
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInitiation reactions in the high temperature decomposition of styrene†
Travis
Sikes
 a,
Colin
Banyon
a,
Rachel A.
Schwind
b,
Patrick T.
Lynch
c,
Andrea
Comandini
d,
Raghu
Sivaramakrishnan
a and
Robert S.
Tranter
*a
a,
Colin
Banyon
a,
Rachel A.
Schwind
b,
Patrick T.
Lynch
c,
Andrea
Comandini
d,
Raghu
Sivaramakrishnan
a and
Robert S.
Tranter
*a
aChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Ave., Lemont, IL 60439, USA. E-mail: tranter@anl.gov; Tel: +1-630-252-6505
bChemical and Environmental Engineering, School of Engineering, Brown University, 184 Hope St., Providence, RI 02912, USA
cDepartment of Mechanical and Industrial Engineering, University of Illinois at Chicago, 842 W. Taylor St., Chicago, IL 60607, USA
dInstitut de Combustion, Aérothermique, Réactivité et Environnement, CNRS-INSIS, 1C, Avenue de la recherche scientifique, 45071 cedex 2 Orléans, France
First published on 23rd August 2021
Abstract
The thermal decomposition of styrene was investigated in a combined experimental, theory and modeling study with particular emphasis placed on the initial dissociation reactions. Two sets of shock tube/time-of-flight mass spectrometry (TOF-MS) experiments were performed to identify reaction products and their order of appearance. One set of experiments was conducted with a miniature high repetition rate shock tube at the Advanced Light Source at Lawrence Berkeley National Laboratory using synchrotron vacuum ultraviolet photoionization. The other set of experiments was performed in a diaphragmless shock tube (DFST) using electron impact ionization. The datasets span 1660–2260 K and 0.5–12 atm. The results show a marked transition from aromatic products at low temperatures to polyacetylenes, up to C8H2, at high temperatures. The TOF-MS experiments were complemented by DFST/LS (laser schlieren densitometry) experiments covering 1800–2250 K and 60–240 Torr. These were particularly sensitive to the initial dissociation reactions. These reactions were investigated theoretically and revealed the dissociation of styrene to be a complex multichannel process with strong pressure and temperature dependencies that were evaluated with multi-well master equation simulations. Simulations of the LS data with a mechanism developed in this work are in excellent agreement with the experimental data. From these simulations, rate coefficients for the dissociation of styrene were obtained that are in good agreement with the theoretical predictions. The simulation results also provide fair predictions of the temperature and pressure dependencies of the products observed in the TOF-MS studies. Prior experimental studies of styrene pyrolysis concluded that the main products were benzene and acetylene. In contrast, this study finds that the majority of styrene dissociates to create five styryl radical isomers. Of these, α-styryl accounts for about 50% with the other isomers consuming approximately 20%. It was also found that C–C bond scission to phenyl and vinyl radicals consumes up to 25% of styrene. Finally the dissociation of styrene to benzene and vinylidene accounts for roughly 5% of styrene consumption. Comments are made on the apparent differences between the results of this work and prior literature.
Introduction
Styrene is a commonly used precursor for many industrial and consumer polymers such as polystyrene, acrylonitrile–butadiene–styrene (ABS), and styrene–butadiene rubber (SBR). Styrene is also formed in significant concentrations during the combustion and pyrolysis of aromatic molecules that are commonly found in fuels including alkylbenzenes1 (e.g. toluene,2–4 ethylbenzene,5–7n-propylbenzene,8–10n-butylbenzene11–14), indane,15 tetralin,16 and biomass.17 The global styrene market has been projected to reach over 33 Mt by the end of 2023, making the disposal of these products18 and waste styrene monomer19 a non-trivial matter. Increasing concerns about pollution from plastic waste shine a spotlight on disposal methods of the materials which are typically via landfill or incineration.20,21 As an alternative, significant attention has recently been focused on discovering better ways of mechanically recycling or chemically upcycling these polymers to higher-value products.22,23 Currently, thermal decomposition of polystyrene is the main route for recycling the material (T ∼ 600–800 K, P ∼ 1 atm), and Li et al.24 have shown that the products are highly dependent on the reaction temperature. A complete understanding of the thermal dissociation and subsequent degradation pathways of monomers, such as styrene, is a foundational prerequisite for designing reactors and reaction conditions to reuse polymers efficiently and sustainably. Lastly, for environmental and health considerations, it is important to know what hazardous species, such as polycyclic aromatic hydrocarbons (PAHs),25 are being formed during pyrolysis and oxidation of styrene. Li et al.24 not only observed that increasingly complex PAHs were produced in the decomposition of polystyrene as the reaction temperature increased, but also that the classes of reactions involving styrene were temperature dependent. Westblad et al.19 studied incineration of styrene in a two-stage laboratory furnace and also observed a strong temperature dependence on the formation of PAHs.There have been a number of experimental studies of styrene oxidation and pyrolysis including in a two stage furnace,19 laminar flame speeds,26–28 flow reactors,5,29,30 and shock tubes.31,32 These data have been used in various combinations to validate models for styrene oxidation and key steps from three of the most recent mechanisms by Narayanaswamy et al.,33 Yuan et al.26 and Comandini et al.28 describing the pyrolysis of styrene are summarized in Table 1. Kinetic data for several of these reactions were obtained from Grela et al.30 and, Wang and Frencklach.34
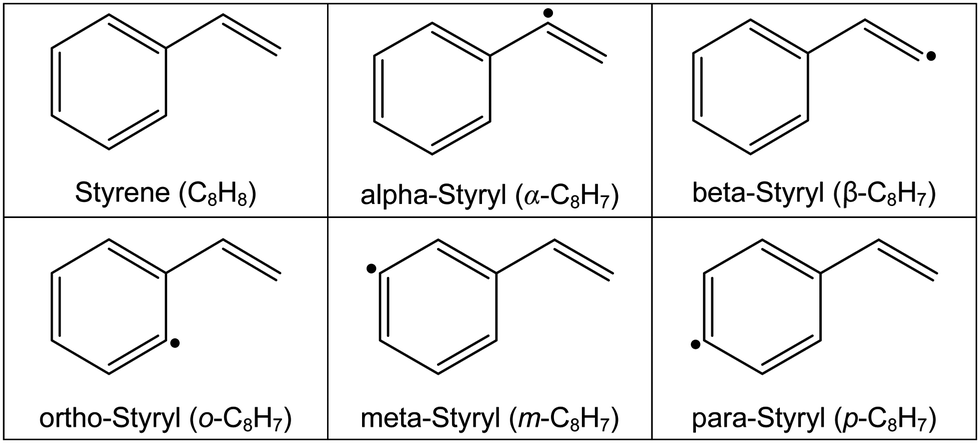
There are a few similarities between these three oxidation mechanisms. First, they all treat dissociation of styrene as a multi-channel problem. The mechanisms have similar pathways for eliminating vinyl (RIV), vinylidene (RV), and for forming styryl radicals through H-elimination (RI–RIII). However, there are significant differences concerning the relative importance of various pathways and which isomers of the styryl radical are formed. The molecular structures of styrene and the various styryl radicals are shown in Table 2. For example, at temperatures relevant to the current study (T > 1700 K) the mechanism of Comandini et al.28 suggests that formation of o-styryl is dominant above 1900 K and elimination of H2CC: is the most important reaction below 1900 K. Yuan et al.26 indicate that ring opening to C4H4 is the main route above 2100 K and a ring contraction to give cyclopentadienyl and propargyl radicals dominates at lower temperatures. Furthermore, there are significant differences, between one to two orders of magnitude over 1700–2100 K, in the total rate coefficients for consumption of styrene as well as in the rate coefficients for identical reactions. At lower temperatures the differences in total rate coefficients reduces to factors of 2–5. Comandini et al.28 and Narayanaswamy et al.33 find the formation of benzene + vinylidene to be the dominant reaction; whereas, Yuan et al. indicates that the ring contraction pathway dominates until T < ∼1150 K. Lastly, pressure dependencies in the rate coefficients are not explicitly accounted for in many of the reactions proposed for styrene dissociation. This is particularly relevant to the present experimental studies that span T > 1500 K and pressures as low as 60 Torr.
|
The literature concerning gas-phase pyrolysis of styrene is quite sparse and appears to be limited to just three studies. Bruinsma et al.29 used a diffusion cell followed by a coiled tube reactor (9 Torr, 950–1100 K); the effluent from the reactor was analyzed by gas chromatography/flame ionization detection and Fourier transform-infrared spectroscopy to estimate total decomposition rates and identify reaction products which included phenylacetylene, benzene, ethylene, and acetylene.
Müller-Markgraf and Troe31 investigated styrene pyrolysis in a shock tube (∼5.5 atm, 2015 K) using UV absorption spectroscopy. The spectra contained signatures not only from styrene but also the decomposition products, and fitting the spectra over a broad wavelength range was necessary to identify products and obtain dissociation rate coefficients. Müller-Markgraf and Troe proposed that styrene decomposes to benzene and acetylene, noting that this was probably by a complex process that required further investigation.
Grela et al.30 used a low pressure flow reactor (∼10 mTorr, 1130–1380 K) coupled by effusive sampling to a mass spectrometer. As the reaction temperature increased, cations appeared at mass to charge ratios, m/z, 26 and 78. These were assigned as C2H2 and C6H6 respectively. Grela et al. proposed that the products were formed directly by elimination of vinylidene which rapidly isomerized to acetylene. Grela et al. measured rate coefficients for the reverse of RV in a separate reactor and obtained values for kV from the equilibrium constant. RRKM analysis showed the derived rate coefficients to be close to the high-pressure limit. Extrapolation to the conditions of Müller-Markgraf and Troe showed reasonable agreement with their results being larger by a factor of 1.4 at 1700 K increasing to 3.6 at 2200 K.
The literature pyrolysis studies indicate that styrene decomposes by a single, possibly multi-step, reaction in contrast to recent oxidation mechanisms that involve multi-channel reactions. Furthermore, the oxidation mechanisms contain significant differences between both the rate coefficients and the reactions involved in styrene pyrolysis. This study resolves these differences through a combination of several different techniques. Electronic structure theory has been used to explore the potential energy surface and characterize dissociation pathways pertinent to the pyrolysis of styrene. The experimental work identifies reaction products and provides targets and constraints for simulations with a new chemical kinetic mechanism that incorporates the theoretical results.
Experimental methods
Experiments were conducted behind incident and reflected shock waves in two diaphragmless shock tubes which are described briefly in the Shock tubes section. Standard shock tube notation is used when referring to temperature, T, and pressure, P, within the shock tubes.35T1 and P1 denote the initial conditions of the driven gas (reagent mixture) and P4 the initial pressure of the driver gas. T2 and P2 refer to the gas state behind the incident shock wave while T5 and P5 denote the gas conditions after the reflected shock wave has passed.The general procedure for all experiments was to fill the driver section to P4 with helium and the driven section to P1 with the reagent mixture, where P4 ≫ P1. A shock wave was initiated by rapidly opening a valve that separated the driver and driven sections. The post-shock conditions were calculated from the ideal shock wave equations35 assuming frozen composition based on the known initial conditions, the incident shock wave velocity, and thermochemical properties of the mixture. The thermochemical properties for these calculations were taken from Goos et al.36 The shock velocity was calculated as Δx/Δt, where Δx is the known distance between pressure transducers. For experiments behind the incident shock wave, Δt was the average time taken for the shock wave to trigger consecutive pressure transducers; whereas, for those behind reflected shock waves, the measured velocities were used to calculate the velocity at the observation point by extrapolation. The shock velocity has an estimated uncertainty of 0.2% resulting in T2 and T5 uncertainties of <0.5%.
Shock tubes
Experiments were performed in two diaphragmless shock tubes that have been fully described previously. One is referred to as the DFST37,38 (diaphragmless shock tube) and the other as the ANL-HRRST39 (Argonne National Laboratory-High Repetition Rate Shock Tube). Both shock tubes are coupled via differentially pumped molecular beam sampling (MBS) systems to time-of-flight mass spectrometers (TOF-MS). The DFST is also equipped for laser schlieren densitometry (LS).The driver section of the DFST is a stainless steel chamber, 78 cm long with an i.d. of 20 cm, and the driven section is a stainless steel tube with an i.d. of 6.35 cm. The driver and driven section of the DFST can be separated by closing a bellows actuated pneumatic valve, and the tube is fired by rapidly opening this valve. A pair of quartz windows for LS experiments are installed in the driven section ∼500 cm downstream of the valve. The driven section extends for approximately 220 cm downstream of the LS windows. A gate valve is positioned in the driven section between the LS windows and the endwall. During LS experiments the gate valve is closed, but it is opened for TOF-MS experiments. The endwall of the driven section contains an orifice, 0.3 mm diameter, that forms the first stage of the MBS interface to the TOF-MS. The DFST is equipped with two sets of piezoelectric pressure transducers for determining shock velocities. Five of these (Dynasen Model CA-1135) are centered around the LS observation location and are spaced 120.0 mm apart. Four (PCB Model 132A35) are spaced 76.2 mm apart with the last transducer 25.4 mm from the driven section endwall.
The ANL-HRRST is a miniature shock tube designed for high pressure experiments in the lab and at synchrotron facilities.39 The driver section is a stainless steel chamber (volume: 20 cm3) equipped with a solenoid actuated valve that separates the driver and driven sections. The driven section has a bore of 6.35 mm and length of 879 mm. The driven section is terminated by a custom-made nozzle that protrudes into the driven section by 0.75 mm and has an included angle of 110°. The nozzle orifice is 0.2 mm, and as in the DFST, the nozzle is the entrance to the MBS. The ANL-HRRST is equipped with six piezoelectric pressure transducers (Dynasen Model CA-1135) that are separated by increments of 50.0 mm. The last pressure transducer is 76.5 mm from the sampling nozzle. The ANL-HRRST is fully automated and it was operated at a rate of one experiment every second. Details of the procedure can be found in prior publications.39–41 For every experiment, initial conditions and considerable data regarding operation of the shock tube are recorded along with the mass spectra. These data permit for post-processing of experiments closely binned by experimental condition.
