
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersion forces in chirality recognition – a density functional and wave function theory study of diols†
Xaiza
Aniban‡
 ,
Beppo
Hartwig‡
,
Axel
Wuttke
and
Ricardo A.
Mata
*
,
Beppo
Hartwig‡
,
Axel
Wuttke
and
Ricardo A.
Mata
*
Institut für Physikalische Chemie, Tammannstrasse 6, Göttingen, Germany. E-mail: rmata@gwdg.de
First published on 14th May 2021
Abstract
In the discussion of chirality recognition, steric considerations and strongly directed interactions such as hydrogen bonds are primarily discussed. However, given the sheer size of biomolecules, it is expected that dispersion forces could also play a determining role for aggregate formation and associated chirality recognition. With the example of diol molecules, we explore different factors in the formation of homo- and hetero-dimers as well as their relative stability. By comparing density functional results with the analysis of local correlation methods, we infer the impact of dispersion not only on the energies but also on the structures of such chiral aggregates. A local orbital based scheme is used to calculate wave function dispersion-free gradients and compare to uncorrected density functional structures.
1 Introduction
The ability of a chiral probe to distinguish between two enantiomers of a chiral molecule, or the so called chirality recognition, is a very important consideration for biochemistry1 and organic chemistry.2,3 The human body has numerous chiral receptors itself. For example, (R)-limonene smells more like lemons while (S)-limonene smells more like oranges.4 In the field of synthesis and drug development as well as toxicology, chirality recognition has also played a very important role.5–9 One crucial historical example is the drug thalidomide which was prescribed to women to alleviate morning sickness. Later it was discovered that the (R)-enantiomer produced the desired effect, while the (S)-enantiomer caused severe birth defects.10–12 With this impact of chirality, understanding its fundamental behavior will allow us to understand biomolecular functions better as well as take advantage of the forces controlling it to lean towards our preferred enantioselectivity.Hydrogen bonding is one of the weak contact pairs that is present in chirality recognition. This is of particular importance especially in supramolecular chirality13 and chirality effects in molecular imprinting.14 However, its transient nature makes it very difficult to identify and characterize in condensed phase. Cold gas-phase experiments, on the other hand, provide a powerful way to examine the chirality recognition without the perturbation brought about by the solvent. Such experiments give information (e.g. structure and dissociation energy) which are helpful to understand the forces at play via theoretical modeling.
Benchmarking by gas phase experiments is highly favorable for quantum mechanical studies because of the absence of perturbations of liquids/solvents and matrices.15 Furthermore, the use of jet cooling allows for low temperatures which more closely coincide with the commonly assumed 0 K for quantum chemical calculations. From the theoretical perspective, one of the important aspects to examine is the relative stability of some local minima representing possible structures that might be present during the gas phase experiments. These structures can be deduced by experiments like IR, Raman and microwave spectroscopy. For the relative stabilities, especially for systems which are governed by non-covalent interactions (e.g. neutral dimers), the choice of electronic structure method plays an important role.
In systems where hydrogen bonding is possible, it is usually assumed that charge transfer and electrostatics effects are the main forces for stabilization of the conformation. While that might often be true, this does not show the whole picture. Other intermolecular forces of attraction such as dispersion are present. Although the latter is considered weaker, its contribution will increase with the size of the system. Current quantum chemical approaches are able to capture this, giving more information about the forces responsible for the structure stability. Dispersion has long been neglected in theoretical treatments because (1) it is a weak force (only true to one pair of interaction) which decays at R−6 and (2) it was thought to have a small effect (percentwise) on the total energy of a system. In recent years, it was shown that this is not as negligible as thought, and in some instances, dispersion is a key force for stabilization16,17 and is an important point to consider in interpreting reactivity.18–24
The importance of hydrogen bonding, and by proxy also dispersion interactions, for chirality recognition in the gas phase has been studied extensively by infrared, Raman, microwave and mass spectroscopy.25–35 Furthermore, Zehnacker and Suhm provide a comprehensive look at literature known chiral recognition phenomena in ref. 36. Most studies are concerned with intermolecular chiral interactions but intramolecular recognition can also occur, for example in the mother of all folding, i.e. n-alkanes where all gauche angles in the kink of the most stable hairpin structure exhibit the same sign.37 Other examples can also be found in ref. 36. The importance extends to solution as well. For instance, it is the driving force for the stereo-specific CH/CC activation for a manganese based catalyst.21 Similarly Schreiner and co-workers23 showed that the enantioselectivity of the Corey–Bakshi–Shibata reduction is governed by London dispersion instead of steric hindrance. By introducing easily polarizable groups for the catalyst as well as the reactant, the enantiomeric excess can be significantly enhanced. Another reaction where dispersion is vital is for catalytic asymmetric Diels–Alder reactions as studied by Bistoni and co-workers.24 It was shown that the chiral ion pair forms a pocket for the diene unit, resulting in the enantioselectivity, which is stabilised by London forces. Dispersion also plays an important role in host–guest interactions, responsible for the chiral separation in chromatographic methods.38–41
The failure of density functional theory (DFT) to describe dispersion interactions has been documented 25 years ago.42–44 The success of some functionals (most notably B3LYP/6-31G(d)) was due to the error compensation between the lack of dispersion and pronounced BSSE.45 London dispersion is a long range interaction while in standard DFT, energies are approximated on the basis of local quantities of the local electron density or the reduced density gradient in GGA functionals.46 This has been remedied by Grimme's popular dispersion correction schemes, which involves a ‘correction’ potential function which is added to the exchange–correlation functional of choice.47–50 However, this does not only capture dispersion interactions but corrects for other shortcomings as well. Wavefunction theory, on the other hand, captures dispersion not as a separate term but as a part of the total energy. To isolate dispersion contributions and non-covalent interactions (NCI) in general, several approaches have been devised. Symmetry Adapted Perturbation Theory (SAPT)51 can be used as a method to compute accurate interaction energies and as an energy decomposition analysis (EDA) scheme. Energy components in SAPT (and its corresponding variants) include the usual terms: electrostatics, exchange and dispersion. Another scheme is the local correlation-based EDA which naturally describes long-range correlation effects, like London dispersion. The latter works by assigning each orbital (occupied and virtual) to the fragment in which it is dominantly localized which allows regrouping of the double excitations (see Fig. 2) into several families corresponding to different physical components of the interactions (e.g. the LMP2 approach).52 Two types of contributions can be obtained: intra and inter-fragment contributions, in which the latter can be further subdivided into several terms, one of which is the dispersion. Lastly, local variants of coupled cluster methods are also used to compute NCIs. PNO-LCCSD(T)-F1253 and DLPNO-CCSD(T)/LED54–56 methods are available, to mention a few.
One interesting mechanism proposed to explain chirality recognition between chiral species is via chirality induced spin selectivity (CISS).57 According to Kumar et al.,58 spin polarization interaction is less repulsive for homo-chiral molecules than that of heterochiral molecules. Thus, the overall interaction between each type of systems is enantiospecific. However, it must be noted that this is a short-range interaction where a significant orbital overlap is relevant. Furthermore, quantum electrodynamics would generally indicate that for self aggregation hetero-chiral pairings are favourable with regards to their dispersion interaction.59 In this work, we investigate if chirality recognition for several molecular systems is driven by dispersion – a long-range type of interaction. We focused on ethanediol (EDO), cyclohexanediol (CHexDO) and pinacol neutral dimer systems. The transiently chiral properties for ethanediol and pinacol as well as the permanently chiral properties for cyclohexanediol are illustrated in Fig. 2. Dispersion contributions were “turned on and off” for DFT and WFT to see how both the energy and structure of the systems were affected. Changes in structure were carefully examined as this aspect is oftentimes neglected. Most works quantify dispersion by examining energy differences on fixed structures. Two important structural parameters were analyzed, i.e. intermolecular hydrogen bonding (O–H bonding) and the distance between the center of masses of each monomer in the dimer, R(CM–CM). These are important considerations in benchmarking. For the energy part, the range of relative stabilities and more importantly, the heterochiral–homochiral (het–hom) energy gap were carefully analyzed. Lastly, we used dispersion interaction density (DID) visualization60,61 to observe which specific parts of the system are interacting due to dispersion.
2 Computational methods
2.1 DFT calculations
Previously the CREST (Conformer-Rotamer Ensemble Sampling Tool) program,62,63 using the semi empirical GFN2-xtb (Geometries Frequencies Noncovalent Interactions version 2 – extended Tight Binding) method,64,65 was employed to tackle the conformational complexity of ethanediol and cyclohexanediol and proved its general reliability.33 Therefore, this combination was again used to explore the conformational landscape of pinacol. Specifically the NCI mode (non-covalent interactions mode) was used which adapts the number of metadynamics runs and the parameters of the Gaussian functions to be better suited for aggregates. Furthermore, a wall potential is applied to prevent dissociation which increases the effectiveness of the sampling. To ensure that no conformers have been overlooked multiple sampling runs were made as well as manual cross-checking.The large amount of conformers generated by CREST were then pre-optimised with the B97-3c66 functional using the ORCA (version 4.2.1) program package67,68 which was also used for all density functional calculations. Subsequently, these structures were optimised with the BP86,69–71 PBE,72 PBE073,74 and B3LYP69,75,76 functionals, followed by an analytical frequency calculation within the double harmonic approximation. To account for London forces Grimme's D3 dispersion correction including three body terms was used together with Becke Johnson damping49,77 (D3(BJ,abc)). For BP86 and PBE density fitting (RI-J), with the corresponding auxiliary basis set78 was used whereas for PBE0 and B3LYP no density fitting was used. For all calculations the ma-def2-TZVP basis set was employed.79,80 In our testing with B3LYP we found that for the dimers of ethanediol relative energies of almost aug-cc-pVQZ81 level (largest basis set tested) can be reached with this basis set but with much lower computational cost (about 90 times faster on average). Basis sets of double up to quadruple zeta level where tested for the Ahlrichs,79,80,82 Dunning81,83 and Jensen84 basis set families in its non augmented and augmented forms. It should be noted though that the largest basis set is not necessarily the best if the method contains empirical parameters adjusted for a finite basis set. An Overview of the results for each basis set can found in the ESI† (Fig. S1).
