
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rovibronic signatures of molecular aggregation in the gas phase: subtle homochirality trends in the dimer, trimer and tetramer of benzyl alcohol†
Robert
Medel
 *a,
Ander
Camiruaga
b,
Rizalina Tama
Saragi
c,
Pablo
Pinacho
de,
Cristóbal
Pérez
de,
Melanie
Schnell
de,
Alberto
Lesarri
c,
Martin A.
Suhm
a and
José A.
Fernández
*b
*a,
Ander
Camiruaga
b,
Rizalina Tama
Saragi
c,
Pablo
Pinacho
de,
Cristóbal
Pérez
de,
Melanie
Schnell
de,
Alberto
Lesarri
c,
Martin A.
Suhm
a and
José A.
Fernández
*b
aInstitute of Physical Chemistry, University of Goettingen, Tammannstr. 6, 37077 Goettingen, Germany. E-mail: rmedel@gwdg.de
bDepartment of Physical Chemistry, Faculty of Science and Technology, University of the Basque Country (UPV/EHU), Barrio Sarriena S/N, 4894 Leioa, Spain. E-mail: josea.fernandez@ehu.es
cDepartamento de Química Física y Química Inorgánica, Facultad de Ciencias – I.U. CINQUIMA, Universidad de Valladolid, Paseo de Belén, 7, 47011 Valladolid, Spain
dDeutsches Elektronen-Synchrotron DESY, Notkestraße 85, 22607 Hamburg, Germany
eInstitute of Physical Chemistry, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 1, 24118 Kiel, Germany
First published on 11th October 2021
Abstract
Molecular aggregation is of paramount importance in many chemical processes, including those in living beings. Thus, characterization of the intermolecular interactions is an important step in its understanding. We describe here the aggregation of benzyl alcohol at the molecular level, a process governed by a delicate equilibrium between OH⋯O and OH⋯π hydrogen bonds and dispersive interactions. Using microwave, FTIR, Raman and mass-resolved double-resonance IR/UV spectroscopic techniques, we explored the cluster growth up to the tetramer and found a complex landscape, partly due to the appearance of multiple stereoisomers of very similar stability. Interestingly, a consistently homochiral synchronization of transiently chiral monomer conformers was observed during cluster growth to converge in the tetramer, where the fully homochiral species dominates the potential energy surface. The data on the aggregation of benzyl alcohol also constitute an excellent playground to fine-tune the parameters of the most advanced functionals.
Introduction
Molecular aggregation is an interesting phenomenon involved in many chemical processes that shape the environment around us. The conditions on Earth, usually close to standard conditions, are optimal for molecules to build weakly-bound complexes that can form and dissociate on a short time scale. In its tremendous ability to exploit every single mechanism that may confer living beings an advantage, life makes extensive use of intermolecular interactions as a convenient way to drive many biological reactions, like signal transduction or host–guest recognition. Therefore, the interest that characterization of the weak intermolecular interactions has attracted is of no surprise, resulting in the development of sophisticated experiments and refined computational procedures. This effort permits extraction of experimental observables from the molecular aggregates and prediction of the aggregation preferences of the molecules under specific circumstances. Still, even with well elaborated methods, intermolecular aggregations are elusive, and their accurate description is a real challenge, even for the most advanced computational protocols.1,2One of the strategies to characterize intermolecular interactions is to study clusters of small and simple molecules, containing only one or two selected chemical groups. For example, the study of water aggregation allows to finely characterize the biologically relevant OH⋯O interaction in absence of any external perturbation, as exceptionally illustrated with the water decamer.3,4 Further introduction of additional chemical groups provides modulation of the OH⋯O hydrogen bonds. In this way, characterization of the OH⋯O interaction has proceeded through systems of increasing complexity, mostly aliphatic5–11 and aromatic alcohols.12–18 This procedure enables investigating the intra- and intermolecular factors affecting intermolecular interactions, which also control large scale or macromolecular aggregation.
One of the properties of the OH group that greatly complicates characterization of its intermolecular interactions is the existence of torsional isomerism. The orientation of the OH group in, for example, ethanol5–7 gives rise to trans and gauche conformational isomers. Furthermore, the latter can be divided into gauche+ and gauche− transient enantiomers, depending on the sign of the HOCαCβ dihedral angle. The barriers for interconversion are low18,19 (3.5–4.5 kJ mol−1) and therefore, population transfer between both stereoisomers occurs almost freely at room temperature.
Although the enantiomerism of alcohols acts as a multiplier of the number of possible conformations for each aggregate of a given stoichiometry, in practice, not all combinations are equally stable, and one can distinguish between homo- and heterochiral aggregates.20 As it happens with biological molecules, the different handedness of enantiomers results in slightly different interaction energies. As demonstrated in the past, prediction of the correct energetic order of enantiomeric aggregates is a real challenge for theory,21–23 asking for assistance from high-resolution spectroscopic experiments for model verification. A second challenge is the extension of the gas-phase studies to larger clusters, normally obfuscated by low signals and spectral congestion. In particular, studies aiming at structural elucidation of clusters in the trimer–pentamer scale are scarce and may be quite useful to refine computational models.
In a previous work, Medel and Suhm explored how transient chirality modulates the aggregation of benzyl alcohol (BnOH) and 1-phenylethanol using a combination of FTIR and Raman spectroscopy.16 The results were interpreted with the aid of density-functional theory (DFT) calculations. The authors concluded that homochiral aggregation was favored over the heterochiral counterparts. The effect was a kind of concerted selection of the relative chirality of the monomers to give rise to a homochiral aggregate. Interestingly, when the two molecules form two OH⋯π interactions instead of one OH⋯π and one OH⋯O hydrogen bond, the most stable aggregate is heterochiral, highlighting the relevance that the OH⋯O bridge has in the selection of the chirality of the monomers. The results also emphasized the difficulties of some DFT functionals to accurately reproduce the experimental observations: only those functionals with the optimal parameterization of the London dispersion forces were able to correctly predict the energetic order of the computed structures.
Here we extend the previous work on BnOH aggregation in two directions. First, thanks to the joint effort of four experimental groups, we revisit the monomer and dimer of BnOH using rotational and electronic spectroscopy techniques in supersonic expansions. Interestingly, the slightly different cooling conditions of each experiment result in the detection of a different number of species for each stoichiometry. Then, we move up in the aggregation ladder to characterize the structure of the BnOH trimer and tetramer, using FTIR, Raman and mass-resolved excitation spectroscopy (MRES). Interpretation of the results in the light of DFT calculations shows that the chirality synchronization is amplified as the cluster grows. As a result, we offer a comprehensive multi-methodological rovibrational description of BnOH aggregation from the dimer to tetramer, building a detailed picture of the aggregation mechanism of this alcohol.
Methods
This work contains results obtained by several experimental groups. Therefore, to keep the manuscript to a reasonable length, only the most relevant details of each experimental set up are included. A thorough description may be found in the references provided within the text.Microwave spectroscopy
The rotational spectroscopy experiments in Valladolid and Hamburg used broadband (chirped-pulse) Fourier transform microwave spectrometers operating in the 2–8 GHz region.24,25 A detailed description of the Hamburg COMPACT25 spectrometer is given in ref. 26, with some recent improvements as described in ref. 4. The Valladolid spectrometer is based on the direct-digital design of ref. 27. In these experiments, a pulsed vertical jet expansion is crossed horizontally by a short (1–5 μs) microwave chirped pulse, inducing a full-band fast-passage excitation and rotational coherence.28 The resulting transient molecular emission or free-induction decay is recorded in the time domain (ca. 40 μs) and Fourier-transformed to the frequency domain. The chirp was synthesized digitally with an arbitrary-waveform generator and followed by power amplification to 20 W in Valladolid and 300 W in Hamburg. The FID signal was captured with a digital oscilloscope. Typical final linewidths (FWHM) in Hamburg and Valladolid were of 65 kHz and 100 kHz, respectively. The jet was formed by thermal vaporization of a commercial BnOH sample (99.8%, Sigma-Aldrich) contained in a heated nozzle reservoir (ca. 313 K). The sample vapor was diluted in a stream of a carrier gas (Ne in Valladolid; Ne or He in Hamburg), at stagnation pressures of 2–3 bar. The pulsed injection system used a Parker (series 9) solenoid valve with a circular nozzle (0.8 mm). The final spectra were averaged for ca. 1 M cycles. The accuracy of the microwave frequency measurements is estimated to be better than 10 kHz.FTIR spectroscopy
The vapor pressure of BnOH (99%, Alfa Aesar) at 343 K was picked up by 1.5 bar of helium (estimated molar fraction of 0.2%)29 and expanded into vacuum through a slightly angled (60 × 0.2 mm2) slit nozzle at 363 K. The pulsed (1 min−1) expansion was probed by synchronized FTIR scans at 2 cm−1 resolution with a Bruker IFS 66 v/S spectrometer (double-sided-fast-return mode). About 300 repetitions were averaged for the spectrum within the filter window of 4000–2900 cm−1. More details on the Göttingen based setup are available in ref. 30 and references therein.Raman spectroscopy
