
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microelectrode-based transient amperometry of O2 adsorption and desorption on a SrTiO3 photocatalyst excited under water†
Takumu
Kosaka
a,
Tomohiro
Ando
b,
Takashi
Hisatomi
 cd,
Hiroshi
Nishiyama
e,
Yuanshu
Zhou
f,
Kazunari
Domen
ce,
Yasufumi
Takahashi
df and
Hiroshi
Onishi
*ag
cd,
Hiroshi
Nishiyama
e,
Yuanshu
Zhou
f,
Kazunari
Domen
ce,
Yasufumi
Takahashi
df and
Hiroshi
Onishi
*ag
aDepartment of Chemistry, School of Science, Kobe University, Kobe 657-8501, Japan. E-mail: oni@kobe-u.ac.jp
bDivision of Electrical Engineering and Computer Science, Kanazawa University, Kanazawa 920-1192, Japan
cResearch Initiative for Supra-Materials, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Nagano 380-8553, Japan
dPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan
eOffice of University Professors, The University of Tokyo, Tokyo 113-8656, Japan
fNano Life Science Institute, Kanazawa University, Kanazawa 920-1192, Japan
gResearch Center for Membrane and Film Technology, Kobe University, Kobe 657-8501, Japan
First published on 2nd September 2021
Abstract
Oxygen evolution at water–solid interfaces is a key reaction for sustainable energy production. Although some intermediate states have been detected in transient absorption spectroscopy, the O2 evolution kinetics after the multi-step, four-electron oxidation of water remain unknown. In this study, transient amperometry with a microelectrode was applied to operando O2 detection over Al-doped SrTiO3 particles doubly loaded with RhCrOx and CoOy cocatalysts, an efficient photocatalyst for the overall water-splitting reaction. Electrochemical O2 detection at intervals of 0.1 s unexpectedly indicated instantaneous O2 adsorption and desorption in addition to steady, photocatalytic O2 evolution on the photocatalyst modified under intense light irradiation. We hypothesized that electrons excited in the conduction band were transferred to O2 in water thorough Ti cations neighboring an oxygen anion vacancy on the modified Al-doped SrTiO3. The negatively charged O2 was then bound to the Ti cations. It was neutralized and released when shaded through electron back-transfer to the conduction band. The hypothesized mechanism for O2 adsorption and desorption was compared with the photoinduced O2 desorption known to occur on anion vacancies of TiO2(110). The microelectrode-based transient amperometry demonstrated in this paper will be applied to many other phenomena at liquid–solid interfaces.
Introduction
Oxygen evolution at water–solid interfaces is a key reaction for sustainable energy production.1–4 In semiconductor photocatalysts and photoelectrodes, the creation and consumption kinetics of bandgap-excited charge carriers5–14 or chemical intermediates15–20 have been traced using transient absorption spectroscopy (TAS) with good time resolutions of femtoseconds to milliseconds. The kinetics observed by TAS for the initial and intermediate species must be compared with O2 evolution kinetics to verify or deny a proposed reaction mechanism. Nevertheless, it is still difficult to experimentally detect O2 with a compatible time resolution.Molecular oxygen released in water is currently accumulated in the gas phase and quantified with gas chromatography once or twice per hour. An improved time resolution on the scale of minutes could be achieved with advanced setups that shorten the gas-sampling intervals.21 The authors have been developing a method using microelectrode-based transient amperometry to further speed up operando O2 detection. Our simple idea is to shorten the physical distance from the place of reaction to the electrode for detection, as was conducted in scanning electrochemical microscopy (SECM) suitable for O2 concentration mapping.22–26 In our latest study,27 we used diffusion simulations and electrochemical detection on a microelectrode to determine the absolute O2 evolution rate with a time resolution of 0.1 s on a SrTiO3 photocatalyst film immersed in a nitrogen-purged, aqueous KCl solution.
In the present study, the photocatalyst film was excited in an aerobic solution exposed to air. Surprisingly, molecular oxygen having been dissolved in the solution was adsorbed on the irradiated film, and it was released when shaded from the irradiation. We hypothesized that bandgap-excited electrons were injected into the O2 in the solution. After receiving the electrons, O2 was negatively charged and bound to the Ti cations neighboring an oxygen anion vacancy on the surface of SrTiO3.
