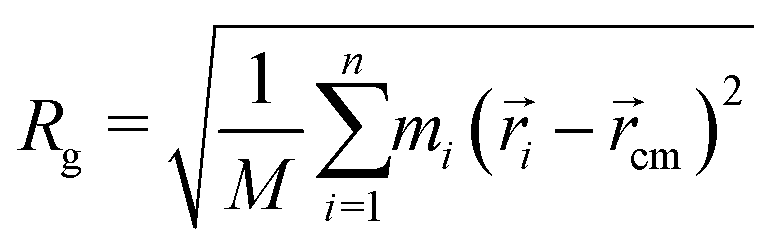Local structuring of diketopyrrolopyrrole (DPP)-based oligomers from molecular dynamics simulations†
Received
16th July 2021
, Accepted 12th August 2021
First published on 12th August 2021
Abstract
The microscopic structure of high mobility semiconducting polymers is known to be essential for their performance but it cannot be easily deduced from the available experimental data. A series of short oligomers of diketopyrrolopyrrole (DPP)-based materials that display high charge mobility are studied by molecular dynamics simulations to understand their local structuring at an atomic level. Different analyses are proposed to compare the ability of different oligomers to form large aggregates and their driving force. The simulations show that the tendency for this class of materials to form aggregates is driven by the interaction between DPP fragments, but this is modulated by the other conjugated fragments of the materials which affect the rigidity of the polymer and, ultimately, the size of the aggregates that are formed. The main structural features and the electronic structure of the oligomers are fairly similar above the glass transition temperature and at room temperature.
Introduction
The use of organic semiconducting polymers (SCPs) in electronic devices is a topic of great interest due to this class of materials displaying properties such as environmental stability, easy solution processability and flexibility1,2 while exhibiting charge carrier mobilities gradually approaching those of their inorganic counterparts. These materials have applications across fields such as the active layer of field-effect transistors (FETs),3,4 organic photovoltaics (OPVs)5,6 and bioelectronics7–9 with reported mobilities as high as ∼10 cm2 V−1 s−1.10,11
Following the analogy with inorganic semiconductors in which crystalline phases have the highest charge mobility, thiophene-based semicrystalline organic semiconductors such as P3HT (poly(3-hexylthiophene)), PQT (poly(3,3′-dialkyl-quaterthiophene)) and PBTTT with a choice of alkyl side chains for solution processability were extensively studied for a long while.12 The experimentally reported higher charge mobility for PBTTT compared to P3HT in thin-film transistors (TFT) resulted in theoretical and computational studies focusing on the morphology of these materials.13,14 A review of many different techniques to analyse the structure of these materials was provided by Do et al.15 The larger free volume between alkyl side chains of PBTTT was shown to facilitate a higher degree of crystallinity by the formation of a lamellar structure of interdigitated side chains as well as the higher degree of planarity of the backbones. Interdigitation of side chains was also experimentally observed using X-ray diffraction (XRD).16,17 Even though a high volume of research was initially carried out for more crystalline SCPs, it became increasingly apparent by collating the experimental evidence18 that long-range ordering was not essential for a high charge carrier mobility. The existence of short-range stacked domains was shown to be sufficient enough as long as there existed long chains connecting ordered domains. Experimental techniques such as grazing incidence X-ray diffraction (GIXRD)19 have provided evidence for the existence of ordered domains in a wide range of different classes of materials from semicrystalline P3HT and regioregular P3HT (Rr-P3HT) to amorphous materials. In understating these materials theoretical approaches have been used as a stand-alone method or in conjunction with experimental data16,20 to investigate the structural properties such as aggregation, the existence of local ordered domains and segregation of different parts of the polymeric chains. The chemical structure of these materials and the endless list of possible combinations of aromatic rings in the backbone and alkyl side chains make it nearly impossible to study all the variations and their effects on morphological properties and, consequently, the device performance.21 Some of these variations are changing the size of the side chains, using branched or linear side chains, the length and planarity of the backbone, and the type of conjugating aromatic rings.4,22–24 Despite many years of investigation, the relationship between the polymer structure and charge transport properties remains unclear.
In the following, the family of DPP-base semiconducting polymers is selected to be investigated for their morphological and structural properties. The rationale behind choosing this family has been the figure of merit, their reported high charge carrier mobility without having an overall crystalline structure.18,25–27 These materials’ paracrystalline properties, meaning they can form short-range ordered domains without having an overall crystalline structure,28 and the strong electron-withdrawing nature of the DPP units make them good candidates for use in ambipolar devices such as organic solar cells, OFETs and OLEDs,29–32 one of the first organic devices to report near-infrared (NIR) light emission.11 Despite many reports of high mobility when these materials are used in an OFET device it is not yet determined how the varying factors contribute to such performance.33 Some of these factors are changing the composition of the conjugated backbone, changing the side chain length and whether a linear or branched side chain is used. Huang et al.34 investigated the effect of changes in the length of conjugated backbones by studying the morphology and structural ordering of DPP-4T and DPP-6T. Using UV-vis measurements, they showed that both oligomers, DPP-4T and DPP-6T, exhibit a red-shifted spectra in solution compared to the solid state due to the higher level of aggregation and decreased intermolecular distances in the solid state.35 Grazing incidence wide-angle X-ray scattering (GIWAXS) and electron diffraction methods were used to study the morphology of crystalline portions of these systems. The π-stacking distance calculated from lattice modelling was reported to be ∼3.2 Å and ∼3.8 Å for DPP-4T and DPP-6T, respectively. It was concluded that the higher level of conformational degrees of freedom for the longer conjugated backbone of DPP-6T can result in the introduction of defects in the crystalline portion and therefore shorten the charge-transporting channel and lower charge mobility. Magic-angle spinning NMR, which is suitable to investigate the local structure in the absence of long range ordering, was used to study bulk and thin film DPP-DTT.36 These measurements in combination with computational quantum and molecular dynamics modelling showed a high level of backbone planarity for DPP-DTT with a donor-on-acceptor stacking arrangement. The preferred conformational stacking was rationalised by the existence of weak hydrogen bonding37 between the thiophene rings and DPP, with a measured π-stacking of 3.67 Å. The aggregation of the same polymer was also verified in solution using small angle neutron scattering (SANS) and light scattering.38
The effect of the chemistry of the backbone was investigated by replacing the T rings with fused aromatic segments such as thienothiophene (TT)39,40 or furan rings,41–43 with both changes reported to increase the planarity and therefore improve the chain stacking and aggregation. The effect of using phenyl rings instead of thiophene was also investigated for single molecules by studying their solid-state features.44 In this case where stronger hydrogen bonds are present and the phenyl is twisted outside the conjugation plane, a face-to-edge packing was observed hindering the π-stacking and lower charge mobility. Zhang et al.45 reported that increasing the number of thiophene and bithiophene rings between DPP units increases the glass transition temperature but does not change appreciably the mechanical properties. Replacing the sulphur atoms of the thiophene rings with larger heteroatoms such as selenium was reported to result in a higher degree of π-stacking caused by the stronger intermolecular interactions between Se atoms, compared to those of S atoms.46 The solid state aggregation properties and chain stacking is reported to improve by random terpolymerisation of DPP-4T with a low amount of isoindigo47 resulting in higher charge mobility due to the enhancements in amorphous features. Wang et al.46,48 measured the changes in the mobility of the DPP-selenephene system where one branched side chain is replaced by a linear side chain.
Different aspects of alkyl side chains can also be studied48 such as the existence of an extra carbon atom before the branching position in a branched side chain which is reported to increase the π–π stacking.49 UV-vis obtained for DPP-4T showed that changing the side chains from linear to branched had subtle effects on the spectra in solution, while in thin films the linear side chains showed stronger intermolecular interactions.50 The effects of different end groups were investigated for different end groups such as hydrogen atoms, thiophene rings or pyrene segments.51–53 Finally, optical spectra have shown evidence of H- and J-aggregates54 in polymers of the DPP family.
The combination of these studies point to a coherent qualitative picture: DPP polymers form short range aggregates with the size of the aggregate being modulated by changes in the polymer backbone and side chain.55–57 The conjugated portions in the more ordered phases are co-planar with a measurable π-stacking distance but there is no other information available on the local structure and therefore on the driving force for aggregation and the microscopic effect of chemical changes.
Classical molecular dynamics (MD) simulations have helped to establish a link between experimental observations and microscopic structures since the beginning of organic electronics. A substantial number of studies have focused on P3HT and PBTTT for many years due to the earlier assumption of these materials being best for exhibiting crystalline nature.58–61 More reports on the simulation of polymer melts have appeared for oligomers such as poly(phenylenevinylene) (PPV),62 Didodecylquaterthiophenes (DDQT),63 and poly(2-vinyl pyridine) (P2PV)64 including recent attempts to collectively compare the performance of different materials for different applications.65–68 A particularly challenging aspect is the generation of equilibrated structures for the melt phase, which requires large computational resources. To overcome this obstacle, different approaches have been followed, such as simulated annealing,69 coarse-grained models70 or implicit solvent models.71 Where possible, the simulation data are checked against the experimental data.72–75 Recent molecular dynamics studies at an atomic level of DPP systems employed single long chains in explicit solvents to understand the conformational and morphological properties71 and small copolymers to investigate the effect of the position of side chain linkers on electronic and optical properties.76 Simulations are used in conjunction with the experimental techniques to compare, validate or isolate the effect of a single change in the chemistry of a material on other properties. The effect of weak hydrogen bonds of DPP polymers on the π-stacking chain conformations showed good agreement between values obtained from NMR spectroscopy and computational simulations (DFT and MD).36 The comparison between experimental data and computational calculations was also reported using coarse-grained methods45 for chain alignments. The effect of changing a bulky branched side chain with a linear chain was studied using MD simulations48 in agreement with experimental data to show that linear side chains result in higher planarity.
In this work, we present MD simulation analyses of three systems belonging to the DPP family with different backbone segments. All three systems were chosen based on their reported high charge carrier mobilities.51,77–80 Studying the family of related compounds helps in discriminating between the general feature of polymers containing the DPP fragment and aspects that are more sensitive to the overall chemistry of the polymer. The alkyl side chains are the same for all the systems, a branched alkyl chain at position 2, which are reported to possess the best solubility.81 Different backbone combinations ranging from 2 thienothiophene (2TT) rings, 4 thiophene rings (4T) and 6 thiophene rings (6T) were studied to analyse the backbone's contribution to the overall morphology of the polymeric systems (see Fig. 1).