Helium (1.0 bar) was enriched with the vapor pressure of BnOH (99%, Alfa Aesar) at 323 K (estimated molar fraction of 0.05%) and the gas mixture expanded through a (4 × 0.15) mm2 slit nozzle at 333 K into vacuum. The continuous expansion was probed at 2 mm distance by a Spectra Physics Millenia eV laser (532 nm, cw, 25 W). The scattered light was collected perpendicular to the laser and the nozzle flow via a camera lens and focused onto a one-meter monochromator (McPherson). Photons from spontaneous Stokes Raman scattering were co-added by a LN2-cooled CCD-camera (Princeton, PyLoN 400) over 3 min exposure time and averaged over 20 repetitions. The combination of laser and monochromator results in a resolution of about 1 cm−1. The spectrum was calibrated with neon vacuum transitions and spans the range 3700–3245 cm−1. More details on the Göttingen based setup are available in ref. 30 and references therein.Mass-resolved excitation spectroscopy
A detailed description of the experimental setup may be found in ref. 31. Briefly, BnOH (Sigma Aldrich, 98% purity) was introduced in a sample holder and heated to 353 K. Then, the sample compartment was connected to a flow of either He, Ne or Ar/He (1/10, 99.995% purity). Typical pressures of 1–3 bar were used. The BnOH/rare gas mixture was injected into the measuring compartment of an in-house designed mass spectrometer using a pulsed valve (Jordan Inc.). The subsequent expansion created at each valve opening was explored using a combination of UV and IR lasers. For REMPI experiments, the output of a Fine Adjustment Pulsare Pro laser operated with Coumarin 500/540A dye mixture was used as first color, while a second Quantel Q-Smart laser with Coumarin 540A dye was used as second (ionization) color. The same set of lasers was used in UV/UV hole burning experiments, but with a delay of 200 ns. In such experiments, the burning laser was operated at half the repetition rate of the probe laser to perform a real-time active subtraction, which substantially reduces the background noise. The output of an OPO (LaserVision, 5 mJ per pulse) was used in the IR/UV double resonance experiments, to scan the OH/CH stretching region. The IR laser was fired 120 ns in advance with respect to the ionization laser. Active subtraction was also used to minimize the signal's background.Computational
The conformational search for the monomer and aggregates of BnOH used manual and automated procedures, using both molecular mechanics (MMFF32) and Grimme's conformer-rotamer ensemble sampling tool (CREST33). The structures obtained were optimized using the B3LYP hybrid method,34 D3 two-body empirical dispersion terms (Becke–Johnson damping function35) and a computationally economic basis set (6-311++G(d,p) or def2-TZVP). Finally, those within 6 kJ mol−1 were re-optimized using the larger minimally augmented may-cc-pVTZ basis set,36 ultrafine integration grid and tight convergence criteria. No density fitting was applied. The vibrational modes were evaluated within the double-harmonic approximation. A full list of used keywords is available in the ESI.† Calculations were implemented in Gaussian 1637 and ORCA.38Results
BnOH monomer
The torsion of the hydroxymethyl group around the Cα–Cipso and Cα–O bonds gives BnOH a rich conformational landscape and internal dynamics (Fig. 1). The two flexible torsional degrees of freedom lead to two conformers. In the global minimum the HOCαCipso and the OCαCipsoCortho dihedrals adopt gauche–gauche arrangements with equal signs. This conformer, denoted for simplicity gauche, is four-fold degenerate with pairs of enantiomers on each side of the phenyl plane. The barrier for the interconversion of the gauche−/gauche+ pair on the same side of the plane is predicted to be shallow (2.0 kJ mol−1, B3LYP-D3(BJ)/may-cc-pVTZ, zero-point energy corrected). The electronic minimum structure of the second conformer with a trans arrangement of the HOCαCipso dihedral is calculated to be slightly asymmetric as well, but here the barrier for racemization vanishes with zero-point correction, so that an effectively Cs-symmetric structure with all heavy-atoms in one plane is expected. This trans conformer is 3.8 kJ mol−1 higher in energy, and the barrier for relaxation into the gauche conformer is only 1.7 kJ mol−1. A full-range two-dimensional relaxed scan of the conformational space is available in the ESI of ref. 16.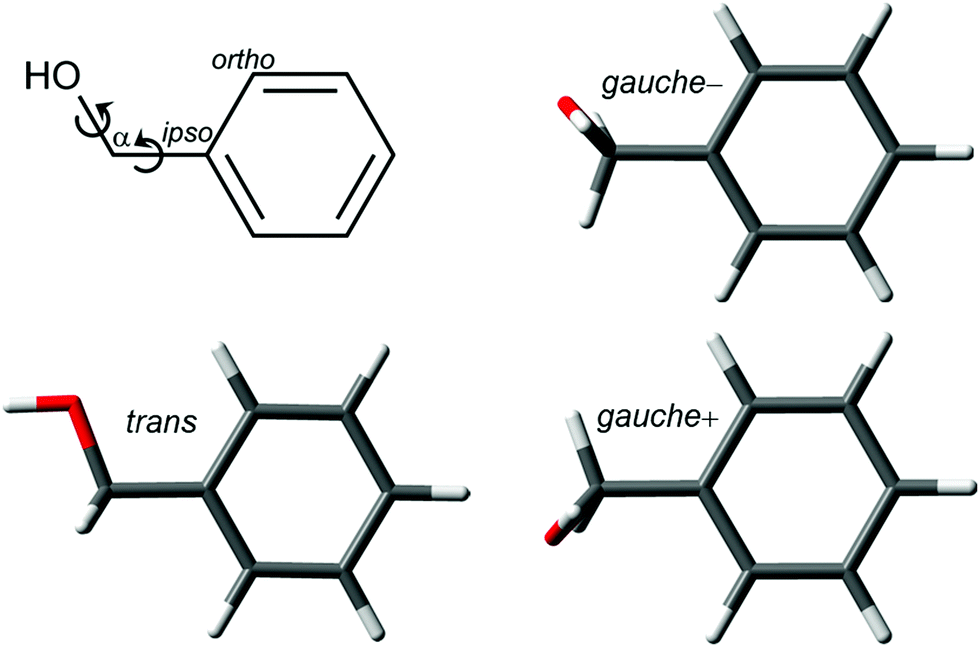 | ||
| Fig. 1 Two-dimensional structural formula of benzyl alcohol with atom labeling and conformational degrees of freedom as well as optimized conformers at the B3LYP-D3(BJ)/may-cc-pVTZ level. | ||
The relative stability of the rotamers was examined in detail in previous publications, and it was demonstrated that only the gauche conformation is detected in a supersonic expansion,13–18 just as in the gas phase at room temperature.39 The four-fold degenerate minima are connected by reflection on the ring plane or through a plane perpendicular to the ring. However, only this second large-amplitude motion creates an experimentally detectable inversion barrier (3.3 kJ mol−1 excluding torsional zero-point energy), since characteristic tunneling doublings can be observed in the rotational spectrum, strongly perturbed by Coriolis interactions.18 Exploration of the electronic spectroscopy of BnOH resulted in the spectrum in Fig. 2. It is a clean trace with very limited vibrational activity, in good agreement with previous publications.13,40 Tuning the UV laser at the wavenumber of the 0–0 transition (37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 528 cm−1) and scanning the IR laser, the spectrum in Fig. S1 (ESI†) was obtained. In that spectrum, the OH stretching vibration is clearly visible at 3644 cm−1. At the red end of the spectrum, several bands corresponding to the CH stretching modes also appear. Comparison with the DFT predictions shows also a good agreement, even for those run within the harmonic approximation and rescaled using a correction factor.
528 cm−1) and scanning the IR laser, the spectrum in Fig. S1 (ESI†) was obtained. In that spectrum, the OH stretching vibration is clearly visible at 3644 cm−1. At the red end of the spectrum, several bands corresponding to the CH stretching modes also appear. Comparison with the DFT predictions shows also a good agreement, even for those run within the harmonic approximation and rescaled using a correction factor.
 | ||
| Fig. 2 2c-REMPI spectra of BnOH and its aggregates up to the tetramer, between 37300 and 37750 cm−1. The ionization laser was tuned at 35714 cm−1. | ||
FTIR and Raman jet spectra of BnOH (available over the full OH stretching range in Fig. S2 of the ESI† and in ref. 16) both show an intense signal at 3648 cm−1 which is assigned to the OH stretching fundamental of the gauche conformer.14,15,17 A second less intense band at 3657 cm−1 in the Raman jet spectrum is attributed to the mostly Raman-active symmetric stretching fundamental of a water impurity. No signal assignable to the trans conformer is observed. According to a model for the prediction of OH stretching fundamentals of alcohol conformers,41 its expected position is 3688 cm−1 based on empirically shifted harmonic B3LYP-D3(BJ) and PBE0-D3(BJ) calculations.
BnOH dimer
The results from the manual conformational search for dimers of BnOH conducted in ref. 16 were verified here with the automatic search procedures. No previously unknown isomers within a potentially relevant range of 5 k J mol−1 were found. Within this span, all dimers are constituted exclusively from molecules in the gauche conformation. Following previous nomenclature,16 their relative chirality is stated as homochiral or heterochiral. This is followed by the acceptors of the formed hydrogen bonds, which can be a π-system, an oxygen atom O, or a ‘void’ V acceptor (equivalent to no acceptor, meaning a dangling hydroxy group). Further distinguished in the O–H⋯O bonded isomers are the two non-equivalent hydrogen bond acceptor oxygen free electron pairs E according to their dihedral EOCαCipso, denoted gauche (Og) or trans (Ot). The position of the isomer in the energy sequence is added with a Roman numeral, with ‘I’ representing the global minimum. The two most stable isomers are the homo- and heterochiral variants of the Ogπ motif. They are trailed by the two variants of the ππ motif. These four structures are shown with their OH stretching vibrational properties in Fig. 3. Further isomers with relative energies between 3 and 5 kJ mol−1 belong to the Otπ, OtV and OgV motifs and are listed in the ESI† (Tables S2–S4).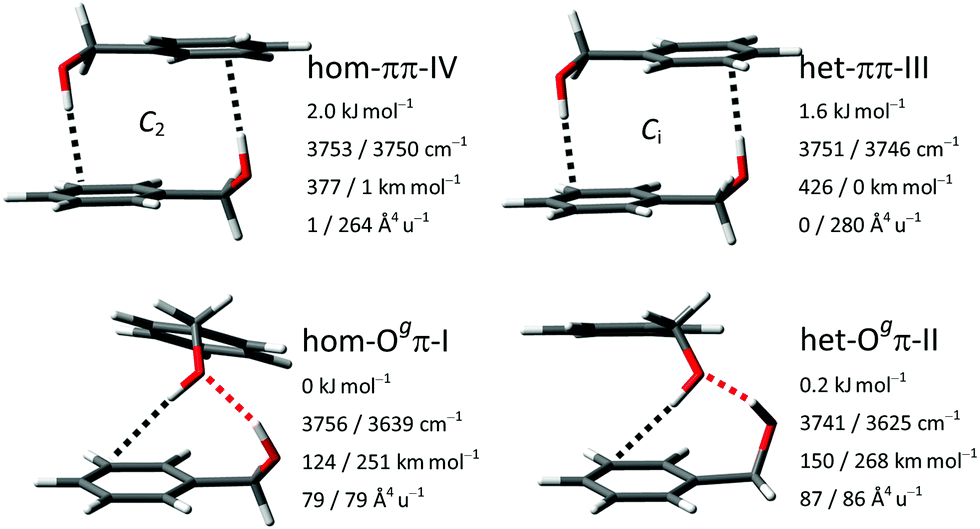 | ||
| Fig. 3 The four most stable BnOH dimer isomers at B3LYP-D3(BJ)/may-cc-pVTZ level with relative zero-point corrected energies, harmonic OH stretching wavenumbers and associated IR band strengths and Raman activities. Symmetry elements are indicated where present. | ||
Previous studies on aggregation of the BnOH dimer using FTIR and Raman spectroscopy demonstrated that at least three of these isomers are formed and detected in the free expansion: hom-Ogπ-I, het-Ogπ-II and het-ππ-III.14–16 Briefly, the assignment included two high-intensity bands at 3582 and 3514 cm−1, attributed to the global minimum hom-Ogπ-I, and two low-intensity bands at 3576 and 3504 cm−1 to het-Ogπ-II. By comparison with the aggregation of the permanently chiral but otherwise analogous 1-phenylethanol, it was verified that harmonic B3LYP-D3(BJ)/may-cc-pVTZ predicts the correct sign of the energy and spectral splittings between the diastereomeric pair. The presence of one band at 3598 cm−1 solely in the FTIR spectrum and of a second band, only in the Raman spectrum, slightly further downshifted to 3594 cm−1, revealed the presence of the symmetric ππ motif. Based on its lower calculated energy, its higher relaxation barriers and the comparison with 1-phenylethanol, het-ππ-III was preferentially assigned over hom-ππ-IV. Unresolved contributions of both ππ-isomers to the same signals could not be excluded, as the spectra of 1-phenylethanol indicate that they might be spectrally even more similar than calculated.
Most of the isomers are connected by low potential energy barriers, reducing the total number of species trapped during the cooling stage of the expansion. The leading interaction is the O–H⋯O hydrogen bond, accompanied by an O–H⋯π interaction of the second hydroxy group with the ring and a secondary C(sp2)–H⋯π interaction between the rings. Perhaps due to the higher tolerance of the O–H⋯π towards the angle of the attack of the donor molecule, or to a strength not far away from that of the O–H⋯O, the isomers formed by two O–H⋯π interactions are not very high in energy, and the heterochiral isomer was also detected using FTIR and Raman spectroscopy.