Experimental section
The photocatalyst was SrTiO3 particles doped with Al cations.28 SrTiO3, Al2O3, and SrCl2 were mixed at a molar ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:10 and heated at 1423 K to produce Al-doped particles that were 200–500 nm in size. A RhCrOx cocatalyst (Rh 0.1 wt% and Cr 0.1 wt%) was loaded onto the particles using impregnation from an aqueous solution of Na3RhCl6 and Cr(NO3)3. A CoOy cocatalyst (Co 0.1 wt%) was additionally loaded by photodeposition in an aqueous solution of CoCl2. The RhCrOx cocatalyst was loaded for assisting electron-driven H2 evolution,29 whereas the CoOy cocatalyst was for hole-driven O2 evolution.18,30 This photocatalyst was active for the overall water-splitting reaction with an apparent quantum yield of 55%. The photocatalyst particles were suspended in water with SiO2 nanoparticles (1:2 weight ratio), dropped on a frosted glass plate, and dried at 323 K to be fixed. Photocatalyst-coated plates prepared in a similar manner had previously been placed in a panel reactor and tested for large-scale water splitting under natural sunlight.28
:2:10 and heated at 1423 K to produce Al-doped particles that were 200–500 nm in size. A RhCrOx cocatalyst (Rh 0.1 wt% and Cr 0.1 wt%) was loaded onto the particles using impregnation from an aqueous solution of Na3RhCl6 and Cr(NO3)3. A CoOy cocatalyst (Co 0.1 wt%) was additionally loaded by photodeposition in an aqueous solution of CoCl2. The RhCrOx cocatalyst was loaded for assisting electron-driven H2 evolution,29 whereas the CoOy cocatalyst was for hole-driven O2 evolution.18,30 This photocatalyst was active for the overall water-splitting reaction with an apparent quantum yield of 55%. The photocatalyst particles were suspended in water with SiO2 nanoparticles (1:2 weight ratio), dropped on a frosted glass plate, and dried at 323 K to be fixed. Photocatalyst-coated plates prepared in a similar manner had previously been placed in a panel reactor and tested for large-scale water splitting under natural sunlight.28
Details of the experimental setup are described in ref. 27 and its ESI. A millimeter-sized photocatalyst-coated glass plate was placed in a KCl aqueous solution (0.1 mol l−1, pH = 7) that was exposed to air in this study. A platinum wire with a 10 μm radius was coated with glass to expose a metal section as the working electrode for O2 detection. The electrode was immersed in the solution perpendicular to the photocatalyst film which was electrically isolated in the solution. The distance between the electrode and film was controlled in the 100–200 μm range with 0.1 μm precision. Molecular oxygen in the solution was electrochemically detected on the electrode with a time resolution of 0.1 s, which was biased at −0.5 V relative to a Ag wire as the counter electrode. A four-electron reduction reaction, O2 + 2H2O + 4e− → 4OH− and/or O2 + 4H+ + 4e− → 2H2O,31 produced an electrode current on the order of nano-amps that was proportional to the O2 concentration. The proportional relation of O2 concentration and electrode current was checked and confirmed.27 The Ag wire served as a quasi Ag/AgCl reference electrode in the KCl solution. On the other hand, the oxidative reaction on the wire, Ag + h+ → Ag+, as the counter electrode caused silver contamination in the solution to a limited extent. A freshly polished microelectrode was used in each set of measurements to minimize silver contamination on the microelectrode apex.
Results and discussion
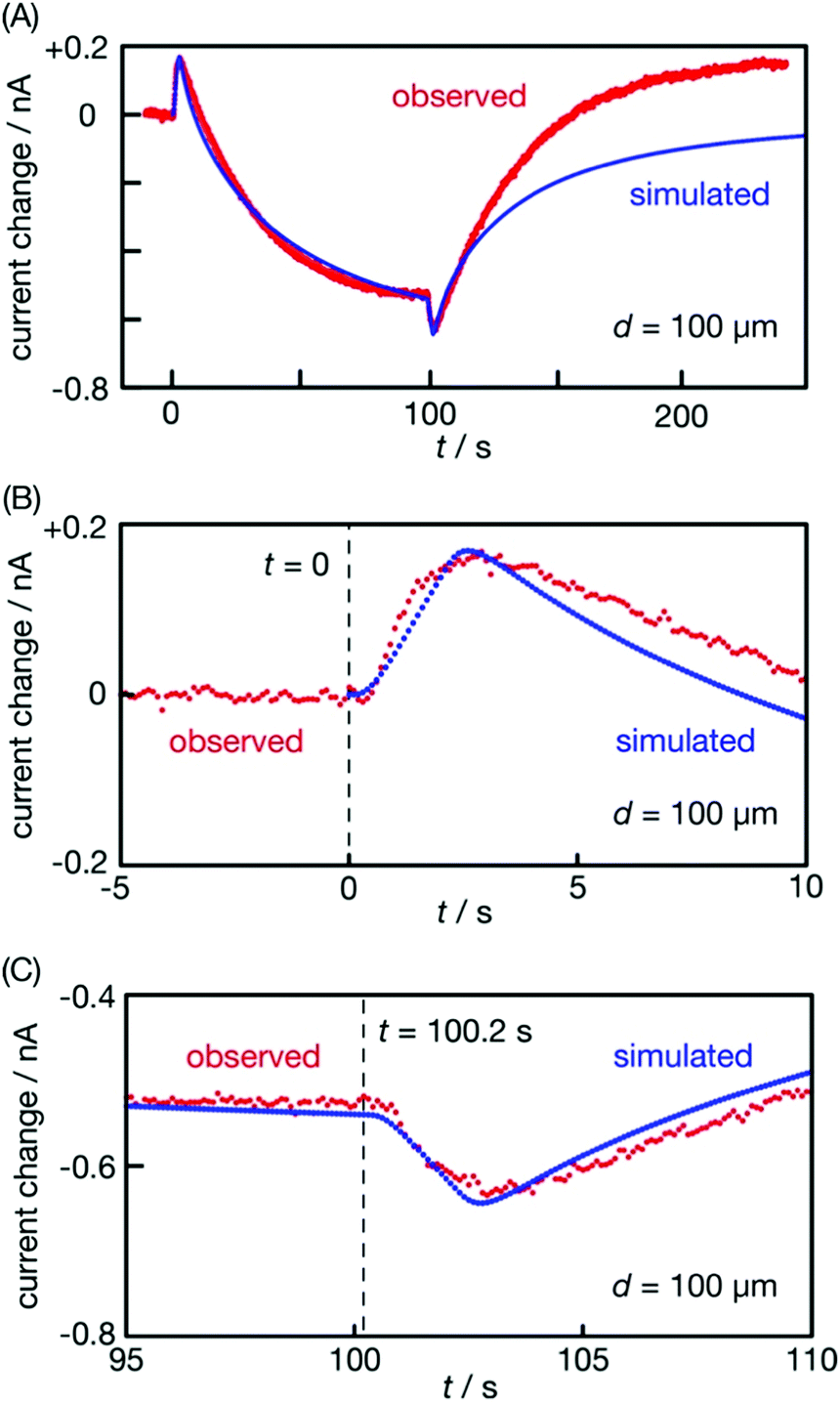
