
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational modelling of Pd-catalysed alkoxycarbonylation of alkenes and alkynes†‡
Shahbaz
Ahmad
and
Michael
Bühl
 *
*
School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: buehl@st-andrews.ac.uk
First published on 22nd July 2021
Abstract
This perspective highlights the computational modelling of alkene and alkyne alkoxycarbonylation at palladium catalysts. We cover studies on Pd-catalysed alkoxycarbonylation of alkenes with bidentate diphosphine ligands, which reveal a hydride pathway is operating with an intermolecular alcoholysis step, where explicit solvation is mandatory to estimate the overall barriers correctly and model alcoholysis/copolymerisation selectivities. Subsequently, we discuss Pd-catalysed alkyne alkoxycarbonylation with P,N-chelating ligands, where an in situ base mechanism is operating involving ketene-type intermediates. We also discuss catalyst poisoning due to allene and designing a potential new catalyst tolerant towards allene poisoning.
Introduction
The design of economic catalysts for the regioselective direct synthesis of fine chemicals from abundant resources is a critical industrial challenge. Homogeneous catalysis at transition metal centres is an important tool toward this goal, because electronic and steric tuning of ligands at the metal centre may control chemo- and regioselectivities and catalytic activities. However, isolation procedures, adequate reaction times and conditions, high atom economy, and broad substrate scope are further challenges. Homogeneous palladium catalysts with suitable ligands play a key role in fundamental transformations of synthetic chemistry (e.g., carbon–carbon bond formation, migratory insertion, oxidative addition, reductive elimination, and β-hydride elimination).1,2 When equipped with phosphine-based ligands, Pd(0) and Pd(II) complexes can facilitate carbonylation reactions, which are essential industrial processes to expand carbon chains with high selectivities.2–11 The substituents around the phosphorus donor atom can modify its electron-donating ability and steric profiles to fine-tune the reactivity of the metal and its surrounding environment.Direct synthesis of acrylate esters is achievable by palladium-catalysed alkoxycarbonylation of widely available unsaturated hydrocarbons with 100% atom economy.12–27 Carbonylations are C–C bond forming reactions using CO as feedstock; alkoxycarbonylations are variants producing esters from CO and alcohols (see Scheme 1(i)). An example is methyl propionate (MePro)—a small feedstock obtained by methoxycarbonylation of ethene and a precursor for methyl methacrylate (MMA).28–33 Poly(methyl methacrylate), also known as Perspex (trade name), which is a polymer of MMA, has a high demand because of its use in liquid crystal display (LCD) screens,34,35 and, more recently, in face shields.36,37
 | ||
| Scheme 1 (i) Methoxycarbonylation of ethene with the Pd–DTBPX catalyst system; (ii) methyl propionate to methyl methacrylate over a heterogeneous catalyst. | ||
At the industrial scale, production of MMA is a two-step process: (i) alkoxycarbonylation of ethene at a homogeneous Pd with 1,2-bis(di-tert-butylphosphinomethyl)benzene (DTBPX), a catalyst that yields methyl propionate (Scheme 1); and (ii) followed by a heterogeneous conversion to MMA.28,31,34,35,38,39 In contrast, under the same reaction conditions, the use of bidentate tertiary phosphines (Ph2P(CH2)n–PPh2) with Pd triggers copolymerisation to produce oligo-/polyketones.40 Reaction rates depend on the backbone spacer's length between the phosphorus atoms (maximum rate and highest molecular weight of the polyketones with 100% selectivity when n = 3, no reaction when n = 1). However, the catalysts’ turnover rate with DTBPX for MePro production is ten times higher than the most efficient oligo/polymer-producing catalyst.28
Widely variable coordination modes of hemilabile Pd(P,N)-type ligands make them very interesting for homogeneous catalysis. Methoxycarbonylation of propyne using a homogeneous hemilabile Pd(P,N) catalyst is another attractive MMA production route.16,17,20,21,41 The reaction produces the branched product (MMA) with high selectivity with only traces of the linear product (methyl coronate). Increasing bulk at the pyridyl moiety further increases the branched selectivity. This homogenously catalysed single-step MMA synthesis with 100% atom economy seems to be very attractive. However, propadiene, an impurity in propyne from industrial waste, instantly poisons the catalyst.16,17,21 Consequently, propyne would need to be purified before its methoxycarbonylation, making this route uncompetitive on an industrial scale with the current two-step process.
The alkoxycarbonylation of terminal alkenes or alkynes either leads to the branched or the linear product. The selectivity towards branched or linear products depends on the catalytic system or the substrate. Many studies support a hydride cycle, and the hydride transfer to produce alkyl or alkenyl palladium intermediate is considered as the selectivity determining step. Scheme 2 shows catalytic cycles for the alkoxycarbonylation of terminal alkenes and alkynes, leading to branched and linear products.
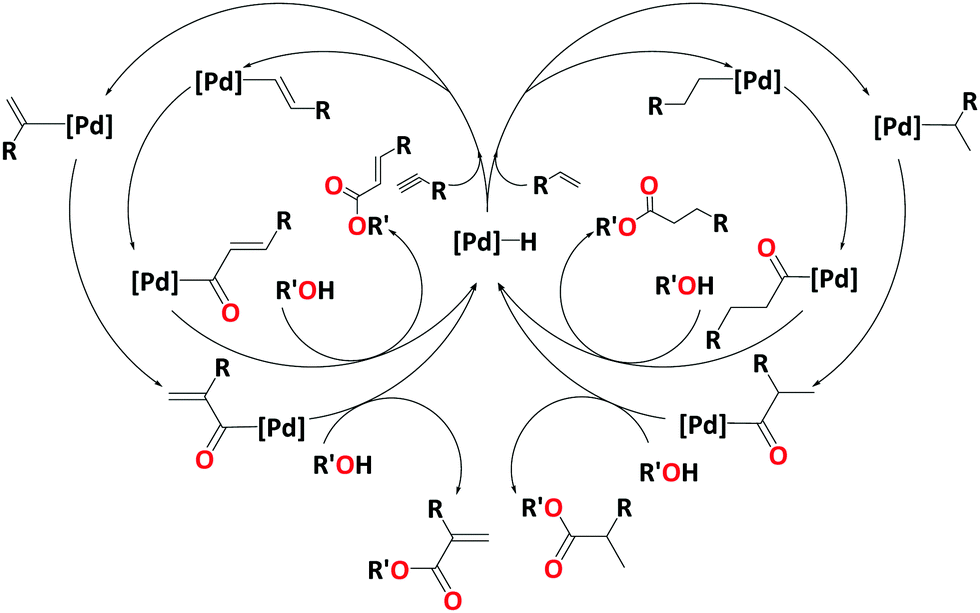 | ||
| Scheme 2 Catalytic cycles for alkoxycarbonylation of alkynes (left) and alkenes (right) emerging from a [Pd]–H complex, showing the formation of branched (outer cycle) and linear products (inner cycle). | ||
Due to the high demand for acrylate esters, the methodologies for their production will always have room for improvement. Mechanistic insights into a catalytic cycle help improve the current catalytic system by designing new catalysts. In a catalytic reaction, there might be more than one pathway. It is rarely possible to strictly prove a particular reaction mechanism with experimental studies; however, computational studies can disprove mechanistic proposals when the calculated barriers are too high to be overcome. By comparing the overall barriers computed for competing pathways, modelling studies can identify plausible pathways with the lowest overall barriers for further rational design. This perspective highlights the computational modelling of alkoxycarbonylation of alkenes and alkynes at homogenous Pd catalysts with phosphine-based ligands to study reaction pathways based on the free energy barriers.