To judge the influence of the dispersion correction all geometries have been reoptimised without D3(BJ,abc) and a frequency calculation was performed. However, for the influence of dispersion itself this approach may lead to inaccurate results. Firstly, density functionals already account for dispersion-like contributions at short distances. Secondly, it is not clear to what extent the empirical correction actually corrects for other shortcomings such as the correlation and exchange part of the functional. In case of hybrid functionals the degree to which the exact Hartree–Fock exchange is mixed in introduces another uncertainty. However, by using a multitude of functionals we are able to obtain a more general idea of how DFT is influenced by D3(BJ,abc). On the other hand, the proposed WFT approach provides a more rigorous distinction between the inclusion or neglect of dispersion interactions. Therefore, we are also able to ascertain the validity of directly taking the D3(BJ,abc) correction value to judge London interactions. The comparison between local correlation analysis and dispersion corrections has over the years shown that the agreement varies, depending on the type of correction and functional applied. In some examples, an extreme variability has been shown,85 while in other cases the D3/D4 corrections are in line, albeit smaller than the local correlation values (see Fig. 10 in ref. 86). A more extensive review of interpretations based on dispersion corrections is provided in ref. 56, for the interested reader. All DFT results with and without D3(BJ,abc) can be found in the ESI† (Fig. S2).
Gibbs energy calculations were also carried out with the ORCA program package which assumes ideal gas statistical mechanics. Low lying frequencies were treated with Grimme's Quasi-RRHO87 approach to compute their contributions to the vibrational entropy. The rotational contributions to the entropy are computed according to Herzberg.88 The influence of the symmetry number (σ) is included. Furthermore, we also take the degeneracy into account which arises from the fact that most dimers are themselves chiral and penalise those that are not chiral.
2.2 WFT calculations
For methods utilizing WFT, optimization and frequency calculations of the molecular systems were done using LMP289 and SCS-LMP290 (basis set: aug-cc-pVTZ, H = cc-pVTZ).81,83 The Pipek–Mezey localization91 scheme was used, as well as density fitting for both reference wavefunction and LMP2 calculations. Geometry optimisations were also carried out removing dispersion contributions with a Molpro 2018.192 development version. To do so, the following procedure was adopted:• An LMP2 energy calculation was carried out at each step. The amplitudes are obtained in an iterative fashion given that one is using non-canonical orbitals.
• The amplitudes for pair excitations describing dispersion (Tijab with i,a ∈ A and j,b ∈ B ≠ A, cf.Fig. 1) were set to zero at the end of the LMP2 iterations.
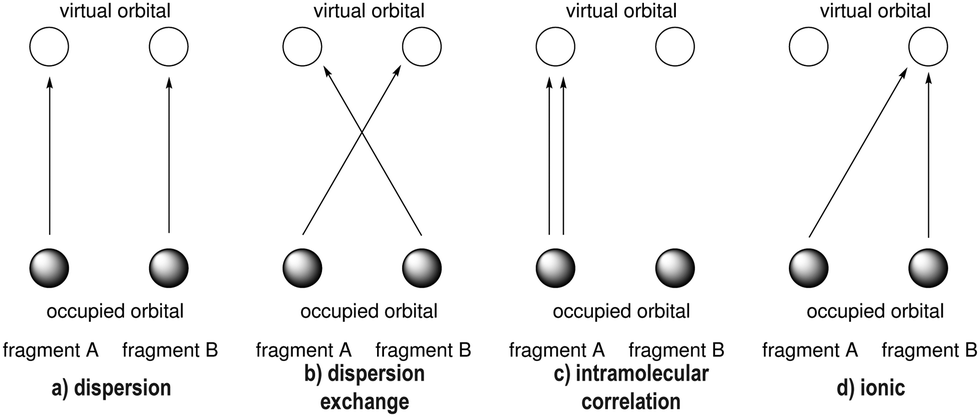 | ||
| Fig. 1 Excitation scheme for the energy decomposition of the local WFT approach between a fragment A and a fragment B. | ||
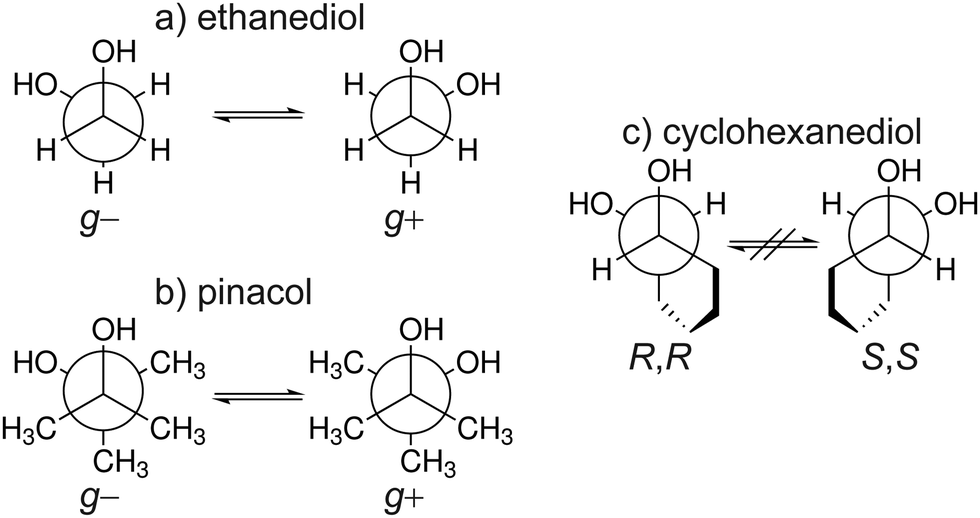 | ||
| Fig. 2 Overview of the three molecular systems studied shown in their respective Newman projections. The transient chiral nature of ethanediol (a) and pinacol (b) between the gauche + and − conformers is shown. Similarly the permanent chirality of cyclohexanediol (c) between the R,R and S,S conformers. | ||
• The coupled perturbed localization equations were solved and the (SCS-)LMP2 gradients computed, all using the altered amplitudes matrix.
• If the Hess-matrix was computed, the same amplitudes matrix was used, consistent with the gradients.
The results were confirmed by numerical gradients.
For WFT methods with dispersion contributions, the Molpro 2018.192 commercial version was utilized. In order to visualize the spatial contributions to the dispersion energy the dispersion interaction density (DID) approach60 was applied. DID calculations were carried out using Molpro 2019.2.93
Unless otherwise stated, all presented energies are zero point corrected.
