
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of methanol on rutile TiO2(110) and tungsten oxide clusters: 2. The role of defects and electron transfer in bifunctional oxidic photocatalysts†
Lars
Mohrhusen
 *,
Jessica
Kräuter
and
Katharina
Al-Shamery
*,
Jessica
Kräuter
and
Katharina
Al-Shamery
Institute of Chemistry, Carl von Ossietzky University of Oldenburg, Carl-von-Ossietzky Strasse 9-11, D-26129 Oldenburg, Germany. E-mail: lars.mohrhusen@uol.de
First published on 17th May 2021
Abstract
The photochemical conversion of organic compounds on tailored transition metal oxide surfaces by UV irradiation has found wide applications ranging from the production of chemicals to the degradation of organic pollutants e.g. in waste water treatment. Here, we present a systematic surface science-based study of the UV photoconversion of methanol on a rutile TiO2(110) surface. Under the used conditions, the dominant photoreaction is the photo-oxidation forming formaldehyde, that is drastically boosted by the presence of adsorbed oxygen as well as (sub-)surface defects such as oxygen vacancies and Ti3+ interstitials. Moreover, a photostimulated and Ti3+ mediated C–C coupling was observed leading to the production of ethene. We have further deposited tungsten oxide clusters on the rutile surface and examined the impact on the methanol photochemistry. In this case, the C–C coupling can be suppressed. Surprisingly, especially for high Ti3+ contents the population of the photochemical pathway is quenched in favor of the population of the thermal reaction yielding more methane from the deoxygenation reaction. So, the common concept that long time charge separation is efficient by combining two photocatalysts with similar band gaps, but different work functions in order to enhance photochemical yields is apparently too naive for certain systems. We attribute the loss of photoproducts with tungsten oxide coadsorption to the “pinning” of Ti3+ centers and a related enhancement of electron density near the oxide clusters which makes a concomitant recombination of the photochemical relevant holes with the excess surface electrons more likely.
1. Introduction
Photochemical reactions on transition metal oxide based semiconductors play nowadays an indisputable role in heterogenous photocatalysis. Particularly titanium dioxide attracted the interest of many researchers, either as rutile, which is the most stable configuration, anatase, or mixtures of both. The probably most popular example is the degradation of organic pollutants by UV photo-oxidation at a mixture of rutile and anatase known as P25.1,2 However, not only total oxidation can occur but a controlled photochemistry yielding valuable products can be realized by adjustment of the reaction properties (such as irradiation wavelength and power, mass transport etc.).1,3 Here, surface science studies under well-defined conditions are of particular interest when aiming for a comprehensive understanding. For instance, we have recently investigated the photo-oxidation of isopropanol towards acetone.3 Several other examples including the photo-oxidation of ethanol to acetaldehyde,4 the photo-oxidation of carbon monoxide,5 and the photochemistry of methanol at TiO2 single crystals6–9 and titania nanomaterials10 are available in the literature.11,12 By now, defects such as oxygen vacancies, Ti3+ interstitials and other reduced sites are recognized to be an essential key to increase and further control the reactivity in thermal reactions towards oxygen and oxygen-containing molecules.13–20 Their role in photochemical reactions, however, has not been studied so intensively in comparison.4,5,21–23 Therefore, we first focus on the methanol photochemistry on a rutile TiO2(110) surface as a function of the Ti3+ content.In a subsequent step, we mimic a mixed oxide–oxide bifunctional catalyst by deposition of tungsten oxide clusters on rutile TiO2(110). Such clusters on various supports were already tested in theoretical and experimental studies for thermal alcohol or aldehyde conversion reactions.24–32 Surprisingly, the photochemistry of these clusters has not been investigated yet although both materials (WO3 and TiO2) are promising candidates for photochemical reactions. Moreover, the band gap of bulk WO3 (2.8 eV) is smaller than the one of rutile TiO2.33 However, as the (WO3)n clusters appear more like molecular quantum dots, they exhibit an expanded band gap (or HOMO–LUMO gap, respectively) of approx. 3.4 eV.30,34,35 Hence, (WO3)3 clusters can be directly excited by UV light (λ ≤ 365 nm) to act as a photocatalyst similar to TiO2.36–38 Due to the relatively high work function of WO3, the LUMO of (WO3)n clusters is expected to be below the conduction band of rutile TiO2.39 Therefore, photogenerated charge carriers are expected to be efficiently separated. Photoelectrons should accumulate at the low-lying LUMOs of tungsten oxide clusters, while holes travel to the TiO2 surface. Indeed, improved photocatalytic properties were reported for real WO3/TiO2 catalysts for degradation of several organic molecules. The authors of these studies, which were performed under technical relevant reaction conditions, commonly refer additionally to the higher surface acidity as well as to beneficial effects on the exciton lifetime.37,40–45
Here, we present a systematic temperature programmed reaction spectroscopy (TPRS) based study of the photo-oxidation of methanol on such surfaces. We highlight the drastically increased photoproduction of formaldehyde in presence of high Ti3+ contents on bare TiO2 and present the photostimulated C–C coupling route forming ethene from two molecules of methanol. After deposition of the tungsten oxide cluster, the thermal chemistry is more dominant than for bare TiO2. We shall combine these results with our recent finding, that an electron transfer between TiO2 point defects and tungsten oxide clusters is possible under accumulation of surface near Ti3+ sites and formation of partially anionic clusters.46
2. Experimental
All results were obtained using a home-built UHV chamber (base pressure below 10−10 mbar) as described elsewhere in detail.47 High purity compounds (here, oxygen (Air liquide, 99.999%), argon (Air liquide, 99.999%) and methanol (Fisher Scientific, 99.99% (HPLC grade)) can be adsorbed onto the sample via a directional pinhole doser or chamber backfilling via a leak valve. Before use, methanol was cleaned by at least five freeze–pump–thaw cycles. All TPR spectra were recorded using a temperature ramp of 2 K s−1.For all experiments, rutile TiO2(110) single crystals (10 × 10 × 1 mm3, Surface Net GmbH) were used. Clean (110) (1 × 1) surfaces were produced by repeated cycles of argon ion bombardment (20 minutes at 300 K, 1 keV, 2 μA cm−2 at 5 × 10−5 mbar argon background pressure) and subsequent annealing at 880 K for 15 minutes in UHV. Since this procedure leads to a partially reduced material, it will be called one “reduction cycle” in the following.47 The surface quality was frequently checked by low energy electron diffraction (LEED).