Computational methodology
In this perspective, we are primarily interested in the overall reaction energy barriers related to a change in Gibbs free energy of the system, denoted as ΔG. ΔG is estimated from quantum chemical simulations based on density functional theory (DFT). Here we have presented several studies that use different DFT levels to explore the mechanisms for palladium-catalysed alkoxycarbonylation of alkenes, alkynes, and allenes based on free energy barriers. For example, studies by Zuidema et al. presented in this perspective use the local density approximation (LDA, in form of the VWN functional)42 to describe the importance of destabilising the most abundant reaction intermediate (MARI) and stabilising the highest energy transition state (HEST) based on their bite angles (for studies related to alkoxycarbonylation of alkenes at Pd catalyst with bidentate diphosphine ligands).39,43 While LDA can provide reasonable molecular structures, it suffers from sever overbinding, rendering energetics less reliable.Some studies in this perspective use functionals based on the generalised gradient approximation (GGA), which is an improvement over LDA.44–47 In this perspective, we have included one study, which uses BP86 functional (exchange and correlation GGA functional).48,49 and one using M06-L (of the Minnesota family),50 an example of a meta-GGA functional.
Most of the studies covered within this perspective use hybrid-GGA functionals, of which B3PW9151–54 has been frequently used. B3PW91 coupled with Grimme's DFT-D355–64 dispersion correction benchmarks well against a highly accurate ab initio correlated CCSD(T)65ref. 66. A few studies also use B3LYP.51,67–69 One study uses the PBE0 functional,45 which is the hybrid version of the popular GGA functional PBE.
To describe the heavy metal, relativistically adjusted effective core potentials (ECPs)70–73 are usually used on the palladium atom, either the small-core SDD74 or LANL2DZ75 ECPs along with their valence double- or triple-ζ basis sets. During optimisations, non-metal atoms are treated with polarised split-valence double-ζ basis sets76–80 such as 6-31G* or 6-31G** from the Pople family. Some studies use the energies obtained from the split-valence double-ζ basis set. In contrast, many refine the energies using a split-valence triple-ζ, 6-311+G**,81,82 basis set, which adds s-type and p-type diffuse functions to the heavy atoms. Some studies also use TZVP83,84 (triple-ζ for valence electrons, with one polarization function) and def2-TZVP85,86 (the newer redefinition of TZVP) basis sets.
All studies covered in this perspective have used analytical harmonic frequency calculations to verify the nature of all possible minima and transition states. Most of these studies evaluate the enthalpic and entropic corrections at the standard conditions (298.15 K, 1 bar), whereas some use the Martin–Hay–Pratt entropic correction (MHP) scheme.87 This scheme uses an elevated pressure to mimic translational entropies in condensed phases, which can become important for associative or dissociative elementary steps.
A catalytical cycle obtained from DFT generates energies of the intermediates and transition states, which can be related to the rate constant, k via transition state theory (TST).88 While absolute rate constants (related to turnover frequencies) are very difficult to compute quantitatively, relative changes between rate constants (e.g. between alternative catalytic cycles or upon branching leading to different products) tend to be more reliable. Most of the work presented in this perspective deals with the reaction rates based on the energy difference between the highest energy transition state (HETS) and the most abundant reaction intermediate (MARI) on the reaction profile, in the spirit of Kozuch and Shaik's energetic span model.89,90 This model attempts to estimate the turnover frequency of a closed catalytic cycle from the (free) energy difference between these key stationary points.
Most studies covered in this perspective use implicit solvation at the single point level to account for the presence of a solvent, typically methanol. Some studies use the conductor-like screening model, COSMO.91 Many use the polarizable continuum model, PCM,92–94 and one study uses the solvent model based on density, SMD.95 Such continuum models can be efficient means to capture bulk solvation effects, but usually fail to capture specific solute–solvent interactions (e.g. H-bonds). Therefore, in some cases a number of solvent molecules have been added explicitly to form microsolvated clusters. In general, free activation barriers are evaluated using methodologies mimicking the actual experimental conditions (temperature, solvent).
Computational mechanistic studies
Pd-catalysed alkoxycarbonylation of alkenes with bidentate diphosphine ligands
Because of their improved performance over monodentate phosphines, this chapter will concentrate on studies of bidentate diphosphines. For a few examples of modelling studies involving monophosphines, see ref. 96 (alkoxycarbonylation of α,β-unsaturated amides with PPh3)96 and ref. 97 (alkoxycarbonylation of alkenes with PPh3 and 1-(2-methoxyphenyl)-1H-pyrrolyl diphenylphosphine).97The alkoxycarbonylation of alkenes may proceed either via an alkoxy or a hydride mechanism. Here we have added an example of the possible mechanistic pathways from our recent work on methoxycarbonylation of ethene.98 The alkoxy cycle begins with the migratory insertion of CO to the Pd–OMe bond followed by insertion of alkene into the ester functionality with alcoholysis as the final step (Scheme 3A). The hydride cycle operates via a Pd–H complex, which begins with an α,β alkene insertion, followed by an α,α insertion of the alkyl group into coordinated CO closing at the alcoholysis step (Scheme 3B). On the hydride cycle, there is also the possibility of alkene insertion to the Pd–acyl complex leading to copolymerisation, but this depends on the nature of the ligand.40 There is also a possibility of the formation of an alkyl ketene at the hydride cycle. The highly reactive alkyl ketene dissociates from the metal to reproduce Pd–H complex and reacts with the solvent to produce the desired product (Scheme 3C).