Methods
Force-field
The molecular model employed in the present work was based on an implementation of All-atom Optimised Potentials for Liquid Simulations82 (OPLS) force-field. While the OPLS mathematical form has been used in this document, the key parameters were derived from density functional theory (DFT) calculations. All the equilibrium bond distances and angles of the conjugated part were taken from the optimisation of trimer chains where values of the middle monomer were used to account for intermonomer connections at the B3LYP/6-311G** level of theory, the atomic point charges were calculated via CHelpG (charges from electrostatic potentials using a grid-based method)83 and the torsional potentials between the conjugated fragments were recomputed through scanning the dihedral potential at the MP2/6-311G** level.84,85 The equilibrium geometry and intermolecular interaction parameters are very similar to those computed by Jiang et al.86 with a force matching procedure and validated through simulations of small molecular crystals. OPLS-AA parameters were used for the alkyl side chains. Non-bonded van der Waals (vdW) parameters based on the 6-12 Lennard-Jones (LJ) potential were taken directly from OPLS-AA for all atom types using the geometric mixing rule as well as long range Coulomb interactions between point charges, as a common practice for the simulation of polymeric semiconductors.86
Simulation details
Attempts have been made to develop a specialised force-field (FF)86–88 for this class of materials; however, due to the complexity of the systems, some parametrisation is needed for each new polymer. In this work, simulations were performed on oligomers of three repeat units of each of the monomers. The choice of the length of oligomers was made having in mind the computational resources and the need for studying long enough chains to account for interactions of several monomers. While the slow-down in simulation production is obvious for longer chains, the choice was made based on our objective of having equilibrated and highly-reproducible simulations to use as a reference for these systems. All the MD simulations were performed using large-scale atomic/molecular massively parallel simulator (LAMMPS).89 The Velocity Verlet algorithm was adopted and periodic boundary conditions (PBCs) were applied in all directions with a time step of 2 fs while using the rattle algorithm to constrain H–X bonds. The Nosé–Hoover isothermal–isobaric barostat and a thermostat were used to control the ensemble properties. The particle–particle particle-mesh (PPPM) summation algorithm was used during production simulations to compute the electrostatic forces with a cut-off of 12 Å.
The oligomers studied here form glassy phases at room temperature that are difficult to equilibrate (and to verify they are equilibrated). The primary focus of this paper is to study the aggregation of DPP polymers in equilibrated and highly reproducible phases above the glass transition temperature (Tg). To establish the simulation temperature, simulated annealings were performed on all three systems from 1000 K to 100 K at a rate of 0.02 K ps−1. This process was repeated three times to make sure that annealings were performed for long enough and at a plausible rate. The three simulations were all performed at the same rate where the last point of one run is taken to start the next run by instantly heating it up to 1000 K and slowly cooling it down to 100 K at the given rate. Annealing processes have been shown to improve the structural ordering in the films as evidenced by X-ray scattering and atomic force microscopy.17,90Fig. 2 represents the inverse density as a function of temperature for the three sets of simulated annealing for each system where each set is presented with a different colour. The average gradient of the three sets is plotted for both the lower range (0 to 300 K) and the higher range (600 to 1000 K) of temperatures in the dashed black line. The intersection of the two lines is considered as Tg. The exact values of Tg are reported in red for each system. The production runs were performed at a temperature above the Tg of all three systems (550 K). Production simulations were performed from the last point of the second set of simulated annealings for the three systems in an NPT ensemble at 550 K for 400 ns with an integration time step of 2 fs. In agreement with ref. 45 the Tg increases with an increase in the number of thiophene rings, although a direct comparison is not possible as our molecular weight is much smaller than the one considered experimentally.
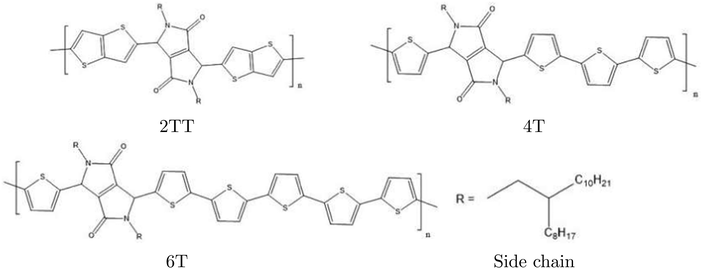 |
| | Fig. 1 Chemical structure of the three systems used in this study. | |
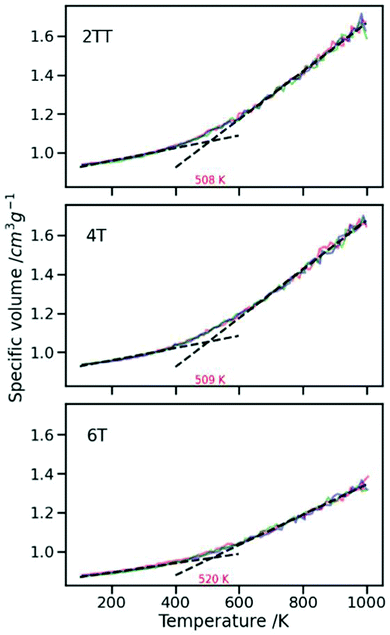 |
| | Fig. 2 Graphs presenting the specific volume as a function of temperature for three sets of simulated annealing in different colours for each system. The glass transition temperature is found by the intersection point between the two linear gradient fits of lower and higher range of temperatures. Dashed lines and the value of Tg in red are the average of the three sets. | |
Results and discussion
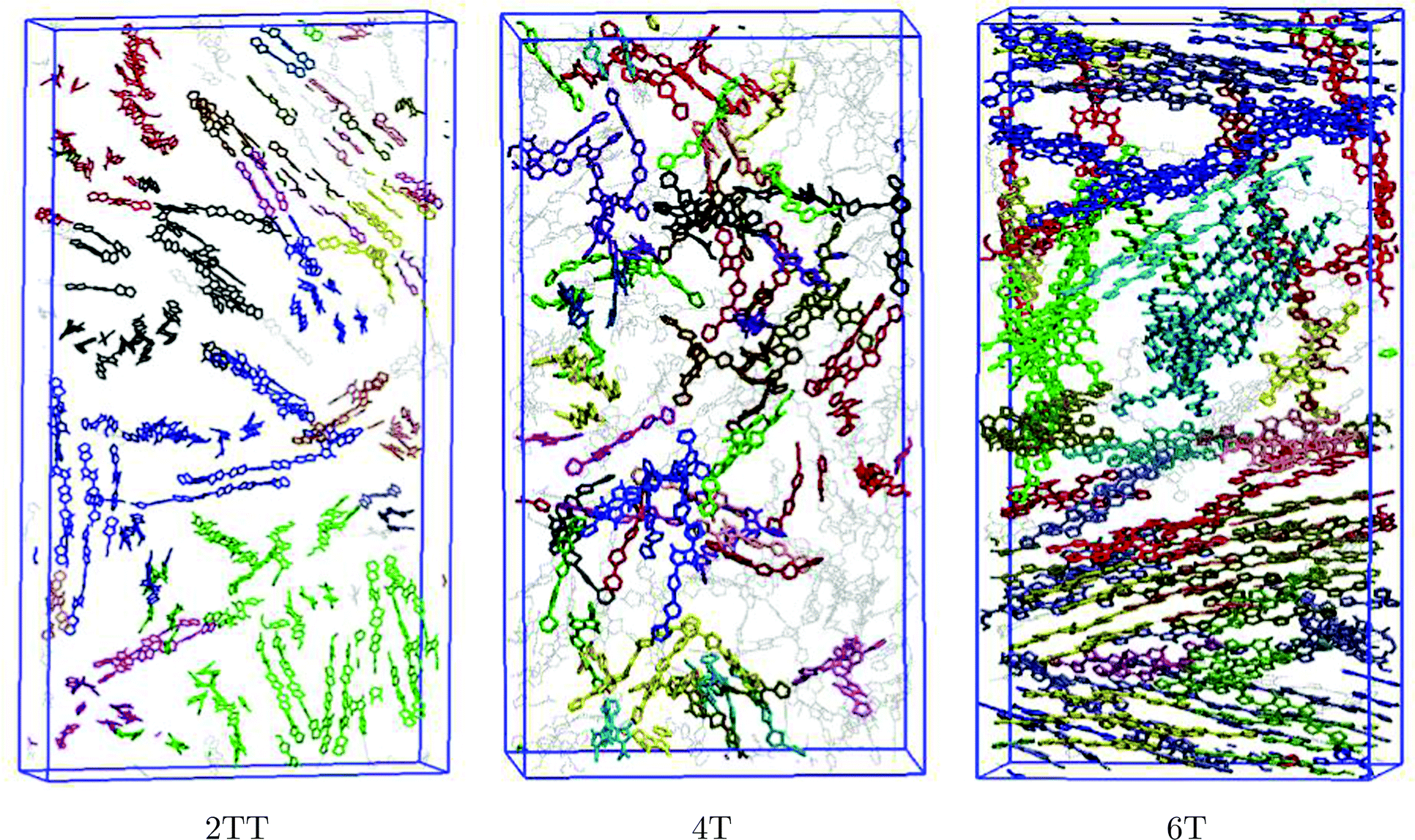
Production simulations were performed on systems of 64 chains of three repeat units for each of the systems, except 2TT, which after equilibration adopted an excessively elongated shape, provoking some undesired interactions between atoms and their PBC images. To avoid this finite size effect, 128 chains were used instead for this particular polymer. The results of the double-sized simulation of 2TT are presented in the following unless otherwise specified. System specifications are provided in Table 1.
