
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLongitudinal strand ordering leads to shear thinning in Nafion†
Nicholas
Michelarakis
a,
Florian
Franz
a,
Konstantinos
Gkagkas
b and
Frauke
Gräter
 *ac
*ac
aMolecular Biomechanics Group, Heidelberg Institute for Theoretical Studies, Schloss-Wolfsbrunnenweg 35, 69118 Heidelberg, Germany
bToyota Motor Europe, Technical Center, Toyota Motor Europe NVSA, Zavente, Belgium
cInterdisciplinary Center for Scientific Computing, Heidelberg University, INF 205, 69120 Heidelberg, Germany
First published on 15th November 2021
Abstract
Proton-exchange membrane fuel cells (PEMFC) offer a promising energy generation alternative for a wide range of technologies thanks to their ecological friendliness and unparalleled efficiency. At the heart of these electrochemical cells lies the membrane electrode assembly with its most important energy conversion components, the Proton Exchange Membrane. This component is created through the use of printing techniques and Nafion inks. The physicochemical properties of the ink, such as its viscosity under shear, are critical for the finished product. In this work we present non-equilibrium Molecular Dynamics simulations using a MARTINI based coarse-grained model for Nafion to understand the mechanism governing the shear viscosity of Nafion solutions. By simulating a Couette flow and calculating density maps of the Nafion chains in these simulations we shed light on the process that leads to the experimentally observed shear thinning effects of Nafion solutions under flow. We observe rod-shaped Nafion microstructures, 3 nm in size on average, when shear flow is absent or low. Higher shear rates instead break these structures and align Nafion strands along the direction of the flow, resulting in lower shear viscosities. Our work paves the way for a deeper understanding of the dynamic and mechanical properties of Nafion including studies of more complex CL and PEM inks.
1 Introduction
Shear flow is recognized as a critical factor for structure formation in various materials, from proteins1 to polymers.2–5 Understanding the molecular mechanism underlying shear-induced assembly is key to many technological applications, as shear flow is present in various processing steps. A prominent example is the deposition of ionomers – or more complex mixtures involving them – for producing proton-exchange membranes (PEMs) as part of fuel cells.PEM fuel cells are electrochemical cells that convert chemical energy into electrical energy. An electric current is generated by feeding hydrogen to an anode, where it is oxidised, and oxygen to a cathode where it is reduced. The protons generated by the hydrogen oxidation are conducted through an electronic non-conductive (proton-exchange) membrane to the cathode. The resulting electrons are rerouted through a bypass, generating an electrical current.6
At the heart of fuel cells, the membrane electrode assembly (MEA) is located. It comprises the gas diffusion layer (GDL), the microporous layer (MPL), the catalyst layer (CL), and the PEM.7 Of these components, the CL and the PEM are the most important for the PEM fuel cell since those are the areas where the chemical reactions and proton transfer occur.6 Currently, the creation of CLs and PEMs involves a variety of ink based deposition methods such as electrospray deposition,8 ultrasonic spraying,9 screen printing,10 brush printing,11 gravure,12 doctor blading,13 slot dying,14 and ink-jet printing.15 In all of the above cases, either the catalyst ink is deposited on a solid material (PEM or GDL) or the PEM solution is deposited on the CL.16 Deposition often involves shearing of the material, but the influence of the involved flow on the resulting structure and properties of the deposited material remains largely elusive.
Typically, catalysts or PEM inks consist of a variety of solvents, ionomers, supports and the catalyst material. Perfluorosulfonic acid (PFSA) based ionomers are currently the preferred choice with Nafion being most commonly used.17 The properties of the resulting CL and/or PME, and as an extension, the performance of the PEMFC, depend on the components of the ink and the applied dispersion system.18,19 The basic rheological properties of the ink, such as viscosity and evaporation, notably influence the deposition process. Consequently, it should come as no surprise that a thorough understanding of those inks is of paramount importance in the commercialisation process of PEMFCs and, hence, an area of intense study.
There have been experimental efforts to rheologically investigate the effects of shearing on the viscosity of PEM inks.20–22 These studies focus on the effect of different preparation and printing techniques on shear viscosities20 or on the effect of the addition of different ink adjuvants such as carbon black or Pt,21 mostly at low Nafion concentration. Interestingly, Gupit et al. recently observed shear-thinning of Nafion solutions, that is, a decrease in viscosity under shear flow.22
The CL and PEM inks are complex, multi-scale systems. The interactions occurring at the nano and micro-scale affect not only the local environment but also the macro-scale rheological and physical properties.23,24 Enormous progress has been made in understanding molecular and material properties of these inks and the Nafion membranes using atomistic and coarse-grained simulations.25–29 Proton transfer across membranes,30–36 intra-membrane water dynamics,31,32,36–40 ionomer assembly and aggregation,36,39–47 confinement effects,48–50 polymer chain length effects,51 solvent sorption and swelling52,53 and tensile strength and nucleation54 are just some of the important aspects of these simulations. More recently, efforts have been made to utilise the power of CG MD simulations and machine learning approaches in predicting the nano-structure of Nafion films at different hydration levels.55
A variety of different force fields have been employed in MD simulations of Nafion. Amongst them, the DREIDING force field56 is one of the most popular choices when it comes to all atom simulations.33,41,44 Of late, the polymer consistent force field57 has also been used48,58 in combination with the COMPASS force field59 to simulate the polymer matrix and assign partial charges, respectively. The parameters developed by Cui et al.60 have also been utilised37,53 in their original or modified versions.
In the case of CG MD Nafion simulations a number of CG based39,43,46,61 and dissipative particle dynamics62,63 force fields have been employed, most of them tailor-made for each study. Lately, studies have been published42,54,64 where the MARTINI force field65,66 is used for coarse-graining.