 | ||
| Scheme 3 Mechanistic possibilities explored by Ahmad et al.98 (A) carbomethoxy pathway, (B) ketene pathway, (C) hydride–hydroxyalkylpalladium pathway. Reprinted from ref. 98 (Copyright 2020 Royal Society of Chemistry). | ||
Spectroscopic studies for methoxycarbonylation of ethene suggest a hydride cycle rather than a methoxy cycle, which fits well with the requirement of highly acidic conditions involving p-toluenesulfonic acid.29–33 No information detailing the cleaving methanolysis step could be furnished due to an incredibly facile reaction (on the order of minutes) of the Pd–acyl complex. Clegg et al. characterised a square planar geometry of 1,2-bis(di-tert-butylphosphinomethyl)benzene palladium(II)propanoyl chloride with the diphosphine coordinating in a cis fashion.30 Based on this geometry, they suggested that methanolysis occurs from a cis-coordinated acyl group and methanol. Supporting Clegg and co-workers’ conclusion, van Leeuwen et al. proposed an intramolecular methanolysis step rather than an intermolecular step.32
Macgregor and co-workers showed that less geometric congestion at the transition state relative to its reactant lowers the barrier.99 At the VWN/TZP level of theory, Zuidema et al. correlated the bite angles in Pd–diphosphine complexes with the reaction rates by stabilising the transition state and destabilising the reactant.43 They also used QM/MM methods to elucidate the substituents' steric and electronic effects on the diphosphine ligands. They concluded that the rate enhancement is electronic rather than steric in origin; however, such a QM/MM model with substituents on the diphosphine just included in the MM part may account only partially for electronic properties of ligands and their derivatives. By implications of steric or electronic effects, if one could stabilise the highest energy transition state (HETS) and destabilise the most abundant reaction intermediate (MARI), the overall barrier can be moderately reduced, accelerating the reaction rate. In follow-up studies, Zuidema et al., using the VWN/TZP/COSMO//VWN/TZP level of theory, studied ester versus polyketone formation at a Pd catalyst with the bis(dimethylphosphino)ethane ligand. As expected, they computed a lower barrier for ethene insertion into the Pd–acyl bond. However, the intramolecular methanolysis barriers were higher, even when increased the phosphine bulk in contradiction to the experimental observations.39
Donald et al. studied Pd–acyl methanolysis at the BP86/6-31G**/SDD level using 1,3-diphosphinopropane as the model system.100 The hemilabile dissociation of one of the phosphine moieties makes the system responsive to intra- and intermolecular methanolysis by increasing the acyl carbon's electrophilicity; however, the calculated barriers showed the possibility of inter- rather than intramolecular methanolysis. These are promising modelling studies for Pd-catalysed alcoholysis but do not cover the nature of alkoxycarbonylation versus copolymerisation and the steric effects of phosphine bulk.
Walther et al. studied Pd-catalysed methoxycarbonylation of cis-3-hexene using the B3LYP/TZVP/LANL2DZ//B3LYP/6-31G*/LANL2DZ level of theory with the 1,2-bis((dimethylphosphaneyl)methyl)benzene (DMBPX) ligand.101 They did not include dispersion or solvation corrections, although these may not be so critical in this case; however, they took the overall barrier, reported as 15.5 kcal mol−1, relative to the reactant complex, which is not appropriate for the whole reaction under turnover. Using Kozuch and Shaik's model,89,90 the Pd–acyl intermediate on their profile can be identified as the MARI and the transition state associated with the intramolecular methanolysis step as the HETS, taking the overall barrier to ∼37.5 kcal mol−1 (see Scheme S1 in the ESI‡ for more details). Such a high (essentially unsurmountable) barrier would indicate this is not the actual pathway followed experimentally.
Roesle et al. reported a Pd-catalysed mechanism for methoxycarbonylation of methyl 4-heptenoate with DTBPX and DMBPX ligands.102 All the energy profiles were calculated at the B3LYP/6-31G*/LANL2DZ level of theory. Although they refined the energies of selected species through single-point calculations with the TZVP basis set, they did not incorporate solvation effects or dispersion corrections, which are potentially important for the bulky ligand. While the relative activities of both ligands were assessed, no comparison between the methanolysis and competitive alkene insertion was made. Furthermore, the overall barrier of 29.1 kcal mol−1 for the intramolecular methanolysis step with the Pd–DTBPX catalyst does not match this system's high reactivity at low temperatures.
Beller et al. also suggested a mechanism for the Pd-catalysed methoxycarbonylation of ethene with DTBPX at the B3PW91-D3/TZVP/LANL2DZ/SMD level of theory.103 The key steps are the formation of an acyl complex followed by methanolysis. The overall barrier of ∼42.4 kcal mol−1 associated with the methanolysis step is insurmountable and contradictory with the experimental conditions.103 They also did not consider the copolymerisation leading to oligo-/polymers. The same study showed a notably lower methanolysis barrier (∼30.2 kcal mol−1) at Pd with 1,1′-bis(tert-butyl(pyridin-2-yl)phosphanyl)ferrocene ligand,103 which arises from hemilabile coordination of nitrogen to the Pd centre acting as a proton relay.
At the PBE0-D3/def2-TZVP/COSMO level of theory, Jameel et al. suggested a higher linear selectivity for Pd-catalysed methoxycarbonylation of methyl 10-undecenoate with the DTBPX ligand system.104 Again the authors report an overall barrier relative to the reactant complex (affording ∼18.9 kcal mol−1). However, when applying the energy span model,89,90 it turns out that the Pd–acyl complex should be taken as the MARI; increasing the overall barrier to ∼40.0 kcal mol−1 (see Scheme S2 in the ESI‡ for more details). The authors showed the effects of explicit solvation on the activation barriers for the methanolysis step in the gas phase, lowering the overall barrier from ∼34.7 kcal mol−1 to ∼29.9 kcal mol−1, still too high to be overcome under the reaction conditions.
Gallarati et al. performed mechanistic studies of Pd-catalysed methoxycarbonylation of styrene with chiral Phanephos diphosphines.105 They demonstrated the effect of explicit solvation on the methanolysis barrier at the B3PW91-D3/6-311+G**/SDD/PCM level of theory. Their methanolysis barriers with similar schemes turned to be much lower than those we recently published for a different system.98 These low barriers come from further destabilisation of the Pd–acyl complex due to greater steric bulk on the phosphines; however, off-cycle intermediates appeared to be the MARIs. In this study the MHP scheme87 was found to be crucial to account for the extent of reversibility of individual reaction steps, in order to account for the observations from deuterium labelling studies. While regio- and stereoselectivities for two ligands with different electronic properties were broadly compatible with experiment, the overall barriers were on the high side, in the range of 28–35 kcal mol−1.