Table 1 Simulation specification for the production runs
| Polymer |
Atoms/trimer chain |
Number of chains |
Number of atoms/simulation |
Density (g cm−3) |
| 2TT |
458 |
128 |
58624 |
0.889 |
| 4T |
482 |
64 |
30848 |
0.882 |
| 6T |
524 |
64 |
33536 |
1.013 |
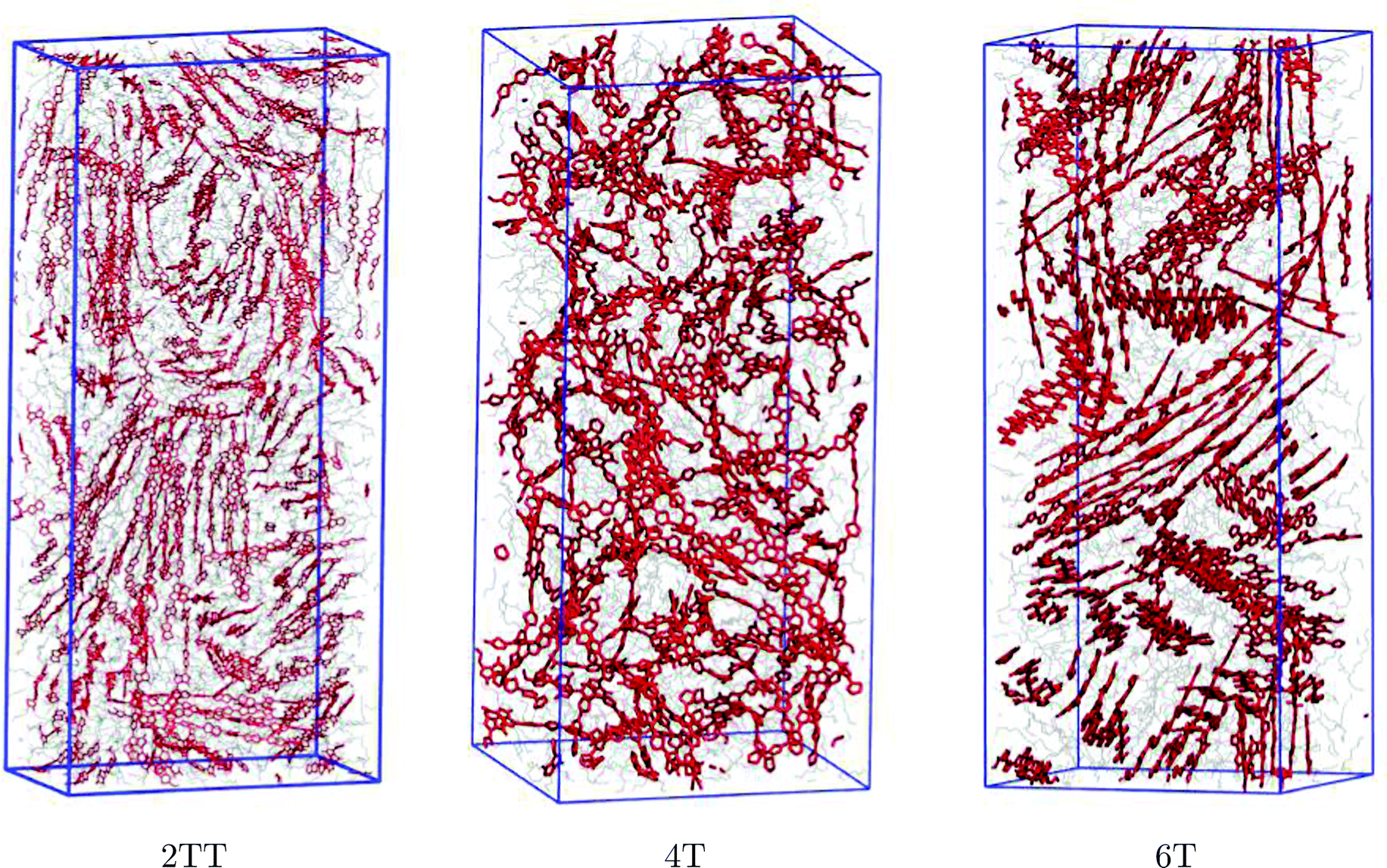
The snapshots of the three simulated systems shown in Fig. 3 already indicate the presence of local ordering in this kind of material. 2TT and 6T appear to show more rigid chains that result in more visible π-stacking. The remainder of the manuscript focuses on the quantitative description of the oligomers’ flexibility and the aggregation properties.
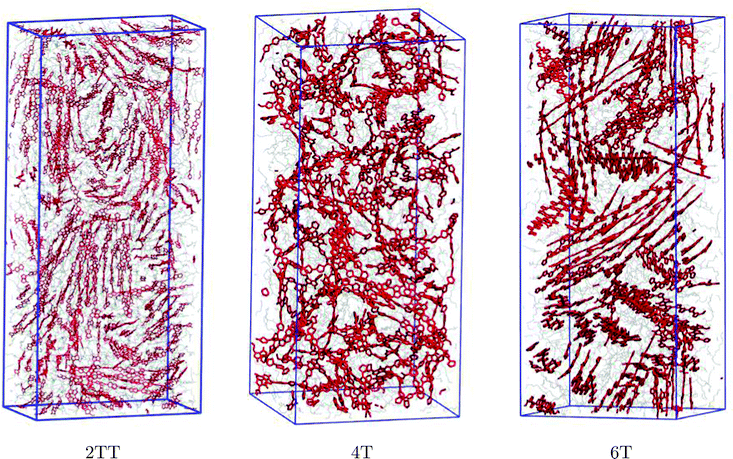 |
| | Fig. 3 Presentation of simulation boxes of the three polymers. The backbones are shown in red and the chain atoms are shown in grey while hydrogen atoms are omitted for simplicity. Snapshots were produced using the visual molecular dynamics (VMD)91 software. Simulation boxes are not shown in the same scale. | |
Radius of gyration (Rg) and end-to-end distance (Dee)
A structural property to determine the flexibility of the polymeric chains and to study their conformation is the radius of gyration (Rg). Rg for a group of n atoms is defined as:92| | 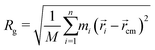 | (1) |
where M is the total mass of the group, ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) cm is the centre of mass position, i is the position of each atom in the group and mi is the mass of each atom. Rg was computed for individual chains as well as an ensemble average for each of the oligomers. Another property of the polymeric chains calculated to determine the rigidity of the chains is the end-to-end distance (Dee). End-to-end distances were computed as the distance between the two closing hydrogen atoms at either end of a chain. Comparing this value to that of an isolated fully elongated chain is an indication of how coiled the chains are. Tables 2 and 3 respectively show comparative values of Rg and Dee both at 550 K and 0 K for the three systems. Comparative representation of the most coiled and the most elongated chains is presented in Fig. 4 for each system, to exemplify the different conformational behaviour of the different systems. Considering the two systems of 2TT and 4T where both have the same number of conjugated moieties along the backbone, the existence of the extra torsional angles between individual thiophene rings provides additional degrees of freedom contributing to the flexibility of the chains and, therefore, a greater standard deviation for both Rg and Dee. When considering the fraction of molecular volume taken up by the free chains, the free volume between the adjacent side chains increases in the order of 2TT, 4T and 6T. The extra free volume for 6T, compared to 2TT, in addition to the rotational degrees of freedom enhances the chance of 6T displaying more flexible and coiled conformations.
cm is the centre of mass position, i is the position of each atom in the group and mi is the mass of each atom. Rg was computed for individual chains as well as an ensemble average for each of the oligomers. Another property of the polymeric chains calculated to determine the rigidity of the chains is the end-to-end distance (Dee). End-to-end distances were computed as the distance between the two closing hydrogen atoms at either end of a chain. Comparing this value to that of an isolated fully elongated chain is an indication of how coiled the chains are. Tables 2 and 3 respectively show comparative values of Rg and Dee both at 550 K and 0 K for the three systems. Comparative representation of the most coiled and the most elongated chains is presented in Fig. 4 for each system, to exemplify the different conformational behaviour of the different systems. Considering the two systems of 2TT and 4T where both have the same number of conjugated moieties along the backbone, the existence of the extra torsional angles between individual thiophene rings provides additional degrees of freedom contributing to the flexibility of the chains and, therefore, a greater standard deviation for both Rg and Dee. When considering the fraction of molecular volume taken up by the free chains, the free volume between the adjacent side chains increases in the order of 2TT, 4T and 6T. The extra free volume for 6T, compared to 2TT, in addition to the rotational degrees of freedom enhances the chance of 6T displaying more flexible and coiled conformations.
Table 2 Calculated average of radius of gyration and its standard deviation during 400 ns production run at 550 K. R0g for a single chain at 0 K is used as an indication of how coiled the chains are
| System |
![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) g/Å
g/Å |
σ R
g/Å |
Min Rg/Å |
Max Rg/Å |
R
0g/Å |
| 2TT |
15.24 |
0.02 |
13.55 |
16.60 |
15.74 |
| 4T |
16.38 |
1.067 |
10.41 |
19.20 |
17.58 |
| 6T |
19.52 |
0.29 |
17.99 |
20.70 |
19.55 |
Table 3 Calculated average of end-to-end distance and its standard deviation during 400 ns production run at 550 K. D0ee for a single chain at 0 K is used as an indication of how coiled the chains are
| System |
![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) ee/Å
ee/Å |
σ D
ee/Å |
Min Dee/Å |
Max Dee/Å |
D
0ee/Å |
| 2TT |
49.90 |
0.26 |
44.37 |
51.52 |
51.07 |
| 4T |
48.82 |
3.52 |
2.94 |
62.28 |
59.16 |
| 6T |
67.24 |
0.46 |
59.73 |
70.12 |
68.12 |
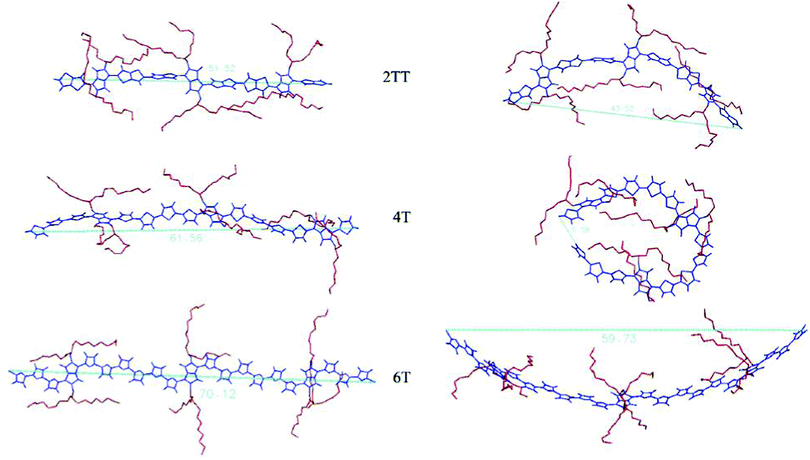 |
| | Fig. 4 Comparison between the most elongated (on the left) and the most coiled (on the right) chains for the three systems. | |
Radial distribution function (RDF)
The radial distribution function (RDF) gab between atoms of class a and b was computed as:93,94| | 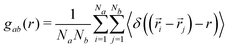 | (2) |
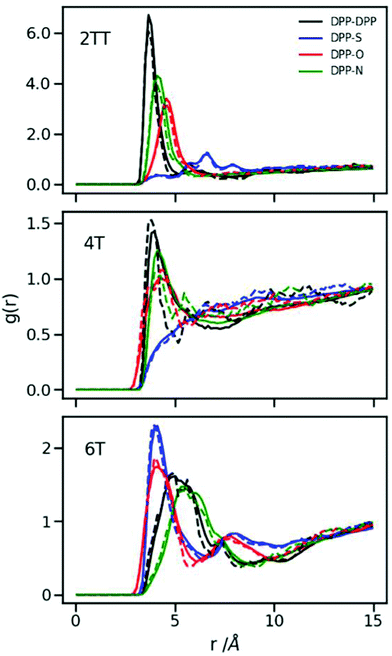
where Na and Nb are number of atoms in each of the classes, ri and rj are the coordinates of atoms belonging to each of the classes and r is the defined radius of a shell for which RDF is defined. Different atom classes were defined to best monitor the variation in structural properties. The DPP group was identified with its two carbon atoms closest to the DPP centre. The S group consists of all the sulphur atoms in the thiophene or thienothiophene units, and the O and N groups contain the oxygen and nitrogen atoms of the DPP, respectively. Interchain pair distribution functions (i.e. excluding atom pairs with both atoms belonging to the same chain) between different atom groups were used to investigate any preferred direction of chain stacking.