The immersed film was side lit with ultraviolet (UV) light (center wavelength: 280 nm) passing through the solution, as illustrated in the table of contents entry. The light intensity was tuned to 3 W m−2 (4 × 1018 photons m−2 s−1) to probe light-triggered O2 concentration changes without producing O2 and H2 bubbles on the irradiated film. Fig. 1(A) presents the electrode current I that responded to light irradiation at an electrode–film distance d = 100 μm. The negative sign of the current indicates that electrons are transferred from the electrode to the solution. Molecular oxygen in the solution produced a background current of −6.5 nA. When the probe light was turned on at time zero (t = 0), the electrode current gradually increased to −9.5 nA; when the light stopped at 100.5 s, the current gradually decreased back to the background. | ||
| Fig. 1 Microelectrode current in response to UV light irradiation. Irradiation of a photocatalyst film with light for probing (intensity: 3 W m−2) at t = 0–100.5 s in a KCl solution resulted in response (A). The film was then irradiated with intense light to modify the photocatalyst (30 W m−2) for 4 h in the solution. Immediate irradiation of the modified film with light for probing (3 W m−2) at t = 0–100.2 s resulted in response (B). The instantaneous current decrease and increase are marked in the panel. The film was removed from the solution and dried in air for 9 days. The dried film showed response (C) when immersed in the solution and irradiated with light for probing (3 W m−2) at t = 0–101.1 s. The electrode–film distance d was set at 100 μm. | ||
We had observed gradual current increases and decreases in response to UV light irradiation on the same photocatalyst,27 where the whole setup was purged with N2 to remove background O2 in the solution. The current changes observed in the N2 atmosphere were quantitatively interpreted with photocatalytic O2 evolution on the irradiated film followed by O2 diffusion into the bulk solution. The electrode current response shown in Fig. 1(A) increased and then decreased in a similar manner to what was reported in ref. 27 Hence, the response in Fig. 1(A) was ascribed to photocatalytic O2 evolution on the irradiated film.
The film examined in Fig. 1(A) was then irradiated with UV light at 10 times the intensity (280 nm, 30 W m−2) for 4 h in the solution. The film was photocatalytically active enough to produce O2 and H2 bubbles during this intense irradiation. The electrode current exceeded the quantification limit, 10.5 nA. When the intense irradiation was suspended, the film was immediately probed with weak light (3 W m−2). Response (B) obtained in this way was qualitatively different from response (A). The electrode current was stable at −7.7 nA, indicating that O2 was dissolved in the air-exposed solution prior to irradiation. When the probe light was switched on, the current instantaneously decreased from −7.7 to −7.5 nA. It then gradually increased, indicating photocatalytic O2 production. When the probe light was stopped at t = 100.2 s, the electrode current instantaneously increased from −8.3 to −8.4 nA and gradually reduced back to the background level. We ascribed the instantaneous current increase coupled with the decrease to O2 adsorption and desorption on the film triggered by light irradiation; background O2 in the solution was adsorbed on the film when irradiated and was desorbed when shaded.
Pretreatment with intense light irradiation (30 W m−2 for 4 h) was required to modify the photocatalyst feasible for probe-light-triggered O2 adsorption and desorption. Immersing the photocatalyst film in the solution for 4 h without intense light irradiation provided no instantaneous current response, as shown in Fig. S1 in the ESI.† Intense irradiation on the microelectrode induced no current response in the absence of the film (Fig. S2, ESI†). In a K2SO4 solution (0.1 mol l−1), the film was modified under intense light to present similar instantaneous current responses (Fig. S3, ESI†) being insensitive to the chemical identity of electrolytes. Oxidation of Cl− anions, if any on the UV-irradiated SrTiO3 photocatalyst, did not matter to make the instantaneous current responses.
