
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater–biomolecule clusters studied by photoemission spectroscopy and multilevel atomistic simulations: hydration or solvation?†
Giuseppe
Mattioli
 a,
Lorenzo
Avaldi
*a,
Paola
Bolognesi
a,
John D.
Bozek
b,
Mattea C.
Castrovilli
a,
Jacopo
Chiarinelli
a,
Alicja
Domaracka
c,
Suvasthika
Indrajith
c,
Sylvain
Maclot
d,
Aleksandar R.
Milosavljević
b,
Chiara
Nicolafrancesco
bc and
Patrick
Rousseau
c
a,
Lorenzo
Avaldi
*a,
Paola
Bolognesi
a,
John D.
Bozek
b,
Mattea C.
Castrovilli
a,
Jacopo
Chiarinelli
a,
Alicja
Domaracka
c,
Suvasthika
Indrajith
c,
Sylvain
Maclot
d,
Aleksandar R.
Milosavljević
b,
Chiara
Nicolafrancesco
bc and
Patrick
Rousseau
c
aCNR-Istituto di Struttura della Materia, Area della Ricerca di Roma 1, CP 10, Monterotondo Scalo, Italy. E-mail: lorenzo.avaldi@ism.cnr.it
bSynchrotron SOLEIL, L’Orme de Merisiers, 91192, Saint Aubin, BP48, 1192, Gif-sur-Yvette Cedex, France
cNormandie Univ., ENSICAEN, UNICAEN, CEA, CNRS, CIMAP, 14000 Caen, France
dPhysics Department, University of Gothenburg, Origovägen 6B, 41296 Göteborg, Sweden
First published on 25th June 2021
Abstract
The properties of mixed water–uracil nanoaggregates have been probed by core electron–photoemission measurements to investigate supramolecular assembly in the gas phase driven by weak interactions. The interpretation of the measurements has been assisted by multilevel atomistic simulations, based on semi-empirical tight-binding and DFT-based methods. Our protocol established a positive-feedback loop between experimental and computational techniques, which has enabled a sound and detailed atomistic description of such complex heterogeneous molecular aggregates. Among biomolecules, uracil offers interesting and generalized skeletal features; its structure encompasses an alternation of hydrophilic H-bond donor and acceptor sites and hydrophobic moieties, typical in biomolecular systems, that induces a supramolecular core–shell-like organization of the mixed clusters with a water core and an uracil shell. This structure is far from typical models of both solid-state hydration, with water molecules in defined positions, or liquid solvation, where disconnected uracil molecules are completely surrounded by water.
1 Introduction
A large variety of relevant bio-processes occur in an aqueous environment and are driven by non-covalent interactions. For example, H-bonds and π-stacking interactions as well as van der Waals forces define the structural arrangement, i.e. the tertiary and quaternary structure, of large biomolecules, such as DNA and proteins. These interactions play also a key role in molecular recognition processes, like protein–protein or protein–ligand interactions. Moreover, biomolecules are highly sensitive to the dynamic interactions with solvent molecules. The water hydrogen-bonded network affects the electronic and geometrical structure of these molecules and even small changes can be essential for biological functions.1 In solution, solvent molecules massively surrounding non-covalently bonded systems hamper direct probe of weak interactions. Water as solvent, in particular, with its strong dipole moment significantly perturbs the distribution of electronic charge of the molecule and affect the bonds to the limit of disruption of the networks of weak interactions in the solvation process. The study of molecular clusters in the gas phase, as prototypes of weakly bonded systems2 hold together by non-covalent weak interactions, can overcome these limitations and provide valuable information on structures and mechanisms at work at the edge between liquid and solid phases. In this regard, some studies3–5 on the fragmentation of nucleobase clusters in collisions with multiply charged ions have been reported in the literature. Although the main goal of such studies was the stability of the clusters against radiation damage, mainly for clinical applications, the interpretation of the observed OH or O + H loss channels lead to the conclusion3 that in small clusters planar structures determined by H-bonds are generally more stable than the stacked ones determined by dispersive forces. Multiphoton ionization of thymine and uracil clusters6 from supersonic beams provided evidence of an alternation of mass spectral intensities between even- and odd-numbered clusters, which has been attributed to a photodimerization reaction. In order to mimic more realistic environments some studies have been also devoted to microhydrated clusters of uracil and its halogenated compounds by multiphoton ionization,7 electron impact8,9 and multiply charged ions.4,5 These studies have outlined that hydration increases the stability of the aggregates, although some other fragmentation channels arise, and a significant energy transfer from the nucleobases to the neighboring water molecules occurs.In a previous work10 we have studied homogeneous uracil clusters in the gas phase and highlighted the key role of H-bonds in the organization of supramolecular aggregates. It was found that the formation of large clusters (up to 50 uracil molecules) is driven by the anisotropic distribution of H-bond donor and acceptor sites, which can be also modulated by weaker dispersion forces, particularly involving π-conjugate charge distributions. These findings overcame previous results involving few uracil molecules, suggesting that dispersion forces have a role in the transition toward the formation of globular clusters when more than ten molecules are involved. Here we study the effect of the environment by considering the aggregation of water-uracil admixtures, which is a major step ahead in the mimicking of a more realistic environment in which biomolecules, like uracil, are embedded and perform their functions. Mixed clusters indeed offer the opportunity to investigate a complex H-bond network in an amorphous cluster, that is a sort of a solution. The clusters contain uracil molecules, which have both hydrophobic and hydrophilic moieties, and water molecules, which have a strong tendency to fourfold tetrahedral coordination through donor and acceptor H-bond sites in bulk. Thus, one may expect that, depending on the relative number of uracil and water molecules, mixed clusters either assume a solid-state-like hydrated structure or approach liquid solvation. In the former case water molecules occupy defined positions in the hydrophilic sites partially replacing H-bond among uracil molecules, where in the latter the uracil molecules are dissolved in water, i.e. fully surrounded by a few hydration shells of water molecules, and minor or no interactions among the uracil molecules exist.
Core-level spectroscopies (X-ray Photoelectron Spectroscopy, XPS, Near Edge X-ray Absorption Spectroscopy, NEXAFS, and Auger Electron Spectroscopy, AES) are well suited probes for the exploration of structures and electronic properties of noncovalent molecular clusters. Among them, XPS has the advantage to be very sensitive to the local surroundings of the ionization site, making therefore affordable the investigation of site-specific properties even related to identical molecules, and to produce XPS spectra less congested than, for example, AES ones. However, assignment of the chemical shifts in core-level spectra, dominated by final-state charge relaxation and modulated by weak non-covalent interactions, meets insurmountable resolution limits in the description of the fine tuning of weak interactions in large, complex aggregates. Such experimental limits can be overcome by close comparison between measurements and atomistic simulations. The simulations, once properly benchmarked, can be indeed used to describe structures and properties of large aggregates at a precision unattainable by experiments, which use beams with a distribution of cluster sizes, as already shown in the case of homogeneous uracil clusters.10 In the present work, the inclusion of water in the formation of mixed water–uracil clusters produced by a gas aggregation cluster source11,12 has been first probed by C, N and O 1s XPS spectra, measured at the PLEIADES beamline of the SOLEIL synchrotron. Beside the comparison with the XPS spectra of uracil clusters and isolated uracil molecules,13 the structure of mixed uracil–water clusters is interpreted via thorough computational simulations. Wide-range structural studies of clusters of different size and composition have been performed using finite-temperature tight-binding molecular dynamics simulations14–16 extended up to 1ns (see ESI†) and completed by the calculation of core-ionization energies of each atom in the cluster obtained by Density Functional Theory, DFT, methods.17,18
Previous works19–26 investigated water arrangement, and structural and electronic properties of water–nucleobase aggregates. In a few cases then molecular dynamics simulations of isolated molecules or dimers in water solution have been performed, but they were extended only up to a few tens of ps. However the supersonic expansion used to produce the beams, lead to the formation of small clusters with a few water molecules. In this work we have used a gas aggregation source allowing the formation of larger and more hydrated clusters.4,5,12 Then at the end of a positive-feedback loop between measurements and simulations, our joint experimental–computational protocol is able to provide a sound and fine description of large, realistic nanoaggregates characterized by a complex network of interactions, illustrating a mixed phase that does resemble neither a hydrated crystal, nor the nucleation of a water solution.