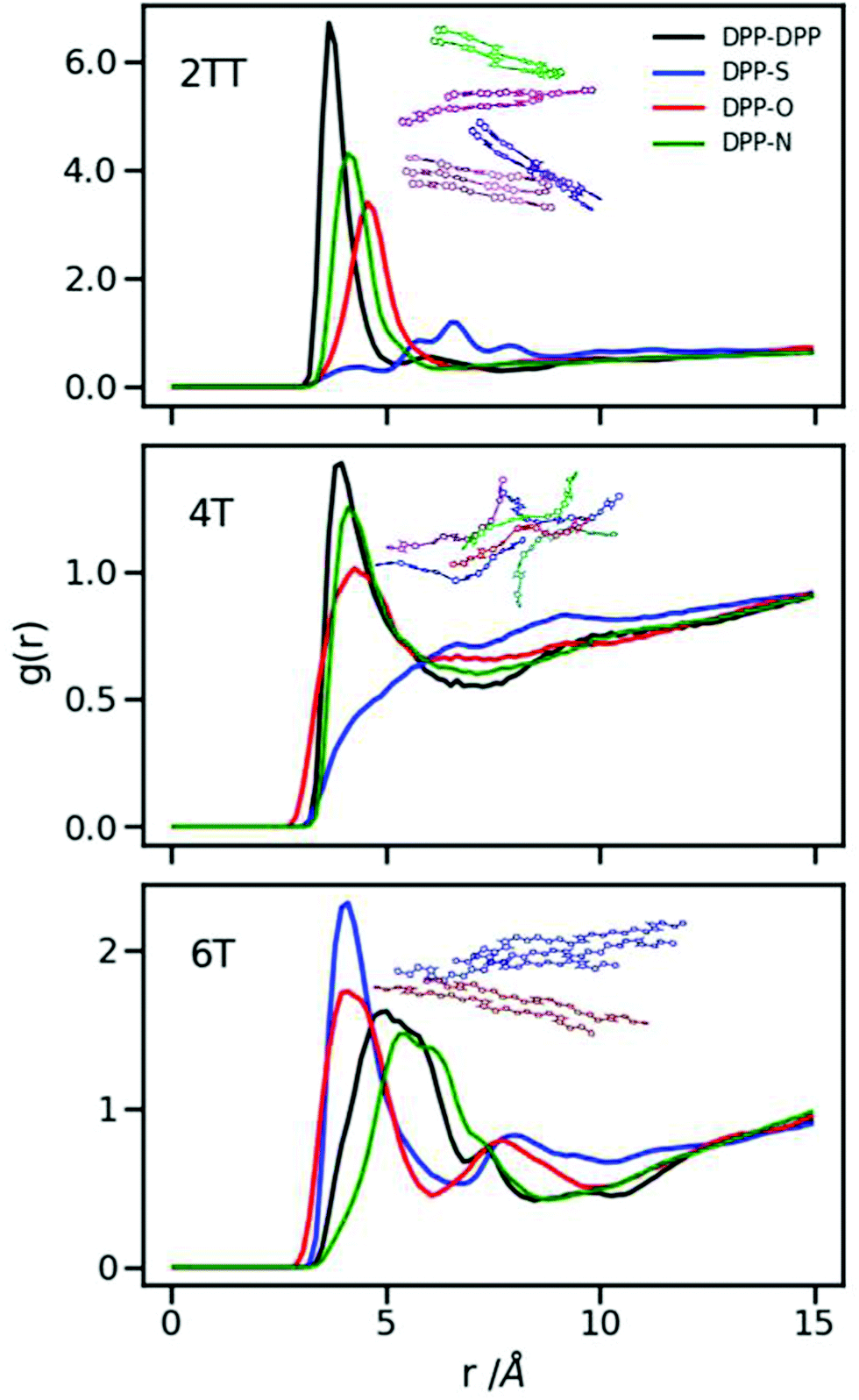
It is evident from Fig. 5 that the dominant interactions for the aggregation are different between oligomers where the DPP–DPP interaction is more likely, for 2TT and 4T, while it is the interaction between DPP and S that seems to dominate the aggregations in 6T, making the DPP unit stacking with the thiophene units result in a shifted chain stacking. Interestingly, despite 4T and 6T oligomers being chemically similar, RDFs show that DPP-S aggregation is mostly non-existent in 4T while it is the main aggregation event in 6T, showing that a small change in the number of backbone repeat units can completely condition the structural properties of the material. The geometry of selected paired chains is depicted as insets of Fig. 5 to illustrate the preferential direction of chain stacking. These insets show that the DPP units are aligned in 2TT while for 6T the alignment of DPP units and thiophenes is preferred resulting in a stepwise stacking. The RDFs provide quite a comprehensive view of the tendency of DPP polymers to aggregate. As one can see from the plots, the height of the RDFs, i.e. the tendency to form stable π-staking interactions, decreases in the order of 2TT > 6T > 4T. On the other hand, the existence of more pronounced second peaks in the order of 6T > 2TT > 4T indicates a different trend in the tendency of these materials to form longer range aggregates. Studying the position of RDFs for the strongest interacting atom group pair indicated that the observed π-stacking for these systems is consistent with the structural information deriving from the study by Huang et al.34 where DPP-4T has a shorter π-stacking distance.
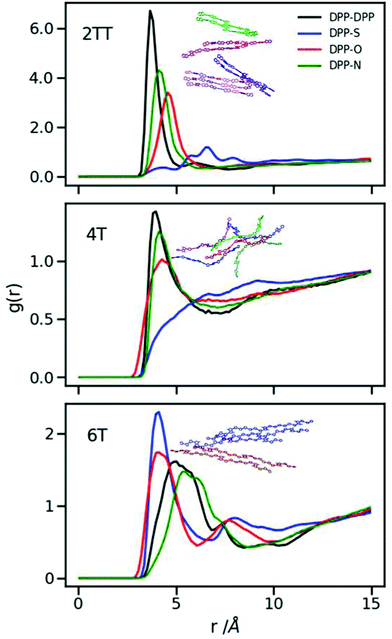 |
| | Fig. 5 Inter-chain RDFs plotted for the atom groups belonging to the conjugated backbone of the systems with an inset of a selection of paired chains. | |
Root mean square displacement
Root mean square displacement (RMSD) per atom type allows the determination of the dynamics of the polymer chains, both as a whole and by looking at each component separately. At short times, different atoms can exhibit a considerable gap in their RMSD, meaning that some parts can be more dynamic with respect to the centre of mass of the molecule than others. In a melt at long enough times, the gap between RMSDs of different atoms should become irrelevant as compared to the dynamics of the molecule as a whole. By contrast, in a solid (or a melt at short times), the molecule might be unable to diffuse but still have moieties with faster dynamics (or more freedom to move) than others. Fig. 6 shows the RMSD calculated for different atom groups as well as all atoms using the equation:95| | 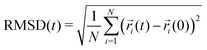 | (3) |
As shown in Fig. 6 middle panel, in the case of 4T, which was previously discussed to be the most disordered and flexible system, all-atom groups seem to be following the same trend with little to no difference between the overall RMSD and the RMSD of different atom groups, indicating that the chain moving as a whole dominates the dynamics in this time range. For 2TT and 6T, as shown in Fig. 6 top and bottom panels, the backbone dynamics is much slower than that of the alkyl-chains, which indicates that the dynamics is dominated by the latter, with the backbone dynamics hampered by the structural micro-domains formed by these molecules. In particular, the faster alkyl chain dynamics observed for 6T is due to the larger free volume in between monomer units caused by the two extra thiophene rings in the backbone. The slow side chain dynamics in 2TT could be a consequence of a higher degree of backbone stacking and segregation of backbone and side chains. It is also worth mentioning that 2TT is the only one exhibiting an increasing backbone RMSD, which indicates that 4T and 6T, regardless of the side chain dynamics, do not behave as a melt in the time-range studied here.
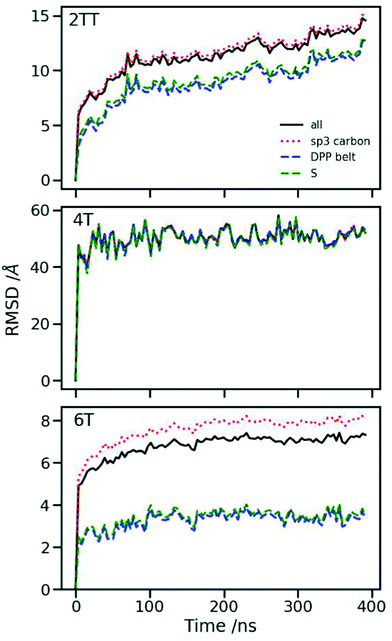 |
| | Fig. 6 RMSD calculations for the three systems showing that there is a greater range of movement associated with the atoms in the side chain for the case of 6T followed by 2TT and 4T. | |
Nematic order parameter
To be able to compute the liquid crystalline ordering across the three systems studied, the nematic liquid crystal order parameter was determined.96 The calculations were carried out by identifying each monomer with a vector connecting the first and last carbon atom of a monomer unit depicted in Fig. 1. A snapshot of these sections (in red) imposed over the atomistic view of each system is depicted in Fig. 7. A unit vector is then assumed at the mid-point of each section to be used in calculations.