Re-examination of the BnOH dimer using REMPI spectroscopy resulted in the spectra collected in Fig. 2 and 4. The 0–0 transition is shifted 71 cm−1 to the red, and there is a significantly larger abundance of lines compared with the spectrum of the monomer. The UV/UV hole burning spectra in Fig. 4(A) demonstrate that at least two different isomers are contributing to the REMPI trace, with origin bands at 37457 and 37482 cm−1. Further search for additional isomers was unsuccessful.
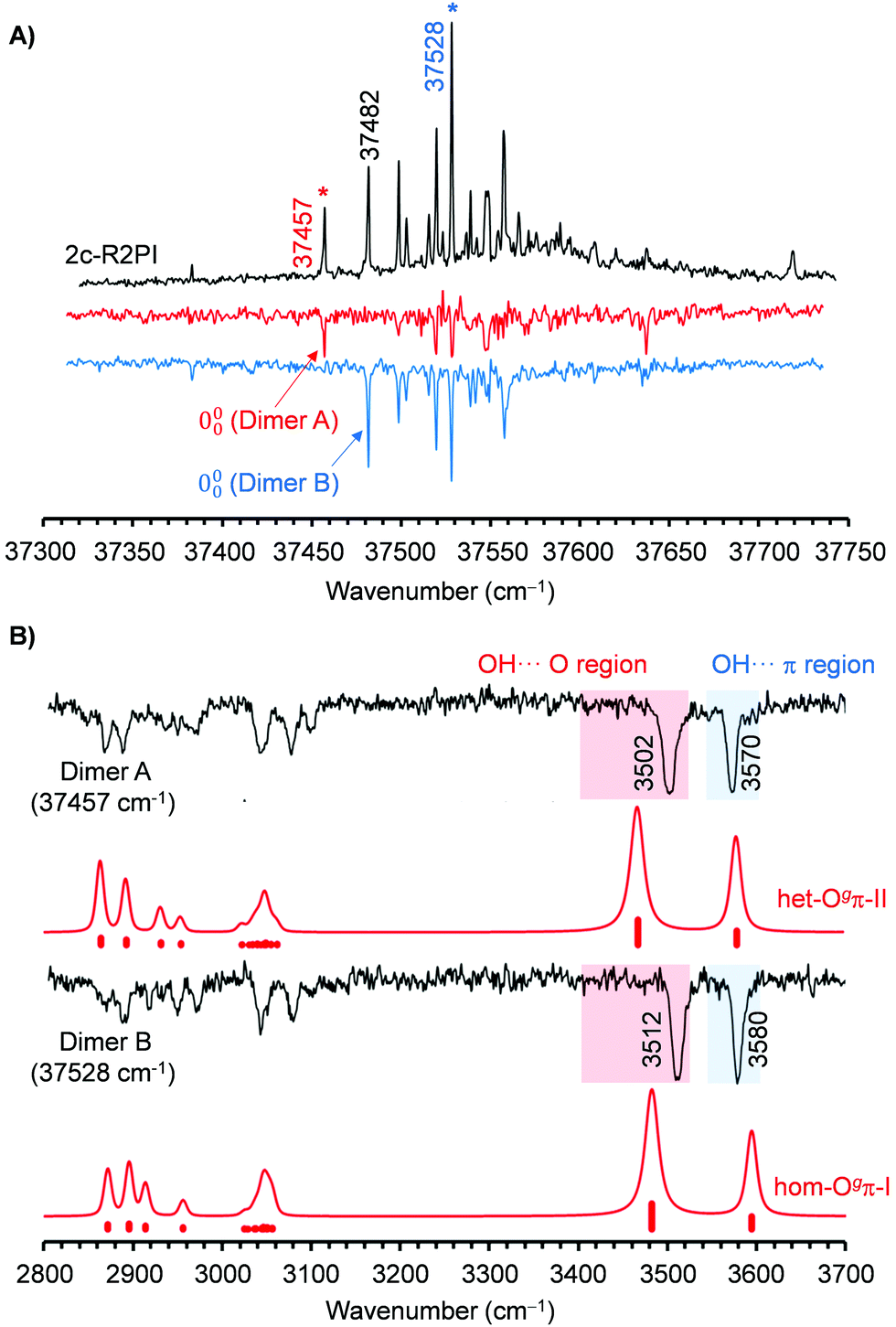 | ||
| Fig. 4 (A) Comparison between the REMPI spectrum of BnOH dimer and the two UV/UV hole burning traces obtained probing the transitions at 37457 and 37528 cm−1. (B) Comparison between the IR/UV spectrum of the two BnOH dimer isomers and the DFT predictions at the B3LYP-D3(BJ)/may-cc-pVTZ level. A factor of 0.957 was used to account for anharmonicity. The procedure used for the simulation, may be found in the ESI.† | ||
Tuning the UV laser to transitions of each isomer and scanning the IR laser resulted in the traces collected in Fig. 4(B). The spectra of the two isomers are very similar and contain a pair of transitions at 3512/3580 cm−1 and 3502/3570 cm−1, respectively. Comparison with the results reported by Medel and Suhm16 shows that the bands in the IR/UV spectrum are substantially broader and present slight shifts, but the general agreement is good enough to correlate the two isomers detected by IR/UV with the two most stable structures, hom-Ogπ-I and het-Ogπ-II, reported in ref. 16. The absence of the third isomer may be due to a reduced population under the conditions of our expansion.
Exploration of the BnOH dimer using MW spectroscopy produced unexpected results. Independent spectral analyses conducted in Hamburg and Valladolid with neon as carrier gas observed only a single isomer. Additional experiments in Hamburg using helium did not report additional species either. The rotational spectrum in Fig. 5 was fitted to a semi-rigid rotor Hamiltonian and delivered the experimental parameters in Table 1. The large number of transitions (332) and the presence of both R- and Q-branches for all (μa, μb, μc) selection rules produced a good determination of the rotational parameters and all quartic centrifugal distortion constants (experimental transitions are listed in Table S1, ESI†). Despite further search, no other isomers could be assigned in the spectrum. Similarly, trimers or tetramers were not observed, so we conclude that their overall numbers are too low. The experimental observation of the homochiral dimer further confirms that the calculations correctly predict this conformer to be the global minimum, despite the small energy difference with the heterochiral dimer.
 | ||
| Fig. 5 Microwave spectrum of BnOH (black positive trace) and simulated rotational transitions (quantum numbers JK−1,K+1) based on the fitted rotational parameters of the gauche monomer (blue negative trace) and the hom-Ogπ-I dimer (red negative trace) given in Table 1. This spectrum was recorded using neon as carrier gas and averaged over 1 M cycles. The simulation used a rotational temperature of 2 K and the predicted electric dipole moments. Panel (a) shows the full 6 GHz bandwidth, while sections of 200 MHz and 20 MHz are shown in panels (b) and (c), respectively. | ||
| Experiment | B3LYP-D3(BJ)/may-cc-pVTZ | ||
|---|---|---|---|
| hom-Ogπ-I | het-Ogπ-II | ||
| a Rotational constants (A, B, C), Watson's S-reduction centrifugal distortion constants (DJ, DJK, DK, d1, d2). b Electric dipole moments (μα, α = a, b, c). c Number of transitions (N) and rms deviation (σ) of the fit. d Relative energies corrected with the zero-point energy (ZPE). e Standard errors in units of the last digit. | |||
| A/MHza | 712.35893(14)e | 697.6 | 697.8 |
| B/MHz | 416.768155(82) | 441.2 | 456.3 |
| C/MHz | 367.109188(87) | 383.0 | 393.7 |
| D J /kHz | 0.14317(40) | 0.1651 | 0.0846 |
| D JK /kHz | −0.2637(11) | −0.375 | 0.012 |
| D K /kHz | 0.3970(15) | 0.518 | 0.052 |
| d 1/kHz | −0.01921(14) | −0.0289 | −0.0135 |
| d 2/kHz | −0.000490(60) | −0.00069 | −0.00153 |
| |μa|/Db | Medium intense | 1.7 | 1.3 |
| |μb|/D | Most intense | 3.0 | 2.4 |
| |μc|/D | Least intense | 0.6 | 1.1 |
| N | 332 | ||
| σ/kHz | 7.0 | ||
| ΔE/kJ mol−1d |
0 | 0.2 | |
The most likely explanation for the detection of a single isomer by MW spectroscopy is either that a different regime of the expansion was probed or that a different final temperature of the beam was achieved. In any case, the positive rotational detection of the dimer global minimum does not exclude the presence of other minor species, which might exist in the jet in smaller proportions. To find the reason for this difference, the REMPI spectrum of the BnOH dimer was recorded using different buffer gases (Fig. 6). Replacing He by Ne improves conformational cooling and may reduce the population of less stable isomers. Using 10% Ar/He instead, the more energetic collisions with Ar are sometimes able to even further transfer population from local minima into more stable isomers.42,43
 | ||
| Fig. 6 Comparison between the REMPI spectra of BnOH dimer recorded using He, Ne or 10% Ar/He as carrier gases. | ||
In the present system, replacing He by Ne strongly favored formation of larger clusters. m/z peaks up to the nonamer were clearly visible in the mass spectrum. However, the spectrum of the dimer did not show relevant differences. There was a variation in the relative intensity of some secondary bands, which were due to fragmentation from the trimer and/or from the BnOH:water 2:1 cluster. A slight reduction in the relative intensity of the 0–0 transition of the less stable isomer was observed, pointing to a lower final isomeric temperature in the beam. The main impact caused by the use of the Ar/He mixture as carrier gas was the increase in fragmentation from BnOH:water clusters. Careful probing of all the bands in the spectrum showed that no new isomers were trapped. There was a notable reduction in the intensity of the 0–0 transition of the less stable isomer, which is now hardly visible over the background created by fragmentation. This observation matches very well with the detection of a single isomer using MW spectroscopy. Therefore, one can conclude that the geometry used in the MW experiments produced an improved beam cooling. The opposite situation was the expansion used in the FTIR and Raman experiments. The higher isomeric temperature of those experiments was somehow an advantage, as it enabled detection of three isomers of BnOH dimer. The intermediate situation was the expansion used in the IR/UV and REMPI experiments, where two isomers were detected.
BnOH trimer
For the large conformational space of the trimers the manual and the two automatic searches provided complementary results, i.e., no single approach yielded all of the pooled isomers within a 5 kJ mol−1 range. Critically, the CREST search missed the global minimum. A similar issue was reported before for the (experimental) global minimum of the heterochiral trimer of propylene oxide.44 Again, no stabilization of the trans conformer by aggregation is found, as all isomers up to 4 kJ mol−1 contain exclusively gauche monomers. We extended the nomenclature established for the dimers by stating the relative chirality along the hydrogen bonding chain (in the direction from donor to acceptor) as well as the nature of the acceptor groups.The six most stable trimer isomers up to 3.3 kJ mol−1 are all variants of the OgOgπ motif, with a third molecule inserted in either hydrogen bond of the most stable Ogπ dimer motif. For the mixed trimer of benzyl alcohol with two water molecules, an analogous motif was concluded.45 The hydrogen bonding topology of the OgOgπ motif is a chain, but the trimer overall is cyclic, due to the donor and acceptor groups at both ends being covalently connected by a methylene linker. The four most stable isomers with relative energies up to 1.9 kJ mol−1 are shown in Fig. 7 and correspond to the four possible permutations of relative chirality along the hydrogen bonding chain: homhom-OgOgπ-I, homhet-OgOgπ-II, hethet-OgOgπ-III and hethom-OgOgπ-IV. The fifth and sixth most stable isomers at 2.5 and 3.3 kJ mol−1, respectively, also belong to the hethom-OgOgπ class but with somewhat different relative orientations of the phenyl rings. Further isomers up to 5 kJ mol−1 and their calculated properties are listed in the ESI.† Among them are isomers at 3.9, 4.5 and 4.7 kJ mol−1 with cyclic networks of three OH⋯O hydrogen bonds, with the last featuring C3 symmetry.