Analysis of the regioselectivities turned out to be complicated by the fact that after olefin insertion the resulting linear and branched Pd–alkyl species (labelled as 3-L and 3-(R)/(S) in the original publication) can interconvert via β-hydride elimination at the Pd/(S)-Xylyl-Phanephos catalyst. Such interconversion is possible via a common intermediate 4-L (Scheme 4).105 The isomerisation barrier of 17.9 kcal mol−1 at the Pd/(S)-Xylyl-Phanephos catalyst is lower than the isomerisation barrier of 21.0 kcal mol−1 at the Pd/(S)-F24-Phanephos catalyst, making the latter more selective than the former.
 | ||
| Scheme 4 Isomerisation mechanism using Pd/(S)-Xylyl-Phanephos catalyst at the B3PW91-D3/6-311+G**/SDD/PCM level of theory. Labels R/S and L denote branched and linear products, respectively Energies are relative to intermediate 1 (not shown here). Adapted from ref. 105, reprinted with permission (Copyright 2020 American Chemical Society). | ||
We recently studied Pd-catalysed methoxycarbonylation of ethene with bidentate diphosphine ligands at the B3PW91-D3/SDD/6-311+G**/PCM//B3PW91/SDD/6-31G** level of theory using methanol as the model solvent.98 We use the same methodology in all work from us presented in this perspective. We explored three pathways, (i) carbomethoxy pathway, (ii) ketene pathway, and (ii) hydride–hydroxyalkylpalladium pathway. The names of these pathways come from the key intermediates appearing on the reaction profiles (see Scheme 3). By comparing the overall barriers of the three pathways, we suggest the hydroxyalkylpalladium cycle to be the one most likely to operate.
The hydroxyalkylpalladium cycle (C) starts with a Pd–H complex with two explicit solvent molecules coordinated to the Pd atom. Ethene uptake results in an ethylpalladium intermediate, which gives a Pd–propionyl intermediate after CO uptake and migratory insertion into the Pd–ethyl bond. Consistent with suggestions from the previous studies discussed above that intramolecular methanolysis is energetically challenging, we find an overall barrier of 38.2 kcal mol−1 for this process. It turned out that a route following an intermolecular methanolysis process via a hydroxyalkylpalladium intermediate (14˙MeOH in Scheme 5) is much more favourable, with an overall barrier of 23.0 kcal mol−1.
 | ||
| Scheme 5 Hydride–hydroxyalkylpalladium pathway (pathway C in the original paper): free energy profile using methanol as the model solvent at the B3PW91-D3/6-311G**/SDD/PCM level. Energies are in kcal mol−1 relative to 7˙2MeOH. Explicit solvent molecules are shown in light grey colour. The ligand DTBPX, is shown in Scheme 1(i). Reprinted from ref. 98 (Copyright 2020 Royal Society of Chemistry). | ||
We also studied the competing co-polymerisation side reaction. Experimentally it is known that bulky ligands are required to suppress this. Adding three explicit solvent molecules not only brings the overall barrier for MePro formation down to 17.8 kcal mol−1, but also improves the selectivity of MePro to 99.9% compared to copolymerization (Scheme 6). We further compared the steric bulk in DTBPX and DMBPX ligands. Less steric bulk at the phosphines of DMBPX stabilises the MARI, which increases the overall barrier, whereas enhanced steric clash in DTBPX destabilises the MARI, thus reducing the overall barrier (Fig. 1). These results are fully consistent with the experimental observations as increasing the bulk at the phosphines increases the selectivity towards esterification and the reaction rates.
 | ||
| Scheme 6 Methanolysis/copolymerisation selectivity: right – pathway C, leading to the production of MePro, left – copolymerisation leading to the formation of oligoketones or polymers. Energies in kcal mol−1 relative to 10˙2MeOH. Explicit solvent molecules are shown in light grey colour. The ΔΔG‡ of the selectivity determining transition states increases to 5.4 kcal mol−1 when an additional explicit methanol molecule is added. The ligand DTBPX, is shown in Scheme 1(i). Reprinted from ref. 98 (Copyright 2020 Royal Society of Chemistry). | ||
 | ||
| Fig. 1 Intermediates 11′ using DTBPX (left) and DMBPX (right) ligands. The enhanced steric clash in DTBPX ligand system is circled. Reprinted from ref. 98 (Copyright 2020 Royal Society of Chemistry). | ||
Palladium-catalysed alkoxycarbonylation of alkynes with chelating phosphine ligands
The modelling studies discussed so far cover alkoxycarbonylation of alkenes with bidentate diphosphine ligands, where bulk and backbone spacer's nature and length play an important role in selectivity towards esterification or copolymerisation. We now turn to alkoxycarbonylation of alkynes.Liu et al. designed a novel class of diphosphine ligands with pyridine substituents.107 Using 1,3-bis(di-tert-butylphosphino)propane ligand they obtained 1,4-dicarboxylic acid diester, with a yield and selectivity of 88%, whereas 1,5-bis(tert-butyl(pyridin-2-yl)phosphanyl)pentane gave a branched acrylate. Zhu et al. have performed mechanistic studies on the methoxycarbonylation of phenylacetylene using the two ligands at the M06-L/6-311+G**/SDD level of theory.108 Their pathways are comparable to those calculated by Ahmad et al., where hemilabile pyridyl rings facilitate the alkyne protonation and the methanolysis steps.109–112 They also identified a ketene intermediate on their path with 1,5-bis(tert-butyl(pyridin-2-yl)phosphanyl)pentane ligand, leading to the branched product previously suggested by us.111,112
In the early 1990s, Drent and co-workers reported that under mild conditions, Pd-catalysed propyne methoxycarbonylation with 2-pyridyldiphenylphosphine (2-PyPPh2) ligands attains a turnover of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mol (mol Pd) h−1 and selectivity of ≈99% towards MMA.20,21 These were encouraging results, promising a one-step rather than the present two-step synthesis of this product. Drent et al. initially proposed a carbomethoxy mechanism involving two 2-PyPPh2 ligands (Scheme 7 – right); however, Scrivanti et al. proposed a completely different mechanism as a result of isotope labelling studies.22 They suggested a proton transfer initiating the catalytic cycle of alkoxycarbonylation of alkynes from a protonated 2-PyPPh2 ligand onto the coordinated alkyne. This proton transfer results in formation of a Pd–vinyl species, which gives an alkyl Pd–acryloyl intermediate after the CO uptake and its migratory insertion into the Pd–vinyl bond. Finally, alcoholysis results in the product formation and regeneration of the catalyst (Scheme 7 – left).
