
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin–spin interactions and spin delocalisation in a doped organic semiconductor probed by EPR spectroscopy†
Claudia E.
Tait
 *ab,
Anna
Reckwitz
a,
Malavika
Arvind
c,
Dieter
Neher
c,
Robert
Bittl
a and
Jan
Behrends
*a
*ab,
Anna
Reckwitz
a,
Malavika
Arvind
c,
Dieter
Neher
c,
Robert
Bittl
a and
Jan
Behrends
*a
aDepartment of Physics, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany. E-mail: claudia.tait@chem.ox.ac.uk; j.behrends@fu-berlin.de
bPhysical and Theoretical Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, OX1 3QZ Oxford, UK
cInstitute of Physics and Astronomy, Universität Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany
First published on 14th June 2021
Abstract
The enhancement and control of the electrical conductivity of organic semiconductors is fundamental for their use in optoelectronic applications and can be achieved by molecular doping, which introduces additional charge carriers through electron transfer between a dopant molecule and the organic semiconductor. Here, we use Electron Paramagnetic Resonance (EPR) spectroscopy to characterise the unpaired spins associated with the charges generated by molecular doping of the prototypical organic semiconductor poly(3-hexylthiophene) (P3HT) with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) and tris(pentafluorophenyl)borane (BCF). The EPR results reveal the P3HT radical cation as the only paramagnetic species in BCF-doped P3HT films and show evidence for increased mobility of the detected spins at high doping concentrations as well as formation of antiferromagnetically coupled spin pairs leading to decreased spin concentrations at low temperatures. The EPR signature for F4TCNQ-doped P3HT is found to be determined by spin exchange between P3HT radical cations and F4TCNQ radical anions. Results from continuous-wave and pulse EPR measurements suggest the presence of the unpaired spin on P3HT in a multitude of environments, ranging from free P3HT radical cations with similar properties to those observed in BCF-doped P3HT, to pairs of dipolar and exchange-coupled spins on P3HT and the dopant anion. Characterisation of the proton hyperfine interactions by ENDOR allowed quantification of the extent of spin delocalisation and revealed reduced delocalisation in the F4TCNQ-doped P3HT films.
1 Introduction
Improvement of the electrical conductivity of organic semiconductors through controlled and stable doping is a crucial requirement for their widespread use in optoelectronic devices.1 The basic idea behind molecular doping of organic semiconductors is the introduction of additional mobile charge carriers by electron transfer from a donor molecule for n-type doping, or to an acceptor molecule for p-type doping. However, the exact mechanistic details of the doping process and their dependence on the properties of the organic semiconductor material and the molecular dopant, as well as on processing conditions, are still not fully understood. Two different mechanisms have been invoked to describe molecular doping of organic semiconductors: integer charge transfer (ICT), involving complete transfer of one electron between host and dopant molecules to generate a charge carrier on the host and an ionised dopant molecule, and charge-transfer complex (CTC) formation, based on an overall electrically neutral ground-state charge-transfer complex of host and dopant that requires additional thermal or optical excitation to lead to charge carriers on the host.2–4 While it was initially believed that doping proceeds either via ICT or CTC formation for a given host-dopant pair, there is now mounting evidence for the concurrent presence of both mechanisms for organic semiconductors based on π-conjugated polymers, with the prevalence of one or the other determined by the local morphology of the polymer chain, and thus dependent on processing conditions.5–7Efficient doping not only requires the ICT mechanism to prevail, but also relies on the separation of the host-dopant ion pair to generate free mobile charge carriers in the host matrix.3,8 In contrast to inorganic semiconductors, organic semiconductors are characterised by a low permittivity and therefore Coulomb interactions between the ionised dopant molecules and the charge generated on the polymer backbone can prevent charge separation and impede charge transport through localisation of charge carriers in the potential well of the dopant ion.9–11
Since successful doping of organic semiconductors leads to an unpaired electron on the host and typically also on the dopant molecule, Electron Paramagnetic Resonance (EPR) spectroscopy can be used for an accurate determination of charge carrier concentrations as well as for the characterisation of the paramagnetic species generated by doping and their interactions with the molecular environment. The potential of this technique in the characterisation of unpaired electrons in conductive polymers has been recognised early on,12–14 and over time the development of more advanced EPR methods has enabled new insights to be gained, for example through the determination of the extent of spin delocalisation on doped polymers.7,15,16
In this work, we report a comparative EPR study of the prototypical electron donor polymer poly(3-hexylthiophene) (P3HT) doped with the widely investigated π-electron acceptor 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ)17 and the Lewis acid tris(pentafluorophenyl)borane (B(C6F5)3, BCF)18–20 (see Fig. 1).
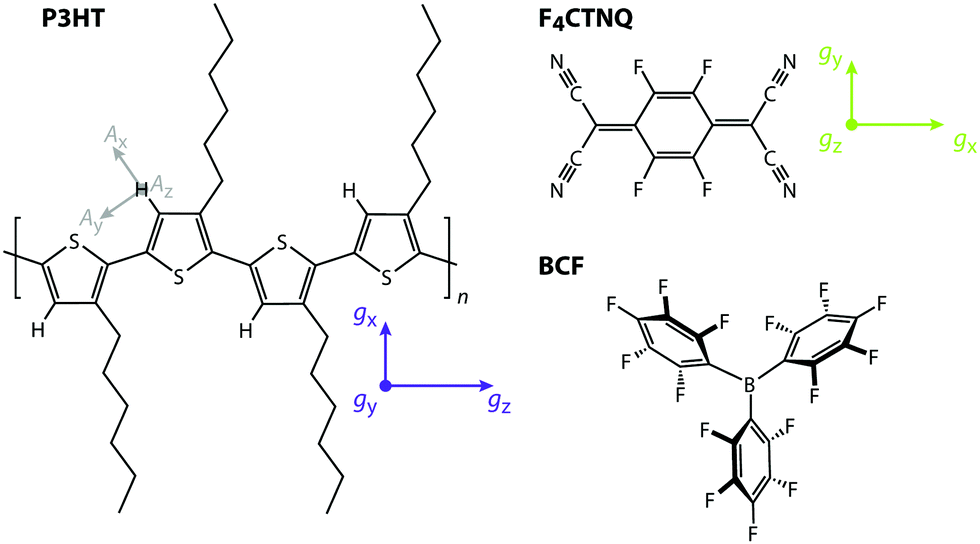 | ||
| Fig. 1 Molecular structures of the polymer poly(3-hexylthiophene) (P3HT) and the dopants 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) and tris(pentafluorophenyl)borane (B(C6F5)3, BCF). The principal axes of the g-matrix are indicated for P3HT and F4TCNQ, as well as the principal hyperfine axes for the proton on the P3HT backbone. | ||
Doping of P3HT with F4TCNQ has previously been demonstrated to occur mostly via ICT with a high ionization efficiency of around 50–75%17,19,21 determined from the F4TCNQ radical anion contribution to the UV-vis absorption spectrum. However, a large discrepancy between the amount of ionised F4TCNQ molecules and the number of mobile charge carriers determined by admittance measurements was observed and attributed to a significant fraction of bound hole-F4TCNQ anion pairs.10,17,22 X-ray diffraction studies revealed structural changes in the crystalline regions of P3HT upon doping that were initially interpreted in terms of intercalation of the planar F4TCNQ molecules between the π–π-stacked thiophene backbones.23,24 Later studies however concluded that the F4TCNQ anions reside between the P3HT side chains instead.25–28 Hamidi-Sakr et al. demonstrated an orthogonal arrangement of the transition dipole moments of P3HT and the F4TCNQ anion by polarised UV-vis-NIR spectroscopy,29 further supporting this conclusion and providing evidence for an arrangement of the F4TCNQ anions with the long molecular axis perpendicular to the polymer backbone. The lower energy polaron absorption peak P1 in optical absorption spectra of doped P3HT, assigned to the transition from the valence band to the lower unoccupied intragap state of a P3HT cation and typically centred in the range from 0.3 to 0.5 eV, is known to be sensitive to changes in polaron delocalisation. Scholes et al. modelled the position and inhomogeneous broadening of this peak as a function of the distance between the polymer backbone and a negative point charge representing the dopant anion and causing localisation of the polaron, and showed that the experimental results were consistent with a distance of 6–8 Å between the F4TCNQ anion and the polymer backbone.25 While doping of P3HT with F4TCNQ proceeds mostly by ICT, several recent studies have provided evidence for the coexistence of ICT and CTC formation, with a ratio depending on the microstructure of the film, and therefore processing conditions. In crystalline regions of the film, doping was shown to proceed preferentially via ICT, while increased CTC formation was found in amorphous regions, where the polymer backbones are twisted out of planarity and characterised by a larger degree of disorder.5,6,30
A significantly different behaviour was observed for P3HT films doped with the bulky Lewis acid tris(pentafluorophenyl)borane (B(C6F5)3, generally referred to as BCF), which, for films prepared by mixed-solution doping, exhibited higher hole mobilities and conductivities compared to the corresponding F4TCNQ-doped films.19 A detailed investigation of the doping of poly[2,6-(4,4-bis(2-hexadecyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,6(2,1,3-benzothiadiazole)] (PCPDTBT) with BCF recently led to the proposal of a multi-step doping mechanism involving formation of a BCF·OH2 adduct, which acts as a Brønsted acid and protonates the polymer backbone at a thiophene ring, followed by electron transfer from a neutral polymer segment to the protonated segment to generate a radical cation on the former and a neutral protonated radical on the latter.20 In a recent paper we have shown that results obtained for BCF-doped P3HT in solution are consistent with this general mechanism, but the absence of any EPR signatures beyond that of the P3HT radical cation suggested further reaction of the radical on the protonated chain, likely leading to elimination of H2 and leaving only neutral P3HT chains in addition to the P3HT radical cation (see Fig. S1 in the ESI† for a schematic representation of the proposed doping mechanism).7 A recent computational study confirmed that H2 loss resulting in the formation of a single spin-carrying charged species is required to drive the doping reaction with BCF to completion, and additionally revealed that in order for the protonation reaction to occur, the resulting counterion needs to be stabilised by formation of a bridged complex involving two BCF molecules ([BCF(OH)BCF]− or [BCF(OH)(OH2)BCF]−).31
The presence of only a single paramagnetic species, the P3HT radical cation, for BCF-doped P3HT, and of both P3HT˙+ and F4TCNQ˙− for F4TCNQ-doped P3HT provides the ideal opportunity for an investigation of the effect of the charged dopant radical on the properties of the spin centres introduced on the organic semiconductor.
In this study, we first identify and assign the spectral signatures of the paramagnetic species generated by doping of P3HT with BCF and F4TCNQ by continuous wave X- and W-band EPR. We investigate the effect of increased spin concentration on the properties of the spin centres on the P3HT backbone by comparing P3HT films with different doping ratios. Additionally, to help distinguish effects due to different dopants from differences resulting solely from changes in morphology, we also compare P3HT films generated by solution processing from two commonly used solvents, o-dichlorobenzene and chloroform. Temperature-dependent cw EPR measurements are performed to investigate dynamics of the spins associated with the charge carriers in the doped P3HT films and to verify the presence of interactions between spin centres. We then go on to use a series of advanced pulse EPR techniques at Q-band to gain additional insights into the interactions of the spins with their molecular environment by characterisation of the electron-nuclear hyperfine interactions. We use 1H ENDOR measurements to quantify the extent of spin delocalisation on the P3HT backbone and 19F ENDOR and 14N ESEEM measurements to identify contributions of the dopant radical anion F4TCNQ˙− to the pulse EPR spectra. Overall, our in-depth characterisation of electron–electron and electron–nuclear hyperfine interactions provides insights into carrier-dopant and carrier–carrier interactions as well as charge carrier delocalisation and attempts to unravel their dependence on the nature of the dopant as well as the microscopic order of the organic semiconductor matrix.