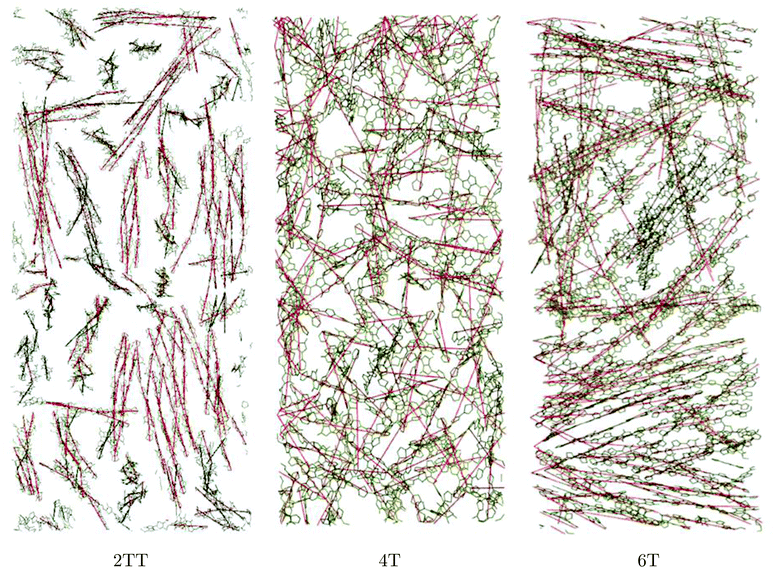 |
| | Fig. 7 Graphical representation of the vectors assigned for each monomer for a snapshot of the simulation for the three systems (side chains are omitted). | |
The order parameters were calculated for each of the systems using the following equation which constructs the Qαβ tensor made up of all the unit vectors ûi with Nm being the number of vectors.97,98
| | 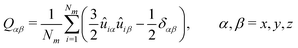 | (4) |
The order parameter P0 is equal to the largest eigenvalue of the Qαβ matrix, taking values between 0 and 1, where 0 indicates a completely isotropic system and 1 a completely aligned system.
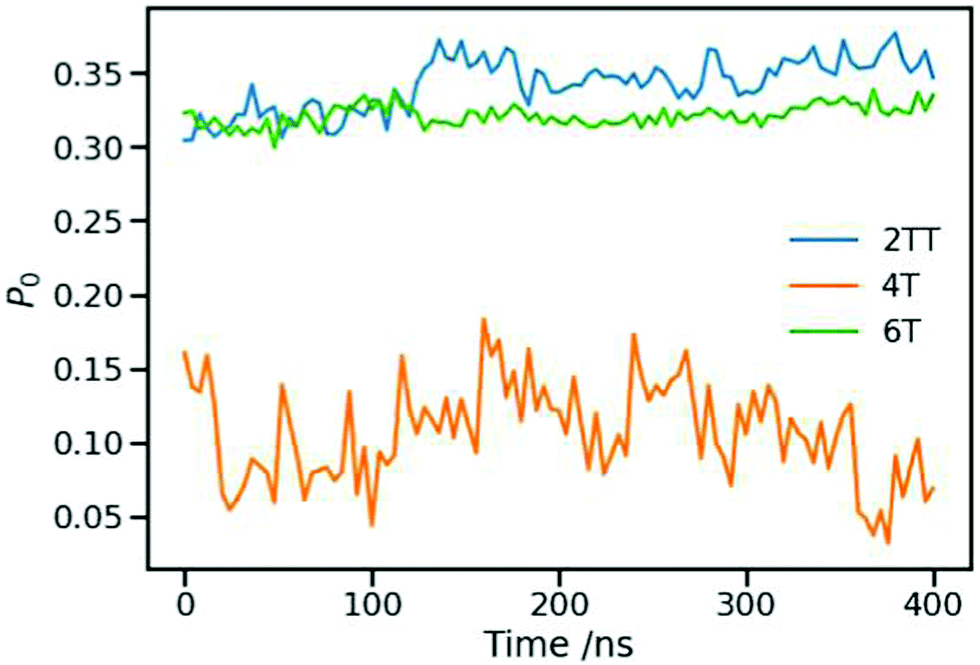
An average order parameter for the three polymers during 400 ns of production simulation at 550 K in an NPT ensemble was calculated and represented in Fig. 8. The average value and their standard deviation are represented in Table 4 to compare the local structure ordering within the three systems.97,99 The larger value of P0 for 2TT followed by 6T and 4T indicates a good agreement of the liquid crystallinity with the conformation analyses. Longer chains of 6T are more susceptible to form longer-range ordered regions while it is the fused rings in the case of 2TT resulting in a high P0 compared to 4T. On comparing between 2TT and 6T, while the value of P0 is similar, there is no indication of different aggregation properties evident from the RDF. We have, therefore, considered an alternative measure of aggregation.
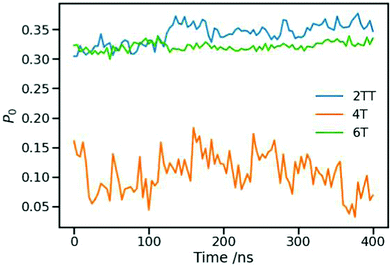 |
| | Fig. 8 Order parameter calculated for the three systems during 400 ns of production simulation. | |
Table 4 Average values calculated for the order parameter of each of the systems throughout 400 ns of simulations
| System |
Order parameter |
Standard deviation |
| 2TT |
0.342 |
0.018 |
| 4T |
0.107 |
0.033 |
| 6T |
0.321 |
0.007 |
Aggregation properties
To define a cluster of aggregates, a set of criteria were defined based on the same representation of trimers as three vectors explained in the previous section. Any two vectors from different oligomers are assumed to belong to the same cluster if they are closer than 10 Å to each other and make an angle smaller than 30°. Distances larger than ∼12 Å resulted in the erroneous selection of chains not in the van der Waals contact while distances shorter ∼8 Å resulted in not including all the good π-stackings. A reasonable angle must be smaller than 45° since angles larger than 45° would include chains in closer-to-perpendicular alignment and we have seen that angles smaller than 20° showed to be too restrictive and excluding well stacked oligomers. The two parameters were chosen as representative of this range, within which the conclusion of analysis remains unchanged. Fig. 9 shows the snapshot of the systems where all monomers belonging to the same cluster are presented in the same colour.
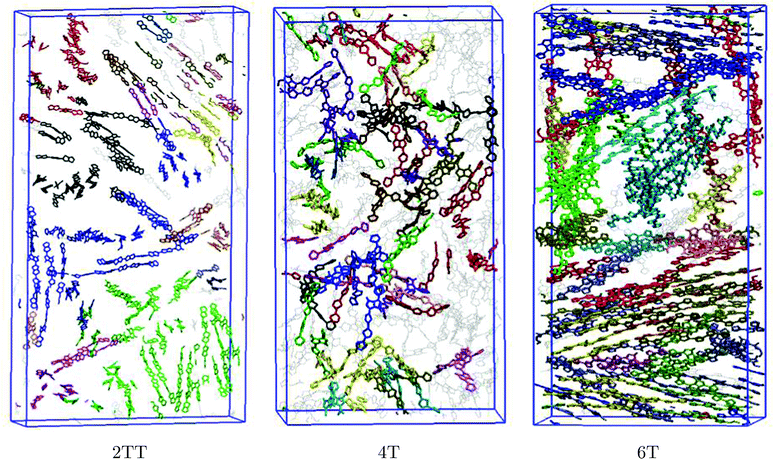 |
| | Fig. 9 Graphical representation of clusters of aggregates. All units belonging to the same aggregate are coloured the same. Units in grey do not belong to any cluster and side chains are omitted for clarity. | |
Fig. 10a represents the number of monomer units in the largest aggregate at different snapshots over 400 ns production simulation at 550 K. In this part of the analysis simulations of the same number of oligomer chains were used for all systems for direct comparison. Consistent with previous analysis of the chain flexibility, RDFs and P0, calculations of aggregate clusters demonstrate the existence of larger aggregates resulting in a higher level of ordering for 2TT and 6T compared to 4T. Unlike P0, which is calculated for the overall simulation box, the calculations of local clusters reveal more dynamical properties of the aggregates. The largest aggregate in 2TT can have a varying number of monomers from 28 to 6, from 49 to 9 for 6T and from 20 to 5 monomer units for 4T. When considered with the P0 order parameter, the time evolution does not suggest an evolution toward a more ordered phase in time, which implies good equilibration and is consistent with the temperature being above the Tg. The greater fluctuation in the size of the largest aggregate for 6T is considered to be caused by the movement of monomers of this naturally longer chain between aggregate clusters. Fig. 10b shows a distribution plot where the y-axis presents the number of times a specifically-sized cluster is formed during the 400 ns of production simulation and the x-axis presents the size of the clusters. It is shown that, for the least ordered system of 4T, the size of the aggregates does not exceed 20 monomers whereas it is increased to 28 for 2TT and 49 for 6T. The cluster size distribution is consistent with the observation made in the previous analyses and considered the most effective measure to differentiate the aggregation properties between oligomers.