These experimental results supported our interpretation that probe-light-triggered O2 adsorption and desorption occurred on the photocatalyst particles modified during the pretreatment.
After the film displayed response (B), it was removed from the solution and dried in air at room temperature for 9 days. This film was then immersed in the KCl solution and examined with probe light (3 W m−2) again. The observed response (C) displayed a gradual current increase and decrease, which were attributed to photocatalytic O2 evolution; no instantaneous response could be assigned to O2 adsorption and desorption. The modified photocatalyst particles returned to their original state when dried in air.
In the descriptions above, we qualitatively characterized the electrode current responses according to the presence or absence of instantaneous responses triggered by probe-light irradiation. We then quantitatively interpreted response (B) in Fig. 1, using a numerical simulation including instantaneous O2 adsorption, photocatalytic O2 evolution, instantaneous O2 desorption, and O2 diffusion in the solution. The simulation space defined in cylindrical coordinates, r and z, is illustrated in Fig. 2(A). The oxygen diffusion driven by a concentration gradient is described by Fick's law:
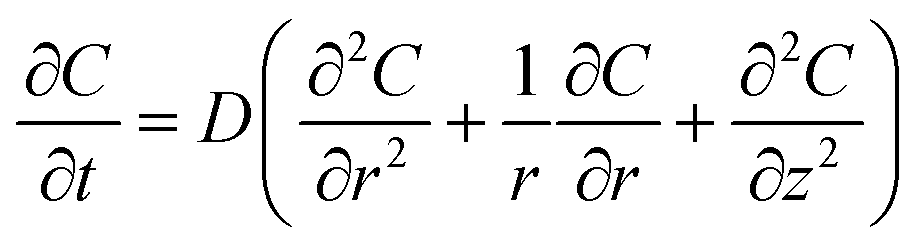
 | ||
| Fig. 2 Finite-element simulation of electrode current. The simulation space, which has cylindrical symmetry around the z-axis, is illustrated in (A). Platinum electrode section radius, a: 10 μm; glass wrap outer radius, Rg: 20 μm; electrode–film distance, d: 100 μm; radius of the irradiated photocatalyst film, Rphoto: fitted to be 800 μm in the simulation (see text); solution dish radius, Rsolution: 13 mm; solution thickness, Zsolution: 10 mm; glass plate thickness, Zphoto: 1 mm. The solid line in (B) depicts the assumed O2 release rate on the film. | ||
The O2 release rate depicted in Fig. 2(B) represented a net release rate. When light-triggered O2 absorption or desorption occurred with photocatalytic O2 evolution in parallel, the net release rate was evaluated by fitting simulated current responses to the experimental result.
The simulated current was fitted to the experimental result in two steps. In the first step, Rphoto and revolution were optimized to follow the gradual current increase during probe-light irradiation, while τ was set to zero. The other quantities (a, Rg, d, Rsolution, Zsolution, and Zphoto) were fixed at experimentally determined values, as described in the caption of Fig. 2. The radius of the irradiated photocatalyst film Rphoto was difficult to quantitatively determine since the light-spot boundary was oval and graded on the film. Thus, Rphoto and revolution were optimized to fit the experimentally observed response. The obtained Rphoto was 800 μm, while revolution was 0.085 μmol m−2 s−1. In the second step, radsorption and τ were fitted to the observed response for t = 0–10 s. Fitting to the response in t = 0–10 s was long enough to evaluate the impulsive O2 suction having occurred at t = 0 − τ, since the time delay for diffusion across the electrode–film gap was characterized by d2/D = 4 s.
When irradiation was stopped, rdesorption and τ were optimized to follow the observed response at t = 100–110 s. The current response simulated in this way was compared with the observed response in Fig. 3, in which the background current (−7.7 nA) was subtracted to deduce the current change induced by probe-light irradiation. The simulated response reproduced the observed current in the three time windows, t = 0–250, 0–10, and 95–110 s, and the optimized parameters were radsorption = −0.35 μmol m−2 s−1, rdesorption = 0.21 μmol m−2 s−1, and τ = 2.0 s. Two more response curves were experimentally observed on the same film upon changing d to 150 and 200 μm (Fig. 4). The two curves were fitted to validate the simulation scheme and the optimized parameters.