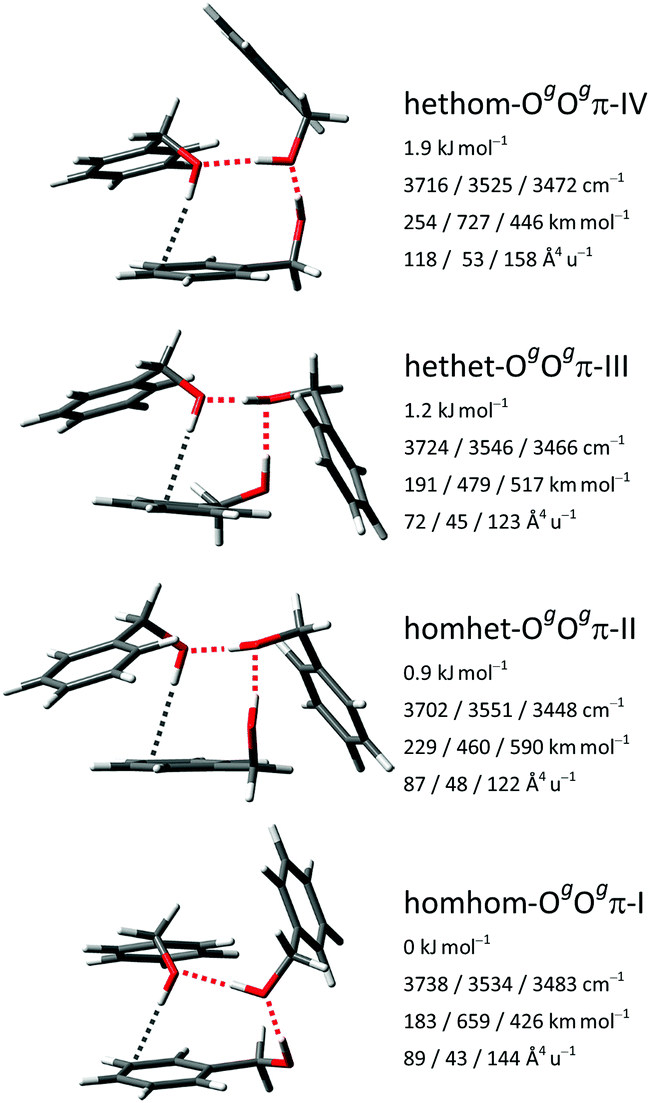 | ||
| Fig. 7 The four most stable BnOH trimer isomers at the B3LYP-D3(BJ)/may-cc-pVTZ level with relative zero-point corrected energies, harmonic OH stretching wavenumbers and associated IR band strengths and Raman activities. | ||
The respective three OH stretching fundamentals of the four energetically relevant OgOgπ isomers fall in three distinct spectral ranges: in a high-wavenumber window for the OH⋯π hydrogen bond at the end of the chain, in a medium-wavenumber region for the OH⋯O hydrogen bond at the start of the chain and finally in a strongly downshifted area for the OH⋯O hydrogen bond cooperatively enhanced from both sides in the middle of the chain. This localized picture is somewhat simplified as the two OH⋯O oscillators are slightly to moderately coupled to each other, depending on their difference in wavenumber in the respective isomer.
Examination of the REMPI spectrum of the trimer in Fig. 8 shows a complex and noisy spectrum that gains intensity as it progresses to the blue. Depending on the conditions of the expansion, formation of larger aggregates and/or clusters with water was favored. However, even under the most favorable conditions fragmentation from BnOH:water 3:1 could not be avoided. Surprisingly, no signal was observed in the mass channel corresponding to the monohydrated trimer.
 | ||
| Fig. 8 2-Color REMPI of the BnOH trimer recorded under different expansion conditions, together with the UV/UV hole burning (HB) traces recorded in the two bands labelled with asterisks. Depending on the conditions, fragmentation from the tetramer and/or from water clusters was observed. | ||
Thus, the trace recorded tuning the UV laser at 37538 cm−1 shows the bands coming from the 3:1 cluster, while the UV spectrum recorded tuning the probe laser at 37639 cm−1 contains the bands due to BnOH trimer. The position of the origin band of this isomer, which presents a red shift of 125 cm−1 with respect to the dimer, indicates that it presents a linear hydrogen bond network as the one predicted for the global minimum. Exploration of the REMPI spectrum in search for other isomers with the 0–0 transition shifted to the blue gave negative results, indicating that against what was observed for other systems, isomers with cyclic hydrogen bond networks were not formed. Formation of a dimer of an aromatic alcohol usually results in the shift of the electronic 0–0 transition to the red: both chromophores can be excited, but the electronic state in which the donor is excited is lower in energy, as excitation increases its acidity, reinforces its proton-donor ability and produces an increase in the stability of the dimer. When a third molecule is added and a cyclic hydrogen bond is formed, the increase in proton-donor ability upon electronic excitation is compensated by a reduction in its proton-acceptor capacity, and the shift in the 0-0 transition is reduced, sometimes moving it to the blue of the monomer's origin band.
Interestingly, probing the transitions at 37332 and 37384 cm−1, two different IR/UV spectra were obtained (Fig. 9), which means that both isomers share transitions in the UV spectrum and, therefore, their respective contributions cannot be separated using the UV/UV-double resonance technique.
 | ||
| Fig. 9 IR/UV spectra obtained tuning the UV laser at (A) 37332 and (B) 37384 cm−1 and comparison with the theoretical predictions for isomers I–IV computed at the at B3LYP-D3(BJ)/may-cc-pVTZ level. A scaling factor of 0.957 was used to account for the anharmonicity. | ||
Comparison of the IR/UV spectrum with the computed predictions for the most stable structures of the BnOH trimer shows a good agreement with the predictions for the two most stable structures. However, the predictions for other structures close in energy are not very different from the experimental spectra.
A more precise attribution of experimental spectra to the computed structures comes from the FTIR and Raman spectra shown in Fig. 10. The superior spectral resolution allowed us to unambiguously identify four different isomers, whose spectra contribute to the broader IR/UV traces. Most of the observed spectral activity, beyond signals already assigned to dimers, can be explained by simulations assuming a Boltzmann distribution of the calculated trimer structures with a conformational temperature of 100 K, as employed before for the simulation of the dimer signals.16 At this temperature, isomers beyond the four most stable are calculated to have no significant contributions and are omitted for clarity. Spectral positions for trimers are predicted within reasonable limits when applying the average corrections from matching experiment and harmonic calculations for the Ogπ dimer signals, separate for OH⋯π and OH⋯O bands.
 | ||
| Fig. 10 Comparison between experimental (second and third trace) and simulated (first and fourth trace) FTIR and Raman jet spectra for BnOH trimers. The simulations assume a conformational temperature of 100 K and identical vibrational and rotational partition functions. Used are a Gaussian FWHM of 3 cm−1 and shifts of −169 cm−1 for OH⋯π and −123 cm−1 for OH⋯O oscillators, which are the respective averages for matching experiment and theory for the Ogπ dimers. Bands assigned before to dimers are marked with ‘D’, to larger clusters with ‘>Trimer’. | ||
Similar to the dimers,16 the downshifts from the monomer are somewhat underestimated for the OH⋯π fundamentals and more strongly overestimated for the OH⋯O fundamentals by the calculations in harmonic approximation. Compared to the dimer signals of the same hydrogen bond type, the trimer bands are more strongly downshifted, reflecting increased cooperativity and/or reduced strain. Similarly enhanced downshifts for OH⋯OH⋯π chains in trimers relative to their OH⋯OH and OH⋯π components in dimers were observed before.46,47
The predicted drastic drop in relative intensity when switching from FTIR to Raman detection for the medium-wavenumber signals (3450–3400 cm−1) is confirmed. Assignments in this central spectral region are somewhat more ambiguous. Those proposed in Fig. 10 are based on the best agreement between experiment and simulation for wavenumber as well as intensity sequences and are compatible with those made with UV/IR for the two most stable isomers (Table 2). However, bands are partially overlapping with FTIR and Raman detection and often broad with UV/IR detection, so that the agreement in positions between the methods is imperfect and the matching of signals therefore not completely unambiguous. Already for the monomer band a discrepancy of 4 cm−1 is noted.
| Cluster size | Isomer | UV 0–0 | IR/UV | FTIR + Raman |
|---|---|---|---|---|
| Monomer | gauche | 37528 |
3644 | 3648 |
| Dimer | hom-Ogπ-I | 37482 |
3580 | 3582 |
| 3512 | 3514 | |||
| het-Ogπ-II | 37457 |
3570 | 3576 | |
| 3502 | 3504 | |||
| het-ππ-III | 3598 | |||
| 3594 | ||||
| Trimer | homhom-OgOgπ-I | 37384 |
3563 | 3561 |
| 3533 | ||||
| 3421 | 3417 | |||
| 3382 | 3374 | |||
| homhet-OgOgπ-II | 37332 |
3533 | 3538 | |
| 3439 | 3441 | |||
| 3389 | ||||
| 3334 | 3333 | |||
| hethet-OgOgπ-III | 3555 | |||
| 3433 | ||||
| 3348 | ||||
| hethom-OgOgπ-IV | 3546 | |||
| 3410 | ||||
| 3364 | ||||
| Tetramer | homhomhom-OgOgOgOg-I | 37476 |
3353 | |
| 3313 | ||||
| 3253 |
A rather intense IR band at 3426 cm−1 (question mark in Fig. 10) remains unexplained with this assignment. It was observed before with similar relative intensity in older spectra with a different BnOH sample,15 so that an impurity seems unlikely. Its spectral position suggests a cooperatively enhanced OH⋯O hydrogen bond and its concentration dependence15 likely another trimer isomer. It is unclear whether this isomer might have been missed by all three conformational search strategies or has a much higher population than expected because of kinetic trapping or theory deficits. However, for a trimer with these properties one expects multiple bands with similar relative intensities (FTIR or/and Raman), even with the highest possible C3 symmetry. Experimental partners for this band are missing, unless one assumes spectral coincidence with one or more of the intense bands of homhom-OgOgπ-I. None of the closer investigated isomers up to 5 kJ mol−1 has a matching spectral signature. Alternatively, the calculated spectral sequence of the four energetically relevant trimer isomers in this spectral region may contain larger uncertainties than expected. If such is the case, the band intensity suggests an assignment to the second most stable isomer. Considering the good agreement for relative band positions and intensities in the other spectral regions, this seems unlikely. Also, it would conflict with the IR/UV assignments for the two most stable isomers.