An individual evaporation chamber allowed deposition of tungsten oxide clusters by electron beam evaporation from bulk WO3 and subsequent sample transfer in vacuo (<10−9 mbar). Again, the system is described elsewhere in more detail.46 Here, identical conditions, devices and evaporating materials were used. Therefore, the surface characterization may be directly transferred from that publication.32,46 Before use in TPRS experiments, the clusters were initially heated to 880 K (2 K s−1, keeping 880 K for 1 minute) to remove possible residual contaminants from the cluster evaporation. Based on XPS and TPRS experiments, a significant role of surface hydroxylation by water adsorption from the chamber background can be excluded.32
For the photochemical reactions, adsorbates were irradiated with UV light at the base temperature (approx. 110 K) and reaction products were analyzed by means of TPRS. In recent reports, monochromatic high-power UV-LED setups in or ex vacuo appear very convenient for this purpose due to their high intensity in combination with low costs, easy operation and small size.48,49 Therefore, we have established a home-built UV-LED device following a design by Brummel et al.49 that can be mounted on the chamber viewports and allows UV irradiation of the sample. Here, a commercial UV-LED (Seoul Viosys CUN6AF4A, nominal wavelength of 365 nm, 2.75 W) was used. We have additionally characterized the system and found good agreement with the LED specifications, see ESI,† Fig. S1. In detail, the used UV light consists of two spectral contributions (λ1 = 368.8 nm, FWHM = 11.7 nm; λ2 = 380.1 nm, FWHM = 27.3 nm, λ1 is dominating). For our setup, the photon flux at the sample position was determined by a THORLABS S130C photodiode-based power meter to be >1.5 × 1016 s−1 cm−2 or >8 mW cm−2, respectively. During UV irradiation, only a slight temperature increase (below 3 K) was detected, and no signs for a significant photodesorption of either oxygen, methanol or other products were observed. Unless stated otherwise, an irradiation time of 30 minutes was used.
3. Results
The photoconversion of methanol adsorbed at a preoxidized rutile TiO2(110) surface was realized by UV irradiation at 110 K and subsequently followed by TPRS. To prepare a mixed oxide–oxide model catalyst, we have deposited tungsten oxide clusters onto the rutile TiO2(110) surface by evaporation of bulk WO3 powder from an electron beam evaporator. WO3 is known to form ring shaped (WO3)n clusters at roughly 1120–1300 K, whereas n = 3 appears as the dominant compound.50–54 We have thoroughly characterized the produced system combining X-ray photoelectron spectroscopy (XPS), LEED and probe molecule temperature-programmed desorption (TPD) spectroscopy in another recent work.46 Since we use the same equipment, materials and devices, we have identical conditions to previous experiments. At this point, we refer to the original publication46 for a detailed description and the accompanying work regarding the thermal chemistry of methanol on this system.32 Here, we just summarize the major findings relevant for this work: up to 7.1 WO3 nm−2, a layer of stoichiometric clusters (W oxidation state +VI) is formed in direct interaction with the titania surface. At higher coverages, also a small fraction of oxygen deficient substoichiometric clusters appear. Up to 28 WO3 nm−2, accessible fivefold-coordinated titanium sites (Ti5c) were found as apparent in water TPD, but the amount decreases with the cluster coverage. So, the clusters appear as an unordered, open layer that grows in a three-dimensional motif. No significant cluster decomposition was found by XPS up to 900 K on TiO2. Exclusively for highly reduced TiO2, Ti3+ sites from the bulk accumulate at the surface near the clusters between 500 and 800 K. Based on W4f spectra, the parallel formation of anionic [(WO3)n]z− like structures is suggested due to electron transfer from defect sites towards the clusters. Such electron rich species have been predicted in a number of DFT-based studies.30,33,34,55All following experiments were performed as a function of the tungsten oxide cluster coverage as well as the amount of bulk Ti3+ interstitials. For this purpose, two different titania substrates were used. One is only slightly reduced and therefore exhibits a small amount of Ti3+ defects (light blue, 10–15 reduction cycles; Ti3+/Ti4+ ratio <4.2%). The second sample contains a large amount of Ti3+ bulk defects (dark blue, >80 reduction cycles; Ti3+/Ti4+ ratio >7.5%). While we have not measured Ti2p XPS for the samples used here due to sample size limitations, we refer to the Ti3+/Ti4+ ratio calculated from Ti2p spectrum deconvolution of samples that were analogously prepared with a similar number of reduction cycles under the same conditions in our group.46
3.1 Methanol photo-oxidation on rutile TiO2 and tungsten oxide clusters
In the following we will take a closer look at the UV photoconversion of methanol in the presence of oxygen on the system introduced above. We observe drastic changes by the presence of Ti3+ interstitials. Therefore, we shall first compare results for low and highly reduced TiO2. In a second step, we discuss the effect of tungsten oxide clusters.Fig. 1 shows selected m/z traces of the recorded TPR spectra after preadsorption of 75 L O2 and a saturation coverage of methanol at 110 K followed by UV irradiation (30 minutes, nominal wavelength 365 nm, photon flux >1.5 × 1016 s−1 cm−2). Here, a rutile sample with a large amount of bulk Ti3+ was used. The corresponding TPR spectra for TiO2 samples with a small bulk Ti3+ content are to be found in the ESI,† Fig. S3. Under the chosen conditions, we expect the maximal conversion for the first oxidation step based on similar experiments for isopropanol photo-oxidation on preoxidized rutile TiO2 with a small amount of Ti3+.3 However, it may not be excluded that extended irradiation times may lead to the formation of subsequent products or increased total oxidation.
 | ||
| Fig. 1 Temperature programmed reaction spectra after UV irradiation (30 minutes, nominal wavelength 365 nm, photon flux > 1.5 × 1016 s−1 cm−2) of a saturation coverage of methanol with preadsorption of 75 L O2 at 110 K. (a) Obtained from the pristine, highly reduced rutile TiO2(110) surface and (b) from the same surface after deposition of 3.5 WO3 nm−2 flashed to 880 K before adsorption of methanol and oxygen at 110 K. | ||
In all spectra the typical desorption signals for unconverted, molecular methanol are observed. They can be easily identified in the m/z = 15, 18, 29, 30, 31 traces with respect to the fragmentation pattern (Table 1 and Fig. S2 in the ESI†). In detail, we observe such features peaking around 140 K, 175 K and 290–300 K. These are well known and can be assigned to desorption of the physisorbed multilayer (140 K), desorption of hydrogen bonded methanol from bridging oxygen (Obr) sites (175 K) and desorption of methanol from Ti5c sites (290–300 K).
| m/z | Major contributor | Fragments | Minor contributor | Fragments |
|---|---|---|---|---|
| a H2O is not a direct fragment of methanol, but can be detected in traces likely arising from recombination in the mass spectrometer. | ||||
| 15 | Methane | CH3, (13CH2) | Ethane | CH3, (13CH2) |
| Methanol | CH3, (13CH2) | Formaldehyde | 13CH2 | |
| 18 | Water | H2O | Methanol | H2Oa |
| 26 | Ethene | HCCH, H2CC | — | — |
| Ethane | HCCH, H2CC | |||
| 29 | Methanol | HCO | Ethene | H213CCH2 |
| Formaldehyde | HCO | |||
| Ethane | H3CCH2, (H213CCH2) | |||
| 30 | Formaldehyde | H2CO | — | — |
| Methanol | H2CO, HCOH | |||
| Ethane | H3CCH3 | |||
| 31 | Methanol | H3CO, H2COH | — | — |
These features are similar for small (Fig. S3 in the ESI†) and large amounts of (bulk) defects. However, in consent with earlier results, the Ti5c desorption feature is somewhat shifted to higher temperatures for the highly reduced TiO2 sample.32 Within this work, a methanol coverage was used that leads to a saturation of all adsorption sites (namely, Obr and Ti5c) and therefore is called saturation coverage in the following. Thus, many of our spectra exhibit small shoulders of the desorption feature at 140 K due to an emerging multilayer, but the percentage is small. All three major molecular desorption features are in well agreement with the literature15,56 and will not be further discussed.