Both shock tubes create very reproducible reaction conditions that permit signal averaging to improve signal/noise.37–40 From the DFST/TOF-MS experiments, 5–10 experiments were ensemble averaged; whereas, 100 experiments for each data point were ensemble averaged in the ANL-HRRST/TOF-MS work.
Time-of-flight mass spectrometry
The shock tubes are connected to the TOF-MSs by differentially pumped MBS interfaces. Gases continuously elute from the reaction zone in the shock tube through a nozzle in the driven section endwall. The expanding gases form a supersonic jet that quenches the reactions, thereby preserving the high temperature composition. The core of the jet passes through a skimmer (Beam Dynamics Model 2, orifice: 0.2 mm ANL-HRRST; 0.5 mm DFST) and forms a molecular beam that flows into the ion source of the TOF-MS (Kasedorf CB-5). The nozzle and skimmer orifices were selected to ensure that the pressure in the TOF-MS ion source remained within safe ranges while maximizing signal levels. The tips of the skimmers were positioned ∼10 nozzle diameters downstream of the nozzles. The TOF-MSs were positioned orthogonal to the molecular beam and ions were extracted in well-defined packets from the ionization zones by applying a 1 μs voltage pulse to the repeller and extractor electrodes. The extraction pulse frequency was 100 kHz giving one mass spectrum every 10 μs. For each experiment, 300 mass spectra were obtained that covered the pre-shock and post-shock regimes. The analog signals from the multi-channel plate detectors were digitized at 1 GS s−1 with a 12-bit data acquisition board (GageApplied PCIe EON series).The DFST/TOF-MS studies used electron impact ionization (EI) whereas the ANL-HRRST/TOF-MS work used synchrotron VUV photoionizaton (PI). EI experiments were conducted with electron energies of 21 eV which minimized the signal from the bath gas neon (IE = 21.56 eV42) and somewhat reduced fragmentation of organic species in the ion source. It was not possible to eliminate neon ions entirely as the electron beam has quite a long high energy tail. However, the neon peaks were small compared to those of interest and provided a known point for calibration of the mass axis. The electron energy is lower than normally employed in shock tube TOF-MS experiments (30 eV43 or ∼50 eV44) and results in reduced signal levels. The main benefits are that the bath gas signal is minimized, and the detector sensitivity can be increased to improve detection of organic species. At higher sensitivities the noise in the detector signal increases but this is easily compensated for with the DFST by averaging over several experiments. By signal averaging spurious peaks, which appear more frequently at higher detector voltages, were removed as well as improving the signal-to-noise ratio.
For the ANL-HRRST/TOF-MS work, experiments were conducted at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory. Highly tunable synchrotron vacuum ultraviolet (VUV) light (8–12 eV) was obtained at the Chemical Dynamics Beamline (9.0.2), T2 end station. By performing experiments at different photon energies (PE) both mass and photoionization spectra were obtained, facilitating the identification of species. The T2 end station is not equipped with a monochromator, so the photon energy was instead controlled by the undulator gap. This had two consequences, one was that the light had a bandwidth of ∼0.3 eV, and the other was that unwanted high-harmonic light was produced. An argon gas filter removed most of this high-energy light; however, some of it escaped the gas filter. For this reason, pairs of experiments were performed with and without a 1 mm thick magnesium fluoride (MgF2) filter. The attenuation due to the filter was ∼50% and increased rapidly to near 100% between 10.8–11.2 eV and blocked photons above 11.2 eV. The filter was located between the TOF-MS ion source and the synchrotron beamline and was ∼150 mm from the ion source. Due to the intensity of the VUV beam, the MgF2 filter degraded and had to be repositioned by 1–2 mm about every 30 min. The PE was incremented between 8–12 eV in 0.25 eV steps for experiments without the MgF2 filter and 8–10.5 eV in 0.5 eV steps for those experiments with the filter. At each PE, 100 shocks were performed. During post-processing of the mass spectra, the 100 shocks were signal averaged for improved signal-to-noise and then converted from intensity vs. flight time to intensity vs. mass-to-charge ratio (m/z) through a calibration based on known standards. The data were then processed through a baseline-correction code to remove background features that would complicate peak identification.45
Laser schlieren densitometry
DFST/LS experiments were performed behind incident shock waves. The LS technique has been described in detail elsewhere.46,47 The principle of laser schlieren densitometry is that density gradients cause a laser beam to be deflected. In these experiments, this deflection correlates to axial density gradients through the mixture molar refractivity, KL.48,49 It is assumed that KL remains constant throughout the experiment, so the deflection correlates directly to the axial density gradient. This assumption is reasonable for the dilute mixtures used herein. Following shock heating, the reagent begins to pyrolyze, and the density gradient (dρ/dx) is established proportionally to the heat release of each reaction, eqn (1).48,49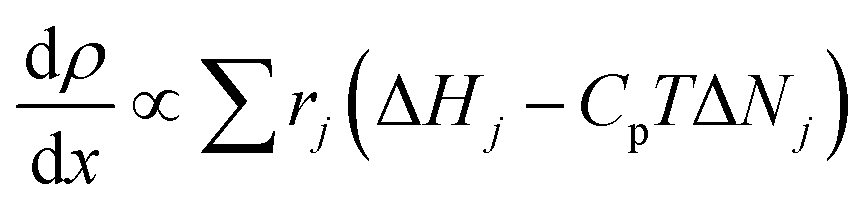 | (1) |
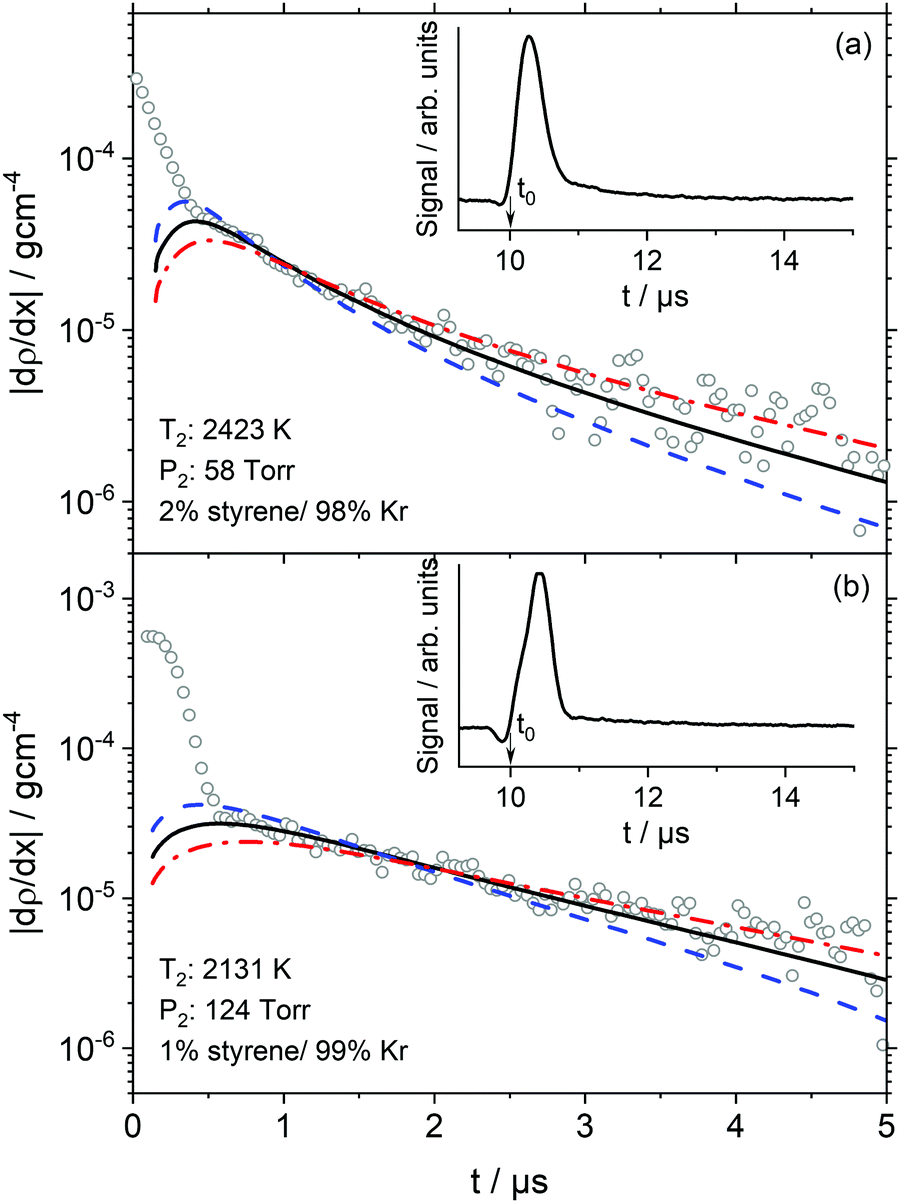 | ||
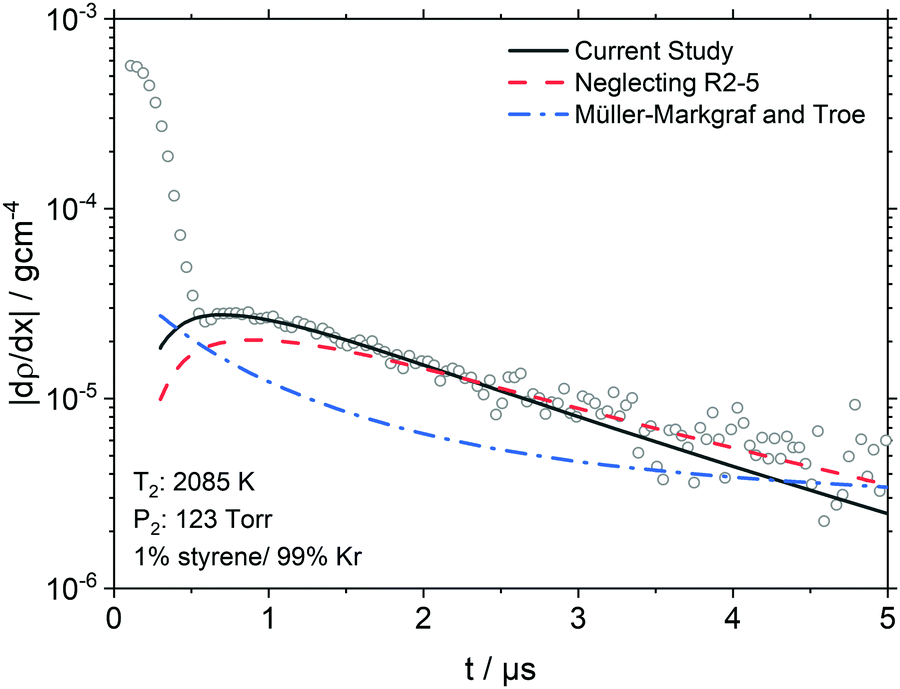
| Fig. 1 Experimental data (symbols) are shown with the best-fit modeling results (solid line) and sensitivity to the total rate of styrene decomposition by k × 1.5 (dashed line) and k/1.5 (dashed-dotted line) based on the best-fit chemical kinetics mechanism of styrene. For clarity, the experimental data has been decimated by 2. The inset plots show the raw LS signal and t0 location. | ||
In the Fig. 1 inset plots, the raw signals show large valleys and peaks that are the result of the interaction of the shock front with the laser beam.48 Following the large peaks are sharp transitions to decaying signals, which are entirely due to chemical reactions. The start of reaction is defined as being at time t0. As shown in Fig. 1 the location of t0 is beneath the large peak masking the onset of chemical reaction. Consequently, t0 is calculated by a well-established method which has an uncertainty of <0.2 μs.48 In some experiments, such as those shown in Fig. 1, a small offset of 0.2 ± 0.1 μs between the simulation and experiment was found. These offsets are similar to the uncertainty in t0 but may also be symptoms of small incubation delay due to vibrational relaxation.
At t0 the only reactions occurring are those by which styrene dissociates. Thus obtaining the density gradient at t0, (dρ/dx)t0, allows the total rate coefficient for a multi-channel reaction such as styrene pyrolysis to be determined by extrapolation to t0 with chemical kinetic simulations of the density gradients. The simulations and mechanism are described in Results and Discussion.
Reagents
The driver gas in all experiments was helium (Airgas 99.999%). For DFST experiments reagent mixtures were prepared manometrically in a 50 L glass vessel and stirred for at least an hour prior to use. Typically, a total pressure of 100 Torr of mixture was prepared. Styrene (Sigma Aldrich, >99.0%) was degassed by repeated freeze/thaw cycles with liquid nitrogen. For the LS experiments the bath gas was krypton (Airgas, 99.999%); whereas, for the DFST/TOF-MS studies the bath gas was neon (Linde, 99.999%). Argon (Airgas 99.999%) was added to the DFST/TOF-MS mixtures at 1% and acted as an internal standard.38Table 3 contains the experimental conditions and mixture compositions for each experimental technique in this study.| Shock tube | Technique | Temperature | Pressure | Composition |
|---|---|---|---|---|
| ANL-HHRST | PI TOF-MS | 1660–2260 K | ∼11 bar | 0.06–0.25% styrene/Ar |
| DFST | EI TOF-MS | 1830 K | 365 Torr | 0.5% styrene/1% Ar/Ne |
| DFST | LS | 1800–2550 K | 60, 120, 240 Torr | 1%, 2%, 4% styrene/Kr |
The consumption of reagent mixture in the ANL-HRRST was sufficiently large that an alternate method of preparation was employed using a novel batch/flow mixing apparatus which has been previously described.39,41,50 In short, reagent mixtures were prepared on demand and delivered via a vacuum regulator to the driven section of the ANL-HRRST. Part of the argon stream entering the mixing apparatus was diverted through a high-pressure bubbler containing liquid styrene at room temperature. The gases exiting the bubbler were a mixture of argon and styrene with the composition determined by the partial pressures of styrene and argon. This mixture flowed through a mass flow controller to a static mixing element where it was blended with additional argon admitted via a second mass flow controller to obtain the desired composition.