In conclusion, four isomers of benzyl alcohol trimer were assigned. The global minimum structure is confirmed to be homochirally synchronized – as it was anticipated already from the similarity between the spectra of benzyl alcohol and enantiopure 1-phenylethanol.16
BnOH tetramer
For the tetramers of benzyl alcohol, the dominance of a cyclic arrangement of four OH⋯O hydrogen bonds and with all phenyl rings being located at the same side of an approximate plane defined by the four oxygen atoms is predicted. The global minimum, isomer homhomhom-OgOgOgOg-I, features C2 symmetry with two pairs of slightly distorted gauche conformers with overall synchronized handedness. A conceivable C4-symmetric structure is a transition state at 4.7 kJ mol−1 for the interconversion between these pairs. Systematic variation of monomer conformers within this hydrogen bonding pattern leads only to a single additional reasonably competitive isomer at 1.5 kJ mol−1, which can be classified as homhomhet-OgOgOgOt-II with the chirality of one gauche conformer being inverted. These two structures and their OH stretching vibrational properties are given in Fig. 11. Both of these low-energy isomers were found by optimization of manual guesses but missed by the automated molecular mechanics conformational search. The third and fourth most stable isomers again retain three homochirally synchronized gauche conformers with the fourth one converted to trans at a cost of 3.7 and 4.8 kJ mol−1, respectively, again similar to the one needed for the isolated monomer. Isomers III and IV share a hydrogen bonding motif but differ in the arrangement of the phenyl rings. An S4-symmetric hethethet-OgOgOgOg isomer with pairs of phenyl rings interacting above and below the oxygen plane has a calculated relative energy of 28 kJ mol−1. Isomers with an OOOπ chain have relative energies not lower than 14 kJ mol−1. With all four OH stretching oscillators being in very similar chemical environments in homhomhom-OgOgOgOg-I, they are strongly coupled to a high-wavenumber overall concerted anti-phase movement, a low-wavenumber concerted in-phase movement and two near-degenerate medium-wavenumber anti-phase movements of the two symmetry-equivalent pairs. Similar to the C2-symmetric hom-ππ-IV dimer, the fundamental transitions are either nearly exclusively infrared (pair movements) or Raman active (fully concerted movements). The mild symmetry reductions in isomers II and IV impact the vibrational properties only slightly, while the infrared band strength is more evenly distributed between the OH stretching modes in the more strongly asymmetric isomer III (Fig. 12). The REMPI spectrum of the tetramer (Fig. 1) shows a 0–0 transition shifted to the blue from that of the trimer, anticipating the formation of cyclic hydrogen bond networks. The whole spectrum contains a limited number of vibronic transitions built on top of a broad absorption that may be due to fragmentation from larger clusters. The weak signal and the presence of the background hampered recording UV/UV hole burning experiments. Therefore, all the discrete transitions in the spectrum were probed, looking for variations in the IR/UV trace. In all cases, the spectrum in Fig. 12 was obtained, which shows a central strong band at 3313 cm−1 containing the contribution from several OH stretches. That band is flanked by a shoulder at 3353 cm−1 and a satellite band at 3253 cm−1. Further to the red, the CH stretches appear. This spectrum clearly is the signature of the formation of a cyclic hydrogen bond network, as all the OH stretches are grouped together, indicating that they are immersed in a similar environment. The calculated spectra of isomers I, II and IV all feature a slightly asymmetric dominant band, but underestimate the observed relative intensities of the shoulder and the satellite. Isomer III could account better for these features; however, its predicted intense high-wavenumber band is not observed. Based on its lowest predicted energy, we very tentatively assign isomer I and speculate that the double-harmonic approximation might be stretched beyond its limits for the finer details of the vibrational coupling pattern.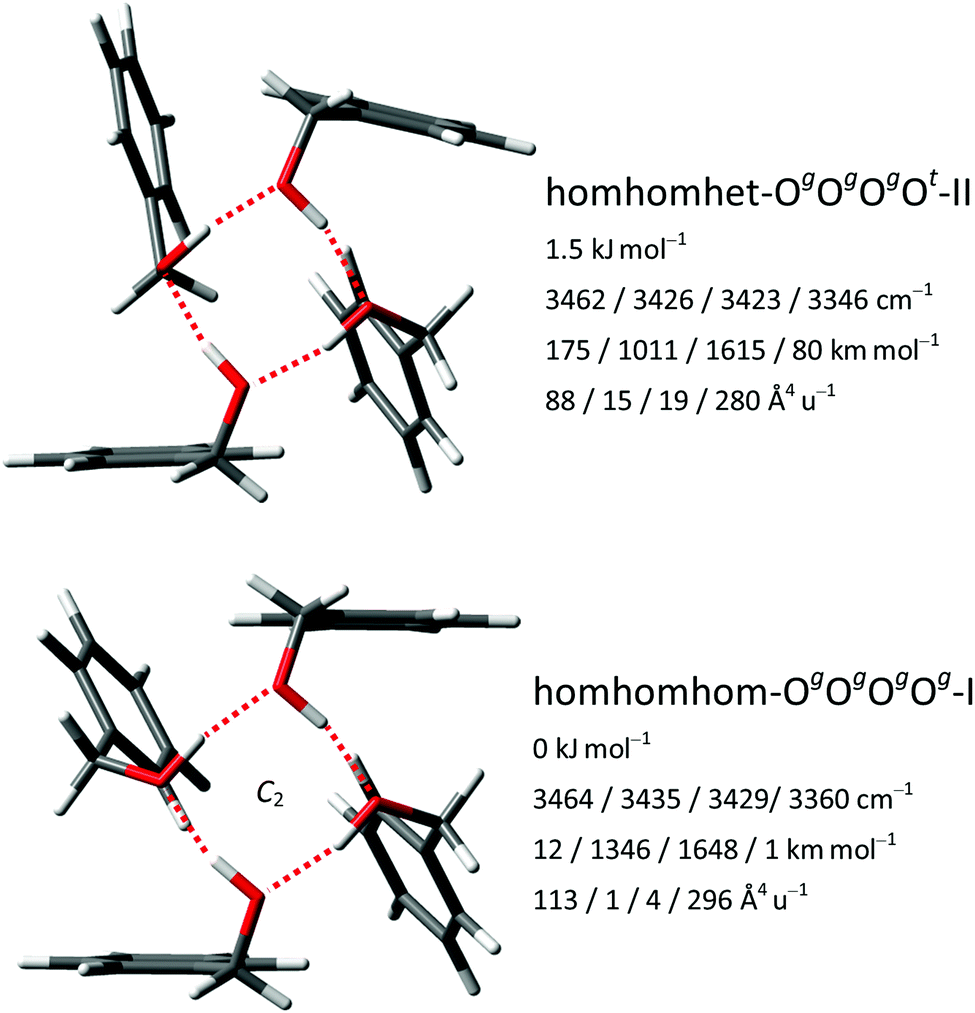 | ||
| Fig. 11 The two most stable BnOH tetramer isomers at the B3LYP-D3(BJ)/may-cc-pVTZ level viewed along the (for II approximate) C2-symmetry axis with relative zero-point corrected energies, harmonic OH stretching wavenumbers and associated IR and Raman activities. | ||
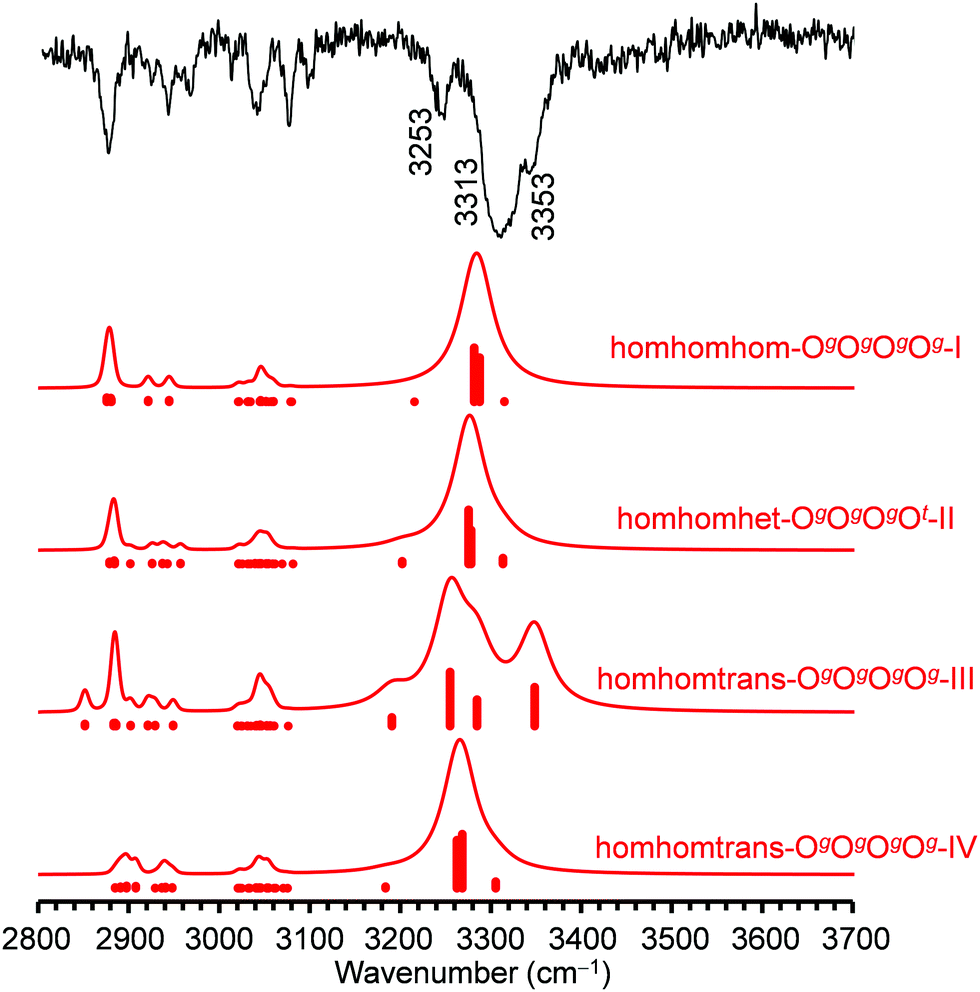 | ||
| Fig. 12 Comparison between the IR/UV spectrum, probed at 37476 cm−1, and the predictions at the B3LYP-D3(BJ)/may-cc-pVTZ level for the four most stable structures of the BnOH tetramer. A scaling factor of 0.957 was used to account for the anharmonicity. | ||
For the FTIR and Raman simulation of the tetramer spectra (Fig. 13), a Lorentzian broadening with a full width at half maximum of 24 cm−1 is used, which was found suitable for describing the tetramer spectra of methanol and ethanol. An empirical shift of the harmonic wavenumber might not be as transferable between alcohols, as it is substantially different for methanol (−82.5 cm−1) and ethanol (−103 cm−1).48 Here, we use again the same correction as for the trimers, derived from the OH⋯O signals of the dimers (−123 cm−1). Comparison with the experimental FTIR spectra makes an assignment to the broad band formation centered at about 3310 cm−1 plausible, which overlaps on the high-wavenumber side with the trimer bands and on the low-wavenumber side with absorption likely caused by even larger clusters. The absence of this band formation in the Raman spectrum agrees with the prediction for the tetramer. The concerted out-of-phase vibrations expected at roughly 3340 cm−1 for both considered isomers with some Raman activity might be obstructed by the trimer bands in this region, especially the trimer band at 3333 cm−1 appears noticeably broadened compared to the one at 3374 cm−1. Part of the concerted in-phase fundamentals with higher Raman activity might be visible at the edge of the monochromator range in the 3260–3240 cm−1 region. In the same area a broad scattering for racemic 1-phenylethanol is observed, while it is upshifted by 30 cm−1 for the enantiopure compound.16 These assignments are very tentative due to the lack of conformational and size resolution. Such selectivity could be achieved for the further investigation of these weakly IR but more strongly Raman active bands with ionization loss (or gain) stimulated Raman spectroscopy,49 in analogy to the IR/UV technique used here.