2 Methods
2.1 Experimental methods
The experiments have been performed at the PLEIADES beamline at the French national synchrotron radiation facility SOLEIL, where a cluster source has been mounted in the dedicated Multi-Purpose Source Chamber (MPSC).27 To produce a beam of neutral molecular clusters in the gas phase a newly designed gas aggregation cluster source was used.12 The design was inspired by a cluster source used in the CIMAP Laboratory. In a gas aggregation source, clusters are formed in a liquid nitrogen-cooled condensation channel via three body collisions in the cold buffer gas, leading to the production of clusters with internal energies of about 80 K. The buffer gas also acts as a carrier, guiding the cluster beam through a skimmer towards the collision chamber where it crosses the X-ray beam at right angle. Both the source and the skimmer position can be remotely controlled to align the cluster beam to the photon beam. The molecular uracil vapor is produced by evaporation of a commercial powder (Sigma Aldrich; purity 99%) in a resistively heated oven placed inside the source filled with He as buffer gas at the pressure of a few mbar. The temperature of the oven was 183 °C. In order to produce hydrated species, the He buffer gas flows inside a Nafion tube immersed in ultra pure water as proposed by Kočišek et al.8,9 Nafion membranes are permeable to water, thus water molecules can pass through the membrane and humidify the buffer gas. The photon beam is produced by a permanent magnet APPLE II undulator (80 mm period) in combination with 600 L mm−1 grating of the modified Peterson plane grating monochromator. The ejected photoelectrons have been detected by a Scienta R4000 electron energy analyzer equipped with a wide-angle entrance lens (±30° aperture). In the present experiment, the analyzer has been operated at 200 meV energy resolution.The C, N and O 1s XPS spectra were recorded at photon energies about 60 eV above their respective ionization thresholds. The monochromator slit was set to 200, 250 and 100 μm for respectively C 1s, N 1s and O 1s, which in combination with the analyzer pass energy of 0.1 eV and the appropriate slit, resulted in an overall spectral resolution of 0.5, 0.8 and 0.6 eV, respectively. The energy spectra were calibrated using a mixture of the molecule under study and a calibration gas (separately introduced as diffuse gas) with known XPS spectra, i.e. CO2 (C 1s, v = 0 and O 1s)28,29 and N2 (N 1s).30 Note that the two-dimensional XPS imaging allows to distinguish XPS lines produced by a focused cluster beam, from those produced by the calibration gas that fill the entire interaction region.31
2.2 Theoretical methods
Atomistic simulations of hydrated uracil clusters have been carried out by following a multilevel protocol: large scale exploration of the potential energy surfaces and long (up to ns) molecular dynamics simulations have been performed by using a semiempirical tight-binding approach. We have reported elsewhere that tight binding simulations of uracil aggregates provide results in very good agreement with those obtained by using ab initio simulations.10 Accurate thermodynamic and spectroscopic properties of a wide subset of systems and configurations have been then calculated by using ab initio simulations based on density functional theory. In the former case, the GFN2-xTB Hamiltonian14 has been used either to perform molecular dynamics simulations, or as engine for the search of minimum energy configurations through an automated conformer-rotamer ensemble sampling tool (CREST).15,16 In the latter case, DFT simulations at the dispersion-corrected B3LYP-D3 level of theory have been performed in a plane-wave/pseudopotential framework by using the Quantum ESPRESSO suite of programs.17,19 A complete account of our computational setup, including all the investigated structures and configurations, is provided in the ESI.†3 Results and discussion
The XPS spectra of mixed water–uracil clusters, measured at around 60 eV above the ionization thresholds of the different atomic species, are reported in Fig. 1 together with those measured for the uracil clusters and isolated uracil molecules.13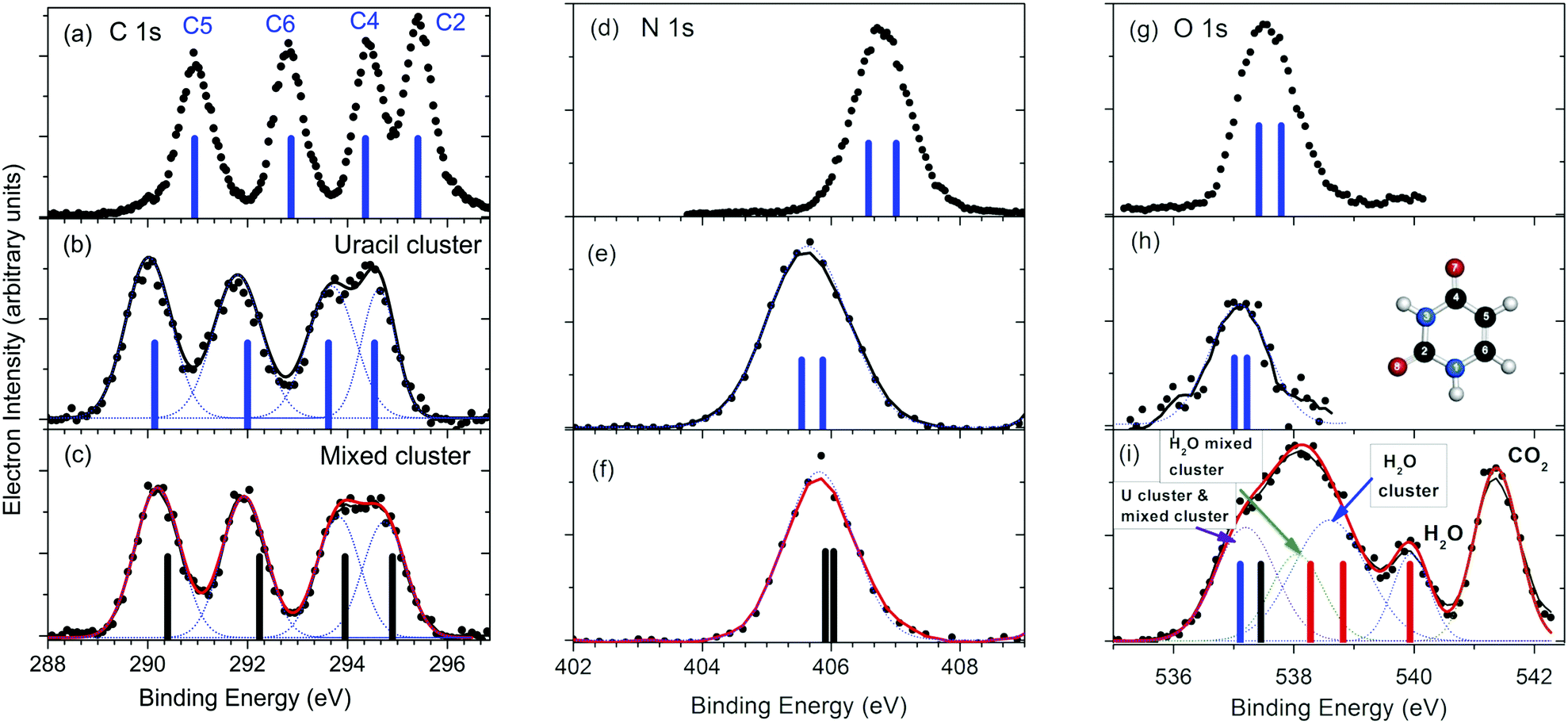 | ||
| Fig. 1 Comparison between the C, N and O 1s XPS spectra for the isolated uracil molecule (a, d and g),13 uracil clusters (b, e and h) and mixed water–uracil clusters (c, f and i). The bars in the spectra of the uracil molecule and uracil cluster correspond to the average calculated BEs values,10 (see Table S1 of ESI†), while the ones in the spectra of the mixed clusters represent the present calculated values (see Table S1 of ESI†). In the case of the O 1s spectrum of the mixed water–uracil cluster (i) the contributions of the different components, including the one due to CO2 used for the calibration of the BE scale, are also shown. The blue and black bars assigned to the combined O 1s contribution of the uracil and mixed clusters, respectively, correspond to the average of the calculated BEs for O7 and O8 in both species, while the red bars correspond to the water O 1s BE in water molecules, water clusters and mixed water–uracil clusters. In panel (h) the scheme of the uracil molecule is shown. | ||
The uracil ring is formed by two nitrogen atoms in positions 1 and 3 and four carbon atoms (see inset in Fig. 1h). The C2 and C4 atoms form carbonyl groups with O8 and O7 atoms, respectively, while C5 and C6 are conjugated methine units. In the isolated molecule (Fig. 1a), the resulting C 1s XPS spectrum displays four distinct features due to the different chemical connections, with the two at higher binding energy (BE) slightly overlapping. The C2 atom has the highest BE among the four non-equivalent C atoms, because the larger electronegativity of the neighboring O and N atoms induces the strongest charge depletion. Vice versa the peak at the lowest BE has been assigned to C5,13 situated in the middle of the conjugated C4–C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6 moiety. The two central peaks have been assigned to C4 and C6,13 whose shifts depend on the neighboring O and N or only N atom, respectively. All of these assignments have been confirmed by our ab initio calculations (see Table S1 in ESI†). In the case of the homogeneous uracil clusters, the whole C 1s spectrum (Fig. 1b) is broadened and shifted by about 0.9 eV towards lower BE, with the C4 and C2 features almost overlapping.