 | ||
| Fig. 3 Electrode current response shown in Fig. 1(B) fitted with the finite-element simulation of O2 adsorption, evolution, and desorption on the photocatalyst film. The experimentally observed (red) and simulated (blue) current changes are shown in time windows of (A) 0–250, (B) 0–10, and (C) 95–110 s. Electrode–film distance d = 100 μm. UV light (3 W m−2) was irradiated for probing on the photocatalyst film at t = 0–100.2 s. Five fitting parameters were optimized: Rphoto = 800 μm, revolution = 0.085 μmol m−2 s−1, radsorption = −0.35 μmol m−2 s−1, rdesorption = 0.21 μmol m−2 s−1, and τ = 2.0 s. | ||
 | ||
| Fig. 4 Current changes at d = (left panel) 150 and (right panel) 200 μm. The experimentally observed (red) and simulated (blue) current changes are shown in time windows of (A) 0–250, (B) 0–10, and (C) 95–110 s. UV light for probing (3 W m−2) was irradiated on the film at t = (left panel) 0–101.8 and (right panel) t = 0–101.3 s. The five fitting parameters (Rphoto, revolution, radsorption, rdesorption, and τ) were fixed at values optimized in the simulation at d = 100 μm. | ||
A negative release rate of −0.35 μmol m−2 s−1 was evaluated in the first 2.0 s of probe-light irradiation. The integrated quantity of adsorbed O2, which was calculated as the product of radsorption and τ, had a value of 4.2 × 1017 molecules m−2. The integrated quantity corresponded to 6% of the surface unit cell density of crystalline SrTiO3, 6.6 × 1018 m−2 on its (100) truncation.33 The integrated quantity suggests O2 adsorption on SrTiO3 rather than on the RhCrOx and/or CoOy cocatalysts loaded by 0.1 wt%. Partial contribution of the cocatalysts cannot be excluded, though. Since the film was composed of particulate materials, the unit cell density exposed to the solution should be larger than this number. Hence, the integrated quantity of 6% relative to the unit cell density provides the maximum estimate.
The integrated quantity of desorbed O2, which was calculated as the product of rdesorption and τ, had a value of 2.5 × 1017 molecules m−2. The desorbed quantity was 40% smaller than the adsorbed quantity. The decremental difference was ascribed to O2 desorption during probe-light irradiation or to the conversion of the adsorbed O2 to resident species on the shaded film.
Finally, we considered the mechanism through which O2 in the solution was adsorbed on the Al-doped SrTiO3 particles. Several peroxotitanate complexes having Ti metal centers coordinated by O22− ligands were previously reported.34 We therefore hypothesize that negatively charged O2 species, possibly O22−, were bound to Ti cations on the surface of the SrTiO3 during probe-light irradiation. Bandgap excitation by probe light provided two electrons, which were transferred to an impinging O2 molecule. By shading the particles, the electrons were back-transferred to the photocatalyst particle, resulting in the release of the neutralized O2 into the solution.
Titanium cations exposed on pristine particles were unable to capture O2 even under the probe light, as was evidenced by the absence of the instantaneous current response in Fig. 1(A). The surface of the particles was modified during the pretreatment under intense light irradiation (30 W m−2) to enable the capture of negatively charged O2. The modified surface returned to its original state when the film was dried in air at room temperature. The reversible state changes suggested that a limited number of oxygen anions were removed during the pretreatment and resultant anion vacancies served as active site for O2 adsorption. The vacancies were healed on the dried film. If metal (Sr, Ti, Al, Cr, Rh or Co) cations were removed under irradiation and induced the light-triggered adsorption, then they could not be restored when the surface was dried in air.
An analogous mechanism for O2 adsorption and desorption is known to occur on TiO2(110) wafers placed in vacuum. Experimental35,36 and theoretical37 studies have concluded that O2 was adsorbed on Ti3+ cations that neighbored an oxygen anion vacancy when the vacancy was thermally created on the wafer surface. The adsorbed O2 receives two excess electrons that were originally localized on the Ti3+ cations. When the wafer is bandgap excited, two holes attached to the negatively charged O2 and the neutralized O2 escaped as gas. Electron transfer and back-transfer switched the O2 adsorption and desorption on the anion vacancy.
We further hypothesized how oxygen anions were removed from Al-doped SrTiO3 particles during the pretreatment. Upon capture of two holes, an oxygen anion is neutralized and leave the surface. This process is known as photocorrosion;38 however, SrTiO3 is recognized as a stable semiconductor against photocorrosion. The neutralization of an oxygen anion leading to vacancy creation thermodynamically and kinetically competes with water oxidation, when a metal oxide particle is excited under water. The electrochemical potential for this phenomenon, i.e., corrosion potential, is known to be 1.42 eV vs. NHE on TiO2 in water having a pH of 0. On the other hand, the potential for water oxidation is 1.23 eV at pH 0. When we assume an identical corrosion potential on the surface of Al-doped SrTiO3, photoexcited holes are thermodynamically transferred to water and not to surface oxygen anions.
A finite number of holes are still feasible for neutralizing oxygen anions when water oxidation is kinetically limited. The Al-doped SrTiO3 photocatalyst examined in this study was doubly loaded with the CrRhOx cocatalyst for electron-driven H2 production29 and the CoOy cocatalyst for hole-driven O2 production.28 A finite number of holes could have been present in Al-doped SrTiO3 particles even modified with the CoOy cocatalyst for water oxidation.
Thus, the authors hypothesized that a small number of oxygen anions were neutralized and removed during the pretreatment under intense light irradiation. Indeed, a previous study39 reported photoetching of TiO2 electrodes which is known to be thermodynamically stable oxide against photocorrosion. The self-oxidation of the cocatalysts, if any during the pretreatment, may have created oxygen vacancies on the cocatalysts and contributed to the probe-light induced O2 adsorption and desorption.