3 Results and discussion
The nomenclature used is the same that was used in ref. 33. It distinguishes between dimers of the same chirality (homo = hom) and different chirality (hetero = het). Furthermore, the conformers are characterised by the amount of intermolecular hydrogen bonds and how the terminal hydrogen bond (including only intermolecular hydrogen bonds) is oriented. If the terminal O–OH angle is below 120° a′ is added. This is shown in Fig. 3 which illustrates the difference between hom3 and hom3′. To retain uniqueness of each label an a is added if any label reoccurs. It also represents its own structural motif, where the C–C backbone is oriented parallel whereas it is more orthogonal otherwise (see ESI,† Fig. S7 for a direct comparison). Additionally the subscript b is added for bifurcated hydrogen bond arrangements. In the bifurcated arrangement shown in Fig. 3 it becomes necessary to account for two different O–OH angles which overall results in hom3b′ as its name. het4 on the other hand has no terminal/dangling OH groups and therefore no more specification is needed. All conformers with the same name of the different compounds share a close structural similarity.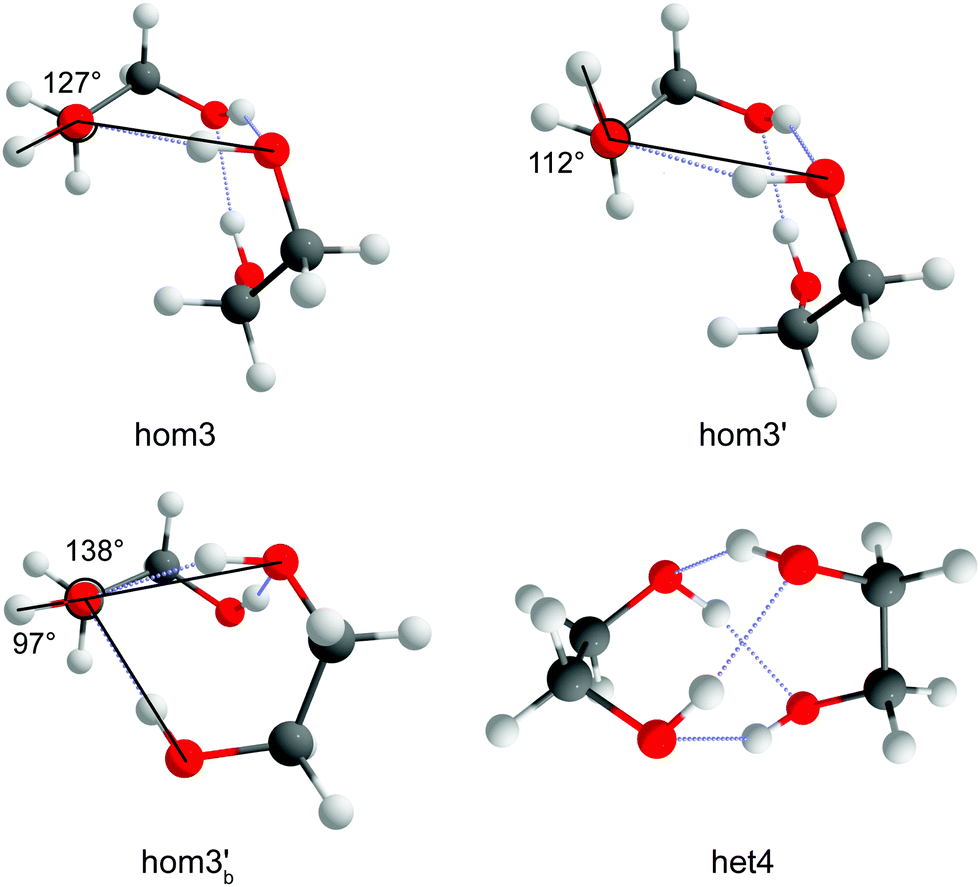 | ||
| Fig. 3 Illustration of the reference O–OH angle and how it determines the different specifiers. An example of bifurcation (hom3b′) is also shown as well as a structure with 4 intermolecular hydrogen bonds (het4) which requires no additional specification since no terminal/dangling OH groups exist. | ||
3.1 Structure
Optimization with and without dispersion, using WFT and DFT, has an impact on the optimized structures of the different conformers. One of the significant features that is noteworthy to analyze is the intermolecular O–H distance. This is important because simulated frequency calculations are highly dependent on the structure, so care must be taken in benchmarking signature peaks (e.g. hydrogen bond stretching). All intermolecular O–H distances can be found in the ESI† (see Chapter 3).Fig. 5 shows the density distribution of the O–H bond distances when the dispersion is accounted for and when it is removed. The distributions have been scaled uniformly to make the comparisons easier. For EDO, the shift in average O–H bond distances is around 0.20 Å while SCS-LMP2 indicated a 0.17 Å shift. B3LYP has a very slight shift of 0.05 Å. The same pattern is observed in CHexDO. The mean values of O–H bonding shifted by 0.18 and 0.14 Å for LMP2 and SCS-LMP2, respectively, while B3LYP only showed a 0.06 Å shift. There is an increase in the shifts for the pinacol system. B3LYP is showing a shift of 0.11 Å. Once dispersion is removed, the shift in the average O–H bond distances doubles, i.e. 0.27 Å for LMP2 and 0.22 Å for SCS-LMP2.
In general, WFT methods indicate a more pronounced shift in peaks once dispersion is removed. There is also an evident broadening alongside with the change in shift. DFT, on the other hand, shows a slight shift once the D3 correction was not included in the optimization. The change in shift as well as the broadening in peaks indicate that the removal of dispersion contribution upon optimization impacts the intermolecular O–H bonding. Structurally, we observe longer hydrogen bonds once we neglect dispersion which results in weaker hydrogen bonds. The larger impact in (SCS)-LMP2 is easy to explain. With uncorrected DFT, short-range dispersion is still included so that the effect is smaller. This is a common issue when interpreting the impact of dispersion solely from the D3 correction.
Another interesting structural feature to analyze is the distance between the center of masses of the two monomers in a dimer, denoted as R(CM–CM). The change in R(CM–CM) indicates whether the monomers move towards or far from each other. Here we discuss the average increase in the R(CM–CM) once dispersion is removed.
Similar trends for R(CM–CM) (Fig. 4) can be observed when contrasted with the intermolecular O–H bond mapping (Fig. 5). For EDO, removal of dispersion caused the monomers to move away from each other by 0.22 Å (SCS-LMP2) to 0.27 Å (LMP2). B3LYP showed a very small increase in the separation of monomers, almost half from the WFT methods, with 0.12 Å. The trend is similar between EDO and pinacol. For the latter, B3LYP only resulted for 0.16 Å but the R(CM–CM) doubled, 0.32 Å, when LMP2 was used. SCS-LMP2 had a 0.10 Å increase from that of B3LYP. On the other hand, CHexDO has a similar trend to the other two systems but the differences are not as high as the others. LMP2 results indicated a 0.30 Å average separation of R(CM–CM) while B3LYP results are just 0.10 Å lower. SCS-LMP2 results are closer to B3LYP results, with only 0.23 Å increase in R(CM–CM) when dispersion was removed. The explanation would be similar as in the H-bond distances.
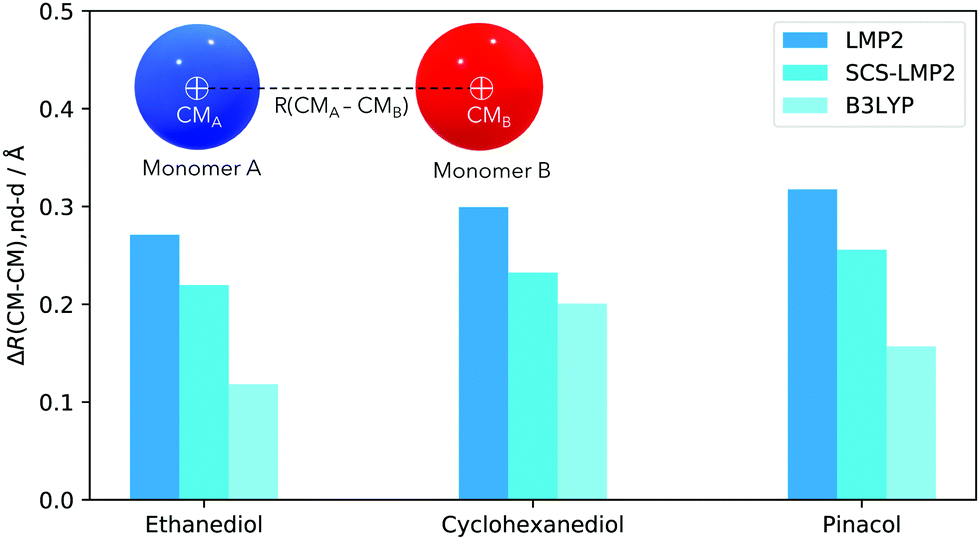 | ||
| Fig. 4 Difference of the center of mass, ΔR(CM–CM), of each monomer in a dimer. Values are always positive, indicative of the increase of the distances of the monomers, without dispersion/dispersion correction. | ||
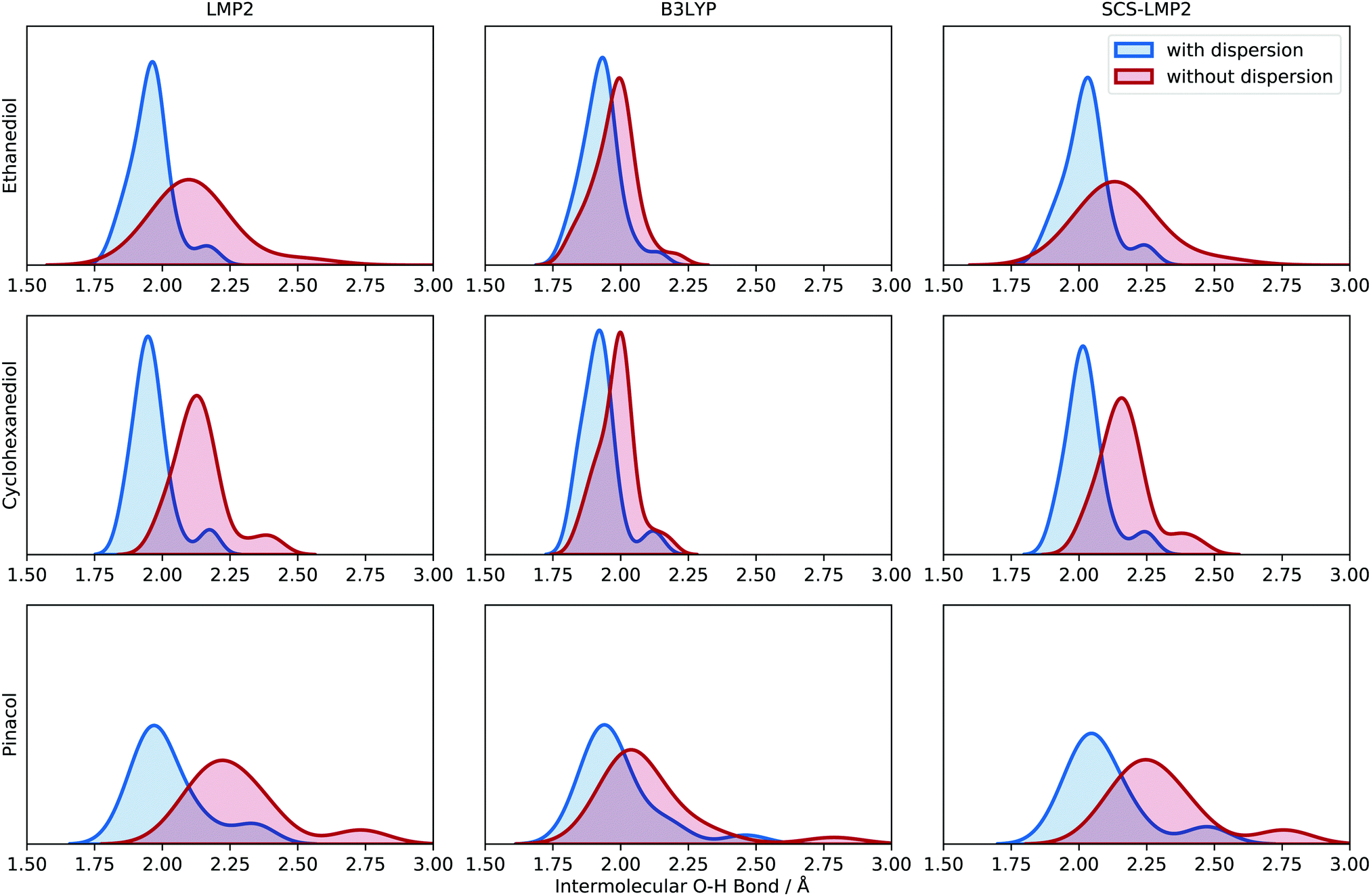 | ||
| Fig. 5 Kernel density estimate (KDE) plot of the intermolecular O–H bond distance for the three molecular systems (top to bottom) evaluated at different levels of theory (left to right). Blue indicates results with dispersion while red shows the results without dispersion. Each O–H bond distance is represented in the density plot to illustrate the frequency and distribution. See Chapter 3 in the ESI† for more information about the generation of the density plots. | ||
Overall, Fig. 4 shows that once dispersion contributions are neglected during optimization, the effect on the structures is significant – the monomers generally move away from each other. WFT indicates a more pronounced separation of the monomers in the dimer compared to DFT since the latter does not fully capture the effect.