000 mol (mol Pd) h−1 and selectivity of ≈99% towards MMA.20,21 These were encouraging results, promising a one-step rather than the present two-step synthesis of this product. Drent et al. initially proposed a carbomethoxy mechanism involving two 2-PyPPh2 ligands (Scheme 7 – right); however, Scrivanti et al. proposed a completely different mechanism as a result of isotope labelling studies.22 They suggested a proton transfer initiating the catalytic cycle of alkoxycarbonylation of alkynes from a protonated 2-PyPPh2 ligand onto the coordinated alkyne. This proton transfer results in formation of a Pd–vinyl species, which gives an alkyl Pd–acryloyl intermediate after the CO uptake and its migratory insertion into the Pd–vinyl bond. Finally, alcoholysis results in the product formation and regeneration of the catalyst (Scheme 7 – left).

Crawford et al. studied the mechanistic details of homogeneously catalysed methoxycarbonylation of propyne at the B3PW91-D3/6-311+G**/SDD/PCM level of theory.109,110 The proposed mechanism involving proton shuffling, with the hemilabile pyridyl groups, acting as in situ base113 in the initiation and termination steps (mechanism D, Scheme 8), appeared to be consistent with observed activities and selectivities instead of the initially suggested carbomethoxy mechanism. Mechanism D has three crucial stages, (i) the protonation of coordinated propyne, followed by (ii) thermodynamically favoured CO insertion, and (iii) terminating methanolysis. At the protonation step, the steric interaction between the aromatic ring of the ligand and the propyne's methyl group should govern the regioselectivity (pathway D).
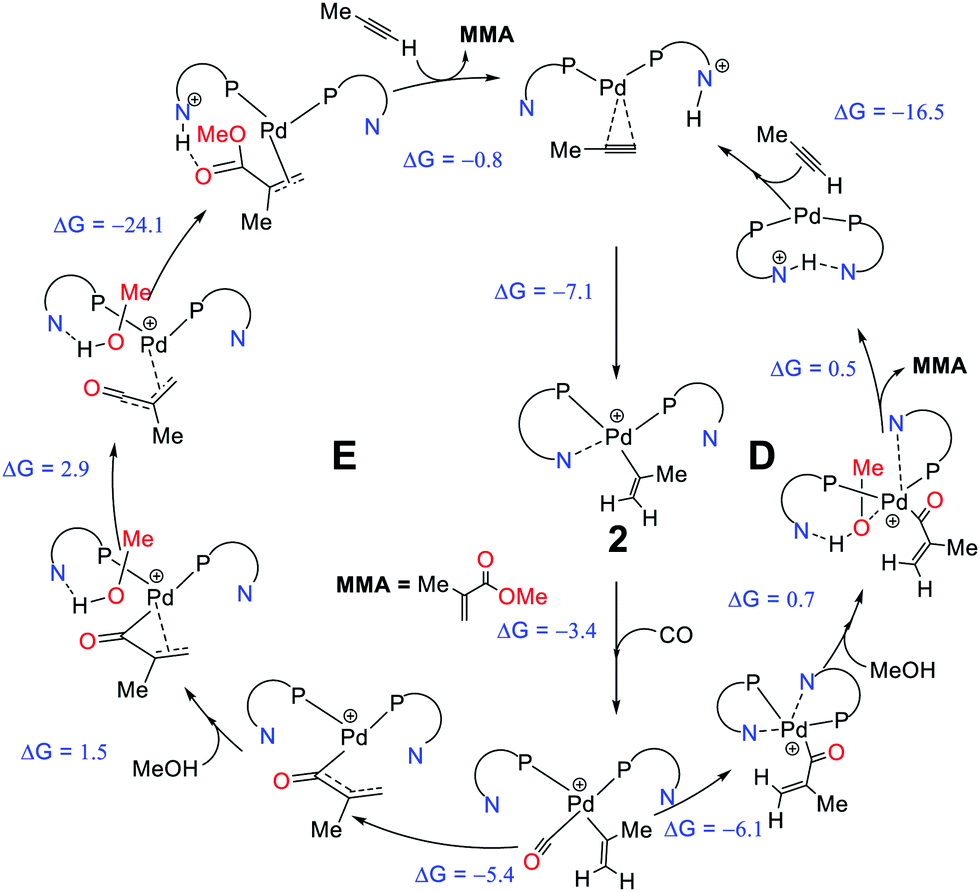 | ||
| Scheme 8 Comparison of the two pathways, pathway D109,110 and pathway E111 including the computed driving forces for elementary steps (ΔG298K in kcal mol−1). Reprinted from ref. 111 (Copyright 2019 Royal Society of Chemistry). | ||
Based on mechanism D, 2-(4-NMe2)-PyPPh2 ligand was predicted to increase the catalyst activity, but when it was synthesised and tested experimentally, it failed to produce the expected rate enhancement.111 Revisiting and revising the mechanism D at the same DFT level, ee identified new highly reactive acryloyl and ketene-type intermediates (affording new pathway E, Scheme 8). The presence of such intermediates on the reaction profile gave very low alcoholysis and low overall kinetic barriers (ΔG‡ = 16.8 kcal mol−1, overall barrier).111 Furthermore, this revised mechanism is now able to account for the reduced, rather than enhanced activity of the 2-(4-NMe2)-PyPPh2 ligand, because the proton-shuffling step that would have benefitted from a stronger in situ base is not rate-limiting.