Given the importance of Nafion as the current material of choice in PEM inks,17 a number of studies have examined its properties under shear stress, both in silico and in vitro. In the past, emphasis was placed on elucidating the relationship between the morphology of Nafion structures under shear and its effect on proton conductivity. It was determined, both through the use of experiments67–69 and molecular simulations,62,70 that when shear stress is applied to Nafion membranes it results in the alignment of the Nafion strands along the flow or electrical field direction, leading to the formation of water channels which might facilitate proton hopping and enhance PEM proton conductivity.
A direct relationship between ink viscosities and the underlying molecular structure of the Nafion solution has remained elusive. While shear-induced structural changes have been previously observed in pioneering molecular simulations by Metatla et al. using a highly simplified Dissipative Particle Dynamics (DPD) model,62 a link to the viscosity of PEM inks and their peculiar shear-thinning remains to be established. We believe that a computational model investigating shear-dependent structure formation and resulting rheological properties of PEM inks would be of paramount importance in covering this gap and advancing our understanding of PEM and CL inks and their rheological properties.
In this work we present a coarse-grained model to study Nafion solutions under shear flow in Molecular Dynamics simulations. We observe a drop in viscosity at higher shear rates for highly concentrated Nafion solutions (equal or above 15 wt% Nafion117 in water/propanol), in close agreement with experiments.22 We attribute this shear thinning to the shear-promoted ordering of Nafion strands along the flow direction, according to our analysis of spatial correlations of densities. At lower shear rates, instead, Nafion assembles into globular structures with high chain entanglement.
To the best of our knowledge, this is the first CG-MD based study examining Nafion in this environment and at such scale. The results of this study shed light on the connection between the nano-scale ionomer interactions and their effect on the macro-scale rheological properties which in turn will inform the manufacturing process of PEMFC components. The Nafion MARTINI-based CG model also can serve as a starting point to computationally study more complex PEM inks.
2 Methods
2.1 Nafion model
The systems simulated in this work consist of Nafion, water and propanol. The Nafion chains used are made up of four sub-units (Fig. 1 top) with the l and m parts forming the backbone and the remainder forming the side chain. The equivalent weight (EW), which is defined as the ratio between the molecular weight and the number of sulfonate groups, is known to affect the properties of Nafion. The most frequently used Nafion in commercial fuel cell PEMs is Nafion 117 which translates to a membrane of 0.007 inches in thickness made of Nafion with an EW of 1100 g mol−1 SO3−. Nafion is a stochastic polymer and as result its molecular weight can vary between different solutions and manufacturing processes.71,72 The l and m values shown in Fig. 1 can therefore vary within the molecule whereas n usually has a value of one. In this work we have selected l, m and n values equal to seven, one and one, respectively. This represents a simpler case of a regular polymeric structure where the side chains are equally spaced on the backbone. The resulting monomer can be seen in the middle of Fig. 1. Each chain consists of 20 monomers which corresponds to an EW close to that of Nafion 117.73 In this study, we additionally employed chains of a higher EW, consisting of 50 monomers, to assess the effect of the EW on the Nafion viscosity. | ||
| Fig. 1 Structure of Nafion. Shown are the chemical formula of Nafion 117 modelled in this work (top), and the all-atom structure of a Nafion 117 monomer (middle) overlayed with its CG model (bottom). The color code of the CG model specifies the MARTINI bead type (red: negatively charged, purple: polar, green: hydrophobic) and is adopted in the chemical structure. | ||
2.2 Coarse-grained model
The coarse-graining of the system is based on the MARTINI 2.2 force field.65,66 In this model, four heavy atoms are coarse-grained into one MARTINI bead, on average. There are four different types of beads: apolar (C), non-polar (N), polar (P) and charged (Q). Each bead type is also characterised by a subtype letter (d, a, da, 0) or number (1 to 5), denoting the hydrogen bonding capabilities or the degree of polarity of the bead, respectively. Beads with the S-prefix, such as SP1, denote the “smaller” size beads employed when a lower number of heavy atoms, such as two or three, are coarse-grained into one MARTINI bead.Fig. 1 (middle) shows the all-atom representation of the Nafion monomer employed in this work while the bottom shows its coarse-grained representation. On average, three heavy atoms were coarse-grained into one MARTINI bead. As a result, and in accordance with previous works using the MARTINI forcefield to model polymers,74 the S type beads are used. The Nafion backbone is modelled using the SC1 bead type (Fig. 1 bottom green) given the hydrophobic nature of the backbone. The first and second parts of the side chain are modelled using the SN0 bead type (purple) in an effort to maintain the original MARTINI mapping of ether groups. The last part of the side chain is coarse-grained using the SQa bead type (red) to represent a charged sulfonate group since Nafion side chains have been shown to be completely deprotonated at high hydration levels.58 The standard MARTINI models for water and propanol are used, where four water or propanol molecules are coarse-grained into one P4 and one P1 bead respectively.