C6 moiety. The two central peaks have been assigned to C4 and C6,13 whose shifts depend on the neighboring O and N or only N atom, respectively. All of these assignments have been confirmed by our ab initio calculations (see Table S1 in ESI†). In the case of the homogeneous uracil clusters, the whole C 1s spectrum (Fig. 1b) is broadened and shifted by about 0.9 eV towards lower BE, with the C4 and C2 features almost overlapping.
Each one of the N and O 1s spectra in the molecule (Fig. 1d and g), is expected to contain two non-equivalent contributions, with predicted separation of 0.43 eV and 0.37 eV, respectively, in the isolated molecule. Similarly to the C 1s spectrum also the N and O 1s features (Fig. 1e and h) in the uracil clusters are broadened and shifted to lower BE with respect to the molecular case. In the case of mixed uracil–water clusters, the features of the C 1s spectrum (Fig. 1c) display a small, but clear shift (≤200 meV) (see Fig. 3 in ESI†) towards higher BE with respect to the homogeneous uracil clusters. A small variation in shape is observed in the region of the C4 and C2 components, the two atoms involved in the formation of the CO carbonyl groups which experience more changes in the local environment due to the formation of H-bonds with water molecules. In the case of the N 1s spectrum (Fig. 1f) the peak appears to be slightly shifted towards higher BE (200 meV) and narrower than the one of the uracil clusters. In the region of the O 1s binding energy (Fig. 1i), the XPS spectrum is quite different from the uracil cluster one. It is characterized by a broad feature and a peak on the high binding energy flank of the feature, likely ascribed to the sum of a few contributions: isolated water molecules, clearly identified by the peak at 539.9 eV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 accompanied by the unresolved contributions of pure water clusters and of water and uracil molecules of the mixed clusters. In detail, we assign the O 1s contribution of pure water clusters at about 1.3 eV below the molecular peak, in agreement with previous results reported by Örhwall et al.,34 who determined a peak-to-peak difference in the molecular and cluster binding energies of 1.3 and 1.6 eV for small and large water clusters, respectively. The peak shifted around 0.5 eV to lower binding energy is attributed to water molecules in mixed clusters. In this case, the observed shift is due to the direct connection of water molecules with CO acceptor and N–H donor partners in H-bonds, with the electron-rich, conjugated uracil molecules offering a better screening of the water core hole than water molecules themselves. Then, a further feature, which can be assigned to the O 1s of the uracil molecules in both the uracil and mixed water–uracil clusters, contributes to the low BE part the broad structure. The centroid of this feature is at about 100 meV higher BE than the O 1s peak in the uracil cluster (see Table S2 in ESI†). Attempts to account for the two separate contributions in this region of the spectrum produce a fit with undetermined positions of the peaks, even though the clearly identified contribution of water oxygen in mixed water/uracil clusters confirms the formation of mixed clusters in the sample.
33 accompanied by the unresolved contributions of pure water clusters and of water and uracil molecules of the mixed clusters. In detail, we assign the O 1s contribution of pure water clusters at about 1.3 eV below the molecular peak, in agreement with previous results reported by Örhwall et al.,34 who determined a peak-to-peak difference in the molecular and cluster binding energies of 1.3 and 1.6 eV for small and large water clusters, respectively. The peak shifted around 0.5 eV to lower binding energy is attributed to water molecules in mixed clusters. In this case, the observed shift is due to the direct connection of water molecules with CO acceptor and N–H donor partners in H-bonds, with the electron-rich, conjugated uracil molecules offering a better screening of the water core hole than water molecules themselves. Then, a further feature, which can be assigned to the O 1s of the uracil molecules in both the uracil and mixed water–uracil clusters, contributes to the low BE part the broad structure. The centroid of this feature is at about 100 meV higher BE than the O 1s peak in the uracil cluster (see Table S2 in ESI†). Attempts to account for the two separate contributions in this region of the spectrum produce a fit with undetermined positions of the peaks, even though the clearly identified contribution of water oxygen in mixed water/uracil clusters confirms the formation of mixed clusters in the sample.
The analysis of the measured XPS spectra has been performed through a careful computational protocol, based on a multilevel theoretical approach and thoroughly detailed in the Methods section and in the ESI.† First, a wide-ranging investigation based on tight-binding molecular dynamics simulations using the xTB suite of programs14–16 has been performed to explore and find stable configurations of clusters at finite temperature and across different sizes and uracil/water compositional ratios. Then, a selected, but still very wide subset of such configurations has been studied by DFT simulations using the Quantum ESPRESSO suite of programs17,18 in a plane-wave/pseudopotential framework, to obtain refined structural properties of the clusters and to calculate core ionization energies of all involved atoms. This approach has been successfully employed in the XPS studies of several molecules35–37 and clusters,10 in particular in the case of uracil and its halogenated derivatives.35 This theoretical approach provides results comparable with calculations performed using all-electron ΔSCF approach38 or the fourth-order algebraic-diagrammatic construction (ADC(4)) approximation scheme for the one-particle Greens’ function adapted for the case of K-shell ionization.39 To understand how the different interactions in the cluster affect the observed binding energies, a bottom-up approach has been followed. In detail, several different cluster sizes (from one to twelve uracil molecules, interacting with a number of water molecules variable between one and thirty-six) have been considered and their structures fully optimized in the gas phase, with the BE of each atom calculated, as reported in Table S1 of ESI.†
In Fig. 2, we summarize the average calculated BE of the different atoms in the uracil molecules of the cluster as a function of the number of uracil and water molecules. Water and uracil O 1s BE values are displayed using red and black open circles, respectively. The addition of water molecules generally results in a shift of the BE with respect to that of the corresponding uracil clusters, also reported as blue bars in the same figure.