Theory
The potential energy surface (PES) for styrene decomposition was characterized using electronic structure theory. The rovibrational properties of the minima and transition states (for the molecular channels) for the relevant molecular and radical processes in styrene were determined using the M06-2X/cc-pVTZ hybrid functional which has been shown51 to provide reliable geometries and rovibrational properties for use in kinetics calculations. Higher level energy estimates for these stationary points were obtained using the CCSD(T)/cc-pV∞Z method, where the infinite basis set limits are estimated from an extrapolation of results52,53 obtained from sequences of cc-pVnZ where n = (D, T, Q) basis sets. More specifically for the CCSD(T)/cc-pV∞Z method, the results use a CCSD(T)/cc-pVnZ n = (D, T) extrapolation, and two sets of MP2/cc-pVnZ n = (D, T) and n = (T, Q) extrapolations to converge at the CCSD(T)/cc-pV∞Z limit.54Fig. 2 represents a simplified PES for styrene dissociation. Additionally, higher energy level estimates for all relevant stationary points on this simplified PES were also computed using the G455 composite method. The Gaussian 09 software package56 was used to perform the electronic structure calculations reported here.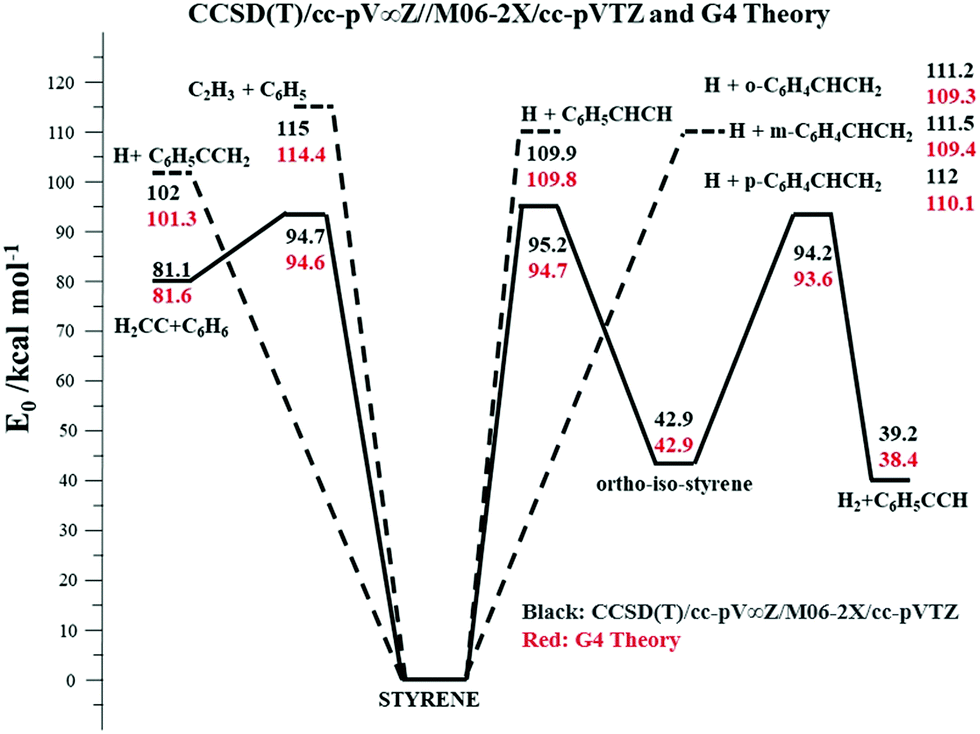 | ||
| Fig. 2 Simplified PES of styrene dissociation. The CCSD(T)/cc-pV∞Z//M06-2X/cc-pvtz results are shown in black text and the G4 results in red text. The units of the values are in kcal mol−1. The solid black lines represent molecular processes while the dashed lines represent bond-fissions. | ||
The lowest energy process is a molecular channel leading to the formation of benzene and vinylidene (H2CC:) with a barrier ∼95 kcal mol−1 (Fig. 2). An alternative molecular channel involving the migration of an H-atom leading to the formation of ortho-iso-styrene has a nearly similar barrier as that of the H2CC: + C6H6 channel. This ring-walk mechanism was previously investigated in toluene57 and ethylbenzene54 dissociation, but there were no energetically accessible exit channels from these ring isomers in either molecule. Consequently, dissociation ultimately proceeds from the more stable toluene and ethylbenzene minima on the C7H8 and C8H10 PES's respectively. In the present case though, an energetically accessible six-center elimination channel forms H2 and phenylacetylene from the ortho-iso-styrene isomer. The barrier for this reaction is ∼1 kcal mol−1 lower than isomerization of styrene to ortho-iso-styrene, Fig. 2. Subsequent isomerizations of ortho-iso-styrene to its meta-isomer and meta to the para isomer have barriers that are 3–5 kcal mol−1 higher than this H2 elimination from the ortho-isomer and therefore the meta- and para-isomers are expected to play an insignificant role in the dissociation kinetics of styrene. Pyrolysis30 and photodissociation studies58,59 on styrene indicate that H2, C2H2, and C6H6 are important products. Direct pathways for H2, C2H2, and C2H4 elimination were also characterized, but these molecular channels were found to have barriers larger than 110 kcal mol−1 and are therefore not expected to be relevant in styrene dissociation. In the absence of active molecular channels with barriers <90 kcal mol−1 and coupled with the shock tube results indicating noticeable styrene dissociation only at T > 1700 K, bond-fission channels are expected to assume importance. C–H fission to form the resonantly stabilized C6H5CCH2 radical (α-C8H7) is the lowest energy radical channel, with C–H fissions from the terminal CH2 group and from the aromatic ring being ∼9–10 kcal mol−1 endothermic relative to the lowest energy radical pathway. C–C fission to form C2H3 + C6H5 has a bond energy ∼114 kcal mol−1, but entropic considerations allow us to consider the role of this higher energy channel in interpreting the present high temperature shock tube experiments.
Master equation calculations were performed with the VARIFLEX code60 using the ab initio based energetics and molecular properties to obtain theoretical rate constants for the radical and molecular channels. VARIFLEX is a group of programs to calculate rate constants for gas-phase reactions using applications of Transition State Theory61 among a suite of other methods. It includes methods to treat pressure-dependence of reactions that occur over multiple, interconnected potential wells by solving the time-dependent multiple-well Master equation.62 In the present case, the transition state partition functions are evaluated using Phase Space Theory (PST)63 for the C–H and C–C bond fissions in styrene whereas a conventional transition state theory (TST) treatment was employed for treating the kinetics of the molecular channels. The coefficient for the inverse sixth power potential in the PST calculations was calibrated to match the capture rate constants for H + C6H5CHCH3,62 H + C6H5CH2,61 and H + C6H564 recombinations, which are reasonable approximations to the H + C6H5CCH2 (α-C8H7), H + C6H5CHCH (β-C8H7), and H + o,m,p-C6H4CHCH2 (o,m,p-C8H7) capture rate constants respectively forming styrene. There are no measurements for the capture rate for C6H5 + C2H3. However, known capture rate constants for self-reactions of C2H365 and C6H543 have been used to calculate the capture rate constants for the cross-reactions assuming the validity66 of the geometric mean rule. Over the T-range (1500–2500 K) of the present studies, the cross-reaction (C2H3 + C6H5) capture rate constants exhibit a small negative T-dependence (they vary from 1.3 × 1013 to 1.6 × 1013 cm3 mol−1 s−1). Consequently, the coefficient for the inverse sixth power potential in the PST calculations was calibrated to match the capture rate constants for C2H3 + C6H5 that were assumed to be temperature independent (over 1500–2750 K) and ∼1.5 × 1013 cm3 mol−1 s−1. Lennard-Jones parameters for Kr and estimates for styrene were taken from the literature.67 Pressure dependent rate constants were calculated over the temperature range 1500–2750 K for the multi-channel dissociation processes involving the lower energy molecular processes and the various C–H and C–C bond fissions. An exponential down model was used for energy transfer. Using the barriers and energetics at the G4 level of theory and a temperature dependent 〈ΔEdown〉 = 150(T/298)0.85 cm−1, which is a reasonable estimate61 for such aromatics, provided a best fit with the present experimentally determined total rate constants at 60–240 Torr (see Laser schlieren densitometry). The calculations indicate that the lowest energy bond fission channel (forming H + α-C8H7) accounts for ∼60–45% of the total reaction flux 1500–2750 K and 60–240 Torr. Despite C–C fission being ∼13 kcal mol−1 endothermic to this lowest energy C–H fission, the present calculations indicate that C–C bond breaking is the second most important process and accounts for 16–25% of the reaction flux over the present experimental conditions. The other C–H bond fissions cumulatively account for ∼15–40% of the reaction flux over 1500–2750 K with their contributions increasing at higher temperatures. Elimination to H2CC: + C6H6 involves tight transition states and accounts for only 6% of the reaction flux at 1500 K, with its relevance diminishing further at higher temperatures. The present calculations also indicate that H2 + C8H6 formation from ortho-iso-styrene is expected to be a negligible process since the rate constants for its rate-limiting process (isomerization of styrene to ortho-iso-styrene) are at least an order of magnitude lower than the molecular channel leading to H2CC: + C6H6. This is not surprising given that the barrier for the rate limiting step is a little higher than molecular elimination to H2CC: + C6H6. However, due to the tight nature of this isomerization process, kinetics calculations indicate that it does not assume importance even when its barrier is artificially lowered by 1 kcal mol−1 (to 93.7 kcal mol−1 at the G4 level of theory) and thereby making this the lowest energy molecular process in styrene dissociation.
Results and discussion
Mass spectrometry
The purpose of the TOF-MS experiments was to determine the radical and stable species formed during the early stages of styrene pyrolysis to help guide development of a reaction mechanism. The majority of the experiments were performed in the ANL-HRRST; more than 7000 shocks (T5 = 1660–2260 K, P5 ≈ 11 bar, 0.06–0.25% styrene/Ar). For the purposes of this work, the most useful mass spectra were those collected with the MgF2 filter in place as these minimized dissociative ionization and greatly simplified species identification through a combination of mass-to-charge ratio and ionization energy. These mass spectra become weak above 10.8 eV and species with ionization energies greater than ∼11 eV, for example acetylene, cannot be seen at all. However, the experiments without the MgF2 filter and the DFST/TOF-MS experiments provide complementary data obtained at higher ionization energies, and species such as acetylene are easily observed, although products from dissociative ionization are more prevalent and must be accounted for.The full data set from these experiments has three independent variables: mass-to-charge ratio (m/z), reaction time (t), photoionization energy (PE), and one dependent variable: signal intensity (S). The measured m/z vs. t mass spectra at each photon energy are essentially slices from a cuboid of data that was reassembled in post processing. Integrating segments of the data along the various axes allows information about the m/z vs. t and m/z vs. PE responses to be extracted. Details of the methods of analyzing and processing these data are described elsewhere.41,45 For the following discussion, the data are reduced to ‘classical’ mass spectra (m/z vs. S), photoionization spectra (PE vs. S, for a particular m/z), and ‘concentration’ species time history (t vs. S, for a particular m/z). Often these datasets are formed from subsets of the total range of data to reveal particular features e.g. mass spectra at discrete photon energies, photo ionization spectra at various reaction times etc.
Example mass spectra from the highest and lowest temperature experiments are shown in Fig. 3. The uncertainties in this and subsequent TOF-MS figures refer to the standard deviation of the experiments which comprise the signal-averaged results shown. To allow peak sizes of different magnitudes to be easily seen the mass spectra were scaled using a bi-symmetric log68 form. Unlike simple log scaling, the bi-symmetric log scaling accommodates positive and negative values which are encountered in the mass spectral datasets and creates an adjustable region around zero where the gradient remains finite. The unscaled data are shown in the ESI† (Fig. S1).
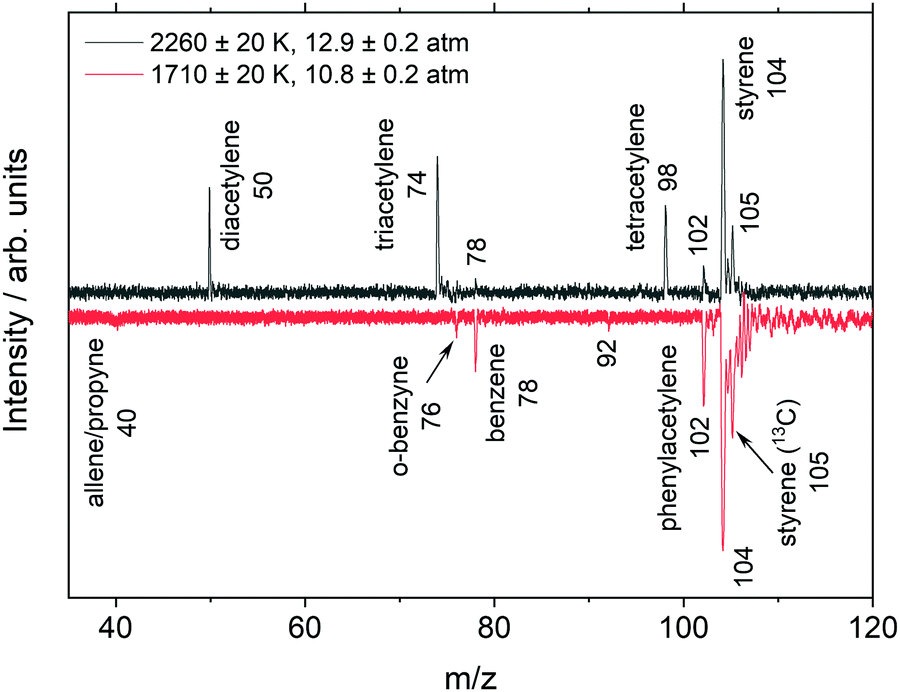 | ||
| Fig. 3 Mass spectra on a bi-symmetric log scale68 taken with the MgF2 filter ∼150 μs after the shock front arrival in a 0.06% C8H8/99.94% Ar mixture. Mass spectra are integrated from 8–10.5 eV. | ||
Fig. 3 (upper) shows that at higher temperatures the aromatic ring is not preserved but quickly opens, resulting in the formation of linear products including diacetylene, triacetylene, and tetracetylene. These polyacetylenes are not necessarily formed in elementary reactions but may be the result of sequences of reactions. The ionization energy threshold of acetylene (m/z 26) is 11.4 eV69 and is above the cutoff of the MgF2 filter. Similar mass spectra obtained without the MgF2 filter or with EI, discussed later, contain peaks at m/z 26 in addition to the polyacetylenes. The lower plot in Fig. 3 shows a markedly different set of species where the aromatic ring is preserved and polyacetylenes are not observed. The two datasets in Fig. 3 represent the extremes of the temperature range, and as might be expected, mass spectra from experiments at intermediate temperatures contain both aromatic species and polyacetylenes.