Compared to previous studies of Nafion which also employed the MARTINI forcefield42,54,64 our model differs in the type and number of MARTINI beads used to represent the Nafion chains. Our three-to-one coarse-graining scheme and the use of the S type beads results in Nafion chains consisting of eight beads instead of the six employed in these studies.42,54,64 Furthermore, the aforementioned studies also employ a C1 type bead to represent the ether groups, whereas our models uses two SN0 type beads. Despite these minor differences, both models are able to recapture the behaviour of Nafion in water/alcohol solutions, such as the formation of the cylindrical, bundle like aggregates.42
The non-bonded interactions between two beads are modelled according to the MARTINI force field using a shifted Lennard-Jones (LJ) 12-6 potential (eqn (1)),
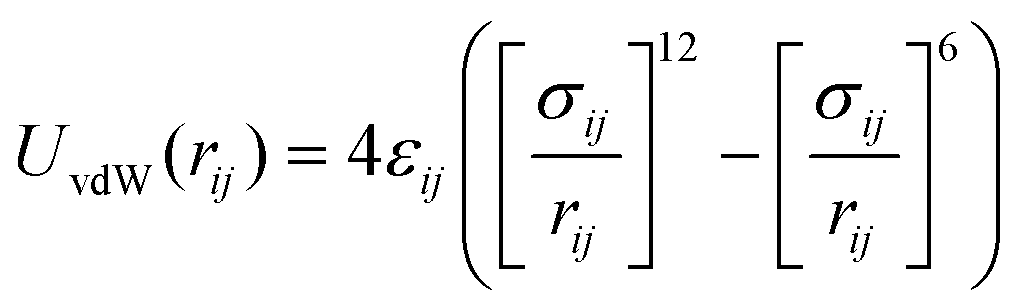 | (1) |
 | (2) |
The bonded interactions are described by the following potentials:
 | (3) |
 | (4) |
2.3 Simulation set up
Experimental studies have shown that Nafion chains can consist of approximately 100 sulfonate groups.64 In our current study we use Nafion chains consisting of 20 (Fig. 2A) and 50 sulfonate groups to achieve a balance between the size of the systems studied and the need for computational resources. | ||
| Fig. 2 (A) A Nafion chain consisting of 20 monomers (top) and an example for a Nafion aggregate structure after equilibration, in this case consisting of three Nafion chains (bottom); (B) simulation system after 400 ns of shearing at a shear rate of 4 × 108 s−1. Shearing along the indicated direction is achieved by moving the bottom wall at a constant velocity. The inter-surface distance d was 25 nm in the shown example. Color code as in Fig. 1. | ||
Inspired by the work of Savio et al.76 we designed a simulation set up as shown in Fig. 2B in order to model a Couette flow. The system consists of two surfaces along the Y-axis of the simulation box. The top wall is immovable (via Gromacs freeze groups), while the bottom wall coordinates can be translated along the pulling direction. A pulling force is applied to the bottom wall until it achieves a constant velocity. The viscosity of the system is then calculated by the relationship
 | (5) |
 corresponds to the rate at which momentum is supplied to the system per unit area along the pulling direction, d corresponds to the distance between the two surfaces, υ0 is the constant velocity of the moving surface and η is the viscosity coefficient under shear stress. This method assumes that the velocity of the solvent adjacent to the wall is the same as the one of the wall, meaning that there is no slip. However, Thompson and Trojan77 have shown that there is wall slip for nano-channel flows and that the velocity profile is not linear. Nevertheless, this method is still used since it is one of the simplest approximations for MD and is consistent with viscosity measurements taken using a surface force apparatus.78,79 The interactions of the walls with the rest of the system were set to mimic those of water to minimise any slip effects at the wall/solvent boundaries.
corresponds to the rate at which momentum is supplied to the system per unit area along the pulling direction, d corresponds to the distance between the two surfaces, υ0 is the constant velocity of the moving surface and η is the viscosity coefficient under shear stress. This method assumes that the velocity of the solvent adjacent to the wall is the same as the one of the wall, meaning that there is no slip. However, Thompson and Trojan77 have shown that there is wall slip for nano-channel flows and that the velocity profile is not linear. Nevertheless, this method is still used since it is one of the simplest approximations for MD and is consistent with viscosity measurements taken using a surface force apparatus.78,79 The interactions of the walls with the rest of the system were set to mimic those of water to minimise any slip effects at the wall/solvent boundaries.
Tables S1 and S2 of the ESI,† show a summary of the simulations run. For each system, we randomly inserted the Nafion chains into the simulation box and solvated the system until the desired concentration was reached. The solvent consists of a water/propanol mixture at a 70/30 percentage weight ratio, in an effort to reproduce the experimental set up of Gupit et al.22 as closely as possible. We then ran a 20 ns equilibration in an NPT ensemble without walls after which the walls were introduced by transforming the top and bottom solvent layers to the desired wall particles. Following this, we ran a further 40 ns equilibration in an NVT ensemble. The systems were then sheared in an NVT ensemble until they reached constant velocities of 10 m s−1, 1 m s−1 and 0.1 m s−1 corresponding to shear rates of 4 × 108 s−1, 4 × 107 s−1 and 4 × 106 s−1 respectively. The distance between the walls was set at d = 25 nm. In addition, we also simulated a Nafion solution of 20 wt% in an NPT ensemble to be used as a control. We also ran a set of control simulations at the aforementioned shear rates, using a 10 wt% Nafion solution and varying the inter-surface distance between 15 and 45 nm (Fig. S2, ESI†). We observe that the shear viscosity calculated is independent of the inter-surface distance.
The temperature was maintained at 323 K through the use of the velocity-rescale thermostat.80 The pressure was kept constant at 1 atm through the use of the Parrinello–Rahman barostat.81,82 The GROMACS 2019.483 molecular dynamics package was consistently used for the simulations of all systems with an integration time step of 10 fs.