3.2 Energy
Although structure is a very important aspect to examine as dispersion contributions are removed, chirality recognition should be discussed in terms of relative energies, which will determine the populations of the different conformers. One noticeable effect of dispersion is the energy range of the relative stability in every molecular system being examined, which can be seen in Fig. 6 by the spread of the conformers. In EDO for example, an energy range of 8.4–11 kJ mol−1 is observed in calculations with dispersion. However, removal of dispersion caused a decrease in the relative energies span: 3.4–6.1 kJ mol−1. Calculations with dispersion resulted in a 6.1–9.1 kJ mol−1 relative stability range for CHexDO, while removal of dispersion lowered the energy range, i.e. 4.1–6.4 kJ mol−1. For pinacol, the energy range of optimization with dispersion is 5.5–9.6 kJ mol−1 and the absence of which resulted in at most 5.5 kJ mol−1.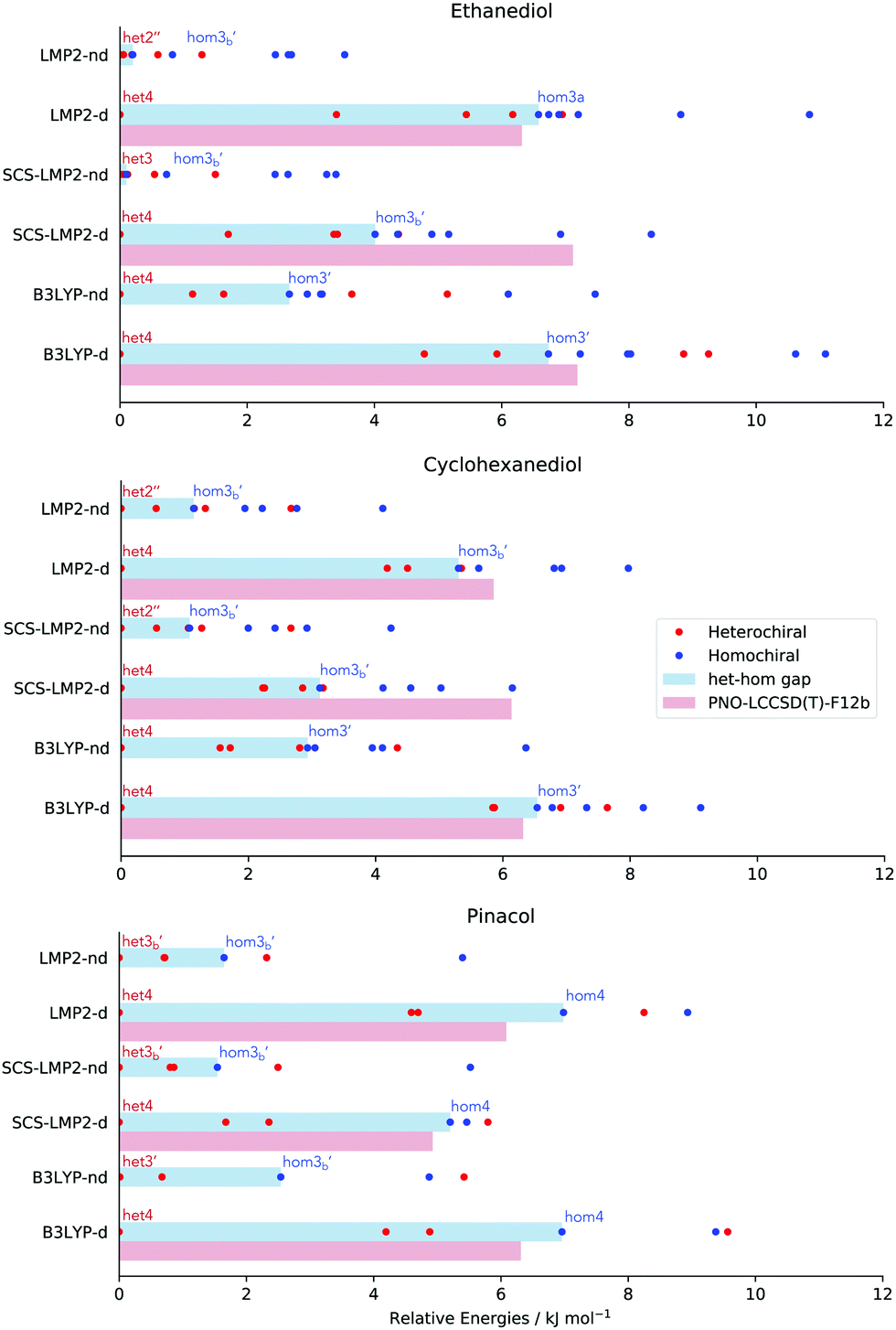 | ||
| Fig. 6 Relative stability (red dots: heterochiral, blue dots: homochiral) and het–hom gap (blue bar) of different molecular systems, with (−d) and without dispersion (−nd). Furthermore, the red bar represents het–hom gap calculated at PNO-LCCSD(T)-F12 using the ZPVE of the respective methods. | ||
Along with the decrease in the relative stability range of every molecular system is also the decrease in the het–hom gap once dispersion is removed. Het–hom gap refers to the difference of the energy between the most stable heterochiral and homochiral structure. This is the single most important value for chirality recognition. A significant decrease in the het–hom gap once dispersion is ‘turned off’ is an indication that dispersion is the driving force for the chirality recognition. The blue bar in Fig. 6 indicates the het–hom gap calculated from both DFT and WFT.
In EDO, it is very evident that chirality recognition is driven by dispersion. A dramatic decrease in the het–hom gap is observed using LMP2 with no dispersion leading to iso-energetic hetero and homo structures (6.6 down to 0.2 kJ mol−1), while SCS-LMP2's het–hom gap went down to 0.1 kJ mol−1 from 4.0 kJ mol−1, a 3.9 kJ mol−1 difference. A decrease of more than half of the value (6.7 down to 2.7 kJ mol−1) when D3 correction was removed in the case of B3LYP is also an evidence for the relevance of dispersion in the het–hom gap for EDO.
There is also a significant decrease in the het–hom gap of CHexDO. The LMP2 gap was lowered by 4.2 kJ mol−1. The SCS-LMP2 value also went down by 1/3 of its original het–hom gap (3.1 to 1.1 kJ mol−1) while B3LYP's decrease was more than half, i.e. 3.6 kJ mol−1. Similar to EDO, chirality recognition in CHexDO appears to be driven by dispersion.
Finally, we have the case of pinacol. The difference in the het–hom gap in LMP2 is 5.3 kJ mol−1, while SCS-LMP2 has 3.7 kJ mol−1. Lastly, the B3LYP het–hom gap was lowered by almost 1/3 when D3 correction was removed.
It is interesting to note in Fig. 6 that the structures responsible for the het–hom gaps change depending on (1) method of choice and (2) the presence of dispersion. The only trend observed in here is that for structure optimizations with dispersion, it is always the het4 conformation that is most stable. For the homochiral species, it varies depending on the method and the system. For EDO and CHexDO, the most stable homochiral species is a variant of hom3, while for pinacol, hom4 is the most stable if dispersion is accounted for.
To benchmark how the B3LYP, LMP2 and SCS-LMP2 methods fare with the prediction of the het–hom gap energies, PNO-LCCSD(T)-F12 energy calculations were done on the structures responsible for the het–hom gap of each method. For example, we took het4 and hom4 structures optimized at B3LYP-d and recalculated their electronic energies. Note that all ZPVEs used were those of the structures at which they were optimized. For pinacol, the trend is clear – LMP2, SCS-LMP2 and B3LYP slightly overestimated the het–hom gap compared to the ones predicted by PNO-LCCSD(T)-F12. For EDO and CHexDO, it is a mix of slight overestimation and underestimation relative to the coupled cluster energy. An outstanding observation for both systems is with SCS-LMP2. According to the PNO-LCCSD(T)-F12 calculations, SCS-LMP2 significantly underestimated the het–hom gap for these two systems (3 kJ mol−1).
