
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of aqueous peptide solutions in folded and disordered states examined by dynamic light scattering and dielectric spectroscopy†
Jorge H.
Melillo
 a,
Jan Philipp
Gabriel
bc,
Florian
Pabst
c,
Thomas
Blochowicz
c and
Silvina
Cerveny
*ad
a,
Jan Philipp
Gabriel
bc,
Florian
Pabst
c,
Thomas
Blochowicz
c and
Silvina
Cerveny
*ad
aCentro de Física de Materiales (CSIC-UPV/EHU)-Material Physics Centre (MPC), Paseo Manuel de Lardizabal 5 (20018), San Sebastián, Spain. E-mail: scerveny@ctq.csic.es; silvina.cerveny@ehu.es
bSchool for Molecular Sciences, Arizona State University, Tempe, 85287, USA
cInstitute for Condensed Matter Physics, Technical University of Darmstadt, 64289 Darmstadt, Germany
dDonostia International Physics Center, Paseo Manuel de Lardizabal 4 (20018), San Sebastián, Spain
First published on 24th June 2021
Abstract
Characterizing the segmental dynamics of proteins, and intrinsically disordered proteins in particular, is a challenge in biophysics. In this study, by combining data from broadband dielectric spectroscopy (BDS) and both depolarized (DDLS) and polarized (PDLS) dynamic light scattering, we were able to determine the dynamics of a small peptide [ε-poly(lysine)] in water solutions in two different conformations (pure β-sheet at pH = 10 and a more disordered conformation at pH = 7). We found that the segmental (α-) relaxation, as probed by DDLS, is faster in the disordered state than in the folded conformation. The water dynamics, as detected by BDS, is also faster in the disordered state. In addition, the combination of BDS and DDLS results allows us to confirm the molecular origin of water-related processes observed by BDS. Finally, we discuss the origin of two slow processes (A and B processes) detected by DDLS and PDLS in both conformations and usually observed in other types of water solutions. For fully homogeneous ε-PLL solutions at pH = 10, the A-DLS process is assigned to the diffusion of individual β-sheets. The combination of both techniques opens a route for understanding the dynamics of peptides and other biological solutions.
1 Introduction
Proteins are macromolecules present in living systems that perform functions in virtually all biological processes. Aside from the huge number and variety of chemical reactions in which they participate, proteins transmit nerve impulses, provide immune protection, control the flow of material through membranes, generate movement, and transport and store other molecules such as oxygen. Since 1958, when the 3D structure of myoglobin was solved,1 it had been assumed that the biological functions of proteins were encoded in their 3D structures. However, later, it was observed that some segments of proteins are composed of elongated chains and that some other proteins lack a well-structured three-dimensional fold. These intrinsically disordered proteins (IDPs), which are abundant in nature (30–50% in eukaryotic cells2,3), are still functional,4 challenging the traditional structure-function paradigm.5 The dynamics of proteins (structured or unstructured) is closely tied to the properties of the solvent.6 In fact, hydration is responsible for the stabilization of the protein structure: it mediates binding through hydrogen bonds and contributes to their catalytic properties. In well-folded proteins (for instance, globular proteins), water plays an important role in biological activity, and it has been proposed that water determines their structures and dynamics.6 There is a remarkable coupling between the motions of a solvent and a solute,7–9 and this seems to be valid for IDS proteins10–12 as well as non-biological solutions.13–15The timescales of the motions of proteins span several orders of magnitude (from picoseconds to seconds16), and broadband dielectric spectroscopy17 (BDS), among other techniques, is appropriate for analysing these motions because of its extremely broad frequency range. However, in the case of biological solutions, the heterogeneities and interfaces between the solvent and the solute (which lead to Maxwell–Wagner polarization effects) along with their strong conductivity, prevent accurate analysis of the dynamics of proteins and of the α-relaxation in particular, which is the main relaxation process that defines the transition from a solid to a liquid. In contrast, dynamic light scattering18 (DLS) is not sensitive to charge transport,19 and therefore it is a much-used technique for studying the dynamics of proteins in solutions. Nevertheless, DLS requires semi-transparent solutions (materials through which some light can pass), and hydrated protein powders or concentrated solutions of proteins are therefore not suitable for this technique. While very dilute solutions of proteins can be examined using DLS, such solutions crystallize on cooling, preventing the determination of the α-relaxation at lower temperatures.
In recent years, we analyzed the dynamics of ε-poly(lysine) (ε-PLL) using BDS13,20 and nuclear magnetic resonance21 (NMR) at pH = 10. Compared with proteins and other peptides, ε-PLL has the advantages that it can be completely dissolved in water, its concentrated solution is semi-transparent, and crystallization is avoided at all temperatures for high pH values (≥10). Due to these characteristics, an ε-PLL water solution at pH = 10 is an appropriate system for analysis using both, DLS and BDS. Moreover, this system is folded in a β-sheet conformation, and it therefore presents an ideal means for studying the characteristics of the α-relaxation of a protein in a solution. This is in contrast to hydrated protein powders,22–25 for which the diffusive dynamics of the entire protein is suppressed. The dynamical behaviour of this system is complex and, in particular, it is laborious to determine the segmental (α-) relaxation using BDS,20 and it cannot be directly observed using NMR.21
In this work, we analysed the dynamics of aqueous solutions of ε-PLL in two different conformations (a pure β-sheet conformation (at pH = 10) and a more elongated chain (at pH = 7)) using DDLS, PDLS and BDS. The combination of these techniques allows us to analyse the dynamics of the solute and the solvent; we can determine both the α-relaxation and the water dynamics around the peptide in the two conformations. In addition, using DLS measurements we can analyse whether our solutions are essentially homogeneous at length scales larger than the dimensions of a β-sheet, or whether instead they present some aggregations apart from some degree of short local structuring such as molecular clusters or solvation shells on the sub-nanometre scale.
2 Experimental methods
2.1 Samples
The ε-PLL (average molecular weight Mw = 4.700 g mol−1 and polydispersity index Mw/Mn = 1.15) was supplied by JNC Corporation (Japan). This was purified using an ion-exchange resin (AG 501 X8, Bio-Rad Laboratories) to remove all ionic species from the solution. After this, ε-PLL was lyophilized and reserved in a glove box. Water (w4502) was purchased from Merck.To prepare the aqueous solutions, water was added to concentrations (cw) of 35 and 40 wt%, and the two mixtures were sealed for at least three months to achieve a good water distribution. The pH value of the solutions was 10 without the addition of any buffer or salt. To lower the pH, we added HCl (Merck) to the sample with cw = 35 wt% to reach a total water content of 40 wt%. Thus, both samples had the same water content. In addition, we also prepared dilute solutions (cw = 95%) at the two pH values, for which different protocols of preparation are described later.
Differential scanning calorimeter (DSC) measurements were carried out using a Q2000 (TA Instruments) in the standard mode with cooling and heating rates of 10 K min−1. Infrared spectra were recorded using a Jasco 6500 equipped with an attenuated total reflectance unit at low temperatures (from 170 to 300 K). Viscosity measurements were made using a Malvern SV-10 Vibro Viscometer.