Before we explain the desorption of valuable products (formaldehyde, methane and ethene), we shall first discuss the desorption trace of water (m/z = 18). The formation of methoxy species and therefore water removal is one of the central steps not only for the thermal, but especially for the photochemical conversion as we show at a later point. After dissociation of OH and CH bonds by either bridging oxygen atoms or other oxygen species, the desorption of water is the most important channel to remove hydrogen from the surface. This also involves the disproportionation of two surface hydroxyls forming one molecule of water and a remaining oxygen adatom. Moreover, water can either be released by simple desorption of a molecular adsorbate (observed in water TPD at 178 K for hydrogen bonded to Obr and 262 K for desorption from Ti5c sites46) or during a reaction at the surface, for example in case of C–H dissociation forming formaldehyde. One should note, that based on the fragmentation pattern, small contributions in the m/z = 18 trace can also originate from molecular methanol desorption (Fig. S2, ESI†). Moreover, artefact signals can occur due to thermal changes of equipment such as the Feulner cup. Indeed, the recorded m/z = 18 traces are superpositions of all these contributions. The possible assignment of an example water trace is further given in the ESI,† Fig. S4.
Although a significant amount of methanol desorbs unreacted, we will now focus on additional desorption features which arise from methanol photoconversion products. The dominant photochemical reaction is the photo-oxidation under formation of formaldehyde, that desorbs at 252 K (Fig. 1a) as seen in m/z = 29 and 30, but not significantly in 31 or 15 (see also Table 1). This is labeled as LT (low-temperature) formaldehyde in the following. Also, the concomitant desorption of water (m/z = 18) can be seen at that temperature. Comparative experiments without oxygen preadsorption did yield by far less formaldehyde (see Fig. S5 in the ESI†). Furthermore, it is important to note that the formation of formaldehyde increases substantially on titania with a high Ti3+ density (+120%) compared to low reduced TiO2 (Fig. 2 and Fig. S3, ESI†). In comparison to the thermal formaldehyde formation (dark reaction), the formaldehyde desorption is strongly enhanced after UV irradiation and peaking at slightly lower temperature (see Fig. 2; −11 K). This shift may be due to a higher density of the product in the photochemical case. Hence, repulsive intermolecular interactions become more important, similar to classical coverage-dependent peak shifts in TPD spectra.22
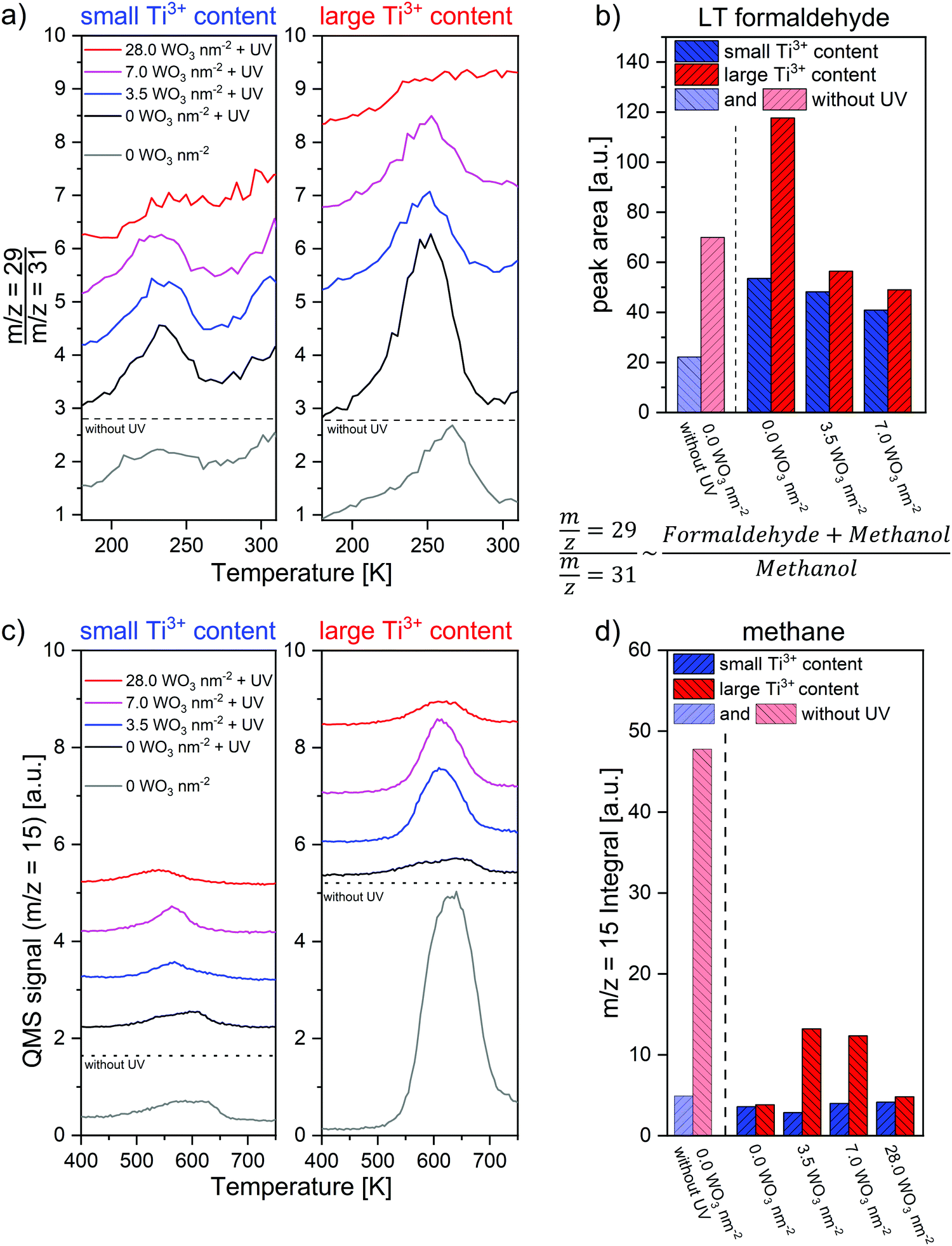 | ||
| Fig. 2 Overview on photoreaction products. (a) Low temperature desorption of formaldehyde after photo-oxidation, demonstrated by the ratio of m/z = 29 to m/z = 31, that is proportional to the desorption of formaldehyde under the given conditions. The original TPR spectra were obtained after UV irradiation (30 minutes, nominal wavelength 365 nm, photon flux > 1.5 × 1016 s−1 cm−2) of a saturation coverage of methanol with preadsorption of 75 L O2 at 110 K. (b) Integral area of the formaldehyde desorption as shown in a. (c) Methane desorption features at elevated temperatures in the m/z = 15 trace. (d) Integral area of the methane desorption features in c (m/z = 15). | ||
In the high temperature region above 400 K, no formaldehyde desorption was observed for both TiO2 substrates after UV irradiation. Two smaller features for the desorption of methane (m/z = 15) around 540–580 K and 600–640 K are present. Compared to the dark reaction, the formation of methane (m/z = 15) is drastically reduced by UV irradiation (see Fig. 2c). Together with our earlier finding, that the methane formation in the deoxygenation route proceeds via an alkoxy (here methoxy) intermediate,15 this suits perfectly with a systematic methanol photo-oxidation study by the Henderson group, showing that only the methoxy-species can be photo-oxidized on rutile TiO2(110).7,8 Hence, methoxy-fragments are widely photoconverted to yield formaldehyde and are therefore missing after UV irradiation to serve as the relevant methane precursor.