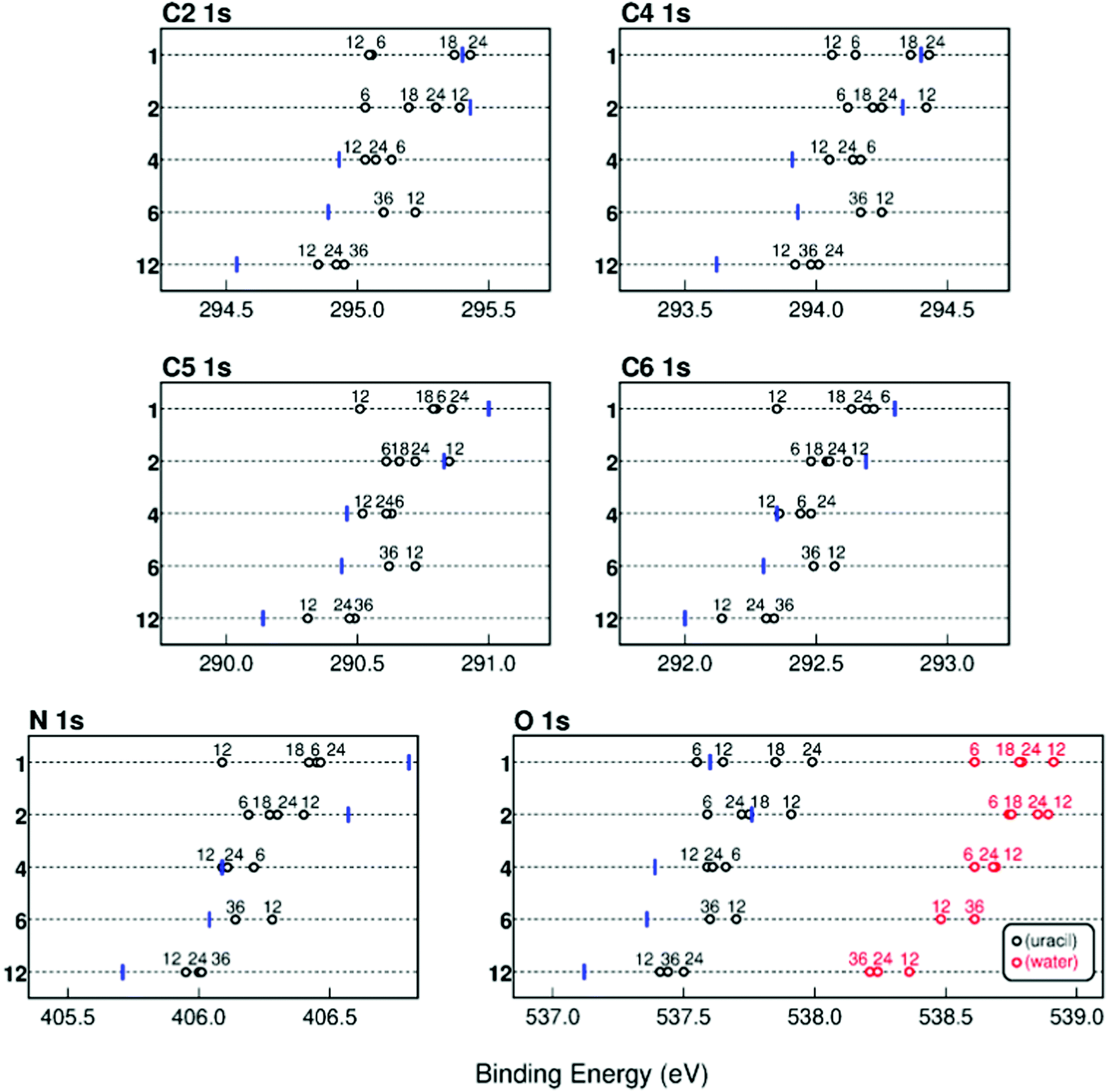 | ||
| Fig. 2 Average calculated BEs of the different atoms in the uracil molecule in mixed clusters when the number of uracil (reported on the vertical axis)32 and water (numbers on the top of the open circles) molecules is varied from one to twelve (blue bars) and from six to thirty six (black open circles), respectively. In the case of both the N and O 1s atoms the average position of the two components is reported. In the case of the O atom also the value of the BE of the O 1s from the water molecules (red open circles) in the mixed clusters is shown. | ||
It stands out that the calculated shifts do not follow a monotonic behavior with respect to the number of water molecules, but for larger clusters the BE of all the atoms decreases with respect to the one of the uracil cluster. This is a clear indication that the addition of water affects the original structure of the uracil cluster changing the connectivity among the molecules. To explain more clearly such behavior in Fig. 3 the formation energy per mass unit of a uracil dimer, to which up to twenty-four water molecules are added, is reported and selected stable structures are shown. Starting from the most stable planar structure of the uracil dimer with a bidirectional and symmetric H-bond,40,41 we see that the most stable structure of the dimer is still planar with a few water molecules around (e.g., two water molecules in structure 1 in Fig. 3), but then it becomes stacked when four water molecules are added (structure 2 in Fig. 3). Addition of further water molecules results in the building up of a 3D network of water molecules, with the two uracil molecules in a stacked configuration on a side (16 water molecules, structure 3 in Fig. 3).21
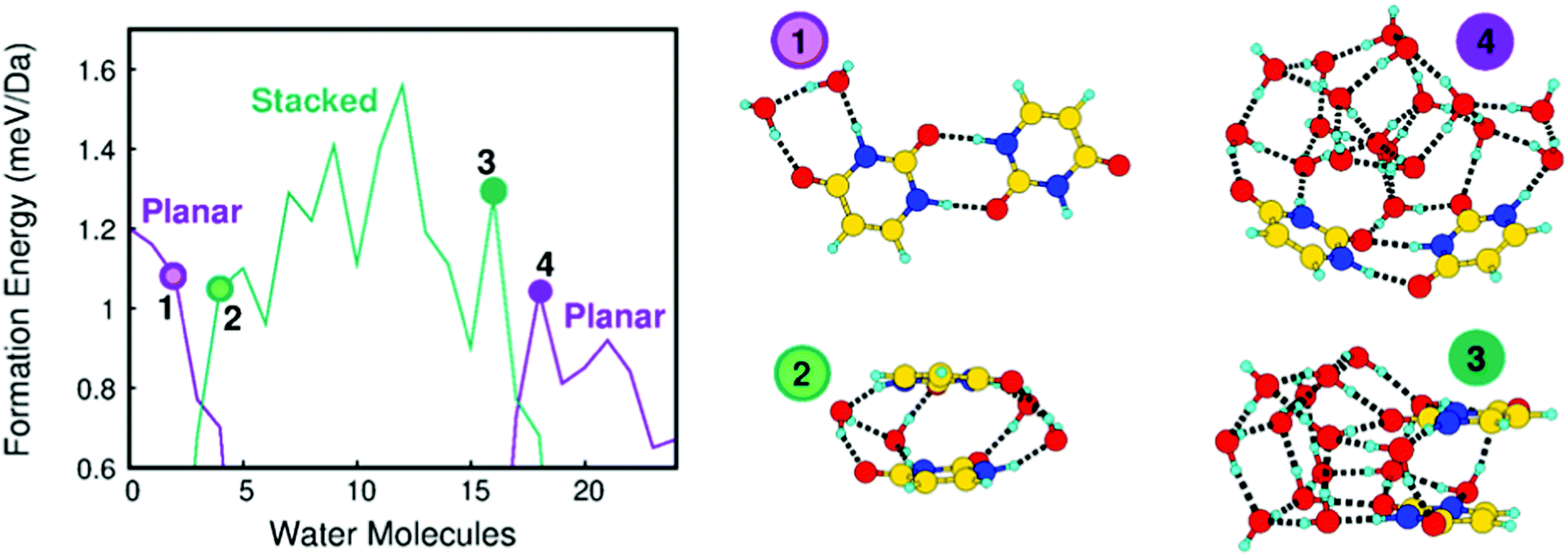 | ||
| Fig. 3 Formation energy per mass unit versus number of water molecules in a mixed cluster with two uracil molecules (left panel). The most stable structures of mixed clusters with two uracil molecules and n (n = 2, 4, 16 and 18) water molecules (right panel). | ||
This situation changes when the number of water molecules becomes larger than sixteen. In this case a planar-like configuration of the two uracil molecules is restored on the external surface of the large water cluster (18 water molecules, structure 4 in Fig. 3). The trend of the formation energy of such a relatively simple system already outlines five properties of mixed clusters that can be found also in larger system, albeit not so clearly spotted: (i) a few water molecules tend to saturate first CO and N–H uracil sites, which can behave as H-bond donor or acceptors; (ii) further water molecules added to the system tend to maximize their connectivity by forming the maximum number of inter-water H-bonds, thus leading to an inversion of the stability of planar and stacked dimers with respect to uracil dimers; (iii) the strong cohesion of water molecules in the gas phase leads to the formation of nearly separated water and uracil subsystems, connected through H-bonds; (iv) large water clusters activate a stronger dispersion interaction with a uracil dimer which recovers a (almost) planar structure and wraps the water cluster; (v) water molecules tend to maximize connectivity, thus resulting in an oscillating trend of the formation energy corresponding to particularly (un)favorable numbers of molecules. This complex behavior is manifested by the predicted values of the BE, as observed in Fig. 2 where the shift introduced by the variation of the number of water molecules in the cluster changes sign going from 6 to 24 water molecules.