We identified the protonation of coordinated propyne as the linear/branched selectivity determining step (see Scheme 9), similar to that proposed by Crawford et al.109,110 There are some minor changes in the conformational preferences. Still, these changes have no overall effect on the selectivity determining step. We found a ΔΔG‡ of 3.7 kcal mol−1 between the two kinetic barriers leading to the isomeric products. Since the reaction conditions are highly acidic, it is possible that a significant fraction of the catalyst has both the ligands protonated, rather than just one (as implicated in Schemes 7 and 8). We also considered such a dicationic pathway, where the calculated selectivity was slightly reduced, i.e., ΔΔG‡ = 2.9 kcal mol−1.111
 | ||
| Scheme 9 Selectivity determining steps on pathway E: pathways for formation of branched (right) and linear (left) products. The free energy difference between TS1a-1 and TS1-2L (ΔΔG‡) governs the selectivity. Adapted from ref. 111. | ||
In pathway E, we find the barriers controlling branched/linear selectivity comparable to pathway D (Scheme 9). On changing ligand from 2-PyPPh2 to 2-(6-Me)-PyPPh2 and 2-(6-Cl)-PyPPh2, the barrier for proton transfer to the central carbon of propyne increases which enhances the selectivity towards the branched product, i.e., MMA. On the other hand, theoretically, the 2-(6-Cl)-PyPPh2 ligand system decreases the overall barrier from 16.8 kcal mol−1 to 15.9 kcal mol−1, consistent with the experimental observations.111
 | ||
| Scheme 10 The proposed basis for ligand design: tolerance of the catalyst toward propadiene is expected if entry into the pathway leading to the formation of π-allyl palladium complex is blocked, i.e., if the ratio of kbr′/klin′ is maximised.112 Copyright Wiley-VCH, from Bühl et al. 2020.112 | ||
In line with these findings, the design of new, improved ligands should aim at maximising the branched: linear selectivity in the reaction with propadiene because it should only be the pathway leading to the linear product (which is the undesired one anyway) that is responsible for catalyst deactivation. Based on the (Py)PPh2 scaffold, we have explored a number of possible ligand variations, placing Cl, Me, Me2N, and NO2 substituents at various positions of the Py moiety (see ESI‡). Unfortunately, none of the derivatives considered so far is predicted to surpass the (6-Cl-3-Me-Py)PPh2 ligand in its branched: linear selectivity in the reaction with propadiene (see Table S1 in the ESI‡). However, the search for different ligand motifs could be rewarding. The (6-Cl-3-Me-Py)PPh2 ligand increases the difference of energies between the selectivity determining transition states (ΔΔG‡) from 1.3 kcal mol−1 (see ΔΔG‡ values in Table S1 in the ESI‡) to 4.0 kcal mol−1. It also decreases the overall barrier from 16.8 kcal mol−1 to 9.1 kcal mol−1. Accordant with these results, the (6-Cl-3-Me-Py)PPh2 ligand system is predicted to be more tolerant to propadiene and extremely efficient for alkoxycarbonylation of alkynes and allenes (Fig. 2). These findings are consistent with reports in the context of a patent.114 It appears, however, that even better selectivities will be required to produce catalysts with lifetimes long enough (i.e., turnover numbers high enough) to make them economically viable.
 | ||
| Fig. 2 Steric clash in TS9-10L+ (left) gives greater tolerance against allene poisoning. Copyright Wiley-VCH, from Bühl et al. 2020.112 | ||
Current challenges and outlook
In this perspective, we have discussed several DFT models used to study Pd-catalysed alkoxycarbonylation of alkenes and alkynes, where hybrid functionals, particularly B3LYP and B3PW91, tend to perform better than other, non-hybrid functionals. The results obtained including treatment of dispersion for bulky systems and continuum solvation models as well as inclusion explicit solvent molecules, are more realistic, i.e., close to the experiment.The modelling studies compare the overall barriers of the computed catalytic cycles to identify plausible pathways. This perspective includes a range of studies using different substrates and catalysts. The overall barriers from these studies support a hydride pathway for alkoxycarbonylation of alkenes with bidentate diphosphine ligands. There was a challenge to identify viable pathways for the final alcoholysis step (i.e., with barriers that would not be prohibitively high). The overall barrier associated with intramolecular alcoholysis is always unsurmountable at the turnover conditions. Pioneered by Macgregor and co-workers,99 several studies employed intermolecular alcoholysis to achieve lower overall barriers than intramolecular alcoholysis.
Computed regio and chemo-selectivities are essential indicators to gauge the credibility of a computed pathway. Because slight energy differences are usually involved, computation and eventual prediction of such selectivities remain a challenge. It is usually compounded by the conformational flexibility of realistic catalyst models. Intermolecular alcoholysis helps to lower the overall barrier; explicit inclusion of additional solvent molecules is usually required to explain regio and chemo-selectivities when modelling alkoxycarbonylation of alkenes with bidentate diphosphine ligands.
Studies on Pd-catalysed alkoxycarbonylation of alkynes with 2-PyPPh2 ligands still do not support a carbomethoxy mechanism. However, instead of a hydride pathway, in situ base mechanism is shown to be operating. In this mechanism, one or two Py moieties are protonated rather than the involvement of a Pd–H complex. Also, the latter complex is less stable than the former. Using the energy difference between the two transition states leading to branched and linear products, we designed new catalysts.111 Out of these, some catalysts have been tested experimentally, confirming the precision of our predictions.111 In this perspective, we further incorporated our study on the poisoning of Pd-catalyst with 2-PyPPh2 ligand system with allene. This study predicted the Pd-catalyst with (6-Cl-3-Me-Py)PPh2 ligand system as a highly active catalyst and tolerant towards allene poisoning. This work still has the scope to model several new catalysts, which could be further tolerant towards allene poisoning.
As the ligands get larger and more flexible, sampling the conformational phase space can become an issue. In principle, extensive conformational analysis should be carried out at each step of the reaction profile. or at least for the crucial steps, i.e., steps determining the overall barriers and selectivities. Attempts to automate such conformational searches are promising,115 but may reach their limits for shallow potential energy surfaces where transition states are notoriously difficult to locate. Another option for potentially unbiased conformational analysis is molecular dynamics (MD) simulations.116–119 Because all or part of the system will have to be described quantum-mechanically, such simulations will be limited by the short time scales that are usually affordable. Enhanced sampling techniques can be used,120,121 but will require bespoke set-up of the simulations for each new system (e.g. finding appropriate collective variables for metadynamics). Because rather small energetic differences can decide whether a catalyst is active or selective enough to be useful on a laboratory or even industrial scale, there will always be room for improvement on the computational modelling side.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MB gratefully acknowledges the collaborations with experimental colleagues from St Andrews and beyond in the field of homogeneous catalysis, which have inspired much of the work presented here, including (but not limited to) Matthew L. Clarke, D. J. Cole-Hamilton, Eite Drent, Paul. C. J. Kamer, Steve P. Nolan, Paul G. Pringle, Andrew D. Smith, and Robert Tooze. The hard work of many postgraduate students and postdoctoral fellows dedicated to modelling of homogeneous catalysis is also thankfully acknowledged, with special thanks (besides Shahbaz Ahmad, first author of this article) to L. Ellis Crawford, Maicon Delarmelina, Simone Gallarati, and Nicolas Sieffert. The work was supported by the School of Chemistry and EaStCHEM. Calculations were performed on a local compute cluster maintained by Dr H. Früchtl. The research data supporting this publication can be accessed at https://doi.org/10.17630/1ddd6203-1b51-457b-acf9-4a77ae2e93f5.Notes and references
- S. Caddick, Palladium reagents and catalysts: Innovation on organic synthesis, Wiley, Chichester, 1997, vol. 527 Search PubMed.
