ only in the number of hydrogen bonds formed between the ammonia and the water molecules, see Fig. 12. Supposedly, there is a small energy barrier between the different hydrogen-bond networks, and more than two possible conformations with similar energies are possible. However, since the mapping of these networks is beyond the scope of this investigation, we did not proceed further.
| Notation | ΔE [kcal mol−1] | Notation | ΔE [kcal mol−1] |
|---|---|---|---|
| 0-2H2O | 0.0 | 0-3H2O | 0.0 |
| TS0-2-2H2O | 28.9 | TS0-2-3H2O | 27.6 |
| 1-2H2O | 10.0 | 1-3H2O | 6.7 |
| 1′-2H2O | 11.7 | 1′-3H2O | 14.8 |
| TS1′-2-2H2O | 30.3 | TS1′-2-3H2O | 32.9 |
| 2-2H2O | −6.8 | 2-3H2O | −4.9 |
 | ||
| Fig. 12 Ball and stick representations of two conformationally different intermediate structures with three water molecules (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
In conclusion, we found no evidence that water molecules would make the ammonia addition less likely. Instead, the presence of one water facilitated the reaction. Adding more water molecules did not change the reaction mechanism qualitatively and caused only minor changes in the energies. Therefore, we concluded that only one water molecule plays an active role in the ammonia addition reaction.
D. Interaction of two ammonia molecules with PRGO models
Another amine-containing molecule can also fulfil the role of proton donor in nucleophilic substitution, see Fig. 1c.34 We considered this third scenario by modelling the ammonia addition reaction on a PRGO model with two ammonia molecules.Assuming that both ammonia molecules are positioned on the same side of the PRGO flake, the calculated reaction route is similar to the previously established reaction routes, which involve the migration of the epoxy group as a first step. However, by comparing the actual energy profiles with the previous nucleophilic attack scenarios, we found that the second ammonia molecule does not have the same reaction determining role in the reaction as a water molecule. The two-ammonia reaction mechanism, summarised in Fig. 13 and Table 1, is almost identical to the single ammonia reaction route in Fig. 8. Only a slight decrease in the second activation barrier from 34.0 to 31.1 kcal mol−1 is found, accompanied by a slight increase in the energy of the final product from −8.4 to −6.3 kcal mol−1.
 | ||
| Fig. 13 Reaction mechanism of binding an ammonia molecule in the presence of another ammonia molecule on the same side on the ovalene model. Relative energies are employed to illustrate the height of activation barriers. The basis of the comparison is the starting arrangement (0-2NH3). The optimised geometry of each reaction step is shown with the ball and stick images (red: oxygen, white: hydrogen, orange: carbon, blue: nitrogen). | ||
The first difference can be explained by comparing the natural charges within TS1-2 and TS1-2-2NH3. The charge of the reacting ammonia molecule is more positive in the latter one than in the former one (0.39 a.u. vs. 0.25 a.u., see Table S12 in ESI†), suggesting that the ammonia molecule is more reactive in TS1-2-2NH3. The difference between final product stabilities might also be the result of a more reactive final amine group, evidenced by the shortened N1–H1 distances (1.751 Å vs. 1.815 Å) and lengthened H1–O1 bonds (0.994 Å vs. 0.986 Å) comparing the 2-2NH3 structure to 2. In both cases, the increased reactivity is most likely caused by the weak secondary interaction between the two ammonia molecules.
Based on our findings, we concluded that the proposed reaction mechanism for a nucleophilic substation with a second amine group (Fig. 1c) is not likely to take place in our model. In contrast to a water molecule, which donated its proton in the process by forming a trimolecular state in TS2-3-H2O, a second ammonia molecule does not play an active role in the amine addition.
4. Conclusions
In this present work, we investigated the possible reactions of ammonia on a partially reduced graphene oxide model by optimising possible reaction pathways for three basic ammonia-epoxy nucleophilic substitution scenarios on an ovalene based four-epoxy group-containing polycyclic aromatic hydrocarbon.Based on our findings, instead of the supposed one-step nucleophilic substitution between the partially reduced graphene oxide model and the ammonia, the overall reaction mechanism consists of two steps: the migration of an epoxy group and the nucleophilic addition of the ammonia molecule. The displacement of the reacting epoxy group is a prerequisite of the following nucleophilic addition since it converts the stable epoxy group formation to a more reactive one. Interestingly, the forming amine group does not bond to the surface through the carbon atom of the opening epoxy ring but rather via a neighbouring one. Looking at the calculated bond order of each carbon atom in the PAH molecule, we concluded that instead of positioning over a more partially positive carbon, the amine is bonding to the one with the smaller bond order.