A similar situation is observed also in the case of larger clusters, as shown in Fig. 4 for the mixed clusters built on twelve uracil molecules. In the uracil cluster, 12 uracil molecules are in number large enough to form a stable globular cluster (blue arrow 1 in Fig. 4c). The total energy is minimized by the maximization of the intermolecular connectivity through the formation of CO⋯H–N hydrogen bonds. In Fig. 4a, we show the changes in the distribution of such inter-uracil hydrogen bonds across 0.5 ns of molecular dynamics simulations caused by the insertion of water molecules. The main features of the distribution, peaked at around 2.8 Å and 4.9 Å and indicated by magenta and light-blue dots in the figure, correspond to the H-bond descriptor between directly connected molecules (AB distances in Fig. 4b) and to the first O⋯N distance not corresponding to the formation of H-bonds (AC distances), respectively. Thus, the higher the contribution of the 2.8 Å peak the larger is the number of nearby uracil molecules. Increasing the number of water molecules up to 36, the inter-uracil connectivity drops to the minimum of AB distances (12u + 36w yellow curve in Fig. 4a). In terms of cluster shape, this drop starts with sparse water molecules at the surface of the uracil cluster saturating dangling H bonds (red arrow 1, 12u + 6w in Fig. 4c). Water molecules immediately tend to assemble in core water clusters within uracil shells (red arrows 2 and 3; 12u + 12w and 12u + 24w), with a substantial drop of inter-uracil connectivity, which reaches its minimum at 12u + 36w. In this cluster, water molecules are still inside the cluster, but completely split the uracil cluster (red arrow 4) in poorly connected units. A complementary tendency clearly emerges (blue arrows 2) with the orientation of the hydrophobic –CHCH– side of uracil outside the cluster. The addition of further water molecules leads to the segregation of a compact, globular water cluster (red arrows 5 and 6; 12u + 48w and 12u + 60w) connected to a thin, almost hemispherical uracil wrapper (blue arrow 3), which recovers part of the initial connectivity (compare, e.g. the 12u + 36w yellow curve with the 12u + 60w red curve).
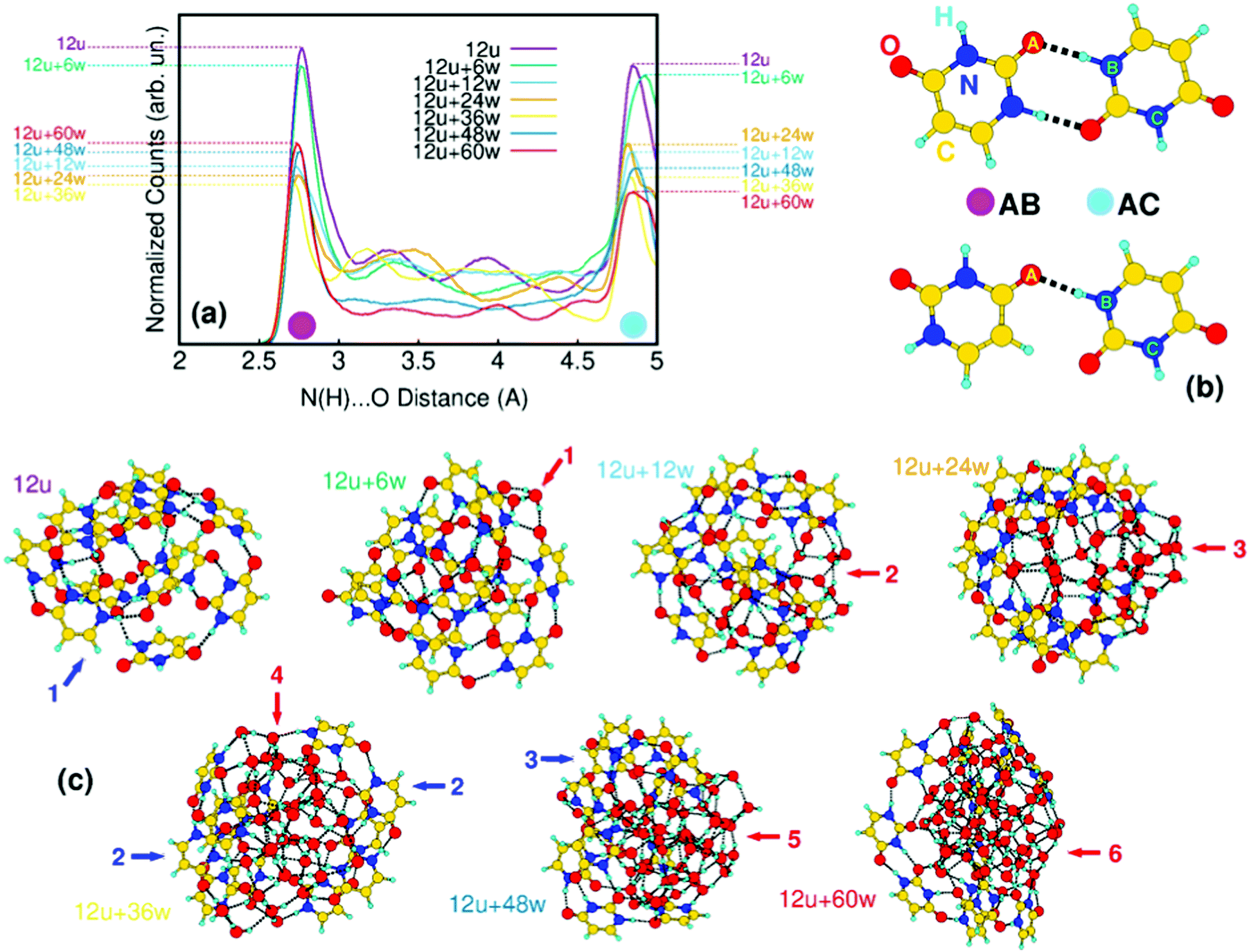 | ||
| Fig. 4 (a) Normalized distributions of inter-uracil (N(H)⋯OC) distances averaged along 0.5 ns of molecular dynamics simulations of mixed clusters containing 12-uracil molecules and variable amounts (0–60) of water molecules. (b) Most relevant interaction patterns between uracil molecules, represented by bidirectional (upper) and monodirectional (lower) (N(H)⋯OC) hydrogen bonds; AB and AC distances represent the main peaks of panel (a). (c) Most stable structures found by applying the CREST algorithm in the investigated 12u + nw mixed cluster. Blue and red arrows refer to the evolution of water and uracil connectivity along the structures, as discussed in the text. | ||
The loop is closed by comparing XPS lines calculated in the case of selected uracil-water structures with measured data. 12 uracil molecules with the addition of a few dozens of water molecules are indeed a sensible upper limit for this kind of calculations, yet clusters of such size provide an already high compatibility with measurements, representing therefore an educated guess of real hydrated uracil clusters. The average calculated BEs are represented by black and red bars in the bottom panels of Fig. 1(c, f and i). A further view of the comparison between the calculated and measured BEs is given in Fig. 5, where the shifts between the XPS peaks in the uracil molecule and uracil cluster (Fig. 5a) and in the mixed and uracil cluster (Fig. 5b) are shown. As already observed,10 an excellent agreement between calculation and measurements is found in the case of uracil cluster. An overall satisfactory agreement with an average difference between theory and experiment of 200 meV is found also in the case of the mixed clusters. In the latter case, for all the atoms calculations appear to predict a BE higher than the measured one. This might be due to a not negligible content of uracil clusters in the beam. Here we note that in the case of the uracil clusters the existence of different types of O⋯H–N hydrogen bonds (mono- and bidirectional with different orientations)10 can lead to different shifts of the BE of the C2 and C4 peaks depending on the chemical environment of each atom. This manifests in a broadening of the peaks, which are not anymore resolved as in the uracil molecule. The situation gets worse in the case of the mixed cluster, where the presence of water molecules increases the difference in the chemical environments of the C2 and C4 sites with respect to the ones of the C5 and C6 sites. By comparing the structure of the cluster of 12 uracil molecules with the one of the mixed cluster it is clear that the number of the NH groups not involved in the H-bond decreases as the number of water molecules increases. This produces the observed narrowing of the N 1s peak. Finally, the predicted values of the average O 1s BE in the uracil molecules of mixed and uracil clusters are about 300 meV apart. Considering the expected broadening of the features this value hampers the separation of the two features in the experiment.