Species were mainly identified by m/z and the observed ionization threshold which was obtained from photoionization (PI) spectra. An example PI spectrum is shown in Fig. 4 from the low temperature, 1710 K dataset without the MgF2 filter. The ionization thresholds for each species show good agreement with the literature values but are all about 0.2 eV lower than the literature values. This discrepancy is consistent with the ∼0.3 eV bandwidth of the VUV beam. The PI spectra for two polyacetylenes are shown in Fig. 4; although, the species were not observed in the corresponding plot of Fig. 3. This apparent discrepancy arises because the data in Fig. 4 are from integration over all reaction times whereas Fig. 3 represents a snapshot at one time, 150 μs.
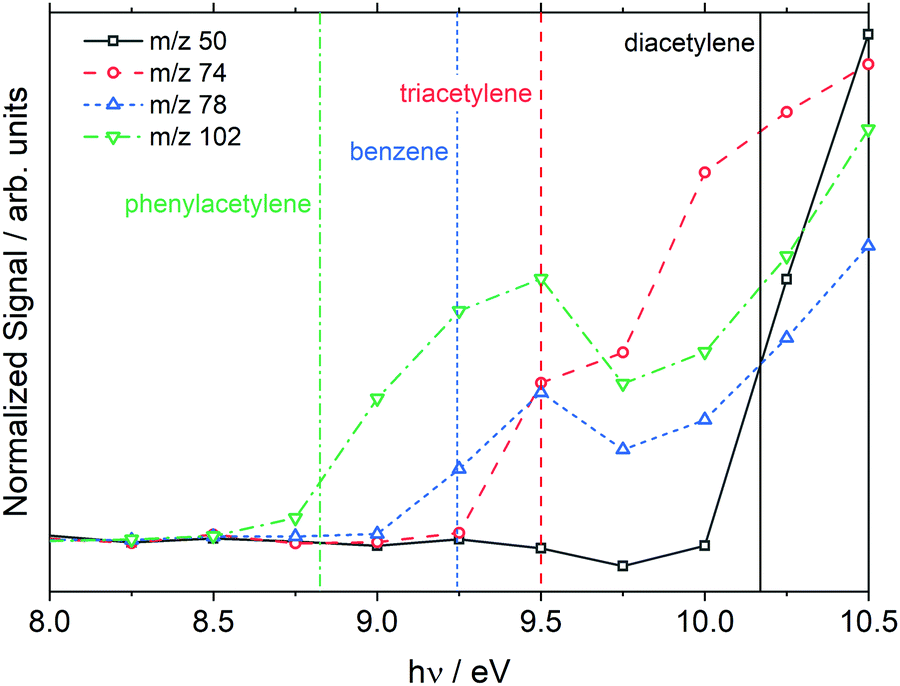 | ||
| Fig. 4 Photoionization spectra of products formed from dissociation of styrene at 1710 ± 20 K, 10.8 ± 0.2 atm. The data were taken without the MgF2 filter and extend to 12.0 eV. Vertical lines are the literature photoionization thresholds.69 | ||
Fig. 5 shows the time histories of benzene, phenylacetylene, diacetylene and triacetylene from the same experiment in Fig. 4. Some caution must be exercised when interpreting plots such as Fig. 5 at long reaction times. It has previously been determined that the post-shock reaction conditions are well-defined up to 300 μs in the ANL-HRRST.39 At longer times there is greater uncertainty in the thermodynamic state of the gases due to complex wave behavior in the miniature tube.
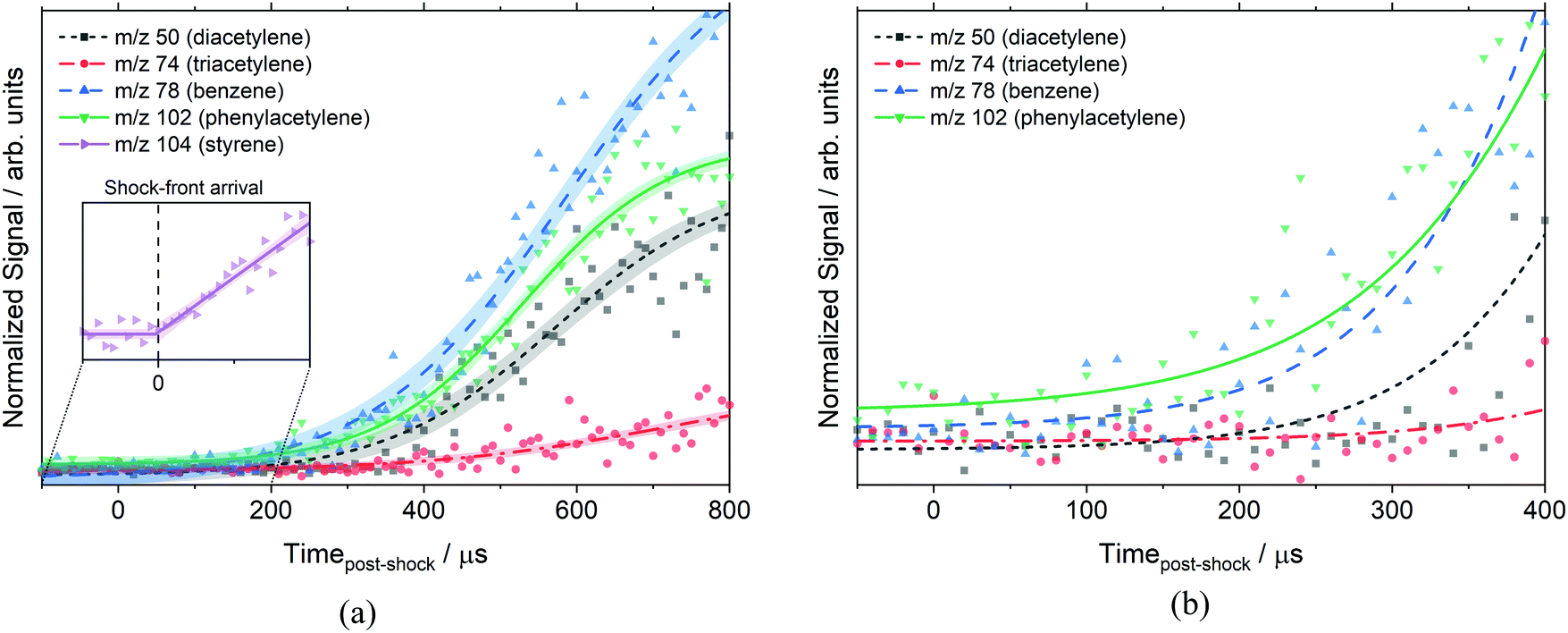 | ||
| Fig. 5 Species time histories of m/z 50, 74, 78, and 102 at 1710 ± 20 K, 10.8 ± 0.2 atm, and 10.5 eV. Reaction conditions are the same as Fig. 4. Signals are normalized based on their photoionization cross-section (diacetylene,70 triacetylene71 (estimate), benzene,70 phenylacetylene72). (a) Shows the profiles out to 800 μs whereas (b) focuses on a short time and more clearly shows the early formation of benzene and phenylacetylene. The inset in (a) shows the sharp change in the styrene signal on reflection of the shock wave that is used to determine the start of reaction. Symbols represent the data, lines are experimental fits for clarity only, and shading is the 95% confidence interval of the experimental fit. | ||
The data in Fig. 5 have been scaled by the photoionization cross-section for each species (σ) at 10.5 eV to allow for direct comparison of relative concentrations, [X] ∝ S/σ. The assumption has been made that mass discrimination effects are small.41 The photoionization cross-sections at 10.5 eV used for normalization were: diacetylene (23.8 × 10−18 cm2),70 triacetylene (23.8 × 10−18 cm2),71 tetracetylene (10.0 × 10−18 cm2),71 benzene (31.8 × 10−18 cm2),70 phenylacetylene (62.4 × 10−18 cm2),72 and styrene (43.2 × 10−18 cm2).72 Most of these were direct measurements, but the cross-sections for triacetylene and tetracetylene were estimated values. Fig. 5b shows that the two aromatic species are formed first, followed by the polyacetylenes. Triacetylene is clearly not formed as a primary product from styrene as it does not reach a measurable concentration until ∼350 μs after the start of reaction. Likewise, diacetylene does not begin to rise until ∼100 μs.
From the 2260 K mass spectra the following ratios were obtained for polyacetylene concentrations at ∼150 μs: [C4H2]/[C6H2] ≈ 0.65 and [C6H2]/[C8H2] ≈ 1.4. However, the simulated values with the current model at these conditions are significantly different, [C4H2]/[C6H2] ≈ 4.0 and [C6H2]/[C8H2] ≈ 0.5. The differences could be due to the conversion of the experimental signal to relative concentrations which is dependent on the photoionization cross-sections, two of which are estimates with unknown accuracy. While the absolute values of the cross-sections may have significant uncertainties the ratios of these values will be rather less uncertain, particularly for such similar species. On the other hand, the simulated concentration ratios depend on the accuracy of rate constants in the model and the accurate description of the pathways creating and destroying a species. The experimental polyacetylene ratios were also different than the exponential decrease with increasing carbon atoms that was seen by Li et al.73 in acetylene flames. However, as the mass spectra indicate, polyacetylenes are not formed from styrene directly, and the uncertainties in the detailed chemistry of polyacetylenes do not hinder the primary goal of this work, the dissociation of styrene.
The data in Fig. 3 were obtained with the MgF2 filter and acetylene was not observed. However, the non-MgF2 and DFST/TOF-MS experiments, do show acetylene. Fig. 6 shows acetylene, diacetylene, and triacetylene obtained from the DFST/TOF-MS experiments. It is evident that acetylene is formed followed by diacetylene and later triacetylene.