2 Results
2.1 Quantitative X-band cw EPR at room temperature
P3HT films doped with either BCF or F4TCNQ, deposited on the inner walls of an EPR tube by solvent evaporation from mixed solutions of host and dopant in o-dichlorobenzene (oDCB) or in chloroform (CF), were initially characterised by room temperature continuous wave (cw) EPR spectroscopy. The EPR spectra obtained for doping ratios in the range from 10−3 to 10−1 dopant molecules per thiophene unit are compared in Fig. 2 and show clear differences for films obtained using different dopants and solvents (see also Fig. S2 in the ESI† for a more detailed comparison of the spectral shapes).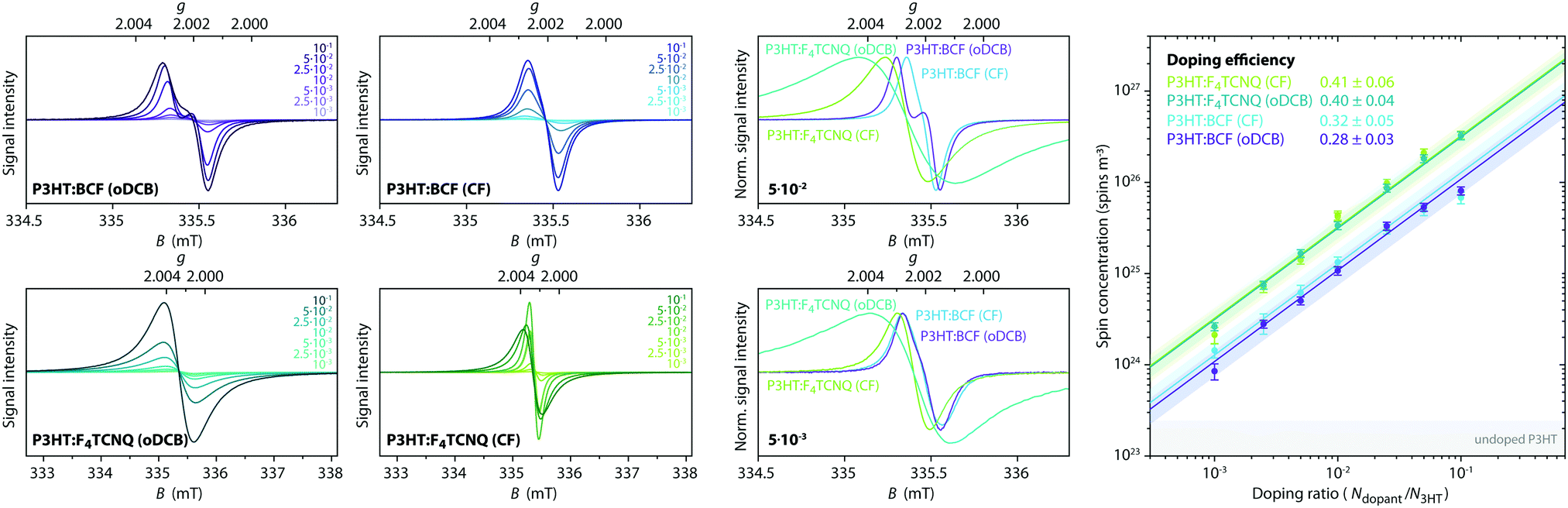 | ||
| Fig. 2 X-band room temperature cw EPR data for BCF- and F4TCNQ-doped P3HT films prepared by solution processing from o-dichlorobenzene (oDCB) or chloroform (CF). (left) Comparison of EPR spectra for each dopant–solvent pair at different doping ratios (note the difference by a factor three in the width of the displayed field range for the BCF- and F4TCNQ-doped samples) and direct comparison of normalised EPR spectra for different dopant–solvent combinations at a low and a high doping ratio. (right) Spin concentration as a function of doping ratio, for BCF-doped P3HT this corresponds to the concentration of P3HT radical cations, for F4TCNQ-doped P3HT it includes the contribution from F4TCNQ˙− (see text for details). The doping efficiencies are determined from the displayed fits. The grey shaded region indicates typical spin concentrations determined for undoped P3HT films. | ||
The BCF-doped P3HT films are characterised by EPR spectra centred at a g-value of 2.0021. For films prepared from CF, the spectra consist of a single approximately Gaussian line with a peak-to-peak linewidth that decreases from 0.25 mT for the lowest doping ratio to 0.18 mT for the highest. The spectra for BCF-doped P3HT films prepared from oDCB are almost identical to those of the films prepared from CF for doping ratios below 10−2, while for more highly doped films an increasingly pronounced additional feature appears close to the zero-crossing point.
The spectra recorded for the F4TCNQ-doped P3HT films are characterised by predominantly Lorentzian lineshapes and, in the case of the films prepared from oDCB as well as highly doped films prepared from CF, are significantly broader compared to those of BCF-doped P3HT films. For films prepared from both solvents, the position of the EPR line shifts from a g-value of about 2.0022 to about 2.0027 for increasing doping ratios. The spectra of F4TCNQ-doped P3HT films prepared from CF initially narrow for increasing doping ratios up to 10−2 and then broaden significantly from a peak-to-peak linewidth of 0.15 mT to 0.30 mT. The films prepared from oDCB broaden from a peak-to-peak linewidth of about 0.30 mT for the lowest doping ratio up to 0.50 mT for a doping ratio of 5⋅10−2.
The cw EPR spectra of doped P3HT films show strong similarities with the spectra obtained for the corresponding solutions (see Fig. S3 in the ESI†). In a previous investigation of P3HT doping in solution, we assigned the EPR spectra observed for BCF-doped P3HT to the P3HT radical cation, while the observed shift in g-value for F4TCNQ-doped P3HT was attributed to exchange interactions between P3HT radical cations and F4TCNQ radical anions.7
The spin concentration determined from the double integral of the recorded EPR spectra is shown as a function of doping ratio in Fig. 2 and increases linearly across almost the complete range of investigated doping concentrations. The doping efficiency η, defined as the number of radical cations generated on P3HT per added dopant molecule was determined from a linear fit within this region (NP3HT spins = η Ndopants). Assuming that the P3HT radical cation is the only contribution to the EPR spectra recorded for the BCF-doped P3HT films, doping efficiencies of 0.28 ± 0.03 and 0.32 ± 0.05 are obtained for films prepared from oDCB and CF, respectively. Based on the assignment of the EPR spectra obtained for F4TCNQ-doped P3HT films to exchange-coupled P3HT and F4TCNQ, the spin concentration determined for the F4TCNQ-doped P3HT films was assumed to be due in equal parts to the P3HT and F4TCNQ radicals. This assumption was previously validated by the excellent agreement of ionisation efficiencies determined by UV-vis-NIR spectroscopy with doping efficiencies measured by EPR for doped P3HT solutions.7 The resulting doping efficiencies for the F4TCNQ-doped P3HT films prepared from oDCB and CF are therefore 0.40 ± 0.04 and 0.41 ± 0.06, respectively. The determined doping efficiencies are in good agreement with previous results from optical spectroscopy, admittance measurements and quantitative EPR on BCF- and F4TCNQ-doped P3HT films that found 20–70% of dopants leading to P3HT polarons and 5–18% leading to mobile holes, with some dependence on the P3HT batch used.17,19
A qualitative comparison of the conductivity for the different samples based on the reduction in the resonator quality factor (Q) for increasing doping concentrations is also consistent with trends in conductivity reported in the literature (see Fig. S4 in the ESI†). Lower Q-values observed for BCF-doped P3HT films compared to F4TCNQ-doped films with similar concentrations of P3HT radical cations indicate increased conductivity for the BCF-doped films, in agreement with previous observations of ≈3 times higher conductivity for BCF-compared to F4TCNQ-doped films,19 and the lower Q-values for F4TCNQ-doped films prepared from CF compared to oDCB are in line with the increased conductivity previously determined for films prepared from CF compared to chlorobenzene.32
2.2 W-band cw EPR measurements
The increased g-resolution for EPR measurements at W-band frequencies was exploited to gain additional insight into the nature of the paramagnetic species observed in the doped P3HT films and to provide further evidence for the spectral assignments.The P3HT radical cation is known to be characterised by an orthorhombic g-matrix15,33 and W-band measurements on BCF-doped P3HT films prepared from oDCB (Fig. 3, left) clearly display features at the g-values reported in the literature for I2-doped drop-cast P3HT films (gx = 2.0029, gy = 2.0019 and gz = 2.0011).15 However, the relative intensities of the observed features do not match the spectrum expected in the presence of randomly oriented P3HT radical cations, but indicate partial alignment of the polymer backbones in the film samples. This was confirmed by comparison to measurements on powders obtained from drop-cast films, shown in grey in Fig. 3.
 | ||
| Fig. 3 (top) X- and W-band cw EPR spectra for films of BCF-doped P3HT prepared from oDCB at doping ratios of 10−2 and 2.5⋅10−2 and corresponding simulations performed assuming partial alignment of the P3HT backbones (dashed lines). Spectra recorded on the corresponding powders are shown in grey in the background. The distribution of gz orientations with respect to the sample frame considered in the simulations are shown in the insets. (bottom) Schematic representation of the doped P3HT films deposited on the inside of an EPR tube and orientation of the P3HT g-matrix with respect to the sample tube for two possible edge-on orientations of the P3HT film. | ||
Solution processed P3HT films consist of crystalline domains embedded in an amorphous matrix with a preference for an edge-on orientation of the crystallites with respect to the substrate.25,34 The reduced intensity of the features at gy and gz with respect to the feature corresponding to gx, which based on DFT calculations33 corresponds to the direction of the alkyl chains, would be in agreement with an edge-on orientation of P3HT with respect to the walls of the EPR tube (Fig. 3, bottom). This is confirmed by simulations of the W-band EPR spectra of the BCF-doped P3HT films that further indicate a slight preferential orientation of the gz axis, corresponding to the long axis of the P3HT backbone,15 along the sample tube axis (see insets in Fig. 3).
The presence of partial alignment also explains the feature appearing at the centre of the EPR line in the X-band spectra of highly BCF-doped P3HT films prepared from oDCB. The orientational distribution functions obtained in this case show some similarities with the ones obtained for the W-band data, in particular at the higher doping ratio; they are however significantly narrower and the spectra contain reduced contributions from the extreme values of the g-matrix (leading to distributions with gz centred at around 40° from the sample axis for both the W-band data at the doping ratio of 2.5⋅10−2 and the X-band data). These differences are likely a result of motional averaging due to hopping of the hole across crystallites with different orientations with respect to the magnetic field at a rate that leads to increased averaging at X- compared to W-band (due to the different relative magnitude of the inverse of the correlation time compared to the spectral anisotropy at the two frequency bands35), as well as more significant averaging at higher doping concentrations due to increased mobility (vide infra).