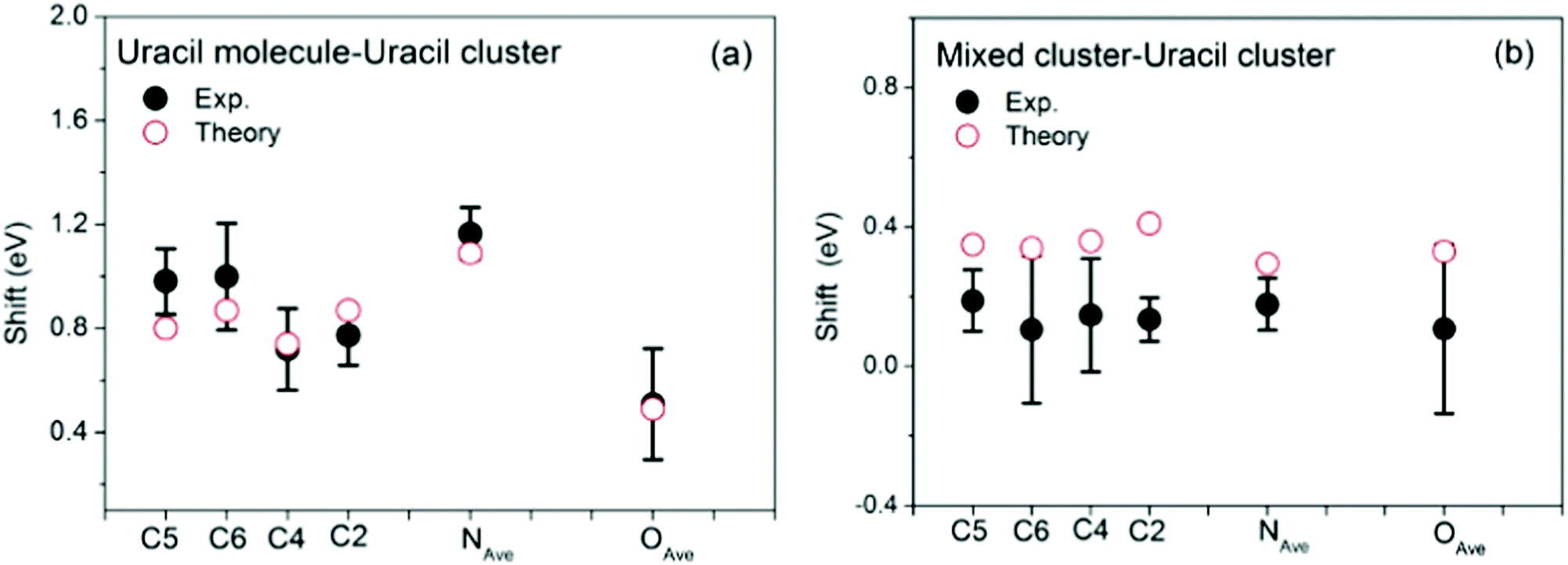 | ||
| Fig. 5 Comparison of the measured (full dots) and calculated (open circles) BE shifts between the molecule and the uracil cluster (a) and between the mixed water-uracil and uracil clusters (b). The theoretical average BEs (see Table S1 in ESI†) have been used. In the case of N and O 1s the experiment does not resolve the two components, thus in the comparison the average value of the theoretical N1 and N3 and O7 and O8 BEs is used. | ||
To shed light on the role of intermolecular interactions in the screening of core holes and to the primary role of noncovalent connectivity in the determination of electronic properties of hydrated clusters, we have calculated charge difference-density maps like those shown in Fig. 6 in the case of a homogeneous uracil tetramer (4u) and a hydrated tetramer (4u + 6w). The maps show the electronic valence density difference between ground and core-ionized states, allowing to follow the displacement of charge density (from blue to red regions) induced by the formation of a core hole. In particular, we focus on the ionization of one of the O8 uracil oxygen atoms, and we compare it to the ionization of a water oxygen in the case of a mixed cluster. These examples have been chosen because they allow a relatively simple graphical representation as well as the comparison between uracil and water behavior. As a general feature of such kind of non-covalent aggregates, one can clearly see that in the homogenous uracil cluster the major contribution to the screening of the core hole comes from an intramolecular contraction of the charge density, highlighted by blue arrows in Fig. 6. The main contribution of the surrounding molecules to the screening of the core hole, highlighted by the leftmost green arrow, acts through the CO⋯H–N hydrogen bond. Further details appear when a few water molecules are mixed into the homogeneous system. In the case of the uracil core–hole (middle part of Fig. 6), the sp2 hybridization of O is clearly visible as the screening along the H bonds involves one water molecule and one N–H moiety of a neighboring uracil (green ellipses and arrows). Moreover, the tendency of water to form tight H-bonded aggregates can be acknowledged by observing that all the water molecules react to the formation of the core hole on uracil by promoting small rearrangements of sigma charge along the dense network of H bonds. When the core hole is induced on a water O atom (right part of the figure), the sp3 hybridization becomes clearly visible (green ellipses and arrows) through tetrahedral connectivity of the water molecules involving two donor (H atoms of neighboring water molecules) and two acceptor (CO moieties of neighboring uracil molecules) H-bond partners. If we extrapolate the screening patterns to larger clusters (e.g., those shown in Fig. 4) to the interpretation of the crowded O 1s XPS lines shown in Fig. 1i), we note that two opposite tendencies act in mixed clusters. Water tends to strongly aggregate inside mixed clusters, almost inducing a core–shell organization of the system when suitable amounts of molecules are used. This is compatible with an extra-screening of O 1s core holes in water molecules, particularly induced by proximity with the electron-rich CO groups and compatible with an average red shift of around 400 meV for the O 1s peak of water in mixed cluster with respect to homogeneous water clusters. On the contrary, water has a twofold, if tiny, effect on O 1s uracil lines: water molecules reduce CO⋯H–N connections and substitute partially conjugated N–H groups35 with more electronegative and non-conjugated O–H groups, leading to the observed blue shift of the uracil O 1s line, compared with homogeneous clusters.