- P. W. N. M. van Leeuwen, Homogeneous catalysis: understanding the art, Kluwer, Dordrecht, 2005, vol. 2005 Search PubMed.
- C. Amatore, E. Carré, A. Jutand, M. A. M’Barki, B. O. Kneisel, R. Herbst-Irmer, M. A. Pellinghelli and A. Tiripicchio, Organometallics, 1995, 14, 1818–1826 CrossRef CAS.
- A. Flahaut, S. Roland and P. Mangeney, J. Organomet. Chem., 2007, 692, 5754–5762 CrossRef CAS.
- A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243–8246 CrossRef CAS PubMed.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- A. Jutand, J. Pytkowicz, S. Roland and P. Mangeney, Pure Appl. Chem., 2010, 82, 1393–1402 CAS.
- S. Demir, I. Özdemir, H. Arslan and D. VanDerveer, J. Organomet. Chem., 2011, 696, 2589–2593 CrossRef CAS.
- X. F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229–241 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed.
- G. M. Roberts, P. J. Pierce and L. K. Woo, Organometallics, 2013, 32, 2033–2036 CrossRef CAS.
- G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed.
- A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef.
- A. A. Núñez Magro, L. M. Robb, P. J. Pogorzelec, A. M. Z. Slawin, G. R. Eastham and D. J. Cole-Hamilton, Chem. Sci., 2010, 1, 723–730 RSC.
- C. Jimenez Rodriguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720–1721 RSC.
- A. Dervisi, P. G. Edwards, P. D. Newman, R. P. Tooze, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1999, 1113–1120 RSC.
- A. Dcrvisi, P. G. Edwards, P. D. Newman and R. P. Tooze, J. Chem. Soc., Dalton Trans., 2000, 523–528 RSC.
- C. S. Consorti, G. Ebeling and J. Dupont, Tetrahedron Lett., 2002, 43, 753–755 CrossRef CAS.
- A. Scrivanti, M. Bertoldini, V. Beghetto, U. Matteoli and A. Venzo, J. Organomet. Chem., 2009, 694, 131–136 CrossRef CAS.
- E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1993, 455, 247–253 CrossRef CAS.
- E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57–63 CrossRef CAS.
- A. Scrivanti, V. Beghetto, E. Campagna, M. Zanato and U. Matteoli, Organometallics, 1998, 17, 630–635 CrossRef CAS.
- D. Zargarian and H. Alper, Organometallics, 1991, 10, 2914–2921 CrossRef CAS.
- D. Zargarian and H. Alper, Organometallics, 1993, 12, 712–724 CrossRef CAS.
- M. Akao, S. Sugawara, K. Amino and Y. Inoue, J. Mol. Catal. A: Chem., 2000, 157, 117–122 CrossRef CAS.
- B. El Ali, J. Tijani and A. M. El-Ghanam, Tetrahedron Lett., 2001, 42, 2385–2387 CrossRef CAS.
- C. Grosjean, K. Novakovic, S. K. Scott, A. Whiting, M. J. Willis and A. R. Wright, J. Mol. Catal. A: Chem., 2008, 284, 33–39 CrossRef CAS.
- W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. L. Wang and K. Whiston, Chem. Commun., 1999, 1877–1878 RSC.
- G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609–610 RSC.
- W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Organometallics, 2002, 21, 1832–1840 CrossRef CAS.
- W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, J. Chem. Soc., Dalton Trans., 2002, 3300–3308 RSC.
- P. W. N. M. Van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523–5539 CrossRef CAS PubMed.
- J. Liu, B. T. Heaton, J. A. Iggo, R. Whyman, J. F. Bickley and A. Steiner, Chem. – Eur. J., 2006, 12, 4417–4430 CrossRef CAS PubMed.
- B. Harris, Ingenia, 2010, 18–23 Search PubMed.
- M. Moukwa, Chem. World, 2010, 50–52 Search PubMed.
- GlobeNewswire, https://www.globenewswire.com/news-release/2020/06/04/2044075/0/en/Amid-the-COVID-19-crisis-and-the-looming-economic-recession-the-Methyl-Methacrylate-MMA-market-worldwide-will-grow-by-a-projected-US-2-5-Billion-during-the-analysis-period.html, accessed 25 February 2021.
- Bulletin Line, https://bulletinline.com/2020/05/30/potential-impact-of-covid-19-on-a-latest-research-provides-insights-about-polymethyl-methacrylate-pmma-market/, accessed 25 February 2021.
- G. R. Eastham, R. P. Tooze, M. Kilner, D. F. Foster and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 8, 1613–1617 RSC.
- E. Zuidema, C. Bo and P. W. N. M. Van Leeuwen, J. Am. Chem. Soc., 2007, 129, 3989–4000 CrossRef CAS PubMed.
- E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663–681 CrossRef CAS PubMed.
- A. Dervisi, P. G. Edwards, P. D. Newman, R. P. Tooze, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1998, 3771–3776 RSC.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- E. Zuidema, P. W. N. M. Van Leeuwen and C. Bo, Organometallics, 2005, 24, 3703–3710 CrossRef CAS.
- J. P. Perdew and K. Burke, Int. J. Quantum Chem., 1996, 57, 309–319 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. Jacoby, in Chemical & Engineering News, ed. P. Kamer, D. Vogt and J. Thybaut, Royal Society of Chemistry, 2001, vol. 79, p. 11 Search PubMed.
- J. P. Perdew and A. Ruzsinszky, Int. J. Quantum Chem., 2010, 110, 2801–2807 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- O. A. von Lilienfeld, I. Tavernelli, U. Rothlisberger and D. Sebastiani, Phys. Rev. Lett., 2004, 93, 153004 CrossRef PubMed.
- U. Zimmerli, M. Parrinello and P. Koumoutsakos, J. Chem. Phys., 2004, 120, 2693–2699 CrossRef CAS PubMed.