Further evidence for the presence of motional averaging is provided by doping ratio and temperature-dependent W-band EPR measurements on powders of BCF-doped P3HT films prepared from oDCB (see Fig. 4). At low doping concentrations, the spectral shapes with principal g-values corresponding to gx = 2.0029, gy = 2.0020 and gz = 2.0010 are independent of temperature and in excellent agreement with literature on I2-doped P3HT (gx = 2.0029, gy = 2.0019 and gz = 2.0011 for drop-cast films at 100 K).15 At doping ratios above 10−2, the room temperature spectra clearly show partial averaging of the g-anisotropy, suggesting fast hole hopping at rates exceeding the difference in electron Zeeman interaction between P3HT chains with different orientations with respect to the magnetic field. The g-anisotropy is almost fully recovered at 100 K, indicating a decrease in mobility with decreasing temperature, in agreement with a corresponding decrease in conductivity inferred from the changes in resonator Q-values with decreasing temperature (Fig. S4 in the ESI†).
 | ||
| Fig. 4 W-band cw EPR spectra recorded at room temperature, 200 K and 100 K for powders of BCF- and F4TCNQ-doped P3HT films at different doping ratios (prepared from oDCB). Reference spectra for the P3HT radical cation (BCF-doped P3HT with a doping ratio of 5⋅10−3 at 100 K) and for F4TCNQ˙− (film sample prepared from a solution of F4TCNQ in THF) are shown at the bottom. The grey vertical lines indicate the principal g-values determined for P3HT by simulation. | ||
Comparison of the W-band EPR data for BCF-doped and F4TCNQ-doped P3HT films provides clear evidence for the correct interpretation of the X-band data in terms of exchange-coupled P3HT and F4TCNQ. Fig. 4 shows the W-band cw EPR spectra of F4TCNQ-doped P3HT powder samples prepared from oDCB as well as reference spectra for P3HT˙+ (obtained for BCF-doped P3HT) and F4TCNQ˙− (measured on a film of partially ionised F4TCNQ prepared from THF). In contrast to the results for BCF-doped P3HT films, the spectra are characterised by a single line centred at a g-value of about 2.0027 independent of doping ratio and on whether the sample consists of a powder or a film. An averaging of the g-matrices characteristic of two different spin centres is expected in the presence of an exchange interaction J exceeding the difference in the electron Zeeman interactions giμBB, i.e. |J| ≫ |g1 − g2|μBB,36 and has previously been reported for organic charge-transfer salts.37,38 The observation of an EPR line centred at a g-value intermediate between those of P3HT˙+ (gx = 2.0029, gy = 2.0020, gz = 2.0010) and F4TCNQ˙− (gx = 2.0030, gy = 2.0030, gz = 2.0024, see Section S2.7 in the ESI†) both at X- and W-band supports the presence of exchange interactions between the spins on host and dopant and places a lower limit of about 100 MHz on J.
2.3 Temperature-dependent X-band cw EPR measurements
In order to gain further insights into spin–spin interactions in doped P3HT, temperature-dependent X-band cw EPR measurements were performed and the results are shown in Fig. 5. A comparison of EPR spectra recorded at different temperatures reveals increasing line narrowing for increasing temperatures for almost all of the considered samples. This would suggest motional and/or exchange narrowing to be the main mechanism determining the observed linewidth. An exception to this is the BCF-doped P3HT film prepared from oDCB with the highest doping concentration (0.1 dopants per thiophene unit), where the EPR line broadens and becomes less structured as the temperature increases, likely indicating a contribution of lifetime broadening due to fast relaxation. A different behaviour is also displayed for the F4TCNQ-doped P3HT films prepared from oDCB, where the linewidth remains almost invariant across the whole temperature range from 6 K to room temperature. The Lorentzian lineshape of these spectra, as well as the significant broadening compared to the other film samples, might in this case be determined by lifetime broadening.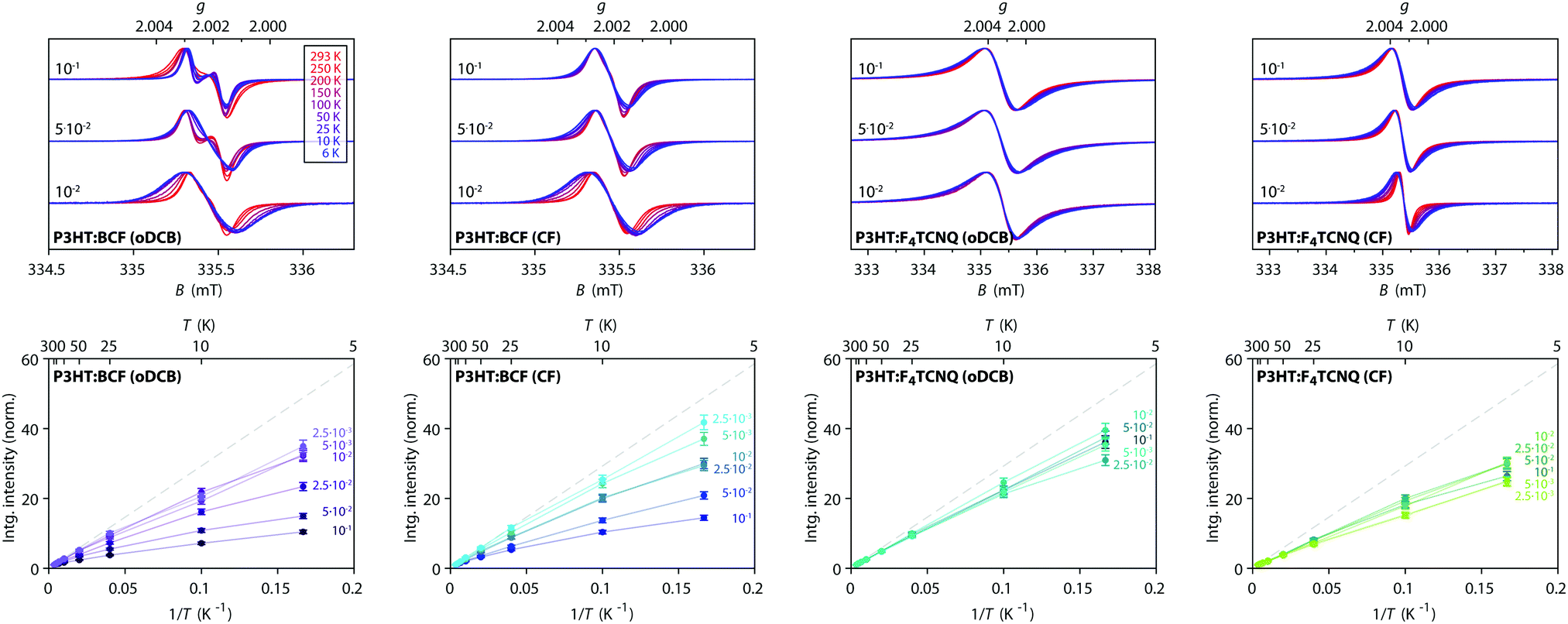 | ||
Fig. 5 Temperature-dependent X-band cw EPR measurements on BCF- and F4TCNQ-doped P3HT films. (top) Comparison of EPR spectra recorded at different temperatures within the range from 6 K to room temperature for selected doping ratios (the displayed spectra are normalised to the maximum to highlight changes in spectral shape, see Fig. S5 in the ESI† for the full set of spectra). (bottom) Relative integrated EPR intensity (double integral, normalised to the room temperature value) as a function of temperature for samples with different doping ratios. The dashed line indicates the Curie law dependence  expected for isolated expected for isolated  spin centres. spin centres. | ||
The change in EPR signal intensity as a function of temperature deviates from the Curie-law behaviour expected for isolated spin centres  for all of the measured samples and indicates the presence of significant spin–spin interactions. The relative change in integrated intensity for decreasing temperatures is displayed with respect to the inverse of the measurement temperature in Fig. 5.
for all of the measured samples and indicates the presence of significant spin–spin interactions. The relative change in integrated intensity for decreasing temperatures is displayed with respect to the inverse of the measurement temperature in Fig. 5.
The reversible decrease in spin concentration with decreasing temperatures observed for the BCF-doped P3HT films indicates formation of spin pairs with an antiferromagnetically coupled ground state. The stronger deviations from the Curie behaviour observed for increasing doping ratios suggest increased spin pairing at higher spin concentrations. The spin concentration dependence and the observation of the P3HT radical cation as the only paramagnetic species present in the BCF-doped P3HT films indicate that the observed temperature dependence is due to interactions between spins on the P3HT backbone.
Deviations from the Curie law are also observed for the temperature-dependence of the EPR signal intensities for F4TCNQ-doped P3HT films, where they are however less pronounced and show no clear systematic dependence on doping ratio. Similar temperature-dependences of the EPR intensity and EPR spectra determined by an averaged g-matrix have previously been observed in charge transfer molecular crystals consisting of parallel alternating stacks of donor and acceptor molecules37–39 and have been interpreted in terms of exchange coupled spins with a diamagnetic singlet ground state and a thermally accessible triplet state, with a singlet–triplet energy gap corresponding to the exchange coupling constant J. The EPR signal intensity for the thermally excited triplet state is expected to vary with temperature as40–42
 | (1) |
In summary, the cw EPR data reveals significant differences for P3HT films doped with BCF and F4TCNQ, with clear evidence for the P3HT radical cation being the only paramagnetic species present in the BCF-doped P3HT films, while the spectra for F4TCNQ-doped P3HT films indicate the presence of exchange interactions between P3HT radical cations and dopant radical anions. The temperature-dependence of the EPR intensity suggests the formation of antiferromagnetically coupled spin pairs in both BCF- and F4TCNQ-doped P3HT films, with pairing between spins on P3HT for the BCF-doped films and between spins on P3HT and on the dopant anion for F4TCNQ-doped P3HT.
2.4 Q-band pulse EPR spectra
For a more in-depth characterisation, pulse EPR measurements for the determination of the relaxation properties and the hyperfine interactions of the spin centres in doped P3HT were performed at Q-band.The echo-detected Q-band pulse EPR spectra recorded at 10 K for the different samples are compared in Fig. 6 and 7. Fig. 6 also shows the dependence of the signal intensities, the phase memory times determined from two-pulse echo decay measurements and the spin–lattice relaxation times from inversion recovery measurements on the doping ratio for the different doped films. The dependence of the echo intensity on the doping ratio shows an initial increase followed by a steep decrease determined by shorter phase memory times for increasing spin concentrations. The trends observed for BCF-doped P3HT films prepared from both oDCB and CF and for F4TCNQ-doped films prepared from CF are very similar, however, the echo intensities observed for the F4TCNQ-doped films prepared from oDCB are significantly lower across all doping ratios despite longer spin–spin and spin–lattice relaxation times. The comparable spin concentrations determined by cw EPR for all samples, combined with the significant differences in echo intensities, suggest that only a small fraction of the spins detected by cw EPR contribute to the pulse EPR data for F4TCNQ-doped P3HT films prepared from oDCB. This is in agreement with the observation of cw EPR spectra with Lorentzian lineshapes and widths that translate to spin–spin relaxation times T2 in the range of 10 to 20 ns  for these samples. For F4TCNQ-doped films prepared from CF with doping ratios above 2.5⋅10−2, the cw EPR spectra are also Lorentzian with widths corresponding to T2 relaxation times of 20 to 30 ns and again no corresponding echo signals could be observed.
for these samples. For F4TCNQ-doped films prepared from CF with doping ratios above 2.5⋅10−2, the cw EPR spectra are also Lorentzian with widths corresponding to T2 relaxation times of 20 to 30 ns and again no corresponding echo signals could be observed.