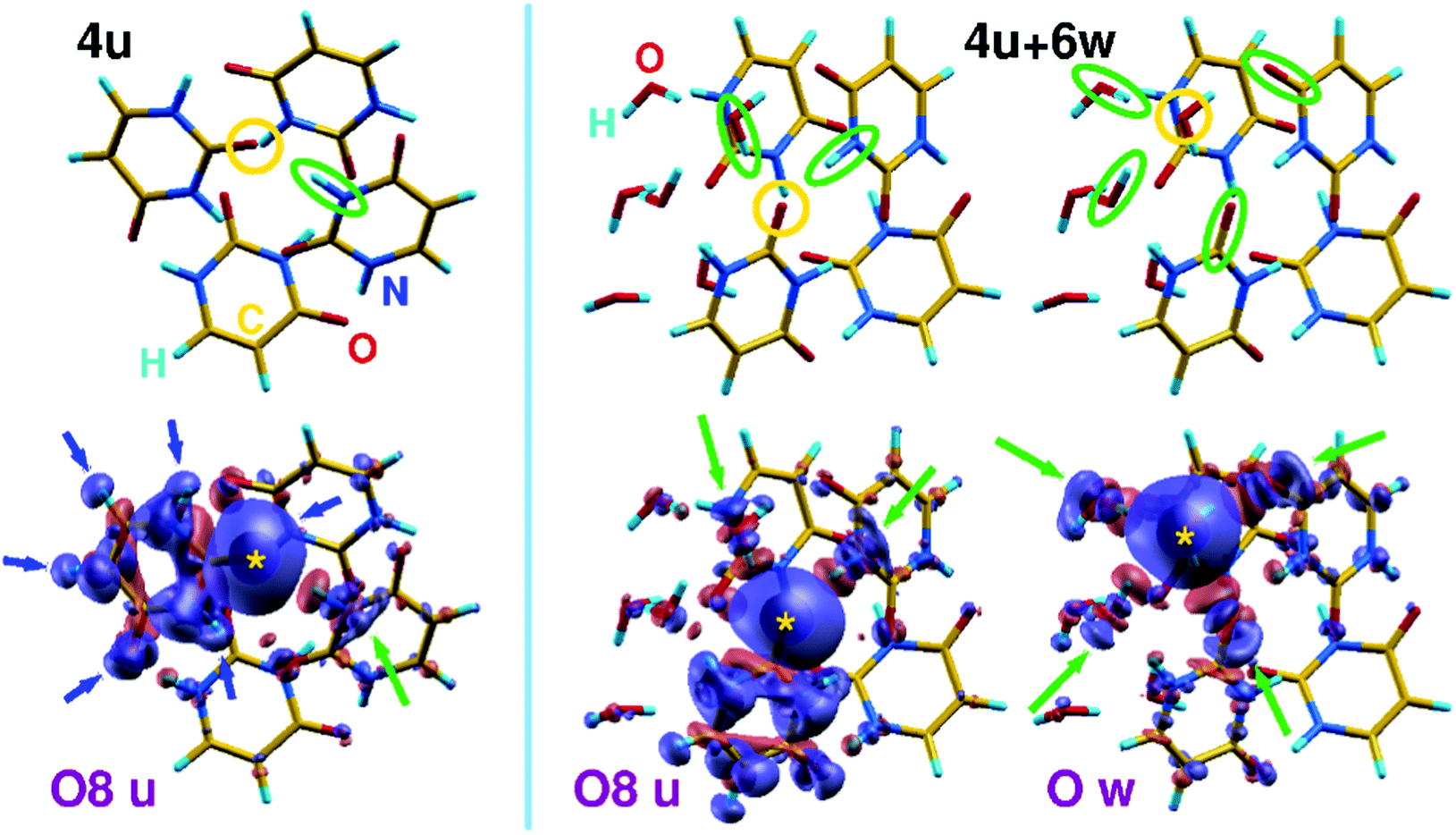 | ||
| Fig. 6 Charge difference-density plots between ground- and core-ionized electronic states of (left) uracil tetramer (4u) and (right) uracil tetramer plus six water molecules (4u + 6w). Charge accumulation (in red) and depletion (in blue) regions are sampled by using 0.0002 a.u. bohr−3 isosurfaces. For the sake of clarity, O atoms where the core hole is localized are indicated by yellow circles in upper skeletal models and by yellow stars in lower density-difference maps. Green ellipses and arrows indicate intermolecular contributions to the screening of the core hole, as discussed in the text. | ||
4 Conclusion
A very close link between experiment and simulation has been established in the investigation of mixed water–uracil clusters in the gas phase. Our results rely on the facts that photoemission measurements of clusters formed by unknown admixtures of uracil, a prototypical biological molecule, and water, solvent of choice of biological environment, molecules strictly require theoretical interpretation, and that theoretical simulations can claim to have identified realistic structures only when they closely reproduce measurements. Such a link has been shaped through the application of a careful computational protocol based on multilevel atomistic simulations. First, a series of large scale explorations conducted by finite temperature molecular (meta)dynamics simulations has allowed to discover the severe variation of the structure of the clusters as the number of water molecules increases. Then, refined structural properties and XPS spectra have been computed using ab initio calculations and compared with measurements until a satisfactory agreement was reached, and the electronic properties of selected systems have been explored to shed light on quantum mechanical effects which accompany the formations of core hole in non-covalent heterogeneous systems. Finally, long molecular dynamics simulation of the most realistic structures have been performed to describe in detail the effect of water added to the clusters on the intramolecular connectivity of uracil molecule, ultimate responsible of changes in XPS spectra. The results focus with unmatched resolution upon the interesting evolution of such prototypical system of small biomolecular nanoaggregates in contact with water molecules outside of a water environment. The presence of hydrophobic moieties, which are ubiquitous even in water-soluble organic molecules, leads in clusters of increasing size to the formation of a core–shell-like supramolecular organization. This peculiar phase resembles neither the uniform mixing typical of hydrated crystals, nor the sharp separation of solute molecules by means of solvent molecules. In this phase, water exploits its strong, four-fold donor–acceptor H-bond coordination in the smallest cluster to form tightly packed cores, while uracil is better suited to form a shell around the core, with its hydrophilic side bonded to water and its hydrophobic side stabilizing the cluster against vacuum. Such a configuration is unprecedent, to the best of our knowledge, for an organic molecule with both hydrophilic and hydrophobic sites, rather resembling a few examples of completely hydrophobic organic species, such as adamantane, surrounding small water clusters transported by helium nanodroplets42 or the core–shell structures found in the mixed water-ammonia clusters.43 New self-organization patterns, such as those unraveled here, may disclose new routes for controlling the deposition or delivery of precise amount of molecular blends by means of their insertion in a controlled environment, preserving their size and composition. This is at variance with the most common deposition methods, like for example molecular beam epitaxy, where the patterns of the deposited molecules depend on the interaction with the surface and temperature. Of course this needs the capability to produce and control clusters of selected composition. The recent successful attempts of producing micro-hydrated nucleobases reported in the literature8,9 may pave the way towards this development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is based upon work from COST Action CA18212 – Molecular Dynamics in the GAS phase (MD-GAS), supported by COST (European Cooperation in Science and Technology) and it has been partially supported by CNRS PICS Project BIFACE, and International Associated Laboratory ‘DYNAMO’, the PRIN 20173B72NB ‘Predicting and controlling the fate of bio-molecules driven by extreme-ultraviolet radiation’. C. N. is co-funded by Région Normandie and Synchrotron SOLEIL. SOLEIL support is acknowledged under project no. 20171346.References
- N. Ottosson, K. J. Børve, D. Spângberg, H. Bergersen, L. J. Sæthre, M. Faubel, W. Pokapanich, G. Öhrwall, O. Bjölrneholm and B. Winter, J. Am. Chem. Soc., 2011, 133, 3120–3130 CrossRef CAS PubMed.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1–18 RSC.
- T. Schlathölter, F. Alvarado, S. Bari, A. Lecointre, R. Hoekstra, V. Bernigaud, B. Manil, J. Rangama and B. Huber, ChemPhysChem, 2006, 7, 2339–2345 CrossRef PubMed.