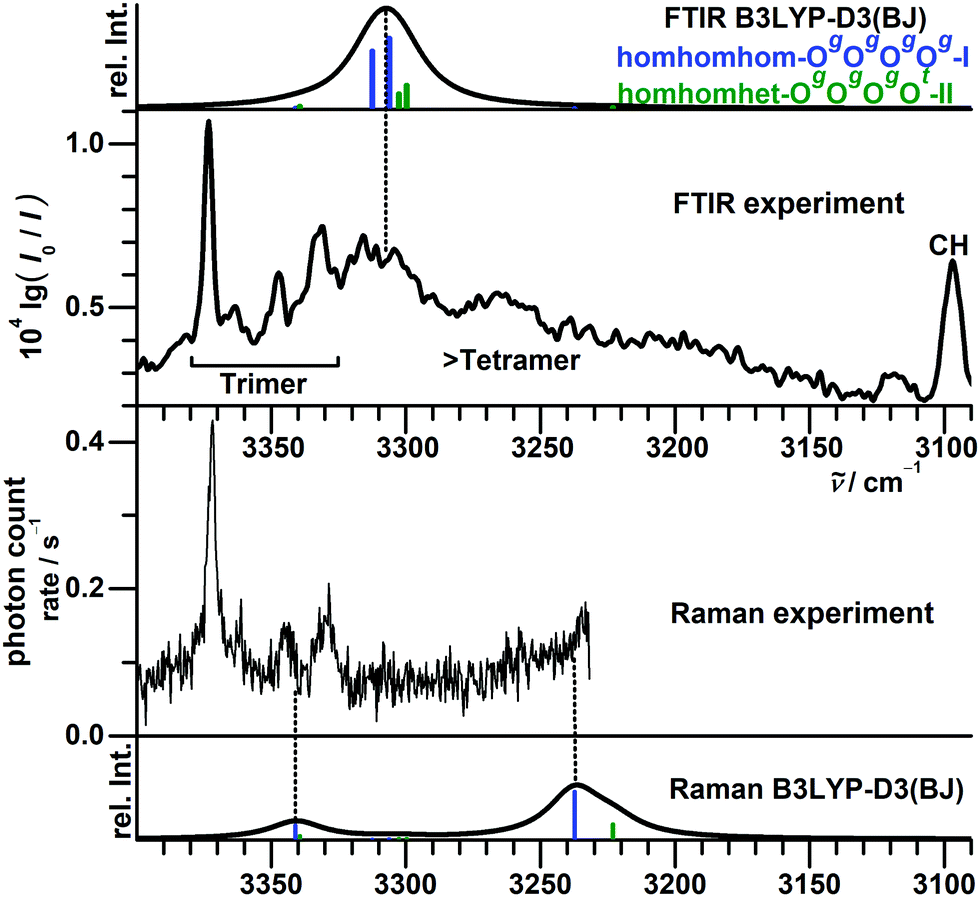 | ||
| Fig. 13 Comparison between experimental (second and third trace) and simulated (first and fourth trace) FTIR and Raman jet spectra of BnOH tetramers. The simulations assume a conformational temperature of 100 K and identical vibrational and rotational partition functions except for the rotational symmetry number. Used are a Lorentzian FWHM of 24 cm−1 and a uniform shift of −123 cm−1. The latter is the average value needed for matching the OH⋯O signals of the BnOH dimers. | ||
Discussion
The signature intermolecular interaction in homomolecular dimers of simple compounds containing hydroxy groups is the OH⋯OH hydrogen bond, which dominates the association in water and small saturated alcohols.50 The presence of other functional groups can modify the molecular interplay, e.g. by acting as an additional or alternative acceptor for a hydrogen bond. Due to the relative torsional rigidity of phenol there is only a single classical OH⋯OH hydrogen bond in its dimer, but dispersion interactions between the phenyl groups already deform the geometry substantially. The exact structure is therefore a challenge for electronic structure methods.12 For benzyl alcohol the flexibility introduced by the methylene linker between the hydroxy and the phenyl group enables the additional formation of an OH⋯π hydrogen bond. This cooperative pattern is also found in the most stable dimers of 1-phenylethanol,16 1-indanol47 and propargyl alcohol.51,52 Analogous motifs are experimentally observed when the additional acceptor is a halogen atom, as in fluorinated23,53,54 and chlorinated53 derivatives of ethanol, and occasionally also with an ether55 or carbonyl56,57 oxygen atom.An additional acceptor group also enables the competition with an often symmetric motif with two isolated hydrogen bonds, with the balance between the motifs depending strongly on the strength of the acceptor. The topology with isolated hydrogen bonds is not viable with a halogen acceptor, is typically moderately disfavored with a π acceptor16,47 (as observed for benzyl alcohol), is moderately to strongly favored with an oxygen acceptor10,56,58,59 and dominates with a nitrogen acceptor.60–62
Strong dispersion interaction between the residues, conformational rigidity and/or steric crowding can also lead to further motifs in which hydrogen bonds are reduced in number and/or strength. Further examples, beyond the already mentioned phenol dimer, are the dimers of 1-naphthol,63 propofol,64 3-chlorophenol65 and racemic (but not enantiopure) methyl mandelate.66 For benzyl alcohol such structures are calculated to be either high in energy or no minimum at all, and are not observed.
In the trimers of water67 and saturated alcohols48,68 a cyclic hydrogen bonding topology is formed with two of the residues on the same side of the plane spanned by the oxygen atoms and the third one on the opposite side (‘chair’ arrangement). This orientational preference is already pronounced for sterically undemanding water and increases only moderately when moving to methanol and tert-butyl alcohol (calculated advantages of 2.4, 2.8 and 3.0 kJ mol−1, respectively, at B3LYP-D3(BJ)/may-cc-pVTZ level). The ROO′R′ dihedral angle between molecules with the residues on opposite sides of the OOO plane resembles the one in the dimer (about 120°), suggesting a mainly electrostatic explanation. This preference can be inverted if attractive interactions between the residues become strong enough. Examples for the realization of this alternative 'bowl' arrangement with all three residues on the same side are the trimers of phenol,12 2,2,2-trifluoroethanol,69 1,1,1,3,3,3-hexafluoro-propan-2-ol70 and enantiopure methyl lactate.71 This preference is also predicted for benzyl alcohol. However, if an alternative hydrogen bond acceptor is present, the cyclic hydrogen bonding topology might be replaced with a chain. This is the global minimum structure for the trimers of benzyl alcohol, 1-indanol47 and 2-fluoroethanol.72 Even though the chain topology involves a loss in cooperativity and the replacement of an OH⋯O hydrogen bond with a typically weaker OH⋯π or OH⋯F hydrogen bond, it is apparently enabled by the release of ring strain.
Accordingly, the reduced ring strain in the tetramer strongly encourages the cyclic hydrogen bonding pattern. Again, different relative arrangements of the residues on the two sides of the (approximate) oxygen plane are possible. For water73 and small saturated alcohols the orientation of the residues alternates along the cycle, typically resulting in S4-symmetry. In contrast, in the benzyl alcohol tetramer the dispersion interaction between the phenyl rings brings them to the same side of the plane. Similar C2-symmetric structures were found before for the tetramers of 1-indanol,47 2-fluoroethanol74 and enantiopure methyl lactate.75 Nevertheless, for the latter compound a spectacular chirality-induced switch to four isolated hydrogen bonds occurs in the racemic tetramer,75 probably enabled by the superior acceptor strength of the carbonyl over the hydroxy group and the compactness of the structure.
According to a survey by Taylor and Macrae,76 primary monohydric alcohols have a strong tendency (73% of the 15 compounds in the sample) to form infinite chains of OH⋯O hydrogen bonds in the crystal structure, followed by cyclic (13%) and isolated (7%) arrangements and the absence (7%) of OH⋯O hydrogen bonds. The crystal structure of benzyl alcohol can be thus classified as a typical case with hom-Ot chains in our nomenclature.77 Yet again, the molecules adopt homochirally synchronized gauche conformations − like the respective most stable isomer of the dimer, trimer and tetramer, as concluded here. One difference relative to the small clusters is a switch from Og to Ot coordination. While in this crystal structure all molecules and hydrogen bonds are equivalent, an earlier FTIR and Raman study of solid BnOH concluded in contrast two different types of hydrogen bonds.78
2,2,2-Trifluoroethanol prefers as well a homochiral synchronization of its gauche conformers in the dimer23,79 and trimer,69 but there is also the participation of an achiral trans conformer in the latter. It features heterochirality in the crystal, but a switch back to homochirality can be induced by pressure.80 2-Fluoroethanol instead has a heterochiral preference in its dimer54,81 and trimer,72 but a homochiral one in its tetramer.74 Its crystal structure appears to be yet undetermined. For ethanol, the dimer6 is homochirally synchronized, but for the trimer48 and the tetramer48 a heterochiral preference is predicted – as it is observed in the crystal structure.82 There is a one-third participation of the trans conformer in the trimer and one-half in the crystal.
The results from a SAPT2(+3)/aug-cc-pVDZ//B3LYP-D3(BJ)/may-cc-pVTZ analysis of the interaction energy for the four most stable BnOH dimers are summarized in Table 3.
| Energetic term | hom-Ogπ-I | het-Ogπ-II | het-ππ-III | hom-ππ-IV |
|---|---|---|---|---|
| Electrostatic | −58.2 | −59.2 | −37.8 | −36.5 |
| + Induction | −18.6 | −20.2 | −17.9 | −16.4 |
| + Dispersion | −54.2 | −55.2 | −52.6 | −52.4 |
| + Exchange | 89.1 | 94.0 | 69.3 | 68.0 |
| = Total interaction | −41.9 | −40.8 | −39.0 | −37.4 |
| + Deformation | 1.6 | 1.3 | 4.7 | 2.8 |
| + Zero-point change | 6.5 | 6.5 | 3.9 | 4.3 |
| = Total binding | −33.8 | −33.0 | −30.3 | −30.3 |
Both the absolute values of the electrostatic and the inductive terms are found to be larger for het-Ogπ-II than for hom-Ogπ-I. This well correlates with the lower OH stretching wavenumbers and higher IR and Raman activities for het-Ogπ-II at B3LYP-D3(BJ) level, indicating stronger hydrogen bonds. The London dispersion interaction is larger as well for het-Ogπ-II, which in turn correlates with all of its three calculated rotational constants being larger, reflecting a more compact structure. However, this comes at the price of substantially higher exchange repulsion, which ultimately tips the scale to the homochiral isomer being the global minimum. Deformation and zero-point vibrational energy changes relative to two relaxed gauche monomers at B3LYP-D3(BJ) level are very similar and thus only slightly impact the chirodiastaltic energy.