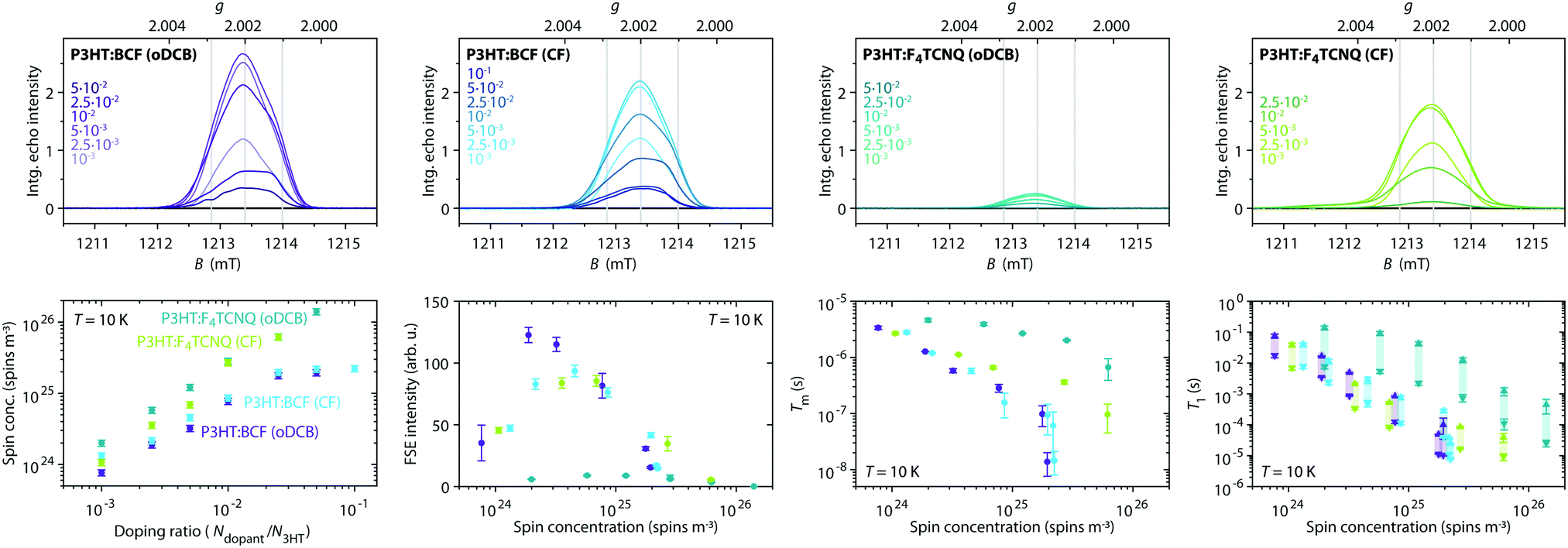 | ||
| Fig. 6 Q-band pulse EPR measurements (T = 10 K) on BCF- and F4TCNQ-doped P3HT films prepared from solutions in oDCB or CF at different doping ratios. (top) Echo-detected field-swept spectra (tπ = 2tπ/2 = 64 ns, τ = 400 ns). The spectra are displayed with the actual relative integrated echo intensities to allow a semiquantitative comparison of the intensities. The grey lines indicate the principal g-values determined for the P3HT radical cation. (bottom, left to right) Spin concentration determined at 10 K from the X-band cw EPR data as a function of doping ratio, integrated intensities of the echo-detected field-swept spectrum, phase memory times (from a stretched exponential fit to echo decay traces) and spin–lattice relaxation times T1 (both values obtained from a biexponential fit of inversion recovery traces are shown to indicate the range of what is likely a distribution of relaxation times, see Section S2.3 in the ESI† for details) as a function of spin concentration for the different dopant and solvent combinations. (See Fig. S6 in the ESI† for the experimental echo decay and inversion recovery traces and fits.) | ||
The spectral shapes of the Q-band echo-detected pulse EPR spectra, compared in Fig. 7, show no significant dependence on type of dopant, solvent or doping concentration and are assigned to the P3HT radical cation based on agreement with the g-values determined by W-band cw EPR as well as ENDOR spectra showing hyperfine interactions with the protons on the P3HT backbone (vide infra). For F4TCNQ-doped P3HT films prepared from CF, a weak and broad underlying signal with clear contributions at lower fields is also observed. The strong similarities of the echo-detected pulse EPR spectra for films doped with BCF or F4TCNQ despite significant differences in the cw EPR spectra indicate a difference in the species observed with the two methods. In particular, for the F4TCNQ-doped P3HT films, the exchange-coupled pairs of P3HT and F4TCNQ observed by cw EPR appear to relax too quickly to give a measurable echo signal even at low temperatures. In contrast to the results obtained for the film samples, measurements on frozen solutions of F4TCNQ-doped P3HT clearly reveal a contribution at g-values corresponding to F4TCNQ˙− in addition to the P3HT˙+ signal also observed in films (see Section S2.4 for details, ESI†).
 | ||
| Fig. 7 (top) Q-band pulse EPR spectra (T = 10 K) for BCF- and F4TCNQ-doped P3HT films at different doping ratios (prepared from oDCB or CF). Echo-detected or echo skew projection spectra43 are shown as solid lines and FID skew projection spectra as dashed lines. The FID skew projection spectra are shown for highly F4TCNQ-doped P3HT samples for which no echo signal could be detected. (bottom) Echo and FID transients as a function of magnetic field for F4TCNQ-doped P3HT films (prepared from CF) at doping ratios of 10−2, 2.5⋅10−2 and 5⋅10−2. Positive signal intensities are shown in blue, negative signal intensities in red. Echoes were recorded for a pulse sequence with tπ = 2tπ/2 = 64 ns and τ = 400 ns, FIDs were recorded after a tπ/2 = 200 ns pulse. The intensities are scaled to match the experimentally observed relative intensities of the FID and echo signals for each doping ratio (see Section S2.5 in the ESI† for additional data). The grey and green vertical lines indicate the principal g-values determined for P3HT˙+ and F4TCNQ˙−, respectively. | ||
Pulse EPR with echo detection therefore appears to be selective for isolated P3HT radical cations and F4TCNQ radical anions, while the faster relaxation induced by spin–spin interactions for exchange coupled spins prevents detection of an echo signal. However, for samples with high doping concentrations, strong FID signals were observed in addition to or in the absence of echo signals. Echo and FID transients recorded as a function of magnetic field are compared in Fig. 7 for F4TCNQ-doped P3HT films at three different doping ratios (see Section S2.5 in the ESI† for results on films with other dopant–solvent combinations and for additional details). Fourier transform of the echo or FID signals followed by projection onto a magnetic field axis and summation results in pulse EPR spectra with increased sensitivity, and in some cases increased resolution, compared to standard pulse measurements performed by echo integration.43 For F4TCNQ-doped P3HT films at a doping ratio of 10−2, both the echo as well as the FID after a selective pulse (tπ/2 = 200 ns) give the same spectrum as obtained by echo integration. At a doping ratio of 2.5⋅10−2, the echo intensity has significantly decreased compared to the FID intensity on account of the faster spin–spin relaxation, and an additional contribution to the FID transients becomes evident. This contribution is centred at a lower magnetic field compared to the echo signal and is characterised by alternating positive and negative transients depending on field position. For a doping ratio of 5⋅10−2, this is the only contribution present in the FID measurements and no echo signal could be detected. The spectrum resulting from this FID signal consists of a relatively narrow line centred at a g-value of about 2.0027 (shown as dashed line in Fig. 7). This signal is clearly shifted with respect to the signal of the P3HT radical cation and centred at the same g-value as the X- and W-band cw EPR spectra for the F4TCNQ-doped P3HT films. A similar FID contribution is also observed for F4TCNQ-doped films prepared from oDCB, and FID-detected inversion recovery measurements for films with doping ratios of 2.5⋅10−2 and 5⋅10−2 show spin–lattice relaxation times of the order of tens of microseconds, compared to hundreds of microseconds determined for the corresponding echo signals (see Fig. S11 in the ESI†). The FID signals observed for the F4TCNQ-doped films are therefore assigned to fast-relaxing exchange-coupled P3HT and F4TCNQ spins that constitute the main contribution to the cw EPR spectra. Unfortunately, the fast relaxation times prevented measurements for further characterisation and confirmation of this assignment.
2.5 Characterisation of the proton hyperfine interactions
While the nearly identical echo-detected Q-band pulse EPR spectra for all investigated combinations of dopants and solvents clearly indicate that the P3HT radical cation is detected in all cases, an additional characterisation of the hyperfine interactions to magnetic nuclei is necessary to resolve any dopant- or solvent-dependent changes in the molecular environment of the unpaired electron spin on the P3HT backbone. The measurement of hyperfine couplings to protons on polymer backbones by Electron Nuclear DOuble Resonance (ENDOR), combined with simulations based on quantum chemical calculations, can be used to estimate the extent of spin delocalisation.The ENDOR spectra of P3HT radical cations are mainly determined by the hyperfine couplings of the protons on the thiophene rings of the P3HT backbone. The principal hyperfine values for aromatic α-protons can be estimated from the spin density on the adjacent carbon atom, ρC, using the following equations:44,45
 | (2) |
Fig. 8 shows the results of low temperature 1H Davies ENDOR measurements for BCF- and F4TCNQ-doped P3HT films prepared from oDCB and CF with different doping ratios, as well as for the corresponding frozen solutions at a doping ratio of 10−2. The ENDOR spectra are all characterised by a relatively featureless absorption centred at the proton Larmor frequency, as expected for a distribution of proton hyperfine couplings with varying strengths. Extensive spin delocalisation over multiple thiophene units leads to a large number of weakly coupled protons, while fewer contributions from more strongly coupled protons are expected for localisation of the spin density on a small number of thiophene units. Since the overall width of the ENDOR spectrum is determined by the largest hyperfine couplings, narrower spectra therefore indicate increased spin density delocalisation. The widths of the experimental ENDOR spectra for the different investigated samples vary from about 3 MHz for the spectra recorded on both BCF- and F4TCNQ-doped P3HT in oDCB solution to between 5 and 6 MHz for F4TCNQ-doped P3HT films, indicating differences in the extent of spin delocalisation.
 | ||
| Fig. 8 (left) Q-band 1H Davies ENDOR spectra recorded at 10 K for BCF- and F4TCNQ-doped P3HT films prepared from oDCB or CF. The ENDOR spectra are compared for each dopant–solvent pair at different doping ratios and for film samples (solid lines) and the corresponding frozen solutions (dashed lines). All ENDOR spectra were recorded at a field position corresponding to the maximum of the P3HT radical cation EPR spectrum (ca. 1213.4 mT). (right) Comparison of experimental ENDOR spectra recorded on films (solid lines) and frozen solutions (dashed lines) for selected doping ratios and of the distributions of spin density derived from simulation and fitting as described in Section S1.4.2 in the ESI.† The shaded regions cover the range of distributions in agreement with the experimental data (see Fig. S14 in the ESI†). | ||
In order to estimate the extent of spin delocalisation for the different samples investigated here, ENDOR spectral simulations were performed based on eqn (2) for a range of model spin density distributions, and compared to the experimental data (see Section S1.4.2 in the ESI† for details). The resulting estimated distributions of spin densities are shown in Fig. 8.
Comparison of the ENDOR results for frozen solutions of BCF- and F4TCNQ-doped P3HT in oDCB and CF shows a clear difference in the extent of spin delocalisation on the P3HT backbone between the two types of solvents, while the spectra obtained for the two different dopants are almost identical. Doped P3HT in frozen oDCB solution is characterised by the largest extent of delocalisation, over about 18 thiophene units, while delocalisation is reduced to about 12 thiophene units in frozen CF solution. The absence of a dependence of the extent of delocalisation on the type of dopant indicates that, in solution, the molecular environment experienced by the spin on P3HT is similar despite the different nature of the counter ion.