In the presence of a high amount of Ti3+, photo-stimulated reductive C–C coupling under the formation of ethene desorbing at 576 K (m/z = 26) becomes a noticeable contribution. As m/z = 29 and 30 do not exhibit any feature at this temperature, ethane formation can be excluded. It is noteworthy, that several other photochemical products such as dimethyl ether or methyl formate10 were not observed here, while the authors are not aware of a study demonstrating photo-stimulated C–C coupling starting from methanol under ethene production. Interestingly, the ethene formation is not accompanied by a significant water desorption. Assuming that water is the only possibility to remove hydrogen from the C–H dissociation from the surface implies that the C–H dissociation step must have been accomplished already at lower temperatures and here only the remaining (dioxo-)methylene fragments recombine under the desorption of ethene. Hence, a two-step reaction is observed here involving C–H dissociation followed by reductive coupling. This was not yet reported for ethene production starting from methanol. However, experimental (TPRS, HREELS, XPS and FTIR) as well as theoretical studies already showed the coupling of two dioxomethylene moieties forming ethene after formaldehyde adsorption on the rutile (110) surface.22,23,58,59 While in thermal (dark) reactions only a small fraction (approx. 2%)22 of formaldehyde undergoes this reaction, the ethene yield was found to be increased by UV irradiation.58,59 Wöll and coworkers indicated that the thermal reaction of formaldehyde towards ethene appears defect-related, but in contrast to this work they have also found the absence of ethene formation in the presence of additional oxygen adsorbed on the surface.22,23 The oxygen preadsorption in turn was mandatory for this pathway in this work. Moreover, for the photochemical reaction starting from methanol, the impact of defects was not observed yet. The role of Ti3+ may be expanded by the fact that Ti3+ sites can easily abstract or dissociate available oxygen. This was for example shown by the group of Besenbacher in a STM based study in which Ti3+ sites were observed to dissociate oxygen under subsequent formation of TiOx islands.60 Analogously, Ti3+ sites are expected to facilitate the C–O dissociation, suiting the trend for the thermal deoxygenation reaction forming methane.15,17 One should note that Ti3+ appears involved in almost all steps of the ethene formation starting from methanol (O–H dissociation, C–H dissociation, C–O dissociation). Therefore, the ethene yield is likely amplified not in a linear way but drastically more. So, UV excitation appears as a promising approach to induce such Ti3+ mediated C–C coupling reactions in order to utilize methanol as a building block for larger valuable compounds.
After deposition of 3.5 WO3 nm−2 on the TiO2 surface, the TPR spectrum reveals that the thermal chemistry becomes more dominant especially for large amounts of Ti3+ (Fig. 1b). Similar to the results of bare TiO2, a low temperature partial oxidation forming formaldehyde (desorption feature around 246 K in m/z = 29 and 30, not 31 and 15) was detected, but in smaller amounts. On the other hand, the thermal products (particularly methane) are more important. Here, the high-temperature (HT) desorption of methane (612 K, m/z = 15) after UV irradiation is more pronounced compared to the absence of clusters. No significant production of ethene via C–C coupling was observed. Also a weak signal of formaldehyde desorption at 642 K was found. For small Ti3+ contents (ESI,† Fig. S3), this HT formaldehyde desorption is fully absent, while only one relatively broad desorption feature of methane was found (567 K). In all cases, no significantly enhanced formation of total oxidation products such as CO or CO2 was detected. These would be expected to desorb around 135 K (CO)61,62 as well as 137 K and 166 K (CO2).63 Since we do not observe significant signals at these temperatures in the m/z = 28 and m/z = 44 trace, we exclude total oxidation to be of importance here (see ESI,† Fig. S6–S8).
3.2 Analysis of the product distribution
In the following, we want to analyze trends for the two dominant products (methane and formaldehyde) as a function of cluster coverage and the bulk Ti3+ content. For methane this is quite straightforward, since in all our spectra, methane is the only desorbing compound above 400 K that exhibits a m/z = 15 fragment. So, we can simply integrate the desorption feature. In case of formaldehyde, this is a bit more challenging, because the m/z = 29 trace, that is the most intense fragment for formaldehyde, also contains contributions from methanol. For this reason, we follow the m/z = 29 to m/z = 31 ratio to monitor the formaldehyde formation (Fig. 2a and b). Based on the fragmentation pattern, this ratio is proportional to the sum of methanol and formaldehyde partial pressures divided by the one of methanol and therefore measures the formaldehyde production. We have additionally tried to extract the actual methanol fragmentation pattern from the multilayer desorption features and thereby tried to correct the m/z = 29 trace from methanol contributions, but this was found to be inconsistent with the results visible in the raw spectra probably due to the complex spectra and background correction issues.Fig. 2 illustrates the trend for the formaldehyde (a and b) and methane (c and d) production. Here, the trends shown before become more evident: the LT formaldehyde yield is always higher for high Ti3+ contents. It is furthermore roughly doubled by UV irradiation. However, deposition of tungsten oxide clusters results in a decrease of the formaldehyde formation in comparison to the pristine TiO2 in this temperature regime, particularly drastic for high Ti3+ contents. In all cases, the LT formaldehyde yield after UV irradiation drops with increasing tungsten oxide coverage. For 28 WO3 nm−2, no significant LT formaldehyde desorption was measured at all. The methane formation shows reverse trends (Fig. 2c and d). In case of the highest formaldehyde yield, the methane production is amongst the lowest one within our experiments. While the formaldehyde desorption after UV irradiation decreases from 0 WO3 nm−2 to 7.0 WO3 nm−2 for titania with a large bulk Ti3+ content, the methane formation increases concordantly. In case of a low amount of bulk Ti3+, the effects of cluster deposition are smaller, but still some anticorrelation between methane and formaldehyde production is visible. Hence, against intuition, the deposition of submonolayer tungsten oxide clusters can surprisingly modify the methanol photo-oxidation in favor of the thermal deoxygenation at titania.