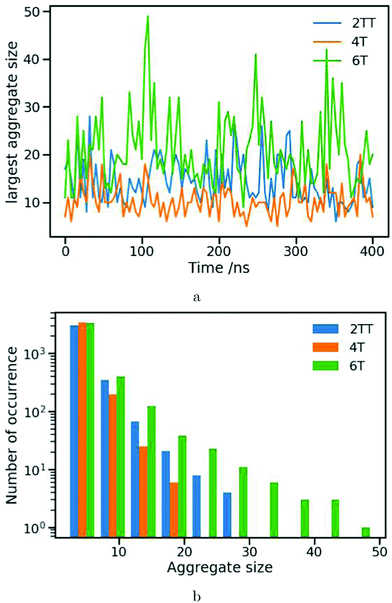 |
| | Fig. 10 Plots showing (a) the size of the largest aggregate cluster for snapshots at 4 ns interval during 400 ns of production simulation at 550 K (b) the count of different sized aggregate clusters for each of the systems. | |
Extension to 300 K
In this work we have focused on the structural characterisation of semiconducting polymers at a temperature above Tg, where equilibration is easier to achieve and the results are highly reproducible. It is nevertheless instructive to compare the main RDFs with a simulation at 300 K to establish the changes that could be expected. This method of comparison allows determining if the observations made prior can be transferred to lower temperatures. To achieve simulations at 300 K, starting from the last snapshot of the equilibrated simulation at 550 K, a simulated annealing to 300 K was performed for each of the systems using the same cooling rate as the initial annealing (0.02 K ps−1). A further 50 ns equilibration simulation was then performed on each of the simulations at 300 K. Fig. 11 shows a comparison plot of RDFs between the simulations at 550 K (solid) and 300 K (dashed lines). Comparing the RDFs between the two temperatures, there are minimal differences in the intensity and the position of peaks for all important interactions. The increased noise in the RDFs for 300 K is likely caused by a poorer configuration sampling below the Tg in what is, effectively, a quenched phase. The RDFs of all systems have a better overlap at shorter ranges with the biggest difference between 300 and 550 K at longer ranges for 4T (middle panel in Fig. 11), the least ordered of the three systems.
 |
| | Fig. 11 Comparison between RDFs of different atom groups between an equilibrated simulation at 550 K in solid lines and 300 K in dashed lines. The definition of atom groups is the same as mentioned in the previous sections. | |
Electronic structure of the ensemble of oligomers
The electronic structure of semiconducting polymers is often described in terms of the density of states (DOS) of the valence band, which provides an indication of the energetic position and disorder of the states occupied by the positive charge carriers.100,101 Energetically narrower bands are associated with better transport properties and are thought to originate from more ordered conjugated backbones.102 The interaction between chains was shown to influence the conformation and, therefore, the DOS but the electronic structure of the bulk polymer is to a great extent dictated by the electronic structure of individual chains.62
In this section we compute the DOS for individual chains extracted from 4 snapshots of the MD simulations separated by 15 ns at 300 and 550 K. A total of 64 chains for each of the 6 systems have been used. The electronic structure calculation was performed at the B3LYP/3-21G* level, suitable for this kind of large scale electronic structure calculation and well correlated with calculation with higher basis sets.62 The DOS, was obtained from broadening the orbital energies with a normalised Gaussian function of standard deviation 0.03 eV and the results are collected in Fig. 12 which focuses on the region immediately below the band gap. The choice of the Gaussian broadening parameter is not very critical for the discussion in this section as the features reported in Fig. 12 are much wider than this broadening (>0.1 eV), i.e. are due to the distribution of the energy levels rather than the broadening parameter. The DOS is approximately constant for broadening in the range 0.01–0.04 eV and would become too noisy if the broadening parameter is set to a value much smaller than the energy difference between orbital energies, e.g. <0.005 eV for the sample size considered here. The electronic structure calculations have been performed for the model system where the side chains have been shortened to −CH3.
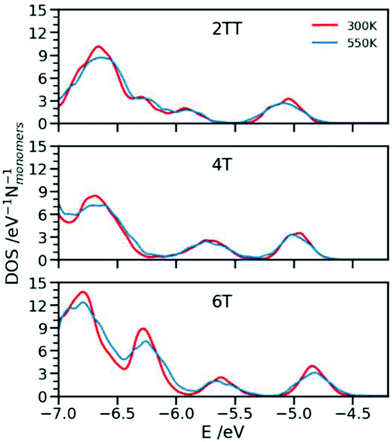 |
| | Fig. 12 Density of States (DOS) calculated for the three systems at 300 and 550 K. | |
The first interesting observation is that there is no substantial change in the DOS shape in going from 550 to 300 K except for the (expected) slightly larger broadening of the peaks seen at 550 K where a greater range of inter-monomer dihedral angles is explored. This is useful in practice because simulations at higher temperatures are more easily equilibrated and it is possible to use them as a guide if the electronic structure of many polymers is to be considered. The most relevant feature of the DOS is the peak at higher energy. This is dominated by the DPP units which has the largest HOMO energy among the fragments with the position of the peak modulated by the remaining constituents of the chains. The polymer with the greatest order (6T) displays the narrower 300 K peak, which is a combined result of the greater structural order discussed in the previous sections and the greater distance between DPP units which tends to narrow down the highest energy band (i.e. to decrease the interaction between units).103 As shown in a recent work, changing the composition of the backbone leads to the effect of changing both the conformational flexibility and the sensitivity of the electronic structure to the conformational flexibility.104
Conclusion
Atomic MD simulations were employed to study the local structuring and morphological characteristics of three oligomers belonging to the same family of SCPs based on the DPP monomer and different conjugated units constituting the backbone unit. The corresponding polymers of the systems studied here have displayed high charge carrier mobility, which is generally attributed to the ability of these materials to form partially ordered aggregates. An important difference among the polymers studied is their rigidity which depends on the chemistry of the constituents and also the ratio between the volume of the side chain and that of the conjugated part. The polymer containing 6 thiophene rings between the DPP units is more rigid than the one containing 4 thiophene units suggesting a role on the side chain in disrupting the ordering. Strong aggregation at short distances was observed for all three systems and was quantitatively analysed using RDFs that showed a higher degree of short-range stacking for 2TT followed by 6T and 4T. A stronger interaction between the DPP units and sulphur atoms for 6T compared to DPP units for 2TT points to the different reasons for the chain stacking across the oligomers. The dynamics of the side chains, identical for the three systems studied, is seen to be affected by the nature of the conjugated portion, with greater flexibility observed for 6T (where there is more space between side chains) and 2TT (the most rigid oligomer). The existence and the size of aggregate clusters were studied using the nematic liquid crystalline order parameter and the distribution of cluster sizes. The main structural features and the electronic structure of the oligomers are fairly similar above the glass transition temperature and at room temperature. In essence, polymers containing DPP fragments tend to form aggregates driven by the π-stacking of the conjugated units, however, this tendency is highly modulated by the polymer composition, which can influence the flexibility and the tendency to form extended stacks.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to PRACE for providing computational resources for this work (Project number: 2018184449). Rex Manurung is grateful for financial support from the Leverhulme Trust via the Leverhulme Research Centre for Functional Materials Design (Grant No. Leverhulme Trust RC-2015-036).
References
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Mechanical properties of organic semiconductors for stretchable, highly exible, and mechanically robust electronics, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- G. Gelinck, P. Heremans, K. Nomoto and T. D. Anthopoulos, Organic transistors in optical displays and microelectronic applications, Adv. Mater., 2010, 22, 3778–3798 CrossRef CAS PubMed.
- W. Wu, Y. Liu and D. Zhu, π-Conjugated molecules with fused rings for organic fielde effect transistors: design, synthesis and applications, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- Z. Yi, S. Wang and Y. Liu, Design of high-mobility diketopyrrolopyrrole-based π-conjugated copolymers for organic thin-film transistors, Adv. Mater., 2015, 27, 3589–3606 CrossRef CAS PubMed.
- M. Yoneya, S. Matsuoka, J. Tsutsumi and T. Hasegawa, Self-assembly of donor-acceptor semiconducting polymers in solution thin films: a molecular dynamics simulation study, J. Mater. Chem. C, 2017, 5, 9602–9610 RSC.
- Y. Kawanabe, A. J. Moulé and R. Faller, Molecular dynamics study of the local structure of photovoltaic polymer PCDTBT, J. Chem. Eng. Data, 2014, 59, 2982–2986 CrossRef CAS.
- T. Sun, J. Han, S. Liu, X. Wang, Z. Y. Wang and Z. Xie, Tailor-made semiconducting polymers for second near-infrared photothermal therapy of orthotopic liver cancer. Am. Chem, Soc. Nano., 2019, 13, 7345–7354 CAS.
- G.-J. N. Wang, A. Gasperini and Z. Bao, Stretchable polymer semiconductors for plastic electronics, Adv. Electron. Mater., 2018, 4, 1700429 CrossRef.
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig and J. Lopez, Skin electronics from scalable fabrication of an intrinsically stretchable transistor array, Nature, 2018, 555, 83 CrossRef CAS PubMed.
- H.-J. Yun, G. B. Lee, D. S. Chung, Y.-H. Kim and S.-K. Kwon, Novel Diketopyrroloppyrrole Random Copolymers: High Charge-Carrier Mobility From Environmentally Benign Processing, Adv. Mater., 2014, 26, 6612–6616 CrossRef CAS PubMed.
- A. Facchetti, π-Conjugated polymers for organic electronics and photovoltaic cell applications, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, High-performance semiconducting polythiophenes for organic thin-film transistors, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS.
- C. Poelking, E. Cho, A. Malafeev, V. Ivanov, K. Kremer, C. Risko, J.-L. Brédas and D. Andrienko, Characterization of charge-carrier transport in semicrystalline polymers: electronic couplings, site energies, and charge-carrier dynamics in poly (bithiophene-alt-thienothiophene)[PBTTT], J. Phys. Chem. C, 2013, 117, 1633–1640 CrossRef CAS.
- T. Liu and A. Troisi, Understanding the microscopic origin of the very high charge mobility in PBTTT: tolerance of thermal disorder, Adv. Funct. Mater., 2014, 24, 925–933 CrossRef CAS.
- K. Do, M. K. Ravva, T. Wang and J.-L. Brédas, Computational methodologies for developing structure-morphology-performance relationships in organic solar cells: A protocol review, Chem. Mater., 2016, 29, 346–354 CrossRef.
- P. Brocorens, Van Vooren, A. Chabinyc, M. L. Toney, M. F. Shkunov, M. Heeney, M. McCulloch, I. Cornil, J. Lazzaroni and R. Solid-State, Supramolecular Organization of Polythiophene Chains Containing Thienothiophene Units, Adv. Mater., 2009, 21, 1193–1198 CrossRef CAS.
- R. J. Kline, D. M. DeLongchamp, D. A. Fischer, E. K. Lin, M. Heeney, I. McCulloch and M. F. Toney, Significant dependence of morphology and charge
carrier mobility on substrate surface chemistry in high performance polythiophene semiconductor films, Appl. Phys. Lett., 2007, 90, 062117 CrossRef.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, A general relationship between disorder, aggregation and charge transport in conjugated polymers, Nat. Mater., 2013, 12, 1038 CrossRef CAS PubMed.
- O. Alexiadis and V. G. Mavrantzas, All-atom molecular dynamics simulation of temperature effects on the structural, thermodynamic, and packing properties of the pure amorphous and pure crystalline phases of regioregular P3HT, Macromolecules, 2013, 46, 2450–2467 CrossRef CAS.