The low temperature ENDOR results on the doped films show only minor differences for films prepared from different solvents, but a clear dependence on the dopant, with broader spectra observed for F4TCNQ-doped films. Compared to the frozen solution ENDOR results, the film spectra are characterised by a more gradual decrease in intensity in the wings of the spectrum that can only be modelled by considering contributions for different extents of delocalisation, including more localised spin density distributions, in agreement with the expected greater heterogeneity of molecular environments. The dependence of the ENDOR spectra on the doping ratio is relatively minor, except for a decrease in delocalisation for highly BCF-doped P3HT films prepared from oDCB. The ENDOR spectrum for F4TCNQ-doped P3HT films prepared from oDCB at a doping ratio of 10−3 is also characterised by a clearly different shape that appears to result from a combination of contributions from P3HT in environments similar to those of the corresponding BCF-doped film as well as those observed for the F4TCNQ-doped films at higher doping concentrations. The spin density distributions determined for BCF-doped P3HT films extend over about 12 thiophene units, in agreement with a delocalisation over 12–13 units previously determined for I2-doped P3HT films15 and for BCF- and F4TCNQ-doped P3HT in CF solutions.7 In F4TCNQ-doped P3HT films, the extent of delocalisation is reduced to about 8–10 thiophene rings.
The observation of the narrowest ENDOR spectra for doped P3HT in oDCB solutions is consistent with observations from UV-vis-NIR spectroscopy indicating the presence of isolated solvated P3HT chains in chlorobenzene,7 which would allow for increased spatial separation from the counter ion and facilitate increased delocalisation of the unpaired spin on the P3HT backbone. The determined extent of delocalisation over about 18 thiophene units closely matches the one reported for light-induced polarons in P3HT:PC61BM blends, where the more extensive delocalisation has been attributed to the absence of counter ions in the vicinity of the polymer backbone.33 The UV-vis-NIR spectra for doped P3HT in chloroform, on the other hand, are characteristic of P3HT aggregates,7 which explains the strong similarities between the ENDOR data for BCF-doped solutions and films prepared from this solvent and the decreased delocalisation. The broader ENDOR spectra observed for F4TCNQ-compared to BCF-doped P3HT films suggest a role of the F4TCNQ molecule in limiting delocalisation, either through changes in film morphology or through localisation of the hole on P3HT as a result of a nearby dopant anion.
The absence of any features that can be attributed to fluorine hyperfine couplings in the ENDOR spectra recorded at the field position corresponding to the maximum of the P3HT radical cation signal in F4TCNQ-doped P3HT films (the 19F Larmor frequency is about 3 MHz lower than the 1H Larmor frequency at the magnetic field of the experiment) indicates either the absence of F4TCNQ molecules in the direct vicinity of the detected P3HT radical cations or their random distribution with respect to the P3HT backbone, leading to extensive signal broadening that prevents detection.
2.6 Contributions from F4TCNQ in pulse EPR of doped P3HT solutions and films
The Q-band echo-detected pulse EPR spectra for the doped P3HT films were shown to be dominated by the signal of the P3HT radical cation, however, the spectra recorded for F4TCNQ-doped P3HT in solution additionally contain a signal at g-values in agreement with those determined for the F4TCNQ radical anion. Additionally, the spectra for the F4TCNQ-doped films prepared from CF also contain a weak, broad signal with contributions at lower magnetic fields compared to P3HT. This prompted a more detailed investigation of the origin of the additional signals observed when F4TCNQ is used as the dopant.Attempts to separate different signal contributions based on differences in relaxation times were not successful, as field-dependent inversion recovery measurements did not show significant differences in spin–lattice relaxation times across the spectrum (Fig. S7 in the ESI†). However, the F4TCNQ molecule contains four fluorine and four nitrogen nuclei, which provides the opportunity for distinguishing any spectral contributions from F4TCNQ based on the presence of the corresponding hyperfine couplings. As a reference, the g-values, 19F hyperfine interaction parameters and 14N hyperfine and nuclear quadrupole interaction parameters of the isolated F4TCNQ radical anion were initially determined by X-, Q- and W-band cw EPR, Q- and W-band 19F ENDOR and Q-band 14N ESEEM measurements and are reported in Section S2.7 in the ESI.†
Fig. 9 shows the Q-band echo-detected EPR spectra for F4TCNQ-doped P3HT in CF solution and for the corresponding film as well as the results of magnetic field-dependent ENDOR and three-pulse ESEEM measurements.
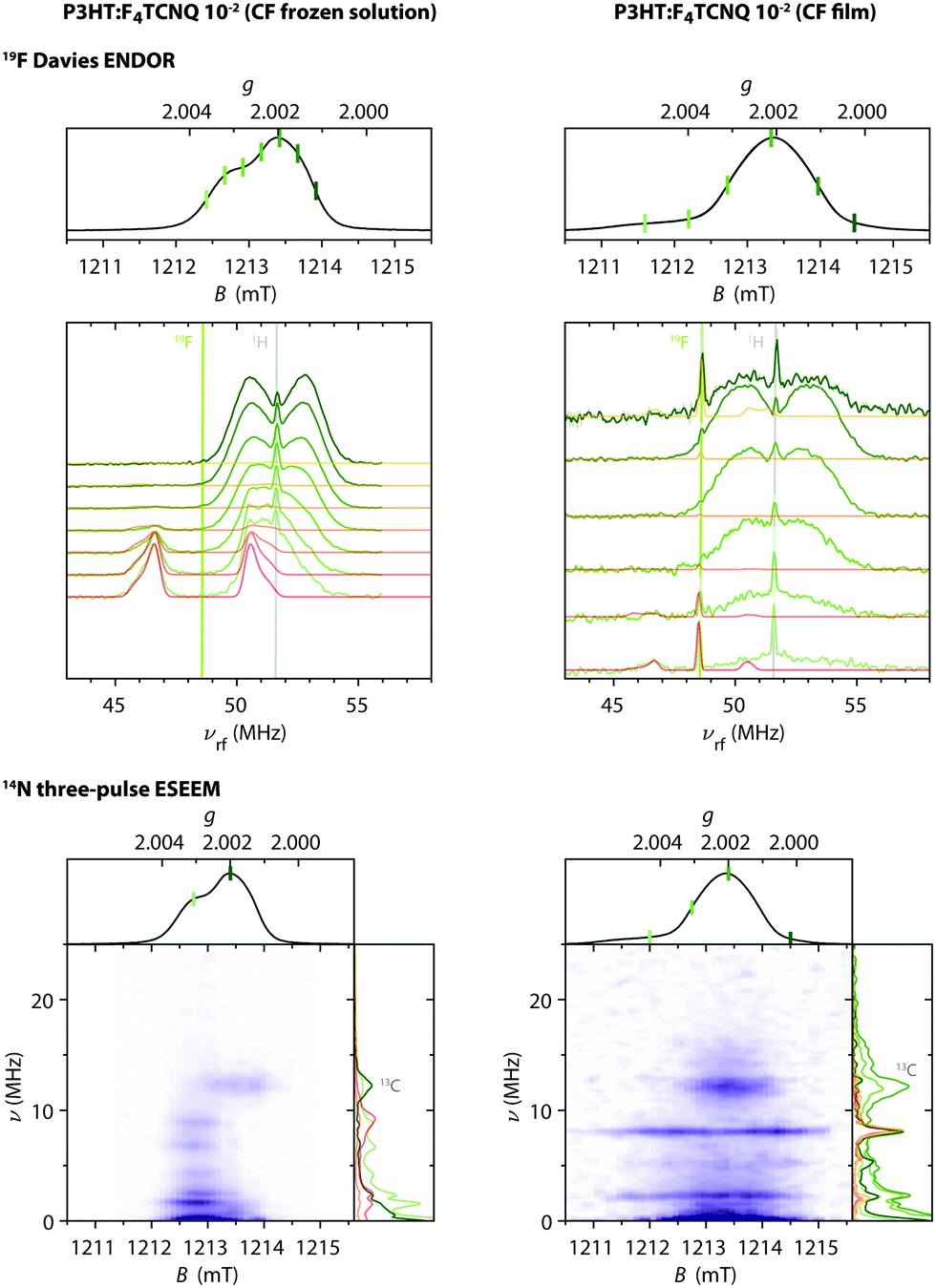 | ||
| Fig. 9 Q-band 19F ENDOR and 14N ESEEM data for F4TCNQ-doped P3HT in CF solution and for the corresponding film (T = 10 K). (top) Echo-detected pulse EPR and Davies ENDOR spectra recorded at different magnetic fields. The experimental ENDOR data is compared to 19F ENDOR simulations (in red-orange) for F4TCNQ˙− (left) and for a coupled pair of P3HT˙+ and F4TCNQ˙− with D = 80 MHz and J ≥ 500 MHz (right). The principal values of the 19F hyperfine matrix used in the simulations are Ax = 3.75 MHz, Ay = 3.67 MHz and Az = 6.00 MHz and Ax = 3.62 MHz, Ay = 3.54 MHz and Az = 5.86 MHz, respectively. (bottom) Three-pulse ESEEM spectra as a function of magnetic field. ESEEM spectra at selected field positions are compared to simulations for F4TCNQ˙− and a coupled spin pair in the side panels. The principal values of the 14N hyperfine matrix used in the simulations are Ax = −1.17 MHz, Ay = −1.87 MHz and Az = 12.43 MHz with the nuclear quadrupole interaction parameters Q = −1.00 MHz and η = 0.33 (principal axes orientations are displayed in Fig. S15 in the ESI†). | ||
For the frozen solution sample, the additional signal observed at lower fields compared to the P3HT radical cation is associated with peaks in Davies ENDOR spectra centred at the fluorine Larmor frequency. Field-dependent ENDOR measurements allow clear separation of this contribution from the P3HT radical cation spectrum (see Fig. S16 in the ESI†). The 19F hyperfine couplings determined by simulation of the experimental data are in close agreement with those determined for F4TCNQ˙−, confirming assignment of this signal. Three-pulse ESEEM measurements on the frozen solutions of F4TCNQ-doped P3HT also show modulations due to nitrogen hyperfine couplings that closely resemble the results obtained for the isolated F4TCNQ radical anion (see Fig. S17 in the ESI†). Field-dependent ESEEM measurements again allow a clear distinction between the F4TCNQ˙− contribution with 14N hyperfine couplings and the P3HT˙+ contribution with a weak ESEEM signal due to natural abundance 13C (Fig. 9).
For the F4TCNQ-doped P3HT films prepared from CF, Davies ENDOR measurements show a sharp peak at the fluorine Larmor frequency both at lower and higher fields compared to the P3HT spectrum. The ENDOR spectrum recorded at about 1211.7 mT, corresponding to the weak shoulder observed on the lower field end of the EPR spectrum, additionally includes a signal at frequencies lower than the fluorine Larmor peak with a position and shape that closely resembles the fluorine ENDOR contribution for the frozen solution sample. The field-dependent ESEEM measurements show a contribution around the 13C Larmor frequency for field positions corresponding to P3HT, as already observed in the corresponding frozen solution spectra. However, instead of the nitrogen ESEEM peaks at about 1.6, 2.4, 6.6 and 8.8 MHz observed at field positions corresponding to the F4TCNQ˙−g-values for the frozen solution sample, the ESEEM data for the film sample is characterised by two pronounced and one slightly weaker peak at frequencies of 2.2, 5.3 and 8.0 MHz spanning the field range from about 1211 to 1215 mT and extending to both sides of the P3HT spectrum.