4. Discussion
Besides unconverted desorption of molecular methanol, three important (photo)conversion products are identified from our results: first, formaldehyde as the most important photo-oxidation product which desorbs around 250 K. On the way towards formaldehyde, the C–H dissociation is the important reaction step, that can be done by photochemical reactants (namely photogenerated holes and radical species produced thereby).1 In consent with our results, Henderson et al. found, that only methoxy-fragments can participate in this photoreaction.7,8 Methoxy moieties, that do not undergo this step during the UV irradiation, are converted in the second important reaction route: the thermal deoxygenation reaction forming methane.15 Therefore, we observe anticorrelation between the deoxygenation and partial photo-oxidation reaction yields. Moreover, after C–H dissociation, the remaining fragments can react with each other forming ethene in a reductive coupling reaction. Nevertheless, a large amount of methanol still desorbs unconverted.4.1 Photochemistry at bare TiO2
First, we shall discuss results for pristine TiO2 in the absence of tungsten oxide, starting with formaldehyde as the most important photochemical partial oxidation product of methanol coadsorbed with oxygen. Based on our own, recent FT-IRRAS results,15 we assume, that formaldehyde is not present in its molecular form but desorbs from a dioxomethylene-like precursor species. This is consistent with the findings within this work, because based on theoretical and experimental studies, remaining methanol is claimed to react with molecular formaldehyde via hemiacetal intermediates forming methyl formate. This was not observed here.9,10,64 Independent on the reduction degree, the formaldehyde yield is approximately doubled after UV irradiation compared to the corresponding dark reaction. Remarkably, the partial photo-oxidation appears very sensitive to the presence of Ti3+ sites. In case of high Ti3+ amounts present, the photochemical formaldehyde yield is more than doubled compared to the sample with a small concentration of defects. While we cannot rule out other options, some possible reasons shall be discussed at this point:(1) Based on our own water desorption traces and concordant FT-IRRAS and TPRS experiments available in the literature, the presence of Ti3+ and oxygen boosts the formation of methoxy-species by O–H dissociation due to the available charge with a certain mobility.7,15,65,66 In turn, the formaldehyde yield is increased because methoxy-fragments were identified by Henderson as the photoactive species.7,8 This also includes the impact of electron-rich oxygen species, which can be formed due to charge transfer from reduced titania sites towards adsorbed oxygen according to recent results from the Diebold group.67 In that AFM based study they further showed, that such species are similar to photogenerated oxygen species and concordantly acting as Lewis bases. Hence, protons can be easily abstracted to form surface hydroxyl species. Indeed, the interaction of oxygen with the TiO2 surface was found to be stronger for high Ti3+ contents in a STM and O2 TPD based study by the group of Besenbacher.16
(2) An enhanced light absorption causing a better photon efficiency would be another option. This may be due to populated states in the band gap (approx. 1 eV below the Fermi level46,68) which lower the effective band gap. Therefore, the lower energetic parts of our UV irradiation (see ESI,† Fig. S1 for details) may additionally contribute to the formaldehyde yield.
(3) Another possible option is an improved exciton lifetime due to the higher conductivity of the TiO2 substrate in case of higher Ti3+ contents. After exciton generation, a prompt recombination is commonly the most important photoinhibition pathway.1 This may be prevented, if a fast electron–hole separation can be realized, for example due to a high conductivity throughout the bulk crystal. Similarly, concomitant hole trapping at surface defect sites may be beneficial for the photocatalytic efficiency.1,21 Indeed, photogenerated holes exhibited longer lifetimes on anatase TiO2 nanoparticles due to interaction with adsorbed oxygen in combination with Ti3+ defect sites.69 This matches our finding that the presence of oxygen appears crucial for the photo-oxidation. Also defect induced localized surface states may contribute to this effect. However, these are only a few possible options, which we cannot further judge from our data.
4.2 Impact of tungsten oxide clusters on photochemical reactions
In contrast to bare TiO2, for tungsten oxide cluster coverages up to 7.0 WO3 nm−2 still the thermal chemistry is more dominant after UV irradiation. Compared to the photo-stimulated reaction without any clusters, the photo-oxidation yield is drastically attenuated for high bulk Ti3+ contents for coverages up to 7.0 WO3 nm−2. At high coverages (28 WO3 nm−2), no photo-oxidation at all was observed.It is striking, that the photo-oxidation quenching is drastically more important for high Ti3+ contents, while it is only mildly observed for low Ti3+ densities. This is surprising, since both semiconductors (TiO2 and WO3) are actually photocatalysts themselves and the TPRS results with bare TiO2 show, that reduced states can improve the photo-oxidation yields. Hence, possible causes for the low activity of tungsten oxide clusters on rutile TiO2 have to be discussed. One possible reason may be, that Ti3+ sites become “pinned” near the tungsten oxide clusters due to their electronic interaction. This is in line with an accumulation of surface near Ti3+ as reported before.46 So, after exciton generation, the holes, that are known to travel towards the TiO2 surface due to band bending effects,1 should easily recombine with these surface near sites. Therefore, virtually no surface-near holes would be available for the oxidation reaction. The same would apply in case of exciton generation in partially anionic tungsten oxide clusters. Further, we want to mention selected experimental reports dealing with wet-chemically synthesized WO3/TiO2 composite materials. Based on UV-Vis absorption and photo-electron spectroscopy experiments combined with measurements of degradation rates of organic pollutants, it was claimed that photogenerated electrons can be trapped at tungsten oxide (under generation of W+V) while holes favor the titania substrate, likely because the conduction band edge of WO3 is expected slightly below the one of rutile TiO2.39,70,71 Consequently, the partial covering of accessible Ti5c sites may also contribute to the photo-oxidation deactivation especially in case of high coverages. Finally, we cannot rule out other effects such as band bending, that also can drastically change the photochemical efficiency.1
We likely exclude other reasons for the depleted LT formaldehyde formation: As only the methoxy-fragment is photoactive according to Shen and Henderson,7,8 one could think of a lack of methoxy adsorbates after tungsten oxide cluster deposition. We exclude this possibility, because a significant formation of methane instead of formaldehyde is observed in presence of the clusters. As we have shown recently, this reaction also proceeds via methoxy intermediates, that are, thus, sufficiently available.15 We also rule out the possible oligomerization of formaldehyde, that was reported at WO3 catalysts below 100 K.29–31 We do not expect this to be limiting here because the methane desorption proves that remaining methoxy sites are present which indicates that less formaldehyde was produced than on bare TiO2. Moreover, Dohnálek et al. point out, that at least two monolayers of formaldehyde are required to yield reasonable amounts of oligomers.29,31 Even if all formaldehyde would be present at the surface in its molecular form (which is not expected), the coverage is much less than two monolayers. Finally, we exclude a possible UV shielding of the TiO2 surface by tungsten oxide material, because it is well known that the penetration depth of UV light (at least 160 nm) is much higher than the expected tungsten oxide layer thickness even for high coverages.1,72
5. Conclusion
The photochemical conversion of methanol with oxygen preadsorption was examined at the rutile TiO2(110) surface as a function of the Ti3+ content. The partial oxidation of methanol forming formaldehyde is the relevant photochemical reaction on bare TiO2 for the used conditions (30 minutes UV irradiation at 110 K, nominal wavelength 365 nm, photon flux >1.5 × 1016 s−1 cm−2). Within this time regime, total oxidation appears not to be of significant importance. High bulk Ti3+ densities do not only enhance the partial photo-oxidation efficiency significantly, but also yield fair amounts of C–C coupling products (here ethene) implying a certain mobility of adsorbates at the TiO2 surface.In a second step, a mixed oxide–oxide bifunctional catalyst was mimicked by deposition of tungsten oxide clusters on top of the TiO2 substrate. Surprisingly, the deposition of tungsten oxide clusters inhibits the photo-oxidation compared to bare TiO2 and therefore favors the thermal conversion routes. This photo-oxidation quenching is drastically more important for high Ti3+ densities. Besides covering of the Ti5c sites by the clusters, one suggested reason for this is the accumulation of charge near the surface in form of “pinned” Ti3+ sites appearing next to the tungsten oxide clusters and partially anionic clusters itself. Both electron rich species may act as recombination sites for holes.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Thanks to Ralf Nustedt for support with the technical construction. Olaf Brummel and Jörg Libuda are acknowledged for fruitful discussions during the construction of the UV LED irradiation setup. The funding of the DFG research training group GRK 2226 “Chemical Bond Activation” is appreciated. L. M. acknowledges the funding of the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes).References
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Understanding TiO2 Photocatalysis: Mechanisms and Materials, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- Z. Shayegan, C.-S. Lee and F. Haghighat, TiO2 Photocatalyst for Removal of Volatile Organic Compounds in Gas Phase – A Review, Chem. Eng. J., 2018, 334, 2408–2439 CrossRef CAS.