2.2 BDS experiments
To measure the complex dielectric permittivity, ε*(ω) = ε′(ω) − iε′′(ω), we used a Novocontrol Alpha-S analyser in the frequency range 10−2 to 106 Hz and an Agilent 4192B RF impedance analyzer over the frequency range 106 to 109 Hz.Isothermal frequency scans were performed every 2.5 K in the temperature range 140 to 300 K (temperature stability 0.1 K). The samples were prepared by forming a parallel-plate capacitor between gold-plated electrodes with diameters of 20 and 10 mm for the low- and high-frequency ranges, respectively. Further details of the fitting procedures can be found in previous works.13,20
To analyse the complex permittivity (ε*), simultaneous fitting of both the real (ε′) and imaginary (ε′′) components were performed by the use of a symmetric Cole–Cole function. However, when experiments are dominated by conductivity (as in the case of biological solutions), ε′′ can be approximated from the derivative of ε′.
 | (1) |
2.3 DLS experiments
In our concentrated solutions (cw = 40 wt%), we were interested in analysing the dynamics in terms of rotational correlation functions and density–density correlation functions across a broad temperature range including low temperatures. Therefore, light scattering measurements were performed using two different setups depending on the temperature range we were seeking to access.The reorientation of the solute molecules can be measured in a DDLS experiments in vertical–horizontal (VH) polarization mode with a coherent Verdi solid-state laser (wavelength, λ = 532 nm). The depolarized (VH) light-scattering experiment relates the intensity fluctuations of the depolarized component of the scattered light with the rotation of the molecular optical anisotropy tensor through the second Legendre polynomial.26 The scattered light was collected in 90° scattering geometry. More details of this setup have been described elsewhere.27,28 The concentrated low-temperature samples (cw = 40 wt%) were mounted in a Cryo Vac cryostat and measured in the temperature range 220 to 300 K for the sample pH = 10 sample and 255 to 300 K for the pH = 7 sample. Each temperature was measured for 10 h after 90 min of thermal stabilization at each temperature.
To examine the density–density correlation function the vertical–vertical (VV) polarization geometry was measured26 for both concentrations (cw = 40 and 95 wt%) in a PDLS experiment. In this case, we used an air-cooled helium–neon laser with a wavelength of 632 nm and an output power of approximately 30 mW in VV polarization geometry. The detector was mounted on a goniometer to cover angles from 30° to 150°, which provided a q-range of between 0.00515 and 0.0192 nm−1.
The ε-PLL solutions were transparent and could be examined using light scattering, despite being slightly yellowish. Dust particles were removed by filtering the sample with 0.2 μm Merck Millipore syringe filters into pre-cleaned sample cells with approximately 3 ml volume. In addition to filtering, extra-diluted samples were prepared by degassing in a vacuum oven at 0.1 bar for 24 h.
3 Results
3.1 Infrared spectra and calorimetric results
Fig. 1 shows the amide I band of the infrared spectra for ε-PLL (cw = 40%) at pH = 10 and 7 for different temperatures (indicated in the plot) after subtraction of the buffer spectrum. For pH = 10, the amide I band is found at 1630 cm−1, which indicates a pure β-sheet conformation. In addition, it has been proposed29 that the main chain takes a parallel β-sheet form similar to that of Nylon-6. For pH = 7, we found two peaks, at 1628 and 1665 cm−1, and these correspond with a more disordered conformation (a mix between β-sheets and turns). As can be seen in the figure, these conformations do not change at low temperatures. In addition, Fig. 1c shows the calorimetric responses of the two samples. For pH = 10, there is no crystallization, whereas the pH = 7 sample presents crystallization in both the cooling and heating cycles. This restricts the temperature range in which this sample with pH = 7 can be studied by DDLS without crystallization (above 250 K). The glass transition temperature (Tg) for the pH = 10 sample was found to be (217 ± 1) K whereas for the partially crystallized sample at pH = 7 this was (195 ± 1) K. | ||
| Fig. 1 (a) Amide I band (1630 cm−1) of the infrared spectra of ε-PLL, pH = 10 sample and amide I bands (1630 and 1655 cm−1) for pH = 7 sample at different temperatures indicated in the figure. (c) DSC scans measured at 5 K min−1 for both pH values; for the pH = 7 sample, the temperature range over which the DDLS measurements were made is shown. | ||
3.2 BDS results
The dynamics of ε-PLL aqueous solutions at pH = 10 have been characterized by dielectric spectroscopy across a broad temperature range when the water remains amorphous (pH = 10).13 Dielectric data for ε-PLL solutions at temperatures above Tg, as is common for other hydrated proteins,23,30,31 are dominated by both polarization and conductivity effects, which prevents a clear observation of the glass transition-related relaxation, even when the bio-solutions are dialyzed and filtered. As analyzed in previous works,13,14,20,21 the dielectric response of ε-PLL reveals three main relaxations (called fast-water relaxation, slow-water relaxation and α-relaxation) as seen in Fig. 2. | ||
Fig. 2 Real (a and c) and Imaginary (b and d) parts of the complex dielectric permittivity (ε*(f) = ε′(f) − iε′′(f)) of ε-PLL water solutions at pH = 10 at temperatures below and above Tg. In (d) the derivative of the real part of the permittivity  is also displayed. The solid black curves indicate the fitting results. The pink, orange, and green solid lines indicate the α-relaxation, the slow water relaxation, and the fast water relaxation, respectively. is also displayed. The solid black curves indicate the fitting results. The pink, orange, and green solid lines indicate the α-relaxation, the slow water relaxation, and the fast water relaxation, respectively. | ||
The real ε′(f) and imaginary ε′′(f) parts of the complex permittivity ε*(f) of ε-PLL with 40 wt% of water at pH = 10 are displayed in Fig. 2 for two temperatures, below (T = 210 K) and above (T = 245 K) the glass transition temperature, respectively. For T < Tg, the fast- and slow-water relaxations are observed whereas for T > Tg, the relaxations are masked by conductivity and electrode polarization but two clear steps appear in the real part of the dielectric permittivity. Using derivative analysis of the permittivity32 (eqn (1)), we can estimate the slow water and the α-relaxations as shown in 2d.
The molecular origin of each of the relaxation processes observed in Fig. 2 was also determined in previous works analysing mixtures of ε-PLL at different water contents.13,14 Fig. S1 in ESI,† shows the concentration dependence of the relaxation strength for the three processes (fast-, slow- and α-relaxations) as a function of water content. As seen in that figure, both the relaxation strength of the slow- and fast-processes increases with water content. This is an indication that both processes originate from the motion of water molecules although the slow water relaxation is affected by the solute.14 Moreover, as shown below, these two relaxations are not detected by DDLS, a technique insensitive to the relaxation of water molecules. For the α-relaxation, the relaxation strength decreases with water content and, in addition, the extrapolation of their relaxation time to 100 s agree with the calorimetric Tg. Therefore, this relaxation is related to the solute molecules. For the pH = 7 solution, by quenching, we can access the low temperature region without crystallization; however, due to the re-crystallization of water, it is challenging to detect the α-relaxation because of its overlap with the ice relaxation.33,34
Fig. 3 shows the temperature dependence of the relaxation times, which displays the three main relaxations20 of ε-PLL solution at pH = 10. As shown in Fig. 2, below Tg we found the fast-water relaxation with an Arrhenius temperature dependence with an activation energy of Ea = 0.53 and 0.57 eV for pH = 10 and 7, respectively. These values are characteristic for hydrogen-bond breaking.35 Above Tg, two relaxations can be seen, one due to the relaxation of the solute affected by water (α-relaxation) and a water relaxation affected by the solute (slow-water relaxation).13 These two processes have the same non-Arrhenius temperature dependence at different time scale (slaving phenomenon).20 For solutions at pH = 7, we can only observe the fast water relaxation because, as mentioned, crystallization as well as conductivity prevents the observation of other relaxations.