The hypothesized mechanism is depicted in Fig. 5. In panel (A), a small number (6% or less relative to surface lattice density) of surface oxygen anions of Al-doped SrTiO3 are neutralized and removed during the pretreatment. The large number of holes excited under intense light irradiation drive this step. Excited electrons corresponding to the holes that neutralize the anions are consumed in the H2 evolution reaction, 2H2O + 2e− → H2 + 2OH−. The OH anions left in the reaction can be released in the solution. The ionic charges in the particle are balanced without need of creating Ti3+ cations, as a result. (B) O2 cannot be adsorbed on the anion vacancy when shaded. (C) Under probe-light irradiation, electrons excited in the particle are transferred to an impinging O2 through Ti cations neighboring the vacancy. (D) The negatively charged O2 is bound to the vacancy. (E) When the probe light is turned off, the excited electrons leave the adsorbed O2, and the neutralized O2 is released into the solution. The electrons leaving the adsorbed O2 are consumed in the H2 evolution reaction. (F) The anion vacancies were slowly healed after the film was dried in air.
 | ||
| Fig. 5 A possible mechanism for light-triggered O2 adsorption on the Al-doped SrTiO3 photocatalyst particles. (A) A surface oxygen anion neutralized and removed during intense light irradiation (30 W m−2). (B) No O2 adsorption to the vacancy in the electronic ground state. (C) O2 adsorption to the vacancy, assisted by electrons excited under probe-light irradiation (3 W m−2). (D) Adsorption of a negatively charged O2 on the vacancy. (E) O2 desorption under shaded conditions, with electrons leaving. (F) The anion vacancy slowly healed in air at room temperature. | ||
O2 desorption leaving an oxygen anion vacancy, which was hypothesized in Fig. 5(E), is identical to the O2 release step in the lattice oxygen evolution reaction (LOER), e.g., Fig. 4 of ref. 3 LOER has been recently proposed as an electrochemical cycle of the O2 evolution reaction on perovskite-structured metal oxide electrodes; it is different from the proton-coupled electron transfer4 that was previously assumed as the mechanism.
To investigate our hypothesis, we compared the photocatalyst films before and after the pretreatment using X-ray photoelectron spectroscopy (Fig. S5, ESI†) and optical absorption (Fig. S6, ESI†). The results show no sign of reduced Ti cations. This is consistent with the proposed scheme (Fig. 5), which involves Ti cations only in the 4+ valence state.
The SrTiO3 particles were doped with Al cations in this study to enhance water-splitting efficiency by limiting electron–hole recombination. Transient absorption studies40,41 showed recombination rate in SrTiO3 particles decreased by Na doping, whereas recombination rate was not yet examined on Al-doped SrTiO3. It is possible that the Al cations embedded in SrTiO3 lattice played a positive or negative role in removing oxygen anions and in the instantaneous O2 responses. This possibility should be examined in the future.
The light-triggered O2 adsorption and desorption found in this study open an additional path for electron–hole recombination. Two excited electrons can be located in an O2 adsorbed on the vacancy. These electrons recombine with two holes, thus releasing the adsorbed O2 under steady light irradiation. Two electrons and holes are consumed to catch and release an O2 molecule in the solution without splitting water.
In addition, the hypothesized hole-induced vacancy creation is a side-reaction that is problematic for photocatalyst durability. Durability of the Al-doped SrTiO3 photocatalyst was enhanced over a span of 1300 h under simulated sunlight irradiation by loading the CoOy cocatalyst.30 The limited durability in the absence of CoOy was interpreted with hole-induced degradation of the RhCrOx cocatalyst. The hole-induced vacancy creation hypothesized in this study may have been an additional reason of the limited durability.
A semiconductor photocatalyst that efficiently drives the water-splitting reaction is potentially modified by the hole-induced creation of anion vacancies since electron–hole recombination is limited to increased quantum yields. Note that the light intensity for vacancy creation was 30 W m−2 near UV intensity in natural sun light (40 W m−2 for wavelengths shorter than 400 nm). When this occurs on a photocatalyst under development, the tuning of cocatalysts to accelerate water-induced, hole-consuming reactions can be a beneficial way to optimize the number of holes present in the semiconductor particles under excitation light of a given intensity.