Is the het–hom gap in Fig. 6 a structural effect or solely an electron correlation effect? To clarify this matter, we picked the LMP2 optimized structures responsible for the het–hom gap (as indicated in 6) and did a series of single point energy calculations. The energies were analyzed with regards to the het–hom gap as is shown in Fig. 7. Note that zero point vibrational energy (ZPVE) of the optimization procedure (LMP2) was added into the total electronic energy of the respective conformation. The bars in the shade of blue show the final electronic energy with dispersion while bars in red stand for the final electronic energy without dispersion. The shift of energy from dark blue to light blue and dark red to light red reflects a structural effect. The shift from blue to red indicates the correlation effect.
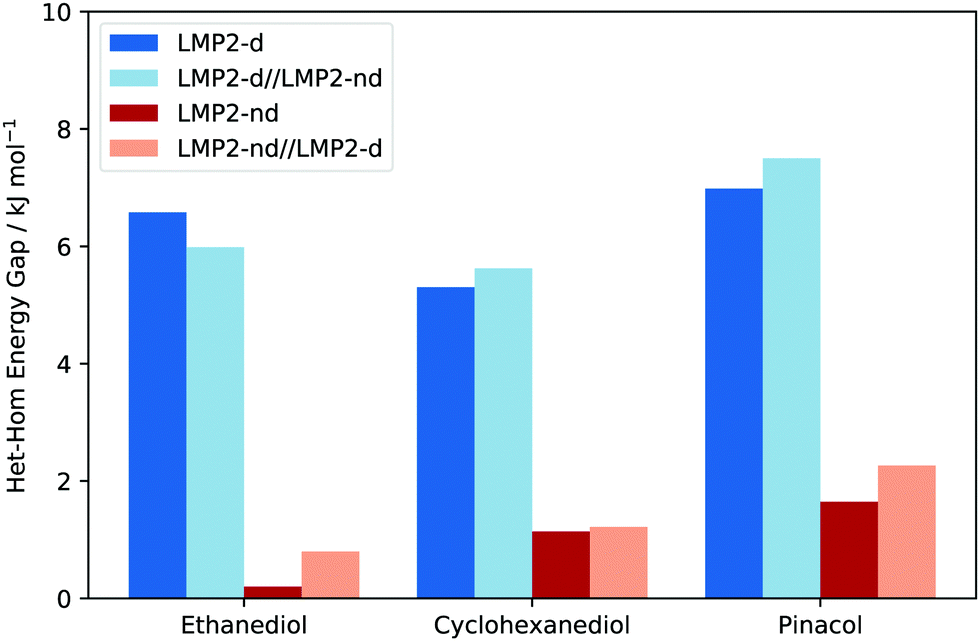 | ||
| Fig. 7 Het–hom gaps of different systems using different LMP2 approaches to evaluate if the chirality recognition due to dispersion is a structural or correlation effect. Dark blue to light blue as well as dark red to light red represents structural effects while any change from blue to red indicates an electron correlation effect. | ||
Results suggest that there is an insignificant structural effect on the het–hom gap, as can be seen from the small difference between the dark and light colours in Fig. 7. Recall that dark blue represents LMP2 with dispersion, light blue is single point energy calculation with dispersion on top of structures optimized without dispersion. Similarly, dark red is LMP2 without dispersion and light red is LMP2 single point calculation without dispersion on top of structures optimized with dispersion. These results support the common approach to quantify dispersion effects, taking fixed structures and observing solely differences in energy.
To identify the behaviour of commonly used functionals we compared the results of BP86, PBE and PBE0 to B3LYP and the WFT ones. Looking at Fig. 8 (left row) it immediately becomes apparent that the neglect of dispersion/dispersion correction varies greatly with the used functional. B3LYP from all tested functionals is especially sensitive to the use of D3(BJ). PBE0 on the other hand depends significantly less on this correction. A similar trend can be observed for the GGAs where BP86 dependence on the correction is larger than that of PBE. This indicates that judging dispersion by the use or lack of D3(BJ) on fully optimised structures may lead to different conclusions regarding the impact of London forces. However, interpretations based on B3LYP would generally be in line with the WFT results.
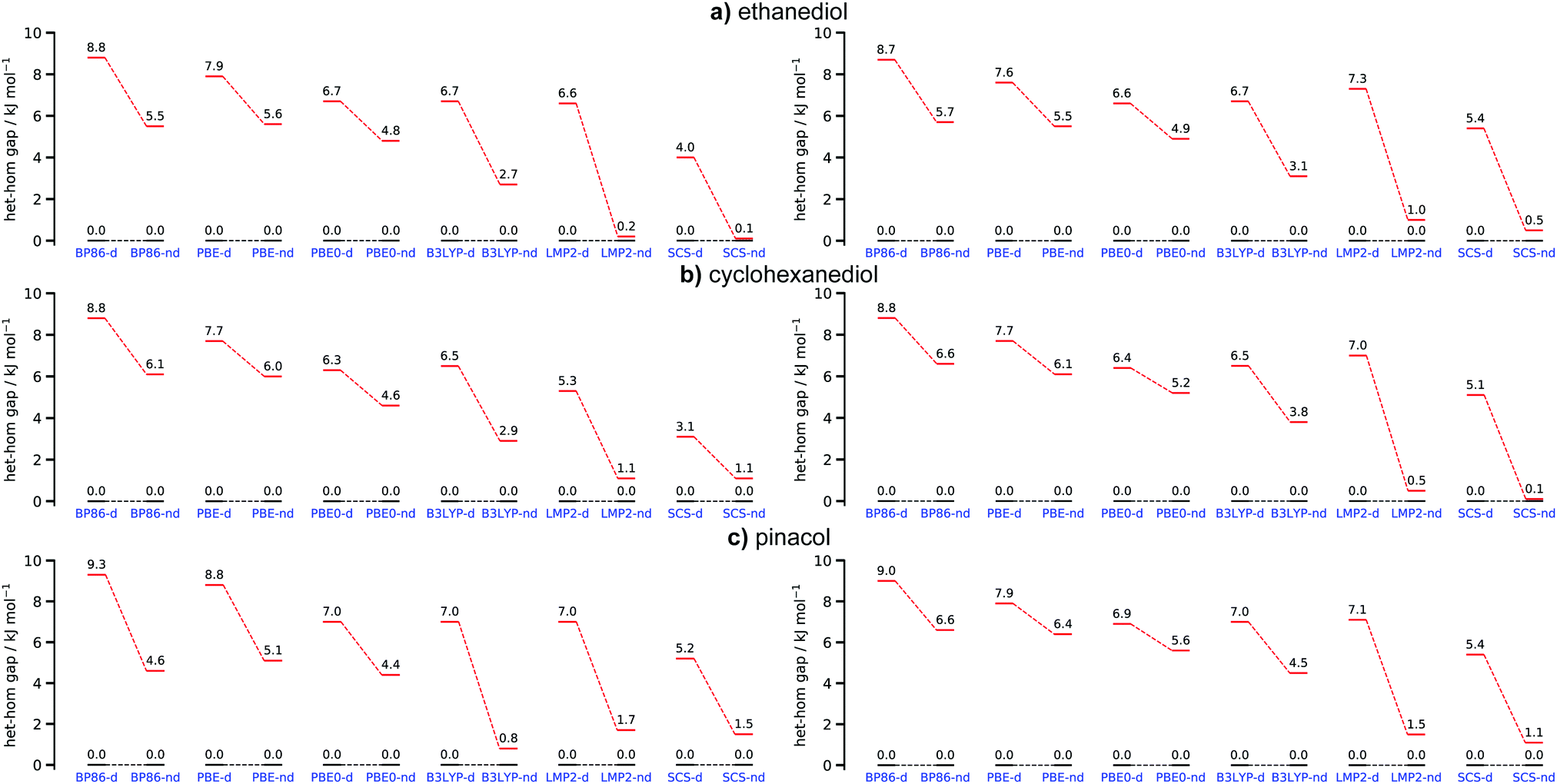 | ||
| Fig. 8 Overview of the hetero–homo energy gap for (a) ethanediol, (b) cyclohexanediol and (c) pinacol for all tested methods with (d) and without (nd) dispersion/dispersion correction. Hetero energy levels are coloured in black and homo levels in red. The graphs on the left hand side show results based on their respective optimised structures whereas the right hand side shows results based on the most stable hetero and homo structures of B3LYP-D3(BJ,abc) as a reference. | ||
In order to assess how sensitive DFT and WFT approaches are in examining the impact of dispersion in chirality recognition from an energy standpoint, single point energy calculations were performed on the optimized B3LYP-d structures. ZPVE from the B3LYP calculations were added into the total electronic energies. The structures responsible for the het–hom gap at the B3LYP level were chosen as a baseline. The most stable hetero conformer remains het4 for all systems whereas hom3′ is the most stable homo species for EDO and CHexDO. The hom4 is the most stable one for pinacol. Examining Fig. 8, it becomes apparent that the het–hom gap decreases for all functionals. The deviations are especially large for pinacol where in case of B3LYP the dispersion induced gap changes from 6.2 kJ mol−1 for the fully optimised case to just 2.5 kJ mol−1 for the B3LYP-d baseline results. This indicates that one should be mindful when trying to judge the influence of London forces by doing single point calculations without dispersion correction on structures optimised with dispersion correction. This example shows that such an approach may lead to a significant underestimation of the importance of dispersion. The LMP2 results remain largely unchanged whereas the het–hom gap for the SCS-LMP2 results generally increases. In terms of the general trends the WFT methods would lead to the same conclusions in either case.