 | ||
| Fig. 3 Temperature dependence of the relaxation times of ε-PLL solutions obtained from BDS at two pH values. The vertical dashed lines indicate the glass transition temperature Tg = 217 K for pH = 10 and 195 K for pH = 7. | ||
3.3 DDLS results
Fig. 4 shows the DDLS autocorrelation function g1(t) obtained in VH geometry at 90° for selected temperatures of an ε-PLL–water solution with pH = 7. The details of the calculation of g1(t) were reported by Pabst et al.19 The results for pH = 10 (Fig. S2, ESI†) are similar to those observed for pH = 7. We can detect two decays (called α-relaxation in agreement with the relaxation observed in Fig. 2 and 3, and the A-DLS process), followed by a strong decay at longer times (B-DLS process). Therefore, we fitted the ε-PLL response to a weighted sum of two single exponentials (β = 1) and one stretched exponential: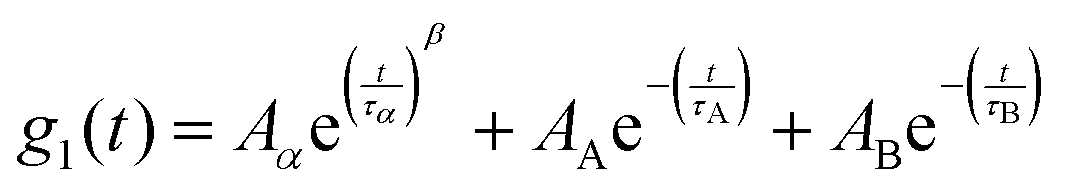 | (2) |
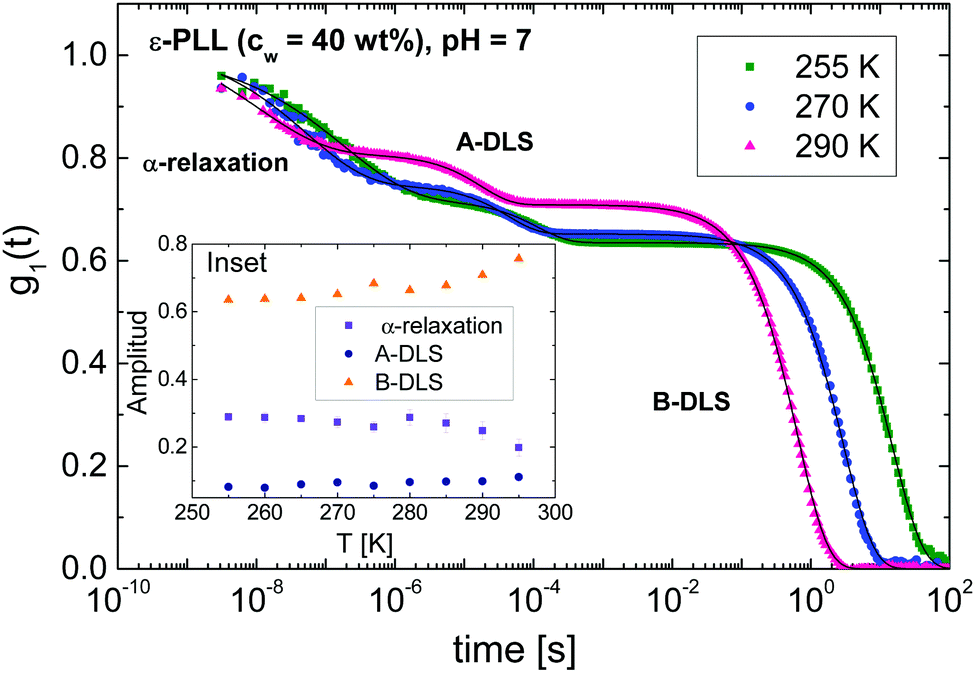 | ||
| Fig. 4 Normalized DDLS electric field correlation function (g1(t)) for an ε-PLL–water solution with 40 wt% of water and pH = 7 at different temperatures and scattering angle of θ = 90° in VH geometry. The A-DLS and B-DLS processes as well as the α-relaxation are displayed. The solid lines are the fits using two single exponentials decays for A- and B-DLS processes and an extended exponential for the α-relaxation. The inset shows the amplitude as a function of the temperature for the three mentioned processes. | ||
To compare the DDLS field correlation function g1(t) with BDS, it is useful to calculate a generalized DDLS susceptibility,28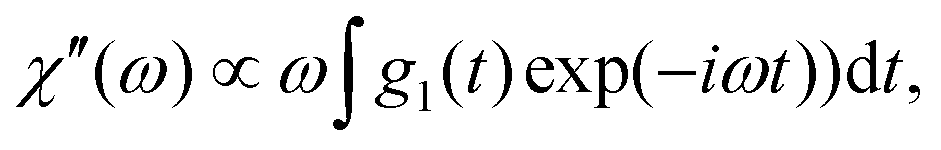 which is proportional to the Fourier transform of g1(t) multiplied by ω. This was previously established for ionic liquids,19 glass-forming liquids,37 and alcohols.28 The resulting susceptibilities are shown in Fig. 5 for samples at both pH values with selected temperatures covering the above-mentioned three processes.
which is proportional to the Fourier transform of g1(t) multiplied by ω. This was previously established for ionic liquids,19 glass-forming liquids,37 and alcohols.28 The resulting susceptibilities are shown in Fig. 5 for samples at both pH values with selected temperatures covering the above-mentioned three processes.