Conclusion
In this study, we found that the O2 adsorption and desorption on the Al-doped SrTiO3 photocatalyst particles doubly loaded with RhCrOx and CoOy cocatalysts were triggered by photoexcitation in water. Electrochemical detection on the microelectrode combined with diffusion simulations allowed us to quantitatively determine the rates of O2 adsorption, O2 desorption, and photocatalytic O2 evolution. The time resolution for O2 detection, 0.1 s, was critical for recognizing the instantaneous O2 adsorption and desorption that occurred during steady O2 evolution. The transient amperometry using a microelectrode can be applied to many other phenomena at liquid–solid interfaces.The light-triggered O2 adsorption and desorption were interpreted as the excited-electron transfer to and back-transfer from the adsorbed O2. Upon receiving electrons, O2 in the solution was negatively charged and could bind to the surface Ti4+ cations, while shaded particles could neutralize and release O2 by back-transferring the electrons. Intense light irradiation (30 W m−2) was required to modify the particle to enable O2 adsorption. We hypothesized that a small number of lattice oxygen anions were neutralized by receiving holes to leave the surface under intense irradiation. Anion vacancies created in this way served as O2 adsorption sites. Future studies that examine the role of the Al cations, RhCrOx cocatalyst, and CoOy cocatalyst are needed to verify the hypothesized scheme.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Yasuko Kuromiya, Yoshie Nagatuma, and Masaharu Yamaguchi are thanked for their technical assistance. This work was supported by JSPS KAKENHI (grant numbers: JP19H00915 and 18KK0161). T. H. and Y. T. acknowledge support from PRESTO (JPMJPR20T9 and JPMJPR18T8) respectively from the Japan Science and Technology Agency.References
- H. Inoue, T. Shimada, Y. Kou, Y. Napitane, D. Masui, S. Takagi and H. Tachibana, The Water Oxidation Bottleneck in Artificial Photosynthesis: How can We Get Through It? An Alternative Route Involving a Two-electron Process, ChemSusChem, 2011, 4, 173–179 CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, Transition Metal Oxides as Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Solutions: An Application-inspired Renaissance, J. Am. Soc. Chem., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- E. Fabbri and T. J. Schmidt, Oxygen Evolution Reaction—The Enigma in Water Electrolysis, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, Electrolysis of Water on Oxide Surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- C. Kranz and M. Wächtler, Characterizing photocatalysts for water splitting: from atoms to bulk and from slow to ultrafast processes, Chem. Soc. Rev., 2021, 50, 1407–1437 RSC.
- Y. Paz, Transient IR spectroscopy as a tool for studying photocatalytic materials, J. Phys.: Condens. Matter, 2019, 31, 503004 CrossRef CAS PubMed.
- K. Takanabe, Photocatalytic Water Splitting: Quantitative Approaches toward Photocatalyst by Design, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- A. Yamakata, T. Ishibashi and H. Onishi, Kinetics of the Photocatalytic Water-Splitting Reaction on TiO2 and Pt/TiO2 Studied by Time-Resolved Infrared Absorption Spectroscopy, J. Mol. Catal. A: Chem., 2003, 199, 85–94 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, Identification of Reactive Species in Photoexcited Nanocrystalline TiO2 Films by Wide-Wavelength-Range (400–2500 nm) Transient Absorption Spectroscopy, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- J. Tang, J. R. Durrant and D. R. Klug, Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- M. Maruyama, A. Iwase, H. Kato, A. Kudo and H. Onishi, Time-Resolved Infrared Absorption Study of NaTaO3 Photocatalysts Doped with Alkali Earth Metals, J. Phys. Chem. C, 2009, 113, 13918–13923 CrossRef CAS.
- Y. Ma, S. R. Pendlebury, A. Reynal, F. Le Formal and J. R. Durrant, Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation, Chem. Sci., 2014, 5, 2964–2973 RSC.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, Rate Law Analysis of Water Oxidation on a Hematite Surface, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS.
- T. J. Miao and J. Tang, Characterization of charge carrier behavior in photocatalysis using transient absorption spectroscopy, J. Chem. Phys., 2020, 152, 194201 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Generation and Detection of Reactive Oxygen Species in Photocatalysis, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- R. Nakamura and Y. Nakato, Primary Intermediates of Oxygen Photoevolution Reaction on TiO2 (Rutile) Particles, Revealed by in Situ FTIR Absorption and Photoluminescence Measurements, J. Am. Chem. Soc., 2004, 126, 1290–1298 CrossRef CAS.
- N. Sivasankar, W. W. Weare and H. Frei, Direct Observation of a Hydroperoxide Surface Intermediate upon Visible Light-Driven Water Oxidation at an Ir Oxide Nanocluster Catalyst by Rapid-Scan FT-IR Spectroscopy, J. Am. Chem. Soc., 2011, 133, 12976–12979 CrossRef CAS.
- M. Zhang, M. de Respinis and H. Frei, Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst, Nat. Chem., 2014, 6, 362–367 CrossRef CAS.
- D. M. Herlihy, M. M. Waegele, X. Chen, C. D. Pemmaraju, D. Prendergast and T. Cuk, Detecting the oxyl radical of photocatalytic water oxidation at an n-SrTiO3/aqueous interface through its subsurface vibration, Nat. Chem., 2016, 8, 549–555 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- K. Han, T. Kreuger, B. Mei and G. Mul, Transient behavior of Ni@NiOx functionalized SrTiO3 in overall water splitting, ACS Catal., 2017, 7, 1610–1614 CrossRef CAS PubMed.
- J. Lee, H. Ye, S. Pan and A. J. Bard, Screening of Photocatalysts by Scanning Electrochemical Microscopy, Anal. Chem., 2008, 80, 7445–7450 CrossRef CAS PubMed.
- M. Plaza, X. Huang, J. Y. P. Ko, M. Shen, B. H. Simpson, J. Rodríguez-López, N. L. Ritzert, K. Letchworth-Weaver, D. Gunceler, D. G. Schlom, T. A. Arias, J. D. Block and H. D. Abruña, Structure of the Photo-catalytically Active Surface of SrTiO3, J. Am. Chem. Soc., 2016, 138, 7816–7819 CrossRef CAS PubMed.