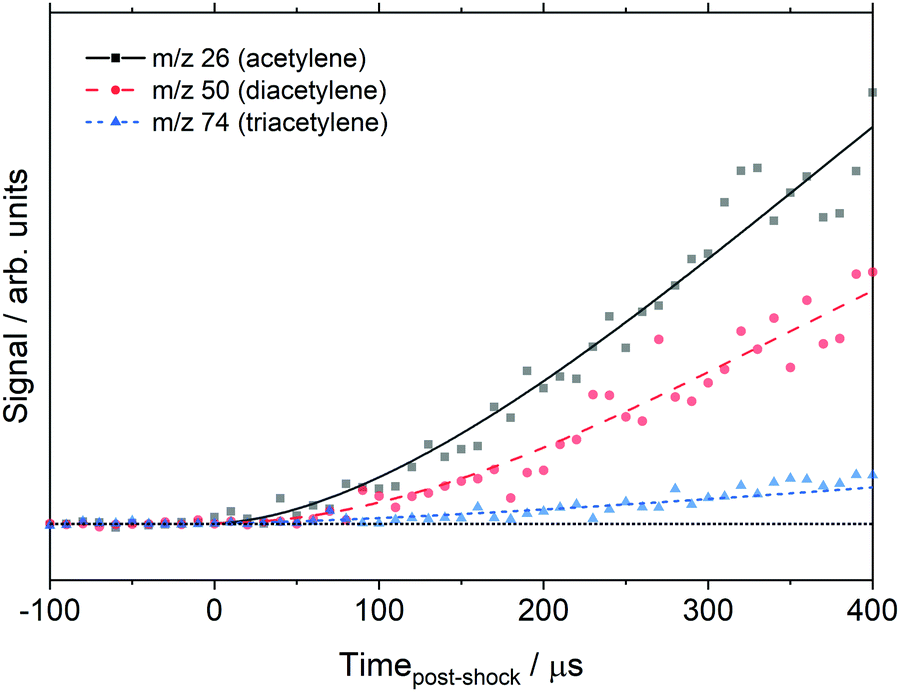 | ||
| Fig. 6 EI time history of the polyacetylenes at 1830 ± 25 K, 368 ± 7 Torr, 0.5% C8H8/1.0% Ar/98.5% Ne and 21 eV. Symbols represent the data and lines are experimental fits for clarity only. | ||
An example of the non-MgF2 and EI mass spectra are shown in Fig. 7. Comparisons to the pre-shock, non-reacting signals, Fig. 7a, show that m/z 26 is not formed by dissociative ionization of styrene due to high energy photons in the non-MgF2 work or in the EI experiments. In Fig. 7 several peaks are observed that are not present in Fig. 3. These are at m/z 18, 28 and 32 and are due to background gases in the TOF-MS chamber being ionized when the filter is removed, similar effects were observed in other studies.41,45
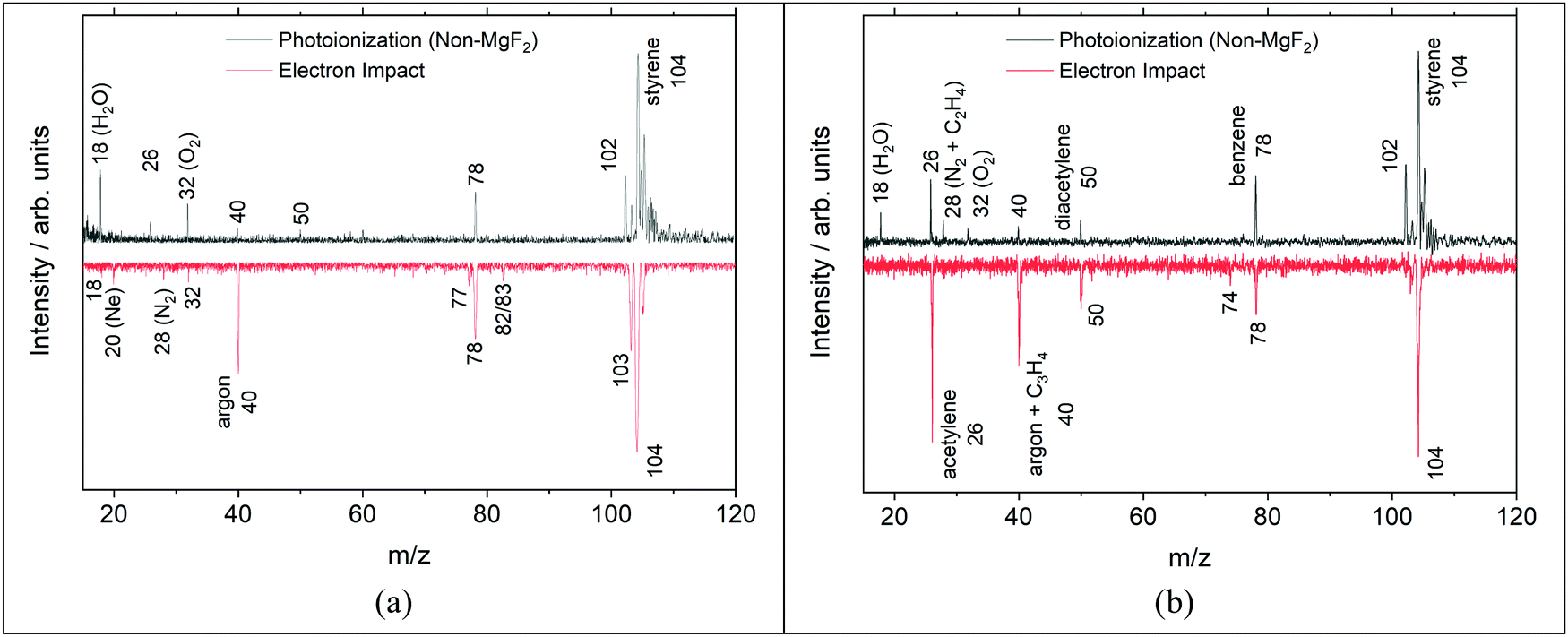 | ||
| Fig. 7 Mass spectra on a bi-symmetric log scale68 from photoionization (top) and electron impact (bottom) TOF-MS experiments (a) pre-shock and (b) ∼150 μs post-shock. PI spectra were taken at 1875 ± 25 K, 11.3 ± 0.2 atm, 0.25% C8H8/99.755 Ar, and integrated from 8–10.5 eV. EI spectra were taken at 1830 ± 25 K, 368 ± 7 Torr, 0.5% C8H8/1.0% Ar/98.5% Ne and 21 eV. | ||
In Fig. 7bm/z 74 (triacetylene) is not observed in the PI mass spectrum and m/z 102 (phenylacetylene) is absent from the EI mass spectrum. The m/z 74 peak in the EI MS of Fig. 7 is very small in this snapshot at 150 μs. However, at longer times the peak becomes significant in size as shown in the ESI† (Fig. S4). It is likely that the m/z 74 peak in the non-MgF2 PI experiment, shown in Fig. 7, is simply below the detection limit as at longer times it is also evident in the corresponding MgF2 PI experiment at the same conditions in the ESI† (Fig. S5). For phenylacetylene, the differences between the EI and PI mass spectra in Fig. 7b are almost certainly due to changes in the reaction chemistry occurring at the different reaction pressures (EI: 0.5 atm; PI: 11.3 atm). Simulations for the two sets of reaction conditions qualitatively show a decrease in the ratio of [phenylacetylene]/[styrene] as pressure decreases. The simulation results are shown in the ESI† (Fig. S6) and predict that the ratio of [C8H6]/[C8H8] is 16 times smaller at the EI conditions compared to the PI conditions.
At 1710 K in Fig. 3 (lower), there are prominent peaks at m/z 102 (phenylacetylene), and 78 (benzene). Phenylacetylene is expected to be produced by subsequent C–H fissions in the sequence styrene → α/β-styryl → phenylacetylene. Benzene production is slightly more complicated. There is a direct path from styrene → benzene through the loss of vinylidene, cf. Grela et al.;30 however, there are also pathways through H + styrene = benzene + vinyl and H + α-styryl = benzene + vinylidene. When taking σ into consideration for benzene (31.8 × 10−18 cm2)70 and phenylacetylene (62.4 × 10−18 cm2),72 the ratio of [C8H6]/[C6H6] ≈ 0.9; whereas, the simulation predicts [C8H6]/[C6H6] ≈ 3.8. It is currently unknown if this discrepancy is mechanistic or experimental in nature. The principle source of experimental error is the uncertainty in photoionization cross-sections. However, these are well-defined for benzene and phenylacetylene. This suggests the source of the discrepanccy is more likely to reside in the secondary chemistry of the mechanism.
The peaks at m/z 105 and m/z 103 are likely to be styrene and phenylacetylene 13C isotopes respectively. The evidence for this is that the m/z 103 and 105 peak heights mirror those of m/z 102 and 104, the parent peaks, through all PE and t. Furthermore, the S for m/z 103 and 105 are roughly 9% of the m/z 102 and 104 peaks which is to be expected since 1.109% of C atoms are 13C. Styryl radicals are also m/z 103; however, as the magnitude of the m/z 103 peak is consistent with the 13C isotope it suggests that styryl radicals are not contributing to the m/z 103 peak. This could be due to dissociative ionization of the radicals. However, preliminary results from Jasper74 indicate that a stable α-styryl cation exists at ∼6.77 eV and it should not dissociatively ionize below ∼10.5 eV. The simulations that are discussed later indicate that reactions forming styryl radicals are important in the pyrolysis of styrene but that they are short lived. The mass spectra indicate that the concentration of styryl radicals never rises above the detection limits.
Mechanism development
The styrene pyrolysis mechanism was developed by combining parts from prior mechanisms and results from the theoretical studies described earlier. The TOF-MS experiments provided guidance to the species produced and the temporal and temperature dependencies of their concentrations. The mechanism was tested by simulating the density gradients from the LS experiments which are sensitive in the first few microseconds to the initial dissociation reactions. The mechanism was subsequently refined in an iterative process which also involved refinement of the theoretical study. The full mechanism is presented in the ESI.† It has 66 species and 247 reactions and is intended for the early stages of styrene pyrolysis. A subset of reactions that are important to the initial dissociation of styrene are given in Table 4. All reactions are treated as reversible, unless stated otherwise, with the reverse rate coefficients calculated from known thermochemistry. Most species’ thermochemical properties were taken from the ANL Core Mechanism, although some were obtained from Glarborg et al.75 and RMG's group additivity.76 The details of species’ and reactions’ provenance are provided in the full mechanism.| Reactiona | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ab Ab |
n | E a | ΔHr,298Kb | Ref. | |
|---|---|---|---|---|---|---|
|
a Full mechanism, available in the ESI with reaction references, consists of 66 species and 247 reactions.
b
k = AT
n
exp(−Ea/RT). Units: mol, cm, s, kcal.
c Pressure-dependent rate defined in ESI.
d
k
13 × branching ratio.13
e
k
16 × branching ratio.79
f See ESI for more details.
g Analogy to R16.
|
||||||
| R1c | C8H8 → α-C8H7 + H | 145.427 | −35.82 | 211.79 | 104.3 | PW, theory |
| R2c | C8H8 → β-C8H7 + H | 150.723 | −37.47 | 221.78 | 110.5 | PW, theory |
| R3c | C8H8 → o-C8H7 + H | 121.245 | −29.42 | 192.05 | 110.3 | PW, theory |
| R4c | C8H8 → m-C8H7 + H | 115.033 | −27.74 | 185.75 | 110.5 | PW, theory |
| R5c | C8H8 → p-C8H7 + H | 114.841 | −27.83 | 186.40 | 111.2 | PW, theory |
| R6c | C8H8 → C6H5 + C2H3 | 152.001 | −37.60 | 221.88 | 117.9 | PW, theory |
| R7c | C8H8 → benzene + H2CC: | 137.973 | −34.21 | 202.61 | 82.6 | PW, theory |
| R8 | C8H8 + H = α-C8H7 + H2 | 3.788 | 2.86 | 8.00 | 0.1 | PW, theory |
| R9–11 | C8H8 + H = β,o,m-C8H7 + H2 | 5.588 | 2.47 | 12.24 | ∼6.3 | PW, theoryf |
| R12 | C8H8 + H = p-C8H7 + H2 | 6.719 | 2.36 | 16.92 | 7.0 | 33 |
| R13c | C8H8 + H = C6H5 + C2H4 | 38.410 | −6.53 | 35.00 | 7.1 | 77 |
| R14c | C8H8 + H = benzene + C2H3 | 25.947 | −3.22 | 23.16 | 3.0 | PWd |
| R15c | β-C8H7 = α-C8H7 | 39.773 | −8.29 | 52.03 | −6.2 | PWe |
| R16c | β-C8H7 = o-C8H7 | 37.393 | −8.04 | 33.79 | −0.2 | 78 |
| R17c | m-C8H7 = o-C8H7 | 37.393 | −8.04 | 33.79 | −0.1 | PW, analogyg |
| R18c | p-C8H7 = m-C8H7 | 37.393 | −8.04 | 33.79 | −0.7 | PW, analogyg |
| R19–20c | α-C8H7 + H = β,o-C8H7 + H | — | — | — | ∼6.1 | PW, theoryf |
| R21–25c | α,β,o,m,p-C8H7 + H = C6H5 + C2H3 | 63.667 | −13.05 | 64.68 | ∼8.5 | PW, theoryf |
| R26–30c | α,β,o,m,p-C8H7 + H = benzene + H2CC: | 68.070 | −14.63 | 66.33 | ∼−26.8 | PW, theoryf |
| R31–32c | α,β-C8H7 + H = C8H6 + H2 | 67.388 | −14.77 | 68.33 | ∼−67.1 | PW, theoryf |
| R33–35c | C8H6 + H = α,β,o-C8H7 | — | — | — | ∼−36.1 | 78 |
| R36–38c | C6H5 + C2H2 = α,β,o-C8H7 | — | — | — | ∼−45.0 | 78 |
| R39 | C8H6 = C6H4C2H + H | 16.699 | 0.00 | 113.30 | 109.8 | 28 |
| R40c | C8H6 = C6H5 + C2H | 135.097 | −34.27 | 171.21 | 142.3 | 26 |
The base of the styrene pyrolysis mechanism is the ANL Core Mechanism80,81 with reactions involving oxygen-containing species removed. The acetylene chemistry was updated with reactions from Zádor et al.82 Extensions to the acetylene chemistry included C4H5/acetylene reactions from Huang et al.83 and acetylene/polyacetylene chemistry from Kiefer et al.84 and Raman.85 As an important product, the phenylacetylene sub-mechanism was taken from Sun et al.86 The Comandini et al. styrene mechanism28 provided the base of the styrene chemistry with additions from the theoretical work.
At the high temperatures of the shock tube study styrene can decompose by several channels. Based on the pyrolysis and oxidation literature a number of permutations of initiation reactions were tested. However, styrene dissociation is best described as a multi-channel unimolecular reaction with R1–7 (Table 4) being active. R1–5 form styryl radical isomers. R1–3 were previously investigated computationally by Wang and Frenklach;34 their RRKM calculations for k1 and k2 fall within a factor of 3–4 of the values in this study. The rate coefficient of R3 however appears to be an analogy to phenyl + H = benzene and is significantly different than the calculated values here. In addition to these three isomers, the reactions R4 and R5 forming the m,p-C8H7 isomers (Table 2) were also investigated in the theoretical portion of this work. The master equation results indicate that R1, forming α-C8H7 is the dominant channel accounting for 50% of the total styrene decomposition rate coefficient, ktot (ktot = k1 + k2 + k3 + k4 + k5 + k6 + k7), between 1800–2500 K (Fig. 9a) and R2–5 cumulatively account for 20% of ktot. To demonstrate the importance of the styryl isomers other than α-C8H7, an example is given in Fig. 8 showing a comparison with and without inclusion of R2–5 in the simulation of a LS experiment.
 | ||
| Fig. 8 LS data (symbols) and simulations at the specified conditions (lines). The solid line shows the current mechanism. The dashed line shows the effect of neglecting the minor styryl channels (R2–5). The dash-dot line assumes that styrene only decomposes via R7 per Müller-Markgraf and Troe31 and Grela et al.30 For clarity, the LS data has been decimated by 2. | ||
Isomerization of styryl radicals (R15–18) was previously studied by Mebel et al.78 and, Tokmakov and Lin.79 R15, isomerization of β-styryl to α-styryl, plays an key role in the mechanism and k15 was estimated as follows: Mebel et al. had rigorously calculated k16, β-C8H7 isomerization to o-C8H7, but not isomerization to α-C8H7. Tokmakov and Lin79 on the other hand, calculated a more complete PES for isomerization of styryl radicals, but they did not report pressure-dependent rate coefficients. Here k15 was estimated by deriving temperature-dependent branching ratios from the work of Tokmakov and Lin79 and combining this with k16 from Mebel et al.78 to get rate coefficients for β-C8H7's isomerization to α-C8H7, R15. Isomerization rate coefficients for m-styryl, k17, and p-styryl, k18, were obtained by analogy with k16.
As noted earlier in the TOF-MS section, styryl radicals were not observed and it was suggested that the concentration of styryl radicals was below the detection limit of the TOF-MS. The simulations show that the total concentration of styryl radicals is approximately two order of magnitudes lower than ortho-benzyne, which can barely be distinguished above noise, Fig. 3 (lower). The lack of buildup of styryl radicals is due to their fast dissociation at the high temperatures ( >1500 K) of this work. For example, k34, dissociation of α-styryl the dominant isomer, is at least 106 s−1. Thus, it is not incongruent that the pathways through styryl radicals are major routes in the decomposition of styrene but that the radicals are not observed in the TOF-MS data. Another important radical producing channel that was also considered previously by Wang and Frenklach.34 is R6: styrene → phenyl + vinyl. R6 accounts for 25% of styrene consumption, making it the second most important reaction after R1.