2.4 Autocorrelation analysis
For the analysis of the production and control runs, we split the trajectories at 10 ns intervals keeping the last frame of each interval. From these forty snapshots we then calculated the density maps of the Nafion chains using the GROma ρs analysis tool84 (ESI,† Fig. S1). Using the density maps we computed the autocorrelation of the densities along the axis of shear flow, with a lag of up to 12 nm. We also calculated the rate of decay of the autocorrelation during the simulation time through the use of an in-house script to fit an exponential decay function to the autocorrelation calculated from each snapshot described previously.3 Results
3.1 Viscosity
To characterize the rheological properties of the Nafion solutions under shear flow, we calculate their viscosities at different shear rates. We considered Nafion chains made of 20 and 50 Nafion monomers, each at four different concentrations (5, 10, 16, and 20 wt%) and at three different shear rates, resulting in 24 systems. Viscosities η were obtained from the average forces observed during the last 100 ns of the MD simulations using eqn (5) and are shown in Fig. 3. We find that at lower Nafion concentrations (5 and 10 wt%), the solutions show Newtonian fluid properties, with their viscosities exhibiting no dependence on the applied shear rate. For the higher concentrations (16 and 20 wt%) we observe a shear thinning effect with the viscosity of the solution decreasing as the shear rate increases. The trends we observe here are in qualitative agreement with the experimental results published by Gupit et al.22 for Nafion solutions of similar water/alcohol ratios. On the absolute scale, the computed viscosities are smaller by a factor of approximately 7–10, and the reasons for this discrepancy are potentially two-fold: first, our shear rates are significantly higher compared to experiments (0.1–700 s−1) in order to observe the rheological response at the MD-accessible time scale. Secondly, coarse-graining always involves smoothing of the energy lanscape so that a lower resistance to shearing can be expected. However, the overall very good agreement with the trends observed in the experiment and the recovery of the expected loss of viscosity at high concentrations and shear rates suggest that we capture the major properties of the Nafion solution in shear. | ||
| Fig. 3 Viscosities of Nafion solutions at different concentrations and different shear rates, with Nafion chains consisting of 20 monomers (orange) and 50 monomers (green). | ||
Comparing the shear viscosities calculated for Nafion chains consisting of 20 and 50 monomers, for all different Nafion concentrations, we see higher values for longer chains when keeping the concentration the same. This agrees with published literature and experimental results for polymer chains.85–87 Longer chains show stronger entanglement, leading to a higher resistance against shearing. In the case of the 10 wt% Nafion solution there is a significant difference between the viscosities of the shorter and longer chains, with the viscosity of the longer chain solution at the lower shear rates of 4 × 106 and 4 × 107 s−1 being approximately two-fold the value calculated for the chains consisting of 20 monomers at these rates. Furthermore, the viscosities of the longer chain solutions exhibit a shear thinning effect with the values decreasing as the shear rate increases, similar to the solutions of higher concentrations. In contrast, the shorter chain solution exhibits a Newtonian fluid behaviour as described previously. These observations indicate the existence of a transition point at this concentration upon which the chain length starts to play a dominant role in the micro-structures formed by the Nafion particles.
Our nano-scale capillary, albeit at the larger side of Nafion systems simulated to date to our knowledge, induces boundary effects such as lifting off of chains from the wall, which are absent in mascroscopic-scale experiments. In order to ensure that the distance between the shearing surfaces (d in Fig. 2B) has no effect on the shear viscosities calculated, we run a set of simulations at the same three different shear rates with d varying from 15 to 45 nm, as summarised in the ESI,† Table S2. The solution with a 10 wt% Nafion concentration, and chains made of 20 monomers, was used as a mid-point example of the different concentrations examined. We see that the effect of the wall distance is negligible, as none of the calculated values show significant variation with d. Moreover, we observe that the solutions retain their Newtonian liquid properties irrespective of wall distance, as seen previously for the case of the 10 wt%, 20-monomer chains.
3.2 Shear-induced strand alignment
Nafion self-assembles into nano-scale domains in which the hydrophobic fluorinated carbon chains form a core and the charged sulfonate functions point toward the hydrophilic water/ethanol solvent.47,88 These rod-like assemblies have been observed previously42,88 and in the absence of flow take up various shapes and orientations (Fig. 2A). Flow leads to an alignment of Nafion strands and their multi-strand assemblies along the flow direction, leading to the formation of bands or lamellae, visible as stripes in Fig. 2B. To quantify this conformational transition, we calculated maps of Nafion densities along the simulated trajectories (see Methods section and ESI,† Fig. S1). For each of these maps, we then calculated the autocorrelation along the flow axis with a lag of up to 12 nm as a measure for the strand alignment (Fig. 4). | ||
| Fig. 4 Shear-flow induces strand alignment at high shear rates and high Nafion concentrations. Shown is the (A) autocorrelation along the flow (X) axis of the density maps, for different shear rates and along the simulated time (0–400 ns at 10 ns intervals, color-coded from black to bronze), for 20-monomer Nafion chains. Top, middle and bottom graphs show results for the highest, intermediate, and lowest shear rate, respectively. (B) Autocorrelation along the X, Y and Z axis of the equilibrium simulations. Results for the 50-monomer Nafion chains are shown in the ESI,† Fig. S3. | ||
For the highest shear rate and the higher Nafion concentrations of 20 and 16 wt%, there is a clear increase in the autocorrelation along the flow axis as the simulation progresses (black to light brown/copper). This trend continues for the higher shear rate and the 10 wt% Nafion concentration while there is no significant change in the autocorrelation for the lowest, 5 wt% concentration. For the lower shear rates and the higher concentrations we also observe a much smaller change for the autocorrelation compared to the highest shear rate. Finally, for the 10 wt% concentration, remarkably, even the lower shear rates suffice to increase the density autocorrelation during the simulation time.
In order to compare the shear-induced strand ordering more quantitatively to the observed changes in viscosity we calculated the rate of decay of the autocorrelation for every time frame featured in Fig. 4. Fig. 5 shows that once the desired velocity has been achieved (within 100 ns or less), the highest shearing rate leads to the lowest rate of autocorrelation decay. Therefore, at high shear rates, Nafion structures prevail along the flow direction as bands, in contrast to the rod-like structures which alternate with solvent and thus show faster decays in the density correlations. Taken together with the autocorrelations calculated in Fig. 4, our data support the argument of the breakage of Nafion microstructures and alignment of the Nafion strands at the highest shear rate. The microstructures formed by the Nafion strand for the 10, 16 and 20 wt% cases break apart and align along the shear direction once a certain amount of shear is applied while a concentration of 5 wt% is apparently not high enough to lead to the formation of these structures. Structural changes along the flow direction are absent, and the assemblies remain randomly oriented.
It is interesting to note that even in the case of the 5 wt% concentration, where no such microstructures were forming, the higher shear rates still lead to a lower rate of decay compared to the slower rates. Moreover, the initial steep decrease of the rate of decay before reaching a plateau that is present at higher Nafion concentrations is not observed here. This can be explained by a possible alignment or even stretching of the long rod-like structures known to be formed by Nafion at lower concentrations in polar solvents,88,89 an example of which can be seen in Fig. 2A (middle).