Magnetic-field-dependent nutation experiments were performed to provide additional insights into the origin of this signal. The results of phase-inverted echo-amplitude detected nutation (PEANUT) experiments for F4TCNQ-doped P3HT films prepared from oDCB, where only the P3HT signal is detected, and for films prepared from CF, where the additional broad underlying contribution characterised by both fluorine and nitrogen hyperfine couplings is present, are compared in Fig. 10. The spectrum for the film prepared from CF clearly contains contributions at a nutation frequency of  at both lower and higher fields compared to the intense P3HT˙+ signal at ν1, where ν1 corresponds to the nutation frequency of an
at both lower and higher fields compared to the intense P3HT˙+ signal at ν1, where ν1 corresponds to the nutation frequency of an 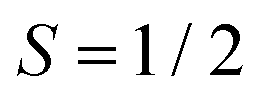 spin. The contribution at
spin. The contribution at  could only be identified in the PEANUT spectra for F4TCNQ-doped P3HT films prepared from CF, while it is absent for films prepared from oDCB and for BCF-doped films (see Fig. S19 and S20 in the ESI†). The nutations observed at
could only be identified in the PEANUT spectra for F4TCNQ-doped P3HT films prepared from CF, while it is absent for films prepared from oDCB and for BCF-doped films (see Fig. S19 and S20 in the ESI†). The nutations observed at  are an indication of the presence of dipolar coupled spin pairs that can be described by the following Hamiltonian
are an indication of the presence of dipolar coupled spin pairs that can be described by the following Hamiltonian
 | (3) |
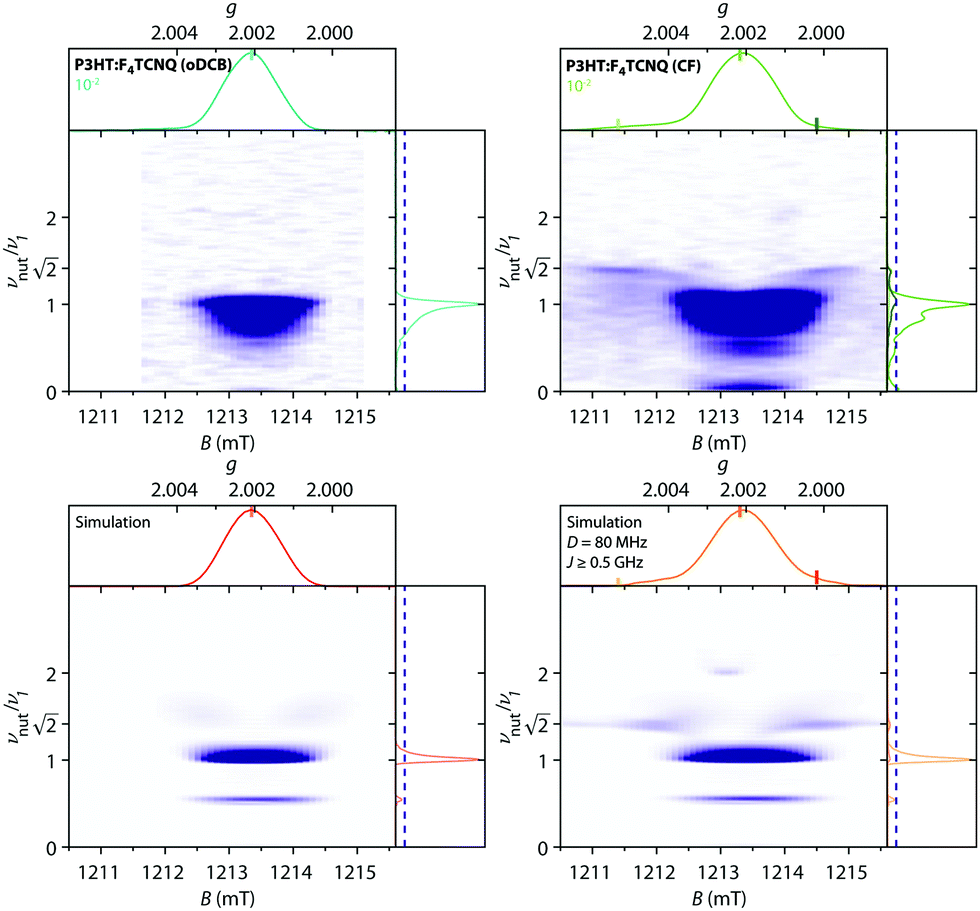 | ||
Fig. 10 Experimental two-dimensional PEANUT data recorded at Q-band (T = 10 K) for F4TCNQ-doped P3HT films prepared from oDCB and CF at a doping ratio of 10−2 (top) and corresponding simulations (bottom). For a better display of the weak contributions arising from the broad underlying signal in the film prepared from CF, the intensities in the 2D plots are clipped at 10% of the maximum intensity (marked by a dashed line in the side panels). The experimental nutation frequency for an  spin centre corresponds to ν1 ≈ 21 MHz. The simulation shown on the left was performed for the P3HT radical cation, the simulation on the right contains contributions from the P3HT radical cation simulation and from a pair of coupled P3HT and F4TCNQ radicals with D = 80 MHz and J ≥ 500 MHz (with relative weights of 0.95 spin centre corresponds to ν1 ≈ 21 MHz. The simulation shown on the left was performed for the P3HT radical cation, the simulation on the right contains contributions from the P3HT radical cation simulation and from a pair of coupled P3HT and F4TCNQ radicals with D = 80 MHz and J ≥ 500 MHz (with relative weights of 0.95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05). :0.05). | ||
Numerical simulations for PEANUT on coupled pairs of F4TCNQ and P3HT radicals were performed assuming the F4TCNQ molecules to be oriented with the long axis perpendicular to the P3HT backbone as proposed by Hamidi-Sakr et al.29 and considering varying dipolar and exchange coupling constants (see Fig. S21 in the ESI†). The results for D = 80 MHz without exchange coupling and with an exchange coupling constant J > D are compared in Fig. 11. The general features of the simulated 2D PEANUT spectra, with nutation frequencies sloping upwards from ν1 at the centre of the spectrum to 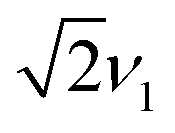 towards the wings, match the features observed in the experimental spectra recorded for the F4TCNQ-doped P3HT films prepared from CF. A comparison of the simulations in Fig. 11 shows that in the presence of exchange coupling the main features on either side of the centre flatten, with the highest intensities observed at nutation frequencies of
towards the wings, match the features observed in the experimental spectra recorded for the F4TCNQ-doped P3HT films prepared from CF. A comparison of the simulations in Fig. 11 shows that in the presence of exchange coupling the main features on either side of the centre flatten, with the highest intensities observed at nutation frequencies of  , while in the absence of exchange coupling the maximum intensities are reached at slightly lower nutation frequencies. In both cases, the distance between the main peaks corresponds to D. As previously observed,46 in the absence of exchange coupling, weaker contributions at nutation frequencies lower than ν1 are also present at the centre of the spectrum. These contributions disappear for increasing exchange couplings and a contribution at 2ν1 at the centre of the spectrum gains in intensity, but is still significantly weaker compared to the main features at
, while in the absence of exchange coupling the maximum intensities are reached at slightly lower nutation frequencies. In both cases, the distance between the main peaks corresponds to D. As previously observed,46 in the absence of exchange coupling, weaker contributions at nutation frequencies lower than ν1 are also present at the centre of the spectrum. These contributions disappear for increasing exchange couplings and a contribution at 2ν1 at the centre of the spectrum gains in intensity, but is still significantly weaker compared to the main features at 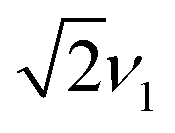 .
.
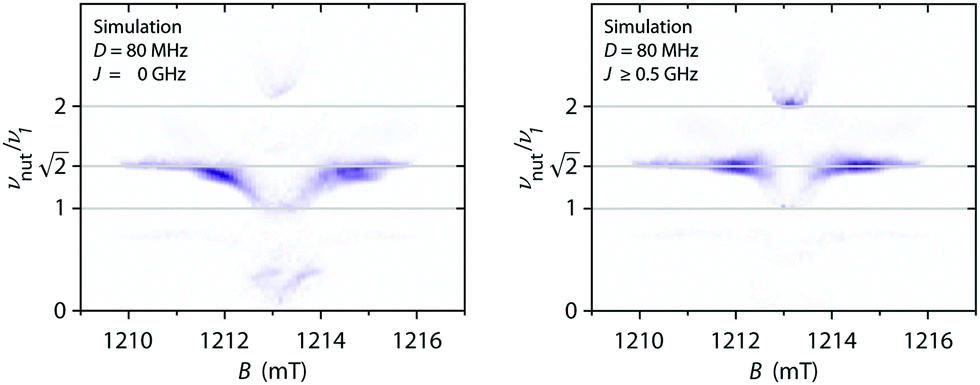 | ||
| Fig. 11 Simulations of two-dimensional Q-band PEANUT spectra for a pair of coupled P3HT and F4TCNQ radicals with a dipolar coupling of D = 80 MHz and with an exchange coupling constant J = 0 (left) and J = 500 MHz (right). The principal g-values used for the simulations were gx = 2.0029, gy = 2.0020 and gz = 2.0010 for P3HT and gx = 2.0030, gy = 2.0030 and gz = 2.0024 for F4TCNQ. See Section S1.4.3 in the ESI† for further details. | ||
The simulations for D-values in the range from about 60 to 100 MHz reproduce the features observed experimentally around  . The partially overlapping and significantly stronger contribution at ν1 from the isolated P3HT radical cation, combined with the likely presence of a distribution of coupling strengths due to the intrinsic heterogeneity of the film samples, complicates a precise determination of the dipolar and exchange couplings. However, simulations for J > D appear to provide a better match for the experimental data. A feature at 2ν1, which is typically considered a clear indication of the presence of strong exchange coupling,46 appears to be present but is too faint to allow any definite conclusions. The simulations for isolated P3HT˙+ and for a linear combination of isolated P3HT˙+ and a small fraction of coupled P3HT-F4TCNQ spin pairs with D = 80 MHz and J > D shown in Fig. 10 are in reasonable agreement with the experimental results for F4TCNQ-doped P3HT films prepared from oDCB and CF, respectively.
. The partially overlapping and significantly stronger contribution at ν1 from the isolated P3HT radical cation, combined with the likely presence of a distribution of coupling strengths due to the intrinsic heterogeneity of the film samples, complicates a precise determination of the dipolar and exchange couplings. However, simulations for J > D appear to provide a better match for the experimental data. A feature at 2ν1, which is typically considered a clear indication of the presence of strong exchange coupling,46 appears to be present but is too faint to allow any definite conclusions. The simulations for isolated P3HT˙+ and for a linear combination of isolated P3HT˙+ and a small fraction of coupled P3HT-F4TCNQ spin pairs with D = 80 MHz and J > D shown in Fig. 10 are in reasonable agreement with the experimental results for F4TCNQ-doped P3HT films prepared from oDCB and CF, respectively.