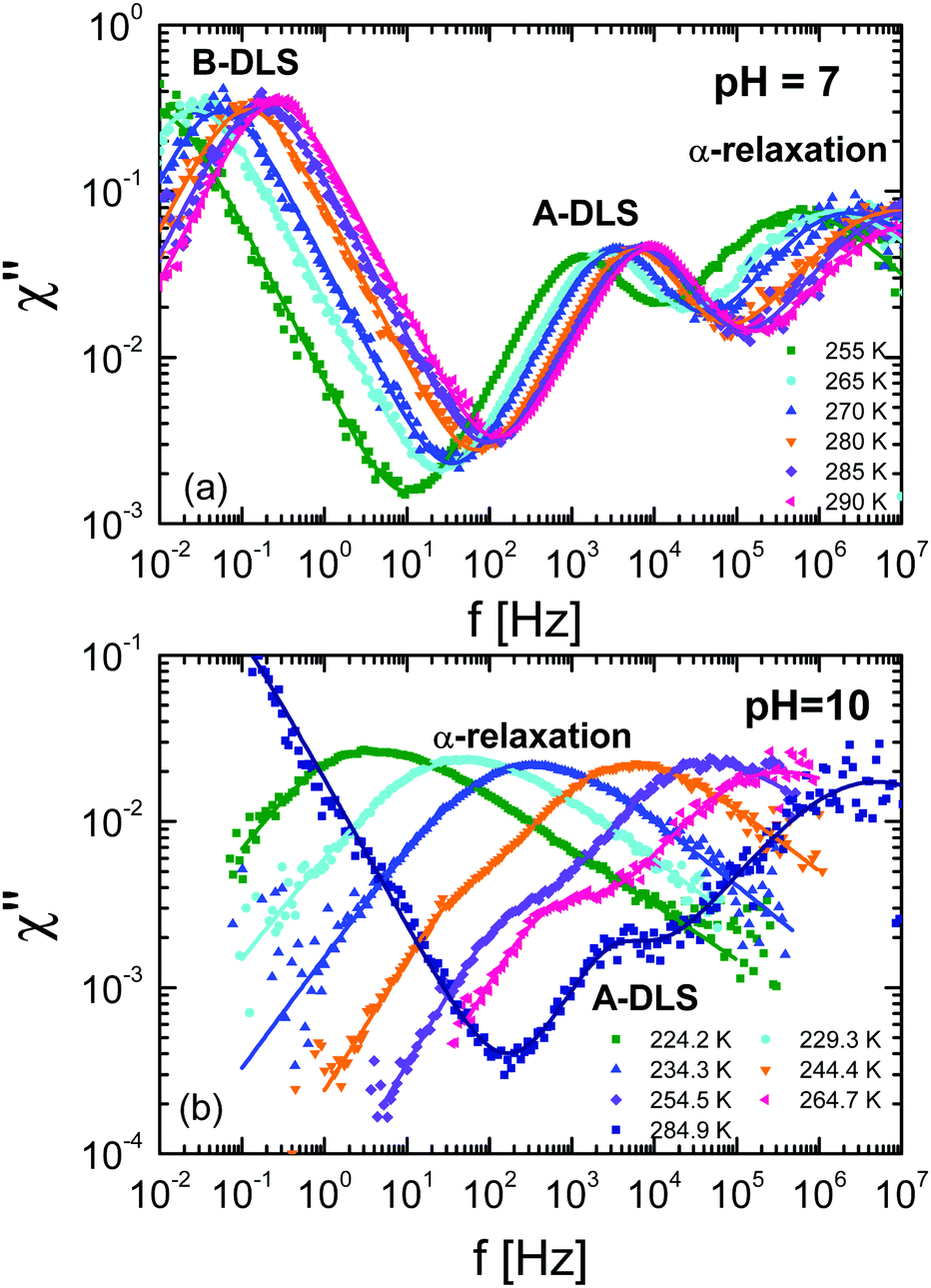 | ||
| Fig. 5 Generalized DDLS susceptibilities (χ) for ε-PLL–water solutions with 40 wt% of water at (a) pH = 7 and (b) pH = 10. The data were taken at a scattering angle of 90°. | ||
3.4 A-DLS and B-DLS processes
To analyse the origin of the A- and B-DLS processes observed in concentrated solutions of ε-PLL, we measured samples subject to different preparation procedures. Fig. 6 shows the field autocorrelation function for VV geometry and a scattering angle of 90° for concentrated samples (blue circles), diluted and repeatedly filtered (0.02 μm) samples (orange diamonds), and samples diluted and placed in a vacuum oven for 24 h (this removed several bubbles; magenta triangles). At room temperature, we can observe the A- and B-DLS processes, but the α-relaxation is outside of the time window. It is clear that the relative intensity of the B-DLS process decreases when the samples are diluted, and it is entirely removed after the samples are maintained under vacuum (see insets of Fig. 6, where the ratio of the amplitudes AB/AA decreases significantly over the whole q range). Thus, even after repeated cycles of filtration (0.2 and 0.02 μm), the B-DLS process still maintains a certain level of intensity (see Fig. 6a), while it vanishes completely after degassing the sample. Therefore, we can conclude that the scattering centers responsible for the B-DLS process are neither solid particles nor aggregates of the solute but rather gaseous nanobubbles. This is supported by the observation that the B-DLS process reappears when previously degassed solutions are vigorously shaken. We also note that we observed a similar behavior in bulk water during the present study (Fig. S3, ESI†) and mention that similar phenomena were reported in the literature for bulk water and solutions created using different sample preparation protocols.38–42 Thus, the B-DLS process is not discussed any further in the following. | ||
| Fig. 6 Normalized PDLS electric field correlation function (g1(t)) for ε-PLL–water solution of (a) pH = 7 and (b) pH = 10 at room temperature and a scattering angle of θ = 90°, showing the B-DLS and A-DLS processes. The insets show the ratio of the amplitude of both processes (AB/AA) as a function of the q value. | ||
Fig. 7 shows the 1/q2 dependence of the relaxation times of concentrated ε-PLL solutions at pH = 10 and pH = 7 for the A-DLS process. A linear relationship between τ and 1/q2 is found, as expected for long-range translational diffusion motions. The same linear relation is observed after the samples are diluted, filtered, and vacuum treated [see Fig. 7(b) and (c)]. In addition, the A-DLS process at pH = 10 does hardly change the relaxation time for the different samples at pH = 10 (only ∼0.3 decade) whereas for pH = 7 it becomes faster (∼0.8 decade).
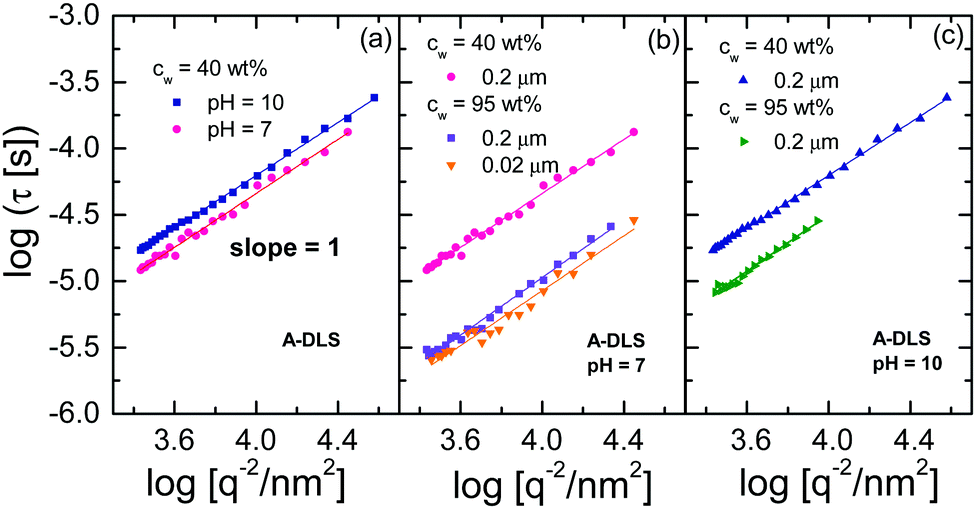 | ||
| Fig. 7 (a) 1/q2 dependence of the relaxation times for the A-DLS process of ε-PLL solutions at pH = 10 and 7; the asterisks represent the relaxation times obtained by DDLS under VH geometry at 90°. The same representation in (a) is shown for diluted, filtered and vacuum-dried samples of ε-PLL solutions at (b) pH = 10 and (c) pH = 7. | ||
Referring to the observed τ ∝ 1/q2 dependence one could tentatively estimate the hydrodynamic radius RH of a self-diffusing particle using the Stokes–Einstein relation
 | (3) |
| Sample | Filtered (μm) | c w (wt%) | R H (nm)-DLS process |
|---|---|---|---|
| pH = 10 | 0.2 | 40 | 1.36 ± 0.01 |
| pH = 10 | 0.2 | 95 | 0.78 ± 0.01 |
| pH = 10 | 0.02 + degassing | 95 | 0.65 ± 0.01 |
| pH = 7 | 0.2 | 40 | 0.94 ± 0.02 |
| pH = 7 | 0.2 | 95 | 0.21 ± 0.01 |
| pH = 7 | 0.02 + degassing | 95 | 0.18 ± 0.01 |
4 Discussion
4.1 Comparison of BDS and DDLS-α-relaxation results
The BDS and DDLS methods can be compared in terms of reorientation correlation functions. Whereas dielectric spectroscopy is sensitive to the reorientation of dipole moments (in both water and ε-PLL molecules), depolarized light scattering is sensitive to the reorientation of the optical anisotropy tensor of the ε-PLL molecules but not to the almost isotropic tensor of water. While BDS quantifies reorientation as being proportional to the first Legendre polynomial (Φl=1(θ) ∝ cos(θ)), DDLS quantifies it as proportional to the second Legendre polynomial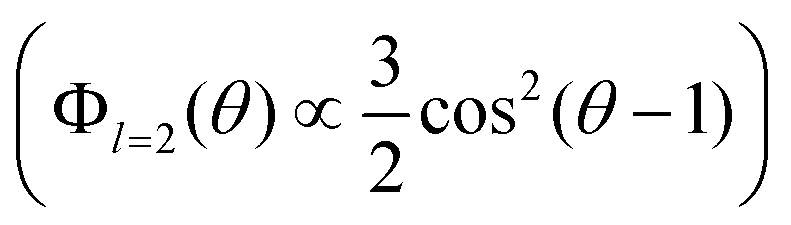 . In this way, although both techniques measure collective quantities, DDLS probes the reorientation of the optical anisotropy tensor (l = 2) of ε-PLL molecules whereas BDS probes the reorientation of both water and ε-PLL dipoles (l = 1).26