For the 20 wt% Nafion concentration, we notice that the decay rate of the autocorrelation is similar for both of the lower shear rates, with the absence of an initial steep decrease. Again, this confirms that those shear rates are not high enough to cause the breakage of the Nafion microstructures. At a concentration of 10 wt%, the lower shear rates lead to a similar decrease in the rate of decay of the autocorrelation and also include an initial steep decrease. Again, this corroborates the notion that the rod-like structures, with more solvent being present, can break and align more readily at these shear rates compared to higher wt% solutions.
We also calculated the autocorrelation along the X, Y and Z axes of the 20 wt% simulation ran with no shear in an NPT ensemble (Fig. 4B) as a control. We see that after the initial frame there is no change in the autocorrelation for the duration of the simulation. Furthermore, we also calculated the decay rate for the autocorrelation along the same axes (Fig. 5B). As before, we see that after the initial frame there is no significant difference between the axes, excluding the possibility of the effects observed in the shearing simulations being due to internal Nafion particle rearrangements.
 | ||
| Fig. 5 (A) Rate of autocorrelation decay along the flow (X) axis of the density maps of the different 20 monomer Nafion chain solutions for the different shear rates. (B) Rate of autocorrelation decay along the X, Y and Z axis for the control simulations under equilibrium. Blue corresponds to the highest shear rate of 4 × 108 s−1, orange to 4 × 107 s−1 and green to the lowest 4 × 106 s−1. | ||
3.3 Shear-thinning and strand alignment correlate
We next brought the observed structural transition into context with the shear-thinning. As a measure for strand alignment, we used the average rate of decay of the density autocorrelation, once a plateau has been reached. Fig. 6 shows this structure observable as a function of the viscosity calculated from each simulation. | ||
| Fig. 6 Strand ordering correlates with shear thinning. The average rate of autocorrelation decay along the flow (X) axis of the density maps versus the shear viscosity calculated for each simulation system (varying in Nafion concentration and shear rate). The 5 and 10 wt% solutions are shown in red and green, respectively, while the 16 and 20 wt% cases are shown in orange and blue (top). Two snapshots corresponding to a higher (1) and lower (2) viscosity (bottom). | ||
Nafion solutions at 5 and 10 wt% show minor changes in the strand alignment (as reflected by the decay rate) while the shear viscosity is unaffected. For both, 5 (red) and 10 (green) wt%, as the rate of decay decreases, the viscosity remains approximately the same at around 4 and 6.5 mPa s, respectively. As seen before, at lower concentrations the Nafion microstructures appear to break and align at all shear rates, and the resistance to shearing as measured by the viscosity remains comparably low. For the higher concentrations of 16 (orange) and 20 (blue) wt%, instead, there is a clear effect of the rate of decay of the autocorrelation on the viscosity: the higher the rate of decay, the higher the viscosity. Shearing strongly restructures the Nafion assemblies from randomly oriented rods into highly aligned bands, resulting in lower resistance against shearing. An example snapshot of the highly aligned Nafion strands (leading to lower viscosity) and the randomly oriented rods (higher viscosity) can be seen in Fig. 6 1 and 2 respectively.
4 Discussion
In this work we shed light on the mechanism governing the shear viscosity and the shear thinning effects observed in Nafion solutions with concentrations higher than 10 wt% through the use of a non equilibrium CG-MD based approach. Our data suggest that the mechanism behind shear thinning is based on the strong alignment of the Nafion strands along the shear flow. As a result, when the shear rate is high enough, or the concentration is low enough for the Nafion microstructures to break and alignment to occur, then this mechanism leads to a lower viscosity. If shear rates are not high enough to break the physical cross-linking of the Nafion strands, the viscosity remains high.Our simulations qualitatively reproduce the trends exhibited by Nafion solutions sheared in experiments22 and allow us to study the structural properties that underlie this mechanism. Our results suggest that the described shear thinning effect is due to the alignment of the Nafion strands along the direction of the flow. At high enough shear rates (4 × 107 s−1 and beyond), the rod-like microstructures formed by entanglements or cross-linking of the Nafion strands orient and deform. The alignment of the polymer strands, results in a decrease in viscosity. Shearing involves primarily a sliding of the aligned and highly solvated Nafion bands against each other. In contrast, at lower shear rates (4 × 106 s−1 or lower), microstructures remain largely unaffected. This results in shear viscosities largely independent of the flow rate, as shearing requires relative motions within the microstructures, which span a larger region vertical to the flow. Moreover, in the case of very dilute Nafion solutions, where the concentration is too low to lead to the formation of the aforementioned Nafion microstructures, we observe a Newtonian-liquid like behaviour, that is, the shear viscosity is independent of the shear rate. This further supports our conclusions and those of Gupit et al.
Our work addresses the need for computational studies examining the rheological and structural effects of shear flow on the viscosities and structures of Nafion solutions. Although the alignment of Nafion strands along the flow or the electric field direction has been observed previously62,67–70 it was interpreted within the context of water channel formation, proton hopping and enhanced membrane conductivity.
In this study we demonstrate for the first time the ability of our model to capture the experimental trends of the viscosity of Nafion solutions under shear. We then offer an insight into the mechanisms behind the effects observed in Nafion solutions under shear, more specifically shear thinning. We demonstrate that the aforementioned Nafion strand alignment, observed in ours and the previously mentioned studies, is responsible for the shear thinning of Nafion solutions under flow. Furthermore, the coarse-grained nature of our model allows to reach time and length scales not assessed previously in simulations, and to thus directly and quantitatively predict the shear response and solvent effects of the ink.