Despite the fact that the het–hom gaps are significantly impacted by dispersion, whether or not chirality recognition absolutely depends on these forces cannot easily be answered. Giving an energy threshold is difficult since in principle any energy difference would lead to chirality recognition at sufficiently low temperatures in terms of the homo and hetero populations. Instead we examine when chirality recognition would no longer manifest itself. This point would be reached when both hetero and homo conformers are populated. For this purpose we calculated the Gibbs energies at different temperatures ranging from 0–400 K which can be seen in Fig. 9 for EDO at the B3LYP-d and B3LYP-nd level of theory. Corrections due to the symmetry number (σ = 2 for het4 S4 and hom4 C2) and the fact that some dimers are themselves not chiral (het4 S4 and het2′′ Ci) are included. For a simple and qualitative measure we compare the results to the thermal energy RT and look for the point where the thermal energy (green line) first crosses with the lowest lying homo chiral species (red lines) as an indication when chirality recognition would no longer manifest. The point at which chirality recognition would be no longer present is reached much earlier without dispersion correction (ca. 160 K) in comparison to the corrected results (ca. 270 K).
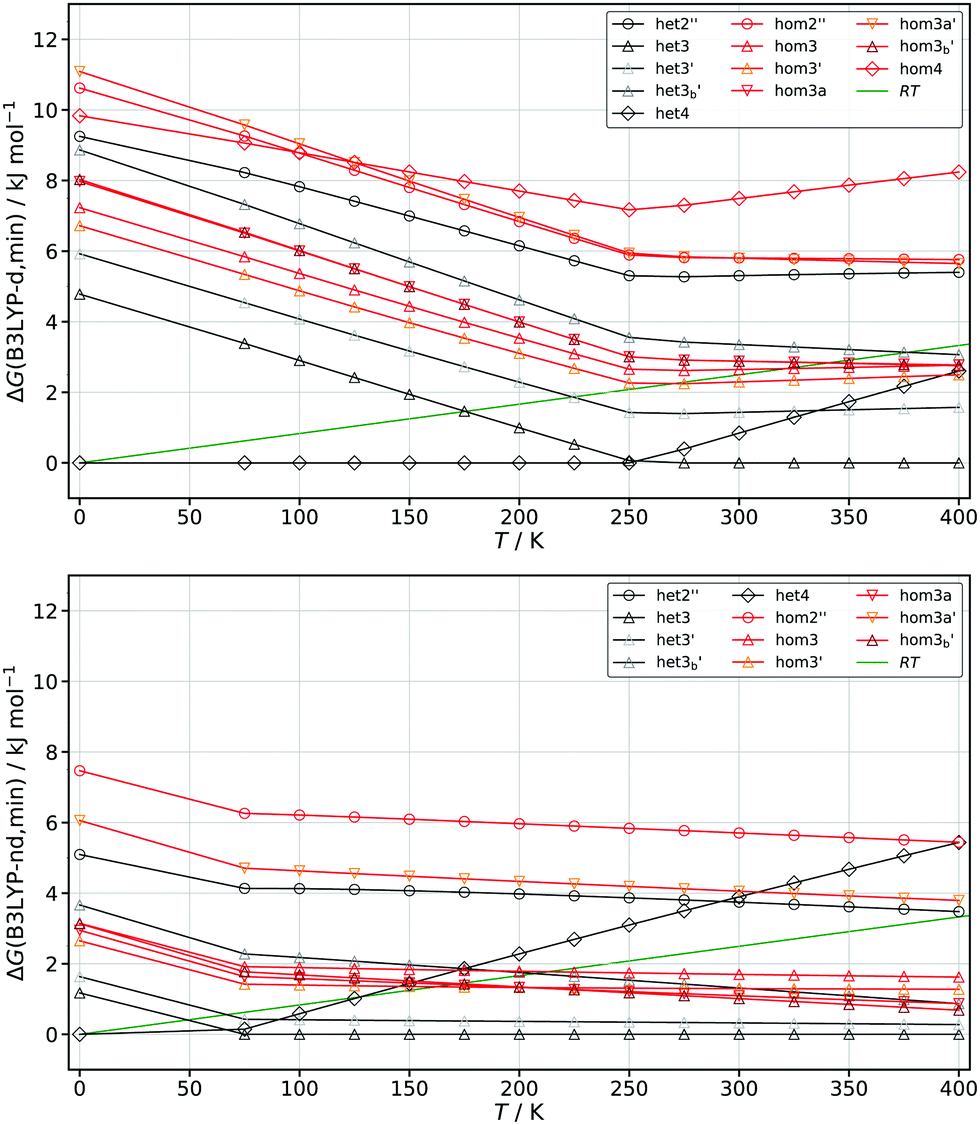 | ||
| Fig. 9 Calculated Gibbs-energies relative to the minimum at different temperatures based on the B3LYP-d (top) and B3LYP-nd (bottom) results for EDO. Black lines represent hetero dimers and red line homo dimers. The thermal energy (RT) is shown in green. | ||
A figure comparing the results for ethanediol, cyclohexanediol and pinacol can be found in the ESI† (Fig. S3). Similar results to EDO were found for cyclohexanediol going from about 230 K for B3LYP-d to about 140 K for B3LYP-nd. The increased sensitivity of pinacol with regards to dispersion correction can also be seen in this analysis. This is clearly indicated by a downshift of ca. 260 K (B3LYP-nd: 60 K; B3LYP-d: 320 K) in comparison to about 100 K in the case of EDO and CDO. This simple but generally applicable analysis confirms that dispersion is indeed a driving force for chirality recognition or rather its manifestation. The results also suggest that without dispersion, chirality recognition would be a low temperature phenomenon in these instances. Since B3LYP showed very similar behaviour as the WFT methods we also expect similar results for this kind of analysis in those cases.
3.3 DID visualization: which moiety has the largest dispersion contribution?
Analysis from the previous sections show the impact of dispersion in the chirality recognition, both in energies and structures of the conformations investigated. Using DID visualization,60 we can pinpoint which particular part of the dimer has a larger dispersion contribution. Density contribution to the interaction was calculated using SCS-LMP2. In this section, we looked at the conformations which are responsible for the het–hom energy gap.The most stable heterochiral and homochiral conformations for the three systems examined are shown in Fig. 10, except for pinacol where we chose hom3b′ for the homochiral species so as to be consistent with the other two molecular systems. A consistent range of density (i.e. 3.0 × 10−10 to 1.5ea0−3) was chosen for the visualization of all the species to ensure comparability. The DID plot shows that the OH moiety of each monomer contributes the most in terms of dispersion interaction with one another, as indicated by the yellow to red density range. More specifically, more pronounced dispersion contributions are coming from the oxygen atom, and an observable dispersion density on the hydrogen atom. This is due to the increase in polarizability of oxygen when bonded to the hydrogen atom.
Het4 systems in Fig. 10 show larger dispersion interaction density compared to its homochiral counterpart, which meant that dispersion as a stabilizing force is stronger in these set of conformations. This is due to the nature of the molecular geometry of the system itself. Het4 contains 4 hydrogen bonds while hom3b′ only has 3 hydrogen bonds. When comparing the DID, it becomes apparent that in the case of het4, OH groups are strongly interacting. For hom3b′, density in the OHs are not as strong as the het4 counterpart. Furthermore, the hydrogen in the free OH group (see hom3b′) does not significantly contribute to the DID. It is notable that the density is symmetric among het4 structures due to its S4 symmetry. Fig. S8 in the ESI† further illustrates the symmetry for EDO as an example. The intermolecular hydrogen bonds make the intermolecular distance of oxygen atoms from each monomer shorter, leading to stronger dispersion interactions.