- J. Kräuter, L. Mohrhusen, F. Waidhas, O. Brummel, J. Libuda and K. Al-Shamery, Photoconversion of 2-Propanol on Rutile Titania: A Combined Liquid-Phase and Surface Science Study, J. Phys. Chem. C, 2021, 125, 3355–3367 CrossRef.
- C. A. Walenta, S. L. Kollmannsberger, J. Kiermaier, A. Winbauer, M. Tschurl and U. Heiz, Ethanol Photocatalysis on Rutile TiO2(110): The Role of Defects and Water, Phys. Chem. Chem. Phys., 2015, 17, 22809–22814 RSC.
- M. Wagstaffe, H. Noei and A. Stierle, Elucidating the Defect-Induced Changes in the Photocatalytic Activity of TiO2, J. Phys. Chem. C, 2020, 124, 12539–12547 CrossRef CAS.
- M. Shen, D. P. Acharya, Z. Dohnálek and M. A. Henderson, Importance of Diffusion in Methanol Photochemistry on TiO2(110), J. Phys. Chem. C, 2012, 116, 25465–25469 CrossRef CAS.
- M. Shen and M. A. Henderson, Role of Water in Methanol Photochemistry on Rutile TiO2(110), J. Phys. Chem. C, 2012, 116, 18788–18795 CrossRef CAS.
- M. Shen and M. A. Henderson, Identification of the Active Species in Photochemical Hole Scavenging Reactions of Methanol on TiO2, J. Phys. Chem. Lett., 2011, 2, 2707–2710 CrossRef CAS.
- K. R. Phillips, S. C. Jensen, M. Baron, S.-C. Li and C. M. Friend, Sequential Photo-oxidation of Methanol to Methyl Formate on TiO2(110), J. Am. Chem. Soc., 2013, 135, 574–577 CrossRef CAS PubMed.
- A. S. Crampton, L. Cai, N. Janvelyan, X. Zheng and C. M. Friend, Methanol Photo-Oxidation on Rutile TiO2 Nanowires: Probing Reaction Pathways on Complex Materials, J. Phys. Chem. C, 2017, 121, 9910–9919 CrossRef CAS.
- C. A. Walenta, M. Tschurl and U. Heiz, Introducing catalysis in photocatalysis: What can be understood from Surface Science Studies of Alcohol Photoreforming on TiO2, J. Phys.: Condens. Matter, 2019, 31, 473002 CrossRef CAS PubMed.
- M. A. Henderson and I. Lyubinetsky, Molecular-Level Insights into Photocatalysis from Scanning Probe Microscopy Studies on TiO2(110), Chem. Rev., 2013, 113, 4428–4455 CrossRef CAS PubMed.
- S. C. Jensen and C. M. Friend, The Dynamic Roles of Interstitial and Surface Defects on Oxidation and Reduction Reactions on Titania, Top. Catal., 2013, 56, 1377–1388 CrossRef CAS.
- P. M. Clawin, C. M. Friend and K. Al-Shamery, Defects in Surface Chemistry-Reductive coupling of Benzaldehyde on Rutile TiO2(110), Chem. – Eur. J., 2014, 20, 7665–7669 CrossRef CAS PubMed.
- M. Osmić, L. Mohrhusen and K. Al-Shamery, Bulk Defect Dependence of Low-Temperature Partial Oxidation of Methanol and High-Temperature Hydrocarbon Formation on Rutile TiO2(110), J. Phys. Chem. C, 2019, 123, 7615–7626 CrossRef.
- E. Lira, S. Wendt, P. Huo, J. Ø. Hansen, R. Streber, S. Porsgaard, Y. Wei, R. Bechstein, E. Laegsgaard and F. Besenbacher, The Importance of Bulk Ti3+ Defects in the Oxygen Chemistry on Titania Surfaces, J. Am. Chem. Soc., 2011, 133, 6529–6532 CrossRef CAS PubMed.
- J. Kräuter, L. Mohrhusen, T. Thiedemann, M. Willms and K. Al-Shamery, Activation of Small Organic Molecules on Ti2+-Rich TiO2 Surfaces: Deoxygenation vs. C–C Coupling, Z. Naturforsch., A: Phys. Sci., 2019, 74, 697–707 CrossRef.
- N. G. Petrik, Z. Zhang, Y. Du, Z. Dohnálek, I. Lyubinetsky and G. A. Kimmel, Chemical Reactivity of Reduced TiO2(110): The Dominant Role of Surface Defects in Oxygen Chemisorption, J. Phys. Chem. C, 2009, 113, 12407–12411 CrossRef CAS.
- W. Göpel, G. Rocker and R. Feierabend, Intrinsic defects of TiO2(110): Interaction with chemisorbed O2, H2, CO and CO2, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 3427–3438 CrossRef.
- Y. Yoon, Y. Du, J. C. Garcia, Z. Zhu, Z.-T. Wang, N. G. Petrik, G. A. Kimmel, Z. Dohnalek, M. A. Henderson, R. Rousseau, N. A. Deskins and I. Lyubinetsky, Anticorrelation between Surface and Subsurface Point Defects and the Impact on the Redox Chemistry of TiO2(110), ChemPhysChem, 2015, 16, 313–321 CrossRef CAS PubMed.
- J. Yan, G. Wu, N. Guan, L. Li, Z. Li and X. Cao, Understanding the Effect of Surface/Bulk Defects on the Photocatalytic Activity of TiO2: Anatase versus Rutile, Phys. Chem. Chem. Phys., 2013, 15, 10978 RSC.
- X. Yu, Z. Zhang, C. Yang, F. Bebensee, S. Heissler, A. Nefedov, M. Tang, Q. Ge, L. Chen, B. D. Kay, Z. Dohnálek, Y. Wang and C. Wöll, Interaction of Formaldehyde with the Rutile TiO2(110) Surface: A Combined Experimental and Theoretical Study, J. Phys. Chem. C, 2016, 120, 12626–12636 CrossRef CAS.
- H. Qiu, H. Idriss, Y. Wang and C. Wöll, Carbon–Carbon Bond Formation on Model Titanium Oxide Surfaces: Identification of Surface Reaction Intermediates by High-Resolution Electron Energy Loss Spectroscopy, J. Phys. Chem. C, 2008, 112, 9828–9834 CrossRef CAS.
- Z. Li, Z. Fang, M. S. Kelley, B. D. Kay, R. Rousseau, Z. Dohnalek and D. A. Dixon, Ethanol Conversion on Cyclic (MO3)3 (M = Mo, W) Clusters, J. Phys. Chem. C, 2014, 118, 4869–4877 CrossRef CAS.