Müller-Markgraf and Troe,31 and Grela et al.30 assumed that the only initiation step in styrene pyrolysis is R7: styrene → benzene + C2H2. The experimental activation energy for R7 from Grela et al. was consistent with the ejection of vinylidene H2CC,58 rather than acetylene. However, vinylidene rapidly isomerizes to acetylene, the observed product.30,31 From the point of view of an LS experiment isomerization of vinylidene is sufficiently fast that the net enthalpy of reaction is the same regardless of whether R7 yields acetylene or vinylidene. To test if R7 could be the main dissociation path a model was constructed that replaced R1 to R7 with a reaction directly forming acetylene and benzene from styrene with rate coefficients taken from Müller-Markgraf and Troe.31 An example simulation with this model is shown in Fig. 8 (blue dash-dot line). The simulation results predict an entirely different shape for the density gradient profile to that observed experimentally. Any alteration of the rate coefficients for the initial dissociation step results in an equally poor fit confirming that styrene does not simply dissociate to acetylene and benzene at the conditions of the LS work. Prior literature mechanisms, relying on the Müller-Markgraf and Troe, and Grela et al. studies, have viewed the benzene route as an important primary channel of styrene decomposition, but in our mechanism, it contributes only 5% to ktot at the conditions of the LS experiments. Although R7 is a minor channel at the conditions of this study, at lower temperatures it becomes more important, Fig. 9d. However, even under the conditions of Grela et al. it only reaches about 10% of ktot, and it is only at low temperatures and high pressures that it becomes a major channel.
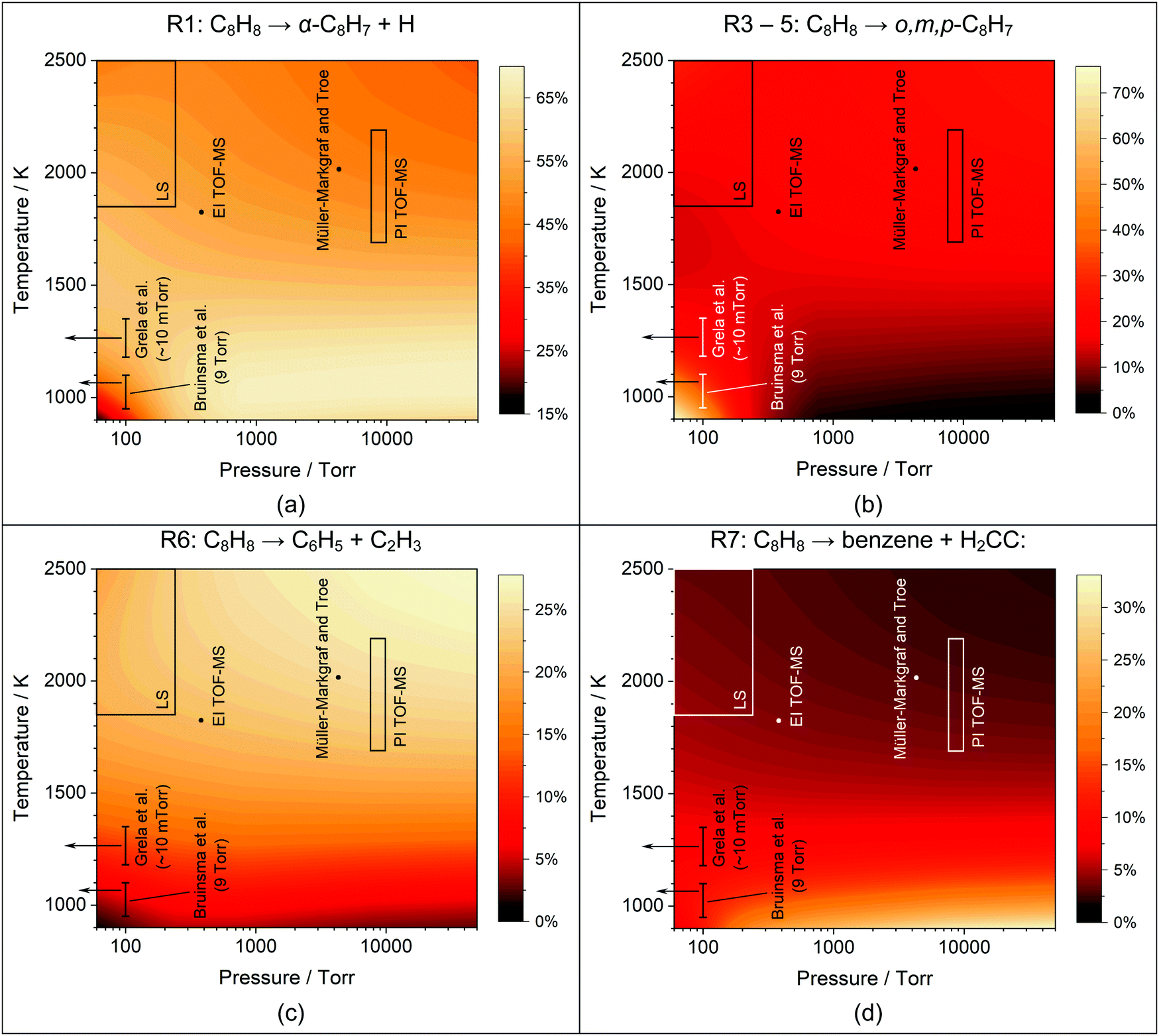 | ||
| Fig. 9 Percentage that (a) R1, (b) R3–5, (c) R6, and (d) R7 comprise of the total styrene decomposition rate based upon the current mechanism. The conditions of the current study and literature studies29–31 are indicated. | ||
The simulations indicate that there are strong temperature and pressure dependencies that influence the branching ratios between R1–7. Consequently, the apparent differences between various experimental studies may be due to changes in dominant reaction paths. Fig. 9 shows the variations in branching fractions for several key channels as functions of temperature and pressure. The regions relevant to each experiment set are marked on the figures. These figures show for example, the importance of R1 and the relative lack of importance of R7 in the literature studies,29–31 and that under the conditions of Bruinsma et al.29 R3–5 becomes more important than R1. Thus, styryl radical formation remains important but the dominant isomers switch from α-styryl to a mixture of o,m,p-styryl radicals.
As discussed in the Theory section, the molecular channel through styrene → phenylacetylene + H2 has been extensively explored both as a direct reaction and through various intermediates such as ortho-iso-styrene. We have concluded that this reaction is of minimal importance because it contributes <0.5% to ktot.
There is a potential pathway for styrene to decompose by a ring contraction mechanism, styrene → cyclopentadienyl + propargyl, which was proposed by Slavinskaya and Frank.87 At these temperatures and pressures, cyclopentadienyl radical dissociates effectively instantaneously (k > 1011 s−1),88 so it would not be observed in the TOF-MS data. Decomposition of styrene to cyclopentadienyl and propargyl radicals has not been included as a decomposition step because c-C5H5 + C3H3 is >110 kcal mol−1 endothermic relative to styrene and is very likely to be formed via a ring contraction mechanism that is unlikely to compete with the lowest energy direct C–H bond fission from styrene.
Similarly, ortho-benzyne + ethylene = styrene, as considered by Friedrichs et al.,89 is another potential pathway; although, in this study, the reaction is viewed in the reverse direction. Note that in the Friedrichs et al.89 study this was only a potential product of this reaction, and at their conditions bicyclic benzocyclobutene was considered the more likely product. This reaction has not been included because: (1) there is a substantial barrier (∼120 kcal mol−1) for the direct elimination of C2H4 from styrene, (2) there is little evidence for ethylene in the mass spectra at short reaction times, and (3) a mechanism based upon this reaction could not simulate the observed density gradients in the present LS data.
Laser schlieren densitometry
DFST/LS experiments were performed at 60, 120, and 240 Torr between 1800–2550 K with 1%, 2%, 4% styrene/Kr mixtures. Representative LS signals and density gradients are shown in Fig. 1. The density gradient simulations were performed with Frhodo, a newly developed, open source program to simulate experimental data and optimize chemical kinetics mechanisms.90 In this case, the in-house, Cantera-based incident shock reactor derived from first principles was used to simulate the DFST/LS experiments.91 Generally the relevant rate constants would be best fit for each experiment so that the simulation overlays the experimental density gradients, such as the black lines in Fig. 1, but in this study individually best-fitting R1–7 was infeasible given the difficulties in applying proper constraints to the total rate of decomposition and the branching ratios of the various channels. Consequently, ktot for decomposition of styrene were adjusted to obtain the best fit to the experimental data to the LS data while allowing the results of theoretical calculations to dictate the branching ratios. A new modified Arrhenius fit was then made to the experimentally derived ktot, and then the process was repeated until convergence, Fig. 10. Additionally, the model simulations needed to qualitatively match the results of the TOF-MS data. Significant use of Frhodo's ability to optimize mechanisms was used to quickly explore a number of variations of the mechanism.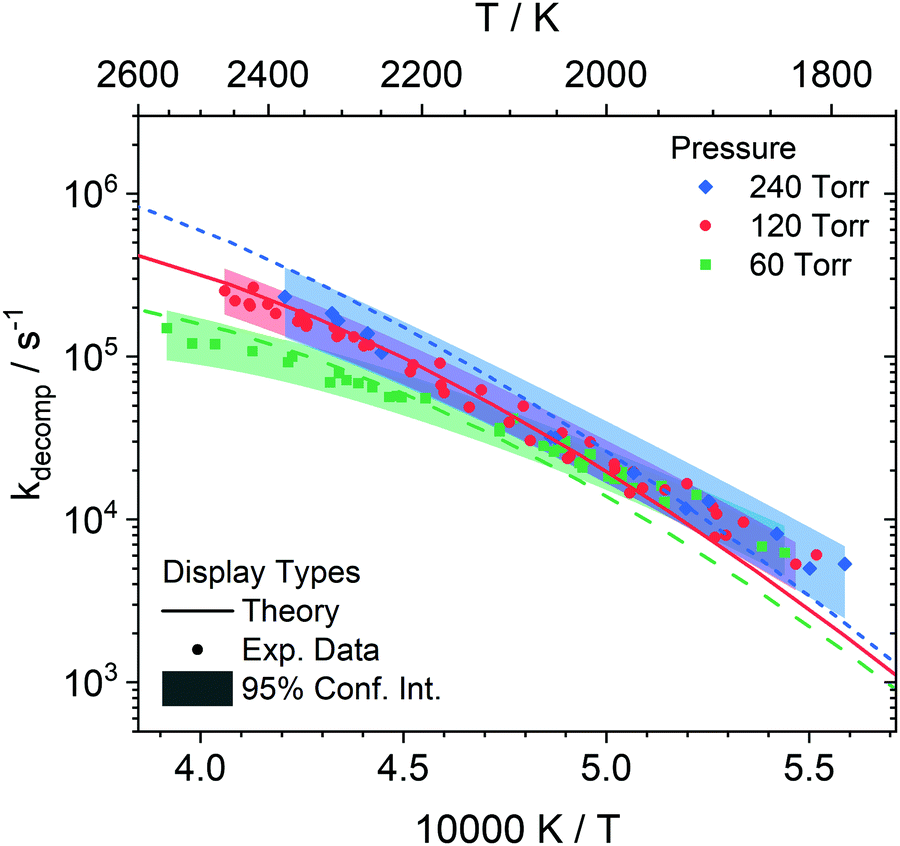 | ||
| Fig. 10 Arrhenius plot showing rate coefficients (symbols) derived from LS experiments with values from the theoretical study (lines) and 95% confidence intervals (shaded) of the total decomposition at 60, 120 and 240 Torr. The uncertainties are roughly 30% for 60 and 120 Torr and ∼45% for 240 Torr. | ||
From eqn (1) the enthalpy of reaction and rate of reaction are the two factors that mainly determine a reaction's contribution to the observed density gradient. The ΔHr,298K for R1–R5 forming styryl radical isomers are similar, Table 4. R1 is smaller by ∼6 kcal mol−1 than R2–R5 but it is still very large at 104.3 kcal mol−1 and the differences in enthalpy of reaction are minimal for these channels. R6 has ΔHr,298K = 117.9 kcal mol−1 and for R7 it is 82.6 kcal mol−1. For all the ΔHr,298K for R7 appears small relative to R1–R6 it is still a large enthalpy of reaction. Consequently, the dominant factor in influencing the density gradients in styrene pyrolysis is the rate of each reaction. As expected from the data in Fig. 9 in combination with the enthalpies of reaction R1 is the main contributor to the density gradients. The combination of the largest enthalpy of reaction and second largest rate coefficient give R6 a large influence on the observed density gradient. Individually, R2–R5 make small contributions due to their relatively small rate coefficients and R7 is of minor significance with respect to the observed density gradient. The simulations were optimized to fit each experiment by fixing all rate coefficients apart R1–R7 which were adjusted to obtain the best fit. This was done by adjusting ktot and obtaining k1 to k7 from the theoretical branching fractions. The ktot obtained reflect the experimental uncertainties and are shown in Fig. 10. The shaded bands show the uncertainties and are based upon the fit uncertainty of the experimentally derived rate coefficients as well as the uncertainty due to error in evaluating the rate coefficients for each experiment and uncertainty in the rate coefficients for the individual H + styrene/styryl reactions.