The activation barriers of both reaction steps are generally above 30 kcal mol−1, which is high enough to suggest that the reaction would not occur spontaneously at room temperature. The presence of an additional water molecule lowers the energies of these transition states by around 5 kcal mol−1. It modifies the ammonia addition by involving an extra step via an ionic intermediate and forming a trimolecular chain during the epoxy reduction. Introducing more explicit water molecules resulted in slight changes of energies due to the many different possible conformations. However, it did not cause any significant difference in the reaction pathway. In contrast to the inclusion of a water molecule, introducing a second ammonia molecule in the model has no significant effect on the reaction mechanism and its energy profile.
In conclusion, one ammonia molecule will interact with the epoxy groups of a reduced graphene oxide sheet by forming an amine group on the surface and reducing the epoxy to a hydroxyl group. The reaction mechanism might differ in an aqueous medium by involving a trimolecular state during the nucleophilic addition, but the final products will be the same.
Our results can be limited by the size of our chosen RGO model, as the energies might be affected by the finite size and, therefore, the sterically hindered graphene plate. Also, fewer or more epoxy groups on the surface can result in different outcomes, although the overall reaction mechanism possibly stays the same. However, further investigation of larger systems for the same reaction could give more insight into the possible ammonia interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of the National Research Development and lnnovation Office (NKTH) grant number K 128410 and the helpful comments of Professor Krisztina Laszlo is gratefully acknowledged.References
- A. K. Geim and K. S. Novoselov, The rise of graphene, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Two-dimensional atomic crystals, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov and D. Jiang, Electric field effect in atomically thin carbon films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Two-dimensional gas of massless Dirac fermions in graphene, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Experimental observation of the quantum Hall effect and Berry's phase in graphene, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, The electronic properties of graphene, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Chemically derived, ultrasmooth graphene nanoribbon semiconductors, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- X. Wang, Y. Ouyang, X. Li, H. Wang, J. Guo and H. Dai, Room-temperature all-semiconducting sub-10-nm graphene nanoribbon field-effect transistors, Phys. Rev. Lett., 2008, 100, 206803 CrossRef PubMed.
- C. Petit and T. J. Bandosz, MOF-graphite oxide composites: Combining the uniqueness of graphene layers and metal–organic frameworks, Adv. Mater., 2009, 21, 4753–4757 CrossRef CAS.
- D. Selvakumar, H. Sivaram, A. Alsalme, A. Alghamdi and R. Jayavel, Freestanding flexible, pure and composite form of reduced graphene oxide paper for ammonia vapor sensing, Sci. Rep., 2019, 9(8749), 1–8 CAS.
- K. Toda, R. Furue and S. Hayami, Recent progress in applications of graphene oxide for gas sensing: A review, Anal. Chim. Acta, 2015, 878, 43–53 CrossRef CAS PubMed.
- N. Joshi, T. Hayasaka, Y. Liu, H. Liu, O. N. Oliveira Jr and L. Lin, A review on chemiresistive room temperature gas sensors based on metal oxide nanostructures, graphene and 2D transition metal dichalcogenides, Microchim. Acta, 2018, 185(213), 1–16 CAS.
- C. Mackin, V. Schroeder, A. Zurutuza, C. Su, J. Kong, T. M. Swager and T. Palacios, Chemiresistive graphene sensors for ammonia detection, ACS Appl. Mater. Interfaces, 2018, 10, 16169–16176 CrossRef CAS.
- R. Ghosh, A. Midya, S. Santra, S. K. Ray and P. K. Guha, Chemically reduced graphene oxide for ammonia detection at room temperature, ACS Appl. Mater. Interfaces, 2013, 5, 7599–7603 CrossRef CAS PubMed.
- H. Liu, Y. Liu and D. Zhu, Chemical doping of graphene, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- H. E. Romero, P. Joshi, A. K. Gupta, H. R. Gutierrez, M. W. Cole, S. A. Tadigadapa and P. C. Eklund, Adsorption of ammonia on graphene, Nanotechnology, 2009, 20, 245501 CrossRef PubMed.
- C. Uthaisar, V. Barone and B. D. Fahlman, On the chemical nature of thermally reduced graphene oxide and its electrochemical Li intake capacity, Carbon, 2013, 61, 558–567 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Structural evolution during the reduction of chemically derived graphene oxide, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- S. Böttcher, H. Vita, M. Weser, F. Bisti, Y. S. Dedkov and K. Horn, Adsorption of water and ammonia on graphene: Evidence for chemisorption from X-ray absorption spectra, J. Phys. Chem. Lett., 2017, 8, 3668–3672 CrossRef PubMed.