The approach presented here is based on the MARTINI force field. Transferring it to Nafion was straightforward, and our CG model recovered the expected structural and rheological behavior. Compared to earlier CG studies that explored the structural properties of Nafion42,54,64 using the MARTINI force field, our model was able to recoup the structural features of Nafion strands in water/alcohol mixtures reported in those studies. Studying the assembly behavior of Nafion and its response to shear required system sizes comprising many Nafion chains (here: 490 20-monomer chains) that can not be easily handled at the all-atom level, in particular when it comes to the shear response. The production simulations (400 ns each, 400 thousand atoms) required approximately 24 hours on three nodes of an in-house high-performance computing cluster, consisting of six Intel Haswell (E5-2630v3) CPUs and 24 cores in total. The MARTINI model for Nafion that we have used in this study can easily be employed for bigger and more complex systems. The top-down and bottom-up parametrization approaches followed by the MARTINI force field allow for more intricate set ups, an example being the modeling of the manufacturing process suggested by Yang et al.,16 in which case the simulation of the drying of the CL and PEM inks would be necessary. For the former case, the inclusion of C and Pt particles is also required.
Obviously, our coarse-grained model cannot capture all chemical details. For example, MARTINI cannot differentiate between solvents such as 1- or 2-propanol due to the reduced chemical representation that results from four heavy atoms being mapped onto one larger MARTINI bead. More importantly, a distinction between experiment and MD is the difference in the shear rates accessible. In experimental viscometry set ups, the maximum shear rates available are in the range of 2 × 105 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 90 (although shear rates up to 7 × 106 s−1 have been reported91) but to the best of our knowledge we could not find viscosity measurements of Nafion solutions at shear rates higher than 103 s−1. At the same, it is difficult to reach values lower than 4 × 105 s−1 in MD simulations. On the other hand, the MARTINI model yields a smoother energy landscape resulting in an effective four-fold speed-up of the sampled dynamics,65 bringing the simulated shear rates slightly closer to the experimental range. In addition, while we observe a structural relaxation of the MARTINI system upon shearing on the time scale of 100 ns or less (Fig. 5), all-atom simulations would require roughly 400 ns until the Nafion solution restructured as a response to the shear flow.
90 (although shear rates up to 7 × 106 s−1 have been reported91) but to the best of our knowledge we could not find viscosity measurements of Nafion solutions at shear rates higher than 103 s−1. At the same, it is difficult to reach values lower than 4 × 105 s−1 in MD simulations. On the other hand, the MARTINI model yields a smoother energy landscape resulting in an effective four-fold speed-up of the sampled dynamics,65 bringing the simulated shear rates slightly closer to the experimental range. In addition, while we observe a structural relaxation of the MARTINI system upon shearing on the time scale of 100 ns or less (Fig. 5), all-atom simulations would require roughly 400 ns until the Nafion solution restructured as a response to the shear flow.
Our computed viscosities are on average 7 to 10 orders of magnitude lower than the experimentally determined values. The reasons for this discrepancy in absolute values are (i) two to three orders of magnitude higher shear rates, which is only partly balanced by the faster dynamics of MARTINI systems, (ii) the limited nm-scale system size, and (iii) the coarse-grained nature of the model that lacks degrees of freedom and chemical details. Yet, we are able to reproduce the overall experimental trends.
Lastly, another interesting observation is that at a Nafion concentration of 10 wt%, longer chain lengths lead to significantly higher viscosity values and shear thinning effect. This indicates that the chain length can strongly affect the shear viscosity, and by extension other physicochemical and structural properties as well. As most computational works utilise Nafion chains of short lengths to maximise computational efficiency, it is an area which merits further examination.
5 Conclusions
In this work we utilise non-equilibrium CG-MD simulations to understand the mechanism governing the shear viscosity of Nafion solutions. Our simulations allow the calculation of the shear viscosities of Nafion solutions and their comparison to structural changes measured by spatial correlations of Nafion densities. Our results recover the experimentally observed shear thinning and provide an underlying mechanism on a molecular level. Nafion strands align along the direction of the flow, and rod-like assemblies orient and transform into band-like structures along the flow direction. This conformational transition reduces resistance to shear. The Nafion strand alignment is in agreement with previously published experimental and computational results while the observed shear thinning and the mechanism behind it that is proposed in this work offers new insights into the interplay between structural and rheological properties of Nafion structures in solutions under shear flow, underlining the need for computational studies focusing on the subject. Finally, the protocol we present here offers an excellent starting point for the simulation of even more complex PEM or CL inks which in turn can further inform the manufacturing process, leading to improved PEMFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the members of the Molecular Biomechanics Group at the Heidelberg Institute for Theoretical Studies for helpful discussions. We would like to thank Camilo Aponte-Santamaria in particular for help with the GROma ρs analysis tool. We would like to thank the Klaus Tschira Foundation and Toyota Europe for their generous financial support. F.G. acknowledges funding through the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – 2082/1 – 390761711.References
- A. M. Herrera-Rodríguez, V. Miletić, C. Aponte-Santamaría and F. Gräter, Biophys. J., 2019, 116, 1579–1585 CrossRef PubMed.
- D. E. Smith, H. P. Babcock and S. Chu, Science, 1999, 283, 1724–1727 CrossRef CAS PubMed.
- M. Q. Tu, M. Lee, R. M. Robertson-Anderson and C. M. Schroeder, Macromolecules, 2020, 53, 9406–9419 CrossRef CAS.
- A. Korolkovas, P. Gutfreund and M. Wolff, J. Chem. Phys., 2018, 149, 074901 CrossRef PubMed.
- J. Fidalgo, K. Zografos, L. Casanellas, A. Lindner and M. S. N. Oliveira, Biomicrofluidics, 2017, 11, 064106 CrossRef.
- S. Litster and G. McLean, J. Power Sources, 2004, 130, 61–76 CrossRef CAS.
- Y. Guo, F. Pan, W. Chen, Z. Ding, D. Yang, B. Li, P. Ming and C. Zhang, Electrochem. Energy Rev., 2021, 4, 67–100 CrossRef CAS.
- R. Liu, W. Zhou, L. Wan, P. Zhang, S. Li, Y. Gao, D. Xu, C. Zheng and M. Shang, Curr. Appl. Phys., 2020, 20, 11–17 CrossRef.