- Z. Fang, Z. Li, M. S. Kelley, B. D. Kay, S. Li, J. M. Hennigan, R. Rousseau, Z. Dohnálek and D. A. Dixon, Oxidation, Reduction, and Condensation of Alcohols over (MO3)3 (M = Mo, W) Nanoclusters, J. Phys. Chem. C, 2014, 118, 22620–22634 CrossRef CAS.
- X. Tang, D. Bumueller, A. Lim, J. Schneider, U. Heiz, G. Ganteför, D. H. Fairbrother and K. H. Bowen, Catalytic Dehydration of 2-Propanol by Size-Selected (WO3)n and (MoO3)n Metal Oxide Clusters, J. Phys. Chem. C, 2014, 118, 29278–29286 CrossRef CAS.
- Y. K. Kim, R. Rousseau, B. D. Kay, J. M. White and Z. Dohnálek, Catalytic Dehydration of 2-Propanol on (WO3)3 Clusters on TiO2(110), J. Am. Chem. Soc., 2008, 130, 5059–5061 CrossRef CAS PubMed.
- R. Rousseau, D. A. Dixon, B. D. Kay and Z. Dohnalek, Dehydration, dehydrogenation, and condensation of alcohols on supported oxide catalysts based on cyclic (WO3)3 and (MoO3)3 clusters, Chem. Soc. Rev., 2014, 43, 7664–7680 RSC.
- Z. Li, Z. Zhang, B. D. Kay and Z. Dohnálek, Polymerization of Formaldehyde and Acetaldehyde on Ordered (WO3)3 Films on Pt(111), J. Phys. Chem. C, 2011, 115, 9692–9700 CrossRef CAS.
- C. Di Valentin, M. Rosa and G. Pacchioni, Radical versus Nucleophilic Mechanism of Formaldehyde Polymerization Catalyzed by (WO3)3 Clusters on Reduced or Stoichiometric TiO2(110), J. Am. Chem. Soc., 2012, 134, 14086–14098 CrossRef CAS PubMed.
- J. Kim, B. D. Kay and Z. Dohnálek, Formaldehyde Polymerization on (WO3)3/TiO2(110) Model Catalyst, J. Phys. Chem. C, 2010, 114, 17017–17022 CrossRef CAS.
- L. Mohrhusen and K. Al-Shamery, Conversion of Methanol on Rutile TiO2(110) and Tungsten Oxide Clusters: 1. Population of Defect-Dependent Thermal Reaction Pathways, Phys. Chem. Chem. Phys., 2021 10.1039/d1cp01175h.
- C. Di Valentin, F. Wang and G. Pacchioni, Tungsten Oxide in Catalysis and Photocatalysis: Hints from DFT, Top. Catal., 2013, 56, 1404–1419 CrossRef CAS.
- X. Huang, H.-J. Zhai, B. Kiran and L.-S. Wang, Observation of d-Orbital Aromaticity, Angew. Chem., Int. Ed., 2005, 44, 7251–7254 CrossRef CAS PubMed.
- X. Huang, H.-J. Zhai, J. Li and L.-S. Wang, On the Structure and Chemical Bonding of Tri-Tungsten Oxide Clusters W3On− and W3On (n = 7–10): W3O8 As A Potential Molecular Model for O-Deficient Defect Sites in Tungsten Oxides, J. Phys. Chem. A, 2006, 110, 85–92 CrossRef CAS PubMed.
- J. Papp, S. Soled, K. Dwight and A. Wold, Surface Acidity and Photocatalytic Activity of TiO2, WO3/TiO2, and MoO3/TiO2 Photocatalysts, Chem. Mater., 1994, 6, 496–500 CrossRef CAS.
- I. Shiyanovskaya and M. Hepel, Bicomponent WO3/TiO2 Films as Photoelectrodes, J. Electrochem. Soc., 1999, 146, 243–249 CrossRef CAS.
- S. Prabhu, A. Nithya, S. C. Mohan and K. Jothivenkatachalam, Synthesis, Surface Acidity and Photocatalytic Activity of WO3/TiO2 Nanocomposites – An Overview, Mater. Sci. Forum, 2014, 781, 63–78 CAS.
- L. Zhang, M. Qin, W. Yu, Q. Zhang, H. Xie, Z. Sun, Q. Shao, X. Guo, L. Hao, Y. Zheng and Z. Guo, Heterostructured TiO2/WO3 Nanocomposites for Photocatalytic Degradation of Toluene under Visible Light, J. Electrochem. Soc., 2017, 164, H1086–H1090 CrossRef CAS.
- W. Huang, J. Wang, L. Bian, C. Zhao, D. Liu, C. Guo, B. Yang and W. Cao, Oxygen Vacancy Induces Self-Doping Effect and Metalloid LSPR in Non-Stoichiometric Tungsten Suboxide Synergistically Contributing to the Enhanced Photoelectrocatalytic performance of WO3−x/TiO2−x heterojunction, Phys. Chem. Chem. Phys., 2018, 20, 17268–17278 RSC.
- V. Keller, P. Bernhardt and F. Garin, Photocatalytic oxidation of butyl acetate in vapor phase on TiO2, Pt/TiO2 and WO3/TiO2 catalysts, J. Catal., 2003, 215, 129–138 CrossRef CAS.
- W. Lee, C. Lai and S. Hamid, One-Step Formation of WO3-Loaded TiO2 Nanotubes Composite Film for High Photocatalytic Performance, Materials, 2015, 8, 2139–2153 CrossRef CAS.
- Y. Tae Kwon, K. Yong Song, W. I. Lee, G. J. Choi and Y. R. Do, Photocatalytic Behavior of WO3-Loaded TiO2 in an Oxidation Reaction, J. Catal., 2000, 191, 192–199 CrossRef.
- B. Gao, Y. Ma, Y. Cao, W. Yang and J. Yao, Great Enhancement of Photocatalytic Activity of Nitrogen-Doped Titania by Coupling with Tungsten Oxide, J. Phys. Chem. B, 2006, 110, 14391–14397 CrossRef CAS PubMed.
- J. H. Pan and W. I. Lee, Preparation of Highly Ordered Cubic Mesoporous WO3/TiO2 Films and Their Photocatalytic Properties, Chem. Mater., 2006, 18, 847–853 CrossRef CAS.
- L. Mohrhusen, M. Grebien and K. Al-Shamery, Electron Transfer in Oxide–Oxide Cocatalysts: Interaction of Tungsten Oxide Clusters with Ti3+ States in Rutile TiO2, J. Phys. Chem. C, 2020, 124, 23661–23673 CrossRef CAS.
- L. Mohrhusen, J. Kräuter, M. Willms and K. Al-Shamery, Argon Embedded by Ion Bombardment: Relevance of Hidden Dopants in Rutile TiO2, J. Phys. Chem. C, 2019, 123, 20434–20442 CrossRef CAS.
- M. Schwarz, C. Schuschke, T. N. Silva, S. Mohr, F. Waidhas, O. Brummel and J. Libuda, A simple high-intensity UV-photon source for photochemical studies in UHV: Application to the photoconversion of norbornadiene to quadricyclane, Rev. Sci. Instrum., 2019, 90, 024105 CrossRef PubMed.