. In this way, although both techniques measure collective quantities, DDLS probes the reorientation of the optical anisotropy tensor (l = 2) of ε-PLL molecules whereas BDS probes the reorientation of both water and ε-PLL dipoles (l = 1).26
Fig. 8a shows a comparison between the DDLS and BDS data from ε-PLL solutions at pH = 10 for temperatures between 225 and 245 K. To establish this comparison, the DDLS data were vertically shifted to overlap with the BDS data in the high-frequency flank of the α-relaxation.‡ At higher frequencies, BDS data show a water relaxation (called “slow water relaxation”13) whereas DDLS, which is insensitive to water, does not show any contribution. This fact confirms that the relaxation observed by BDS is mainly due to the reorientation of water molecules with little influence of the solute. In spite of the fact that determining the spectral shape of the α-relaxation in BDS experiments (especially at low frequency) is difficult due to the strong conductivity, it is obvious that BDS data are systematically slower and broader than DDLS data as can be seen in Fig. 8(b).
 | ||
| Fig. 8 (a) Comparison of dielectric data (lines) and DDLS data (symbols) at different temperatures for ε-PLL, pH = 10. In (b) we compare the α-relaxation obtained from the fitting of the dielectric data with the susceptibilities at different temperatures as indicated in the figure. | ||
Fig. 9(a) and (b) show the temperature dependence of the relaxation times obtained from DDLS (data were taken in VH geometry at a 90° scattering angle) and BDS at both pH values, showing the α-relaxation and A-DLS processes. We note that in order to compare time constants of different model functions we used 1/(2πνp) with νp being the loss peak frequency, as a measure for the most probable relaxation time in all cases. From Fig. 9(a), it is evident that the α-relaxation times detected by BDS are slower but close to those determined by DDLS, and with the same fragility in both cases. Assuming that the dipole moment and optical anisotropy probe the same entity, the relaxation times obtained by both techniques will depend on the type of motions detected.19,26 For the limit of rotational diffusion, the ratio between the dielectric and DDLS relaxation times (τBDS/τDDLS) is 3, whereas in the case of random angle jumps it should be unity.26 It is likely that a continuous diffusion rather than a jump process is involved in the α-relaxation, and a factor of 3 is therefore expected. At high temperatures, the ratio τBDS/τDDLS [see inset of Fig. 9(a)] is approximately 3 within experimental uncertainty, whereas at low temperatures, the ratio increases. For pure liquids,28,46 the relaxation times of the α-relaxation measured by both techniques are very similar, but this is not the case for our water mixtures. The origin of this discrepancy could be related to the way in which self-correlations and cross-correlations (intermolecular solute–solute and solute–water) are detected in the different experiments. In fact, for water solutions, we expect a major role of cross-correlations in the first hydration shell. However, a more detailed analysis of this effect requires the calculation of the Kirkwood correlation factor gk in the static case, which is difficult for α-PLL solutions and goes beyond the purpose of the present work. This will be a future task.
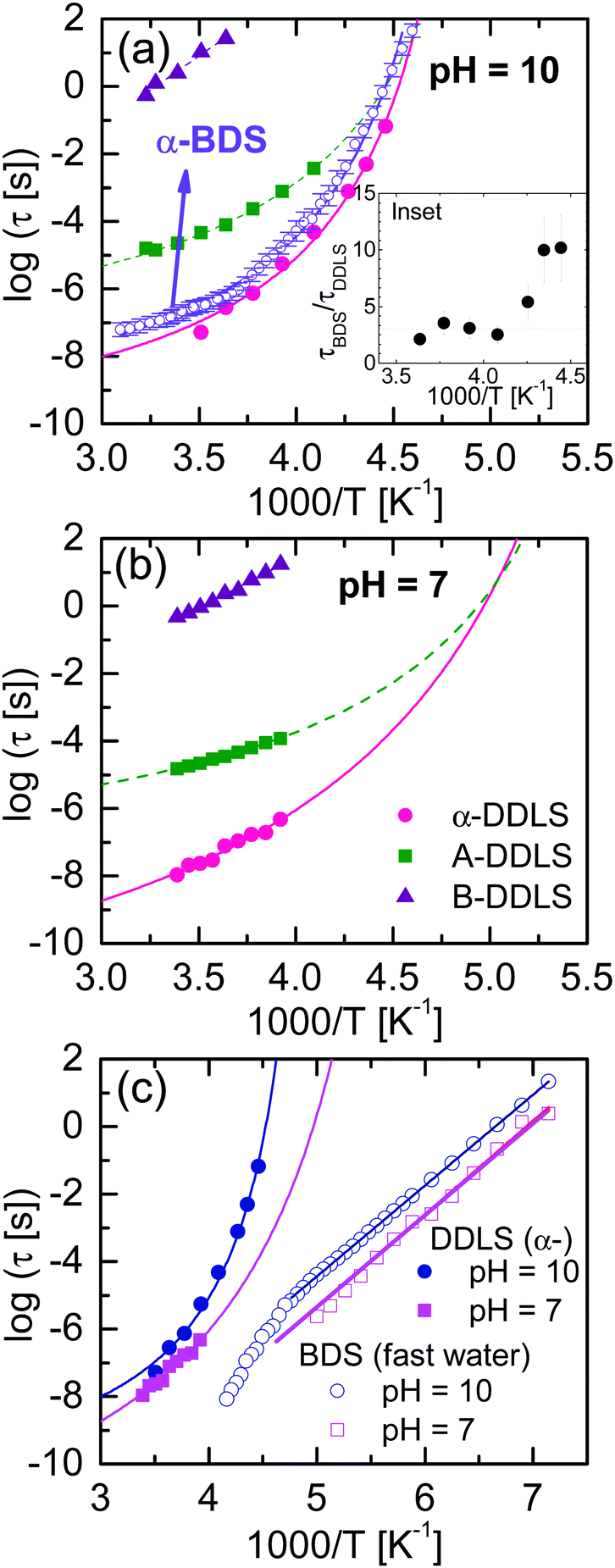 | ||
| Fig. 9 Temperature dependence of the relaxation times for ε-PLL–water solutions with (a) pH = 10 and (b) pH = 7. (c) Comparison of the relaxation times for samples at pH = 10 and 7. α-Relaxation times were obtained by DDLS, whereas the fast-water relaxation times by BDS (the slow-water relaxation is omitted for clarity). Inset: Ratios of the timescales of the α-relaxation obtained by DDLS and BDS. The dashed line indicates a factor of three (rotational diffusion; see text). | ||
Based on the data in Fig. 9, Table 2 shows the Vogel–Fulcher–Tammann (VFT) parameters corresponding to the α-relaxation for both samples. The temperature at which the structural (α-) relaxation time extrapolates to 100 s defines a “dielectric” or “DDLS” glass transition temperature, Tg,100s. For the sample at pH = 10, τα-BDS reaches 100 s at T = 218.6 K, and τα-DDLS reaches 100 s at T = 216.5 K. These values are in good agreement with the calorimetric Tg value (217 K), and both processes can therefore be related to the glass transition phenomenon.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(DT0/(T − T0))) for the α-relaxation determined by BDS and DDLS for the pH = 10 sample and by DDLS for the pH = 7 sample