- M. B. Sassin, Y. Garsany, B. D. Gould and K. E. Swider-Lyons, Anal. Chem., 2017, 89, 511–518 CrossRef CAS PubMed.
- W. Wang, S. Chen, J. Li and W. Wang, Int. J. Hydrogen Energy, 2015, 40, 4649–4658 CrossRef CAS.
- L. Xiong and A. Manthiram, Electrochim. Acta, 2005, 50, 3200–3204 CrossRef CAS.
- S. A. Mauger, K. C. Neyerlin, A. C. Yang-Neyerlin, K. L. More and M. Ulsh, J. Electrochem. Soc., 2018, 165, F1012–F1018 CrossRef CAS.
- I.-S. Park, W. Li and A. Manthiram, J. Power Sources, 2010, 195, 7078–7082 CrossRef CAS.
- X. Ding, S. Didari, T. F. Fuller and T. A. L. Harris, J. Electrochem. Soc., 2012, 159, B746–B753 CrossRef CAS.
- C. A. Gomes Bezerra, L. J. Deiner and G. Tremiliosi-Filho, Adv. Eng. Mater., 2019, 21, 1900703 CrossRef CAS.
- C. Yang, N. Han, Y. Wang, X.-Z. Yuan, J. Xu, H. Huang, J. Fan, H. Li and H. Wang, ACS Sustainable Chem. Eng., 2020, 8, 9803–9812 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- K. Karan, Curr. Opin. Electrochem., 2017, 5, 27–35 CrossRef CAS.
- T. Ioroi, Z. Siroma, S. Yamazaki and K. Yasuda, Adv. Energy Mater., 2019, 9, 1801284 CrossRef.
- S. Du, W. Li, H. Wu, P.-Y. Abel Chuang, M. Pan and P.-C. Sui, Int. J. Hydrogen Energy, 2020, 45, 29430–29441 CrossRef CAS.
- S. Khandavalli, J. H. Park, N. N. Kariuki, D. J. Myers, J. J. Stickel, K. Hurst, K. C. Neyerlin, M. Ulsh and S. A. Mauger, ACS Appl. Mater. Interfaces, 2018, 10, 43610–43622 CrossRef CAS PubMed.
- C. I. Gupit, X. Li, R. Maekawa, N. Hasegawa, H. Iwase, S. Takata and M. Shibayama, Macromolecules, 2020, 53, 1464–1473 CrossRef CAS.
- M. B. Dixit, B. A. Harkey, F. Shen and K. B. Hatzell, J. Electrochem. Soc., 2018, 165, F264–F271 CrossRef CAS.
- K. B. Daly, A. Z. Panagiotopoulos, P. G. Debenedetti and J. B. Benziger, J. Phys. Chem. B, 2014, 118, 13981–13991 CrossRef CAS PubMed.
- M. So, T. Ohnishi, K. Park, M. Ono, Y. Tsuge and G. Inoue, Int. J. Hydrogen Energy, 2019, 44, 28984–28995 CrossRef CAS.
- T. Mashio, A. Ohma and T. Tokumasu, Electrochim. Acta, 2016, 202, 14–23 CrossRef.
- K. Morohoshi and T. Hayashi, Polymers, 2013, 5, 56–76 CrossRef.
- P. Frühwirt, A. Kregar, J. T. Törring, T. Katrašnik and G. Gescheidt, Phys. Chem. Chem. Phys., 2020, 22, 5647–5666 RSC.
- E. J. F. Dickinson and G. Smith, Membranes, 2020, 10, 310 CrossRef CAS PubMed.
- Y.-K. Choe, E. Tsuchida, T. Ikeshoji, S. Yamakawa and S.-A. Hyodo, Phys. Chem. Chem. Phys., 2009, 11, 3892–3899 RSC.
- P. V. Komarov, P. G. Khalatur and A. R. Khokhlov, Beilstein J. Nanotechnol., 2013, 4, 567–587 CrossRef PubMed.
- S. Akbari, M. T. H. Mosavian, A. Ahmadpour and F. Moosavi, ChemPhysChem, 2017, 18, 3485–3497 CrossRef CAS PubMed.
- J. Karo, A. Aabloo, J. O. Thomas and D. Brandell, J. Phys. Chem. B, 2010, 114, 6056–6064 CrossRef CAS PubMed.
- T. Mabuchi and T. Tokumasu, J. Nanosci. Nanotechnol., 2015, 15, 2958–2963 CrossRef CAS PubMed.
- J. A. Clark, E. E. Santiso and A. L. Frischknecht, J. Chem. Phys., 2019, 151, 104901 CrossRef PubMed.
- A. Vishnyakov, R. Mao, M.-T. Lee and A. V. Neimark, J. Chem. Phys., 2018, 148, 024108 CrossRef PubMed.
- K. B. Daly, J. B. Benziger, A. Z. Panagiotopoulos and P. G. Debenedetti, J. Phys. Chem. B, 2014, 118, 8798–8807 CrossRef CAS PubMed.
- M. Ozmaian and R. Naghdabadi, Phys. Chem. Chem. Phys., 2014, 16, 3173–3186 RSC.
- K. Malek, M. Eikerling, Q. Wang, Z. Liu, S. Otsuka, K. Akizuki and M. Abe, J. Chem. Phys., 2008, 129, 204702 CrossRef PubMed.
- A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2014, 118, 11353–11364 CrossRef CAS PubMed.
- J. H. Lee, G. Doo, S. H. Kwon, S. Choi, H.-T. Kim and S. G. Lee, Sci. Rep., 2018, 8, 10739 CrossRef PubMed.
- T. Mabuchi, S.-F. Huang and T. Tokumasu, Macromolecules, 2020, 53, 3273–3283 CrossRef CAS.
- M. Ghelichi, K. Malek and M. H. Eikerling, Macromolecules, 2016, 49, 1479–1489 CrossRef CAS.