- M. Piacenza and S. Grimme, J. Am. Chem. Soc., 2005, 127, 14841–14848 CrossRef CAS PubMed.
- M. Piacenza and S. Grimme, ChemPhysChem, 2005, 6, 1554–1558 CrossRef CAS PubMed.
- J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 5287–5293 RSC.
- R. Peverati and K. K. Baldridge, J. Chem. Theory Comput., 2008, 4, 2030–2048 CrossRef CAS PubMed.
- M. Pavone, N. Rega and V. Barone, Chem. Phys. Lett., 2008, 452, 333–339 CrossRef CAS.
- Y. S. Lin, G. De Li, S. P. Mao and J. Da Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- M. K. Kesharwani and J. M. L. Martin, Theor. Chem. Acc., 2014, 133, 1–14 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- G. Frenking, I. Antes, M. Böhme, S. Dapprich, A. W. Ehlers, V. Jonas, A. Neuhaus, M. Otto, R. Stegmann, A. Veldkamp and S. F. Vyboishchikov, Rev. Comput. Chem., 2007, 63–144 Search PubMed.
- M. Dolg and X. Cao, Chem. Rev., 2012, 112, 403–480 CrossRef CAS PubMed.
- P. O. Löwdin, Phys. Rev., 1955, 97, 1509–1520 CrossRef.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 720–723 CrossRef.
- W. J. Hehre, K. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921–2923 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. L. Martin, P. J. Hay and L. R. Pratt, J. Phys. Chem. A, 1998, 102, 3565–3573 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 63–71 CrossRef.
- S. Kozuch, S. E. Lee and S. Shaik, Organometallics, 2009, 28, 1303–1308 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Chem. Phys., 2001, 114, 5691–5701 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. Suleiman, V. Polo and B. El Ali, RSC Adv., 2016, 6, 8440–8448 RSC.
- H. Li, K. Dong, H. Jiao, H. Neumann, R. Jackstell and M. Beller, Nat. Chem., 2016, 8, 1159–1166 CrossRef CAS PubMed.
- S. Ahmad, L. E. Crawford and M. Bühl, Phys. Chem. Chem. Phys., 2020, 22, 24330–24336 RSC.
- S. A. Macgregor, G. W. Neave and C. Smith, Faraday Discuss., 2003, 124, 111–127 RSC.
- S. M. A. Donald, S. A. Macgregor, V. Settels, D. J. Cole-Hamilton and G. R. Eastham, Chem. Commun., 2007, 562–564 RSC.
- G. Walther, L. R. Knöpke, J. Rabeah, M. P. Checiński, H. Jiao, U. Bentrup, A. Brückner, A. Martin and A. Köckritz, J. Catal., 2013, 297, 44–55 CrossRef CAS.
- P. Roesle, L. Caporaso, M. Schnitte, V. Goldbach, L. Cavallo and S. Mecking, J. Am. Chem. Soc., 2014, 136, 16871–16881 CrossRef CAS PubMed.
- K. Dong, R. Sang, Z. Wei, J. Liu, R. Dühren, A. Spannenberg, H. Jiao, H. Neumann, R. Jackstell, R. Franke and M. Beller, Chem. Sci., 2018, 9, 2510–2516 RSC.
- F. Jameel, E. Kohls and M. Stein, ChemCatChem, 2019, 11, 4894–4906 CrossRef CAS.
- S. Gallarati, P. Dingwall, J. A. Fuentes, M. Bühl and M. L. Clarke, Organometallics, 2020, 39, 4544–4556 CrossRef CAS.
- R. Suleiman, A. Ibdah and B. El Ali, J. Organomet. Chem., 2011, 696, 2355–2363 CrossRef CAS.
- J. Liu, K. Dong, R. Franke, H. Neumann, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2018, 140, 10282–10288 CrossRef CAS PubMed.
- L. Zhu, L. J. Liu, Y. Y. Jiang, P. Liu, X. Fan, Q. Zhang, Y. Zhao and S. Bi, J. Org. Chem., 2020, 85, 7136–7151 CrossRef CAS PubMed.
- L. E. Crawford, D. J. Cole-Hamilton, E. Drent and M. Bühl, Chem. – Eur. J., 2014, 20, 13923–13926 CrossRef CAS PubMed.
- L. E. Crawford, D. J. Cole-Hamilton and M. Bühl, Organometallics, 2015, 34, 438–449 CrossRef CAS.
- S. Ahmad, A. Lockett, T. A. Shuttleworth, A. M. Miles-Hobbs, P. G. Pringle and M. Bühl, Phys. Chem. Chem. Phys., 2019, 21, 8543–8552 RSC.
- S. Ahmad and M. Bühl, Chem. – Eur. J., 2019, 25, 11625–11629 CrossRef CAS PubMed.
- Under the acidic reaction conditions the “in situ base” is protonated preferentially. After transferring the proton to the substrate, the base is ready to accept a proton from the incoming methanol during the methanolysis step.
- E. Drent, W. W. Jager and J. C. L. J. Suykerbuyk, WO 95/05357, 1995.
- Y. Guan, V. M. Ingman, B. J. Rooks and S. E. Wheeler, J. Chem. Theory Comput., 2018, 14, 5249–5261 CrossRef CAS PubMed.
- R. J. Meier, O. M. Aagaard and F. Buda, J. Mol. Catal. A: Chem., 2000, 160, 189–197 CrossRef CAS.
- P. Vidossich, A. Lledós and G. Ujaque, Acc. Chem. Res., 2016, 49, 1271–1278 CrossRef CAS PubMed.
- Y. Luo, Z. Huang, Z. Chen, Z. Xu, J. Meng, H.-Y. Li, Q. Meng and D. Tang, J. Org. Chem., 2020, 85, 11626–11634 CrossRef CAS PubMed.
- M. S. Teynor, R. Carlsen and D. H. Ess, Organometallics, 2020, 39, 1393–1403 CrossRef CAS.
- A. Urakawa, M. Iannuzzi, J. Hutter and A. Baiker, Chem. – Eur. J., 2007, 13, 6828–6840 CrossRef CAS PubMed.
- M. Schilling, R. A. Cunha and S. Luber, ACS Catal., 2020, 10, 7657–7667 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Prof. Paul C. J. Kamer. |
| ‡ Electronic supplementary information (ESI) available: Computational details and results of previously unpublished calculations. See DOI: 10.1039/d1cp02426d |
| This journal is © the Owner Societies 2021 |