exp(DT0/(T − T0))) for the α-relaxation determined by BDS and DDLS for the pH = 10 sample and by DDLS for the pH = 7 sample
| Sample | pH | D | T 0 (K) | log(τ0/s) | T g,100s (K) |
|---|---|---|---|---|---|
| ε-PLL-BDS | 10.0 | 3.4 | 193.6 | −9.5 | 218.6 |
| ε-PLL-DDLS | 10.0 | 3.4 | 192.6 | −10.0 | 216.5 |
| ε-PLL-DDLS | 7.4 | 8.1 | 155 | −11.8 | 194.6 |
The α-relaxation times of the sample at pH = 10 are slower than those at pH = 7 [see Fig. 9(c)]. When ε-PLL is in a pure β-sheet conformation, several hydrogen bonds are established between the carboxyl (COOH) and amine (NH2) groups located on the backbone of the polypeptide chain promoting the peptide hydrophobic collapse in a well-folded structure. However, at pH = 7, when there are several disordered regions (the backbone is more extended and flexible because of the partial lack of the hydrophobic core), these H-bonds no longer exist and the dynamics is consequently faster in this case.
Fig. 10 shows a comparison of DDLS spectra at a fixed temperature (T = 275 K) for samples at different pH values. The shape of the α-relaxation is the same for both samples independent of the conformation (see inset of Fig. 10). As we have indicated, at pH = 7 the chain is disordered while at pH = 10 it is folded. This means that if we look at an area several monomers away from a given monomer, the environments in each case will be somewhat different. However, the spectral shape of the α-relaxation does not change significantly, indicating that the environments are not very different.36 We conclude that very few monomers are involved in the α-relaxation, i.e., that the correlation length must be very small and limited to the direct neighbours of a given monomer.
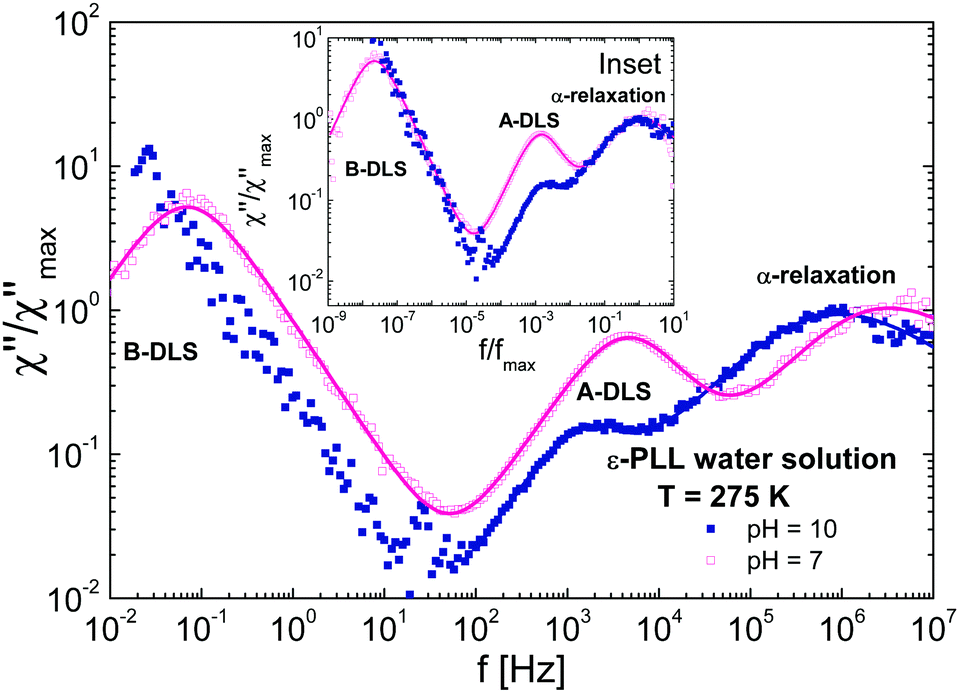 | ||
| Fig. 10 Normalized DDLS susceptibilities (χ) for ε-PLL–water solutions with 40 wt% water at 275 K and (a) pH = 7 (b) pH = 10. Inset: The same data but frequency normalized. | ||
4.2 A-DLS process
Now we focus on the origin of the A-DLS process observed in the solutions at both pH values. It is well known that many liquids, or binary mixtures of liquids, show a process slower than the α-relaxation detectable by DLS. In some cases, this process approaches the α-relaxation at temperatures close to Tg. In addition, as shown in Fig. 7, the A-DLS process shows a 1/q2 dependence, which indicates diffusive dynamics. With these characteristics, this process could be related to thermally driven concentration fluctuations.47,48 However, to classify this process as concentration fluctuations, the presence of some inhomogeneities in the sample is necessary. Based on previous analyses using small-angle X-ray scattering,14 no structural inhomogeneity on a length scale from 20 to 80 nm for the sample with pH = 10 samples can be detected. Even more, this sample does not present any crystallization on heating or cooling detected by DSC. This implies that, on a scale above ∼1 or 2 nm, the sample is homogeneous, as otherwise water molecules would crystallize. Molecular simulations on smaller 1- and 4-lysine molecules have shown that no aggregation is observed at high pH values.49,50 These facts strongly suggest that the origin of the A-DLS cannot be due to concentration fluctuations. Additionally, a process observed by dynamic light scattering with the same phenomenological behaviour as observed here for ε-PLL, has previously been found for other aqueous solutions (such as water mixtures of xylitol,43 glucose,51 or α-cyclodextrin52). In all these cases, this process was attributed to the diffusion of an individual solute molecule or very small clusters of solute molecules,43 as in the present case.The A-DLS process of the ε-PLL solutions at pH = 10 is not affected by dilution, filtration, or degassing (see inset of Fig. 6). The diameters (2RH) obtained from PDLS (see 1) were 2.72 and 1.56 nm for the concentrated and diluted samples, respectively. Considering that ε-PLL has 32 residues and that it is uncommon to find fewer than five strands in a parallel β-sheet,53 the average length of an ε-PLL β-sheet should be about 2.2 nm. Comparing this value with the diameter (2RH) obtained from PDLS, we can assign this process to the diffusion of individual β-sheets in ε-PLL solutions, possibly each surrounded by a layer of water.