At low to mid temperatures in Fig. 10 (1800–2000 K), the model calculations fall below the experimental data. This can be explained because the LS signal magnitudes at low temperatures can be small, amplifying both experimental and simulation errors. At higher temperatures, the model predictions fall within the experimental data. There is more influence at high temperatures by secondary chemistry, such as C8H8 + H and C8H7 + H, due to the faster consumption of styrene rapidly creating a radical pool which dominates the observed chemistry. For example, at 120 Torr by normalizing the rate coefficients for the bimolecular reactions, R8–14, by the limiting reactant concentration, H, the branching ratios between the unimolecular decomposition reactions and the bimolecular reactions can be compared. At 1800 K, the bimolecular reactions become more important than the unimolecular reactions at 1.1 μs whereas at 2400 K they do so at 0.4 μs. However, R8–14 have very small enthalpies of reaction making their overall contributions to the net density gradient small, and it is only subsequent reactions of their products that have a significant impact on the observed density gradients. Overall, the current mechanism does a reasonable job at predicting the LS data. To better understand the initial decomposition steps, a reaction path analysis was performed, shown in Fig. 11.
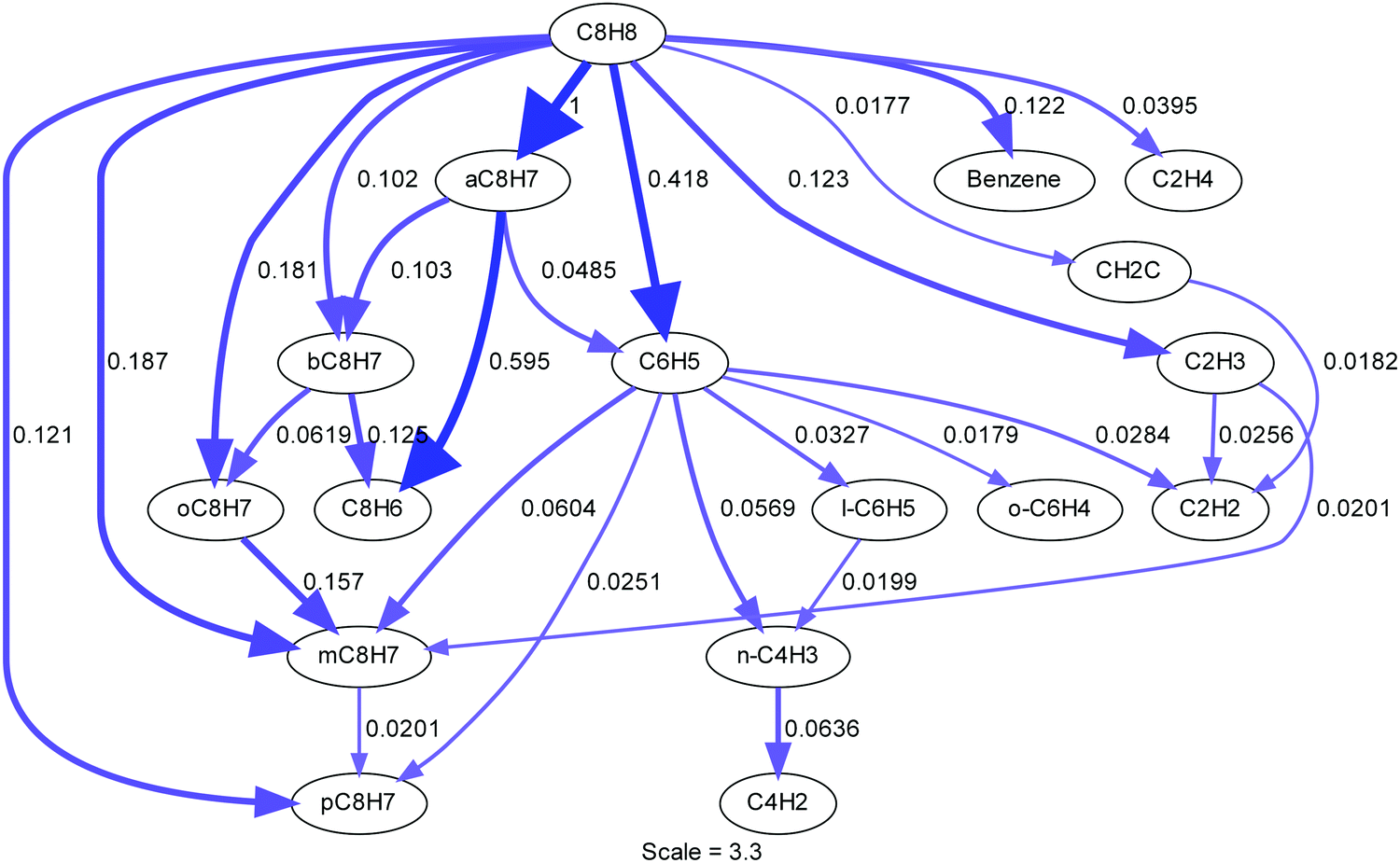 | ||
| Fig. 11 Reaction path analysis at 2% consumption of styrene (2100 K, 120 Torr) in a constant volume reactor. Arrows are proportional to the normalized molar flux of the carbon-containing species, the exact value is given next to the arrows. | ||
Fig. 11 is based on the state of the system at 2% consumption of styrene. Consequently, several pathways involving styrene are shown that have not yet been discussed. These arise from reactions of styrene with radicals that are formed from the initial dissociation pathways and represent secondary reactions rather than dissociation of styrene which is the focus of this work. In terms of the LS experiments, they are much less important than the initial decompositions reactions, but they are important for simulating the latter part of the LS profiles and predicting species concentrations at longer times such as in the TOF-MS studies.
Phenylacetylene is a key species in the early stages of styrene pyrolysis. It is not formed directly but through elimination of an H-atom from α-styryl and β-styryl. The primary reactions consuming phenylacetylene, R39–40, yield C6H4C2H and C6H5 both of which eventually form diacetylene, see the full mechanism in the ESI.† These routes to diacetylene increase the concentration of diacetylene beyond what would be expected for sequential addition from acetylene. From diacetylene the rest of the polyacetylenes can be formed through sequential addition. This touches upon one of the deficiencies of this mechanism, the inability to simulate the TOF-MS polyacetylene ratios as previously discussed in the TOF-MS section. This indicates that the current description of polyacetylene chemistry is inadequate, but the uncertainty introduced into the LS simulations is small and affects the later part of the density gradient. The LS results are primarily sensitive to the unimolecular styrene decomposition reactions and H + styrene/styryl reactions. The o-styryl radical has an exit channel to phenylacetylene, benzene, and phenyl. The m,p-styryl radicals have exit channels to benzene, phenyl and can isomerize to o-styryl. The importance of phenylacetylene, the primary product of the styryl isomers, is shown to be an important species from the TOF-MS spectra to the reaction path analysis shown here. It is clear that to understand and model styrene pyrolysis, styryl isomers must play a key role, as they do in the present study.
Conclusions
A combination of time-of-flight mass spectrometry (TOF-MS) in a miniature high repetition rate shock tube (ANL-HRRST), in a diaphragmless shock tube (DFST), and laser schlieren densitometry (LS) experiments were used to study the pyrolysis of styrene diluted in inert gases (Ar, Ne, Kr). The TOF-MS experiments show a large difference in the dissociation pathways depending upon temperature. At low temperatures (1710 K) the benzene ring remains mostly intact, whereas, at high temperatures (2260 K) the ring breaks and significant amounts of polyacetylenes are generated. A styrene pyrolysis mechanism was developed with a combination of the current data, theoretical calculations, and prior literature submechanisms. The full mechanism is provided in the ESI.† Simulations of the TOF-MS data predict the separate aromatic ring/polyacetylene pathways but fail to capture the specific polyacetylene ratios. This would be an interesting area of future research that has shown to be important in styrene decomposition. Simulations of the LS data are particularly sensitive to the initial dissociation reactions of styrene. The simulations show excellent agreement with the experimental data. It was found that the primary dissociation reactions were (in descending order of importance): C8H8 → α-C8H7 + H, C8H8 → C6H5 + C2H3, and the decomposition to the other four styryl radicals. The present study concludes that the initial dissociation mechanism is more complex than prior studies indicate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the auspices of the Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, U.S. Department of Energy, under contract number DE-AC02-06CH11357. The LS and modeling work were conducted in the Chemical Dynamics in the Gas Phase Program. The ANL-HRRST experiments were conducted in the Argonne/Sandia Consortium on High Pressure Combustion Chemistry. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The experiments were performed at beamline 9.0.2. RAS was supported through the NSF INTERN program supplementary to NSF award 1701343. PTL acknowledges support from the NSF award 1747774. AC acknowledges funding from the European Research Council (ERC) (grant agreement no. ERC-2017-STG 756785).References
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, A comparative kinetic study of C8–C10 linear alkylbenzenes pyrolysis in a single-pulse shock tube, Combust. Flame, 2020, 221, 136–149 CrossRef CAS.
- Y. Li, J. Cai, L. Zhang, T. Yuan, K. Zhang and F. Qi, Investigation on chemical structures of premixed toluene flames at low pressure, Proc. Combust. Inst., 2011, 33, 593–600 CrossRef CAS.
- W. Yuan, Y. Li, P. Dagaut, J. Yang and F. Qi, Investigation on the pyrolysis and oxidation of toluene over a wide range conditions. I. Flow reactor pyrolysis and jet stirred reactor oxidation, Combust. Flame, 2015, 162, 3–21 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Probing PAH formation chemical kinetics from benzene and toluene pyrolysis in a single-pulse shock tube, Proc. Combust. Inst., 2021, 38, 891–900 CrossRef CAS.
- T. A. Litzinger, K. Brezinsky and I. Glassman, The oxidation of ethylbenzene near 1060 K, Combust. Flame, 1986, 63, 251–267 CrossRef CAS.
- Y. Li, J. Cai, L. Zhang, J. Yang, Z. Wang and F. Qi, Experimental and modeling investigation on premixed ethylbenzene flames at low pressure, Proc. Combust. Inst., 2011, 33, 617–624 CrossRef CAS.
- A. Ergut, Y. A. Levendis and J. Carlson, Emissions from the combustion of polystyrene, styrene and ethylbenzene under diverse conditions, Fuel, 2007, 86, 1789–1799 CrossRef CAS.
- T. A. Litzinger, K. Brezinsky and I. Glassman, Reactions of n-propylbenzene during gas phase oxidation, Combust. Sci. Technol., 1986, 50, 117–133 CrossRef CAS.
- P. Dagaut, A. Ristori, A. El Bakali and M. Cathonnet, Experimental and kinetic modeling study of the oxidation of n-propylbenzene, Fuel, 2002, 81, 173–184 CrossRef CAS.
- Z. Wang, Y. Li, F. Zhang, L. Zhang, W. Yuan, Y. Wang and F. Qi, An experimental and kinetic modeling investigation on a rich premixed n-propylbenzene flame at low pressure, Proc. Combust. Inst., 2013, 34, 1785–1793 CrossRef CAS.
- K. Brezinsky, G. T. Linteris, T. A. Litzinger and I. Glassman, High temperature oxidation of n-alkyl benzenes, Symp. Combust., 1988, 21, 833–840 CrossRef.
- E. Pousse, P. A. Glaude, R. Fournet and F. Battin-Leclerc, A lean methane premixed laminar flame doped with components of diesel fuel. I. n-Butylbenzene, Combust. Flame, 2009, 156, 954–974 CrossRef CAS.
- P. Diévart and P. Dagaut, The oxidation of n-butylbenzene: Experimental study in a JSR at 10 atm and detailed chemical kinetic modeling, Proc. Combust. Inst., 2011, 33, 209–216 CrossRef.
- B. Husson, R. Bounaceur, K. Tanaka, M. Ferrari, O. Herbinet, P. A. Glaude, R. Fournet, F. Battin-Leclerc, M. Crochet, G. Vanhove, R. Minetti, C. J. Tobin, K. Yasunaga, J. M. Simmie, H. J. Curran, T. Niass, O. Mathieu and S. S. Ahmed, Experimental and modeling study of the oxidation of n-butylbenzene, Combust. Flame, 2012, 159, 1399–1416 CrossRef CAS.
- E. Pousse, Z. Y. Tian, P. A. Glaude, R. Fournet and F. Battin-Leclerc, A lean methane premixed laminar flame doped with components of diesel fuel part III: Indane and comparison between n-butylbenzene, n-propylcyclohexane and indane, Combust. Flame, 2010, 157, 1236–1260 CrossRef CAS.
- Y. Li, L. Zhang, Z. Wang, L. Ye, J. Cai, Z. Cheng and F. Qi, Experimental and kinetic modeling study of tetralin pyrolysis at low pressure, Proc. Combust. Inst., 2013, 34, 1739–1748 CrossRef CAS.
- B. Gullett, A. Touati and L. Oudejans, PCDD/F and aromatic emissions from simulated forest and grassland fires, Atmos. Environ., 2008, 42, 7997–8006 CrossRef CAS.
- Williams & Marshall Strategy, The Global Styrene Market, 2019.
- C. Westblad, Y. A. Levendis, H. Richter, J. B. Howard and J. Carlson, A study on toxic organic emissions from batch combustion of styrene, Chemosphere, 2002, 49, 395–412 CrossRef CAS PubMed.
- L. Jia, S. Evans and S. van der Linden, Motivating actions to mitigate plastic pollution, Nat. Commun., 2019, 10, 1–3 CrossRef PubMed.
- A. H. Tullo, Plastic has a problem; is chemistry the solution?, C&EN Glob. Enterp., 2019, 97, 29–34 Search PubMed.
- V. G. Pol, Upcycling: Converting waste plastics into paramagnetic, conducting, solid, pure carbon microspheres, Environ. Sci. Technol., 2010, 44, 4753–4759 CrossRef CAS PubMed.
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. M. Van Steenberge, D. R. D’hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, The chemistry of chemical recycling of solid plastic waste via pyrolysis and gasification: State-of-the-art, challenges, and future directions, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef.
- J. Li, J. Cai, T. Yuan, H. Guo and F. Qi, A thermal decomposition study of polymers by tunable synchrotron vacuum ultraviolet photoionization mass spectrometry, Rapid Commun. Mass Spectrom., 2009, 23, 1269–1274 CrossRef CAS PubMed.
- K. H. Kim, S. A. Jahan, E. Kabir and R. J. C. Brown, A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects, Environ. Int., 2013, 60, 71–80 CrossRef CAS PubMed.