- C. Petit, M. Seredych and T. J. Bandosz, Revisiting the chemistry of graphite oxides and its effect on ammonia adsorption, J. Mater. Chem., 2009, 19, 9176–9185 RSC.
- M. Seredych and T. J. Bandosz, Combined role of water and surface chemistry in reactive adsorption of ammonia on graphite oxides, Langmuir, 2010, 26, 5491 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, Simultaneous nitrogen doping and reduction of graphene oxide, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y. Peng and J. Li, Ammonia adsorption on graphene and graphene oxide: A first-principles study, Front. Environ. Sci. Eng., 2013, 7, 403–411 CrossRef CAS.
- S. Tang and Z. Cao, Adsorption and dissociation of ammonia on graphene oxides: A first-principles study, J. Phys. Chem. C, 2012, 116, 8778–8791 CrossRef CAS.
- L. Huang and K. E. Gubbins, ammonia dissociation on graphene oxide: An ab initio density functional theory calculation, Z. Phys. Chem., 2015, 229, 1211–1223 CrossRef CAS.
- E. C. Mattson, K. Pande, M. Unger, S. Cui, G. Lu, M. Gajdardziska-Josifovska, M. Weinert, J. Chen and C. J. Hirschmugl, exploring adsorption and reactivity of NH3 on reduced graphene oxide, J. Phys. Chem. C, 2013, 117, 10698–10707 CrossRef CAS.
- A. Nancy Anna Anasthasiya, M. Khaneja and B. G. Jeyaprakash, Electronic structure calculations of ammonia adsorption on graphene and graphene oxide with epoxide and hydroxyl groups, J. Electron. Mater., 2017, 46, 5642–5656 CrossRef CAS.
- R. B. Sunoj and M. Anand, Microsolvated transition state models for improved insight into chemical properties and reaction mechanisms, Phys. Chem. Chem. Phys., 2012, 14, 12715–12736 RSC.
- Y. Zhang and S. Vyazovkin, Effect of substituents in aromatic amines on the activation energy of epoxy-amine reaction, J. Phys. Chem. B, 2007, 111, 7098–7104 CrossRef CAS PubMed.
- J. Y. Lee, M. J. Shim and S. W. Kim, Synthesis of liquid crystalline epoxy and its mechanical and electrical characteristics-curing reaction of LCE with diamines by DSC analysis, J. Appl. Polym. Sci., 2002, 83, 2419–2425 CrossRef CAS.
- I. T. Smith, The mechanism of the crosslinking of epoxide resins by amines, Polymer, 1961, 2, 95–108 CrossRef CAS.
- L. Shechter, J. Wynstra and R. P. Kurkjy, glycidyl ether reactions with amines, Ind. Eng. Chem., 1956, 48, 94–97 CrossRef CAS.
- B. A. Rozenberg, Epoxy Resins and Composites II, Berlin, 1986 Search PubMed.
- B. Barkakaty, K. Morino, A. Sudo and T. Endo, Amidine-mediated delivery of CO2 from gas phase to reaction system for highly efficient synthesis of cyclic carbonates from epoxides, Green Chem., 2010, 12, 42–44 RSC.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Graphite oxide: Chemical reduction to graphite and surface modification with primary aliphatic amines and amino acids, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, Structure of graphite oxide revisited, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- D. Pandey, R. Reifenberger and R. Piner, Scanning probe microscopy study of exfoliated oxidised graphene sheets, Surf. Sci., 2008, 602, 1607–1613 CrossRef CAS.
- V. B. Shenoy, M. Chhowalla, Y. J. Chabal, M. Acik, C. Mattevi and A. Bagri, Structural evolution during the reduction of chemically derived graphene oxide, Nat. Chem., 2010, 2, 581–587 CrossRef PubMed.
- E. Makkos and D. Szieberth, Budapest University of Technology and Economics, MSc thesis, 2013.
- D. W. Boukhvalov and M. I. Katsnelson, Modeling of graphite oxide, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush and M. Llunell, C. Science, A. Technologies, Crystal09 2.0.1 User's Manual, 2013, pp. 1–307 Search PubMed.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr. – Cryst. Mater., 2005, 220, 571 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Dovesi, M. Causa’, R. Orlando, C. Roetti and V. R. Saunders, Ab initio approach to molecular crystals: A periodic Hartree–Fock study of crystalline urea, J. Chem. Phys., 1990, 92, 7402–7411 CrossRef CAS.