In light of this assignment of the broad signal observed in F4TCNQ-doped P3HT films prepared from CF to a pair of coupled spins on F4TCNQ and P3HT, the ENDOR and ESEEM results of Fig. 9 can be revisited. The sharp peaks at the 19F and 1H Larmor frequencies observed in the ENDOR data at field positions with relatively small overlap with the P3HT signal are reminiscent of the contributions from the mS = 0 manifold typically observed in triplet (S = 1) ENDOR spectra. Simulations of 19F ENDOR spectra for P3HT-F4TCNQ spin pairs with an exchange coupling leading to a clear separation of the singlet and triplet levels and for fluorine hyperfine couplings almost identical to those determined for F4TCNQ˙− indeed provide good agreement with the experimental spectrum recorded at a field position of about 1211.7 mT. The ENDOR spectra recorded at most of the other field positions are dominated by the 1H contributions from P3HT˙+, making the contribution at the 19F Larmor frequency the only indication of the presence of the coupled spin pair. The intense 1H Larmor peaks at the lowest and highest field positions used for the ENDOR measurements are attributed to the contribution of P3HT in the spin pair, however a determination of the corresponding hyperfine couplings was prevented by the overlap with the 1H ENDOR spectrum of the isolated P3HT radical cation and significant signal broadening.
In the three-pulse ESEEM data, the positions of the three prominent peaks contributing to the ESEEM spectrum across the field range covered by the signal attributed to coupled P3HT and F4TCNQ pairs are in excellent agreement with simulations including the nitrogen hyperfine and nuclear quadrupole parameters determined for F4TCNQ˙− for one component of the spin pair. These features arise from the mS = 0 manifold and are determined solely by the nuclear Zeeman and nuclear quadrupole interactions, contributions from the other manifolds that would provide information on the strength of the hyperfine coupling are unfortunately too weak to be unequivocally identified in the recorded spectra.
The results from the magnetic-field dependent PEANUT, ENDOR and ESEEM measurements thus clearly identify a spectral contribution from dipolar and exchange-coupled pairs of P3HT and F4TCNQ radicals in the doped films prepared from CF. The signal intensity of this contribution is significantly reduced compared to the main contribution from the P3HT radical cation, which is the only contribution present in the pulse EPR data for the other investigated P3HT films.
3 Discussion
The EPR measurements on BCF- and F4TCNQ-doped P3HT films presented in this study reveal significant differences in the molecular environment of the spins introduced on the P3HT backbone with the two different dopants. Since the appearance of EPR spectra is influenced by both static and dynamic interactions of the electron spin with its surroundings, the spin is used as a probe for gaining molecular level insights into doped organic semiconductors that can be used to rationalise observed differences in macroscopic film properties, such as conductivity.The first clear indication for an influence of the F4TCNQ dopant anion on the spin introduced on P3HT was revealed by the appearance of the W-band cw EPR spectra. For BCF-doped P3HT films, they contain the characteristic signature of the P3HT radical cation with an orthorhombic g-matrix and thus confirm previous results indicating a doping mechanism that leads to P3HT radical cations as the only paramagnetic species formed upon doping with BCF.7,31 In contrast to this, the observation of a single EPR line centred at a g-value intermediate between those of the P3HT radical cation and F4TCNQ radical anion for the F4TCNQ-doped P3HT films indicates the presence of exchange interaction between the spins on the P3HT backbone and on the dopant molecule. This exchange interaction exceeds the difference in electron Zeeman interactions at both X- and W-band frequencies and the appearance of the spectra suggests any dipolar interactions between the spins are likely averaged out. In the case of BCF-doped P3HT, the cw EPR spectra also revealed ordering effects for doped films, whereas spectra characteristic of randomly oriented paramagnetic centres were observed for the corresponding powders. P3HT is known to form semicrystalline films characterised by crystalline and amorphous domains with charge transport occurring mainly along the P3HT backbone and π–π stacking direction within the crystalline domains and along tie chains bridging neighbouring crystallites.25,34 The observation of EPR spectral shapes determined by partial ordering and consistent with the known preferential edge-on orientation of the P3HT molecules with respect to the substrate therefore indicates that the observed paramagnetic species reside within the ordered crystalline regions of the P3HT films relevant for charge transport. The observation of more pronounced ordering effects for films prepared from oDCB compared to CF is in agreement with previous literature reporting increased crystallinity for films prepared from higher boiling point solvents.47
Further evidence for the detection of mobile charge carriers in the BCF-doped P3HT films by cw EPR was provided by the temperature-dependent partial averaging of the g-anisotropy observed in the W-band cw EPR spectra of the highly doped films and the narrowing of the X-band cw EPR linewidths both for increasing temperatures as well as increasing doping ratios. Both observations indicate that, at room temperature, the spins experience an averaged molecular environment due to rapid hopping on the EPR time scale (ca. 10−8–10−6 s), which is consistent with a charge transport mechanism determined by thermally activated hopping as proposed for organic semiconducting polymers.48
Temperature-dependent cw EPR measurements were performed to provide additional information on the strength of the spin–spin interactions in the doped P3HT films, which determine the appearance of the cw EPR spectra in the case of F4TCNQ-doped P3HT films. Interestingly, spin–spin interactions were found to be present also in the BCF-doped films, where the temperature-dependent change in spin concentration revealed formation of antiferromagnetically coupled pairs of spins on the P3HT backbone, with increased spin pairing at increasing spin concentrations and decreasing temperatures. In a previous combined in situ EPR and optical spectroscopic study of iodine doping of P3HT films, a decrease in spin concentration observed for high doping levels at room temperature was also attributed to antiferromagnetically coupled P3HT polarons.49 EPR thus provides experimental evidence for carrier–carrier interactions in doped P3HT films, which have recently been invoked in a theoretical study to explain decreases in conductivity observed for doped organic semiconductors at high carrier densities.50
For the F4TCNQ-doped P3HT films, the decrease in spin density with decreasing temperature was found to be less pronounced and no clear systematic dependence on the doping ratio could be identified. This observation, combined with the appearance of the EPR spectra, indicates that in this case the temperature dependence of the EPR intensity can mostly be attributed to spin–spin interactions between P3HT and F4TCNQ. The observed behaviour likely results from a combination of contributions with different exchange coupling strengths as well as contributions from free P3HT radical cations, as evidenced from the pulse EPR data. The EPR results on the F4TCNQ-doped P3HT films therefore provide clear experimental evidence for interactions between the spins on the dopant and host, in agreement with the presence of Coulomb interactions between the hole on the semiconductor and dopant anions that has previously been proposed to be the cause of low mobilities observed in films doped with small molecular dopants such as F4TCNQ.17,19,51 The estimated strength of the exchange interaction between spins on P3HT and F4TCNQ is smaller than typical values reported for π-stacked radicals,37,38 in agreement with literature demonstrating that dopant anions in P3HT films are located within the alkyl side chain regions of P3HT crystallites rather than intercalated between the P3HT backbones.25,29,52
In contrast to the observation of clearly different spectral signatures for BCF- and F4TCNQ-doped P3HT films by cw EPR, the echo-detected pulse EPR spectra appear almost identical for all dopant–solvent combinations and can be unequivocally assigned to the P3HT radical cation based on the proton ENDOR data. In the case of F4TCNQ-doped films prepared from oDCB in particular, there is also a significant discrepancy between the signal intensities observed by cw and pulse EPR. This suggests that the main signals observed in the cw EPR spectra, in particular in the case of F4TCNQ-doped films, are characterised by relaxation times that prevent detection of echo signals by pulse EPR. The absence of echo signals, or the detection of only very weak echo signals, for samples with cw EPR spectra characterised by significantly broadened Lorentzian lines with peak-to-peak linewidths suggesting relaxation times on the order of tens of nanoseconds seems to support this conclusion. The fast-relaxing exchange-coupled P3HT and F4TCNQ radicals dominating the cw EPR spectra do however appear to be associated with strong FID signals giving a spectrum centred at the same g-value as the corresponding cw EPR spectra. These results highlight that in general the spins detected by pulse EPR do not necessarily correspond to the dominant fraction of spins contributing to the cw EPR spectrum, with spins characterised by fast relaxation not visible by pulse EPR, in particular if only echo detection is used.
P3HT films are characterised by a relatively large degree of structural disorder due to their semicrystalline nature and doping likely leads to spins on P3HT backbones within crystalline domains as well as within amorphous domains or at the interface between them. The degree of crystallinity of P3HT films is known to differ significantly depending on film preparation conditions.25 Different possible positions of dopant anions with respect to the P3HT polaron further increase the range of molecular environments experienced by the spins on P3HT. While the cw EPR spectra contain contributions of unpaired spins in the full range of different environments, pulse EPR, and in particular echo-detected pulse EPR, selectively detects spins in a subset of environments, characterised by sufficiently long relaxation times. The properties determined by advanced pulse EPR methods are therefore only reflective of this subset of spins. The echo-detected pulse EPR spectra appear to only show contributions from “free” P3HT radical cations for both BCF- and F4TCNQ-doped P3HT films, as suggested by the agreement with the orthorhombic g-matrix of P3HT˙+ determined from the cw EPR spectra for BCF-doped P3HT, the identical spectral shapes for both dopants and the absence of any corresponding ENDOR signals that could be assigned to fluorine hyperfine couplings.
A characterisation of the delocalisation of the spin on P3HT associated with the observed echo signals for all investigated films revealed reduced spin delocalisation in the F4TCNQ-doped P3HT films compared to the BCF-doped films, demonstrating that the presence of F4TCNQ, even when not directly detectable, does still have an appreciable effect on the ability of the spin to delocalise on the P3HT backbone.
The echo-detected EPR spectra of F4TCNQ-doped P3HT films prepared from CF also revealed the presence of a third contribution, in addition to the FID-detected spectrum of exchange-coupled P3HT and F4TCNQ and the echo-detected spectrum due to the P3HT radical cation. Based on field-dependent ENDOR and ESEEM spectra, this contribution is characterised by fluorine and nitrogen hyperfine couplings, and PEANUT experiments further show that it is associated with a nutation frequency corresponding to  times the nutation frequency of an
times the nutation frequency of an 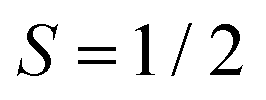 system. The signal was therefore assigned to dipolar- and exchange-coupled pairs of P3HT and F4TCNQ molecules. Neglecting spin delocalisation on P3HT, an estimate of the average spin–spin distances corresponding to the dipolar coupling strengths determined from the width of the echo-detected spectrum and the 2D PEANUT data yields 0.8 ± 0.1 nm. This very rough estimate would be close to the average dopant anion to polymer backbone distances of 0.6 to 0.8 nm previously proposed based on theoretical modelling of polaron absorption features25 and expected for F4TCNQ anions located within the side chain regions of P3HT crystallites. In contrast to the contribution of coupled spins on P3HT and F4TCNQ leading to the spectra observed by cw EPR, in this minor contribution observed by pulse EPR, the dipolar interaction not washed out by spin exchange, suggesting the observed coupled spin pair is in a region of relatively low local spin concentration.
system. The signal was therefore assigned to dipolar- and exchange-coupled pairs of P3HT and F4TCNQ molecules. Neglecting spin delocalisation on P3HT, an estimate of the average spin–spin distances corresponding to the dipolar coupling strengths determined from the width of the echo-detected spectrum and the 2D PEANUT data yields 0.8 ± 0.1 nm. This very rough estimate would be close to the average dopant anion to polymer backbone distances of 0.6 to 0.8 nm previously proposed based on theoretical modelling of polaron absorption features25 and expected for F4TCNQ anions located within the side chain regions of P3HT crystallites. In contrast to the contribution of coupled spins on P3HT and F4TCNQ leading to the spectra observed by cw EPR, in this minor contribution observed by pulse EPR, the dipolar interaction not washed out by spin exchange, suggesting the observed coupled spin pair is in a region of relatively low local spin concentration.