- J. H. Bae, A. B. Nepomnyashchii, X. Wang, D. V. Potapenko and M. V. Mirkin, Photo-scanning Electrochemical Microscopy on the Nanoscale with Through-tip Illumination, Anal. Chem., 2019, 91, 12601–12605 CrossRef CAS PubMed.
- X. Zhou, Z. T. Gossege, B. H. Simpson, J. Hui, Z. J. Barton and J. Rodríguez-López, Electrochemical Imaging of Photoanodic Water Oxidation Enhancements on TiO2 Thin Films Modified by Subsurface Aluminum Nanodimers, ACS Nano, 2016, 10, 9346–9352 CrossRef CAS PubMed.
- K. Ikeda, H. Sakai, R. Baba, K. Hashimoto and A. Fujishima, Photocatalytic Reactions Involving Radical Chain Reactions Using Microelectrodes, J. Phys. Chem. B, 1997, 101, 2617–2620 CrossRef CAS.
- T. Kosaka, Y. Teduka, T. Ogura, Y. Zhou, T. Hisatomi, H. Nishiyama, K. Domen, Y. Takahashi and H. Onishi, Transient Kinetics of O2 Evolution in Photocatalytic Water-Splitting Reaction, ACS Catal., 2020, 10, 13159–13164 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, A particulate photocatalyst water-splitting panel for large-scale solar hydrogen generation, Joule, 2018, 2, 509–520 CrossRef CAS.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, Role and Function of Noble-Metal/Cr-Layer Core/Shell Structure Cocatalysts for Photocatalytic Overall Water Splitting Studied by Model Electrodes, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- H. Lyu, T. Hisatomi, Y. Goto, M. Yoshida, T. Higashi, M. Katayama, T. Takata, T. Minegishi, H. Nishiyama, T. Yamada, Y. Sakata, K. Asakura and K. Domen, An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity for over 1000 h of constant illumination, Chem. Sci., 2019, 10, 3196–3201 RSC.
- E. Yeager, Electrocatalysts for O2 reduction, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- Handbook of Chemistry: Pure Chemistry, Chemical Society of Japan, Maruzen, Tokyo, 4th edn, 1993, p. II-63 Search PubMed.
- The surface unit cell density was calculated with the cubic unit cell length (0.390 nm) reported in Y. A. Abramov, V. G. Tsirelson, V. E. Zavodnik, S. A. Ivanov and I. D. Brown, The Chemical Bond and Atomic Displacements in SrTiO3 from X-ray Diffraction Analysis, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 942–951 CrossRef.
- M. Kakihana, M. Tada, M. Shiro, V. Petrykin, M. Osada and Y. Nakamura, Structure and Stability of Water Soluble (NH4)8[Ti4(C6H4O7)4(O2)4]·8H2O, Inorg. Chem., 2001, 40, 891–894 CrossRef CAS and references therein.
- G. Lu, A. Linsebigler and J. T. Yates, Jr., The photochemical identification of two chemisorption states for molecular oxygen on TiO2(110), J. Chem. Phys., 1995, 102, 3005–3008 CrossRef CAS.
- N. G. Petrik and G. A. Kimmel, Electron- and Hole-Mediated Reactions in UV-Irradiated O2 Adsorbed on Reduced Rutile TiO2(110), J. Phys. Chem. C, 2011, 115, 152–164 CrossRef CAS.
- M. P. de Lara-Castells and J. L. Krause, Theoretical study of the UV-induced desorption of molecular oxygen from the reduced TiO2(110) surface, J. Chem. Phys., 2003, 118, 5098–5105 CrossRef CAS.
- H. Gerischer, Solar Photoelectrolysis with Semiconductor Electrodes, in Solar Energy Conversion, Solid-State Physics Aspects, ed. B. O. Seraphin, Springer-Verlag, Berlin, 1979, p. 141 Search PubMed.
- A. Tsujiko, T. Kisumi, Y. Magari, K. Kurakoshi and Y. Nakatao, Selective Formation of Nanoholes with (100)-Face Walls by Photoetching of n-TiO2 (Rutile) Electrodes, Accompanied by Increases in Water-Oxidation Photocurrent, J. Phys. Chem. B, 2000, 104, 4873–4879 CrossRef CAS.
- K. Kato, J. Jiang, Y. Sakata and A. Yamakata, Effect of Na-Doping on Electron Decay Kinetics in SrTiO3 Photocatalyst, ChemCatChem, 2019, 11, 6349–6354 CrossRef CAS.
- J. Jiang, K. Kato, H. Fujimori, A. Yamakata and Y. Sakata, Investigation on the highly active SrTiO3 photocatalyst toward overall H2O splitting by doping Na ion, J. Catal., 2020, 390, 81–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reagents, current responses, simulation details, X-ray photoelectron spectra, and optical absorption spectra. See DOI: 10.1039/d1cp03264j |
| This journal is © the Owner Societies 2021 |