- W. Yuan, Y. Li, P. Dagaut, J. Yang and F. Qi, Experimental and kinetic modeling study of styrene combustion, Combust. Flame, 2015, 162, 1868–1883 CrossRef CAS.
- X. Meng, E. Hu, X. Li and Z. Huang, Experimental and kinetic study on laminar flame speeds of styrene and ethylbenzene, Fuel, 2016, 185, 916–924 CrossRef CAS.
- A. Comandini, G. Pengloan, S. Abid and N. Chaumeix, Experimental and modeling study of styrene oxidation in spherical reactor and shock tube, Combust. Flame, 2016, 173, 425–440 CrossRef CAS.
- O. S. L. Bruinsma and R. S. Geertsma, P. Bank and J. A. Moulijn, Gas phase pyrolysis of coal-related aromatic compounds in a coiled tube flow reactor. 1. Benzene and derivatives, Fuel, 1988, 67, 327–333 CrossRef CAS.
- M. A. Grela, V. T. Amorebieta and A. J. Colussi, Pyrolysis of styrene. Kinetics and mechanism of the equilibrium styrene ↔ benzene + acetylene, J. Phys. Chem., 1992, 96, 9861–9865 CrossRef CAS.
- W. Müller-Markgraf and J. Troe, Thermal decomposition of ethylbenzene, styrene, and bromophenylethane: UV absorption study in shock waves, J. Phys. Chem., 1988, 92, 4914–4922 CrossRef.
- G. Pengloan, P. Dagaut, N. Djebaili-Chaumeix, C. É. Paillard and M. Cathonnet, Colloque Combustion Propre, Combustion Institute, French Section, Orléans, 2001, p. 9 Search PubMed.
- K. Narayanaswamy, G. Blanquart and H. Pitsch, A consistent chemical mechanism for oxidation of substituted aromatic species, Combust. Flame, 2010, 157, 1879–1898 CrossRef CAS.
- H. Wang and M. Frenklach, A detailed kinetic modeling study of aromatics formation in laminar premixed acetylene and ethylene flames, Combust. Flame, 1997, 110, 173–221 CrossRef CAS.
- A. G. Gaydon and I. R. Hurle, The shock tube in high-temperature chemical physics, Chapman and Hall, 1963 Search PubMed.
- E. Goos, A. Burcat and B. Ruscic, Extended Third Millenium Ideal Gas Thermochemical Database with updates from Active Thermochemical Tables, 2021, http://garfield.1605 chem.elte.hu/Burcat/THERM.DAT, accessed 6th January 2021 Search PubMed.
- J. B. Randazzo and R. S. Tranter, Note: An improved driver section for a diaphragmless shock tube, Rev. Sci. Instrum., 2015, 86, 016117 CrossRef CAS PubMed.
- R. S. Tranter and B. R. Giri, A diaphragmless shock tube for high temperature kinetic studies, Rev. Sci. Instrum., 2008, 79, 094103 CrossRef PubMed.
- R. S. Tranter and P. T. Lynch, A miniature high repetition rate shock tube, Rev. Sci. Instrum., 2013, 84, 094102 CrossRef CAS PubMed.
- R. S. Tranter and T. Sikes, Solenoid actuated driver valve for high repetition rate shock tubes, Rev. Sci. Instrum., 2020, 91, 056101 CrossRef CAS PubMed.
- J. B. Randazzo, R. Sivaramakrishnan, A. W. Jasper, T. Sikes, P. T. Lynch and R. S. Tranter, An experimental and theoretical study of the high temperature reactions of the four butyl radical isomers, Phys. Chem. Chem. Phys., 2020, 22, 18304–18319 RSC.
- D. R. Lide, Ionization potentials of atoms and atomic ions, Handbook of Chemistry and Physics, 1992, pp. 10–211 Search PubMed.
- R. S. Tranter, S. J. Klippenstein, L. B. Harding, B. R. Giri, X. Yang and J. H. Kiefer, Experimental and theoretical investigation of the self-reaction of phenyl radicals, J. Phys. Chem. A, 2010, 114, 8240–8261 CrossRef CAS PubMed.
- S. H. Dürrstein, M. Aghsaee, L. Jerig, M. Fikri and C. Schulz, A shock tube with a high-repetition-rate time-of-flight mass spectrometer for investigations of complex reaction systems, Rev. Sci. Instrum., 2011, 82, 084103 CrossRef PubMed.
- C. Banyon, T. Sikes and R. S. Tranter, Reactions of propyl radicals: A shock tube–VUV photoionization mass spectrometry study, Combust. Flame, 2021, 224, 14–23 CrossRef CAS.
- J. H. Kiefer and A. C. Manson, Refractive index change and curvature in shock waves by angled beam refraction, Rev. Sci. Instrum., 1981, 52, 1392–1396 CrossRef CAS.
- J. H. Kiefer, M. Z. Al-Alami and J.-C. Hajduk, Physical optics of the laser-schlieren shock tube technique, Appl. Opt., 1981, 20, 221 CrossRef CAS PubMed.
- J. H. Kiefer, in Shock Waves in Chemistry, ed. A. Lifshitz, Marcel Dekker, New York, 1981, pp. 219–277 Search PubMed.
- J. H. Kiefer, M. Z. Al-Alami and K. A. Budach, A shock tube, laser-schlieren study of propene pyrolysis at high temperatures, J. Phys. Chem., 1982, 86, 808–813 CrossRef CAS.
- P. T. Lynch, T. P. Troy, M. Ahmed and R. S. Tranter, Probing combustion chemistry in a miniature shock tube with synchrotron VUV photo ionization mass spectrometry, Anal. Chem., 2015, 87, 2345–2352 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. M. L. Martin, Ab initio total atomization energies of small molecules - Towards the basis set limit, Chem. Phys. Lett., 1996, 259, 669–678 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, From the multiple-well master equation to phenomenological rate coefficients: Reactions on a C3H4 potential energy surface, J. Phys. Chem. A, 2003, 107, 2680–2692 CrossRef CAS.
- L. Ye, L. Xing, W. Yuan, Y. Li, L. Zhang and F. Qi, Predictive kinetics on the formation and decomposition of ethylbenzene, Proc. Combust. Inst., 2017, 36, 533–542 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, Gaussian-4 theory, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. J. Klippenstein, L. B. Harding and Y. Georgievskii, On the formation and decomposition of C7H8, Proc. Combust. Inst., 2007, 31 I, 221–229 CrossRef.
- C. F. Yu, F. Youngs, R. Bersohn and N. J. Turro, Photodissociation at 193 nm of cyclooctatetraene and styrene into benzene and acetylene, J. Phys. Chem., 1985, 89, 4409–4412 CrossRef CAS.
- Y. R. Lee, C. C. Chen and S. M. Lin, Photoelimination of C2H2 and H2 from styrene at 193 nm, J. Chem. Phys., 2003, 118, 9073–9076 CrossRef CAS.
- S. J. Klippenstein, A. F. Wagner, R. C. Dunbar, D. M. Wardlaw, S. H. Robertson and J. A. Miller, VARIFLEX 2.03m, 2010, unpublished work.
- A. Fernández-Ramos, J. A. Miller, S. J. Klippenstein and D. G. Truhlar, Modeling the kinetics of bimolecular reactions, Chem. Rev., 2006, 106, 4518–4584 CrossRef PubMed.
- J. A. Miller and S. J. Klippenstein, Master equation methods in gas phase chemical kinetics, J. Phys. Chem. A, 2006, 110, 10528–10544 CrossRef CAS PubMed.
- P. Pechukas and J. C. Light, On detailed balancing and statistical theories of chemical kinetics, J. Chem. Phys., 1965, 42, 3281–3291 CrossRef CAS.
- L. B. Harding, Y. Georgievskii and S. J. Klippenstein, Predictive theory for hydrogen atom-hydrocarbon radical association kinetics, J. Phys. Chem. A, 2005, 109, 4646–4656 CrossRef CAS PubMed.
- H. Ismail, P. R. Abel, W. H. Green, A. Fahr, L. E. Jusinski, A. M. Knepp, J. Zador, G. Meloni, T. M. Selby, D. L. Osborn and C. A. Taatjes, Temperature-dependent kinetics of the vinyl radical (C 2H 3) self-reaction, J. Phys. Chem. A, 2009, 113, 1278–1286 CrossRef CAS PubMed.
- S. J. Klippenstein, Y. Georgievskii and L. B. Harding, Predictive theory for the combination kinetics of two alkyl radicals, Phys. Chem. Chem. Phys., 2006, 8, 1133–1147 RSC.
- B. E. Poling, J. M. Prausnitz and J. P. O’Connell, Properties of Gases and Liquids, McGraw-Hill Education, New York, Chicago, San Francisco, Athens, London, Madrid, Mexico City, Milan, New Delhi, Singapore, Sydney, Toronto, 5th edn, 2001 Search PubMed.
- J. B. W. Webber, A bi-symmetric log transformation for wide-range data, Meas. Sci. Technol., 2013, 24, 27001 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD-20899, 2021, https://webbook.nist.gov/chemistry, accessed 3rd Mach 2021 Search PubMed.
- T. A. Cool, J. Wang, K. Nakajima, C. A. Taatjes and A. Mcllroy, Photoionization cross sections for reaction intermediates in hydrocarbon combustion, Int. J. Mass Spectrom., 2005, 247, 18–27 CrossRef CAS.
- J. Yang, Y. Li and Z. Cheng, Photonionization Cross Section Database (Version 2.0), http://flame.nsrl.ustc.edu.cn/database.
- Z. Zhou, M. Xie, Z. Wang and F. Qi, Determination of absolute photoionization cross-sections of aromatics and aromatic derivatives, Rapid Commun. Mass Spectrom., 2009, 23, 3994–4002 CrossRef CAS PubMed.
- Y. Li, L. Zhang, Z. Tian, T. Yuan, K. Zhang, B. Yang and F. Qi, Investigation of the rich premixed laminar acetylene/oxygen/argon flame: Comprehensive flame structure and special concerns of polyynes, Proc. Combust. Inst., 2009, 32, 1293–1300 CrossRef CAS.
- A. W. Jasper, 2020, private communication.
- P. Glarborg, J. A. Miller, B. Ruscic and S. J. Klippenstein, Modeling nitrogen chemistry in combustion, Prog. Energy Combust. Sci., 2018, 67, 31–68 CrossRef.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- D. Belisario-Lara, A. M. Mebel and R. I. Kaiser, Computational study on the unimolecular decomposition of JP-8 jet fuel surrogates III: butylbenzene isomers (n-, s-, and t-C14H10), J. Phys. Chem. A, 2018, 122, 3980–4001 CrossRef CAS PubMed.
- A. M. Mebel, Y. Georgievskii, A. W. Jasper and S. J. Klippenstein, Temperature- and pressure-dependent rate coefficients for the HACA pathways from benzene to naphthalene, Proc. Combust. Inst., 2017, 36, 919–926 CrossRef CAS.
- I. V. Tokmakov and M. C. Lin, Reaction of phenyl radicals with acetylene: Quantum chemical investigation of the mechanism and master equation analysis of the kinetics, J. Am. Chem. Soc., 2003, 125, 11397–11408 CrossRef CAS PubMed.
- A. Krisman, C. Mounaïm-Rousselle, R. Sivaramakrishnan, A. J. Miller and H. J. Chen, Reference natural gas flames at nominally autoignitive engine-relevant conditions, Proc. Combust. Inst., 2019, 37, 1631–1638 CrossRef CAS.
- J. A. Miller, R. Sivaramakrishnan, Y. Tao, C. F. Goldsmith, M. P. Burke, A. W. Jasper, N. Hansen, N. J. Labbe, P. Glarborg and J. Zádor, Combustion chemistry in the twenty-first century: Developing theory-informed chemical kinetics models, Prog. Energy Combust. Sci., 2021, 83, 100886 CrossRef.
- J. Zádor, M. D. Fellows and J. A. Miller, Initiation reactions in acetylene pyrolysis, J. Phys. Chem. A, 2017, 121, 4203–4217 CrossRef PubMed.
- C. Huang, B. Yang, F. Zhang and G. Tian, Quantification of the resonance stabilized C4H5 isomers and their reaction with acetylene, Combust. Flame, 2018, 198, 334–341 CrossRef CAS.
- J. H. Kiefer, S. S. Sidhu, R. D. Kern, K. Xie, H. Chen and L. B. Harding, The homogeneous pyrolysis of acetylene II: The high temperature radical chain mechanism, Combust. Sci. Technol., 1992, 82, 101–130 CrossRef CAS.
- A. Raman, Role of Diacetylene in Soot Formation – Experimental and Modeling Study, Doctoral thesis, University of Illinois at Chicago, 2007 Search PubMed.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, An experimental and kinetic modeling study of phenylacetylene decomposition and the reactions with acetylene/ethylene under shock tube pyrolysis conditions, Combust. Flame, 2020, 220, 257–271 CrossRef CAS.
- N. A. Slavinskaya and P. Frank, A modelling study of aromatic soot precursors formation in laminar methane and ethene flames, Combust. Flame, 2009, 156, 1705–1722 CrossRef CAS.
- R. D. Kern, Q. Zhang, J. Yao, B. S. Jursic, R. S. Tranter, M. A. Greybill and J. H. Kiefer, Pyrolysis of cyclopentadiene: Rates for initial C–H bond fission and the decomposition of c-C5H5, Symp. Combust., 1998, 27, 143–150 CrossRef.
- G. Friedrichs, E. Goos, J. Gripp, H. Nicken, J.-B. Schönborn, H. Vogel and F. Temps, The products of the reactions of o-benzyne with ethene, propene, and acetylene: A combined mass spectrometric and quantum chemical study, Z. Phys. Chem., 2009, 223, 387–407 CrossRef CAS.
- T. Sikes and R. S. Tranter, Frhodo, https://github.com/Argonne-National-Laboratory/Frhodo.
- D. G. Goodwin, R. L. Speth, H. K. Moffat and B. W. Weber, Cantera: An object-oriented software toolkit for chemical kinetics, thermodynamics, and transport processes, https://www.cantera.org/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02437j |
| This journal is © the Owner Societies 2021 |