4 Conclusions
In conclusion, our multifrequency cw and pulse EPR study of doped P3HT films provides a complex picture of spins in a range of different molecular environments reflective of the heterogeneity of solution-processed films of organic semiconductors containing both amorphous regions as well as interconnected crystallites. The comparison of results obtained for a dopant that generates P3HT radical cations as the only paramagnetic species, BCF, and a dopant forming a radical anion after electron transfer from P3HT, F4TCNQ, greatly aided interpretation of the EPR results and helped unravel the different contributions.In the F4TCNQ-doped P3HT films, we identified contributions from P3HT radicals characterised by different interactions with their molecular environment and with distinct EPR spectral properties:
• exchange-coupled P3HT and F4TCNQ dominating the X- and W-band cw EPR data with featureless spectra centred at an averaged g-value and characterised by short relaxation times that prevent echo detection and only allow observation in the form of an FID by pulse EPR;
• dipolar- and exchange-coupled pairs of P3HT and F4TCNQ radicals detected as a weak contribution by pulse EPR with a spectral width determined by the dipolar interaction;
• uncoupled P3HT radical cations, seemingly not interacting with F4TCNQ radical anions, constituting the main contribution to the echo-detected pulse EPR data with spectra identical to those observed for BCF-doped P3HT.
The detection of dipolar and exchange interactions between spins on P3HT and F4TCNQ by EPR provides direct experimental evidence for the presence of bound pairs of charges on the organic semiconductor and dopant, in addition to the desired mobile charges required to increase the electrical conductivity in P3HT films. The observation of a reduced spin delocalisation in F4TCNQ-doped compared to BCF-doped P3HT films even for spins on P3HT with no F4TCNQ radical anions detected in close proximity suggests that F4TCNQ still plays a role in limiting delocalisation. Since charge delocalisation in π-conjugated polymers correlates with mobility, this points to a further limitation in the conductivity of P3HT doped with the small molecular dopant F4TCNQ. This study therefore allowed us to link differences at the microscopic level, probed through the interactions of the spin associated with charge carriers with the molecular environment, to different macroscopic conductivities in solution-processed BCF- and F4TCNQ-doped P3HT films.
Overall, our results provide a new perspective in the on-going investigation of molecular doping of organic semiconductors and suggest that the often distinguished mechanisms of doping by integer charge transfer and formation of a ground state charge-transfer complex are merely two extremes of a range of dopant-host configurations with varying interaction strengths present even within the same doped film.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Felix Kraffert for helpful discussions and the HPC Service of ZEDAT, Freie Universität Berlin, for computing time.53 C. E. T. would like to acknowledge financial support through a Marie Curie Individual Fellowship (H2020-MSCA-IF-743419) and currently through a Royal Society University Research Fellowship (URF\R1\201071). This work was in part supported by the German Research Foundation (DFG) through the project “FoMEDOS” (Project number: 286798544).References
- B. Lüssem, M. Riede and K. Leo, Phys. Solid State, 2013, 210, 9–43 Search PubMed.
- I. Salzmann and G. Heimel, J. Electron. Spectrosc. Relat. Phenom., 2015, 204, 208–222 CrossRef CAS.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed.
- B. Neelamraju, K. E. Watts, J. E. Pemberton and E. L. Ratcliff, J. Phys. Chem. Lett., 2018, 9, 6871–6877 CrossRef CAS PubMed.
- I. E. Jacobs, C. Cendra, T. F. Harrelson, Z. I. Bedolla Valdez, R. Faller, A. Salleo and A. J. Moulé, Mater. Horiz., 2018, 5, 655–660 RSC.
- M. Arvind, C. E. Tait, M. Guerrini, J. Krumland, A. M. Valencia, C. Cocchi, A. E. Mansour, N. Koch, S. Barlow, S. R. Marder, J. Behrends and D. Neher, J. Phys. Chem. B, 2020, 124, 7694–7708 CrossRef CAS PubMed.
- M. L. Tietze, J. Benduhn, P. Pahner, B. Nell, M. Schwarze, H. Kleemann, M. Krammer, K. Zojer, K. Vandewal and K. Leo, Nat. Commun., 2018, 9, 1182 CrossRef PubMed.
- A. Abate, D. R. Staff, D. J. Hollman, H. J. Snaith and A. B. Walker, Phys. Chem. Chem. Phys., 2014, 16, 1132–1138 RSC.
- A. R. Chew and A. Salleo, MRS Commun., 2017, 7, 728–734 CrossRef CAS.
- T. J. Aubry, J. C. Axtell, V. M. Basile, K. J. Winchell, J. R. Lindemuth, T. M. Porter, J.-Y. Liu, A. N. Alexandrova, C. P. Kubiak, S. H. Tolbert, A. M. Spokoyny and B. J. Schwartz, Adv. Mater., 2019, 31, 1805647 CrossRef PubMed.
- J. Tang, C. P. Lin, M. K. Bowman, J. R. Norris, J. Isoya and H. Shirakawa, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 2845–2847 CrossRef CAS.
- C. L. Young, D. Whitney, A. I. Vistnes and L. R. Dalton, Annu. Rev. Phys. Chem., 1986, 37, 459–491 CrossRef CAS.
- K. Mizoguchi, Jpn. J. Appl. Phys., 1995, 34, 1–19 CrossRef CAS.
- A. Aguirre, P. Gast, S. Orlinskii, I. Akimoto, E. J. J. Groenen, H. El Mkami, E. Goovaerts and S. Van Doorslaer, Phys. Chem. Chem. Phys., 2008, 10, 7129–7138 RSC.
- R. Steyrleuthner, Y. Zhang, L. Zhang, F. Kraffert, B. P. Cherniawski, R. Bittl, A. L. Briseno, J.-L. Brédas and J. Behrends, Phys. Chem. Chem. Phys., 2017, 19, 3627–3639 RSC.
- P. Pingel and D. Neher, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 115209 CrossRef.
- G. C. Welch and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 4632–4644 CrossRef CAS PubMed.
- P. Pingel, M. Arvind, L. Kölln, R. Steyrleuthner, F. Kraffert, J. Behrends, S. Janietz and D. Neher, Adv. Electron. Mater., 2016, 2, 1600204 CrossRef.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T.-Q. Nguyen, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- C. Wang, D. T. Duong, K. Vandewal, J. Rivnay and A. Salleo, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 085205 CrossRef.
- P. Pingel, R. Schwarzl and D. Neher, Appl. Phys. Lett., 2012, 100, 143303 CrossRef.
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef CAS.
- H. Méndez, G. Heimel, S. Winkler, J. Frisch, A. Opitz, K. Sauer, B. Wegner, M. Oehzelt, C. Röthel, S. Duhm, D. Többens, N. Koch and I. Salzmann, Nat. Commun., 2015, 6, 8560 CrossRef PubMed.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef CAS PubMed.
- E. Lim, K. A. Peterson, G. M. Su and M. L. Chabinyc, Chem. Mater., 2018, 30, 998–1010 CrossRef CAS.
- W. Liu, L. Müller, S. Ma, S. Barlow, S. R. Marder, W. Kowalsky, A. Köhn and R. Lovrincic, J. Phys. Chem. C, 2018, 122, 27983–27990 CrossRef CAS.
- A. Hamidi-Sakr, L. Biniek, J. L. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, Adv. Funct. Mater., 2017, 27, 1700173 CrossRef.
- D. A. Stanfield, Y. Wu, S. H. Tolbert and B. J. Schwartz, Chem. Mater., 2021, 33, 2343–2356 CrossRef CAS.
- P. S. Marqués, G. Londi, B. Yurash, T.-Q. Nguyen, S. Barlow, S. R. Marder and D. Beljonne, Chem. Sci., 2021, 12, 7012–7022 RSC.
- L. Müller, D. Nanova, T. Glaser, S. Beck, A. Pucci, A. K. Kast, R. R. Schröder, E. Mankel, P. Pingel, D. Neher, W. Kowalsky and R. Lovrincic, Chem. Mater., 2016, 28, 4432–4439 CrossRef.
- J. Niklas, K. L. Mardis, B. P. Banks, G. M. Grooms, A. Sperlich, V. Dyakonov, S. Beaupré, M. Leclerc, T. Xu, L. Yu and O. G. Poluektov, Phys. Chem. Chem. Phys., 2013, 15, 9562–9574 RSC.
- M. Brinkmann, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1218–1233 CrossRef CAS.
- P. Gast and E. J. J. Groenen, eMagRes, 2016, 5, 1435–1444 CAS.
- R. Calvo, Appl. Magn. Reson., 2007, 31, 271–299 CrossRef CAS.
- P. L. Nordio, Z. G. Soos and H. M. McConnell, Annu. Rev. Phys. Chem., 1966, 17, 237–260 CrossRef CAS.
- B. M. Hoffman and R. C. Hughes, J. Chem. Phys., 1970, 52, 4011–4023 CrossRef CAS.
- R. C. Hughes and Z. G. Soos, J. Chem. Phys., 1968, 48, 1066–1076 CrossRef CAS.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. A, 1952, 214, 451–465 CAS.
- D. Bijl, H. Kainer and A. C. Rose-Innes, J. Chem. Phys., 1959, 30, 765–770 CrossRef CAS.
- J. A. Weil, J. R. Bolton and J. E. Wertz, Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, John Wiley & Sons, Inc., New York, 1994 Search PubMed.
- M. K. Bowman, M. D. Krzyaniak, A. A. Cruce and R. T. Weber, J. Magn. Reson., 2013, 231, 117–125 CrossRef CAS PubMed.
- H. M. McConnell and D. B. Chesnut, J. Chem. Phys., 1958, 28, 107–117 CrossRef CAS.
- J. R. Morton, Chem. Rev., 1964, 64, 453–471 CrossRef CAS.
- M. E. Limes, J. Wang, W. J. Baker, S. Y. Lee, B. Saam and C. Boehme, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165204 CrossRef.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2012, 94, 163306 CrossRef.
- H. Bässler, Phys. Status Solidi B, 1993, 175, 15–56 CrossRef.
- Y. Tsutsui, H. Okamoto, D. Sakamaki, K. Sugiyasu and M. Takeuchi, J. Phys. Chem. Lett., 2018, 9, 3639–3645 CrossRef CAS PubMed.
- M. Koopmans, M. A. T. Leiviska, J. Liu, J. Dong, L. Qiu, J. C. Hummelen, G. Portale, M. C. Heiber and L. J. A. Koster, ACS Appl. Mater. Interfaces, 2020, 12, 56222–56230 CrossRef CAS PubMed.
- T. J. Aubry, K. J. Winchell, C. Z. Salamat, V. M. Basile, J. R. Lindemuth, J. M. Stauber, J. C. Axtell, R. M. Kubena, M. D. Phan, M. J. Bird, A. M. Spokoyny, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2020, 30, 2001800 CrossRef CAS PubMed.
- V. Untilova, T. Biskup, L. Biniek, V. Vijayakumar and M. Brinkmann, Macromolecules, 2020, 53, 2441–2453 CrossRef CAS.
- L. Bennett, B. Proppe and B. Melchers, CURTA – A General-purpose High-Performance Computer, Freie Universität Berlin Technical Report, 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and experimental methods, additional experimental data and simulations. See DOI: 10.1039/d1cp02133h |
| This journal is © the Owner Societies 2021 |