- H. Sirringhaus, 25th anniversary article: organic field-effect transistors: the path beyond amorphous silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- M. Selivanova, C.-H. Chuang, B. Billet, A. Malik, P. Xiang, E. Landry, Y.-C. Chiu and S. Rondeau-Gagné, Morphology and electronic properties of semiconducting polymer and branched polyethylene blends, Am. Chem. Soc. Appl. Mater. Interfaces, 2019, 11, 12723–12732 CrossRef CAS.
- S. B. Darling and M. Sternberg, Importance of side chains and backbone length in defect modeling of poly(3-alkylthiophenes), J. Phys. Chem. B, 2009, 113, 6215–6218 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, Record high hole mobility in polymer semiconductors via side-chain engineering, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- G. H. Heintges, K. H. Hendriks, F. J. Colberts, M. Li, J. Li and R. A. Janssen, The in uence of siloxane side-chains on the photovoltaic performance of a conjugated polymer, RSC Adv., 2019, 9, 8740–8747 RSC.
- T. L. Nelson, T. M. Young, J. Liu, S. P. Mishra, J. A. Belot, C. L. Balliet, A. E. Javier, T. Kowalewski and R. D. McCullough, Transistor paint: high mobilities in small bandgap polymer semiconductor based on the strong acceptor, diketopyrrolopyrrole and strong donor, dithienopyrrole, Adv. Mater., 2010, 22, 4617–4621 CrossRef CAS PubMed.
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald and H.-J. Kirner, Winnewisser, C. High-mobility ambipolar near-infrared light-emitting polymer field-effect transistors, Adv. Mater., 2008, 20, 2217–2224 CrossRef.
- J. S. Lee, S. K. Son, S. Song, H. Kim, D. R. Lee, K. Kim, M. J. Ko, D. H. Choi, B. Kim and J. H. Cho, Importance of solubilizing group and backbone planarity in low band gap polymers for high performance ambipolar field-effect transistors, Chem. Mater., 2012, 24, 1316–1323 CrossRef CAS.
- C. R. Snyder, D. M. DeLongchamp, R. C. Nieuwendaal and A. A. Herzing, Semicond. Polym., 2016, 219–274 Search PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. Janssen, Diketopyrrolopyrrole polymers for organic solar cells, Acc. Chem. Res., 2015, 49, 78–85 CrossRef PubMed.
- C. Zhao, Y. Guo, Y. Zhang, N. Yan, S. You and W. Li, Diketopyrrolopyrrole-based conjugated materials for non-fullerene organic solar cells, J. Mater. Chem. A, 2019, 7, 10174–10199 RSC.
- K. Sim, A. K. Palai, G. Tarsoly, H. Na and S. Pyo, Polymer binder assisted, solution processed cyanophenyl functionalized diketopyrrolopyrrole microwire for n-channel field-effect transistors, Synth. Met., 2019, 250, 152–160 CrossRef CAS.
- A. Negash, Z. Genene, R. T. Eachambadi, J. Kesters, N. Van den Brande, J. D’Haen, H. Penxten, B. A. Abdulahi, E. Wang and K. Vandewal, Diketopyrrolopyrrole-based terpolymers with tunable broad band absorption for fullerene and fullerene-free polymer solar cells, J. Mater. Chem. C, 2019, 7, 3375–3384 RSC.
- J. C. Bijleveld, A. P. Zoombelt, S. G. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. Janssen, Poly [diketopyrrolopyrrole- terthiophene] for Ambipolar Logic and Photovoltaics, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- Y.-F. Huang, S.-T. Chang, K.-Y. Wu, S.-L. Wu, G.-T. Ciou, C.-Y. Chen, C.-L. Liu and C.-L. Wang, In uences of conjugation length on organic field-effect transistor performances and thin film structures of diketopyrrolopyrrole-oligomers, ACS Appl. Mater. Interfaces, 2018, 10, 8869–8876 CrossRef CAS.
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, Design, synthesis, and self-assembly of oligothiophene derivatives with a diketopyrrolopyrrole core, J. Phys. Chem. C, 2008, 112, 15543–15552 CrossRef CAS.
- S. R. Chaudhari, J. M. Griffin, K. Broch, A. Lesage, V. Lemaur, D. Dudenko, Y. Olivier, H. Sirringhaus, L. Emsley and C. P. Grey, Donor-acceptor stacking arrangements in bulk and thin-film high-mobility conjugated polymers characterized using molecular modelling and MAS and surface-enhanced solid-state NMR spectroscopy, Chem. Sci., 2017, 8, 3126–3136 RSC.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Effect of uorination on the properties of a donor-acceptor copolymer for use in photovoltaic cells and transistors, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- C. Liu, W. Hu, H. Jiang, G. Liu, C. C. Han, H. Sirringhaus, F. Boué and D. Wang, Chain Conformation and Aggregation Structure Formation of a High Charge Mobility DPP-Based Donor-Acceptor Conjugated Polymer, Macromolecules, 2020, 53, 8255–8266 CrossRef CAS.
- S. Zhang, M. U. Ocheje, L. Huang, L. Galuska, Z. Cao, S. Luo, Y.-H. Cheng, D. Ehlenberg, R. B. Goodman and D. Zhou, The Critical Role of Electron-Donating Thiophene Groups on the Mechanical and Thermal Properties of Donor-Acceptor Semiconducting Polymers, Adv. Electron. Mater., 2019, 5, 1800899 CrossRef.
- Y. Li, S. P. Singh and P. Sonar, A High Mobility P-Type DPP-Thieno [3, 2-b] thiophene Copolymer for Organic Thin-Film Transistors, Adv. Mater., 2010, 22, 4862–4866 CrossRef CAS.
- S. P. Ponnappa, S. Arumugam, H. J. Spratt, S. Manzhos, A. P. O’Mullane, G. A. Ayoko and P. Sonar, A comparative study of electrochemical, optical properties and electropolymerization behavior of thiophene-and furan-substituted diketopyrrolopyrrole, J. Mater. Res., 2017, 32, 810–821 CrossRef CAS.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. Fréchet, Side-chain tunability of furan-containing low-band-gap polymers provides control of structural order in efficient solar cells, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS PubMed.
- D. A. Warr, L. M. Perdigão, H. Pinfold, J. Blohm, D. Stringer, A. Leventis, H. Bronstein, A. Troisi and G. Costantini, Sequencing conjugated polymers by eye, Sci. Adv., 2018, 4, eaas9543 CrossRef PubMed.
- J. Humphreys, F. Pop, P. A. Hume, A. S. Murphy, W. Lewis, E. S. Davies, S. P. Argent and D. B. Amabilino, Solid state structure and properties of phenyl diketopyrrolopyrrole derivatives, CrystEngComm., 2021, 23, 1796–1814 RSC.
- S. Zhang, A. Alesadi, G. T. Mason, K.-L. Chen, G. Freychet, L. Galuska, Y.-H. Cheng, P. B. J. St. Onge, M. U. Ocheje and G. Ma, Molecular Origin of Strain- Induced Chain Alignment in PDPP-Based Semiconducting Polymeric Thin Films, Adv. Funct. Mater., 2021, 31, 2100161 CrossRef CAS.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen and H. Sirringhaus, Chalcogenophene comonomer 35 comparison in small band gap diketopyrrolopyrrole-based conjugated polymers for high-performing field-effect transistors and organic solar cells, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- Y.-C. Lin, Y.-W. Huang, C.-C. Hung, Y.-C. Chiang, C.-K. Chen, L.-C. Hsu, C.-C. Chueh and W.-C. Chen, Backbone Engineering of Diketopyrrolopyrrole-Based Conjugated Polymers through Random Terpolymerization for Improved Mobility- Stretchability Property, ACS Appl. Mater. Interfaces, 2020, 12, 50648–50659 CrossRef CAS.
- Z. Wang, Z. Liu, L. Ning, M. Xiao, Y. Yi, Z. Cai, A. Sadhanala, G. Zhang, W. Chen and H. Sirringhaus, Charge mobility enhancement for conjugated DPPselenophene polymer by simply replacing one bulky branching alkyl chain with linear one at each DPP unit, Chem. Mater., 2018, 30, 3090–3100 CrossRef CAS.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo and J. Nelson, Photocurrent enhancement from diketopyrrolopyrrole polymer solar cells through alkyl-chain branching point manipulation, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS.
- B. W. Saes, M. Lutz, M. M. Wienk, S. C. Meskers and R. A. Janssen, Tuning the Optical Characteristics of Diketopyrrolopyrrole Molecules in the Solid State by Alkyl Side Chains, J. Phys. Chem. C, 2020, 124, 25229–25238 CrossRef CAS.
- Q. V. Hoang, C. E. Song, I.-N. Kang, S.-J. Moon, S. K. Lee, J.-C. Lee and W. S. Shin, Low band gap diketopyrrolopyrrole-based small molecule bulk heterojunction solar cells: in uence of terminal side chain on morphology and photovoltaic performance, RSC Adv., 2016, 6, 28658–28665 RSC.
- K. Zhang, P. Wucher, T. Marszalek, M. Babics, A. Ringk, P. W. Blom, P. M. Beaujuge and W. Pisula, Long-Range Molecular Self-Assembly from π-Extended Pyrene- Functionalized Diketopyrrolopyrroles, Chem. Mater., 2018, 30, 5032–5040 CrossRef CAS.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Design of diketopyrrolopyrrole (DPP)-Based small molecules for organic-solar-cell applications, Adv. Mater., 2017, 29, 1600013 CrossRef PubMed.
- M. Tang, S. Wu, W. Xing, H. Shen, L. Xiang, Y. Liang, W. Xu and D. Zhu, Diketopyrrolopyrrole based small molecular semiconductors containing thiazole units for solution-processed n-channel thin-film transistors, Dyes Pigm., 2019, 163, 707–714 CrossRef CAS.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch and S. C. Hayes, Exploring the origin of high optical absorption in conjugated polymers, Nat. Mater., 2016, 15, 746–753 CrossRef CAS PubMed.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, Significant improvement of semiconducting performance of the diketopyrrolopyrrole-quaterthiophene conjugated polymer through side-chain engineering via hydrogen-bonding, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- P. J. Leenaers, H. Van Eersel, J. Li, M. M. Wienk and R. A. Janssen, In uence of regioregularity on the optoelectronic properties of conjugated diketopyrrolopyrrole polymers comprising asymmetric monomers, Macromolecules, 2020, 53, 7749–7758 CrossRef CAS PubMed.
- D. L. Cheung, D. P. McMahon and A. Troisi, Computational study of the structure and charge-transfer parameters in low-molecular-mass P3HT, J. Phys. Chem. B, 2009, 113, 9393–9401 CrossRef CAS PubMed.