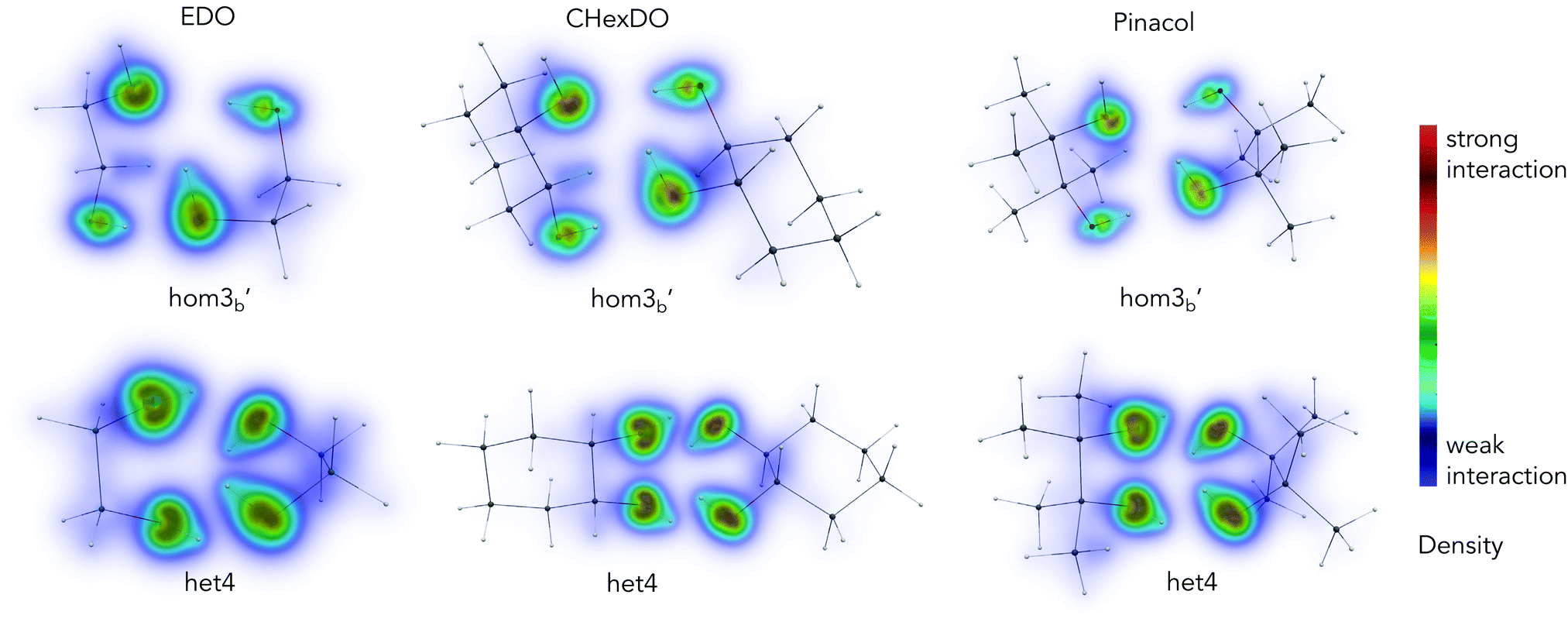 | ||
| Fig. 10 Dispersion interaction density (DID) visualization of the structures responsible for the het–hom gap, results from SCS-LMP2/aug-cc-pVTZ, H = cc-pVTZ optimization. Density range: 3.0 × 10−10 to 1.5ea0−3. | ||
To gain some further insight into the individual contributions of the OH groups and the backbone we further fragment the dispersion energy (EDisp). The backbone consists of everything besides the OH moieties. Specifically we look at the interaction of the OH groups and the backbone of monomer A with the entirety of monomer B, which is illustrated in Fig. 11. The sum of the two contributions is equivalent to the total dispersion energy. The results of this analysis for SCS-LMP2-d are shown in Fig. 12. Dispersion energies of zero are given when a conformer converges to a different one or shows an imaginary frequency. It immediately becomes apparent that het4 always has the highest (more negative) dispersion energy, which is also always the most stable conformer overall. Furthermore, the OH contributions dominate the dispersion energy, which can also be seen from the DID (see Fig. 10). It should be noted though that when going from ethanediol to cyclohexanediol to pinacol the contributions of the backbone become more and more important. For instance, in case of het2′′, a conformer with a generally high influence of the backbone, the contribution of the backbone to the dispersion energy increases from 30% to 36% for cyclohexanediol up to 41% for pinacol. Additionally, despite the fact that cyclohexanediol (C6H12O2) and pinacol (C6H14O2) hardly vary in terms of their number of electrons, pinacol profits more from dispersion than cyclohexanediol which can be attributed to the more compact structure of pinacol.
 | ||
| Fig. 11 Sketch of the energy fragmentation of the three studied molecular systems. Dark blue represents the OH group interaction with the entirety of the other monomer fragment in black and light blue the interaction of the backbone respectively. | ||
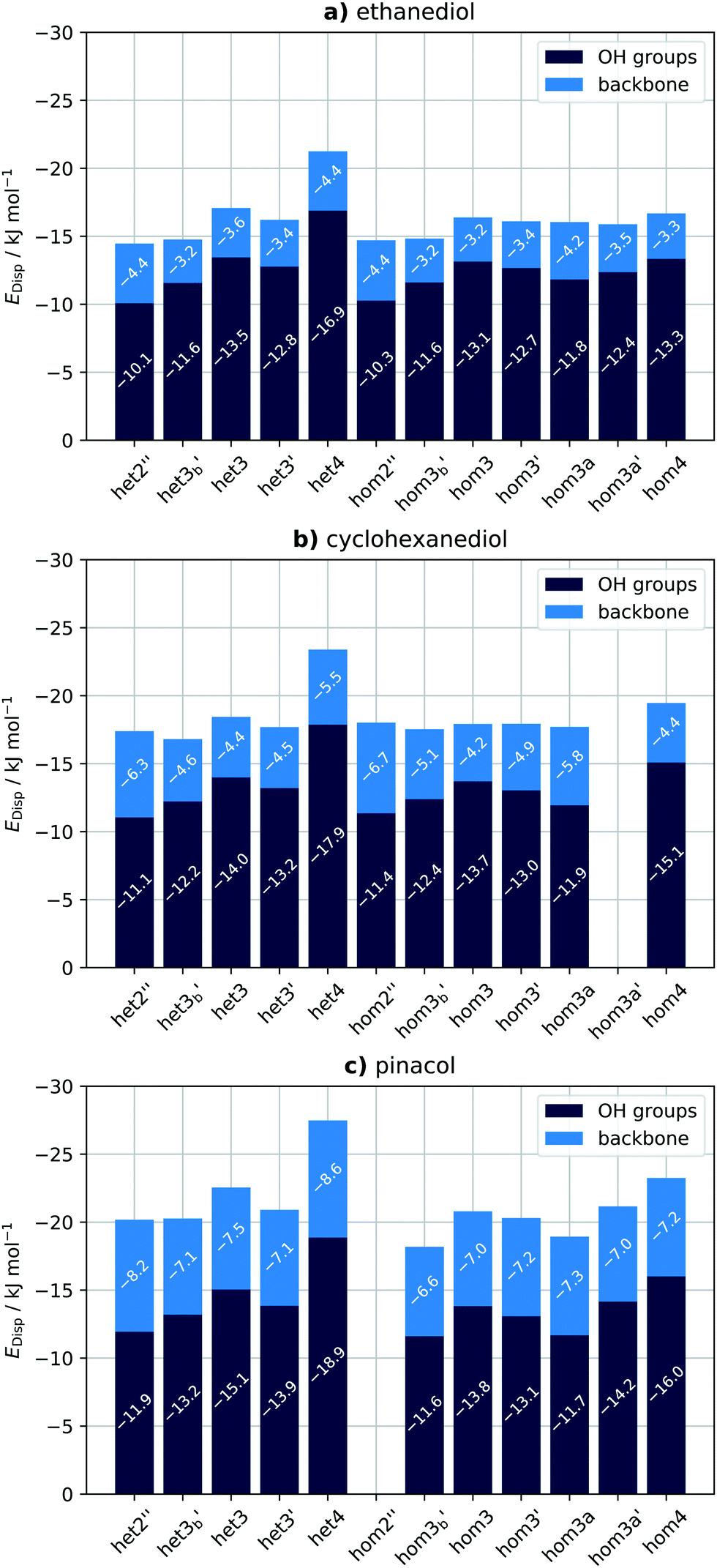 | ||
| Fig. 12 Overview of the intermolecular dispersion contributions of the OH groups (dark blue) and the backbone (light blue) for (a) ethanediol, (b) cyclohexanediol and (c) pinacol for the SCS-LMP2-d results. Zero values indicate that a conformer either converges to a different one or exhibits an imaginary frequency. | ||
Interestingly the same analysis for LMP2-d results shows qualitatively similar results (see ESI,† Fig. S5), with the major difference that for EDO and CHexDO hom4 is much more competitive to het4 with regards to its dispersion energy. Pinacol on the other hand behaves much more similar to the SCS-LMP2 results although het3 no longer remains a stable conformer. This difference can also be pictured by looking at the het–hom gap purely based on the dispersion energies and the actual LMP2-d and SCS-LMP2-d results (see ESI,† Fig. S4). For LMP2 there is a major difference in case of EDO and CHexDO which would only indicate a het–hom gap of about 2 kJ mol−1 based on EDisp. In contrast to the previously mentioned ones pinacol at the LMP2 level and all systems at the SCS-LMP2 show much more similar results between each other and between the differently computed het–hom gaps. Overall for these systems simply looking at the dispersion energies would already be sufficient to account for the het–hom gap. It also becomes apparent that LMP2 yields about 13% higher dispersion energies on average, for each system respectively, than SCS-LMP2. This indicates that LMP2 overestimates the London forces, which is well known for π–π interactions and mostly remedied by the SCS approach.90
4 Conclusions
In this paper we carefully analyzed the impact of dispersion in both structure and energy of EDO, CHexDO and pinacol. We also examined if dispersion is a relevant driving force for the chirality recognition of these diols. In addition, we looked into which moiety of the dimer contributes the most dispersion. DFT and local correlation methods were used.A significant impact on the structure of the molecular aggregates was observed once dispersion was removed. An evident increase in the intermolecular O–H bonding as well as the shift of the monomers away from each other as shown by the analysis of their ΔR(CM–CM) were observed.
Dispersion is also found to be a relevant driving force in the differential chiral binding of these molecular systems. The removal of dispersion showed a significant decrease in the het–hom gap of EDO, CHexDO and pinacol. Furthermore, dispersion is crucial for the manifestation of chirality recognition. Our results also indicate that without dispersion (correction) chirality recognition would be a low temperature phenomenon for the investigated diols. In addition, chiral recognition in these molecular systems are more affected by electron correlation and only slightly by structural effects. Both WFT and some functionals clearly demonstrate this, but a more obvious het–hom gap lowering is observed from WFT methods. This also further emphasizes the importance of adding D3 corrections (in cases where DFT is preferred) in systems with non-covalent interactions.
The DFT results also show that one should be cautious when trying to gauge the impact of London forces by doing single point calculations without dispersion correction on structures that were previously optimised including such effects. This is especially true for pinacol where differences of up to 3.7 kJ mol−1 can be found which would lead to widely different interpretations. Furthermore, although all tested functionals show a decrease of the het–hom gap when no D3(BJ) was used B3LYP and BP86 are more sensitive than PBE0 and PBE which should be kept in mind when judging the influence of dispersion. B3LYP also clearly matches the WFT results the best.