When the sample is in a disordered state, the chains are extended in a polymer-like structure. In this case, the susceptibility calculated from DDLS measurements in Fig. 10 shows a more pronounced and faster process than that for the sample at pH = 10. Since this process is faster than that at pH = 10, it is unlikely to represent the diffusive motion of the whole molecule as in the previous case. This process for the sample at pH = 7, also has a diffusive q-dependency, and may therefore be related to segmental relaxation consisting of several monomers.
Since the DDLS experiment measures the reorientation of the optical anisotropy tensors of the ε-PLL molecule, the α-relaxation can be identified as a reorientation of monomer segments of the ε-PLL molecule in the β-sheet conformation and in the unfolded structure. But obviously the reorientation of the optical anisotropy tensor of α-PLL during the α process is incomplete, and it is terminated by the q-dependent density–density correlation of the A-DLS process. As the intensity of the A-DLS process is slightly larger in the pH = 7 case, the α-relaxation in the β-sheet conformation leaves less relaxation intensity for the final A-DLS process and thus seems to be less restricted than in the more unfolded conformation. While it is likely that the A-process is related to the whole ε-PLL molecule in the solution with pH = 10, it is unclear how to attribute our findings to a more concrete molecular picture for the sample at pH = 7.
4.3 Fast-water relaxation
Finally, we focus on the water relaxation observed by BDS [see Fig. 9(c)]. As established in previous works,13,14,20 the fast-water relaxation for the sample with pH = 10 is universal for all confined water and aqueous solutions, and it can therefore be considered as bulk-like water54 (i.e., with minor influence from interactions with the solute). This can be seen in Fig. 11b where we show a comparison of the relaxation times for solutions that present two water related relaxations such as poly(vinyl pyrrolidone) or dextran. Independent on the type of solute, the fast water relaxation times (at high enough water contents) are very similar. However, the fast water relaxation becomes one decade faster at low pH values when the main chain is unfolded, as shown in Fig. 9(c). The ε-PLL in the disordered state has a larger content of polar and non-polar residues exposed to water than ε-PLL in the folded state. In addition, carboxyl and amine groups are protonated at pH = 7, and this could affect the concentration of free ions in the solution. In fact, previous studies on the properties of the hydration water in disordered proteins reveal a non-uniform distribution and higher hydration water density,55 as well as a faster dynamics11 around these proteins as compared to the globular case, in agreement with our findings. | ||
| Fig. 11 Temperature dependence of the relaxation times of the fast-water relaxations in different systems (ε-PLL at pH = 10, low density amorphous ice (LDA),56 poly(vinyil pyrrolidone)13 and dextran14). | ||
5 Conclusions
We combined DLS and BDS approaches studying the dynamics of a fully homogeneous aqueous ε-PLL solution at pH = 10 with the ε-PLL in β-sheet conformation. In addition, we examined the dynamics of the same solution at pH = 7, when the ε-PLL chain is in a disordered state.As previously concluded from studies using BDS and NMR, the dynamics of this system is complex. Due to the particular combination of techniques in the present study, the segmental ε-PLL α-relaxation was clearly identified in both solutions. Moreover it turned out that the segmental and fast water relaxations are slower when ε-PLL is in a folded state.
In addition to the α-relaxation, by both DDLS and PDLS we find two slower processes (A-DLS and B-DLS processes) that cannot be observed in BDS experiments. The B-DLS process is due to the presence of bubbles and not solid particles in the solutions as determined by both diluting and degassing the sample. The A-DLS process has a 1/q2 dependent relaxation time, which indicates a diffusive dynamics. For the homogeneous sample at pH = 10, this DLS process is assigned to the diffussion of single-molecule ε-poly(lysine) whereas in the polymer-like conformation (pH = 7), the picture is less clear and needs more detailed investigation.
Author contributions
J. H. M. and J. P. G. contributed equally, J. H. M. and J. P. G. analyzed the data, J. H. M., J. P. G. and F. P. performed the PCS measurements, J. H. M., J. P. G., T. B. and S. C. wrote the manuscript, T. B. and S. C. provided the resources, funding, and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support through the Grant No. PID2019-104650GB-C21 (Spanish Government “Ministerio de Ciencia, Innovacion y Universidades”), LINKB20012 (CSIC), Project No. IT-1175-19 (Basque Government) are gratefully acknowledged.Notes and references
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef CAS PubMed.
- J. Ward, J. Sodhi, L. McGuffin, B. Buxton and D. Jones, J. Mol. Biol., 2004, 337, 635–645 CrossRef CAS PubMed.
- L. M. Iakoucheva, C. J. Brown, J. Lawson, Z. Obradović and A. Dunker, J. Mol. Biol., 2002, 323, 573–584 CrossRef CAS PubMed.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS PubMed.
- A. K. Dunker and Z. Obradovic, Nat. Biotechnol., 2001, 19, 805–806 CrossRef CAS PubMed.
- M.-C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef CAS PubMed.
- H. Frauenfelder, P. Fenimore and B. McMahon, Biophys. Chem., 2002, 98, 35–48 CrossRef CAS PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14408–14413 CrossRef CAS PubMed.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129–5134 CrossRef CAS PubMed.
- F.-X. Gallat, A. Laganowsky, K. Wood, F. Gabel, L. van Eijck, J. Wuttke, M. Moulin, M. Härtlein, D. Eisenberg, J.-P. Colletier, G. Zaccai and M. Weik, Biophys. J., 2012, 103, 129–136 CrossRef CAS PubMed.
- G. Schirò, Y. Fichou, F.-X. Gallat, K. Wood, F. Gabel, M. Moulin, M. Härtlein, M. Heyden, J.-P. Colletier and A. Orecchini, et al. , Nat. Commun., 2015, 6, 1–8 Search PubMed.
- W. Adamski, N. Salvi, D. Maurin, J. Magnat, S. Milles, M. R. Jensen, A. Abyzov, C. J. Moreau and M. Blackledge, J. Am. Chem. Soc., 2019, 141, 17817–17829 CrossRef CAS PubMed.
- I. Combarro Palacios, C. Olsson, C. S. Kamma-Lorger, J. Swenson and S. Cerveny, J. Chem. Phys., 2019, 150, 124902 CrossRef PubMed.
- S. Cerveny and J. Swenson, J. Chem. Phys., 2019, 150, 234904 CrossRef PubMed.
- L. M. Sáiz and S. Cerveny, J. Non-Cryst. Solids, 2015, 407, 486–493 CrossRef.
- M. Grimaldo, F. Roosen-Runge, F. Zhang, F. Schreiber and T. Seydel, Q. Rev. Biophys., 2019, 52, e7 CrossRef.