The isomers of the ππ motif have smaller inductive and far lesser electrostatic terms than their Ogπ competitors, reflecting the absence of an OH⋯O hydrogen bond. Dispersive terms are also somewhat smaller. This is mostly but not entirely compensated by much lower exchange repulsion. Overall, the ππ motif has much weaker overall interaction between its constituent molecules, both attractive and repulsive. Higher deformation and lower zero-point vibrational energies roughly compensate each other. Again, higher electrostatic and inductive terms for the heterochiral isomer correlate with lower calculated OH stretching wavenumbers and higher spectral activities. The dispersive terms for het-ππ-III and hom-ππ-IV are very similar, so are their rotational constants (within ±4 MHz of each other, detailed values in the ESI†).
Visualization with non-covalent interaction (NCI) analysis of the spatial distribution confirms the localized hydrogen bonds and the more widespread weak dispersive attraction between the phenyl rings, with details given in the ESI.† The gauche monomer conformation appears to be not stabilized by an intramolecular OH⋯π interaction.
Conclusions
Here we presented a detailed spectroscopic and computational analysis of the first instants of the aggregation of benzyl alcohol. Using MW spectroscopy, accurate structural parameters of the monomer and the dimer were obtained, confirming that the most stable structure predicted by the DFT calculations is, indeed, the global minimum. Careful analysis of FTIR, Raman and IR/UV data allowed us to confirm previously published data and to, in addition, characterize the trimer and tetramer stages of the aggregation. Up to that size, the cluster growth is guided by a combination of hydrogen bond and dispersive forces between the aromatic rings. While OH⋯OH⋯π and OH⋯OH⋯OH⋯π are the leading topologies in the dimer and trimer, respectively, addition of the fourth monomer collapses the structure to form a cyclic, symmetric and elegant isomer. This resembles and bridges the previously observed preferences of the dimer and the crystal towards a homochiral synchronization of exclusively gauche conformers. The complex conformational landscape, with numerous minima of very similar stability and the abundant experimental data provided in this work, make of benzyl alcohol aggregation an excellent system to test and fine tune the most advanced functionals.Author contributions
Robert Medel: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review and editing; Ander Camiruaga: formal analysis, investigation, methodology, visualization, writing – original draft, writing – review; Rizalina Tama Saragi: formal analysis, investigation, methodology, visualization, writing – original draft, writing – review; Pablo Pinacho: formal analysis, investigation, writing – review; Cristóbal Pérez: formal analysis, investigation; Melanie Schnell: funding acquisition, methodology, supervision, writing – review and editing; Alberto Lesarri: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review and editing; Martin A. Suhm: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review and editing; José A. Fernández: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Göttingen part of the project was partly funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 271107160/SPP1807. We thank M. Lange and E. K. M. M. Sennert for the measurement of the FTIR spectrum and E. Meyer for help with the measurement of the Raman spectrum. Computational resources from the GWDG and the Göttingen Faculty of Chemistry (DFG – 405832858/INST 186/1294-1 FUGG) are acknowledged. We thank the Göttingen chemistry workshops for valuable support. This publication was supported financially by the Open Access Grant Program of the DFG and the Open Access Publication Fund of the University of Göttingen. The Bilbao and Valladolid groups acknowledge funding from the Spanish Ministerio de Ciencia e Innovación (MICINN-FEDER PGC2018-098561-B-C21 and PGC2018-098561-B-C22). Bilbao's group also thank the SGIKER (UPV/EHU, MICIU-FEDER) for the computational and laser resources. The Hamburg part of this work was financially supported by the Deutsche Forschungsgemeinschaft (SCHN1280/4-2, project number 271359857) in the context of the priority program SPP 1807 “Control of London dispersion interactions in molecular chemistry”. P. Pinacho would like to thank the Alexander von Humboldt Foundation for a postdoctoral fellowship.Notes and references
- P. Hobza and K. Müller-Dethlefs, Non-Covalent Interactions, Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- Noncovalent Forces, ed. S. Scheiner, Springer International Publishing, Cham, Switzerland, 2015, vol. 19 Search PubMed.
- C. Pérez, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Hydrogen bond cooperativity and the three-dimensional structures of water nonamers and decamers, Angew. Chem., Int. Ed., 2014, 53, 14368–14372 CrossRef PubMed.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Broadband Fourier transform rotational spectroscopy for structure determination: The water heptamer, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- J. P. I. Hearn, R. V. Cobley and B. J. Howard, High-resolution spectroscopy of induced chiral dimers: A study of the dimers of ethanol by Fourier transform microwave spectroscopy, J. Chem. Phys., 2005, 123, 1–7 CrossRef PubMed.
- T. N. Wassermann and M. A. Suhm, Ethanol monomers and dimers revisited: A Raman study of conformational preferences and argon nanocoating effects, J. Phys. Chem. A, 2010, 114, 8223–8233 CrossRef CAS PubMed.
- D. Loru, I. Peña and M. E. Sanz, Ethanol dimer: Observation of three new conformers by broadband rotational spectroscopy, J. Mol. Spectrosc., 2017, 335, 93–101 CrossRef CAS.
- M. S. Snow, B. J. Howard, L. Evangelisti and W. Caminati, From transient to induced permanent chirality in 2-propanol upon dimerization: A rotational study, J. Phys. Chem. A, 2011, 115, 47–51 CrossRef CAS PubMed.
- A. K. King and B. J. Howard, A microwave study of the hetero-chiral dimer of butan-2-ol, Chem. Phys. Lett., 2001, 348, 343–349 CrossRef CAS.
- A. Maris, B. M. Giuliano, D. Bonazzi and W. Caminati, Molecular recognition of chiral conformers: A rotational study of the dimers of glycidol, J. Am. Chem. Soc., 2008, 130, 13860–13861 CrossRef CAS PubMed.
- M. Juanes, I. Usabiaga, I. León, L. Evangelisti, J. A. Fernández and A. Lesarri, The six isomers of the cyclohexanol dimer: A delicate test for dispersion models, Angew. Chem., Int. Ed., 2020, 59, 14081–14085 CrossRef CAS PubMed.
- N. A. Seifert, A. L. Steber, J. L. Neill, C. Pérez, D. P. Zaleski, B. H. Pate and A. Lesarri, The interplay of hydrogen bonding and dispersion in phenol dimer and trimer: Structures from broadband rotational spectroscopy, Phys. Chem. Chem. Phys., 2013, 15, 11468–11477 RSC.
- H. S. Im, E. R. Bernstein, H. V. Secor and J. I. Seeman, Supersonic jet studies of benzyl alcohols: Minimum energy conformations and torsional motion, J. Am. Chem. Soc., 1991, 113, 4422–4431 CrossRef CAS.
- M. Mons, E. G. Robertson and J. P. Simons, Intra- and intermolecular π-type hydrogen bonding in aryl alcohols: UV and IR-UV ion dip spectroscopy, J. Phys. Chem. A, 2000, 104, 1430–1437 CrossRef CAS.
- J. Altnöder, S. Oswald and M. A. Suhm, Phenyl- vs. cyclohexyl-substitution in methanol: Implications for the OH conformation and for dispersion-affected aggregation from vibrational spectra in supersonic jets, J. Phys. Chem. A, 2014, 118, 3266–3279 CrossRef PubMed.
- R. Medel and M. A. Suhm, Understanding benzyl alcohol aggregation by chiral modification: The pairing step, Phys. Chem. Chem. Phys., 2020, 22, 25538–25551 RSC.
- C. E. H. Dessent, W. D. Geppert, S. Ullrich and K. Müller-Dethlefs, Ionization-induced conformational changes: REMPI and ZEKE spectroscopy of salicyl and benzyl alcohol, Chem. Phys. Lett., 2000, 319, 375–384 CrossRef CAS.
- K. A. Utzat, R. K. Bohn, J. A. Montgomery, H. H. Michels and W. Caminati, Rotational spectrum, tunneling motions, and potential barriers of benzyl alcohol, J. Phys. Chem. A, 2010, 114, 6913–6916 CrossRef CAS PubMed.
- C. R. Quade, M. Liu and C. F. Liu, An IAM calculation of the torsional–rotational term values for selected species of –OH and –OD ethyl alcohol, J. Mol. Spectrosc., 2000, 201, 319–320 CrossRef CAS PubMed.
- A. Zehnacker and M. A. Suhm, Chirality recognition between neutral molecules in the gas phase, Angew. Chem., Int. Ed., 2008, 47, 6970–6992 CrossRef CAS PubMed.
- I. Alkorta and J. Elguero, Discrimination of hydrogen-bonded complexes with axial chirality, J. Chem. Phys., 2002, 117, 6463–6468 CrossRef CAS.
- N. Borho and Y. Xu, Tailoring the key in a molecular lock-and-key model system: The propylene oxide⋯2-fluoroethanol complex, J. Am. Chem. Soc., 2008, 130, 5916–5921 CrossRef CAS PubMed.
- J. Thomas and Y. Xu, Chirality synchronization in trifluoroethanol dimer revisited: The missing heterochiral dimer, J. Phys. Chem. Lett., 2014, 5, 1850–1855 CrossRef CAS PubMed.
- S. T. Shipman and B. H. Pate, in Handbook of High-resolution Spectroscopy, ed. F. Merkt and M. Quack, John Wiley & Sons, Ltd, New York, 2011, pp. 801–828 Search PubMed.
- J.-U. Grabow, in Handbook of High-resolution Spectroscopy, ed. F. Merkt and M. Quack, John Wiley & Sons, Ltd, New York, 2011, pp. 723–799 Search PubMed.
- D. Schmitz, V. Alvin Shubert, T. Betz and M. Schnell, Multi-resonance effects within a single chirp in broadband rotational spectroscopy: The rapid adiabatic passage regime for benzonitrile, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS.
- J. L. Neill, S. T. Shipman, L. Alvarez-Valtierra, A. Lesarri, Z. Kisiel and B. H. Pate, Rotational spectroscopy of iodobenzene and iodobenzene-neon with a direct digital 2–8 GHz chirped-pulse Fourier transform microwave spectrometer, J. Mol. Spectrosc., 2011, 269, 21–29 CrossRef CAS.
- J. C. McGurk, T. G. Schmalz and W. H. Flygare, Fast passage in rotational spectroscopy: Theory and experiment, J. Chem. Phys., 1974, 60, 4181–4188 CrossRef CAS.
- Vapor Pressure and Antoine Constants for Oxygen Containing Organic Compounds, ed. K. R. Hall, Springer-Verlag, Berlin/Heidelberg, 2000, vol. 20B Search PubMed.
- B. Hartwig, M. Lange, A. Poblotzki, R. Medel, A. Zehnacker and M. A. Suhm, The reduced cohesion of homoconfigurational 1,2-diols, Phys. Chem. Chem. Phys., 2020, 22, 1122–1136 RSC.
- I. León, E. J. Cocinero, J. Millán, S. Jaeqx, A. M. Rijs, A. Lesarri, F. Castaño and J. A. Fernández, Exploring microsolvation of the anesthetic propofol, Phys. Chem. Chem. Phys., 2012, 14, 4398–4409 RSC.
- T. A. Halgren, MMFF VI. MMFF94s option for energy minimization studies, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, Perspectives on basis sets beautiful: Seasonal plantings of diffuse basis functions, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- F. Neese, Software Update: the ORCA Program System, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1 Search PubMed.