- P. Markush, P. Bolognesi, A. Cartoni, P. Rousseau, S. Maclot, R. Delaunay, A. Domaracka, J. Kočišek, M. C. Castrovilli and B. A. Huber, et al. , Phys. Chem. Chem. Phys., 2016, 18, 16721–16729 RSC.
- M. Castrovilli, P. Markush, P. Bolognesi, P. Rousseau, S. Maclot, A. Cartoni, R. Delaunay, A. Domaracka, J. Kočišek and B. Huber, et al. , Phys. Chem. Chem. Phys., 2017, 19, 19807–19814 RSC.
- N. J. Kim, H. Kang, G. Jeong, Y. S. Kim, K. T. Lee and S. K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4841–4843 CrossRef CAS PubMed.
- B. Barc, M. Ryszka, J. Spurrell, M. Dampc, P. Limão-Vieira, R. Parajuli, N. Mason and S. Eden, J. Chem. Phys., 2013, 139, 244311 CrossRef CAS PubMed.
- J. Kočišek, A. Pysanenko, M. Fárnk and J. Fedor, J. Phys. Chem. Lett., 2016, 7, 3401–3405 CrossRef PubMed.
- J. Poštulka, P. Slaviček, J. Fedor, M. Fárnk and J. Kočišek, J. Phys. Chem. B, 2017, 121, 8965–8974 Search PubMed.
- G. Mattioli, L. Avaldi, P. Bolognesi, J. D. Bozek, M. C. Castrovilli, J. Chiarinelli, A. Domaracka, S. Indrajith, S. Maclot and A. R. Milosavljević, et al. , Sci. Rep., 2020, 10, 1–6 CrossRef PubMed.
- H. Haberland, Clusters of atoms and molecules: theory, experiment, and clusters of atoms, Springer Science & Business Media, 2013, vol. 52 Search PubMed.
- C. Nicolafrancesco, S. Hartweg, J.-F. Gil, E. Robert, J.-M. Ramillon, C. Nicolas, S. Indrajith, J. Bozek, L. Nahon and A. R. Milosavljević, et al. , Eur. Phys. J. D, 2021, 75, 1–10 CrossRef.
- V. Feyer, O. Plekan, R. Richter, M. Coreno, G. Vall-Llosera, K. C. Prince, A. B. Trofimov, I. L. Zaytseva, T. E. Moskovskaya and E. V. Gromov, et al. , J. Phys. Chem. A, 2009, 113, 5736–5742 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, et al. , Phys. Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al. , J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- M.-P. Gaigeot and M. Ghomi, J. Phys. Chem. B, 2001, 105, 5007–5017 CrossRef CAS.
- X. Bao, H. Sun, N.-B. Wong and J. Gu, J. Phys. Chem. B, 2006, 110, 5865–5874 CrossRef CAS PubMed.
- M.-C. Bacchus-Montabonel and F. Calvo, Phys. Chem. Chem. Phys., 2015, 17, 9629–9633 RSC.
- A. Furmanchuk, O. Isayev, O. V. Shishkin, L. Gorb and J. Leszczynski, Phys. Chem. Chem. Phys., 2010, 12, 3363–3375 RSC.
- A. Furmanchuk, O. V. Shishkin, O. Isayev, L. Gorb and J. Leszczynski, Phys. Chem. Chem. Phys., 2010, 12, 9945–9954 RSC.
- A. Semmeq, S. Ouaskit, A. Monari and M. Badawi, Phys. Chem. Chem. Phys., 2019, 21, 4810–4821 RSC.
- A. Semmeq, M. Badawi, A. Ouaskit, O. Said and S. Ouaskit, Chem. – Eur. J., 2020, 26, 11340–11344 CrossRef CAS PubMed.
- B. Milovanović, M. Kojić, M. Petković and M. Etinski, J. Chem. Theory Comput., 2018, 14, 2621–2632 CrossRef PubMed.
- A. Lindblad, J. Söderström, C. Nicolas, E. Robert and C. Miron, Rev. Sci. Instrum., 2013, 84, 113105 CrossRef PubMed.
- T. Hatamoto, M. Matsumoto, X.-J. Liu, K. Ueda, M. Hoshino, K. Nakagawa, T. Tanaka, H. Tanaka, M. Ehara and R. Tamaki, et al. , J. Electron Spectrosc. Relat. Phenom., 2007, 155, 54–57 CrossRef CAS.
- T. D. Thomas and R. W. Shaw Jr, J. Electron Spectrosc. Relat. Phenom., 1974, 5, 1081–1094 CrossRef CAS.
- B. Kempgens, A. Kivimäki, M. Neeb, H. Köppe, A. Bradshaw and J. Feldhaus, J. Phys. B: At. Mol. Phys., 1996, 29, 5389 CrossRef CAS.
- D. Danilović, D. K. Božanić, R. Dojćilović, N. Vukmirović, P. Sapkota, I. Vukašinović, V. Djoković, J. Bozek, C. Nicolas and S. Ptasinska, et al. , J. Phys. Chem. C, 2020, 124, 23930–23937 CrossRef.
- At variance with the results reported in ref. 10 the calculated C2 and O 1s BEs of the uracil dimer are higher than the ones of the molecule. This is because here the BE of the most stable structure has been calculated, while in ref. 10 the optimized structure of a dimer cut from the crystallographic structure of uracil were used.
- R. Sankari, M. Ehara, H. Nakatsuji, Y. Senba, K. Hosokawa, H. Yoshida, A. De Fanis, Y. Tamenori, S. Aksela and K. Ueda, Chem. Phys. Lett., 2003, 380, 647–653 CrossRef CAS.
- G. Öhrwall, R. Fink, M. Tchaplyguine, L. Ojamäe, M. Lundwall, R. Marinho, A. Naves de Brito, S. Sorensen, M. Gisselbrecht and R. Feifel, et al. , J. Chem. Phys., 2005, 123, 054310 CrossRef PubMed.
- M. Castrovilli, P. Bolognesi, E. Bodo, G. Mattioli, A. Cartoni and L. Avaldi, Phys. Chem. Chem. Phys., 2018, 20, 6657–6667 RSC.
- F. Rondino, D. Catone, G. Mattioli, A. A. Bonapasta, P. Bolognesi, A. R. Casavola, M. Coreno, P. O'Keeffe and L. Avaldi, RSC Adv., 2014, 4, 5272–5282 RSC.
- P. Bolognesi, G. Mattioli, P. O'Keeffe, V. Feyer, O. Plekan, Y. Ovcharenko, K. Prince, M. Coreno, A. Amore Bonapasta and L. Avaldi, J. Phys. Chem. A, 2009, 113, 13593–13600 CrossRef CAS PubMed.
- G. t. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- L. S. Cederbaum, W. Domcke and J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1980, 22, 206 CrossRef CAS.
- J. A. Frey, A. Müller, M. Losada and S. Leutwyler, J. Phys. Chem. B, 2007, 111, 3534–3542 CrossRef CAS PubMed.
- M. Pitoňák, K. E. Riley, P. Neogrády and P. Hobza, ChemPhysChem, 2008, 9, 1636–1644 CrossRef PubMed.
- L. Kranabetter, P. Martini, N. Gitzl, M. Kuhn, F. Saleem, B. Rasul, M. M. Darian, E. J. Al Maalouf, I. Sukuba and A. Kaiser, et al. , Phys. Chem. Chem. Phys., 2018, 20, 21573–21579 RSC.
- B. Oostenrijk, D. Barreiro, N. Walsh, A. Sankari, E. P. Månsson, S. Maclot, S. L. Sorensen, S. Daz-Tendero and M. Gisselbrecht, Phys. Chem. Chem. Phys., 2019, 21, 25749–25762 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Details of the theoretical methods; experimental evaluation of the shift between mixed and uracil clusters. See DOI: 10.1039/d1cp02031e |
| This journal is © the Owner Societies 2021 |