Lastly, the dispersion forces in these diols are primarily contributed by the O–H moiety. The polarizability of oxygen and its close proximity to other polarizable atoms with instantaneous multipoles results in the significant dispersion interaction in these systems. The backbone also becomes increasingly important when moving from ethanediol to cyclohexanediol to pinacol while the larger dispersion energies for pinacol highlight the importance of compact structures rather than extended ones.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455 (XA and BH) and 405832858 for computational resources. AW gratefully acknowledges the SPP1807 “Control of London dispersion interactions in molecular chemistry” (MA5063/3-2) for funding.Notes and references
- A. Pietropaolo, Chirality in Biochemistry: A Computational Approach for Investigating Biomolecule Conformations, John Wiley & Sons, Ltd, 2010, ch. 12, pp. 291–312 Search PubMed.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- M. Hu, Y.-X. Yuan, W. Wang, D.-M. Li, H.-C. Zhang, B.-X. Wu, M. Liu and Y.-S. Zheng, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- R. Bentley, Chem. Rev., 2006, 106, 4099–4112 CrossRef CAS PubMed.
- P. Cintas, Biochirality, Springer Berlin Heidelberg, 2013 Search PubMed.
- F. Carey, Advanced Organic Chemistry, Springer, New York, NY, 2007 Search PubMed.
- E. Francotte and W. Lindner, Chirality in Drug Research, Wiley-VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed.
- B. Testa, Chirality, 1989, 1, 7–9 CrossRef CAS PubMed.
- S. W. Smith, Toxicol. Sci., 2009, 110, 4–30 CrossRef CAS PubMed.
- W. Lenz, Teratology, 1988, 38, 203–215 CrossRef CAS PubMed.
- W. Heger, H.-J. Schmahl, S. Klug, A. Felies, H. Nau, H.-J. Merker and D. Neubert, Teratog., Carcinog. Mutagen., 1994, 14, 115–122 CrossRef CAS PubMed.
- T. Eriksson, S. Björkman, B. Roth and P. Höglund, J. Pharm. Pharmacol., 2000, 52, 807–817 CrossRef CAS PubMed.
- L. J. Prins, J. Huskens, F. de Jong, P. Timmerman and D. N. Reinhoudt, Nature, 1999, 398, 498–502 CrossRef CAS.
- M. Kempe and K. Mosbach, J. Chromatogr. A, 1995, 694, 3–13 CrossRef CAS.
- R. A. Mata and M. A. Suhm, Angew. Chem., Int. Ed., 2017, 56, 11011–11018 CrossRef CAS PubMed.
- J. Echeverra, G. Aullón, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. Dahl, R. M. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef CAS PubMed.
- I. Fenández López, M. Solà i Puig and F. M. Bickelhaupt, Chem. – Eur. J., 2013, 19, 7416–7422 CrossRef PubMed.
- E. Lyngvi, I. A. Sanhueza and F. Schoenebeck, Organometallics, 2015, 34, 805–812 CrossRef CAS.
- M. S. G. Ahlquist and P.-O. Norrby, Angew. Chem., Int. Ed., 2011, 50, 11794–11797 CrossRef CAS PubMed.
- T. H. Meyer, W. Liu, M. Feldt, A. Wuttke, R. A. Mata and L. Ackermann, Chem. – Eur. J., 2017, 23, 5443–5447 CrossRef CAS PubMed.
- E. Detmar, V. Müller, D. Zell, L. Ackermann and M. Breugst, Beilstein J. Org. Chem., 2018, 14, 1537–1545 CrossRef CAS PubMed.
- C. Eschmann, L. Song and P. R. Schreiner, Angew. Chem., Int. Ed., 2021, 60, 4823–4832 CrossRef CAS PubMed.
- D. Yepes, F. Neese, B. List and G. Bistoni, J. Am. Chem. Soc., 2020, 142, 3613–3625 CrossRef CAS PubMed.
- F. Winkler, R. Medina, J. Winkler and H. Krause, J. Chromatogr. A, 1994, 666, 549–556 CrossRef CAS.
- N. Seurre, K. Le Barbu-Debus, F. Lahmani, A. Zehnacker, N. Borho and M. A. Suhm, Phys. Chem. Chem. Phys., 2006, 8, 1007–1016 RSC.
- A. Mahjoub, A. Chakraborty, V. Lepere, K. Le Barbu-Debus, N. Guchhait and A. Zehnacker, Phys. Chem. Chem. Phys., 2009, 11, 5160–5169 RSC.
- D. Scuderi, K. Le Barbu-Debus and A. Zehnacker, Phys. Chem. Chem. Phys., 2011, 13, 17916–17929 RSC.
- J. Altnöder, A. Bouchet, J. J. Lee, K. E. Otto, M. A. Suhm and A. Zehnacker-Rentien, Phys. Chem. Chem. Phys., 2013, 15, 10167–10180 RSC.
- K. Schwing and M. Gerhards, Int. Rev. Phys. Chem., 2016, 35, 569–677 Search PubMed.
- F. Kollipost, K. E. Otto and M. A. Suhm, Angew. Chem., Int. Ed., 2016, 55, 4591–4595 CrossRef CAS PubMed.
- J. Klyne, A. Bouchet, S.-I. Ishiuchi, M. Fujii, M. Schneider, C. Baldauf and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 28452–28464 RSC.
- B. Hartwig, M. Lange, A. Poblotzki, R. Medel, A. Zehnacker and M. A. Suhm, Phys. Chem. Chem. Phys., 2020, 22, 1122–1136 RSC.
- F. Xie, M. Fusè, A. S. Hazrah, W. Jäger, V. Barone and Y. Xu, Angew. Chem., Int. Ed., 2020, 59, 22427–22430 CrossRef CAS PubMed.
- K. Le Barbu-Debus, D. Scuderi, V. Lepère and A. Zehnacker, J. Mol. Struct., 2020, 1205, 127583 CrossRef CAS.
- A. Zehnacker and M. Suhm, Angew. Chem., Int. Ed., 2008, 47, 6970–6992 CrossRef CAS PubMed.
- N. O. B. Lüttschwager, T. N. Wassermann, R. A. Mata and M. A. Suhm, Angew. Chem., Int. Ed., 2013, 52, 463–466 CrossRef PubMed.
- K. B. Lipkowitz, B. Coner, M. A. Peterson and A. Morreale, J. Phys. Org. Chem., 1997, 10, 311–322 CrossRef CAS.
- K. B. Lipkowitz, R. Coner, M. A. Peterson, A. Morreale and J. Shackelford, J. Org. Chem., 1998, 63, 732–745 CrossRef CAS PubMed.
- V. Schurig, J. Chromatogr. A, 2001, 906, 275–299 CrossRef CAS PubMed.
- C. Cagliero, B. Sgorbini, C. Cordero, E. Liberto, P. Rubiolo and C. Bicchi, Isr. J. Chem., 2016, 56, 925–939 CrossRef CAS.
- S. Kristyán and P. Pulay, Chem. Phys. Lett., 1994, 229, 175–180 CrossRef.
- J. Pérez-Jordá and A. D. Becke, Chem. Phys. Lett., 1995, 233, 134–137 CrossRef.
- P. Hobza, J. Šponer and T. Reschel, J. Comput. Chem., 1995, 16, 1315–1325 CrossRef CAS.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- M. Schütz, G. Rauhut and H.-J. Werner, J. Phys. Chem. A, 1998, 102, 5997–6003 CrossRef.
- Q. Ma and H.-J. Werner, J. Chem. Theory Comput., 2019, 15, 1044–1052 CrossRef PubMed.
- W. B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2016, 12, 4778–4792 CrossRef CAS PubMed.
- A. Altun, F. Neese and G. Bistoni, J. Chem. Theory Comput., 2018, 15, 215–228 CrossRef PubMed.
- G. Bistoni, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1442 CAS.
- R. Naaman and D. H. Waldeck, J. Chem. Phys. Lett., 2012, 3, 2178–2187 CrossRef CAS PubMed.
- A. Kumar, E. Capua, M. K. Kesharwani, J. M. Martin, E. Sitbon, D. H. Waldeck and R. Naaman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2474–2478 CrossRef CAS PubMed.
- D. Craig and T. Thirunamachandran, Molecular Quantum Electrodynamics, Dover Publications, 2012 Search PubMed.
- A. Wuttke and R. A. Mata, J. Comput. Chem., 2017, 38, 15–23 CrossRef CAS PubMed.
- A. Wuttke, M. Feldt and R. A. Mata, J. Phys. Chem. A, 2018, 122, 6918–6925 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- J. G. Brandenburg, C. Bannwarth, A. Hansen and S. Grimme, J. Chem. Phys., 2018, 148, 064104 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2014, 10, 1074–1085 CrossRef CAS PubMed.
- U. Ryde, R. A. Mata and S. Grimme, Dalton Trans., 2011, 40, 11176–11183 RSC.
- M. Bursch, E. Caldeweyher, A. Hansen, H. Neugebauer, S. Ehlert and S. Grimme, Acc. Chem. Res., 2019, 52, 258–266 CrossRef CAS PubMed.
- S. Grimme, Theory, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- G. Herzberg, Infrared and Raman Spectra of Polyatomic Molecules, Van Nostrand Reinhold, 1945 Search PubMed.
- P. Pulay and S. Saebø, Theor. Chim. Acta, 1986, 69, 357–368 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, et al., MOLPRO, version 2018.1, a package of ab initio programs, 2018, see http://www.molpro.net Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, et al., MOLPRO, version 2019.2, a package of ab initio programs, 2019, see http://www.molpro.net Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01225h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2021 |