- B. J. Miller, H. G. Kjaergaard, K. Hattori, S. Ishiuchi and M. Fujii, The most stable conformer of benzyl alcohol, Chem. Phys. Lett., 2008, 466, 21–26 CrossRef CAS.
- N. Guchhait, T. Ebata and N. Mikami, Discrimination of rotamers of aryl alcohol homologues by infrared-ultraviolet double-resonance spectroscopy in a supersonic jet, J. Am. Chem. Soc., 1999, 121, 5705–5711 CrossRef CAS.
- R. Medel and M. A. Suhm, Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: different correction patterns for density functional and wave-function-based methods, Phys. Chem. Chem. Phys., 2021, 23, 5629–5643 RSC.
- P. Felder and H. H. Günthard, Conformational interconversions in supersonic jets: Matrix IR spectroscopy and model calculations, Chem. Phys., 1982, 71, 9–25 CrossRef CAS.
- R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, Relaxation of conformers and isomers in seeded supersonic jets of inert gases, J. Chem. Phys., 1990, 93, 3142–3150 CrossRef CAS.
- F. Xie, M. Fusè, A. S. Hazrah, W. Jäger, V. Barone and Y. Xu, Discovering the elusive global minimum in a ternary chiral cluster: Rotational spectra of propylene oxide trimer, Angew. Chem., Int. Ed., 2020, 59, 22427–22430 CrossRef CAS PubMed.
- N. Guchhait, T. Ebata and N. Mikami, Structures of hydrogen-bonded clusters of benzyl alcohol with water investigated by infrared-ultraviolet double resonance spectroscopy in supersonic jet, J. Chem. Phys., 1999, 111, 8438–8447 CrossRef CAS.
- R. Medel, M. Heger and M. A. Suhm, Molecular docking via olefinic OH⋯π interactions: A bulky alkene model system and its cooperativity, J. Phys. Chem. A, 2015, 119, 1723–1730 CrossRef CAS PubMed.
- J. Altnöder, A. Bouchet, J. J. Lee, K. E. Otto, M. A. Suhm and A. Zehnacker-Rentien, Chirality-dependent balance between hydrogen bonding and London dispersion in isolated (±)-1-indanol clusters, Phys. Chem. Chem. Phys., 2013, 15, 10167–10180 RSC.
- R. Medel, Schwingungsspektroskopische Untersuchungen zur Chiralitätserkennung und Torsionsdynamik bei Alkoholen, PhD dissertation, Georg-August-Universität Göttingen, 2020 Search PubMed.
- D. Maué, P. H. Strebert, D. Bernhard, S. Rösel, P. R. Schreiner and M. Gerhards, Dispersion-bound isolated dimers in the gas phase: Observation of the shortest intermolecular cH⋯H–C distance via stimulated Raman spectroscopy, Angew. Chem., Int. Ed., 2021, 60, 11305–11309 CrossRef PubMed.
- M. A. Suhm, in Advances in Chemical Physics, Volume 142, ed. S. A. Rice, John Wiley & Sons, Inc., Hoboken, NJ, 2008, pp. 1–57 Search PubMed.
- D. Mani and E. Arunan, Rotational spectra of propargyl alcohol dimer: A dimer bound with three different types of hydrogen bonds, J. Chem. Phys., 2014, 141, 164311 CrossRef PubMed.
- J. Saini and K. S. Viswanathan, Discerning near-isoergic isomers. A matrix isolation infrared and ab initio study of the propargyl alcohol dimers, J. Phys. Chem. A, 2017, 121, 1448–1459 CrossRef CAS PubMed.
- T. Scharge, T. N. Wassermann and M. A. Suhm, Weak hydrogen bonds make a difference: Dimers of jet-cooled halogenated ethanols, Z. Phys. Chem., 2008, 222, 1407–1452 CrossRef CAS.
- X. Liu, N. Borho and Y. Xu, Molecular self-recognition: Rotational spectra of the dimeric 2-fluoroethanol conformers, Chem. – Eur. J., 2009, 15, 270–277 CrossRef CAS PubMed.
- N. Borho and M. A. Suhm, Glycidol dimer: Anatomy of a molecular handshake, Phys. Chem. Chem. Phys., 2002, 4, 2721–2732 RSC.
- J. Altnöder, J. J. Lee, K. E. Otto and M. A. Suhm, Molecular recognition in glycolaldehyde, the simplest sugar: Two isolated hydrogen bonds win over one cooperative pair, ChemistryOpen, 2012, 1, 269–275 CrossRef PubMed.
- N. Borho and M. A. Suhm, Self-organization of lactates in the gas phase, Org. Biomol. Chem., 2003, 1, 4351 RSC.
- J. A. Davies, M. W. D. Hanson-Heine, N. A. Besley, A. Shirley, J. Trowers, S. Yang and A. M. Ellis, Dimers of acetic acid in helium nanodroplets, Phys. Chem. Chem. Phys., 2019, 21, 13950–13958 RSC.
- A. Longarte, C. Redondo, J. A. Fernández and F. Castaño, IR/UV and UV/UV double-resonance study of guaiacol and eugenol dimers, J. Chem. Phys., 2005, 122, 164304 CrossRef PubMed.
- P. Asselin, B. Madebène, P. Soulard, R. Georges, M. Goubet, T. R. Huet, O. Pirali and A. Zehnacker-Rentien, Competition between inter- and intra-molecular hydrogen bonding: An infrared spectroscopic study of jet-cooled amino-ethanol and its dimer, J. Chem. Phys., 2016, 145, 224313 CrossRef PubMed.
- F. Lahmani, M. Broquier and A. Zehnacker-Rentien, The o-cyanophenol dimer as studied by laser-induced fluorescence and IR fluorescence dip spectroscopy: A study of a symmetrical double hydrogen bond, Chem. Phys. Lett., 2002, 354, 337–348 CrossRef CAS.
- N. Seurre, K. Le Barbu-Debus, F. Lahmani, A. Zehnacker-Rentien and J. Sepioł, Electronic and vibrational spectroscopy of jet-cooled m-cyanophenol and its dimer: Laser-induced fluorescence and fluorescence-dip IR spectra in the S0 and S1 states, Chem. Phys., 2003, 295, 21–33 CrossRef CAS.
- N. A. Seifert, A. S. Hazrah and W. Jäger, The 1-naphthol dimer and its surprising preference for π–π stacking over hydrogen bonding, J. Phys. Chem. Lett., 2019, 10, 2836–2841 CrossRef CAS PubMed.
- I. León, J. Millán, F. Castaño and J. A. Fernández, A spectroscopic and computational study of propofol dimers and their hydrated clusters, ChemPhysChem, 2012, 13, 3819–3826 CrossRef PubMed.
- S. Nakagawa, Y. Matsushita, T. Suzuki and T. Ichimura, Resonance enhanced multiphoton ionization and time-of-flight mass spectra of jet-cooled 3-chlorophenol dimer, J. Mol. Struct., 2005, 779, 68–71 CrossRef CAS.
- M. Albrecht, A. Borba, K. Le Barbu-Debus, B. Dittrich, R. Fausto, S. Grimme, A. Mahjoub, M. Nedić, U. Schmitt, L. Schrader, M. A. Suhm, A. Zehnacker-Rentien and J. Zischang, Chirality influence on the aggregation of methyl mandelate, New J. Chem., 2010, 34, 1266 RSC.
- F. N. Keutsch, J. D. Cruzan and R. J. Saykally, The water trimer, Chem. Rev., 2003, 103, 2533–2577 CrossRef CAS PubMed.
- R. W. Larsen, P. Zielke and M. A. Suhm, Hydrogen-bonded OH stretching modes of methanol clusters: A combined IR and Raman isotopomer study, J. Chem. Phys., 2007, 126, 194307 CrossRef PubMed.
- J. Thomas, N. A. Seifert, W. Jäger and Y. Xu, A direct link from the gas to the condensed phase: A rotational spectroscopic study of 2,2,2-trifluoroethanol trimers, Angew. Chem., Int. Ed., 2017, 56, 6289–6293 CrossRef CAS PubMed.
- S. Oswald, N. A. Seifert, F. Bohle, M. Gawrilow, S. Grimme, W. Jäger, Y. Xu and M. A. Suhm, The chiral trimer and a metastable chiral dimer of achiral hexafluoroisopropanol: A multi-messenger study, Angew. Chem., Int. Ed., 2019, 58, 5080–5084 CrossRef CAS PubMed.
- M. Fárník, M. Weimann, C. Steinbach, U. Buck, N. Borho, T. B. Adler and M. A. Suhm, Size-selected methyl lactate clusters: Fragmentation and spectroscopic fingerprints of chiral recognition, Phys. Chem. Chem. Phys., 2006, 8, 1148 RSC.
- J. Thomas, X. Liu, W. Jäger and Y. Xu, Unusual H-bond topology and bifurcated H-bonds in the 2-fluoroethanol trimer, Angew. Chem., Int. Ed., 2015, 54, 11711–11715 CrossRef CAS PubMed.
- J. D. Cruzan, L. B. Braly, K. Liu, M. G. Brown, J. G. Loeser and R. J. Saykally, Quantifying hydrogen bond cooperativity in water: VRT spectroscopy of the water tetramer, Science, 1996, 271, 59–62 CrossRef CAS PubMed.
- N. A. Seifert, J. Thomas, W. Jäger and Y. Xu, Rotational spectra and theoretical study of tetramers and trimers of 2-fluoroethanol: Dramatic intermolecular compensation for intramolecular instability, Phys. Chem. Chem. Phys., 2018, 20, 27630–27637 RSC.
- T. B. Adler, N. Borho, M. Reiher and M. A. Suhm, Chirality-induced switch in hydrogen-bond topology: Tetrameric methyl lactate clusters in the gas phase, Angew. Chem., Int. Ed., 2006, 45, 3440–3445 CrossRef CAS PubMed.
- R. Taylor and C. F. Macrae, Rules governing the crystal packing of mono- and dialcohols, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 815–827 CrossRef CAS PubMed.
- S. K. Nayak, R. Sathishkumar and T. N. G. Row, Directing role of functional groups in selective generation of C–H⋯π interactions: In situ cryo-crystallographic studies on benzyl derivatives, CrystEngComm, 2010, 12, 3112–3118 RSC.
- D. A. Prystupa, A. Anderson and B. H. Torrie, Raman and infrared study of solid benzyl alcohol, J. Raman Spectrosc., 1994, 25, 175–182 CrossRef CAS.
- T. Scharge, T. Häber and M. A. Suhm, Quantitative chirality synchronization in trifluoroethanol dimers, Phys. Chem. Chem. Phys., 2006, 8, 4664–4667 RSC.
- S. A. Barnett and D. R. Allan, The high-pressure and low-temperature structural behaviour of 2,2,2-trifluoroethanol, CrystEngComm, 2019, 21, 4501–4506 RSC.
- T. Scharge, C. Emmeluth, T. Häber and M. A. Suhm, Competing hydrogen bond topologies in 2-fluoroethanol dimer, J. Mol. Struct., 2006, 786, 86–95 CrossRef CAS.
- P. G. Jönsson, Hydrogen bond studies. CXIII. The crystal structure of ethanol at 87 K, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 232–235 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional spectra, calculational results for isomers up to 5 kJ mol−1, NCI results. See DOI: 10.1039/d1cp03508h |
| This journal is © the Owner Societies 2021 |