- F. Kremer and A. Schönhals, Broadband dielectric spectroscopy, Springer, 2003 Search PubMed.
- J. Gabriel, F. Pabst, A. Helbling, T. Böhmer and T. Blochowicz, in Depolarized Dynamic Light Scattering and Dielectric Spectroscopy: Two Perspectives on Molecular Reorientation in Supercooled Liquids, ed. F. Kremer and A. Loidl, Springer International Publishing, Cham, 2018, pp. 203–245 Search PubMed.
- F. Pabst, J. Gabriel, P. Weigl and T. Blochowicz, Chem. Phys., 2017, 494, 103–110 CrossRef CAS.
- S. Cerveny, I. Combarro-Palacios and J. Swenson, J. Phys. Chem. Lett., 2016, 7, 4093–4098 CrossRef CAS PubMed.
- M. Weigler, I. Combarro-Palacios, S. Cerveny and M. Vogel, J. Chem. Phys., 2020, 152, 234503 CrossRef CAS PubMed.
- S. Magazù, F. Mezei, P. Falus, B. Farago, E. Mamontov, M. Russina and F. Migliardo, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3504–3512 CrossRef PubMed.
- S. Khodadadi and A. P. Sokolov, Soft Matter, 2015, 11, 4984–4998 RSC.
- K. Sasaki, I. Popov and Y. Feldman, J. Chem. Phys., 2019, 150, 204504 CrossRef PubMed.
- Y. Kurzweil-Segev, A. Greenbaum (Gutina), I. Popov, D. Golodnitsky and Y. Feldman, Phys. Chem. Chem. Phys., 2016, 18, 10992–10999 RSC.
- B. J. Berne and R. Pecora, Dynamic light scattering: with applications to chemistry, biology, and physics, Dover Publications, 1976 Search PubMed.
- T. Blochowicz, E. Gouirand, S. Schramm and B. Stühn, J. Chem. Phys., 2013, 138, 114501 CrossRef PubMed.
- J. Gabriel, F. Pabst and T. Blochowicz, J. Phys. Chem. B, 2017, 121, 8847–8853 CrossRef CAS PubMed.
- S. Maeda, K.-K. Kunimoto, C. Sasaki, A. Kuwae and K. Hanai, J. Mol. Struct., 2003, 655, 149–155 CrossRef CAS.
- K. L. Ngai, S. Capaccioli and N. Shinyashiki, J. Phys. Chem. B, 2008, 112, 3826–3832 CrossRef CAS PubMed.
- A. Panagopoulou, A. Kyritsis, N. Shinyashiki and P. Pissis, J. Phys. Chem. B, 2012, 116, 4593–4602 CrossRef CAS PubMed.
- M. Wübbenhorst and J. van Turnhout, J. Non-Cryst. Solids, 2002, 305, 40–49 CrossRef.
- K. Sasaki, R. Kita, N. Shinyashiki and S. Yagihara, J. Phys. Chem. B, 2016, 120, 3950–3953 CrossRef CAS PubMed.
- K. Sasaki, A. Panagopoulou, R. Kita, N. Shinyashiki, S. Yagihara, A. Kyritsis and P. Pissis, J. Phys. Chem. B, 2017, 121, 265–272 CrossRef CAS PubMed.
- S. Cerveny, J. Colmenero and Á. Alegría, Eur. Phys. J. Spec. Top., 2007, 141, 49–52 CrossRef.
- F. Pabst, J. P. Gabriel, T. Böhmer, P. Weigl, A. Helbling, T. Richter, P. Zourchang, T. Walther and T. Blochowicz, J. Phys. Chem. Lett., 2021, 12, 3685–3690 CrossRef CAS PubMed.
- F. Pabst, A. Helbling, J. Gabriel, P. Weigl and T. Blochowicz, Phys. Rev. E, 2020, 102, 010606 CrossRef CAS PubMed.
- F. Eklund and J. Swenson, Langmuir, 2018, 34, 11003–11009 CrossRef CAS PubMed.
- M. Alheshibri, J. Qian, M. Jehannin and V. S. J. Craig, Langmuir, 2016, 32, 11086–11100 CrossRef CAS PubMed.
- Z. Fang, L. Wang, X. Wang, L. Zhou, S. Wang, Z. Zou, R. Tai, L. Zhang and J. Hu, J. Phys. Chem. C, 2018, 122, 22418–22423 CrossRef CAS.
- J. Qiu, Z. Zou, S. Wang, X. Wang, L. Wang, Y. Dong, H. Zhao, L. Zhang and J. Hu, ChemPhysChem, 2017, 18, 1345–1350 CrossRef CAS PubMed.
- J. Tian, F. Yang, H. Cui, Y. Zhou, X. Ruan and N. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 26579–26584 CrossRef CAS PubMed.
- K. Elamin, S. Cazzato, J. Sjöström, S. M. King and J. Swenson, J. Phys. Chem. B, 2013, 117, 7363–7369 CrossRef CAS PubMed.
- D. L. Sidebottom and T. D. Tran, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 051904 CrossRef CAS PubMed.
- K. Elamin and J. Swenson, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 032306 CrossRef PubMed.
- J. Gabriel, F. Pabst, A. Helbling, T. Böhmer and T. Blochowicz, Phys. Rev. Lett., 2018, 121, 035501 CrossRef CAS PubMed.
- R. Kant, S. K. Kumar and R. H. Colby, Macromolecules, 2003, 36, 10087–10094 CrossRef CAS.
- S. Schramm, T. Blochowicz, E. Gouirand, R. Wipf, B. Stühn and Y. Chushkin, J. Chem. Phys., 2010, 132, 224505 CrossRef PubMed.
- A. Nasedkin, S. Cerveny and J. Swenson, J. Phys. Chem. B, 2019, 123, 6056–6064 CrossRef CAS PubMed.
- A. Henao, G. N. Ruiz, N. Steinke, S. Cerveny, R. Macovez, E. Guàrdia, S. Busch, S. E. McLain, C. D. Lorenz and L. C. Pardo, Phys. Chem. Chem. Phys., 2020, 22, 6919–6927 RSC.
- D. L. Sidebottom, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 011505 CrossRef CAS PubMed.
- F. Jin, X. Ye and C. Wu, J. Phys. Chem. B, 2007, 111, 13143–13146 CrossRef CAS PubMed.
- J. S. Richardson, Nature, 1977, 268, 495–500 CrossRef CAS PubMed.
- S. Cerveny, F. Mallamace, J. Swenson, M. Vogel and L. Xu, Chem. Rev., 2016, 116, 7608–7625 CrossRef CAS PubMed.
- L. Aggarwal and P. Biswas, J. Phys. Chem. B, 2018, 122, 4206–4218 CrossRef CAS PubMed.
- K. Amann-Winkel, C. Gainaru, P. H. Handle, M. Seidl, H. Nelson, R. Böhmer and T. Loerting, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17720–17725 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Relaxation strength versus water concentration, normalized DDLS electric field correlation function for ε-PLL solution at pH = 10, Intensity correlation function in VV geometry for water. See DOI: 10.1039/d1cp01893k |
| ‡ We note that in contrast to BDS the DDLS data originate from a normalized correlation function and therefore do not contain information on the absolute amplitude. |
| This journal is © the Owner Societies 2021 |