- O. Brummel, F. Waidhas, U. Bauer, Y. Wu, S. Bochmann, H.-P. Steinrück, C. Papp, J. Bachmann and J. Libuda, Photochemical Energy Storage and Electrochemically Triggered Energy Release in the Norbornadiene–Quadricyclane System: UV Photochemistry and IR Spectroelectrochemistry in a Combined Experiment, J. Phys. Chem. Lett., 2017, 8, 2819–2825 CrossRef CAS PubMed.
- J. Kim, O. Bondarchuk, B. D. Kay, J. M. White and Z. Dohnálek, Preparation and Characterization of Monodispersed WO3 Nanoclusters on TiO2(110), Catal. Today, 2007, 120, 186–195 CrossRef CAS.
- M. Wagner, S. Surnev, M. G. Ramsey, G. Barcaro, L. Sementa, F. R. Negreiros, A. Fortunelli, Z. Dohnalek and F. P. Netzer, Structure and Bonding of Tungsten Oxide Clusters on Nanostructured Cu–O Surfaces, J. Phys. Chem. C, 2011, 115, 23480–23487 CrossRef CAS.
- Z. Li, Z. Zhang, Y. K. Kim, R. S. Smith, F. Netzer, B. D. Kay, R. Rousseau and Z. Dohnálek, Growth of Ordered Ultrathin Tungsten Oxide Films on Pt(111), J. Phys. Chem. C, 2011, 115, 5773–5783 CrossRef CAS.
- J. Berkowitz, W. A. Chupka and M. G. Inghram, Polymeric Gaseous Species in the Sublimation of Tungsten Trioxide, J. Chem. Phys., 1957, 27, 85–86 CrossRef CAS.
- O. Bondarchuk, X. Huang, J. Kim, B. D. Kay, L.-S. Wang, J. M. White and Z. Dohnálek, Formation of Monodisperse (WO3)3 Clusters on TiO2(110), Angew. Chem., Int. Ed., 2006, 45, 4786–4789 CrossRef CAS PubMed.
- X. Huang, H.-J. Zhai, T. Waters, J. Li and L.-S. Wang, Experimental and Theoretical Characterization of Superoxide Complexes [W2O6(O2−)] and [W3O9(O2−)]: Models for the Interaction of O2 with Reduced W Sites on Tungsten Oxide Surfaces, Angew. Chem., Int. Ed., 2006, 45, 657–660 CrossRef CAS PubMed.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, The Chemistry of Methanol on the TiO2(110) Surface: The Influence of Vacancies and Coadsorbed Species, Faraday Discuss., 1999, 114, 313–329 RSC.
- NIST Mass Spectrometry Data Center and W. E. Wallace, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 2018 Search PubMed.
- C. Xu, W. Yang, Q. Guo, D. Dai, T. K. Minton and X. Yang, Photoinduced Decomposition of Formaldehyde on a TiO2(110) Surface, Assisted by Bridge-Bonded Oxygen Atoms, J. Phys. Chem. Lett., 2013, 4, 2668–2673 CrossRef CAS.
- Q. Yuan, Z. Wu, Y. Jin, F. Xiong and W. Huang, Surface Chemistry of Formaldehyde on Rutile TiO2(110) Surface: Photocatalysis vs Thermal-Catalysis, J. Phys. Chem. C, 2014, 118, 20420–20428 CrossRef CAS.
- Z. Zhang, J. Lee, J. T. Yates, R. Bechstein, E. Lira, J. Ø. Hansen, S. Wendt and F. Besenbacher, Unraveling the Diffusion of Bulk Ti Interstitials in Rutile TiO2(110) by Monitoring Their Reaction with O Adatoms, J. Phys. Chem. C, 2010, 114, 3059–3062 CrossRef CAS.
- A. Linsebigler, G. Lu and J. T. Yates, CO chemisorption on TiO2(110): Oxygen vacancy site influence on CO adsorption, J. Chem. Phys., 1995, 103, 9438–9443 CrossRef CAS.
- Z. Dohnálek, J. Kim, O. Bondarchuk, J. M. White and B. D. Kay, Physisorption of N2, O2, and CO on Fully Oxidized TiO2(110), J. Phys. Chem. B, 2006, 110, 6229–6235 CrossRef.
- M. A. Henderson, Evidence for Bicarbonate Formation on Vacuum Annealed TiO2(110) resulting from a Precursor-Mediated Interaction between CO2 and H2O, Surf. Sci., 1998, 400, 203–219 CrossRef CAS.
- X. Lang, B. Wen, C. Zhou, Z. Ren and L.-M. Liu, First-Principles Study of Methanol Oxidation into Methyl Formate on Rutile TiO2(110), J. Phys. Chem. C, 2014, 118, 19859–19868 CrossRef CAS.
- E. Farfan-Arribas and R. J. Madix, Different Binding Sites for Methanol Dehydrogenation and Deoxygenation on Stoichiometric and Defective TiO2(110) Surfaces, Surf. Sci., 2003, 544, 241–260 CrossRef CAS.
- E. Farfan-Arribas and R. J. Madix, Role of Defects in the Adsorption of Aliphatic Alcohols on the TiO2(110) Surface, J. Phys. Chem. B, 2002, 106, 10680–10692 CrossRef CAS.
- M. Setvin, J. Hulva, G. S. Parkinson, M. Schmid and U. Diebold, Electron Transfer between Anatase TiO2 and an O2 Molecule Directly Observed by Atomic Force Microscopy, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2556–E2562 CrossRef CAS PubMed.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. Li, J. O. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Laegsgaard, B. Hammer and F. Besenbacher, The Role of Interstitial Sites in the Ti3d Defect State in the Band Gap of Titania, Science, 2008, 320, 1755–1759 CrossRef CAS PubMed.
- J. Gao, J. Lyu, J. Li, J. Shao, Y. Wang, W. Ding, R. Cheng, S. Wang and Z. He, Localization and Stabilization of Photogenerated Electrons at TiO2 Nanoparticle Surface by Oxygen at Ambient Temperature, Langmuir, 2018, 34, 7034–7041 CrossRef CAS PubMed.
- C. Martín, G. Solana, V. Rives, G. Marcì, L. Palmisano and A. Sclafani, Physico-Chemical Properties of WO3/TiO2 Systems Employed for 4-Nitrophenol Photodegradation in Aqueous Medium, Catal. Lett., 1997, 49, 235–243 CrossRef.
- G. Marcì, L. Palmisano, A. Sclafani, A. M. Venezia, R. Campostrini, G. Carturan, C. Martin, V. Rives and G. Solana, Influence of Tungsten Oxide on Structural and Surface Properties of Sol–Gel Prepared TiO2 Employed for 4-Nitrophenol Photodegradation, J. Chem. Soc., Faraday Trans., 1996, 92, 819–829 RSC.
- D. M. Eagles, Polar Modes of Lattice Vibration and Polaron Coupling Constants in Rutile (TiO2), J. Phys. Chem. Solids, 1964, 25, 1243–1251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Spectroscopic characterization of the used UV source; Fig. S2: Fragmentation patterns of methanol and possible reaction products; Fig. S3: Additional TPR spectra for small amount of Ti3+; Fig. S4: Deconvolution of m/z = 18 trace; Fig. S5: Additional TPR spectra without oxygen coadsorption; Fig. S6–S8: Additional TPR traces for m/z = 28 and 44. See DOI: 10.1039/d1cp01176f |
| This journal is © the Owner Societies 2021 |