- C. K. Knox and G. A. Voth, J. Phys. Chem. B, 2010, 114, 3205–3218 CrossRef CAS PubMed.
- R. Wang, S. Liu, L. Wang, M. Li and C. Gao, Nanomaterials, 2019, 9, 869 CrossRef PubMed.
- W. Chen, F. Cui, L. Liu and Y. Li, J. Phys. Chem. B, 2017, 121, 9718–9724 CrossRef CAS PubMed.
- A. Tarokh, K. Karan and S. Ponnurangam, Macromolecules, 2020, 53, 288–301 CrossRef CAS.
- S. Sengupta and A. V. Lyulin, J. Phys. Chem. B, 2018, 122, 6107–6119 CrossRef CAS PubMed.
- D. Sun and J. Zhou, AIChE J., 2013, 59, 2630–2639 CrossRef CAS.
- P. Vanya, J. Sharman and J. A. Elliott, J. Chem. Phys., 2017, 147, 214904 CrossRef CAS PubMed.
- S. Sengupta and A. Lyulin, Polymers, 2020, 12, 907 CrossRef CAS PubMed.
- M. Soniat and F. A. Houle, J. Phys. Chem. B, 2018, 122, 8255–8268 CrossRef CAS PubMed.
- K. B. Daly, J. B. Benziger, P. G. Debenedetti and A. Z. Panagiotopoulos, J. Phys. Chem. B, 2013, 117, 12649–12660 CrossRef CAS PubMed.
- W. Gonçalves, T. Mabuchi and T. Tokumasu, J. Phys. Chem. C, 2019, 123, 28958–28968 CrossRef.
- L. Dumortier and S. Mossa, J. Phys. Chem. B, 2020, 124, 8918–8927 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- H. Sun, S. J. Mumby, J. R. Maple and A. T. Hagler, J. Am. Chem. Soc., 1994, 116, 2978–2987 CrossRef CAS.
- S. Sengupta and A. V. Lyulin, J. Phys. Chem. B, 2019, 123, 6882–6891 CrossRef CAS PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- S. Cui, J. Liu, M. E. Selvan, D. J. Keffer, B. J. Edwards and W. V. Steele, J. Phys. Chem. B, 2007, 111, 2208–2218 CrossRef CAS PubMed.
- A.-T. Kuo, S. Urata, K. Nakabayashi, H. Watabe and S. Honmura, Macromolecules, 2021, 54, 609–620 CrossRef CAS.
- N. Metatla, S. Palato and A. Soldera, Soft Matter, 2013, 9, 11093–11097 RSC.
- J. A. Elliott, D. Wu, S. J. Paddison and R. B. Moore, Soft Matter, 2011, 7, 6820–6827 RSC.
- T. Mabuchi, S.-F. Huang and T. Tokumasu, J. Polym. Sci., 2020, 58, 487–499 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- D. H. de Jong, G. Singh, W. F. D. Bennett, C. Arnarez, T. A. Wassenaar, L. V. Schäfer, X. Periole, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2013, 9, 687–697 CrossRef CAS PubMed.
- Q. Duan, H. Wang and J. Benziger, J. Membr. Sci., 2012, 392-393, 88–94 CrossRef CAS.
- X. Kong and K. Schmidt-Rohr, Polymer, 2011, 52, 1971–1974 CrossRef CAS.
- J. Li, J. K. Park, R. B. Moore and L. A. Madsen, Nat. Mater., 2011, 10, 507–511 CrossRef CAS PubMed.
- E. Allahyarov and P. L. Taylor, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 80, 020801 CrossRef PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- C. Heitner-Wirguin, J. Membr. Sci., 1996, 120, 1–33 CrossRef CAS.
- J. Lucid, S. Meloni, D. MacKernan, E. Spohr and G. Ciccotti, J. Phys. Chem. C, 2013, 117, 774–782 CrossRef CAS.
- F. Grunewald, G. Rossi, A. H. de Vries, S. J. Marrink and L. Monticelli, J. Phys. Chem. B, 2018, 122, 7436–7449 CrossRef CAS PubMed.
- A.-T. Kuo, S. Okazaki and W. Shinoda, J. Chem. Phys., 2017, 147, 094904 CrossRef PubMed.
- D. Savio, L. Pastewka and P. Gumbsch, Sci. Adv., 2016, 2, e1501585 CrossRef PubMed.
- P. A. Thompson and S. M. Troian, Nature, 1997, 389, 360–362 CrossRef CAS.
- R. Balasundaram, S. Jiang and J. Belak, Chem. Eng. J., 1999, 74, 117–127 CrossRef CAS.
- A. Jabbarzadeh, J. D. Atkinson and R. I. Tanner, J. Non-Newtonian Fluid Mech., 1998, 77, 53–78 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- R. Briones, C. Blau, C. Kutzner, B. L. D. Groot and C. Aponte-Santamaría, Biophys. J., 2019, 116, 4–11 CrossRef CAS PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1940, 62, 1057–1070 CrossRef CAS.
- V. Tirtaatmadja, K. C. Tam and R. D. Jenkins, Macromolecules, 1997, 30, 3271–3282 CrossRef CAS.
- A. J. Tsamopoulos, A. F. Katsarou, D. G. Tsalikis and V. G. Mavrantzas, Polymers, 2019, 11, 1194 CrossRef PubMed.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75–83 CrossRef CAS PubMed.
- M. Yamaguchi, T. Matsunaga, K. Amemiya, A. Ohira, N. Hasegawa, K. Shinohara, M. Ando and T. Yoshida, J. Phys. Chem. B, 2014, 118, 14922–14928 CrossRef CAS PubMed.
- X. Wang, W. W. Carr, D. G. Bucknall and J. F. Morris, Rev. Sci. Instrum., 2010, 81, 065106 CrossRef PubMed.
- T. W. Selby and G. C. Miiller, 5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02024b |
| This journal is © the Owner Societies 2021 |