- D. P. McMahon, D. L. Cheung, L. Goris, J. Dacuna, A. Salleo and A. Troisi, Relation between microstructure and charge transport in polymers of different regioregularity, J. Phys. Chem. C, 2011, 115, 19386–19393 CrossRef CAS.
- T. Liu, D. L. Cheung and A. Troisi, Structural variability and dynamics of the P3HT/PCBM interface and its effects on the electronic structure and the chargetransfer rates in solar cells, Phys. Chem. Chem. Phys., 2011, 13, 21461–21470 RSC.
- K. Do, D. M. Huang, R. Faller and A. J. Moulé, A comparative MD study of the local structure of polymer semiconductors P3HT and PBTTT, Phys. Chem. Chem. Phys., 2010, 12, 14735–14739 RSC.
- T. Qin and A. Troisi, Relation between structure and electronic properties of amorphous MEH-PPV polymers, J. Am. Chem. Soc., 2013, 135, 11247–11256 CrossRef CAS PubMed.
- I. Yavuz, L. Zhang, A. L. Briseno and K. Houk, Simulations of Molecular Ordering and Charge-Transport of Oligo-Didodecylquaterthiophenes [DDQT], J. Phys. Chem. C, 2015, 119, 158–165 CrossRef CAS.
- S. Caputo, De Nicola, A. Donati, G. David, A. Raos, G. Milano and G. All-Atom, Model of Atactic 2-Vinyl Pyridine Polymer: Structural Properties Investigation by Molecular Dynamics Simulations, J. Electrochem., 2019, 166, B3309–B3315 CAS.
- N. B. Kotadiya, A. Mondal, P. W. Blom, D. Andrienko and G.-J. A. Wetzelaer, A window to trap-free charge transport in organic semiconducting thin films, Nat. Mater., 2019, 18, 1182–1186 CrossRef CAS PubMed.
- D. Abbaszadeh, A. Kunz, N. B. Kotadiya, A. Mondal, D. Andrienko, J. J. Michels, G.-J. A. Wetzelaer and P. W. Blom, Electron trapping in conjugated polymers, Chem. Mater., 2019, 31, 6380–6386 CrossRef CAS.
- S. M. Swick, W. Zhu, M. Matta, T. J. Aldrich, A. Harbuzaru, J. T. L. Navarrete, R. P. Ortiz, K. L. Kohlstedt, G. C. Schatz and A. Facchetti, Closely packed, low reorganization energy π-extended postfullerene acceptors for efficient polymer solar cells, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8341–E8348 CrossRef CAS.
- C. M. Wolf, K. H. Kanekal, Y. Y. Yimer, M. Tyagi, S. Omar-Diallo, V. Pakhnyuk, C. K. Luscombe, J. Pfaendtner and L. D. Pozzo, Assessment of molecular dynamics simulations for amorphous poly [3-hexylthiophene] using neutron and X-ray scattering experiments, Soft Matter, 2019, 15, 5067–5083 RSC.
- A. Baggioli, M. Casalegno, G. Raos, L. Muccioli, S. Orlandi and C. Zannoni, Atomistic Simulation of Phase Transitions and Charge Mobility for the Organic Semiconductor Ph-BTBT-C10, Chem. Mater., 2019, 31, 7092–7103 CrossRef CAS.
- R. Alessandri, J. J. Uusitalo, A. H. de Vries, R. W. Havenith and S. J. Marrink, Bulk heterojunction morphologies with atomistic resolution from coarse-grain solvent evaporation simulations, J. Am. Chem. Soc., 2017, 139, 3697–3705 CrossRef CAS PubMed.
- N. E. Jackson, K. L. Kohlstedt, B. M. Savoie, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, Conformational order in aggregates of conjugated polymers, J. Am. Chem. Soc., 2015, 137, 6254–6262 CrossRef CAS.
- M. M. Henry, M. L. Jones, S. D. Oosterhout, W. A. Braunecker, T. W. Kemper, R. E. Larsen, N. Kopidakis, M. F. Toney, D. C. Olson and E. Jankowski, Simplified Models for Accelerated Structural Prediction of Conjugated Semiconducting Polymers, J. Phys. Chem. C, 2017, 121, 26528–26538 CrossRef.
- M. Casalegno, T. Nicolini, A. Famulari, G. Raos, R. Po and S. V. Meille, Atomistic modelling of entropy driven phase transitions between different crystal modifications in polymers: the case of poly [3-alkylthiophenes], Phys. Chem. Chem. Phys., 2018, 20, 28984–28989 RSC.
- S. A. Mollinger, B. A. Krajina, R. Noriega, A. Salleo and A. J. Spakowitz, Percolation, tie-molecules, and the microstructural determinants of charge transport in semicrystalline conjugated polymers, ACS Macro Lett., 2015, 4, 708–712 CrossRef CAS.
- P. Carbone and A. Troisi, Charge diffusion in semiconducting polymers: analytical relation between polymer rigidity and time scales for intrachain and interchain hopping, J. Phys. Chem. Lett., 2014, 5, 2637–2641 CrossRef CAS.
- D. Raychev, R. D. Méndez López, A. Kiriy, G. Seifert, J.-U. Sommer and O. Guskova, Copolymers of diketopyrrolopyrrole and benzothiadiazole: design and function from simulations with experimental support, Macromolecules, 2019, 52, 904–914 CrossRef.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins and Y. Geerts, Thieno [3, 2-b] thiophenediketopyrrolopyrrole- containing polymers for high-performance organic field-effect transistors and organic photovoltaic devices, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- M. H. Hoang, D. N. Nguyen, T. T. Ngo, H. A. Um, M. J. Cho and D. H. Choi, Molecular-weight engineering of high-performing diketopyrrolopyrrole-based copolymer bearing high π-extended long donating units, Polymer, 2016, 83, 77–84 CrossRef CAS.
- C. E. Song, Y. J. Kim, S. R. Suranagi, G. P. Kini, S. Park, S. K. Lee, W. S. Shin, S.-J. Moon, I.-N. Kang and C. E. Park, Impact of the crystalline packing structures on charge transport and recombination via alkyl chain tunability of DPP-based small molecules in bulk heterojunction solar cells, Am. Chem. Soc. Appl. Mater. Interfaces, 2016, 8, 12940–12950 CrossRef CAS.
- J. S. Ha, K. H. Kim and D. H. Choi, 2, 5-Bis [2-octyldodecyl] pyrrolo [3, 4-c] pyrrole-1, 4-(2 H, 5 H)-dione-Based Donor-Acceptor Alternating Copolymer Bearing 5, 5′-Di (thiophen-2-yl)-2, 2′-biselenophene Exhibiting 1.5 cm2 V−1 s−1 Hole Mobility in Thin-Film Transistors, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. Janssen, Diketopyrrolopyrrole polymers for organic solar cells, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis, J. Comput. Chem, 1990, 11, 361–373 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, A direct MP2 gradient method, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Semi-direct algorithms for the MP2 energy and gradient, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- L. Jiang, D. M. Rogers, J. D. Hirst and H. Do, Force fields for macromolecular assemblies containing Diketopyrrolopyrrole and Thiophene, J. Chem. Theory Comput., 2020, 16, 5150–5162 CrossRef CAS.
- K. Broch, D. Venkateshvaran, V. Lemaur, Y. Olivier, D. Beljonne, M. Zelazny, I. Nasrallah, D. J. Harkin, M. Statz and R. D. Pietro, Measurements of Ambipolar Seebeck Coefficients in High-Mobility Diketopyrrolopyrrole Donor-Acceptor Copolymers, Adv. Electron. Mater., 2017, 3, 1700225 CrossRef.
- V. Sundaram, A. V. Lyulin and B. Baumeier, Development and Testing of an All-Atom Force Field for Diketopyrrolopyrrole Polymers with Conjugated Substituents, J. Phys. Chem. B, 2020, 124, 11030–11039 CrossRef CAS PubMed.
- S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner and W. Zhang, Liquid-crystalline semiconducting polymers with high charge-carrier mobility, Nat. Mater., 2006, 5, 328 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD - Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
-
M. Rubinstein and R. H. ColbyPolymer Physics, Oxford University Press, New York, 2003; vol. 23 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, MDAnalysis: a toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
-
R. J. Gowers; M. Linke; J. Barnoud; T. J. E. Reddy; M. N. Melo; S. L. Seyler; J. Domanski; D. L. Dotson; S. Buchoux and I. M. Kenney MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations; 2019.
- J. J. Burgos-Mármol and A. Patti, Unveiling the impact of nanoparticle size dispersity on the behavior of polymer nanocomposites, Polymer, 2017, 113, 92–104 CrossRef.
-
G. Vertogen and W. H. De Jeu, Thermotropic liquid crystals, fundamentals, Springer Science & Business Media, 2012; vol. 45 Search PubMed.
-
D. L. G. Cheung, Structures and properties of liquid crystals and related molecules from computer simulation, PhD thesis, Durham University, 2002.
- J. J. Burgos-Mármol, O. Álvarez-Machancoses and A. Patti, Modeling the effect of polymer chain stiffness on the behavior of polymer nanocomposites, J. Phys. Chem. B, 2017, 121, 6245–6256 CrossRef PubMed.
- A. Saupe, Recent results in the field of liquid crystals, Angew. Chem., 1968, 7, 97–112 CrossRef CAS.
- M. Mladenović and N. Vukmirović, Charge carrier localization and transport in organic semiconductors: Insights from atomistic multiscale simulations, Adv. Funct. Mater., 2015, 25, 1915–1932 CrossRef.
- D. Cheung, D. P. McMahon and A. Troisi, A realistic description of the charge carrier wave function in microcrystalline polymer semiconductors, J. Am. Chem. Soc., 2009, 131, 11179–11186 CrossRef CAS PubMed.
- R. P. Fornari and A. Troisi, Theory of charge hopping along a disordered polymer chain, Phys. Chem. Chem. Phys., 2014, 16, 9997–10007 RSC.
- R. P. Fornari and A. Troisi, Narrower bands with better charge transport: The counterintuitive behavior of semiconducting copolymers, Adv. Mater., 2014, 26, 7627–7631 CrossRef CAS PubMed.
- R. Manurung, P. Li and A. Troisi, Rapid Method for Calculating the Conformationally Averaged Electronic Structure of Conjugated Polymers, J. Phys. Chem. B, 2021, 125, 6338–6348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03257g |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
J. Javier
Burgos-Mármol
,
J. Javier
Burgos-Mármol