- J. Scaranto and S. Giorgianni, A quantum-mechanical study of CO adsorbed on TiO2: A comparison of the Lewis acidity of the rutile(110) and the anatase(101) surfaces, THEOCHEM, 2008, 858, 72–76 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- D. J. Frisch, M. J. Trucks, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Mennucci, B. Petersson, G. A. Nakatsuji, H. Caricato, M. Li, X. Hratchian, H. P. Izmaylov, A. F. Bloino, J. Zheng and G. Sonnenb, Gaussian09, Revision A, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- G. Schaftenaar and J. H. Noordik, Molden: A pre- and post-processing program for molecular and electronic structures, J. Comput. Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, 6-31G* basis set for atoms K through Zn, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarisation functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- D. Y. Zubarev, X. You, J. McClean, W. A. Lester, Jr. and M. Frenklach, Patterns of local aromaticity in graphene oxyradicals, J. Mater. Chem., 2011, 21, 3404 RSC.
- J. R. Dias, Aromaticity and cyclic conjugation of three pairs of strongly subspectral series of conjugated molecules as models for graphene armchair and zigzag edges, Mol. Phys., 2010, 108, 3425–3429 CrossRef CAS.
- Z. Li, W. Zhang, Y. Luo, J. Yang and J. G. Hou, How graphene is cut upon oxidation?, J. Am. Chem. Soc., 2009, 131, 6320–6321 CrossRef CAS PubMed.
- J.-L. Li, K. Kudin, M. McAllister, R. Prud’homme, I. Aksay and R. Car, Oxygen-driven unzipping of graphitic materials, Phys. Rev. Lett., 2006, 96, 176101 CrossRef PubMed.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, A low-temperature method to produce highly reduced graphene oxide, Nat. Commun., 2013, 4, 1537–1539 CrossRef PubMed.
- W. Liu and G. Speranza, Tuning the oxygen content of reduced graphene oxide and effects on its properties, ACS Omega, 2021, 6, 6195–6205 CrossRef CAS PubMed.
- Q. Tang, Z. Zhou and Z. Chen, Graphene-related nanomaterials: tuning properties by functionalisation, Nanoscale, 2013, 5, 4541–4583 RSC.
- J. Ito, J. Nakamura and A. Natori, Semiconducting nature of the oxygen-adsorbed graphene sheet, J. Appl. Phys., 2008, 103, 113712 CrossRef.
- Z. Xu and K. Xue, Engineering graphene by oxidation: A first-principles study, Nanotechnology, 2010, 21, 045704 CrossRef PubMed.
- M. A. Palafox, M. Gill, N. J. Nunez, V. K. Rastogi, L. Mittal and R. Sharma, Scaling factors for the prediction of vibrational spectra. II. The aniline molecule and several derivatives, Int. J. Quantum Chem., 2005, 103, 394–421 CrossRef CAS.
- M. Sala, O. M. Kirkby, S. Guérin and H. H. Fielding, New insight into the potential energy landscape and relaxation pathways of photoexcited aniline from CASSCF and XMCQDPT2 electronic structure calculations, Phys. Chem. Chem. Phys., 2014, 16, 3122–3133 RSC.
- L. Cesari, L. Canabady-Rochelle and F. Mutelet, Computational study on the molecular conformations of phenolic compounds, Struct. Chem., 2018, 29, 179–194 CrossRef CAS.
- A. Melnichuk and R. J. Bartlett, Gas phase solvatochromic effects of phenol and naphthol photoacids, J. Chem. Phys., 2011, 134, 244303 CrossRef PubMed.
- L. Pardo, R. Osman, H. Weinstein and J. R. Rabinowitz8, Mechanisms of nucleophilic addition to activated double bonds: 1,2-and 1,4-Michael addition of ammonia, J. Am. Chem. Soc., 1993, 115, 8263–8269 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02320a |
| ‡ The formation energy is calculated by comparing the energy of the epoxy containing PRGO model to the sum of the energy of the used PAHs and two oxygen molecules. |
| § We note that the graphene/PAH plane has mirror symmetry, therefore formations containing epoxy groups at the same positions relative to each other but opposite relative to the plane are interchangeable. For example, the structure with up-facing epoxy groups O1, O2 and down-facing epoxy groups O3, O4 on Fig. 4 OV is the same as the ovalene molecule with down-O1, O2 and up-facing O3, O4 oxygens. |
| ¶ We note, that applying IEFPCM solvent model on the ionic structures of 2-H2O and TS2-3–H2O decreased their energies with around 5 kcal mol−1. However, the overall reaction mechanism did not change and there was negligible difference in the energy of the final product (see Table S3, ESI†). |
| This journal is © the Owner Societies 2021 |